WO2001062760A2 - A NOVEL CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR - Google Patents
A NOVEL CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR Download PDFInfo
- Publication number
- WO2001062760A2 WO2001062760A2 PCT/US2001/001229 US0101229W WO0162760A2 WO 2001062760 A2 WO2001062760 A2 WO 2001062760A2 US 0101229 W US0101229 W US 0101229W WO 0162760 A2 WO0162760 A2 WO 0162760A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- heptahydrate
- pyrrolo
- oxo
- benzoyl
- dihydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention provides an improved crystalline form of the antifolate compound N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]pyrirnidin-5- yl)ethyl]benzoyl]-L-glutamic acid, or pemetrexed disodium, also known as multitargeted antifolate or MTA (hereinafter pemetrexed), as well as the process for its preparation.
- MTA multitargeted antifolate
- Pemetrexed is a 5-substituted pyrrolo[2,3-d]pyrimidine disodium salt. Extensive research and evaluation has revealed that pemetrexed is a potent inhibitor of several folate-requiring enzymes, including thymidine synthase, dihydrofolate reductase and glycinamide ribonucleotide formyltransferase. Pemetrexed disodium is currently in clinical trials for treatment of patients with a wide variety of solid tumors.
- pemetrexed can exist in the form of a heptahydrate which is much more stable than the previously known 2.5 hydrate.
- One method by which the heptahydrate is formed is when the product is recrystallized from a volatile, water miscible solvent, such as acetone.
- the primary advantage of the heptahydrate crystalline form over the 2.5 hydrate crystal form is stability with respect to solvent content.
- the heptahydrate crystalline form is also more stable with respect to growth of related substances.
- the discovery of the present invention is that the heptahydrate crystalline form is what crystallizes from water/acetone.
- the key to isolating it is in how it is dried.
- the heptahydrate is subjected to elevated temperatures, low humidity, and/or vacuum, it converts to the 2.5 hydrate crystal form by loss of water.
- a disadvantage of prior art water/ethanol procedure is that there are no known conditions that successfully remove ethanol or isopropyl alcohol from the wetcake without also losing water.
- the prior art water/ethanol procedure as discussed by Barnett, et al., first produces the 7.0 ethanolate and then this is converted to the 2.5 hydrate upon evaporation of the ethanol.
- U.S. Patents 5,416,211, 5,344,932 and 5,539,113 disclose processes for preparing certain substituted pyrrolo[2,3-d]pyrimidine based antifolate derivatives, including pemetrexed.
- a number of pyrrolo[2,3-d]pyrimidine based antifolates, including pemetrexed, are described in U.S. Patents 4,966,206; 5,106,974; 4,997,838; and 5,106,974.
- the present invention provides a novel hydrate crystal form of disodium N-[4-[2-
- the invention further provides a method of use of the heptahydrate crystalline form for the manufacture of a medicament for the treatment of cancer.
- the invention further provides for a process for preparing a medicament comprising combining the heptahydrate crystalline form in an aqueous solution.
- the invention further provides for a formulation comprising the heptahydrate crystalline form in association with a pharmaceutically acceptable carrier.
- the invention further provides for a process for the preparation of the heptahydrate crystalline form comprising crystallizing disodium N-[4-[2-(2-amino-4,7- dihydro-4-oxo-3H- ⁇ yrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic acid salt from an appropriate solvent.
- the invention further provides an article of manufacture comprising packaging material and a composition comprising the heptahydrate crystalline form contained within said packaging material, wherein said crystalline salt is effective in the treatment of cancer and a label which indicates that said crystalline salt can be used in the treatment of cancer.
- the present invention further provides the heptahydrate crystalline salt of a compound of formula I:
- the present invention also provides a method for preparing such compounds by recrystallizing a compound of the formula I from a water miscible solvent.
- Figure 1 Brief Description of the Figure Figure 1 is a representative XRD pattern for the heptahydrate crystalline salt taken at 25 ⁇ 2°C and ambient relative humidity.
- the compound of formula I can exist in tautomeric equilibrium with the corresponding 4(3H)-oxo compound.
- the equilibrium for the pyrrolopyrimidine ring system and the numbering thereof, are shown below:
- the pH of the crystallization of pemetrexed is between 2.5 and 3.5.
- At least 5 volumes of water are used as a wash during the filtration to collect pemetrexed.
- pemetrexed 2Na
- pemetrexed 2Na
- pemetrexed 2Na
- N-[4 2-(2-Ardno-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]pyrimidin-5- yl)ethyl]benzoyl]-L-glutamic acid diethyl ester p-toluenesulfonic acid salt and aqueous sodium hydroxide (4 to 6 equivalents) are combined and stirred at 0 to 30 °C.
- the solution may be filtered. Water (to a total of between 10 and 16 volumes) and denatured ethanol (5 to 8 volumes) are added. Dilute hydrochloric acid and dilute sodium hydroxide solution (if needed) are added to adjust the pH to between 2.5 and 3.5.
- the slurry is warmed to between 60 °C and reflux (approximately 78 °C), then cooled to between 0 and 30 °C.
- Pemetrexed is collected by filtration, and washed with water (not less than 2.5 volumes).
- Purified water (265 L), 50% sodium hydroxide (22 kg, 4.5 equivalents), and N-[4- [2-(2-Amino-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-L- glutamic acid diethyl ester p-toluenesulfonic acid salt (40 kg) were combined and stirred at between 20 and 30 °C until no solids were visible. The resulting solution was filtered. Purified water (270 L) and denatured ethanol (270 L) were added.
- the pH was adjusted to between 2.8 and 3.2 using dilute hydrochloric acid (106 L of 2N first, then 0.5 N HC1 and/or 0.5N NaOH to hit the pH (63.8 kg of 0.5 N HC1 and 5.34 kg of 0.5 N NaOH were required)).
- the slurry was adjusted to between 60 and 70 °C, then cooled slowly to between 20 and 25 °C. Pemetrexed was collected by filtration, and washed with purified water.
- Pemetrexed, purified water (271 L), and 50% sodium hydroxide (10.2 kg) were combined at between 20 and 30 °C.
- the pH was adjusted to between 7.5 and 8.5 using 0.5 N HC1 (8.0 L were required).
- the resulting slurry was adjusted to between 45 and 55 °C and acetone (1300 L) was added.
- the mixture was cooled to between 20 and 25 °C.
- Pemetrexed*2Na was collected by filtration, and washed with acetone. Residual acetone was removed at less than 35 °C using a stream of water-saturated nitrogen.
- the slurry was heated to 65 °C and then allowed to cool to room temperature.
- the slurry was filtered and the wetcake was transferred to a 1 L Erlenmeyer.
- the wetcake was dissolved in 84 ml of 0.5 N NaOH and the pH adjusted to around 8 using dilute acid.
- the resulting solution was warmed to 45-50 °C and then 400 ml of acetone were added. Crystallization began after about 250 ml has been added.
- the slurry was cooled to room temperature and filtered.
- the solids were washed with acetone and dried in a vacuum oven at 25 °C under a slight vacuum (-700 mm Hg). A stream of damp air was passed through the oven during the 2 hour drying time.
- X-ray powder diffraction analysis can be readily performed as follows. After lightly grinding the sample with an agate mortar and pestle, the sample is loaded into a sample holder for the ⁇ - r ay powder diffraction measurement.
- Disodium MTA Hydrate Form I has a typical XRD pattern with interplanar spacings (d) in Angstroms and typical relative intensities (I/I 0 ) as shown in Tables I and II. The error of measurement is +/- 0.04 A. X-ray peaks with I/I 0 of 10% or greater were reported in the Tables below. The cutoff was chosen arbitrarily. The intensities are reported normalized to the most intense line. The effects of preferred orientation can be greatly reduced using a sample that is prepared in a manner that minimizes this effects, such as the use of a well ground sample.
- the 2.5 hydrate crystalline form is characterized by an X-ray diffraction pattern which comprises intensities corresponding to the following d spacings: 18.66 and or 9.33 +/-0.04 A when obtained at 22 ⁇ 2°C and at ambient % relative humidity using a copper radiation source.
- a properly prepared sample of the 2.5 hydrate crystalline form may be characterized as having an X-ray diffraction pattern which comprises other strong, characteristic peaks corresponding to the following d spacings: 18.66, 9.33 and or 4.92 +/- 0.04 A when obtained at 22 ⁇ 2°C and 31 ⁇ 10% relative humidity from a copper radiation source.
- the heptahydrate crystalline form is characterized by an X-ray diffraction pattern which comprises intensities corresponding to the following d spacing: 7.78 +/-0.04 A when obtained at 22 ⁇ 2°C and at ambient % relative humidity using a copper radiation source.
- a properly prepared sample of the heptahydrate crystalline form may be characterized as having an X-ray diffraction pattern which comprises other strong, characteristic peaks corresponding to the following d spacings: 21.60, 7.78, 5.26 and 3.22 +/- 0.04 A when obtained at 22 ⁇ 2°C and 31 ⁇ 10% relative humidity from a copper radiation source.
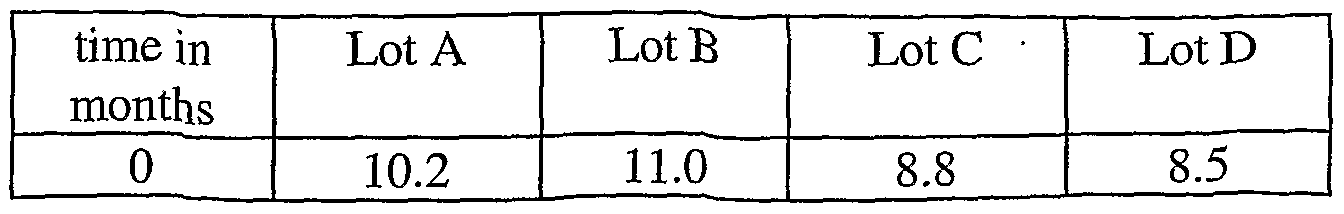
- the solvents content in lots A, B and C includes water and ethanol.
- Water content in the 2.5 hydrate crystalline form is theoretically 8.7%; the ethanol content, however, is only 0.06%, 0.08%, and 0.1% respectively for these lots and is not a significant contribution to the total solvents.
- the solvents content for lot D is for water only.
- the ethanol level in lot D is 0.08%, and is not a significant contribution to the total solvents.
- the solvents (mainly water) content for all lots stored at 5 °C increases over time to approximately 21% which indicates the material is in the heptahydrate form. However, for lot C, only the last time point showed the material was in the heptahydrate form.
- the accelerated conditions for lots A, B and C are 30°C, and for lot D is 25°C, 60% relative humidity. Only the D lot shows an increase in water content to the heptahydrate form. The other lots vary in water content over the time of the study.
- the change in the percent total related substances for lots A, B and D which were stored at 5°C did not change significantly over time.
- the percent total related substances for lot C did increase somewhat over time.
- the change in the percent total related substances for lots A, B and C which were stored at accelerated conditions did change significantly over time.
- the percent total related substances for lot D did not increase somewhat over time.
- heptahydrate crystalline form results The data for the 2.5 hydrate crystalline form give an indication that material that is in the heptahydrate form shows less degradation over time as compared to material that is not in the heptahydrate form.
- a laboratory lot of the 2.5 hydrate crystalline form was converted to the heptahydrate form by placing the material in an 85% relative humidity chamber for 3 days. X-ray powder diffraction data was used to confirm that the material was in the heptahydrate form before the stability study was initiated. This material was put on a laboratory stability study and the results are shown below.
- the water content for this lot stored at 5°C is approximately 21%, which indicates the material is in the heptahydrate form. This value does not change significantly over time and indicates that the heptahydrate material is stable with respect to water content at 5°C.
- the accelerated conditions for this lot are 25°C/60% relative humidity and room temperature/uncontrolled humidity.
- the water content for material stored at these conditions is approximately 21%, which indicates the material is in the heptahydrate form. This value does not change significantly over time and indicates that the heptahydrate material is stable with respect to water content at both of these accelerated conditions.
- Solvents water results for the heptahydrate lot stored at 5°C, 25°C/60% relative humidity, and 30°C/uncontrolled relative humidity. .
- Total Related Substances results for the heptahydrate lot stored at 5°C, 25°C/60% relative humidity, and 30°C/uncontrolled relative humidity.
- the present invention also includes methods employing pharmaceutical formulations which contain, as the active ingredient, the heptahydrate crystalline form, in association with pharmaceutical carriers.
- pharmaceutical formulations which contain, as the active ingredient, the heptahydrate crystalline form, in association with pharmaceutical carriers.
- a skilled artisan would know of such formulations and their manufacture, see, e.g., REMINGTON'S PHARMACEUTICAL SCIENCES, (16th ed. 1980).
- the formulations are preferably formulated in a unit dosage form, each dosage containing from about 5 to about 100 mg, more usually about 10 to about 30 mg, of the active ingredient.
- unit dosage form refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of active material calculated to produce the desired therapeutic effect, in association with a suitable pharmaceutical excipient.
- the crystals are effective over a wide dosage range. For example, dosages per day normally fall within the range of about 0.5 to about 30 mg/kg of body weight.
- the amount of the crystal actually administered will be determined by a physician, in the light of the relevant circumstances, including the condition to be treated, the chosen route of administration, the actual crystal administered, the age, weight, and response of the individual patient, and the severity of the patient's symptoms, and therefore the above dosage ranges are not intended to limit the scope of the invention in any way. In some instances dosage levels below the lower limit of the aforesaid range may be more than adequate, while in other cases still larger doses may be employed without causing any harmful side effect, provided that such larger doses are first divided into several smaller doses for administration throughout the day.
- crystals of the present invention can be administered alone or in the form of a pharmaceutical composition in combination with pharmaceutically acceptable carriers or excipients, the proportion and nature of which are determined by the solubility and chemical properties of the compound selected, the chosen route of administration, and standard pharmaceutical practice.
- the present invention provides pharmaceutical compositions comprising a therapeutically effective amount of the crystal in admixture or otherwise in association with one or more pharmaceutically acceptable carriers or excipients.
- the pharmaceutical compositions are prepared in a manner well known in the pharmaceutical art.
- the carrier or excipient may be a solid, semi-solid, or liquid material, which can serve as a vehicle or medium for the active ingredient. Suitable carriers or excipients are well known in the art.
- the pharmaceutical composition may be adapted for oral, inhalation, parenteral, or topical use and may be administered to the patient in the form of tablets, capsules, aerosols, inhalants, suppositories, solution, suspensions, or the like.
- the crystals of the present invention may be administered orally, for example, with an inert diluent or with an edible carrier. They may be enclosed in gelatin capsules or compressed into tablets.
- the crystals may be incorporated with excipients and used in the form of tablets, troches, capsules, elixirs, suspensions, syrups, wafers, chewing gums and the like.
- These preparations should contain at least 4% of the compound of the present invention, the active ingredient, but may be varied depending upon the particular form and may conveniently be between 4% to about 70% of the weight of the unit.
- the amount of the crystal present in compositions is such that a suitable dosage will be obtained.
- compositions and preparations according to the present invention may be determined by someone skilled in the art.
- the tablets, pills, capsules, troches and the like may also contain one or more of the following adjuvants: binders such as microcrystalline cellulose, gum tragacanth or gelatin; excipients such as starch or lactose, disintegrating agents such as alginic acid, Primogel, corn starch and the like; lubricants such as magnesium stearate or Sterotex; glidants such as colloidal silicon dioxide; and sweetening agents such as sucrose or saccharin may be added or a flavoring agent such as peppermint, methyl salicylate or orange flavoring.
- binders such as microcrystalline cellulose, gum tragacanth or gelatin
- excipients such as starch or lactose, disintegrating agents such as alginic acid, Primogel, corn starch and the like
- lubricants such as magnesium stearate or Sterotex
- glidants such as coll
- the dosage unit form When the dosage unit form is a capsule, it may contain, in addition to materials of the above type, a liquid carrier such as polyethylene glycol or a fatty oil.
- a liquid carrier such as polyethylene glycol or a fatty oil.
- Other dosage unit forms may contain other various materials, which modify the physical form of the dosage unit, for example, as coatings.
- tablets or pills may be coated with sugar, shellac, or other enteric coating agents.
- a syrup may contain, in addition to the present compounds, sucrose as a sweetening agent and certain preservatives, dyes and colorings and flavors. Materials used in preparing these various compositions should be pharmaceutically pure and non-toxic in the amounts used.
- the crystals of the present invention may be incorporated into a solution or suspension.
- compositions and preparations should contain at least 0.1% of a crystal of the invention, but may be varied to be between 0.1 and about 50% of the weight thereof.
- the amount of the heptahydrate crystalline form present in such compositions is such that a suitable dosage will be obtained.
- Preferred compositions and preparations are able to be determined by one skilled in the art.
- the crystals of the present invention may also be administered by inhalation, such as by aerosol or dry powder. Delivery may be by a liquefied or compressed gas or by a suitable pump system, which dispenses the compounds of the present invention or a formulation thereof. Formulations for administration by inhalation of compounds of formula (1) may be delivered in single phase, bi -phasic, or tri -phasic systems. A variety of systems are available for the administration by aerosol of the compounds of formula (1). Dry powder formulations are prepared by either pelletizing or milling the compound of formula (1) to a suitable particle size or by admixing the pelletized or milled compound of formula (1) with a suitable carrier material, such as lactose and the like. Delivery by inhalation includes the necessary container, activators, valves, subcontainers, and the like. Preferred aerosol and dry powder formulations for administration by inhalation can be determined by one skilled in the art.
- the crystals of the present invention may also be administered topically, and when done so the carrier may suitably comprise a solution, ointment or gel base.
- the base for example, may comprise one or more of the following: petrolatum, lanolin, polyethylene glycols, bee wax, mineral oil, diluents such as water and alcohol, and emulsifiers and stabilizers.
- Topical formulations may contain a concentration of the formula (1) or its pharmaceutical salt from about 0.1 to about 10% w/v (weight per unit volume).
- the solutions or suspensions may also include one or more of the following adjuvants: sterile diluents such as water for injection, saline solution, such as sodium chloride and mannitol, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl paraben; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylene diaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose.
- the parenteral preparation can be enclosed in ampoules, disposable syringes or multiple dose vials mane of glass or plastic.
- Active ingredient means the heptahydrate crystalline form.
- Example 1 active ingredient 4% (total solution)
- the pH of the solution was adjusted to 8.5 using sodium hydroxide.
- the pH adjusted solution was protected from light.
- the solution was purged with nitrogen for twenty minutes and then sterile filtered.
- the formulation was dispensed into prewashed, depyrogenated vials and then stoppered with a prewashed, presterilized teflon coated stopper. Caps were attached using a crimper.
- the sterile filtration and dispensing steps were conducted using a nitrogen isolator (5% v/v Oxygen).
- the solution filled vials were heat sterlized.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicinal Preparation (AREA)
Abstract
Description









Claims
Priority Applications (23)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60100750T DE60100750T2 (en) | 2000-02-25 | 2001-02-12 | NEW CRYSTALLINE FORM OF N- [4- [2- (2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO [2,3-d] -PYRIMIDIN-5-YL) ETHYL] BENZOYL] -L -Glutamic acid and process for its preparation |
| PL356423A PL208061B1 (en) | 2000-02-25 | 2001-02-12 | A NOVEL CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| CA002400155A CA2400155C (en) | 2000-02-25 | 2001-02-12 | A novel crystalline form of n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid and process therefor |
| EA200200905A EA004684B1 (en) | 2000-02-25 | 2001-02-12 | NOVEL CRYSTALLINE FORM OF N-[4-[2(2-AMONO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| SI200130032T SI1259513T1 (en) | 2000-02-25 | 2001-02-12 | A NOVEL CRYSTALLINE FORM OF N- 4- 2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO 2,3-d)PYRIMIDIN-5-YL)ETHYL)BENZOYL)-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| HU0204232A HU229704B1 (en) | 2000-02-25 | 2001-02-12 | A novel crystalline form of n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid and process for its preparation |
| IL15048001A IL150480A0 (en) | 2000-02-25 | 2001-02-12 | A NOVEL-CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4, 7-DIHYDRO-4-OXO-3H-PYRROLO[2, 3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| EP01906554A EP1259513B1 (en) | 2000-02-25 | 2001-02-12 | A NOVEL CRYSTALLINE FORM OF N- 4- 2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO 2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| SK1186-2002A SK287375B6 (en) | 2000-02-25 | 2001-02-12 | A crystalline form of N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H- pyrrolo[2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-L-glutamic acid and process for its preparation |
| HK03102573.8A HK1051856B (en) | 2000-02-25 | 2001-02-12 | A novel crystalline form of n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo [2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid and process therefor |
| BR0108604-9A BR0108604A (en) | 2000-02-25 | 2001-02-12 | Crystalline form of n- [4- [2- (2-amino-4,7-dihydro-4-oxo-3hpirrolo [2-3-d] pyrimidin-5-yl) ethyl] benzoyl] -l-glutamic and process for its preparation |
| AU34451/01A AU777047B2 (en) | 2000-02-25 | 2001-02-12 | A novel crystalline form of N-(4-(2-(2-amino-4,7-dihydro-4- oxo-3H-pyrrolo(2,3-d)pyrimidin-5-YL)ethyl)benzoyl)-L- glutamic acid and process therefor |
| NZ519796A NZ519796A (en) | 2000-02-25 | 2001-02-12 | A crystalline form of N-[4-[2-(2-amino-4,7-dihydro-4-oxo-3H-pyrrolo[2,3-d]pyrimidin-5-YL)ethyl]benzoyl]-L-glutamic acid and process therefor |
| DK01906554T DK1259513T3 (en) | 2000-02-25 | 2001-02-12 | New crystalline form of N- [4- [2- (2-amino-4,7-dihydro-4-oxo-3H-pyrrolo [2,3-d] pyrimidin-5-yl) ethyl] benzoyl] -L- glutamic acid and methods for its preparation |
| MXPA02008242A MXPA02008242A (en) | 2000-02-25 | 2001-02-12 | A novel crystalline form of n-[4-[2- (2-amino-4, 7-dihydro- 4-oxo -3h-pyrrolo [2, 3-d] pyrimidin -5-yl) ethyl]benzoyl] -l-glutamic acid and process therefor. |
| DZ013283A DZ3283A1 (en) | 2000-02-25 | 2001-02-12 | NOVEL CRYSTALLINE FORM OF N- [4- [2- (2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO [2,3-D] PYRIMIDIN-5-YL) ETHYL] ACID] -L-GLUTAMINIQUE AND PROCESS FOR THE PREPARATION THEREOF |
| AT01906554T ATE249462T1 (en) | 2000-02-25 | 2001-02-12 | NEW CRYSTALLINE FORM OF N-(4-(2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO(2,3-D)-PYRIMIDINE-5- YL)ETHYL)BENZOYL)-L -GLUTAMIC ACID AND PROCESS FOR THE PRODUCTION THEREOF |
| US10/182,991 US7138521B2 (en) | 2000-02-25 | 2001-02-12 | Crystalline of N-[4-[2-(2-Amino-4,7-dihydro-4oxo-3H-pyrrolo[2,3-D]pyrimidin-5-YL)ethyl]benzoyl]-L-glutamic acid and process therefor |
| HR20020701A HRP20020701B1 (en) | 2000-02-25 | 2001-02-12 | A NOVEL CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
| JP2001562542A JP4846158B2 (en) | 2000-02-25 | 2001-02-12 | Novel crystals of N- [4- [2- (2-amino-4,7-dihydro-4-oxo-3H-pyrrolo [2,3-d] pyrimidin-5-yl) ethyl] benzoyl] -L-glutamic acid Shape and manufacturing method thereof |
| UA2002086886A UA72791C2 (en) | 2000-02-25 | 2001-12-02 | A CRYSTALLINE FORM N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3N-PYRROLO[2,3-d]PYRIMIDINE-5-YL)ETHYL]BENZOYL]-L-GLUTAMINIC ACID AND A METHOD FOR THE PREPARATION THEREOF |
| IL150480A IL150480A (en) | 2000-02-25 | 2002-06-27 | CRYSTALLINE FORM OF N - [4 - [2 - (2 - AMINO - 4,7 - DIHYDRO - 4 - OXO - 3H - PYRROLO [2,3-d] PYRIMIDIN-5 - YL) ETHYL] BENZOYL] - L - GLUTAMIC ACID AND PROCESS THEREFOR |
| NO20023974A NO323422B1 (en) | 2000-02-25 | 2002-08-21 | New crystalline form of N- [4- [2- (2-amino-4,7-dihydro-4-oxo-3H-pyrrolo [2,3-d] pyrimidin-5-yl) ethyl] benzoyl] -L- glutamic acid and process for its preparation |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US18496400P | 2000-02-25 | 2000-02-25 | |
| US60/184,964 | 2000-02-25 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001062760A2 true WO2001062760A2 (en) | 2001-08-30 |
| WO2001062760A3 WO2001062760A3 (en) | 2001-12-06 |
Family
ID=22679017
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/001229 Ceased WO2001062760A2 (en) | 2000-02-25 | 2001-02-12 | A NOVEL CRYSTALLINE FORM OF N-[4-[2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO[2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR |
Country Status (35)
| Country | Link |
|---|---|
| US (1) | US7138521B2 (en) |
| EP (1) | EP1259513B1 (en) |
| JP (1) | JP4846158B2 (en) |
| KR (1) | KR100744917B1 (en) |
| CN (1) | CN1183135C (en) |
| AR (1) | AR029471A1 (en) |
| AT (1) | ATE249462T1 (en) |
| AU (1) | AU777047B2 (en) |
| BR (1) | BR0108604A (en) |
| CA (1) | CA2400155C (en) |
| CO (1) | CO5261585A1 (en) |
| CZ (1) | CZ303772B6 (en) |
| DE (1) | DE60100750T2 (en) |
| DK (1) | DK1259513T3 (en) |
| DZ (1) | DZ3283A1 (en) |
| EA (1) | EA004684B1 (en) |
| EG (1) | EG24073A (en) |
| ES (1) | ES2206403T3 (en) |
| HR (1) | HRP20020701B1 (en) |
| HU (1) | HU229704B1 (en) |
| IL (2) | IL150480A0 (en) |
| MX (1) | MXPA02008242A (en) |
| MY (1) | MY124784A (en) |
| NO (1) | NO323422B1 (en) |
| NZ (1) | NZ519796A (en) |
| PE (1) | PE20011082A1 (en) |
| PL (1) | PL208061B1 (en) |
| PT (1) | PT1259513E (en) |
| SI (1) | SI1259513T1 (en) |
| SK (1) | SK287375B6 (en) |
| SV (1) | SV2002000321A (en) |
| TW (1) | TWI237024B (en) |
| UA (1) | UA72791C2 (en) |
| WO (1) | WO2001062760A2 (en) |
| ZA (1) | ZA200205265B (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100364993C (en) * | 2003-05-30 | 2008-01-30 | 江苏恒瑞医药股份有限公司 | Peimequizane salt and its preparation |
| WO2008124485A3 (en) * | 2007-04-03 | 2009-01-15 | Reddys Lab Ltd Dr | Solid forms of pemetrexed |
| EP2213674A4 (en) * | 2007-10-24 | 2010-12-29 | Chongqing Pharm Res Inst Co | PROCESS FOR THE PURIFICATION OF SALTS, SODIUM SALTS AND DISSODIC SALTS OF PEMETREXED |
| WO2011064256A1 (en) | 2009-11-24 | 2011-06-03 | Azad Pharmaceutical Ingredients Ag | A new crystalline form of pemetrexed disodium |
| EP2334685A4 (en) * | 2008-09-08 | 2011-10-26 | Reddys Lab Ltd Dr | Amorphous pemetrexed disodium |
| WO2012134392A1 (en) * | 2011-03-25 | 2012-10-04 | Scinopharm Taiwan Ltd | Process for the production of a pemetrexed salt |
| WO2012121523A3 (en) * | 2011-03-10 | 2012-11-01 | Kuhnil Pharm. Co., Ltd. | Process for preparing pharmaceutical formulation in form of antioxidant-free solution for injection containing pemetrexed or its salt |
| US8324382B2 (en) | 2008-09-22 | 2012-12-04 | Chongqing Pharmaceutical Research Institute Co., Ltd. | Crystalline forms of Pemetrexed diacid, and preparations thereof |
| WO2014060959A1 (en) * | 2012-10-17 | 2014-04-24 | Shilpa Medicare Limited | Crystalline pemetrexed dipotassium process |
| WO2015075601A1 (en) * | 2013-11-25 | 2015-05-28 | Shilpa Medicare Limited | Process for crystalline pemetrexed dipotassium salt |
| US9051322B2 (en) | 2011-03-23 | 2015-06-09 | Scinopharm Taiwan, Ltd. | Process for the production of a pemetrexed salt |
| WO2016068796A1 (en) * | 2014-10-30 | 2016-05-06 | Scinopharm Taiwan, Ltd. | Crystalline forms of pemetrexed diacid and manufacturing processes therefor |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100364994C (en) * | 2004-11-25 | 2008-01-30 | 重庆医药工业研究院有限责任公司 | A new crystal form of pemetrexed disodium and its preparation method |
| JP2009514891A (en) * | 2005-11-04 | 2009-04-09 | メルク エンド カムパニー インコーポレーテッド | Methods using SAHA and erlotinib for treating cancer |
| CA2636596A1 (en) * | 2005-11-04 | 2007-05-18 | James Pluda | Method of treating cancers with saha and pemetrexed |
| EP1957497B1 (en) * | 2006-08-14 | 2015-09-30 | Sicor, Inc. | Processes for preparing intermediates of pemetrexed |
| ATE492547T1 (en) * | 2006-08-14 | 2011-01-15 | Sicor Inc | METHOD FOR PRODUCING LIPOPHILIC PHARMACEUTICALLY ACCEPTABLE SALTS FROM PEMETREXED DISACID |
| WO2008021405A1 (en) * | 2006-08-14 | 2008-02-21 | Sicor Inc. | Crystalline forms of pemetrexed diacid and processes for the preparation thereof |
| KR20090052355A (en) * | 2006-08-14 | 2009-05-25 | 시코르, 인크. | Highly pure pemetrexed diacids and methods for their preparation |
| CN101528037A (en) * | 2006-11-03 | 2009-09-09 | 默克公司 | Methods of using SAHA and Bortezomib for treating multiple myeloma |
| EP2072518A1 (en) | 2007-12-23 | 2009-06-24 | Sun Pharma Advanced Research Company Limited | Stable Amorphous Form of Pemextred Disodium |
| WO2010030598A2 (en) * | 2008-09-11 | 2010-03-18 | Dr. Reddy's Laboratories Limited | Pharmaceutical formulations comprising pemetrexed |
| CN102050825B (en) * | 2009-11-05 | 2014-12-17 | 上海创诺制药有限公司 | Method for preparing pemetrexed disodium 2.5 water crystal |
| CN102372719B (en) * | 2010-08-26 | 2013-10-30 | 齐鲁制药有限公司 | Pemetrexed methyl ester p-toluenesulfanate crystal form and preparation method thereof |
| ITRM20120398A1 (en) | 2012-08-08 | 2014-02-09 | Berlin Chemie Ag | PEMETREXED SYNTHESIS PROCESS AND ITS LYSINE SALT. |
| CN102911176B (en) * | 2012-10-10 | 2015-07-22 | 德州德药制药有限公司 | Preparation method of pemetrexed disodium |
| WO2014122460A2 (en) | 2013-02-06 | 2014-08-14 | Cipla House | Pemetrexed complexes and pharmaceutical compositions containing pemetrexed complexes |
| US20160051679A1 (en) | 2013-04-12 | 2016-02-25 | Actavis Group Ptc Ehf. | Pemetrexed Formulation |
| KR101485243B1 (en) | 2013-05-08 | 2015-01-21 | 씨제이헬스케어 주식회사 | A stabilized pemetrexed preparation |
| JP6094388B2 (en) * | 2013-06-07 | 2017-03-15 | ニプロ株式会社 | Injectable composition comprising pemetrexed |
| ES2657944T3 (en) | 2013-06-14 | 2018-03-07 | Synthon B.V. | Stable pemetrexed arginine salt and compositions comprising it |
| US9688682B2 (en) | 2013-07-16 | 2017-06-27 | Dr. Reddy's Laboratories Limited | Crystalline forms of pemetrexed tromethamine salts |
| EP3040074B1 (en) | 2013-10-03 | 2018-07-25 | Fujifilm Corporation | Injection preparation and method for producing same |
| NZ630299A (en) | 2014-06-30 | 2014-11-28 | Shilpa Medicare Ltd | Pemetrexed dipotassium formulations |
| CN105566328B (en) * | 2014-11-06 | 2018-04-24 | 博瑞生物医药(苏州)股份有限公司 | The polymorphous preparation method of pemetrexed diacid |
| EP3804686B1 (en) * | 2017-10-10 | 2023-08-30 | Sun Pharmaceutical Industries Ltd | Intravenous infusion dosage form for pemetrexed |
| CN111954531A (en) | 2018-02-07 | 2020-11-17 | L.E.A.F.控股集团公司 | Alpha polyglutamate pemetrexed and uses thereof |
| EP3749311A4 (en) | 2018-02-07 | 2022-07-06 | L.E.A.F Holdings Group LLC | GAMMA POLYGLUTAMATED PEMETREXED AND USES THEREOF |
| WO2025244944A1 (en) * | 2024-05-20 | 2025-11-27 | Opna Bio SA | Crystal forms of an agent |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NO169490C (en) | 1988-03-24 | 1992-07-01 | Takeda Chemical Industries Ltd | ANALOGY PROCEDURE FOR THE PREPARATION OF THERAPEUTIC ACTIVE PYRROLOPYRIMIDINE DERIVATIVES |
| KR0162654B1 (en) | 1989-12-11 | 1998-11-16 | 알렌 제이. 시니스갤리 | N-[pyrrolo (2, 3-d) pyrimidin-3yl acryl]-glutamic acid derivatives |
| US5416211A (en) | 1992-09-25 | 1995-05-16 | Eli Lilly And Company | Process for preparing 5-substituted pyrrolo-[2,3-d]pyrimidines |
| EP0923287A4 (en) * | 1996-08-30 | 2001-08-01 | Lilly Co Eli | Nonclassical pyrrolo 2,3-d]pyrimidine antifolates |
| BR9812524A (en) * | 1997-09-26 | 2000-07-25 | Lilly Co Eli | Processes and intermediates usable to produce antifolates |
| ZA987550B (en) * | 1997-09-26 | 2000-02-21 | Lilly Co Eli | Processes and intermediates useful to make antifolates. |
| EP1212325A2 (en) * | 1999-08-23 | 2002-06-12 | Eli Lilly And Company | A novel crystalline form of disodium n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid salt and processes therefor |
-
2001
- 2001-02-12 PT PT01906554T patent/PT1259513E/en unknown
- 2001-02-12 IL IL15048001A patent/IL150480A0/en active IP Right Grant
- 2001-02-12 JP JP2001562542A patent/JP4846158B2/en not_active Expired - Fee Related
- 2001-02-12 CN CNB01805627XA patent/CN1183135C/en not_active Expired - Fee Related
- 2001-02-12 AU AU34451/01A patent/AU777047B2/en not_active Ceased
- 2001-02-12 NZ NZ519796A patent/NZ519796A/en not_active IP Right Cessation
- 2001-02-12 EA EA200200905A patent/EA004684B1/en not_active IP Right Cessation
- 2001-02-12 PL PL356423A patent/PL208061B1/en unknown
- 2001-02-12 SK SK1186-2002A patent/SK287375B6/en not_active IP Right Cessation
- 2001-02-12 US US10/182,991 patent/US7138521B2/en not_active Expired - Lifetime
- 2001-02-12 HU HU0204232A patent/HU229704B1/en unknown
- 2001-02-12 EP EP01906554A patent/EP1259513B1/en not_active Expired - Lifetime
- 2001-02-12 AT AT01906554T patent/ATE249462T1/en active
- 2001-02-12 ES ES01906554T patent/ES2206403T3/en not_active Expired - Lifetime
- 2001-02-12 CA CA002400155A patent/CA2400155C/en not_active Expired - Fee Related
- 2001-02-12 SI SI200130032T patent/SI1259513T1/en unknown
- 2001-02-12 HR HR20020701A patent/HRP20020701B1/en not_active IP Right Cessation
- 2001-02-12 DZ DZ013283A patent/DZ3283A1/en active
- 2001-02-12 DK DK01906554T patent/DK1259513T3/en active
- 2001-02-12 BR BR0108604-9A patent/BR0108604A/en not_active Application Discontinuation
- 2001-02-12 CZ CZ20022875A patent/CZ303772B6/en not_active IP Right Cessation
- 2001-02-12 WO PCT/US2001/001229 patent/WO2001062760A2/en not_active Ceased
- 2001-02-12 DE DE60100750T patent/DE60100750T2/en not_active Expired - Lifetime
- 2001-02-12 KR KR1020027011122A patent/KR100744917B1/en not_active Expired - Fee Related
- 2001-02-12 MX MXPA02008242A patent/MXPA02008242A/en active IP Right Grant
- 2001-02-21 EG EG20010167A patent/EG24073A/en active
- 2001-02-23 PE PE2001000194A patent/PE20011082A1/en not_active Application Discontinuation
- 2001-02-23 CO CO01014634A patent/CO5261585A1/en not_active Application Discontinuation
- 2001-02-23 SV SV2001000321A patent/SV2002000321A/en unknown
- 2001-02-23 AR ARP010100844A patent/AR029471A1/en unknown
- 2001-02-23 TW TW090104119A patent/TWI237024B/en not_active IP Right Cessation
- 2001-02-23 MY MYPI20010823A patent/MY124784A/en unknown
- 2001-12-02 UA UA2002086886A patent/UA72791C2/en unknown
-
2002
- 2002-06-27 IL IL150480A patent/IL150480A/en not_active IP Right Cessation
- 2002-07-01 ZA ZA200205265A patent/ZA200205265B/en unknown
- 2002-08-21 NO NO20023974A patent/NO323422B1/en not_active IP Right Cessation
Non-Patent Citations (2)
| Title |
|---|
| DATABASE CHEMABS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; CALVERT, A.H. ET AL. : "Clinical studies with MTA" retrieved from STN Database accession no. 129:310246 XP002160356 & BR. J. CANCER (1998) 78 (SUPPL. 3, NEW ANTIMETABOLITES IN CANCER CHEMOTHERAPY AND THEIR CLINICAL IMPACT), 35-40, 1998, * |
| GANGJEE ET AL.: "Design, synthesis, and X-ray crystal structure of a potent dual inhibitor of thymidylate synthase and dihydrofolate reductase as an antitumor agent" JOURNAL OF MEDICINAL CHEMISTRY., vol. 43, no. 21, 2000, pages 3837-3851, XP002173880 AMERICAN CHEMICAL SOCIETY. WASHINGTON., US ISSN: 0022-2623 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100364993C (en) * | 2003-05-30 | 2008-01-30 | 江苏恒瑞医药股份有限公司 | Peimequizane salt and its preparation |
| US8507508B2 (en) | 2007-04-03 | 2013-08-13 | Dr. Reddy's Laboratories Limited | Solid forms of pemetrexed |
| WO2008124485A3 (en) * | 2007-04-03 | 2009-01-15 | Reddys Lab Ltd Dr | Solid forms of pemetrexed |
| JP2010523589A (en) * | 2007-04-03 | 2010-07-15 | ドクター レディズ ラボラトリーズ リミテッド | Solid form of pemetrexed |
| EP2213674A4 (en) * | 2007-10-24 | 2010-12-29 | Chongqing Pharm Res Inst Co | PROCESS FOR THE PURIFICATION OF SALTS, SODIUM SALTS AND DISSODIC SALTS OF PEMETREXED |
| US8686140B2 (en) | 2007-10-24 | 2014-04-01 | Chongqing Pharmaceutical Research Institute Co., Ltd. | Method of purifying a salt, sodium salt and disodium salt of pemetrexed |
| AU2008318137B2 (en) * | 2007-10-24 | 2012-04-19 | Chongqing Pharmaceutical Research Institute Co., Ltd. | Purification method of pemetrexed salts,sodium salts and disodium salts |
| EP2334685A4 (en) * | 2008-09-08 | 2011-10-26 | Reddys Lab Ltd Dr | Amorphous pemetrexed disodium |
| US8324382B2 (en) | 2008-09-22 | 2012-12-04 | Chongqing Pharmaceutical Research Institute Co., Ltd. | Crystalline forms of Pemetrexed diacid, and preparations thereof |
| WO2011064256A1 (en) | 2009-11-24 | 2011-06-03 | Azad Pharmaceutical Ingredients Ag | A new crystalline form of pemetrexed disodium |
| US9174991B2 (en) | 2009-11-24 | 2015-11-03 | Azad Pharmaceutical Ingredients Ag | Crystalline form of pemetrexed disodium |
| WO2012121523A3 (en) * | 2011-03-10 | 2012-11-01 | Kuhnil Pharm. Co., Ltd. | Process for preparing pharmaceutical formulation in form of antioxidant-free solution for injection containing pemetrexed or its salt |
| US9051322B2 (en) | 2011-03-23 | 2015-06-09 | Scinopharm Taiwan, Ltd. | Process for the production of a pemetrexed salt |
| KR101767713B1 (en) | 2011-03-25 | 2017-08-11 | 시노팜 타이완 리미티드 | Process for the production of a pemetrexed salt |
| WO2012134392A1 (en) * | 2011-03-25 | 2012-10-04 | Scinopharm Taiwan Ltd | Process for the production of a pemetrexed salt |
| JP2014508805A (en) * | 2011-03-25 | 2014-04-10 | サイノファーム タイワン リミテッド | Method for producing pemetrexed salt |
| WO2014060959A1 (en) * | 2012-10-17 | 2014-04-24 | Shilpa Medicare Limited | Crystalline pemetrexed dipotassium process |
| WO2015075601A1 (en) * | 2013-11-25 | 2015-05-28 | Shilpa Medicare Limited | Process for crystalline pemetrexed dipotassium salt |
| US9604990B2 (en) | 2014-10-30 | 2017-03-28 | Scinopharm Taiwan, Ltd. | Crystalline forms of pemetrexed diacid and manufacturing processes therefor |
| WO2016068796A1 (en) * | 2014-10-30 | 2016-05-06 | Scinopharm Taiwan, Ltd. | Crystalline forms of pemetrexed diacid and manufacturing processes therefor |
| US9765079B2 (en) | 2014-10-30 | 2017-09-19 | Scintopharm Taiwan, Ltd. | Crystalline forms of pemetrexed diacid and manufacturing processes therefor |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1259513B1 (en) | A NOVEL CRYSTALLINE FORM OF N- 4- 2-(2-AMINO-4,7-DIHYDRO-4-OXO-3H-PYRROLO 2,3-d]PYRIMIDIN-5-YL)ETHYL]BENZOYL]-L-GLUTAMIC ACID AND PROCESS THEREFOR | |
| SU799664A3 (en) | Method of preparing 9-(2-oxyethoxymethyl)guanine or its salts | |
| EA000364B1 (en) | Guanine derivative | |
| EP1212325A2 (en) | A novel crystalline form of disodium n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid salt and processes therefor | |
| US20090181990A1 (en) | Stable amorphous form of pemetrexed disodium | |
| RU2394038C2 (en) | Organic compounds | |
| EP0944612B1 (en) | N-(4-acetyl-1-piperazinyl)-4-fluorobenzamide hydrate | |
| HK1051856B (en) | A novel crystalline form of n-[4-[2-(2-amino-4,7-dihydro-4-oxo-3h-pyrrolo [2,3-d]pyrimidin-5-yl)ethyl]benzoyl]-l-glutamic acid and process therefor | |
| NL1014634C1 (en) | Zolpidem salts. | |
| EP0586298B1 (en) | Inhibitor of herpes simplex viral thymidine kinase | |
| EP3845537B1 (en) | Crystal of pyrazolo[3,4-d]pyrimidine | |
| TW202308646A (en) | Crystal form of a compound for treating influenza and use thereof | |
| MXPA99005427A (en) | N-(4-acetyl-1-piperazinyl)-4-fluorobenzamide hydrate | |
| MX2011009395A (en) | Pemetrexed crystallines shapes and process for obtaining the same. | |
| MXPA97005462A (en) | Derived from guan | |
| HK1199026B (en) | Tenofovir alafenamide hemifumarate | |
| HK1003055A1 (en) | Guanine derivative | |
| HK1003055B (en) | Guanine derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IN/PCT/2002/845/KOL Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 519796 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 150480 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002/05265 Country of ref document: ZA Ref document number: 200205265 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 34451/01 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001906554 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2400155 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11862002 Country of ref document: SK |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2002000207 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200200765 Country of ref document: VN |
|
| ENP | Entry into the national phase |
Ref document number: 2001 562542 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2002/008242 Country of ref document: MX Ref document number: PV2002-2875 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020027011122 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20020701A Country of ref document: HR Ref document number: 01805627X Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200200905 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020027011122 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10182991 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001906554 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 519796 Country of ref document: NZ |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2002-2875 Country of ref document: CZ |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 519796 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001906554 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 34451/01 Country of ref document: AU |