WO2003040116A1 - A process for the preparation of cephalosporins side chains - Google Patents
A process for the preparation of cephalosporins side chains Download PDFInfo
- Publication number
- WO2003040116A1 WO2003040116A1 PCT/EP2002/012328 EP0212328W WO03040116A1 WO 2003040116 A1 WO2003040116 A1 WO 2003040116A1 EP 0212328 W EP0212328 W EP 0212328W WO 03040116 A1 WO03040116 A1 WO 03040116A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- residue
- nitrogen
- salts
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(C(C1=NC(N)=S*1)=NO*)=O Chemical compound *C(C(C1=NC(N)=S*1)=NO*)=O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/40—Unsubstituted amino or imino radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/587—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with aliphatic hydrocarbon radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms, said aliphatic radicals being substituted in the alpha-position to the ring by a hetero atom, e.g. with m >= 0, Z being a singly or a doubly bound hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
Definitions
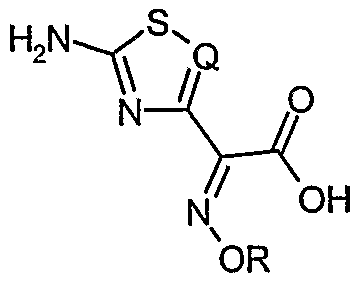
- the present invention relates to a process for the preparation of salts of organic nitrogen bases with carboxylic acids of formula (I)
- - Q is nitrogen, a hydrocarbyl (CH) or chlorocarbyl (CC1) residue;
- the salts of acids of formula (I) with organic nitrogen bases can be easily transformed into the corresponding free acids (I) or into solvates thereof, or into compounds of general formula (III), which can either be isolated or used in situ, for the preparation of third and fourth generation cephalosporanic antibiotics, such as Cefdinir and Cefdaloxime
- Q and R are as defined above, and Z is a carboxy-activating group conventionally used in the synthesis of cephalosporanic antibiotics, such as an anhydride, an ester or an acyl halide.
- Carboxylic acids of formula (I) are important syntons for the preparation of third and fourth generation cephalosporins having wide spectrum activity and high potency against Gram-positive and Gram-negative microorganisms .
- Said salts can be advantageously used for the preparation of intermediates, which can be isolated or used in situ, for the preparation of third and fourth generation cephalosporins.
- these salts allow faster reaction rates with advantages in terms of both yield and purity of the obtained products.
- the present invention relates to a process for the preparation of salts of organic nitrogen bases with carboxylic acids of general formula (I)
- - Q is nitrogen, a hydrocarbyl (CH) or chlorocarbyl (CC1) residue, preferably nitrogen;
- - R is trityl, benzhydryl or para-methoxy benzyl, preferably trityl; which process comprises reacting a carboxylic acid of general formula
- halide of formula (V) (V) RX wherein R has the meaning defined above and X is a halogen selected from chlorine, bromine and iodine, in the presence of a nitrogen organic base and of an organic inert solvent.
- the nitrogen organic base can be selected from tertiary amines, preferably triethylamine, tributylamine, N-ethyl diisopropylamine, N-methyl morpholine, N-methyl pyrrolidine, N-methyl piperidine, trioctylamine; amidines, preferably diazabicyclononene (DBN) and diazabicycloundecene
- the organic inert solvent can be selected from: halogenated hydrocarbons, preferably methylene chloride and dichloroethane; carboxylic acid esters, preferably ethyl acetate and butyl acetate; ketones, preferably acetone, diethyl ketone and methyl ethyl ketone; amides, for example N,N-dimethylformamide, N,N- dimethylacetamide, N-methylpyrrolidone; aromatic hydrocarbons, preferably benzene, toluene and xylene; ethers, preferably tetrahydrofuran, dioxane or ethylene glycol dimethyl ether; sulfoxides or sulfones, preferably dimethylsulfoxide, dimethyl sulfone and sulfolane; or mixtures thereof.
- halogenated hydrocarbons preferably methylene chloride and dichloroethane
- carboxylic acid esters preferably ethy
- the reaction can be carried out at temperatures ranging from -50° to 200°C, preferably from 0° to 100°C.
- the halide (N) is used typically in stoichiometric amounts to compound of formula (II) or in a slight molar excess, whereas the organic base can be present in a ratio ranging from 1 : 1 to 1 :10, preferably from 1 :2 to 1 :5.
- the halide which is usually added to a mixture consisting of compound of formula (II), base and organic inert solvent, can be added directly in a single or more portions, or it can be dissolved in a suitable organic solvent, for example methylene chloride, dichloroethane, toluene or xylene, and then added to the reaction mixture in a time ranging from a few minutes to some hours.
- a suitable organic solvent for example methylene chloride, dichloroethane, toluene or xylene
- the reaction is usually considered complete when the residual compound (II) is lower than 3% (HPLC analysis).
- the salts which will hereinafter be referred to as compounds (IA), usually crystallize during the reaction or upon cooling the mixture and can therefore be filtered off easily.
- salts (IA) can be further purified from traces of the starting compound (II) and any water-soluble amines hydrochlorides present by treatment with water or an aqueous solvent.
- Drying of salts (IA) does not require particular procedures and can be carried out, for example, under vacuum or by ventilation at temperatures from 30° to 100°C.
- Salts (IA) can be used for the preparation of the corresponding free acids of formula (I) and the solvates thereof, for example with formamide, dimethylformamide, dimethylacetamide or N-methylpyrrolidone, according to conventional methods reported in literature (Liebigs Ann. 1996, 1743 - 1749).
- salts (IA) can be used for the preparation of reactive derivatives of general formula (III), which can be isolated or used in situ in the acylation reactions to obtain cephalosporanic antibiotics, such as Cefdinir, Cefdaloxime and other third and fourth generation cephalosporins, according to the procedures described in literature (US 6,093,814, Organic Process Research & Development 1997, 1 121-123).
- thioester of formula -C(0)-S-R 3 wherein R 3 is a heterocyclic residue selected from 2-pyridyl, benzothiazol-2-yl, benzoxazol-2-yl, benzimidazol-2-yl; preferred is the thioester with 2- mercaptobenzothiazole (EP 0037380, EP 0849269);
- NR 3 R 1 is the residue of a nitrogen heterocycle selected from imidazolyl, 1,2,4- triazolyl, tetrazolyl, benzotriazolyl; preferred is the amide with benzotriazol-3-oxide-l-yl (The Journal of Antibiotics , 1993, 46(2), 359 - 361);
- salts (IA) are usually more soluble in the organic solvents traditionally used for the activation reactions, such as methylene chloride, ethyl acetate and tetrahydrofuran, and the reactions are usually faster, give higher yields and yield compounds of formula (III) having higher purity.
- Salts (IA) allow therefore to carry out a process for the preparation of cephalosporins of general formula (IX),
- Q is as defined above and A is a typical residue of cephalosporins chemistry, preferably vinyl, (-CH ⁇ CH ⁇ or methoxymethylene (-CH 2 -0-CH 3 ).
- Example 1 Preparation of (Z)-2-(2-aminothiazoI-4-yI)-2- trityloxyimino acetic acid triethylammonium salt.
- the suspension was heated to 50°C and a further 1100 ml of ethanol was added in 30 minutes.
- the mixture was gradually cooled to 10°C in an hour, acidified to pH 4.0 by addition of 20% hydrochloric acid, then kept for an hour at 10°C and filtered.
- the solid was washed with water and dried to obtain 366 g of (Z)-2-(2-aminothiazol-4-yl)-2-trityloxyimino acetic acid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Cephalosporin Compounds (AREA)
- Thiazole And Isothizaole Compounds (AREA)
Abstract
Description



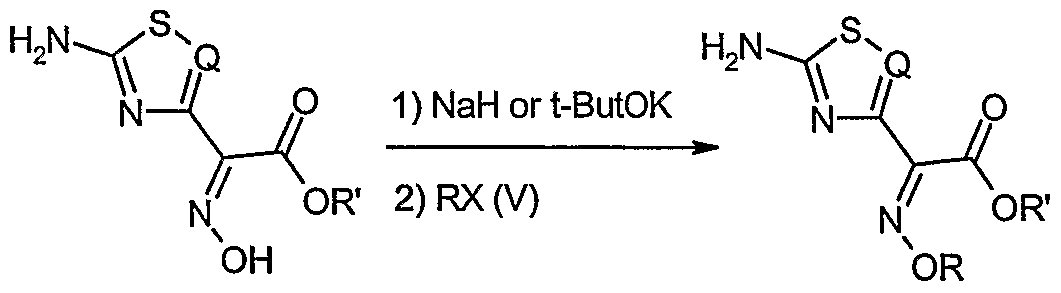





Claims





Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2004-7006840A KR20040064270A (en) | 2001-11-09 | 2002-11-05 | A process for the preparation of cephalosporins side chains |
| JP2003542162A JP2005508385A (en) | 2001-11-09 | 2002-11-05 | Method for producing cephalosporin side chain |
| EP02802646A EP1442029A1 (en) | 2001-11-09 | 2002-11-05 | A process for the preparation of cephalosporins side chains |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IT2001MI002363A ITMI20012363A1 (en) | 2001-11-09 | 2001-11-09 | METHOD FOR THE SYNTHESIS OF SIDE CHAINS OF CEPHALOSPORINE |
| ITMI2001A002363 | 2001-11-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2003040116A1 true WO2003040116A1 (en) | 2003-05-15 |
| WO2003040116A8 WO2003040116A8 (en) | 2004-06-17 |
Family
ID=11448586
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2002/012328 Ceased WO2003040116A1 (en) | 2001-11-09 | 2002-11-05 | A process for the preparation of cephalosporins side chains |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP1442029A1 (en) |
| JP (1) | JP2005508385A (en) |
| KR (1) | KR20040064270A (en) |
| CN (1) | CN1309713C (en) |
| IT (1) | ITMI20012363A1 (en) |
| WO (1) | WO2003040116A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011029596A1 (en) * | 2009-09-11 | 2011-03-17 | Lonza Ltd | Process for preparing 2-aminothiazol-4-yl-acetic acid derivates |
| US9139542B2 (en) | 2013-03-13 | 2015-09-22 | Theravance Biopharma Antibiotics Ip, Llc | Crystalline form of a substituted thiazolylacetic acid triethylamine salt |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102659713B (en) * | 2012-05-07 | 2014-03-05 | 山东金城柯瑞化学有限公司 | Preparation method for cefdinir side-chain acid active ester |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4935508A (en) * | 1988-08-23 | 1990-06-19 | Bristol-Myers Company | Process for cephem prodrug esters |
| WO1992007840A1 (en) * | 1990-11-02 | 1992-05-14 | Taisho Pharmaceutical Co., Ltd. | Thiazole thioester derivative |
| JPH0797368A (en) * | 1993-09-29 | 1995-04-11 | Tokuyama Corp | Process for producing protected hydroxyl group-containing heterocyclic compound |
| JPH0841041A (en) * | 1994-08-01 | 1996-02-13 | Tokuyama Corp | Method for producing 2- (2-aminothiazol-4-yl) -2-acetoxyiminoacetic acid or its derivative |
| US5589594A (en) * | 1992-02-14 | 1996-12-31 | Hoechst Aktiengesellschaft | Process for the preparation of cephem prodrug esters |
| EP0849269A1 (en) * | 1996-12-19 | 1998-06-24 | F. Hoffmann-La Roche Ag | Vinyl pyrrolidine cephalosporins with basic substituents |
-
2001
- 2001-11-09 IT IT2001MI002363A patent/ITMI20012363A1/en unknown
-
2002
- 2002-11-05 KR KR10-2004-7006840A patent/KR20040064270A/en not_active Withdrawn
- 2002-11-05 JP JP2003542162A patent/JP2005508385A/en active Pending
- 2002-11-05 CN CNB028222148A patent/CN1309713C/en not_active Expired - Fee Related
- 2002-11-05 WO PCT/EP2002/012328 patent/WO2003040116A1/en not_active Ceased
- 2002-11-05 EP EP02802646A patent/EP1442029A1/en not_active Withdrawn
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4935508A (en) * | 1988-08-23 | 1990-06-19 | Bristol-Myers Company | Process for cephem prodrug esters |
| WO1992007840A1 (en) * | 1990-11-02 | 1992-05-14 | Taisho Pharmaceutical Co., Ltd. | Thiazole thioester derivative |
| US5589594A (en) * | 1992-02-14 | 1996-12-31 | Hoechst Aktiengesellschaft | Process for the preparation of cephem prodrug esters |
| US5637721A (en) * | 1992-02-14 | 1997-06-10 | Hoechst Aktiengesellschaft | Process for the preparation of cephem prodrug esters |
| JPH0797368A (en) * | 1993-09-29 | 1995-04-11 | Tokuyama Corp | Process for producing protected hydroxyl group-containing heterocyclic compound |
| JPH0841041A (en) * | 1994-08-01 | 1996-02-13 | Tokuyama Corp | Method for producing 2- (2-aminothiazol-4-yl) -2-acetoxyiminoacetic acid or its derivative |
| EP0849269A1 (en) * | 1996-12-19 | 1998-06-24 | F. Hoffmann-La Roche Ag | Vinyl pyrrolidine cephalosporins with basic substituents |
Non-Patent Citations (4)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 123, no. 13, 25 September 1995, Columbus, Ohio, US; abstract no. 169430, KOYANAGI S. ET AL: "Preparation of heterocycles for side chains of cephems" XP002234210 * |
| CHEMICAL ABSTRACTS, vol. 124, no. 25, 17 June 1996, Columbus, Ohio, US; abstract no. 343292, IWASAKI F. ET AL: "Method for producing 2-(2-aminothiazol-4-yl)-2-acetoxyiminoacetic acid and its derivative" XP002234212 * |
| CHEMICAL ABSTRACTS, vol. 127, no. 24, 15 December 1997, Columbus, Ohio, US; abstract no. 331464, ZHU Y ET AL: "Improved synthesis of (Z)-2--(2-aminothiazol-4-yl)-2-trityloxyiminoacetic acid" XP002234211 * |
| ZHONGGUO YIYAO GONGYE ZAZHI, vol. 28, no. 6, 1997, CHINA, pages 270 - 271 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011029596A1 (en) * | 2009-09-11 | 2011-03-17 | Lonza Ltd | Process for preparing 2-aminothiazol-4-yl-acetic acid derivates |
| US9139542B2 (en) | 2013-03-13 | 2015-09-22 | Theravance Biopharma Antibiotics Ip, Llc | Crystalline form of a substituted thiazolylacetic acid triethylamine salt |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1585758A (en) | 2005-02-23 |
| EP1442029A1 (en) | 2004-08-04 |
| ITMI20012363A1 (en) | 2003-05-09 |
| KR20040064270A (en) | 2004-07-16 |
| JP2005508385A (en) | 2005-03-31 |
| WO2003040116A8 (en) | 2004-06-17 |
| CN1309713C (en) | 2007-04-11 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6388070B1 (en) | Thioester derivatives of thiazolyl acetic acid and their use in the preparation of cephalosporin compounds | |
| JP3948628B2 (en) | Method for producing cefdinir | |
| CA2326441C (en) | Process for purification of a cephalosporin derivative | |
| EP0175814B1 (en) | Process for preparing cephem derivatives | |
| JP2939129B2 (en) | Intermediates in the synthesis of cephalosporins | |
| FI109127B (en) | Process for the preparation of cefepime dihydrochloride hydrate antibiotic | |
| KR100342600B1 (en) | New Thiazole compounds and their preparations | |
| WO2003040116A1 (en) | A process for the preparation of cephalosporins side chains | |
| AU690482B2 (en) | Process for producing cephalosporin antibiotics | |
| HU213267B (en) | Process for producing stereospecific cefepime-dihydrochloride-hydrate at ph 5-7,5 | |
| KR890002107B1 (en) | Method for preparing cephalosporin derivative | |
| US5594130A (en) | Preparation of a cephalosporin antibiotic using the syn-isomer of a thiazolyl intermediate | |
| JPH064641B2 (en) | Method for producing cefalosporin derivative | |
| KR930007260B1 (en) | Process for preparing cephalosporin derivatives | |
| EP0613480A1 (en) | Process for the preparation of cephem derivatives | |
| KR100355115B1 (en) | New cephem compouns for preparation of ceftiofur | |
| EP0528343A2 (en) | New process for the production of cephalosporines and novel intermediates in this process | |
| KR0174432B1 (en) | Novel crystalline Cefdinir intermediate and preparation method thereof | |
| JPS6242989A (en) | Production of 7alpha-methoxycephalosporin compound | |
| KR100531669B1 (en) | Processes for the preparation of cephem derivatives | |
| HU197579B (en) | Process for production of derivatives of cefem carbonic acid | |
| JPH02270882A (en) | Production of 7alpha-methoxycephalosporin compound | |
| JPH0331287A (en) | Synthesis of cephalosporin compound | |
| JPH0623189B2 (en) | Process for producing azetidinone derivative | |
| WO2002008184A1 (en) | Process for preparing distamycin derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SD SE SG SI SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR IE IT LU MC NL PT SE SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003542162 Country of ref document: JP Ref document number: 1020047006840 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20028222148 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2002802646 Country of ref document: EP |
|
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: IN PCT GAZETTE 20/2003 UNDER (72, 75) REPLACE "POZZI, GIOVANNI, VIA BELVEDERE, 19/A, I-20045 BESANA BRIANZA" BY "POZZI, GIOVANNI, VIA BELVEDERE, 19/F, I-20045 BESANA BRIANZA" |
|
| WWP | Wipo information: published in national office |
Ref document number: 2002802646 Country of ref document: EP |