SYNTHESIS OF BENZONITRILES AND BENZIMIDATES
RELATED APPLICATIONS This application claims the benefit of U.S. Provisional Application Nos. 60/381,012,
60/381,021, 60/380,894, 60/380,910, 60/380,880, 60/381,017, 60/380,895, 60/380,903, 60/381,013, 60/380,878 and 60/380,909, all of which were filed May 15, 2002. This application also claims the benefit of U.S. Provisional Application No. 60/392,833, filed June 27, 2002. The entire teachings of the above-referenced applications are incorporated herein by reference.
BACKGROUND OF THE INVENTION
Nitrile-containing compounds are highly in demand because nitrile moieties are versatile reagents for organic synthesis, as exemplified in their applications in the preparation of thiazoles, chrial 2-oxazolines, tetrazoles, 1,2-diarylimidazoles, triazolo[l,5-c]pyrimidines, and benzamidines. Compounds prepared from nitriles have properties including superoxide inhibition, ferrielectric liquid crystal dopants, antipicornaviral agents, anti-inflammatory agents, anti-asthma agents, and fibrinogen antagonists.
An imidate moiety is strongly electrophilic, and as such, represents an important functional group in organic synthesis. The imidate moiety can be transformed into a wide variety of products, by virtue of undergoing reaction with a large number of nucleophiles. In addition, the imidate moiety can serve as a free radical scavenger. For example, ethyl 3,4,5- trihydroxybenzimidate blocks free radical generation from NADPH oxidase. This free radical scavenging activity decreases the amount of tissue damage, such as limiting damage to the heart following an infarction or other ischemic episode.
The use of nitriles and aryl imidates in the preparation of thiazoles, or when reduced, thiazolines and thiazolidines, is of particular interest. Compounds such as desferrithiocin and structural analogues contain a thiazoline ring, and these compounds represent an advance in
iron chelation therapy for subjects suffering from iron overload diseases. Present therapeutic agents such as desferroxamine require parenteral administration and have a very short half-life in the body, so that patient compliance and treatment cost are serious problems for subjects receiving long-term chelation therapy. Desferrithiocin and related compounds are effective when orally administered, thereby reducing patient compliance issues.
Unfortunately, 2,4-dihydroxybenzonitrile, which is a precursor to the potent, less toxic form of desferrithiocin known as 4'-hydroxydesazadesferrithiocin, remains a synthetic challenge. At this time, 2,4-dihydroxybenzonitrile and alkyl 2,4-dihydroxybenzimidates are not commercially available and the related 2,4-dimethoxybenzonitrile is expensive. Therefore, there is a need for novel methods of producing 2,4-dihydroxybenzonitrile and alkyl 2,4- dihydroxybenzimidates at a reasonable cost.
SUMMARY OF THE INVENTION
The invention includes a method of preparing a substituted benzonitrile represented by Structural Formula (I):
where Rj and R
2 are each independently -H, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group; comprising the steps of: a.) amidating a substituted benzoic acid represented by Structural Formula (II):
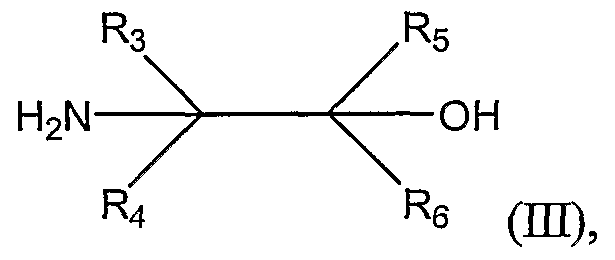
where Ri and R
2 are as defined above; by reacting the substituted benzoic acid with an activating agent and an α,β-aminoalcohol represented by Structural Formula (H-I):
where R
3, R^ R
5, and R
6 are each -H or substituted or unsubstituted alkyl groups, thereby forming a substituted 2-phenyloxazoline represented by Structural Formula (IV):
b.) reacting the substituted 2-phenyloxazoline with phosphorus oxychloride, thereby forming the substituted benzonitrile represented by Structural Formula (I). The invention also includes a method of preparing a substituted benzonitrile represented by Structural Formula (V):
where R
7 and R
8 are each -H or a substituted or unsubstituted aryl group; comprising the steps of:
a.) protecting hydroxyl groups of 2,4-dihydroxybenzoic acid with one or more substituted or unsubstituted arylalkyl protecting groups, thereby forming a protected 2,4-dihydroxybenzoic acid; b.) amidating the protected 2,4-dihydroxybenzoic acid, by reacting the protected 2,4-dihydroxybenzoic acid with an activating agent and an α,β-aminoalcohol represented by Structural Formula (VI):
where R
9, Rio, Rπ, and Rι
2 are each -H or substituted or unsubstituted alkyl groups, thereby forming a substituted 2-phenyloxazoline represented by Structural Formula (VII):
c) reacting the substituted 2-phenyloxazoline with phosphorus oxychloride, thereby forming the substituted benzonitrile represented by Structural Formula (V). In another embodiment, the invention provides a method of preparing a compound represented by Structural Formula (VIII):
comprising the steps of: a.) amidating a substituted benzoic acid represented by Structural Formula (II):
where Ri and R
2 are each independently -H, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group; by reacting the substituted benzoic acid with an activating agent and an α,β-aminoalcohol represented by Structural Formula (Dl):
where R
3, R
4, R
5, and Re are each -H or substituted or unsubstituted alkyl groups, thereby forming a substituted 2-phenyloxazoline represented by Structural Formula (IV):
b.) reacting the substituted 2-phenyloxazoline with phosphorus oxychloride, thereby forming a substituted benzonitrile; c) if Ri and R
2 are not each -H, cleaving ether groups in the product of step (b.), thereby forming 2,4-dihydroxybenzonitrile; and
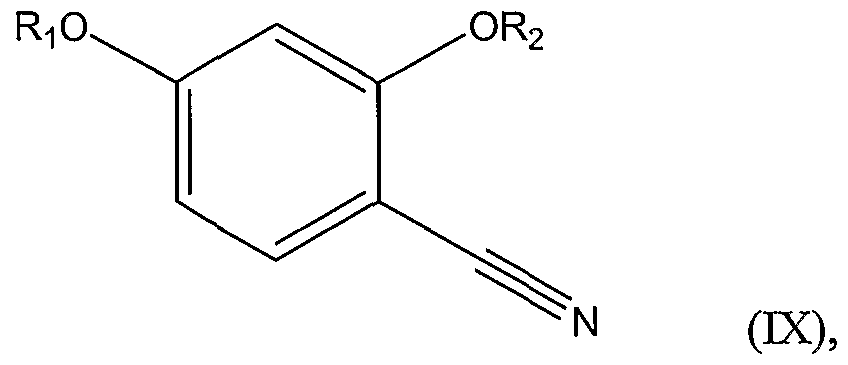
d.) coupling (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (VM). The invention includes a method of preparing a substituted benzonitrile represented by Structural Formula (DC):
wherein Ri and R
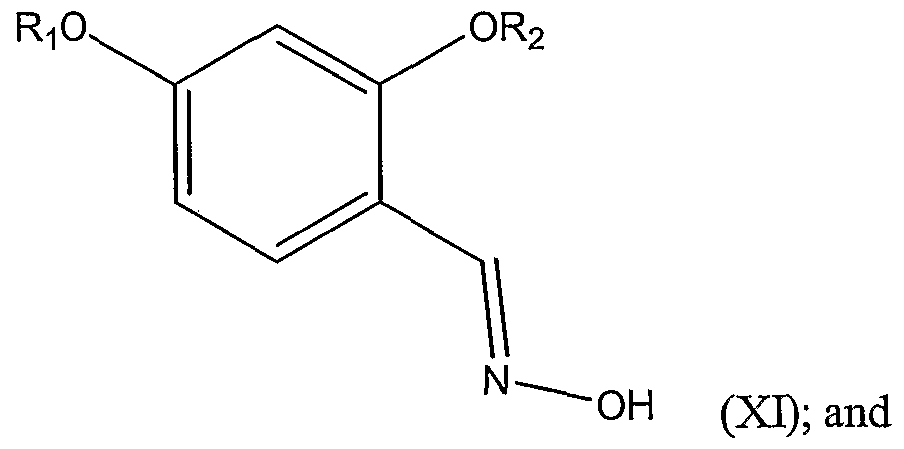
2 are each independently -H, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group; comprising the steps of: a.) reacting hydroxylamine or a protected derivative or a salt thereof and a disubstituted benzaldehyde represented by Structural Formula (X):
wherein Ri and R
2 are as defined above, thereby forming a substituted benzaldoxime represented by Structural Formula (XI):
b.) reacting the substituted benzaldoxime with diphosphorus pentoxide, thereby forming the substituted benzonitrile represented by Structural Formula (DC).
In another embodiment, the invention is a method of preparing a compound represented by Structural Formula (XH):
comprising the steps of: a.) reacting hydroxylamine or a protected derivative or a salt thereof and a disubstituted benzaldehyde represented by Structural Formula (X):
wherein Ri and R
2 are each independently -H, a substituted or unsubstituted alkyl group, or a substituted or unsubstituted aryl group; thereby forming a substituted benzaldoxime represented by Structural Formula (Dl):
b.) reacting the substituted benzaldoxime with diphosphorus pentoxide, thereby forming the substituted benzonitrile represented by Structural Formula (I):
c.) if Ri and R
2 are not each -H, reacting the product of step (b.) with a deprotecting agent, thereby forming 2,4-dihydroxybenzonitrile; and d.) coupling (S)-2~ιnethylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (XII)- In one embodiment, the invention is a method of preparing a substituted benzimidate represented by Structural Formula (XIII):
wherein: Ri and R
2 are each independently -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, or a substituted or unsubstituted arylalkyl group; and
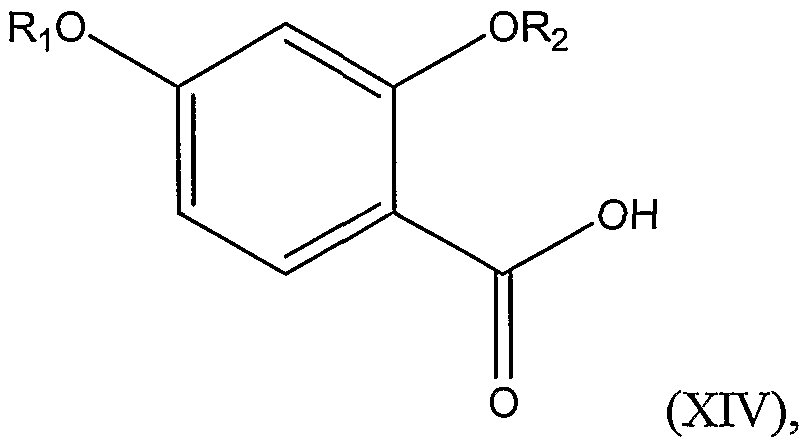
R3 is a substituted or unsubstituted alkyl group; comprising the steps of: a.) reacting a chlorinating agent and a disubstituted benzoic acid represented by
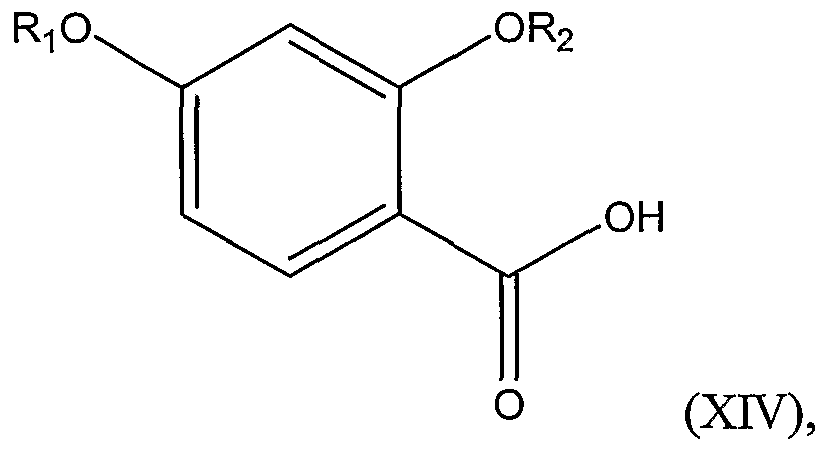
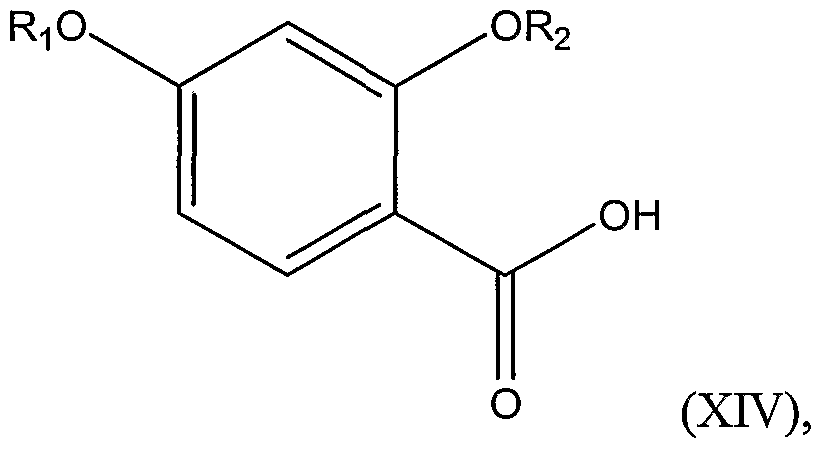
Structural Formula (XIV):
wherein Ri and R
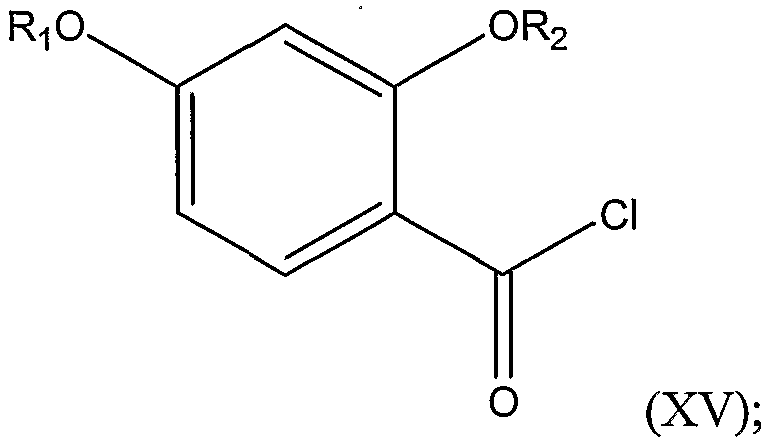
2 are as defined above, thereby forming a substituted benzoyl chloride represented by Structural Formula (XV):
b.) reacting the substituted benzoyl chloride with ammonia or a salt thereof, thereby forming a substituted benzamide represented by Structural Formula (XVT):
c.) reacting the substituted benzamide with a trialkyloxonium hexafluorophosphate of the formula (R
3)
3OPF
6, wherein R
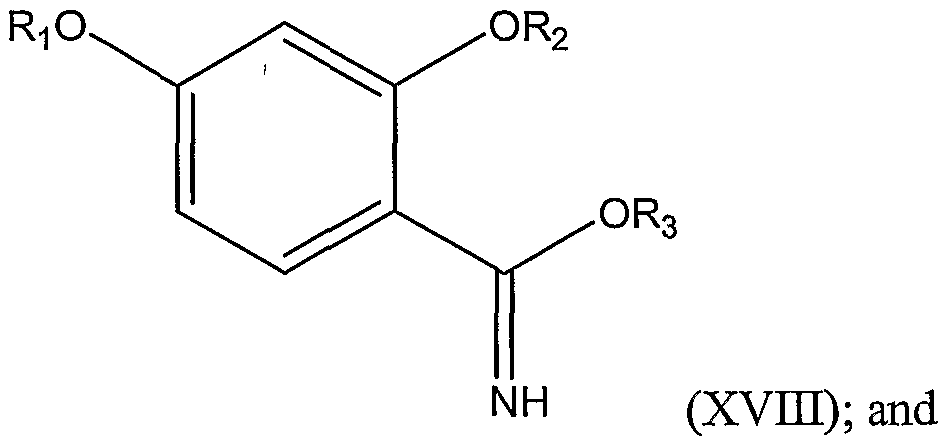
3 is as represented above, thereby forming the substituted benzimidate represented by Structural Formula (XIII). In another embodiment, the invention is a method of preparing a 2,4- dihydroxybenzimidate represented by Structural Formula (XVII):
wherein R
3 is a substituted or unsubstituted alkyl group; comprising the steps of: a.) protecting 2,4-dihydroxybenzoic acid with protecting groups, thereby forming protected benzoic acid represented by Structural Formula (XTV):
wherein Ri and R
2 are each independently a substituted or unsubstituted alkyl group, a substituted or unsubstituted aryl group, or a substituted or unsubstituted arylalkyl group; b.) reacting a chlorinating agent and the protected benzoic acid, wherein Ri and R
2 are as defined above, thereby forming a protected benzoyl chloride represented by Structural Formula (XV):
c) reacting the protected benzoyl chloride with ammonia or a salt thereof, thereby forming a protected benzamide represented by Structural Formula (XVT):
d.) reacting the protected benzamide with a trialkyloxonium hexafluorophosphate of the formula (R
3)
3OPF
6, wherein R
3 is as defined above, thereby forming a protected benzimidate represented by Structural Formula (XVTJI):
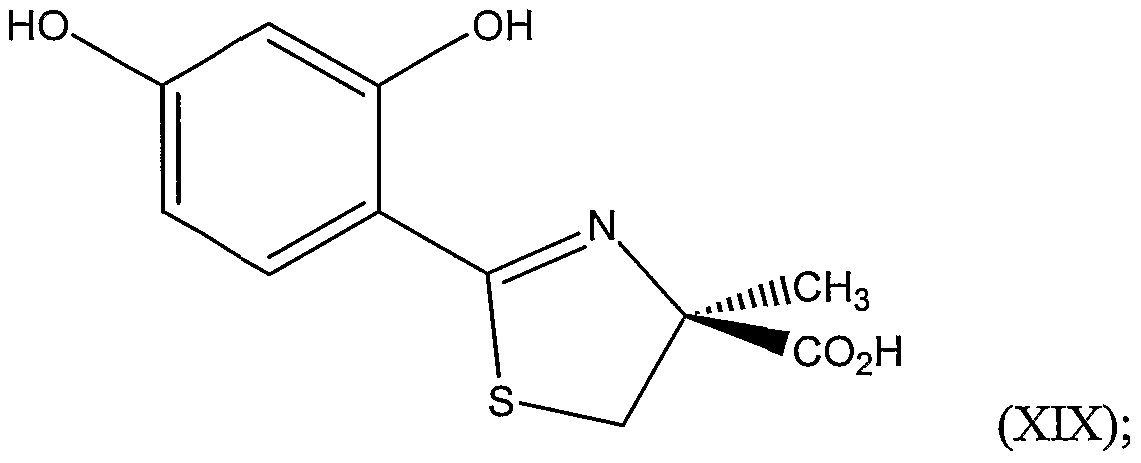
e.) deprotecting the protected benzimidate, thereby forming the 2,4- dihydroxybenzimidate. hi another embodiment, the invention is a method of preparing a compound represented by Structural Formula (XDC):
which is prepared by coupling (S)-2-methylcysteine and a 2,4-dihydroxybenzimidate, the preparation of which is described above.
Advantages of the present invention include the facile synthesis of 2,4- dihydroxybenzonitrile, or an ether or diether thereof, and alkyl 2,4-dihydroxybenzimidates from inexpensive and readily available starting materials. 2,4-Dihydroxybenzonitrile and alkyl 2,4-dihydroxybenzimidates prepared by the method of the present invention can be coupled to (S)-2-methylcysteine to form 4'-hydroxydesazadesferrithiocin, also referred to as 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4(S)-carboxylic acid, an iron chelating agent.
DETAILED DESCRIPTION OF THE INVENTION
A first useful and efficient method of preparing 2,4-dihydroxybenzonitrile, or an ether or diether thereof, involves reacting 2,4-dihydroxybenzoic acid (or an ether or diether thereof) with an α,β-aminoalcohol to form a 2-(2',4'-dihydroxyphenyl)-oxazoline (or an ether or diether
thereof). The 2-(2',4'-dihydroxyphenyl)-oxazoline can subsequently be reacted with phosphorus oxychloride to obtain 2,4-dihydroxybenzonitrile or a related compound. For ethers and diethers of 2,4-dihydroxybenzonitrile, additional steps may be desirable to cleave the ether moieties and obtain 2,4-dihydroxybenzonitrile. hi one example, it is desirable to protect one, or preferably, both of the hydroxyl groups of 2,4-dihydroxybenzoic acid before proceeding with the conversion to 2,4- dihydroxybenzonitrile. A preferred protecting group is a substituted or unsubstituted arylalkyl group such as a benzyl group. Protecting groups can be added, for example, by reacting 2,4- dihydroxybenzoic acid, a base, and a benzyl compound having a leaving group (e.g., benzyl tosylate, a benzyl halide such as benzyl chloride or benzyl bromide) in a polar solvent and refluxing the mixture for several hours, typically 1 or more hours, 1 to 12 hours, 2 to 8 hours, or 3 to 6 hours. The amount of the benzyl compound depends, in part, on the number of hydroxyl groups to be protected and is generally one or more (e.g., to protect one hydroxyl group) or two or more equivalents, such as about 1 to about 10 equivalents, about 2 to about 8 equivalents, or about 3 to about 5 equivalents. The reaction temperature typically depends on the solvent, and is selected such that the reaction mixture refluxes at the chosen temperature(s), which is generally at or greater than room temperature. Suitable solvents in the present reaction are typically polar, aprotic solvents such as acetone, tetrahydrofuran, dimethylformamide, acetonitrile, ethyl acetate, ethyl ether, dioxane, and hexamethylphosphoramide. Suitable bases for the present reaction typically include alkali metal and alkaline earth metal hydroxides, alkoxides, and carbonates, including sodium hydroxide, sodium methoxide, sodium ethoxide, sodium carbonate, potassium hydroxide, potassium methoxide, potassium ethoxide, cesium carbonate, calcium carbonate, and potassium carbonate.
It may be advantageous to protect both the hydroxyl groups and the carboxylic acid of 2,4-dihydroxybenzoic acid, and the carboxylic acid can be protected before, simultaneously with or after the hydroxyl groups. The fully protected product is generally a solid, and can be filtered and concentrated by suitable methods.
In order to deprotect the carboxylic acid moiety, the solid can be reacted at, for example, room temperature or greater (e.g., 20-100°C, 25-80°C, 30-60°C, 35-50°C) with abase, such as
those listed above, and a polar, protic solvent (e.g., methanol, ethanol, propanol, isopropanol, water, formamide, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal) for several hours (e.g. one or more hours, 1-12 hours, 2-10 hours, 3-8 hours, 4-6 hours). The amount of base can be catalytic or stoichiometric, but is preferably stoichiometric, such that there are one or more equivalents (e.g., about 1 to about 10 equivalents, about 2 to about 8 equivalents, about 3 to about 6 equivalents) of base. The deprotected carboxylate acid (carboxylate) moiety can be neutralized with an excess of a dilute acid such as hydrochloric acid, hydrobromic acid, nitric acid, or sulfuric acid. The neutralized acid often forms a solid, where only the hydroxyl groups of 2,4-dihydroxybenzoic acid are protected. This solid can be filtered and optionally recrystallized from a solvent mixture, such as a methanol-chloroform mixture.
In the next step of the reaction, either 2,4-dihydroxybenzoic acid or one of the protected species described above can be reacted with an activating agent such as a chlorinating agent, for example oxalyl chloride, phosphorus trichloride, or preferably thionyl chloride, in a nonpolar solvent such as pentane, heptane, octane, hexane(s), cyclohexane, carbon tetrachloride, toluene, xylenes, or preferably benzene, to form an acid chloride. The acid chloride can be dissolved in a polar, aprotic solvent such as those listed above, and optionally cooled below room temperature (e.g., about 15°C to about -35°C, about 10°C to about -20°C, 5°C to about -5°C). Then, an α,β-aminoalcohol can be added, followed by a base. Alternatively, 2,4- dihydroxybenzoic acid or the protected species described above can be reacted with an activating agent such as hydroxybenzotriazole (HOBt), imidazole, or 1,3- dicyclohexylcarbodiimide (DCC) and an α,β-aminoalcohol to produce an N- hydroxyethylarnide. α,β-Aminoalcohols are typically represented by Structural Formula (III) or Structural Formula (VI):
Preferably, R
3, R4, R , and Rι
0 are each independently an unsubstituted alkyl group, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, or t-butyl, and R
5, R
6, Rπ, and Rι
2 are each -H. Even more preferably, R
3, R , R
9, and Rio are each methyl. Suitable bases include dialkylamines and trialkylamines, preferably dimethylamine, diethylamine, diphenylamine, triphenylamine, trimethylamine, diisopropylamine, diisopropylethylamine, 1,4- diazabicyclo[2.2.2]octane (DABCO), l,5-diazabicyclo[4.3.0]non-5-ene (DBN), or triethylamine. The mixture of acid chloride and α,β-aminoalcohol are typically stirred at, for example, room temperature or greater, for at least about 15 minutes. Typically, the reaction continues for 15 minutes to 6 hours, 30 minutes to 3 hours, 45 minutes to 2 hours, or 60 to 90 minutes. After a desired amount of time, the mixture can be washed with an aqueous basic salt solution such as sodium hydroxide, potassium hydroxide, sodium carbonate, potassium carbonate, sodium bicarbonate, or potassium bicarbonate, and subsequently dried over a hydroscopic substance such as potassium carbonate, sodium carbonate, potassium sulfate, or sodium sulfate, and concentrated. The product of this step is an N-hydroxyethylamide. The N-hydroxyethylamide and one or more equivalents of an activating agent such as thionyl chloride (e.g, about 1 to about 10 equivalents, about 2 to about 8 equivalents, about 3 to about 6 equivalents) are generally stirred for at least 10 minutes (e.g., 10 minutes to 200 minutes, 20 minutes to 100 minutes, 30 minutes to 50 minutes) at, for example, about 0°C to about 90°C, about 10°C to about 60°C, about 15°C to about 40°C, or about 20°C to about 30°C. The mixture of N-hydroxyethylamide and thionyl chloride is neutralized with an aqueous base such as potassium hydroxide, sodium hydroxide, potassium carbonate, sodium carbonate, potassium bicarbonate, or sodium bicarbonate. The mixture can then be extracted with a polar, aprotic solvent, as listed above, and washed with water. The mixture can be dried over a hydroscopic substance and concentrated to obtain a 2-aryloxazoline. A mixture of the 2-aryloxazoline, an organic base, and phosphorus oxychloride are typically heated together. Suitable organic bases include piperidine, pyrrolidine, and preferably pyridine, which are present in a stoichiometric or a catalytic amount. When the above mixture is heated, the temperature is generally 60°C or greater, such as about 60°C to about 150°C,
about 70°C to about 130°C, about 80°C to about 120°C, or about 90°C to about 110°C. Preferably, the above mixture is heated for at least about 30 minutes, such as about 30 minutes to about 6 hours, about 1 hour to about 4 hours, or about 2 hours to about 3 hours. The mixture can be cooled to about room temperature (e.g., about 20°C to about 40°C or about 20°C to about 30°C), and then ice-cold water can be added. The mixture can be extracted with a polar, aprotic solvent, preferably ethyl acetate. The extracted mixture can be washed with a basic aqueous salt solution, preferably a sodium bicarbonate or potassium bicarbonate solution, before water is evaporated to give either 2,4-dihydroxybenzonitrile or the protected form, 2,4- dibenzyloxybenzonitrile. In one embodiment, protecting groups of 2,4-dihydroxybenzonitrile are cleaved. The protecting groups are typically bonded to 2,4-dihydroxybenzonitrile through an ether linkage (e.g., 2,4-dibenzyloxybenzonitrile). Ether linkages can be cleaved, for example, by methods described on pages 433-434 and 1012-1014 of "Advanced Organic Chemistry, Fourth Edition," by Jerry March, Wiley-Interscience, 1992 and references therein, all of which are incorporated by reference. Typically, ether linkages are cleaved by reaction with a mineral acid (e.g, HBr, HI) or a Lewis acid. Suitable Lewis acids include BF
3, BC1
3, (CH
3)
2BBr, BBr
3, A1C1
3, (CH
3)
3SiI, SiC /Nal, SiH
2I
2, LiI, NaI/BF
3, and (CH
3)
3SiCl/NaI.
In another embodiment, the invention includes a method of preparing 2,4- dihydroxybenzonitrile, or an ether or diether thereof, involves reacting 2,4- dihydroxybenzaldehyde or a diether thereof, such as 2,4-dimethoxybenzaldehyde, with hydroxylamine or a protected derivative or a salt thereof, to form an oxime. The oxime is typically dehydrated with diphosphorus pentoxide to form 2,4-dihydroxybenzonitrile or an ether or diether thereof. For ethers and diethers of 2,4-dihydroxybenzonitrile, additional steps may be desirable to cleave the ether moieties and obtain 2,4-dihydroxybenzonitrile. In examples where Ri and R2 are each -H, Ri and R2 can be protected by protecting groups, prior to reaction of the aldehyde moiety. A preferred protecting group is a substituted or unsubstituted alkyl group such as a methyl group. Protecting groups can be added, for example, by reacting 2,4-dihydroxybenzaldehyde, a base, and CH3-Z, where Z is a leaving group (e.g., tosylate, halide such as chloride or bromide) in a polar, aprotic solvent. Suitable
bases include sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, sodium carbonate, calcium carbonate, cesium carbonate, and potassium carbonate. Polar, aprotic solvents include acetone, acetonitrile, dimethylformamide, dioxane, ethyl acetate, ethyl ether, tetrahydrofuran, and hexamethylphosphoramide. Other suitable protecting group can be found in "Protective Groups in Organic Synthesis," by Peter G. M. Wuts and Theodora W. Greene, Wiley-hiterscience, 1990, the teachings of which are incorporated herein by reference in their entirety.
The first step of the reaction involves reacting a compound represented by Structural Formula (X):
with hydroxylamine or a protected derivative or a salt thereof (e.g., hydroxylammonium sulfate), to form an oxime. The hydroxyl moiety of hydroxylamine can be protected as benzyl ether, t-butyl ether, 2,6-dichlorobenzyl ether, 2-bromobenzyl ether, and 3,5-dibromobenzyl ether. In a preferred embodiment, Ri and R
2 are each independently a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group. In a more preferred embodiment, Ri and R
2 are each methyl. Typically, the compound represented by Structural Formula (X) is reacted with one or more equivalents (e.g., about 1 to about 10 equivalents, about 2 to about 8 equivalents, about 3 to about 6 equivalents) of hydroxylamine. Suitable conditions for reacting an aldehyde and hydroxylamine can be found, for example, on pages 906-907 of "Advanced Organic Chemistry, Fourth Edition," by Jerry March, Wiley-
Interscience, 1992, and references therein, all of which are incorporated by reference. In reactions of an aldehyde and hydroxylamine, the pH of the solvent (e.g., water or a mixture of water and a water-miscible organic solvent) is preferably about 4, or is in a range from about 3.5 to about 4.5, about 3 to about 5, or about 2 to about 6.
The second step of the reaction involves reacting the oxime with diphosphorus pentoxide. Typically, P2O5 is heated with the oxime for 1 or more hours (e.g., about 1 to about 12 hours, about 2 to about 8 hours, about 3 to about 6 hours) at room temperature or greater (e.g., about 20°C to about 200°C, about 40°C to about 150°C, about 60°C to about 100°C). One or more equivalents, such as about 1 to about 6 equivalents, about 1.5 to about 5 equivalents, or about 2 to about 4 equivalents, of P2O5 are generally required for the reaction.
Following the second step, when Ri and R2 are not each -H, it is often advantageous to remove and R2, otherwise known as deprotecting the ether groups of a nitrile product. Typically, the product of the second step is isolated before proceeding with deprotecting. Deprotecting an ether group can be achieved by reacting a protected ether with a deprotecting agent. Preferred deprotecting agents include boron trihalides such as boron trifluoride, boron trichloride, and boron tribromide. Additional deprotecting methods can be found in "Protective Groups in Organic Synthesis," which was previously incorporated by reference.
Diphosphorus pentoxide is a dehydrating agent. Other dehydrating agents include acetic anhydride, ethyl ortho formate in an acidic solution, triphenylphosphine in carbon tefrachloride, trichloromethylchloroformate, methyl cyanoformate, ethyl cyanoformate, trifluoromethane sulfonic anhydride, P2I , SeO2, trichloroformyl chloride in triethylamine, and chloromethylene dimethylammonium chloride.
In yet another embodiment, the invention includes a method of preparing ethyl 2,4- dihydroxybenzimidate, or an ether or diether thereof, involves amidating 2,4-dihydroxybenzoic acid or a diether thereof, such as 2,4-dibenzyloxybenzaldehyde. The amide is typically reacted with a trialkyloxonium salt to form an imidate. Functional groups protecting hydroxyl groups, such as benzyl groups, can be removed, for example, by hydrogenation.
In examples where Ri and R2 of Structural Formula (XIII) are each -H, Ri and R2 can be protected by protecting groups. A preferred protecting group is a substituted or unsubstituted arylalkyl group such as a benzyl group. Protecting groups can be added, for example, by reacting 2,4-dihydroxybenzoic acid, a base, and a benzyl compound substituted with a leaving group (e.g., benzyl tosylate, a benzyl halide such as benzyl chloride or benzyl bromide) in a polar, aprotic solvent (e.g., acetone, acetonitrile, dimethylformamide, dioxane, ethyl acetate,
ethyl ether, hexamethylphosphoramide, tetrahydrofuran). Suitable bases include sodium hydride, potassium hydride, sodium amide, potassium amide, and lithium diisopropylamide. In one example, 2,4-dihydroxybenzoic acid is reacted with sodium hydride and benzyl bromide in dimethylformamide to yield a compound with protected hydroxyl and carboxylic acid groups. The carboxylic acid can be deprotected by refluxing the compound in a basic dioxane solution. Suitable bases include sodium hydroxide and potassium hydroxide. The solution is acidified after refluxing. Additional protecting groups can be found, for example, in "Protective Groups in Organic Synthesis, Third Edition," by Peter G. M. Wuts and Theodora W. Greene, Wiley- hiterscience, 1999.
The first step of the reaction involves reacting a compound represented by Structural Formula (XIV):
with a chlorinating agent in a polar, aprotic solvent (as defined above) or a mixture of a polar, aprotic solvent and a nonpolar solvent, in order to form an acid chloride. Suitable chlorinating agents include thionyl chloride, phosphorus trichloride, or, preferably, oxalyl chloride. Suitable nonpolar solvents include pentane, heptane, octane, hexane(s), cyclohexane, carbon tefrachloride, toluene, xylenes, and benzene. Typically, the reaction is carried out at or below 30°C, such as from about -50°C to about 30°C, about -30°C to about 25°C, or about 0°C to about 25°C. If the carboxylic acid is protected (e.g., as a result of protecting the hydroxyl groups), the acid is preferably deprotected prior to reaction with a chlorinating agent.
The acid chloride can be reacted with ammonia or a salt thereof (e.g., NH OH) to form an amide (e.g., a benzamide). For example, the acid chloride can be reacted with an aqueous ammonia solution in a polar, aprotic solvent such as methylene chloride or another of those listed above to form the amide.
The amide is typically reacted with a trialkyloxonium hexafluorophosphate of the formula (R3)3OPF6 to form a benzimidate. Preferred R3 groups are C1-C4 alkyl groups such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, and t-butyl. Ethyl is an especially preferred R3. In one example, the amide formed above is reacted with triethyloxonium hexafluorophosphate in a polar, aprotic solvent such as methylene chloride to form an ethyl benzimidate.
In examples where K\ and R2 are not each -H in the benzimidate formed above, it may be desirable to cleave Ri and R2 to deprotect hydroxyl groups. A suitable method to deprotect the hydroxyl groups includes reacting the protected benzimidate with hydrogen. For example, a protected benzimidate can be hydrogenated by reacting the benzimidate with 1 atmosphere of hydrogen in the presence of a palladium-carbon catalyst in a polar, protic solvent such as methanol or ethanol. The hydrogenation yields a deprotected benzimidate, such as ethyl 2,4- dihydroxybenzimidate .
Although a benzamide is preferably reacted with a trialkyloxonium hexafluorophosphate, benzamide can also be reacted with trialkyloxoxium tetrafluoroborate salts.
Common Definitions and Techniques
The section below applies to the invention as a whole, such that these definitions and techniques can generally be applied to the various methods and embodiments of the invention described above. Under circumstances where discussion in the individual sections above duplicates the discussion in this section, the discussion in the individual section should be considered as a preferred embodiment of that method. Unless otherwise indicated, these common definitions and techniques are applicable to the entire invention.
Cysteine or a 2-alkylcysteine such as (S)-2-methylcysteine can be coupled with 2,4- dihydroxybenzonitrile, or an ether or diether thereof. Cysteine and related compounds can also be coupled with other substituted and unsubstituted aryl nitriles. In a preferred embodiment, (S)-2-methylcysteine is coupled to 2,4-dihydroxybenzonitrile to form 4,5-dihydro-2-(2,4- dihydroxyphenyl)-4-methylthiazole-4(S)-carboxylic acid (also known as 4'- hydroxydesazadesferrithiocin) .
Cysteine or a 2-alkylcysteine such as (S)-2-methylcysteine can be coupled with 2,4- dihydroxybenzimidate, or an ether or diether thereof. In a preferred embodiment, (S)-2- methylcysteine is coupled to 2,4-dihydroxybenzimidate to form 4,5-dihydro-2-(2,4- dihydroxyphenyl)-4-methylthiazole-4(S)-carboxylic acid (also known as 4'- hydroxydesazadesferrithiocin).
Syntheses of cysteine and cysteine derivatives suitable for coupling can be found in U.S. Application Nos. 60/381,012, 60/381,021, 60/380,894, 60/380,910, 60/380,880, 60/381,017, 60/380,895 and 60/380,903, filed May 15, 2002, and U.S. Application No. 60/392,833, filed June 27, 2002; the entire teachings of which are incorporated herein by reference. Typically, coupling of cysteine or a 2-alkylcysteine and a substituted benzonitrile includes converting the benzonitrile into a benzimidate. The benzimidate can be formed, for example, by reacting the benzonitrile with an alcohol such as methanol, ethanol, n-propanol, or isopropanol in the presence of an acid such as hydrochloric acid. The benzimidate (obtained from a benzonitrile or from the method disclosed herein) is then reacted with the cysteine (or related compound) under basic conditions. Acceptable bases include trimethylamine, triethylamine, triphenylamine, diisopropylamine, diisopropylethylamine, diethylamine, dimethylamine, diphenylamine, DABCO, DBN, and the like. The reaction between the benzimidate and the cysteine results in the thiazoline (or 4,5-dihydrothiazole) containing product. When forming the benzimidate from a hydroxylated benzonitrile (e.g., 2,4- dihydroxybenzonitrile), the hydroxyl groups are advantageously protected (e.g., with a substituted or unsubstituted alkyl or arylalkyl group such as a benzyl group). The protecting groups are subsequently cleaved, typically by catalytic hydrogenation.
Products of the above methods can be purified by methods known in the art, such as emulsion crystallization. The methods of the claimed invention can be used to manufacture other related desferrithiocin analogs and derivatives. Examples of such analogs include those described in U.S. Patent Nos. 5,840,739, 6,083,966, 6,159,983, 6,521,652 and 6,525,080 to Raymond J. Bergeron, Jr., the contents of which are incorporated herein by reference. Additional examples
can be found in PCT/US93/10936, PCT/US97/04666, and PCT/US99/ 19691, the contents of which are incorporated by reference.
An alkyl group is a hydrocarbon in a molecule that is bonded to one other group in the molecule through a single covalent bond from one of its carbon atoms. Alkyl groups can be cyclic or acyclic, branched or unbranched, and saturated or unsaturated. Typically, an alkyl group has one to about 24 carbons atoms, or one to about 12 carbon atoms. Lower alkyl groups have one to four carbon atoms and include methyl, ethyl, 77-propyl, ώo-propyl, «-butyl, sec-butyl and tert-butyl.
Aromatic (or aryl) groups include carbocyclic aromatic groups such as phenyl, p-tolyl, 1 -naphthyl, 2-naphthyl, 1-anthracyl and 2-anthracyl. Aromatic groups also include heteroaromatic groups such as N-imidazolyl, 2-imidazole, 2-thienyl, 3 -thienyl, 2-furanyl, 3- fiiranyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 2-pyranyl, 3-pyranyl, 3- pyrazolyl, 4-pyrazolyl, 5-pyrazolyl, 2-pyrazinyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2- oxazolyl, 4-oxazolyl and 5-oxazolyl. Aromatic groups also include fused polycyclic aromatic ring systems in which a carbocyclic, alicyclic, or aromatic ring or heteroaryl ring is fused to one or more other heteroaryl or aryl rings. Examples include 2-benzothienyl, 3 -benzothienyl, 2-benzofuranyl, 3- benzofuranyl, 2-indolyl, 3-indolyl, 2-quinolinyl, 3-quinolinyl, 2-benzothiazole, 2- henzooxazole, 2-benzimidazole, 2-quinolinyl, 3-quinolinyl, 1-isoquinolinyl, 3-quinolinyl, 1- isoindolyl and 3-isoindolyl.
Suitable substituents for alkyl groups include -OH, halogen (-Br, -CI, -I and -F), -O(R'), -0-CO-(R '), -CΝ, -Ν02, -COOH, =O, -NH2, -NH(R'), -N(R')2, -COO(R'), -CONH2, -CONH(R'), -CON(R')2, -SH, -S(R'), and guanidine. Each R' is independently an alkyl group or an aryl group. Alkyl groups can additionally be substituted by an aryl group (e.g. an alkyl group can be substituted with an aromatic group to form an arylalkyl group). A substituted alkyl group can have more than one substituent.
Suitable substituents for aryl groups include -OH, halogen (-Br, -CI, -I and -F), -0(R'), -0-CO-(R '), -CN, -N02, -COOH, =O, -NH2, -NH(R'), -N(R') , -COO(R'), -CONH2,
-CONH(R'), -CON(R')2, -SH, -S(R'), and guanidine. Each R' is independently an alkyl group or an aryl group. Aryl groups can additionally be substituted by an alkyl or cycloaliphatic group (e.g. an aryl group can be substituted with an alkyl group to form an alkylaryl group such as tolyl). A substituted aryl group can have more than one substituent.
Boron trihalides are acceptable deprotecting agents (i.e., for hydrolysis of ethers) for use in the present invention. Other deprotecting agents include (CH3)2BBr, A1C13, (CH3)3SiI, SiCl4/NaI, SiH2I2, Lil, NaI/BF3, and (CH3)3SiCl/NaI.
EXEMPLIFICATION
EXAMPLE 1
Synthesis of 2,4-Benzyloxybenzonitrile
A. 2,4-Dibenzyloxy Benzoic Acid A solution of 2,4-dihydroxybenzoic acid (5 g), anhydrous K C03 (40 g) and benzyl bromide (16 mL) in acetone (100 mL) was refluxed for 4 hours. After filtration of solid, the filtrate was concentrated. The residue was stirred at room temperature with KOH (6 g), and methanol (20 mL) for 4 hours, and neutralized with dilute HC1 (pH 2). The solid thus formed was filtered and recrystallized from a methanol-chloroform mixture (3:1) to get 3.5 g of the product. Concentration of mother liquor gave an additional 1 g of the product.
B . Preparation of Oxazoline
The suspension of the acid from Part A (3.2 g), SOCl2 (2 mL) in dry benzene (10 mL) was heated at reflux for 8 hours, concentrated and co-distilled with benzene. The resulting acid chloride was dissolved in CH2C12 (8 L), cooled in ice water and then 2-amino-2- methylpropanol (2.2 g) was added, followed by friethylamine (1.4 mL). The resulting mixture was stirred at room temperature for 1 hour and washed with a sodium bicarbonate solution, water, dried over K2C03 and concentrated to give an amide (3.3 g) as a solid.
The above amide (1 g) and SOCl2 (1 mL) were stirred at room temperature for 0.5 hours, neutralized with 20% aqueous NaOH. The reaction mixture was extracted with CHC1 , washed with water, and dried over K2C03 and concentrated to give an oxazoline (0.88 g) as a solid.
C. Preparation of 2,4-Dibenzyloxy Benzonitrile
The oxazoline (0.75 g), pyridine (2 mL) and POCl3 (1 mL) were heated at 90°C for 2 hours, cooled to room temperature, and decomposed with ice cold water and extracted with ethyl acetate. The organic layer was washed with saturated sodium bicarbonate, and the water was evaporated to give 2,4-dibenzyloxybenzonitrile (0.45 g) as a solid.
EXAMPLE 2
35 mg of R- and S-4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4- carboxylic acid were dissolved in 1 ml of a mixture of 9% N-methyl-pyrrolidone, 9% v/v 2- hexanol, 10% v/v Rhodafac RE 610, 5% v/v Soprophor FL and 68% v/v water by heating to 50°C in a 5 mL vial. After the product was completely dissolved, the microemulsion was cooled down to room temperature and agitated with a shaking machine (350 rpm). During two hours, no spontaneous crystallisation was observed. The mixture was then seeded with two drops of a dilute, finely ground suspension of pure S-product crystals grown under similar conditions. After two hours of shaking, the resulting crystals were filtered off, washed with water and dried in a gentle nitrogen stream. The procedure yielded 5.4 mg (15.4%) of colorless crystals, with a greater than 90% purity of the S enantiomer.
EXAMPLE 3
4.00 g (S)-2-methylcysteine hydrochloride (23.3 mmol,1.0 meq) and 3.14 g 2,4- dihydroxy benzonitrile (23.3 mmol, 1.0 meq) were suspended in 40 mL ethanol. After
degassing this mixture with nitrogen (30 min) 4.95 g triethylamine (6.8 mL, 48.9 mmol, 2.05 meq) were added. The obtained suspension was heated under reflux in an atmosphere of nitrogen for 20 hours and then cooled to room temperature. From this suspension ethanol was evaporated under reduced pressure until an oil (20 % of the initial volume) was obtained. This oil was dissolved in 50 mL water. The solution was adjusted to pH 7.5 with 1.20 ml 20 % KOH and was extracted two times each with 20 mL methyl t-butyl ether (MTBE). The aqueous layer was separated, adjusted with 20 % KOH to pH 11 and again extracted two times each with 20 mL MTBE. After separating the aqueous layer the pH was set with concentrated HCl to 7.5 and traces of MTBE were distilled off. Then the aqueous solution was acidified with 1.50 L concentrated HCl to pH 1.5. The product precipitated. This suspension was stirred at 4 °C for 1 hour. Then the precipitate was filtered, washed two times each with 10 mL water (5°C) and dried at 45 °C under vacuum. The reaction yielded 5.17 g (87.6 %) of crude 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4(S)- carboxylic acid product. 'H-NMR showed no significant impurity.
EXAMPLE 4
2,4-Dimethoxybenzoic acid is reacted with hydroxylamine to form 2,4- dimethoxybenzaldoxime. 2,4-Dimethoxybenzaldoxime is reacted with diphosphorus pentoxide to form 2,4-dimethoxybenzonitrile. 2,4-Dimethoxybenzonitrile is reacted with boron trichloride to form 2,4-dihydroxybenzonitrile.
EXAMPLE 5
Synthesis of 2,4-Dihydroxybenzonitrile
In a double walled reactor 50.0 g (0.362 mol, 1.0 meq) 2,4-dihydroxy-benzaldehyde were added to 180 mL formic acid, which resulted in a brown suspension at room temperature. Then 45.8 g (0.673 mol, 1.8 meq) sodium formate were added
over 2 min, and the temperature increased to 33°C. After the temperature decreased to 30 °C, 35.6 g (0.217 mol, 1.2 meq) hydroxyl ammonium sulfate were added during 3 min to give a thick brown suspension which became a brown solution after stirring 10 min at 30-32 °C. While heating the mixture to 100°C, crystallization occurred at 38°C and stirring was interrupted. At 70 °C the reaction mixture became a thin suspension, which was easy to stir. This reaction mixture was stirred for 2 hours at 100°C. The color turned dark brown. TLC (silica gel 60 F254, acetone:n-hexane:water 20:20:1) showed an almost complete reaction. Formic acid (170 mL) was evaporated under reduced pressure (60°C, 10 mbar). The solid dark brown residue was stirred with 400 mL MTBE at 40 °C for 1 hour (incomplete dissolution). The insoluble residue (62.5 g) was filtered and washed two times each with 50 mL MTBE. To the mother liquor 10 g activated carbon (Norit CA 5) were added and this mixture was refluxed for 1 hour and filtered at 40°C by Celite Super Hyflow (washing with 2x50 mL portions of MTBE). The MTBE-mother liquor was washed three times each with 100 mL water. After removing the water from this MTBE solution with azeotropic distillation (water separator) it was concentrated under reduced pressure to 20% of the starting volume and 500 mL toluene were added. Then the MTBE was distilled off under reduced pressure. During this process a brown residue began to precipitate, which was filtered. The toluene mother liquor was concentrated to 150 mL and 2,4- dihydroxybenzonitrile precipitated, which was filtered and washed two times each with 30 mL toluene. The pale tan product was dried under reduced pressure (45°C, 20 mbar). The reaction yielded 34.5 g of 2,4-dihydroxybenzonitrile (70.5%, purity 97% (HPLC)).
EXAMPLE 6
2,4-Dihydroxybenzoic acid was reacted with sodium hydride and benzyl bromide in dimethylformamide (DMF) to give a compound protected with benzyl groups at hydroxyl and carboxylic acids moieties in a 91% yield. The benzyl group was removed from the carboxylic acid moiety by refluxing it with 2 N sodium hydroxide in dioxane, followed by acidifying the mixture, to give 2,4-dibenzyloxybenzoic acid in 83% yield.
2,4-Dibenzyloxybenzoic acid was reacted with oxalyl chloride in toluene and DMF at 0- 25°C to give an acid chloride. The acid chloride was reacted with aqueous ammonia in methylene chloride to yield 87% (over 2 steps) of 2,4-dibenzyloxybenzamide. 2,4- Dibenzyloxybenzamide was reacted with triethyloxonium hexafluorophosphate in methylene chloride to yield 69% of ethyl 2,4-dibenzyloxybenzimidate. The ethyl 2,4- dibenzyloxybenzimidate was hydrogenated over a palladium/carbon catalyst with 1 atmosphere of hydrogen gas in ethanol, to yield 75% of ethyl 2,4-dihydroxybenzimidate. The overall yield for the reaction sequence was 34% for six steps.
EXAMPLE 7
2,4-Dibenzyloxybenzonixrile (0.121 mol) was dissolved in 5.85 g (0.127 mol) ethanol and 19.4 ml 1,2-dimethoxyethane in a double walled reactor. This solution was cooled to -5 °C, stirred and saturated with dry HCl gas over 5 hours at 0-3 °C. The reaction mixture was stirred overnight at 2-4 °C under nitrogen. During this time, a product crystallized. The white crystals were filtered off, washed with 1,2-dimethoxyethane (5 °C, three times each with 13 ml) and dried. A total of 30 of the protected ethyl benzimidate was isolated (Yield 88.4%, purity 98.9%).
The protected ethyl benzimidate described above was dissolved in methanol to generate a 10% solution and was catalytically hydrogenated at room temperature using 5% Pd/C as a catalyst. The reaction was completed after 8 hours. The solution was filtered and the solvent evaporated to yield the deprotected product as an orange-yellow solid. The reaction yielded 19.6 g (94%) of product.
In contrast, the formation of the imidate with 2,4 dihydroxybenzonitrile was a low yielding process, generating the desired product in only 20% yield and with less than desired purity.
While this invention has been particularly shown and described with references to preferred embodiments thereof, it will be understood by those skilled in the art that various
changes in form and details may be made therein without departing from the scope of the invention encompassed by the appended claims.