0-FLUORO-17 (20) -VINYL STEROIDS AS INHIBITORS OF C17-20-LYASE AND 5-ALPHA REDUCTASE
BACKGROUND OF THE INVENTION
1. Field of the Invention:
The present invention relates to 20-fluoropregna-5,17(20)-diene-3/3,21-diol, 20-fluoro-pregna-5, 17(20)- dien-3β-ol and related compounds, to processes for their preparation, and to compositions incorporating these compounds as well as the use of these compounds in the treatment of conditions which would be affected by inhibition of Ci7;20 lyase and/or 5α-reductase, including androgen and estrogen mediated or dependent disorders, such as, for example benign prostatic hyperplasia; dihydrotestosterone-mediated disorders such as, for example, acne; estrogen dependent breast cancer and androgen mediated prostatic cancer. The present invention provides a novel series of compounds which also disable the operation of Ciγ-hydroxylase; thus, disorders that are characterized by an oversynthesis of cortisol can also be treated by the compounds of the invention. For example, hypokalemia, metabolic alkalosis, polydipsia, polyurea, Cushing's syndrome and hypertensive conditions.
2. Description of the Art:
The enzyme steroid C1 20 lyase cleaves the 17-20 carbon-carbon bond in steroids having a two carbon side chain at the 17β-carbon position to form important precursor molecules for the formation of testosterone, 5 -dihydrotestosterone and the estrogens, principally estrone and estradiol. Compounds which inhibit this enzyme would thus serve to inhibit the formation of the indicated precursors and thereby be useful in the treatment of various androgenic as well as estrogenic disorders. A treatment incorporating such enzymatic inhibitors is not limited to the origin of the precursor molecule, such as various organ ablation techniques which are currently known. For example, while orchiectomy will effectively reduce gonadal androgen, it will have not have significant effect upon adrenal androgen production. Moreover, such an enzymatic treatment is a much more focused treatment in that it is directed to the immediate hormonal imbalance believed responsible for the condition, as opposed to a broad spectrum remedy which not only affects the particular symptom, but causes permanent endocrine deficits necessitating life-long dependency on replacement therapy.
It is further known that certain types of breast cancers are estrogen dependent. Adrenalectomy, ovariectomy and hypophysectomy have been employed as well as non-surgical techniques resulting in tumor regressions. It has been shown that human patients with advanced breast cancer, who are administered estrogen biosynthesis enzyme inhibitors, show dramatically reduced plasma estradiol levels and improved therapeutic effects, at least as effective as adrenalectomy. [Van Wauve, J. and Janssen, P.A.J., J. Med. Chem. 1989, 32, 2231-2239].
Prostatic cancer, or neoplastic tissue disorders which originate in the parenchymal epithelium of the prostate, is one of the most common malignancies among men, and exhibits one of the highest cancer-specific deaths of all malignant carcinomas. It is known that patients with metastatic prostate cancer respond positively to hormonal therapy. It is reported by Cookson and Sarosdy that androgen ablation has had a positive, beneficial response for as high as 60% to 80% of all patients tested. [Cookson, CS. and Sarosdy, M.F., South Med. J. 1994, 87, 1-6].
More specifically, Cι7)20 lyase inhibitors would be useful in the treatment of hormonal dependent prostatic carcinoma, prostatic hyperplasia, virilism, congenital adrenal hyperplasia due to 21 -hydroxylase deficiency, hirsutism, hormonal dependent breast cancer, polycystic ovarian syndrome correlated with elevated
C 17,20 lyase activity as well as other neoplastic tissue disorders such as endometrial, hepatocellular and adrenal carcinomas.
The enzyme steroid 5α-reductase, present in mammalian tissues including skin, male genitalia and the prostate, catalyzes the conversion of testosterone (17β-hydroxyandrost-4-en-3-one) into dihydrotestosterone or DHT (17β-hydroxy-5α-androstan-3-one), which is also known as stanolone. DHT is a more potent androgen than testosterone, and acts as an end-organ effector in certain tissues, particularly in mediating growth. DHT formation can occur in certain tissues themselves by the action of 5α-reductase. The conversion of testosterone to DHT itself can be associated with various androgenic disorders, especially when DHT levels build up to excessive amounts. For example, high levels of DHT in the skin has been associated in the pathogenesis of acne, including acne vulgaris. In the treatment of androgen mediated or androgen dependent disorders, such as acne, benign prostatic hyperplasia and prostatic cancer, including hormonal dependent carcinoma, the inhibition of DHT would be highly desirable.
Agents that have the ability to inhibit both Cι7;20 lyase and 5α-reductase would not only inhibit DHT production, but also testosterone formation. In inhibiting the principal androgenic steroidal hormones, such compounds would have enhanced utility in the treatment of androgen mediated or dependent disorders.
The enzyme C\η hydroxylase catalyzes the Cι7 hydroxylation of steroid substrates during the biosynthesis of cortisol. As C1 ;20 lyase and Cj7 hydroxylase are the same active site of the same enzyme, the inhibition of one usually results in the inhibition of the other. Cortisol excess results in a syndrome characterized by
hypokalemia, metabolic alkalosis, polydipsia, polyuria, Cushing's syndrome and hypertensive conditions. Inhibition of cortisol synthesis via Cj7 hydroxylase would, therefore, have a beneficial therapeutic effect for the treatment of these disorders or conditions.
SUMMARY OF THE INVENTION
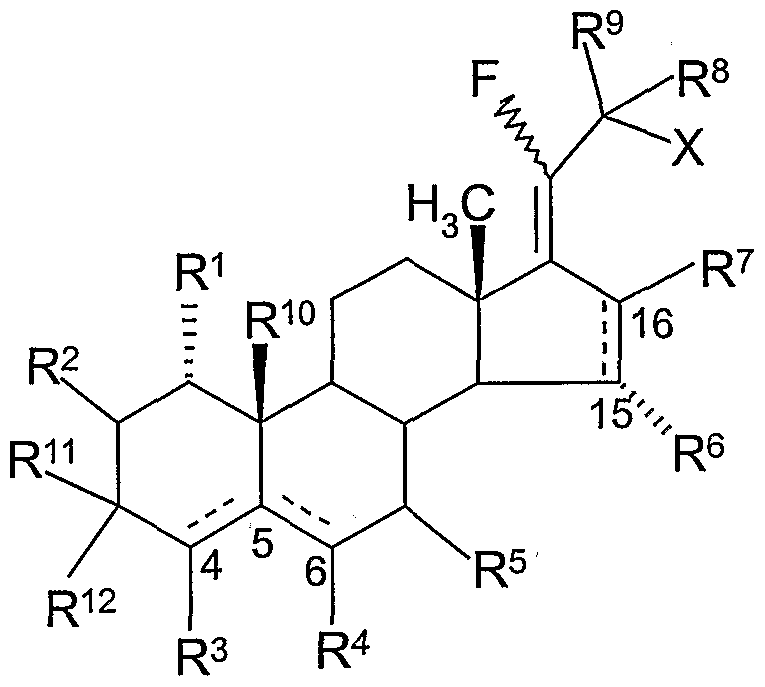
More particularly, the present invention is directed to a group of compounds, and to their pharmaceutically acceptable salts, of the formula:
Rl is H or C ι_4 alkyl;
R2 is H or C i-4 alkyl;
R3 is H, chloro, nirro, amino or Cι-4 alkyl;
R4 is H or C i-4 alkyl;
R5 is H or C i-4 alkyl;
R6 is H or methyl;
R7 is H or methyl;
R8 is H or methyl ;
R9 is H or methyl; or R8 and R9 taken together is oxo;
R10 is H or methyl;
Rn is H;
R12 is hydroxy; or Rn and R12 taken together is oxo; X is H, hydroxy or methoxy; with the proviso that when: a) Ru is H and R12 is hydroxy, bond C ι5 is a single bond, bond C5,6 is a double bond and bond Cι5>ι6 is optionally a single bond or a double bond , and
b) Ru and R12 taken together is oxo, bond C^5 is a double bond, bond C5j6 is a single bond, and bond Cι5)i6 is a single bond.
Another embodiement of the invention provides use of the compounds of the invention as inhibitors of Ci7,2o lyase and 5α-reductase for the treatment of androgen or estrogen mediated or dependent disorders such as breast cancer, polycystic ovarian syndrome, prostatic hyperplasia, prostatic cancer, virilism, hirsutism, and acne.
In another embodiement, the invention provides use of the compounds of the invention for the treatment of Cushing's syndrome.
In yet another embodiement, the invention provides a pharmaceutical composition comprising a compound of the invention and a pharmaceutically acceptable carrier.
In another embodiment, the compounds of the invention may be administered in combination with other effective treatments for enhanced therapeutic effect. For example, in the treatment of androgen-dependent disorders, including prostatic cancer, flutamide, a known androgen receptor antagonist, may be used in combination with compounds of the invention.
A preferred embodiement of the invention are compounds wherein R1, R2, R3, R4, R5, Rδ, R7, R8, R9, Ru and X are hydrogen, R10 is methyl, R12 is hydroxy, bond C4j5 and bond C15ιl6 are each a single bond and bond C5]6 is a double bond.
A most preferred embodiement of the invention are compounds wherein R1, R2, R3, R4, R5, R6, R7, R8, R9 and Rn are hydrogen, R10 is methyl, X and R12 are each hydroxy, bond C^s and bond C15 6 are each a single bond and bond Cs^ is a double bond.
DETAILED DESCRIPTION OF THE INVENTION
As used herein, the term "C1.4 alkyl" means any straight or branched chain alkyl radical of one to four carbon atoms, for example, methyl, ethyl, n-propyl, z'-propyl, «-butyl, s-butyl, or f-butyl.
As used herein, the following structural designations as used in the formulas shall have the following meanings: is defined as a bond below the plane of the steroid (the α-face).
,^— is defined as a bond above the plane of the steroid (the β-face). v Λ" is defined as a cis or trans bond (or mixture of the two) whose stereochemistry is not defined. is defined as an optional double bond.
As used herein, the term "pharmaceutically acceptable salt" is intended to apply to any salt, whether previously known or future discovered, that is used by one skilled in the art that is a non-toxic organic or inorganic addition salt which is suitable for use as a pharmaceutical. Illustrative bases which form suitable salts include alkali metal or alkaline-earth metal hydroxides such as sodium, potassium, calcium or magnesium hydroxides; ammonia and aliphatic, cyclic or aromatic amines such as me ylamine, dimethylamine, triemylarnine, diethylamine, isopropyldiefhylamine, pyridine and picoline. Illustrative acids which form suitable salts include inorganic acids such as, for example, hydrochloric, hydrobro ic, sulfuric, phosphoric and like acids, and organic carboxylic acids such as, for example, acetic, propionic, glycolic, lactic, pyruvic, malonic, succinic, fumaric, malic, tartaric, citric, ascorbic, maleic, hydroxymaleic and dihydroxymaleic, benzoic, phenylacetic, 4-aminobenzoic, 4-hydroxybenzoic, anthranilic, cinnamic, salicylic, 4-aminosalicylic, 2- phenoxybenzoic, 2-acetoxybenzoic, mandelic and like acids, and organic sulfonic acids such as methanesulfonic and p-toluenesulfonic acids.
As used herein "stereoisomer" is a general term used for all isomers of individual molecules that differ only in the orientation of their atoms in space. The term stereoisomer includes mirror image isomers (enantiomers), geometric (cis/trans or E/Z) isomers, and isomers of compounds with more than one chiral center that are not mirror images of one another (diastereoisomers).
As used herein, the term "effective inhibitory amount," is such an amount wherein an enzyme inhibitory effect is achieved to cause a therapeutic effect in a patient. The exact amount of compound to be administered can be readily determined by the attending diagnostician, as one skilled in the art, by the use of conventional techniques and by observing the results obtained under analogous circumstances. Factors significant in determining the dose include: the species of animal, the animal's size, age and general health; the specific disease or disorder involved, the degree of involvement or the severity of the disease; the response of the individual patient; the particular compound administered; the mode of administration; the bioavailability characteristics of the preparation administered; the dose regimen selected; the use of concomitant medication; and other relevant circumstances. That said, the exact amount employed may vary over a wide range, for example, from about 0.625 to 200 mg/kg of body weight per day, preferably from about 5 to 100 mg/kg of body weight per day.
"Treat" or "treating" means any treatment, including but not limited to, alleviating symptoms, eliminating the causation of the symptoms either on a temporary or permanent basis, or to preventing or slowing the appearance of symptoms and progression of the named disease, disorder or condition.
As described herein, the term "patient" refers to a warm blooded animal such as a mammal which is afflicted with a particular disease, disorder or condition. It is explicitly understood that guinea pigs, dogs, cats, rats, mice, horses, cattle, sheep, and humans are examples of animals within the scope of the meaning of the term.
In practicing the methods of this invention, the active ingredient is preferably incorporated into a composition containing a pharmaceutical carrier, although the compounds are effective and can be administered,
in and of themselves. The term "pharmaceutical carrier" refers to known pharmaceutical excipients useful in formulating pharmaceutically active compounds for administration, and which are substantially nontoxic and nonsensitizing under conditions of use. The exact proportion of these excipients are determined by the solubility and chemical properties of the active compound, the chosen route of adniinistration as well as standard pharmaceutical practice. That said, the proportion of active ingredient can vary from about 5 to 90% by weight.
FORMULATIONS
The pharmaceutical compositions of the invention are prepared in a manner well known in the pharmaceutical arts. The carrier or excipients may be a solid, semisolid or liquid material which can serve as a vehicle or medium for the active ingredient. Suitable carriers or excipients are well known in the art. The pharmaceutical composition may be adapted for oral, inhalation, parenteral or topical use, and may be administered to the patient in the form of tablets, capsules, suspensions, syrups, aerosols, inhalants, suppositories, salves, powders, solutions and the like. As used herein, the term "pharmaceutical carrier" means one or more excipients.
In preparing formulations of the compounds of the invention, care should be taken to ensure bioavailability of an effective inhibitory amount, including oral, parenteral and subcutaneous routes. For example, effective routes of administration may include subcutaneously, intravenously, transdermally, intranasally, rectally, vaginally and the like including release from implants as well as direct injection of the active ingredient and/or composition directly into the tissue or tumor sites. Suitable pharmaceutical carriers and formulation techniques are found in standard texts, such as Remington: The Science and Practice of Pharmacy, 19th edition, Volumes 1 and 2, 1995, Mack Publishing Co., Easton, Pennsylvania, U.S.A., which is herein incorporated by reference.
For oral administration, the compounds can be formulated into solid or liquid preparations, with or without inert diluents or edible carriers, such as capsules, pills, tablets, troches, powders, solutions, suspensions or emulsions. The capsules, pills, tablets, troches and the like may also contain one or more of the following adjuvants: binders such as microcrystalline cellulose, gum tragacanth; excipients such as starch or lactose, disintegrating agents such as alginic acid, corn starch and the like; lubricants such as stearic acid, magnesium stearate or Sterotex®, (Stokely-Van Camp Inc., Indinapolis, Indiana) glidants such as colloidal silicon dioxide; sweetening agents such as sucrose or saccharin; and flavoring agents such as peppermint, methyl salicylate or fruit flavoring. When the dosage unit form is a capsule, it may also contain a liquid carrier such as polyethylene glycol or a fatty oil. Materials used should be pharmaceutically pure and nontoxic in the amounts used.
For parenteral administration, the compound may be administered as injectable dosages of a solution or suspension of the compound in a physiologically acceptable diluent with a pharmaceutical carrier which can be a sterile liquid such as water-in-oil or without the addition of a surfactant and other pharmaceutically acceptable excipients. Illustrative oils which can be employed in the preparations are those of petroleum, animal, vegetable or synthetic origin such as, for example, peanut oil, soybean oil and mineral oil. In general, water, saline,
aqueous dextrose and related sugar solutions, ethanol and glycols, such as propylene glycol are preferred liquid carriers, particularly for injectable solutions. The parenteral preparation can be enclosed in ampoules, disposable syringes or multiple dose vials made of inert plastic or glass.
The solutions or suspensions described above may also include one or more of the following adjuvants: sterile diluents such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerin, propylene glycol or other synthetic solvents, antibacterial agents such as ascorbic acid or sodium bisulfite; chelating agents such as efhylenediaminetetra-acetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose.
The compounds can be administered in the form of a cutaneous patch, a depot injection or implant preparation which can be formulated in such a manner as to permit a sustained release of the active ingredient. The active ingredient can be compressed into pellets or small cylinders and implanted subcutaneously or intramuscularly as depot injections or implants. Implants may employ inert materials such as biodegradable polymers and synthetic silicones. Further information on suitable pharmaceutical carriers and formulation techniques are found in standard texts such as Remington: The Science and Practice of Pharmacy, 19 edition, Volumes 1 and 2, 1995, Mack Publishing Co., Easton, Pennsylvania, U.S.A.
CHEMICAL SYNTHESES The compounds of the present invention can be prepared by processes analogous to those known in the art. Reaction schemes A to O and the corresponding descriptive text describe the preparation of the various compounds of the invention. The methods disclosed and examples are provided for illustration purposes and in no way limit the scope of the present invention. Alternative reagents, reaction conditions, and other combinations and permutations of the steps herein described to arrive at individual compounds are readily apparent to one of ordinary skill in the art.
List of Abbreviations
DIBALH = diisobutylalurninum hydride; DMAP = 4- dimethylaminopyridine; DMF = dimethylformamide; LAH = lithium aluminum hydride; LHMDS = lithium hexamethyldisilazide; NBS = N-bromosuccinimide; PCC = pyidinium chlorochromate; PDC = pyridinium dichromate; Pyr-S03 = sulfur trioxide pyridine complex; TBAF = tetrabutylammorrium fluoride; TBDMS = t-butyldimethyl-silyl; TEA = triethylamine; THF = tetrahydrofuran; Ac20 = acetic anhydride; TsOH = tosic acid (p-toluenesulphonic acid); ξ = designation for undefined geometry about a double bond, g = grams; mmol = millimole, mL = milliliters; bp = boiling point; mp = melting point; °C = degrees Celsius; mm Hg = millimeters of mercury; μL = microliters, . μg = micrograms; μM = micromolar; mM = millimolar; μCi = microcurie; M = molar; NADPH = hydrogenated nicotinamide adenine dinucleotide phosphate; DMSO = dimethylsulfoxide; EDTA = emylenediaminetetraacetic acid; HPLC = high performance liquid chromatography.
Scheme A. Unsubstituted Steroid-5-en-3-ols
A4 (E-isomer)
Scheme B. C
2ι Substituted Steroid-5-en-3-oIs
B17
The unsubstituted steroid-5-en-3-ols of this invention may be prepared by as depicted in Scheme A. Protecting the hydroxyl group of dehydroepiandrosterone (Al) by reaction with t-butyldimethylsilyl chloride gives silyl ether A2. Wittig reaction on the Cι ketone of A2 with the ylid formed by reaction of triethyl 2- fluoro-2-phosphonoacetate with a suitable base such as lithium hexamethyldisilazide gives vinyl fluoride ester A3 as a mixture of E- and Z-isomers. A suitable base in this instance is any base that will function to form a ylid by reaction with triethyl 2-fluoro-2-phosphonoacetate such as, for example, lithium hexamethyldisilazide, alkyl lithium bases such as t-butyllitbium, potassium t-butoxide and the like. Separation of the isomers is possible, but difficult at this point, so it is usually done after the next step. Reduction of ester A3 is accomplished with a suitable reducing agent such as diisobutylaluminum hydride in dichloromethane to give a mixture of hydroxymethyl vinyl fluorides which are separated into the individual E- and Z-isomers A4 and A5, respectively. Removal of the silyl protecting group of E-olefin A4 with tetrabutylammonium fluoride gives diol A6. Similarly, the silyl protecting group of Z-olefin A5 is removed to yield diol A9. Further reduction of alcohol A4 or A5 using sulfur trioxide pyridine complex in tetrahydrofuran followed by treatment with lithium aluminum hydride gives the corresponding C21 deoxy derivatives A7 and AlO, respectively. Removal of the silyl
protecting groups from A7 and AlO as described above gives alcohols A8 and All, respectively.
The C2ι substituted steroid-5-en-3-ols of this invention may be prepared following the methodology depicted in Scheme B. Using the mixture of vinyl fluoride esters A3 as starting material, the following transformations can be accomplished. The silyl group of A3 is removed using tetrabutylammonium fluoride giving alcohol B12. The latter is oxidized with pyridinium chlorochromate (see Parish, E.J. and Honda, H. Syn. Commun., 1990, 20, 1167-1174) to give conjugated ketone B13. Reduction of the ester group of compound B13 required a two step sequence. Thus, treating B13 with trimethyl orthoformate in the presence of tosic acid provides dienol ether B14, and then the ester of B14 is reduced with DIBALH in methylene chloride (CH2C12). If the work-up involves treatment with dilute hydrochloric acid, hydroxy-enone B15 is isolated. Addition of excess methyl Grignard to A3 gives tertiary alcohol B16. Careful removal of the silyl protecting group from B16 with tetrabutylammonium fluoride in tetrahydrofuran gives the desired diol B17.
Scheme C. Ci Substituted Steroid-4-en-3-ones
(EtO)2 POCHFC02 Et LHDMS, THF
Scheme D. Ci Substituted Steroid-5-en-3-ols
The Ci substituted steroid-4-en-3-ones of this invention may be prepared as depicted in Scheme C. The starting lα-alkylandrost-5-ene-3,17-diones (C19) are prepared from androsta-l,4-diene-3,17-dione (C18) according to Westermann and Nickisch (Westermann, J. and Nickisch, K., 1993, Angew. Chem. Int. Ed. Engl, 32, 1389-1370). The enone C19 is then protected as a dienol ether by treating C19 with trimethyl orthoformate in the presence of tosic acid. The resulting dienol ether €20 is reacted with the ylid formed by reaction of triethyl 2-fluoro-2-phosphonoacetate with base to give the vinyl fluoride C21. Diisobutylaluminum hydride reduction of the ester group in C21, followed by acid catalyzed hydrolysis of the dienol ether gives the desired 21-hydroxy-20ξ-fluoro-lα-methylpregna-4,17(20)-dien-3-one (C23). Similar hydrolysis of the dienol ether €21 gives the corresponding 20ξ-fluoro-lα-methyl-pregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (C24).
The Ci substituted steroid-5-en-3-ols of this invention may be prepared as depicted in Scheme D. The starting material, 20ξ-fluoro-lα-methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (C24a), is first converted to the 3,5-dienol acetate D25a using acetic anhydride in refluxing toluene with a strong acid such as perchloric or tosic acid as a catalyst. Reduction of the 3,5-dienol acetate moiety with sodium borohydride is known to give the corresponding 5-en-3-/3ol, which, in this case, affords compound D26a. Further reduction of D26a with diisobutylaluminum hydride gives 20ξ-fluoro-l -methylpregna-5,17(20)-diene-3β,21-diol (D27a). Compounds D27b and D27c are prepared in similar manner
The C2 substituted steroid-4-en-3-ones of this invention may be prepared as depicted in Scheme E. The known 2α-methylandrost-5-ene-3,17-dione (E29a, Iriarte, J. and Ringold, H.J., 1958, Tetrahedron, 3, 28-36) and 2 -ethylandrost-5-ene-3,17-dione (E29b, prepared by the methods of Ringold, H.J. and Rosenkranz, G., 1976, J.
Org. Chem., 1976, 21, 1333-1335), serve as starting materials. Enone E29 is first protected as a dienol ether by treating E29 with trimethyl orthoformate in the presence of tosic acid. The resulting dienol ether E30 is reacted with the ylid formed by reaction of triethyl 2-fluoro-2-phosphonoacetate with base to give the vinyl fluoride ester E31. Diisobutylaluminum hydride reduction of the ester group, followed by acid catalyzed hydrolysis of the dienol ether gives the desired 21-hydroxy-20ξ-fluoro-2α-alkylpregna-4,17(20)-dien-3-ones (E33a and E33b). Similar hydrolysis of the dienol ethers E31 gives the corresponding 20ξ-fluoro-2 -alkylpregna-4,17(20)-dien-3- on-21-oic acid ethyl esters E34a and E34b.
Scheme E. C2 Substituted 4-en-3-ones
HC(OCH3)3 TsOH
The C2 substituted steroid-5-en-3-ols of this invention may be prepared as depicted in Scheme F. The starting 20ξ-fluoro-2α-alkylpregna-4,17(20)-dien-3-on-21-oic acid ethyl esters (E34a-c) are converted to the 3,5- dienol acetates F35 using acetic anhydride in refluxing toluene with a strong acid such as perchloric or tosic acid as a catalyst. Reduction of F35 with sodium borohydride gives the corresponding steroid-5-en-3-ols F36. Further reduction of F36 with diisobutylaluminum hydride and acid hydrolysis gives 20ξ-fluoro-2α-methyl-
pregna-5,17(20)-diene-3β,21-diol (F37a), 20ξ-fluoro-2α-ethylpregna-5,17(20)-diene-3β,21-diol (F37b), 20ξ- fluoro-2α-propylρregna-5,17(20)-diene-3β,21-diol (F37c).
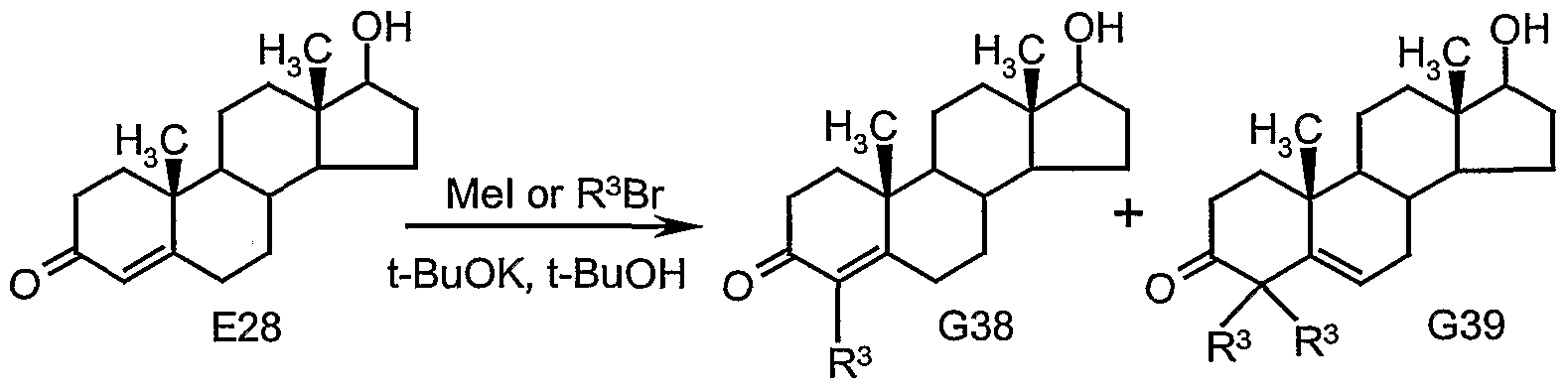
The C4 substituted steroid-4-en-3-ones of this invention may be prepared as depicted in Scheme G2. The starting point for the synthesis of each of these compounds is the appropriately substituted 4-alkytestosterone derivatives whose syntheses are detailed in Scheme Gl. We found the most convenient route to these starting materials (G38) to be direct alkylation of testosterone (E28) by slow addition of alkyl iodide or alkyl bromide to a reluxing solution of testosterone and potassium t-butylate in t-butanol as described by Atwater (Atwater, N.W., J. Am. Chem. Soc, 1960, 82, 2847-2852). These compounds are also prepared by addition of an appropriate Grignard reagent to enol lactone G40 followed by Robinson annelation (see Ringold, H.J. and Rosenkranz, G., 1957, J. Org. Chem., 22, 602-605). By this latter process the branched alkyl substituted steroid 17β-hydroxy-4- (2-propyl)androst-4-en-3-one (G38c) is prepared. Jones oxidation of G38a-d provides 4-substituted steroid 4- en-3-ones G41a-d, respectively. 4-Chloroandrost-4-ene-3,17-dione (G41e) is prepared by reaction of androstenedione (G42) with sulfuryl chloride in pyridine as previously described (Kirk, D.N., Patel, D.K. and Petrow, V., J. Chem. Soc, 1956, 1184-1186; Mori, H., Chem. Pharm. Bull, 1962, 10, 429-432).
Transformation of the various 4-substituted steroid 4-en-3-ones G41a-e to vinyl fluorides G44a-e is shown in Scheme G2 and follows the general strategy previously developed. The steroid 4-en-3-one C41 is first protected as a dienol ether treating G41 with trimethyl orthoformate in the presence of tosic acid. The protected steroid G43 is then reacted with the ylid formed by reaction of triethyl 2-fluoro-2-phosphonoacetate with base to give the vinyl fluoride ester G44. Diisobutylaluminum hydride reduction of the ester group of G44, followed by acid catalyzed hydrolysis of the dienol ether gives the desired 21-hydroxy-20ξ-fluoro-4-substituted-pregna- 4,17(20)-dien-3-ones (G46a-e). Similar acid hydrolysis of the dienol ether moiety of vinyl fluoride esters G44a- e gives the corresponding 20ξ-fluoro-4-substituted-pregna-4,17(20)-dien-3-on-21-oic acid ethyl esters (G47a-e).
The 4-nitro- and 4-aminosteroids (Scheme G3) are prepared using methodologies developed by Curran et al. (Curran, T.T., Flynn, G.A., Rudisill, D.E. and Weintraub, P.M., 1995, Tetrahedron Lett, 36, 4761-4764). By way of example, 20ξ-fluoro-21-hydroxy-l -methylpregna-4,17(20)-dien-3-one (G48Aa) is reacted with t- butylate in t-butanol to form the thermodynamic enolate which then is reacted with i-propyl nitrate to give 20ξ- fluoro-21 -hydroxy- 1 -methyl-4-nitropregna-4, 17(20)-dien-3-one (G49Aa). 20ξ-Fluoro-2 l-hydroxy-7α-methyl- 4-nitropregna-4,17(20)-dien-3-one (G49Ab) and 20ξ-fluoro-21-hydroxy-15α-mefhyl-4-nitropregna-4,17(20)- dien-3-one (G49Ac) are prepared in an analogous manner. Chemoselective reduction of the nitro groups in G49Aa-c is accomplished by catalytic hydrogenation over Lindlar catalyst giving the corresponding amines: 4- amino-20ξ-fluoro-21 -hydroxy- 1 α-methylpregna-4, 17(20)-dien-3-one (G50 Aa), 4-amino-20ξ-fluoro-21 -hydroxy- 7α-methylpregna-4,17(20)-dien-3-one (G50Ab) and 4-amino-20ξ-fluoro-21 -hydroxy- 15α-methylpregna- 4,17(20)-dien-3-one (GSOAc), respectively. In the fashion just described, the 4-nitro-C2i -esters G49Ba-c are prepared and transformed into the 4-amino-C2i -esters G50Ba-c.
Scheme F. C2 Substituted 5-en-3-ols
NaBH4 EtOH
F37 F36 a. R2 =CH3, b. R2 =C2H5, c. R2 =C3H7
Scheme Gl. C4 Alkylandrost-4-ene-3,17-diones
1. 03/CH2CI2/MeOH a. R3=CH3
2. Ac20 b. R3=C2H5 c. R3=i-C3H7 d. R3=C4H9
The C4 substituted steroid-5-en-3-ols described in this invention may be prepared as depicted in Scheme H. Starting materials are the 20ξ-fluoro-4-alkylpregna-4,17(20)-dien-3-on-21-oic acid ethyl esters (G47a-e) described in Scheme G2. As in the previous examples, the steroid 4-en-3-ones G47 are first converted to the 3,5- dienol acetates H51 using acetic anhydride in refluxing toluene with a strong acid such as perchloric or tosic acid as a catalyst. Reduction of the 3,5-dienol acetates H51 to the corresponding 5-en-3-ols H52 is effected with sodium borohydride. Further reduction of H52 with diisobutylaluminum hydride gives the corresponding 20ξ- fluoro-4-substituted ρregna-5,17(20)-diene-3β,21-diols (H53a-e).
Scheme G2. C4 Substituted Steroid-4-en-3~ones
The C6-alkylandrost-4-ene-3,17-diones (156) which serve as starting material for the C6 substituted steroids of this invention are synthesized in five steps (the latter three are shown in Scheme I starting from 154) from androst-4-ene-3,17-dione using a method previously reported (Numazawa, M. and Oshibe, M. J. Med. Chem., 1994, 37, 1312-1319). After the C3 carbonyls of I56a-c are protected as dienol ethers I57a-c, a Wittig reaction is performed on the C17 ketones with the ylid formed by reaction of triethyl 2-fluoro-2- phosphonoacetate with base to give the vinyl fluoride esters I58a-c as mixtures of E- and Z-isomers. Diisobutylaluminum hydride reduction of the ester group of I58a-c followed by mild acid hydrolysis of the dienol ether protecting group of I59a-c affords the 20ξ-fluoro-6-substituted-pregna-4,17(20)-dien-21-ol-3-one
(I60a-c). Acid catalyzed unmasking of the C3 carbonyl of I58a-c gives the corresponding 20ξ-fluoro-6- substitutedpregna-4,17(20)-dien-3-on-21-oic acid ethyl esters (I61a-c).
Scheme G3. C4-Nitro and C4-Amino Steroids
Scheme H. C4 Substituted Steroid-5-en-3-ols
Scheme I. Cβ Substituted Steroid-4-en-3-ones
Scheme J. Cβ Substituted Steroid-5-en-3-ols
The C6 substituted steroid-5-en-3-ols described in this invention may be prepared as depicted in Scheme J. Starting materials are the 20ξ-fluoro-6-alkylpregna-4,17(20)-dien-3-on-21-oic acid ethyl esters (I61a-c) described in Scheme I. As in the previous examples, the steroid 4-en-3-ones (161 a-c) are first converted to the 3,5-dienol acetates J62a-c using acetic anhydride in refluxing toluene with a strong acid such as perchloric or tosic acid as a catalyst. Reduction of the 3,5-dienol acetates J62a-c to the corresponding 5-en-3-ols J63a-c is effected with sodium borohydride. After further reduction of J63a-c with diisobutylaluminum hydride there is obtained 20ξ-fluoro-6-subsrituted-pregna-5,17(20)-diene-3β,21-diols J64a-c.
The starting C7α-alkylandrost-4-ene-3,17-diones (K67a-d) and C β-alkylandrost-4-ene-3,17-diones (K67e-h) are synthesized starting from the known C substituted steroids K66a-h (Grunwell, J.E., Benson, H.D., Johnston, J.O. and Petrow, V. Steroids, 1976, 27, 760-771) which in turn are prepared from 6- dehydrotestosterone (K65) by the method of Benson and coworkers (loc. cit). The C3 carbonyls are protected as dienol ethers K68a-h (see Scheme K). Wittig reaction on the Cι ketones of dienol ethers K68a-h with the ylid formed by reaction of triethyl 2-fiuoro-2-phosphonoacetate with base gives the vinyl fluoride esters K69a-h as mixtures of E- and Z-isomers. Reduction of the dienol esters K69a-h with diisobutylaluminum hydride and acidic removal of the C3 protecting group gives 20ξ-fluoro-7-substituted-pregna-4,17(20)-dien-21-ol-3-ones K71a-h. Finally, acid catalyzed unmasking of the C3 carbonyl of K69a-h gives the desired 20ξ-fluoro-7- substitute-dpregna-4,17(20)-dien-3-on-21-oic acid ethyl esters K72a-h.
Scheme K. Cη Substituted Steroid-4-en-3-ones
Scheme L outlines the syntheses of C7 substituted steroid-5-en-3-ols. The selectively C3 protected C - alkylandrost-5-en-3,17-diols (L76a-d) and C7β-alkylandrost-5-en-3,17-diols (L76e-h) are synthesized starting from the known C7 substituted steroids L74a-h (Grunwell, J.E., Benson, H.D., Johnston, J.O. and Petrow, V. Steroids, 1976, 27, 760-771) by borohydride reduction of L74a-h to give L75a-h. Protection of the C3 hydroxyl group as the ^butyldimethylsilyl ether with i-butyldimethylsilyl chloride in dimethylformamide and removal of the acetate moiety by saponification with lithium hydroxide in aqueous methanol/terrahydrofuran gives 3β- [[(l,l-dimethylethyl)-dimethylsilyl]oxy]-7-substituted androst-5-en-17β-ols (L76a-h). Oxidation of C17 alcohols L76a-h with Jones reagent gives the corresponding ketones L77a-h. The latter undergo Wittig reaction at the Cι7 ketone with the ylid formed by reaction of triethyl 2-fluoro-2-phosphonoacetate with base to afford the vinyl fluoride esters L78a-h as mixtures of E- and Z-isomers. Reduction of the ester groups with diisobutylaluminum hydride and tetrabutylammonium fluoride catalyzed removal of the C3 silyl group gives the
20ξ-fluoro-7-substituted-pregna-5, 17(20)-diene-3 β,21 -diols L80a-h.
Scheme L. C Substituted Steroid-5-en-3-ols
(EtO)
2POCHFC0
2Et LHDMS, THF
3β-Hydroxyandrosta-5,15-dien-17-one (Scheme M, M81) is prepared by the method of Reeder and Joannou (Reeder, A.Y. and Joannou, G.E., Steroids, 1996, 61, 74-81) and used as starting material for the preparation steroids containing an additional double bond at C15 as shown in Scheme M. Alcohol M81 is first silylated by reaction with f-butyldimethylsilyl chloride to give silyl ether M82. Wittig reaction on the Cj7 ketone with the ylid formed by reaction of triethyl 2-fluoro-2-phosphonoacetate with base gives vinyl fluoride ester M83 as a mixture of E- and Z-isomers. Reduction of ester M83 with diisobutylaluminum hydride in dichloromethane gives a mixture of alcohols which are separated by flash chromatography into the individual E- and Z-isomers M85 and M84, respectively. Removal of the silyl protecting group of Z-olefin M84 with tetrabutylammonium fluoride gives diol M86. Similar removal of the silyl protecting group of E-olefin M85 gives diol M87.
Scheme M. C15 Unsaturated Steroid-5-en-3-ols
M81 M82
(EtO)2PO0HFCO2Et LHMDS, THF
M87 M86
The Ci5-alkyl-androst-5-en-17-ones (Scheme N, N88a-c), which serve as starting materials for the 5 substituted steroids of this invention, are synthesized in two steps from 3β-hydroxyandrost-5,15-dien-17-ones M81 as shown in Scheme N.. Wittig reaction on C17 ketones N88a-c with the ylid formed by reaction of triethyl 2-fluoro-2-ρhosphonoacetate with base gives vinyl fluoride esters N89a-c as mixtures of E- and Z-isomers. Diisobutyl aluminum hydride reduction of the ester group of N89a-c gives alcohols N90a-c that on subsequent tetrabutylammom'um fluoride catalyzed removal of the silyl protecting groups affords the 20ξ-fluoro-15- substituted-pregna-5, 17(20)-diene-3 β,21 -diols N91a-c. Silyl protected 19-nordehydroepiandrosterone (Scheme O, 097) is prepared in five steps from the known
19-nortestosterone (092) by modification of the method of Campbell and Babcock (Campbell, J.A. and Babcock, J.C, 1971, U.S. 3,597,418) wherein the C3 hydroxyl group is protected with a ϊ-butyldimethylsilyl group rather than a tetrahydropyranyl group. Thus, alcohol 094 is prepared as described in U.S. 3,597,418 and is treated with ?-butyldimethylsilyl chloride as previously described herein to give silyl ether 095. The C] acetate is hydrolyzed with potassium carbonate in aqueous methanol, and resulting alcohol 096 is oxidized with pyridinium clrromate to C]7 ketone 097. Introduction of the vinyl fluoride via Wittig reaction as previously described herein affords vinyl fluoride ester 098. Reduction of 098 with diisobutylaluminum hydride and fluoride catalyzed removal of the silyl protecting group of 099 provides the desired diol O100 as a mixture of E and Z isomers.
Scheme N. C15 Substituted Steroid-5-en-3-ols
Scheme O. Ci9-Norsteroid 5-en-3-ols
General Experimental Conditions
Melting points were determined with a Thomas-Hoover capillary melting point apparatus and are uncorrected. TLC analyses were performed with Merck DC-F254 or Analtech GHLF silica gel plates, with visualization by alkaline permanganate and UV irradiation. Flash chromatography was performed with Merck silica gel 60 (0.040-0.063 mm). NMR spectra were recorded on Varian VXR-300, Unity 300, Unity 400 or
Gemini-300 spectrometers in CDC13, unless otherwise stated. *H and 13C NMR signals are reported in ppm from tetramethylsilane, ^F NMR signals are reported in ppm from CFC13 and coupling constants are reported in Hertz (Hz). IR spectra were recorded on a Perkin-Elmer Model 1800 or Mattson Galaxy 5020 FT-IR spectrophotometer. MS data were collected at 70 eV on a Finnigan MAT 4600, Mat TSQ-700 or VG Analytical
Limited ZAB2-SE mass spectrophotometer and computerized peak matching with perfluorokerosene as the reference is utilized for HRMS. Combustion analysis was performed using a Perkin-Elmer Model 2400 elemental analyzer and the results were within ± 0.4% of the calculated values. Organic extracts were dried over anhydrous MgS04 or Na2S04 prior to solvent removal on a rotary evaporator. Celite® (diatomaceous earth)
(Celite Corporation, 137 West Central Avenue, Lompor, California 93436) was used as a filtering aid unless otherwise indicated.
EXAMPLE la 3β-[[(l, l-Dimethylethyl)dimethylsilyl]oxy]androst-5-en-17-one (A2)
Add t-butyldimethylsilyl chloride (10.97 g, 72.8 mmol), 4-dimethylaminopyridine (0.42 g, 3.47 mmol) and triemylamine (10.63 mL, 76.27 mmol) to a stirred solution of dehydroepiandrosterone (Al, 20.00 g, 69.3 mmol) in anhydrous DMF (350 mL) under nitrogen. Stir the resultant suspension at room temperature for 3 days and then pour into rapidly stirred water (1.5 L). Filter the resultant suspension and recrystallize the white solid from aqueous acetone to give A2 (24.64 g, 88%) as a white crystalline solid: mp 146-148°C. TLC Rf 0.78, ethyl acetate/hexane (1:1); JH NMR δ 5.38-5.32 (m, IH), 3.55-3.43 ( , IH), 1.03 (s, 3H), 0.89 (s, 9H), 0.88(s, 3H), 0.06(s, 6H); MS (CI, CH4) m/z (rel intensity) 403 (MH+ 3), 401 (5), 387 (9), 345 (18), 271 (100). Analysis. Calculated for C25H42Siθ2: C, 74.57;H, 10.51. Found: C, 74.89; H, 10.84.
EXAMPLE lb
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (A3) Add lithium hexamethyldisilazide (3.50 mL of a 1.0M solution in THF, 3.50 mmol) to a stirred solution of triethyl 2-fluoro-2-phosphonoacetate (0.76 mL, 3.75 mmol) in THF (15 mL) under nitrogen. After 1 hour, add a solution of A2 (1.01 g, 2.50 mmol) in THF (5 mL) and heat the reaction mixture to reflux. After 2.5 hours, allow the reaction mixture to cool to room temperature and concentrate. Partition the residue between diethyl ether (40 mL) and 0.4M aqueous hydrochloric acid (40 mL). Separate the layers and wash the organic layer with 0.5M aqueous hydrochloric acid (20 mL), saturated aqueous sodium bicarbonate (20 mL), and brine (20 mL). Dry the organic phase, filter, and concentrate to give crude A3. Purify the material by flash chromatography (6x17 cm column) and elute with ethyl acetate/ hexane (2:98) to give A3 (mixture of E and Z isomers, 0.83 g, 67%) as a
white solid. TLC Rf 0.41 and 0.51, ethyl acetate/ hexane (3:97); 19F NMR δ -121.59 (s, E isomer) and -135.49
(s, Z isomer); MS (CI, CH4) m/z (rel intensity) 491 (MH+ 97), 475 (59), 445 (24), 433 (65), 359 (100), 339 (24). Analysis. Calculated for Qs^FOsSi: C, 70.97; H, 9.65. Found: C, 71.32; H, 10.02.
EXAMPLE lc
(17JB)-3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20-fluoropregna-5,17(20)-dien-21-ol (A4) and (17Z)-3β-
[[(1,1 -Dimethylethyl)dimethylsilyl]oxy]-20-fluoropregna-5, 17(20)-dien-21 -ol (A5)
Add diisobutylaluminum hydride (6.55 mL of a 1.0M solution in dichloromethane, 65.5 mmol) to a stirred solution of A3 (7.29 g, 14.86 mmol) in dichloromethane (135 mL) under nitrogen and cool slowly to -78°C. After 1 hour, quench the reaction with a solution of glacial acetic acid (3.8 mL) in dichloromethane (9 mL) and pour the reaction mixture into dichloromethane (250 mL)/saturated aqueous potassium sodium tartrate (250 mL).
Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the layers. Wash the organic layer with saturated aqueous potassium sodium tartrate (130 mL), saturated aqueous sodium bicarbonate (250 mL), and brine (100 mL). Dry, filter, and concentrate the organic phase to give the crude product. Purify by flash chromatography (2 equal batches, 8x20 cm column) and elute with a gradient (10 to 15%) of ethyl acetate in hexane to give A4 (3.80 g, 57%) as a white solid: mp 144-147°C.
TLC Rf 0.37, ethyl acetate/ hexane (15:85); !H NMR (400 MHz) δ 5.34-5,29 (m, IH), 4.29 (ddd, IH, J = 21.4, 13.4, 6.4 Hz), 4.20 (ddd, IH, J = 21.4, 13.4, 6.4 Hz), 3.53-3.44 (m, IH), 1.01 (s, 3H), 0.91 (d, 3H, J = 1.1 Hz), 0.89 (s, 9H), 0.054 (s, 6H); 19F NMR δ -114.32 (br t, J = 24 Hz); MS (CI, CH4) m/z (rel intensity) 449 (MH+, 3), 448 (5), 447 (19), 431(10), 429 (5), 391 (32), 299 (100), 297 (17). Analysis. Calculated for C27H45Fθ2Si: C, 72.27; H, 10.11. Found: C, 72.18; H, 10.28.
Also, isolate A5 (1.10 g, 16%) as a white solid: mp 174-176°C. TLC Rf 0.29, ethyl acetate/ hexane
(15:85); !H NMR δ 5.36-5.31 (m, IH), 4.15 (dd, 2H, J = 21.2, 6.1 Hz), 3.56-3.44 (m, IH), 1.03 (s, 3H), 0.94 (s, 3H), 0.91 (s, 9H), 0.075 (s, 6H); 19F NMR δ -128.10 (t, J = 21.1 Hz); MS (CI, CH4) m/z (rel intensity) 449
(MH+, 2), 448 (4), 447 (15), 431 (10), 429 (5), 391 (26), 317 (47), 299 (100) 297 (15);.Analysis. Calculated for C27H45F02Si: C, 72.27; H, 10.11. Found: C, 72.06; H, 10.22.
EXAMPLE Id (17£)- 20-Fluoropregna-5,17(20)-diene-3β,21-diol (A6)
Add tetrabutylammonium fluoride (3.0 mL of a 1.0M solution in THF, 3.0 mmol) to compound A4 (307 mg, 0.68 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (50 mL), filter the resultant suspension and dry the filter cake to give A6 (218 mg, 95%) as a white solid: mp = 214-216°C. TLC Rf 0.15, ethyl acetate/ hexane (45:55); !H NMR (DMSO-d6) δ 5.30-5.25 (m, IH), 4.92 (t, IH, J = 5.5 Hz), 4.60 (d, IH, J = 4.8 Hz), 4.15-3.89 ( , 2H),
3.35-3.19 (m), 0.95 (s, 3H), 0.85 (d, 3H, J = 1.1Hz); 19F NMR (DMSO-d6) δ -108.66 (dd, J = 28.0, 24.1 Hz),
MS (CI, CH4) .m z (rel intensity) 335 (MH+, 4), 334 (9), 333 (18), 317 (100), 299 (93), 297 (28). Analysis.
Calculated for C2iH31F02: C, 75.41; H, 9.34. Found: C, 75.61; H, 9.50.
EXAMPLE le
(17£)-3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20-fluoropregna-5,17(20)-diene (A7) Add sulfur rrioxide pyridine complex (0.84 g, 5.27 mmol) to a stirred solution of A4 (1.35 g, 3.00 mmol) in THF (30mL), under nitrogen with cooling in an ice water bath. Stir the resultant suspension at ice bath temperature for 3 hours, and then store in a refrigerator overnight. Carefully add lithium aluminum hydride (0.80 g, 21.08 mmol) in portions to the stirred suspension. Quench the reaction by cautiously adding 0.6 mL of water,
0.6 mL of l.ON aqueous sodium hydroxide and, finally, another 0.6 mL of water. Dilute the resultant suspension with diethyl ether (80 mL) and stir vigorously for several minutes. Filter the suspension and concentrate the filtrate to give crude A7. Purify by flash chromatography (5x14 cm column) and elute with ethyl acetate/ hexane
(5:95) to give A7 (0.99 g, 76%) as a white solid. Recrystallize a portion of A7 from aqueous acetone to give a white crystalline solid: mp 128-130°C. TLC Rf 0.52, ethyl acetate/ hexane (2:98); lR NMR δ 5.35-5.31 (m, IH), 3.55-3.42 (m, IH), 1.92 (dt, 3H, J = 18.9, 1.9 Hz) 1.01 (s, 3H) 0.89 (s, 9H), 0.86 (d, 3H, J = 1.3 Hz), 0.06 (s, 6H); 19F NMR δ -95.86 (q, J = 18.9 Hz); MS (CI,CH4) m/s (rel intensity) 433 (MH+, 9), 432 (10), 431 (38), 417 (43) 413 (70), 375 (55), 301 (100). Analysis. Calculated for C27H45FOSi: C, 74.94; H, 10.48. Found: C, 75.16; H, 10.46.
EXAMPLE If (17£)- 20-fluoropregna-5,17(20)-dien-3β-ol (A8)
Prepare A8 from A7 in a manner analogous to the preparation of A6 from A4 to give A8 (299 mg, 94%) as a white solid: mp 129-133°C. TLC Rf 0.25, ethyl acetate/ hexane (1:3); lH NMR δ 5.38-5.34 (m, IH), 3.60-
3.47 (m, IH), 1.92 (dt, 3H, J = 18.9, 1.9 Hz), 1.02 (s, 3He), 0.86 (d, 3H, J = 1.2 Hz); 19F NMR δ -95.79 (q, J =
18.8 Hz); MS (CI, CH4) m/z ( rel intensity) 319 (MH+, 9), 318 (17), 317 (33), 301 (100), 299 (65), 281 (9). Analysis. Calculated for C21H31FO: C, 79.20; H, 9.81. Found: C, 79.10, H, 9.81.
EXAMPLE lg (17Z)- 20-Fluoropregna-5,17(20)-diene-3β,21-diol (A9) Prepare A9 from A5 in a manner analogous to the preparation of A6 from A4 to give A9 (299 mg, 94%) as a white solid: mp 198-203°C. TLC Rf 0.19, ethyl acetate (45:55); !H NMR δ 5.29-5.25 (m, IH), 4.91 (t, IH, J = 5.7 Hz), 4.60 (d, IH, J = 4.5 Hz), 3.88 (dd, IH, J = 23.1, 5.7 Hz), 3.34-3.19 (m, IH), 0.95 (s, 3H), 0.86 (s, 3H); 19F NMR δ -123.18 (t, J = 23.1 Hz); MS (CI, CH4) m/z (rel intensity) 335 (MH+, 4), 334 (6), 333( 15) 317 (100) 299 (46). Analysis. Calculated for C2ιH31F02: C, 75.41; H, 9.34. Found: C, 75.37; H,9.43.
EXAMPLE lh
(17Z)-3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20-fluoropregna-5,17(20)-dien-3β-ol (A10) Prepare AlO (0.59 g, 91%) from A5 in a manner analogous to the preparation of A7 from A4.
Crystallization from acetone gives AlO as a white crystalline solid: mp 138-140°C. TLC Rf 0.52, ethyl acetate/ hexane (2:98); *H NMR δ 5.34-5.30 (m IH), 3.54-3.42 (rn, IH), 1.79 (dt, 3H, J = 17.2, 1.4 Hz), 1.01 (s, 3H), 0.89 (s, 12H), 0.057 (s); 19F NMR δ -110.32 (q of q, J = 17.1, 2.0 Hz); MS (CI, CH4) m/z (rel intensity) 433 (MH+ 10), 432 (12), 431 (45), 417 (58), 413 (45), 375 (58), 301 (100). Analysis. Calculated for C27H45FOSi: C, 74.94; H, 10.48. Found: C, 75.20; H, 10.43.
EXAMPLE li (17Z)-20-Fluoropregna-5,17(20)-dien-3β-ol (All) Prepare All from AlO in a manner analogous to the preparation of A8 from A7 to give All (204 mg, 64%) as a white crystalline solid, after crystallization from methanol: mp 153-155°C. TLC Rf 0.27, ethyl acetate/ hexane (1:3); !H NMR δ 5.37-5.33 (m, IH), 3.59-3.46 (m, IH), 3.49 (s, 0.6H, MeOH solvate), 1.79 (dt, 3H, J = 17.2, 1.4 Hz, 21-Me), 1.02 (s, 3H, 19-Me), 0.89 (s, 3H, ); 19F NMR δ -110.27 (q of q, J = 17.2, 2.0 Hz); MS (CI, CH4) m/z (rel intensity) 319 (MH+ 7), 318 (15), 317 (28), 301 (100), 299 (39), 281 (8). Analysis. Calculated for C21H31FO-0.2 MeOH: C, 78.38; H,9.87. Found: C, 78.40; H, 9.82.
EXAMPLE 2a 20ξ-Fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (B12) Add tetrabutylammonium fluoride (45.0 mL of a 1.0M solution in THF, 45.0 mmol) to compound A3 (7.3 g, 14.87 mmol) and stir the resultant solution at room temperature for 30 hours. Slowly add the reaction solution to vigorously stirred cold water (750 mL), filter the resultant suspension and dry to give crude B12. Purify B12 by flash chromatography.
EXAMPLE 2b 20ξ-Fluoropregna-4,17(20)-dien-3-on-21-oic Acid, Ethyl Ester (B13) Dissolve alcohol B12 (7.14 g, 18.96 mmol) in benzene (200 mL) and add 3A molecular sieves (1 g). Add pyridinium chlorochromate (81.7 g, 0.379 mol) and reflux the mixture under an argon atmosphere for 5 hours with mechanical stirring. Decant the benzene solution, and wash the residue with ether (4x200 mL). Combine the organic layers, wash with saturated brine, dry, filter, concentrate and purify the residue by flash chromatography to give B13.
EXAMPLE 2c 20ξ-Fluoro-21-hydroxypregna-4,17(20)-dien-3-one (B15) Add methyl orthoformate (6 g) andj?-tolenesulphonic acid (0.3 g) to a solution of ketone B13 (5.0 g, 18.2 mmol) in dioxane (50 mL) and stir for two hours. Add pyridine (1.2 mL), dilute the reaction with water (60 mL) and extract with ether (3x30 mL). Dry the combined ether extracts, treat with charcoal, filter and evaporate to give B14 which is used without further purification for the next step.
Slowly and cautiously add diisobutylaluminum hydride (8.0 mL, 80.2 mmol) to a stirred cooled (-78°C)
solution of the above crude B14 and dichloromethane (250 mL). Quench the reaction after 1 hour with a solution of acetic acid (5 mL) in dichloromethane (10 mL). Dilute the reaction mixture with dichloromethane (300 mL) and shake with saturated potassium sodium tartrate (300 mL). Filter the resultant emulsion through a Celite® pad, separate the organic layer and wash sequentially with saturated potassium sodium tartrate (150 mL), saturated sodium bicarbonate (150 mL), and brine (150 mL). Dry, filter and concentrate the organic phase. Dissolve the residue in dichloromethane, place atop a column of silica gel and purify by flash chromatography to afford pure 20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (B15).
EXAMPLE 2d 21,21-Dimemyl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-ol (B16)
Dissolve ester A3 (5.0 g, 10.2 mmol) in anhydrous diethyl ether (200mL) and THF (100 mL). Cool the resulting solution in an ice-water bath and treat with methylmagnesium bromide (13.5 mL of a 3.0M solution in ether, 40.5 mmol). Quench the reaction after 4 hours by pouring the reaction mixture into cold water (200 mL) containing acetic acid (10 mL). Separate the aqueous layer and extract with ether (2x200 mL). Wash the combined organic layer and ether extract with water, dry, filter and concentrate to give a solid. Purify the solid by flash chromatography to afford B16 as a mixture of stereoisomers.
EXAMPLE 2e
21,21 -Dimethyl-20ξ-fluoropregna-5, 17(20)-diene-3 β,21 -diol (B17) Add tetrabutyl-ammonium fluoride (3.0 mL of a 1.0M solution in THF, 3.0 mmol) to compound B16 (500 mg, 1.05 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (50 mL) and filter the resultant suspension. Dry the filter cake to give crude B17 and purify B17 by flash chromatography.
EXAMPLE 3a lα-propylandrosta-3,17-dione (C19c)
Add a 10% solution of tri-propylaluminum in toluene (42.6 mL, 49.5 mmol) to a solution of androsta-1,4- diene-3,17-dione (C18, 12.38 g, 45 mmol) and cuprous bromide (129 mg, 0.9 mmol) dissolved in tetrahydrofuran (200 mL) under a nitrogen atmosphere. Add trimethylsilyl chloride (5.88 g, 54 mmol) dropwise to the solution. After 2 hours, cautiously add water (5 mL). Filter the solids off and wash. Purify the crude product by flash chromatography to give lα-propylandrosta-3,17-dione (C19c).
Similarly prepared are the known compounds (Westermann, J. and Nickisch, K., 1993, Angew. Chem. Int. Ed. Engl, 32, 1368-1370): l -methylandrosta-3,17-dione (C19a) l -ethylandrosta-3,17-dione (C19b)
EXAMPLE 3b Experimental procedures for the synthesis of these compounds and their intermediates can be found in General Procedures 1.
3-Methoxy-l α-methylandrost-5-en-17-one (C20a) lα-Ethyl-3-methoxyandrost-5-en-17-one (C20b) 3-Methoxy-l α-propylandrost-5-en-17-one (C20c)
EXAMPLE 3c
Experimental procedures for the synthesis of these compounds and their intermediates can be found in General Procedures 2.
20ξ-Fluoro-3-methoxy-lα-methylpregna-3,5,17(20)-trien-21-oic Acid Ethyl Ester (C21a) lα-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid Ethyl Ester (C21b)
20ξ-Fluoro-3-methoxy-lα-propylpregna-3,5,17(20)-trien-21-oic Acid Ethyl Ester (C21c)
EXAMPLE 3d
Experimental procedures for the synthesis of compounds C23 from €21 and their intermediates €22 can be found in General Procedures 4.
20ξ-Fluoro-21-hydroxy-lα-methylpregna-4,17(20)-dien-3-one (C23a); lα-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (C23b) 20ξ-Fluoro-21 -hydroxy- 1 α-propylpregna-4, 17(20)-dien-3-one (C23c)
EXAMPLE 3e Experimental procedures for the synthesis of compounds €24 from C21 can be found in General Procedures 3.
20ξ-Fluoro-lα-methylpregna-4,17(20)-dien-3-on-21-oic Acid Ethyl Ester (C24a) lα-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid Ethyl Ester (C24b)
20ξ-Fluoro-lα-propylpregna-4,17(20)-dien-3-one-21-oic Acid Ethyl Ester (C24c)
EXAMPLE 4a 3-Acetoxy-20ξ-fluoro-lα-methylpregna-3,5,17(20)-triene-21-oic Acid Ethyl Ester (D25a) Stir a solution of ethyl 20ξ-fluoro-lα-methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (C24a, 2.5 g, 6.43 mmol) in ethyl acetate (250 mL), acetic anhydride (25 mL) and 70% perchloric acid (0.10 mL) at room temperature for 1 hour. Extract the solution with saturated sodium bicarbonate (100 mL) and brine (100 mL). Dry and concentrate to give 3-acetoxy-20ξ-fluoro-lα-methylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (D25a) which one may purify by crystallization or use directly in the next step.
By this means the following compounds may be prepared:
3-acetoxy-lα-ethyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (D25b) 3-acetoxy-20ξ-fluoro-l -propylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (D25c)
EXAMPLE 4b 20ξ-Fluoro-3β-hydroxy-lα-methylpregna-5,17(20)-dien-21-oic Acid Ethyl Ester (D26a) Add sodium borohydride (0.40 g, 10.6 mmol) to a solution of 3-acetoxy-20ξ-fluoro-lα-methylpregna- 3,5,17(20)-triene-21-oic acid ethyl ester (D25a, 2.23 g, 5.18 mmol) in ethanol (400 mL) and tetrahydrofuran (just enough to effect solution). Stir overnight and add formic acid dropwise until gas evolution ceases. Concentrate to remove the solvents. Dissolve the residue in ethyl acetate (300 mL), wash with water (3x100 mL), dry and concentrate. Purify the residue by flash chromatography to give 20ξ-fluoro-3β-hydroxy-lα-methylpregna- 5,17(20)-dien-21-oic acid ethyl ester (D26a).
By this means the following compounds are prepared: lα-Ethyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic Acid Ethyl Ester (D26b) 20ξ-Fluoro-3β-hydroxy-lα-propylpregna-5,17(20)-dien-21-oic Acid Ethyl Ester (D26c)
EXAMPLE 4c The experimental procedure for the reduction of D26a-c to D27a-c can be found in General Procedures 4 wherein the final acid hydrolysis step is omitted.
20ξ-Fluoro-l -methylpregna-5,17(20)-diene-3β,21-diol (D27a) 1 α-Ethyl-20ξ-fluoro-pregna-5, 17(20)-diene-3β,21-diol (D27b) 20ξ-Fluoro-lα-propylpregna-5,17(20)-diene-3β,21-diol (D27c)
EXAMPLE 5a 2α-Ethylandrost-4-ene-3-dione (E28b) Add ethyl formate (3.03 g, 40.9 mmol) to a mixture of testosterone (E28 R = H, 1.92 g, 6.66 mmol) in toluene (125 mL). Add sodium ethylate (3.40 g, 50 mmol), stopper the mixture and allow to stand at room temperature for 5 days. Remove the solids by filtration, wash with ether, suspend in ether (200 mL) and make acidic with 10% aq hydrochloric acid. Separate the organic layer, wash with brine, dry, and concentrate to give crude 2-hydroxymethylenetestosterone.
Dissolve the above crude material in acetone (10 mL), add iodoethane (1.0 mL, 9.60 mmol) and potassium carbonate (0.60 g, 4.34 mmol), and reflux the mixture overnight. Cool the reaction; dilute with ether (150 mL); wash with water (1x25 mL), 10% aq sodium hydroxide (3x25 mL), and water (1x25 mL); and concentrate. Dissolve the residue in acetone (50 mL), treat with 1 N hydrochloric acid (25 mL) and strr overnight at room temperature. Remove the acetone on a rotary evaporator and extract the residue with ether (3x100 mL). Combine the ether extracts, dry, and concentrate to give crude 2α-ethyl-17β-hydroxyandrost-4-en- 3-one (E28b) which is purified by flash chromatography.
Add a solution of chromium trioxide (1.67 g, 16.7 mmol) in water (10 mL) and acetic acid (50 mL) to a solution of 2α-ethyltestosterone (E28b, 3.49 g, 11.0 mmol) in acetic acid (100 mL). Stir at room temperature for 1 hour, pour the reaction into water (200 mL) and collect the solids by filtration. Wash the filter cake with water,
dry and purify by flash chromatography to give 2α-ethylandrost-4-ene-3,17-dione (E29b).
EXAMPLE 5b The experimental procedure for the synthesis of E30a-b from steroid-4-en-3-ones E29a-b can be found in General Procedures 1.
3-Methoxy-2α-methylandrost-5-en-17-one (E30a) 2α-Ethyl-3-methoxy-androst-5-en-17-one (E30b)
EXAMPLE 5c The experimental procedure for the synthesis of E31a-b from ketones E30a-b can be found in General
Procedures 2.
20ξ-Fluoro-3-methoxy-2α-methylρregna-3,5,17(20)-trien-21-oic acid ethyl ester (E31a) 2α-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (E31b)
EXAMPLE 5d
The experimental procedure for the synthesis of E33a-b from esters E31a-b can be found in General Procedures 4.
20ξ-Fluoro-2α-methylpregna-4, 17(20)-diene-3 β,21 -diol (E33a) 2α-Ethyl-20ξ-fluoro-pregna-4,17(20)-diene-3β,21-diol (E33b)
EXAMPLE 5e
The experimental procedure for the synthesis of E34a-b from esters E31a-b can be found in General Procedures 3.
20ξ-Fluoro-2α-methylρregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (E34a) 2α-Ethyl-20ξ-fluoropregna-4, 17(20)-dien-3 -one-21 -oic acid ethyl ester (E34b)
EXAMPLE 6a 3-Acetoxy-20ξ-fluoro-2α-methylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (F35a) Stir a solution of 20ξ-fluoro-2α-methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (E34a, 2.5 g, 6.43 mmol) in ethyl acetate (250 mL), acetic anhydride (25 mL) and 70% perchloric acid (0.10 mL) at room temperature for 1 hour. Extract the solution with saturated sodium bicarbonate (100 mL) and brine (100 mL). Dry and concentrate to give F35a which one can purify by crystallization or use directly in the next step. By this procedure the following compounds may be prepared: 3-acetoxy-2α-ethyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (F35b) 3-acetoxy-20ξ-fluoro-2α-proρylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (F35c)
EXAMPLE 6b 20ξ-Fluoro-3β-hydroxy-2α-methylpregna-5,17(20)-dien-21-oic acid ethyl ester (F36a) Add sodium borohydride (0.40 g, 10.6 mmol) to a solution of 3-acetoxy-20ξ-fluoro-lα-methylpregna-
3,5,17(20)-triene-21-oic acid ethyl ester (F35a, 2.23 g, 5.18 mmol) in ethanol (400 mL) and tetrahydrofuran (just enough to effect solution). Stir overnight and add formic acid dropwise until gas evolution ceases. Remove the solvents, dissolve the residue in ethyl acetate (300 mL), wash with water (3x100 mL), dry and concentrate. Purify the residue by flash chromatography to give 20ξ-fluoro-3β-hydroxy-2α-methylpregna-5,17(20)-dien-21- oic acid ethyl ester (F36a).
By this means the following compounds may be prepared:
2α-Ethyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (F36b) 20ξ-Fluoro-3β-hydroxy-2α-ρropylpregna-5,17(20)-dien-21-oic acid ethyl ester (F36c)
EXAMPLE 6c
The experimental procedure for the synthesis of F37a-c starting from esters F36a-c can be found in General Procedures 4.
20ξ-Fluoro-2α-methylpregna-5,17(20)-diene-3β,21-diol (F37a)
2α-Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (F37b) 20ξ-Fluoro-2α-propylpregna-5,17(20)-diene-3β,21-diol (F37c)
EXAMPLE 7a 17β-Hydroxy-4-(2-propyl)androst-4-en-3-one (G38c) Stir and treat a cooled (0°C in a salt-ice bath) solution of 4-nor-4-oxasteroid G40 (7g, 21.1 mmol) in ether (100 mL) and tetrahydrofuran (30 mL) with 2N isopropyl-magnesium bromide in ether (15 mL, 30 mmol). Stir the reaction for 18 hours and pour into cold water (500 mL): Acidify the mixture by addition of 10% hydrochloric acid. Remove the aqueous layer and extract with ether (3x150 mL). Wash the combined ether layer and extracts with 20% hydrochloric acid (150 mL), water (150 mL), saturated sodium bicarbonate (150 mL), and brine (150 mL), and then dry and concentrate. Dissolve the residue in methanol (500 mL), add a solution of sodium hydroxide (21 g) in water (100 mL) and reflux the reaction for 6 hours. Cool to room temperature, acidify the reaction with acetic acid and concentrate the solution is to a volume of approximately 50 mL. Pour into water and extract with ethyl acetate (3x150 mL). Wash the combined organic extract with water (150 mL), dry and concentrate to a gum. Purify the gum by flash chromatography to give 17β-hydroxy-4-(2- propyl)androst-4-en-3-one (G38c).
EXAMPLE 7b 4-(2-propyl)androst-4-ene-3, 17-dione (G41c) Cool a solution of alcohol G38c (2.20 g, 6.70 mmol) in acetone (200 mL) to 3 °C in an ice-water bath. Add Jones reagent (ca 3 mL, Djerassi, C, Engle, R.R. and Bowers, A., J. Org. Chem., 1956, 21, 1547) until the greenish color persists. Decompose excess reagent by addition of isopropanol. Remove the solids by filtration and wash with acetone. Combine the filtrate, wash and concentrate to a greenish gum. Purify the gum by flash chromatography to give 4-(2-propyl)androst-4-ene-3, 17-dione (G41c).
By this procedure the following compounds may be prepared: 4-methylandrost-4-ene-3, 17-dione (G41a)
4-ethylandrost-4-ene-3, 17-dione (G41b) 4-butylandrost-4-ene-3,17-dione (G41d)
EXAMPLE 7c The experimental procedure for the syntheses of G43a-e starting from the steroid-4-en-3-ones G41a-e can be found in General Procedures 1.
3-Methoxy-4-methylandrosta-3,5-dien-17-one (G43a) 4-Ethyl-3-methoxyandrosta-3,5-dien-17-one (G43b) 4-Isoρropyl-3-methoxyandrosta-3,5-dien-17-one (G43c) 4-Butyl-3-methoxyandrosta-3,5-dien-17-one (G43d)
4-Chloro-3-methoxyandrosta-3,5-dien-17-one (G43e)
EXAMPLE 7d The experimental procedure for the syntheses of G44a-e starting from the C]7 ketones G43a-e can be found in General Procedures 2.
20ξ-Fluoro-3-methoxy-4-methylpregna-3,5,17(20)-trien-21-oic acid ethyl ester (G44a) 4-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (G44b) 4-Isopropyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid, ethyl ester (G44c) 4-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (G44d) 4-Chloro-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (G44e)
EXAMPLE 7e The experimental procedure for the syntheses of G46a-e starting from esters G44a-e can be found in General Procedures 4. 20ξ-Fluoro-21-hydroxy-4-methylpregna-4,17(20)-diene-3-one (G46a)
4-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (G46b) 4-Isopropyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (G46c) 4-Butyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (G46d) 4-Chloro-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-diene-3-one (G46e)
EXAMPLE 7f The experimental procedure for the syntheses of G47a-e starting from esters G44a-e can be found in General Procedures 3.
20ξ-Fluoro-4-methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (G47a) 4-Ethyl-20ξ-fluoropregna-4,17(20)-dien-on-21-oic acid ethyl ester (G47b)
4-Isopropyl-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (G47c) 4-Butyl-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (G47d) 4-Cl loro-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (G47e)
EXAMPLE 7g 20ξ-Fluoro-21 -hydroxy- 1 α-methyl-4-nitropregna-4, 17(20)-diene-3-one (G49Aa) Stir a mixture of potassium t-butylate (1.70 g, 15 mmol), and 20ξ-fluoro-21 -hydroxy- lα-methylpregna- 4,17(20)-diene-3-one (G48Aa, 1.73 g, 5.0 mmol) in t-butanol (25 mL) at room temperature for 2.5 hours under a nitrogen atmosphere. Add to the resulting yellow solution isopropyl nitrate (0.51 mL, 5.0 mmol). The reaction becomes deep violet in color. Stir the reaction overnight. Acidify fhe reaction mixture with acetic acid and dilute the mixture with dichloromethane. Remove the solids by filtration and wash with the same solvent. Combine the filtrate, wash and concentrate to a red-brown semi-solid. Purify the semi-solid by flash chromatography to give 20ξ-fluoro-21-hydroxy-l -methyl-4-nitropregna-4,17(20)-diene-3-one (G49Aa).
By this procedure the following compounds may be prepared:
20ξ-Fluoro-21-hydroxy-7α-methyl-4-nitropregna-4,17(20)-diene-3-one (G49Ab)
20ξ-Fluoro-21-hydroxy-15α-methyl-4-nitropregna-4,17(20)-diene-3-one (G49Ac)
EXAMPLE 7h
4-Arnino-20ξ-fluoro-21 -hydroxy- 1 -methylpregna-4, 17(20)-diene-3-one (G50Aa)
Treat a solution of 20ξ-fluoro-21 -hydroxy- lα-methyl-4-nitropregna-4,17(20)-diene-3-one (G49Aa, 2.09 g, 5.35 mmol) in absolute ethanol (30 mL) with Lindlar's catalyst (0.81 g) and quinoline (37 μL). and shake
(Paar shaker) under 1 atmosphere of hydrogen for 24 hours. Filter the mixture through a Celite® pad and concentrate the filtrate to a yellow solid. Purify the solid by flash chromatography to afford 4-amino-20ξ-fluoro-
21 -hydroxy- 1 α-methylpregna-4, 17(20)-diene-3-one (G50 Aa).
By this procedure the following compounds may be prepared: 4-amino-20ξ-fluoro-21-hydroxy-7α-methylρregna-4,17(20)-diene-3-one (G50Ab) 4-amino-20ξ-fluoro-21-hydroxy-15α-methylpregna-4,17(20)-diene-3-one (G50Ac).
EXAMPLE 7i Utilizing the procedures described in examples 7g and 7h, one may prepare G50Ba-c from G48Ba-c. 4-Amino-20ξ-fluoro-lα-methylpregna-4,17(20)-diene-3-one (G50Ba) 4-Amino-20ξ-fluoro-7α-methylpregna-4,17(20)-diene-3-one (G50Bb)
4-Amino-20ξ-fluoro- 15α-methyIpregna-4, 17(20)-diene-3-one (G50Bc)
EXAMPLE 8a 3-Acetoxy-20ξ-fluoro-4-methylpregna-3,5,17(20)-triene-21-oic Acid Ethyl Ester (H51a) Stir a solution of 20ξ-fluoro-4-methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (G47a, 2.5 g, 6.43 mmol) in ethyl acetate (250 mL), acetic anhydride (25 mL) and 70% perchloric acid (0.10 mL) at room temperature for 1 hour. Extract the solution with saturated sodium bicarbonate (100 mL) and brine (100 mL). Dry and concentrate the organic phase to give 3-acetoxy-20ξ-fluoro-4-methylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (H51a). Purify by crystallization or use directly in the next step.
By this procedure the following compounds may be prepared: 3-acetoxy-4-ethyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (H51b) 3-acetoxy-4-isopropyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (H51c) 3-acetoxy-4-buryl-20ξ-fluoropregna-3,5,17(20)-rriene-21-oic acid ethyl ester (H51d)
3-acetoxy-4-chloro-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (H51e)
EXAMPLE 8b
20ξ-Fluoro-3β-hydroxy-4-methylpregna-5,17(20)-dien-21-oic Acid Ethyl Ester (H52a) Add sodium borohydride (0.40 g, 10.6 mmol) to a solution of 3-acetoxy-20ξ-fluoro-4-methylpregna-
3,5,17(20)-triene-21-oic acid ethyl ester (H51a, 2.23 g, 5.18 mmol) in ethanol (400 mL) and tetrahydrofuran (just enough to effect solution). Stir overnight, add formic acid dropwise until gas evolution ceases. Remove the solvents, dissolve the residue in ethyl acetate (300 mL) and wash with water (3x100 mL). Dry and concentrated the organic phase. Purify the residue by flash chromatography to give 20ξ-fluoro-3β-hydroxy-4-methylpregna- 5,17(20)-dien-21 -oic acid ethyl ester (H52a).
By this procedure the following compounds may be prepared: 4-Ethyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (H52b) 4-Isopropyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (H52c) 4-Butyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (H52d)
4-Chloro-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (H52e)
EXAMPLE 8c 20ξ-Fluoro-4-methylpregna-5,17(20)-diene-3β,21-diol (H53a) The experimental procedure for the synthesis of H53a-e starting from esters H52a-e can be found in
General Procedures 4.
20ξ-Fluoro-4-methylpregna-5,17(20)-diene-3β,21-diol (H53a) 4-Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (H53b) 20ξ-Fluoro-4-isopropyl-pregna-5,17(20)-diene-3β,21-diol (H53c) 4-Butyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (H53d)
4-Chloro-20ξ-fluoropregna-5, 17(20)-diene-3 β,21 -diol (H53e)
EXAMPLE 9a The experimental procedure for the syntheses of I57a-c starting from the steroid-4-en-3-ones I56a-c can be found in General Procedures 1.
3-Methoxy-6-methylpregna-3,5-dien-17-one (I57a) 6-Ethyl-3-methoxypregna-3,5-dien-17-one (I57b) 6-Butyl-3-methoxypregna-3,5-dien-17-one (I57c)
EXAMPLE 9b The experimental procedure for the syntheses of I58a-c starting from the Cj ketones I57a-c can be found in General Procedures 2.
20ξ-Fluoro-3-methoxy-6-methylpregna-3,5,17(20)-trien-21-oic acid ethyl ester (I58a) 6-Ethyl-20ξ-fluoro-3-methoxypregna-3,5, 17(20)-trien-21 -oic acid ethyl ester (I58b)
6-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (I58c)
EXAMPLE 9c The experimental procedure for the syntheses of I60a-c starting from the esters I58a-c can be found in General Procedures 4.
20ξ-Fluoro-21 -hydroxy-6-methylpregna-4, 17(20)-dien-3-one (I60a) 6-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (I60b) 6-Butyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (I60c)
EXAMPLE 9d
The experimental procedure for the syntheses of 161 a-c starting from the esters I60a-c can be found in General Procedures 3.
20ξ-Fluoro-6-methylpregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (161 a) 6-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (I61b) 6-Butyl-20ξ-fluoropregna-4, 17(20)-dien-3 -one-21 -oic acid ethyl ester (161 c)
Example 10a 3-Acetoxy-20ξ-fluoro-6-methylρregna-3,5,17(20)-triene-21-oic Acid Ethyl Ester (J62a) Stir a solution of 20ξ-fluoro-6 -methylpregna-4,17(20)-dien-3-on-21-oic acid ethyl ester (I61a, 2.5 g, 6.43 mmol) in ethyl acetate (250 mL), acetic anhydride (25 mL) and 70% perchloric acid (0.10 mL) at room temperature for 1 hour. Extract the solution with saturated sodium bicarbonate (100 mL) and brine (100 mL). Dry and concentrate the organic phase to give 3-acetoxy-20ξ-fluoro-6-methylpregna-3,5,17(20)-triene-21-oic acid ethyl ester (J62a). Purify the material by crystallization or use it directly in the next step.
By this procedure the following compounds may be prepared:
3-acetoxy-6-ethyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (J62b) 3-acetoxy-6-butyl-20ξ-fluoropregna-3,5,17(20)-triene-21-oic acid ethyl ester (J62c)
EXAMPLE 10b 20ξ-Fluoro-3 β-hydroxy-6-methylpregna-5, 17(20)-dien-21 -oic Acid Ethyl Ester (J63a)
Add sodium borohydride (0.40 g, 10.6 mmol) to a solution of 3-acetoxy-20ξ-fluoro-6-methylpregna- 3,5,17(20)-triene-21-oic acid ethyl ester (J62a, 2.23 g, 5.18 mmol) in ethanol (400 mL) and tetrahydrofuran (just enough to effect solution). Stir overnight and add formic acid dropwise until gas evolution ceases. Remove the solvents and dissolve the residue in ethyl acetate (300 mL). Wash with water (3x100 mL), dry and concentrate
the organic phase. Purify the residue by flash chromatography to give 20ξ-fluoro-3β-hydroxy-6-methylpregna- 5,17(20)-dien-21-oic acid ethyl ester (J63a).
By this procedure the following compounds may be prepared: 4-ethyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (J63b)
4-butyl-20ξ-fluoro-3β-hydroxypregna-5,17(20)-dien-21-oic acid ethyl ester (J63c)
EXAMPLE 10c The experimental procedure for the synthesis of J64a-c starting from esters J63a-c can be found in General Procedures 4.
20ξ-Fluoro-6-methylpregna-5,17(20)-diene-3β,21-diol (J64a) 6-Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (J64b) 6-Butyl-20ξ-fluoropregna-5, 17(20)-diene-3 β,21-diol (J64c)
EXAMPLE 11a
The experimental procedure for the syntheses of K68a-h starting from the steroid-4-en-3-ones K67a-h can be found in General Procedures 1.
3-methoxy-7α-methylpregna-3,5-dien-17-one (K68a)
7α-Ethyl-3-methoxypregna-3,5-dien-17-one (K68b) 3-methoxy-7α-propylpregna-3,5-dien-17-one (K68c)
7α-Butyl-3-methoxypregna-3,5-dien-17-one (K68d)
3-methoxy-7 β-methylpregna-3 ,5-dien- 17-one (K68e)
7β-Ethyl-3-methoxypregna-3,5-dien-17-one (K68f)
3-methoxy-7 β-ρropylpregna-3 ,5-dien- 17-one (K68g) 7β-Butyl-3-methoxypregna-3,5-dien-17-one (K68h)
EXAMPLE l ib The experimental procedure for the syntheses of K69a-h starting from the C] ketones K68a-h can be found in General Procedures 2. 20ξ-Fluoro-3-methoxy-7α-methylpregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69a)
7α-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69b) 20ξ-Fluoro-3-methoxy-7α-ρropylρregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69c) 7α-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69d) 20ξ-Fluoro-3-methoxy-7β-methylpregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69e) 7β-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69f)
20ξ-Fluoro-3-methoxy-7β-propylpregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69g) 7β-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic acid ethyl ester (K69h)
EXAMPLE l lc The experimental procedure for the syntheses of K71a-h starting from the esters K69a-h can be found in General Procedures 4. 20ξ-Fluoro-21 -hydroxy-7 -methylpregna-4, 17(20)-dien-3-one (K71 a)
7α-Ethyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (K71b) 20ξ-Fluoro-2 l-hydroxy-7α-propylpregna-4, 17(20)-dien-3-one (K71c) 7α-Butyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (K71d) 20ξ-FIuoro-21 -hydroxy-7β-methylpregna-4, 17(20)-dien-3-one (K71 e) 7β-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (K71f)
20ξ-Fluoro-21 -hydroxy-7β-ρropylpregna-4, 17(20)-dien-3-one (K71 g) 7β-Butyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (K71h)
EXAMPLE l id The experimental procedure for the syntheses of K72a-h starting from the esters K69a-h can be found in
General Procedures 3.
20ξ-Fluoro-7α-methylpregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72a) 7α-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72b) 20ξ-Fluoro-7α-propylpregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72c) 7α-Butyl-20ξ-fluoropregna-4, 17(20)-dien-3-one-21 -oic acid ethyl ester (K72d)
20ξ-Fluoro-7β-methylpregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72e) 7β-Ethyl-20ξ-fluoroρregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72f) 20ξ-Fluoro-7β-propylpregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72g) 7β-Buryl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic acid ethyl ester (K72h)
EXAMPLE 12a 7α-Methylandrost-5-ene-3β,17β-diol 17 Acetate (L75a) Add slowly a solution of 7α-methylandrost-5-en-3-on-17j3-ol 17 acetate (L74a, 6.3 g, 18.9 mmol) in tetrahydrofuran (50 mL) to a solution of sodium borohydride (0.72 g, 18.9 mmol) in 95% ethanol (200 mL) cooled to -3°C in a salt/ice bath with stirring. Stir at this temperature for 3 hours and decompose the excess reagent by cautious addition of acetic acid (10 mL). Remove the solvents and purify the resulting crude product by flash chromatography to give 7α-methylandrost-5-ene-3β,17β-diol 17 acetate (L75a) as a white solid. Similarly prepare the following analogs:
7α-Ethylandrost-5-ene-3β,17β-diol 17 Acetate (L75b) 7α-Propylandrost-5-ene-3β,17β-diol 17 Acetate (L75c)
7α-Butylandrost-5-ene-3β,17β-diol 17 Acetate (L75d) 7β-Methylandrost-5-ene-3β,17β-diol 17 Acetate (L75e) 7β-Ethylandrost-5-ene-3β,17β-diol 17 Acetate (L75f)
7β-Propylandrost-5-ene-3β,17β-diol 17 Acetate (L75g) 7β-Butylandrost-5-ene-3β,17β-diol 17 Acetate (L75h)
EXAMPLE 12b 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7 -methylandrost-5-en-17-ol (L76a)
Add to a stirred solution of 7α-methylandrost-5-ene-3β,17β-diol 17 acetate (L75a, 3.20 g, 9.57 mmol) in anhydrous DMF (50 mL), under nitrogen, t-butyldimethylsilyl chloride (1.51 g, 10.0 mmol), 4- dimethylaminopyridine (0.06 g, 0.48 mmol) and triethylamine (1.5 mL, 10.6 mmol). Stir the resultant suspension at room temperature for 3 days and then pour into rapidly stirred water (200 mL). Filter the resultant suspension and crystallize the white solid from aqueous acetone to give L76a. Prepare the following compounds by this procedure:
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7α-ethylandost-5-en-17-ol (L76b) 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7α-propylandost-5-en-17-ol (L76c) 7α-Butyl3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]androst-5-en-17-ol (L76d) 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-methylandrost-5-en-17-ol (L76e)
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-ethylandrost-5-en-17-ol (L76f) 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-propylandrost-5-en-17-ol (L67g) 7β-Butyl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]androst-5-en-17-ol (L76h)
EXAMPLE 12c
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7α-methylandrost-5-en-17-one (L77a) Cool a solution of alcohol L76a (5.23 g, 12.49 mmol) in acetone (350 mL to 3 °C in an ice-water bath and add Jones reagent until the greenish color persists. Decompose the excess reagent by addition of isopropanol. Remove the solids by filtration and wash with acetone. Combine the filtrate and wash, and concentrate to a greenish gum. Purify the gum by flash chromatography to give 3β-[[(l,l- dimethylethyl)dimethylsilyl]-oxy]-7α-methylandrost-4-en-17-one (L77a). By this procedure the following compounds may be prepared:
3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-7α-ethylandrost-5-en-17-one (L77b);
3β-[[(l,l-dime ylemyl)dimethylsilyl]oxy]-7α-propylandrost-5-en-17-one (L77c); 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-7α-butylandrost-5-en-17-one (L77d)
3 β-[[( 1 , 1 -dimethylethyl)dimethylsilyl]-oxy]-7 β-mefhylandrost-5-en- 17-one (L77e)
3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-7β-ethylandrost-5-en-17-one (L77f);
3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-7β-propylandrost-5-en-17-one (L77g);
3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-7β-butylandrost-5-en-17-one (L77h)
EXAMPLE 12d The experimental procedure for the syntheses of L78a-h starting from the Cj7 ketones L77a-h can be found in General Procedures 2.
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7 -methylpregna-5,17 (20)-dien-21-oic acid ethyl ester (L78a)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7α-ethyl-20ξ-fluoropregna-5,17 (20)-dien-21-oic acid ethyl ester (L78b) 3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7α-propylpregna-5,17 (20)-dien-21-oic acid ethyl ester (L78c)
7α-Butyl-3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-3-methoxy-pregna-5,17(20)-dien-21-oic acid ethyl ester (L78d)
3-[[(l,l-Dimethylemyl)dimethylsilyl]oxy]-20ξ-fluoro-7β-methylpregna-5,17 (20)-dien-21-oic acid ethyl ester (L78e)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-ethyl-20ξ-fluoropregna-5,17 (20)-dien-21-oic acid ethyl ester (L78f)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7β-propylpregna-5,17 (20)-dien-21-oic acid ethyl ester (L78g) 7β-Butyl-3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17 (20)-dien-21-oic acid ethyl ester (L78h)
EXAMPLE 12e 3-[[(l,l-Dimethyletlιyl)dimethylsilyl]oxy]-20ξ-fluoro-7α-methylpregna-5,17(20)-dien-21-ol (L79a) Slowly add diisobutylaluminum hydride (6.75 mL of a 1.OM solution in dichloromethane, 67.5 mmol) to a stirred solution of ester L78a (7.57 g, 15 mmol) in dichloromethane (135 mL) under nitrogen and cooled to - 78°C. After 1 hour, quench the reaction with a solution of glacial acetic acid (3.8 mL) in dichloromethane (9 mL) and pour the reaction mixture into dichloromethane (250 mL)/saturated aqueous potassium sodium tartrate (250 mL). Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the layers. Wash the organic layer with saturated aqueous potassium sodium tartrate (130 mL), saturated aqueous sodium bicarbonate (250 mL), and brine (100 mL). Dry the organic phase, filter, and concentrate to give crude 3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7 -methylpregna-5,17 (20)-dien- 21-ol (L79a) and purify by flash chromatography.
By this procedure the following compounds are prepared: 3-[[(l,l-Dimethyl-ethyl)dimetlιylsilyl]oxy]-7α-ethyl-20ξ-fluoropregna-5,17(20)-dien-21-ol (L79b)
7α-Buryl-3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-ol (L79c) 3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7α-ρropylpregna-5,17(20)-dien-21-ol (L79d) 3-[[(l,l-Dime lemyl)α methylsilyl]oxy]-20ξ-fluoro-7β-methyl-pregna-5,17(20)-dien-21-ol (L79e) 3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-ethyl-20ξ-fluoropregna-5,17(20)-dien-21-ol (L79f) 7β-Butyl-3-[[(l,l-drmethylethyl)dimethyl-silyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-ol (L79g)
3-[[(l,l-Dimethylethyl)-dimethylsilyl]oxy]-20ξ-fluoro-7β-propylpregna-5,1720)-dien-21-ol (L79h)
EXAMPLE 12f 20ξ-Fluoro-7α-mefhylpregna-5,17(20)-diene-3β,21-diol (L80a) Add tetrabutylammonium fluoride (9.0 mL of a 1.0M solution in THF, 9.0 mmol) to t-butyldimethylsilyl ether L79a (923 mg, 2.0 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (150 mL). Filter the resultant suspension and dry to give 20ξ-fluoro-7α-methylpregna-5,17(20)-diene-3β,21-diol (L80a) as a white solid. Purify the solid by flash chromatography.
By this procedure the following compounds are prepared: 7 -Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (L80b)
20ξ-Fluoro-7α-propylpregna-5,17(20)-diene-3β,21-diol (L80c) 7α-Butyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (L80d) 20ξ-Fluoro-7β-methylpregna-5,17(20)-diene-3β,21-diol (L80e) 7β-Ethyl-20ξ-fluoropregna-5, 17(20)-diene-3 β,21 -diol (L80f) 20ξ-Fluoro-7β-propylpregna-5,17(20)-diene-3β,21-diol (L80g)
7β-Butyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (L80h)
EXAMPLE 13a 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]androst-5,15-dien-17-one (M82) Add t-butyldimethylsilyl chloride (5.49 g, 36.4 mmol), 4-dimethylaminopyridine (0.21 g, 1.74 mmol) and, triethylamine (5.32 mL, 38.14 mmol) to a stirred solution of 3β-hydroxyandrosta-5,15-dien-17-one (M81, 10.00 g, 34.8 mmol) in anhydrous DMF (175 mL) under nitrogen. Stir the resultant suspension at room temperature for 3 days and then pour into rapidly stirred water (750 mL). Filter the resultant suspension and crystallize the white solid to give 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]androst-5,15-dien-17-one (M82).
EXAMPLE 13b 3β-[[(l,l-Dime lethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,15,17(20)-trien-21-oic Acid Ethyl Ester (M83)
Add lithium hexamethyldisilazide (7.0 mL of a 1.0M solution in THF, 7.0 mmol) to a stirred solution of triethyl 2-fluoro-2-phosphonoacetate (1.52 mL, 7.50 mmol) in THF (30 mL) under nitrogen. Add after 1 hour, a solution of M82 (2.01 g, 5.0 mL) in THF (10 mL) and heat he reaction mixture to reflux. After 2.5 hours, allow the reaction mixture to cool to room temperature and concentrate. Partition the residue between diethyl ether (80 mL) and 0.4M aqueous hydrochloric acid (80 mL). Separate the layers and wash the organic layer with 0.5M aqueous hydrochloric acid (40 mL), saturated aqueous sodium bicarbonate (40 mL), and brine (40 mL). Dry, filter , and concentrate the organic phase to give crude 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ- fluoropregna-5,15,17(20)-trien-21-oic acid ethyl ester (M83). Purify the material by flash chromatography.
EXAMPLE 13c (17Z)-3β-[[(l,l-Dιrmemylethyl)dimethylsilyl]oxy]-20-fluoropregna-5,15,17(20)-trien-21-ol (M84) an^ [[(l,l-Dimetlιyle1±ιyl)d]rmethylsilyl]oxy]-20-fluoropregna-5,15,17(20)-trien-21-ol (M85)
Slowly add diisobutylaluminum hydride (21.8 mL of a 1.0M solution in dichloromethane, 21.8 mmol) to a stirred, cooled (-78°C) solution of M83 (2.43 g, 4.95 mmol) in dichloromethane (50 mL) under nitrogen. After 1 hour quench the reaction with a solution of glacial acetic acid (1.3 mL) in dichloromethane (3 mL) and pour the reaction mixture into dichloromethane (100 mL)/saturated aqueous potassium sodium tartrate (100 mL). Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the layers. Wash the organic layer with saturated aqueous potassium sodium tartrate (130 mL), saturated aqueous sodium bicarbonate (250 mL), and brine (100 mL). Dry, filter, and concentrate the organic phase to give the crude product. Purification by flash chromatography gives M84 and M85.
EXAMPLE 13d
(17Z)- 20-Fluoropregna-5,15,17(20)-triene-3β,21-diol (M86) Add tetrabutylammomum fluoride (0.85 mL of a 1.0M solution in THF, 0.85 mmol) to compound M84 (lOOmg, 0.192 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (20 mL) and filter the resultant suspension. Dry the filter cake to give (17Z)- 20-fluoropregna-5,15,17(20)-triene-3β,21-diol (M86) a white solid.
EXAMPLE 13e (17E)- 20-Fluoropregna-5,15,17(20)-triene-3β,21-diol (M87) Add tetrabutylammomum fluoride (6.0 mL of a 1.0M solution in THF, 6.0 mmol) to compound M85 (704 mg, 1.35 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (100 mL) and filter the resultant suspension. Dry the filter cake to give (17E)- 20-fluoropregna-5,15,17(20)-triene-3β,21-diol (M87) as a white solid.
EXAMPLE 14a 3β-[[(l,l-Dimefhylethyl)dimethylsilyl]oxy]-15α-methylandrost-5-en-17-one (N88a)
Prepare M82 by treating a solution of M81 in DMF with t-butyldimethylsilyl chloride following the procedure described in example 13a. To a solution of 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]androst-5,15- dien-17-one (M82) (10.0 g, 24.96 mmol) and cuprous bromide (71.5 mg, 0.5 mmol) dissolved in tetrahydrofuran (100 mL) under a nitrogen atmosphere, add a 2M solution of tri-methylaluminum in toluene (13.8 mL, 27.6 mmol). Add trimethylsilyl chloride (5.88 g, 54 mmol) dropwise to the solution. After 2 hours, add water (5 mL) cautiously. Remove the solids by filtration and wash. Purify the crude product by flash chromatography to give 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-15 -methylandrost-5-en-17-one (N88a). By this procedure the following compounds are prepared: 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-15α-ethylandrost-5-en-17-one (N88b)
15α-Butyl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]androst-5-en-17-one (N88c)
EXAMPLE 14b The experimental procedure for the syntheses of N89a-c starting from the C17 ketones N88a-h can be found in General Procedures 2.
3β-[[(l,l-Dimethylethyl)dimetlιylsilyl]oxy]-15α-methylpregna-5-en-21-oic acid ethyl ester (N89a) 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-15α-ethylpregna-5-en-21-oic acid ethyl ester (N89b) 15α-Butyl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]ρregna-5-en-21-oic acid ethyl ester (N89c)
EXAMPLE 14c 3β-[[(l,l-Dime le l)dimethylsilyl]oxy]-20ξ-fluoro-15α-methylpregna-5,17(20)-dien-21-ol (N90a) Slowly add diisobutylaluminum hydride (9.34 mL of a 1.0M solution in dichloromethane, 9.34 mmol) to a stirred, cooled (-78°C) solution of N89a (1.07 g, 2.12 mmol) in dichloromethane (25 mL) under nitrogen. Quench the reaction after 1 hour with a solution of glacial acetic acid (0.6 mL) in dichloromethane (2 mL), and pour the reaction mixture into dichloromethane (50 mL)/saturated aqueous potassium sodium tartrate (50 mL). Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the layers Wash the organic layer with saturated aqueous potassium sodium tartrate (60 mL), saturated aqueous sodium bicarbonate (100 mL), and brine (50 mL). Dry, filter, and concentrate the organic phase to give the crude product. Purify the material by flash chromatography to give 3β-[[(l,l-dimethylethyl)- dimethylsilyl]oxy]-20ξ-fluoro-15α-methylpregna-5,17(20)-dien-21-ol (N90a). By this procedure the following compounds are prepared:
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-15α-ethyl-20ξ-fluoropregna-5,17(20)-dien-21-ol (N90b) 15α-Butyl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-ol (N90c)
EXAMPLE 14d 20ξ-fluoro-15 -methylρregna-5,17(20)-dien-3β,21-ol (N91a) Add tetrabutylammonium fluoride (0.85 mL of a 1.0M solution in THF, 0.85 mmol) to compound N90a (lOOmg, 0.192 mmol), under nitrogen, and stir the resultant solution at room temperature for 23 hours. Add the reaction solution dropwise to vigorously stirred water (20 mL) and filter the resultant suspension. Dry the filter cake to give 20ξ-fluoro-15 -methylρregna-5,17(20)-dien-3β,21-ol (N91a) a white solid.
By this procedure the following compounds are prepared: 15α-Ethyl-20ξ-fluoropregna-5,17(20)-dien-3β,21-ol (N91b) 15 -Butyl-20ξ-fluoropregna-5,17(20)-dien-3β,21-ol N91c)
EXAMPLE 15a 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-19-norpregna-5,17(20)-dien-21-oic Acid, Ethyl Ester
(098) Prepare 093 from 092 by treating with acetic anhydride as described for the synthesis of J62 in example
10a, and then treat 093 with sodium borohydride to provide 094 as described for the preparation of J63 in example 10b. Add -butyldimethylsilyl chloride (16.44 g, 109.1 mmol), 4-dimethylaminopyridine (0.62 g, 5.20 mmol) and triethylamine (15.9 mL, 114.3 mmol) to a stirred solution of estr-5-ene-3β,17β-diol 17-acetate (094, 33.1 g, 103.9 mmol, Campbell, J.A. and Babcock, J.C, 1971, U.S. 3,597,418) in anhydrous DMF (350 mL) under nitrogen. Stir the resultant suspension at room temperature for 3 days and then pour into vigorously stirred water (2.5 L). Filter the resultant suspension to give 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]estr-5-en-17|8-ol 17 acetate (095) as an off-white solid.
Add methanol (150 mL) to a solution of acetate 095 (38.2 g, 88.3 mmol) in tetrahydrofuran (350 mL) and then add a solution of potassium carbonate (15 g, 109 mmol) in water (150 mL). Heat the resulting solution to reflux for 2.5 hours. Remove the organic solvents and partition the remaining mixture between dichloromethane
(500 mL) and water (200 mL). Separate the organic layer, dry and concentrate to give 3β-[[(l,l- dimethylethyl)dimethylsilyl]oxy]estr-5-en-17|8-ol (096).
Add pyridinium dichromate (90 g, 239 mmol) to a solution of the above Cj7 alcohol 096 (30.6 g, 81.3 mmol) dissolved in pyridine (500 mL). Stir the mixture at room temperature for 2 days. Dilute the reaction with ether (1 L) and toluene (1 L), and filter the resulting mixture through a pad of Celite®. Wash the filtrate with water (3x750 mL) and then with saturated brine. Dry the organic phase and concentrate. Purify the residue by flash chromatography to give 3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-estr-5-en-17-one (097).
Added lithium hexamethyldisilazide (80 mL of a 1.0M solution in THF, 80 mmol) to a stirred solution of triethyl 2-fluoro-2-phosρhonoacetate (17.3 mL, 85.4 mmol) in THF (350 mL) under nitrogen. After 1 hour, add a solution of ketone 097 (21.3 g, 56.9 mmol) in THF (100 mL) and heat the reaction mixture to reflux. After 2.5 hours, cool the reaction mixture to room temperature and concentrate. Partition the residue between diethyl ether (750 mL) and 0.4M aqueous hydrochloric acid (750 mL). Separate the layers and wash the organic layer with 0.5M aqueous hydrochloric acid (400 mL), saturated aqueous sodium bicarbonate (400 mL), and brine (400 mL). Dry, filter, and concentrate the organic phase to give crude 3β-[[(l,l-dimethylethyl)-dimethylsilyl]oxy]-20ξ- fluoro-19-norpregna-5,17(20)-dien-21-oic acid ethyl ester (098). Purify the crude material by flash chromatography.
EXAMPLE 15b 3β-[[(l,l-Dime ylethyl)dimethylsilyl]oxy]-20ξ-fluoro-19-norpregna-5,17(20)-dien-21-ol (O99) Slowly add diisobutylaluminum hydride (2.18 mL of a 1.0M solution in dichloromethane, 2.18 mmol) to a stirred, cooled (-78°C) solution of 098 (2.36 g, 4.95 mmol) in dichloromethane (50 mL) under nitrogen. After 1 hour, quench the reaction with a solution of glacial acetic acid (1.3 mL) in dichloromethane (3 mL) and pour the reaction mixture into dichloromethane (100 mL)/saturated aqueous potassium sodium tartrate (100 mL). Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the
layers. Wash the organic layer with saturated aqueous potassium sodium tartrate (45 mL), saturated aqueous sodium bicarbonate (100 mL), and brine (50 mL). Dry, filter, and concentrate the organic phase to give crude 099. Purify the crude material by flash chromatography or use the crude material directly in the next step.
EXAMPLE 15c 20ξ-Fluoro-l 9-norpregna-5, 17(20)-diene-3 β,21 -diol (O100) Add tetrabutylammonium fluoride (14.0 mL of a 1.0M solution in THF, 14.0 mmol) to compound 099 (0.652 g, 1.50 mmol) and stir the resultant solution at room temperature for 30 hours. Add the reaction solution slowly to vigorously stirred cold water (250 mL), filter the resultant suspension and dry the filter cake. Purify the crude material by flash chromatography to give 20ξ-fluoro-19-norpregna-5,17(20)-diene-3β,21-diol (O100).
General Procedure 1 : Protection of the C ketones Method A. (After the method described in Friedlander, M., J. Org. Chem., 1962, 27, 4046 and Burn, D., Cooley, G., Davies, M.T., Ducker, J.W., Ellis, B., Feather, P., Hiscock, A.K., Kirk, D.N., Leftwick, A.P., Petrow, V. and Williamson, D.M., 1964, Tetrahedron, 20, 597-609) Add methyl orthoformate (12 mL) and p- tolenesulphonic acid (0.3 g) to a solution of Δ4-3-ketosteroid (5.0 g) in dioxane (50 mL) and stir the resulting solution for 2 hours. Add pyridine (1.2 mL) and pour the reaction into water (500 mL). Collect the solids by filtration, wash with water and dry to give the crude material. Purify the dienol ethers by crystallization. Method B. Following the method of Broess et al. (Broess, A.I.A., van Vliet, N.P., Groen, M.B. and
Hamersma, H. Steroids, 1992, 57, 514-521) cool a suspension of Δ4-3-ketosteroid (3.30 g) in a mixture of anhydrous methanol (10 mL) and trimethyl orthoformate (5 mL) to 0°C. Add to this suspension p- toluenesulfonic acid (50 mg) and stir the mixture for 7 hours. Basify the reaction mixture by addition of triethylamine (1 mL) and collect the resulting solids by filtration to give the desired dienol ether.
3-Methoxy-lα-methylandrost-5-en-17-one (C20a) (Method B) lα-Ethyl-3-methoxy-androst-5-en-17-one (C20b) (Method B) 3-Methoxy-lα-propylandrost-5-en-17-one (C20c) (Method B) 3-Methoxy-2α-methylandrost-5-en-17-one (E30a) (Method B) 2α-Ethyl-3-methoxyandrost-5-en-17-one (E30b) (Method B)
3-Methoxy-4-methylandrosta-3,5-dien-17-one (G43a) (Method B) 4-Ethyl-3-methoxyandrosta-3,5-dien-17-one (G43b) (Method B) 4-Isoproρyl-3-methoxyandrosta-3,5-dien-l 7-one (G43c) (Method B) 4-Butyl-3-methoxyandrosta-3,5-dien-17-one (G43d) (Method B) 4-Chloro-3-methoxyandrosta-3,5-dien-17-one (G43e) (Method A)
3-Methoxy-6-methylρregna-3,5-dien-17-one (I57a) (Method A) 6-Ethyl-3-methoxypregna-3,5-dien-17-one (I57b) (Method A) 6-Butyl-3-methoxypregna-3,5-dien-l 7-one (I57c) (Method A) 3-methoxy-7α-methylpregna-3,5-dien-17-one (K68a) (Method B)
7α-Ethyl-3-methoxypregna-3,5-dien-17-one (K68b) (Method B) 3-methoxy-7α-propylpregna-3,5-dien-17-one (K68c) (Method B) 7α-Butyl-3-methoxypregna-3,5-dien-17-one (K68d) (Method B) 3-methoxy-7β-methylpregna-3,5-dien-17-one (K68e) (Method B) 7β-Ethyl-3-methoxypregna-3,5-dien-17-one (K68f) (Method B)
3-methoxy-7β-propylpregna-3,5-dien-17-one (K68g) (Method B) 7β-Butyl-3-methoxypregna-3,5-dien-17-one (K68h) (Method B)
General Procedure 2: Wittig reaction of Cyj ketones Add lithium hexamethyldisilazide (3.50 mL of a 1.0M solution in THF, 3.50 mmol) to a stirred solution of triethyl 2-fluoro-2-phosphonoacetate (0.76 mL, 3.75 mmol) in THF (15 mL) under nitrogen. After 1 hour, add a solution of the Cπ ketone (1.01 g) in tetrahydrofuran (5 mL) and heat the reaction mixture to reflux. After 2.5 hours, cool the reaction mixture to room temperature and concentrate. Partition the residue between diethyl ether (40 mL) and 0.4M aqueous hydrochloric acid (40 mL). Separate the layers and wash the organic layer washed with 0.5M aqueous hydrochloric acid (20 mL), saturated aqueous sodium bicarbonate (20 mL), and brine (20 mL). Dry, filter, and concentrate the organic phase to give the crude vinyl fluoride. Normal phase flash chromatography gives pure vinyl fluoride. Prepare the following C3 protected C20-fluoro-C2ι-carboxylates by this method:
20ξ-Fluoro-3-methoxy-lα-methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (C21a) lα-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (C21b)
20ξ-Fluoro-3-methoxy-lα-propylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (C21c) 20ξ-Fluoro-3-methoxy-2 -methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (E31a) 2α-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (E31b) 20ξ-Fluoro-3-methoxy-4-methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (G44a) 4-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (G44b)
4-Isopropyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (G44c) 4-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (G44d) 4-Chloro-20ξ-fluoro-3-methoxypregna-3,5,17(20)-rrien-21-oic Acid, Ethyl Ester (G44e) 20ξ-Fluoro-3-methoxy-6-methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (I58a) 6-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (I58b)
6-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (I58c) 20ξ-Fluoro-3-methoxy-7α-methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69a) 7α-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69b) 20ξ-Fluoro-3-methoxy-7α-ρropylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69c) 7α-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69d)
20ξ-Fluoro-3-methoxy-7β-methylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69e) 7β-Ethyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69f) 20ξ-Fluoro-3-methoxy-7β-propylpregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69g)
7β-Butyl-20ξ-fluoro-3-methoxypregna-3,5,17(20)-trien-21-oic Acid, Ethyl Ester (K69h) 3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7α-mefhylpregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78a)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7α-ethyl-20ξ-fluoropregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78b)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7α-propylpregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78c)
7α-Butyl-3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-3-mefhoxypregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78d) 3-[[(l,l-Dime lemyl)dimethylsilyl]oxy]-20ξ-fluoro-7β-methylpregna-5,17(20)-dien-21-oic Acid, Ethyl
Ester (L78e)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-7β-ethyl-20ξ-fluoropregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78f)
3-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoro-7β-propylpregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78g)
7β-Butyl-3-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-20ξ-fluoropregna-5,17(20)-dien-21-oic Acid, Ethyl Ester (L78h)
3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-15α-methylpregna-5-en-21-oic Acid Ethyl Ester (N89a) 3β-[[(l,l-Dimethylethyl)dimethylsilyl]oxy]-15α-ethylpregna-5-en-21-oic Acid Ethyl Ester (N89b) 15α-Buryl-3β-[[(l,l-dimethylethyl)dimethylsilyl]oxy]pregna-5-en-21-oic Acid Ethyl Ester (N89c)
General Procedure 3: Hydrolysis of the dienol ethers
Dissolve the dienol ether (5 g) in THF (lOOmL). Add 0.1 N aq hydrochloric acid (10 mL) and stir the solution for 3 hours at room temperature. Basify the reaction by addition of solid sodium bicarbonate and remove the solvent keeping the bath temperature below 30°C. Dissolve the residue in dichloromethane, place atop a column of silica gel and purify by flash chromatography to give the pure steroid 4-en-3-one. Prepare the following steroid 4-en-3-ones are prepared by this means:
20ξ-Fluoro-lα-methylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (C24a) lα-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (C24b) 20ξ-Fluoro-lα-propylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (C24c)
20ξ-Fluoro-2α-methylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (E34a) 2α-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (E34b) 20ξ-Fluoro-4-methylpregna-4,17(20)-dien-3-on-21-oic Acid, Ethyl Ester (G47a) 4-Ethyl-20ξ-fluoropregna-4,17(20)-dien-on-21-oic Acid, Ethyl Ester (G47b) 4-Isopropyl-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic Acid, Ethyl Ester (G47c)
4-Butyl-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic Acid, Ethyl Ester (G47d) 4-Chloro-20ξ-fluoropregna-4,17(20)-dien-3-on-21-oic Acid, Ethyl Ester (G47e) 20ξ-Fluoro-6-methylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (I61a)
6-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (I61b) 6-Butyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (I61c) 20ξ-Fluoro-7α-methylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72a) 7α-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72b) 20ξ-Fluoro-7α-propylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72c)
7α-Butyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72d) 20ξ-Fluoro-7β-methylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72e) 7β-Ethyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72f) 20ξ-Fluoro-7β-propylpregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72g) 7β-Butyl-20ξ-fluoropregna-4,17(20)-dien-3-one-21-oic Acid, Ethyl Ester (K72h)
General Procedure 4: Reduction of C?1 carboxylates and Hydrolysis of the Dienol Ethers
Slowly add diisobutylaluminum hydride (6.75 mL of a 1.0M solution in dichloromethane, 67.5 mmol) to a stirred, cooled (-78°C) solution of the steroid ester (15 mmol) in dichloromethane (135 mL) under nitrogen. After 1 hour, quench the reaction with a solution of glacial acetic acid (3.8 mL) in dichloromethane (9 mL) and pour the reaction mixture into dichloromethane (250 mL)/saturated aqueous potassium sodium tartrate (250 mL).
Filter the resultant emulsion through a Celite® pad (3 cm), transfer the filtrates to a separatory funnel, and separate the layers. Wash the organic layer with saturated aqueous potassium sodium tartrate (130 mL), saturated aqueous sodium bicarbonate (250 mL), and brine (100 mL). Dry, filter, and concentrate the organic phase to give the crude C2ι hydroxy dienol ether product. Compounds D27a-c do not require subsequent acidic hydrolysis.
Dissolve the above C2ι hydroxy dienol ether (1 g) in THF (20mL), add 0.1 N aqueous hydrochloric acid (2 mL) and stir the solution 3 hours at room temperature. Basify the reaction by addition of solid sodium bicarbonate and remove the solvent keeping the bath temperature below 30°C. By careful flash chromatography, the C2o double bond stereoisomers are generally more separable at this point. If not, isolate the mixture of cis and trans isomers and use as is. Prepare by this means the following 3-keto-20-fluoro-21-hydroxy compounds: 20ξ-Fluoro-21-hydroxy-lα-methylpregna-4,17(20)-dien-3-one (C23a) 1 α-Ethyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (C23b) 20ξ-Fluoro-21 -hydroxy- 1 α-ρropylpregna-4, 17(20)-dien-3-one (C23c) 20ξ-Fluoro-lα-methylpregna-5,17(20)-diene-3β,21-diol (D27a)
1 α-Ethyl-20ξ-fluoro-pregna-5, 17(20)-diene-3 β,21 -diol (D27b) 20ξ-Fluoro-l -propylpregna-5,17(20)-diene-3β,21-diol (D27c) 20ξ-Fluoro-21-hydroxy-2 -methylpregna-4,17(20)-dien-3-one (E33a) 2 -Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (E33b) 20ξ-Fluoro-2α-methylpregna-5,17(20)-diene-3β,21-diol (F37a)
2α-Ethyl-20ξ-fluoro-pregna-5,17(20)-diene-3β,21-diol (F37b) 20ξ-Fluoro-21 -hydroxy-4-methylpregna-4, 17(20)-diene-3-one (G46a) 4-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (G46b)
4-Isopropyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-diene-3-one (G46c)
4-Butyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-diene-3-one (G46d)
4-Chloro-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-diene-3-one (G46e)
20ξ-Fluoro-4-methylpregna-5,17(20)-diene-3β,21-diol (H53a) 4-Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (H53b)
20ξ-Fluoro-4-isopropyl-pregna-5 , 17(20)-diene-3 β,21 -diol (H53c)
4-Butyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (H53d)
4-Chloro-20ξ-fluoropregna-5, 17(20)-diene-3 β,21-diol (H53e)
20ξ-Fluoro-21 -hydroxy-6-methylpregna-4, 17(20)-dien-3-one (I60a) 6-Efhyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (I60b)
6-Butyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (I60c)
20ξ-Fluoro-6-methylpregna-5,17(20)-diene-3β,21-diol (J64a)
6-Ethyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (J64b)
6-Butyl-20ξ-fluoropregna-5,17(20)-diene-3β,21-diol (J64c) 20ξ-Fluoro-21-hydroxy-7α-methylpregna-4,17(20)-dien-3-one ( 71a)
7α-Ethyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (K71b)
20ξ-Fluoro-21 -hydroxy-7α-propylpregna-4, 17(20)-dien-3-one (K71 c)
7α-Butyl-20ξ-fluoro-2 l-hydroxypregna-4, 17(20)-dien-3-one (K71d)
20ξ-Fluoro-21 -hydroxy-7β-methylpregna-4, 17(20)-dien-3-one (K71 e) 7β-Ethyl-20ξ-fluoro-21-hydroxypregna-4,17(20)-dien-3-one (K71f)
20ξ-Fluoro-21-hydroxy-7β-propylpregna-4,17(20)-dien-3-one (K71g)
7β-Butyl-20ξ-fluoro-21 -hydroxypregna-4, 17(20)-dien-3-one (K71h)
Biological Methods and Results In vitro G?™ lyase assay: Compounds were tested for inhibition of cynomologous monkey Cι7j2o lyase in vitro using microsomal preparations of the enzyme from testicular tissue. Testes were removed from anesthetized animals and flash frozen in liquid nitrogen. Microsomes were isolated as described in Schatzman et al., Anal. Biochem., 175. 219-226 (1988). The compound to be tested was dissolved in DMSO and diluted in 0.05M potassium phosphate buffer, pH 7.4, to give the desired concentrations of test compound, in an amount which contributes 0.1 % v/v DMSO to the total assay volume. Assays contained 0.05M potassium phosphate, pH 7.4, an NADPH regenerating system (1 mM NADPH, 5 mM glucose-6-phosphate, 1 IU/mL glucose-6-phosphate dehydrogenase), test compound, substrate and microsomal protein in a total volume of 0.2 mL. Control assays contained all components, including DMSO, but no test compound. All assays were performed in duplicate. The reaction was initiated by the addition of substrate, 7-3H-17α-hydroxypregnenolone (11.2 mCi/mmole; 0.20 mCi per assay) plus unlabeled 17α-hydroxypregnenolone dissolved in DMSO, contributing 2.5% v/v to the final assay mix, and phosphate buffer, yielding a final concentration of 0.05 mM 17α-hydroxypregnenolone (ca. equal to the
Km value) to the other assay components. The complete assay was incubated at 34°C for 6 minutes. Each assay was terminated by addition of 5 mL of chloroform:methanol (2:1) and 0.9 mL water. Carrier steroids representing substrates and products (2.5μg each of 17c-hydroxypregnenolone, dehydroepiandrosterone, and androst-5-ene-3|3,17j3-dioι) and 0.8 mL of distilled, deionized water were added. The steroids were then extracted by the method of Moore and Wilson (Methods in Enzymol., eds, O. Malley, B.W. and Hardman, J.G. 36, 466-474 (1975). The organic phase containing the steroids was evaporated using nitrogen gas, the residues were dissolved in 18% tetrahydrofuran (v/v) in hexane, and the steroids were separated by HPLC on a Si60 (5 mm) column (4x250 mm) using a gradient of 18-22% tetrahydrofuran (v/v) in hexane. Radioactivity in the steroid peaks was measured using a Radiometric Model HS or Model A515 Flo-One detector. The enzyme activity for each assay was calculated from the percent conversion of substrate to products, and the results expressed as percent inhibition of control.
Z-isomer E-isomer

In vitro 5α-reductase assay: The activity of the present compounds as inhibitors of steroid 5α-reductase was determined using microsomal preparations of the 5α-reductase enzyme from human or laboratory animal prostate tissue. Specifically, prostate tissue was rapidly frozen in liquid nitrogen following removal from the human patient or from a cynomologus monkey (Macaca fascicularis) and subsequently stored at -80°C. The tissue was thawed, minced, and then homogenized in 5 volumes of 0.05M potassium phosphate buffer, pH 7.0, using a Tekmar homogenizer with 3x5 second bursts, followed by 10 strokes in a Dounce homogenizer. The homogenate was sonified in three pulses of 5 seconds each at 50% of maximum power. The homogenate was subjected to differential centrifugation with each supernatant successively centrifuged at 600xG and 9000xG in a Beckman J21 centrifuge followed by 120,000xG in a Beckman Model L5-75 ultracentrifuge. The final pellet, containing the microsomal fraction, was reserved and resuspended in 0.05M potassium phosphate buffer, pH 7.0, containing 25% (w/v) glycerol equal to 1 mL per 3 g wet tissue. The suspension was divided into aliquots, flash frozen using dry ice in methanol, and stored at -80°C. Enzyme activity was stable for at least 1 year under these conditions. Rat prostate tissue was treated in a similar manner as described above except that fresh prostate tissue was removed from male Sprague-Dawley rats (Charles Rivers), 0.05M potassium phosphate buffer, pH 6.6, was used for homogenization and 0.05M potassium phosphate buffer, pH 6.6, containing 25% (w/v) glycerol was used for storage. Protein concentration was determined by the BioRad dye binding method (BioRad, Richmond, California, USA).
Assays of human, cynomologus monkey and rat prostatic 5oj-reductase contained 100 mM potassium phosphate-sodium citrate buffer (pH 5.6), 0.1% bovine serum albumin (w/v, Sigma Chemicals), 1.0 mM sodium EDTA, 7-96 μg of microsomal protein, 1.0 mM NADPH; 5.0 mM glucose-6-phosphate, 1 IU/mL glucose-6- phosphate dehydrogenase, [l,2-3H]-testosterone (0.15μCi, DuPont-New England Nuclear,), unlabeled testosterone to yield the desired concentration of substrate, and test compound which was dissolved in DMSO and then diluted in 100 mM potassium phosphate-sodium citrate buffer (pH 6.5) to yield a final assay concentration of 0.1% (v/v) DMSO. The same buffer and DMSO without test compound were used in control assays. Background radioactivity was determined from assays containing all components except enzyme. Assays were performed in duplicate. The reaction was initiated by the addition of testosterone and incubated for 30 minutes at 25°C in a Dubnoff shaker incubator. The compound to be tested for inhibition was added simultaneously with testosterone. The total volume of the assay was 100 μL. The assay was linear with time to 30 minutes under these conditions. For IC50 determinations, a single concentration of testosterone at the Km level was used. Testosterone concentration was varied over a range of 0.5Km to 8Km for determination of inhibition mechanism and K; values. The Km values of testosterone, determined in multiple experiments, ranged from 0.125-0.273 μM for human 5α-reductase, 0.025-0.091 μM for cynomolgus monkey 5α-reductase, and 0.74-0.90 μM for rat 5α-reductase.
Each assay was terminated by addition of 5 mL of chloroform:methanol (2:1) and 0.9 mL of water. Carrier steroids representing substrates and products (2.5 μg each of testosterone, 5α-dihydrotestosterone, and 3,17-androstenediol) were added. The steroids were extracted by the method of Moore and Wilson (Methods in Enzymol., eds, O. Malley, B.W. and Hardman, J.G. 36, 466-474 (1975).
The organic phase containing the steroids was evaporated using nitrogen gas, the residues were dissolved in 3% isopropanol (v/v) in hexane, and the steroids were separated by normal phase HPLC (LiCrosorb® DIOL derivatized silica gel column (lOμM; 4x250 mm; EM Sciences, Gibbstown, New Jersey). After injection of sample, the steroids were separated with a 3% to 7.5% isopropanol in hexane gradient over 24 minutes, and then under isocratic conditions for 2 minutes at 75% (v/v) isopropanol in hexane using a flow rate of 1 mL per minute. The column was re-equilibrated with 3% (v/v) isopropanol in hexane prior to the next injection. The retention times were: 5α-dihydrotestosterone, 10.1-11.2 minutes; testosterone, 14.2-16.1 minutes; and 3/3,17/3- androstanediol, 17.1-20.2 minutes. The HPLC system used to separate the steroid components of the human and rat 5α-reductase assays consisted of Beckman 114M pumps and a 421A controller, a Waters WISP 710B autosampler, a Kratos Spectraflow 783 UV detector (wavelength set at 238 nm) and a Radiomatic model HS radioactivity analyzer. The HPLC system used for analysis of the cynomolgus monkey 5α-reductase assay was composed of a Waters 600E controller and dual pump unit, a Waters WiSP model 715 autosampler, a Waters 486 UV detector (wavelength, 238 nm), and a Radiomatic A515 radioactivity analyzer. FloScint II was used at a ratio of 1.6:1 (scrntillant to column effluent) for detection of [3H]-dihydrotestosterone. FloOne HS radioactivity data from the human and rat 5α-reductase assays were analyzed using the Beckman Data Transporter (Beckman Instruments, Fullerton, CA), which transferred integrated data collected from the FloOne HS to a mainframe computer and data were analyzed using RSI (BBN Software Products Corp., Cambridge, MA). Radioactivity data from the FloOne arising from the cynomolgus monkey 5α-reductase assays were analyzed using Waters Millenium software. Reaction rates were determined by multiplying the initial testosterone concentration by the percent of dihydrotestosterone and 3 , 17-androstenediol formed.
Concentrations of 10 μM and 1 μM of test compound were used to evaluate inhibitory activity. IC50 values were obtained using 6 concentrations of inhibitor. The data from these experiments were fitted to Equation 1 using a Vax computer. f(x) = 100/{1 + [X/B3]**[(B2 * B3)/-25]} Eq. 1
where B2 is the slope at IC50 and B3 is the IC50 value. Inhibition constants (K; ) were determined by fitting the data to the competitive inhibition model using Equation 2 (W.W. Cleland (1963) Biochim. Biophys. Ada, 67, 188-196) by nonlinear regression on a Compaq 386s using Dexter Northrop's MegaBasic program which was adapted from R. Duggleby's procedure (R. Duggleby, (1984) Comput. Biol. Med., 14, 447-455.). v = VmaxA/[Km(l + I/KO + A] Eq. 2 where Vmax is the maximal velocity, A is the concentration of testosterone, I is the test compound concentration and K; is the inhibition constant. The enzyme activity for each assay was calculated from the percent conversion of substrate to products, and the results were expressed as percent inhibition of control.