WO2002060492A1 - Methods of inhibiting kinases - Google Patents
Methods of inhibiting kinases Download PDFInfo
- Publication number
- WO2002060492A1 WO2002060492A1 PCT/AU2002/000089 AU0200089W WO02060492A1 WO 2002060492 A1 WO2002060492 A1 WO 2002060492A1 AU 0200089 W AU0200089 W AU 0200089W WO 02060492 A1 WO02060492 A1 WO 02060492A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- heterocyclyl
- alkyl
- halo
- amino
- heterocycle
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *Nc1nc(Br)c[n]2c1ncc2 Chemical compound *Nc1nc(Br)c[n]2c1ncc2 0.000 description 2
- UQCZZGIPIMJBCL-UHFFFAOYSA-N Brc(nc1Br)c[n]2c1ncc2 Chemical compound Brc(nc1Br)c[n]2c1ncc2 UQCZZGIPIMJBCL-UHFFFAOYSA-N 0.000 description 1
- WLUZPVKNVNHRCE-RGURZIINSA-N C[C@@H](C1C=CC=CC1)Nc1nc(Cl)cnc1 Chemical compound C[C@@H](C1C=CC=CC1)Nc1nc(Cl)cnc1 WLUZPVKNVNHRCE-RGURZIINSA-N 0.000 description 1
- UVAZCJYSUJAXIZ-ZDUSSCGKSA-N C[C@@H](c1ccccc1)Nc1nc(-c(cc2)cnc2OC)cnc1 Chemical compound C[C@@H](c1ccccc1)Nc1nc(-c(cc2)cnc2OC)cnc1 UVAZCJYSUJAXIZ-ZDUSSCGKSA-N 0.000 description 1
- QIIGNRQSZCAWBS-CQSZACIVSA-N C[C@H](c1ccccc1)Nc1cncc(-c(cc2)ccc2NC(C)=O)n1 Chemical compound C[C@H](c1ccccc1)Nc1cncc(-c(cc2)ccc2NC(C)=O)n1 QIIGNRQSZCAWBS-CQSZACIVSA-N 0.000 description 1
- JZKMKUMKDDCQRK-SECBINFHSA-N C[C@H](c1ccccc1)Nc1cncc(Cl)n1 Chemical compound C[C@H](c1ccccc1)Nc1cncc(Cl)n1 JZKMKUMKDDCQRK-SECBINFHSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/443—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/14—Decongestants or antiallergics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
Definitions
- the present invention relates generally to the field of non-peptidyl inhibitors of protein tyrosine kinases. More particularly, the present invention concerns methods of inhibiting specific protein tyrosine kinases, including members of the JAK family of protein tyrosine kinases.
- cytokines play a pivotal role in the regulation of the immune system, they are appropriately considered to be key targets for therapeutic intervention in immune pathologies.
- the intracellular signal transduction pathways that are regulated by cytokines are potential points of therapeutic intervention in diseases that involve overproduction of cytokine signalling.
- protein kinases There are many different types of protein kinases. Each type has the ability to add a phosphate group to an amino acid in a target protein. The phosphate is provided by hydrolyzing ATP to ADP.
- a protein kinase has an ATP-binding site and a catalytic domain that can bind to the target protein molecule.
- the JAK family of protein tyrosine kinases play a central role in the cytokine dependent regulation of the proliferation and end function of several important cell types of the immune system. play a central role in the cytokine dependent regulation of the proliferation and end function of several important cell types of the immune system.
- PTKs protein tyrosine kinases
- a direct comparison of the four currently known mammalian JAK family members reveals the presence of seven highly conserved domains (Harpur et al , 1992).
- the classification used was guided by the approach of Pawson and co-workers (Sadovski et al, 1986) in their treatment of the SRC homology (SH) domains.
- JAK Homology domain 1 JAK Homology domain 1
- JH2 domain The next domain N-terminal to JHI is the kinase-related domain, designated here as the JH2 domain.
- JH7 The high degree of conservation of these JAK homology (JH) domains suggests that they are each likely to play an important role in the cellular processes in which these proteins operate.
- JAK homology domains are arbitrary, and may or may not define functional domains. Nonetheless, their delineation is a useful device to aid the consideration of the overall structural similarity of this class of proteins.
- JHI JAK1
- JH2 JAK1
- JHI putative PTK domain of JAKl
- JHI contains highly conserved motifs typical of PTK domains, including the presence of a tyrosine residue at position 1022 located 11 residues C-terminal to sub-domain VII that is considered diagnostic of membership of the tyrosine-specific class of protein kinases.
- JAKl PTK domain 255 amino acids
- other members of the PTK class of proteins revealed homology with other functional PTKs (for example, 28% identity with c-fes (Wilks and Kurban, 1988) and 37% homology to TRK (Kozma et al, 1988).
- the JHI domains of each of the JAK family members possess a interesting idiosyncrasy within the highly conserved sub-domain Vm motif (residues 1015 to 1027 in JAK2) that is believed to lie close to the active site, and define substrate specificity.
- the phenylalanine and tyrosine residues flanking the conserved tryptophan in this motif are unique to the JAK family of PTKs.
- JHI domains of each of the members of the JAK family are typical PTK domains.
- the central role played by the JAK family of protein tyrosine kinases in the cytokine dependent regulation of the proliferation and end function of several important cell types means that agents which inhibit JAK are useful in the prevention and chemotherapy of disease states dependent on these enzymes.
- Potent and specific inhibitors of each of the currently known four JAK family members will provide a means of inhibiting the action of those cytokines that drive immune pathologies, such as asthma (e.g. IL-13; JAKl, JAK2), and leukemia/lymphoma (e.g. IL-2: JAKl and JAK3).
- cancers such as prostate cancer develop autocrine production of certain cytokines as a selectable mechanism of developing growth and/or metastatic potential.
- An example of this is cancer of the prostate, where IL-6 is produced by and stimulates the growth of prostate cancer cell lines such as TSU and TC3 (Spiotto MT, and Chung TD, 2000).
- levels of IL-6 are elevated in sera of patients with metastatic prostate cancer.
- cytokine signalling A great deal of literature covers the area of cytokine signalling.
- the present inventors have focussed on the JAK/STAT pathway that is involved in the direct connection of cytokine receptor to target genes (such as cell cycle regulators (e.g. p21) and anti-apoptosis genes (such as Bcl-X L )).
- target genes such as cell cycle regulators (e.g. p21) and anti-apoptosis genes (such as Bcl-X L )).
- a cytokine receptor chain such as the Interleukin-4 receptor or the Interferon ⁇ receptor
- a member or members of the JAK family of PTKs
- a member(s) of the STAT family of transcription factors and (iv) a sequence specific DNA element to which the activated STAT will bind.
- JAKs In addition to the diseases listed in Tables 1 and 2, inhibitors of JAKs could be used as immunosuppresive agents for organ transplants and autoimmune diseases such as lupus, multiple sclerosis, rheumatoid arthritis, Type I diabetes, autoimmune thyroid disorders, Alzheimer's disease and other autoimmune diseases. Additionally, treatment of cancers such as prostate cancer by JAK inhibitors is indicated.
- the present inventors have found that a group of compounds based either upon a 2-amino-6-carba-disubstituted pyrazine scaffold or a 2-amino-6- carba-disubstituted pyridine scaffold are JAK inhibitors.
- the present invention consists in a method of inhibiting JAK in a cell, the method comprising administering to the cell an effective amount of a composition comprising a carrier and a compound of the general formula I:
- X is either carbon or nitrogen
- RI is C 1 0 Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R2 is selected from C wo Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, Halo, OH, or 6-7 membered Heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S.
- the present invention consists in a method of inhibiting JAK in a cell, the method comprising administering to the cell an effective amount of a composition comprising a carrier and a compound of the general formula TJ:
- R6 is C LU , Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R7 is C ⁇ o Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, Halo, OH, or Heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S.
- the present invention also includes the use of compounds of formula I or II is the prophylaxis and/or treatment of JAK-associated disease states.
- the present invention consists in a method of inhibiting JAK in a cell, the method comprising administering to the cell an effective amount of a composition comprising a carrier and a compound of the general formula I: or pharmaceutically acceptable salts, hydrates, solvates, crystal forms or diastereomers thereof, wherein
- X is either carbon or nitrogen
- RI is C ⁇ o Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R2 is selected from C l W Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, Halo, OH, or 6-7 membered Heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S.
- R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the heterocycle includes two heteroatoms, preferably two nitrogen atoms.
- X is nitrogen ox carbon
- Rl is C 2 . 10 Alkylphenyl, Phenyl, or Heterocyclyl, wherein the Alkyl, Phenyl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 member ring and in which the hetero atom is O, N or S;
- R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the compound is selected from the compounds set out in Table 4.
- the present invention consists in a method of inhibiting JAK in a cell, the method comprising administering to the cell an effective amount of a composition comprising a carrier and a compound of the general formula II:
- R6 is Cj.i o Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 member ring and in which the hetero atom is O, N or S;
- R7 is C J . JO Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, Halo, OH, or Heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 member ring and in which the hetero atom is O, N or S.
- R8, R9 and RIO are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the heterocycle includes two heteroatoms, preferably two nitrogen atoms.
- R6 is C 2 . 10 Alkylphenyl, Phenyl, or Heterocyclyl, wherein the Alkyl, Phenyl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S; R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the compound is selected from the compounds set out in Tables 6 and 7.
- the method is conducted in vivo. It is also preferred that the JAK is JAKl, JAK2, JAK3 or TYK2.
- the present invention consists in a method of treating an individual suffering from a JAK-associated disease state, the method comprising administering to the individual a composition comprising a pharmaceutically acceptable carrier and a compound of the general formula:
- X is either carbon or nitrogen
- Rl is C x . 10 Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R2 is selected from C WD Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, Halo, OH, or 6-7 membered Heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 member ring and in which the hetero atom is O, N or S.
- R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the heterocycle includes two heteroatoms, preferably two nitrogen atoms.
- Rl is C 2 . 10 Alkylphenyl, Phenyl, or Heterocyclyl, wherein the Alkyl, Phenyl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of chloro, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the compound is selected from the compounds set out in Table 4.
- the present invention consists in a method of treating an individual suffering from a JAK-associated disease state, the method comprising administering to the individual a composition comprising a pharmaceutically acceptable carrier and a compound of the general formula:
- R6 is C J . JO Alkyl, C 2 . 10 Alkenyl, C 2 . 10 Alkynyl, C 2 . 10 Alkylaryl, Aryl, or Heterocyclyl, or Rl with N may form a substituted or unsubstituted heterocyclyl, wherein the Alkyl, Alkenyl, Alkynyl, Alkylaryl, Aryl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 member ring and in which the hetero atom is O, N or S; R7 is G t .
- R8, R9 and R10 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the heterocycle includes two heteroatoms, preferably two nitrogen atoms.
- R6 is C 2 . 10 Alkylphenyl, Phenyl, or Heterocyclyl, wherein the Alkyl, Phenyl, and Heterocyclyl, is optionally substituted with one to three members selected from the group consisting of halo, amino, hydroxy, hydroxyalkyl, alkylamide, arylamide, hydroxyalkylamide, nitrilo, aminoalkylamide, nitriloaryl, alkoxy (in particular methoxy), heterocyclic alkyl in which heterocycle is a 5-7 membered ring and in which the hetero atom is O, N or S;
- R3, R4 and R5 are the same or different and are H, halo, OH, hydroxyamide, amino, hydroxyalkyl, aminoalkylamide, alkylamide, arylamide or alkoxy.
- the compound is selected from the compounds set out in Tables 6 and 7.
- the disease state involves JAKl, JAK2, JAK3 or TYK2.
- the disease state is selected from the group consisting of Atopy, such as Allergic Asthma, Atopic Dermatitis (Eczema), and Allergic Rhinitis; Cell Mediated Hypersensitivity, such as Allergic Contact Dermatitis and Hypersensitivity Pneumonitis; Rheumatic Diseases, such as Systemic Lupus Erythematosus (SLE), Rheumatoid Arthritis, Juvenile Arthritis, Sj ⁇ gren's Syndrome, Scleroderma, Polymyositis, Ankylosing Spondylitis, Psoriatic Arthritis; Other autoimmune diseases such as Type I diabetes, autoimmune thyroid disorders, and Alzheimer's disease; Viral Diseases, such as Epstein Barr Virus (EBV),
- EBV Epstein Barr Virus
- Hepatitis B Hepatitis C
- HTV Hepatitis HTLV 1
- VZV Varicella-Zoster Virus
- HPV Human Papilloma Virus
- Cancer such as Leukemia, Lymphoma and Prostate Cancer.
- the present invention provides the use of the compounds described in the preparation of medicaments for the treatment of JAK-associated disease states.
- JAK As used herein the term "JAK”, “JAK kinase” or “JAK family” refers to protein tyrosine kinases which possess the characterizing features of JAKl, JAK2, JAK3 and TYK as described herein.
- JAK-associated disease state refers to those disorders which result from aberrant JAK activity, and/or which are alleviated by inhibition of one or more of these enzymes.
- compositions comprising at least one of the compounds of the formula I or TJ capable of treating a JAK-associated disorder in an amount effective therefor, and a pharmaceutically acceptable vehicle or diluent.
- the compositions of the present invention may contain other therapeutic agents as described below, and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (for example, excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation.
- the compounds of the formula I or II may be administered by any suitable means, for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; buccally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intracisternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non- toxic, pharmaceutically acceptable vehicles or diluents.
- suitable means for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; buccally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intracisternal injection or infusion techniques (e.g., as sterile injectable
- the compounds may, for example, be administered in a form suitable for immediate release or extended release. Immediate release or extended release may be achieved by the use of suitable pharmaceutical compositions comprising the present compounds, or, particularly in the case of extended release, by the use of devices such as subcutaneous implants or osmotic pumps.
- the compounds may also be administered liposomally.
- primates such as humans
- a variety of other mammals can be treated according to the method of the present invention. For instance, mammals including, but not limited to, cows, sheep, goats, horses, dogs, cats, guinea pigs, rats or other bovine, ovine, equine, canine, feline, rodent or murine species can be treated.
- the method can also be practiced in other species, such as avian species (e.g., chickens).
- the disease or condition is one in which the actions of eosinophils and/or lymphocytes are to be inhibited or promoted, in order to modulate the inflammatory response.
- the subjects treated in the above methods, in whom which JAK inhibition is desired are mammals, including, but not limited to, cows, sheep, goats, horses, dogs, cats, guinea pigs, rats or other bovine, ovine, equine, canine, feline, rodent or murine species, and preferably a human being, male or female.
- terapéuticaally effective amount means the amount of the subject composition that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment.
- compositions for the administration of the compounds of this invention may conveniently be presented in dosage unit form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients.
- the pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- the pharmaceutical compositions containing the active ingredient may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate may be employed. They may also be coated to form osmotic therapeutic tablets for control release.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydroxy-propylmethylcellulose, sodium alginate, polyvinyl-pyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate.
- dispersing or wetting agents may be a naturally-occurring phosphatide, for example lecithin, or
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n-propyl, p- hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- preservatives for example ethyl, or n-propyl, p- hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerin, glycerin, glycerin, glycerin, glycerin, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, sorbitol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol, glycerol
- the pharmaceutical compositions of the invention may also be in the form of oil-in- water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or arachis oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally- occurring gums, for example gum acacia or gum tragacanth, naturally- occurring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol anhydrides, for example sorbitan monooleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- sweetening agents for example glycerol, propylene glycol, sorbitol or sucrose.
- Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example as a solution in 1,3-butane diol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds of the present invention may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non- irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- a suitable non- irritating excipient which is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- creams, ointments, jellies, solutions or suspensions, etc., containing the compounds of the present invention are employed. (For purposes of this application, topical application shall include mouthwashes and gargles.)
- compositions and method of the present invention may further comprise other therapeutically active compounds as noted herein which are usually applied in the treatment of the above mentioned pathological conditions.
- cyclosporins e.g., cyclosporin A
- CTLA4-Ig antibodies such as ICAM-3, anti-IL-2 receptor (Anti-Tac), anti-CD45RB, anti-CD2, anti-CD3 (OKT-3), anti- CD4, anti-CD80, anti-CD86
- agents blocking the interaction between CD40 and gp39 such as antibodies specific for CD40 and/or gp39 (i.e., CD154), fusion proteins constructed from CD40 and gp39 (CD401g and CD8gp39), inhibitors, such as nuclear translocation inhibitors, of NF-kappa B function, such as deoxyspergualin (DSG), cholesterol biosynthesis inhibitors such as HMG CoA reductase inhibitors (lovastatin and simvastatin), non-steroidal antiinflammatory drugs (NSAIDs) such as ibuprofen and cyclooxygenase inhibitors such as rofecoxib, steroids such as pred
- NSAIDs non-
- an appropriate dosage level will generally be about 0.01 to 500 mg per kg patient body weight per day which can be administered in single or multiple doses.
- the dosage level will be about 0.1 to about 250 mg/kg per day; more preferably about 0.5 to about 100 mg/kg per day.
- a suitable dosage level may be about 0.01 to 250 mg/kg per day, about 0.05 to 100 mg/kg per day, or about 0.1 to 50 mg kg per day. Within this range the dosage may be 0.05 to 0.5, 0..5 to 5 or 5 to 50 mg/kg per day.
- compositions are preferably provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, particularly 1.0, 5.0, 10.0, 15.0. 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0, 400.0, 500.0, 600.0, 750.0, 800.0, 900.0, and 1000.0 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- All compounds may be prepared in a 2-step process starting from a dihalogenated heterocycle.
- the dihalogenated heterocyclic starting materials 2,6-dichloropyrazine and 2,6-dibromopyridine are obtained commercially.
- 6,8-Dibromo-imidazo-[l,2- ⁇ ]-pyrazine can be prepared following the literature route (see for example, Sablayrolles, C. et al, J. Med. Chem., 1984, 27, 206).
- the first step is a nucleophilic aromatic substitution to generate a monoamino-monohalo intermediate. (Scheme 1).
- the nucleophilic aromatic substitution is typically carried out by addition of a primary amine to the di-halogenated heterocycle in a solvent such as ethanol, isopropanol, ferf-butanol, dioxane, THF, DMF, toluene or xylene.
- a solvent such as ethanol, isopropanol, ferf-butanol, dioxane, THF, DMF, toluene or xylene.
- the reaction is typically performed at elevated temperature in the presence of excess amine or a non-nucleophilic base such as triethylamine or diisopropylethylamine, or an inorganic base such as potassium carbonate or sodium carbonate.
- the second step of the synthesis typically involves a palladium mediated cross-coupling of the monoamino-monohalo intermediate with a suitably functionalised coupling partner.
- Typical coupling partners are boronic acids (Suzuki coupling: see for example Miyaura, N. and Suzuki, Chem Rev. 1995, 95 2457) or stannanes (Stille coupling: see for example Stille, J.K., Angew. Chem., Int. Ed. Engl, 1986, 25, 508) (Scheme 2).
- the Suzuki coupling is the preferred coupling method and is typically performed in a solvent such as DME, THF, DMF, ethanol, toluene, or 1,4- dioxane in the presence of a base such as potassium carbonate, lithium hydroxide, caesium carbonate, sodium hydroxide, potassium fluoride or potassium phosphate.
- a base such as potassium carbonate, lithium hydroxide, caesium carbonate, sodium hydroxide, potassium fluoride or potassium phosphate.
- the reaction may be carried out at elevated temperatures and the palladium catalyst employed may be selected from [Pd(PPh 3 ) 4 ], Pd(OAc) 2 , [PdCl 2 (dppf)], Pd 2 (dba) 3 /P(f-Bu) 3 .
- JAK kinase domains were produced in the following manner:
- the kinase domain of humanJAKl was amplified from U937mRNA using the polymerase chain reaction with the following primers: XHOI-J1 5'-CCG CTC GAG ACT GAA GTG GAC CCC ACA CAT-3' Jl-KPNI 5'-CGG GGT ACC TTA TTT TAA AAG TGC TTC AAA-3'
- JAKl PCR products were cloned into the pFastBac HTb expression vector (Gibco) via the Xho I and Kpn I sites.
- the JAKl plasmid was then transformed into competent DHlOBac cells (Gibco), and the recombinant baculovirus produced prepared for transfection into Sf9 insect cells.
- the kinase domain of humanJAK2 was amplified from U937mRNA using the polymerase chain reaction with the following primers: SALI-jk2 5'-ACG CGT CGA CGG TGC CTT TGA AGA CCG GGA T-3' jk2-NOTI 5'-ATA GTT TAG CGG CCG CTC AGA ATG AAG GTC ATT T-3'
- JAK2 PCR products were cloned into the pFastBac HTc expression vector (Gibco) via the Sal I and Not I sites.
- the JAK2 plasmid was then transformed into competent DHlOBac cells (Gibco), and the recombinant baculovirus produced prepared for transfection into Sf9 insect cells.
- the kinase domain of humanJAK3 was amplified from U937mRNA using the polymerase chain reaction with the following primers: XHOI-J3 5'-CCG CTC GAG TAT GCC TGC CAA GAC CCC ACG-3' J3-KPNI 5'-CGG GGT ACC CTA TGA AAA GGA GAG GGA GTG-3' JAK3 PCR products were cloned into the pFastBac HTb expression vector (Gibco) via the Xho I and Kpn I sites. The JAK3 plasmid was then transformed into competent DHlOBac cells (Gibco), and the recombinant baculovirus produced prepared for transfection into Sf9 insect cells.
- the kinase domain of humanTYK2 was amplified from A549 mRNA using the polymerase chain reaction with the following primers: HT2EK 5'-GGA GCA CTC GAG ATG GTA GCA CAC AAC CAG GTG-3' ITY2.2R 5'-GGA GCA GGA ATT CCG GCG CTG CCG GTC AAA TCT GG-3'
- TYK2 PCR products were cloned into pBlueBacHis2A (Invitrogen) via the EcoRI site.
- the recombinant TYK2 baculovirus produced was prepared for transfected into Sf9 insect cells.
- JAK kinase domains were purified by affinity chromatography on a Probond (Invitrogen) nickel chelate affinity column.
- Kinase assays were performed in a 96 well capture-based ELISA assay, using approximately 1.5 ⁇ g of affinity purified PTK domain in the presence of 50mM HEPES, pH 7.5, 10mM MgCl 2 , 150m NaCl and 10-20 ⁇ M ATP.
- the biotinylated substrate biotin-EGPWLEEEEEAYGWMDF-NH 2 (final concentration 5 ⁇ M) was used as substrate, and tyrosine phosphorylation was quantitated following transfer to an avidin coated ELISA plate using peroxidase-linked anti-phospho-tyrosine antibody PY20. Inhibitors were added to the assays fifteen minutes prior to the addition of ATP.
- Inhibitors were added in aqueous DMSO, with DMSO concentrations never exceeding 1%.
- Cellular assays were performed as follows: Cell suspensions were prepared by harvesting cells from culture. Cell used in this test should be in later log phase growth and high viability. Cells were diluted in correct growth medium to l.lx final concentration (from 50000 cell/ml to 200,000 cell/ml, depending on cell line). 90 ⁇ L was added to samples, diluted in PBS to lOx final concentration in flat-bottom 96-well plates (lO ⁇ L). After incubation for 40 hr in 37 °C 5% C0 2 incubator, MTT 5mg/ml (in PBS, filter sterile) 20 ⁇ l per well was added. The plates were returned to incubator for another 6 hours. Lysis Buffer (10% SDS, 0.01N HCl) 100 ⁇ l per well was added and the plate put back in incubator overnight. The plate was then read at 590 nm.
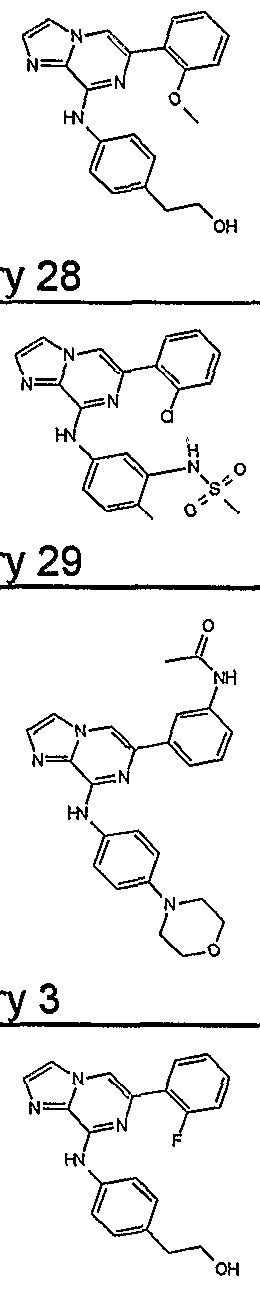
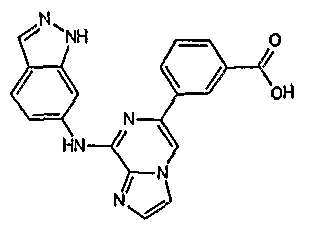
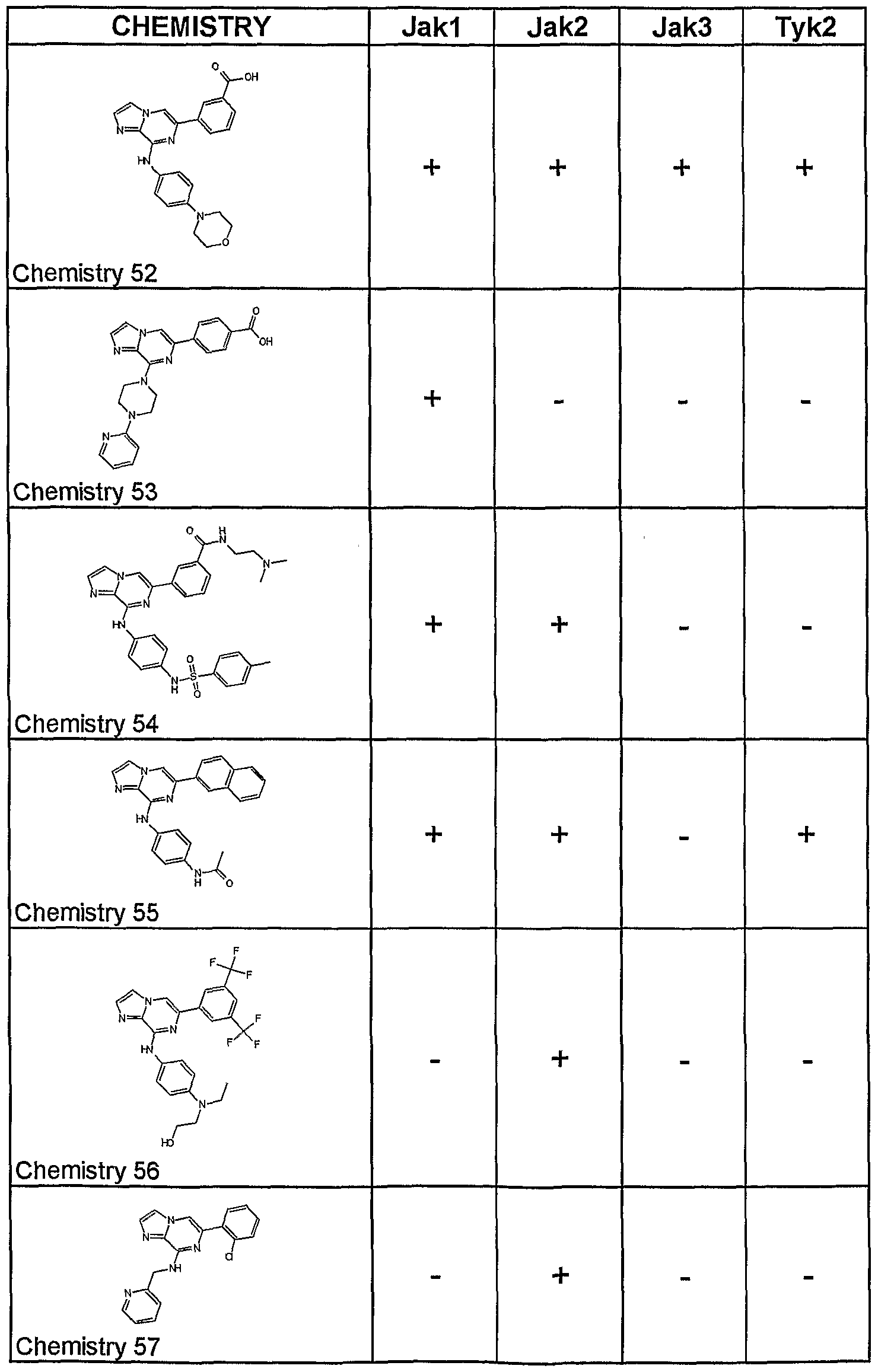
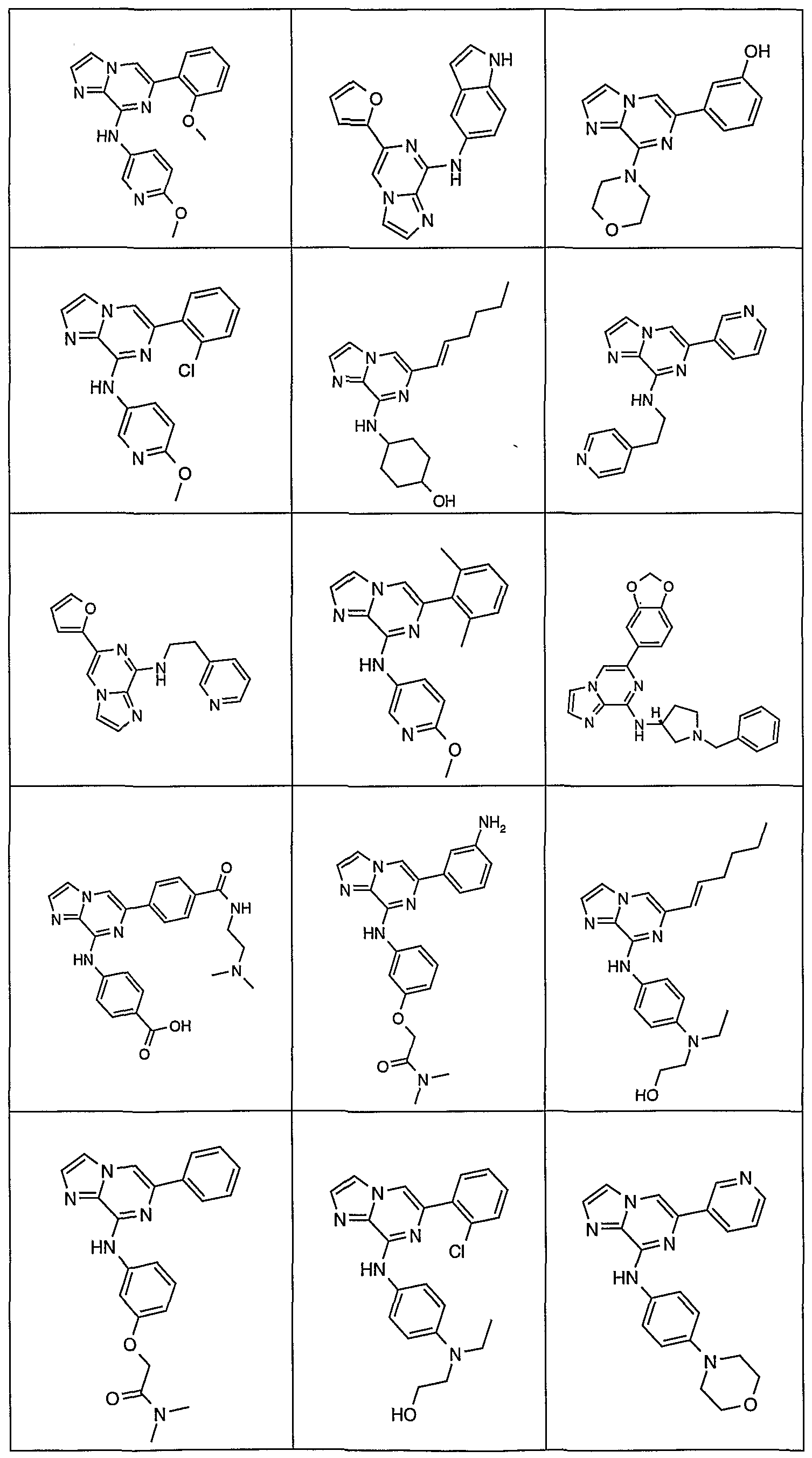
- Table 4 Selected 2-amino-6-carba-disubstituted pyrazines and 2-amino- 6-carba-disubstituted pyridines possessing JAK inhibitory activity
- JAK2 a third member of the JAK family of protein tyrosine kinases. Oncogene; 7 1347-53.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Virology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Communicable Diseases (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Molecular Biology (AREA)
- Obesity (AREA)
- Hospice & Palliative Care (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- AIDS & HIV (AREA)
- Ophthalmology & Optometry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Tropical Medicine & Parasitology (AREA)
- Dermatology (AREA)
Abstract
Description
























































Claims































Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002560683A JP2004528295A (en) | 2001-01-30 | 2002-01-30 | Kinase inhibition method |
| AU2002226197A AU2002226197B2 (en) | 2001-01-30 | 2002-01-30 | Methods of inhibiting kinases |
| US10/470,955 US20040102455A1 (en) | 2001-01-30 | 2002-01-30 | Method of inhibiting kinases |
| CA002436487A CA2436487A1 (en) | 2001-01-30 | 2002-01-30 | Methods of inhibiting kinases |
| EP02715984A EP1363702A4 (en) | 2001-01-30 | 2002-01-30 | Methods of inhibiting kinases |
| US11/223,633 US20060069084A1 (en) | 2001-01-30 | 2005-09-09 | Methods of inhibiting kinases |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AUPR2792 | 2001-01-30 | ||
| AUPR2793 | 2001-01-30 | ||
| AUPR2792A AUPR279201A0 (en) | 2001-01-30 | 2001-01-30 | Jak inhibitors |
| AUPR2793A AUPR279301A0 (en) | 2001-01-30 | 2001-01-30 | Method of inhibiting jak |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/223,633 Division US20060069084A1 (en) | 2001-01-30 | 2005-09-09 | Methods of inhibiting kinases |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2002060492A1 true WO2002060492A1 (en) | 2002-08-08 |
Family
ID=25646570
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/AU2002/000089 Ceased WO2002060492A1 (en) | 2001-01-30 | 2002-01-30 | Methods of inhibiting kinases |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US20040102455A1 (en) |
| EP (1) | EP1363702A4 (en) |
| JP (1) | JP2004528295A (en) |
| CA (1) | CA2436487A1 (en) |
| WO (1) | WO2002060492A1 (en) |
Cited By (121)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003027295A3 (en) * | 2001-09-24 | 2003-10-16 | Univ Aarhus | Methods for diagnosis and treatment of diseases associated with altered expression of jak1 |
| WO2003099811A1 (en) * | 2002-05-23 | 2003-12-04 | Cytopia Pty Ltd | Kinase inhibitors |
| WO2003099796A1 (en) * | 2002-05-23 | 2003-12-04 | Cytopia Pty Ltd | Protein kinase inhibitors |
| WO2003089434A3 (en) * | 2002-04-19 | 2004-01-15 | Cellular Genomics Inc | IMIDAZO[1,2-a]PYRAZIN-8-YLAMINES METHOD OF MAKING AND METHOD OF USE THEREOF |
| WO2004022562A1 (en) * | 2002-09-09 | 2004-03-18 | Cellular Genomics, Inc. | 6-ARYL-IMIDAZO[1,2-a]PYRAZIN-8-YLAMINES, METHOD OF MAKING, AND METHOD OF USE THEREOF |
| WO2004026877A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| WO2004026310A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Novel imidazopyrazines as cyclin dependent kinase inhibitors |
| WO2004004730A3 (en) * | 2002-07-06 | 2004-04-29 | Astex Technology Ltd | 2-aminopyrazine derivatives as inhibitors of cyclin dependent kinases for the treatment of proliferative disorders |
| WO2004026229A3 (en) * | 2002-09-04 | 2004-06-17 | Schering Corp | Pyrazolo[1,5-a]pyrimidines compounds as cyclin dependent kinase inhibitors |
| WO2004052868A1 (en) * | 2002-12-11 | 2004-06-24 | Cytopia Pty Ltd | Pyrazine-based tubulin inhibitors |
| WO2004072081A1 (en) * | 2003-02-10 | 2004-08-26 | Cellular Genomics, Inc. | Certain 8-heteroaryl-6-phenyl-imidazo[1,2-a]pyrazines as modulators of kinase activity |
| WO2004074289A1 (en) * | 2003-02-18 | 2004-09-02 | Altana Pharma Ag | 6-substituted imidazopyrazines |
| WO2005005429A1 (en) * | 2003-06-30 | 2005-01-20 | Cellular Genomics, Inc. | Certain heterocyclic substituted imidazo[1,2-a]pyrazin-8-ylamines and methods of inhibition of bruton’s tyrosine kinase by such compounds |
| WO2005014599A1 (en) * | 2003-06-04 | 2005-02-17 | Cellular Genomics, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of bruton’s tyrosine kinase by such compounds |
| WO2005019220A3 (en) * | 2003-08-11 | 2005-03-24 | Cellular Genomics Inc | Substituted imidazo[1,2-a]pyrazines as modulators of kinase activity |
| WO2005037836A3 (en) * | 2003-10-15 | 2005-06-09 | Osi Pharm Inc | Imidazo ‘1, 5 - a ! pyrazine tyrosine kinase inhibitors |
| WO2005054199A1 (en) * | 2003-12-03 | 2005-06-16 | Cytopia Research Pty Ltd | Tubulin inhibitors |
| WO2005033105A3 (en) * | 2003-09-30 | 2005-06-23 | Amgen Inc | Vanilloid receptor ligands and their use in treatments |
| WO2005058876A1 (en) * | 2003-12-16 | 2005-06-30 | Gpc Biotech Ag | Pyrazine derivatives as effective compounds against infectious diseases |
| WO2005047290A3 (en) * | 2003-11-11 | 2005-08-11 | Cellular Genomics Inc | Imidazo[1,2-a] pyrazin-8-ylamines as kinase inhibitors |
| WO2005085252A1 (en) * | 2004-03-04 | 2005-09-15 | Biofocus Discovery Limited | Imidazo ‘1,2-a’ pyrazine compounds which interact with protein kinases |
| WO2005121126A1 (en) | 2004-04-13 | 2005-12-22 | Icagen, Inc. | Polycyclic pyrazines as potassium ion channel modulators |
| JP2006505571A (en) * | 2002-10-15 | 2006-02-16 | リゲル ファーマシューテイカルズ、インコーポレイテッド | Substituted indoles and their use as HCV inhibitors |
| US7015227B2 (en) | 2002-06-21 | 2006-03-21 | Cgi Pharmaceuticals, Inc. | Certain amino-substituted monocycles as kinase modulators |
| JP2006520794A (en) * | 2003-03-21 | 2006-09-14 | スミスクライン ビーチャム コーポレーション | Compound |
| JP2006523238A (en) * | 2003-04-09 | 2006-10-12 | エクセリクシス, インク. | TIE-2 modulator and usage |
| JP2006524682A (en) * | 2003-03-19 | 2006-11-02 | エクセリクシス, インク. | TIE-2 modulator and usage |
| US7157460B2 (en) | 2003-02-20 | 2007-01-02 | Sugen Inc. | Use of 8-amino-aryl-substituted imidazopyrazines as kinase inhibitors |
| US7186832B2 (en) | 2003-02-20 | 2007-03-06 | Sugen Inc. | Use of 8-amino-aryl-substituted imidazopyrazines as kinase inhibitors |
| US7205308B2 (en) | 2002-09-04 | 2007-04-17 | Schering Corporation | Trisubstituted 7-aminopyrazolopyrimidines as cyclin dependent kinase inhibitors |
| WO2007098507A2 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| WO2007126964A3 (en) * | 2006-03-31 | 2007-12-21 | Schering Corp | Kinase inhibitors |
| WO2007138072A3 (en) * | 2006-05-31 | 2008-02-14 | Galapagos Nv | Triazolopyrazine compounds useful for the treatment of degenerative & inflammatory diseases |
| US7335667B2 (en) | 2004-12-22 | 2008-02-26 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-4-yl-amines as Janus kinase inhibitors |
| WO2008022164A3 (en) * | 2006-08-16 | 2008-04-17 | Boehringer Ingelheim Int | Pyrazine compounds, their use and methods of preparation |
| US7435814B2 (en) | 2002-02-01 | 2008-10-14 | Rigel Pharmaceuticals, Inc. | 2,4-Pyrimidinediamine compounds and their uses |
| US7452879B2 (en) | 2003-07-30 | 2008-11-18 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2008138842A1 (en) * | 2007-05-10 | 2008-11-20 | Galapagos N.V. | Imidazopyrazines and triazolopyrazine for the treatment of joint degenerative and inflammatory diseases |
| US7491732B2 (en) | 2005-06-08 | 2009-02-17 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| WO2009024585A3 (en) * | 2007-08-21 | 2009-08-06 | Biofocus Dpi Ltd | Imidazo [1,2-a] pyrazine compounds for treatment of viral infections such as hepatitis |
| WO2009102468A1 (en) | 2008-02-13 | 2009-08-20 | Cgi Pharmaceuticals, Inc. | 6-aryl-imidaz0[l, 2-a] pyrazine derivatives, method of making, and method of use thereof |
| WO2009095399A3 (en) * | 2008-02-01 | 2009-10-01 | Akinion Pharmaceuticals Ab | Pyrazine derivatives and their use as protein kinase inhbitors |
| US7598257B2 (en) | 2005-12-13 | 2009-10-06 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US7709468B2 (en) | 2005-09-02 | 2010-05-04 | Abbott Laboratories | Imidazo based heterocycles |
| US7737152B2 (en) | 2004-12-22 | 2010-06-15 | The Wellcome Trust Limited | 6-carboaryl-oxy-pyrazin-2-yl-carboaryl-amines and compositions comprising said compounds |
| WO2010069684A1 (en) * | 2008-12-17 | 2010-06-24 | Biomarin Iga, Ltd. | Compounds for treatment of duchenne muscular dystrophy |
| WO2010088368A2 (en) | 2009-01-29 | 2010-08-05 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
| US7777040B2 (en) | 2005-05-03 | 2010-08-17 | Cgi Pharmaceuticals, Inc. | Certain substituted ureas, as modulators of kinase activity |
| US7812029B1 (en) | 2002-07-29 | 2010-10-12 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2010119264A1 (en) * | 2009-04-16 | 2010-10-21 | Centro Nacional De Investigaciones Oncólogicas (Cnio) | Imidazopyrazines for use as kinase inhibitors |
| US7820662B2 (en) | 2004-04-02 | 2010-10-26 | Osi Pharmaceuticals, Inc. | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US7834022B2 (en) | 2007-06-13 | 2010-11-16 | Incyte Corporation | Metabolites of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| EP1689739A4 (en) * | 2003-12-03 | 2010-12-22 | Ym Biosciences Australia Pty L | INHIBITORS OF AZOLE-BASED KINASES |
| US7893058B2 (en) * | 2006-05-15 | 2011-02-22 | Janssen Pharmaceutica Nv | Imidazolopyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| WO2011056916A1 (en) * | 2009-11-05 | 2011-05-12 | Lexicon Pharmaceuticals, Inc. | Tryptophan hydroxylase inhibitors for the treatment of cancer |
| WO2011080176A1 (en) * | 2009-12-31 | 2011-07-07 | Novartis Ag | Pyrazine derivatives and their use in the treatment of neurological disorders |
| WO2011141713A1 (en) * | 2010-05-13 | 2011-11-17 | Centro Nacional De Investigaciones Oncologicas (Cnio) | New bicyclic compounds as pi3-k and mtor inhibitors |
| US8138339B2 (en) | 2008-04-16 | 2012-03-20 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| WO2012041476A1 (en) * | 2010-09-30 | 2012-04-05 | Almirall, S.A. | Pyridine and isoquinoline derivatives as syk- and jak-kinase inhibitors |
| US8158616B2 (en) | 2008-03-11 | 2012-04-17 | Incyte Corporation | Azetidine and cyclobutane derivatives as JAK inhibitors |
| EP2444084A1 (en) * | 2010-10-21 | 2012-04-25 | Centro Nacional de Investigaciones Oncológicas (CNIO) | Use of PI3K inibitors for the treatment of obesity |
| WO2012052745A1 (en) | 2010-10-21 | 2012-04-26 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Combinations of pi3k inhibitors with a second anti -tumor agent |
| WO2012061428A2 (en) | 2010-11-01 | 2012-05-10 | Portola Pharmaceuticals, Inc. | Nicotinamides as jak kinase modulators |
| EP2373318A4 (en) * | 2008-12-08 | 2012-05-30 | Gilead Connecticut Inc | IMIDAZOPYRAZINE SYK INHIBITORS |
| EP2373169A4 (en) * | 2008-12-08 | 2012-08-29 | Gilead Connecticut Inc | IMIDAZOPYRAZINE SYK INHIBITORS |
| US8258144B2 (en) | 2008-04-22 | 2012-09-04 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US8309566B2 (en) | 2008-02-15 | 2012-11-13 | Rigel Pharmaceuticals, Inc. | Pyrimidine-2-amine compounds and their use as inhibitors of JAK kinases |
| CN102791715A (en) * | 2009-12-31 | 2012-11-21 | 西班牙国家癌症研究中心 | Tricyclic compounds for use as kinase inhibitors |
| WO2013004332A1 (en) * | 2011-07-07 | 2013-01-10 | Merck Patent Gmbh | Substituted azaheterocycles for the treatment of cancer |
| US8450321B2 (en) | 2008-12-08 | 2013-05-28 | Gilead Connecticut, Inc. | 6-(1H-indazol-6-yl)-N-[4-(morpholin-4-yl)phenyl]imidazo-[1,2-A]pyrazin-8-amine, or a pharmaceutically acceptable salt thereof, as a SYK inhibitor |
| US8513270B2 (en) | 2006-12-22 | 2013-08-20 | Incyte Corporation | Substituted heterocycles as Janus kinase inhibitors |
| US8563541B2 (en) | 2005-09-22 | 2013-10-22 | Incyte Corporation | Azepine inhibitors of Janus kinases |
| US8691807B2 (en) | 2011-06-20 | 2014-04-08 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US8722693B2 (en) | 2007-06-13 | 2014-05-13 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| WO2014106606A1 (en) * | 2013-01-05 | 2014-07-10 | F. Hoffmann-La Roche Ag | Nove phenyl/pyridine series substitued by hydroxyethylamino for the treatment of cancer |
| CN104080769A (en) * | 2011-04-13 | 2014-10-01 | Epizyme股份有限公司 | Aryl or heteroaryl substituted benzene compounds |
| US8933085B2 (en) | 2010-11-19 | 2015-01-13 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US8952027B2 (en) | 2008-04-16 | 2015-02-10 | Portola Pharmaceuticals, Inc. | Inhibitors of syk and JAK protein kinases |
| US8987443B2 (en) | 2013-03-06 | 2015-03-24 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| WO2015100217A1 (en) * | 2013-12-23 | 2015-07-02 | Gilead Sciences, Inc. | Syk inhibitors |
| US9073927B2 (en) | 2010-01-22 | 2015-07-07 | Fundacion Centro Nacional De Investigaciones Oncologicas Carlos Iii | Inhibitors of PI3 kinase |
| WO2015158283A1 (en) * | 2014-04-17 | 2015-10-22 | Abbvie Inc. | Heterocyclic kinase inhibitors |
| WO2015166370A1 (en) | 2014-04-28 | 2015-11-05 | Pfizer Inc. | Heteroaromatic compounds and their use as dopamine d1 ligands |
| US9193733B2 (en) | 2012-05-18 | 2015-11-24 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9216984B2 (en) | 2009-05-22 | 2015-12-22 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9233934B2 (en) | 2007-03-12 | 2016-01-12 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| US9249145B2 (en) | 2009-09-01 | 2016-02-02 | Incyte Holdings Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US9290505B2 (en) | 2013-12-23 | 2016-03-22 | Gilead Sciences, Inc. | Substituted imidazo[1,2-a]pyrazines as Syk inhibitors |
| US9334274B2 (en) | 2009-05-22 | 2016-05-10 | Incyte Holdings Corporation | N-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines and pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US9359358B2 (en) | 2011-08-18 | 2016-06-07 | Incyte Holdings Corporation | Cyclohexyl azetidine derivatives as JAK inhibitors |
| US9359308B2 (en) | 2011-11-23 | 2016-06-07 | Portola Pharmaceuticals, Inc. | Pyrazine kinase inhibitors |
| US9358229B2 (en) | 2011-08-10 | 2016-06-07 | Novartis Pharma Ag | JAK PI3K/mTOR combination therapy |
| US9382256B2 (en) | 2013-07-30 | 2016-07-05 | Gilead Connecticut, Inc. | Formulation of Syk inhibitors |
| US9464088B2 (en) | 2010-03-10 | 2016-10-11 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US9487521B2 (en) | 2011-09-07 | 2016-11-08 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| US9512161B2 (en) | 2009-10-09 | 2016-12-06 | Incyte Corporation | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US9562056B2 (en) | 2010-03-11 | 2017-02-07 | Gilead Connecticut, Inc. | Imidazopyridines Syk inhibitors |
| US9593082B2 (en) | 2005-06-08 | 2017-03-14 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US9655854B2 (en) | 2013-08-07 | 2017-05-23 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US9657023B2 (en) | 2013-07-30 | 2017-05-23 | Gilead Connecticut, Inc. | Polymorph of Syk inhibitors |
| US9676756B2 (en) | 2012-10-08 | 2017-06-13 | Portola Pharmaceuticals, Inc. | Substituted pyrimidinyl kinase inhibitors |
| US9687492B2 (en) | 2013-12-04 | 2017-06-27 | Gilead Sciences, Inc. | Methods for treating cancers |
| US9707236B2 (en) | 2014-07-14 | 2017-07-18 | Gilead Sciences, Inc. | Combination methods for treating cancers |
| US9815815B2 (en) | 2013-01-10 | 2017-11-14 | Pulmokine, Inc. | Non-selective kinase inhibitors |
| US9925184B2 (en) | 2013-10-11 | 2018-03-27 | Pulmokine, Inc. | Spray-dry formulations |
| US9993480B2 (en) | 2011-02-18 | 2018-06-12 | Novartis Pharma Ag | mTOR/JAK inhibitor combination therapy |
| US10166191B2 (en) | 2012-11-15 | 2019-01-01 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US10231966B2 (en) | 2016-10-27 | 2019-03-19 | Pulmokine, Inc. | Combination therapy for treating pulmonary hypertension |
| US10596161B2 (en) | 2017-12-08 | 2020-03-24 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US10758543B2 (en) | 2010-05-21 | 2020-09-01 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| USRE48285E1 (en) | 2014-06-12 | 2020-10-27 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| US10899736B2 (en) | 2018-01-30 | 2021-01-26 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| WO2021249417A1 (en) * | 2020-06-09 | 2021-12-16 | 赛诺哈勃药业(成都)有限公司 | Heterocyclic compound and derivative thereof |
| US11304949B2 (en) | 2018-03-30 | 2022-04-19 | Incyte Corporation | Treatment of hidradenitis suppurativa using JAK inhibitors |
| US11339168B2 (en) | 2019-02-22 | 2022-05-24 | Kronos Bio, Inc. | Crystalline forms of 6-(6-aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine as Syk inhibitors |
| US11384082B2 (en) | 2017-08-25 | 2022-07-12 | Kronos Bio, Inc. | Hydrates of polymorphs of 6-(1H-indazol-6-YL)-N-(4-morpholinophenyl)-2,3-dihydroimidazo[1,2-A]pyrazin-8-amine bisemsylate as Syk inhibitors |
| US11642348B2 (en) | 2012-10-15 | 2023-05-09 | Epizyme, Inc. | Substituted benzene compounds |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| WO2024026260A1 (en) * | 2022-07-25 | 2024-02-01 | Celgene Corporation | Substituted imidazopyrazine compounds as irak3 binders |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101103018A (en) * | 2004-11-16 | 2008-01-09 | 神经化学(国际)有限公司 | Compounds for the treatment of CNS and amyloid-related diseases |
| AU2005309019A1 (en) * | 2004-11-24 | 2006-06-01 | Novartis Ag | Combinations of JAK inhibitors and at least one of Bcr-Abl, Flt-3, FAK or RAF kinase inhibitors |
| US20070117804A1 (en) * | 2005-11-10 | 2007-05-24 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
| US8575164B2 (en) * | 2005-12-19 | 2013-11-05 | OSI Pharmaceuticals, LLC | Combination cancer therapy |
| MX2009013213A (en) * | 2007-06-08 | 2010-03-30 | Abbott Lab | 5-heteroaryl substituted indazoles as kinase inhibitors. |
| DE102007032349A1 (en) * | 2007-07-11 | 2009-01-15 | Bayer Healthcare Ag | Imidazo, pyrazolopyrazines and imidazotriazines and their use |
| US20110046144A1 (en) * | 2008-01-18 | 2011-02-24 | Mulvihill Mark J | Imidazopyrazinol derivatives for the treatment of cancers |
| US20090275529A1 (en) * | 2008-05-05 | 2009-11-05 | Reiss Allison B | Method for improving cardiovascular risk profile of cox inhibitors |
| ES2396613T3 (en) * | 2008-05-19 | 2013-02-22 | OSI Pharmaceuticals, LLC | Imidazopyrazines and substituted imidazotriazines |
| US8513415B2 (en) | 2009-04-20 | 2013-08-20 | OSI Pharmaceuticals, LLC | Preparation of C-pyrazine-methylamines |
| EP2427192A1 (en) * | 2009-05-07 | 2012-03-14 | OSI Pharmaceuticals, LLC | Use of osi-906 for treating adrenocortical carcinoma |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| WO2014013014A1 (en) | 2012-07-18 | 2014-01-23 | Fundació Privada Centre De Regulació Genòmica (Crg) | Jak inhibitors for activation of epidermal stem cell populations |
| WO2014074421A1 (en) * | 2012-11-07 | 2014-05-15 | Merck Sharp & Dohme Corp. | Prodrug bipyridylaminopyridines as syk inhibitors |
| WO2018041989A1 (en) | 2016-09-02 | 2018-03-08 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for diagnosing and treating refractory celiac disease type 2 |
| EP3947737A2 (en) | 2019-04-02 | 2022-02-09 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods of predicting and preventing cancer in patients having premalignant lesions |
| EP3955920A1 (en) | 2019-04-16 | 2022-02-23 | Institut National de la Santé et de la Recherche Médicale (INSERM) | Use of jak inhibitors for the treatment of painful conditions involving nav1.7 channels |
| WO2023222565A1 (en) | 2022-05-16 | 2023-11-23 | Institut National de la Santé et de la Recherche Médicale | Methods for assessing the exhaustion of hematopoietic stems cells induced by chronic inflammation |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3821225A (en) * | 1971-05-14 | 1974-06-28 | Science Union & Cie | Pyridyl piperazines |
| EP0340836A2 (en) * | 1988-04-29 | 1989-11-08 | Merck & Co. Inc. | Alkylpiperazinylpyridines as hypoglycemic agents |
| JPH09132529A (en) * | 1995-11-09 | 1997-05-20 | Ono Pharmaceut Co Ltd | Nitric oxide synthase inhibitor |
| AU7351700A (en) * | 1999-09-10 | 2001-04-10 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5494908A (en) * | 1992-11-23 | 1996-02-27 | Hoechst-Roussel Pharmaceutical Incorporated | Substituted 3-(aminoalkylamino)-1,2-benzisoxazoles and related compounds |
| US5654307A (en) * | 1994-01-25 | 1997-08-05 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6231833B1 (en) * | 1999-08-05 | 2001-05-15 | Pfizer Inc | 2,7-substituted octahydro-1H-pyrido[1,2-A]pyrazine derivatives as ligands for serotonin receptors |
| US5958919A (en) * | 1996-09-20 | 1999-09-28 | Washington University | Treatment of presymptomatic alzheimer's disease to prevent neuronal degeneration |
| CA2322311C (en) * | 1998-03-04 | 2009-10-13 | Bristol-Myers Squibb Company | Heterocyclo-substituted imidazopyrazine protein tyrosine kinase inhibitors |
| SK286640B6 (en) * | 1998-06-19 | 2009-03-05 | Pfizer Products Inc. | Pyrrolo[2,3-d]pyrimidine compounds, the use thereof for producing a medicament, pharmaceutical composition containing the same, the use of its combination with additional agents and kits containing the same for producing a medicament |
| EP0982030A3 (en) * | 1998-08-17 | 2000-05-10 | Pfizer Products Inc. | 2,7-substituted octahydro-pyrrolo 1,2-a]pyrazine derivatives as 5ht 1a ligands |
| DE69914357T2 (en) * | 1998-11-04 | 2004-11-11 | Smithkline Beecham Corp. | PYRIDIN-4-YL OR PYRIMIDIN-4-YL SUBSTITUTED PYRAZINE |
| CN1660840A (en) * | 1999-03-30 | 2005-08-31 | 诺瓦提斯公司 | Phthalazine derivatives for treating inflammatory diseases |
| JP2003516974A (en) * | 1999-12-17 | 2003-05-20 | カイロン コーポレイション | Pyrazine-based inhibitors of glycogen synthase kinase 3 |
| US6335342B1 (en) * | 2000-06-19 | 2002-01-01 | Pharmacia & Upjohn S.P.A. | Azaindole derivatives, process for their preparation, and their use as antitumor agents |
| EP1317266A2 (en) * | 2000-08-08 | 2003-06-11 | Ortho-McNeil Pharmaceutical, Inc. | Neuroprotective 2-pyridinamine compositions and related methods |
| DE60118025T2 (en) * | 2000-10-24 | 2006-11-23 | Merck & Co., Inc. | Dibenzoxazepine ALPHA-V integrin receptor ANTAGONIST |
| CA2450769A1 (en) * | 2001-06-15 | 2002-12-27 | Vertex Pharmaceuticals Incorporated | 5-(2-aminopyrimidin-4-yl) benzisoxazoles as protein kinase inhibitors |
-
2002
- 2002-01-30 US US10/470,955 patent/US20040102455A1/en not_active Abandoned
- 2002-01-30 JP JP2002560683A patent/JP2004528295A/en active Pending
- 2002-01-30 CA CA002436487A patent/CA2436487A1/en not_active Abandoned
- 2002-01-30 WO PCT/AU2002/000089 patent/WO2002060492A1/en not_active Ceased
- 2002-01-30 EP EP02715984A patent/EP1363702A4/en not_active Withdrawn
-
2005
- 2005-09-09 US US11/223,633 patent/US20060069084A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3821225A (en) * | 1971-05-14 | 1974-06-28 | Science Union & Cie | Pyridyl piperazines |
| EP0340836A2 (en) * | 1988-04-29 | 1989-11-08 | Merck & Co. Inc. | Alkylpiperazinylpyridines as hypoglycemic agents |
| JPH09132529A (en) * | 1995-11-09 | 1997-05-20 | Ono Pharmaceut Co Ltd | Nitric oxide synthase inhibitor |
| AU7351700A (en) * | 1999-09-10 | 2001-04-10 | Merck & Co., Inc. | Tyrosine kinase inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| BONNET P.A. ET AL: "Synthesis and antibronchospastic activity of 8-alkoxy- and 8-(alkylamino)imidazo(1,2-a)pyrazines", JOURNAL OF MEDICINAL CHEMISTRY, vol. 35, no. 18, 1992, pages 3353 - 3358, XP002971280 * |
| DATABASE WPI Week 199730, Derwent World Patents Index; Class B03, AN 1997-328451, XP002971279 * |
Cited By (340)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003027295A3 (en) * | 2001-09-24 | 2003-10-16 | Univ Aarhus | Methods for diagnosis and treatment of diseases associated with altered expression of jak1 |
| US7557210B2 (en) | 2002-02-01 | 2009-07-07 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| US7655797B2 (en) | 2002-02-01 | 2010-02-02 | Rigel Pharmaceuticals, Inc. | Intermediates for making 2,4-pyrimidinediamine compounds |
| US7820819B2 (en) | 2002-02-01 | 2010-10-26 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| US7803939B2 (en) | 2002-02-01 | 2010-09-28 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| US7435814B2 (en) | 2002-02-01 | 2008-10-14 | Rigel Pharmaceuticals, Inc. | 2,4-Pyrimidinediamine compounds and their uses |
| US8334296B2 (en) | 2002-02-01 | 2012-12-18 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| AU2003221731B2 (en) * | 2002-04-19 | 2010-04-15 | Cgi Pharmaceuticals, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines method of making and method of use thereof |
| WO2003089434A3 (en) * | 2002-04-19 | 2004-01-15 | Cellular Genomics Inc | IMIDAZO[1,2-a]PYRAZIN-8-YLAMINES METHOD OF MAKING AND METHOD OF USE THEREOF |
| US6919340B2 (en) | 2002-04-19 | 2005-07-19 | Cellular Genomics, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines, method of making, and method of use thereof |
| US7122550B2 (en) | 2002-05-23 | 2006-10-17 | Cytopia Pty Ltd | Protein kinase inhibitors |
| US7259179B2 (en) | 2002-05-23 | 2007-08-21 | Cytopia Research Pty Ltd | Kinase inhibitors |
| GB2392154B (en) * | 2002-05-23 | 2005-01-19 | Cytopia Pty Ltd | Protein Kinase Inhibitors |
| US7511047B2 (en) | 2002-05-23 | 2009-03-31 | Cytopia Research Pty Ltd | Kinase inhibitors |
| GB2392154A (en) * | 2002-05-23 | 2004-02-25 | Cytopia Pty Ltd | Substituted pyrazines useful as inhibitors of protein kinases |
| WO2003099796A1 (en) * | 2002-05-23 | 2003-12-04 | Cytopia Pty Ltd | Protein kinase inhibitors |
| WO2003099811A1 (en) * | 2002-05-23 | 2003-12-04 | Cytopia Pty Ltd | Kinase inhibitors |
| US7598272B2 (en) | 2002-05-23 | 2009-10-06 | Cytopia Research Pty Ltd | Kinase inhibitors |
| US7015227B2 (en) | 2002-06-21 | 2006-03-21 | Cgi Pharmaceuticals, Inc. | Certain amino-substituted monocycles as kinase modulators |
| WO2004004730A3 (en) * | 2002-07-06 | 2004-04-29 | Astex Technology Ltd | 2-aminopyrazine derivatives as inhibitors of cyclin dependent kinases for the treatment of proliferative disorders |
| US8158621B2 (en) | 2002-07-29 | 2012-04-17 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| US7812029B1 (en) | 2002-07-29 | 2010-10-12 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| US7067661B2 (en) | 2002-09-04 | 2006-06-27 | Schering Corporation | Pyrazolopyrimidines as cyclin dependent kinase inhibitors |
| WO2004026229A3 (en) * | 2002-09-04 | 2004-06-17 | Schering Corp | Pyrazolo[1,5-a]pyrimidines compounds as cyclin dependent kinase inhibitors |
| US7205308B2 (en) | 2002-09-04 | 2007-04-17 | Schering Corporation | Trisubstituted 7-aminopyrazolopyrimidines as cyclin dependent kinase inhibitors |
| US7514442B2 (en) | 2002-09-04 | 2009-04-07 | Schering Corporation | Trisubstituted 4-aminopyrazolopyrimidines as cyclin dependent kinase inhibitors |
| WO2004022562A1 (en) * | 2002-09-09 | 2004-03-18 | Cellular Genomics, Inc. | 6-ARYL-IMIDAZO[1,2-a]PYRAZIN-8-YLAMINES, METHOD OF MAKING, AND METHOD OF USE THEREOF |
| US7312341B2 (en) | 2002-09-09 | 2007-12-25 | Cgi Pharmaceuticals, Inc. | 6-aryl-imidazo[1,2-a] pyrazin-8-ylamines, method of making, and method of use thereof |
| AU2003275031B2 (en) * | 2002-09-23 | 2006-08-17 | Schering Corporation | Novel imidazopyrazines as cyclin dependent kinase inhibitors |
| JP2006503838A (en) * | 2002-09-23 | 2006-02-02 | シェーリング コーポレイション | A novel imidazopyrazine as a cyclin-dependent kinase inhibitor |
| WO2004026310A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Novel imidazopyrazines as cyclin dependent kinase inhibitors |
| US7186740B2 (en) | 2002-09-23 | 2007-03-06 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| US6919341B2 (en) | 2002-09-23 | 2005-07-19 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| US7432265B2 (en) | 2002-09-23 | 2008-10-07 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| WO2004026877A1 (en) * | 2002-09-23 | 2004-04-01 | Schering Corporation | Imidazopyrazines as cyclin dependent kinase inhibitors |
| JP2006505571A (en) * | 2002-10-15 | 2006-02-16 | リゲル ファーマシューテイカルズ、インコーポレイテッド | Substituted indoles and their use as HCV inhibitors |
| WO2004052868A1 (en) * | 2002-12-11 | 2004-06-24 | Cytopia Pty Ltd | Pyrazine-based tubulin inhibitors |
| GB2412372B (en) * | 2002-12-11 | 2007-02-21 | Cytopia Pty Ltd | Pyrazine-based tubulin inhibitors |
| US8084456B2 (en) | 2002-12-11 | 2011-12-27 | Ym Biosciences Australia Pty Ltd | Pyrazine-based tubulin inhibitors |
| GB2412372A (en) * | 2002-12-11 | 2005-09-28 | Cytopia Pty Ltd | Pyrazine-based tubulin inhibitors |
| AU2003285211B8 (en) * | 2002-12-11 | 2015-04-16 | Ym Biosciences Australia Pty Ltd | Pyrazine-based tubulin inhibitors |
| AU2003285211B2 (en) * | 2002-12-11 | 2009-03-26 | Ym Biosciences Australia Pty Ltd | Pyrazine-based tubulin inhibitors |
| WO2004072080A1 (en) * | 2003-02-10 | 2004-08-26 | Cellular Genomics, Inc. | Certain 8-heteroaryl-6-phenyl-imidazo[1,2-a]pyrazines as modulators of hsp90 complex activity |
| US7160885B2 (en) | 2003-02-10 | 2007-01-09 | Cgi Pharmaceuticals, Inc. | Certain 6, 8-(heteroaryl or aryl) disubstituted imidazo[1,2-a]pyrazines as modulators of Hsp90 complex activity |
| US7189723B2 (en) | 2003-02-10 | 2007-03-13 | Cgi Pharmaceuticals, Inc. | Certain 8-heteroaryl-6-phenyl-imidazo[1,2-a]pyrazines as modulators of kinase activity |
| WO2004072081A1 (en) * | 2003-02-10 | 2004-08-26 | Cellular Genomics, Inc. | Certain 8-heteroaryl-6-phenyl-imidazo[1,2-a]pyrazines as modulators of kinase activity |
| WO2004074289A1 (en) * | 2003-02-18 | 2004-09-02 | Altana Pharma Ag | 6-substituted imidazopyrazines |
| US7186832B2 (en) | 2003-02-20 | 2007-03-06 | Sugen Inc. | Use of 8-amino-aryl-substituted imidazopyrazines as kinase inhibitors |
| US7157460B2 (en) | 2003-02-20 | 2007-01-02 | Sugen Inc. | Use of 8-amino-aryl-substituted imidazopyrazines as kinase inhibitors |
| US8013156B2 (en) | 2003-03-19 | 2011-09-06 | Exelixis, Inc. | Tie-2 modulators and methods of use |
| JP2006524682A (en) * | 2003-03-19 | 2006-11-02 | エクセリクシス, インク. | TIE-2 modulator and usage |
| JP2006520794A (en) * | 2003-03-21 | 2006-09-14 | スミスクライン ビーチャム コーポレーション | Compound |
| US7459454B2 (en) | 2003-03-21 | 2008-12-02 | Smithkline Beecham Corporation | Aminopyrazine derivatives and compositions |
| US7598251B2 (en) | 2003-03-21 | 2009-10-06 | Smithkline Beecham Corporation | Aminopyrazine derivatives and compositions |
| EP1606266A4 (en) * | 2003-03-21 | 2008-06-25 | Smithkline Beecham Corp | Chemical compounds |
| JP4895806B2 (en) * | 2003-04-09 | 2012-03-14 | エクセリクシス, インク. | TIE-2 modulator and usage |
| JP2006523238A (en) * | 2003-04-09 | 2006-10-12 | エクセリクシス, インク. | TIE-2 modulator and usage |
| US7405295B2 (en) | 2003-06-04 | 2008-07-29 | Cgi Pharmaceuticals, Inc. | Certain imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of Bruton's tyrosine kinase by such compounds |
| WO2005014599A1 (en) * | 2003-06-04 | 2005-02-17 | Cellular Genomics, Inc. | Imidazo[1,2-a]pyrazin-8-ylamines and method of inhibition of bruton’s tyrosine kinase by such compounds |
| WO2005005429A1 (en) * | 2003-06-30 | 2005-01-20 | Cellular Genomics, Inc. | Certain heterocyclic substituted imidazo[1,2-a]pyrazin-8-ylamines and methods of inhibition of bruton’s tyrosine kinase by such compounds |
| US7393848B2 (en) | 2003-06-30 | 2008-07-01 | Cgi Pharmaceuticals, Inc. | Certain heterocyclic substituted imidazo[1,2-A]pyrazin-8-ylamines and methods of inhibition of Bruton's tyrosine kinase by such compounds |
| US8178671B2 (en) | 2003-07-30 | 2012-05-15 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2, 4-pyrimidinediamine compounds |
| US7452879B2 (en) | 2003-07-30 | 2008-11-18 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| US7582648B2 (en) | 2003-07-30 | 2009-09-01 | Rigel Pharmaceuticals, Inc. | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2005019220A3 (en) * | 2003-08-11 | 2005-03-24 | Cellular Genomics Inc | Substituted imidazo[1,2-a]pyrazines as modulators of kinase activity |
| US7259164B2 (en) | 2003-08-11 | 2007-08-21 | Cgi Pharmaceuticals, Inc. | Certain substituted imidazo[1,2-a]pyrazines, as modulators of kinase activity |
| US7390907B2 (en) | 2003-09-30 | 2008-06-24 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
| WO2005033105A3 (en) * | 2003-09-30 | 2005-06-23 | Amgen Inc | Vanilloid receptor ligands and their use in treatments |
| WO2005037836A3 (en) * | 2003-10-15 | 2005-06-09 | Osi Pharm Inc | Imidazo ‘1, 5 - a ! pyrazine tyrosine kinase inhibitors |
| RU2405784C2 (en) * | 2003-10-15 | 2010-12-10 | Оси Фармасьютикалз, Инк. | Imidazopyrazines as tyrosine kinase inhibitors |
| WO2005047290A3 (en) * | 2003-11-11 | 2005-08-11 | Cellular Genomics Inc | Imidazo[1,2-a] pyrazin-8-ylamines as kinase inhibitors |
| WO2005054199A1 (en) * | 2003-12-03 | 2005-06-16 | Cytopia Research Pty Ltd | Tubulin inhibitors |
| EP1689739A4 (en) * | 2003-12-03 | 2010-12-22 | Ym Biosciences Australia Pty L | INHIBITORS OF AZOLE-BASED KINASES |
| US9139560B2 (en) | 2003-12-03 | 2015-09-22 | Ym Biosciences Australia Pty Ltd. | Substituted pyrazines as tubulin inhibitors |
| JP4772690B2 (en) * | 2003-12-03 | 2011-09-14 | ワイエム・バイオサイエンシズ・オーストラリア・ピーティーワイ・リミテッド | Tubulin inhibitor |
| AU2004294354B2 (en) * | 2003-12-03 | 2009-02-19 | Ym Biosciences Australia Pty Ltd | Tubulin inhibitors |
| JP2007513093A (en) * | 2003-12-03 | 2007-05-24 | サイトピア・リサーチ・ピーティーワイ・リミテッド | Tubulin inhibitor |
| KR101211514B1 (en) | 2003-12-03 | 2012-12-12 | 와이엠 바이오사이언시즈 오스트레일리아 피티와이 엘티디 | Azole kinase inhibitors |
| US7981900B2 (en) | 2003-12-03 | 2011-07-19 | Ym Biosciences Australia Pty Ltd | 2-phenyl pyrimidines which are tubulin inhibitors |
| JP2011093930A (en) * | 2003-12-03 | 2011-05-12 | Ym Biosciences Australia Pty Ltd | Tubulin inhibitor |
| EP2277865A1 (en) * | 2003-12-03 | 2011-01-26 | YM BioSciences Australia Pty Ltd | Phenyl-substituted 6-ring nitrogen-heterocycles as microtubule polymerisation inhibitors |
| US8394806B2 (en) | 2003-12-03 | 2013-03-12 | Ym Biosciences Australia Pty Ltd | 2-phenyl pyrazines as tubulin inhibitors |
| US9732046B2 (en) | 2003-12-03 | 2017-08-15 | Ym Biosciences Australia Pty Ltd. | Substituted 1,2,4-triazines as tubulin inhibitors |
| WO2005058876A1 (en) * | 2003-12-16 | 2005-06-30 | Gpc Biotech Ag | Pyrazine derivatives as effective compounds against infectious diseases |
| WO2005085252A1 (en) * | 2004-03-04 | 2005-09-15 | Biofocus Discovery Limited | Imidazo ‘1,2-a’ pyrazine compounds which interact with protein kinases |
| US7820662B2 (en) | 2004-04-02 | 2010-10-26 | Osi Pharmaceuticals, Inc. | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US8735405B2 (en) | 2004-04-02 | 2014-05-27 | OSI Pharmaceuticals, LLC | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US8101613B2 (en) | 2004-04-02 | 2012-01-24 | OSI Pharmaceuticals, LLC | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US8367826B2 (en) | 2004-04-02 | 2013-02-05 | OSI Pharmaceuticals, LLC | 6,6-bicyclic ring substituted heterobicyclic protein kinase inhibitors |
| US7642354B2 (en) | 2004-04-13 | 2010-01-05 | Icagen, Inc. | Polycyclic pyrazines as potassium ion channel modulators |
| WO2005121126A1 (en) | 2004-04-13 | 2005-12-22 | Icagen, Inc. | Polycyclic pyrazines as potassium ion channel modulators |
| US7737152B2 (en) | 2004-12-22 | 2010-06-15 | The Wellcome Trust Limited | 6-carboaryl-oxy-pyrazin-2-yl-carboaryl-amines and compositions comprising said compounds |
| US8053433B2 (en) | 2004-12-22 | 2011-11-08 | Ineyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-5-yl-amines as janus kinase inhibitors |
| US8741895B2 (en) | 2004-12-22 | 2014-06-03 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-5-yl-amines as Janus kinase inhibitors |
| US8445488B2 (en) | 2004-12-22 | 2013-05-21 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-5-yl-amines as Janus kinase inhibitors |
| US9879010B2 (en) | 2004-12-22 | 2018-01-30 | Incyte Holdings Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b] pyrimidin-5-yl-amines as Janus kinase inhibitors |
| US7335667B2 (en) | 2004-12-22 | 2008-02-26 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-4-yl-amines as Janus kinase inhibitors |
| US9580419B2 (en) | 2004-12-22 | 2017-02-28 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-5-yl-amines as Janus kinase inhibitors |
| EP2671882B1 (en) | 2004-12-22 | 2018-02-21 | Incyte Holdings Corporation | pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2m3-b]pyrimidin-4-yl-amines as janus kinase inhibitors |
| US9090611B2 (en) | 2004-12-22 | 2015-07-28 | Incyte Corporation | Pyrrolo[2,3-b]pyridin-4-yl-amines and pyrrolo[2,3-b]pyrimidin-5-yl-amines as janus kinase inhibitors |
| US7777040B2 (en) | 2005-05-03 | 2010-08-17 | Cgi Pharmaceuticals, Inc. | Certain substituted ureas, as modulators of kinase activity |
| US9593082B2 (en) | 2005-06-08 | 2017-03-14 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US10421752B2 (en) | 2005-06-08 | 2019-09-24 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US7491732B2 (en) | 2005-06-08 | 2009-02-17 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US9248190B2 (en) | 2005-06-08 | 2016-02-02 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US9732073B2 (en) | 2005-06-08 | 2017-08-15 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US11198689B2 (en) | 2005-06-08 | 2021-12-14 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US11827628B2 (en) | 2005-06-08 | 2023-11-28 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US8415365B2 (en) | 2005-06-08 | 2013-04-09 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US8399472B2 (en) | 2005-06-08 | 2013-03-19 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US7709468B2 (en) | 2005-09-02 | 2010-05-04 | Abbott Laboratories | Imidazo based heterocycles |
| US8563541B2 (en) | 2005-09-22 | 2013-10-22 | Incyte Corporation | Azepine inhibitors of Janus kinases |
| US8835423B2 (en) | 2005-09-22 | 2014-09-16 | Incyte Corporation | Azepine inhibitors of janus kinases |
| US7598257B2 (en) | 2005-12-13 | 2009-10-06 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US8946245B2 (en) | 2005-12-13 | 2015-02-03 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US9662335B2 (en) | 2005-12-13 | 2017-05-30 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| US8933086B2 (en) | 2005-12-13 | 2015-01-13 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US10639310B2 (en) | 2005-12-13 | 2020-05-05 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US9079912B2 (en) | 2005-12-13 | 2015-07-14 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase inhibitors |
| US10398699B2 (en) | 2005-12-13 | 2019-09-03 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US9206187B2 (en) | 2005-12-13 | 2015-12-08 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase |
| US11331320B2 (en) | 2005-12-13 | 2022-05-17 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US11744832B2 (en) | 2005-12-13 | 2023-09-05 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US9974790B2 (en) | 2005-12-13 | 2018-05-22 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| US9814722B2 (en) | 2005-12-13 | 2017-11-14 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| WO2007098507A2 (en) | 2006-02-24 | 2007-08-30 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the jak pathway |
| US8962643B2 (en) | 2006-02-24 | 2015-02-24 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US11667611B2 (en) | 2006-02-24 | 2023-06-06 | Rigel Pharmaceuticals, Inc. | Compositions and methods for inhibition of the JAK pathway |
| US7700601B2 (en) | 2006-03-31 | 2010-04-20 | Schering Corporation | Substituted indazoles of formula 1.0 that are kinase inhibitors |
| WO2007126964A3 (en) * | 2006-03-31 | 2007-12-21 | Schering Corp | Kinase inhibitors |
| US8173656B2 (en) * | 2006-05-15 | 2012-05-08 | Janssen Pharmaceutica Nv | Imidazolopyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| US7893058B2 (en) * | 2006-05-15 | 2011-02-22 | Janssen Pharmaceutica Nv | Imidazolopyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| WO2007138072A3 (en) * | 2006-05-31 | 2008-02-14 | Galapagos Nv | Triazolopyrazine compounds useful for the treatment of degenerative & inflammatory diseases |
| WO2008022164A3 (en) * | 2006-08-16 | 2008-04-17 | Boehringer Ingelheim Int | Pyrazine compounds, their use and methods of preparation |
| US8318723B2 (en) | 2006-08-16 | 2012-11-27 | Boehringer Ingelheim International Gmbh | Pyrazine compounds, their use and methods of preparation |
| US8841318B2 (en) | 2006-12-22 | 2014-09-23 | Incyte Corporation | Substituted heterocycles as janus kinase inhibitors |
| US8513270B2 (en) | 2006-12-22 | 2013-08-20 | Incyte Corporation | Substituted heterocycles as Janus kinase inhibitors |
| US12570614B2 (en) | 2007-03-12 | 2026-03-10 | Glaxosmith Kline Llc | Phenyl amino pyrimidine compounds and uses thereof |
| US9238628B2 (en) | 2007-03-12 | 2016-01-19 | YM Biosicences Australia PTY LTD | Phenyl amino pyrimidine compounds and uses thereof |
| US9233934B2 (en) | 2007-03-12 | 2016-01-12 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| US9006237B2 (en) | 2007-05-10 | 2015-04-14 | Janssen Pharmaceutica Nv | Fused pyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| US8148369B2 (en) | 2007-05-10 | 2012-04-03 | Janssen Pharmaceutica Nv | Fused pyrazine compounds useful for the treatment of degenerative and inflammatory diseases |
| WO2008138842A1 (en) * | 2007-05-10 | 2008-11-20 | Galapagos N.V. | Imidazopyrazines and triazolopyrazine for the treatment of joint degenerative and inflammatory diseases |
| US9376439B2 (en) | 2007-06-13 | 2016-06-28 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US10016429B2 (en) | 2007-06-13 | 2018-07-10 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US10463667B2 (en) | 2007-06-13 | 2019-11-05 | Incyte Incorporation | Metabolites of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US10610530B2 (en) | 2007-06-13 | 2020-04-07 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8829013B1 (en) | 2007-06-13 | 2014-09-09 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8889697B2 (en) | 2007-06-13 | 2014-11-18 | Incyte Corporation | Metabolites of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8822481B1 (en) | 2007-06-13 | 2014-09-02 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8722693B2 (en) | 2007-06-13 | 2014-05-13 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US11213528B2 (en) | 2007-06-13 | 2022-01-04 | Incyte Holdings Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US7834022B2 (en) | 2007-06-13 | 2010-11-16 | Incyte Corporation | Metabolites of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| EP2567960A3 (en) * | 2007-08-21 | 2013-06-05 | Biofocus DPI Limited | Imidazo[1,2-a]pyrazine compounds for treatment of viral infections such as hepatitis |
| WO2009024585A3 (en) * | 2007-08-21 | 2009-08-06 | Biofocus Dpi Ltd | Imidazo [1,2-a] pyrazine compounds for treatment of viral infections such as hepatitis |
| US8362018B2 (en) | 2007-08-21 | 2013-01-29 | Biofocus Dpi, Ltd. | Substituted imidazo[1,2-a]pyrazine compounds useful for the treatment of viral infections |
| US8436171B2 (en) | 2008-02-01 | 2013-05-07 | Akinion Pharmaceuticals Ab | Amino substituted pyrazines as inhibitors or protein kinases |
| WO2009095399A3 (en) * | 2008-02-01 | 2009-10-01 | Akinion Pharmaceuticals Ab | Pyrazine derivatives and their use as protein kinase inhbitors |
| EP2671876A3 (en) * | 2008-02-01 | 2014-02-05 | Akinion Pharmaceuticals AB | Pyrazine derivatives and their use as protein kinase inhbitors |
| RU2493152C2 (en) * | 2008-02-01 | 2013-09-20 | Акинион Фармасьютикалз Аб | Novel compounds, use and production thereof |
| US8697699B2 (en) | 2008-02-13 | 2014-04-15 | Gilead Connecticut, Inc. | Imidazopyrazine SYK inhibitors |
| WO2009102468A1 (en) | 2008-02-13 | 2009-08-20 | Cgi Pharmaceuticals, Inc. | 6-aryl-imidaz0[l, 2-a] pyrazine derivatives, method of making, and method of use thereof |
| JP2011511835A (en) * | 2008-02-13 | 2011-04-14 | シージーアイ ファーマシューティカルズ,インコーポレーテッド | 6-Aryl-imidazo [1,2-a] pyrazine derivatives, methods for their preparation, and methods for their use |
| US9624229B2 (en) | 2008-02-15 | 2017-04-18 | Rigel Pharmaceuticals, Inc. | Pyrimidine-2-amine compounds and their use as inhibitors of JAK kinases |
| US8735418B2 (en) | 2008-02-15 | 2014-05-27 | Rigel Pharmaceuticals, Inc. | Pyrimidine-2-amine compounds and their use as inhibitors of JAK kinases |
| US8309566B2 (en) | 2008-02-15 | 2012-11-13 | Rigel Pharmaceuticals, Inc. | Pyrimidine-2-amine compounds and their use as inhibitors of JAK kinases |
| US8420629B2 (en) | 2008-03-11 | 2013-04-16 | Incyte Corporation | Azetidine and cyclobutane derivatives as JAK inhibitors |
| US8158616B2 (en) | 2008-03-11 | 2012-04-17 | Incyte Corporation | Azetidine and cyclobutane derivatives as JAK inhibitors |
| US8501944B2 (en) | 2008-04-16 | 2013-08-06 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US8952027B2 (en) | 2008-04-16 | 2015-02-10 | Portola Pharmaceuticals, Inc. | Inhibitors of syk and JAK protein kinases |
| US10533001B2 (en) | 2008-04-16 | 2020-01-14 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US11414410B2 (en) | 2008-04-16 | 2022-08-16 | Alexion Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US8937070B2 (en) | 2008-04-16 | 2015-01-20 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US8138339B2 (en) | 2008-04-16 | 2012-03-20 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US9868729B2 (en) | 2008-04-16 | 2018-01-16 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US9579320B2 (en) | 2008-04-16 | 2017-02-28 | Portola Pharmaceuticals, Inc. | Inhibitors of syk and JAK protein kinases |
| US8258144B2 (en) | 2008-04-22 | 2012-09-04 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| US9139581B2 (en) | 2008-04-22 | 2015-09-22 | Portola Pharmaceuticals, Inc. | Inhibitors of protein kinases |
| TWI478922B (en) * | 2008-12-08 | 2015-04-01 | Gilead Connenticut Inc | Imidazopyrazine syk inhibitors |
| EP2373318A4 (en) * | 2008-12-08 | 2012-05-30 | Gilead Connecticut Inc | IMIDAZOPYRAZINE SYK INHIBITORS |
| CN104059073B (en) * | 2008-12-08 | 2017-04-12 | 吉利德康涅狄格公司 | Imidazopyrizine Syk Inhibitors |
| CN104744476A (en) * | 2008-12-08 | 2015-07-01 | 吉利德康涅狄格股份有限公司 | IMIDAZOPYRAZINE Syk INHIBITORS |
| AU2016202807B2 (en) * | 2008-12-08 | 2017-10-12 | Gilead Connecticut, Inc. | Imidazopyrazine Syk inhibitors |
| EP2373169A4 (en) * | 2008-12-08 | 2012-08-29 | Gilead Connecticut Inc | IMIDAZOPYRAZINE SYK INHIBITORS |
| US9796718B2 (en) | 2008-12-08 | 2017-10-24 | Gilead Connecticut, Inc. | 6-(benzo[d]thiazol-5-yl)-n-(3,4-dimethoxyphenyl)imidazo[1,2-a]pyrazin-8-amine |
| US9120811B2 (en) | 2008-12-08 | 2015-09-01 | Gilead Connecticut, Inc. | 6-(1H-indazol-6-YL)-N-(4-morpholinophenyl)imidazo[1,4-A]pyrazin-8-amine for treating leukemia or lymphoma |
| EP3123864A1 (en) * | 2008-12-08 | 2017-02-01 | Gilead Connecticut, Inc. | Imidazopyrazine syk inhibitors |
| US10093684B2 (en) | 2008-12-08 | 2018-10-09 | Gilead Connecticut, Inc. | Substituted imidazo[1,2-a]pyrazines as Syk inhibitors |
| US8440667B2 (en) | 2008-12-08 | 2013-05-14 | Gilead Connecticut, Inc. | Imidazopyrazine Syk inhibitors |
| US8765761B2 (en) | 2008-12-08 | 2014-07-01 | Gilead Connecticut, Inc. | 6-(1H-indazol-6-yl)-N-{4-[2-methyl-1-(morpholin-4-yl)propan-2-yl]phenyl}imidazo[1,2-A]pyrazin-8-amine, or a pharmaceutically acceptable salt thereof, as a syk inhibitor |
| EP3552607A3 (en) * | 2008-12-08 | 2020-01-15 | Gilead Connecticut, Inc. | Imidazopyrazine syk inhibitors |
| KR101748925B1 (en) | 2008-12-08 | 2017-06-19 | 질레드 코네티컷 인코포레이티드 | Imidazopyrazine syk inhibitors |
| US8455493B2 (en) | 2008-12-08 | 2013-06-04 | Gilead Connecticut, Inc. | Imidazopyrazine Syk inhibitors |
| US9212191B2 (en) | 2008-12-08 | 2015-12-15 | Gilead Connecticut, Inc. | 6-(2,3-dihydro-1H-pyrido-[2,3-b][1,4]oxazin-7-yl)-N-(4-morpholinophenyl)imidazo[1,2-a] pyrazin-8-amine as a spleen tyrosine kinase inhibitor |
| US8796270B2 (en) | 2008-12-08 | 2014-08-05 | Gilead Connecticut, Inc. | N-[6-(1H-indazol-6-yl)imidazo[1,2-A]pyridin-8-yl]-6-(morpholin-4-yl)pyridazin-3-amine, or a pharmaceutically acceptable salt thereof, as a SYK inhibitor |
| CN102307581B (en) * | 2008-12-08 | 2016-08-17 | 吉利德康涅狄格股份有限公司 | imidazopiperazine SYK inhibitors |
| US8748607B2 (en) | 2008-12-08 | 2014-06-10 | Gilead Connecticut, Inc. | Imidazopyrizine syk inhibitors |
| US8962835B2 (en) | 2008-12-08 | 2015-02-24 | Gilead Connecticut, Inc. | Imidazopyrazine Syk inhibitors |
| US8450321B2 (en) | 2008-12-08 | 2013-05-28 | Gilead Connecticut, Inc. | 6-(1H-indazol-6-yl)-N-[4-(morpholin-4-yl)phenyl]imidazo-[1,2-A]pyrazin-8-amine, or a pharmaceutically acceptable salt thereof, as a SYK inhibitor |
| US9567348B2 (en) | 2008-12-08 | 2017-02-14 | Gilead Connecticut, Inc. | Substituted pyrazolo[1,5-a]pyrimidines as Syk inhibitors |
| WO2010069684A1 (en) * | 2008-12-17 | 2010-06-24 | Biomarin Iga, Ltd. | Compounds for treatment of duchenne muscular dystrophy |
| WO2010088368A2 (en) | 2009-01-29 | 2010-08-05 | Schering Corporation | Imidazopyrazines as protein kinase inhibitors |
| US8778935B2 (en) | 2009-04-16 | 2014-07-15 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Imidazopyrazines for use as kinase inhibitors |
| WO2010119264A1 (en) * | 2009-04-16 | 2010-10-21 | Centro Nacional De Investigaciones Oncólogicas (Cnio) | Imidazopyrazines for use as kinase inhibitors |
| EA022629B1 (en) * | 2009-04-16 | 2016-02-29 | Фундасьон Сентро Насиональ Де Инвестигасьонес Онколохикас Карлос Iii | Imidazopyrazines for use as kinase inhibitors |
| AU2010238361B2 (en) * | 2009-04-16 | 2015-08-06 | Fundacion Centro Nacional De Investigaciones Oncologicas Carlos Iii | Imidazopyrazines for use as kinase inhibitors |
| CN102428087B (en) * | 2009-04-16 | 2015-06-17 | 卡洛斯三世国家癌症研究中心基金会 | Imidazopyrazines for use as kinase inhibitors |
| KR101792837B1 (en) | 2009-04-16 | 2017-11-02 | 푼다시온 센트로 나시오날 드 인베스티가시오네스 온콜로기카스 카를로스Ⅲ | Imidazopyrazines for use as kinase inhibitors |
| CN102428087A (en) * | 2009-04-16 | 2012-04-25 | 西班牙国家癌症研究中心 | Imidazopyrazines for use as kinase inhibitors |
| US9334274B2 (en) | 2009-05-22 | 2016-05-10 | Incyte Holdings Corporation | N-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines and pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US9623029B2 (en) | 2009-05-22 | 2017-04-18 | Incyte Holdings Corporation | 3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane- or heptane-nitrile as JAK inhibitors |
| US9216984B2 (en) | 2009-05-22 | 2015-12-22 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9249145B2 (en) | 2009-09-01 | 2016-02-02 | Incyte Holdings Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US9512161B2 (en) | 2009-10-09 | 2016-12-06 | Incyte Corporation | Hydroxyl, keto, and glucuronide derivatives of 3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| WO2011056916A1 (en) * | 2009-11-05 | 2011-05-12 | Lexicon Pharmaceuticals, Inc. | Tryptophan hydroxylase inhibitors for the treatment of cancer |
| CN102791715B (en) * | 2009-12-31 | 2016-04-27 | 卡洛斯三世国家癌症研究中心基金会 | As the tricyclic compound of kinase inhibitor |
| WO2011080176A1 (en) * | 2009-12-31 | 2011-07-07 | Novartis Ag | Pyrazine derivatives and their use in the treatment of neurological disorders |
| AU2010338365B2 (en) * | 2009-12-31 | 2014-01-30 | Novartis Ag | Pyrazine derivatives and their use in the treatment of neurological disorders |
| CN102791715A (en) * | 2009-12-31 | 2012-11-21 | 西班牙国家癌症研究中心 | Tricyclic compounds for use as kinase inhibitors |
| US9073927B2 (en) | 2010-01-22 | 2015-07-07 | Fundacion Centro Nacional De Investigaciones Oncologicas Carlos Iii | Inhibitors of PI3 kinase |
| US9999619B2 (en) | 2010-03-10 | 2018-06-19 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US11285140B2 (en) | 2010-03-10 | 2022-03-29 | Incyte Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US9464088B2 (en) | 2010-03-10 | 2016-10-11 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US10695337B2 (en) | 2010-03-10 | 2020-06-30 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US10092583B2 (en) | 2010-03-11 | 2018-10-09 | Gilead Connecticut, Inc. | Imidazopyridines Syk inhibitors |
| US9562056B2 (en) | 2010-03-11 | 2017-02-07 | Gilead Connecticut, Inc. | Imidazopyridines Syk inhibitors |
| US10842803B2 (en) | 2010-03-11 | 2020-11-24 | Kronos Bio, Inc. | Imidazopyridines Syk inhibitors |
| WO2011141713A1 (en) * | 2010-05-13 | 2011-11-17 | Centro Nacional De Investigaciones Oncologicas (Cnio) | New bicyclic compounds as pi3-k and mtor inhibitors |
| US11571425B2 (en) | 2010-05-21 | 2023-02-07 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US11219624B2 (en) | 2010-05-21 | 2022-01-11 | Incyte Holdings Corporation | Topical formulation for a JAK inhibitor |
| US10869870B2 (en) | 2010-05-21 | 2020-12-22 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US10758543B2 (en) | 2010-05-21 | 2020-09-01 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US11590136B2 (en) | 2010-05-21 | 2023-02-28 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US12226419B2 (en) | 2010-05-21 | 2025-02-18 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US12544381B2 (en) | 2010-05-21 | 2026-02-10 | Incyte Corporation | Topical formulation for JAK inhibitor |
| US12564593B2 (en) | 2010-05-21 | 2026-03-03 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| WO2012041476A1 (en) * | 2010-09-30 | 2012-04-05 | Almirall, S.A. | Pyridine and isoquinoline derivatives as syk- and jak-kinase inhibitors |
| EP2441755A1 (en) * | 2010-09-30 | 2012-04-18 | Almirall, S.A. | Pyridine- and isoquinoline-derivatives as Syk and JAK kinase inhibitors |
| US9993554B2 (en) | 2010-10-21 | 2018-06-12 | Centro Nacional De Investigaciones Oncologicas (Cnio) | Use of P13K Inhibitors for the Treatment of Obesity, Steatosis and ageing |
| EP2444084A1 (en) * | 2010-10-21 | 2012-04-25 | Centro Nacional de Investigaciones Oncológicas (CNIO) | Use of PI3K inibitors for the treatment of obesity |
| WO2012052745A1 (en) | 2010-10-21 | 2012-04-26 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Combinations of pi3k inhibitors with a second anti -tumor agent |
| WO2012052730A1 (en) * | 2010-10-21 | 2012-04-26 | Centro Nacional De Investigaciones Oncológicas (Cnio) | Use of pi3k inibitors for the treatment of obesity, steatosis and ageing |
| EP2975027A1 (en) | 2010-11-01 | 2016-01-20 | Portola Pharmaceuticals, Inc. | Nicotinamides as jak kinase modulators |
| US9102625B2 (en) | 2010-11-01 | 2015-08-11 | Portola Pharmaceuticals, Inc. | Nicotinamides as JAK kinase modulators |
| WO2012061428A2 (en) | 2010-11-01 | 2012-05-10 | Portola Pharmaceuticals, Inc. | Nicotinamides as jak kinase modulators |
| US10640506B2 (en) | 2010-11-19 | 2020-05-05 | Incyte Holdings Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidines derivatives as JAK inhibitors |
| US8933085B2 (en) | 2010-11-19 | 2015-01-13 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US9993480B2 (en) | 2011-02-18 | 2018-06-12 | Novartis Pharma Ag | mTOR/JAK inhibitor combination therapy |
| US12168015B2 (en) | 2011-04-13 | 2024-12-17 | Epizyme, Inc. | Aryl-or heteroaryl-substituted benzene compounds |
| US12168016B2 (en) | 2011-04-13 | 2024-12-17 | Epizyme, Inc. | Aryl-OR heteroaryl-substituted benzene compounds |
| US12161645B2 (en) | 2011-04-13 | 2024-12-10 | Epizyme, Inc. | Aryl-or heteroaryl-substituted benzene compounds |
| US9855275B2 (en) | 2011-04-13 | 2018-01-02 | Epizyme, Inc. | Aryl-or heteroaryl-substituted benzene compounds |
| US10420775B2 (en) | 2011-04-13 | 2019-09-24 | Epizyme, Inc. | Aryl-or heteroaryl-substituted benzene compounds |
| US11052093B2 (en) | 2011-04-13 | 2021-07-06 | Epizyme, Inc. | Aryl-or heteroaryl-substituted benzene compounds |
| CN104080769A (en) * | 2011-04-13 | 2014-10-01 | Epizyme股份有限公司 | Aryl or heteroaryl substituted benzene compounds |
| CN104080769B (en) * | 2011-04-13 | 2017-06-20 | Epizyme股份有限公司 | Aryl or heteroaryl substituted benzene compounds |
| US9090562B2 (en) | 2011-04-13 | 2015-07-28 | Epizyme, Inc. | Aryl- or heteroaryl-substituted benzene compounds |
| US10155002B2 (en) | 2011-04-13 | 2018-12-18 | Epizyme, Inc. | Aryl- or heteroaryl-substituted benzene compounds |
| US9522152B2 (en) | 2011-04-13 | 2016-12-20 | Epizyme, Inc. | Aryl- or heteroaryl-substituted benzene compounds |
| US9549931B2 (en) | 2011-04-13 | 2017-01-24 | Epizyme, Inc. | Aryl- or heteroaryl-substituted benzene compounds |
| US11214573B2 (en) | 2011-06-20 | 2022-01-04 | Incyte Holdings Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US8691807B2 (en) | 2011-06-20 | 2014-04-08 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9611269B2 (en) | 2011-06-20 | 2017-04-04 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US10513522B2 (en) | 2011-06-20 | 2019-12-24 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9023840B2 (en) | 2011-06-20 | 2015-05-05 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| AU2012280725B2 (en) * | 2011-07-07 | 2017-02-02 | Merck Patent Gmbh | Substituted azaheterocycles for the treatment of cancer |
| WO2013004332A1 (en) * | 2011-07-07 | 2013-01-10 | Merck Patent Gmbh | Substituted azaheterocycles for the treatment of cancer |
| US9199962B2 (en) | 2011-07-07 | 2015-12-01 | Merck Patent Gmbh | Substituted azaheterocycles for the treatment of cancer |
| US9358229B2 (en) | 2011-08-10 | 2016-06-07 | Novartis Pharma Ag | JAK PI3K/mTOR combination therapy |
| US9359358B2 (en) | 2011-08-18 | 2016-06-07 | Incyte Holdings Corporation | Cyclohexyl azetidine derivatives as JAK inhibitors |
| US9718834B2 (en) | 2011-09-07 | 2017-08-01 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9487521B2 (en) | 2011-09-07 | 2016-11-08 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9359308B2 (en) | 2011-11-23 | 2016-06-07 | Portola Pharmaceuticals, Inc. | Pyrazine kinase inhibitors |
| US9193733B2 (en) | 2012-05-18 | 2015-11-24 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9676756B2 (en) | 2012-10-08 | 2017-06-13 | Portola Pharmaceuticals, Inc. | Substituted pyrimidinyl kinase inhibitors |
| US11642348B2 (en) | 2012-10-15 | 2023-05-09 | Epizyme, Inc. | Substituted benzene compounds |
| US10874616B2 (en) | 2012-11-15 | 2020-12-29 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11576864B2 (en) | 2012-11-15 | 2023-02-14 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US10166191B2 (en) | 2012-11-15 | 2019-01-01 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11896717B2 (en) | 2012-11-15 | 2024-02-13 | Incyte Holdings Corporation | Sustained-release dosage forms of ruxolitinib |
| US11576865B2 (en) | 2012-11-15 | 2023-02-14 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11337927B2 (en) | 2012-11-15 | 2022-05-24 | Incyte Holdings Corporation | Sustained-release dosage forms of ruxolitinib |
| WO2014106606A1 (en) * | 2013-01-05 | 2014-07-10 | F. Hoffmann-La Roche Ag | Nove phenyl/pyridine series substitued by hydroxyethylamino for the treatment of cancer |
| US9815815B2 (en) | 2013-01-10 | 2017-11-14 | Pulmokine, Inc. | Non-selective kinase inhibitors |
| US10532994B2 (en) | 2013-01-10 | 2020-01-14 | Pulmokine, Inc. | Non-selective kinase inhibitors |
| US10246438B2 (en) | 2013-01-10 | 2019-04-02 | Pulmokine, Inc | Non-selective kinase inhibitors |
| US8987443B2 (en) | 2013-03-06 | 2015-03-24 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9714233B2 (en) | 2013-03-06 | 2017-07-25 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9221845B2 (en) | 2013-03-06 | 2015-12-29 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9382256B2 (en) | 2013-07-30 | 2016-07-05 | Gilead Connecticut, Inc. | Formulation of Syk inhibitors |
| US9949932B2 (en) | 2013-07-30 | 2018-04-24 | Gilead Connecticut, Inc. | Formulation of syk inhibitors |
| US9918939B2 (en) | 2013-07-30 | 2018-03-20 | Gilead Connecticut, Inc. | Formulation of Syk inhibitors |
| US10266539B2 (en) | 2013-07-30 | 2019-04-23 | Gilead Connecticut, Inc. | Polymorph of Syk inhibitors |
| US9657023B2 (en) | 2013-07-30 | 2017-05-23 | Gilead Connecticut, Inc. | Polymorph of Syk inhibitors |
| US9655854B2 (en) | 2013-08-07 | 2017-05-23 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US10561616B2 (en) | 2013-08-07 | 2020-02-18 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US11045421B2 (en) | 2013-08-07 | 2021-06-29 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US12151026B2 (en) | 2013-08-07 | 2024-11-26 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US9925184B2 (en) | 2013-10-11 | 2018-03-27 | Pulmokine, Inc. | Spray-dry formulations |
| US9974792B2 (en) | 2013-12-04 | 2018-05-22 | Gilead Sciences, Inc. | Methods for treating cancers |
| US9687492B2 (en) | 2013-12-04 | 2017-06-27 | Gilead Sciences, Inc. | Methods for treating cancers |
| US12263163B2 (en) | 2013-12-23 | 2025-04-01 | Kronos Bio, Inc. | Crystalline succinate salt of 6-(6-aminopyrazin-2-yl)-n-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine as a Syk inhibitor |
| US9290505B2 (en) | 2013-12-23 | 2016-03-22 | Gilead Sciences, Inc. | Substituted imidazo[1,2-a]pyrazines as Syk inhibitors |
| US10342794B2 (en) | 2013-12-23 | 2019-07-09 | Gilead Sciences, Inc. | Substituted imidazo[1,2-a]pyrazines as Syk inhibitors |
| CN105934434B (en) * | 2013-12-23 | 2017-12-12 | 吉利德科学公司 | SYK inhibitor |
| CN107746405A (en) * | 2013-12-23 | 2018-03-02 | 吉利德科学公司 | Syk inhibitor |
| CN107746405B (en) * | 2013-12-23 | 2020-09-22 | 吉利德科学公司 | SYK inhibitor |
| EA031131B1 (en) * | 2013-12-23 | 2018-11-30 | Джилид Сайэнс, Инк. | Syk INHIBITORS |
| CN105934434A (en) * | 2013-12-23 | 2016-09-07 | 吉利德科学公司 | SYK inhibitors |
| US10828299B2 (en) | 2013-12-23 | 2020-11-10 | Kronos Bio, Inc. | Crystalline monomesylate salt of 6-(6-aminopyrazin-2-yl)-n-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine |
| US9968601B2 (en) | 2013-12-23 | 2018-05-15 | Gilead Sciences, Inc. | Substituted imidazo[1,2-a]pyrazines as Syk inhibitors |
| WO2015100217A1 (en) * | 2013-12-23 | 2015-07-02 | Gilead Sciences, Inc. | Syk inhibitors |
| EP3342773A1 (en) * | 2013-12-23 | 2018-07-04 | Gilead Sciences, Inc. | Syk inhibitors |
| US11517570B2 (en) | 2013-12-23 | 2022-12-06 | Kronos Bio, Inc. | Crystalline succinate salt of 6-(6-aminopyrazin-2-yl)-n-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine |
| CN106459045A (en) * | 2014-04-17 | 2017-02-22 | 艾伯维公司 | Heterocyclic kinase inhibitors |
| US10280184B2 (en) | 2014-04-17 | 2019-05-07 | Abbvie Inc. | Heterocyclic kinase inhibitors |
| WO2015158283A1 (en) * | 2014-04-17 | 2015-10-22 | Abbvie Inc. | Heterocyclic kinase inhibitors |
| WO2015166370A1 (en) | 2014-04-28 | 2015-11-05 | Pfizer Inc. | Heteroaromatic compounds and their use as dopamine d1 ligands |
| US9856263B2 (en) | 2014-04-28 | 2018-01-02 | Pfizer Inc. | Heteroaromatic compounds and their use as dopamine D1 ligands |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| USRE48285E1 (en) | 2014-06-12 | 2020-10-27 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| USRE49445E1 (en) | 2014-06-12 | 2023-03-07 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| USRE50497E1 (en) | 2014-06-12 | 2025-07-22 | Glaxosmithkline Llc | (N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| US9707236B2 (en) | 2014-07-14 | 2017-07-18 | Gilead Sciences, Inc. | Combination methods for treating cancers |
| US10080756B2 (en) | 2014-07-14 | 2018-09-25 | Gilead Sciences, Inc. | Combination methods for treating cancers |
| US10231966B2 (en) | 2016-10-27 | 2019-03-19 | Pulmokine, Inc. | Combination therapy for treating pulmonary hypertension |
| US11364238B2 (en) | 2016-10-27 | 2022-06-21 | Pulmokine, Inc. | Combination therapy for treating pulmonary hypertension |
| US12122781B2 (en) | 2017-08-25 | 2024-10-22 | Kronos Bio, Inc. | Hydrates of polymorphs of 6-(1H-indazol-6-yl)-N-(4-morpholinophenyl)-2,3-dihydroimidazo[1,2-a]pyrazin-8-amine bis-mesylate as Syk inhibitors |
| US11384082B2 (en) | 2017-08-25 | 2022-07-12 | Kronos Bio, Inc. | Hydrates of polymorphs of 6-(1H-indazol-6-YL)-N-(4-morpholinophenyl)-2,3-dihydroimidazo[1,2-A]pyrazin-8-amine bisemsylate as Syk inhibitors |
| US11278541B2 (en) | 2017-12-08 | 2022-03-22 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US10596161B2 (en) | 2017-12-08 | 2020-03-24 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US10899736B2 (en) | 2018-01-30 | 2021-01-26 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US12280054B2 (en) | 2018-03-30 | 2025-04-22 | Incyte Corporation | Treatment of hidradenitis suppurativa using JAK inhibitors |
| US11304949B2 (en) | 2018-03-30 | 2022-04-19 | Incyte Corporation | Treatment of hidradenitis suppurativa using JAK inhibitors |
| US11339168B2 (en) | 2019-02-22 | 2022-05-24 | Kronos Bio, Inc. | Crystalline forms of 6-(6-aminopyrazin-2-yl)-N-(4-(4-(oxetan-3-yl)piperazin-1-yl)phenyl)imidazo[1,2-a]pyrazin-8-amine as Syk inhibitors |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| US12440495B2 (en) | 2020-06-03 | 2025-10-14 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| WO2021249417A1 (en) * | 2020-06-09 | 2021-12-16 | 赛诺哈勃药业(成都)有限公司 | Heterocyclic compound and derivative thereof |
| WO2024026260A1 (en) * | 2022-07-25 | 2024-02-01 | Celgene Corporation | Substituted imidazopyrazine compounds as irak3 binders |
Also Published As
| Publication number | Publication date |
|---|---|
| US20060069084A1 (en) | 2006-03-30 |
| US20040102455A1 (en) | 2004-05-27 |
| EP1363702A1 (en) | 2003-11-26 |
| CA2436487A1 (en) | 2002-08-08 |
| JP2004528295A (en) | 2004-09-16 |
| EP1363702A4 (en) | 2007-08-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2002060492A1 (en) | Methods of inhibiting kinases | |
| CA2486187C (en) | Kinase inhibitors | |
| RU2632900C2 (en) | Heterocyclic amines and their application | |
| KR102835667B1 (en) | Quinazolinones as PARP14 inhibitors | |
| EP1689739B1 (en) | Azole-based kinase inhibitors | |
| EP4061812A1 (en) | Compounds useful as inhibitors of helios protein | |
| KR20210086674A (en) | Amide-substituted heterocyclic compounds for the treatment of conditions associated with modulation of IL-12, IL-23 and/or IFN-alpha | |
| RS64285B1 (en) | NAPHTHYRIDINONE COMPOUNDS USEFUL AS T CELL ACTIVATORS | |
| JP5897566B2 (en) | Cyclic N, N'-diarylthiourea and N, N'-diarylurea-androgen receptor antagonists, anticancer agents, methods for their preparation and uses | |
| IL175572A (en) | Condensed heterocycles, jak selective kinase inhibitors and pharmaceutical compositions comprising them | |
| JP2023520759A (en) | Substituted oxoisoindoline compounds for cancer therapy | |
| WO2018019222A1 (en) | Heterocyclic compound as jak inhibitor, and salts and therapeutic use thereof | |
| CZ20032287A3 (en) | Treatment methods of inflammatory and immune diseases by making use of IkB kinase inhibitors (IKK) | |
| JP2013515766A (en) | Imatinib dichloroacetate and anticancer composition containing the same | |
| JP7597710B2 (en) | Substituted indole dimer compounds | |
| KR20250068738A (en) | Substituted oxoisoindolinyl piperidine-2,6-dione compounds | |
| AU2002226197B2 (en) | Methods of inhibiting kinases | |
| AU2002226197A1 (en) | Methods of inhibiting kinases | |
| ZA200409346B (en) | Kinase inhibitors. | |
| US20230036867A1 (en) | N-(3-(5-(pyrimidin-4-yl)thiazol-4-yl)phenyl)sulfonamide compounds and their uses as braf inhibitors | |
| CN121627720A (en) | Preparation and application of macrocyclic compound | |
| AU2005203919B2 (en) | Selective kinase inhibitors | |
| AU2003291839B2 (en) | Nicotinamide-based kinase inhibitors | |
| WO2026041115A1 (en) | Double bond substituent containing compound serving as protein tyrosine phosphatase inhibitor, and use thereof | |
| AU2004294355A1 (en) | Azole-based kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002560683 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2436487 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10470955 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002226197 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002715984 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002715984 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |