WO2003091213A1 - Novel amide derivatives or salts thereof - Google Patents
Novel amide derivatives or salts thereof Download PDFInfo
- Publication number
- WO2003091213A1 WO2003091213A1 PCT/JP2003/005198 JP0305198W WO03091213A1 WO 2003091213 A1 WO2003091213 A1 WO 2003091213A1 JP 0305198 W JP0305198 W JP 0305198W WO 03091213 A1 WO03091213 A1 WO 03091213A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lower alkyl
- aryl
- alkylene
- lower alkylene
- ring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/30—Indoles; Hydrogenated indoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to carbon atoms of the hetero ring
- C07D209/42—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
Definitions
- the present invention relates to a novel amide derivative or a salt thereof, particularly a novel amide derivative or a amide derivative in which an indole ring or a chenobilol ring is bonded to an aryl or aromatic hetero ring having a hydroxyethylene moiety as a substituent via an amide bond. About its salt. Further, the present invention relates to a medicine, particularly to an amide derivative or a salt thereof which is a glycogen phosphorylase inhibitor. The compound of the present invention is useful for treating and preventing insulin-dependent diabetes (type 1 diabetes), non-insulin-dependent diabetes (type 2 diabetes), insulin resistance disease, and obesity. Background art
- the global diabetes population is projected to exceed 135 million (1995) and to increase to 300 million by 2025 (King HJ, Diabetes Care 21; 1414). -1431, 1998), and the social demands for progress in the treatment of this disease are very large.
- the mainstay of diabetes treatment is the application of hypoglycemic drugs, which may result in lowering the rate of glycemic control, leading to diabetic neuropathy, retinopathy, nephropathy, and other diabetic complications and mortality. It has been revealed.
- hypoglycemic drugs which may result in lowering the rate of glycemic control, leading to diabetic neuropathy, retinopathy, nephropathy, and other diabetic complications and mortality. It has been revealed.
- Glucose release from the liver is strictly controlled by the relative regulation of glucagon and insulin, but in diabetic conditions, the absolute lack of insulin ( Insufficient relative action (type 1 diabetes mellitus: insulin-dependent diabetes mellitus) and increased relative hepatic glucose output due to lack of relative action (type 2 diabetes mellitus: non-insulin-dependent diabetes mellitus), resulting in a hyperglycemic state.
- Hepatic glucose release is expressed as the sum of two pathways: hepatic glycogen degradation and gluconeogenesis.
- hepatic glycogen decomposition system is enhanced (Tayek JA, Am. J. Physio, 270; E709-E717, 1996, Diraison F, Diabeto). Iogia 41; 212-220, 1998).
- suppression of hepatic glycogen degradation normalizes the enhancement of hepatic glucose release in diabetic patients (Hell réelle in MK, J. Clin. Invest. 100; 1305-1319, 1997, Pimenta, Diabeto Iogia 37; 697-702, 1994) 0 From these facts, it is considered that the enhancement of hepatic glycogen degradation contributes to the diabetic condition.
- glycogen phosphorylase EG 2.4.1.1
- glucose glycemia
- the glycogen phosphorylase inhibitor inhibits the above-mentioned glycogen decomposition reaction and suppresses glucose release (sugar release) from the liver.
- glucose release sucrose release
- various compounds described in Patent Documents 1 to 3 below are known as glycogen phosphorylase inhibitors.
- Patent Document 1 describes that a compound represented by the following general formula is used as a glycogen phosphorylase inhibitor in the treatment of diabetes and the like.
- the feature of the compound is that a chenobilol ring or the like is bonded to an ethylene moiety through an amide bond.
- Patent Document 2 describes that a compound represented by the following general formula is used as a glycogen phosphorylase inhibitor for the treatment of a glycogen phosphorylase-dependent disease.
- the feature of the compound is that an indole ring or the like is bonded to an ethylene moiety through an amide bond.
- Patent Document 3 describes that a compound represented by the following general formula is used as a glycogen phosphorylase inhibitor for the treatment of diabetes and the like.
- the feature of the compound is that an indole ring or the like is bonded to a methylene moiety via an amide bond.
- the compound described in Patent Document 4 is known.
- the use of the compound is a PDEIV (phosphodiesterase IV) inhibitor, and there is no disclosure or suggestion of the use as a glycogen phosphorylase inhibitor.
- the features of the compound are that an indole ring or the like is bonded to an aryl moiety directly via an amide bond or across methylene, and that the indole ring or the like has a substituent such as alkoxy or alkylene-aryl.
- Glycogen phosphorylase inhibitors are excellent treatment and prevention agents for insulin-dependent diabetes mellitus (type 1 diabetes), non-insulin-dependent diabetes mellitus (type 2 diabetes), insulin resistance disease, and obesity. It is expected as. Therefore, there is a strong demand for the creation of a compound having a glycogen phosphorylase inhibitory action, which has a different chemical structure from the above-mentioned known compounds, and which has a more excellent effect.
- Patent Document 1 Japanese Patent Application Laid-Open No. 2000-13-1131
- Patent Literature 2 Tokiohei 1 1-5 0 0 4 4 5
- Patent Literature 3 Japanese Patent Publication No. Hei 10-0-5 1 1 6 8 7
- Patent Document 4 International Publication No. 0 1/6 4 6 3 9 Pamphlet Disclosure of the Invention
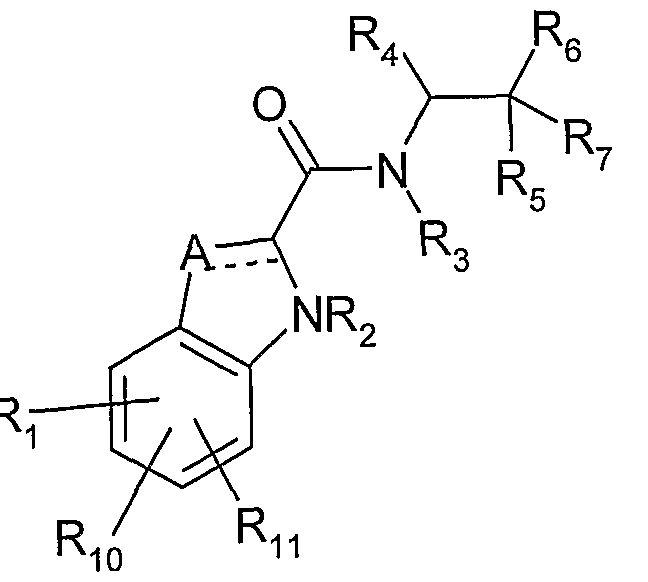
- the present invention relates to an amide derivative represented by the following general formula (I) or a salt thereof, and a dalycogen phosphorylase inhibitor containing the same as an active ingredient, particularly an agent for treating or preventing diabetes.
- the compound of the present invention and the compounds described in Patent Documents 1 to 4 They differ from the compounds described in Patent Documents 1 to 4 in that they have an A ring (aryl or aromatic hetero ring) having a ethylene moiety as a substituent. Further, the compound of Patent Document 4 differs from the compound of the present invention also in that it has a substituent such as alkoxy or alkylene-aryl on the indole ring or the like.
- Ring A Aryl, or 5 to 1 to 4 heteroatoms selected from N, S, O
- Ring B benzene or thiophene
- R 10 hydrogen atom or lower alkyl
- R 11 hydrogen atom, lower alkyl, or -lower alkylene-aryl
- the ring A is benzene, thiazol, pyrazine, pyrimidine or pyridine.
- an amide derivative represented by the following general formula (II) or a salt thereof is more preferable.
- a ring aryl, or a 5- or 6-membered aromatic hetero ring having 1 to 4 hetero atoms selected from N, S, O,
- ring A is benzene, thiazole, pyrazine, pyrimidine or pyridine. Ring A is most preferably benzene.
- lower means a straight or branched carbon chain having 1 to 6 carbon atoms, unless otherwise specified.
- lower alkyl includes, for example, straight chain or branched such as methyl, ethyl, propyl, isopropyl, butyl, isoptyl, sec-butyl, tert-butyl, pentyl, isopentyl, hexyl and isohexyl. And alkyl. Among them, those having 1 to 3 carbon atoms are preferred, and methyl and ethyl are particularly preferred.
- “Lower alkylene” includes methylene, ethylene, propylene, butylene and the like, It may be a branched lower alkylene. Methylene and ethylene are particularly preferred.
- aryl means an aromatic hydrocarbon ring containing a condensed ring, and includes aryl having 6 to 14 carbon atoms. Benzene, naphthalene and anthracene are preferred.
- “-Lower alkylene-aryl” means a compound in which the above alkylene is bonded to the above lower alkylene, and specific examples include benzyl, phenyl and the like. Benzyl is particularly preferred.
- ⁇ 5- or 6-membered aromatic heterocyclic ring having 1-4 heteroatoms selected from N, S, O '', 1-3 heteroatoms selected from N, S, O It means a 5- or 6-membered aromatic hetero ring having 1 to 4 in total.
- Specific examples include furan, thiophene, pyrrole, pyridine, oxazole, thiazole, isothiazole, imidazole, virazole, tetrazole, pyrazine, pyrimidine, pyridazine, and triazine.
- Thiazole, pyrazine, pyrimidine and pyridine are particularly preferred.
- substituent in the formula o, it may mean a heterocycle in which a nitrogen atom has been oxidized, such as pyridineoxide ⁇ pyrimidineoxide.
- halogen atom examples include a fluorine atom, a chlorine atom, a bromine atom, and an iodine atom. Fluorine, chlorine, and bromine are preferred.
- Halogen-substituted lower alkyl means the above-mentioned lower alkyl substituted with the above-mentioned halogen atom. Particularly, lower alkyl substituted with a fluorine atom is preferable. Further the preferred - is CF 3.
- Car mouth alkyl means cycloalkyl having 3 to 8 carbon atoms.
- “Lower alkynyl” means those having 2 to 6 carbon atoms, preferably acetylinyl.
- ring B or A ring is a 5-membered ring, each R 5 and R 9, or R 2 4 may that we do not exist.
- the compound of the present invention includes a mixture of various stereoisomers such as tautomers and optical isomers, and an isolated compound.
- the compound of the present invention may form an acid addition salt.
- a salt with a base may be formed.
- Specific examples of such salts include mineral acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, nitric acid, and phosphoric acid, formic acid, acetic acid, propionic acid, oxalic acid, malonic acid, succinic acid, Organic acids such as fumaric acid, maleic acid, lactic acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, aspartic acid, glutamic acid Acid addition salts with acidic amino acids such as acid, inorganic bases such as sodium, potassium, magnesium, calcium and aluminum; organic bases such as methylamine, ethylamine and ethanolamine; basic amino acids such as lysine and orditin And ammonium salts.
- the compounds of the present invention also include hydrates, various pharmaceutically acceptable solvates and polymorphs.
- the compound of the present invention is not limited to the compounds described in Examples below, but may be a derivative represented by the general formula (I) or (II) and a pharmaceutically acceptable derivative thereof. It includes all salts.
- the compounds of the present invention also include all compounds that are metabolized in vivo and converted into the above general formula (I) or (II), or compounds converted into salts thereof, so-called prodrugs.
- Examples of the group that forms a prodrug of the compound of the present invention include those described in Prog. Med. 5:21 57-21 61 (1 985) and Hirokawa Shoten, 1990, “Development of Drugs”.
- the groups described in Vol. 7 , Molecular Design, pp. 163-198 are listed.
- the compounds of the present invention and their pharmaceutically acceptable salts can be produced by applying various known synthetic methods, utilizing characteristics based on the basic structure or the types of substituents.
- an appropriate protecting group that is, a group that can be easily converted to the functional group at the stage of a raw material or an intermediate. May be.
- the desired compound can be obtained by removing the protecting group, if necessary.
- Examples of such a functional group include a hydroxyl group and a carboxyl group.
- Examples of such a protective group include, for example, Greene and Wuts, “Protective Groups in Organic Synthesis” No. 2
- the protecting groups described in the edition can be mentioned.
- X is meant hydroxyl, a leaving group or a leaving atom such as a lower alkoxy group, or a halogen atom
- This reaction is a reaction in which the compound (II) and the compound (III) are reacted as they are or in a solvent, and then, if necessary, the protecting group is removed to obtain the compound of the present invention.
- the solvent include aromatic hydrocarbons such as toluene and xylene, ketones such as methyl ethyl ketone and acetate, ethers such as dioxane, tetrahydrofuran and diglyme, methanol and ethanol. And alcohols such as isopropanol, acetonitrile, dimethylformamide, dimethylsulfoxide, water, and a mixed solvent thereof, which are appropriately selected according to various reaction conditions.
- X is a hydroxyl group
- a method of reacting in the above solvent in the presence of a condensing agent can be applied.
- the condensing agent include N, N'-dicyclocarbodiimide, 1-ethyl-3- (3'-dimethylaminopropyl) carbodiimide, 1,1, -carbonyldiimidazole, diphenylphosphoryl azide and the like.
- X is a halogen atom
- a method of reacting in the solvent in the presence of a base can be applied.
- the base include sodium carbonate, potassium carbonate such as potassium carbonate, sodium hydrogen carbonate, potassium carbonate such as potassium hydrogen carbonate, triethylamine, diisopropylethylamine, pyridine, and the like. Organic amines and the like. Manufacturing method 2
- This reaction is a reaction in which R 12 is added to compound (IV) as it is or after addition in a solvent, and the protecting group is removed as required to obtain the compound of the present invention.
- R 1 2 is hydrogen atom, lithium borohydride, hydrogen Kaboku Increment butoxy aluminum lithium and diisobutylaluminum hydride, lithium, sodium, by reaction with a - reducing agent such as zinc An inventive compound can be obtained.
- R 12 is an alkyl group or the like
- the compound of the present invention can be obtained by a reaction using an alkyl metal or the like.
- the solvent include aromatic hydrocarbons such as toluene and xylene, ethers such as dioxane, tetrahydrofuran and diglyme, alcohols such as methanol, ethanol and isopropanol, and acetates.
- aromatic hydrocarbons such as toluene and xylene
- ethers such as dioxane, tetrahydrofuran and diglyme
- alcohols such as methanol, ethanol and isopropanol
- acetates examples include tolyl, dimethylformamide, dimethylsulfoxide, water, and a mixed solvent thereof, which is appropriately selected according to various reaction conditions.
- This reaction is a reaction in which CR 13 R 14 R 15 is added to compound (V) as it is or after it is added in a solvent, followed by removal of a protecting group as required to obtain the compound of the present invention.
- CR R 1 4 R 15 is an alkyl group can be obtained the compound of the present invention by reaction with a an alkyl metal.
- the solvent include aromatic hydrocarbons such as toluene and xylene, ethers such as dioxane, tetrahydrofuran, and diglyme, acetonitrile, dimethylformamide, and a mixed solvent thereof. Is appropriately selected according to the conditions.
- This reaction is a reaction for oxidizing compound (VI) in a solvent and then removing a protecting group as required to obtain a compound of the present invention in which R 15 is a hydroxyl group.
- the solvent include aromatic hydrocarbons such as toluene and xylene, ketones such as methyl ethyl ketone and acetone, ethers such as dioxane, tetrahydrofuran and diglyme, and methanol and ethanol and isopropanol.
- Alcohols, acetonitrile, dimethylformamide, dimethylsulfoxide, water, or a mixed solvent thereof may be mentioned, but may be appropriately selected according to various reaction conditions.
- examples of the oxidizing agent include osmium tetroxide, hydrogen peroxide, potassium permanganate, and the like. If necessary, a co-oxidizing agent such as N-methylmorpholine-N-oxide-trimethylamine-N-oxide is used. Can also be added.
- the starting compound (II) is commercially available or can be obtained by a known method, for example,
- This reaction is a reaction in which R 12 is added to compound (VII) as it is or after addition in a solvent, followed by removal of a protecting group as required to obtain a starting compound (Ml).
- the reaction conditions are the same as in the reaction for producing the compound of the present invention from compound (IV) (Production method 2). Manufacturing method 3
- R 6 to R 9 , R 11 to R 15 , and A ring are the same as those described above, and Y represents a halogen atom, a trifluoromethanesulfonyl group, or an acyl group
- This reaction is a synthesis method particularly when R 15 in the intermediate (III) is a hydroxyl group.
- compound (IX) when Y is a halogen atom or a trifluoromethanesulfonyl group, Angew. Chem. Int. Ed. Engl., 25, 508 (1986) and J. Am. Chem. Soc., 106, 4630 ( The compound can be converted to the corresponding compound (X) by the method described in 1984) or a method analogous thereto.
- Y is an acyl group in the compound (IX), the method described in Org. Synth., 751 (1973) or Am. Chem. Soc., 90, 6816 (1968) or a method analogous thereto.
- Compound (IX) when Y is a halogen atom or a trifluoromethanesulfonyl group, Angew. Chem. Int. Ed. Engl., 25, 508 (1986) and J. Am. Che
- This compound (X) is compounded by the oxidation reaction described above.
- the nitro group of the compound (XI) can be led to the intermediate (III) by catalytic hydrogenation or reduction reaction such as metal reduction.
- This reaction is a reaction in which the compound (VIII) is condensed with the compound (II), and then, if necessary, the protecting group is removed to obtain the starting compound (V).
- the reaction conditions are the same as in the reaction (Production method 1) for obtaining the compound of the present invention from compounds (II) and (III).
- This reaction is a reaction in which the compound (XII) is condensed with the compound (II) and, if desired, the protecting group is removed to obtain the starting compound (VI).
- the reaction conditions are the same as in the reaction (Production method 1) for obtaining the compound of the present invention from compounds (II) and (III). Furthermore, some ⁇ : compounds contained in the compound of the present invention are compounds obtained by the above method.
- the compound of the present invention thus produced can be isolated and purified by a known method, for example, extraction, precipitation, fractional chromatography, fractional crystallization, recrystallization and the like.
- a known method for example, extraction, precipitation, fractional chromatography, fractional crystallization, recrystallization and the like.
- optical isomers exist. These optical isomers can be separated by a conventional method such as fractional crystallization for recrystallization with an appropriate salt or column chromatography.
- the compound of the present invention has a glycogen phosphorylase inhibitory action, and its action is adapted to diseases such as diabetes (insulin-dependent diabetes (type 1 diabetes) and insulin-independent diabetes (type 2 diabetes)). , Insulin resistance disease, and obesity.
- diabetes insulin-dependent diabetes (type 1 diabetes) and insulin-independent diabetes (type 2 diabetes)
- Insulin resistance disease Insulin resistance disease
- obesity Insulin resistance disease
- the excellent glycogen phosphorylase inhibitory activity of the compound of the present invention was confirmed by the following test methods.
- the procedure for measuring the GP activity is as follows. The reaction was performed using a 96-well plate. 45 mM potassium phosphate, 0.24% glycogen, 1.6 mM magnesium chloride, 120 iM ethylenediaminetetraacetic acid trisodium, 22.5 M S-NADP, 4 10 _4 % -glucose 1,6-diphosphate, glucose-6-phosphate mixing the aqueous solution of acid dehydrogenase one peptidase 385Un it / and phosphonium Darco Mutter peptidase 77Unit / L, and a pH of 6.8 (Reaction Cocktai l) 0 measurement Weng fruits, by averaging the values of 3 Ueru identical conditions Calculated.
- Each compound subjected to the reaction was dissolved in dimethyl sulfoxide, and added at a rate of 10 L per 1 ⁇ : c. After adding 26.5 iL of the above-mentioned aqueous solution (Reaction Cocktail) to each well, the human liver-type GP protein solution (GP protein is dissolved in 40 mM; 8 glycerol phosphate, 80 mM cysteine (pH 6.8)) 23.5 L each, and reacted at room temperature. The GP enzyme reaction was detected by the increase in absorbance at 340 nm (SPEGTRAmax, Molecular Device, Sunnyvale, GA).
- the GP inhibitory activity of the test compound was evaluated by the percentage (%) of the reaction in the absence of the compound added to the reaction in the L (control). The concentration of the test compound that inhibited the control reaction by 50% (IC 50 value) ). As a result, the compound of the present invention exhibited a strong glycogen phosphorylase (GP) inhibitory action equal to or higher than that of a conventional glycogen phosphorylase inhibitor.
- the IC 50 values of representative compounds of the present invention are shown in Table 1 below. table 1
- Example 57 0.25
- mice were allocated to each group (6 animals each) so as to be even. The next day, the test compound was orally administered to each of the assigned groups using a probe. At a certain time after the oral administration, blood was collected in the same manner as described above, and the blood glucose level was measured. The hypoglycemic effect of the test compound was evaluated by measuring the blood glucose level of the test compound administration group relative to the blood glucose level of the solvent group. As a result, the compound of the present invention showed a strong hypoglycemic effect.
- the compound of the present invention exhibits a strong glycogen phosphorylase (GP) inhibitory action at least as high as that of a conventional glycogen phosphorylase inhibitor, and has a strong hypoglycemic action, so that it can be used as a pharmaceutical composition, particularly as an antidiabetic agent Useful.
- Pharmaceutical compositions containing one or more of the compound of the present invention or a pharmaceutically acceptable salt thereof as an active ingredient may be used as carriers, excipients, and other additives commonly used for pharmaceuticals. It is prepared into tablets, powders, fine granules, granules, capsules, pills, solutions, injections, suppositories, ointments, patches, etc., and administered orally or parenterally.
- the parenteral dose is 0.01 to 100 mg, which can be administered once or in several divided doses. Since the dose varies under various conditions, an amount smaller than the above dose range may be sufficient.
- the one or more active substances include at least one inert diluent, such as lactose, mannitol, glucose, hydroxypropyl cellulose, microcrystalline cellulose, starch, polyvinyl. Pyrrolidone, mixed with magnesium aluminate metasilicate.
- composition should be prepared according to the usual methods Additives other than 3 05198, for example, lubricants such as magnesium stearate and disintegrants such as calcium cellulose glycolate; It may contain a stabilizing agent such as acetic acid, a solubilizing agent such as glutamic acid or aspartic acid or a solubilizing agent. If necessary, tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate or the like, or with a film of gastric or enteric substance.
- lubricants such as magnesium stearate and disintegrants such as calcium cellulose glycolate
- a stabilizing agent such as acetic acid, a solubilizing agent such as glutamic acid or aspartic acid or a solubilizing agent.
- tablets or pills may be coated with sugar coating such as sucrose, gelatin, hydroxypropylcellulose, hydroxypropylmethylcellulose phthalate or the like, or with a film of gastric
- Liquid compositions for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, elixirs and the like, and commonly used inert diluents such as Contains purified water and ethyl alcohol.
- the composition may contain, in addition to the inert diluent, solubilisers, solubilizing agents, wetting agents, auxiliary agents such as suspending agents, sweetening agents, flavoring agents, fragrances, and preservatives.
- Injections for parenteral administration include sterile aqueous or non-aqueous solutions, suspensions, and emulsions.
- Diluents for aqueous solutions and suspensions include, for example, distilled water for injections and physiological saline.
- examples of diluents for water-insoluble solutions and suspensions include propylene glycol, polyethylene glycol, vegetable oils such as olive oil, alcohols such as ethyl alcohol, and polysorbate 80 (trade name).
- compositions may further comprise additives such as tonicity agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), solubilizers or solubilizers. .
- additives such as tonicity agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), solubilizers or solubilizers.
- additives such as tonicity agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), solubilizers or solubilizers.
- additives such as tonicity agents, preservatives, wetting agents, emulsifiers, dispersants, stabilizers (eg, lactose), solubilizers or solubilizers.
- N- (4-Promo-2--2-butanol) -5-coguchi-1H-indole-2-caproloxamide 2.55 g, iron powder 1.76 g and ammonium chloride 0.34 in a mixture of ethanol 150 m I And 25 ml of water were added, and the mixture was heated under reflux for 12 hours. After concentrating the reaction mixture, dimethylformamide was added, the insolubles were removed by filtration, the solvent was distilled off under reduced pressure, the residue obtained was washed with getyl ether, dried and dried to give N- (2-amino-4-bromophenyl). 1.36 g of -5-Chloro-1H-indole-2-force lipoxamide were obtained.
- Example 57 the compounds of Examples 58 to 59 were obtained.
- Example 60 In the same manner as in Example 60, the compound of Example 61 was obtained.
- Example 62 In the same manner as in Example 62, the compounds of Examples 63 to 95 were obtained.
- Example 101 In the same manner as in Example 100, the compounds of Examples 101 to 103 were obtained.
- Example 104 In the same manner as in Example 104, the compounds of Examples 105 to 110 were obtained.
- AD-mix dissolve 8700 mg in ⁇ -butanol-water (1: 1, 5 ml) and add 5-chloro-N-[(2,3,5,7-tetrafluoro- mouth-4-vinyl) phenyl ] -1H-indole-2-carboxamide (184 mg) was added, and the mixture was stirred at 0 ° C for 24 hours. Further, methanesulfonamide (48 mg) was added, and the mixture was stirred at 0 ° C for 28 hours.
- AD-mix-j 81.05g and ⁇ -butanol-water (1: 1, 5ml) were added, and the mixture was stirred at 0 ° C. for 3 hours, and then 5.5 g of sodium sulfite was added.
- AD-mix- ⁇ .05 g and ⁇ -butanol-water (1: 1, 5 ml) stir at 0 ° C for 89 hours, add AD-mix- 1.75 g, and add 0 ° C For 23 hours.
- AD-mix- 1.75 g and add 0 ° C For 23 hours.
- 1.75 g of AD-mix and -butanol-water (1: 1, 5 ⁇ ) were added, and the mixture was stirred at 0 ° G for 48 hours, and then 5.5 g of sodium sulfite was added.
- R f. Reference example number
- Ex . Example number
- Structure Structural formula
- Data Physical property data
- NMR Nuclear magnetic resonance spectrum (TMS internal standard)
- S Mass spectrometry value
- Ac Acetyl
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Diabetes (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description






































Claims


Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2003227360A AU2003227360A1 (en) | 2002-04-25 | 2003-04-23 | Novel amide derivatives or salts thereof |
| JP2003587778A JPWO2003091213A1 (ja) | 2002-04-25 | 2003-04-23 | 新規なアミド誘導体又はその塩 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002-123926 | 2002-04-25 | ||
| JP2002123926 | 2002-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003091213A1 true WO2003091213A1 (en) | 2003-11-06 |
Family
ID=29267507
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2003/005198 Ceased WO2003091213A1 (en) | 2002-04-25 | 2003-04-23 | Novel amide derivatives or salts thereof |
Country Status (3)
| Country | Link |
|---|---|
| JP (1) | JPWO2003091213A1 (ja) |
| AU (1) | AU2003227360A1 (ja) |
| WO (1) | WO2003091213A1 (ja) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004092158A1 (en) * | 2003-04-17 | 2004-10-28 | Pfizer Products Inc. | Carboxamide derivatives as anti-diabetic agents |
| EP1532980A1 (en) * | 2003-11-24 | 2005-05-25 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| WO2006059163A1 (en) * | 2004-12-02 | 2006-06-08 | Prosidion Limited | Treatment of diabetes with glycogen phosphorylase inhibitors |
| WO2006059164A3 (en) * | 2004-12-02 | 2006-08-17 | Prosidion Ltd | Pyrrolopyridine-2-carboxylic acid amides |
| US7115648B2 (en) | 2002-03-06 | 2006-10-03 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7122567B2 (en) | 2002-03-06 | 2006-10-17 | Astrazeneca Ab | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity |
| US7129249B2 (en) | 2002-03-06 | 2006-10-31 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phoshorylase |
| US7138415B2 (en) | 2002-03-06 | 2006-11-21 | Astrazeneca Ab | Indolamid derivatives which possess glycogenphosphorylase inhibitory activity |
| US7166636B2 (en) | 2002-03-06 | 2007-01-23 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| US7169927B2 (en) | 2002-03-06 | 2007-01-30 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7214704B2 (en) | 2004-11-15 | 2007-05-08 | Bristol-Myers Squibb Company | 2-Amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7223786B2 (en) | 2004-11-15 | 2007-05-29 | Bristol-Myers Squibb Company | 2-aminonaphthalene derivatives and related glycogen phosphorylase inhibitors |
| US7226942B2 (en) | 2004-11-15 | 2007-06-05 | Bristol-Myers Squibb Company | 2-amino-4-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| US7307174B2 (en) | 2002-10-03 | 2007-12-11 | Astrazeneca Ab | Process and intermediates for the preparation of thienopyrrole derivatives |
| WO2008047821A1 (en) * | 2006-10-18 | 2008-04-24 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| US7365061B2 (en) | 2004-11-15 | 2008-04-29 | Bristol-Myers Squibb Company | 2-Amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7411074B2 (en) | 2002-10-03 | 2008-08-12 | Astrazeneca Ab | Process and intermediates for the preparation of the thienopyrrole derivatives |
| WO2008074755A3 (en) * | 2006-12-18 | 2008-10-02 | Neurosearch As | Novel cinnamic amide derivatives useful as ion channel modulators |
| WO2008122329A1 (en) * | 2007-03-19 | 2008-10-16 | Dsm Ip Assets B.V. | Uv-filter compounds |
| EP1732566A4 (en) * | 2004-04-05 | 2010-01-13 | Takeda Pharmaceutical | 6-azaindole COMPOUND |
| WO2009080821A3 (en) * | 2007-12-21 | 2010-01-14 | Giuliani International Limited | Multitarget compounds active at a ppar and cannabinoid receptor |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| JP2010540443A (ja) * | 2007-09-21 | 2010-12-24 | アレイ バイオファーマ、インコーポレイテッド | 糖尿病治療のためのグルコキナーゼ活性化剤としてのピリジン−2−イル−アミノ−1,2,4−チアジアゾール誘導体 |
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| JP2015527388A (ja) * | 2012-09-07 | 2015-09-17 | ノバルティス アーゲー | インドールカルボキサミド誘導体およびその使用 |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996039384A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer, Inc. | Substituted n-(indole-2-carbonyl)-glycinamides and derivatives as glycogen phosphorylase inhibitors |
| WO1996039385A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
| EP1088824A2 (en) * | 1999-09-30 | 2001-04-04 | Pfizer Products Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
-
2003
- 2003-04-23 JP JP2003587778A patent/JPWO2003091213A1/ja not_active Withdrawn
- 2003-04-23 WO PCT/JP2003/005198 patent/WO2003091213A1/ja not_active Ceased
- 2003-04-23 AU AU2003227360A patent/AU2003227360A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996039384A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer, Inc. | Substituted n-(indole-2-carbonyl)-glycinamides and derivatives as glycogen phosphorylase inhibitors |
| WO1996039385A1 (en) * | 1995-06-06 | 1996-12-12 | Pfizer Inc. | Substituted n-(indole-2-carbonyl-) amides and derivatives as glycogen phosphorylase inhibitors |
| EP1088824A2 (en) * | 1999-09-30 | 2001-04-04 | Pfizer Products Inc. | Bicyclic pyrrolyl amides as glycogen phosphorylase inhibitors |
Cited By (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7169927B2 (en) | 2002-03-06 | 2007-01-30 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7332515B2 (en) | 2002-03-06 | 2008-02-19 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| US7115648B2 (en) | 2002-03-06 | 2006-10-03 | Astrazeneca Ab | Indole-amide derivatives and their use as glycogen phosphorylase inhibitors |
| US7122567B2 (en) | 2002-03-06 | 2006-10-17 | Astrazeneca Ab | Heterocyclic amide derivatives having glycogen phosphorylase inhibitory activity |
| US7129249B2 (en) | 2002-03-06 | 2006-10-31 | Astrazeneca Ab | Heterocyclic amide derivatives as inhibitors of glycogen phoshorylase |
| US7138415B2 (en) | 2002-03-06 | 2006-11-21 | Astrazeneca Ab | Indolamid derivatives which possess glycogenphosphorylase inhibitory activity |
| US7166636B2 (en) | 2002-03-06 | 2007-01-23 | Astrazeneca Ab | Indole-amid derivatives which possess glycogen phosphorylase inhibitory activity |
| US7411074B2 (en) | 2002-10-03 | 2008-08-12 | Astrazeneca Ab | Process and intermediates for the preparation of the thienopyrrole derivatives |
| US7307174B2 (en) | 2002-10-03 | 2007-12-11 | Astrazeneca Ab | Process and intermediates for the preparation of thienopyrrole derivatives |
| US6992092B2 (en) | 2003-04-17 | 2006-01-31 | Pfizer Inc. | Anti-diabetic agents |
| WO2004092158A1 (en) * | 2003-04-17 | 2004-10-28 | Pfizer Products Inc. | Carboxamide derivatives as anti-diabetic agents |
| US8148413B2 (en) | 2003-11-24 | 2012-04-03 | Transtech Pharma, Inc. | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| US7812043B2 (en) | 2003-11-24 | 2010-10-12 | Transtech Pharma, Inc. | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| JP2007512264A (ja) * | 2003-11-24 | 2007-05-17 | ノボ ノルディスク アクティーゼルスカブ | 肥満症の治療においてグルコキナーゼ活性化剤として使用するための、ヘテロアリールインドールカルボキサミドおよびその類似体 |
| WO2005049019A1 (en) * | 2003-11-24 | 2005-06-02 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| EP1532980A1 (en) * | 2003-11-24 | 2005-05-25 | Novo Nordisk A/S | N-heteroaryl indole carboxamides and analogues thereof, for use as glucokinase activators in the treatment of diabetes |
| US7893098B2 (en) | 2003-12-29 | 2011-02-22 | Sepracor Inc. | Pyrrole and pyrazole DAAO inhibitors |
| EP1732566A4 (en) * | 2004-04-05 | 2010-01-13 | Takeda Pharmaceutical | 6-azaindole COMPOUND |
| US7365061B2 (en) | 2004-11-15 | 2008-04-29 | Bristol-Myers Squibb Company | 2-Amino-3-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7226942B2 (en) | 2004-11-15 | 2007-06-05 | Bristol-Myers Squibb Company | 2-amino-4-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| US7223786B2 (en) | 2004-11-15 | 2007-05-29 | Bristol-Myers Squibb Company | 2-aminonaphthalene derivatives and related glycogen phosphorylase inhibitors |
| US7214704B2 (en) | 2004-11-15 | 2007-05-08 | Bristol-Myers Squibb Company | 2-Amino-1-functionalized tetralin derivatives and related glycogen phosphorylase inhibitors |
| WO2006059164A3 (en) * | 2004-12-02 | 2006-08-17 | Prosidion Ltd | Pyrrolopyridine-2-carboxylic acid amides |
| WO2006059163A1 (en) * | 2004-12-02 | 2006-06-08 | Prosidion Limited | Treatment of diabetes with glycogen phosphorylase inhibitors |
| US10562878B2 (en) | 2006-01-06 | 2020-02-18 | Sunovion Pharamceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US9868718B2 (en) | 2006-01-06 | 2018-01-16 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US8877975B2 (en) | 2006-01-06 | 2014-11-04 | Sunovion Pharmaceuticals Inc. | Cycloalkylamines as monoamine reuptake inhibitors |
| US8053603B2 (en) | 2006-01-06 | 2011-11-08 | Sunovion Pharmaceuticals Inc. | Tetralone-based monoamine reuptake inhibitors |
| US8097760B2 (en) | 2006-03-31 | 2012-01-17 | Sunovion Pharmacuticals Inc. | Preparation of chiral amides and amines |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| EP2351568A2 (de) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Verwendungen von dpp iv Inhibitoren |
| US7884124B2 (en) | 2006-06-30 | 2011-02-08 | Sepracor Inc. | Fluoro-substituted inhibitors of D-amino acid oxidase |
| JP5306818B2 (ja) * | 2006-10-18 | 2013-10-02 | 武田薬品工業株式会社 | 縮合複素環化合物 |
| EP2298772A1 (en) * | 2006-10-18 | 2011-03-23 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| WO2008047821A1 (en) * | 2006-10-18 | 2008-04-24 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compound |
| US8492405B2 (en) | 2006-10-18 | 2013-07-23 | Takeda Pharmaceutical Company Limited | Glucokinase-activating fused heterocyclic compounds and methods of treating diabetes and obesity |
| WO2008074755A3 (en) * | 2006-12-18 | 2008-10-02 | Neurosearch As | Novel cinnamic amide derivatives useful as ion channel modulators |
| US7902252B2 (en) | 2007-01-18 | 2011-03-08 | Sepracor, Inc. | Inhibitors of D-amino acid oxidase |
| WO2008122329A1 (en) * | 2007-03-19 | 2008-10-16 | Dsm Ip Assets B.V. | Uv-filter compounds |
| US8669291B2 (en) | 2007-05-31 | 2014-03-11 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| US9586888B2 (en) | 2007-05-31 | 2017-03-07 | Sunovion Pharmaceuticals Inc. | Phenyl substituted cycloalkylamines as monoamine reuptake inhibitors |
| JP2010540443A (ja) * | 2007-09-21 | 2010-12-24 | アレイ バイオファーマ、インコーポレイテッド | 糖尿病治療のためのグルコキナーゼ活性化剤としてのピリジン−2−イル−アミノ−1,2,4−チアジアゾール誘導体 |
| JP2011506581A (ja) * | 2007-12-21 | 2011-03-03 | ジュリアーニ インターナショナル リミテッド | Ppar及びカンナビノイド受容体において活性である多標的化合物 |
| WO2009080821A3 (en) * | 2007-12-21 | 2010-01-14 | Giuliani International Limited | Multitarget compounds active at a ppar and cannabinoid receptor |
| WO2010047982A1 (en) | 2008-10-22 | 2010-04-29 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2010051206A1 (en) | 2008-10-31 | 2010-05-06 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2011106273A1 (en) | 2010-02-25 | 2011-09-01 | Merck Sharp & Dohme Corp. | Novel cyclic benzimidazole derivatives useful anti-diabetic agents |
| WO2012116145A1 (en) | 2011-02-25 | 2012-08-30 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| EP3243385A1 (en) | 2011-02-25 | 2017-11-15 | Merck Sharp & Dohme Corp. | Novel cyclic azabenzimidazole derivatives useful as anti-diabetic agents |
| WO2014022528A1 (en) | 2012-08-02 | 2014-02-06 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| JP2015527388A (ja) * | 2012-09-07 | 2015-09-17 | ノバルティス アーゲー | インドールカルボキサミド誘導体およびその使用 |
| US20160355477A1 (en) * | 2012-09-07 | 2016-12-08 | Novartis Ag | Indole carboxamide derivatives and uses thereof |
| WO2014130608A1 (en) | 2013-02-22 | 2014-08-28 | Merck Sharp & Dohme Corp. | Antidiabetic bicyclic compounds |
| WO2014139388A1 (en) | 2013-03-14 | 2014-09-18 | Merck Sharp & Dohme Corp. | Novel indole derivatives useful as anti-diabetic agents |
| WO2015051725A1 (en) | 2013-10-08 | 2015-04-16 | Merck Sharp & Dohme Corp. | Antidiabetic tricyclic compounds |
| WO2018106518A1 (en) | 2016-12-06 | 2018-06-14 | Merck Sharp & Dohme Corp. | Antidiabetic heterocyclic compounds |
| WO2018118670A1 (en) | 2016-12-20 | 2018-06-28 | Merck Sharp & Dohme Corp. | Antidiabetic spirochroman compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2003227360A1 (en) | 2003-11-10 |
| JPWO2003091213A1 (ja) | 2005-09-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2003091213A1 (en) | Novel amide derivatives or salts thereof | |
| CN101048402B (zh) | 咔唑衍生物、其溶剂合物或其药学上允许的盐 | |
| CN1176915C (zh) | 炔基苯基芳香杂环的菊糖激酶激活剂 | |
| CN1040324C (zh) | 新的噻唑烷二酮及含有这些化合物的药剂 | |
| KR20080040046A (ko) | 티아졸 유도체 | |
| EP1530568A1 (de) | INDOL-ODER BENZIMIDAZOLDERIVATE ZUR MODULATION DER IkB-KINASE | |
| EP2875010B1 (fr) | Derives de thiophenes utiles dans le traitement du diabete | |
| JP2004196702A (ja) | 新規なアミド誘導体又はその塩 | |
| WO1998022459A1 (fr) | Derives de la pyridin-2-yl-methylamine, leur procede de preparation et leur application comme medicaments | |
| JP2009013065A (ja) | 縮合へテロ環化合物 | |
| WO2008016175A1 (en) | Activator for peroxisome proliferator activated receptor | |
| CN117083275A (zh) | 心脏肌节抑制剂 | |
| WO2012020725A1 (ja) | Npy y5受容体拮抗作用を有するヘテロ環誘導体 | |
| JP2009506127A (ja) | 糖尿病の処置に有用なアニリノピラゾール誘導体 | |
| WO2012038904A1 (fr) | Derives de nicotinamide, leur preparation et leur application en therapeutique | |
| TWI857698B (zh) | 作為組蛋白去乙醯酶6抑制劑之1,3,4-二唑三唑化合物及包含其之醫藥組合物 | |
| CA2108064C (fr) | Nouveaux composes de thiazolidine dione, leur procede de preparation et les compositions pharmaceutiques les contenant | |
| WO2007068815A2 (fr) | Derives heterocycliques, leur preparation et leur application en therapeutique. | |
| CN110105286B (zh) | 一种含有脲素骨架的取代杂环类化合物及其制备方法和用途 | |
| JP5782032B2 (ja) | 神経疾患治療薬 | |
| CN103649059B (zh) | 具有npy y5受体拮抗作用的五元环芳香族杂环衍生物 | |
| WO2007043652A1 (ja) | 2-チエニルウレア誘導体 | |
| JPWO2007004733A1 (ja) | ペルオキシソーム増殖剤活性化受容体δの活性化剤 | |
| CN1107844A (zh) | 二氢化茚衍生物和其制法及合成该衍生物的中间体 | |
| CN1938020A (zh) | 离子通道调节剂 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003587778 Country of ref document: JP |
|
| 122 | Ep: pct application non-entry in european phase |