WO2001098270A2 - N-ureidoalkyl-piperidines as modulators of chemokine receptor activity - Google Patents
N-ureidoalkyl-piperidines as modulators of chemokine receptor activity Download PDFInfo
- Publication number
- WO2001098270A2 WO2001098270A2 PCT/US2001/019752 US0119752W WO0198270A2 WO 2001098270 A2 WO2001098270 A2 WO 2001098270A2 US 0119752 W US0119752 W US 0119752W WO 0198270 A2 WO0198270 A2 WO 0198270A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- substituted
- occurrence
- cycloalkyl
- chr
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 **C1=C2OC(*)(*)c3ccc[n]3C2=*=*C=C1 Chemical compound **C1=C2OC(*)(*)c3ccc[n]3C2=*=*C=C1 0.000 description 39
- VSDWXIVREUGXLT-UHFFFAOYSA-N C=CCC1=CC=C(CC2CCN(CC(C(Cc3ccccc3)NC(Nc3cccc(C#C)c3)=O)O)CC2)CC1 Chemical compound C=CCC1=CC=C(CC2CCN(CC(C(Cc3ccccc3)NC(Nc3cccc(C#C)c3)=O)O)CC2)CC1 VSDWXIVREUGXLT-UHFFFAOYSA-N 0.000 description 1
- RIFXIGDBUBXKEI-UHFFFAOYSA-N CC(C)(C)OC(N(CCC1)CC1=O)=O Chemical compound CC(C)(C)OC(N(CCC1)CC1=O)=O RIFXIGDBUBXKEI-UHFFFAOYSA-N 0.000 description 1
- YJTHFKMWAUAXDX-UHFFFAOYSA-N CC(C)(C)OC(N1CCC(Cc2ccccc2)CC1)=O Chemical compound CC(C)(C)OC(N1CCC(Cc2ccccc2)CC1)=O YJTHFKMWAUAXDX-UHFFFAOYSA-N 0.000 description 1
- MXNDNEMGIMDRSU-INIZCTEOSA-N CC(C)(C)OC(NC1(CC1)C(N1C[C@H](Cc(cc2)ccc2F)CCC1)=O)=O Chemical compound CC(C)(C)OC(NC1(CC1)C(N1C[C@H](Cc(cc2)ccc2F)CCC1)=O)=O MXNDNEMGIMDRSU-INIZCTEOSA-N 0.000 description 1
- YMHNHVRBSISYNT-UHFFFAOYSA-N NC(Cc1ccccc1)C(CN1CCC(CC(CC2)=CC=C2F)CC1)O Chemical compound NC(Cc1ccccc1)C(CN1CCC(CC(CC2)=CC=C2F)CC1)O YMHNHVRBSISYNT-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/06—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with radicals, containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/52—Oxygen atoms attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention relates generally to modulators of chemokine receptor activity, pharmaceutical compositions containing the same, and methods of using the same as agents for treatment and prevention of inflammatory diseases such as asthma and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis. •
- Chemokines are chemotactic cytokines, of molecular weight 6-15 kDa, that are released by a wide variety of cells to attract and activate, among other cell types, macrophages, T and B lymphocytes, eosinophils, basophils and neutrophils (reviewed in Luster, New Eng. J Med., 338, 436-445 (1998) and Rollins, Blood, 90, 909-928
- CXC interleukin-8
- NAP-2 neutrophil-activating protein-2
- MGSA melanoma growth stimulatory activity protein
- lymphotactin-1 lymphotactin-1
- lymphotactin-2 both C chemokines
- fractalkine a CXXXC chemokine
- chemokines bind to specific cell-surface receptors belonging to the family of G-protein-coupled seven-transmembrane-domain proteins (reviewed in Horuk, Trends Pharm. Sci., 15, 159-165 (1994)) which are termed "chemokine receptors.”
- chemokine receptors On binding their cognate ligands, chemokine receptors transduce an intracellular signal through the associated trimeric G proteins, resulting in, among other responses, a rapid increase in intracellular calcium concentration, changes in cell shape, increased expression of cellular adhesion molecules, degranulation, and promotion of cell migration.
- CCR-1 or "CKR-1” or "CC-CKR-1" [MlP-l , MCP-3, MCP-4, RANTES] (Ben-Barruch, et al., Cell, 72, 415-425 (1993), Luster, New Eng. J. Med., 338, 436-445 (1998)); CCR-2A and CCR-2B (or "CKR- 2A”/"CKR-2B” or “CC-CKR-2A” / "CC-CKR-2B” ) [MCP-1, MCP-2, MCP-3, MCP-4, MCP-5] (Charo et al .
- CCR-4 or "CKR-4" or "CC-CKR-4" [TARC, MlP-l ⁇ , RANTES, MCP-1] (Power et al . , J. Biol. Chem., 270, 19495-19500 (1995), Luster, New Eng. J. Med., 338, 436-445 (1998)); CCR-5 (or "CKR-5" OR “CC-CKR-5”) [MIP-lCC, RANTES, MIP- I ⁇ ] (Sanson, et al .
- CCR-6 or "CKR-6” or "CC-CKR-6" [LARC] (Baba et al., J. Biol. Chem., 272, 14893-14898 (1997)); CCR-7 (or “CKR-7” or “CC-CKR-7”) [ELC] (Yoshie et al . , J. Leukoc. Biol. 62, 634-644 (1997)); CCR-8 (or “CKR-8” or "CC-CKR-8”) [1-309, TARC, MlP-l ⁇ ] (Napolitano et al . , J.
- mammalian cytomegaloviruses In addition to the mammalian chemokine receptors, mammalian cytomegaloviruses, herpesviruses and poxviruses have been shown to express, in infected cells, proteins with the binding properties of chemokine receptors (reviewed by Wells and Schwartz, Curr. Opin.
- Human CC chemokines such as RANTES and MCP-3, can cause rapid mobilization of calcium via these virally encoded receptors. Receptor expression may be permissive for infection by allowing for the subversion of normal immune system surveillance and response to infection. Additionally, human chemokine receptors, such as CXCR4, CCR2 , CCR3 , CCR5 and CCR8, can act as co-receptors for the infection of mammalian cells by microbes as with, for example, the human immunodeficiency viruses (HIV) .
- HIV human immunodeficiency viruses
- Chemokine receptors have been implicated as being important mediators of inflammatory, infectious, and immunoregulatory disorders and diseases, including asthma and allergic diseases, as well as autoimmune pathologies such as* rheumatoid arthritis and atherosclerosis.
- the chemokine receptor CCR-3 plays a pivotal role in attracting eosinophils to sites of allergic inflammation and in subsequently activating these cells .
- the chemokine ligands for CCR-3 induce a rapid increase in intracellular calcium concentration, increased expression of cellular adhesion molecules, cellular degranulation, and the promotion of eosinophil migration. Accordingly, agents which modulate chemokine receptors would be useful in such disorders and diseases. In addition, agents which modulate chemokine receptors would also be useful in infectious diseases such as by blocking infection of CCR3 expressing cells by HIV or in preventing the manipulation of immune cellular responses by viruses such as cytomegaloviruses .
- Ri is C ⁇ Q alkyl, optionally substituted with functional groups such as -NR 6 CONHR 7 , wherein R 6 and R 7 may be phenyl further substituted with hydroxy, alkyl, cyano, halo and haloalkyl.
- WO 95/13069 is directed to certain piperidine, pyrrolidine, and hexahydro-lH-azepine compounds of general formula :
- Compounds of this type are claimed to promote the release of growth hormone in humans and animals .
- WO 93/06108 discloses pyrrolobenzoxazine derivatives as 5-hydroxytrypta ine (5-HT) agonists and antagonists :
- A is lower alkylene and R 4 may be phenyl optionally substituted with halogen.
- NPY Neuropeptide Y
- U.S. Pat. No. 5,668,151 discloses Neuropeptide Y (NPY) antagonists comprising 1, 4-dihydropyridines with a piperidinyl or tetrahydropyridinyl-containing moiety attached to the 3-position of the 4-phenyl ring:
- B may be NH, NR 1 , 0, or a bond
- R 7 may be substituted phenyl, benzyl, phenethyl and the like.
- one object of the present invention is to provide novel agonists or antagonists of CCR-3, or pharmaceutically acceptable salts or prodrugs thereof. It is another object of the present invention to provide pharmaceutical compositions comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of at least one of the compounds of the present invention or a pharmaceutically acceptable salt or prodrug form thereof.
- the present invention provides novel N-ureidoalkyl-piperidines for use in therapy.
- the present invention provides the use of novel N-ureidoalkyl-piperidines for the manufacture of a medicament for the treatment of allergic disorders.
- E, Z, M, J, K, L, Q, R 1 , R 2 , R 3 , and R 4 are effective modulators of chemokine activity.
- the present invention provides novel compounds of formula (I) :
- M is absent or selected from CH2, CHR 5 , CHR 13 , CR 13 R 13 , and CR 5 R 13 ;
- Q is selected from CH2, CHR 5 , CHR 13 , CR 13 R 13 , and
- J, K and L are independently selected from CH2 , CHR 5 , CHR 6 , CR 6 R 6 and CR 5 R 6 ;
- J is selected from CH2, CHR 5 , CHR 13 , and CR 5 R 13 ;
- Z is selected from 0, S, NR l , CHCN, CHNO2 , and C(CN) 2 ;
- R la is selected from H, Ci-6 alkyl, C3--6 cycloalkyl, C0NR lb R lb , 0R lb , CN, NO2 , and (CH2) phenyl;
- R lb is independently selected from H, C ⁇ _3 alkyl, C3--5 cycloalkyl, and phenyl;
- E is selected from:
- ring A is a C3-6 carbocyclic residue, provided that the C3-6 carbocyclic residue in Ring A is not phenyl;
- R 1 and R-2 are independently selected from H, Ci-6 alkyl, C3-8 alkenyl, and C3-8 alkynyl;
- R 3 is selected from a Ci-io alkyl substituted with 0-5 R 3 9, C3-10 alkenyl substituted with 0-5 R 3 9, and C3-10 alkynyl substituted with 0-5 R 9 ;
- R 9, at each occurrence, is independently selected from Cl, Br, I, F, 02 , CN, NR 3a R 3a P OH, 0(CHR') r R 3d ,
- R 3 is selected from C - ⁇ alkyl, C3-8 alkenyl, C3--8 alkynyl, a (CH2)r _c 3-6 carbocyclic residue substituted with 0-3 R 3e , and (CH2) _5- 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, O, and S, substituted with 0-2 R 3e ;
- R 3d is selected from C3-8 alkenyl, C3-8 alkynyl, methyl, CF 3 , C 2 -6 alkyl substituted with 0-3 R e , a (CH2)r _ 3-io carbocyclic residue substituted with 0-3 R 3e , and a (CH2)r 5_ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, O, and S, substituted with 0-3 R 3e.
- R e at each occurrence, is selected from C ⁇ -6 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)r 3-6 cycloalkyl, Cl, F, Br, I, CN, N02 , (CF2) r CF3, (CH2) r OCi_5 alkyl, OH, SH, (CH2) rS (0) p Ci-5 alkyl,
- R 3 f is selected from H, Ci-6 alkyl, C3-6 cycloalkyl, and phenyl;
- R 4 is absent, taken with the nitrogen to which it is attached to form an N-oxide, or selected from Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, (CH2) r C3-6 cycloalkyl, (CH2) qC (O) R 4b , (CH2) qC (0)NR a R a P (CH2)qC(0)OR 4b , and a (CH2)r" 3-10 carbocyclic residue substituted with 0-3 R 4c ;
- R 4a and R 4a ' are selected from H, C ⁇ _6 alkyl, (CH2)r 3-6 cycloalkyl, and phenyl;
- R 4b at each occurrence, is selected from Ci-6 alkyl
- R 4c at each occurrence, is selected from Ci-6 alkyl
- R 4 joins with R "7 , R9, R 11 , or R 4 to form a 5, 6 or 7 membered piperidinium spirocycle or pyrrolidinium spirocycle substituted with 0-3 R a ;
- R 5 is selected from a (CR 5 'R 5 ") _ 3-io carbocyclic residue substituted with 0-5 R 16 and a (CR 5 'R 5 ")t _ 5-10 membered heterocyclic system containing 1-4 heteroatoms selected from N, O, and S, substituted with 0-3 R 6 ;
- R 5 ' and R 5 ", at each occurrence, are selected from H, Ci-6 alkyl, (CH2)rC3-6 cycloalkyl, and phenyl;
- R 6 at each occurrence, is selected from Ci-6 alkyl
- R 6a and R 6a ' are selected from H, Cl-6 alkyl, C3-6 cycloalkyl, and phenyl substituted with
- R 6b at each occurrence, is selected from Ci-6 alkyl, C3-6 cycloalkyl, and phenyl substituted with 0-3 R 6c ;
- R 6c is selected from C ⁇ s alkyl, C3-6 cycloalkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, (CH2) r OCi-5 alkyl, (CH2) r OH, (CH2) r SC ⁇ _5 alkyl, and
- R 6 at each occurrence, is selected from H, Ci-6 alkyl, and C3-6 cycloalkyl; with the proviso that when any of J, K or L is CR 6 R6 and
- R 6 is bonded to the carbon to which it is attached through a heteroatom, the other R 6 is not bonded to the carbon to which it is attached through a heteroatom;
- R 7 is selected from H, C ⁇ _6 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH )qOH, (CH )qSH, (CH 2 )qOR 7d , (CH )qSR 7d , (CH2)qNR 7a R 7a P (CH2 ) r C (O) OH, (CH2) r C(0)R 7b , (CH2) r C (0)NR 7a R 7 P
- R 7a and R 7a ' are selected from H, Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, (CH2) r C3-6 cycloalkyl, a (CH2)r ⁇ 3-io carbocyclic residue substituted with 0-5 R 7e , and a (CH2) r - 5 - 0 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 7e ;
- R 7a and R 7a ' along with the N to which they are attached, are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 7 ⁇ , O, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 7b is selected from Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2) r -C3-6 carbocyclic residue substituted with 0-2 R e , and a (CH2)r -5 ⁇ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 7e ;
- R 7c is selected from Ci-6 alkyl
- R 7 at each occurrence, is selected from methyl, CF 3 ,
- R 7e is selected from Ci-6 alkyl
- R 7 ⁇ is selected from H, Ci- ⁇ alkyl, and C3-5 cycloalkyl;
- R 7 9 is selected from H, C ⁇ -6 alkyl, C3-6 cycloalkyl, (CH2)rPhenyl, C(0)R 7f , C(0)OR 7f , and S ⁇ 2R 7f ;
- R8 is selected from H, Ci-6 alkyl, C3-6 cycloalkyl, and (CH2) tPhenyl substituted with 0-3 R 8a ;
- R 8a at each occurrence, is selected from C ⁇ _6 alkyl
- R 8b is selected from H, Ci-g alkyl, C3-6 cycloalkyl, OH, CN, and (CH2) r -phenyl;
- R9 is selected from H, Ci- ⁇ alkyl, C2-8 alkenyl, C2-8 alkynyl, F, Cl, Br, I, NO2, CN, (CH2) r OH, (CH2) r SH,
- R 9a and R 9a ' are selected from H, Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2)r _
- R 9a and R a ' are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 9 ⁇ , 0, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 9b is selected from Ci-6 alkyl, C3-8 alkenyl, C3--8 alkynyl, a (CH2) r ⁇ C3-6 carbocyclic residue substituted with 0-2 R 9e , and a (CH2)r _5_ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 9e ;
- R 9c is selected from Ci-6 alkyl
- R 9d is selected from Ci-6 alkyl, C3-6 alkenyl, C3-6 alkynyl, a C3-10 carbocyclic residue substituted with 0-3 R 9c , and a 5-6 membered heterocyclic system containing 1-4 heteroatoms selected from the group consisting of N, O, and S substituted with 0-3 R 9c ;
- R e at each occurrence, is selected from C - ⁇ alkyl
- R 9 f at each occurrence, is selected from H, Ci-6 alkyl, and C3_6 cycloalkyl;
- R 9 9 is selected from H, C ⁇ _6 alkyl, C3--6 cycloalkyl, (CH2)rPhenyl, C(0)R 9f , C(0)OR 9f , and S ⁇ 2R 9f ;
- R 10 is selected from H, Ci-6 alkyl, C2-8 alkenyl, C2-8 alkynyl, F, Cl, Br, I, NO2 , CN, (CH2) r 0H, (CH2) r OR 10d , (CH2) r SR 10d , (CH2) r NR 10a R 10a ' , (CH2) r C(0)OH, (CH2) rC (0) R 10b , (CH2) r C (O)NR 10a R 10a P (CH2) r NR 10a C(O)R 10a , (CH2) r NR 10a C(O)H, (CH2 ) r C (0) OR 10b , (CH2 ) r OC (0) R 10 , (CH2) r OC(O)NR 10a R 10a P (CH2) r NR 10a C (0) OR 10b , (CH 2 ) r S(O)pR 10b , (CH2)rS(O) 2 ,
- RlOa and R- ⁇ -O ' are selected from H, Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2)r- C3-10 carbocyclic residue substituted with 0-5
- R 1 ⁇ e and a (CH2) r -5-1 0 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 10e ;
- R 1 ⁇ 3 - and R ( ⁇ a ' , along with the N to which they are attached, are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 10 9, 0, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle,-
- R 1 ⁇ is selected from Ci-6 alkyl
- R 1 ⁇ d / at each occurrence is selected from C ⁇ _6 alkyl, C3-6 alkenyl, C3-6 alkynyl, a C3-10 carbocyclic residue substituted with 0-3 R 1 ⁇ 0 , and a 5-6 membered heterocyclic system containing 1-4 heteroatoms selected from the group consisting of N, 0, and S substituted with 0-3 R 10c ;
- R 1 ⁇ a each occurrence, is selected from C ⁇ _g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)rC3-6 cycloalkyl, Cl, F, Br, I, CN, N02 , (CF2) r CF 3 , (CH ) r OCl-5 alkyl, OH, SH, (CH2) r SC ⁇ _5 alkyl, (CH2) r NR 10f R 10f , and (CH2 ) rphenyl ;
- R- ⁇ -Of . t each occurrence, is selected from H, C ⁇ _5 alkyl, and C3--6 cycloalkyl; RiOg is selected from H, Ci-6 alkyl, C3.-6 cycloalkyl, (CH2)rPhenyl, C(O)R 10f , C(O)OR 10n , and S ⁇ 2R 10h ;
- RIOI I ⁇ a t e ch occurrence is selected from C1-5 alkyl, and C3-6 cycloalkyl;
- R 9 or R 10 when either of R 9 or R 10 is halogen, cyano, nitro, or bonded to the carbon to which it is attached through a heteroatom, the other of R 9 or R 10 is not bonded to the carbon to which it is attached through a heteroatom;
- R 1 is selected from H, Ci-6 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)q0H, (CH2)qSH, (CH2) q 0R lld , (CH2)qSR lld , (CH )qNR ll R lla P (CH 2 ) r C (0) OH, (CH2) r C(0)R ll , (CH2) r C (0)NR l a R ll ' , (CH2)qNR lla C(0)R lla , (CH2 ) qOC (0) NR ll R lla P (CH2)qNR lla C(0)0R llb , (CH2) q NR lla C (0)NHR lla ,
- R-LJ a an ⁇ 5[ plla' # at each occurrence, are selected from H, C ⁇ _g alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2)r _ C3-10 carbocyclic residue substituted with 0-5
- R le and a (CH2) r ⁇ 5-1 0 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R lle ;
- R lla and R l a are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 11 ⁇ , O, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R llb at each occurrence, is selected from Ci-g alkyl, C3--8 alkenyl, C3-8 alkynyl, a (CH2)r ⁇ C3-6 carbocyclic residue substituted with 0-2 R le , and a (CH2) r -5 ""6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R lle ;
- R c is selected from Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2) r C3-6 cycloalkyl, Cl, Br, I, F, (CF2) r CF3, NO2 , CN, (CH2) rNR llf R llf , (CH2) r OH, (CH2) r OCi_4 alkyl, (CH2) r SC ⁇ _4 alkyl,
- R lld is selected from methyl, CF 3 ,
- R lle at each occurrence, is selected from Ci-g alkyl
- R 1 - ⁇ at each occurrence, is selected from H, Ci-g alkyl, and C3- cycloalkyl;
- R 11 -? is selected from H, Ci-g alkyl, C3-g cycloalkyl, (CH2)rPhenyl, C(0)R llf , C(0)OR llh , and S ⁇ 2R llh ;
- R lln at each occurrence, is selected from C ⁇ _5 alkyl, and C3-g cycloalkyl;
- R 12 is selected from H, Ci-g alkyl, (CH2)qOH, (CH2) r C3-6 cycloalkyl, and (CH2)tphenyl substituted with 0-3 Ri2a ;
- R 1 2 / at each occurrence is selected from C ⁇ _g alkyl, C2-8 alkenyl, C2-8 alkynyl, C3-g cycloalkyl, Cl, F, Br, I, CN, N02, (CF 2 ) r CF3, (CH2) r OCl-5 alkyl, OH,
- R 11 and R- ⁇ -2 join to form a C3_ ⁇ o cycloalkyl, a 5-6-membered lactone or lacta , or a 4-6-membered saturated heterocycle containing 1-2 heteroatoms selected from 0, S, and R 11 ⁇ and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 13 is selected from C ⁇ _g alkyl, C2-8 alkenyl, C2-8 alkynyl, C3-g cycloalkyl, (CF ) CF3, (CH2)NR 13a R 13a P (CH 2 ) q OH, (CH ) q OR 13b , (CH2) q SH, (CH2) q SR 13b , (CH ) W C (0) OH, (CH 2 ) C(0)R 13b , (CH )wC(0)NR 13a R 13a P (CH2 ) q NR 13d C (O) R 13a , (CH2 ) C (0) 0R 13b , (CH ) q 0C (0) Rl 3b , (CH ) W S (0) p R 13b , (CH2) w S(0)2NR 13a R 13a P (CH2 ) q NR 13d S (0) 2R 13b , and (CH2)w"Phenyl substituted
- R 13a and R 13a ' are selected from H, Ci-g alkyl, C3_g cycloalkyl, and phenyl substituted with 0-3 R 13c ;
- R 1 b is selected from Ci-g alkyl, C3-6 cycloalkyl, and phenyl substituted with 0-3 R 13c ;
- R 13c is selected from Ci-g alkyl, C3-6 cycloalkyl, Cl, F, Br, I, CN, NO2 , (CF 2 ) r CF3, (CH 2 )rOCi-5 alkyl, (CH ) r OH, (CH ) r SCi-5 alkyl, and
- Rl3d / a t each occurrence is selected from H, C ⁇ _g alkyl, and C3-g cycloalkyl;
- R 14 at each occurrence, is selected from Ci-g alkyl, (CH2) r C3-6 cycloalkyl, Cl, Br, I, F, NO2 , CN,
- R at each occurrence is selected from H, Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)rC3-6 cycloalkyl, and (CH2 ) rPheny1 substituted with R 14e ;
- R 14a and R 14a P at each occurrence are selected from H, Ci-g alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2) ⁇ C3-IO carbocyclic residue substituted with 0-5 R 1 e , and a (CH2)r ⁇ 5 ⁇ 10 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 14e ;
- R 1 at each occurrence, is selected from C ⁇ _g alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2) r -C3-6 carbocyclic residue substituted with 0-3 R 14e , and (CH2)r ⁇ 5- ⁇ membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 1 e ;
- R 14d is selected from C3--8 alkenyl, C3-8 alkynyl, methyl, CF 3 , C2-6 alkyl substituted with 0-3 R 14e , a (CH2)r ⁇ C3-10 carbocyclic residue substituted with 0-3 R 14e , and a (CH2)r 5 ⁇ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 1 e ;
- R 1 e at each occurrence, is selected from Ci-6 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)r 3-6 cycloalkyl, Cl, F, Br, I, CN, NO2, (CF2) r F3, (CH2) r OCl-5 alkyl, OH, SH, (CH2) r SCi-5 alkyl, (CH2) r NR 14f R 14f , and (CH2 ) rPheny1 ;
- R 14 f at each occurrence, is selected from H, Ci-g alkyl, C3- cycloalkyl, and phenyl;
- R 14 joins with R 4 to form a 5, 6 or 7 membered piperidinium spirocycle or pyrrolidinium spirocycle fused to ring A, the spirocycle substituted with 0-3 R a ;
- R a at each occurrence, is selected from Ci-g alkyl
- R at each occurrence, is selected from H, Ci-g alkyl, C3-6 cycloalkyl, and phenyl;
- R c at each occurrence, is selected from Ci-g alkyl, C3. g cycloalkyl, and phenyl;
- R 15 is selected from C -8 alkyl, (CH2) r C3-6 cycloalkyl, Cl, Br, I, F, N ⁇ 2 , CN,
- R 1 a and R 15aJ are selected from H, Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2)r ⁇ C3-10 carbocyclic residue substituted with 0-5
- R 15e and a (CH2)r ⁇ 5_ 10 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 15e ;
- Rl 5a and R 15a ' along with the N to which they are attached, are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 15 ⁇ , O, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- RlE> b a each occurrence, is selected from Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2) r -C3-6 carbocyclic residue substituted with 0-3 R 1 e , and (CH2)r -5- 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 15e ;
- R 15 is selected from C3-8 alkenyl, C3-8 alkynyl, methyl, CF 3 , C2-6 alkyl substituted with 0-3 R 15e , a (CH2)r ⁇ 3-io carbocyclic residue substituted with 0-3 R 15e , and a (CH2)r 5-6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 15e.
- R 15e is selected from Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)rC3-6 cycloalkyl, Cl, F, Br, I, CN, N02 , (CF ) r CF3, (CH 2 ) r OCi-5 alkyl, OH, SH, (CH ) r SCl-5 alkyl, (CH 2 ) r NR 15f R 15f , and (CH2 ) r phenyl ;
- R 15 f is selected from H, Ci-g alkyl, C3- cycloalkyl, and phenyl;
- R 15 5 is selected from H, Ci-g alkyl, C3-g cycloalkyl,
- R 15h at each occurrence is selected from C1-5 alkyl, and C3-6 cycloalkyl;
- R 16 is selected from Ci-8 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)rC3- cycloalkyl,
- R 16a an( * j l6a' ⁇ a each occurrence, are selected from H, Ci-g alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2)r ⁇ C3-I0 carbocyclic residue substituted with 0-5 R 16e a nd a (CH2)r -5- 10 membered heterocyclic system containing 1-4 heteroatoms selected from N,
- Rl6b is selected from Ci-6 alkyl, C3-8 alkenyl, C3-8 alkynyl, a (CH2) r C3-6 carbocyclic residue substituted with 0-3 R 16e , and a (CH2)r ⁇ 5 ⁇ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 16e ;
- R 16d / a each occurrence is selected from C3-8 alkenyl, C3-8 alkynyl, methyl, CF 3 , C2-6 alkyl substituted with 0-3 R 16e , a (CH2)r ⁇ C3-io carbocyclic residue substituted with 0-3 R 16e , and a (CH2)r- 5 ⁇ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-3 R 16e ;
- R 16e is selected from Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2) r C3-6 cycloalkyl, Cl, F, Br, I, CN, 02 , (CF2) r CF3, (CH2) r OCi-5 alkyl, OH, SH, (CH2) r SCi-5 alkyl, (CH2) r NR 16f R 16f , and (CH2 ) Phenyl ;
- R 16 f at each occurrence, is selected from H, C1-5 alkyl, and C3-g cycloalkyl, and phenyl;
- g is selected from 0, 1, 2, 3, and 4;
- t is selected from 1 and 2;
- w is selected from 0 and 1;
- r is selected from 0, 1, 2, 3, 4, and 5;
- q is selected from 1, 2, 3, 4, and 5;
- p is selected from 0, 1, and 2;
- the compounds of Formula (I) do not include the compounds disclosed in U.S. Patent Application No. 09/466,442 filed December 17, 1999.
- the present invention provides novel compounds of formula (I) : Z is selected from 0, S, NCN, NCONH 2 , CHNO2 , and C(CN) ;
- E is selected from:
- R 4 is absent, taken with the nitrogen to which it is attached to form an N-oxide, or selected from Ci-8 alkyl, (CH2)rC3-6 cycloalkyl, and (CH2) r ⁇ Phenyl substituted with 0-3 R 4c ;
- R 4c at each occurrence, is selected from Ci-g alkyl
- R 4 joins with R 7 or R 9 or R-J- 4 to form a 5, 6 or 7 membered piperidinium spirocycle substituted with 0-3 R a ;
- R 1 and R2 are independently selected from H and C1--4 alkyl ;
- R 6 at each occurrence, is selected from C1-4 alkyl
- R 6a and R 6a at each occurrence are selected from H, Cl-6 alkyl, C3- cycloalkyl, and phenyl substituted with
- R 6 at each occurrence, is selected from C ⁇ _g alkyl
- R6 7 at each occurrence is selected from C ⁇ _g alkyl
- R 6d at each occurrence, is selected from H, Ci-g alkyl, and C3_g cycloalkyl;
- R 7 is selected from H, C1-3 alkyl, (CH2)r 3-6 cycloalkyl, (CH2) q 0H, (CH2)qOR 7d , (CH2) q NR 7a R 7a (CH2) r C(0)R 7b , (CH2) r C(0)NR 7a R 7a P
- R 7a and R 7a ' are selected from H, Ci-6 alkyl, (CH2) r C3-6 cycloalkyl, a (CH2) r phenyl substituted with 0-3 R 7e ;
- R 7 at each occurrence, is selected from Ci-g alkyl
- R 7c at each occurrence, is selected from C1-4 alkyl
- R 7d is selected from Ci-g alkyl, (CH2)r c 3-6 cycloalkyl, (CH2)rPhenyl substituted with 0-3 R 7e ;
- R 7e at each occurrence, is selected from Ci-g alkyl
- R f at each occurrence, is selected from H, C1-5 alkyl, and C3_ cycloalkyl;
- R 11 is selected from H, Ci-g alkyl, (CH2)rC3-6 cycloalkyl, (CH2) q 0H, (CH2) q 0R lld ,
- lla a d Rl la ' at each occurrence, are selected from H, Ci-g alkyl, (CH2)r c 3-6 cycloalkyl, a (CH2)rPhenyl substituted with 0-3 R lle ;
- R lla and R lla - 7 are joined to form a 5-6 membered heterocyclic system containing 1-2 heteroatoms selected from NR 11 ⁇ , 0, and S and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R llb is selected from Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)rC3-6 cycloalkyl, (CH2)rPhenyl substituted with 0-3 R lle ;
- R llc is selected from C1-4 alkyl, C2-8 alkenyl, C2-8 alkynyl, (CH2)r c 3-6 cycloalkyl, Cl, Br, I, F, (CF2) r CF3, N02, CN, (CH2)r-NR llf R llf , (CH2) r OH, (CH ) r OCi-4 alkyl, (CH2) r C (0)R llb , (CH2) r C(0)NR llf R llf , (CH2) r-NR llf C (0) R lla , (CH2)rS(0) p R llb , (CH 2 ) r S
- R lld is selected from Ci-g alkyl, (CH2)rC3-6 cycloalkyl, (CH2)rPhenyl substituted with 0-3 R lle ;
- R lle at each occurrence, is selected from Ci-g alkyl, C2-8 alkenyl, C2-8 alkynyl, C3-g cycloalkyl, Cl, F, Br, I, CN, N02, (CF ) r CF3, (CH2) r OC ⁇ _5 alkyl, OH,
- R-Hf at each occurrence, is selected from H, C1-5 alkyl and C3-g cycloalkyl;
- RH-9 " is selected from H, Ci-g alkyl, C3-g cycloalkyl, (CH2)rPhenyl, C(0)R llf , C(0)0R llf , and S02R llf ;
- R 12 is H
- RH and R 12 join to form a C3-10 cycloalkyl, a 5-6-membered lactone or lactarn, or a 4-6-membered saturated heterocycle containing 1-2 heteroatoms selected from 0, S, and NR 11 ⁇ and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 13 is selected from Ci-4 alkyl, C3-6 cycloalkyl, (CH2)NR 13a R 13a (CH2)0H, (CH 2 )0R 13b , (CH 2 ) C(0)R 13b , (CH 2 )wC(0)NR 13a R 13a P (CH2)NR 13d C(0)R 13a , (CH2)wS(0)2NR 13a R 13a ' , (CH2)NR 13d S(0)2R 13b , and (CH2) "Phenyl substituted with 0-3 R 13c ;
- Rl3a and R 13a ' are selected from H, Ci-g alkyl, C3-g cycloalkyl, and phenyl substituted with 0-3 R 13c ;
- R 13b at each occurrence, is selected from Ci-g alkyl
- R 1 c is selected from Ci-g alkyl, C3-6 cycloalkyl, Cl, F, Br, I, CN, NO2 , (CF2)rCF3, (CH2) r OCi-5 alkyl, (CH2) r OH, and (CH2 ) r NR 13d R 13d ;
- R 1 d at each occurrence, is selected from H, Ci-6 alkyl, and C3-g cycloalkyl; q is selected from 1, 2, and 3; and
- r is selected from 0, 1, 2, and 3.
- ring A is selected from:
- R 5 is selected from (CR ⁇ ) t ⁇ phenyl substituted with 0-5
- R 16 and a (CR 5 ⁇ ) t-heterocyclic system substituted with 0-3 R!6, wherein the heterocyclic system is selected from pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiophenyl, benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, indolinyl, isoindolyl, isothiadiazolyl, isoxazolyl, piperidinyl, pyrrazolyl, 1, 2 , 4-triazolyl, 1, 2 , 3-triazolyl, tetrazolyl, thiadiazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyrimidinyl .
- the heterocyclic system is selected from pyridin
- the present invention provides novel compounds of formula (I-i) :
- R 16 at each occurrence, is selected from Ci-8 alkyl, (CH2) r C3-6 cycloalkyl, CF3 , Cl, Br, I, F,
- R 16a and R 16a ' are selected from H, Ci-6 alkyl, C3-6 cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 16e ;
- Rl6b at each occurrence, is selected from H, Ci-6 alkyl, C3-g cycloalkyl, and (CH2 ) rPhenyl substituted with 0-3 R 16e ;
- Rl6d / at each occurrence is selected from Ci-g alkyl and phenyl;
- R 16e is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, OH, and (CH2) r OC ⁇ _5 alkyl; and
- R 16 f is selected from H, and C1-5 alkyl .
- the present invention provides novel compounds of formula (I-ii) :
- R 16 is selected from Ci-8 alkyl, (CH2)rC3-6 cycloalkyl, CF3 , Cl, Br, I, F, (CH2) r NR 16a R 16a P N02, CN, OH, (CH2) r OR 16d , (CH2 ) r C (0) R 16b , (CH2 ) r (O) NR 16a R 16a ' , (CH2 ) r NR 16f C (0) R 16b , (CH2 ) r S (0) p R 16 , (CH2) r S(0)2NR 16a R 16a P (CH2 ) r NR 16f S (0) 2R 16b , and (CH2)rPhenyl substituted with 0-3 R 16e ;
- Rl6a and R ⁇ 63 -' are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 16e ;
- R 16 at each occurrence, is selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 16e ;
- R 16d is selected from Ci-g alkyl and phenyl
- R 16e is selected from Ci-g alkyl, Cl, F, Br, I, CN, N02, (CF2) r CF3, OH, and (CH2) r OCi-5 alkyl; and
- R 16 f at each occurrence, is selected from H, and C ⁇ _5 alkyl .
- the present invention provides novel compounds of formula (I-i) :
- R 5 is CH2phenyl substituted with 0-3 R!6;
- R 9 is selected from H, Ci-6 alkyl, (CH2) r C3-6 cycloalkyl, F, Cl, CN, (CH2) r OH, (CH2) r OR 9d , (CH2 ) r NR 9a R 9 a (CH2 ) r OC (0) NHR 9a , (CH2 ) rPhenyl substituted with 0-5 R 9e , and (CH2 ) r _ heterocyclic system substituted with 0-2 R 9e , wherein the heterocyclic system is selected from pyridyl, thiophenyl, furanyl, oxazolyl, and thiazolyl;
- R 9a and R 9a P at each occurrence are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2 ) r pheny1 substituted with 0-3 R 9e ;
- R d at each occurrence, is selected from Ci-g alkyl and phenyl ;
- R e at each occurrence, is selected from Ci-g alkyl, Cl, F, Br, I, CN, N02 , (CF2) r F3, OH, and (CH ) r OCi-5 alkyl;
- R 10 is selected from H, Ci-5 alkyl, OH, and CH2OH;
- RlO-3 " is selected from H, Ci-g alkyl, C3-g cycloalkyl, (CH2)rPhenyl, C(O)R 10f , C(O)OR 10f , and S ⁇ 2R 10f ;
- R 9 or R 10 when either of R 9 or R 10 is halogen, cyano, nitro, or bonded to the carbon to which it is attached through a heteroatom, the other of R 9 or R 10 is not bonded to the carbon to which it is attached through a heteroatom;
- R11 J_ S selected from H, Ci-8 alkyl, (CH2 ) rPhenyl substituted with 0-5 R lle , and a (CH2)r ⁇ heterocyclic system substituted with 0-2 R lle , wherein the heterocyclic system is selected from pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiophenyl, benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, isoindolyl, piperidinyl, pyrrazolyl, 1,2,4- triazolyl, 1, 2 , 3-triazolyl, tetrazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyri idinyl; and
- R lle at each occurrence, is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, OH, and (CH2) r OCi-5 alkyl;
- RUST is selected from H, Ci-g alkyl, C3-g cycloalkyl, (CH2)rPhenyl, C(0)R llf , C(0)OR llf , and S ⁇ 2R llf ;
- R 12 is H
- R 11 and R!2 join to form a C3--10 cycloalkyl, a 5-6-membered lactone or lactam, or a 4-6-membered saturated heterocycle containing 1-2 heteroatoms selected from O, S, and R 11 ⁇ and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 14 is selected from Ci-8 alkyl, (CH2)rC3-6 cycloalkyl, CF3 , Cl, Br, I, F, (CH2) r NR 14a R 14a N02, CN, OH, (CH2) r OR 14d , (CH2 ) r C (0) R 14b , (CH2 ) r C (0)NR 1 R 1 ' , (CH2 ) r NR 14f C (0) R 1 , (CH2 ) r S (0) p R 14b , (CH2 ) r S (O) 2NR 1 a R 14a ' , (CH2 ) r NR 14f S (O) 2R 14b (CH2 ) rPheny1 substituted with 0-3 R 14e , and a
- Rl4a an( * j Rl4a' a each occurrence, are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 14e , and a (CH2)r- 5 ⁇ 6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 15e ;
- R 1 b at each occurrence, is selected from H, Ci-g alkyl, C3_g cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 1 e ;
- R 14d is selected from Ci-g alkyl and phenyl
- R 14e at each occurrence, is selected from Ci-6 alkyl, Cl, F, Br, I, CN, N02, (CF2)rCF3, OH, and (CH2)rOC ⁇ _5 alkyl; and R 14r , at each occurrence, is selected from H, and C1-5 alkyl ; and
- r is selected from 0, 1, and 2.
- the present invention provides novel compounds of formula (I-ii) :
- R 5 is CH2phenyl substituted with 0-3 R l ⁇ ;
- R 9 is selected from H, Ci-g alkyl, (CH2)r 3-6 cycloalkyl, F, Cl, CN, (CH2) r OH, (CH2) r OR 9d
- R 9a and R 9a P at each occurrence are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2)rP n enyl substituted with 0-3 R e ;
- R d at each occurrence, is selected from Ci-g alkyl and phenyl ;
- R 9e is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, OH, and (CH )rOCi-5 alkyl;
- R 10 ⁇ is selected from H, Ci-g alkyl, C3-g cycloalkyl, (CH2)rPhenyl, C(O)R 10f , C(O)OR 10f , and S ⁇ 2R 10f ;
- R 9 or R 10 when either of R 9 or R 10 is halogen, cyano, nitro, or bonded to the carbon to which it is attached through a heteroatom, the other of R 9 or R 10 is not bonded to the carbon to which it is attached through a heteroatom;
- R 11 is selected from H, Ci-8 alkyl, (CH2)rPhenyl substituted with 0-5 R lle , and a (CH2)r ⁇ heterocyclic system substituted with 0-2 R lle , wherein the heterocyclic system is selected from pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiophenyl, benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, isoindolyl, piperidinyl, pyrrazolyl, 1,2,4- triazolyl, 1, 2 , 3-triazolyl, tetrazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyrimidinyl; and
- R lle at each occurrence, is selected from Ci-6 alkyl, Cl, F, Br, I, CN, O2 , (CF2) r CF3, OH, and (CH2) r OCi- 5 alkyl;
- R11- ⁇ is selected from H, Ci-g alkyl, C3_g cycloalkyl, (CH2)rPhenyl, C(0)R llf , C(0)0R llf , and S02R llf ;
- R 12 is H
- R 11 and R 12 join to form a C3-10 cycloalkyl, a 5-6-membered lactone or lactarn, or a 4-6-membered saturated heterocycle containing 1-2 heteroatoms selected from 0, S, and NR 11 9 ' and optionally fused with a benzene ring or a 6- membered aromatic heterocycle;
- R 14 is selected from Ci-8 alkyl, (CH2) r C3-6 cycloalkyl, CF3 , Cl, Br, I, F, (CH2)r-NR 14a R 14a O2 , CN, OH, (CH2) r OR 14d , (CH2) r C(0)R 1 b , (CH2) r C(0)NR 1 a R 1 P (CH2 ) r NR 14f C (0) R 1 b , (CH2 ) r S (0) p R 14b , (CH2) r S(0)2NR 1 a R 14a ' , (CH2) r NR 14f S (O) R 14b , (CH2 ) rPhenyl substituted with 0-3 R 14e , and a (CH2)r -5 -6 membered heterocyclic system containing 1-4 heteroatoms selected from N, 0, and S, substituted with 0-2 R 15e ; or two R 14 substituents on adjacent atoms
- R 14a and Rl 4a ' t a each occurrence are selected from H, Ci-6 alkyl, C3_ cycloalkyl, and (CH2 ) rPhenyJ substituted with 0-3 R 1 e ;
- R 14 at each occurrence, is selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2 ) rPhenyl substituted with 0-3 R 14e ;
- R 14d is selected from Ci-g alkyl and phenyl
- R 1 e is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, OH, and (CH ) r OCl-5 alkyl;
- R 14 f at each occurrence, is selected from H, and C1-5 alkyl ;
- r is selected from 0, 1, and 2.
- the present invention provides novel compounds of formula (I-i) :
- J is selected from CH2 and CHR 5 ;
- K is selected from CH2 and CHR 5 ;
- L is selected from CH2 and CHR 5 ;
- R 3 is selected from a Ci-io alkyl substituted with 0-3 R 3 g, C3-10 alkenyl substituted with 0-3 R 3 -?, and
- R 3 *? at each occurrence, is selected from Cl, Br, I, F, N02, CN, NR 3a R a P OH, 0(CHR') r R 3d , SH, C(0)H, S(CHR') r R 3d , C(0)OH, C(O) (CHR') r R 3b , C(0)NR 3a R 3 P
- R 15 wherein the heterocyclic system is selected from pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiophenyl, benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, indolinyl, isoindolyl, isothiadiazolyl, isoxazolyl, piperidinyl, pyrrazolyl, 1, 2 , 4-triazolyl, 1,2,3- triazolyl, tetrazolyl, thiadiazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyrimidinyl, provided that when R 3 9 is a carbocyclic residue or a heterocyclic system, R 3 has at least one other R ⁇ , which is not a carbocyclic
- R 3a and R 3a P at each occurrence are selected from H,
- R 3 at each occurrence, is selected from Ci-g alkyl, and (CH2) r -phenyl substituted with 0-3 R 3e ;
- R 3d is selected from Ci-g alkyl and phenyl substituted with 0-3 R 3e ;
- R 3e at each occurrence, is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r CF3, (CH2) r OCi-5 alkyl, OH;
- R f at each occurrence, is selected from H, C1-5 alkyl;
- R 15 is selected from Ci-8 alkyl, (CH2)rC3-6 cycloalkyl, CF3 , Cl, Br, I, F,
- Rl5a anc j Rl5a' r a t each occurrence are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2 ) Phenyl substituted with 0-3 R 15e ;
- R 1 a and R 1 a# along with the N to which they are attached, are joined to form a morpholine, piperidine, or piperazine ring, and the piperazine optionally substituted with R 15 9;
- R 15b at each occurrence, is selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2 ) rPh ⁇ nyl substituted with 0-3 R 15e ;
- R 15d is selected from C ⁇ _g alkyl and phenyl
- R 1 e is selected from Ci-g alkyl, Cl, F, Br, I, CN, N02, (CF2) r CF3, OH, and (CH2) r OCi_5 alkyl; and
- R 1 ⁇ is selected from H, and C1-5 alkyl .
- the present invention provides novel compounds of formula (I-ii) :
- K is selected from CH2 and CHR 5 ;
- L is selected from CH2 and CHR 5 ;
- R 3 is selected from a Ci-io alkyl substituted with 0-3 R g , C3-10 alkenyl substituted with 0-3 R 3 9, and
- R 3 ST at each occurrence, is selected from Cl, Br, I, F, N02, CN, NR 3a R 3a P OH, 0(CHR') r R 3d , SH, C(0)H, S(CHR') r R 3d C(0)0H, C(O) (CHR') r R 3b / C(0)NR 3a R 3a P NR 3f C(0) (CHR') r R 3 , C (0)0 (CHR') r R 3d OC(O) (CHR') r R 3b , (CH2) r OC(0)NR 3a R 3a P
- R 15 wherein the heterocyclic system is selected from pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiophenyl , benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, indolinyl, isoindolyl, isothiadiazolyl, isoxazolyl, piperidinyl, pyrrazolyl, 1, 2 , 4-triazolyl, 1,2,3- triazolyl, tetrazolyl, thiadiazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyrimidinyl, provided that when R 3 ST is a carbocyclic residue or a heterocyclic system, R 3 has at least one other R 3 -?, which is not phenyl
- R 3a and R a ' are selected from H,
- R 3 at each occurrence, is selected from Ci-6 alkyl, and (CH2)r-phenyl substituted with 0-3 R 3e ;
- R d is selected from Ci-g alkyl and phenyl substituted with 0-3 R 3e ;
- R 3e at each occurrence, is selected from Ci-g alkyl, Cl, F, Br, I, CN, N0 , (CF ) r CF3, (CH ) r OCl_5 alkyl, OH;
- R f at each occurrence, is selected from H, C1-5 alkyl;
- R 15 at each occurrence, is selected from C ⁇ _8 alkyl, (CH2) r C3-6 cycloalkyl, CF3 , Cl, Br, I, F, (CH2)rNR 15a R 15a ' , NO2, CN, OH, (CH2) r OR 15d , (CH2) r C(0)R 15b , (CH2) r C(0)NR 15a R 15a P (CH2) r NR 15f C(0)R 15 , (CH2) r OC (0)NR 15a R 15a (CH2 ) q NR 15a C (0) 0R 15a , (CH 2 ) r S (0) pR 15b , (CH2) r S(0)2NR 15a R 15 ' , (CH2) r NR 15f S (O) 2R 15b , (CH2)rPhenyl substituted with 0-3 R 15e , and a heterocyclic
- R l a and R 15a ' are selected from H, Ci-g alkyl, C3-g cycloalkyl, and (CH2 ) rPhenyl substituted with 0-3 R 15e ;
- R 15a and R1 5&/ along with the N to which they are attached, are joined to form a morpholine, piperidine, or piperazine ring, and the piperazine optionally substituted with R 15 -?;
- Rl5b ? a t each occurrence, is selected from H, Ci-g alkyl, C3- cycloalkyl, and (CH2)rPhenyl substituted with 0-3 R 15e ;
- R 1 at each occurrence, is selected from C ⁇ _g alkyl and phenyl;
- R 15e is selected from Ci-g alkyl, Cl, F, Br, I, CN, NO2 , (CF2) r F3, OH, and (CH2)rOCi-5 alkyl; and
- R 1 f at each occurrence, is selected from H, and C1-5 alkyl .
- the present invention provides novel compounds of formula (I-i) :
- Z is selected from O and N(CN) ;
- R 3 is selected from C 3 _ 8 alkyl wherein the C 3 _ 8 alkyl is selected from methyl, ethyl, propyl, i-propyl, butyl, i-butyl, t-butyl, pentyl, methylpentyl, dimethylpentyl, and trimethylpentyl, and wherein the C 3 _ 8 alkyl is substituted with 0-2 R 3 ⁇ ;
- R 3 s at each occurrence is selected from C(0)OR 3b , OR 3b , OH, OC(0)H, NHC(0)R 3b , CN, NR 3a R 3a P and phenyl;
- R 3a and R 3a P at each occurrence are selected from H and methyl;
- R 3b at each occurrence, is selected from H, methyl, ethyl, propyl, and phenyl; and R 16 is selected from F, Cl, Br, and I.
- the present invention provides novel compounds of formula (I-ii)
- Z is selected from 0 and N(CN) ;
- R 3 is selected from C 3 _ 8 alkyl wherein the C 3 _ 8 alkyl is selected from methyl, ethyl, propyl, i-propyl, butyl, i-butyl, t-butyl, pentyl, methylpentyl , dimethylpentyl , and trimethylpentyl , and wherein the C 3 _ 8 alkyl is substituted with 0-2 R 3 9;
- R 3 ⁇ at each occurrence is selected from C(0)0R 3b , OR 3b , OH, OC(0)H, NHC(0)R 3b , CN, NR 3a R 3a P and phenyl;
- R 3a and R 3a P at each occurrence are selected from H and methyl;
- R 3b at each occurrence, is selected from H, methyl, ethyl, propyl, and phenyl;
- R 16 is selected from F, Cl, Br, and I.
- the present invention provides novel compounds of formula (I) , wherein the compound of formula (I) is selected from: N- (t-butyl) -N'-[ (lR,2S)-2-[[(3S)-3-(4- fluorophenyl)methyl)piperidinyl]methyl] cyclohexy 1] -urea,
- the present invention provides a pharmaceutical composition, comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method for modulation of chemokine receptor activity comprising administering to a patient in need thereof a therapeutically effective amount of the compounds of the present invention.
- the present invention provides a method for treating or preventing inflammatory diseases, comprising administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method for treating or preventing asthma, comprising administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention.
- the present invention provides a pharmaceutical composition, comprising a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method for modulation of chemokine receptor activity comprising administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method for modulation of chemokine receptor activity comprising contacting a CCR3 receptor with an effective inhibitory amount of a compound of the present invention.
- the present invention provides a method for treating inflammatory disorders comprising administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention
- the present invention provides a method for treating or preventing disorders selected from asthma, allergic rhinitis, atopic dermatitis, inflammatory bowel diseases, idiopathic pulmonary fibrosis, bullous pemphigoid, helminthic parasitic infections, allergic colitis, eczema, conjunctivitis, transplantation, familial eosinophilia, eosinophilic cellulitis, eosinophilic pneumonias, eosinophilic fasciitis, eosinophilic gastroenteritis, drug induced eosinophilia, HIV infection, cystic fibrosis, Churg-Strauss syndrome, lymphoma, Hodgkin's disease, and colonic carcinoma.
- disorders selected from asthma, allergic rhinitis, atopic dermatitis, inflammatory bowel diseases, idiopathic pulmonary fibrosis, bullous pemphigoid, helminthic parasitic infections, allergic colitis
- the present invention provides a method for treating or preventing disorders selected from asthma, allergic rhinitis, atopic dermatitis, and inflammatory bowel diseases.
- the present invention provides a method for treating or preventing disorders wherein the disorder is asthma. In a more preferred embodiment, the present invention provides a method for treating or preventing disorders wherein the disorder is allergic rhinitis.
- the present invention provides a method for treating or preventing disorders wherein the disorder is atopic dermatitis .
- the present invention provides a method for treating or preventing disorders wherein the disorder is inflammatory bowel diseases .
- K is selected from CHR 5 or CR 6 R 5 .
- L is selected from CHR 5 or CR 6 R 5 .
- the compounds herein described may have asymmetric centers.
- substituted means that any one or more hydrogens on the designated atom is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- 2 hydrogens on the atom are replaced.
- any variable e.g., R a
- its definition at each occurrence is independent of its definition at every other occurrence.
- R a e.g., R a
- said group may optionally be substituted with up to two R a groups and R a at each occurrence is selected independently from the definition of R a .
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds .
- C ⁇ _ 8 alkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms, examples of which include, but are not limited to, methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, sec-butyl, t-butyl, pentyl, and hexyl.
- C ⁇ _ 8 alkyl is intended to include Ci, C 2 , C 3 , C 4 , C 5 , C ⁇ , C ⁇ , and C 8 alkyl groups.
- Alkenyl is intended to include hydrocarbon chains of either a straight or branched configuration and one or more unsaturated carbon-carbon bonds which may occur in any stable point along the chain, such as ethenyl, propenyl, and the like.
- Alkynyl is intended to include hydrocarbon chains of either a straight or branched configuration and one or more unsaturated triple carbon-carbon bonds which may occur in any stable point along the chain, such as ethynyl, propynyl, and the like.
- C 3 _ 6 cycloalkyl is intended to include saturated ring groups having the specified number of carbon atoms in the ring, including mono-, bi-, or poly-cyclic ring systems, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and cycloheptyl in the case of C 7 cycloalkyl.
- C 3 _ 6 cycloalkyl is intended to include C 3 , C 4 , C 5 , and C 5 cycloalkyl groups
- the compounds of Formula I can also be quaternized by standard techniques such as alkylation of the piperidine or pyrrolidine with an alkyl halide to yield quaternary piperidinium salt products of Formula I .
- Such quaternary piperidinium salts would include a counterion.
- counterion is used to represent a small, negatively charged species such as chloride, bromide, hydroxide, acetate, sulfate, and the like.
- piperidinium spirocycle or pyrrolidinium spirocycle is intented to mean a stable spirocycle ring system, in which the two rings form a quarternary nitrogene at the ring junction.
- 5-6-membered cyclic ketal is intended to mean 2 , 2-disubstituted 1,3- dioxolane or 2 , 2-disubstituted 1,3-dioxane and their derivatives .
- carbocycle or “carbocyclic residue” is intended to mean any stable 3, 4, 5, 6, or 7-membered monocyclic or bicyclic or 7 , 8, 9, 10, 11, 12, or 13-membered bicyclic or tricyclic, any of which may be saturated, partially unsaturated, or aromatic.
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, adamantyl, cyclooctyl,; [3.3.0]bicyclooctane, [4.3.0]bicyclononane,
- heterocycle or “heterocyclic system” is intended to mean a stable 5, 6, or 7-membered monocyclic or bicyclic or 7 , 8, 9, or 10- membered bicyclic heterocyclic ring which is saturated, partially unsaturated or unsaturated (aromatic) , and which consists of carbon atoms and 1, 2, 3, or 4 heteroatoms independently selected from the group consisting of N, NH, 0 and S and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized.
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. If specifically noted, a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and 0 atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another.
- aromatic heterocyclic system is intended to mean a stable 5- to 7- membered monocyclic or bicyclic or 7- to 10-membered bicyclic heterocyclic aromatic ring which consists of carbon atoms and from 1 to 4 heterotams independently selected from the group consisting of N, 0 and S.
- heterocycles include, but are not limited to, lH-indazole, 2-pyrrolidonyl, 2H,6H-1,5,2- dithiazinyl, 2H-pyrrolyl, 3H-indolyl, 4-piperidonyl, 4aH-carbazole, 4H-quinolizinyl, 6H-1, 2 , 5-thiadiazinyl, acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazalonyl, carbazolyl, 4 BB-carbazolyl, ⁇ -carbolinyl, chromanyl, chromenyl , cinnolinyl , decahydroquinol,
- oxazolyl oxazolidinylperimidinyl , phenanthridinyl, phenanthrolinyl, phenarsazinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, pteridinyl, piperidonyl, 4-piperidonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolyl, pyridazinyl, pyridooxazole, pyridoimidazole, pyridothiazole, pyridinyl, pyridyl, pyridyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, pyrrolyl
- Preferred heterocycles include, but are not limited to, pyridinyl, thiophenyl, furanyl, indazolyl, benzothiazolyl, benzimidazolyl, benzothiaphenyl, benzofuranyl, benzoxazolyl, benzisoxazolyl, quinolinyl, isoquinolinyl, imidazolyl, indolyl, isoidolyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pyrrazolyl, 1,2, 4-triazolyl, 1, 2 , 3-triazolyl, tetrazolyl, thiazolyl, oxazolyl, pyrazinyl, and pyrimidinyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles .
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional nontoxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2- acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound which contains a basic or acidic moiety by conventional chemical methods.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred.
- Lists of suitable salts are found in Remington 's Pharmaceutical Sciences, 17th ed. , Mack Publishing Company, Easton, PA, 1985, p. 1418, the disclosure of which is hereby incorporated by reference .
- prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc..) the compounds of the present invention may be delivered in prodrug form.
- the present invention is intended to cover prodrugs of the presently claimed compounds, methods of delivering the same and compositions containing the same.
- Prodrugs are intended to include any covalently bonded carriers which release an active parent drug of the present invention in vivo when such prodrug is administered to a mammalian subject.
- Prodrugs the present invention are prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- Prodrugs include compounds of the present invention wherein a hydroxy, amino, or sulfhydryl group is bonded to any group that, when the prodrug of the present invention is administered to a mammalian subject, it cleaves to form a free hydroxyl, free amino, or free sulfhydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups in the compounds of the present invention.
- Solid compound and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent.
- the compounds of Formula I can be prepared using the reactions and techniques described below. The reactions are performed in a solvent appropriate to the reagents and materials employed and suitable for the transformations being effected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention. It will also be recognized that another major consideration in the planning of any synthetic route in this field is the judicious choice of the protecting group used for protection of the reactive functional groups present in the compounds described in this invention. An authoritative account describing the many alternatives to the trained practitioner is Greene and Wuts (Protective Groups In Organic Synthesis, Wiley and Sons, 1991) .
- halide is not I
- KI can also be added to facilitate the displacement, provided the solvent is suitable, such as an alcohol, 2-butanone, DMF or DMSO, amongst others.
- the displacement can be performed at room temperature to the reflux temperature of the solvent.
- the protecting group is subsequently removed to yield amine 4.
- Protecting groups include phthalimide which can be removed by hydrazine, a reaction familiar to one skilled in the art; bis-BOC which can be removed by either TFA or HCl dissolved in a suitable solvent, both procedures being familiar to one skilled in the art; a nitro group instead of an amine which can be reduced to yield an amine by conditions familiar to one skilled in the art; 2,4-dimethyl pyrrole (S. P.
- Activation of imidazolide intermediates also facilitates urea formation (Bailey, R. A., et al . , Tet. Lett. 1998, 39, 6267-6270) .
- the urea forming reactions are done in a non- hydroxylic inert solvent such as THF, toluene, DMF, etc . , at room temperature to the reflux temperature of the solvent and can employ the use of an acid scavenger or base when necessary such as carbonate and bicarbonate salts, triethylamine, DBU, Hunigs base, DMAP, etc.
- Substituted pyrrolidines and piperidines 1 can either be obtained commercially or be prepared as shown in Scheme 2.
- Commercially available N-benzylpiperid-3- one 16. can be debenzylated and protected with a BOC group employing reactions familiar to one skilled in the art.
- Subsequent Wittig reaction followed by reduction and deprotection yields piperidine 20 .
- Substituted pyrrolidines may be made by a similar reaction sequence.
- Other isomers and analogs around the piperidine ring can also be made by a similar reaction sequence.
- Chiral pyrrolidines/piperidines can be synthesized via asymmetric hydrogenation of 18. using chiral catalysts (see Parshall, G.W.
- a method for introducing substituents in linkage E is that of A. Chesney et al . (Syn. Comm. 1990, 20 (20), 3167-3180) as shown in Scheme 4.
- Michael reaction of pyrrolidine or piperidine 1 with Michael acceptor 26 yields intermediate 27. which can undergo subsequent reactions in the same pot.
- reduction yields alcohol . 28 which can be elaborated to the amine 29 by standard procedures familiar to one skilled in the art .
- Some of these include mesylation or tosylation followed by displacement with NaN 3 followed by reduction to yield amine 29_ .
- Another route as depicted in Scheme 4 involves reaction with diphenylphosphoryl azide followed by reduction of the azide to yield amine 29 . . SCHEME 4
- the mesylate or tosylate can also be displaced by other nucleophiles such as NH 3 , BOC 2 N-, potassium phthalimide, etc., with subsequent deprotection where necessary to yield amines 29 . .
- 29 . can be converted to urea or thiourea 3_0 by procedures discussed for Scheme 1 or to the compounds of this invention by procedures previously discussed.
- aldehyde 27 may be reacted with a lithium or a Grignard reagent 31 to yield alcohol adduct 32 . .
- This in turn can be converted to urea or thiourea 34 in the same way as discussed for the conversion of 28. to 30.
- the OH group can undergo synthetic transformations which are familiar to one skilled in the art and which will be discussed in much detail later on in the application. Chiral auxilliaries can also be used to introduce stereo- and enantioselectivity in these aldol condensations, procedures which are familiar to one skilled in the art.
- ketone 57 which may undergo nucleophilic 1,2-addition with organometallic reagents such as alkyl- or aryllithiums, Grignards, or zinc reagents, with or without CeCl 3 (T. Imamoto, et al . ,
- Epoxides disclosed by structure 54 may be synthesized enantio-selectively from amino acid starting materials by the methods of Dellaria, et al. J Med Chem 1987, 30 (11), 2137, and Luly, et al . J Org Chem 1987, 52 (8), 1487.
- the carbonyl group of ketone 57. in Scheme 8 may undergo Wittig reactions followed by reduction of the double bond to yield alkyl, arylalkyl, heterocyclic- alkyl, cycloalkyl, cycloalkylalkyl, etc. substitution at that position, reactions that are familiar to one skilled in the art.
- Wittig reagents can also contain functional groups which after reduction of the double bond yield the following functionality: esters (Buddrus, J. Angew Chem., 1968, 80), nitriles (Cativiela, C.et al., Tetrahedron 1996, 52 (16), 5881-5888.), ketone (Stork, G.et al .
- Scheme 9 summarizes the displacement chemistry and subsequent elaborations that can be used to synthesize the R 9 groups.
- alcohol 55 or 58 may be tosylated, mesylated, triflated, or converted to a halogen by methods familiar to one skilled in the art to produce compound 59.
- carbon homologs of 55 or 58 where OH can be (CH 2 ) r * H and it is also understood that these carbon homologs may have substituents on the methylene groups as well
- a hydroxyl group may be converted to a bromide by CBr 4 and Ph 3 P (Takano, S.
- Nitrile j50 can be reduced with DIBAL to yield aldehyde .61.
- This aldehyde can undergo reduction to alcohol .62 with, for example, NaBH 4 which in turn can undergo all of the S ⁇ r2 displacement reactions mentioned for alcohol 55 or 58..
- Alcohol .62. is a one carbon homolog of alcohol 55 or 58..
- This alcohol can undergo activation followed by the same S N 2 displacement reactions discussed previously, ad infinitum, to result in 3, 4, 5... etc. carbon homologation products.
- Aldehyde 61. can also be reacted with a lithium or Grignard reagent to form an alcohol 61a which can also undergo the above displacement reactions .
- Oxidation by methods familiar to one skilled in the art yields ketone 61b.
- Displacement by malonate yields malonic ester 63. which can be saponified and decarboxylated to yield carboxylic acid 64, a two carbon homologation product. Conversion to ester 65 (A. Hassner and V. Alexanian, Tet.
- Alcohols may be converted to the corresponding fluoride 70 . by DAST (diethylaminosulfur trifluoride) (Middleton, W. J. ; Bingham, E. M. ; Org. Synth. 1988, VI, pg. 835).
- Sulfones 72 can be converted to the corresponding sulfonamides 73 by the method of H.-C. Huang, E. et al . , Tet. Lett. (1994) 35, 7201-7204 which involves first, treatment with base followed by reaction with a trialkylborane yielding a sulfinic acid salt which can be reacted with hydroxylamine-O-sulfonic acid to yield a sulfonamide.
- Another route to sulfonamides involves reaction of amines with a sulfonyl chloride (G. Hilgetag and A.
- the isothiouronium salt may be synthesized from the corresponding halide, mesylate or tosylate 59. via reaction with thiourea (for a discussion on the synthesis of sulfonyl chlorides see G. Hilgetag and A. Martini, ibid., p. 670).
- the aldehyde 67 obtained from the Weinreb amide reduction can be reduced to the alcohol with NaBH 4 .
- the aldehyde or ketone .67 (or . 61 or 61b for that matter) can undergo Wittig reactions as discussed previously followed by optional catalytic hydrogenation of the olefin.
- This Wittig sequence is one method for synthesizing the carbocyclic and heterocyclic substituted systems at R 9 employing the appropriate carbocyclic or heterocyclic Wittig (or Horner-Emmons ) reagents.
- the Wittig reaction may also be used to synthesize alkenes at R 9 and other functionality as well.
- Ester 65. can also form amides 66 .
- Alcohol 68 can be converted to ether . 69 by procedures familiar to one skilled in the art, for example, NaH, followed by an alkyliodide or by Mitsunobu chemistry (Mitsunobu, 0. Synthesis, 1981, 1-28) .
- acylated can be acylated by procedures familiar to one skilled in the art, for example, by Schotten-Baumann conditions with an acid chloride or by an anhydride with a base such as pyridine to yield 78.
- Halide, mesylate, tosylate or triflate 59 can undergo displacement with azide followed by reduction to yield amine 74 a procedure familiar to one skilled in the art.
- This amine can undergo optional reductive amination and acylation to yield 75 or reaction with ethyl formate (usually refluxing ethyl formate) to yield formamide 75 . .
- Amine 74 can again undergo optional reductive amination followed by reaction with a sulfonyl chloride to yield 76 , for example under Schotten-Baumann conditions as discussed previously. This same sequence may be employed for amine 60a, the reduction product of nitrile 60. Tosylate 5_9 can undergo displacement with cuprates to yield 77 (Hanessian, S.; Thavonekham, B.; DeHoff, B.; J Org. Chem. 1989, 54, 5831).
- Aldehyde 61 or its homologous extensions can be reacted with a carbon anion of an aryl (phenyl, naphthalene, etc.) or heterocyclic group to yield an aryl alcohol or a heterocyclic alcohol.
- CeCl 3 may be added (T. Imamoto, et al., Tet. Lett. 1985, 26, 4763-4766; T. Imamoto, et al., Tet. Lett. 1984, 25, 4233-4236).
- This alcohol may be reduced with Et 3 SiH and TFA (J. Org. Chem. 1969, 34, 4; J. Org. Chem. 1987, 52, 2226) (see discussion of aryl and heterocyclic anions for Schemes 20-22).
- These aryl and heterocyclic anions may also be alkylated by 5_9
- R 9 contains an aryl or heterocyclic group.
- Compound 59 or its carbon homologs may be alkylated by an alkyne anion to produce alkynes at R 9 (see R.C. Larock, Comprehensive Organic Transformations, New York, 1989, VCH Publishers, p 297) .
- carboxaldehyde 61 or its carbon homologs can undergo 1,2 -addition by an alkyne anion
- Nitro groups can be introduced by displacing bromide 59 (or its carbon homologs) with sodium nitrite in DMF (J.K. Stille and E.D. Vessel J. Org. Chem. 1960, 25, 478-490) or by the action of silver nitrite on iodide 59 or its carbon homologs (Org. Syntheses 34, 37-39).
- R 9 is either in its final form or in a suitable protected precursor form.
- This electrophile can be a carbon-based electrophile, some examples being formaldehyde to introduce a CH 2 OH group, an aldehyde or a ketone which also introduces a one- carbon homologated alcohol, ethylene oxide (or other epoxides) which introduces a -CH 2 CH 2 OH group (a two- carbon homologated alcohol), an alkyl halide, etc., all of which can be later elaborated into R 9 .
- It can also be an oxygen-based electrophile such as MCPBA, Davis ' reagent (Davis, F. A.; Haque, M.
- ketone 85 This ketone can also be synthesized in one step directly from 1 and alpha,beta- unsaturated ketone j34 using the same procedure.
- This ketone may be reduced with LAH, aBH 4 or other reducing agents to form alcohol .86.
- ketone 85 can be reacted with an organolithium or Grignard reagents to form tertiary alcohol 87. .
- ester j30 can be directly reduced with LiBH 4 or LAH to yield primary alcohol 88.
- Alcohols 86., 87 , and 8_8 can all be tosylated, mesylated, triflated, or converted to a halogen by methods discussed previously and displaced with an amine nucleophile such as azide, diphenylphosphoryl azide (with or without DEAD and Ph 3 P) , phthalimide, etc. as discussed previously (and which are familiar to one skilled in the art) and after reduction (azide) or deprotection with hydrazine (phthalimide) , for example, yield the corresponding amines . These can then be elaborated into the compounds of this invention as discussed previously.
- an amine nucleophile such as azide, diphenylphosphoryl azide (with or without DEAD and Ph 3 P) , phthalimide, etc.
- Ketone 85 can also be converted into imine j39 which can be reacted with a Grignard reagent or lithium reagent, etc., to form a protected amine 90 which can be deprotected and elaborated into the compounds of this invention as discussed previously.
- Some protecting groups include benzyl and substituted benzyl which can be removed by hydrogenation, and cyanoethyl, which can be removed with aqueous base, etc. It is to be understood that R 7-12 in Scheme 10 can be in their final form or in precursor form which can be elaborated into final form by procedures familiar to one skilled in the art.
- the amino group can be reacted with an isocyanate, an isothiocyanate, a carbamoyl chloride, or any reagent depicted in Scheme 1 to form 95 . which can be alkylated with 1 to form the compounds of this invention.
- amine 1 might have to be activated with Lewis acids in order to open the epoxide ring (Fujiwara, M. ; Imada, M. ; Baba, A.; Matsuda, H .; Tetrahedron Lett 1989, 30, 739; Caron, M. ; Sharpless, K. B.; J Org Chem 1985, 50, 1557) or 1 has to be deprotonated and used as a metal amide, for example the lithium amide (Gorzynski- Smith, J.; Synthesis 1984 (8), 629) or MgBr amide (Carre, M. C . ; Houmounou, J. P. ; Caubere, P. ; Tetrahedron Lett 1985, 26, 3107) or aluminum amide (Overman, L. E.; Flippin, L. A.; Tetrahedron Lett 1981, 22, 195) .
- Lewis acids for example the lithium amide (Gorzynski- Smith, J.; Synthesis 1984 (8)
- the quaternary salts (where R 4 is present as a substituent) of pyrrolidines and piperidines can be synthesized by simply reacting the amine with an alkylating agent, such as methyl iodide, methyl bromide, ethyl iodide, ethyl bromide, ethyl or methyl bromoacetate, bromoacetonitrile, allyl iodide, allylbromide, benzyl bromide, etc. in a suitable solvent such as THF, DMF, DMSO, etc. at room temperature to the reflux temperature of the solvent .
- an alkylating agent such as methyl iodide, methyl bromide, ethyl iodide, ethyl bromide, ethyl or methyl bromoacetate, bromoacetonitrile, allyl iodide, allylbromide, benzyl bromid
- Spiroquaternary salts can be synthesized in a similar manner, the only difference being that the alkylating agent is located intramolecularly as shown in Scheme 12. It is understood by one skilled in the art that functional groups might not be in their final form to permit cyclization to the quaternary ammonium salt and might have to be in precursor form or in protected form to be elaborated to their final form at a later stage.
- the leaving groups represented by X in Scheme 12 may equal those represented in Scheme 1, but are not limited thereto.
- N-oxides of pyrrolidines and piperidines can be made by the procedure of L. W. Deady (Syn. Comm. 1977, 7, 509- 514) . This simply entails reacting the pyrrolidine or piperidine with MCPBA, for example, in an inert solvent such as methylene chloride.
- Multisubstituted pyrrolidines and piperidines may be synthesized by the methods outlined in Scheme 13.
- thermodynamic or kinetic conditions yield regioselectively alkylated products (for a discussion on thermodynamic vs. kinetic alkylations see H. House Modern Synthetic Reactions, W. A. Benjamin, Inc. (Menlo Park, CA: 1972) chapter 9) .
- the amine 109 an then be elaborated into the compounds of this invention by methods discussed previously (Scheme 1) .
- the carbonyl- containing intermediate 107 in Scheme 13 can also be reduced to the methylene analog via a Wolff-Kishner reduction and modifications thereof, or by other methods familiar to one skilled in the art.
- the carbonyl group can also be reduced to an OH group, which can undergo all of the reactions described in Scheme 9 to synthesize the R6 groups .
- This piperidine or pyrrolidine can be deprotected and elaborated to the compounds of this invention by methods discussed earlier.
- R 5 * R 5 or a precursor thereof
- the OH may be reduced by the method of Barton (Barton, D. H. R.; Jaszberenyi, J. C. Tet. Lett. 1989, 30, 2619 and other references therein) .
- the alcohol can also be displaced with dialkyllithium cuprates (not shown) (Hanessian, S.; Thavonekham, B.; DeHoff, B.; J Org. Chem. 1989, 54, 5831).
- Deprotection if necessary yields 120 which may be elaborated as described previously into the compounds of this invention.
- a method for the alkylation of alkyl groups, arylalkyl groups, allylic groups, propargylic groups, etc . , and a variety of other electrophiles onto the pyrrolidinyl and/or piperidinyl alpha-carbons (alpha to the ring nitrogen atom) is represented by the work of Peter Beak, et al . as shown in Scheme 15. It is understood by one skilled in the art that the R 5 and R 13 groups are either in their precursor, protected, or final form. Only one R 5 group is shown to be substituted on piperidine/pyrrolidine 121. However it is understood by one skilled in the art that additional functionality may be present on the ring in either precursor, protected, or final form.
- Scheme 17 describes another method for the synthesis of compounds where R 9 and R 10 are taken together to form cycloalkyl groups.
- alcohols can then be activated either by conversion to a halide or to a mesylate, tosylate or triflate by methods familiar to one skilled in the art and as discussed previously, and then alkylated with pyrrolidine/piperidine by the conditions described in Scheme 1 to yield 135. Subsequent deprotection yields amine 136 which can be elaborated to the compounds of this invention as described previously.
- alcohol 133 can be oxidized to the aldehyde and then reacted with R 7or8 MgBr or R 7or8 Li with or without CeCl 3 to yield the corresponding alcohol 133 where instead of -CH 2 OH, we would have -CHR 7or8 OH.
- This oxidation-1, 2-addition sequence may be repeated to yield a tertiary alcohol.
- the alcohol may then be tosylated, mesylated, triflated, or converted to Cl, Br, or I by procedures familiar to one skilled in the art to yield 134 and then displaced with pyrrolidine/piperidine 1 to yield 135.
- Subsequent deprotection yields 136 which may undergo elaboration to the compounds of this invention as discussed previously.
- a method to introduce cycloalkyl groups at R ⁇ R 1 is shown in Scheme 18. Protection of the nitrogen of compounds 137 which are commercially available yields 138 (the protecting group may be BOC, CBZ, or any other compatible protecting group) by procedures familiar to one skilled in the art. Esterification by any one of a number procedures familiar to one skilled in the art (for example A. Hassner and V. Alexanian, Tet. Lett, 1978, 46, 4475-8) followed by reduction with DIBAL (or alternatively reduction to the alcohol with, for example, LiBH 4 , followed by Swern oxidation (op. cit.)) yields aldehyde 139.
- the protecting group may be BOC, CBZ, or any other compatible protecting group
- aldehyde 141 One carbon homologation via the Wittig reaction followed by hydrolysis of the vinyl ether yields aldehyde 141.
- Reductive amination (Abdel- Magid, A. F., et al. Tet. Lett. 1990, 31, (39) 5595- 5598) yields 142 followed by deprotection yields amine 143 which can be elaborated to the compounds of this invention by the methods previously discussed.
- aldehyde 139 can be reacted with R 9orl0 MgBr or R 9orl0 Li with or without CeCl 3 to yield an alcohol which can be oxidized to a ketone.
- Wittig one-carbon homologation on this ketone as described above followed by hydrolysis yields 141 where the -CH 2 CHO is substituted with one R S or lO gr ⁇ up (-CHR 9or10 CHO) .
- Aldehyde 141 (-CH 2 CHO) or its monosubstituted analog synthesized above (-CHR 9orl0 CHO) can undergo alkylation with R 9orl0 X where X is as defined in Scheme 1 to yield compound 141 containing one or both of the R 9 and R 10 substituents alpha to the aldehyde group.
- Alkylation can be performed using LDA or lithium bistrimethylsilyl amide amongst other bases in an inert solvent such as ether, THF, etc., at -78 °C to room temperature.
- Aldehyde 141 (-CH 2 CHO)or its substituted analogs synthesized above (i.e., -CHR 9 R 10 CHO) can undergo reductive amination with 1 and subsequent elaboration to the compounds of this invention.
- Aldehyde 141 (- CH 2 CHO)or its substituted analogs synthesized above
- -CHR 9 R 10 CHO can also undergo 1,2-addition with R 7or8 MgBr or R 7or8 Li to yield the corresponding alcohol - CH 2 CHR 7or8 OH or -CHR 9 R 10 CHR 7or8 OH.
- the alcohol may then be tosylated, mesylated, triflated, or converted to Cl, Br, or I by procedures familiar to one skilled in the art and displaced with pyrrolidine/piperidine 1 to yield, after subsequent deprotection and elaboration, the compounds of this invention.
- alcohol - CH 2 CHR 7or8 OH or -CR 9 R 10 CHR 7or8 OH can be oxidized (i.e.,
- CR 9 R 10 CHR 7or8 OH can be oxidized (i.e., Swern, op. cit.) to the ketone and reacted once more with R 7or8 MgBr or R 7 °r 8 Li to yield the corresponding alcohol -CHCR 7 R 8 OH or -CR 9 R 10 CR 7 R 8 OH. If the ketone enolizes easily, CeCl 3 may be used together with the Grignard or lithium reagent.
- the alcohol can again be tosylated, mesylated, triflated, or converted to Cl, Br, or I by procedures familiar to one skilled in the art and displaced with pyrrolidine/ piperidine 1 to yield, after subsequent deprotection and elaboration, the compounds of this invention.
- each one of the R 7 , R 8 , R 9 , and R 10 groups may be introduced into compounds 141, 142 and 143 and and, of course, in the compounds of this invention, by the methods discussed above.
- a method for the synthesis of N-substituted heterocycles at R 5 is shown in Scheme 19.
- the heterocycle can be deprotonated with NaH or by other bases familiar to one skilled in the art, in a solvent such as DMF, THF, or another appropriate non-hydroxylic solvent and reacted with piperidine or pyrrolidine 143 at room temperature to the reflux temperature of the solvent .
- a solvent such as DMF, THF, or another appropriate non-hydroxylic solvent
- Deprotection and elaboration as described before yields compounds where R 5 contains an N- substituted heterocycle.
- an acid scavenger such as K 2 C0 3 , KHC0 3 , Na 2 C0 3 , NaHC0 3 , amongst others, can be used in place of NaH, employing THF, DMF, or methyl ethyl ketone as solvents .
- hydroxylic solvents may be used as well, such as methanol, ethanol, etc. from room temperature to the reflux temperature of the solvent.
- Compound 143 as well as its other positional isomers are available, for example, from commercially available 4- hydroxy ethylpiperidine, 2-, 3-, and 4- carboethoxypiperidme, L- or D-proline ethyl ester, or from methyl l-benzyl-5-oxo-3-pyrrolidinecarboxylate by methods familiar to one skilled in the art and as discussed previously in this application. SCHEME 19
- a method for the synthesis of C-substituted heterocycles at R 5 is shown in Scheme 20.
- Many heterocycles such as the ones shown in Scheme 20, but not limited thereto, can be metallated with strong bases such as LDA, n-BuLi, sec-BuLi, t-BuLi, etc. to yield the corresponding anionic species.
- These anions may also be generated via halogen-metal exchange employing n-BuLi , or other alkyllithium reagents .
- These reactions may be performed in THF, ether, dioxane, DME, benzene, etc. at -78 °C to room temperature.
- R suitable protecting -'- - group or functional group etc.
- the anions of the methyl-substituted heterocycles may also be reacted with a BOC-protected piperidone or pyrrolidone (148) to yield alcohols 149 as shown in Scheme 22 (see above reviews on metallations for references) .
- These alcohols may be reduced using Pt ⁇ 2 and TFA (P. E. Peterson and C. Casey, J. Org. Chem. 1964, 29, 2325-9) to yield piperidines and pyrrolidines 150.
- Pt ⁇ 2 and TFA P. E. Peterson and C. Casey, J. Org. Chem. 1964, 29, 2325-9
- the carbonyl group can be located in other positions instead of, for example, the 4-position of piperidine in compound 148 as depicted in Scheme 22.
- other heterocycles may also be used besides the ones shown in Scheme 22.
- R suitable protecting group or functional het group to compounds of by mmeetthhooddss ddee ⁇ scribed previously
- aryl (phenyl, naphthyl, etc.) anions generated either by halogen-metal exchange or by ortho-directed metallation (Snieckus, V. Chem. Rev. 1990, 90, 879-933) using n- or s- or t-BuLi in a non- hydroxylic solvent such as THF, ether, etc., with or without TMEDA and allow them to react with
- R suitable protecting [H] group or functional group heterocycle to compounds by methods described previously
- the carbonyl insertion compounds (158) can also undergo reduction of the carbonyl group to either the CHOH or CH2 linkages by methods already discussed (NaBH 4 or Et 3 SiH, TFA, etc.). These amines can then be converted to isocyanate 5 via the following methods (Nowakowski, J. J Prakt Chem/Chem-Ztg 1996, 338 (7), 667-671; Knoelker, H.-J.et al . , Angew Chem 1995, 107 (22), 2746-2749; Nowick, J. S . et al . , J Org Chem 1996, 61 (11), 3929-3934; Staab, H.
- protected aminobromobenzenes or triflates or protected aminobromoheterocycles or triflates 159 may undergo Suzuki-type couplings with arylboronic acids or heterocyclic boronic acids (160) .
- These same bromides or triflates 159 may also undergo Stille-type coupling (Echavarren, A. M., Stille, J.K. J. Am. Chem. Soc, 1987, 109, 5478-5486) with aryl, vinyl, or heterocyclic stannanes 163.
- Bromides or triflates 159 may also undergo Negishi-type coupling with other aryl or heterocyclic bromides 164 (Negishi E. Accts .
- Compounds b may be also synthesized by reacting iminoyl chloride c with pyrrolidine/piperidine 1 to yield b where R 8 -* 3 is not H (Povazanec, F., et al . , J. J. Heterocycl. Chem., 1992, 29, 6, 1507-1512).
- Iminoyl chlorides are readily available from the corresponding amide via PCI5 or CCl4/PPh3 (Duncia, J.V. et al . , J. Org. Chem., 1991, 56, 2395-2400) . Again, the urea portion may be in final form or in precursor form.
- amines are commercially available and can be used as 9_, 10, or used as precursors to isocyanates or isothiocyanates 5_.
- Ketones and trifluoromethylketones undergo reductive amination in the presence of TiCl 4 followed by NaCNBH 4 to yield amines (Barney, C.L., Huber, E.W., McCarthy, J.R. Tet. Lett. 1990, 31, 5547-5550) .
- Aldehydes and ketones undergo reductive amination with Na(AcO) 3 BH as mentioned previously to yield amines (Abdel-Magid, A. F., et al . Tet. Lett. 1990, 31, (39) 5595-5598) .
- Amines may also be synthesized from aromatic and heterocyclic OH groups (for example, phenols) via the Smiles rearrangement (Weidner, J.J.
- alcohol 198 is quenched with an aldehyde to yield alcohol 198.
- the metallation may also be done enantioselectively using sparteine (P. Beak, S.T. Kerrick, S. Wu, J. Chu J. Am. Chem. Soc. 1994, 116, 3231-3239) .
- This alcohol can be deprotonated with NaH and cyclized to carbamate 198a which permits structural assignments of the erythro and threo isomers.
- Deprotection with base yields aminoalcohol 199.
- Subsequent N-alkylation yields p thalimidoalkylpiperidine 201.
- alkyl chain does not necessarily have to be n- propyl, but that n-propyl was chosen for demonstration purposes only.
- Deprotection of the phthalimido group with hydrazine yields amine 202.
- reaction with an isocyanate or via any of the previously described conditions described in Scheme 1 yields urea 203. If an isocyanate is used, the isocyanate can add twice to yield urea-carbamate 204.
- BOC-1-aminocyclopropane-l-carboxylic acid 216 is coupled to (S) -3- (4-fluorobenzyl) piperidine using a common amide forming reagent such as BOP, HBTU or HATU to furnish the amide tert-l- ⁇ [ (3S) -3- (4-fluorobenzyl) piperidinyl] carbonyl ⁇ cyclopropylcarbamate (217) .
- the amide is reduced to the corresponding amine by a reducing agent such as but not limited to BH 3 in THF at room temperature, followed by the removal of BOC protecting group with TFA and neutralization to afford the free amine 218.
- the free amine is then condensed with an isocyanate or a carbamate to yield the desired urea 219.
- Part B Preparaton of 4-benzyl-l- (3-amino-n-prop-l- yl ) piperidine
- the reaction Upon cooling to 23 °C, the reaction was poured into aqueous hydrogen chloride (0.20 N, 100 L) and diethyl ether (75 mL) . The layers were separated and the aqueous layer was basified with saturated aqueous sodium bicarbonate to pH 9. The aqueous layer was extracted with diethyl ether (4 x 75 mL) , and the combined organic layers were dried over sodium sulfate.
- Part B Preparation of 1, 3-Dihydro-4 ' - [4- fluorophenylmethyl] -4-nitro-spiro [2H-isoindole-2, 1' - piperidinium] bromide
- the filtrate was concentrated in vacuo to a mixture of water and an amber oil .
- the mixture was dissolved in 50 ml of 2-propanol, and concentrated in vacuo to remove excess water.
- the resulting yellow foam was dissolved in methanol and applied to a 3.5 cm x 5 cm quartz column via silica plug.
- the product was eluted with 20% MeOH/CHCl3 to yield 0.81g of a yellow foam.
- Part D Preparation of N- [1, 3-Dihydro-4 ' - [4- fluorophenyl-methyl] spiro [2H-isoindole-2 , 1' -piperdinium- 4-yl] -N' -4-fluorophenylurea bromide
- Part B Preparation of 2-benzyloxycarbonylamino-l- phenyl-3 , 4-epoxy-butane.
- Part C Preparation of 2-benzyloxycarbonylamino-4- [4- (4- fluorophenyl)methyl-1-piperidinyl] -l-phenyl-butan-3-ol .
- Part D Preparation of 2-amino-4- [4- (4- fluorophenyl)methyl-1-piperidinyl] -l-phenyl-butan-3-ol .
- N-Methyl-4-nitro-benzamide (2.25 g, 12.5 mmol, 1 eq.) and PC1 5 (2.60 g, 12.5 mmol, 1 eq. ) were melted together under house vacuum connected to a NaOH trap behind a safety shield. Melting occurred at 100°C. Heated at 130 °C for 1 hour then purified by kugelrohr distillation at 0.1 mmHg at 130°C. CAUTION: THE EXPLOSIVE PROPERTIES OF THIS COMPOUND ARE UNKNOWN) . The iminoyl chloride (12.5 mmol 1 eq. ) in DMF 10 ml was added to NaN 3 in 10 ml of DMF at 25°C and stirred overnight.
- the carbamate 168 can then be reacted with various amines to give the urea 169.
- the aniline 167 can be reacted with the appropriate isocyanates to give the urea 169 directly.
- the saturated ring analogs can also be used.
- 4- benzyl piperidine can be alkylated with the urea mesylate 185 (Scheme 30) to give corresponding cyclohexyl derivative 186.
- 4-benzyl piperidine can also be N-alkylated with the phenacyl bromide 170 to give the nitro ketone 171.
- the nitro group of 171 is then reduced using catalytic hydrogenation to give the corresponding aniline 172.
- the aniline 172 can be reacted with the appropriate isocyanates to give the ketone urea 173.
- the ketone of 173 can be reduced with NaBH 4 to give the alcohol 174.
- the aniline 176 may be treated with various isocyanates to give the urea alcohols 174.
- the 4-benzyl piperidine can also be N-alkylated with 3 -cyanobenzyl bromide (177, Scheme 28) to give the cyano analog 178.
- the cyano group is reduced using Raney nickel to give the corresponding benzyl amine 179.
- Treatment of 179 with isocyanates gives the urea 180.
- the saturated ring analogs can also be synthesized using analogous procedures as outlined in Schemes 30 and 31.
- 4-benzyl piperidine can be alkylated with the urea mesylate 185 (Scheme 29) to give corresponding cyclohexyl derivative 186.
- the enantiomerically pure amino alcohol 187 [J. Am . Chem. Soc . 1996, 118, 5502-5503 and references therein] one can protect the nitrogen to give the N-Cbz alcohol 188. Swern oxidation of the alcohol gives the aldehyde 189.
- Reductive amination with piperidine analogs gives the cyclohexyl methyl-1- piperidinyl analogue 190.
- the Cbz group is removed by catalytic hydrogenation to give the free amine 191, which is treated with a phenylisocyanate to give the desired urea analogue 192.
- Several examples using these synthetic methods are listed in Table 3a and Table 3.1.
- aniline 167 was purified by chromatography (MPLC, 40% ethyl acetate/ hexane; silica gel) to give 2.0 g of aniline 167 as a white solid.
- EXAMPLE 220 N- (2, 5-difluorophenyl ) -N' - [ [3-[ [4- (phenylmethyl) -1- piperidinyl]methyl] phenyl] methyl] -urea .
- aniline 172 was then filter, the solvent removed under vacuum, and the residue purified by chromatography (MPLC, 30% ethyl acetate/hexane; silica gel) to give 1.8 g of aniline 172 as a tan/brown solid.
- a solution of aniline 172 (Scheme 27) (310 mg, 1.0 mmol) in THF is treated with 2, 5-difluoroisocyanate (160 mg, 1.0 mmol) at room temperature for 1 h.
- Step a To a solution of (R,R) amino alcohol 187 [J. Am . Chem. Soc. 1996, 118, 5502-5503 and references therein] (1.9 g, 14.7 mmol) in CH 2 C1 2 (50 mL) is added 50 ml of an aqueous solution of Na 2 C0 3 (2.4 g, 28.9 mmol). While stirring, benzyl chloroformate (2.51 g, 14.7 mmol) is added and the mixture is stirred at room temperature for 1 h. The organic layer is separated and washed with water and brine.
- Step b A solution of DMSO (2.52 g, 30 mmol) in CH 2 CI 2 (50 mL) is cooled to -78°C. To this solution is added drop-wise oxalyl chloride (1.81 g, 14 mmol) and the resulting solution is stirred for an additional 10 min. Then a solution of alcohol 188 (2.5 g, 9.5 mmol) in CH 2 CI 2 (70 ml) is added via an addition funnel and stirred for 10 min. Then Et3N (5.0 g, 50 mmol) is added and the solution is allowed to warm to room temperature. The solution is diluted with water and the organic layer washed with water, 1 N HCl, and brine.
- Step c A solution of aldehyde 189 (2.0 g, 7.7 mmol), 4- (4-fluorophenylmethyl) piperidine hydrochloride (1.8 g, 7.8 mmol) in dichloroethane (80 ml) was treated with Na(OAc) 3 BH (3.23 g, 15 mmol) and 1 ml AcOH and stirred overnight at room temperature. The resulting solution was diluted with methylene chloride and washed with 1 n NaOH, water, and brine. The organic solvents were removed under vacuum and the residue chromatographed on silica gel (50% EtOAc/hex - 100% EtOAc) to give 3.0 g (6.8 mmol) of 190 as an oil.
- Step d A solution of 190 (3.0 g, 6.8 mmol) in MeOH was treated with 1.5 g of 10% Pd/C and hydrogenated at 50 psi overnight in a Parr apparatus. The mixture was filtered and the filtrate concentrated on a rotary evaporator to give 1.8 g (5.9 mmol) of the amine 191 as an oil.
- Step a To a solution of (R,R) amino alcohol 187 [J. Org. Chem . 1996, 61 , 5557-5563; J. Am . Chem . Soc . 1996, 118, 5502-5503] (9.5 g, 73.8 mmol) in CHC1 (200 mL) is added 200 ml of an aqueous solution of Na 2 C0 3 (15 g, 141 mmol). While stirring, benzyl chloroformate (12.6 g, 73.8 mmol) is added slowly and the mixture is stirred at room temperature for 1 h. The organic layer is separated and washed with water and brine. The organic solvent is removed on a rotary evaporator to give a white solid.
- Step b A solution of DMSO (36 g, 430 mmol) in CHC1 (200 mL) is cooled to -78°C. To this solution is added drop-wise oxalyl chloride (27.41 g, 216 mmol) and the resulting solution is stirred for an additional 10 min. A solution of alcohol 188 (38 g, 144 mmol) in CH 2 C1 2 (150 ml) is added via an addition funnel and stirred for
- Step c A solution of aldehyde 189 (19.6 g, 75 mmol) and (3S) -3- (4-fluorophenylmethyl) piperidine (14.5 g, 75 mmol) in dichloroethane (400 ml) was treated with Na(OAc) 3 BH (32 g, 152 mmol) and stirred overnight at room temperature. The resulting solution was poured slowly into a stirred mixture of ice/water/1 N NaOH and stirred for 20 min. The organic layer was separated and washed water, and brine.
- Step d A solution of 193 (32 g, 73 mmol) in MeOH was treated with 8 g of 10% Pd/C and hydrogenated at 50 psi overnight in a Parr apparatus . The mixture was filtered and the filtrate concentrated on a rotary evaporator to give 20 g (65 mmol) of the amine 194, which was used without further purification.
- Step e A solution of amine 194 (10 g, 32.8 mmol) in THF is treated with 3-acetylyphenyl isocyanate (5.3 g, 32.8 mmol) and the mixture is stirred for 30 min. The solvent is removed on a rotary evaporator and the residue is chromatographed on silica gel (0.5:4.5:95 NH 4 ⁇ H/MeOH/CH 2 Cl2) to give 11 g of urea 195 (Example 415) as a solid. Also obtained 2 g of cis isomer (Example 416a) .
- TEDA Tetramethylethylenediamine
- Part G preparation of erythro-cis-1- [3- (3- acetylphenylaminocarbonylamino) -n-prop-1-yl] -4-benzyl- ⁇ - ethylpiperidinemethanol and erythro-cis-1- [3- (3- acetylphenylaminocarbonylamino) -n-prop-1-yl] -2- [1- (3- acetylphenylaminocarbonyloxy) -n-prop-1-yl) -4- benzylpiperidine
- Part B Preparation of 2- [ (3-acetylanilino) ( ⁇ 3- [4- (4-fluorobenzyl) -1-piperidinyl] propyl ⁇ amino)methylene] malononitrile
- Part B Preparation of 1- (3- ⁇ [ (E) -1- ( ⁇ - [4- (4- fluorobenzyl) -1-piperidinyl] propyl ⁇ amino) -2- nitroethylenyl] amino ⁇ phenyl) ethanone
- Example 415, step d) (6 mg, 0.02 mmol) in 1 mL of THF is treated with 2 , 4, 4-trimethyl-2-pentyl isocyanate (3 ⁇ L, 0.03 mmol) at room temperature for 1 h.
- PS-trisamine 33 mg, 0.15 mmol, Argonaut Technologies Inc.
- the reaction mixture was filtered and the polymer was washed with CH 2 C1 2 , and the combined filtrate was concentrated under vacuum.
- the residue is further purified by HPLC, using a VYDAC C18 prepacked column (10 mm, 22 x 250 mm) and UV detection at 214 nm, elution with MeCN-H 2 0-TFA (90:10:0.1-10:90:0.1), flow rate 15 mL/min, to afford 5.6 mg of the urea product as a solid.
- Amine 194 (Scheme 31a) (15 mg, 0.05 mmol) and the product from Part A (117 mg, 0.5 mmol, 10 eq. ) were mixed then stirred overnight . The reaction was stripped and the residue then purified in silica gel in 100% ethyl acetate followed by 9:1 ethyl acetate/methanol . The product was further purified by HPLC, using a VYDAC C18 prepacked column (10 mm, 22 x 250 mm) and UV detection at 214 nm, elution with MeCN-H 2 0-TFA
- Each entry in each table is intended to be paired with each formulae at the start of the table.
- Entry 1 in Table 4 is intended to be paired with each of formulae la-44.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Virology (AREA)
- Oncology (AREA)
- Rheumatology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- AIDS & HIV (AREA)
- Otolaryngology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pain & Pain Management (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Vascular Medicine (AREA)
- Transplantation (AREA)
- Ophthalmology & Optometry (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pyrrole Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
Description
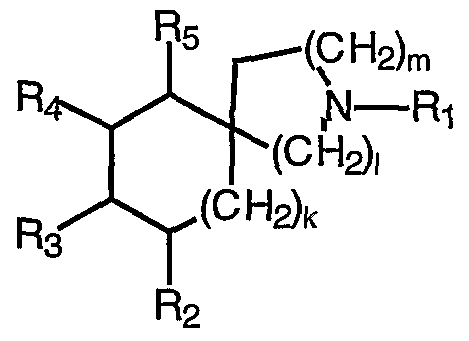
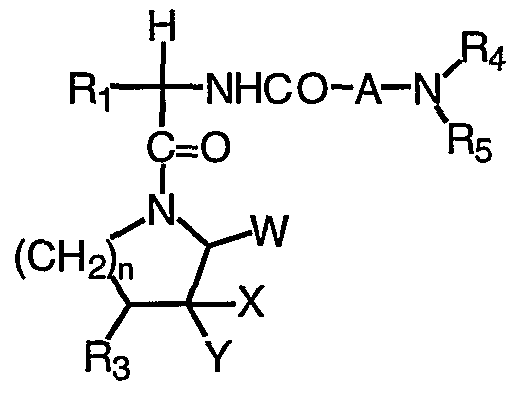
































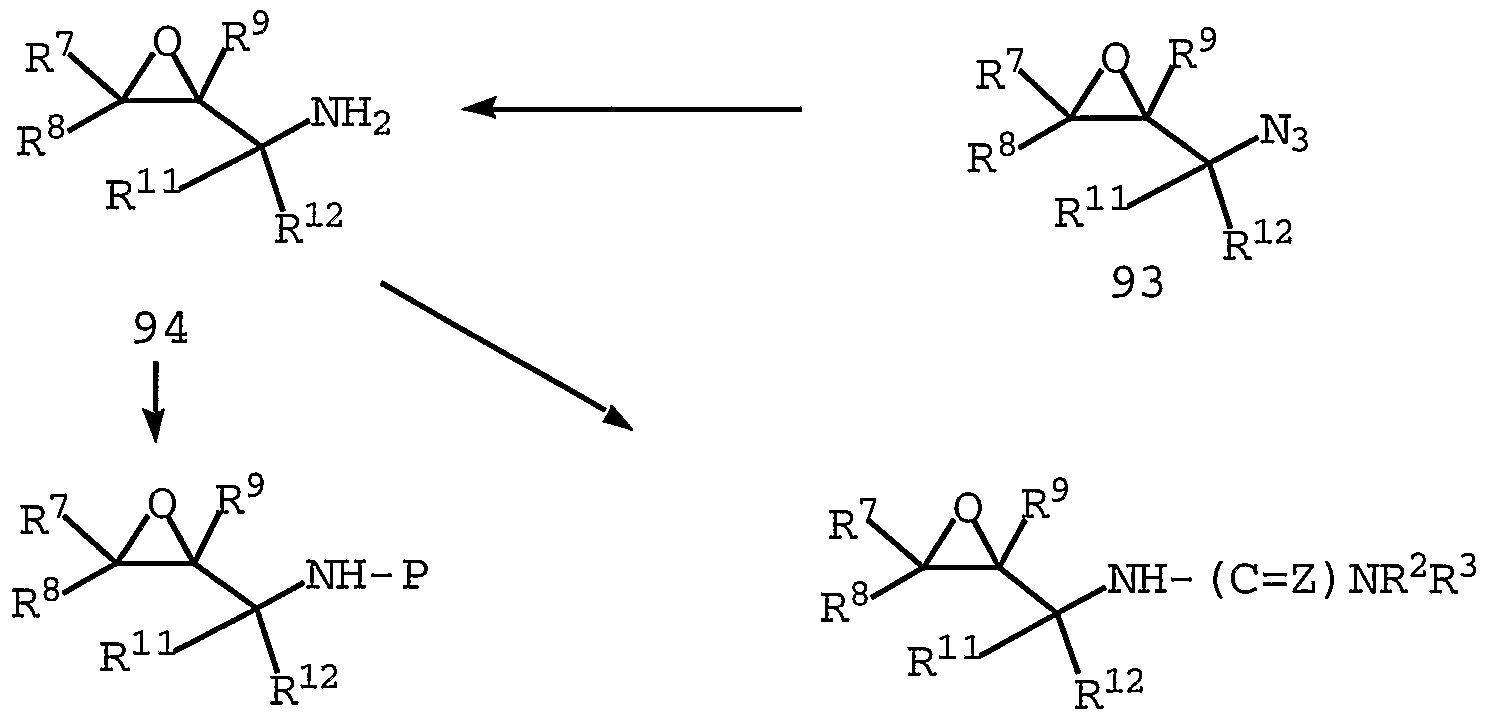


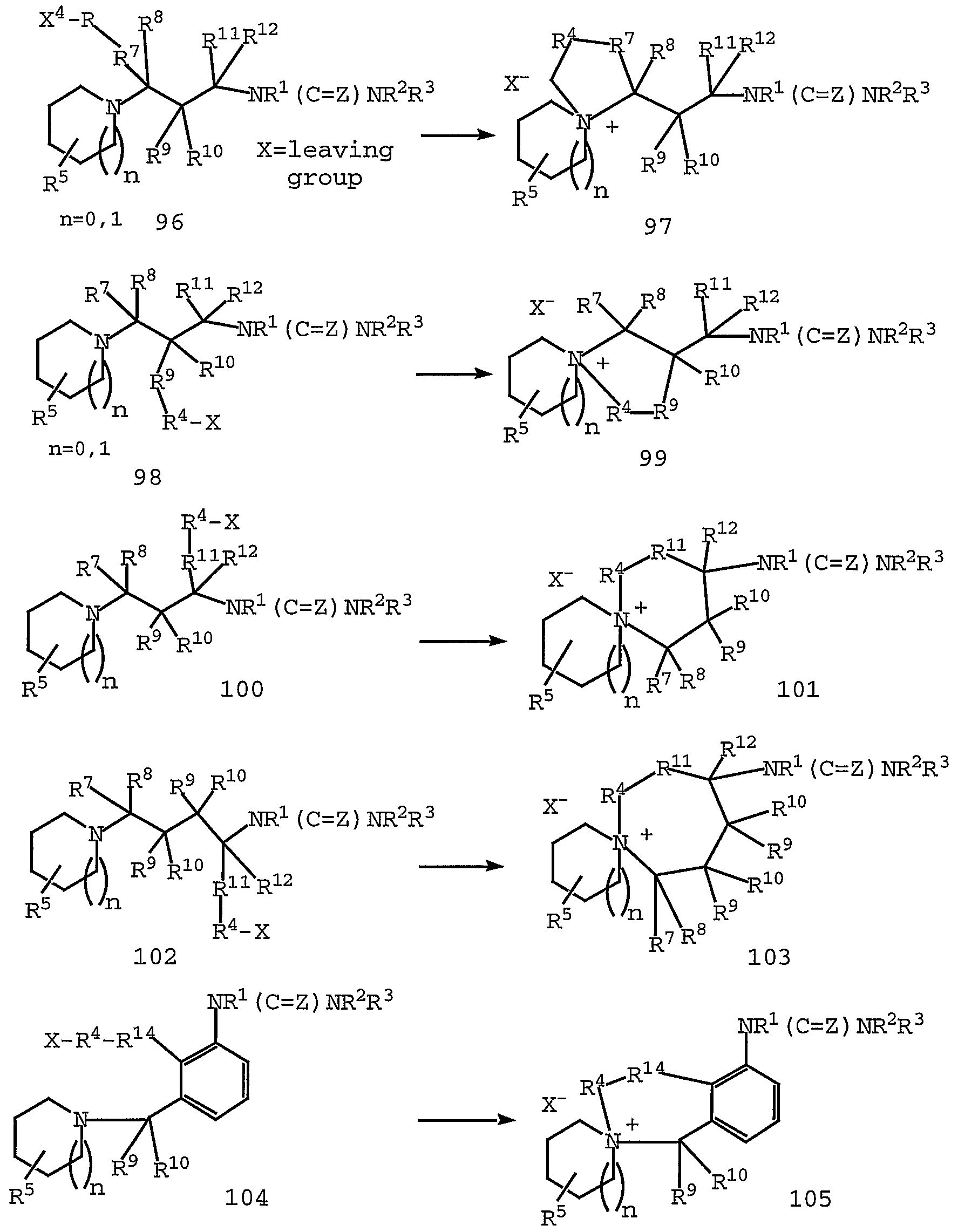






















































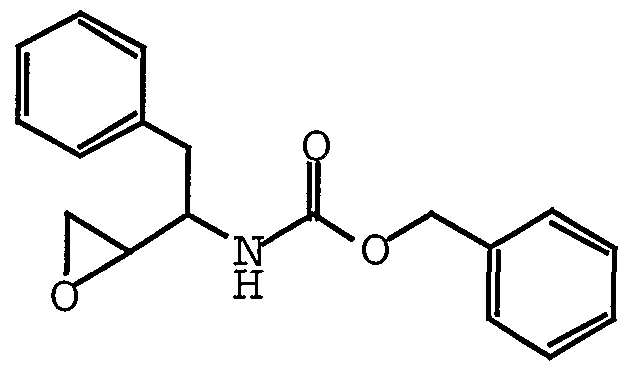

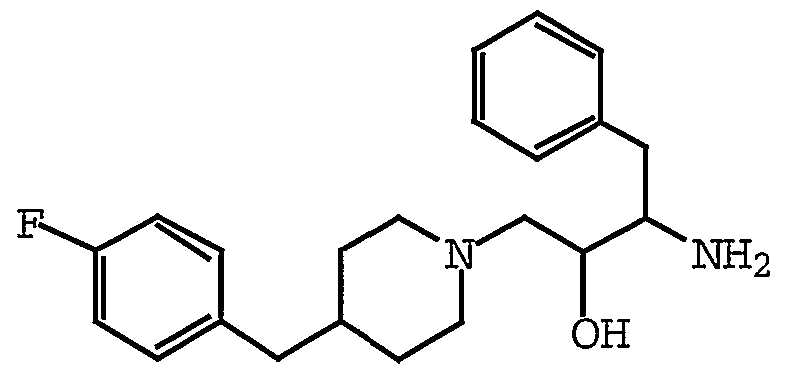













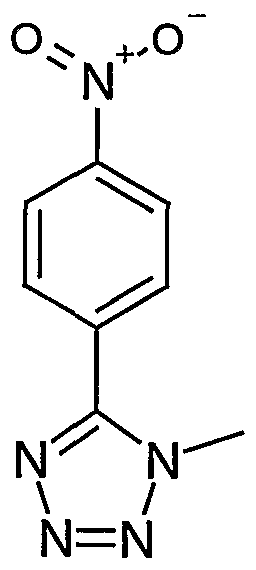





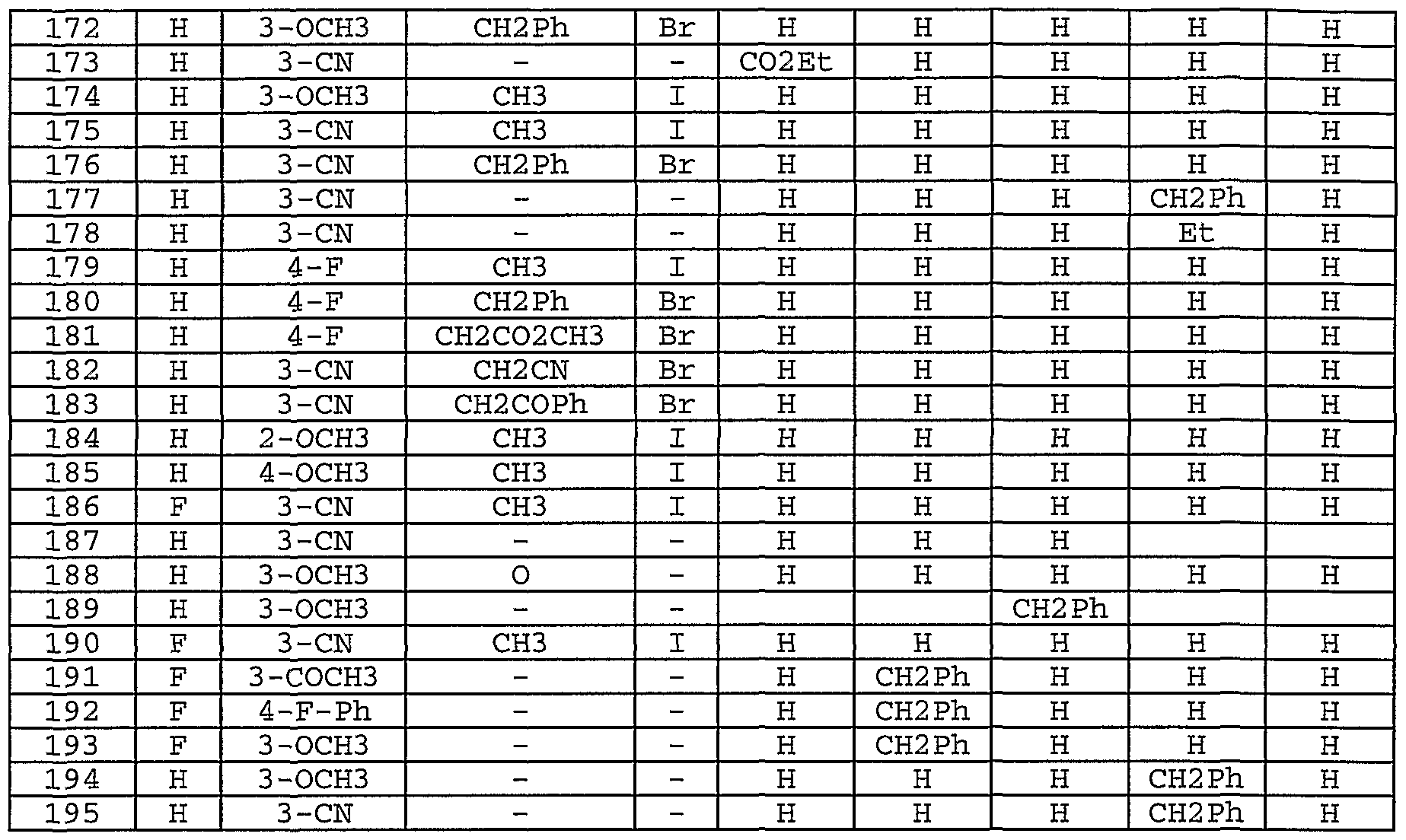















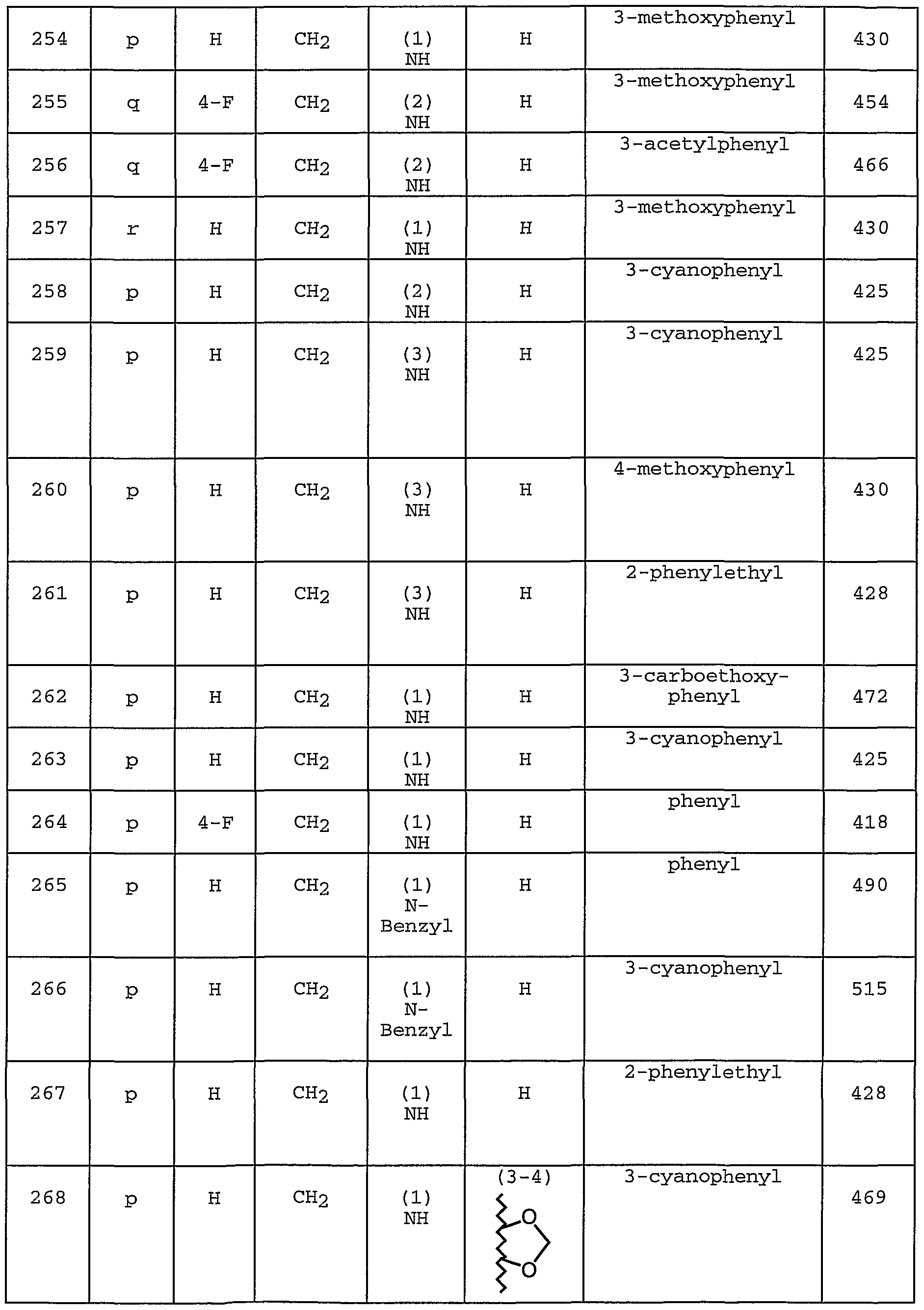




























































































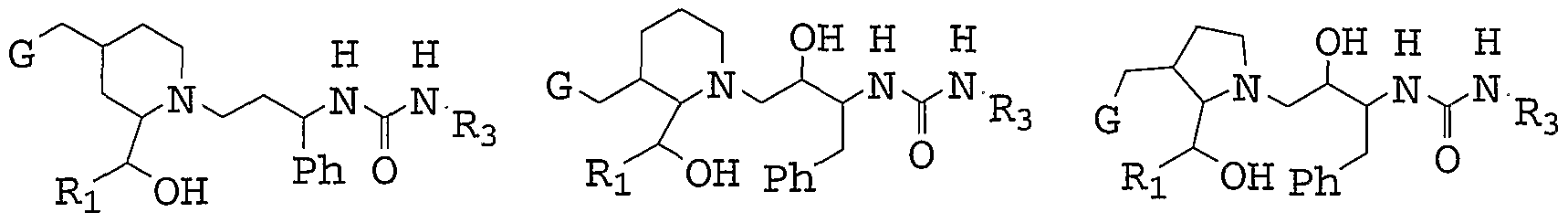

















Claims






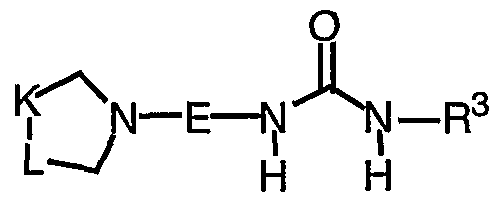


Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002413421A CA2413421A1 (en) | 2000-06-21 | 2001-06-20 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| EP01950360A EP1294690A2 (en) | 2000-06-21 | 2001-06-20 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| JP2002504226A JP2004516238A (en) | 2000-06-21 | 2001-06-20 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| AU7135901A AU7135901A (en) | 2000-06-21 | 2001-07-25 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US21320800P | 2000-06-21 | 2000-06-21 | |
| US60/213,208 | 2000-06-21 | ||
| US09/597,400 US6525069B1 (en) | 1998-12-18 | 2000-06-21 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US09/597,400 | 2000-06-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2001098270A2 true WO2001098270A2 (en) | 2001-12-27 |
| WO2001098270A3 WO2001098270A3 (en) | 2002-05-30 |
Family
ID=26907864
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2001/019752 Ceased WO2001098270A2 (en) | 2000-06-21 | 2001-06-20 | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1294690A2 (en) |
| JP (1) | JP2004516238A (en) |
| AU (1) | AU7135901A (en) |
| CA (1) | CA2413421A1 (en) |
| WO (1) | WO2001098270A2 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6525069B1 (en) | 1998-12-18 | 2003-02-25 | Bristol-Myers Squibb Pharma Co. | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6605623B1 (en) | 1998-12-18 | 2003-08-12 | Bristol-Myers Squibb Pharma Co. | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| WO2004032940A1 (en) * | 2002-10-11 | 2004-04-22 | Alchemia Limited | Classes of compounds that interact with gpcrs |
| WO2004058259A1 (en) * | 2002-12-24 | 2004-07-15 | Biofocus Plc | Compound libraries of n-(aminocarbonyl)-piperidine-4-carboxamide derivatives capable of binding to g-protein coupled receptors |
| WO2005021519A3 (en) * | 2003-08-28 | 2005-05-12 | Novartis Ag | 5-phenyl-4-methyl-thiazol-2-yl-amine derivatives as inhibitors of phosphatidylinositol 3 kinase enzymes (pi3) for the treatment of inflammatory airway diseases |
| WO2006037159A1 (en) * | 2004-10-04 | 2006-04-13 | Alchemia Limited | Selective inhibitors |
| AU2003266858B2 (en) * | 2002-10-11 | 2006-09-14 | Vast Bioscience Pty Limited | Classes of compounds that interact with GPCRs |
| WO2005113534A3 (en) * | 2004-05-12 | 2006-10-26 | Schering Corp | Cxcr1 and cxcr2 chemokine antagonists |
| WO2007024182A1 (en) * | 2005-08-26 | 2007-03-01 | Astrazeneca Ab | A combination of compounds, which can be used in the treatment of respiratory diseases, especially chronic obstructive pulmonary disease (copd) and asthma. |
| WO2007024183A1 (en) * | 2005-08-26 | 2007-03-01 | Astrazeneca Ab | A combination of compounds, which can be used in the treatment of respiratory diseases, especially chronic obstructive pulmonary disease (copd) and asthma |
| US7528156B2 (en) | 2000-06-20 | 2009-05-05 | Astrazeneca Ab | Compounds |
| US7709500B2 (en) | 2002-02-18 | 2010-05-04 | Astrazeneca Ab | Chemical compounds |
| US7956070B2 (en) | 2004-02-02 | 2011-06-07 | Astrazeneca Ab | Piperidines as chemokine modulators (CCR) |
| US8148405B2 (en) | 2005-08-02 | 2012-04-03 | Astrazeneca Ab | Salt I |
| US8222381B2 (en) | 2002-08-08 | 2012-07-17 | Alchemia Limited | Derivatives of monosaccharides for drug discovery |
| US12617796B2 (en) | 2020-05-07 | 2026-05-05 | Janssen Pharmaceutica Nv | Antagonists of the adenosine A2a receptor |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007098826A2 (en) * | 2006-01-18 | 2007-09-07 | Siena Biotech S.P.A. | Modulators of alpha7 nicotinic acetylcholine receptors and therapeutic uses thereof |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2993897A (en) * | 1961-07-25 | S-triazinylmethyl ureas | ||
| US3133061A (en) * | 1962-11-13 | 1964-05-12 | Sterling Drug Inc | Piperidine carboxamides and derivatives thereof |
| NL7800026A (en) * | 1977-01-07 | 1978-07-11 | Acna | PROCESS FOR THE PREPARATION OF SUBSTITUTED AMINO-BENZENES. |
| IL125658A0 (en) * | 1997-08-18 | 1999-04-11 | Hoffmann La Roche | Ccr-3 receptor antagonists |
| SE9802209D0 (en) * | 1998-06-22 | 1998-06-22 | Astra Pharma Inc | Novel compounds |
| UA73492C2 (en) * | 1999-01-19 | 2005-08-15 | Aromatic heterocyclic compounds as antiinflammatory agents | |
| WO2000076512A1 (en) * | 1999-06-11 | 2000-12-21 | Merck & Co., Inc. | Cyclopentyl modulators of chemokine receptor activity |
-
2001
- 2001-06-20 JP JP2002504226A patent/JP2004516238A/en active Pending
- 2001-06-20 WO PCT/US2001/019752 patent/WO2001098270A2/en not_active Ceased
- 2001-06-20 CA CA002413421A patent/CA2413421A1/en not_active Abandoned
- 2001-06-20 EP EP01950360A patent/EP1294690A2/en not_active Withdrawn
- 2001-07-25 AU AU7135901A patent/AU7135901A/en not_active Withdrawn
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6919368B2 (en) | 1998-12-18 | 2005-07-19 | Bristol-Myers Squibb Pharma Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6605623B1 (en) | 1998-12-18 | 2003-08-12 | Bristol-Myers Squibb Pharma Co. | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6525069B1 (en) | 1998-12-18 | 2003-02-25 | Bristol-Myers Squibb Pharma Co. | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US6906066B2 (en) | 1998-12-18 | 2005-06-14 | Bristol-Myers Squibb Pharma Company | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
| US7528156B2 (en) | 2000-06-20 | 2009-05-05 | Astrazeneca Ab | Compounds |
| US7709500B2 (en) | 2002-02-18 | 2010-05-04 | Astrazeneca Ab | Chemical compounds |
| US8222381B2 (en) | 2002-08-08 | 2012-07-17 | Alchemia Limited | Derivatives of monosaccharides for drug discovery |
| WO2004032940A1 (en) * | 2002-10-11 | 2004-04-22 | Alchemia Limited | Classes of compounds that interact with gpcrs |
| AU2003266858B2 (en) * | 2002-10-11 | 2006-09-14 | Vast Bioscience Pty Limited | Classes of compounds that interact with GPCRs |
| US7994140B2 (en) | 2002-10-11 | 2011-08-09 | Alchemia Limited | Classes of compounds that interact with GPCRs |
| WO2004058259A1 (en) * | 2002-12-24 | 2004-07-15 | Biofocus Plc | Compound libraries of n-(aminocarbonyl)-piperidine-4-carboxamide derivatives capable of binding to g-protein coupled receptors |
| US7902375B2 (en) | 2003-08-28 | 2011-03-08 | Novartis Ag | 5-phenyl-4-methyl-thiazol-2-yl-amine derivatives as inhibitors of phosphatidylin ositol 3 kinase enzymes (PI13) for treatment of inflammatory diseases |
| WO2005021519A3 (en) * | 2003-08-28 | 2005-05-12 | Novartis Ag | 5-phenyl-4-methyl-thiazol-2-yl-amine derivatives as inhibitors of phosphatidylinositol 3 kinase enzymes (pi3) for the treatment of inflammatory airway diseases |
| RU2382783C2 (en) * | 2003-08-28 | 2010-02-27 | Новартис Аг | Organic compounds |
| JP2007504109A (en) * | 2003-08-28 | 2007-03-01 | ノバルティス アクチエンゲゼルシャフト | 5-Phenyl-4-methyl-thiazol-2-yl-amine derivatives as inhibitors of phosphatidylinositol 3-kinase enzyme (PI3) for the treatment of inflammatory airway diseases |
| US7956070B2 (en) | 2004-02-02 | 2011-06-07 | Astrazeneca Ab | Piperidines as chemokine modulators (CCR) |
| US7326729B2 (en) | 2004-05-12 | 2008-02-05 | Schering Corporation | CXCR1 and CXCR2 chemokine antagonists |
| WO2005113534A3 (en) * | 2004-05-12 | 2006-10-26 | Schering Corp | Cxcr1 and cxcr2 chemokine antagonists |
| CN1984899B (en) * | 2004-05-12 | 2011-07-27 | 先灵公司 | CXCR1 and CXCR2 chemokine antagonists |
| WO2006037159A1 (en) * | 2004-10-04 | 2006-04-13 | Alchemia Limited | Selective inhibitors |
| US8148405B2 (en) | 2005-08-02 | 2012-04-03 | Astrazeneca Ab | Salt I |
| WO2007024183A1 (en) * | 2005-08-26 | 2007-03-01 | Astrazeneca Ab | A combination of compounds, which can be used in the treatment of respiratory diseases, especially chronic obstructive pulmonary disease (copd) and asthma |
| RU2414222C2 (en) * | 2005-08-26 | 2011-03-20 | Астразенека Аб | Combination of compounds, which can be applied in treatment of respiratory diseases, especially chronical obstructive lung disease (cold) and asthma |
| WO2007024182A1 (en) * | 2005-08-26 | 2007-03-01 | Astrazeneca Ab | A combination of compounds, which can be used in the treatment of respiratory diseases, especially chronic obstructive pulmonary disease (copd) and asthma. |
| US12617796B2 (en) | 2020-05-07 | 2026-05-05 | Janssen Pharmaceutica Nv | Antagonists of the adenosine A2a receptor |
Also Published As
| Publication number | Publication date |
|---|---|
| AU7135901A (en) | 2002-01-02 |
| EP1294690A2 (en) | 2003-03-26 |
| JP2004516238A (en) | 2004-06-03 |
| CA2413421A1 (en) | 2001-12-27 |
| WO2001098270A3 (en) | 2002-05-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1158980B1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6444686B1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6780857B2 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6331541B1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6919368B2 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| EP1156807A1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| EP1140086A1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6605623B1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| WO2001098270A2 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| EP1363881A2 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| US6897234B2 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| AU2001271359A1 (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity | |
| MXPA01006148A (en) | N-ureidoalkyl-piperidines as modulators of chemokine receptor activity |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2413421 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2002 504226 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001271359 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2001950360 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2001950360 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2001950360 Country of ref document: EP |