WO2001079154A2 - Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone - Google Patents
Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone Download PDFInfo
- Publication number
- WO2001079154A2 WO2001079154A2 PCT/EP2001/004348 EP0104348W WO0179154A2 WO 2001079154 A2 WO2001079154 A2 WO 2001079154A2 EP 0104348 W EP0104348 W EP 0104348W WO 0179154 A2 WO0179154 A2 WO 0179154A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dimethoxyphenyl
- azidoethanone
- hci
- compound
- synthesis
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- OHBQPCCCRFSCAX-UHFFFAOYSA-N COc(cc1)ccc1OC Chemical compound COc(cc1)ccc1OC OHBQPCCCRFSCAX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C231/00—Preparation of carboxylic acid amides
- C07C231/14—Preparation of carboxylic acid amides by formation of carboxamide groups together with reactions not involving the carboxamide groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/08—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C247/00—Compounds containing azido groups
- C07C247/02—Compounds containing azido groups with azido groups bound to acyclic carbon atoms of a carbon skeleton
- C07C247/08—Compounds containing azido groups with azido groups bound to acyclic carbon atoms of a carbon skeleton being unsaturated
- C07C247/10—Compounds containing azido groups with azido groups bound to acyclic carbon atoms of a carbon skeleton being unsaturated and containing rings
Definitions
- the present invention relates to the synthesis of midodrine hydrochloride, ⁇ 1-(2',5'- dimethoxyphenyl)-2-glycineamido-ethanol-(1)- HCI , from a novel intermediate, 1 -(2',5'- dimethoxyphenyl)-2-azidoethanone.
- the compound midodrine is part of the class of compounds known as phenylalkanolamine derivatives which have been found to be effective in treating hypertensive conditions due to their long lasting blood pressure increasing effect.
- the objective of the present disclosure is to provide novel synthetic strategies to prepare midodrine HCI from a novel intermediate, 1-(2',5'-dimethoxyphenyl)-2-azidoethanone.
- the synthetic routes of the present invention provide increased yields and minimized by-products, which therefore also minimizes expenses.
- the present invention relates to the synthesis of midodrine hydrochloride, + 1-(2',5'- dimethoxyphenyl)-2-glycineamido-ethanol-(1)- HCI .
- the synthetic route comprises reduction of the intermediate, 1-(2',5'-dimethoxyphenyl)-2-azidoethanone (Compound II) to produce the compound ⁇ 1-(2',5'-dimethoxyphenyl)-2-glycineamido-ethanol-(1)- HCI (Compound I).
- the novel intermediate, 1 -(2',5'-dimethoxyphenyl)-2-azidoethanone (Compound II) is prepared by acylating the compound 1 ,4-dimethoxybenzene (Compound III) by a Friedel-Crafts reaction with haloacetylchloride or haloaceticanhydride and anhydrous aluminum chloride in the presence of a chlorinated organic solvent.
- Halo includes chloro, bromo, and the like.
- Examples of haloacetylchlorides include chloroacetylchloride and bromoacetylchlorides.
- haloacetic anhydrides include chloro-acetic anhydride and bromo-acetic anhdyride.
- chlorinated organic solvents are methylenechloride and dichloroethane. Carbon disulfide may also be used as a solvent.
- the product, 1-(2',5'-dimethoxyphenyl)-2-haloethanone (Compound IV) is obtained in pure form; only one isomer is obtained as all the four positions of benzene nucleus are equivalent. This step is shown schematically as follows, with X representing halo, preferably chloro: Step l
- the intermediate, Compound II is prepared by introducing an azide group to replace halo from the ⁇ -position in Compound IV.
- the halogen containing carbon is highly activated by the presence of the adjacent carbonyl group and is therefore easily replaced.
- This reaction is carried out in nitrogen purged 60% acetone-water mixture. This step is shown schematically as follows:
- This novel ketoazide compound 1-(2',5'-dimethoxyphenyl)-2-azidoethanone (Compound II) is used as an intermediate for the synthesis of 1-(2',5'-dimethoxyphenyl)-2-glycineamido- ethanol-(1) HCI (Compound I).
- the reduction may be by any known method using known reducing agents including reduction by lithium aluminum hydride (LiAIH 4 ) in tetrahydrofuran, or reduction by sodium borohydride in tetrahydrofuran or a mixture of tetrahydrofuran and methanol, or by hydrogenation in the presence of Pd/C (10%) with methanol, ethanol or a mixture of organic solvents.
- LiAIH 4 lithium aluminum hydride
- sodium borohydride in tetrahydrofuran or a mixture of tetrahydrofuran and methanol
- Pd/C platinum/C
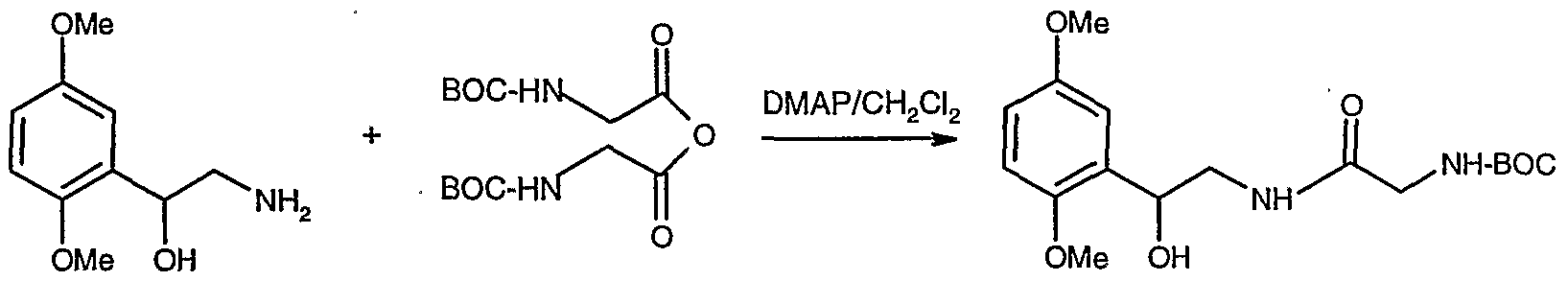
- the aminoethanol, Compound V, produced in the above step may be reacted with N-carbo-t-butoxyglycine-amido (N-BOC-GIycine) in the presence of dicyclohexylcarbodiimide (DCC) to form an amide bond.
- N-BOC-GIycine N-carbo-t-butoxyglycine-amido
- DCC dicyclohexylcarbodiimide
- the yield is poor due to the generation of by-products.
- one of the by-products, dicyclohexylurea, DCU becomes trapped and remains in the product.
- RT signifies ambient temperature
- the 1 -(2',5'-dimethoxyphenyl)-2-(N-carbo-t-butoxyglycine-amido)-ethanol-(1 ) compound, Compound VII, produced in Step 5 is subjected to any polar protic or aprotic solvent to yield the desired ⁇ 1-(2',5'-dimethoxyphenyl)-2-gIycineamido-ethanol-(1)-hydrochloride, Compound I.
- polar protic or aprotic solvents include acetone/aq. HCI or HCI gas/MeOH or 3M HCI-EtOAc.
- the solution is stirred for 4 hours after addition at room temperature and then poured in to a mixture of crushed ice and 126 mL cone. HCI and stirred for 20 minutes again.
- the organic phase is separated and the aqueous phase is extracted with CH 2 CI 2 (100 mL x 3). Total dichloromethane fractions are combined and washed with H 2 0 (100 mL x 2), cold 10% aq. NaOH (200 mL x 1) and H 2 0 (100 mL x 2) and dried over anhydrous sodium sulfate.
- the organic phase is evaporated under reduced pressure and the thick solution is cooled to room temperature yielding a yellow solid (59.0 g, yield 64%).
- the crude product is crystallized from methanol (m.p. 89-91 °C).
- Example 1 A The procedure described in Example 1 A is repeated utilizing chloroacetic anhydride under refluxing conditions instead of chloroacetylchloride.
- the compound 1-(2',5'- dimethoxyphenyl)-2-chloroethanone is obtained as a light grey solid with the same yield.
- the following physical properties are measured: m.p. 89-91 °C (dec) MS (EI-MS): m/z 214 (M + ) 1 H-NMR (CDCI 3 ): ⁇ (ppm):
- the aqueous part is extracted with dichloromethane (30 mL 3) and washed with 5% cold aqueous NaOH solution (20 mLx 1) and saturated brine water (10 mLx 2).
- the aqueous part is dried over anhydrous sodium sulfate, and the solution is treated with 500 mg activated carbon.
- the mixture is warmed to 30°C with a water bath under nitrogen atmosphere and stirred for 20 minutes.
- the solution is filtered and concentrated under reduced pressure.
- the thick yellow colored solution is cooled to room temperature to yield a light yellow color solid which is dried in the vacuum desiccator over P 2 0 5 (5.0 g, yield 82%).
- the solid is crystallized from methanol.
- the following physical properties are measured: m.p. 92-93°C (dec) MS (EI-MS): m/z 221 (M + ) 1 H-NMR (CDCI 3 ): ⁇ (ppm):
- the filtrate is heated to reduce the acetone content and cooled to room temperature to obtain the second crop of solid (0.150 g).
- Total weight of the solid collected is 1.012 g.
- HPLC is used to find out the percentage of the drug substance in the reaction mixture (filtrate).
- Total crude yield is between 80-85% after collection of all crops.
- the solid is crystallized from methanol (m.p. 190-191 °C).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
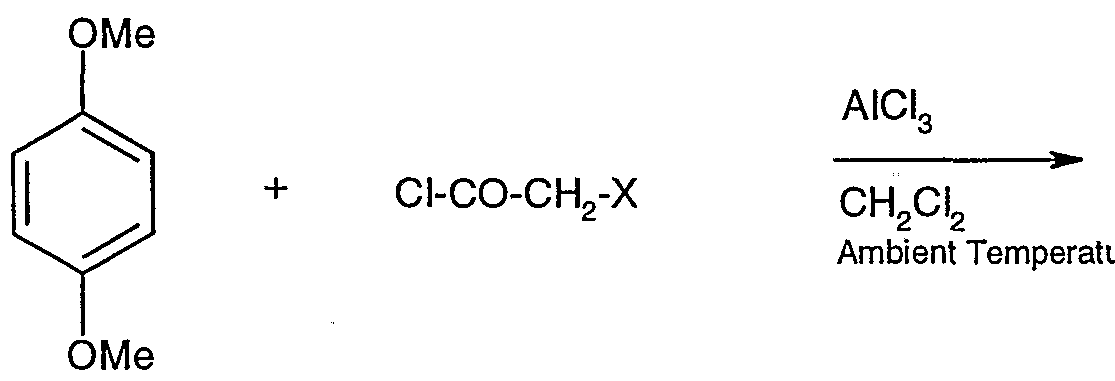






Claims
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01931614A EP1276711B1 (en) | 2000-04-17 | 2001-04-17 | Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone |
| AU2001258344A AU2001258344A1 (en) | 2000-04-17 | 2001-04-17 | Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone |
| DE60101053T DE60101053T2 (en) | 2000-04-17 | 2001-04-17 | SYNTHESIS OF MIDODRIN HYDROCHLORIDE FROM INTERMEDIATE 1- (2'.5'-DIMETHOXYPHENYL) -2-AZIDOETHANONE |
| AT01931614T ATE252545T1 (en) | 2000-04-17 | 2001-04-17 | SYNTHESIS OF MIDODRINE HYDROCHLORIDE FROM THE INTERMEDIATE 1-(2'.5'-DIMETHOXYPHENYL)-2-AZIDOETHANONE |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/550,417 US6201153B1 (en) | 2000-04-17 | 2000-04-17 | Synthesis of midodrine HCI from a novel intermediate 1-(2′,5′-dimethoxyphenyl)-2-azidoethanone |
| US09/550,417 | 2000-04-17 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001079154A2 true WO2001079154A2 (en) | 2001-10-25 |
| WO2001079154A3 WO2001079154A3 (en) | 2002-03-21 |
Family
ID=24197102
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2001/004348 Ceased WO2001079154A2 (en) | 2000-04-17 | 2001-04-17 | Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US6201153B1 (en) |
| EP (1) | EP1276711B1 (en) |
| AT (1) | ATE252545T1 (en) |
| AU (1) | AU2001258344A1 (en) |
| DE (1) | DE60101053T2 (en) |
| ES (1) | ES2210160T3 (en) |
| WO (1) | WO2001079154A2 (en) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2002330731A1 (en) * | 2002-08-21 | 2004-03-11 | Sanmar Speciality Chemicals Ltd | An improved process for the synthesis of (plus or minus) 2-amino-n-(2,(s,5-dimethoxyphenyl)-2-hydroxyethyl) acetamide monohydrochloride |
| CA2421550C (en) * | 2003-03-11 | 2008-06-03 | Brantford Chemicals Inc. | Process for the preparation of midodrine, pharmaceutically-acceptable salts thereof and intermediates |
| CN100434415C (en) * | 2004-11-15 | 2008-11-19 | 天津药物研究院 | Preparation method of midodrine hydrochloride intermediate 2-amino-1-(2.5-dimethoxybenzene)-ethanol |
| KR101356471B1 (en) * | 2011-12-09 | 2014-01-29 | 고려대학교 산학협력단 | Compound for prevention and treatment of hypertension |
| CN115745812A (en) * | 2022-11-11 | 2023-03-07 | 成都沣德煜晟医药科技有限公司 | Preparation method of 2-amino-1- (2,5-dimethoxyphenyl) ethanol |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AT241435B (en) | 1963-06-11 | 1965-07-26 | Chemie Linz Ag | Process for the production of new phenylalkanolamine derivatives and their salts |
| DE2506110C2 (en) * | 1975-02-13 | 1983-04-21 | Lentia GmbH Chem. u. pharm. Erzeugnisse - Industriebedarf, 8000 München | Process for the preparation of 2-amino-N - (β-hydroxy-2,5-dimethoxy-phenethyl) -acetamide (midodrin) |
| JPH07285921A (en) * | 1994-04-15 | 1995-10-31 | Sanyo Kagaku Kenkyusho:Kk | Method for producing 2-amino-N- (β-hydroxyphenethyl) acetamide derivative |
-
2000
- 2000-04-17 US US09/550,417 patent/US6201153B1/en not_active Expired - Lifetime
-
2001
- 2001-04-17 WO PCT/EP2001/004348 patent/WO2001079154A2/en not_active Ceased
- 2001-04-17 AU AU2001258344A patent/AU2001258344A1/en not_active Abandoned
- 2001-04-17 EP EP01931614A patent/EP1276711B1/en not_active Expired - Lifetime
- 2001-04-17 DE DE60101053T patent/DE60101053T2/en not_active Expired - Lifetime
- 2001-04-17 AT AT01931614T patent/ATE252545T1/en active
- 2001-04-17 ES ES01931614T patent/ES2210160T3/en not_active Expired - Lifetime
Also Published As
| Publication number | Publication date |
|---|---|
| ATE252545T1 (en) | 2003-11-15 |
| EP1276711A2 (en) | 2003-01-22 |
| EP1276711B1 (en) | 2003-10-22 |
| DE60101053T2 (en) | 2004-08-19 |
| WO2001079154A3 (en) | 2002-03-21 |
| US6201153B1 (en) | 2001-03-13 |
| ES2210160T3 (en) | 2004-07-01 |
| DE60101053D1 (en) | 2003-11-27 |
| AU2001258344A1 (en) | 2001-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPS63258449A (en) | Novel compound having collagenase inhibiting activity, its production and pharmacological composition containing said compound | |
| AU645935B2 (en) | Production of fluoxetine and new intermediates | |
| CA2455706C (en) | Novel method for synthesis of (2s, 3as, 7as)-1-[(s)-alanyl]-octahydro-1h-indole-2-carboxylic acid derivatives and use for synthesis of perindopril | |
| WO1990012007A1 (en) | New sulfonamides derived from benzocyclic or benzoheterocyclic acids, their preparation and application in thereapeutics | |
| JP2818763B2 (en) | O-alkylated compounds of N- (hydroxy) aralkylphenylethanolamines | |
| JPH07304770A (en) | New benzazepinone derivative | |
| KR920003837B1 (en) | Preparation method of adamantaneamine derivatives | |
| EP1276711B1 (en) | Synthesis of midodrine hci from a novel intermediate 1-(2',5'-dimethoxyphenyl)-2-azidoethanone | |
| JPH11310556A (en) | Novel process for producing 2-amino-2- [2- (4-octylphenyl) ethyl] propane-1,3-diol | |
| KR20040108717A (en) | Method for preparing combretastatins | |
| Murakata et al. | Studies on synthesis of araplysillins via oxidative cyclisation of o-phenolic oxime-acid derivatives using phenyliodonium diacetate | |
| JPH0140823B2 (en) | ||
| CA1338121C (en) | Anticonvulsant and neuroprotective 2-aminoacetamide derivatives | |
| CN111978281A (en) | Method for preparing cyclohexanone/chromene pyrone compound and application of cyclohexanone/chromene pyrone compound | |
| JP2788118B2 (en) | Novel synthesis of tertiary alkyl esters | |
| EP1627881B1 (en) | Process for the preparation of topiramate | |
| WO2000055145A1 (en) | 1,3,4-oxadiazole derivatives and process for producing the same | |
| EP1513868B1 (en) | Process for the production of lisinopril | |
| JP2938193B2 (en) | Novel method for producing nojirimycin and related compounds | |
| CA2236117C (en) | Process for producing optically active cyanohydrins | |
| WO1997017330A1 (en) | Process | |
| JP3110132B2 (en) | Novel β-alanine derivative, method for producing the same, and method for producing glutamate blocker | |
| US5719298A (en) | Methods of producing pantothenic acid derivative and its starting materials for producing the same | |
| JP2745649B2 (en) | Retrohydroxamic acid deferiferioxamine derivatives containing lysine residues | |
| KR100235120B1 (en) | Novel amino acid amide derivatives and process for preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001931614 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001931614 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2001931614 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |