WO2003033475A1 - Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones - Google Patents
Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones Download PDFInfo
- Publication number
- WO2003033475A1 WO2003033475A1 PCT/EP2002/011280 EP0211280W WO03033475A1 WO 2003033475 A1 WO2003033475 A1 WO 2003033475A1 EP 0211280 W EP0211280 W EP 0211280W WO 03033475 A1 WO03033475 A1 WO 03033475A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- malondiimidate
- propan
- preparation
- pyrimidin
- ones
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/52—Two oxygen atoms
Definitions
- the invention relates to a process for the preparation of l-(pyrimidin-2-yl)propan-2-ones of the general formula
- R is in each case a -io-alkyl group, a C 3 _ 8 -cycloal yl group, an allyl group or an aryl-d- -al yl group.
- C ⁇ o-Alkyl groups are understood here and below as meaning all linear or branched primary, secondary or tertiary alkyl groups having 1 to 10 carbon atoms, thus, for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, .sec-butyl, tert-butyl, pentyl, isopentyl, tert-pentyl, neopentyl, hexyl, heptyl, octyl, nonyl or decyl.
- C 3 - 8 -Cycloalkyl are to be understood as meaning, in particular, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- Aryl-Ci- -alkyl groups are the groups composed of an aryl group and an alkyl group having 1 to 4 carbon atoms, aryl groups being understood as meaning, in particular, phenyl or naphthyl groups.
- the aryl groups may also be substituted by one or more d- -alkyl groups,
- Ci- -alkoxy groups or halogen atoms are, in particular, benzyl, 1-phenylethyl, 2-phenylethyl and 3-phenylpropyl.
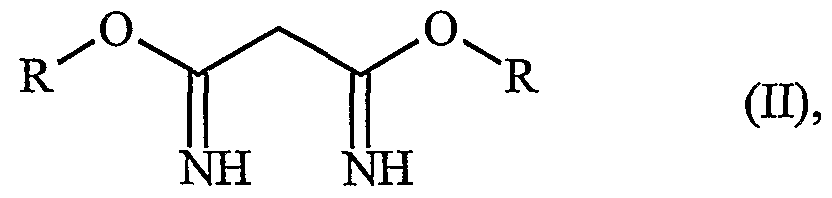
- the object is achieved by the process of Claim 1. It has been found that the malondiimidates, which are readily available from malodinitrile and the corresponding alcohols (DE-A-2426 913, EP-A-0 024 200), of the general formula
- the malondiimidates (H) can either be used without a diluent (as free base) or else be formed in situ from a corresponding salt and a base.
- the latter may be an inorganic base or an organic base such as a tertiary amine.
- the malondiimidates are preferably used without a diluent. For this, they can, for example, be extracted with a solvent of low polarity, such as dichloromethane or diethyl ether, from a neutralized solution of one of their salts and be isolated by evaporating the solvent (EP-A-0 024200).
- the salts of the malondiimidates (II) used are preferably the dihydrochlorides.
- the process according to the invention is advantageously carried out in a solvent which is essentially inert under the reaction conditions, such as, for example, aromatic hydrocarbons like toluene or xylene or ketones like acetone.
- the reaction temperature is advantageously 50 to 150 °C for the aromatic solvents or 0 to 100 °C for ketone solvents.
- Dimethyl malondiimidate dihydrochloride (90.0 kg, 443 mol, 1 eq) and acetone (330 1) were placed under nitrogen in a 630 1 stirred vessel. The suspension was cooled to 0 to 5 °C and triethylamine (98.7 kg, 975 mol, 2.2 eq) was added over the course of 120 min at 0 to 5 °C. The suspension was stirred for another 30 min at 0 to 5 °C and then warmed to 25 °C. Diketene (44.7 kg, 531 mol, 1.2 eq) was added within 90 min at ca. 25 °C.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description


Claims



Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60225578T DE60225578T2 (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of 1- (pyrimidin-2-yl) propan-2-ones |
| JP2003536215A JP4353801B2 (en) | 2001-10-15 | 2002-10-09 | Process for producing 1- (pyrimidin-2-yl) propan-2-ones |
| EP02777297A EP1440063B1 (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones |
| US10/491,281 US7141670B2 (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones |
| HU0401844A HU228816B1 (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of-1(pyrimidin-2-yl)propan-2-ones |
| HK05102899.3A HK1070353B (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP01124587.5 | 2001-10-15 | ||
| EP01124587 | 2001-10-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003033475A1 true WO2003033475A1 (en) | 2003-04-24 |
Family
ID=8178969
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/011280 Ceased WO2003033475A1 (en) | 2001-10-15 | 2002-10-09 | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US7141670B2 (en) |
| EP (1) | EP1440063B1 (en) |
| JP (1) | JP4353801B2 (en) |
| KR (1) | KR100854612B1 (en) |
| CN (1) | CN1262544C (en) |
| AT (1) | ATE388942T1 (en) |
| DE (1) | DE60225578T2 (en) |
| ES (1) | ES2303555T3 (en) |
| HU (1) | HU228816B1 (en) |
| RU (1) | RU2325380C2 (en) |
| WO (1) | WO2003033475A1 (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1935793B (en) * | 2000-10-17 | 2010-06-23 | 庵原化学工业株式会社 | Process for producing substituted aniline compound |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2705562A1 (en) * | 1977-02-10 | 1978-08-17 | Basf Ag | METHOD FOR MANUFACTURING PYRIDONS |
| US4874873A (en) * | 1988-12-27 | 1989-10-17 | Ici Americas Inc. | Process for the preparation of 3-acylpyrrolidones |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2426913A1 (en) | 1974-06-04 | 1975-12-11 | Ferring Arzneimittel Gmbh | Malonic or succinic acid di(imido ester) prodn. - by reacting dicarboxylic acid dinitriles with anhydrous lower alcohols |
| AR225772A1 (en) | 1979-08-14 | 1982-04-30 | Du Pont | NEW 3-AMINO-3-ALCOXI-N-CIANO-2-ALKYL PROPENIMIDATE AND PROCEDURES FOR ITS PREPARATION AND OF PYRIMIDINES STARTING FROM THOSE |
-
2002
- 2002-10-09 AT AT02777297T patent/ATE388942T1/en active
- 2002-10-09 KR KR1020047005534A patent/KR100854612B1/en not_active Expired - Fee Related
- 2002-10-09 WO PCT/EP2002/011280 patent/WO2003033475A1/en not_active Ceased
- 2002-10-09 DE DE60225578T patent/DE60225578T2/en not_active Expired - Lifetime
- 2002-10-09 EP EP02777297A patent/EP1440063B1/en not_active Expired - Lifetime
- 2002-10-09 JP JP2003536215A patent/JP4353801B2/en not_active Expired - Fee Related
- 2002-10-09 US US10/491,281 patent/US7141670B2/en not_active Expired - Fee Related
- 2002-10-09 CN CNB028202295A patent/CN1262544C/en not_active Expired - Fee Related
- 2002-10-09 ES ES02777297T patent/ES2303555T3/en not_active Expired - Lifetime
- 2002-10-09 RU RU2004115022/04A patent/RU2325380C2/en not_active IP Right Cessation
- 2002-10-09 HU HU0401844A patent/HU228816B1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2705562A1 (en) * | 1977-02-10 | 1978-08-17 | Basf Ag | METHOD FOR MANUFACTURING PYRIDONS |
| US4874873A (en) * | 1988-12-27 | 1989-10-17 | Ici Americas Inc. | Process for the preparation of 3-acylpyrrolidones |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1568315A (en) | 2005-01-19 |
| HK1070353A1 (en) | 2005-06-17 |
| KR20050034613A (en) | 2005-04-14 |
| DE60225578T2 (en) | 2009-05-14 |
| ATE388942T1 (en) | 2008-03-15 |
| HUP0401844A3 (en) | 2005-11-28 |
| JP4353801B2 (en) | 2009-10-28 |
| RU2325380C2 (en) | 2008-05-27 |
| JP2005505623A (en) | 2005-02-24 |
| HUP0401844A2 (en) | 2005-01-28 |
| CN1262544C (en) | 2006-07-05 |
| KR100854612B1 (en) | 2008-08-27 |
| US7141670B2 (en) | 2006-11-28 |
| EP1440063A1 (en) | 2004-07-28 |
| DE60225578D1 (en) | 2008-04-24 |
| RU2004115022A (en) | 2005-10-27 |
| HU228816B1 (en) | 2013-05-28 |
| US20040242877A1 (en) | 2004-12-02 |
| ES2303555T3 (en) | 2008-08-16 |
| EP1440063B1 (en) | 2008-03-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1254883B1 (en) | Process for producing substituted 1,1,1-trifluoro-3-butene-2-ones | |
| US7141670B2 (en) | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones | |
| US5266697A (en) | Process for the production of 2-substituted 4,6-dialkoxypyrimidines | |
| US5442076A (en) | Process for the production of 2-substituted-5-chlorimidazole-4-carbaldehydes | |
| HU195773B (en) | Process for preparing alkyl esters of 4-alkoxy-3-pyrrolin-2-on-1-yl-acetic acid | |
| EP0234514A2 (en) | Pyrazolopyrimidines, intermediates thereof and processes for the preparation of them | |
| US6784296B2 (en) | Preparation of terpyridines | |
| US5162529A (en) | Process for the preparation of 5-hydroxy-3,4,5,6-tetrahydro-pyrimidine derivatives | |
| EP1440064B1 (en) | Process for the preparation of (pyrimidin-2-yl)methyl ketones | |
| KR20090116753A (en) | Novel method for preparing [3,4-d] pyridazin-7 (6H) -one with 3-methyl-4-phenylisoxazolo | |
| EP0066440B1 (en) | Chemical process | |
| US5208337A (en) | Process for the preparation of aminopyrimidines | |
| HK1070353B (en) | Process for the preparation of 1-(pyrimidin-2-yl)propan-2-ones | |
| US4536574A (en) | Preparation of 2-alkylthiomethyl-4-hydroxypyrimidines | |
| US4892956A (en) | 5-cyano-4,5-dihydro-3,4-dicarboxypyrazole derivatives | |
| US5262402A (en) | Process for preparing pyrimidinetrione derivatives | |
| US4924001A (en) | Process for preparing 5-cyano-4, 5-dihydro-3,4-dicarboxypyrazole derivatives | |
| EP0484702A1 (en) | 1-Azoniabicyclo[3.3.0]oct-1(5)-ene salt derivatives and process for the preparation of the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BY BZ CA CH CN CO CR CU CZ DE DM DZ EC EE ES FI GB GD GE GH HR HU ID IL IN IS JP KE KG KP KR LC LK LR LS LT LU LV MA MD MG MN MW MX MZ NO NZ OM PH PL PT RU SD SE SG SI SK SL TJ TM TN TR TZ UA UG US UZ VC VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ UG ZM ZW AM AZ BY KG KZ RU TJ TM AT BE BG CH CY CZ DK EE ES FI FR GB GR IE IT LU MC PT SE SK TR BF BJ CF CG CI GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2002777297 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10491281 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 760/CHENP/2004 Country of ref document: IN Ref document number: 20028202295 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003536215 Country of ref document: JP Ref document number: 1020047005534 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002777297 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2002777297 Country of ref document: EP |