WO2001072741A2 - N-benzodioxanylmethyl-1-piperidyl-methylamine compounds for the treatment of central nervous system disorders - Google Patents
N-benzodioxanylmethyl-1-piperidyl-methylamine compounds for the treatment of central nervous system disorders Download PDFInfo
- Publication number
- WO2001072741A2 WO2001072741A2 PCT/EP2001/003463 EP0103463W WO0172741A2 WO 2001072741 A2 WO2001072741 A2 WO 2001072741A2 EP 0103463 W EP0103463 W EP 0103463W WO 0172741 A2 WO0172741 A2 WO 0172741A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- disorders
- formula
- compound
- compounds
- drug
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
Definitions
- the present invention relates to novel therapeutic agents which have affinity for 5-HT ⁇ and/or ct ⁇ and/or D 2 receptors, to processes for their preparation, to pharmaceutical compositions containing them and to their use in the treatment of central nervous system disorders, for example depression, anxiety, psychoses (for example schizophrenia), tardive dyskinesia, Parkinson's disease, obesity, hypertension, Tourette's syndrome, sexual dysfunction, drug addiction, drug abuse, cognitive disorders, Alzheimer's disease, senile dementia, obsessive-compulsive behaviour, panic attacks, eating disorders and anorexia, cardiovascular and cerebrovascular disorders, migraine, non-insulin dependent diabetes mellitus, hyperglycaemia, constipation, arrhythmia, disorders of the neuroendocrine system, stress, prostatic hypertrophy, drug-induced extrapyramidal symptoms and spasticity.
- central nervous system disorders for example depression, anxiety, psychoses (for example schizophrenia), tardive dyskinesia, Parkinson's disease, obesity, hypertension, Tourette's syndrome, sexual dysfunction, drug addiction
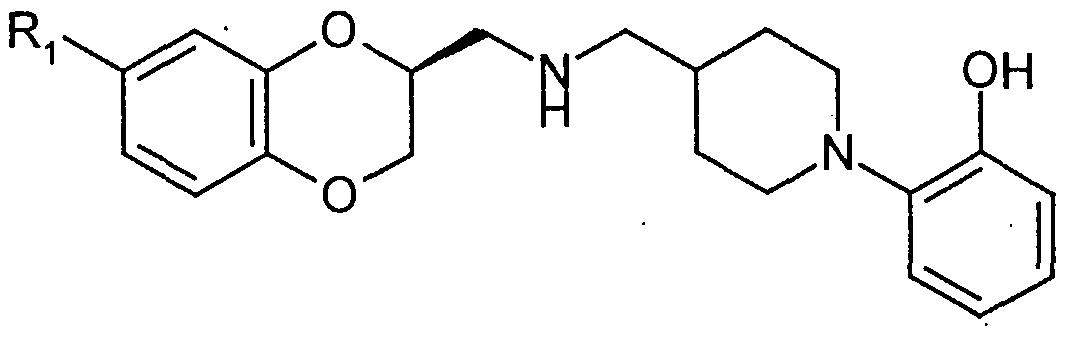
- A is methylene or -O-; B is methylene or -O-; and g is 0, 1 , 2, 3 or 4;
- R-i represents, halo, optionally substituted alkyl, optionally substituted alkoxy, optionally substituted alkylthio, hydroxy, acyloxy, hydroxymethyl, cyano, alkanoyl, alkoxycarbonyl, optionally ⁇ /-substituted carbamoyl, carbamoylmethyl, sulphamoyl or sulphamoylmethyl, an amino group optionally substituted by one or two alkyl groups, or two adjacent R- ⁇ groups together with the carbon atoms to which they are attached form a fused benz ring;
- R 2 is H, aikyl or alkoxy;
- R 3 and R which are the same or different, are H, or alkyl;
- U is analkylene chain optionally substituted by one or more alkyl;
- Q represents a di
- WO99/62902 discloses compounds of formula
- WO99/62902 discloses that (S)-(-)-1-[1-(4-fluoro-2-methoxyphenyl)piperid-4- yl]- ⁇ /-(7-trifluoromethyl-2,3-dihydro-1 ,4-benzodioxin-2-ylmethyl)methylamine and (S)- (-)-1-[1-(2-methoxyphenyl)piperid-4-yl]- ⁇ /-(7-trifluoromethyl-2,3-dihydro-1 ,4-benzo- dioxin-2-ylmethyl)methylamine have unexpectedly superior activity to the compounds disclosed in WO95/07274.
- the present invention provides compounds of formula
- R represents halo or pseudohalo and R 2 represents H or an acyl group derived from a C -C 18 saturated aliphatic carboxylic acid; with the proviso that when R-i is CI or CF 3 and R 2 is H then these compounds are in isolated form.
- pseudohalo includes trifluoromethyl, trifluoromethoxy and trifluoromethylthio and other pharmaceutically acceptable pseudohalo groups known to those skilled in the art.
- the pseudohalo group is trifluoromethyl, trifluoromethoxy and trifluoromethylthio.
- halo includes chloro, bromo and fluoro.
- R 2 represents H, heptanoyl, decanoyl, dodecanoyl, hexadecanoyl or octadecanoyl. More preferably R 2 is H.
- R represents CI or CF 3 , including pharmaceutically acceptable salts thereof, in isolated form.
- the term isolated is used to indicate that the compounds of the invention are in pure form and are not present in a human or an animal either in a free form or in an associated form.
- the compounds of the present invention are advantageous over compounds known in the prior art because of their selectivity in receptor binding assays and their superior oral activity.
- Compounds of formula I may exist as salts with pharmaceutically acceptable acids.
- Such salts include hydrochlorides, hydrobromides, sulphates, methanesulphonates, nitrates, maleates, acetates, citrates, fumarates, tartrates [eg (+)-tartrates, (-)-tartrates or mixtures thereof including racemic mixtures], succinates, benzoates and salts with amino acids such as glutamic acid.
- Compounds of formula I and their salts may exist in the form of soivates (for example hydrates). Certain compounds of formula I and their salts may exist in more than one crystal form and the present invention includes each crystal form and mixtures thereof. Certain compounds of formula I and their salts may also exist in the form of solvates, for example hydrates, and the present invention includes each solvate and mixtures thereof.
- the present invention also includes pharmaceutical compositions containing a therapeutically effective amount of a compound of formula I or a salt thereof together with a pharmaceutically acceptable diluent or carrier.
- the term active compound denotes a compound of formula I or a salt thereof.
- the active compound may be administered orally, rectally, parenterally or topically, preferably orally.
- the therapeutic compositions of the present invention may take the form of any of the known pharmaceutical compositions for oral, rectal, parenteral or topical administration.
- Pharmaceutically acceptable carriers suitable for use in such compositions are well known in the art of pharmacy.
- the compositions of the invention may contain 0.1-99% by weight of active compound.
- the compositions of the invention are generally prepared in unit dosage form. Preferably the unit dosage of active ingredient is 1-500 mg.
- the excipients used in the preparation of these compositions are the excipients known in the pharmacist's art.
- compositions for oral administration are the preferred compositions of the invention and these are the known pharmaceutical forms for such administration, for example tablets, capsules, syrups and aqueous or oil suspensions.
- the excipients used in the preparation of these compositions are the excipients known in the pharmacist's art.
- Tablets may be prepared by mixing the active compound with an inert diluent such as calcium phosphate in the presence of disintegrating agents, for example maize starch, and lubricating agents, for example magnesium stearate, and tableting the mixture by known methods.
- the tablets may be formulated in a manner known to those skilled in the art so as to give a sustained release of the compounds of the present invention.
- Such tablets may, if desired, be provided with enteric coatings by known methods, for example by the use of cellulose acetate phthalate.
- capsules for example hard or soft gelatin capsules, containing the active compound with or without added excipients, may be prepared by conventional means and, if desired, provided with enteric coatings in a known manner.
- the tablets and capsules may conveniently each contain 1 to 500 mg of the active compound.
- Other compositions for oral administration include, for example, aqueous suspensions containing the active compound in an aqueous medium in the presence of a non-toxic suspending agent such as sodium carboxymethyl- cellulose, and oily suspensions containing a compound of the present invention in a suitable vegetable oil, for example arachis oil.
- the active compound may be formulated into granules with or without additional excipients.
- the granules may be ingested directly by the patient or they may be added to a suitable liquid carrier (for example water) before ingestion.
- the granules may contain disintegrants (for example a pharmaceutically acceptable effervescent couple formed from an acid and a carbonate or bicarbonate salt) to facilitate dispersion in the liquid medium.
- compositions of the invention suitable for rectal administration are the known pharmaceutical forms for such administration, for example, suppositories with cocoa butter or polyethylene glycol bases.
- compositions of the invention suitable for parenteral administration are the known pharmaceutical forms for such administration, for example sterile suspensions or sterile solutions in a suitable solvent.
- compositions for topical administration may comprise a matrix in which the pharmacologically active compounds of the present invention are dispersed so that the compounds are held in contact with the skin in order to administer the compounds transdermally.
- a suitable transdermal composition may be prepared by mixing the pharmaceutically active compound with a topical vehicle, such as a mineral oil, petrolatum and/or a wax, for example paraffin wax or beeswax, together with a potential transdermal accelerant such as dimethyl sulphoxide or propylene glycol.
- the active compounds may be dispersed in a pharmaceutically acceptable cream or ointment base.
- the amount of active compound contained in a topical formulation should be such that a therapeutically effective amount of the compound is delivered during the period of time for which the topical formulation is intended to be on the skin.
- the compounds of the present invention may also be administered by continuous infusion either from an external source, for example by intravenous infusion or from a source of the compound placed within the body.
- Internal sources include implanted reservoirs containing the compound to be infused which is continuously released for example by osmosis and implants which may be (a) liquid such as a suspension or solution in a pharmaceutically acceptable oil of the compound to be infused for example in the form of a very sparingly water-soluble derivative such as a dodecanoate salt or ester or (b) solid in the form of an implanted support, for example of a synthetic resin or waxy material, for the compound to be infused.
- the support may be a single body containing all the compound or a series of several bodies each containing part of the compound to be delivered.
- the amount of active compound present in an internal source should be such that a therapeutically effective amount of the compound is delivered over a long period of time.
- the compounds of the present invention in the form of particles of very small size, for example as obtained by fluid energy milling.
- the active compound may, if desired, be associated with other compatible pharmacologically active ingredients.
- compositions containing a therapeutically effective amount of a compound of formula I or a salt thereof may be used to treat depression, anxiety, psychoses (for example schizophrenia), tardive dyskinesia, Parkinson's disease, obesity, hypertension, Tourette's syndrome, sexual dysfunction, drug addiction, drug abuse, cognitive disorders, Alzheimer's disease, senile dementia, obsessive-compulsive behaviour, panic attacks, eating disorders, anorexia, cardiovascular and cerebrovascular disorders, migraine, non-insulin dependent diabetes mellitus, hyperglycaemia, constipation, arrhythmia, disorders of the neuroendocrine system, stress, prostatic hypertrophy, drug-induced extrapyramidal symptoms and spasticity in human beings.
- psychoses for example schizophrenia
- tardive dyskinesia for example schizophrenia
- Parkinson's disease for example schizophrenia
- obesity for example schizophrenia
- hypertension for example schizophrenia
- Tourette's syndrome sexual dysfunction
- drug addiction drug abuse
- cognitive disorders Alzheimer's disease
- senile dementia obsessive
- the amount of active compound administered in such treatment is in the range 1 to 1000 mg preferably 5 to 500 mg given in single or divided doses at one or more times during the day.
- 5-hydroxytryptamine (5- HT) receptors The ability of compounds of formula I to interact with 5-hydroxytryptamine (5- HT) receptors has been demonstrated by the following test which determines the ability of the compounds to inhibit tritiated ligand binding to 5-HT receptors in vitro and in particular to 5-HT 1A receptors.
- Hippocampal tissue from the brains of male Charles River CD rats weighing between 150-250 g were homogenised in ice-cold 50 mM Tris-HCI buffer (pH 7.7) when measured at 25°C, 1 :40 w/v) and centrifuged at 30,000 g at 4°C for 10 minutes. The pellet was rehomogenised in the same buffer, incubated at 37°C for 10 minutes and centrifuged at 30,000 g at 4°C for 10 minutes.
- the final pellet was resuspended in 50 mM Tris-HCI buffer (pH 7.7) containing 4 mM CaCI 2 , 0.1% L- ascorbic acid and 10 ⁇ M pargyline hydrochloride (equivalent to 6.25 mg wet weight of tissue/ml) and used immediately in the binding assay.
- the filters were washed with ice-cold Tris-HCI buffer and dried. The filters were punched out into vials, scintillation fluid added and radioactivity determined by liquid scintillation counting. The percentage displacement of specific binding of the tritiated ligand was calculated for the single concentration (10 '6 M) of test compound. Displacement curves were then produced for those compounds which displaced >50% of specific binding of the tritiated ligand at 10 "6 M using a range of concentrations of the compound. The concentration which gave 50% inhibition of specific binding (IC 50 ) was obtained from the curve. The inhibition coefficient Ki was then calculated using the formula
- [ligand] is the concentration of the tritiated ligand used and K D is the equilibrium dissociation constant for the ligand.
- the final pellet was resuspended in 50 mM Tris-HCI, pH 7.6 (equivalent to 12.5 mg wet weight of tissue/ml) and used immediately in the binding assay. Aliquots (400 ⁇ l; equivalent to 5 mg wet weight of tissue/tube) of this suspension were added to tubes containing the ligand (50 ⁇ l; 0.1 nM) and distilled water (50 ⁇ l; total binding) or phentolamine (50 ⁇ l; 5 ⁇ M; non-specific binding) or test compound (50 ⁇ l; at a single concentration of lO ⁇ M or at 10 concentrations ranging from 10 "11 - 10 '3 M).
- the ligand was [7-methoxy- 3 H]prazosin and the mixture was incubated at 30°C for 30 minutes before the incubation was terminated by rapid filtration.
- the filters were washed with ice-cold Tris-HCI buffer and dried. The filters were punched out into vials, scintillation fluid added and radioactivity determined by liquid scintillation counting. The percentage displacement of specific binding of the tritiated ligand was calculated for the single concentration (10" 6 M) of test compound. Displacement curves were then produced for those compounds which displaced >50% of specific binding of the tritiated ligand at 10 "6 M using a range of concentrations of the compound. The concentration which gave 50% inhibition of specific binding (IC 50 ) was obtained from the curve. The inhibition coefficient Ki was then calculated using the formula
- [ligand] is the concentration of the tritiated ligand used and K D is the equilibrium dissociation constant for the ligand.
- Tris salts buffer (equivalent to 2 mg wet weight of tissue/ml). Aliquots (720 ⁇ l; equivalent to 1.44 mg wet weight of tissue/tube) of this suspension were then added to tubes containing the ligand (40 ⁇ l; 1 nM) and Tris salts buffer (40 ⁇ l; total binding) or spiroperidol (40 ⁇ l; 10 nM; non-specific binding) or test compound (40 ⁇ l; at a single concentration of lO ⁇ M or at 6 concentrations ranging from lO ' ⁇ -IO ⁇ M).
- the ligand was tritiated (S)-sulpiride and the mixture was incubated at 4°C for 40 minutes before the incubation was terminated by rapid filtration.
- the filters were washed with ice-cold Tris-HCI buffer and dried. The filters were punched out in to vials, scintillation fluid added and were left for about 20 hours before being counted by scintillation , spectrophotometry.
- the percentage displacement of specific binding of the tritiated ligand was calculated for the single concentration (10 "6 M) of test compound. Displacement curves were then produced over a range of concentrations for those compounds which displaced >50% of specific binding of the tritiated ligand at 10 "6 M. The concentration which gave a 50% inhibition of specific binding (IC 50 ) was obtained from the curve.
- the inhibition coefficient Ki was then calculated using the formula
- [ligand] is the concentration of the tritiated ligand used and K D is the equilibrium dissociation constant for the ligand.
- mice Groups of 10 male mice weighing 18-35 g (max. range 10 g) were treated with test compound or control vehicle by po administration. 30 minutes later, mice were injected subcutaneously with apomorphine (0.88 mg/kg). Immediately after the apomorphine injection the mice were placed in the test cages and the climbing behaviour of each mouse was assessed at 10 and 20 minutes on a simple 0-2 ranking scale.
- ED 50 values dose causing 50% of the control score
- 95% confidence limits were calculated.
- ED 50 values are calculated as free base equivalents and are given in Table 2 alongside Comparative Example A.
- the compounds of the present invention are more potent orally than compounds previously described. More potent compounds are advantaged as they are less likely to induce systemic toxicological effects on organs which are not the therapeutic target.
- R 2 represents H
- a de-alkylating agent for example hydrobromic acid, pyridine hydrochloride or boron tribromide optionally in the presence of a suitable solvent or mixture of solvents, for example water or glacial acetic acid, or mixtures thereof, at a temperature in the range of 0 - 250°C.
- Compounds of formula I in which R 2 represents an -O-acyl group may be prepared by acylating a compound of formula I in which R 2 represents H by methods known to those skilled in the art, for example by reaction with an anhydride or an acyl chloride, optionally in the presence of a solvent and optionally in the presence of a base.
- a compound of formula I in which R 2 represents H by methods known to those skilled in the art, for example by reaction with an anhydride or an acyl chloride, optionally in the presence of a solvent and optionally in the presence of a base.
- the methylamine nitrogen is protected before the acylation and then deprotected after the acylation by methods known to those skilled in the art.
- the invention is illustrated by the following Examples which are given by way of example only.
- the final product of each of these Examples was characterised by one or more of the following procedures: gas-liquid chromatography; high performance liquid chromatography; elemental analysis, nuclear magnetic resonance spectroscopy and infrared spectroscopy.
- Trifluoroacetic acid (7 ml) was added dropwise with care over a period of 5 minutes to an ice-cold stirred solution of the product from 3b) (3.0 g) in dichloromethane (100 ml). The mixture was allowed to warm to ambient temperature and stirred at this temperature for 24 hours. The mixture was poured into water (300 ml) and then basified with solid sodium bicarbonate. The organic layer was separated and the aqueous layer was extracted with dichloromethane.
- active compound denotes any compound of the invention but particularly any compound which is the final product of one of the preceding Examples.
- capsules 10 parts by weight of active compound and 240 parts by weight of lactose are de-aggregated and blended. The mixture is filled into hard gelatin capsules, each capsule containing a unit dose or part of a unit dose of active compound.
- Tablets are prepared from the following ingredients.
- the active compound, the lactose and some of the starch are de-aggregated, blended and the resulting mixture is granulated with a solution of the polyvinyl- pyrrolidone in ethanol.
- the dry granulate is blended with the magnesium stearate and the rest of the starch.
- the mixture is then compressed in atabletting machine to give tablets each containing a unit dose or a part of a unit dose of active compound.
- Tablets are prepared by the method described in (b) above.
- the tablets are enteric coated in a conventional manner using a solution of 20% cellulose acetate phthalate and 3% diethyl phthalate in ethanol:dichloromethane (1 :1).
- suppositories 100 parts by weight of active compound is incorporated in 1300 parts by weight of triglyceride suppository base and the mixture formed into suppositories each containing a therapeutically effective amount of active ingredient.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Neurology (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description







Claims


Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2001273903A AU2001273903A1 (en) | 2000-03-28 | 2001-03-27 | Therapeutic agents |
| US10/240,180 US20040039023A1 (en) | 2000-03-28 | 2001-03-27 | Therapeutic agents |
| EP01940264A EP1274703A2 (en) | 2000-03-28 | 2001-03-27 | N-benzodioxanylmethyl-1-piperidyl-methylamine compounds for the treament of central nervous system disorders |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0007376.7A GB0007376D0 (en) | 2000-03-28 | 2000-03-28 | Therapeutic agents |
| GB0007376.7 | 2000-03-28 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001072741A2 true WO2001072741A2 (en) | 2001-10-04 |
| WO2001072741A3 WO2001072741A3 (en) | 2002-01-03 |
Family
ID=9888485
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/003463 Ceased WO2001072741A2 (en) | 2000-03-28 | 2001-03-27 | N-benzodioxanylmethyl-1-piperidyl-methylamine compounds for the treatment of central nervous system disorders |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20040039023A1 (en) |
| EP (1) | EP1274703A2 (en) |
| AU (1) | AU2001273903A1 (en) |
| GB (1) | GB0007376D0 (en) |
| WO (1) | WO2001072741A2 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004002483A1 (en) * | 2002-06-27 | 2004-01-08 | Actelion Pharmaceuticals Ltd | Substituted 3- and 4- aminomethylpiperidines for use as beta-secretase in the treatment of alzheimer’s disease |
| US6800635B2 (en) | 2000-03-24 | 2004-10-05 | Pharmacia Italia, S.P.A. | Crystalline form II of cabergoline |
| WO2004082570A3 (en) * | 2003-03-17 | 2005-01-27 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with dopamine receptor d2 (drd2) |
| WO2006116158A1 (en) * | 2005-04-22 | 2006-11-02 | Wyeth | Benzodioxane and benzodioxolane derivatives and uses thereof |
| US7297704B2 (en) | 2005-02-17 | 2007-11-20 | Wyeth | Cycloalkyfused indole, benzothiophene, benzofuran and idene derivatives |
| WO2009133109A1 (en) * | 2008-04-29 | 2009-11-05 | Nsab, Filial Af Neurosearch Sweden Ab, Sverige | Modulators of dopamine neurotransmission |
| US8492372B2 (en) | 2008-04-29 | 2013-07-23 | Integrated Research Laboratories Sweden Ab | Modulators of dopamine neurotransmission |
| US8524766B2 (en) | 2008-04-29 | 2013-09-03 | Nsab, Filial Af Neurosearch Sweden Ab, Sverige | Modulators of dopamine neurotransmission |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9318431D0 (en) * | 1993-09-06 | 1993-10-20 | Boots Co Plc | Therapeutic agents |
| GB9704948D0 (en) * | 1997-03-11 | 1997-04-30 | Knoll Ag | Therapeutic agents |
| JP3955386B2 (en) * | 1998-04-09 | 2007-08-08 | 富士通株式会社 | Semiconductor device and manufacturing method thereof |
| GB9811879D0 (en) * | 1998-06-03 | 1998-07-29 | Knoll Ag | Therapeutic agents |
| US6297063B1 (en) * | 1999-10-25 | 2001-10-02 | Agere Systems Guardian Corp. | In-situ nano-interconnected circuit devices and method for making the same |
-
2000
- 2000-03-28 GB GBGB0007376.7A patent/GB0007376D0/en not_active Ceased
-
2001
- 2001-03-27 EP EP01940264A patent/EP1274703A2/en not_active Withdrawn
- 2001-03-27 WO PCT/EP2001/003463 patent/WO2001072741A2/en not_active Ceased
- 2001-03-27 AU AU2001273903A patent/AU2001273903A1/en not_active Abandoned
- 2001-03-27 US US10/240,180 patent/US20040039023A1/en not_active Abandoned
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6800635B2 (en) | 2000-03-24 | 2004-10-05 | Pharmacia Italia, S.P.A. | Crystalline form II of cabergoline |
| WO2004002483A1 (en) * | 2002-06-27 | 2004-01-08 | Actelion Pharmaceuticals Ltd | Substituted 3- and 4- aminomethylpiperidines for use as beta-secretase in the treatment of alzheimer’s disease |
| WO2004082570A3 (en) * | 2003-03-17 | 2005-01-27 | Bayer Healthcare Ag | Diagnostics and therapeutics for diseases associated with dopamine receptor d2 (drd2) |
| US7297704B2 (en) | 2005-02-17 | 2007-11-20 | Wyeth | Cycloalkyfused indole, benzothiophene, benzofuran and idene derivatives |
| WO2006116158A1 (en) * | 2005-04-22 | 2006-11-02 | Wyeth | Benzodioxane and benzodioxolane derivatives and uses thereof |
| WO2009133109A1 (en) * | 2008-04-29 | 2009-11-05 | Nsab, Filial Af Neurosearch Sweden Ab, Sverige | Modulators of dopamine neurotransmission |
| US8492372B2 (en) | 2008-04-29 | 2013-07-23 | Integrated Research Laboratories Sweden Ab | Modulators of dopamine neurotransmission |
| US8524766B2 (en) | 2008-04-29 | 2013-09-03 | Nsab, Filial Af Neurosearch Sweden Ab, Sverige | Modulators of dopamine neurotransmission |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0007376D0 (en) | 2000-05-17 |
| EP1274703A2 (en) | 2003-01-15 |
| WO2001072741A3 (en) | 2002-01-03 |
| AU2001273903A1 (en) | 2001-10-08 |
| US20040039023A1 (en) | 2004-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU689802B2 (en) | Bicyclic aromatic compounds as therapeutic agents | |
| EP0679642B1 (en) | Condensed heterocyclic compounds, their production and use | |
| EP0876372B1 (en) | Heteroarylcarboxamide derivatives | |
| EP0839144B1 (en) | Piperazine derivatives as therapeutic agents | |
| JPH11512701A (en) | Selective β ▲ 3 ▼ adrenergic agonist | |
| EP0610793A1 (en) | Tetracyclic morpholine derivatives and their use or analgesics | |
| KR20070062609A (en) | Pyrroloindole, pyridoindole and azepineindole as 5-HT2C agonists | |
| JP2001515485A (en) | Dioxino derivatives and their use as therapeutics | |
| US6391891B1 (en) | Bicyclic compounds as ligands for 5-HT1 receptors | |
| JP2007522142A (en) | Benzimidazole-substituted thiophene derivatives having activity against IKK3 | |
| KR0128288B1 (en) | Naphthyloxazolidone derivatives | |
| RU2169147C2 (en) | DERIVATIVES OF CARBOXYLIC ACID AMIDES WITH HETEROCYCLIC SUBSTITUENTS AND COMPOSITION SHOWING ABILITY TO INHIBIT 5-HT1A- AND/OR alpfa- AND/OR alpfa- AND/OR alpfa- AND/OR α1-RECEPTORS | |
| WO2001072741A2 (en) | N-benzodioxanylmethyl-1-piperidyl-methylamine compounds for the treatment of central nervous system disorders | |
| FR2860792A1 (en) | THIOPHENE-2-CARBOXAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE | |
| KR100240242B1 (en) | Optically Active Alkylenedioxybenzene Derivatives and Uses in Therapeutics | |
| US6136825A (en) | Sulfonamide compounds having 5-HT receptor activity | |
| WO1999062902A1 (en) | N-benzodioxanylmethyl-1-piperidyl-methylamine compounds having affinity for 5-ht receptors | |
| JPH09512025A (en) | Tricyclic derivatives as 5HT-lower 2C and 5HT-lower 2B antagonists | |
| KR100437561B1 (en) | Novel Heterocyclic Compounds | |
| McQuaid et al. | Substituted 5-amino-4, 5, 6, 7-tetrahydroindazoles as partial ergoline structures with dopaminergic activity | |
| CA2007401A1 (en) | Bisarylalcene derivatives, process for their preparation and pharmaceutical compositions containing them | |
| CN112457265A (en) | Tetrazole derivative, preparation method thereof, pharmaceutical composition containing tetrazole derivative and application of pharmaceutical composition | |
| US5234948A (en) | Optically active alkylenedioxybenzene derivatives and their use in therapy | |
| MXPA00011523A (en) | N-benzodioxanylmethyl-1-piperidyl-methylamine compounds having affinity for 5-ht receptors | |
| JPH02221274A (en) | New derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001940264 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10240180 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001940264 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001940264 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |