WO2002072858A2 - Degradation of epothilones and ethynyl substituted epothilones - Google Patents
Degradation of epothilones and ethynyl substituted epothilones Download PDFInfo
- Publication number
- WO2002072858A2 WO2002072858A2 PCT/EP2002/002105 EP0202105W WO02072858A2 WO 2002072858 A2 WO2002072858 A2 WO 2002072858A2 EP 0202105 W EP0202105 W EP 0202105W WO 02072858 A2 WO02072858 A2 WO 02072858A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- epothilone
- scheme
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/22—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D277/24—Radicals substituted by oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D493/00—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system
- C07D493/02—Heterocyclic compounds containing oxygen atoms as the only ring hetero atoms in the condensed system in which the condensed system contains two hetero rings
- C07D493/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/188—Preparation; Treatments not provided for in C07F7/20 by reactions involving the formation of Si-O linkages
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
- C07F7/1872—Preparation; Treatments not provided for in C07F7/20
- C07F7/1892—Preparation; Treatments not provided for in C07F7/20 by reactions not provided for in C07F7/1876 - C07F7/1888
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/14—Nitrogen or oxygen as hetero atom and at least one other diverse hetero ring atom in the same ring
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P17/00—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms
- C12P17/18—Preparation of heterocyclic carbon compounds with only O, N, S, Se or Te as ring hetero atoms containing at least two hetero rings condensed among themselves or condensed with a common carbocyclic ring system, e.g. rifamycin
- C12P17/181—Heterocyclic compounds containing oxygen atoms as the only ring heteroatoms in the condensed system, e.g. Salinomycin, Septamycin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P7/00—Preparation of oxygen-containing organic compounds
- C12P7/62—Carboxylic acid esters
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T436/00—Chemistry: analytical and immunological testing
- Y10T436/14—Heterocyclic carbon compound [i.e., O, S, N, Se, Te, as only ring hetero atom]
- Y10T436/141111—Diverse hetero atoms in same or different rings [e.g., alkaloids, opiates, etc.]
Definitions
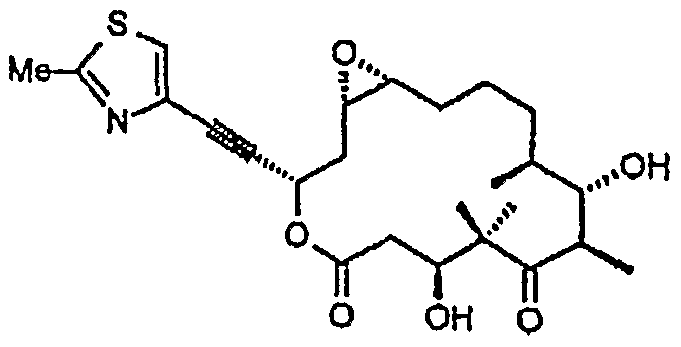
- the invention concerns a process for a degradation of an epothilone C or an epothilone D, wherein an epothilone C or an epothilone D is subjected to an olefin metathesis in the presence of ethylene and subsequently an optional ester hydrolysis (scheme I) .
- the epothilone C or D can be a fermentation product.
- the invention concerns a process for the production of an epothilone of formula 9
- reaction mixture was diluted with a saturated solution of NaHC0 3 (10 mL) .
- the aqueous layer was extracted with ether and the combined organic extracts were washed with a brine and dried over MgS0 4 .
- Concentration under vacuum, and flash column chromatography (silica gel, 10:1 petroleum ether/ethyl acetate), yielded 0.52 g (70.6%) a yellow oil.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Microbiology (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Silicon Polymers (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

Claims

Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA03007466A MXPA03007466A (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones. |
| NZ527557A NZ527557A (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epothilones |
| HU0303895A HUP0303895A3 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones |
| KR10-2003-7011281A KR20030084952A (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epotilones |
| DE60211124T DE60211124T2 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones |
| IL15731202A IL157312A0 (en) | 2001-02-27 | 2002-02-27 | Processes for the preparation of epothilone derivatives and compounds produced thereby |
| US10/468,919 US7157595B2 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones |
| BR0207675-6A BR0207675A (en) | 2001-02-27 | 2002-02-27 | Epothilone Degradation |
| JP2002571909A JP2004522801A (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilone |
| PL02362556A PL362556A1 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epothilones |
| EP02719946A EP1364040B1 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epothilones |
| CA002437707A CA2437707A1 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epothilones |
| NO20033784A NO20033784D0 (en) | 2001-02-27 | 2003-08-26 | Degradation of epothilones |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01104448 | 2001-02-27 | ||
| EP01104448.4 | 2001-02-27 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2002072858A2 true WO2002072858A2 (en) | 2002-09-19 |
| WO2002072858A3 WO2002072858A3 (en) | 2002-12-19 |
Family
ID=8176579
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/002105 Ceased WO2002072858A2 (en) | 2001-02-27 | 2002-02-27 | Degradation of epothilones and ethynyl substituted epothilones |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US7157595B2 (en) |
| EP (2) | EP1564217B1 (en) |
| JP (1) | JP2004522801A (en) |
| KR (1) | KR20030084952A (en) |
| CN (1) | CN1501980A (en) |
| AT (2) | ATE335746T1 (en) |
| BR (1) | BR0207675A (en) |
| CA (1) | CA2437707A1 (en) |
| CZ (1) | CZ20032291A3 (en) |
| DE (2) | DE60213884T2 (en) |
| HU (1) | HUP0303895A3 (en) |
| IL (1) | IL157312A0 (en) |
| MX (1) | MXPA03007466A (en) |
| NO (1) | NO20033784D0 (en) |
| NZ (2) | NZ527557A (en) |
| PL (1) | PL362556A1 (en) |
| RU (1) | RU2003128953A (en) |
| WO (1) | WO2002072858A2 (en) |
| ZA (1) | ZA200306034B (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6204388B1 (en) | 1996-12-03 | 2001-03-20 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| CA2273083C (en) | 1996-12-03 | 2012-09-18 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
| DE19826988A1 (en) * | 1998-06-18 | 1999-12-23 | Biotechnolog Forschung Gmbh | Epothilone minor components |
| US20020058286A1 (en) * | 1999-02-24 | 2002-05-16 | Danishefsky Samuel J. | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US7649006B2 (en) | 2002-08-23 | 2010-01-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| US6921769B2 (en) | 2002-08-23 | 2005-07-26 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto and analogues thereof |
| EP2186811A1 (en) | 2002-08-23 | 2010-05-19 | Sloan-Kettering Institute For Cancer Research | Synthesis of epothilones, intermediates thereto, analogues and uses thereof |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0057736B1 (en) | 1981-02-05 | 1986-11-20 | Phillips Petroleum Company | Olefin disproportionation process and catalyst |
| JP4183099B2 (en) | 1995-11-17 | 2008-11-19 | ゲゼルシャフト・フュア・ビオテヒノロジッシェ・フォルシュング・ミット・ベシュレンクテル・ハフツング(ゲー・ベー・エフ) | Epothilones C and D, production methods and compositions |
| AU716610B2 (en) * | 1996-08-30 | 2000-03-02 | Novartis Ag | Method for producing epothilones, and intermediate products obtained during the production process |
| US5969145A (en) * | 1996-08-30 | 1999-10-19 | Novartis Ag | Process for the production of epothilones and intermediate products within the process |
| ES2312695T3 (en) | 1996-11-18 | 2009-03-01 | Gesellschaft Fur Biotechnologische Forschung Mbh (Gbf) | EPOTILONES E AND F. |
| US6380394B1 (en) * | 1996-12-13 | 2002-04-30 | The Scripps Research Institute | Epothilone analogs |
| US6441186B1 (en) * | 1996-12-13 | 2002-08-27 | The Scripps Research Institute | Epothilone analogs |
| JP2001513098A (en) * | 1997-02-25 | 2001-08-28 | ゲゼルシャフト フュア バイオテクノロギッシェ フォーシュンク エム ベー ハー(ゲー ベー エフ) | Epothilone with modified side chains |
| US6605599B1 (en) | 1997-07-08 | 2003-08-12 | Bristol-Myers Squibb Company | Epothilone derivatives |
| US6365749B1 (en) | 1997-12-04 | 2002-04-02 | Bristol-Myers Squibb Company | Process for the preparation of ring-opened epothilone intermediates which are useful for the preparation of epothilone analogs |
| US6498257B1 (en) | 1998-04-21 | 2002-12-24 | Bristol-Myers Squibb Company | 2,3-olefinic epothilone derivatives |
| US6211412B1 (en) * | 1999-03-29 | 2001-04-03 | The University Of Kansas | Synthesis of epothilones |
| US6518421B1 (en) | 2000-03-20 | 2003-02-11 | Bristol-Myers Squibb Company | Process for the preparation of epothilone analogs |
| US6593115B2 (en) * | 2000-03-24 | 2003-07-15 | Bristol-Myers Squibb Co. | Preparation of epothilone intermediates |
-
2002
- 2002-02-27 NZ NZ527557A patent/NZ527557A/en unknown
- 2002-02-27 AT AT04019590T patent/ATE335746T1/en not_active IP Right Cessation
- 2002-02-27 DE DE60213884T patent/DE60213884T2/en not_active Expired - Lifetime
- 2002-02-27 CA CA002437707A patent/CA2437707A1/en not_active Abandoned
- 2002-02-27 NZ NZ539204A patent/NZ539204A/en unknown
- 2002-02-27 CN CNA028055349A patent/CN1501980A/en active Pending
- 2002-02-27 IL IL15731202A patent/IL157312A0/en unknown
- 2002-02-27 MX MXPA03007466A patent/MXPA03007466A/en unknown
- 2002-02-27 KR KR10-2003-7011281A patent/KR20030084952A/en not_active Withdrawn
- 2002-02-27 AT AT02719946T patent/ATE325201T1/en not_active IP Right Cessation
- 2002-02-27 EP EP04019590A patent/EP1564217B1/en not_active Expired - Lifetime
- 2002-02-27 EP EP02719946A patent/EP1364040B1/en not_active Expired - Lifetime
- 2002-02-27 JP JP2002571909A patent/JP2004522801A/en not_active Withdrawn
- 2002-02-27 BR BR0207675-6A patent/BR0207675A/en not_active IP Right Cessation
- 2002-02-27 WO PCT/EP2002/002105 patent/WO2002072858A2/en not_active Ceased
- 2002-02-27 US US10/468,919 patent/US7157595B2/en not_active Expired - Lifetime
- 2002-02-27 CZ CZ20032291A patent/CZ20032291A3/en unknown
- 2002-02-27 DE DE60211124T patent/DE60211124T2/en not_active Expired - Lifetime
- 2002-02-27 HU HU0303895A patent/HUP0303895A3/en unknown
- 2002-02-27 RU RU2003128953/04A patent/RU2003128953A/en not_active Application Discontinuation
- 2002-02-27 PL PL02362556A patent/PL362556A1/en not_active Application Discontinuation
-
2003
- 2003-08-05 ZA ZA200306034A patent/ZA200306034B/en unknown
- 2003-08-26 NO NO20033784A patent/NO20033784D0/en not_active Application Discontinuation
Also Published As
| Publication number | Publication date |
|---|---|
| BR0207675A (en) | 2004-03-09 |
| NO20033784L (en) | 2003-08-26 |
| RU2003128953A (en) | 2005-03-10 |
| ATE335746T1 (en) | 2006-09-15 |
| HUP0303895A3 (en) | 2007-06-28 |
| WO2002072858A3 (en) | 2002-12-19 |
| NO20033784D0 (en) | 2003-08-26 |
| HUP0303895A2 (en) | 2004-03-29 |
| EP1364040B1 (en) | 2006-05-03 |
| EP1564217A3 (en) | 2005-08-31 |
| DE60213884T2 (en) | 2007-02-22 |
| KR20030084952A (en) | 2003-11-01 |
| IL157312A0 (en) | 2004-02-19 |
| EP1364040A2 (en) | 2003-11-26 |
| DE60213884D1 (en) | 2006-09-21 |
| DE60211124D1 (en) | 2006-06-08 |
| EP1564217A2 (en) | 2005-08-17 |
| US20040092560A1 (en) | 2004-05-13 |
| PL362556A1 (en) | 2004-11-02 |
| CZ20032291A3 (en) | 2004-02-18 |
| NZ539204A (en) | 2005-12-23 |
| MXPA03007466A (en) | 2003-12-08 |
| JP2004522801A (en) | 2004-07-29 |
| ZA200306034B (en) | 2004-08-05 |
| ATE325201T1 (en) | 2006-06-15 |
| EP1564217B1 (en) | 2006-08-09 |
| NZ527557A (en) | 2005-05-27 |
| CN1501980A (en) | 2004-06-02 |
| US7157595B2 (en) | 2007-01-02 |
| DE60211124T2 (en) | 2006-11-30 |
| CA2437707A1 (en) | 2002-09-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6288237B1 (en) | Epothilons C and D, preparation and compositions | |
| RU2201932C2 (en) | Epotilons modified by by-side chain | |
| Schinzer et al. | Total synthesis of (−)‐epothilone A | |
| Nicolaou et al. | Total Synthesis of 16‐Desmethylepothilone B, Epothilone B10, Epothilone F, and Related Side Chain Modified Epothilone B Analogues | |
| US6660758B1 (en) | Epothilone analogs | |
| AU5757798A (en) | Epothilone analogs | |
| EP1564217B1 (en) | Process for the production of epothilones | |
| AU2002251030A1 (en) | Degradation of epothilones and ethynyl substituted epothilones | |
| HK1066569A (en) | Degradation of epothilones and ethynyl substituted epothilones | |
| Zhang et al. | Recent progress in the synthesis of epothilones | |
| US8030503B2 (en) | Process for the preparation of epothilones | |
| US6376230B1 (en) | Stereoselective process for producing intermediates of cryptophycins | |
| EP1127055A2 (en) | Stereoselective process for producing cryptophycins | |
| EP0987268B1 (en) | Pharmaceutical agents containing epothilone A-N-oxide and/or epothilone B-N-oxide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| AK | Designated states |
Kind code of ref document: A3 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT TZ UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A3 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003/06034 Country of ref document: ZA Ref document number: 200306034 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2437707 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 157312 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002571909 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 527557 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2003/007466 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2003-2291 Country of ref document: CZ Ref document number: 2002719946 Country of ref document: EP Ref document number: 028055349 Country of ref document: CN Ref document number: 2002251030 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020037011281 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020037011281 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002719946 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10468919 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2003-2291 Country of ref document: CZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 527557 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 527557 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002719946 Country of ref document: EP |