ANTAGONISTS OF GONADOTROPIN RELEASING HORMONE
The present invention relates to compounds which are antagonists of gonadotropin releasing hormone (GnRH) activity. The invention also relates to pharmaceutical formulations, the use of a compound of the present invention in the manufacture of a medicament, a method of therapeutic treatment using such a compound and processes for producing the compounds.
BACKGROUND TO THE INVENTION
Gonadotropin releasing hormone (GnRH) is a decapeptide that is secreted by the hypothalamus into the hypophyseal portal circulation in response to neural and/or chemical stimuli, causing the biosynthesis and release of luteinizing hormone (LH) and follicle- stimulating hormone (FSH) by the pituitary. GnRH is also known by other names, including gonadoliberin, LH releasing hormone (LHRH), FSH releasing hormone (FSH RH) and LH/FSH releasing factor (LH/FSH RF).
GnRH plays an important role in regulating the action of LH and FSH (by regulation of their levels), and thus has a role in regulating the levels of gonadal steroids in both sexes, including the sex hormones progesterone, oestrogens and androgens. More discussion of GnRH can be found in WO 98/5519 and WO 97/14697, the disclosures of which are incorporated herein by reference.
It is believed that several diseases would benefit from the regulation of GnRH activity, in particular by antagonising such activity. These include sex hormone related conditions such as sex hormone dependent cancer, benign prostatic hypertrophy and myoma of the uterus. Examples of sex hormone dependent cancers are prostatic cancer, uterine cancer, breast cancer and pituitary gonadotrophe adenoma.
The following disclose compounds purported to act as GnRH antagonists: WO 97/44041, WO 98/5519, WO 99/51596 and WO 97/14697.
It would be desirable to provide further compounds, such compounds being GnRH antagonists.
SUMMARY OF THE INVENTION
The present invention accordingly provides a compound of formula I or a pharmaceutically acceptable salt or solvate thereof
wherein:
For RI and R2, either:-
(i) Rl= -C(X)NR5R6; -C(=NCN)NR5R6; -C(=CHNO2)NR5R6; an optionally substituted 5- to 10-membered mono- or bi-cyclic heterocyclic ring structure containing from
1 to 5 heteroatoms independently selected from O, N and S; optionally substituted CI to C8 alkyl; optionally substituted aryl; or optionally substituted aralkyl, where the alkyl moiety is CI to C8; R2 = H; optionally substituted CI to C8 alkyl; optionally substituted aryl; optionally substituted aralkyl; -R7-R8, wherein R7 represents optionally substitutsd CI to
C8 alkyl and R8 represents an optionally substituted 5- to 10-membered mono- or bi-cyclic heterocyclic ring structure containing from 1 to 5 heteroatoms
independently selected from O, N and S; optionally substituted C2 to C12 alkenyl; or optionally substituted alkenylaryl, wherein the alkenyl moiety is C2 to C12; and A = a single bond; optionally substituted CI to C8 alkylene; a C2 to C12 group having at least one alkene double bond; or -R-Ar-R'-, where R and R' are independently selected from a bond, optionally substituted CI to C8 alkylene and a C2 to C12 group having at least one alkene double bond; and Ar represents optionally substituted aryl.
(ii) the structure N-R1R2 represents a 3- to 8- membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S and optionally fused to a C5 to CIO ring structure, N-R1R2 being optionally substituted;
For R3 and R4, either R3 is selected from (iii) and R4 selected from (iv); or R3 is selected from (iv) and R4 selected from (iii):-
(iii) H; -ZR9, halogen; -ZC(O)NR9R10; -ZC(O)OR9; -ZC(O)SR9; -ZC(O)R9; C(R9)=N- OR10; -ZNR9C(O)NR10R11; -ZNR9SO2R10; -ZSO2R9R10; -ZCR9(CN)2; - ZN(R9)CN; or an optionally substituted 3- to 6-membered heterocyclic ring containing from 1 to 3 heteroatoms independently selected from O, N and S;
(iv) -Z'-M, wherein
M represents a mono- or bi-cyclic aromatic ring structure optionally having at least one substituent selected from CN; NR12R13; an optionally substituted CI to C8 alkyl; optionally substituted CI to C8 alkoxy; halogen; (CH2)b-C(O)NR12R13; NR12- C(O)NR13R14; (CH2) -SO2NR12R13; NR12C(O)R13; NR12SO2R13; (CH2)bOH; NR12CN; and CR12(CN)2;
Wherein each R5, R6, RIO, RI 1, R12, R13 and R14 is independently selected from H; optionally substituted CI to C8 alkyl and optionally substituted aryl;
R9 is selected from H; optionally substituted CI to C8 alkyl; optionally substituted aryl; -R-Ar, where R represents CI to C8 alkylene and Ar represents optionally substituted aryl; and optionally substituted 3- to 8- membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S;
X = O; S; or NR" ', where R" ' is H or CI to C8 alkyl;
Y = a bond; CI to C8 alkylene; a C2 to C12 group having at least one alkene double bond; or a C2 to C12 group having at least one alkyne triple bond; Z = a bond; CI to C8 alkylene; a C2 to C12 group having at least one alkene double bond; a C2 to C12 group having at least one alkyne triple bond; or -CR(R'), where R and R' are independently selected from H, CN, halogen, CI to C8 alkyl, CH2F,
CHF2, and C3 to C8 cycloalkyl; Z' = a bond; CI to C8 alkylene; a C2 to C12 group having at least one alkene double bond; a C2 to C12 group having at least one alkyne triple bond; or -CR(R'), where R and R' are independently selected from H, CN, halogen, CI to C8 alkyl, CH2F, CHF2, and C3 to C8 cycloalkyl;
a = zero or an integer from 1 to 8; each b independently represents zero or an integer from 1 to 8;
Wherein ring B is optionally further substituted.
The present invention also provides a pharmaceutical formulation comprising such a compound and a pharmaceutically acceptable diluent or carrier.
Furthermore, the present invention provides the following uses of the compound: -
(a) Use in the manufacture of a composition, for antagonising gonadotropin releasing hormone activity.
(b) Use in the manufacture of a medicament for administration to a patient, for reducing the secretion of luteinising hormone by the pituitary gland of the patient.
(c) Use in the manufacture of a medicament for administration to a patient, for therapeutically treating and/or preventing a sex hormone related condition in the patient.
The present invention also relates to a method of antagonising gonadotropin releasing hormone activity in a patient, comprising administering the compound to the patient.
In addition, the invention provides a process of producing the compound.
DETAILED DESCRIPTION OF THE INVENTION
As discussed above, the present invention provides a compound of formula I or a pharmaceutically acceptable salt or solvate thereof
For RI and R2, either option (i) or (ii) applies:-
Rl= -C(X)NR5R6; -C(=NCN)NR5R6; -C(=CHNO2)NR5R6; an optionally substituted 5- to 10-membered mono- or bi-cyclic heterocyclic ring structure (preferably, a 5- or 6-membered monocyclic ring) containing from 1 to 5 (preferably, 1 or 2) heteroatoms independently selected from O, N and S; optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl); optionally substituted aryl (preferably optionally substituted phenyl); or optionally substituted aralkyl, where the alkyl moiety is CI to C8 (preferably, CI to C4 alkyl, and most preferably methyl) and the aryl moiety is preferably phenyl; R2 = H (which is the most preferred for R2); optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl); optionally substituted aryl (eg, phenyl); optionally substituted aralkyl (eg, phenylalkyl, where the alkyl is CI to C8, preferably CI to C4 alkyl, and most preferably methyl); -R7-R8, wherein R7 represents optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl) and R8 represents an optionally substituted 5- to 10-membered mono- or bi-cyclic heterocyclic ring structure (preferably, a 5- or
6-membered monocyclic ring) containing from 1 to 5 (eg, 1 or 2) heteroatoms independently selected from O, N and S; optionally substituted C2 to C12 alkenyl (preferably, C2 to CIO, more preferably C2 to C6 alkenyl); or optionally substituted alkenylaryl, wherein the alkenyl moiety is C2 to C12 (preferably, C2 to CIO, more preferably C2 to C6 alkenyl), and preferably the aryl is phenyl; and
A = a single bond; optionally substituted CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene); a C2 to C12 group having at least one alkene double bond; or -R-Ar-R'-, where R and R' are independently selected from a bond, optionally substituted CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene) and a C2 to C12 group having at least one alkene double bond; and Ar represents optionally substituted aryl (eg, optionally substituted phenyl).
In the definition of A, where a C2 to C12 group having at least one alkene double bond is mentioned, this may have from 1 to 11 (more preferably 1 to 8) alkene double bonds (eg, 1, 2 or 3 alkene double bonds). The group can be straight chain or branched.
Option (ii) is as follows :-
(ii) the structure N-R1R2 represents a 3- to 8- membered heterocyclic ring (preferably, a 5- or 6-membered monocyclic ring) optionally containing from 1 to 3 (eg, 1) further heteroatoms independently selected from O, N and S and optionally fused to a C4 to CIO (preferably, C4, C5 or C6) ring structure (eg, a carbocyclic ring structure or a ring structure comprising a heteroatom selected from O, N and S), N-R1R2 being optionally substituted.
Preferably, RI is represented by option (i) and X represents S. In addition, or alternatively, preferably, RI is represented by option (i) and R5 and R6 each represent H.
In one embodiment, RI represents optionally substituted pyridyl, eg 2-pyridyl. One example of suitable substitution of the pyridyl is at the 6-position, eg with -NRR', wherein R and R' are independently selected from H and CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl).
In another embodiment, RI represents optionally substituted phenyl and R2 represents H, A being a single bond or methylene.
In a further embodiment, RI represents -(C(=NCN)NH(CH3) and R2 represents H, A being a single bond or methylene.
In yet another embodiment, RI represents optionally substituted phenyl and R2 represents -CH2-phenyl, A being a single bond or methylene.
For R3 and R4, either R3 is selected from (iii) and R4 selected from (iv); or R3 is selected from (iv) and R4 selected from (iii):-
(iii) H; -ZR9 (representing, eg, methyl), halogen (eg F, Br or CI); -ZC(O)NR9R10 (eg, where R9 and RIO are each ethyl); -ZC(O)OR9 (eg, where R9 is ethyl); -ZC(O)SR9; - ZC(O)R9 (preferably where R9 represents optionally substituted phenyl or t-propyl); C(R9)=N-OR10 (preferably where RIO represents optionally substituted methyl); - ZNR9C(O)NR10Rll(preferably, NHC(O)NHRl l); -ZNR9SO2R10; -ZSO2R9R10; - ZCR9(CN) ; -ZN(R9)CN; or an optionally substituted 3- to 6-membered heterocyclic ring containing from 1 to 3 heteroatoms independently selected from O, N and S.
Preferably, the heterocyclic ring is a substituent of formula II or III:-
Option (iv) is as follows:-
(iv) -Z'-M, wherein M represents a mono- or bi-cyclic aromatic ring structure (preferably, phenyl or indolyl, eg 5-indolyl) optionally having at least one (eg,l or 2 substituents) substituent (eg,l or 2 substituents) selected from CN; NR12R13; an optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl); optionally substituted CI to C8 alkoxy (preferably, CI to C8 alkoxy, and most preferably methoxy); halogen (eg, F, Br or CI); CH2)b-C(O)NR12R13 (preferably,
CONH2; CH2CONH2; or CH2CONHCH3); NR12-C(O)NR13R14 (preferably, NH-
C(O)NH2); (CH2)b-SO2NR12R13 (preferably, CH2SO2NHCH3); NR12C(O)R13 (eg, NHC(O)CH3; NR12SO2R13; (CH2)bOH (preferably, OH or CH2OH); NR12CN(preferably, NHCN); and CR12(CN)2 (preferably, CH(CN)2).
In one embodiment, option (iv) represents phenyl substituted with two methyl groups (eg, 3,5-dimethylphenyl.
Each R5, R6, RIO, RI 1, R12, R13 and R14 is independently selected from H; optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl) and optionally substituted aryl (eg, optionally substituted phenyl).
R9 is selected from H; optionally substituted CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl); optionally substituted aryl (eg, optionally substituted phenyl); -R-Ar, where R represents CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene) and Ar represents optionally substituted aryl (eg, optionally substituted phenyl); and optionally substituted 3- to 8- membered heterocyclic ring (eg, a 5- or 6-membered ring) optionally containing from 1 to 3 further heteroatoms (eg, 1 further heteroatom) independently selected from O, N and S.
X represents O; S; or NR'", where R'" is H or CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl).
Y represents a bond; CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene); a C2 to C12 group having at least one alkene double bond; or a C2 to C12 group having at least one alkyne triple bond. Where Y represents a C2 to C12 group having at least one alkene double bond, this may have from 1 to 11 (more preferably 1 to 8) alkene double bonds (eg, 1, 2 or 3 alkene double bonds). The group can be straight chain or branched. Where Y represents a C2 to C12 group having at least one alkyne triple
bond, this may have from 1 to 6 alkyne triple bonds (eg, 1, 2 or 3 alkyne triple bonds). The group can be straight chain or branched.
Z represents a bond; CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene); a C2 to C12 group having at least one alkene double bond; a C2 to C12 group having at least one alkyne triple bond; or -CR(R'), where R and R' are independently selected from H, CN, halogen (eg, F, Br or CI, most preferably F), CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl), CH2F, CHF2, and C3 to C8 cycloalkyl (preferably, C3 to C6 cycloalkyl). Where Z represents a C2 to C12 group having at least one alkene double bond, this may have from 1 to 11 (more preferably 1 to 8) alkene double bonds (eg, 1 , 2 or 3 alkene double bonds). The group can be straight chain or branched. Where Z represents a C2 to C12 group having at least one alkyne triple bond, this may have from 1 to 6 alkyne triple bonds (eg, 1, 2 or 3 alkyne triple bonds). The group can be straight chain or branched.
Z' represents a bond; CI to C8 alkylene (preferably, CI to C4 alkylene, and most preferably methylene); a C2 to C12 group having at least one alkene double bond; a C2 to C12 group having at least one alkyne triple bond; or -CR(R'), where R and R' are independently selected from H, CN, halogen (eg, F, Br or CI, most preferably F), CI to C8 alkyl (preferably, CI to C4 alkyl, and most preferably methyl), CH F, CHF2, and C3 to C8 cycloalkyl (preferably, C3 to C6 cycloalkyl). Where Z' represents a C2 to C12 group having at least one alkene double bond, this may have from 1 to 11 (more preferably 1 to 8) alkene double bonds (eg, 1, 2 or 3 alkene double bonds). The group can be straight chain or branched. Where Z' represents a C2 to C12 group having at least one alkyne triple bond, this may have from 1 to 6 alkyne triple bonds (eg, 1, 2 or 3 alkyne triple bonds). The group can be straight chain or branched.
For the integers a and b, a = zero or an integer from 1 to 8 (preferably, a = zero); and
each b is independently selected from zero or an integer from 1 to 8 (preferably, each b = zero). In one embodiment, a represents zero and Y represents CH2.
The ring B can be optionally further substituted.
In the present specification, unless otherwise indicated, an alkyl, alkylene or alkenyl moiety (eg, the alkyl moiety of an alkylaryl substituent) may be linear or branched.
The term "alkylene" refers to -CH2-. Thus, C8 alkylene for example is -(CH )8-.
Where optional substitution is mentioned at various places above, this refers to one, two, three or more optional substituents. Unless otherwise indicated above (ie, where a list of optional substituents is provided), each substituent can be independently selected from CI to C8 alkyl (preferably C2 to C6 alkyl, and most preferably methyl); O(C3 to C8 cycloalkyl), preferably O-cyclopropyl, or O-cyclobutyl or O-cyclopentyl; O(Cl to C6 alkyl), preferably Omethyl or O(C2 to C4 alkyl); halo, preferably CI or F; CHal3, CHHal2, CH Hal, OCHal3, OCHHal2 or OCH2Hal, wherein Hal represents halogen (preferably F);
CH2OR, NRCOR', NRSO2R' or N-R-R', wherein R and R' independently represent H or
CI to C8 alkyl (preferably methyl or C2 to C6 alkyl or C2 to C4 alkyl) , or N-R-R' represents an optionally substituted C3 to C8, preferably C3 to C6, heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S; H; or COOR" or COR", R" representing H, optionally substituted phenyl or CI to C6 alkyl (preferably methyl, ethyl, /-propyl or t-butyl). For optional substitution of the heterocyclic ring represented by N-R-R', at least one (eg, one, two or three) substituents may be provided independently selected from CI to C6 alkyl (preferably C2 to C4 alkyl, more preferably methyl); phenyl; OCF3; OCHF2; -O(Cl-C8 alkyl), preferably -O-methyl, - O-ethyl or -O(C3 to C6 alkyl); -C(O)O(Cl-C8 alkyl), preferably -C(O)O-methyl, - C(O)O-ethyl, -C(O)O-tert-butyl or -C(O)O(C3 to C6 alkyl); -C(O)O-phenyl; -O-phenyl; - C(O) (C1-C8 alkyl), preferably -C(O)-methyl, -C(O)-ethyl or -C(O)(C3 to C6 alkyl) ; - C(O)OH; -S(C1-C8 alkyl), preferably -S-methyl, -S-ethyl or -S(C3 to C6 alkyl); OH; halogen (eg, F, CI or Br), NRR' where R and R' are independently H or CI to C6 alkyl
(preferably C2 to C4 alkyl, more preferably methyl, most preferably R=R'=methyl); and nitro.
Particularly preferred compounds according to the present invention are:-
N-Benzyl-N-methyl-2-(4-bromophenyl)-3-methylamino-5-methylimidazo[l,2- αjpyridine;
N-Benzyl-N-methyl-2-(4-chlorophenyl)-3-methylamino-5-methylimidazo[l,2- αjpyridine;
N-Benzyl-N-methyl-2-(4-fluorophenyl)-3-methylamino-5-methylimidazo[l,2- ]pyridine;
N-Benzyl-N-methyl-2-(4-cyanophenyl)-3-methylamino-5-methylimidazo[l,2- αjpyridine;
N-Benzyl-N-methyl-2-(4-methoxyphenyl)-3-methylamino-5-methylimidazo[l,2- αjpyridine;
N-B eι_zyl-N-methyl-2-(3 ,4-dimethoxyphenyl)-3 -methylamino-5-methylimidazo [1,2- α]pyridine;
N-B enzyl-N-methyl-2-(3 ,4-dichlorophenyl)-3 -methylamino-5-methylimidazo [ 1 ,2- α]pyridine;
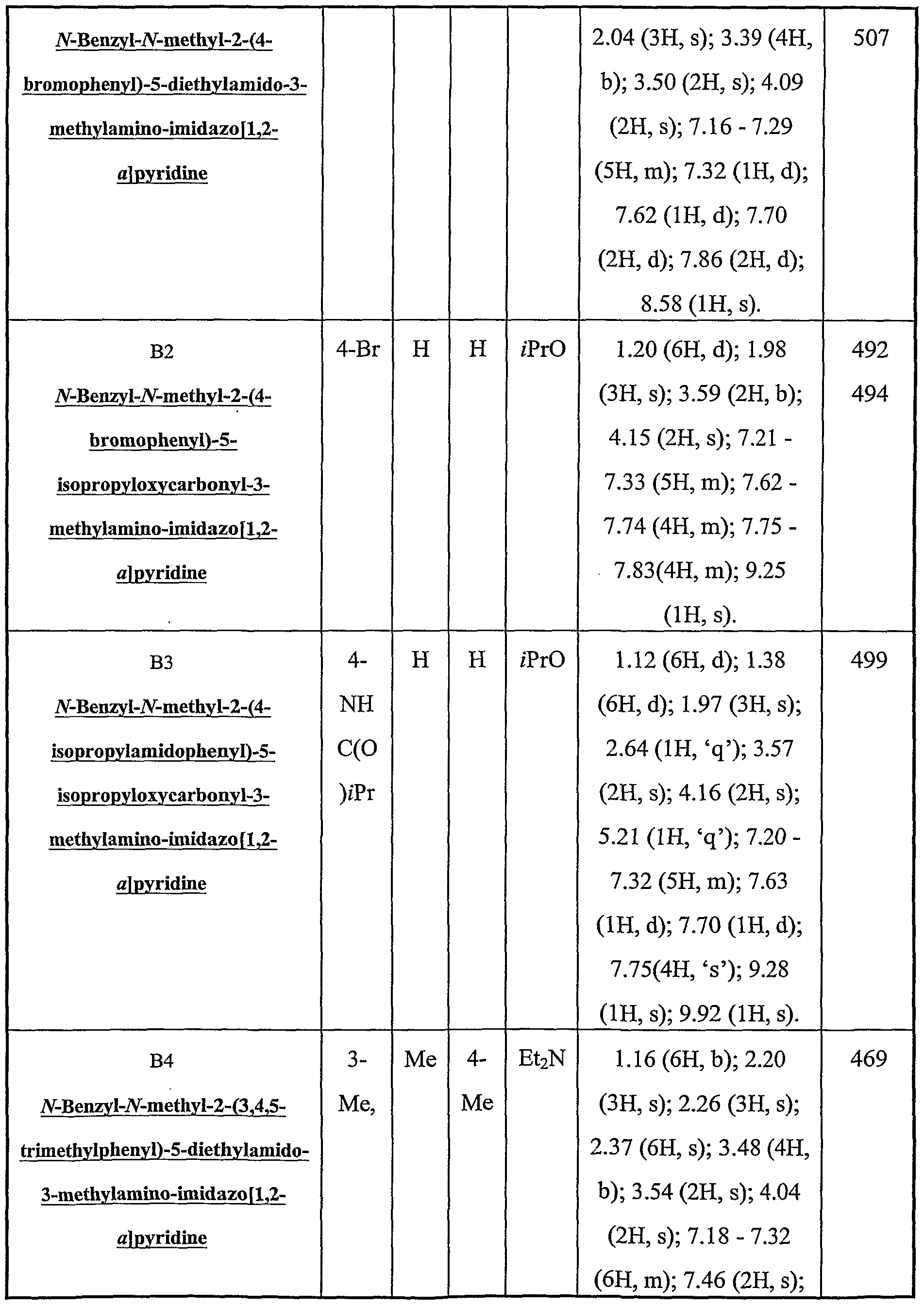
N-Benzyl-N-methyl-2-(4-bromophenyl)-5-diethylamido-3-methylamino-imidazo[l,2- αjpyridine;
N-Benzyl-N-methyl-2-(4-bromophenyl)-5-isopropyloxycarbonyl-3-methylamino- imidazo[ 1 ,2- ]pyridine;
N-Benzyl-N-methyl-2-(4-isoρropylamidophenyl)-5-isopropyloxycarbonyl-3- methylamino-imidazo[l,2-α]pyridine;
N-Benzyl-N-methyl-2-(3,4,5-trimethylphenyl)-5-diethylamido-3-methylamino- imidazo[ 1 ,2-_.]pyridiήe;
Ethyl N-benzyl-N-methyl-5-(3-acetamidophenyl)-3-methylamino-imidazo[ 1 ,2-
_.]pyridine-2-carboxylate;
N-Cyano-N,-[3-(lH-indol-5-yl)-imidazo[l,2-α]pyridin-2-ylmethyl]-N'-methyl- guanidine;
N-Cyano-_\^-[3-(3,4-dime oxyphenyl)-imidazo[l,2- ]ρyridin-2-ylmethyl]-N''- methyl-guanidine;
N-Cyano-N'-[3-(4-cUorophenyl)-imidazo[l,2- ]pyridin-2-ylmethyl]-N''-methyl- guanidine;
N-Cy__no-N'-[3-(3,5-dimemylphenyl)-imidazo[l,2-α]pyridin-2-ylmethyl]-N''-methyl- guanidine;
NN-Dibe____yl-2-methylamino-3-(3,4-dimethoxyphenyl)imidazo[l,2-<3]pyridine;
NN^Diben_^l-2-methylaιnino-3-(4-dimethoxyphenyl)imidazo[ 1 ,2-α]pyridine;
N-ben__yl-N-methyl-2-methylamino-3-(4-chlorophenyl)imidazo[l,2-<2]pyridine;
N-benzyl-N-methyl-2-methylamino-3-(3,5-dimethylphenyl)imidazo[l,2-α]pyridine;
N-B enzyl-5-bromo-3 -(3 -methylpropylamino)-2-(4-methoxyphenyl)imidazo [ 1 ,2- α]pyridine;
N-Ben__yl-N-Methyl-5-methyl-3-propargylamino-2-(4-methoxyphenyl)imidazo[l,2- α]pyridine;
N-(β-methylphenethyl)-N-Methyl-5-methyl-3-ρropargylamino-2-(4- methoxyphenyl)imidazo[ 1 ,2-α]pyridine; and
N-Benzyl-N-methyl-2-(3,5-dimethylphenyl)-3-ethylamino-5-methylimidazo[l,2- αjpyridine.
The invention also contemplates pharmaceutically acceptable salts and solvates of these compounds. Compounds of formula I may be converted to pharmaceutically acceptable salts and solvates thereof, preferably acid addition salts, such as hydrochloride, hydrobromide, phosphate, acetate, fumarate, maleate, tartarate, citrate, oxalate, methanesulphonate or 7-toluenesulphonate, or alkali metal salts such as sodium or potassium salts.
The compounds of formula I can be prepared by a process comprising a step selected from (a) to (y) as follows:-
(a) Reaction of a compound of formula IV as follows
(b) Reaction of a compound of formula IN as follows
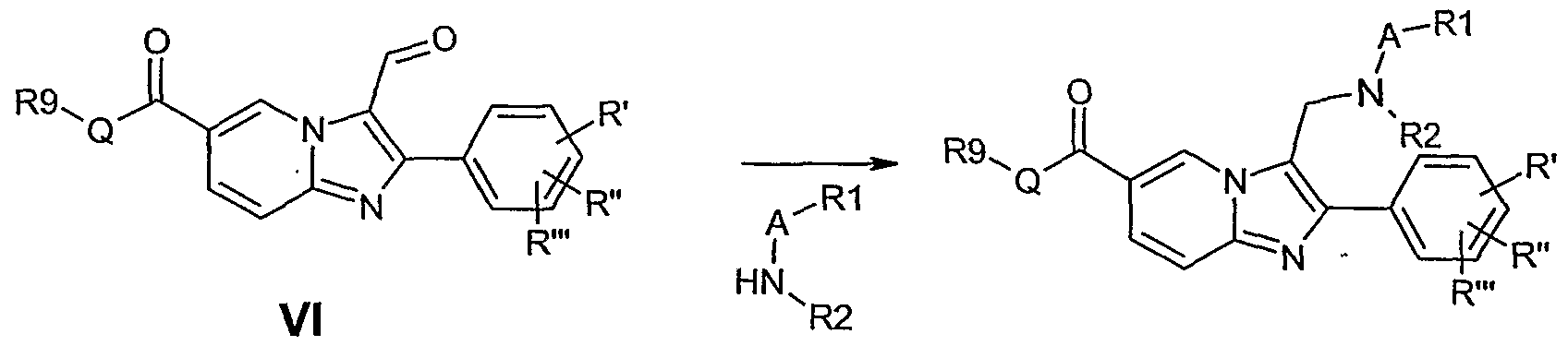
(c) Reaction of a compound of formula VI as follows
(d) Reaction of a compound of formula VII as follows
(e) Reaction of a compound of formula VII as follows
R9. ca-
VII
CO
(f) Reaction of a compound of formula VII as follows
(g) Reaction of a compound of formula Vill as follows
VIII
(h) Reaction of a compound of formula VIII as follows
(i) Reaction of a compound of formula VIII as follows
(j) Reaction of a compound of formula IX as follows
IX
(k) Reaction of a compound of formula XI as follows
XI
(1) Reaction of a compound of formula XI as follows
(m) Reaction of a compound of formula XI as follows
(n) Reaction of a compound of formula XII as follows
XII
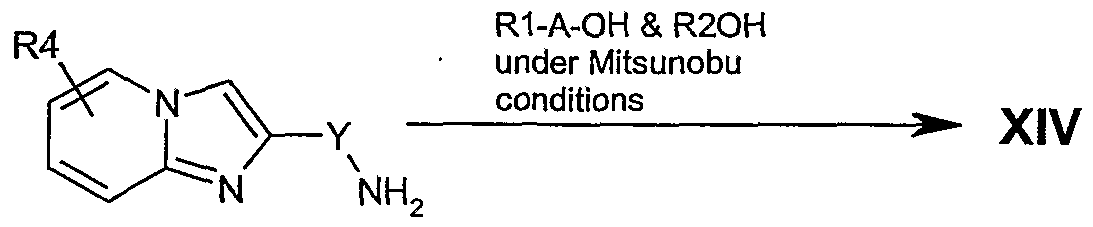
(o) Reaction of a compound of formula XIII as follows
XIV
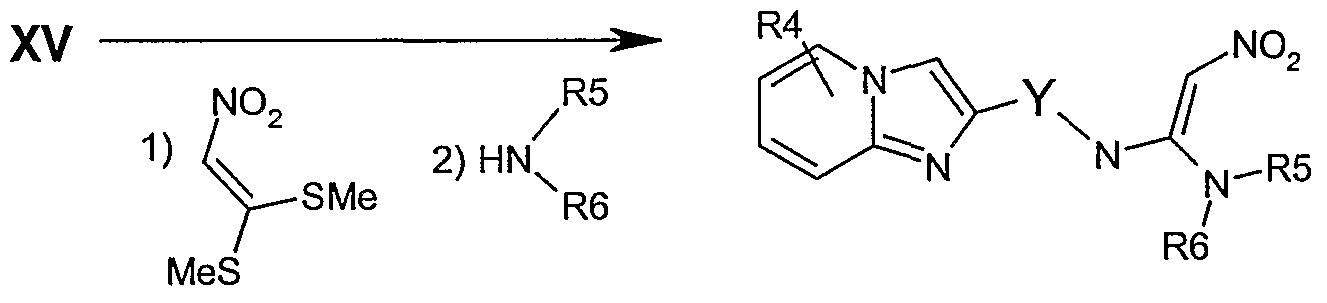
(p) Reaction of a compound of formula XV as follows
XV
(q) Reaction of a compound of formula XV as follows
(r) Reaction of a compound of formula XV as follows
(s) Reaction of a compound of formula XVI as follows
XVI
(t) Reaction of a compound of formula XVI as follows
(u) Reaction of a compound of formula XVI as follows
(v) Reaction of a compound of formula XVI as follows
(w) Reaction of a compound of formula XVII as follows
(x) Reaction of a compound of formula XIX as follows
XIX
(y) Reaction of a compound of formula XX as follows
XX 3) Reduction
Wherein R', R", R'", R*, R** and R*** are independently H or a substituent; L', L", L2 and L* are leaving groups; Q = NR10; S or O; and Q' = NR10; S or O.
It will be appreciated by those skilled in the art that in the processes of the present invention certain functional groups such as hydroxyl or amino groups in the starting reagents or intermediate compounds may need to be protected by protecting groups. Thus, the preparation of the compounds of formula I may involve, at an appropriate stage, the addition and subsequent removal of one or more protecting groups.
The protection and deprotection of functional groups is described in 'Protective Groups in Organic Chemistry', edited by J.W.F. McOmie, Plenum Press (1973) and 'Protective Groups in Organic Synthesis', 2nd edition, T.W. Greene and P.G.M. Wuts, Wiley- Interscience (1991).
EXPERIMENTAL
GENERAL REACTION SCHEMES
solvent, heat Molecular sieves
Scheme a
L=leaving group e.g. CI, Br, I, OMesyl, OTosyl or H, W = epoxide or aziridines
W=group for elaboration into N(-(A)-R1 )R2.
Imidazo[l,2-α]pyridines of the structure (A) can be prepared by the condensation of a suitable substituted 2-aminopyridine 1 and a ketone 2 bearing a leaving group α to the carbonyl group (Br preferred group). Heating at a temperature between 25 °C and 120 °C, preferably 80 °C, in a suitable solvent such as NN-dimethylformamide (DMF), dunethylsulfoxide (DMSO), toluene, xylene, t-BuOH, preferable DMF, with or without the molecular sieves, for a period of 1 to 24 h, effects the condensation. With appropriately substituted groups (e.g. W), the amine group -Ν(-(A)-R1)R2 can then be installed by standard chemistry know to those skilled in the art which is detailed below.
Scheme b
For example, Scheme b shows a general synthesis of 5-carbonyl-2-aryl-3- aminomethylimidazo[l,2-α]pyridine commencing with commercially available 2- aminopyridine and 2'-bromoacetophenone. Thus, condensation of 6-aminonicotinic acid and a 2'-bromoacetophenone 3, under the preferred conditions noted above for the key cyclisation, yields the bicycle 4. Condensation using the coupling reagent l-(3- dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) or 1,3- dicyclohexylcarbodiimide (DCC) or the like, with or without 1-hydroxybenotriazole (HOBt) and suitable amine base, such as triethylamine and the like, in an inert solvent such as methylene chloride, chloroform, DMF, or mixtures thereof, at or near room temperature
for a period of 3 to 24h to provide the corresponding coupled product 5, bearing the substituent N(R9)R10, where R9 and RIO are as defined above. Michael addition reactions can be achieved by the condensation of methyl vinyl ketone with the bicycle 5 by heating in an organic acid, such as acetic acid, to yield a ketone product. Reductive animation under typical conditions of an appropriately substituted amine and a hydride source, such as sodium cyanoborohydride, sodium borohyride, zinc borohyride, lithium borohydride and the like, yields products such as 6. Using classical Mannich chemistry an aminomethyl group can be introduced by treatment of 5 with a mixture of a suitably substituted amine (NH(A-R1)(R2)) and paraformaldehyde. In an organic acid such as acetic acid and the like and stirring at room temperature or heating between 40 and 100°C in this manner compounds such as 8 which correspond to the general structure (A) where Y represents
CH2 are formed. Alternatively, a two step procedure may be employed, where a Vilsmeier reaction, classically employing DMF and phosphorus oxychloride at a temperature between -10 °C and 25 °C, installs a formyl group at the 3-position of the imidazo[l,2- a]pyridine to give 7. Reduction animation employing a suitably substituted amine [HN(- (A)-R1)R2] and a reducing agent such as sodium borohydride, sodium cyanoborohydride, zinc borohydride and the such like, under acid or neutral conditions in a suitable solvent such as methylene chloride, chloroform, benzene, toluene and alcohols such as ethanol and the like, yields the 3-aminomethyl-imidazo[l,2-a]pyridines (8).
As an alternative to N(R9)R10, one can use R9OH in scheme b.
Scheme c
For example, Scheme c shows another general synthesis of 5-carbonyl-2-aryl-3- aminomethylimidazo[ 1 ,2-α]pyridine commencing with commercially available 2- aminopyridines and 2'-bromoacetophenone. Thus, condensation of 2-aminopyridine 9 and a 2'-bromoacetophenone 3 under the preferred conditions noted above for the key cyclisation yields the bicycle 10. The Mannich reaction conditions described above for Scheme b again install the substituted aminomethyl group leading to the bicycle 11. Where L' is chloride, bromide, iodide, O-trifluoromethanesulfonate, trialkyltin or like, 11 can be treated under palladium(O) catalysis with carbon monoxide at 1 atm or higher pressure in the presence of a substituted amine (N(R9) R10 as shown), alcohol (R9OH - not shown) or tliiol (R9SH - not shown) in an inert solvent such as toluene, benzene, dioxane, THF, DMF and the like to yield 5-carbonyl-2-aryl-3-aminomethylimidazo[l,2-α]pyridines such as 8. Where L' is chloride, bromide, iodide, O-trifluoromethanesulfonate, trialkyltin or like 11 can be treated under palladium(O), a weak base such aqueous sodium carbonate and the
like and a substituted aryl boronic acid from commercial sources or prepared (as described in: Gronowitz, S.; Hornfeldt, A.-B.; Yang, Y.,-H Chem. Sci. 1986, 26, 311-314), in an inert solvent such as toluene, benzene, dioxane, THF, DMF and the like, with heating between 25 °C and 100 °C, preferably 80 °C, for a period of 1-12 hours, to give the imidazo[l,2- βjpyridine 12. Similarly coupling of an arylalkylzinc iodide with 11 can be achieved using the methods of Negishi (e.g. Jackson, R. F. W.; James, H.; Wythes, M. J.; Wood, A. J. Chem. Soc. Chem. Commun. 1989, 644) to yield imidazo[l,2-α]pyridines (13).
18
Scheme d
Condensation of a suitable substituted 2-aminopyridine 9 with a bromopyruvate 14 using the condition described above, yields 2-carboxyimidazo[l,2-α]pyridines 15. With same chemical sequences of Mannich reaction then palladium catalysed carbonylation, Suzuki couplings or Negishi couplings the 2-carboxyimidazo[l,2-α]pyrimidines 17 and 18 can be synthesised (Scheme d).
As an alternative to N(R9)R10, one can use R9OH in scheme d.
Scheme e.
Condensation of a suitable 2-aminopyridine 1 and a substituted aryl ketone 20using the conditions described above gives imidazo[l,2-a]pyridines of the type 21 where a 3- position is already substituted with an alkyl chain. The leaving group L* can be converted
to an amine (22) by either the two step route of :- displacement with potassium phthalimide (heating in a suitable inert solvent such as DMSO, DMF or THF and mixtures thereof, and the like) then removal of the phthalimide protecting group (treatment with hydrazine in an inert solvent e.g. methylene chloride, chloroform, THF and mixtures thereof and the like), or displacement with sodium azide (heating in a suitable solvent DMSO, DMF or THF and mixtures thereof and the like) then reduction of the resultant azide (by treatment with hydrogen gas at atmospheric pressure or under high pressure [up to 600 psi] under palladium catalysis, or by Stoedinger reduction with triphenylphosphine). Groups RI and R2 can be introduced by a modified Mitsunobu reaction. Reaction with an arylsufonyl chloride such as 2-nitrobenzenesulphonyl chloride, 4-nitrobenzenesulphonyl chloride, 2,4- nitrobenzenesulphonyl chloride and a hindered amine base such as 2,4,6-collidine, 2,6- lutidine or the like in an inert organic solvent such as methylene chloride, provides the corresponding sulfonamide. Mitsunobu coupling of the sulfonamide and an alcohol (R1OH or R2OH) can be achieved by treatment with an activating agent such as diethyldiazocarboxylate (DEAD), diisopropyldiazocarboxlate or the like with triphenylphosphine, tri-butylphosphine and the like, in an inert solvent such as benzene, toluene, tetrahydrofuran or mixtures thereof to give the dialkylated sulfonamide adduct. Removal of the sulfonamide group is accomplished by treatment with a nucleophilic amine such as ft-propylamine to give substituted amine 23. The primary amine 22 can be converted to a cyano-guanidine (24) by the two step process of reaction with diphenyl cyanocarbonimidate in an inert organic solvent such as methylene chloride, chloroform, benzene, tetrahydrofuran and the like, followed by condensation with an appropriately substituted amine (HNR5R6) in an inert organic from the list above. Similarly, reaction with 1,1 '-bis(methylthio)-2-nitroethylene in an inert solvent such methylene chloride, chloroform, benzene, tetrahydrofuran and the like, followed by condensation with an appropriately substituted amine (HNR5R6) in an inert organic solvent from the list above yields the nitroethyleneimidazo[l,2-_.]pyridine 25.
TFA, anisole
Scheme f
Condensation of 2-aminopyridines 1 with ketones bearing a t-butylcarbamate protected nitrogen atom (26) produces imidazo[l,2-α]pyridines such as 27, where the nitrogen atom is already installed (Scheme f). The t-butylcarbamate protecting group is removed by treatment with an organic acid such as trifluoroacetic acid and the like, in the presence of a carbocation scavenger such as anisole, to yield the same imidazo[l,2- αjpyridines 28 as in scheme f. In the same manner the substitutents on the nitrogen atom can be installed by the Mitsunobu strategy (28— 29) or by condensation with an acid chloride in the presence of a hindered amine base such as triethylamine, in an inert solvent such as methylene chloride, then reduction of this product with lithium aluminium hydride, in an inert solvent such as tetrahydrofuran, or by reduction with borane in a similarly inert solvent (27- 29).
L* leaving group e.g. CI, Br, I.
Pyridinium tribromide
Scheme g.
Substituted ketones (20) can be prepared, as outlined in Scheme g starting from appropriate acid chlorides such as 30. Treatment of the acid chloride with N,N- dimethylhydroxylamine in the presence of an amine base such as triethylamine, and a suitable solvent such as methylene chloride at a temperature of -10 °C to 25 °C, yields the amide 31. Further reaction with a substituted aryl organolithium (prepared essentially as described in Wakefield B, J.; Organolithium Methods Academic Press Limited, 1988, pp. 27-29 and references therein) in an inert solvent such as tetrahydrofuran, diethyl ether, benzene, toluene or mixture thereof and the like, at a temperature between -100 °C and O °C then quenching of the reaction mixture with a mineral acid such as hydrochloric acid, yields the aryl ketone 32. Finally treatment of 32 with a bromine source such as pyridinium tribromide or pyrrolidone hydrobromide in an inert solvent such as chloroform or methylene chloride at -10 °C to 25 °C, yields a bromoketone 20 which is appropriate for the formation of an imidazo[l,2-α]pyridine.
33 34 35
Pyridinium tribromide
Scheme h.
Commencing with a readily available amino acid with a suitable chain length for Y
(33), the nitrogen atom can be installed directly by the route shown in Scheme h. Protection of the amine group of 33 with a tert-butylcarbamate group is achieved by condensation with di-tert-butyl dicarbonate in the presence of an amine base, for example triethylamine, in an inert solvent such as methylene chloride, chloroform, benzene, toluene, tetrahydrofuran and mixtures thereof and the like, at a temperature of -10 °C to 25 °C. Coupling of the acid product with NN-dimethylhydroxylamine in the presence of a coupling reagent l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) or 1,3-dicyclohexylcarbodiimide (DCC) or the like, with or without 1-hydroxybenotriazole (HOBt), and suitable amine base, such as triethylamine and the like, in an inert solvent such as methylene chloride, chloroform, dimethylformamide, or mixture thereof, at or near room temperature for a period of 3 to 24 h provided the corresponding coupled product 34. Following the same route described above for scheme g, the aryl group can then be
installed and subsequently the α-bromo group to give the ketone 26, which is suitable for condensation with a 2-aminopyridine.
Scheme j
An isomeric series of imidazo[l,2- ]pyridines can be synthesised as described in Scheme j. Condensation of a suitably substituted 2-aminopyridine 1, under the general conditions described above, with a ketone bearing two leaving groups α to the carbonyl (36) yields an imidazo[l,2-α]pyridine such as 37. The substituted amino group can be
installed by direct alkylation to yield 39, or by an indirect multistep route as shown above in scheme j (compound 37 to compound 38 via intermediate 39), both routes are analogous to those shown in Schemes e.
In addition the amine group can be elaborated further to a cyano-guanidine 40 or a nifroethylene moiety such as compound 41 by the same methods as described in Scheme e.
Scheme k.
Treatment of the imidazo[l,2-α]pyridine 39 with molecular bromine, pyridinium tribromide, poly (vinyl pyridinium tribromide) or pyrrolidone hydrobromide in an inert solvent such as chloroform or methylene chloride at -10 °C to 25 °C installs a bromo group at the two position (42). The compound QR9 is suitable for palladium (0) catalysed reactions, for example treatment with carbon monoxide at 1 atm or higher pressure in the
presence of a substituted amine (Q=NR10), alcohol (Q=O) or thiol (Q=S) in an inert solvent such as toluene, benzene, dioxane, THF and the like, yields 5-carbonyl-2-aryl-3- aminomethylimidazo[l,2-α]pyridines such as 46. Again, treatment under palladium(O) catalysis with a weak base such aqueous sodium carbonate and the like and a substituted aryl boronic acid from commercial sources or prepared (as described in: Gronowitz, S.; Hornfeldt, A.-B.; Yang, Y.,-H Chem. Sci. 1986, 26, 311-314.) in an inert solvent such as toluene, benzene, dioxane, THF and the like, with heating between 25 °C and 100 °C, preferably 80 °C, for a period of 1-12 hours to give the imidazo[l,2-α]pyridine 44. Similarly coupling of an arylalkylzinc iodide with 42 can be achieved using the method of Negishi (e.g. Jackson, R. F. W.; James, H.; Wythes, M. J.; Wood, A. J. Chem. Soc. Chem. Commun. 1989, 644) yields imidazo[l,2-α]pyridines (45). Finally a Heck coupling reaction can be achieved using palladium (0) and a vinyl substituted aromatic compound in the presence of an organic amine base such as triethylamine and the like, in an inert solvent such as toluene, benzene, dioxane, THF and the like, with heating between 25 °C and 100 °C, preferably 80 °C, for a period of 1-12 hours to give the imidazo[l,2-_z]pyrid_ne 43.
50 51
Scheme m. Condensation of a suitable substituted 2-aminepyridine 1 with a bromoacetophenone 3 under the described conditions above, yields imidazo[l,2-α]pyridines such as 47. A ethylamine group can be installed in several ways to yield compounds described by structure 50. Reaction 47 with oxalylchloride in an inert solvent such as dichloromethane, 1,2-dichloroethane, benzene, toluene, tetrahydrofuran and mixtures thereof and the like, at a temperature between 0°C and 100°C, with or without the presence of an organic base, such as triethylamine, pyridine and the like, yields and acid chloride, which may be reacted in-situ by treatment with and appropriately substituted amine [HN(R2)-A-R1] in the
presence of an organic base such as triethylamine, pyridine and the like. The amide product from this step can then be reduced by an appropriate hydride reducing agent such as lithium aluminium hydride or borane in an appropriate inert solvent such as dichloromethane, 1,2-dichloroethane, benzene, toluene, tetrahydrofuran, thus, a fully. substituted compound such as 48 can be synthesised.
Treatment of 47 with acid chloride 52 [prepared as described by Hubschwerlen, C,; Specklin, J.-L; Org. Synth. 1993, 73, 14] in an inert solvent such as dichloromethane, 1,2- dichloroethane, benzene, toluene, tetrahydrofuran and mixtures thereof and the like, at a temperature between 0°C and 100°C, with or without the presence of an organic base, such as triethylamine, pyridine and the like, yields the imidazo[l ,2- ]pyridines 49. Removal of the phthalimide protecting group by treatment with hydrazine, then reduction of the carbonyl group by an appropriate hydride reducing agent, such as lithium aluminium hydride or borane, in an appropriate inert solvent such as dichloromethane, 1,2- dichloroethane, benzene, toluene, tetrahydrofuran, yields the amine 50, which may then be elaborated as shown in the earlier schemes.
Reaction of imidazo[l,2-α]pyridines 47 with a bromine source such as pyridinium tribromide or pyrrolidone hydrobromide in an inert solvent such as chloroform or methylene chloride at -10 °C to 25 °C, yields 3-bromo substituted imidazo[l,2-α]pyridines, which in turn can be treated with N-vinylphthalimide using Heck coupling conditions of palladium (0) catalysis in the presence of an organic amine base such as triethylamine and the like, in an inert solvent such as toluene, benzene, dioxane, THF and the like, with heating between 25 °C and 100 °C, preferably 80 °C, for a period of 1-12 hours to give the imidazo[l,2-α]pyridine 51. Finally treatment with hydrazine under standard conditions yields the 3-ethylamine imidazo[l,2-α]pyridine 50, which again may be elaborated as shown in the earlier schemes.
EXAMPLES
Example Al - Preparation of N-Benzyl-N-methyI-2-(4-bromophenyl)-3-methylami _.o- 5-methyϋmidazofl,2-fl]pyridine.
Step Al; 2-(4-bromophenyl)-5-methyl-imidazo[l,2-αlpyridine.
A mixture of 2-amino-5-picoline (2.00 g 18.5 mmol) and 2,4'-dibromoacetophenone (5.10 g 18.5 mmol) in DMF (20 mL) was heated at 80 °C for lh 45 min. The mixture was cooled to RT then diluted with water (200 mL) and basified with 2M ΝaOH (aq) (150 mL). The mixture was extracted into EtOAc (2 x 200mL) and the extracts dried (MgSO4) and concentrated in vacuo to give the crude title compound as a yellow solid (5.12 g 96%). Mass Spectrum: m e C14H12BrΝ2 (M+H) 287.37 and 289.38 found.
1H NMR spectrum (DMSO-d6): δ Η NMR (300 MHz, D6 -DMSO) 2.27 (3H, s); 7.11 (IH, d); 7.48 (IH, d); 7.61 (2H, d); 7.90 (2H, d); 7.94 (IH, s); 8.32 (IH, s).
Step A2:N-Ben-_yI-N-methyl-2-(4-bromophenyl)-3-methyla_n_no-5-methyUmidazo[l,2- a] pyridine.
A mixture of 2-(4-bromophenyl)-5-methyl-imidazo[l,2-α]pyridine(4.96 g 17.3 mmol), paraformaldehyde (518 mg 17.3 mmol) and benzylmethylamine(2.23 mL 17.3mmol) in acetic acid (9 mL) was heated for 2h at 60 °C. The majority of the solvent was removed in vacuo and the mixture rediluted with EtOAc (400 mL) and washed with 2M ΝaOH (aq) (2 x 150mL). The solution was dried (MgSO4) and concentrated in vacuo. Flash column chromatograpy (silica gel, slow gradient neat CH2C12 to 6% MeOH) gave the title
compound as an orange oil (3.86 g 53%). HCI salt of title compound was prepared by the addition of l.OM HCI in diethyl ether (23 mL) to a solution of the title compound in EtOAc (4 mL). The salt was precipitated with diethyl ether and collected by centrifuge.
Following a procedure similar to that described in Example 1, the following compounds were prepared.
Example Bl - Preparation of N-Benzyl-N-methyl-2-(4-bromophenyI)-5-diethylamido- 3-methyIanuno-imidazo [ 1,2- l pyridine.
Step Bl : 2-(4-bromophenyl)-5-carboxy-in_idazo[l,2- ]pyridine.
A mixture of 6-aminonicotinic acid (1.00 g 7.24 mmol) and 2,4'-dibromoacetophenone (4.02 g 14.5 mmol) in DMF (10 mL) was heated at 60 °C for 24 h. The mixture was cooled to RT then partitioned between water (200 mL) and EtOAc (250mL). The yellow precipitate between the two layers was removed by filtration and dried by high vacuum to yield the title compound (1.72 g 75%).
Mass Spectrum: m/e Cι H10BrΝ2O2 (M+H) 317.16 and 319.16 found.
Step B2: 2-(4-bromophenyl)-5-diethylamido-imidazo[l,2- lpyridine.
HOBt (426 mg 3.15mmol) was added in one portion to a stirred solution of 2-(4- bromophenyl)-5-carboxylic-imidazo[l,2-a]pyridine (1.00 g 3.15 mmol) and EDC (605 mg 3.15 mmol) in CH2C12 (30 mL) under N2 at O°C. The mixture was stirred for lh then diethylamine (1.63 mL 15.8 mmol) was added and the mixture allowed to stir at RT for 20h. After this time the solvent was removed in vacuo then the mixture rediluted with EtOAc (250 mL) then washed with IM citric acid (200 mL) then saturated NaHCO3 (aq) (200 mL) and brine (200 mL). The organic solution was dried (MgSO4) and concentrated in vacuo to give the title compound as a yellow solid (601 mg 51%). Mass Spectrum: m/e C18H18BrN2O2 (M+H) 272.20 and 274.23 found.
1H NMR spectrum (DMSO-d6): δ Η NMR (300 MHz, D6 -DMSO) 1.15 (6H, t), 3.18 (4H, m); 7.12 (IH, d); 7.30 (IH, d); 7.33 (2H, d); 7.93 (2H, d); 8.44 (IH, s); 8.68 (IH, s).
Step B3: N-Ben__yl-N-methyI-2-(4-bromophenyl)-5-diethylamido-3-methylan_Jno- imidazo [1,2- ] pyridine.
A mixture of 2-(4-bromophenyl)-5-diethylamido-imidazo[l,2-α]pyridine (300 mg 0.806 mmol), paraformaldehyde (26.0 mg 0.812 mmol) and benzylmethylamine(100 μL 0.806 mmol) in acetic acid (2 mL) was heated for lh at 50 °C. The majority of the solvent was removed in vacuo and the mixture rediluted with EtOAc (200 mL) and washed with 2M ΝaOH (aq) (2 x 150mL). The solution was dried (MgSO ) and concentrated in vacuo. Flash column chromatograpy (silica gel, slow gradient neat CH2C12 to 6% MeOH) gave the title compound as an yellow oil (276 mg 68%). HCI salt of title compound was prepared by the addition of l.OM HCI in diethyl ether (150 μL) to a solution of the title compound in EtOAc (500 μL). The salt was precipitated with diethyl ether and collected by centrifuge.
Following a procedure similar to that described in Example 1, the following compounds were prepared.
Example C - Preparation of Ethyl N-benzyl-N-methyl-5-(3-acetamidophenyI)-3- methylamino-imidazo f 1 ,2-a) pyridine-2-carboxyIate.
Step CI : Ethyl 5-bromo-imidazo[l,2-g1pyridine-2-carboxylate.
A mixture of 2-amino-5-bromopyridine (1.00 g 5.78 mmol) and ethyl bromopyruvate
(0.730 mL 5.78 mmol) in DMF (10 mL) was heated at 80 °C for 2 h. The mixture was cooled to RT then partitioned between water (200 mL) and EtOAc (250mL). The aqueous layer was extracted again with EtOAc (2 x lOOmL) and the combined extracts dried
(MgSO4) and concentrated in vacuo to yield the crude title compound as a yellow solid
(1.24 g 80%).
Mass Spectrum: m/e C10H9BrΝ2O2 (EP+) (MH+) 264 and 271 found. *H NMR spectrum (DMSO-d6): δ !H NMR (300 MHz, D6 -DMSO) 1.31 (3H, t), 4.31
(2H, q); 7.46 (IH, d); 7.61 (IH, d); 7.33 (2H, d); 8.45 (IH, s); 8.89 (IH, s).
Step C2: Ethyl N-Ben__yl-N-methyl-5-bromo-3-methylan __no-_n_idazo[l,2-αlpyridine-2- carboxylate.
A mixture of ethyl 5-bromo-imidazo[l,2-_z]pyridine-2-carboxylate (1.24 g 4.61 mmol), paraformaldehyde (138 mg 4.61 mmol) and benzylmethylamine(600 μL 4.61 mmol) in acetic acid (15 mL) was heated for lh at 50°C. The majority of the solvent was removed in vacuo and the mixture rediluted with EtOAc (250 mL) and washed with 2M ΝaOH (aq) (3 x 150mL). The solution was dried (MgSO4) and concentrated in vacuo. Flash column
chromatograpy (silica gel, slow gradient neat CH2C12 to 10% MeOH) gave the title compound as an yellow oil (980 mg 53%).
Mass Spectrum: m e Cι9H20BrN3O2 (EP+) (MH+) 264 and 271 found.
1H NMR spectrum (DMSO-dfi): δ Η NMR (300 MHz, D6 -DMSO) 1.32 (3H, t); 2.08
(3H, s); 3.58 (2H, s); 4.22 (2H, s); 4.33 (2H, q); 7.21 - 7.34 (5H, m); 7.50 (IH, d); 7.59
(lH, d); 8.57 (lH, s).
Step C3: Ethyl N-ben2yl-N-methyl-5-(3-acetamidophenyI)-3-methyIamino- imidazo[l,2-a1pyridine-2-carboxylate.
Tetrakis(triphenylphosphine) palladium(O) (58.0 mg 0.050 mmol) was added in one portion to a degassed mixture of ethyl N-Benzyl-N-methyl-5-bromo-3-methylamino- imidazo[l,2-fl]pyridine-2-carboxylate (200 mg 0.498 mmol) and Ν-acetyl-3- aminobenzeneboronic acid (89.0 mg 0.498 mmol) in toluene (2 mL), ethanol (2 mL) and saturated NaHCO3 (aq) (1 mL). The mixture was heated at 80°C_with vigorous stirring for 4 h then cooled to RT. The mixture was diluted with EtOAc (200 mL) and washed with water (100 mL) and brine (100 mL) then dried (MgSO4). The solution was concentrated in vacuo and the product isolated by flash column chromatography (3 runs on silica slow gradient neat CH2C1 to 20% MeOH). This gave the title compound as a colourless oil (27.0 mg 12%).
Mass Spectrum: m/e C27H28N4O3 (EP+) (MH+) 456 found.
1H NMR spectrum (DMSO-d6): δ Η NMR (300 MHz, CDC13) 1.46 (3H, t); 2.21 (3H, s); 2.25 (3H, s); 3.61 (2H, s); 4.23 (2H, s); 4.48 (2H, q); 7.15 - 7.29 (5H, m); 7.29 - 7.37 (IH, m); 7.38 - 7.49 (3H, m); 7.76 (IH, s); 7.89 (IH, s); 8.39 (IH, s). HCI salt of title compound was prepared by the addition of 1.0M HCI in diethyl ether (177 μL) to a solution of the title compound in EtOAc (300 μL). The salt was precipitated with diethyl ether and collected by centrifuge.
Example Dl - Preparation of N-Cyano-N'- -dH-indol-S-y^-imidazofl^- lpyridin^- ylmethyπ-N* '-methyl-guanidine
Step Dl: Preparation of l-Imidazo[l,2-a]pyridin-2-ylmethyl-3-cyano-2-phenyl- isourea
A mixture of 2-aminomethylimidazo[l,2-α]pyridine (1.20 g, 9.00 mmol) and diphenyl- cyanocarbonimidate (2.35 g, 9.90 mmol) in IP A were stirred at ambient temperature for 4 h. The cloudy reaction gave rise to a precipitate which was filtered. This was washed with ether and dried to yield the title compound as a white solid (1.55 g, 59 %). Mass Spectrum: 292 [MH]+
Step D2: Preparation of N-Cyano-N'-Qmidazo [1,2-α] pyridin^-ylmethy^-N' '-methyl- guanidine
A mixture of l-imidazo[l,2-α]pyridin-2-ylmethyl-3-cyano-2-phenyl-isourea (1.00 g, 3.40 mmol) and excess methylamine in 33% aq. ethanol (5 mL) in IP A were warmed to 70 °C for 2 h. The reaction was concentrated in vacuo, and the residue triturated with ethyl acetate and filtered. The resulting solid was washed with ether and dried to give the title compound as a white solid (0.75 g, 97%). Mass Spectrum: 229 [MH]+
*H ΝMR spectrum (DMSO-d6): 2.72 (3H, s); 4.40 (2H, s); 6.82 (IH, t); 7.17 (IH, brs), 7.19 (IH, t); 7.40 (IH, br s); 7.48 (IH, d); 7.78 (IH, s); 8.48 (IH, d).
Step D3: N-Cyano-N,-(3-bromo-imidazo[l,2-αlpyridin-2-ylmethyl)-N"-methyl- guanidine
A mixture of N-Cyano-N'-(imidazo[ 1 ,2-_z]pyridin-2-ylmethyl)-N' -methyl-guanidine
(0.300 g, 1.30 mmol), poly(4-vinylpyridinium tribromide) (0.467 g, 1.40 mmol), pyridine (2 drops) in CH2C12 were stirred at ambient temperature for 16 h. DMF was added, the solid support filtered off, and the mother liquors concentrated in vacuo. The residue was triturated with CH2C12 and the resulting solid filtered to yield the title compound as a fawn solid (0.390 g, 98%).
Mass Spectrum: 307, 309 [MH]+
XH ΝMR spectrum (DMSO-d6): 2.76 (3H, d); 4.46 (2H, d); 7.21 (IH, d); 7.24 (IH, t);
7.45 (IH, t); 7.58 (IH, t); 7.76 (IH, d); 8.45 (IH, d)
Step D4: Preparation of N-Cyano-N 3-αH-indo_-5-ylHmidazo[l,2-..lpyrid_n-2- ylmethyll-N' '-methyl-guanidine
To a mixture of N-Cyano-N '-(3 -bromo-imidazo[l,2-α]pyridin-2-ylmethyl)-N "-methyl- guanidine (700 mg, 0.230 mmol), 5-indolyl boronic acid (560 mg, 0.345 mmol), saturated Νa2CO3 (1.5 mL), ethanol (0.60 mL) and toluene (3 mL) was added a catalytic amount of Pd(PPh3)4. The reaction was stirred at 80 °C for 16 h. The reaction was poured onto a hydromatrix column and eluted with CH2C12. Flash column chromatography (silica gel, slow gradient, neat CH2C12 to 5% MeOH:CH2Cl2) gave the title compound as a white solid (17.0 mg, 22%). Mass Spectrum: 344 [MH]+
1H NMR spectrum (DMSO-d6): 2.89 (3H, d); 4.02 (2H, d); 5.85 (IH, t); 6.63 (IH, s); 6.88 (IH, t); 7.17- 7.27 (3H, m); 7.36 (IH , m); 7.57 (IH, d); 7.59 (IH, d); 7.66 (IH, s), 8.08 (IH, d); 8.78 (IH, br s)
Following a procedure similar to that described for Example Dl, the following compounds were prepared.
D2 =N-Cyano-N'-[3-(3,4-dimethoxyphenyl)-imidazo[l,2-α]pyridin-2-ylmethyl]-N"- methyl-guanidine
D3 =N-Cyano-N,-[3-(4-chlorophenyI)-imidazo[l,2- ]pyridin-2-yImethyI]-N,,-methyl- guanidine
D4 =N-Cyano-N'-[3-(3,5-dimethylphenyl)-imidazo[l,2- ]pyridin-2-ylmethyl]-N"- methyl-guanidine
Example E - N>N-Dibenzyl-2-methylamino-3-(3,4-dimethoxyphenyI)imidazo[l,2- a\ pyridine
Step El: Preparation of N,N-Dibenzyl-2-methylam_noimidazo f 1,2-g] pyridine
A mixture of 2-aminoethylimidazo[l,2- ]pyridine dihydrochloride (0.500 g, 2.26 mmol), benzyl bromide (0.430 g, 2.50 mmol) and powdered 2CO3 (1.56 g, 11.3 mmol) in DMF
(20 mL) was heated at 100 °C for 2 h. The DMF was removed in vacuo and then residue taken into CH2C12 and filtered to remove inorganics. This was purified by flash column chromatography (silica gel, CH2C12 to 5% MeOH:CH2Cl2) to give the title compound as orange oil (0.405 g, 55%).
Mass Spectrum: 328 [MH]+
1H ΝMR spectrum (CDCI3): 3.68 (4H, s); 3.82 (2H, s); 6.74 (IH, t); 7.10 (IH, t); 7.18- 7.48 (10H, m); 7.55 (IH, d); 7.60 (IH, s); 8.07 (IH, d)
Step E2: Preparation of NN-Dibeiizyl^-methylamino-S-bromoimidazo [1,2- ] pyridine
A mixture ofN,N-Dibenzyl-2-methylanιinoimidazo[l,2-α]pyridine (0.380 g, 1.16 mmol), poly(4-vinylpyridinium tribromide) (0.387 g, 1.16 mmol), pyridine (2 drops) in CH2C12 (20 mL) were stirred at ambient temperature for 16 h. The solid support was filtered off
and the mother liquors concentrated in vacuo. The residue was purified by flash column chromatography (silica gel, slow gradient, CH2C12 to 3:7 EtOAc:Hexane to 6:4
EtOAc:Hexane) to yield the title compound as a yellow oil (0.260 g, 55%).
Mass Spectrum: 406, 408 [MH]+
1H NMR spectrum (CDC13): 3.70 (4H, s); 3.82 (2H, s); 6.90 (IH, t); 7.17-7.48 (1 IH, m);
7.58 (IH, d), 8.08 (IH, d)
Step E3: Preparation of N,N-DibenzyI-2-methylamino-3-(3,4- dimethoxyphenyl)imidazo [1,2- ] pyridine
To a mixture of N,N-Dibenzyl-2-methylamino-3-bromoimidazo[l,2-α]pyridine (150 mg, 0.370 mmol), 3,4-dimethoxybenzene boronic acid (81.0 mg, 0.440 mol), saturated Νa2CO3 (2.5 mL), ethanol (0.90 mL) and toluene (4.50 mL), was added a catalytic amount of Pd(PPh3)4. The reaction was stirred at 80 °C for 3 h. The toluene layer was separated and concentrated in vacuo. Flash column chromatography (silica gel, slow gradient, neat
CH2C12 to 3:7EtOAc:Hexane to 7:3 EtOAσ.Hexane) gave the title compound as an oil. The HCI salt was prepared by addition of IM HCI in diethyl ether to a solution of the compound in EtOAc to give the title compound dihydrochloride as a white solid (98.0 mg, 49%). Mass Spectrum: 464 [MH]+
1H NMR spectrum (DMSO-dtf): 3.75 (3H, s); 3.88 (3H, s); 4.08 (4H, br s); 4.15 (2H, brs); 7.05-7.19 (3H, m); 7.30 (7H, s); 7.46 (4H, s); 7.84 (IH, t); 8.00 (IH, d); 8.43 (IH, d)
Example F - Preparation of N,N-Dibenzyl-2-methyIamino-3-(4- dimethoxyphenyl)imidazo[l,2-a1pyridine
To a mixture of N,N-Dibenzyl-2-methylamino-3-bromoimidazo[l,2-ύ;]pyridine (lOOmg, 0.25mmol), 4-methoxybenzene boronic acid (76.0 mg, 0.500 mol), saturated Νa2CO3 (1.5 mL), ethanol (0.6 mL) and toluene (3 mL), was added a catalytic amount of Pd(PPh3)4. The reaction was stirred at 80 °C for 3 h.
The toluene layer was separated and concentrated in vacuo. Flash column chromatography (silica gel, slow gradient, neat CH2C12 to 3:7 EtOAc:Hexane to 7:3 EtOAc:Hexane) gave the title compound as an oil. The HCI salt was prepared by addition of IM HCI in diethyl ether to a solution of the compound in EtOAc to give the title compound dihydrochloride as a white solid (41.0 mg, 32%) Mass Spectrum: 434 [MH]+
Example G - Preparation of N-benzyl-N-methyl-2-methyIamino-3-(4- chlorophenyl)imidazo f 1,2-aj pyridine
Step Gl: Preparation of 2-chloromethylimidazo[l,2- 1pyridine
A mixture of dichloroacetone (17.8 g, 14.0 mmol) and 2-aminopyridine (10.0 g , 11.0 mmol) in DMF (80 mL) was stirred at ambient temperature for 5 h. The resulting precipitate was filtered off and washed with DMF and then diethyl ether. This solid was then taken up in DMF (100 mL), 4A molecular sieves added and the reaction stirred at 80
°C for 3 h. The resulting precipitate was filtered off and washed with diethyl ether to yield the title compound as a white solid.
Mass Spectrum: 167 [MH]+
1H ΝMR spectrum (DMSO-d ): 5.11 (2H, s); 7.48 (IH, t); 7.95 (2H, s); 8.49 (IH, s); 8.97 (lH, d).
Step G2: Preparation of N-benzyl-N-methyl-2-methylaminoin_idazo[l,2- ]pyridine
A mixture of 2-chloromethylimidazo[l,2- ]pyridine (2.00 g, 12.0 mmol), N- methylbenzylamine (1.75 g, 14.5 mmol) and powdered 2CO3 (3.30 g, 2.40 mmol) in DMF (50 mL) was heated at 90 °C for 4 h. After cooling, the inorganics were filtered off and the filtrate evaporated in vacuo. Purification by flash column chromatography (silica gel,
slow gradient, CH2C12 to 5% MeOH:CH2Cl2) gave the title compound as a yellow oil (1.42 g, 48%).
Mass Spectrum: 252 [MH]+
1H NMR spectrum (DMSO-d6): 2.30 (3H, s); 3.63 (2H, s); 3.79 (2H, s); 6.73 (IH, t); 7.11 (IH, t); 7.20-7.42 (5H, ); 7.55 (IH, s); 7.57 (IH, d); 8.06 (lH,d)
Step G3: Preparation of N-benzyl-N-methyl-2-methylamino-3-bromoimidazo[l,2- a\ pyridine
A mixture of N-benzyl-N-methyl-2-methylaminoimidazo[l,2-α]pyridine (1.30 g, 5.20 mmol), poly(4-vinylpyridinium tribromide) (1.82 g, 5.50 mmol) and pyridine (2 drops) in CH2C12 (20 mL) were stirred at ambient temperature for 16 h. A further portion of poly (4-vinylpyridinium tribromide) (0.400 g, 1.20 mmol) was added and the reaction stirred for a further 16 h. DMF was added, the solid support filtered off, and the mother liquors concentrated in vacuo. The residue was triturated with diethyl ether, and filtered to yield the title compound as a cream solid (1.85 g, 99%). Mass Spectrum: 330, 332 [MH]+ rH ΝMR spectrum (DMSO-d6): 2.77 (3H, s); 4.39 (2H, s); 4.43 (2H, br s); 7.16 (IH, t); 7.42-7.50 (4H, m); 7.55-8.00 (2H, m); 7.69 (IH, d); 8.40 (IH, d)
Step G4: Preparation of N-benzyI-N-methyl-2-methylamino-3-(4- chloropheny_)imidazo[l,2-α1pyridine
To a mixture of N-benzyl-N-methyl-2-methylamino-3-bromoimidazo[l,2-_z]pyridine (200 mg, 0.60 mmol), 4-chlorobenzeneboronic acid (141 mg, 0.910 mmol) and saturated Νa2CO3 (3 mL) in ethanol (1.2 mL) and toluene (6 mL) was added a catalytic amount of Pd(PPh3)4. The reaction was stirred at 80 °C for 16 h. The reaction was poured onto a hydromatrix column and eluted with CH2C12. Flash column chromatography (silica gel, slow gradient, neat CH2C12 to 5% MeOH:CH2Cl2) gave an oil. The HCI salt was prepared by addition of IM HCI in diethyl ether to a solution of the compound in EtOAc to give the title compound as the dihydrochloride as a white solid (26mgs, 10%).
Mass Spectrum: 362 [MH]+
XH NMR spectrum (DMSO-d6): 2.72 (3H, s); 4.28 (2H, brs); 4.38 (2H, s); 7.18 (IH, t);
7.35-7.41 (3H, m); 7.50-7.57 (2H, m); 7.63 (5H ,s); 7.51 (IH, d); 8.34 (lH,d)
Example H: Preparation of N-benzyl-N-methyl-2-methylamino-3-(3,5- dimethylphenyl)imidazo [1 ,2-α] pyridine
To a mixture of N-benzyl-N-methyl-2-methylamino-3-bromoimidazo[l,2-α]pyridine (100 mg, 0.400 mmol), 3,5-dimethylbenzene boronic acid (89.0 mg, 0.600 mmol), and sodium carbonate (2 mL) in dioxan (4 mL) was added a catalytic amount of Pd(PPh3)4. The reaction was stirred at 90 °C for 16 h, then evaporated in vacuo. Purification by flash column chromatography (silica gel, slow gradient, neat CH2CI2 to 5% MeOH:CH2Cl_) gave an oil. The HCI salt was prepared by addition of IM HCI in diethyl ether to give the title compound dihydrochloride as a white solid (50.0 mg, 29%). Mass Spectrum: 356 [MH]+
Example J: Preparation of N-Benzyl-5-bromo-3-(3-methyIpropylamino)-2-(4- methoxyphenyl)imidazo [1,2- l pyridine
Step Jl: 5-bromo-2-(4-methoxyphenyl)imidazo[l,2- lpyridine
2-amino-5-brornopyridine (5.00 g, 28.9 mmol) was added to a solution of 2-bromo-4- methoxyacetophenone (6.62 g, 28.9 mmol) in DMF (50 mL), and the reaction stirred at 80 °C for 2h. The reaction mixture was partitioned between IM NaOH (200 mL) and ethyl acetate (200 mL), upon which the majority of the product precipitated from solution and was filtered under vacuum. The organic layer that remained was extracted, dried over magnesium sulfate and evaporated in vacuo to give a yellow solid, which was combined with the above to give the title compound (6.74 g, 77%). 1H NMR spectrum (DMSO-d6): 3.80 (s, 3H); 7.00 (d, 2H); 7.30 (d, IH); 7.50 (d, IH); 7.85 (d, 2H); 8.25 (s, IH); 8.80 (s, IH). Mass Spectrum: 303, 305 [MH]+
Step J2: 5-bromo-3-(3-oxo-butyl)-2-(4-methoxyphenyl)imidazo[l,2- lpyridine
Methyl vinyl ketone (1.00 mL, 12.0 mmol) was added to a solution of 5-bromo-2-(4- methoxypheny_)imidazo[l,2-α]pyridine (1.00 g 3.32 mmol) in glacial acetic acid (30 mL), followed by the addition of acetic anhydride (10 mL). The reaction was stirred at reflux for 14h. The reaction mixture was evaporated in vacuo and the crude product purified by
flash chromatography (silica gel, eluting from 25% ethyl acetate:hexane to 50% ethyl acetate:hexane) to give the title compound as a yellow solid (1.09 g, 88%). H NMR spectrum (DMSO-dfi): 2.10 (s, 3H); 2.85 (t, 2H); 3.10 (t, 2H); 3.80 (s, 3H); 7.00 (d, 2H); 7.25 (d, IH); 7.50 (d, IH); 7.65 (d, 2H); 8.70 (s, IH). s Mass Spectrum: 373, 375 [MH]+
Step J3: N-Benzyl-5-bromo-3-(3-methylpropylamino)-2-(4- methoxyphenyl)imidazo f 1,2-a] pyridine 0
Under an inert atmosphere, benzylamine (0.090 mL, 0.820 mmol) was added to a solution of 5-bromo-3-(3-oxo-butyl)-2-(4-methoxyphenyl)imidazo[l,2-α]pyridine (300 mg, 0.810 mmol) and toluene sulphonic acid (1 mg, catalytic amount) in anhydrous methanol (10 mL). The reaction mixture was then allowed to reflux over molecular sieves for 16 h. s Sodium borohydride (61.0 mg, 1.61 mmol) was added over a period of 30 min, and the reaction left to stir for 30 min. The reaction mixture was evaporated in vacuo, and purified by flash chromatography (silica gel, eluting with 1% MeOH: CH2C12 to 5% MeOH: CH2C12) to give the title compound as an oil. The hydrochloride salt was formed by addition of HCI in ether, and recrystallised from iso-propanol, to give the title compound 0 hydrochloride as a white solid (40.0 mg, 11%).
1H NMR spectrum (DMSO-dg): 1.35 (d, 3H); 1.80-2.00 (m, 2H); 2.15-2.25 (m, 2H); 3.75 (q, IH); 3.85 (s, 3H); 4.00-4.20 ( , 2H); 7.15 (d, 2H); 7.40 (m, 3H); 7.55 (m, 2H); 7.70 (d, 2H); 7.85-8.00 (dd, 2H); 9.20 (s, IH); 9.45 (s, IH). Mass Spectrum: 464, 466 [MH]+ 5
Example Kl: Preparation of N-Benzyl-N-Methyl-5-methyl-3-propargylan_ino-2-(4- methoxyphenyl)imidazo[l,2-a]pyridine
Step Kl: 5-methyl-2-(4-methoxyphenyl)imidazo[l,2-α]pyridine
2-amino-5-picoline (5.00 g, 46.3 mmol) was added to a solution of 2-bromo-4- methoxyacetophenone (10.6 g, 46.3 mmol) in DMF (50 mL), and the reaction stirred at 80 °C for 2h. The reaction mixture was partitioned between IM ΝaOH (200 mL) and ethyl acetate (200 mL), upon which the majority of the product precipitated from solution and was filtered under vacuum. The organic layer that remained was extracted, dried over magnesium sulfate and evaporated in vacuo to give a yellow solid, which was combined with the above to give the title compound (8.20 g, 74%). XH ΝMR spectrum (DMSO-d6): 2.25 (s, 3H); 3.80 (s, 3H); 6.95 (d, 2H); 7.05 (d, IH); 7.40 (d, IH); 7.85 (d, 2H); 8.15 (s, IH); 8.25 (s, IH). Mass Spectrum: 239 [MH]+
Step K2: 5-methyI-3-bromo-2-(4-methoxyphenyl)in-ldazofl,2- 1pyridine
Poly(4-vinylpyridinium tribromide) (2.80 g, 8.40 mmol) was added to a solution of 5- methyl-2-(4-methoxyρhenyl)imidazo[l,2-α]pyridine (2.00 g, 8.40 mmol) in dichloromethane, which was followed by the addition of a few drops of pyridine. The reaction was allowed to stir at room temperature for 14h. The reaction mixture was filtered by vacuum, washed with water (2 x 75 mL) and the organics separated, dried over
magnesium sulfate, filtered and evaporated in vacuo to give the title compound as a pale yellow solid (1.64 g, 62%).
1H NMR spectrum (DMSO-d6): 2.25 (s, 3H); 3.80 (s, 3H); 6.95 (d, 2H); 7.05 (d, IH); 7.40 (d, IH); 7.85 (d, 2H); 8.15 (s, IH). Mass Spectrum: 317, 319 [MH]+
Step K3: N-Benzyl-N-Methy_-5-methyl-3-propargylamino-2-(4- methoxyphenyl)imidazofl,2-«1pyridine
Pargyline hydrochloride (202 mg, 1.26 mmol) was added to a solution of 5-methyl-3- bromo-2-(4-methoxyphenyl)imidazo[l,2-α]pyridine (20mg, 0.63mmol) in diethylamine (15 mL), and the solution bubbled with nitrogen. Copper Iodide (12.0 mg, 0.060 mmol) was added followed by the addition of bis(triphenylphosphine)palladium dichloride (22.0 mg, 0.030 mmol), and the solution again bubbled with nitrogen. The reaction was then stirred at reflux for 3h, and then at 55 °C for 10 h. The reaction mixture was added to water (50 mL) and extracted with dichloromethane (2 x 50 mL). The organics were dried over magnesium sulfate, filtered, evaporated in vacuo and purified by flash chromatography (silica gel, eluting from 10% ethyl acetate:hexane to 40% ethyl acetate:hexane) to give the product and the hydrochloride salt was formed with HCl/ether to give the title compound as an off white solid (37.0 mg, 14%).
*H ΝMR spectrum (CDC13): 2.40 (s, 3H); 2.50 (s, 3H); 3.75 (s, 2H); 3.80 (s, 2H); 3.85 (s, 3H); 7.00 (d, 2H); 7.10 (d, IH); 7.25-7.70 (m, 6H); 8.10 (s, IH); 8.30 (d, 2H). Mass Spectrum: 396 [MH]+
Example K2: Preparation of N-(yff-methylphenethyl)-N-Methyl-5-methyl-3- propargylamino-2-(4-methoxyphenyl)imidazo [1,2-a] pyridine
L-Deprenyl (592 mg, 3.16 mmol) was added to a solution of 5-methyl-3-bromo-2-(4- methoxyphenyl)imidazo[l,2- ]pyridine (500 mg, 1.58 mmol), and the reaction bubbled with nitrogen. Copper iodide (600 mg, 0.320 mmol) and bis(triphenylphosphine)palladium dichloride (110 mg, 0.160 mmol) were added, and the reaction stirred at reflux for 24h. The reaction mixture was partitioned between water (75 mL) and dichloromethane (75 L), the organics extracted, dried over magnesium sulfate, filtered and evaporated in vacuo. The crude product was purified by flash chromatography (silica gel, eluting with 30% EtOAc:Hexane to 80% EtOAc:Hexane) to give an oil. The hydrochloride salt was formed by addition of HCl/ether to give the title compound dihydrochloride as a yellow solid (63mg, 9%). 1H ΝMR spectrum (DMSO-dfi): 1.20 (m, 3H); 2.40 (s, 3H); 2.75 (m, IH); 2.95 (s, 3H); 3.45 (s, 2H); 3.80 (s, 3H); 4.70 (m, 2H); 7.00 (d, 2H); 7.20-7.65 (m, 7H); 8.20 (d, 2H); 8.70 (s, IH). Mass Spectrum: 424 [MH]+
Example L - Preparation of N-BenzyI-N-methyl-2-(3,5-dimethylphenyl)-3- ethylamino-5-methylimidazo [1 ,2-α] pyridine.
Step Ml: 4-Chloro-2-bromopropyl-3,5-dimethylphenyl ketone.
Pyridinium tribromide (4.56 g 14.3 mmol) was added in one portion to a stirred solution of 4-chloropropyl-3,5-dimethylphenyl ketone (3.00 g 14.3 mmol) [synthesised as described in WO 98/55123] in CH2C12 (30 mL) at RT and the mixture stirred for 2h. The brown solution was then diluted with ether (200 mL) and washed with 20% NaS2O3 (aq) (150 mL), 2M HCI (200 mL) and brine (200 mL). The solution was dried (MgSO4) and concentrated in vacuo to give the crude title compound as a brown oil (4.11 g 99%).
Step M2: 2-(3,5-dimethylphenyl)-3-ethylamino-5-methylin_idazo [1 ,2-αl pyridine.
A mixture of 2-amino-5-methylpyridine (1.08 g 10.0 mmol) and 4-Chloro-2-bromopropyl- 3,5-dimethylphenyl ketone (2.90 g 10.0 mmol) in DMF (15 mL) was heated overnight at 80 °C. The mixture was partitioned EtOAc (50 mL) and saturated NaHCO3 (150mL). The aqueous was extracted with EtOAc (5 x 25 mL) and the combined organics washed with water (25 mL) and brine (25 mL). The solution was dried (MgSO4) and concentrated in vacuo. Flash column chromatograpy (silica gel, slow gradient neat iso-h.exan.QS to 50% EtOAc) gave the title compound as a yellow gum (610 mg 20%). HCI salt of title compound was prepared by the addition of 1.0M HCI in diethyl ether to a solution of the
title compound in EtOAc. The salt was precipitated with diethyl ether and collected by centrifuge.
Mass Spectrum: m/e C18H20C1N2(M+H) 299.16. XH NMR spectrum (CDCI,): δ >H NMR (300 MHz) 2.33 - 2.50 (9H, m), 3.50 - 3.65 (2H, t); 3.70 - 3.80 (2H, t); 6.90 - 7.10 (2H, m); 7.36 (2H, s); 7.55 (IH, d); 7.70 - 7.85 (IH, m).
Step M2: N-Benzyl-N-methyl-2-(3,5-dimethylphenyl)-3-ethylamino-5- methy_imidazo[l,2-α1 pyridine.
N-Methyl-N-benzylamine (95 μL 0.737 mmol) was added in one portion to a stirred solution of 2-(3,5-dimethylphenyl)-3-ethylamino-5-methylimidazo[l,2-α]pyridine (200 mg 0.670 mmol) and di-isσpropylethylamine (128 mL 0.736 mmol) in DMF (25 mL) was heated overnight at 100°C. The mixture was partitioned EtOAc (2 x 50 mL) and saturated ΝaHCO3 (250 L) and the combined organics were dried (MgSO4) and concentrated in vacuo. Flash column chromatograpy (silica gel, slow gradient neat iso-hexanes to 100% EtOAc) gave the title compound as a yellow gum (135 mg 44%). HCI salt of title compound was prepared by the addition of 1.0M HCI in diethyl ether to a solution of the title compound in EtOAc. The salt was precipitated with diethyl ether and collected by centrifuge.
Mass Spectrum: m/e (M+H) 384.67.
1H NMR spectrum (DMSO-d6+ CD3COOD): δ Η NMR (300 MHz) 2.35 (6H, s), 2.75 (3H, s), 3.30 - 3.45 (2H, m); 3.65 - 3.85 (2H, m); 4.20 - 4.50 (2H, m), 7.20 (IH, s); 7.30 (2H, s); 7.35 - 7.45 (3H, m); 7.50 - 7.62 (2H, m); 7.70 - 7.85 (2H, m); 9.05 (IH, s).
THERAPEUTIC USES
Compounds of formula I are provided as medicaments for antagonising gonadotropin releasing hormone (GnRH) activity in men and women. To this end, a compound of formula I can be provided as part of a pharmaceutical formulation which also includes a pharmaceutically acceptable diluent or carrier (eg, water). The formulation may be in the form of tablets, capsules, granules, powders, syrups, emulsions (eg, lipid emulsions), suppositories, ointments, creams, drops, suspensions (eg, aqueous or oily suspensions) or solutions (eg, aqueous or oily solutions). If desired, the formulation may include one or more additional substances independently selected from stabilising agents, wetting agents, emulsifying agents, buffers, lactose, sialic acid, magnesium stearate, terra alba, sucrose, corn starch, talc, gelatin, agar, pectin, peanut oil, olive oil, cacao butter and ethylene glycol.
The compound is preferably orally administered to a patient, but other routes of administration are possible, such as parenteral or rectal administration. For intravenous, subcutaneous or intramuscular administration, the patient may receive a daily dose of O.lmgkg"1 to 30mgkg_1 (preferably, 5mgkg_1 to 20mgkg"1) of the compound, the compound being administered 1 to 4 times per day. The intravenous, subcutaneous and intramuscular dose may be given by means of a bolus injection. Alternatively, the intravenous dose may be given by continuous infusion over a period of time. Alternatively, the patient may receive a daily oral dose which is approximately equivalent to the daily parenteral dose, the composition being administered 1 to 4 times per day. A suitable pharmaceutical formulation is one suitable for oral administration in unit dosage form, for example as a tablet or capsule, which contains between lOmg and lg (preferably, 100 mg and lg) of the compound of the invention.
The following illustrate representative pharmaceutical dosage forms containing a compound of the invention, or a pharmaceutically acceptable salt or solvate thereof (hereafter referred to as "compound X"), for use in humans.
(a)
( )
Buffers, pharmaceutically acceptable cosolvents (eg, polyethylene glycol, propylene glycol, glycerol or EtOH) or complexing agents such as hydroxy-propyl β cyclodextrin may be used to aid formulation.
One aspect of the invention relates to the use of compounds according to the invention for reducing the secretion of LH and/or FSH by the pituitary gland of a patient. In this respect, the reduction may be by way of a reduction in biosynthesis of the LH and FSH and/or a reduction in the release of LH and FSH by the pituitary gland. Thus, compounds according to the invention can be used for therapeutically treating and/or preventing a sex hormone related condition in the patient. By "preventing" we mean reducing the patient's risk of contracting the condition. By "treating" we mean eradicating the condition or reducing its severity in the patient. Examples of sex hormone related conditions are: a sex hormone dependent cancer, benign prostatic hypertrophy, myoma of the uterus, endometriosis, polycystic ovarian disease, uterine fibroids, prostatauxe, myoma uteri, hirsutism and precocious puberty. Examples of sex hormone dependent cancers are: prostatic cancer, uterine cancer, breast cancer and pituitary gonadotrophe adenoma.
ASSAYS
The ability of compounds according to the invention to act as antagonists of GnRH can be determined using the following in vitro assays.
Binding Assay Using Rat pituitary GnRH Receptor
The assay is performed as follows :-
1. Incubate crude plasma membranes prepared from rat pituitary tissues in a Tris.HCl buffer (pH. 7.5, 50 mM) containing bovine serum albumin (0.1%), [I-125]D-t-Bu-Ser6- o Pro9-ethyl amide-GnRH, and the test compound. Incubation is at 4 C for 90 minutes to 2 hours.
2. Rapidly filter and repeatedly wash through a glass fibre filter.
3. Determine the radioactivity of membrane bound radio-ligands using a gamma counter.
From this data, the IC50 of the test compound can be determined as the concentration of the compound required to inhibit radio-ligand binding to GnRH receptors by 50%.
Binding Assay Using Human GnRH Receptor
Crude membranes prepared from CHO cells expressing human GnRH receptors are sources for the GnRH receptor. The binding activity of compounds according to the invention can be determined as an IC50 which is the compound concentration required to
125 125 inhibit the specific binding of [ I]buserelin to GnRH receptors by 50%. [ I]Busereli (a peptide GnRH analogue) is used here as a radiolabelled ligand of the receptor.
Assay to Determine Inhibition of LH release
The LH release assay can be used to demonstrate antagonist activity of compounds, as demonstrated by a reduction in GnRH-induced LH release.
Preparation of Pituitary Glands
Pituitary glands obtained from rats are prepared as follows. Suitable rats are Wistar male rats (150-200g) which have been maintained at a constant temperature (eg, 25°C) on a 12 hour light/ 12 hour dark cycle. The rats are sacrificed by decapitation before the pituitary glands are aseptically removed to tube containing Hank's Balanced Salt Solution (HBSS). The glands are further processed by:-
1. Centrifugation at 250 x g for 5 minutes;
2. Aspiration of the HBSS solution;
3. Transfer of the glands to a petri dish before mincing with a scalpel; 4. Transfer of the minced tissue to a centrifuge tube by suspending the tissue three successive times in 10 ml aliquots of HBSS containing 0.2% collagenase and 0.2% hyaluronidase; 5. Cell dispersion by gentle stirring of the tissue suspension while the tube is kept in a water bath at 37°C; 6. Aspiration 20 to 30 times using a pipette, undigested pituitary fragments being allowed to settle for 3 to 5 minutes;
7. Aspiration of the suspended cells followed by centrifugation at 1200 x g for 5 minutes;
8. Resuspension of the cells in culture medium of DMEM containing 0.37% NaHCO3, 10% horse serum, 2.5% foetal bovine serum, 1% non essential amino acids, 1% glutamine and 0.1 % gentamycin;
9. Treatment of the undigested pituitary fragments 3 times with 30 ml aliquots of the collagenase and hyaluronidase;
10. Pooling of the cell suspensions and dilution to a concentration of 3 x 10 cells/ml;
11. Placing of 1.0ml of this suspension in each of a 24 well tray, with the cells being maintained in a humidified 5% CO2/95% air atmosphere at 37°C for 3 to 4 days
Testing of Compounds
The test compound is dissolved in DMSO to a final concentration of 0.5% in the incubation medium.
1.5 hours prior to the assay, the cells are washed three times with DMEM containing 0.37% NaHCO3, 10% horse serum, 2.5% foetal bovine serum, 1% non essential amino acids (100X), 1% glutamine (100X), 1% penicillin/streptomycin (10,000 units of each per ml) and 25 mM HEPES at pH 7.4. Immediately prior to the assay, the cells are again washed twice in this medium .
Following this, 1ml of fresh medium containing the test compound and 2nM GnRH is added to two wells. For other test compounds (where it is desired to test more than one compound), these are added to other respective duplicate wells. Incubation is then carried out at 37°C for three hours.
Following incubation, each well is analysed by removing the medium from the well and centrifuging the medium at 2000 x g for 15 minutes to remove any cellular material. The supernatant is removed and assayed for LH content using a double antibody radio-immuno assay. Comparison with a suitable control (no test compound) is used to determine whether the test compound reduces LH release. Compounds according to the present invention have activity at a concentration from InM to 30 μM.