WO2003070236A2 - Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents - Google Patents
Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents Download PDFInfo
- Publication number
- WO2003070236A2 WO2003070236A2 PCT/EP2003/001594 EP0301594W WO03070236A2 WO 2003070236 A2 WO2003070236 A2 WO 2003070236A2 EP 0301594 W EP0301594 W EP 0301594W WO 03070236 A2 WO03070236 A2 WO 03070236A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- indazole
- carboxamide
- tetrahydropyrazolo
- phenyl
- dihydropyrazolo
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *[n]1nc(C(N)=O)c2c1C1=CC=CC=CC=C1*2 Chemical compound *[n]1nc(C(N)=O)c2c1C1=CC=CC=CC=C1*2 0.000 description 7
- JILYTRJIQCCTNZ-UHFFFAOYSA-N CCOC(c(c(CC1)c2-c3c1[nH]nc3)n[n]2-c(cc1)ccc1OC)=O Chemical compound CCOC(c(c(CC1)c2-c3c1[nH]nc3)n[n]2-c(cc1)ccc1OC)=O JILYTRJIQCCTNZ-UHFFFAOYSA-N 0.000 description 1
- QKWMIPQJODDALR-UHFFFAOYSA-N COc(cc1)ccc1-[n]1nc(C(N)=O)c(cc2)c1c1c2[nH]nc1 Chemical compound COc(cc1)ccc1-[n]1nc(C(N)=O)c(cc2)c1c1c2[nH]nc1 QKWMIPQJODDALR-UHFFFAOYSA-N 0.000 description 1
- ANVDHIBSYPBETL-UHFFFAOYSA-N COc1ccc(C2c(c3c(cc4)[nH]nc3)c4C(C(O)=O)=NCC2)cc1 Chemical compound COc1ccc(C2c(c3c(cc4)[nH]nc3)c4C(C(O)=O)=NCC2)cc1 ANVDHIBSYPBETL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to tricyclic pyrazole derivatives active as kinase inhibitors and, more in particular, it relates to tricyclic pyrazoles and analogues tricyclic heterocyclic derivatives, to a process for their preparation, to pharmaceutical compositions comprising them and to their use as therapeutic agents, particularly in the treatment of diseases linked to disregulated protein kinases. Discussion of background
- PKs protein kinases
- a large share of the oncogenes and proto-oncogenes involved in human cancers code for PKs.
- the enhanced activities of PKs are also implicated in many non-malignant diseases, such as benign prostate hyperplasia, familial adenomatosis, polyposis, neuro- fibromatosis, psoriasis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary fibrosis, arthritis glomerulonephritis and post-surgical stenosis and restenosis.
- PKs are also implicated in inflammatory conditions and in the multiplication of viruses and parasites. PKs may also play a major role in the pathogenesis and development of neurodegenerative disorders.
- the present inventors have now discovered that the compounds of the invention, hereinafter shortly referred to as tricyclic pyrazole derivatives, are endowed with multiple protein kinase inhibiting activity and are thus useful in therapy in the treatment of diseases associated with disregulated protein kinases.
- the compounds of this invention are useful in the treatment of a variety of cancers including, but not limited to: carcinoma such as bladder, breast, colon, kidney, liver, lung, including small cell lung cancer, esophagus, gall-bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; hematopoietic tumors of lymphoid lineage, including leukemia, acute lymphocitic leukemia, acute lymphoblastic leukemia, B-cell lymphoma, T-cell-lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma and Burkett's lymphoma; hematopoietic tumors of myeloid lineage, including acute and chronic myelogenous leukemias, myelodysplastic syndrome and promyelocytic leukemia; tumors of mesenchymal origin, including fibrosar
- PKs Due to the key role of PKs in the regulation of cellular proliferation, these compounds are also useful in the treatment of a variety of cell proliferative disorders such as, for instance, benign prostate hyperplasia, familial adenomatosis, polyposis, neuro- fibromatosis, psoriasis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary f ⁇ brosis, arthritis glomerulonephritis and post-surgical stenosis and restenosis.
- the compounds of the invention can be useful in the treatment of Alzheimer's disease, as suggested by the fact that cdk5 is involved in the phosphorylation of tau protein (J Biochem., I ll, 741-749, 1995).
- the compounds of the invention are also useful in the treatment and prevention of radiotherapy-induced or chemotherapy-induced alopecia.
- the compounds of this invention may also be useful in the treatment of cancer, viral infections, prevention of AIDS development in HTV-infected individuals, autoimmune diseases and neurodegenerative disorders.
- the compounds of this invention may be useful in inhibiting tumor angiogenesis and metastasis, as well as in the treatment of organ transplant rejection and host versus graft diseases.
- the compounds of the invention are useful as cyclin dependent kinase (cdk) inhibitors and also as inhibitors of other protein kinases such as, for instance, protein kinase C in different isoforms, Met, PAK-4, PAK-5, ZC-1, STLK-2, DDR-2, Aurora 1, Aurora 2, Bub-1, PLK, Chkl, Chk2, HER2, rafl, MEK1, MAPK, EGF-R, PDGF-R, FGF-R, IGF- R, VEGF-R, PI3K, weel kinase, Src, Abl, Akt, ILK, MK-2, IKK-2, Cdc7, Nek, and thus be effective in the treatment of diseases associated with other protein kinases.
- cdk cyclin dependent kinase
- U.S. patent No. 4,734,430 discloses benzo- and cycloheptadipyrazoles as bronchodilators; U.S. Patent No. 3,940,418 describes tricyclic 4,5-dihydrobenz[g]indazoles as anti- inflammatory agents.
- R. Hamilton J. Heterocyclic Chem., 13, 545 (1976)] describes tricyclic 4,5-dihydrobenz[g]indazoles as anti-inflammatory agents.
- U.S. Patent No. 5,134,155 describes fused tricyclic pyrazoles having a saturated ring bridging the pyrazole and a phenyl radical as HMG-CoA reductase inhibitors.
- M. Hashem et al J Med. Chem., 19, 229 (1976)] describes fused tricyclic pyrazoles, having a saturated ring bridging the pyrazole and a phenyl radical, as antibiotics.
- the phytotoxicity of pyrazole derivatives is described [M. Cocco et al, //. Farmaco-Ed. Sci., 40, 272 (1985)], specifically for l-[4-(aminosulfonyl)phenyl]-5-phenyl-lH-pyrazole- 3,4-dicarboxylic acid.
- the use of styryl pyrazole esters for antidiabetes drugs is described [H. Mokhtar et al, Pharmazie, 33, 649-651 (1978)].
- the use of styryl pyrazole carboxylic acids for antidiabetes drugs is described [R. Soliman et al, Pharmazie, 33, 184-5 (1978)].
- a series of 4-[3-substituted methyl-5-phenyl-lH- pyrazol-l-yl]benzenesulfonamides has been prepared as intermediates for anti-diabetes agents, and more specifically, 4-[3-methyl-5-phenyl-lH-pyrazol-l- yljbenzenesulfonamide [ ⁇ . Feid-AUah, Pharmazie, 36, 754 (1981)].
- WO 00/27822 discloses tricyclic pyrazole derivatives
- WO 00/59901 discloses dihydroindeno pyrazoles
- WO 95/15315 discloses diphenyl pyrazole compounds
- WO 95/15317 discloses triphenyl pyrazole compounds
- WO 95/15318 discloses tri-substituted pyrazole compounds
- WO 96/09293 discloses benz[g]indazolyl derivatives.
- WO 95/15316 discloses substituted pyrazolyl benzenesulfamide derivatives. Accordingly, the present invention provides a method for treating diseases caused by and/or associated with an altered protein kinase activity, by administering to a mammal in need thereof an effective amount of a compound represented by formula (I)
- X, Y and Z, being part of an aromatic ring are selected, each independently, from the group consisting of N, NR 1; S, O and CR 1;
- R' and R" are selected, each independently, from the group consisting of hydrido, hydroxy, alkyl, hydroxyalkyl, alkenyl, alkynyl, aryl, arylalkyl, heterocyclyl or heterocyclyl-alkyl;
- B is an aromatic 5 or 6 membered ring having from 0 to 3 heteroatoms selected from S, O and N;
- R z and R y are selected, each independently, from hydrido or lower alkyl; each of the X,Y,Z and B rings being optionally further substituted by one or more -L-R 2 groups, wherein L represents, each independently, a single bond, an alkylidene group or a divalent group selected from NH, NHCO, CONH, NHCONH, SO 2 NH and NHSO ;
- R 2 is, each independently, hydrido, alkyl, 5 to 12 membered mono- or bi-cyclic ring having from 0 to 3 heteroatoms selected from S, O and N, optionally substituted with one or more -(CH 2 ) q -R 3 groups; or R is a group of formula
- W is a 3 to 7 membered ring having one N heteroatom directly linked to Q and from 0 to 2 additional heteroatoms selected from the group consisting of S, SO, SO , O, N and NR 1 , wherein R' is as above defined;
- Q is a divalent group selected from CO, SO 2 and (CH 2 ) n ;
- the disease caused by and/or associated with an altered protein kinase activity is selected from the group consisting of cancer, cell proliferative disorders, Alzheimer's disease, viral infections, auto-immune diseases and neurodegenerative disorders.
- Specific types of cancer that may be treated include carcinoma, squamous cell carcinoma, hematopoietic tumors of myeloid or lymphoid lineage, tumors of mesenchymal origin, tumors of the central and peripheral nervous system, melanoma, seminoma, teratocarcinoma, osteosarcoma, xeroderma pigmentosum, keratoxanthoma, thyroid follicular cancer and Kaposi's sarcoma.
- the cell proliferative disorder is selected from the group consisting of benign prostate hyperplasia, familial adenomatosis polyposis, neuro-fibromatosis, psoriasis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary fibrosis, arthritis glomeralonephritis and post- surgical stenosis and restenosis.
- the method object of the present invention also provides tumor angiogenesis and metastasis inhibition.
- the present invention further provides a compound represented by formula (I)
- X, Y and Z, being part of an aromatic ring are selected, each independently, from the group consisting of N, NR 1? S, O and CR 1;
- R' and R" are selected, each independently, from the group consisting of hydrido, hydroxy, alkyl, hydroxyalkyl, alkenyl, alkynyl, aryl, arylalkyl, heterocyclyl or heterocyclyl-alkyl;
- B is an aromatic 5 or 6 membered ring having from 0 to 3 heteroatoms selected from S, O and N;
- R z and R y are selected, each independently, from hydrido or lower alkyl; each of the X,Y,Z and B rings being optionally further substituted by one or more -L-R 2 groups, wherein L represents, each independently, a single bond, an alkylidene group or a divalent group selected from NH, NHCO, CONH, NHCONH, SO 2 NH and NHSO 2 ;
- R 2 is, each independently, hydrido, alkyl, 5 to 12 membered mono- or bi-cyclic ring having from 0 to 3 heteroatoms selected from S, O and N, optionally substituted with one or more -(CH 2 ) q -R groups; or R 2 is a group of formula
- W is a 3 to 7 membered ring having one N heteroatom directly linked to Q and from 0 to 2 additional heteroatoms selected from the group consisting of S, SO, SO 2 , O, N and NR ⁇ wherein R' is as above defined;
- Q is a divalent group selected from CO, SO 2 and (CH 2 ) n ;
- the present invention includes all of the hydrates, solvates, complexes and prodrugs of the compounds of this invention.
- Prodrugs are any covalently bonded compounds, which release the active parent drug according to formula (I) in vivo.
- a chiral center or another form of an isomeric center is present in a compound of the present invention, all forms of such isomer or isomers, including enantiomers and diastereomers, are intended to be covered herein.
- Compounds containing a chiral center may be used as a racemic mixture, an enantiomerically enriched mixture, or the racemic mixture may be separated using well-known techniques and an individual enantiomer may be used alone, cases in which compounds have unsaturated carbon-carbon double bonds, both the cis (Z) and trans (E) isomers are within the scope of this invention.
- compounds may exist in tautomeric forms, such as keto-enol tautomers, each tautomeric form is contemplated as being included within this invention whether existing in equilibrium or predominantly in one form.
- any substituent at any one occurrence in formula (I) or any sub-formula thereof is independent of its meaning, or any other substituents meaning, at any other occurrence, unless specified otherwise.
- each of X, Y and Z can be independently selected, as formerly indicated, among N, NRi, S, O and
- aromatic ring does not need any further clarification as it refers to any ring which can be conventionally defined as aromatic, such a term being widely used in organic chemistry.
- Non limiting examples of X, Y, Z aromatic rings according to the invention are, for instance, thiophene, furan, furazan, pyrrole, pyrazole, imidazole, thiazole, isothiazole, oxazole or isoxazole.
- hydrido it is intended a single hydrogen atom (H); this hydrido radical may be attached, for example, to an oxygen atom to form a hydroxyl radical or two hydrido radicals may be attached to a carbon atom to form a methylene (-CH 2 -) radical.
- lower alkyl group we intend any straight or branched alkyl group with from
- 1 to 6 carbon atoms such as, for instance, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, n-hexyl, and the like.
- Pefluorinated lower alkyl groups stand for the above lower alkyl groups being further substituted in any of the free positions, at the same or different carbon atom, by more than one fluorine atoms.
- Non limiting examples of perfiuorinated alkyl groups are, for instance, trifluoromethyl, 2,2,2-trifluoroethyl, 1,2-difluoroethyl, 1,1,1,3,3,3- hexafluoropropyl-2-yl, and the like.
- heterocyclyl we intend any 5 or 6 membered heterocyclic radical with from 1 to 3 heteroatoms selected among N, O and S. If not specifically noted otherwise, the said heterocyclic moieties may comprise saturated, partly unsaturated and fully unsaturated heterocycles; these latter, clearly referable to as aromatic heterocycles, are also conventionally known as heteroaromatic or heteroaryl rings.
- Non limiting examples of the said heterocycles of the invention are, for instance, thiophene, furan, furazan, pyran, pyrrole, imidazole, pyrazole, thiazole, isothiazole, oxazole, isoxazole, pyridine, pyrazine, pyrimidine, pyridazine, pyrrolidine, pyrroline, imidazolidine, imidazoline, pyrazolidine, pyrazoline, piperidine, piperazine, morpholine, and the like.
- hydroxyalkyl we intend any of the above straight or branched lower alkyl radicals having from one to six carbon atoms, any one of which may be substituted with one or more hydroxyl radicals.
- halogen atom optionally referable to as "halo" group, herewith intended are fluorine, chlorine, bromine and iodine atoms.
- alkenyl or alkynyl we intend any of the aforementioned lower alkyl groups with from 2 to 6 carbon atoms, bearing a double or triple bond.
- alkenyl or alkynyl groups are thus, for instance, vinyl, allyl, 1-propenyl, isopropenyl, 1- butenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 1-hexenyl, ethynyl, 2-propynyl, 4-pentynyl, and the like.
- aryl we intend, unless otherwise specified, any aromatic ring hence including carbocyclic or 5 or 6 membered heterocyclic rings with from 1 to 3 heteroatoms selected among N, O and S.
- aryl groups are thus phenyl, furyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, and the like.
- arylalkyl or heterocyclyl-alkyl groups we intend any of the above groups being defined according to the single moieties from which they derive. More particularly, arylalkyl and heterocyclyl-alkyl groups stand for the above alkyl groups further substituted by aryl or heterocyclyl groups, respectively, these latter being as above defined.
- B represents a 5 to 6 membered aromatic ring, as formerly indicated, having from 0 to 3 heteroatoms selected from N, O and S. From the above it is clear to the skilled man that B may comprise phenyl, as a 6 membered aromatic ring with 0 heteroatoms, as well as any other 5 or 6 membered aromatic heterocycle with from 1 to 3 heteroatoms, as above defined.
- A represents a divalent linker joining X, Y, Z ring with B ring. According to the meanings provided to A, therefore, it may represent a straight or branched alkylidene group being optionally unsaturated [e.g. -(CR z R y ) p - such as, for instance,
- both B and X, Y, Z rings may be optionally further substituted, each independently, by one or more L-R 2 groups, being the same or different. Substitutions may obviously occur in any of the free positions of both rings, by replacement of one or more hydrogen atoms, otherwise referred to as hydrido.
- L may represent a saturated divalent hydrocarbon group, with from 1 to 6 carbon atoms such as, for instance, a -(CH 2 ) 1-6 - group.
- L may represent a saturated divalent hydrocarbon group, with from 1 to 6 carbon atoms such as, for instance, a -(CH 2 ) 1-6 - group.
- each of the two ring units may be fused to each other or otherwise linked through a single bond.
- Non limiting examples of the above carbocyclic ring systems include, for instance, cyclopentane, cyclopentene, cyclohexane, cyclohexene, cyclohexadiene, benzene, naphthalene and biphenylene.
- Examples of the above heterocylic ring systems may typically include any of the aforementioned 5 or 6 membered, either saturated, partly unsaturated or fully unsaturated heterocycles (see examples above) which may be further condensed to, or linked through a single bond with, any of the aforementioned mono-cyclic carbocyclic or heterocyclic rings themselves.
- the W ring represents a 3 to 7 membered heterocyclic ring at least containing a N nitrogen atom directly linked to Q, as set forth above.
- pharmaceutically acceptable salts embraces salts commonly used to form alkali metal salts and to form addition salts of free acids or free bases. The nature of the salt is not critical, provided that it is pharmaceutically acceptable. Suitable pharmaceutically acceptable acid addition salts of compounds of the present invention may be prepared from an inorganic acid or from an organic acid. Examples of such inorganic acids are hydrochloric, hydrobromic, hydroiodic, nitric, carbonic, sulfuric, and phosphoric acid.
- organic acids may be selected from aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclic, carboxylic and sulfonic classes of organic acids, examples of which are formic, acetic, trifluoroacetic propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic, glucuronic, maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, mesylic, salicyclic, salicyclic, phydroxybenzoic, phenylacetic, mandelic, embonic (pamoic), methanesulfonic, ethanesulfonic, benzenesulfonic, pantothenic, toluenesulfonic, 2-hydroxyethanesulfonic, sulfanilic, stearic, cyclohexylaminosulfonic, algenic, hydroxybutaned
- Suitable pharmaceutically acceptable base addition salts of compounds of the present invention include metallic salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from N,N'- dibenzylethylenediamine, chloroprocaine, choline, diethanolamine, ethylenediamine, meglumine (N-methyl-glucamine) and procaine. All of these salts may be prepared by conventional means from the corresponding compound of the present invention by reacting, for example, the appropriate acid or base.
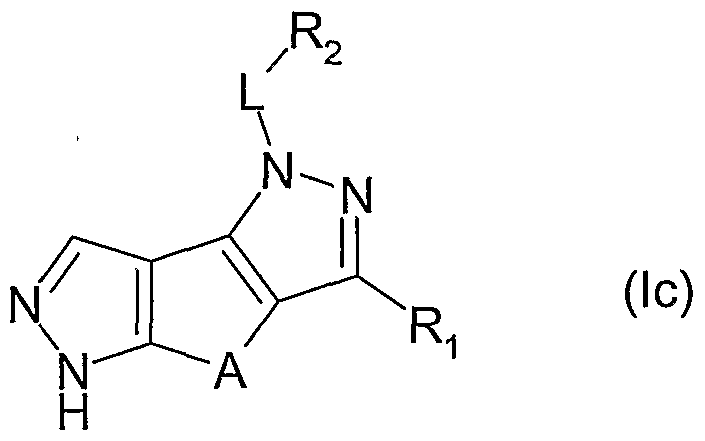
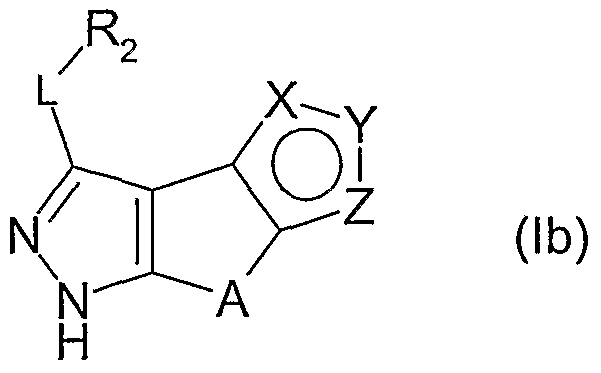
- a class of preferred compounds of the invention is represented by the derivatives of formula (la)
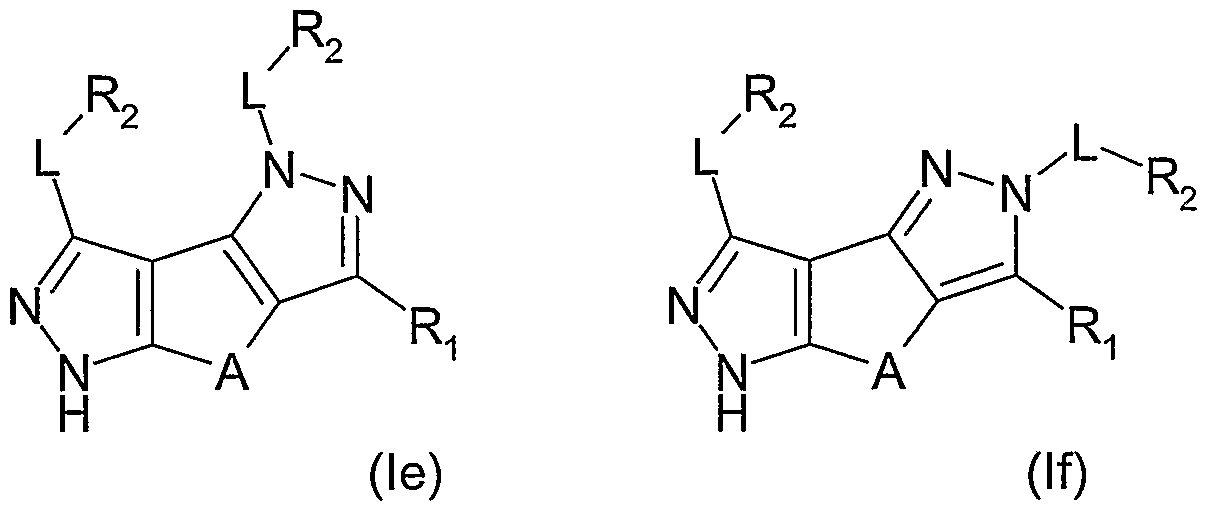
- Another class of preferred compounds of the invention is represented by the derivatives of formulae (le) or (If)
- L and R are, each independently and the same or different in each occasion, as above defined;
- L is methylene or a single bond and R 2 is hydrido, phenyl or a 5 or 6 membered aromatic heterocycle having 1 or 2 heteroatoms selected among N, O and S, the said phenyl or heterocycle being optionally further substituted as above indicated.
- R 2 being optionally further substituted as above indicated, is selected from the group consisting of hydrido, phenyl, pyridyl, pyridazinyl or pyrimidinyl.
- Scheme I describes the synthesis of the pyrazoles of formula (I) with fused heterocycles such as, for instance, substituted pyrimidine and pyrazole derivatives.
- 1,2- cyclohexanedione (1) was refluxed with alcohols such as methanol or ethanol in benzene to provide the desired enone (2).
- enone (2) was treated with a base such as lithium bistrimethylsilylamide, followed by condensation with diethyl oxalate to afford 1,3-diketone (3).
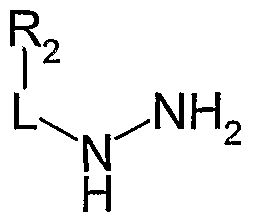
- step three 1,3-diketone was allowed to react with a suitably substituted hydrazine of general formula (8) to form pyrazole (4).
- step four pyrazole was treated with dimethylformamide di-tert-butyl acetal to give enaminone (5).
- step five enaminone was condensed with cyclizing agents such as hydrazine, guanidine, or thiourea derivatives to afford fused pyrazoles and pyrimidines
- the ester was converted to amide (7) by treatment with ammonium hydroxide in methanol, at a temperature ranging from about 25°C to about 70°C, in a sealed tube.
- Hydrazines of general formula (8) are commercially available or can be obtained through synthetic procedures well described in the literature.
- aryl-hydrazines can be conveniently obtained from the corresponding anilines by diazotization, using sodium nitrite, or an alkyl nitrite, followed by catalytic or chemical reduction as described, for example, in J. Med. Chem., 36, 1529 (1993).
- aryl halides suitably activated with electron withdrawing groups can be converted to the corresponding arylhydrazines through displacement of the halogen atom with hydrazine or a carbazate, followed by hydrolysis of the protecting group, for instance as reported in J. Het. Chem., 25, 1543 (1988) or in Tetrah. Lett, 40 (18), 3543 (1999).
- Alkyl-hydrazines can be obtained from alkyl-amines by treatment with hydroxylamine-O- sulfonic acid, for instance as described in JOC, 14, 813 (1949).
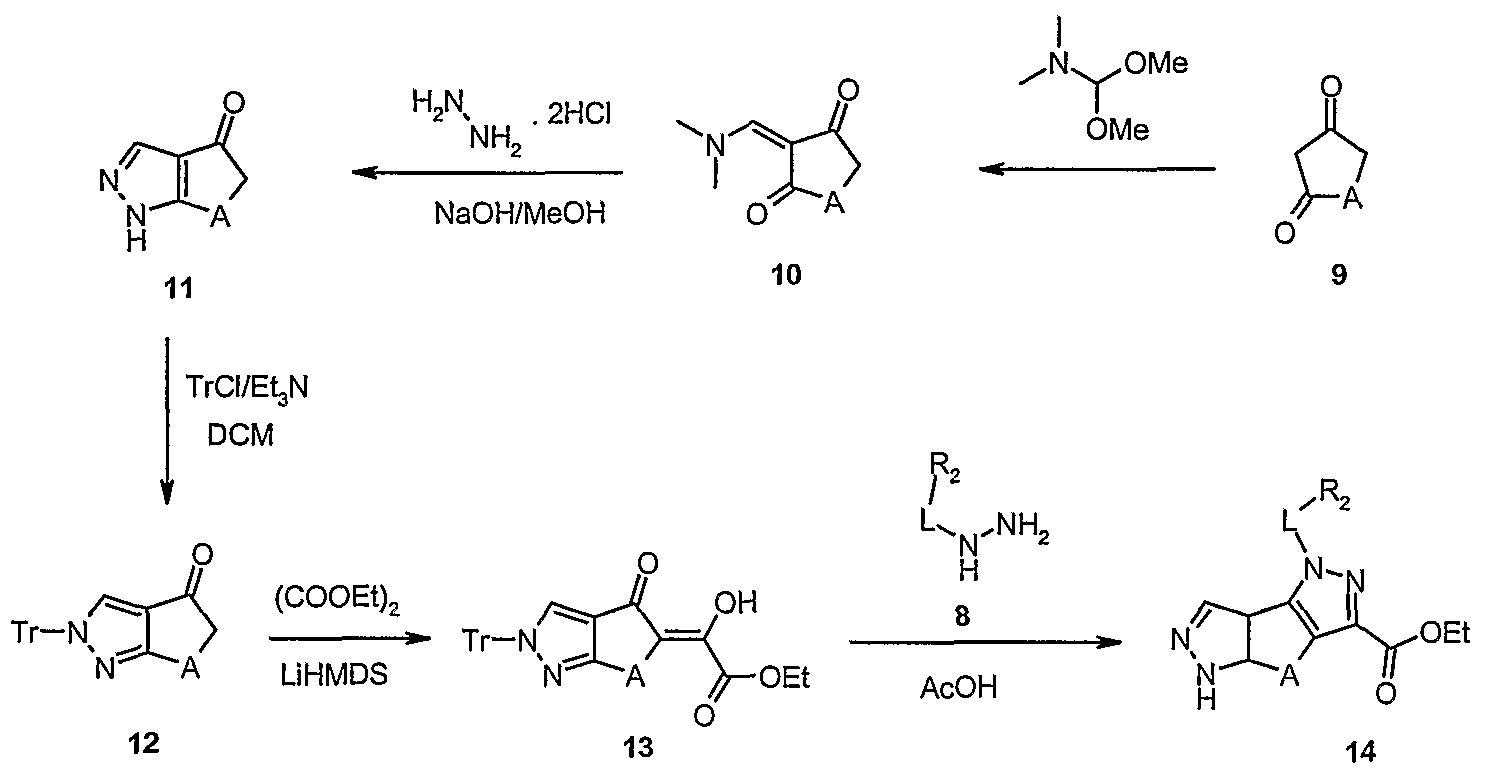
- step one the cyclic diketone (9) was condensed with N,N-dimethylformamide dimethyl acetal to obtain the adduct (10), as described in Heterocycles, 32, 41 (1991).
- step two the adduct (10) was reacted with hydrazine dihydrochloride to obtain the intermediate (11), that was protected with trityl chloride (step three) to give the intermediate (12).
- step four After condensation with oxalyl chloride (step four), the diketoester (13) was allowed to react with a suitably substituted hydrazine (8) (step five) to form the dipyrazole (14). If a salified form of the hydrazine (8) is used (i.e. hydrochloride), the trityl protecting group is normally lost during the cyclization reaction. Optionally, diluted hydrochloric acid can be added to complete the deprotection, once the cyclization has occurred. In step six, the ester was then converted to the amide (15) by treatment with ammonium hydroxide in methanol, at a temperature ranging from about 25°C to about 70°C, in a sealed tube.
- ammonium hydroxide in methanol
- the intermediate compound (11) wherein A is -CH 2 - or -CH 2 -CH 2 -, as well as the intermediate compounds (12) and (13) wherein A is selected from -CH 2 -, -CH 2 -CH 2 - and -CH 2 -C(CH 3 ) 2 - are novel and, hence, represent a further object of the present invention.
- Scheme HI illustrates the general synthetic procedure for the preparation of benzodipyrazole derivatives of general formula (I) wherein B is further substituted by a L-R 2 group wherein L is NH.
- step one the commercially available 3-ethoxy-cyclohex-2-enone (16) is condensed with diethyl oxalate to afford the diketoester (17), which is then reacted, in step two, with a suitably substituted hydrazine (8) to give the pyrazole derivative (18).
- step three the pyrazole (18) is treated in the presence of a base, such as lithium bistrimethylsilylamide, with a suitably substituted isothiocyanate (19) to afford the intermediate (20), which is then converted to the 3-aminobenzodipyrazole ester of formula (21).
- a base such as lithium bistrimethylsilylamide
- isothiocyanate (19) to afford the intermediate (20), which is then converted to the 3-aminobenzodipyrazole ester of formula (21).
- the ester (21) is finally converted to the corresponding amide (22) under standard operative conditions.
- Isothiocyanates of general formula (19) are commercially available or can be obtained through synthetic procedures well described in the literature. SCHEME IV
- Scheme IV describes the general synthetic pathway to obtain compounds of general formula (I) wherein Y and Z are linked so as to form an additional lactamic ring and A is preferably selected from -CH 2 -, -CH 2 -CH 2 - or -CH 2 -C(CH 3 ) -. More generally, scheme IV can also be used to obtain compounds of general formula (I) wherein group L-R is linked to Y.
- the intermediate compound (23) is reacted with hydrazine to form the pyrazole derivative (24).
- step two This is then alkylated, in step two, using an alkyl halide bearing a protected amino group, for instance as tert-butoxy-carbonyl (BOC) amino group, hi step three, after removal of the protecting group, the intermediate (25) is allowed to cyclize so to form the final compound (26) under standard operative conditions.
- a protected amino group for instance as tert-butoxy-carbonyl (BOC) amino group
- Scheme V refers to some examples describing the possibility of obtaining compounds of general formula (I), differently substituted in R .
- the oxidation of the central ring can be accomplished according to conventional techniques, for instance by using activated quinone derivatives, e.g. 2,3-dichloro-5,6-dicyano-l,4-benzoquinone or, alternatively, palladium on charcoal in a suitable solvent such as decalin, at high temperatures.
- any compound of formula (I) of the invention may be prepared by working in analogy to what reported in any one of schemes I to VI and, perhaps, by optionally providing any required modification to the above reactions, on a case by case.
- the said reactions are however known and conventionally adopted when preparing tricyclyc heterocyclic derivatives of formula (I) and substituted compounds thereof.
- the inhibiting activity of putative Cdk/Cychn inhibitors and the potency of selected compounds was determined through a method of assay based on the use of the SPA technology (Amersham Pharmacia Biotech).
- the assay consists of the transfer of radioactivity labelled phosphate moiety by the kinase to a biotinylated substrate.
- a volume of 110 ⁇ l is transferred to Optiplate. After 20 min. incubation for substrate capture, 100. ⁇ l 5M CsCl were added to allow statification of beads to the top of the plate and let stand 4 hours before radioactivity counting in the Top-Count instrument IC50 determination: inhibitors were tested at different concentrations ranging from 0.0015 to 10 ⁇ M.
- Kinetic parameter estimates were estimated by simultaneous nonlinear least-square regression using [Eq.l] (competitive inhibitor respect to ATP, random mechanism) using the complete data set (80 points):
- the selected compounds have been characterized on a panel of ser/threo kinases strictly related to cell cycle (Cdk2/Cyclin E, Cdkl/cyclin Bl, Cdk5/p25, Cdk4/Cyclin Dl), and also for specificity on MAPK, PKA, EGFR, IGF1-R, Aurora-2 and Akt.
- kinase reaction 0,4 uM ⁇ M mouse GST-Rb (769-921) (# sc-4112 from Santa Cruz) substrate, 10 ⁇ M ATP (0.5 ⁇ Ci P 33 ⁇ -ATP), 100 ng of baculovirus expressed GST- Cdk4/Cyclin Dl, suitable concentrations of inhibitor in a final volume of 50 ⁇ l buffer (TRIS HCl 10 mM pH 7.5, MgCl 2 10 mM, 7.5 mM DTT+ 0.2mg/ml BSA) were added to each well of a 96 U bottom well plate. After 40 min at 37 °C incubation, reaction was stopped by 20 ⁇ l EDTA 120 mM.
- Capture 60 ⁇ l were transferred from each well to MultiScreen plate, to allow substrate binding to phosphocellulose filter. Plates were then washed 3 times with 150 ⁇ l/well PBS Ca ⁇ /Mg " free and filtered by MultiScreen filtration system. Detection: filters were allowed to dry at 37°C, then 100 ⁇ l/well scintillant were added and 33 P labeled Rb fragment was detected by radioactivity counting in the Top-Count instrument.
- kinase reaction 10 ⁇ M in house biotinylated MBP (Sigma # M-1891) substrate, 2 ⁇ M ATP (0.04 microCi P 33 ⁇ -ATP), 36 ng insect cell expressed GST-EGFR, inhibitor in a final volume of 30 ⁇ l buffer (Hepes 50 mM pH 7.5, MgCl 2 3 mM, MnCl 2 3 mM, DTT 1 mM, NaVO 3 3 ⁇ M + 0.2 mg/ml BSA) were added to each well of a 96 U bottom. After 20 min at r.t.
- reaction was stopped by 100 ⁇ l PBS + 32 mM EDTA + 0.1% Triton X-100 + 500 ⁇ M ATP, containing 1 mg SPA beads. Then a volume of 110 ⁇ l is transferred to Optiplate.
- the inhibition assay of Cdc7/dbf4 activity was performed according to the following protocol.
- Biotin-MCM2 substrate is trans-phosphorylated by the Cdc7/Dbf4 complex in the presence of ATP traced with ⁇ 33 -ATP.
- the phosphorylated Biotin-MCM2 substrate is then captured by Streptavidin-coated SPA beads and the extent of phosphorylation evaluated by ⁇ counting.
- the inhibition assay of Cdc7/dbf4 activity was performed in 96 wells plate according to the following protocol. To each well of the plate were added:
- test compound (12 increasing concentrations in the nM to ⁇ M range to generate a dose-response curve) - 10 ⁇ l of a mixture of cold ATP (10 ⁇ M final concentration) and radioactive ATP
- the compounds of formula (I) of the present invention suitable for administration to a mammal, e.g. to humans, can be administered by the usual routes and the dosage level depends upon the age, weight, conditions of the patient and the administration route.
- a suitable dosage adopted for oral administration of a compound of formula (I) may range from about 10 to about 500 mg pro dose, from 1 to 5 times daily.
- the compounds of the invention can be administered in a variety of dosage forms, e.g. orally, in the form of tablets, capsules, sugar or film coated tablets, liquid solutions or suspensions; rectally in the form of suppositories; parenteraUy, e.g. intramuscularly, or by intravenous and/or intrathecal and/or intraspinal injection or infusion.
- the compounds of the invention can be administered either as single agents or, alternatively, in combination with known anticancer treatments such as radiation therapy or chemotherapy regimen in combination with cytostatic or cytotoxic agents, antibiotic-type agents, alkylating agents, antimetabolite agents, hormonal agents, immunological agents, interferon-type agents, cyclooxygenase inhibitors (e.g.
- COX-2 inhibitors COX-2 inhibitors
- metallomatrixprotease inhibitors telomerase inhibitors
- tyrosine kinase inhibitors anti-growth factor receptor agents
- anti-HER agents anti-EGFR agents
- anti- angiogenesis agents farnesyl transferase inhibitors
- ras-raf signal transduction pathway inhibitors cell cycle inhibitors, other cdks inhibitors, tubulin binding agents, topoisomerase I inhibitors, topoisomerase II inhibitors, and the like.
- the compounds of the invention can be administered in combination with one or more chemotherapeutic agents such as, for instance, exemestane, formestane, anastrozole, letrozole, fadrozole, taxane, taxane derivatives, encapsulated taxanes, CPT- 11, camptothecin derivatives, anthracycline glycosides, e.g., doxorubicin, idarubicin, epirubicin, etoposide, navelbine, vinblastine, carboplatin, cisplatin, estramustine phosphate, celecoxib, tamoxifen, raloxifen, Sugen SU-5416, Sugen SU-6668, Herceptin, and the like, optionally within liposomal formulations thereof.
- chemotherapeutic agents such as, for instance, exemestane, formestane, anastrozole, letrozole, fadrozole, taxane, taxane
- compositions comprising a compound of formula (1) or a pharmaceutically acceptable salt thereof in association with a pharmaceutically acceptable excipient (which can be a carrier or a diluent).
- a pharmaceutically acceptable excipient which can be a carrier or a diluent.
- the pharmaceutical compositions containing the compounds of the invention are usually prepared following conventional methods and are administered in a pharmaceutically suitable form.
- the solid oral forms may contain, together with the active compound, diluents, e.g. lactose, dextrose, saccharose, sucrose, cellulose, corn starch or potato starch; lubricants, e.g. silica, talc, stearic , magnesium or calcium stearate, and/or polyethylene glycols; binding agents, e.g. starches, arabic gum, gelatin, methylcellulose, carboxymethylcellulose or polyvinyl pyrrolidone; disaggregating agents, e.g.
- diluents e.g. lactose, dextrose, saccharose, sucrose, cellulose, corn starch or potato starch
- lubricants e.g. silica, talc, stearic , magnesium or calcium stearate, and/or polyethylene glycols
- binding agents e.g. starches, arabic gum, gelatin, methylcellulose, carboxymethylcellulose or polyviny
- a starch alginic, alginates or sodium starch glycolate
- effervescing mixtures dyestuffs
- sweeteners wetting agents such as lecithin, polysorbates, laurylsulfates
- wetting agents such as lecithin, polysorbates, laurylsulfates
- non-toxic and pharmacologically inactive substances used in pharmaceutical formulations.
- Said pharmaceutical preparations may be manufactured in known mariner, for example, by means of mixing, granulating, tabletting, sugar-coating, or film-coating processes.
- the liquid dispersions for oral administration may be e.g. syrups, emulsions and suspensions.
- the syrups may contain as carrier, for example, saccharose or saccharose with glycerin and/or mannitol and/or sorbitol.
- the suspensions and the emulsions may contain as carrier, for example, a natural gum, agar, sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl alcohol.
- the suspension or solutions for intramuscular injections may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g. sterile water, olive oil, ethyl oleate, glycols, e.g. propylene glycol, and, if desired, a suitable amount of lidocaine hydrochloride.
- the solutions for intravenous injections or infusions may contain as carrier, for example, sterile water or preferably they may be in the form of sterile, aqueous, isotonic saline solutions or they may contain as a carrier propylene glycol.
- carrier for example, sterile water or preferably they may be in the form of sterile, aqueous, isotonic saline solutions or they may contain as a carrier propylene glycol.
- the suppositories may contain together with the active compound a pharmaceutically acceptable carrier, e.g. cocoa butter, polyethylene glycol, a polyoxyethylene sorbitan fatty ester surfactant or lecithin.
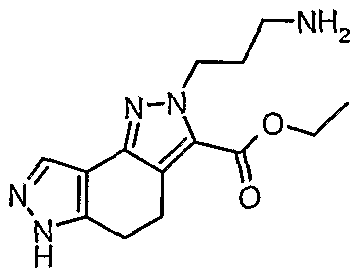
- Step 2 To a suspension of l,5,6,7-tetrahydro-4H-indazol-4-one (8 g, 58.75 mmols) and trityl chloride (18.02 g, 64.64 mmols) in dichloromethane (160 ml), triethylamine (9.8 ml, 70.50 mmols) was added dropwise. The reaction was slightly exothermic.
- Step 3 To a suspension of 2-trityl-2,5,6,7-tetrahydro-4H-indazol-4-one (20 g,
- Step 4 A suspension of ethyl oxo(4-oxo-2-trityl-4,5,6,7-tetrahydro-2H-indazol-5- yl)acetate (400 mg, 0.84 mmols) and (4-methoxyphenyl)-hydrazine hydrochloride (164 mg, 0.94 mmols) in acetic acid (4 ml) was stirred at 65°C for 3 hours.
- Step 1 A solution of 3-Ethoxy-cyclohex-2-enone (4.65 ml, 31.92 mmols) and diethyl oxalate (6.49 ml, 47.89 mmols) in anhydrous ethyl ether (50 ml) is treated dropwise with a IM solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (47.9 ml, 47.9 mmols) under argon atmosphere. After standing at room temperature overnight, the mixture is poured into a 20% NaH2PO4 solution (150 ml) and extracted with ethyl acetate (100 ml x2).
- Step 2 (4-Ethoxy-2-oxo-cyclohex-3-enyl)-oxo-acetic acid ethyl ester (8 g, 31.92 mmols theoretically) is treated with methylhydrazine (1.69 ml, 31.92 mmol) in EtOH (75 ml) and AcOH (5 ml) at room temperature. After 3 hours the solution was concentrated 03/07023
- Step 3 Ethyl 6-ethoxy-l-methyl-4,5-dihydro-lH-indazole-3-carboxylate (7.59 g,
- Example 6 1 - [l-(4-methylphenyl)-l ,6-dihydropyrazo ⁇ o [3 ,4-e] indazol-3-yl] ethanone
- Triethylamine (0.82 ml; 6 mmols) was added to a 3M solution of EtMgCl in THF (0.67 ml; 2 mmols) at 0°C under argon atmosphere. After 10 min, a solution of ethyl l-(4- methylphenyl)-7-trityl-l,7-dihydropyrazolo[3,4-e]indazole-3-carboxylate (564 mg; 1 mmol) in anhydrous THF (6 ml) was added dropwise. After leaving at 0°C for 1 hour and 30 min at room temperature the resulting mixture was poured into a 20% NaH 2 PO 3 solution and extracted with ethyl acetate.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Urology & Nephrology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Epidemiology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Virology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Transplantation (AREA)
- Vascular Medicine (AREA)
- Psychiatry (AREA)
- Communicable Diseases (AREA)
- Pulmonology (AREA)
- Hospice & Palliative Care (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description





























Claims







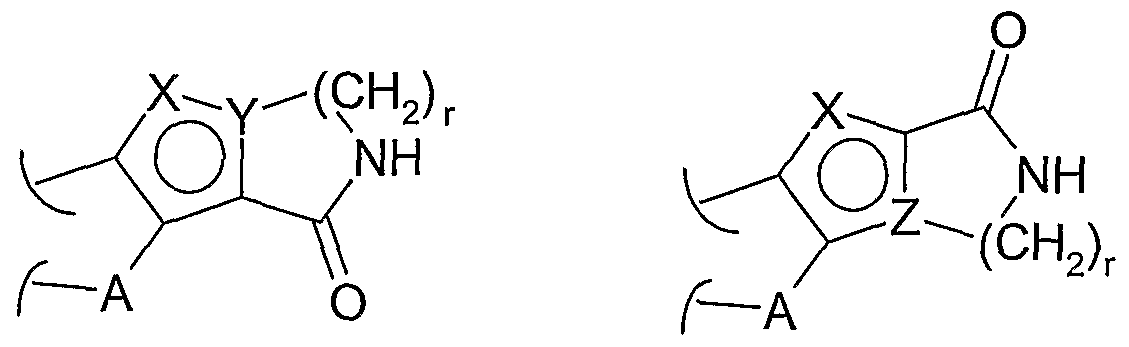










Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA04008680A MXPA04008680A (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents. |
| DE60311567T DE60311567T2 (en) | 2002-02-19 | 2003-02-18 | TRICYCLIC PYRAZOLE DERIVATIVES, METHOD FOR THE PRODUCTION THEREOF AND THEIR USE AS ANTITUMOR AGENTS |
| US10/505,200 US20050176796A1 (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
| JP2003569192A JP2005529850A (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, their preparation and their use as antitumor agents |
| BR0307819-1A BR0307819A (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for preparing them as antitumor agents |
| EP03714744A EP1478357B1 (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
| AU2003218989A AU2003218989A1 (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
| CA002476822A CA2476822A1 (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US35791802P | 2002-02-19 | 2002-02-19 | |
| US60/357,918 | 2002-02-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2003070236A2 true WO2003070236A2 (en) | 2003-08-28 |
| WO2003070236A3 WO2003070236A3 (en) | 2004-02-12 |
Family
ID=27757676
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/001594 Ceased WO2003070236A2 (en) | 2002-02-19 | 2003-02-18 | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US20050176796A1 (en) |
| EP (1) | EP1478357B1 (en) |
| JP (1) | JP2005529850A (en) |
| AT (1) | ATE353013T1 (en) |
| AU (1) | AU2003218989A1 (en) |
| BR (1) | BR0307819A (en) |
| CA (1) | CA2476822A1 (en) |
| DE (1) | DE60311567T2 (en) |
| ES (1) | ES2281633T3 (en) |
| MX (1) | MXPA04008680A (en) |
| WO (1) | WO2003070236A2 (en) |
Cited By (70)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014922A1 (en) * | 2002-08-10 | 2004-02-19 | Astex Technology Limited | 3-(carbonyl) 1h-indazole compounds as cyclin dependent kinases (cdk) inhibitors |
| WO2005095387A1 (en) | 2004-03-24 | 2005-10-13 | Abbott Laboratories | Tricyclic pyrazole kinase inhibitors |
| EP1602658A1 (en) * | 2004-05-24 | 2005-12-07 | Neuroscienze Oharmaness S.c.a.r.l. | Tricyclic pyrazole derivatives as cannabinoid receptor antagonists |
| WO2007022268A3 (en) * | 2005-08-16 | 2007-04-12 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2007060198A1 (en) * | 2005-11-25 | 2007-05-31 | Palau Pharma, S. A. | Pyrazoloisoquinoline derivatives |
| JP2007521331A (en) * | 2003-11-19 | 2007-08-02 | シグナル ファーマシューティカルズ,エルエルシー | Methods of treating diseases and disorders by targeting multiple kinases |
| US7320986B2 (en) | 2003-03-07 | 2008-01-22 | Abbott Labortories | Fused tri and tetra-cyclic pyrazole kinase inhibitors |
| WO2008074788A1 (en) | 2006-12-21 | 2008-06-26 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US7456169B2 (en) | 2003-02-27 | 2008-11-25 | Abbott Laboratories Inc. | Heterocyclic kinase inhibitors |
| US7601847B2 (en) | 2004-10-26 | 2009-10-13 | Wyeth | Preparation and purification of 4-(indazol-3-yl)phenols |
| US7947707B2 (en) | 2005-10-07 | 2011-05-24 | Kissei Pharmaceutical Co., Ltd. | Nitrogenated heterocyclic compound and pharmaceutical composition comprising the same |
| US8053440B2 (en) | 2007-02-01 | 2011-11-08 | Resverlogix Corporation | Compounds for the prevention and treatment of cardiovascular diseases |
| US8114995B2 (en) | 2008-06-26 | 2012-02-14 | Resverlogix Corp. | Methods of preparing quinazolinone derivatives |
| US8252812B2 (en) | 2009-08-10 | 2012-08-28 | Samumed, Llc | Indazole inhibitors of the WNT signal pathway and therapeutic uses thereof |
| US8410109B2 (en) | 2005-07-29 | 2013-04-02 | Resverlogix Corp. | Pharmaceutical compositions for the prevention and treatment of complex diseases and their delivery by insertable medical devices |
| US8450340B2 (en) | 2009-12-21 | 2013-05-28 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US8507541B2 (en) | 2006-09-19 | 2013-08-13 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| CN103387543A (en) * | 2013-07-20 | 2013-11-13 | 李佰林 | Synthesis method of tetrahydropyrazolone derivative |
| US8618128B1 (en) | 2012-05-04 | 2013-12-31 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US8673936B2 (en) | 2012-04-04 | 2014-03-18 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US8796319B2 (en) | 2008-07-08 | 2014-08-05 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US8846726B2 (en) | 2005-05-10 | 2014-09-30 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| EP2789619A1 (en) | 2013-04-12 | 2014-10-15 | Kemotech S.r.l. | Pharmaceutical compounds wiht angiogenesis inbhibitory activity |
| US8951536B2 (en) | 2005-12-20 | 2015-02-10 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US8952021B2 (en) | 2009-01-08 | 2015-02-10 | Resverlogix Corp. | Compounds for the prevention and treatment of cardiovascular disease |
| US9073878B2 (en) | 2012-11-21 | 2015-07-07 | Zenith Epigenetics Corp. | Cyclic amines as bromodomain inhibitors |
| US9238640B2 (en) | 2009-03-18 | 2016-01-19 | Resverlogix Corp. | Anti-inflammatory agents |
| US9271978B2 (en) | 2012-12-21 | 2016-03-01 | Zenith Epigenetics Corp. | Heterocyclic compounds as bromodomain inhibitors |
| US9321755B2 (en) | 2013-11-08 | 2016-04-26 | Incyte Corporation | Process for the synthesis of an indoleamine 2,3-dioxygenase inhibitor |
| CN105916848A (en) * | 2013-12-31 | 2016-08-31 | 山东轩竹医药科技有限公司 | Kinase inhibitors and uses thereof |
| US9610251B2 (en) | 2011-11-01 | 2017-04-04 | Resverlogix Corp. | Pharmaceutical compositions for substituted quinazolinones |
| US9657016B2 (en) | 2014-09-08 | 2017-05-23 | Samumed, Llc | 3-(1h-benzo[d]imidazol-2-yl)-1h-pyrazolo[3,4-c]pyridine and therapeutic uses thereof |
| US9738638B2 (en) | 2014-09-08 | 2017-08-22 | Samumed, Llc | 2-(1H-indazol-3-yl)-3H-imidazo[4,5-B]pyridine and therapeutic uses thereof |
| US9757368B2 (en) | 2009-04-22 | 2017-09-12 | Resverlogix Corp. | Anti-inflammatory agents |
| US9758531B2 (en) | 2014-09-08 | 2017-09-12 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US9765039B2 (en) | 2012-11-21 | 2017-09-19 | Zenith Epigenetics Ltd. | Biaryl derivatives as bromodomain inhibitors |
| US9763951B2 (en) | 2014-09-08 | 2017-09-19 | Samumed, Llc | 3-(3H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US9802916B2 (en) | 2011-09-14 | 2017-10-31 | Samumed, Llc | Indazole-3-carboxamides and their use as Wnt/beta-catenin signaling pathway inhibitors |
| US9844536B2 (en) | 2014-09-08 | 2017-12-19 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| WO2018005883A1 (en) * | 2016-06-29 | 2018-01-04 | Novira Therapeutics, Inc. | Diazepinone derivatives and their use in the treatment of hepatitis b infections |
| WO2018005881A1 (en) * | 2016-06-29 | 2018-01-04 | Novira Therapeutics, Inc. | Oxadiazepinone derivatives and their use in the treatment of hepatitis b infections |
| US9889140B2 (en) | 2014-09-08 | 2018-02-13 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US9908867B2 (en) | 2013-01-08 | 2018-03-06 | Samumed, Llc | 3-(benzoimidazol-2-yl)-indazole inhibitors of the Wnt signaling pathway and therapeutic uses thereof |
| US9949976B2 (en) | 2013-12-31 | 2018-04-24 | Xuanzhu Pharma Co., Ltd. | Kinase inhibitor and use thereof |
| US10072004B2 (en) | 2016-06-01 | 2018-09-11 | Samumed, Llc | Process for preparing N-(5-(3-(7-(3-fluorophenyl)-3H-imidazo [4,5-C]pyridin-2-yl)-1H-indazol-5-yl)pyridin-3-yl)-3-methylbutanamide |
| US10081631B2 (en) | 2014-09-08 | 2018-09-25 | Samumed, Llc | 2-(1H-indazol-3-yl)-1H-imidazo[4,5-C]pyridine and therapeutic uses thereof |
| US10111885B2 (en) | 2015-03-13 | 2018-10-30 | Resverlogix Corp. | Compositions and therapeutic methods for the treatment of complement-associated diseases |
| US10166218B2 (en) | 2015-08-03 | 2019-01-01 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10188634B2 (en) | 2015-08-03 | 2019-01-29 | Samumed, Llc | 3-(3H-imidazo[4,5-C]pyridin-2-yl)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10195185B2 (en) | 2015-08-03 | 2019-02-05 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10206909B2 (en) | 2015-08-03 | 2019-02-19 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10206908B2 (en) | 2015-08-03 | 2019-02-19 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10226453B2 (en) | 2015-08-03 | 2019-03-12 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10226448B2 (en) | 2015-08-03 | 2019-03-12 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridines and therapeutic uses thereof |
| US10231956B2 (en) | 2015-08-03 | 2019-03-19 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10285982B2 (en) | 2015-08-03 | 2019-05-14 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10285983B2 (en) | 2015-08-03 | 2019-05-14 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[3,4-B] pyridines and therapeutic uses thereof |
| US10329309B2 (en) | 2015-08-03 | 2019-06-25 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10350199B2 (en) | 2015-08-03 | 2019-07-16 | Samumed, Llc | 3-(1h-pyrrolo[2,3-b]pyridin-2-yl)-1h-indazoles and therapeutic uses thereof |
| US10383861B2 (en) | 2015-08-03 | 2019-08-20 | Sammumed, LLC | 3-(1H-pyrrolo[2,3-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10392383B2 (en) | 2015-08-03 | 2019-08-27 | Samumed, Llc | 3-(1H-benzo[d]imidazol-2-yl)-1H-pyrazolo[4,3-b]pyridines and therapeutic uses thereof |
| US10463651B2 (en) | 2015-08-03 | 2019-11-05 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1H-indazoles and therapeutic uses thereof |
| US10519169B2 (en) | 2015-08-03 | 2019-12-31 | Samumed, Llc | 3-(1H-pyrrolo[2,3-C]pyridin-2-yl)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10533020B2 (en) | 2014-09-08 | 2020-01-14 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1 H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10544139B2 (en) | 2015-11-06 | 2020-01-28 | Samumed, Llc | Treatment of osteoarthritis |
| US10604512B2 (en) | 2015-08-03 | 2020-03-31 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-indazoles and therapeutic uses thereof |
| US10758523B2 (en) | 2016-11-07 | 2020-09-01 | Samumed, Llc | Single-dose, ready-to-use injectable formulations |
| US10806726B2 (en) | 2016-10-21 | 2020-10-20 | Samumed, Llc | Methods of using indazole-3-carb oxamides and their use as Wnt/B-catenin signaling pathway inhibitors |
| US12281097B2 (en) | 2016-04-27 | 2025-04-22 | Biosplice Therapeutics, Inc. | Isoquinolin-3-yl carboxamides and preparation and use thereof |
| WO2025157165A1 (en) * | 2024-01-23 | 2025-07-31 | 杭州中美华东制药有限公司 | Compound having pkmyt1 inhibition effect |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2002215053A1 (en) * | 2000-11-27 | 2002-06-24 | Pharmacia Italia S.P.A. | Phenylacetamido- pyrazole derivatives and their use as antitumor agents |
| US20060115453A1 (en) * | 2004-11-12 | 2006-06-01 | Yaffe Michael B | Methods and compositions for treating cellular proliferative diseases |
| US8440610B2 (en) * | 2004-11-12 | 2013-05-14 | Massachusetts Institute Of Technology | Mapkap kinase-2 as a specific target for blocking proliferation of P53-defective cells |
| WO2007081572A2 (en) * | 2006-01-04 | 2007-07-19 | Merck & Co., Inc. | Inhibitors of checkpoint kinases |
| CN101379060B (en) | 2006-02-10 | 2012-05-23 | 转化技术制药公司 | Benzoxazole Derivatives, Compositions and Methods of Use as Aurora Kinase Inhibitors |
| JP5330990B2 (en) * | 2007-04-11 | 2013-10-30 | キッセイ薬品工業株式会社 | Nitrogen-containing heterocyclic compound and pharmaceutical composition containing the same |
| MX2009014247A (en) * | 2007-07-16 | 2010-01-28 | Novartis Ag | Heterocyclic compounds useful as mk2 inhibitors. |
| AU2008286879B2 (en) | 2007-08-13 | 2013-08-22 | Monsanto Technology Llc | Compositions and methods for controlling nematodes |
| CN104592231A (en) * | 2008-06-10 | 2015-05-06 | Abbvie公司 | Novel Tricyclic Compounds |
| US7741350B1 (en) | 2009-01-28 | 2010-06-22 | Cara Therapeutics, Inc. | Bicyclic pyrazolo-heterocycles |
| WO2010088050A2 (en) | 2009-01-28 | 2010-08-05 | Cara Therapeutics, Inc. | Bicyclic pyrazolo-heterocycles |
| RU2711869C3 (en) | 2009-12-01 | 2022-02-02 | Эббви Инк. | Imidazopyrrolopyrazine derivatives useful in the treatment of diseases caused by abnormal activity of Jak1, Jak3 or Syk protein kinases |
| PH12012501000A1 (en) * | 2009-12-01 | 2013-02-11 | Abbvie Inc | Novel tricyclic compounds |
| JP5730896B2 (en) * | 2009-12-04 | 2015-06-10 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclopyrazole derivatives |
| MX338327B (en) * | 2010-10-25 | 2016-04-12 | G1 Therapeutics Inc | Cdk inhibitors. |
| WO2015077572A1 (en) | 2013-11-22 | 2015-05-28 | CL BioSciences LLC | Gastrin antagonists (eg yf476, netazepide) for treatment and prevention of osteoporosis |
| US10550126B2 (en) | 2015-10-16 | 2020-02-04 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-A]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11773106B2 (en) | 2015-10-16 | 2023-10-03 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11524964B2 (en) | 2015-10-16 | 2022-12-13 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US11365198B2 (en) | 2015-10-16 | 2022-06-21 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-N-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| SG10201913990RA (en) | 2015-10-16 | 2020-03-30 | Abbvie Inc | PROCESSES FOR THE PREPARATION OF (3S,4R)-3-ETHYL-4-(3H-IMIDAZO[1,2-a]PYRROLO[2,3-e]-PYRAZIN-8-YL)-N-(2,2,2-TRIFLUOROETHYL)PYRROLIDINE-1-CARBOXAMIDE AND SOLID STATE FORMS THEREOF |
| US11512092B2 (en) | 2015-10-16 | 2022-11-29 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| US12365689B2 (en) | 2015-10-16 | 2025-07-22 | Abbvie Inc. | Processes for the preparation of (3S,4R)-3-ethyl-4-(3H-imidazo[1,2-a]pyrrolo[2,3-e]-pyrazin-8-yl)-n-(2,2,2-trifluoroethyl)pyrrolidine-1-carboxamide and solid state forms thereof |
| AR111419A1 (en) * | 2017-04-27 | 2019-07-10 | Novartis Ag | INDAZOL PIRIDONA FUSIONED COMPOUNDS AS ANTIVIRALS |
| CN109574936B (en) * | 2018-11-23 | 2022-02-22 | 沈阳药科大学 | A kind of hydroxamic acid compound with HDAC6 inhibitory activity and its application |
| EP3827816A1 (en) | 2019-11-28 | 2021-06-02 | Universitätsmedizin der Johannes Gutenberg-Universität Mainz | Prooxidative chain-transfer agents for use in the treatment of malignant tumour or infectious diseases |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR880009024A (en) * | 1987-01-27 | 1988-09-13 | 게리 디.스트리트 | Dipyrazoles for Bronchodilators |
| US4734429A (en) * | 1987-01-27 | 1988-03-29 | Merrell Dow Pharmaceuticals Inc. | Cycloalkane[1,2-c:4,3-c']dipyrazoles and their use as bronchodilators |
| US6046208A (en) * | 1996-01-11 | 2000-04-04 | Smithkline Beecham Corporation | Substituted imidazole compounds |
| US6218136B1 (en) * | 1996-03-12 | 2001-04-17 | Smithkline Beecham Corporation | Methods of the identification of pharmaceutically active compounds |
| US6514977B1 (en) * | 1997-05-22 | 2003-02-04 | G.D. Searle & Company | Substituted pyrazoles as p38 kinase inhibitors |
| US6251914B1 (en) * | 1997-07-02 | 2001-06-26 | Smithkline Beecham Corporation | Cycloalkyl substituted imidazoles |
| ES2339196T3 (en) * | 1997-07-18 | 2010-05-17 | Novo Nordisk Health Care Ag | USE OF FVIIA OR FVIIAI FOR THE TREATMENT OF ENDOTELIAL DYSFUNCTION AND FOR THE INHIBITION OF ANGIOGENESIS, RESPECTIVELY. |
| AU1924699A (en) * | 1997-12-19 | 1999-07-12 | Smithkline Beecham Corporation | Compounds of heteroaryl substituted imidazole, their pharmaceutical compositionsand uses |
| CN1311678A (en) * | 1998-04-30 | 2001-09-05 | 巴斯福股份公司 | Substituted tricyclic pyrazole derivatives with protein kinase activity |
| AU762992B2 (en) * | 1998-11-06 | 2003-07-10 | Abbott Gmbh & Co. Kg | Tricyclic pyrazole derivatives |
| US6462036B1 (en) * | 1998-11-06 | 2002-10-08 | Basf Aktiengesellschaft | Tricyclic pyrazole derivatives |
| US6143892A (en) * | 1998-11-20 | 2000-11-07 | G. D. Searle & Co. | Process for making 5-substituted pyrazoles |
| PL350891A1 (en) * | 1999-04-06 | 2003-02-10 | Knoll Gmbh | Substituted 1,4-dichlorindene[1,2-c]pyrazoles as inhibitors of tyrosine kinase |
| HUP0301932A3 (en) * | 2000-05-22 | 2007-09-28 | Leo Pharma As | Benzophenones as inhibitors of il-1 beta and tnf-alpha, pharmaceutical compositions containing them and use of them for producing pharmaceutical compositions |
-
2003
- 2003-02-18 US US10/505,200 patent/US20050176796A1/en not_active Abandoned
- 2003-02-18 AU AU2003218989A patent/AU2003218989A1/en not_active Abandoned
- 2003-02-18 WO PCT/EP2003/001594 patent/WO2003070236A2/en not_active Ceased
- 2003-02-18 BR BR0307819-1A patent/BR0307819A/en not_active IP Right Cessation
- 2003-02-18 JP JP2003569192A patent/JP2005529850A/en not_active Withdrawn
- 2003-02-18 ES ES03714744T patent/ES2281633T3/en not_active Expired - Lifetime
- 2003-02-18 DE DE60311567T patent/DE60311567T2/en not_active Expired - Fee Related
- 2003-02-18 MX MXPA04008680A patent/MXPA04008680A/en unknown
- 2003-02-18 EP EP03714744A patent/EP1478357B1/en not_active Expired - Lifetime
- 2003-02-18 US US10/369,055 patent/US20040127492A1/en not_active Abandoned
- 2003-02-18 CA CA002476822A patent/CA2476822A1/en not_active Abandoned
- 2003-02-18 AT AT03714744T patent/ATE353013T1/en not_active IP Right Cessation
Cited By (144)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014922A1 (en) * | 2002-08-10 | 2004-02-19 | Astex Technology Limited | 3-(carbonyl) 1h-indazole compounds as cyclin dependent kinases (cdk) inhibitors |
| US7456169B2 (en) | 2003-02-27 | 2008-11-25 | Abbott Laboratories Inc. | Heterocyclic kinase inhibitors |
| US7320986B2 (en) | 2003-03-07 | 2008-01-22 | Abbott Labortories | Fused tri and tetra-cyclic pyrazole kinase inhibitors |
| JP2007521331A (en) * | 2003-11-19 | 2007-08-02 | シグナル ファーマシューティカルズ,エルエルシー | Methods of treating diseases and disorders by targeting multiple kinases |
| EP1791831A4 (en) * | 2003-11-19 | 2009-07-08 | Signal Pharm Llc | Methods of treating diseases and disorders by targeting multiple kinases |
| WO2005095387A1 (en) | 2004-03-24 | 2005-10-13 | Abbott Laboratories | Tricyclic pyrazole kinase inhibitors |
| US7468371B2 (en) | 2004-03-24 | 2008-12-23 | Abbott Laboratories Inc. | Tricyclic pyrazole kinase inhibitors |
| US8106218B2 (en) | 2004-05-24 | 2012-01-31 | Neuroscienze Pharmaness S.C. A R.L. | Pharmaceutical compounds |
| EP1602658A1 (en) * | 2004-05-24 | 2005-12-07 | Neuroscienze Oharmaness S.c.a.r.l. | Tricyclic pyrazole derivatives as cannabinoid receptor antagonists |
| JP2005350457A (en) * | 2004-05-24 | 2005-12-22 | Neuroscienze Pharmaness Scarl | Pharmaceutical compounds |
| US7485730B2 (en) | 2004-05-24 | 2009-02-03 | Neuroscienze Pharmaness S.C. A R.L. | Pharmaceutical compounds |
| US7601847B2 (en) | 2004-10-26 | 2009-10-13 | Wyeth | Preparation and purification of 4-(indazol-3-yl)phenols |
| US10208002B2 (en) | 2005-05-10 | 2019-02-19 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| US11192868B2 (en) | 2005-05-10 | 2021-12-07 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| US8846726B2 (en) | 2005-05-10 | 2014-09-30 | Incyte Corporation | Modulators of indoleamine 2,3-dioxygenase and methods of using the same |
| US8410109B2 (en) | 2005-07-29 | 2013-04-02 | Resverlogix Corp. | Pharmaceutical compositions for the prevention and treatment of complex diseases and their delivery by insertable medical devices |
| WO2007022268A3 (en) * | 2005-08-16 | 2007-04-12 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| JP2009504757A (en) * | 2005-08-16 | 2009-02-05 | アイアールエム・リミテッド・ライアビリティ・カンパニー | Compounds and compositions as protein kinase inhibitors |
| US7947707B2 (en) | 2005-10-07 | 2011-05-24 | Kissei Pharmaceutical Co., Ltd. | Nitrogenated heterocyclic compound and pharmaceutical composition comprising the same |
| WO2007060198A1 (en) * | 2005-11-25 | 2007-05-31 | Palau Pharma, S. A. | Pyrazoloisoquinoline derivatives |
| US8951536B2 (en) | 2005-12-20 | 2015-02-10 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| US8507541B2 (en) | 2006-09-19 | 2013-08-13 | Incyte Corporation | N-hydroxyamidinoheterocycles as modulators of indoleamine 2,3-dioxygenase |
| WO2008074788A1 (en) | 2006-12-21 | 2008-06-26 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US8889698B2 (en) | 2007-02-01 | 2014-11-18 | Resverlogix Corp. | Compounds for the prevention and treatment of cardiovascular diseases |
| US10532054B2 (en) | 2007-02-01 | 2020-01-14 | Resverlogix Corp. | Compounds for the prevention and treatment of cardiovascular diseases |
| US8053440B2 (en) | 2007-02-01 | 2011-11-08 | Resverlogix Corporation | Compounds for the prevention and treatment of cardiovascular diseases |
| US9199990B2 (en) | 2007-02-01 | 2015-12-01 | Resverlogix Corp. | Compounds for the prevention and treatment of cardiovascular diseases |
| US8114995B2 (en) | 2008-06-26 | 2012-02-14 | Resverlogix Corp. | Methods of preparing quinazolinone derivatives |
| US8796319B2 (en) | 2008-07-08 | 2014-08-05 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US9789094B2 (en) | 2008-07-08 | 2017-10-17 | Incyte Holdings Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US8822511B2 (en) | 2008-07-08 | 2014-09-02 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US10369137B2 (en) | 2008-07-08 | 2019-08-06 | Incyte Corporation | 1,2,5-Oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US10034864B2 (en) | 2008-07-08 | 2018-07-31 | Incyte Holdings Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US10653677B2 (en) | 2008-07-08 | 2020-05-19 | Incyte Holdings Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US8993605B2 (en) | 2008-07-08 | 2015-03-31 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US9320732B2 (en) | 2008-07-08 | 2016-04-26 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US11207302B2 (en) | 2008-07-08 | 2021-12-28 | Incyte Corporation | 1,2,5-oxadiazoles as inhibitors of indoleamine 2,3-dioxygenase |
| US8952021B2 (en) | 2009-01-08 | 2015-02-10 | Resverlogix Corp. | Compounds for the prevention and treatment of cardiovascular disease |
| US9238640B2 (en) | 2009-03-18 | 2016-01-19 | Resverlogix Corp. | Anti-inflammatory agents |
| US10882828B2 (en) | 2009-03-18 | 2021-01-05 | Resverlogix Corp. | Anti-inflammatory agents |
| US10131640B2 (en) | 2009-03-18 | 2018-11-20 | Resverlogix Corp. | Anti-inflammatory agents |
| US11407719B2 (en) | 2009-03-18 | 2022-08-09 | Resverlogix Corp. | Anti-inflammatory agents |
| US9757368B2 (en) | 2009-04-22 | 2017-09-12 | Resverlogix Corp. | Anti-inflammatory agents |
| US10016406B2 (en) | 2009-08-10 | 2018-07-10 | Samumed, Llc | Indazole inhibitors of the WNT signal pathway and therapeutic uses thereof |
| US9090613B2 (en) | 2009-08-10 | 2015-07-28 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US9763927B2 (en) | 2009-08-10 | 2017-09-19 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US8252812B2 (en) | 2009-08-10 | 2012-08-28 | Samumed, Llc | Indazole inhibitors of the WNT signal pathway and therapeutic uses thereof |
| US8604052B2 (en) | 2009-08-10 | 2013-12-10 | Samumed, Llc | Indazole inhibitors of the WNT signal pathway and therapeutic uses thereof |
| US9067939B2 (en) | 2009-12-21 | 2015-06-30 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US10105370B2 (en) | 2009-12-21 | 2018-10-23 | Samumed, Llc | 1H-pyrazolo[3,4-B]pyridines and therapeutic uses thereof |
| US9855272B2 (en) | 2009-12-21 | 2018-01-02 | Samumed, Llc | 1H-pyrazolo[3,4-B]pyridines and therapeutic uses thereof |
| US8450340B2 (en) | 2009-12-21 | 2013-05-28 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US10464924B2 (en) | 2011-09-14 | 2019-11-05 | Samumed, Llc | Indazole-3-carboxamides and their use as Wnt/β-catenin signaling pathway inhibitors |
| US11066388B2 (en) | 2011-09-14 | 2021-07-20 | Biosplice Therapeutics, Inc. | Indazole-3-carboxamides and their use as WNT/B-catenin signaling pathway inhibitors |
| US9802916B2 (en) | 2011-09-14 | 2017-10-31 | Samumed, Llc | Indazole-3-carboxamides and their use as Wnt/beta-catenin signaling pathway inhibitors |
| US11780823B2 (en) | 2011-09-14 | 2023-10-10 | Biosplice Therapeutics, Inc. | Indazole-3-carboxamides and their use as Wnt/β-catenin signaling pathway inhibitors |
| US9610251B2 (en) | 2011-11-01 | 2017-04-04 | Resverlogix Corp. | Pharmaceutical compositions for substituted quinazolinones |
| US10016426B2 (en) | 2011-11-01 | 2018-07-10 | Resverlogix Corp. | Pharmaceutical compositions for substituted quinazolinones |
| US10947228B2 (en) | 2012-04-04 | 2021-03-16 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US8673936B2 (en) | 2012-04-04 | 2014-03-18 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US10407425B2 (en) | 2012-04-04 | 2019-09-10 | Samumed, Llc | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US11697649B2 (en) | 2012-04-04 | 2023-07-11 | Biosplice Therapeutics, Inc. | Indazole inhibitors of the Wnt signal pathway and therapeutic uses thereof |
| US9994563B2 (en) | 2012-04-04 | 2018-06-12 | Samumed, Llc | Indazole inhibitors of the wnt signal pathway and therapeutic uses thereof |
| US10071086B2 (en) | 2012-05-04 | 2018-09-11 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US10342788B2 (en) | 2012-05-04 | 2019-07-09 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US8618128B1 (en) | 2012-05-04 | 2013-12-31 | Samumed, Llc | 1H-pyrazolo[3,4-b]pyridines and therapeutic uses thereof |
| US9765039B2 (en) | 2012-11-21 | 2017-09-19 | Zenith Epigenetics Ltd. | Biaryl derivatives as bromodomain inhibitors |
| US9073878B2 (en) | 2012-11-21 | 2015-07-07 | Zenith Epigenetics Corp. | Cyclic amines as bromodomain inhibitors |
| US9278940B2 (en) | 2012-11-21 | 2016-03-08 | Zenith Epigenetics Corp. | Cyclic amines as bromodomain inhibitors |
| US9271978B2 (en) | 2012-12-21 | 2016-03-01 | Zenith Epigenetics Corp. | Heterocyclic compounds as bromodomain inhibitors |
| US10654832B2 (en) | 2013-01-08 | 2020-05-19 | Samumed, Llc | 3-(benzoimidazol-2-YL)-indazole inhibitors of the Wnt signaling pathway and therapeutic uses thereof |
| US9908867B2 (en) | 2013-01-08 | 2018-03-06 | Samumed, Llc | 3-(benzoimidazol-2-yl)-indazole inhibitors of the Wnt signaling pathway and therapeutic uses thereof |
| US10183929B2 (en) | 2013-01-08 | 2019-01-22 | Samumed, Llc | 3-(benzoimidazol-2-yl)-indazole inhibitors of the Wnt signaling pathway and therapeutic uses thereof |
| US9181196B2 (en) | 2013-04-12 | 2015-11-10 | Kemotech S.R.L. | Pharmaceutical compounds |
| EP2789619A1 (en) | 2013-04-12 | 2014-10-15 | Kemotech S.r.l. | Pharmaceutical compounds wiht angiogenesis inbhibitory activity |
| CN103387543A (en) * | 2013-07-20 | 2013-11-13 | 李佰林 | Synthesis method of tetrahydropyrazolone derivative |
| CN103387543B (en) * | 2013-07-20 | 2015-08-05 | 李佰林 | A kind of synthetic method of tetrahydropyrazolonederivative derivative |
| US9321755B2 (en) | 2013-11-08 | 2016-04-26 | Incyte Corporation | Process for the synthesis of an indoleamine 2,3-dioxygenase inhibitor |
| US10280157B2 (en) | 2013-11-08 | 2019-05-07 | Incyte Corporation | Process for the synthesis of an indoleamine 2,3-dioxygenase inhibitor |
| US9873688B2 (en) | 2013-11-08 | 2018-01-23 | Incyte Holdings Corporation | Process for the synthesis of an indoleamine 2,3-dioxygenase inhibitor |
| CN105916848B (en) * | 2013-12-31 | 2018-01-09 | 山东轩竹医药科技有限公司 | Kinase inhibitors and uses thereof |
| US9949976B2 (en) | 2013-12-31 | 2018-04-24 | Xuanzhu Pharma Co., Ltd. | Kinase inhibitor and use thereof |
| CN105916848A (en) * | 2013-12-31 | 2016-08-31 | 山东轩竹医药科技有限公司 | Kinase inhibitors and uses thereof |
| US9796701B2 (en) | 2013-12-31 | 2017-10-24 | Xuanzhu Pharma Co., Ltd. | Kinase inhibitor and use thereof |
| US10202377B2 (en) | 2014-09-08 | 2019-02-12 | Samumed, Llc | 3-(1H-benzo[D]imidazol-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10526347B2 (en) | 2014-09-08 | 2020-01-07 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US10052331B2 (en) | 2014-09-08 | 2018-08-21 | Samumed, Llc | 3-(3H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10023572B2 (en) | 2014-09-08 | 2018-07-17 | Samumed, Llc | 2-(1h-indazol-3-yl)-3h-imidazo[4,5-b]pyridine and therapeutic uses thereof |
| US10131677B2 (en) | 2014-09-08 | 2018-11-20 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US9758531B2 (en) | 2014-09-08 | 2017-09-12 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US9844536B2 (en) | 2014-09-08 | 2017-12-19 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US9889140B2 (en) | 2014-09-08 | 2018-02-13 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridine and therapeutic uses thereof |
| US10596154B2 (en) | 2014-09-08 | 2020-03-24 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10081631B2 (en) | 2014-09-08 | 2018-09-25 | Samumed, Llc | 2-(1H-indazol-3-yl)-1H-imidazo[4,5-C]pyridine and therapeutic uses thereof |
| US10280166B2 (en) | 2014-09-08 | 2019-05-07 | Samumed, Llc | 2-(1H-indazol-3-yl)-3H-imidazo[4,5-B]pyridine and therapeutic uses thereof |
| US9738638B2 (en) | 2014-09-08 | 2017-08-22 | Samumed, Llc | 2-(1H-indazol-3-yl)-3H-imidazo[4,5-B]pyridine and therapeutic uses thereof |
| US10533020B2 (en) | 2014-09-08 | 2020-01-14 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1 H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10206929B2 (en) | 2014-09-08 | 2019-02-19 | Samumed, Llc | 3-(1H-imidazo[4,5-c]pyridin-2-yl)-1H-pyrazolo[3,4-b]pyridine and therapeutic uses thereof |
| US9657016B2 (en) | 2014-09-08 | 2017-05-23 | Samumed, Llc | 3-(1h-benzo[d]imidazol-2-yl)-1h-pyrazolo[3,4-c]pyridine and therapeutic uses thereof |
| US9763951B2 (en) | 2014-09-08 | 2017-09-19 | Samumed, Llc | 3-(3H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridine and therapeutic uses thereof |
| US10111885B2 (en) | 2015-03-13 | 2018-10-30 | Resverlogix Corp. | Compositions and therapeutic methods for the treatment of complement-associated diseases |
| US10772894B2 (en) | 2015-03-13 | 2020-09-15 | Resverlogix Corp. | Compositions and therapeutic methods for the treatment of complement-associated diseases |
| US10392383B2 (en) | 2015-08-03 | 2019-08-27 | Samumed, Llc | 3-(1H-benzo[d]imidazol-2-yl)-1H-pyrazolo[4,3-b]pyridines and therapeutic uses thereof |
| US10206909B2 (en) | 2015-08-03 | 2019-02-19 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10350199B2 (en) | 2015-08-03 | 2019-07-16 | Samumed, Llc | 3-(1h-pyrrolo[2,3-b]pyridin-2-yl)-1h-indazoles and therapeutic uses thereof |
| US10463651B2 (en) | 2015-08-03 | 2019-11-05 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1H-indazoles and therapeutic uses thereof |
| US10329309B2 (en) | 2015-08-03 | 2019-06-25 | Samumed, Llc | 3-(3H-imidazo[4,5-B]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10519169B2 (en) | 2015-08-03 | 2019-12-31 | Samumed, Llc | 3-(1H-pyrrolo[2,3-C]pyridin-2-yl)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10166218B2 (en) | 2015-08-03 | 2019-01-01 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10285983B2 (en) | 2015-08-03 | 2019-05-14 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[3,4-B] pyridines and therapeutic uses thereof |
| US10285982B2 (en) | 2015-08-03 | 2019-05-14 | Samumed, Llc | 3-(1H-pyrrolo[2,3-B]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10188634B2 (en) | 2015-08-03 | 2019-01-29 | Samumed, Llc | 3-(3H-imidazo[4,5-C]pyridin-2-yl)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10195185B2 (en) | 2015-08-03 | 2019-02-05 | Samumed, Llc | 3-(1H-imidazo[4,5-C]pyridin-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10604512B2 (en) | 2015-08-03 | 2020-03-31 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-indazoles and therapeutic uses thereof |
| US10383861B2 (en) | 2015-08-03 | 2019-08-20 | Sammumed, LLC | 3-(1H-pyrrolo[2,3-C]pyridin-2-yl)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10231956B2 (en) | 2015-08-03 | 2019-03-19 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1 H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10226448B2 (en) | 2015-08-03 | 2019-03-12 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-yl)-1H-pyrazolo[3,4-B]pyridines and therapeutic uses thereof |
| US10206908B2 (en) | 2015-08-03 | 2019-02-19 | Samumed, Llc | 3-(1H-pyrrolo[3,2-C]pyridin-2-YL)-1H-pyrazolo[3,4-C]pyridines and therapeutic uses thereof |
| US10226453B2 (en) | 2015-08-03 | 2019-03-12 | Samumed, Llc | 3-(1H-indol-2-yl)-1H-pyrazolo[4,3-B]pyridines and therapeutic uses thereof |
| US10544139B2 (en) | 2015-11-06 | 2020-01-28 | Samumed, Llc | Treatment of osteoarthritis |
| US10882860B2 (en) | 2015-11-06 | 2021-01-05 | Samumed, Llc | Treatment of osteoarthritis |
| US10899757B2 (en) | 2015-11-06 | 2021-01-26 | Samumed, Llc | 2-(1H-indazol-3-yl)-3H-imidazo[4,5-C]pyridines and their anti-inflammatory uses thereof |
| US11667632B2 (en) | 2015-11-06 | 2023-06-06 | Biosplice Therapeutics, Inc. | 2-(1H-indazol-3-yl)-3H-imidazo[4,5-C]pyridines and their anti-inflammatory uses thereof |
| US11560378B2 (en) | 2015-11-06 | 2023-01-24 | Biosplice Therapeutics, Inc. | Treatment of osteoarthritis |
| US12281097B2 (en) | 2016-04-27 | 2025-04-22 | Biosplice Therapeutics, Inc. | Isoquinolin-3-yl carboxamides and preparation and use thereof |
| US10633380B2 (en) | 2016-06-01 | 2020-04-28 | Samumed, Llc | Process for preparing N-(5-(3-(7-(3-fluorophenyl)-3H-imidazo[4,5-C]pyridin-2-yl)-1H-indazol-5-yl)pyridin-3-yl)-3-methylbutanamide |
| US12012401B2 (en) | 2016-06-01 | 2024-06-18 | Biosplice Therapeutics, Inc. | Process for preparing N-(5-(3-(7-(3-fluorophenyl)-3H-imidazo[4,5-c]pyridin-2-yl)-1H-indazol-5-yl)pyridin-3-yl)-3-methylbutanamide |
| US10072004B2 (en) | 2016-06-01 | 2018-09-11 | Samumed, Llc | Process for preparing N-(5-(3-(7-(3-fluorophenyl)-3H-imidazo [4,5-C]pyridin-2-yl)-1H-indazol-5-yl)pyridin-3-yl)-3-methylbutanamide |
| CN109843892A (en) * | 2016-06-29 | 2019-06-04 | 诺维拉治疗公司 | Dislike diazepine ketone derivatives and its purposes in treatment hepatitis B infection |
| AU2017290755B2 (en) * | 2016-06-29 | 2021-07-01 | Novira Therapeutics, Inc. | Diazepinone derivatives and their use in the treatment of hepatitis B infections |
| AU2017286890B2 (en) * | 2016-06-29 | 2021-09-09 | Novira Therapeutics, Inc. | Oxadiazepinone derivatives and their use in the treatment of hepatitis B infections |
| US10975077B2 (en) | 2016-06-29 | 2021-04-13 | Novira Therapeutics, Inc. | Diazepinone derivatives and their use in the treatment of hepatitis B infections |
| CN109843892B (en) * | 2016-06-29 | 2022-01-25 | 诺维拉治疗公司 | Oxadiazepine derivatives and their use in treating hepatitis B infection |
| CN109641896A (en) * | 2016-06-29 | 2019-04-16 | 诺维拉治疗公司 | Diazepinone derivatives and their use in the treatment of hepatitis b infections |
| WO2018005881A1 (en) * | 2016-06-29 | 2018-01-04 | Novira Therapeutics, Inc. | Oxadiazepinone derivatives and their use in the treatment of hepatitis b infections |
| CN109641896B (en) * | 2016-06-29 | 2021-09-21 | 诺维拉治疗公司 | Diazepinone derivatives and their use in the treatment of hepatitis b infections |
| US10987359B2 (en) | 2016-06-29 | 2021-04-27 | Novira Therapeutics, Inc. | Oxadiazepinone derivatives and methods of treating hepatitis B infections |
| WO2018005883A1 (en) * | 2016-06-29 | 2018-01-04 | Novira Therapeutics, Inc. | Diazepinone derivatives and their use in the treatment of hepatitis b infections |
| US11684615B2 (en) | 2016-10-21 | 2023-06-27 | Biosplice Therapeutics, Inc. | Methods of using indazole-3-carboxamides and their use as Wnt/β-catenin signaling pathway inhibitors |
| US10806726B2 (en) | 2016-10-21 | 2020-10-20 | Samumed, Llc | Methods of using indazole-3-carb oxamides and their use as Wnt/B-catenin signaling pathway inhibitors |
| US11446288B2 (en) | 2016-11-07 | 2022-09-20 | Biosplice Therapeutics, Inc. | Single-dose, ready-to-use injectable formulations |
| US11819499B2 (en) | 2016-11-07 | 2023-11-21 | Biosplice Therapeutics, Inc. | Single-dose, ready-to-use injectable formulations |
| US10758523B2 (en) | 2016-11-07 | 2020-09-01 | Samumed, Llc | Single-dose, ready-to-use injectable formulations |
| WO2025157165A1 (en) * | 2024-01-23 | 2025-07-31 | 杭州中美华东制药有限公司 | Compound having pkmyt1 inhibition effect |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2476822A1 (en) | 2003-08-28 |
| MXPA04008680A (en) | 2004-12-06 |
| US20040127492A1 (en) | 2004-07-01 |
| DE60311567D1 (en) | 2007-03-22 |
| WO2003070236A3 (en) | 2004-02-12 |
| EP1478357A2 (en) | 2004-11-24 |
| ES2281633T3 (en) | 2007-10-01 |
| US20050176796A1 (en) | 2005-08-11 |
| EP1478357B1 (en) | 2007-01-31 |
| JP2005529850A (en) | 2005-10-06 |
| AU2003218989A1 (en) | 2003-09-09 |
| DE60311567T2 (en) | 2007-10-31 |
| ATE353013T1 (en) | 2007-02-15 |
| BR0307819A (en) | 2005-04-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1478357B1 (en) | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents | |
| KR101084871B1 (en) | Pyrazolo-quinazolin derivatives, methods for their preparation and their use as kinase inhibitors of said derivatives | |
| EP1177185B1 (en) | 4, 5, 6, 7-tetrahydroindazole derivatives as antitumor agents | |
| AU2002313633B2 (en) | Substituted 3-aryl-5-aryl-[1,2,4]-oxadiazoles and analogs | |
| EP2118102B1 (en) | Tricyclic indoles and (4,5-dihydro) indoles | |
| JP5330498B2 (en) | Hydroxamate-based inhibitors of deacetylase B | |
| JP2003513091A (en) | Pyrazolones substituted with heterocycles | |
| WO2005005414A2 (en) | Pyrazolyl-indole derivatives active as kinase inhibitors, process for their preparation and pharmaceutical compositions comprising them | |
| WO2010060854A1 (en) | Bicyclic pyrazole and isoxazole derivatives as antitumor and antineurodegenerative agents | |
| IL172923A (en) | PYRROLO [3,4-c] PYRAZOLE DERIVATIVES AND PHARMACEUTICAL COMPOSITIONS COMPRISING THEM | |
| CN103459396B (en) | As [1,2,4] triazolo [4,3-b] pyridazine compound of c-Met tyrosine kinase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003714744 Country of ref document: EP Ref document number: 2476822 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003569192 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/008680 Country of ref document: MX |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003714744 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10505200 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003714744 Country of ref document: EP |