SYNTHESIS OF 2-ALKYLCYSTEINES, 2-(HYDROXYLATED PHENYL)-4- ALKYLTHIAZOL--- E-4-CA-RBOXYLIC ACIDS AND DERIVATIVES THEREOF
RELATED APPLICATIONS This application claims the benefit of U.S. Provisional Application Nos. 60/381,012,
60/381,021, 60/380,894, 60/380,910, 60/380,880, 60/381,017, 60/380,895, 60/380,903, 60/381,013, 60/380,878 and 60/380,909, all of which were filed May 15, 2002. This application also claims the benefit of U.S. Provisional Application No. 60/392,833, filed June 27, 2002. The entire teachings of the above-referenced applications are incorporated herein by reference.
BACKGROUND OF THE INVENTION
Alpha-amino acids are useful starting materials in the synthesis of peptides, as well as non-peptidal, peptidomimetic pharmaceutically active agents, hi order to enable the synthesis of a large number of compounds from an amino acid precursor, it is advantageous to have naturally occurring and non-naturally occurring amino acids. Non-naturally occurring amino acids typically differ from natural amino acids by their stereochemistry (e.g., enantiomers), by the addition of alkyl groups or other functionalities, or both. At this time, the enantiomers of naturally occurring amino acids are much more expensive than the naturally occurring amino acids. In addition, there are only a limited number of commercially available amino acids that are functionalized or alkylated at the alpha-carbon, and often syntheses involve the use of pyrophoric or otherwise hazardous reagents. Moreover, the syntheses are often difficult to scale up to a commercially useful quantity. Consequently, there is a need for new methodologies of producing such non-naturally occurring amino acids. Non-naturally occurring amino acids of interest include the (R)- and (S)-isomers of 2-methylcysteine, which are used in the design of pharmaceutically active moieties. Several natural products derived from these isomers have been discovered in the past few years. These natural products include desferrithiocin, from Streptomyces antibioticus; as well as tantazole A, mirabazole C, and thiangazole, all from blue-green algae. These compounds have diverse biological activities ranging from iron chelation to murine solid tumor-selective cytotoxicity to inhibition of HIN-1 infection.
Desferrithiocin, deferiprone, and related compounds represent an advance in iron chelation therapy for subjects suffering from iron overload diseases. Present therapeutic agents such as desferroxamine require parenteral administration and have a very short half- life in the body, so that patient compliance and treatment cost are serious problems for subjects receiving long-term chelation therapy. Desferrithiocin and related compounds are effective when orally administered, thereby reducing patient compliance issues. Unfortunately, (S)-2-methylcysteine, which is a precursor to the more active and/or less toxic forms of desferrithiocin and related compounds, remains a synthetic challenge. Therefore, there is a need for novel methods of producing 2-methylcysteine at a reasonable cost, and means of isolating the desired enantiomer.
SUMMARY OF THE INVENTION
Methods For Preparing 2-Alkyl Cysteine Via Phase Transfer Catalysis
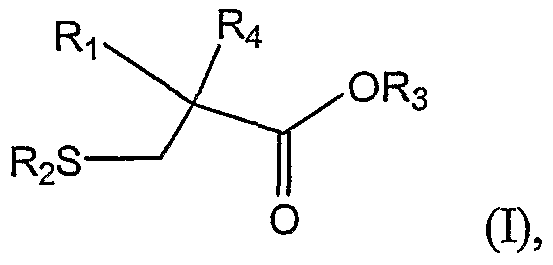
In one aspect, the present invention relates to a method of preparing a 2- alkylcysteine derivative represented by Structural Formula (I) :
or a salt thereof, wherein
Ri is -NH2; -N(R5)(R6) ; -NHR7; or -N=R8; wherein R5, R6, R7, and R8 are, independently, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; R2 and R3 are, independently, -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; and
R is a substituted or unsubstituted alkyl group.
In one embodiment, the method comprises reacting a cysteine derivative represented by Structural Formula (II):
or a salt thereof, wherein Rι,R2 and R3 are defined as above, with a compound having the formula R4-L, wherein R4 is defined as above and L is a leaving group, in the presence of a phase transfer catalyst thereby forming the 2-alkylcysteine derivative represented by Structural Formula (I). Typically, this reaction is carried out in the presence of a base.
The above-described methods may additionally comprise the step of purifying or ultrapurifying the synthesis products by resolving enantiomers or diastereomers of the products. The cysteine derivative formed can be the (R) or (S)-isomer or a mixture thereof. Additionally, the methods can comprise the isolation of the enantiomers of the synthesis products. In a preferred embodiment, the methods of the present invention comprise isolating the (S)-enantiomer of 2-alkylcysteine.
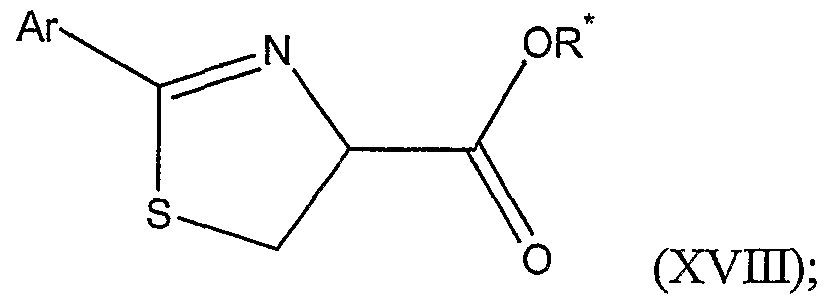
The present invention also relates to a method of preparing a substituted thiazoline carboxylic acid represented by Structural Formula (VII):
or a salt thereof. hi one embodiment, the method comprises:
(a) reacting, in the presence of a phase transfer catalyst, an
(R)-cysteine derivative represented by Structural Formula (NUT):
or a salt thereof, wherein
R20 is -NH2; -N(R25)(R26); -NHR26; or -N=R27, wherein R24, R25,
R26, and R27 are, independently, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; and R2ι and R22 are, independently, -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; with a compound having the formula CH3 -L, wherein L is a leaving group, thereby forming a 2-methylcysteine derivative represented by Structural Formula (DC):
or a salt thereof;
(b) optionally, purifying the (S)-isomer of the 2-methylcysteine derivative;
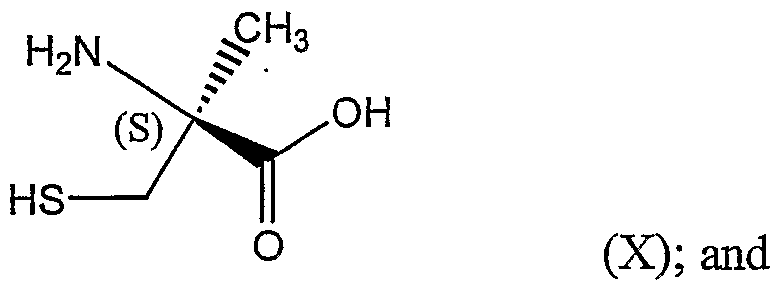
(c) reacting the (S)-isomer of the 2-methylcysteine derivative with acid to form a (S)-2-methylcysteine represented by Structural Formula (X):
(d) coupling the (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile thereby forming the substituted thiazoline carboxylic acid represented by Structural Formula (VH).
Methods of Preparing 2-Alkyl Cysteine Via Chiral Ester
Another useful and efficient method of preparing 2-alkylcysteine involves condensing cysteine with an aryl nitrile to form a 2-arylthiazoline-4-carboxylic acid, esterifying the 2-arylthiazoline-4-carboxylic acid using a substituted or unsubstituted alcohol group comprising one or more chiral carbons, and alkylating at the 4-position of the thiazoline ring to form a 2-aryl-4-alkyl-thiazoline-4-carboxylic acid ester. Esterifying a carboxylic acid with a chiral alcohol results in the formation of a chiral template. The chiral templates present in the thiazoline carboxylic acid ester can provide face selectivity, and consequently stereochemical control, during the delivery of an alkyl group to the 4-position of the thiazoline ring. The chiral templates present in the thiazoline carboxylic acid ester produces an enantiomeric excess of one isomer.
In one aspect, the invention relates to a method of preparing an optically active 2- alkylated cysteine represented by Structural Formula (XV):
or a salt thereof, wherein Ri is a substituted or unsubstituted alkyl group, the method comprising:
(a) coupling a compound (which may be an (R) or (S)-isomer or a mixture thereof) represented by Structural Formula (XVI):
or a salt thereof, with a substituted or unsubstituted aryl nitrile of the formula Ar-CN, wherein Ar is a substituted or unsubstituted aryl group; thereby forming a substituted thiazoline carboxylic acid represented by Structural Formula (XVE):
(b) esterifying the substituted thiazoline carboxylic acid with R -OH , wherein R is a substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms; thereby forming a substituted thiazoline carboxylic acid ester represented by Structural Formula (XVIH-):
(c) alkylating the substituted thiazolme carboxylic acid ester with one or more bases and RiX, wherein X is a leaving group and Ri is as defined above; thereby forming an alkylated substituted thiazoline carboxylic acid ester represented by Structural Formula (XIX):
(d) hydrolyzing the alkylated substituted thiazoline carboxylic acid ester with base or acid, thereby forming an alkylated substituted thiazoline carboxylic acid represented by Structural Formula (XX):
(e) reacting the alkylated substituted thiazoline carboxylic acid with acid, thereby forming the 2-alkylated cysteine represented by Structural Formula
(XV).
The methods described above may additionally comprise the step of purifying or ultrapurifying the alkylated substituted thiazoline carboxylic acid or the alkylated substituted thiazoline carboxylic acid ester. Purifying the ester or the acid can comprise further resolving the enantiomers or diasteromers of the alkylated substituted thiazoline carboxylic acid or the alkylated substituted thiazoline carboxylic acid ester. Additionally, the methods can comprise the isolation of the enantiomers of the synthesis products. Preferably, the (S)- enantiomer of 2-alkylcysteine is isolated (or the corresponding 4-alkyl-2-arylthiazoline carboxylic acid or salt or ester thereof), for example, (S)-2-methylcysteine.
In another aspect, the method relates to a method of preparing a compound represented by Structural Formula (XXI):
or a salt thereof, the method comprising:
(a) coupling a compound (which may be an (R) or (S)-isomer or a mixture thereof) represented by Structural Formula (XXII):
or a salt thereof, with a substituted or unsubstituted aryl nitrile of the formula Ar-CN, wherein Ar is a substituted or unsubstituted aryl group; thereby
forming a substituted thiazoline carboxylic acid represented by Structural Formula (XXIII):
( ) esterifying the substituted thiazoline carboxylic acid with R -OH , wherein R is a substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms; thereby forming a substituted thiazoline carboxylic acid ester represented by Structural Formula (XXIV):
(c) alkylating the substituted thiazoline carboxylic acid ester with one or more bases and CH3X, wherein X is a leaving group; thereby forming an alkylated substituted thiazoline carboxylic acid ester represented by Structural Formula (XXV):
(d) hydrolyzing the alkylated substituted thiazoline carboxylic acid ester with base or acid, thereby forming an alkylated substituted thiazoline carboxylic acid represented by Structural Formula (XXVI):
(e) optionally, purifying the (S)-isomer of the alkylated substituted thiazoline carboxylic acid;
(f) reacting the (S)-isomer of the alkylated substituted thiazolme carboxylic acid with acid, thereby forming (S)-2-methylcysteine; and
(g) coupling (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (XXI).
Methods of Preparing 2-Substituted Amino Acids Via Aziridination
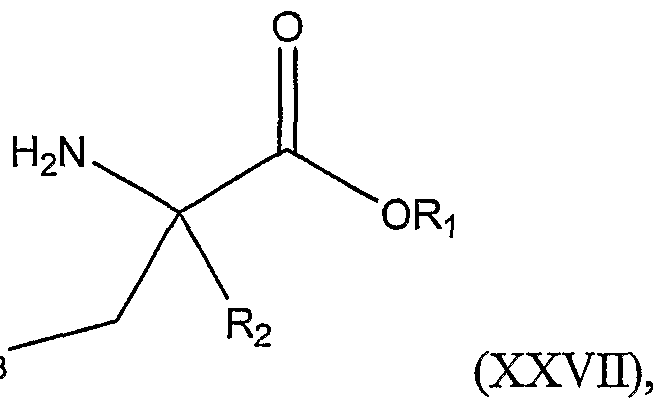
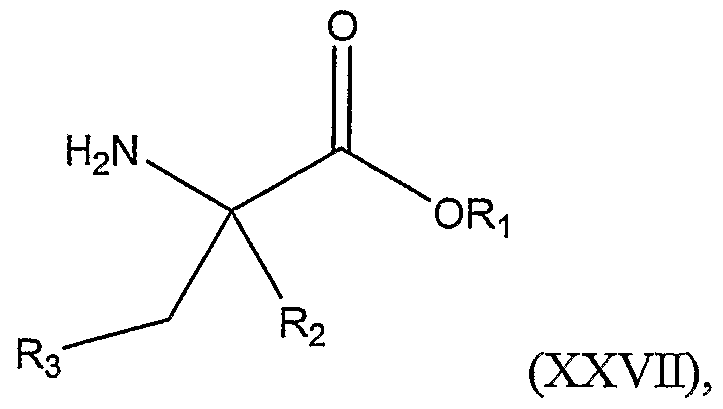
The present invention includes another method of preparing a compound represented by Structural Formula (XXVII):
or a salt thereof; wherein:
Ri and R
2 are, independently, -H or a substituted or unsubstituted alkyl group; R
3 is -H, -(CH
2)
xS(CH
2)
yH, -(CH
2)
xO(CH
2)
yH, -(CH
2)
xNH(CH
2)
yH, -COOH, -CONH
2, -NHC(NH)NH
2, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloahphatic group, a substituted or unsubstituted heterocyclic group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heteroaromatic group, wherein R
3 optionally comprises a protecting group; x is an integer from 0-12; and y is an integer from 0-4; comprising the steps of: a.) reacting the compound represented by Structural Formula (XXEX):
wherein:
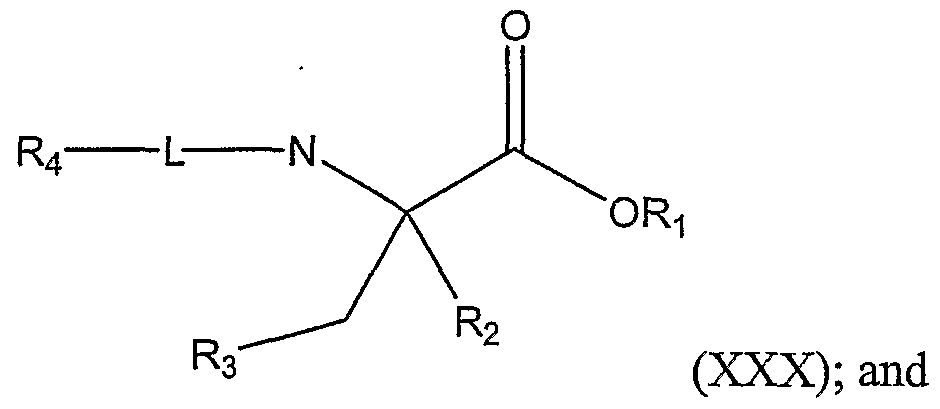
L is a bond, a sulfoxide (-S(O)-), or a sulfone (-S(O)(O)-); R4 is a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group; and Ri and R2 are as defined above; with a nucleophile of the formula A-R , wherein A is -H, -Li, -MgCl, -MgBr, or -Mgl, provided that A and R are not each -H; and R3 is as defined above; thereby forming a compound represented by Structural Formula (XXX):
b.) cleaving L-R-j and optionally the protecting group of R
3 from the compound represented by Structural Formula (XXX), thereby forming the compound represented by Structural Formula (XXVII). h a first preferred embodiment, the aziridine represented by Structural Formula (XXIX) is prepared by aziridinating a compound represented by Structural Formula (XXVH :
by reacting said compound with a source of nitrogen and an aziridination catalyst, thereby forming a compound represented by Structural Formula (XXIX):
In a second preferred embodiment, the compound represented by Structural Formula (XXDC) is prepared by reacting an epoxide represented by Structural Formula (XXVUIa):
(XXNiπa), wherein Ri and R
2 are as defined above, with a nucleophilic nitrogen compound, followed by a hyd oxyl activating agent and a base. Functional groups, other than the epoxide moiety, which can react with the nucleophilic nitrogen compound, the hydroxyl activating agent and/or the base are preferably protected. Typically, this reaction occurs stereospecifically.
Either of these methods of preparing aziridines are suitable for use in the additional embodiments involving an aziridine intermediate. Methods specifically including aziridination of an alkene can have the alternative first step of converting an epoxide moiety into an aziridine moiety.
One embodiment includes a method of preparing a compound represented by Structural Formula (XXXII):
or a salt thereof; where Rg and R
7 are, independently, -H or a substituted or unsubstituted alkyl group; comprising the steps of:
a) aziridinating a compound represented by Structural Formula (XXXIII):
by reacting said compound with a source of nitrogen and an aziridination catalyst, thereby forming a compound represented by Structural Formula (XXXIV):
(XXXIV), wherein L is a bond, sulfoxide (-S(O)-), or sulfone (-S(O)(O)-); R
9 is a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group; and R
6 and R
7 are as defined above;
b) reacting the compound represented by Structural Formula (XXXTV) with a nucleophile, A-S-Z, wherein A is -H; and Z is -H or a protecting group; thereby forming a compound represented by Structural Formula (XXXV):
c) cleaving Z and L-R
9 from the compound represented by Structural Formula
(XXXV), thereby forming the compound represented by Structural Formula (XXXII). Another embodiment includes a method of preparing a compound represented by Structural Formula (XXXVΗ):
(xxxvπ), or a salt thereof; where:
Ru and Rι are, independently, -H or a substituted or unsubstituted alkyl group;
Rι3 is -H, -(CH2)XSH, -(CH2)xOH, -(CH2)XNH2, -COOH, -CONH2, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heteroaromatic group; and x is an integer from 0-12; comprising the steps of: a.) aziridinating a compound represented by Structural Formula (-XXXVTfl):
(xxxvπi), by reacting said compound with a source of mtrogen and a stereospeciflc aziridination catalyst, thereby forming a compound represented by Structural Formula (XXX C):
where L is a bond, sulfoxide (-S(O)-), or sulfone (-S(O)(O)-); Rι is a substituted or unsubstituted alkyl group or a substituted or unsubstituted aryl group; and Ru and Rι
2 are as defined above; b.) reacting the compound represented by Structural Formula (XJS) with a nucleophile, A-R
)3, where A is -H, -Li, -MgCl, -MgBr, or -Mgl, provided
that A and R
13 are not each -H; and Rι
3 is as defined above; thereby forming a compound represented by Structural Formula (XL):
c.) cleaving L-Rι
4 and optionally the protecting group of Rι
3 from the compound represented by Structural Formula (XL), thereby forming the compound represented by Structural Formula (XXXVII). The above methods preferably comprise the additional step of resolving enantiomers or diasteromers of a 2-alkyl amino acid (or an ester or a salt thereof). Synthetic methods leading to a substantial excess of an enantiomer or diastereomer (e.g., asymmetric syntheses producing >85% ee, >90% ee, or >95% ee) can be purified or ultrapurified by an additional resolution step. More preferably, methods of the present invention comprise isolating the (R)- and (S)-enantiomers of 2-alkyl amino acids, or esters or salts thereof.
The invention also includes a method of preparing a compound represented by Structural Formula (XLII):
comprising the step of coupling (S)-2-methylcysteine or a salt thereof, as prepared by a method described above, to 2,4-dihydroxybenzonitrile. Alternatively, an analogous compound can be synthesized by coupling 2-hydroxybenzonitrile and (S)-2-methylcysteine or a salt or an ester thereof. Similar syntheses can be conducted with other substituted benzonitriles.
Methods of Preparing 2-Substituted Amino Acids Via Michael Addition
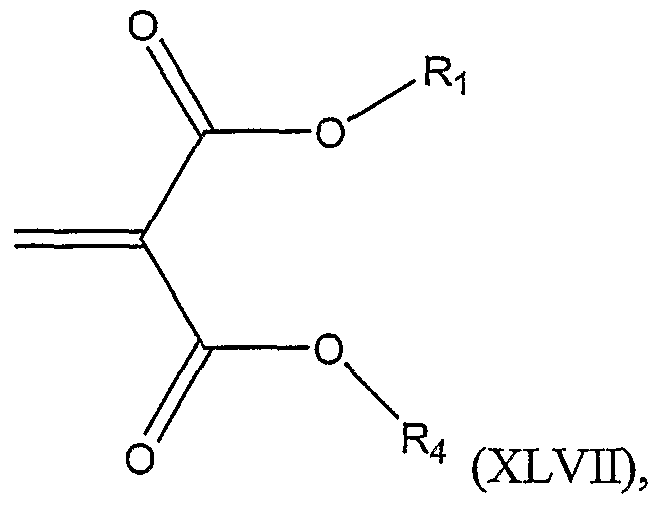
The invention includes a method of preparing a compound represented by Structural Formula (XLVI):
or a salt thereof; wherein:
Ri is -H or a substituted or unsubstituted alkyl group; R2 is a substituted or unsubstituted alkyl group; and
R3 is -H, -SH, -OH, -ΝH2, -CO2H, -CONH2, -NHC(NH)NH2, a substituted or unsubstituted alkyl group, a substituted or unsubstituted cycloahphatic group, a substituted or unsubstituted heterocyclic group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heteroaromatic group, wherein R3 optionally comprises a protecting group; comprising the steps of: a.) reacting a nucleophile of the formula A-R or A-(R3)2, with a compound represented by Structural Formula (XLVH):
A is -H, -Li, -CuLi, -MgCl, -MgBr, or -Mgl, provided that A and R3 are not each -H;
R4 is -H or a substituted or unsubstituted alkyl group; and
Ri and R
3 are as defined above; thereby forming a compound represented by Structural Formula (XLVIII):
b.) reacting the product of step (a.) with one or more bases, R
2X, and a phase transfer catalyst, wherein X is a leaving group; and Ri, R
2, R
3, and R
4 are as defined above; thereby forming a compound represented by Structural Formula (XLIX):
c.) converting the product of step (b.) into a compound represented by Structural Formula (L):
d.) optionally cleaving the protecting group of R
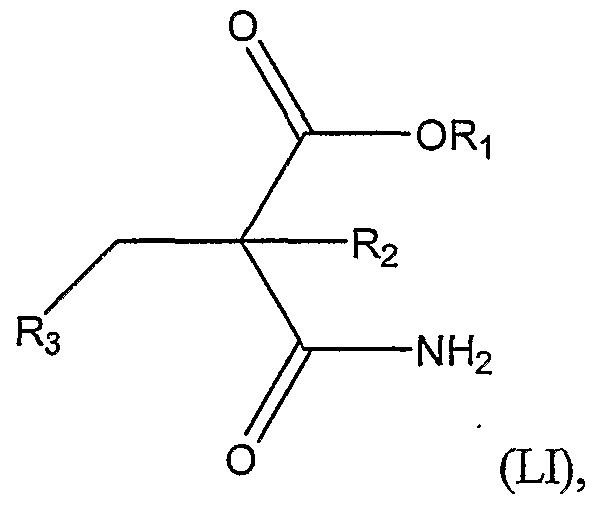
3, thereby forming the compound represented by Structural Formula (XLNI). In another embodiment, the invention is a method of preparing a compound represented by Structural Formula (LIT):
or a salt thereof; wherein:
R6 is -H or a substituted or unsubstituted alkyl group; and
R7 is a substituted or unsubstituted alkyl group, comprising the steps of:
a.) reacting a nucleophile, A-S-Z, with a compound represented by Structural Formula (LID):
R8 is — H or a substituted or unsubstituted alkyl group; Z is a protecting group; and R6 is as defined above; thereby forming a compound represented by Structural Formula (LIN):
b.) reacting the product of step (a.) with one or more bases, R X, and a phase transfer catalyst,
wherein X is a leaving group; and Rό, R , R
8, and Z are as defined above; thereby forming a compound represented by Structural Formula (LV):
c.) converting the product of step (b.) into a compound represented by Structural Formula (LNI):
d.) removing Z from the product of step (c), thereby forming the compound represented by Structural Formula (LIT). Preferably, Ri and J β are methyl and R and R
8 are t-butyl. The above methods can additionally comprise the step of resolving enantiomers or diastereomers of a 2-alkyl amino acid (or an ester or a salt thereof). Preferably, the method comprises isolating the (R)- and (S)-enantiomers of 2-alkyl amino acids, or esters or salts thereof.
The invention also includes a method of preparing a compound represented by Structural Formula (LXI) :
comprising the step of coupling (S)-2-methylcysteine or a salt thereof, as prepared by a method described herein, to 2,4-dihydroxybenzonitrile. Alternatively, an analogous compound can be synthesized by coupling 2-hydroxybenzonitrile and (S)-2-methylcysteine
or a salt or an ester thereof. Similar syntheses can be conducted with other substituted benzonitriles.
Methods of Preparing 2-Alkyl Cysteine Via Oxazolidinone Amide Intermediate
The invention includes a method of preparing a compound represented by Structural Formula ( XJS):
or salts thereof; wherein, Ri is -H or a substituted or unsubstituted alkyl group; and R
2 is a substituted or unsubstituted alkyl group; comprising the steps of: a.) reacting a compound represented by Structural Formula (LXπi):
wherein R
3 is -OH, a substituted or unsubstituted alkyloxy group, or a halogen; with a substituted or unsubstituted aryl carboxylic acid, thereby forming a substituted thiazoline represented by Structural Formula (LXIN):
wherein Ar is a substituted or unsubstituted aryl group and R
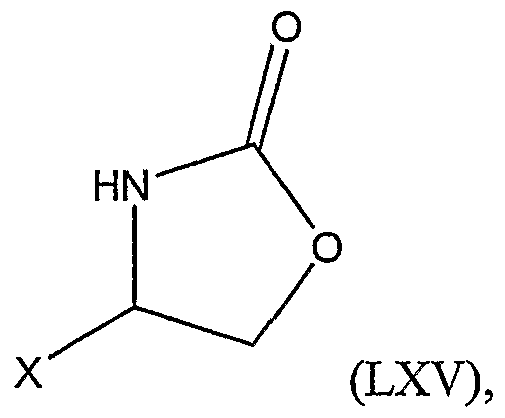
3 is as defined above; b.) reacting the substituted thiazoline with a substituted oxazolidinone represented by Structural Formula (LXN):
wherein X is an aryl or an arylalkyl group, thereby forming a compound represented by Structural Formula (LXNI):
c.) alkylating the product of step (b.) with R
2Y, wherein R
2 is as defined above and Y is a leaving group; thereby forming a compound represented by Structural Formula (LXVD):
wherein R
2 is as defined above; and d.) hydrolyzing the product of step (c), thereby forming the compound represented by Structural Formula (LXII). In one embodiment, the mvention is a method of preparing a compound represented tural Formula (LXVI ):
or salts thereof; where Ri is -H or a substituted or unsubstituted alkyl group; and R
2 is a substituted or unsubstituted alkyl group; comprising the steps of: a) reacting a compound represented by Structural Formula (LXDC):
wherein R
3 is -OH, a substituted or unsubstituted alkyloxy group, or a halogen; with a substituted or unsubstituted aryl carboxylic acid, thereby forming a substituted thiazoline represented by Structural Formula (LXX):
wherein Ar is a substituted or unsubstituted aryl group and R
3 is as defined above; b) reacting the substituted thiazoline with a substituted oxazolidinone represented by Structural Formula (LXXI):
wherein X is an aryl or an arylalkyl group, thereby forming a compound represented by Structural Formula (LXXII):
C) alkylating the product of step (b.) with R
2Y, wherein R
2 is as defined above and Y is a leaving group; thereby forming a compound represented by Structural Formula (LXXHI):
wherein R
2 is as defined above; and d) hydrolyzing the product of step (c), thereby forming the compound represented by Structural Formula (LXVIH). The invention also includes method of preparing a compound represented by Structural Formula (LXXIV):
or salts thereof; where Ri is -H or a substituted or unsubstituted alkyl group; and R
2 is a substituted or unsubstituted alkyl group; comprising the steps of: a) reacting a compound represented by Structural Formula (LXXV) :
wherein R
3 is -OH, a substituted or unsubstituted alkyloxy group, or a halogen; with a substituted or unsubstituted aryl carboxylic acid, thereby forming a substituted thiazoline represented by Structural Formula (LXXVI):
(LXXVI),
wherein Ar is a substituted or unsubstituted aryl group and R
3 is as defined above; b) reacting the substituted thiazoline with a substituted oxazolidinone represented by Structural Formula (LXXND):
wherein X is an aryl or an arylalkyl group, thereby forming a compound represented by Structural Formula (LXXV-HI):
(LXXVm); c) alkylating the product of step (b.) with R
2Y, wherein R
2 is as defined above and Y is a leaving group; thereby forming a compound represented by Structural Formula (LXXDC):
(LXXDC), wherein R
2 is as defined above; and d) hydrolyzing the product of step (c), thereby forming the compound represented by Structural Formula (LXX-N). Preferably, the starting material for the above method is (S)-cysteine, and the product is (S)-2-methylcysteine methyl ester. Alternatively, the starting material for the above method is (R)-cysteine, and the product is (R)-2-methylcysteine methyl ester. The
starting material for the above method can also be a mixture of (R)- and (S)-cysteine, such as the racemate, and the product is a mixture of (R)- and (S)-2-methylcysteine methyl ester.
In other embodiments of the present method, the stereochemistry at the 4-position of the thiazoline ring (i.e., where the amide is attached) may invert during the alkylation in step (c). This is dependent, for example, upon the ability of the amide group to exchange position with the electron pair formed after base deprotonates the thiazoline ring and upon the ability of the 2-oxazolidinone to selectively block the alkylating agent (R2Y) from approaching a face of the thiazoline and prevent alkylation from occurring on that face. Under circumstances when stereochemical inversion occurs, to obtain an (S)-alkylated cysteine it may be advantageous to use (R)-cysteine or a derivative thereof as the starting material.
In another embodiment, the invention is a method of preparing a compound represented by Structural Formula (LXNIH):
(LXVIIT); comprising the steps of: a.) reacting a compound represented by Structural Formula (LXHT):
wherein R
3 is -OH, a substituted or unsubstituted alkyloxy group, or a halogen; with a substituted or unsubstituted aryl carboxylic acid, thereby forming a substituted thiazolme represented by Structural Formula (LXIN):
wherein Ar is a substituted or unsubstituted aryl group and R
3 is as defined above; b.) reacting the substituted thiazoline with a substituted oxazolidinone represented by Structural Formula (LXV):
wherein X is an aryl or an arylalkyl group, thereby forming a compound represented by Structural Formula (LXNI):
c.) alkylating the product of step (b.) with R
2Y, wherein R
2 is as defined above and Y is a leaving group; thereby forming a compound represented by Structural Formula (LXVH):
wherein R
2 is as defined above; and d.) hydrolyzing the product of step (c), thereby forming 2-methylcysteine or a salt thereof, and neutralizing the salt, if present; and e.) coupling (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (LXV-HT).
Alternative forms of the previous embodiment involve coupling 2- hyάroxybenzonitrile and (S)-2-methylcysteine or a salt or an ester thereof. Similar syntheses can be conducted with other substituted benzonitriles or benzimidates.
Methods of Preparing 2-Alkyl Cysteine From Non-Esterified Cysteine
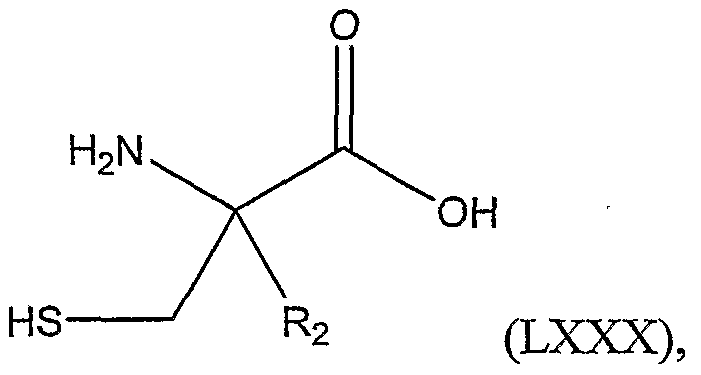
The invention includes a method of preparing a 2-alkylated cysteine represented by Structural Formula (LXXX) :
or salts thereof; wherein R
2 is a substituted or unsubstituted alkyl group; comprising the steps of: a) reacting a compound represented by Structural Formula (LXXXI):
(LXXXI), with a substituted or unsubstituted aryl nitrile of the formula Ar-CN, wherein Ar is a substituted or unsubstituted aryl group; thereby forming a substituted thiazoline represented by Structural Formula (LXXXII):
b) alkylating the substituted thiazoline with one or more bases and R
2X, wherein X is a leaving group and R
2 is as defined above; thereby forming an alkylated substituted thiazoline represented by Structural Formula (LXXXIII):
c) reacting the alkylated substituted thiazoline with acid, thereby forming the 2- alkylated cysteine represented by Structural Formula (LXXX). The invention also includes a method of preparing a compound represented by Structural Formula (LXXXIV):
comprising the steps of: a.) reacting a compound represented by Structural Formula (LXXXI):
with a substituted or unsubstituted aryl nitrile of the formula Ar-CN, wherein Ar is a substituted or unsubstituted aryl group; thereby forming a substituted thiazoline represented by Structural Formula (LXXXII):
(LXXXD); b.) alkylating the substituted thiazoline with one or more bases and CH
3X, wherein X is a leaving group; thereby forming an alkylated substituted thiazoline represented by Structural Formula (LXXXIII):
c.) resolving the alkylated substituted thiazoline into (R)-4-methyl-2- arylthiazoline-4-carboxylic acid and (S)-4-methylthiazoline-4-carboxylic acid; d.) isolating (S)-4-methyl-2-aryltlιiazoline-4-carboxylic acid; e.) reacting (S)-4-methyl-2-arylthiazoline-4-carboxylic acid with acid, thereby forming (S)-2-methylcysteine; and f.) coupling (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (LXXXIV). In another embodiment, an analogous compound to that shown in the previous embodiment can be synthesized by coupling 2-hydroxybenzonitrile and (S)-2- methylcysteine or a salt or an ester thereof. Similar syntheses can be conducted with other substituted benzonitriles.
Methods of Preparing 2-Alkyl Cysteine Via Chiral Amide Intermediate Another useful and efficient method of preparing a 2-alkylcysteine involves condensing cysteine with an aryl nitrile to form a 2-arylthiazoline-4-carboxylic acid, forming a 2-arylthiazoline-4-carboxamide using an amine group comprising at least one substituted or unsubstituted alkyl group that comprises one or more chiral carbon atoms, and alkylating at the 4-position of the thiazolme ring to form a 2-aryl-4-alkyl-thiazoline-4-carboxamide. The thiazoline amide has chiral templates, which can provide face selectivity and consequently desired stereochemistry, during the delivery of an alkyl group to the 4-position of the thiazoline ring. The chiral template present in the thiazoline amide preferably produces an enantiomeric excess of one isomer.
In one embodiment, the invention relates to a method of preparing a 2-alkylated cysteine represented by Structural Formula (LXXXN) :
or a salt thereof, wherein Ri is a substituted or unsubstituted alkyl group, the method comprising:
(a) coupling a compound (which may be an (R) or (S)-isomer or a mixture thereof) represented by Structural Formula (LXXXNI):
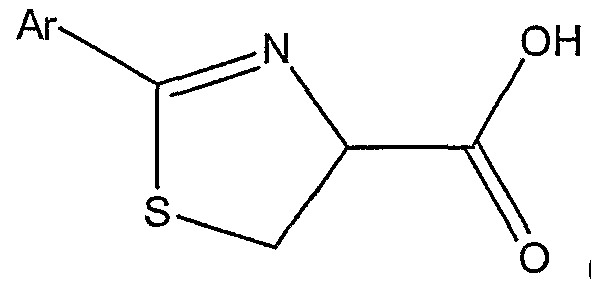
(LXXXVI) with a substituted or unsubstituted aryl nitrile of the formula Ar-CΝ, wherem Ar is a substituted or unsubstituted aryl group; thereby forming a substituted thiazoline carboxylic acid represented by Structural Formula (LXXXVH):
(b) reacting the substituted thiazoline carboxylic acid with an amine represented by Structual Formula (LXXXVϋl):
H
R*- -Ν- ■R? (LXXXXVΠT),
wherein R
* is a substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms and R
2 is a substituted or unsubstituted alkyl or aryl group (optionally with one or more chiral carbons); thereby forming a substituted thiazoline amide represented by Structural Formula (LXXX C):
(LXXXIX);
(c) alkylating the substituted thiazoline amide with one or more bases and RiX, wherein X is a leaving group and Ri is as defined above; thereby forming an alkylated substituted thiazolme amide represented by Structural Formula (XC):
(d) hydrolyzing the alkylated substituted thiazoline amide, thereby forming an alkylated substituted thiazoline carboxylic acid or a salt thereof, the anion of which is represented by Structural Formula (XCI):
(e) reacting the alkylated substituted thiazoline carboxylic acid with acid
(preferably an inorganic acid such as HCl, HBr or sulfuric acid), thereby forming the 2-alkylated cysteine represented by Structural Formula (LXXXV).
The methods described above may additionally comprise the step of purifying or ultrapurifying the alkylated substituted thiazoline carboxylic acid or the alkylated substituted
thiazoline amide. Purifying or ultrapurifying the acid or ester can comprise further resolving the enantiomers or diastereomers of the alkylated substituted thiazoline carboxylic acid or the alkylated substituted thiazoline amide. Alternatively, the 2-alkylated cysteine itself can be resolved. Additionally, the methods can comprise the isolation of the enantiomers of the synthesis products. Preferably, the (S)-enantiomer of 2-alkylcysteine is isolated, for example, (S)-2-methylcysteine.
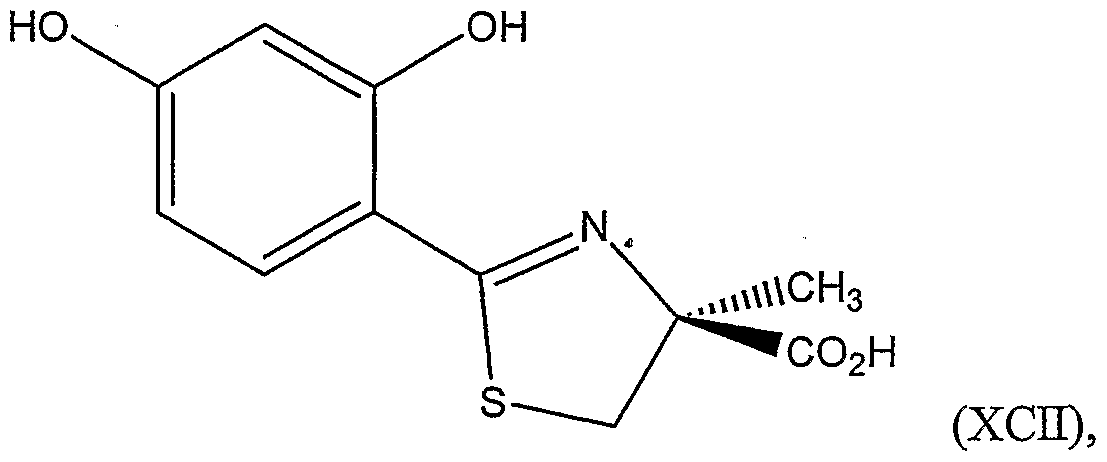
In another aspect, the invention relates to a method of preparing a compound represented by Structural Formula (XCH):
or a salt thereof, the method comprising:
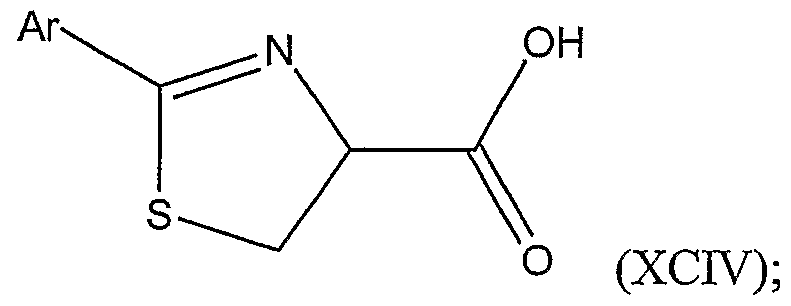
(a) coupling a compound (which may be an (R) or (S)-isomer or a mixture thereof) represented by Structural Formula (XCiπ):
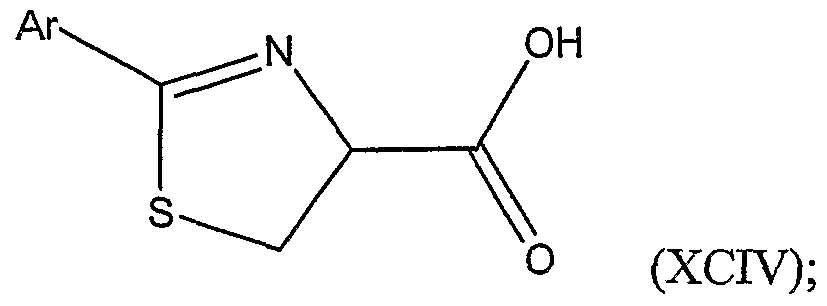
with a substituted or unsubstituted aryl nitrile of the formula Ar-CN, wherein Ar is a substituted or unsubstituted aryl group; thereby forming a substituted thiazoline carboxylic acid represented by Structural Formula (XCIV):
(b) reacting the substituted thiazoline carboxylic acid with an amine represented by Structual Formula (XCV):
H R* — N — R2 (XCV),
wherein R is a substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms and R2 is a substituted or unsubstituted alkyl or aryl group; thereby forming a substituted thiazoline amide represented by Structural Formula (XCVI):
(c) alkylating the substituted thiazoline amide with one or more bases and CH3X, wherein X is a leaving group; thereby forming an alkylated substituted thiazoline amide represented by Structural Formula (XCVH):
(d) hydrolyzing the alkylated substituted thiazoline amide, thereby forming an alkylated substituted thiazoline carboxylic acid or a salt thereof, the anion of which is represented by Structural Formula (XCVIH):
(xcvππ);
(e) optionally, purifying the (S)-isomer of the alkylated substituted thiazoline carboxylic acid; (f) reacting the (S)-isomer of the alkylated substituted thiazoline carboxylic acid with acid, thereby forming (S)-2-methylcysteine; and (g) coupling (S)-2-methylcysteine with 2,4-dihydroxybenzonitrile, thereby forming the compound represented by Structural Formula (XCΗ).
Methods of Alkylating Substituted Thiazolines
In one aspect, the mvention relates to a method of preparing an alkylated thiazoline carboxylic acid or a derivative thereof represented by Structural Formula (XCDC):
or a salt thereof, wherein Ri is a substituted or unsubstituted alkyl group; each R2 is, independently, -H or a substituted or unsubstituted alkyl group; R3 is -H, a substituted or unsubstituted alkyl group, or a carboxyl protecting group; and n is an integer from 1 to 5, the method comprising:
(a) coupling a compound represented by Structural Formula (C):
wherein each -Rj is, independently, a substituted or unsubstituted alkyl group and n is an integer from 1 to 5, with a cysteine ester represented by Structural Formula (Cl):
wherein A is an anion, preferably a halogen such as chloride, bromide or iodine, and R5 is a substituted or unsubstituted alkyl group, thereby forming a substituted thiazoline carboxylic acid ester represented by Structural Formula (CH):
(b) optionally, hydrolyzing the substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxylic acid represented by Structural Formula (CBS):
(c) optionally, protecting the carboxyl group of the substituted thiazoline carboxylic acid to form a protected thiazoline carboxylic acid represented by Structural Formula (CIV):
wherein R
6 is a carboxyl protecting group;
(d) alkylating the optionally protected thiazoline carboxylic acid represented by Structural Formula (CV):
wherein R and n are as defined above and R
7 is — H, R
5 or R
6 (preferably Re or an R
5 that is a carboxyl protecting group), with a compound having the formula Ri-L, wherein Ri is defined as above and L is a leaving group, in the presence of a phase transfer catalyst to form an optionally alkylated protected thiazoline carboxylic acid represented by Structural Formula (CVI):
(e) optionally, hydrolyzing the optionally protected alkylated thiazoline carboxylic acid and cleaving ether groups represented by R to form an alkylated thiazoline carboxylic acid.
The alkylated thiazoline carboxylic acid or the protected alkylated thiazoline carboxylic acid can be the (R) or (S)-isomer or a mixture thereof. The above methods can additionally comprise the step of purifying or ultrapurifying the product by further resolving the enantiomers or diastereomers of the alkylated thiazoline carboxylic acid or a derivative thereof. Additionally, the methods can comprise the isolation of the enantiomers of the synthesis products. Preferably, the (S)-enantiomer of the alkylated thiazoline carboxylic acid, or derivative thereof, is isolated. hi another aspect, the invention relates to a method of preparing a compound represented by Structural Formula (CIX) :
or a salt thereof, the method comprising:
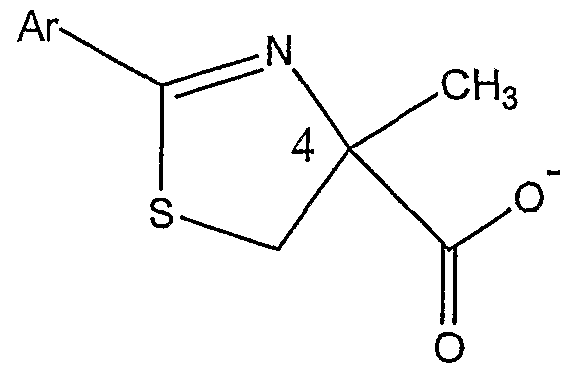
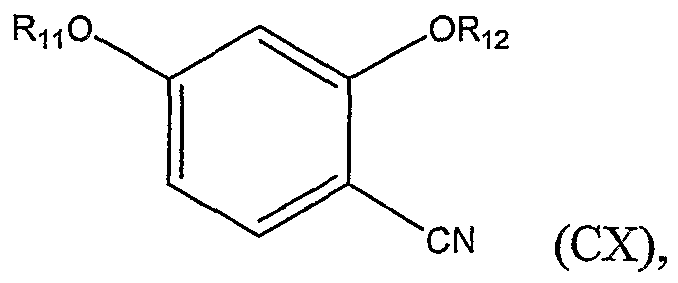
(a) coupling a compound represented by Structural Formula (CX):
wherein Ru and Rι2 are independently, a Cl to C4 substituted or unsubstituted alkyl group, with a cysteine ester represented by Structural Formula (CXI):
wherein A is an anion, preferably a halide such as chloride, bromide or iodine, and Rι3 is a substituted or unsubstituted alkyl group; thereby forming a substituted thiazoline carboxylic acid ester represented by Structural Formula (CXJl):
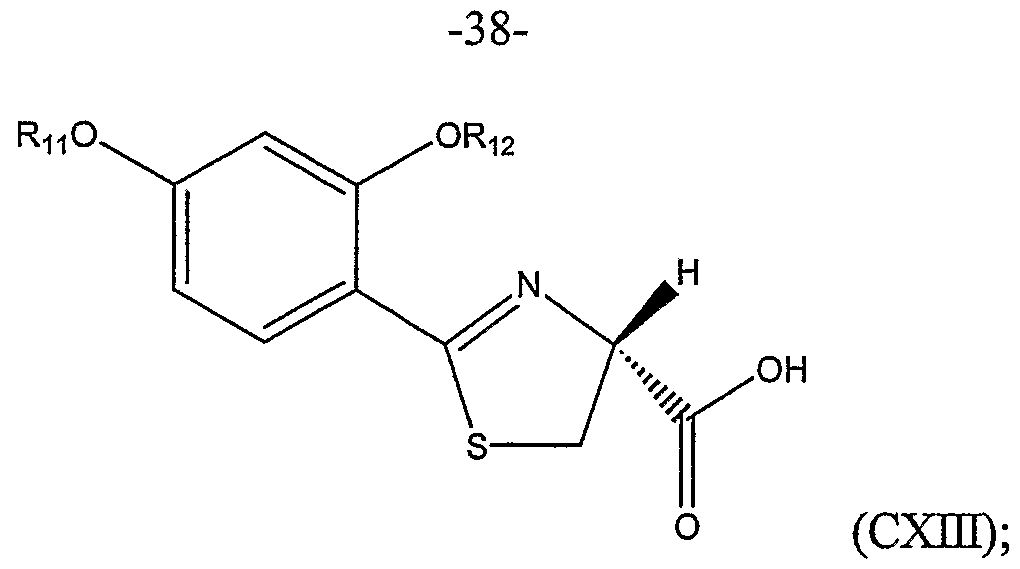
(b) optionally, hydrolyzing the substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxylic acid represented by Structural Formula (CXm):
(c) optionally, protecting the carboxyl group of the substituted thiazoline carboxylic acid to form a protected thiazoline carboxylic acid represented by Structural Formula (CXIV):
wherein R]4 is a carboxyl protecting group;
(d) alkylating the optionally protected thiazoline carboxylic acid represented by Structural Formula (CXV):
wherein Rι5 is -H, Rι3 or Rι4 (preferably Rι4 or an Rj3 that is a carboxyl protecting group), with a compound having the formula CH3-L, wherein L is a leaving group, in the presence of a phase transfer catalyst represented by Structural Formula
(CXVI):
wherein X is a halogen, thereby forming an alkylated protected thiazoline carboxylic acid represented by Structural Formula (CXNII):
(e) hydrolyzing the protected alkylated thiazoline carboxylic acid and cleaving ether groups represented by Ru and Rι2 to form the compound represented by Structural Formula (CXNm):
(f) optionally, purifying the (S)-isomer of the compound represented by Structural Formula (CXVffl).
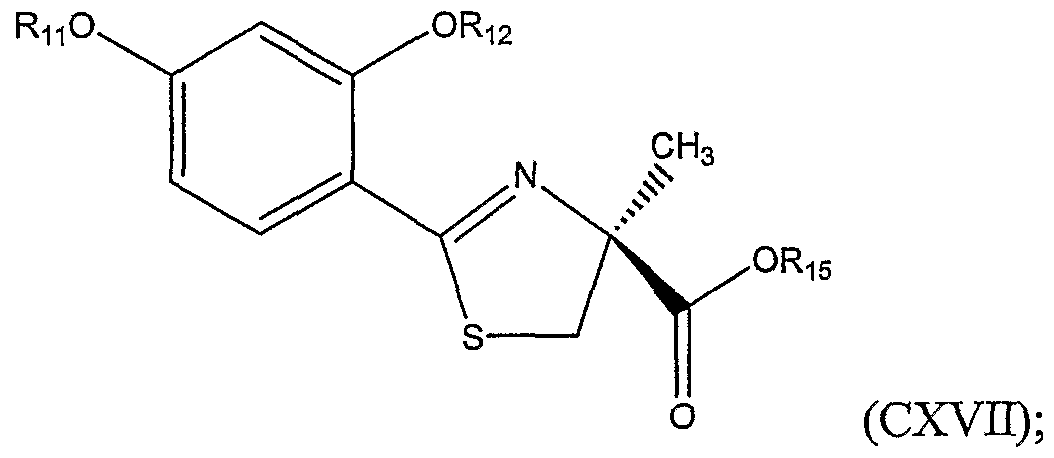
In yet another aspect, the invention includes a method of preparing an alkylated thiazoline carboxylic acid represented by Structural Formula (CXDC):
or a salt thereof, wherein Rι is -H or a substituted or unsubstituted alkyl group; Rπ is a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; and Rι8 is -H, a substituted or unsubstituted alkyl group, or a carboxyl protecting group, the method comprising: (a) coupling a compound represented by Structural Formula (CXX):
wherein Rι is a Cl to C4 substituted or unsubstituted alkyl group, with a cysteine ester represented by Structural Formula (CXXI):
wherein A is an anion (e.g., carboxylates, sulfonates), preferably a halide such as chloride, bromide or iodide, and R
20 is a substituted or unsubstituted alkyl group; thereby forming a substituted thiazoline carboxylic acid ester represented by Structural Formula (CXXH):
(b) optionally, hydrolyzing the substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxylic acid represented by Structural Formula (CXXm):
(c) optionally, protecting the carboxyl group of the substituted thiazoline carboxylic acid to form a protected thiazoline carboxylic acid represented by Structural Formula (CXXIV):
(cxxrv)
wherein R
2ι is a carboxyl protecting group;
(d) alkylating the optionally protected thiazoline carboxylic acid represented by Structural Formula (CXXN):
wherein R22 is — H, R21 or R22 (preferably R22 or an R2ι that is a carboxyl protecting group), with a compound having the formula Rι7 -L, wherein Rι7 is defined as above and L is a leaving group, in the presence of a phase transfer catalyst thereby forming an alkylated optionally protected thiazolme carboxylic acid represented by Structural Formula (CXXNI):
(e) optionally, hydrolyzing the optionally protected alkylated thiazoline carboxylic acid and cleavmg ether groups to form the compound represented by Structural Formula (CXXVH):
(cxxvπ).
A further embodiment of the invention includes reacting a cysteine or derivative thereof, including ester and amide derivatives, with a benzonitrile to form a 2-phenyl thiazoline. Suitable cysteines are preferably substantially enantiomerically pure. Suitable cysteines can also be substituted at the 2- and 3-positions, preferably alkylated. Prefened cysteines include, separately, the (R)- and (S)-enantiomers of 2-methylcysteine, 3,3- dimethylcysteine and 2,3,3-trimethylcysteine, along with esters (e.g., methyl, ethyl) thereof. Benzonitrile are preferably substituted, such as 2,4-dihydroxybenzonitrile, 2- hydroxybenzonitrile, 2,4-dibenyzloxybenzonitrile and 2-benzyloxybenzonitrile. The reaction involves reacting the cysteine and the benzonitrile with a trialkylamine (e.g., trimethylamine, triethylamine, tripropylamine) in an alcoholic solvent (e.g., methanol, ethanol, n-propanol, isopropanol). Preferably, the trialkylamine is triethylamine and the solvent is ethanol. The reaction mixture is advantageously heated to a temperature from about 50 degrees C to about 150 degrees C, where the mixture refluxes. Also, the reaction is preferably conducted under an inert atmosphere (e.g., nitrogen, argon, mixtures thereof).
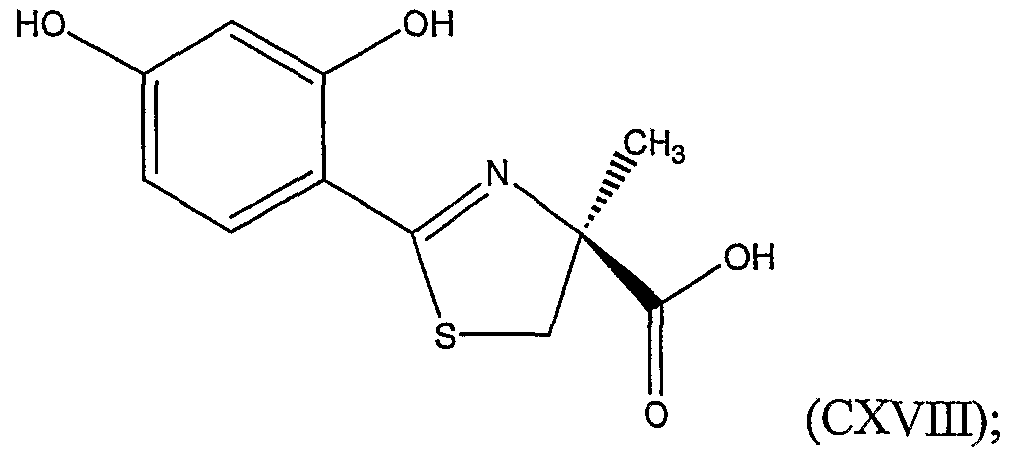
Advantages of the present invention, as a whole, include the facile synthesis of a 2- alkylcysteine and/or a 2-aryl-4-alkylthiazole-4-carboxylic acid from inexpensive and readily available starting materials. 2-Methylcysteine prepared by the method of the present invention can be coupled to 2,4-dihydroxybenzonitrile to form 4'- hydroxydesazadesferrithiocin, also refened to as 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4- methylthiazole-4(S)-carboxylic acid, an iron chelating agent.
DETAILED DESCRIPTION OF THE INVENTION
Methods for Preparing 2- Alkyl Cysteine Via Phase Transfer Catalysis
The invention provides useful and efficient methods of preparing 2-alkylcysteine derivatives. The methods include forming a 2-alkylcysteine derivative from a cysteine
derivative in the presence of a phase transfer catalyst. Additionally, the invention relates to the preparation of 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-alkyl-thiazole-4-carboxylic acid. In particular, the invention provides methods for preparing 2-methylcysteine derivatives as well as 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4-carboxylic acid.
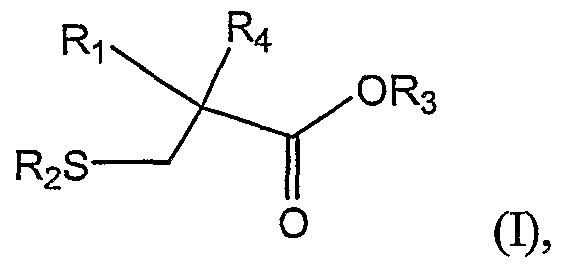
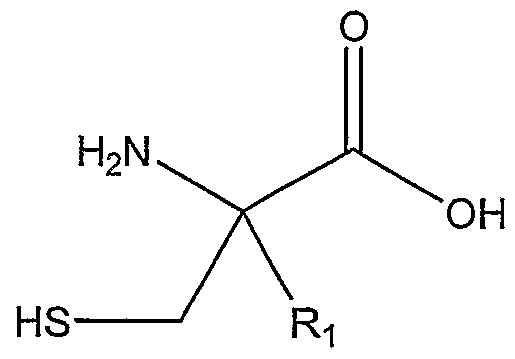
In one aspect, the invention relates to a method of preparing a 2-alkylcysteine derivative represented by Structural Formula (I):
or a salt thereof, wherein
Ri is -NH2; -N(R5)(R6) ; -NHR7; or -N=R8; wherein R5, R6, R7, and R8 are, independently, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; R2 and R3 are, independently, -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; and -Ri is a substituted or unsubstituted alkyl group.
In one embodiment, the method comprises reacting a cysteine derivative represented by Structural Formula (IT) :
or a salt thereof, wherein Rι,R2 and R3 are defined as above, with a compound having the formula R4-L, wherein -Rj. is defined as above and L is a leaving group, in the presence of a phase transfer catalyst thereby forming the 2-alkylcysteine derivative represented by Structural Formula (I).
hi a prefened embodiment, the cysteine derivative reacted is the (R) isomer, represented by Structural Formula (IV):
or a salt thereof, wherein Rl5 R2, and R3 are as defined above, h an especially prefened embodiment, the cysteine derivative reacted is a protected (R)-cysteine and the 2- alkylcysteine derivative thereby formed is a protected 2-methylcysteine. Either the (R)- or the (S)-enantiomer of the 2-alkylcysteine derivative maybe formed in enantiomeric excess. Preferably, the (S)-isomer of a 2-alkylcysteine derivative is formed in enantiomeric excess. More preferably, the (S)-isomer of a protected 2-methylcysteine is formed in enantiomeric excess.
The resulting enantiomers of the product can be further resolved and isolated into pure or substantially pure enantiomer components. Functional groups in compounds can be protected with protecting groups.
Preferably, the cysteine derivative is protected at any reactive site, for example, at the amino, -SH, and/or carboxyl sites of cysteine. A protecting group reduces or eliminates the ability of a functional group to react under certain conditions. For example, a thiol or an alcohol can be protected with an acyl group. Similarly, an alcohol or a thiol can be protected by a trityl, a benzyloxymethyl, a tetrahydropyranyl or a trimethylsilyl group. An amine can, for example, be protected by an Fmoc group or a Boc group. An acid group can be protected, for example, by forming an ester or a carboxamide group. Additional protecting groups, methods of adding a protecting group, and methods of removing a protecting group rd are taught in "Protective Groups in Organic Synthesis, 3 Edition" by Peter G. M. Wuts and Theodora W. Greene, Wiley-Interscience, 1999, the entire contents of which are incorporated herein by reference.
Prefened protecting groups for acidic nitrogen atoms include formyl; 4- toluenesulfonyl; t-butyloxycarbonyl; 2,4-dinitrophenyl; benzyloxymethyl; t-butoxymethyl; 2-chlorobenzyloxy-carbonyl; allyloxycarbonyl; benzyloxycarbonyl (Z); mesitylene-2-
sulfonyl; 4-methyloxy-2,3,6-trimethyl-benzyenesulfonyl; 2,2,5,7,8-pentamethyl-chroma n-6- sulfonyl; 9-xanthenyl; and 2,4,6-trimethoxybenzyl.
In one embodiment, Ri is a protected amino group such as -N=C(Ar)2 wherein each Ar is, independently, a substituted or unsubstituted aryl group. For example, Ri can be a benzophenone imine represented by Structural Formula (III) :
Prefened protecting groups for acidic sulfur groups include 4-methylbenzyl, 3-nitro- 2-pyridinesulfenyl; trityl; 2,4,6-trimethoxybenzyl; acetamidomethyl; trimethylacetaminomethyl; t-butylsulfonyl; and sulfoxide.
In one embodiment, R2 is a protecting group protecting the cysteine -SH group. For example R2 can be -C(Ar)3 wherein each Ar is, independently, a substituted or unsubstituted aryl group. Preferably, R2 is trityl. Prefened protecting groups for acidic oxide groups include benzyl ether; t-butyl ether; benzyl ether; 2,6-dichlorobenzyl ether; 2-bromobenzyl ether; and 3,5-dibromobenzyl ether.
Carboxyl groups can be protected, for example, as esters or as carboxamides. For example, when a carboxyl group is protected as an ester, it takes the fonn of-COOR wherein R is a substituted or unsubstituted Cl to CIO alkyl group, a substituted or unsubstituted up to C30 aryl group, or a substituted or unsubstituted alkyl-aryl group wherein the alkyl group is Cl to C5 and the aryl group is up to C30. When a carboxyl group is protected as a carboxamide, it takes the form of -CONR'R' ' wherem R' and R' ' are, independently, — H, a substituted or unsubstituted Cl to CIO alkyl group, a substituted or unsubstituted up to C30 aryl group, or a substituted or unsubstituted alkyl-aryl group wherein the alkyl group is Cl to C5 and the aryl group is up to C30.
For example, R3 can be a carboxyl protecting group such as a substituted or unsubstituted Cl to CIO alkyl group. In a prefened embodiment, R3 is t-butyl.
In one incarnation of the invention, as illustrated below, the cysteine derivative is 2- (R)-( enzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert-butyl ester. 2-(R)- (Benzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert-butyl ester can be formed by the following process: (1) 2-(R)-(9H-Fluoren-9-ylmethoxycarbonylamino)-3-rritylsulfanyl- propionic acid (i.e., (R)-cysteine with Fmoc a protected amino group and with a trityl protected -SH group), is reacted with t-butyl alcohol and dicyclohexyl carbodiimide (DCC) in 4-(dimethylamino)pyridine (DMAP) and tetrahydrofuran (THF) at room temperature to form 2-(R)-(9H-fluoren-9-ylmethoxycarbonylamino)-3 -tritylsulfanyl-propionic acid tert-butyl ester; (2) the Fmoc group is removed from the 2-(R)-(9H-fluoren-9- ylmethoxycarbonylamino)-3 -tritylsulfanyl-propionic acid tert-butyl ester using either diethylamine in dichloromethane or piperidine in dichloromethane to form 2-(R)-amino-3- tritylsulfanyl-propionic acid tert-butyl ester; and (3) the 2-(R)-ammo-3-tritylsulfanyl- propionic acid tert-butyl ester is reacted with benzhydrylideneamine in dichloromethane at room temperature to form 2-(R)-(benzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert- butyl ester.
The following sequence illustrates the method described above of forming 2-(R)- (benzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert-butyl ester from 2-(R)-(9H- Fluoren-9-ylmethoxycarbonylamino)-3-tritylsulfanyl-propionic acid:
2(R)-(Ben2-hydrylidene-a-n-uno)-3-lritylsulfanyl-propionic acid tert-butyl ester In one embodiment, the cysteine derivative represented by Structural Formula (IT) can be alkylated in the presence of one or more bases, an alkylating agent, and a phase transfer catalyst. For example, 2-(benzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert-butyl ester is reacted with cesium hydroxide monohydrate and excess methyl iodide in dichloromethane at about -80° to -60°C and in the presence of a phase transfer catalyst. Preferably, the cysteine derivative is alkylated using a phase transfer catalyst such that an enantiomeric excess of either the (R)- or (S)-isomer is produced (i.e., the alkylation is stereoselective). Alkylating agents can have the formula R4-L, where R4 is a substituted or unsubstituted alkyl group and L is a leaving group. Prefened R4 groups include substituted or unsubstituted C1-C4 alkyl groups; methyl and benzyl are especially prefened R4 groups. The leaving group L is typically a weak base. Suitable leaving groups include halogen, tosyl, mesyl, triflyl, brosyl, p-nitrophenyl, and 2,4-dinitrophenyl groups. Halogens include bromine, chlorine, and iodine. Iodine is a prefened leaving group. Suitable amounts of alkylating agent can include about 1 to 20, about 2 to 15, about 3 to 10, or, preferably, about 5 equivalents, relative to the amount of cysteine derivative.
Prefened bases include alkali or alkaline earth metal hydroxides, alkoxides, amides, or carbonates or their combinations. Available bases include potassium t-butoxide, sodium methoxide, sodium ethoxide, sodium amide, calcium carbonate, cesium carbonate, and the alkali metal salts of hexamethyl disilazide (HMDS). Prefened bases include potassium hydroxide, sodium hydroxide, and cesium hydroxide monohydate. Suitable amounts of base include about 5 to 25, about 10 to 20, about 10 to 15, or, preferably, about 10 equivalents, relative to the amount of cysteine derivative. A phase transfer catalyst functions at the boundary between two solvents or mixtures of solvents, at least one of which is an organic solvent. The organic phase of the process
can include any organic solvent which is substantially inert to the catalyst, reactants and products. The organic phase may comprise a combination of two or more solvents. Solvents generally include, but are not limited to, aprotic solvents such as acetonitrile, acetone, dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, and hexamethylphosphoramide. In a prefened embodiment, the organic phase comprises dichloromethane.
The alkylation of the cysteine derivative can be performed at temperatures ranging from about -80°C to about room temperature such as between about -80° and 0°C. In a prefened embodiment, the alkylation is performed at temperatures of between about -80° and -40°C, for example, at about -60°C. In one aspect of the invention, a cinchona-alkaloid derived phase transfer catalyst is used to alkylate a cysteine derivative. In one particular embodiment, a cinchona-alkaloid derived phase transfer catalyst is used to stereoselectively alkylate a 2-(benzhydrylidene- amino)-3-tritylsulfanyl-propionic acid tert-butyl ester at the 2-carbon position. The phase transfer catalyst can be derived from cinchonine or from cinchonidine. Use of one of these catalysts in the alkylation reaction can yield enantiomeric excesses of either the (R)- or (S)- enantiomer of the alkylated cysteine derivative, while use of an enantiomer of that catalyst can yield enantiomeric excesses of the other enantiomer of the alkylated cysteine derivative. Thus by selecting the phase transfer catalyst used, one can direct which enantiomer of the alkylated cysteine derivative will form. In a prefened embodiment, the phase transfer catalyst used is derived from cinchonidine and is represented by Structural Formula (V):
wherein
R
9 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; Rio and Ru are, independently, -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group; and X is a halogen. R
9 can be, for example, substituted or unsubstituted napthyl, anthracenyhnethyl, or benzyl. Preferably, R
9 is 9-anthracenylmethyl as represented by Structural Formula (NI):
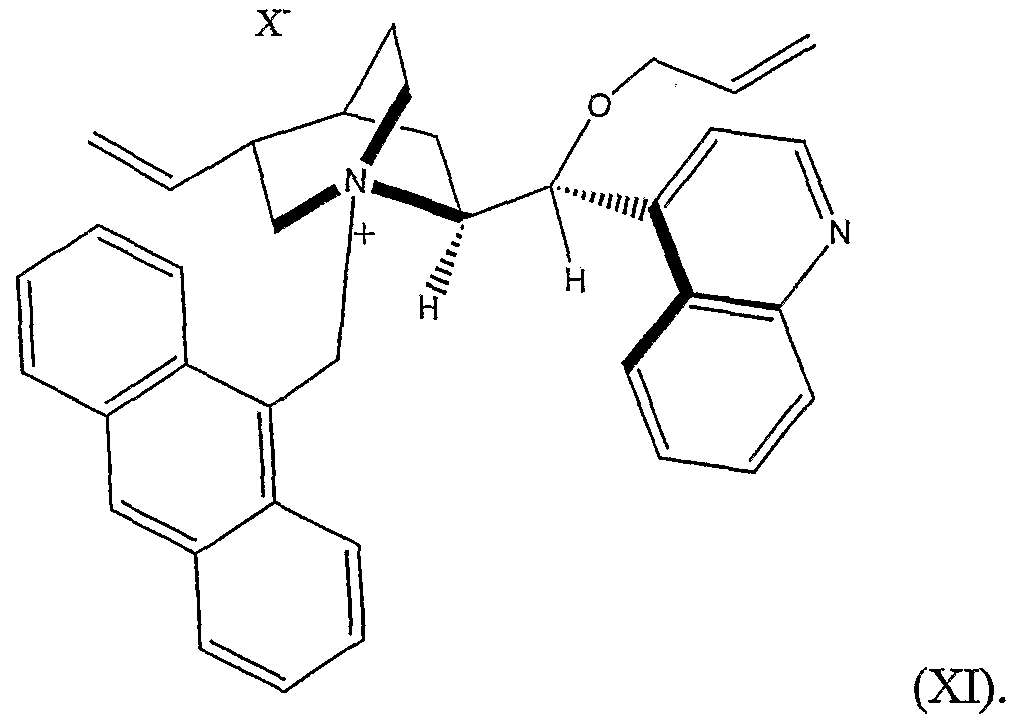
Rio can be, for example, substituted or unsubstituted allyl or benzyl. Preferably, Rio is substituted or unsubstituted allyl. In another prefened embodiment, Ru is substituted or unsubstituted ethenyl. In another, X is chlorine or bromine. Thus the phase transfer catalyst can be represented by Structural Formula (XI):
Additional examples of phase transfer catalysts suitable for use in the present invention are described in U.S. Patent No. 5,554,753 issued to O'Donnell, et al. , the entire teachings of which are incorporated herein by reference.
The phase transfer catalyst represented by Structural Formula (XT) is preferably prepared using the following method as described by Corey, et al, in "A Rational Approach to Catalytic Enantioselective Enolate Alkylation Using a Structurally Rigidified and Defined Chiral Quaternary Ammonium Salt Under Phase Transfer Conditions" (J. Am. Chem. Soc. 119, 12414-12415 and Corey Supplemental therein 1-25 (1997)), the entire contents of which are incorporated by reference herein by reference. In that method, cinchonidine, represented by Structural Formula (XII):
is suspended in toluene and 9-(chloromethyl)anthracene, represented by Structural Fonnula
(Xm):
is added. The mixture is stined at reflux for about 2 hours. The product, N-9- anthracenylmethylcinchonidinium chloride represented by Structural Formula (XIN) :
is collected as a light yellow solid. The N-9-anthracenylmethylcinchonidinium chloride is then suspended in dichloromethane. To this suspension is added 50% KOH and allyl bromide. The resulting mixture is then stined for about 4 hours at about 23 °C. The product, O(9)-allyl-N-9-anthracenylmethylcinchonidium bromide represented by Structural Formula (XI), is collected as a light orange solid.
The use of O(9)-allyl-N-9-anthracenylmethylcinchonidium bromide as a phase transfer catalyst is also described in co-pending U.S. Patent Application Nos. 60/380,903, filed May 15, 2002 and 60/392,833, filed June 27, 2002, the entire contents of which are incorporated herein by reference.
Examples of other phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, triethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2- chloroethylamine chloride HCl, bis(2-chloroethyl)amine HCl, 2-dimethylaminoethyl chloride HCl, 2-ethylaminoethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, methylamine HCl, dimethylamine HCl, trimethylamine HCl, monoethylamine HCl, diethylamine HCl, triethylamine HCl, ethanolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamme HCl, dicyclohexylamine HCl, cyclohexylamme HCl, diisopropylethylamine HCl, ethylenediamine HCl, aniline HCl, methyl salicylate, ethyl salicylate, butyl salicylate amyl salicylate, isoamyl salicylate, 2-ethylsalicylate, and benzyl salicylate.
In one form of the mvention, the phase transfer catalyst, such as O(9)-allyl-N-9- anthracenylmethylcinchonidium bromide, is present in an amount of about 0.05 to 0.4 equivalents relative to the amount of cysteine derivative. Alternatively, the phase transfer catalyst can be present between about 0.05 and 0.25 equivalents, between about 0.1 and 0.15 equivalents, or, preferably, at about 0.1 equivalents (relative to the amount of cysteine derivative).
In a prefened embodiment, (R)-2-(benzhydrylidene-amino)-3-tritylsulfanyl-propiorιic acid tert-butyl ester is reacted with cesium hydroxide monohydrate and excess methyl iodide in dichloromethane at about -60°C and in the presence of O(9)-allyl-N-9-
anthracenylmethylcinchonidium bromide thereby forming (S)-2-(benzhydrylidene-amino)-2- methyl-3 -tritylsulfanyl-propionic acid tert-butyl ester.
Protecting groups, if present, can be removed from the 2-alkylcysteine derivative. Methods of removing a protecting group are well known in the art and taught in "Protective rd Groups in Organic Synthesis, 3 Edition" by Wuts and Greene, incorporated by reference above. For example, 2(S)-(benzhydrylidene-amiιιo)-2-methyl-3-tritylsulfanyl-ρropionic acid tert-butyl ester can be reacted with acid thereby forming (S)-2-methylcysteine.
Methods of Preparing 2-Alkyl Cysteine Via Chiral Ester Another useful and efficient method of preparing 2-alkylcysteine involves condensing cysteine with an aryl nitrile to form a 2-arylthiazoline-4-carboxylic acid, esterifying the 2- arylthiazoline-4-carboxylic acid using a substituted or unsubstituted alcohol group comprising one or more chiral carbons, and alkylating at the 4-position of the thiazoline ring to form a 2- aryl-4-alkyl-thiazolme-4-carboxylic acid ester. The chiral templates present in the thiazoline carboxylic acid ester can provide face selectivity, and consequently desired stereochemistry, during the delivery of an alkyl group to the 4-position of the thiazoline ring. The resulting enantiomers of the product can be further purified and isolated into pure or substantially pure enantiomer components by a number of methods.
The condensation of an aryl nitrile and cysteine typically occurs in a polar, protic solvent in the presence of an excess of base. Typically, the aryl nitrile and cysteine are reflux ed together for several hours, such as 1-20 hours, 2-15 hours, 4-10 hours, or 6-8 hours. Refluxing preferably occurs in an inert atmosphere, such as nitrogen or argon. Prefened aryl nitriles include aryl nitriles where the aryl group is a substituted or unsubstituted phenyl group. Unsubstituted phenyl and substituted phenyl containing such groups as -OH or alkyl are prefened. Suitable polar, protic solvents include, but are not limited to, water, methanol, ethanol, formic acid, acetic acid, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal, and long chain alcohols (e.g., propanol and isopropanol). An alcohol, such as methanol or ethanol, is a prefened solvent. Suitable bases include secondary and tertiary amines such as dimethylamine, diethylamine, methyl-imine, triethylamine,
> diisopropylamine, and diisopropyle ylamine. The base can be added in excess, such as one or more equivalents relative to the amount of cysteine. Suitable amounts of base have at least about one equivalent of base, and range from about 1 to about 10, about 1 to about 5, about 1
to about 3, and about 1 to about 2 equivalents, relative to the amount of cysteine. In one example, cysteine, benzonitrile, and 5 equivalents of triethylamine are refluxed in ethanol for about 6-8 hours to obtain a 2-phenylthiazoline-4-carboxylic acid.
Alternatively, an aryl imidate (e.g., a benzimidate, where the benzene ring can have one or more substituents, as described below) can be condensed with cysteine to form a substituted thiazoline carboxylic acid. The substituted thiazoline carboxylic acid can be formed by coupling an aryl imidate, such as benzimidate, with a cysteine, such as the cysteine represented by Structural Fonnula (XVI). Typically, coupling of a cysteine or a 2- alkylcysteine with an aryl imidate includes reacting a cysteine (or a related compound) with the aryl imidate under basic conditions. Acceptable bases include trimethylamine; triethylamine; dimethylamine; diethylamine; diphenylamine; diisopropylamine; diisopropylethylamine; l,4-diazabicyclo[2.2.2]octane (DABCO); l,5-diazabicyclo-[4.3.0]- non-5-ene (DBN); and the like.
Aryl imidates can be prepared, for example, for aryl nitriles, aryl carboxylic acids, and aryl amides. Methods of forming aryl imidates are discussed in co-filed U.S. Patent Application No. 60/380,909, filed May 15, 2002, the entire contents of wliich are incorporated herein by reference. In one example, an aryl carboxylic acid (e.g., benzoic acid) is converted into an acid chloride, then an amide, followed by reaction with a trialkyloxonium hexafluorophosphate or a trialkyloxonimii tetrafluoroborate to form the aryl imidate. In a second example, an aryl nitrile is converted into an aryl imidate through reaction with an alcohol in the presence of an acid, as is described below.
An ester of a carboxylic acid can be produced using, for example, an alcohol, one embodiment of the present invention, a substituted thiazoline carboxylic acid ester represented by Structural Formula (XVIII) is formed with a chiral alcohol represented by R -OH wherein R is a substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms. Preferably, R is a primary or secondary substituted or unsubstituted alkyl group. More preferably, R is a substituted or unsubstituted cyclic or polycyclic (e.g., bicyclic tricyclic) alkyl group. Preferably, the chiral alcohol used is substantially optically pure (IR, 2R, 3R, 5S)-(-)-isopinocamphenol or (IS, 2R, 5R)-(+)-isomenthol. The substituted thiazoline carboxylic acid ester may be formed through various means. In one embodiment, a substituted thiazoline carboxylic acid ester is produced through the acid catalyzed reaction of a substituted thiazoline carboxylic acid with a chiral alcohol. Common acid catalysts include
sulfuric acid and p-toluenesulfonic acid. A substituted thiazoline carboxylic acid ester can also be produced via intermediate reactive acid derivatives such as acid chlorides. Alternatively, a substituted thiazoline carboxyhc acid is treated with a chiral alcohol in the presence of a coupling agent. Coupling agents include, but are not limited to dicyclohexylcarbodiimide (DCC); alkyl chloroformate and triethylamine; pyridinium salts and tributylamine; Amberlyst-15; phenyl dichlorophosphate; diethyl azodicarboxylate and triphenyl phosphme; DCC and an aminopyridine; 2-chloro-l,3,5-trinitrobenzene and pyridine; l, -carbonylbis(3-methylimidazolium) triflate; di-2-pyridyl carbonate, polystyryl diphenylphosphine; (trimethylsilyl)ethoxyacetylene; chlorosulfonyl isocyanate; chlorosilanes, MeSO2Cl-Et3N; Ph3P-CCL4-Et3N; and N,N'-carbonyldiimidazole. Preferably, dicyclohexylcarbodiimide (DCC) is the coupling agent. As an example, a substituted thiazoline carboxylic acid may be treated with a chiral alcohol, DCC, and 4- (dimethylamino)pyridine (DMAP) in tetrahydrofirran (THF) at room temperature or higher to fonn a substituted thiazoline carboxylic acid ester. The 2-arylthiazoline-4-carboxylic acid ester can be alkylated in the presence of one or more bases, an alkylating agent, and optionally a phase transfer catalyst. Typically, the 2- arylthiazoline-4 carboxylic acid ester is reacted with one or more equivalents (e.g., about 1 to 10, about 1 to 5, about 1 to 3, or about 1.5 to 2.5 equivalents) of base and one or more equivalents (e.g., about 1 to 5, about 1 to 2, about 1 to 1.5, or about 1 to 1.1 equivalents) of an alkylating agent in a polar, aprotic solvent at about -80 to 40°C, about -50 to 25 °C, about -20 to 10°C, or about -5 to 5°C.
Alkylating agents are of the formula RjX, where Ri and X are as defined above. Prefened Ri groups include substituted or unsubstituted C1-C4 alkyl groups, for example, methyl or benzyl. The leaving group X is typically a weak base. Suitable leaving groups include halogen, tosyl, mesyl, triflyl, brosyl, p-nitrophenyl, and 2,4-dinitrophenyl groups. Halogens include bromine, chlorine, aid iodine. Iodine is a prefened leaving group. Prefened bases include potassium t-butoxide, sodium methoxide, sodium ethoxide, and sodium amide. Suitable polar, aprotic solvents include, but are not limited to, dimethylformamide, dimethyl sulfoxide, acetonitrile, acetone, tetrahydrofuran (THF), and hexamethylphosphoramide. Tetrahydrofuran (THF) is a prefened solvent.
In one example, a 2-phenylthiazoline-4 carboxylic acid ester (e.g., the ethyl, methyl, t-butyl, or isopropyl ester) is reacted with about 2 equivalents of base and about 1 equivalent
of methyl iodide in tetrahydrofuran (THF) at 0°C to form a C4 position n-alkylated 2- phenyl-4-carboxylic acid ester (e.g., the ethyl, methyl, t-butyl, or isopropyl ester).
Alternatively, the 2-arylthiazoline-4-carboxylic acid ester can be alkylated in the presence of a phase transfer catalyst. Examples of phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, triethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2-chloroethylamine chloride HCl, bis(2- chloroethyl)amine HCl, 2-dimethylaminoethyl chloride HCl, 2-ethylamino ethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, methylamine HCl, dimethylamine HCl, trimethylamine HCl, monoethylamine HCl, diethylamine HCl, triethylamine HCl, ethanolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamme HCl, dicyclohexylamine HCl, cyclohexylamme HCl, diisopropylethylamine HCl, ethylenediamine HCl, aniline HCl, methyl salicylate, ethyl salicylate, butyl salicylate amyl salicylate, isoa yl salicylate, 2- ethylsalicylate, and benzyl salicylate.
The 2-aryl-4-alkyl-thiazoline-4-carboxylic acid ester can be hydrolyzed with either base or acid to form a 2-aryl-4-alkyl-thiazoline-4-carboxylic acid. The 2-aryl-4-alkyl- thiazoline-4-carboxylic acid then can be reacted with acid to form a 2-alkylcysteine, such as 2-methylcysteine.
Methods of Preparing 2-Substituted Amino Acids Via Aziridination
Another useful and efficient method of preparing 2-alkyl amino acids involves the aziridination of alkylacrylates. The aziridinated alkylacrylate can be further reacted to form 2-alkyl amino acids having a wide variety of side chains.
A first type of aziridination of the present invention typically includes reacting an alkylacrylate with a source of nitrogen. Aziridinations are typically conducted under a nitrogen or other inert atmosphere, often at ambient pressure. Suitable solvents for an aziridination include acetonitrile; acetonitrile in 5-15% water, methanol, ethanol, or t-butanol; dimethylformamide (DMF); dimethylformamide (DMSO); tetrahydrofuran (THF); and acetonitrile in 5-25% DMF, DMSO, or THF. Reaction temperatures are typically about 0°C to about 100°C, about 20°C to about 80°C, about 25°C to about 60°C, or about 30°C to about
50°C. Aziridinations are further described in U.S. Pat. Nos. 5,929,252 and 5,789,599, which are incorporated herein by reference. Aziridinations can be conducted in a continuous process, such that no intennediate purifications are required, although such purifications are optional.
Prefened sources of nitrogen include compounds represented by the Structural Formulas (XXXI), (XXXVI), and (XLI):
(XLI), where M is an alkali metal, X is a halide, and R
5, Rio, and R
15 are each a substituted or unsubstituted alkyl group or substituted or unsubstituted aryl group. Preferably, M is sodimn and X is chloride or bromide. Prefened R
5, Rio, and Rι
5 groups include phenyl, tolyl, p-nitrophenyl, n-butyl, t-butyl, and methyl. An especially prefened R
5, Rio, and Rι
5 is p-tolyl.
Prefened aziridination catalysts include transition metal halides, alkaline earth metal halides, Rh2(acetate)4, a dihalogen, phenyltrimethlammomum tribromide, and pyridinium hydrobromide. Copper halides are especially prefened aziridination catalysts.
Prefened stereospecific aziridination catalysts include copper 4,4'-disubstituted bis(oxazolines). Examples of 4,4'-disubstituted bis(oxazolines) are represented by the structural fonnula:
where R is an alkyl or an arylalkyl group. Preferably, R is -CH(CH
3)
2, -C
6H
5, -C(CH
3)
3, -C(CH
3)
2(C
6H
5), or -C(CH
3)(C
6H
5)
2. Copper 4,4'-disubstituted bis(oxazolines) can be fonned, for example, by reacting a copper(I) or copper(II) salt with a 4,4'-disubstituted bis(oxazoline). Acceptable copper(I) and copper(-Q) salts include copper(I) triflate, copper(II) triflate, copρer(I) chloride, and copper(T) bromide. Stereospecific aziridination catalysts are further described in Evans, et al, J. Am. Chem. Soc. 116: 2742-2753 (1994); Evans, et al, J.
Am. Chem. Soc. 115: 5328-5329 (1993); and Johnson et al, Aec. Chem. Res. 33: 325-335 (2000); which are incorporated herein by reference.
Additional stereospecific aziridination catalysts include zeolites. Suitable zeolites typically comprise a transitional metal such as copper in copper-exchanged zeolites (e.g., copper-exchanged zeolite Y, obtained from Union Carbide as ultrastabilized EL'Υ zeolite) described in publications by Langham et al. , Applied Catalysis A 182: 85-89 (1999); Langham et al, J. Chem. Soc, Perkin Trans. 2: 1043-1049 (1999); and Langham et al., J. Chem. Soc. Chem. Commun.1601-1602 (1998); Gullick, et al, J. Mol. Catalysis A - Chem. 180: 85-89 (2002); Gullick, et al, Catalysis Lett., 75: 151-154 (2001); and Taylor, et al, J. Chem. Soc. Perkins Trans. 2: 1714-1723 (2001); the entire contents of which are incorporated herein by reference.
Reaction of an aziridine with a nucleophile is conducted in an appropriate solvent and at appropriate temperature. Typically, the solvent is an aprotic solvent such as acetonitrile, d iiethylformamide, dioxane, ethyl acetate, ethyl ether, hexamethylphosphorainide, and tetraliydrofuran. Appropriate temperatures are typically about 0°C to about 90°C, about 20°C to about 70°C, or about 30°C to about 60°C.
Acceptable nucleophiles typically have the formula A-R3, where A is -H, -Li, -MgCl, -MgBr, or -Mgl; and R3 is as defined above. Nucleophiles typically have a heteroatom such as N, O, or S, or a metal-carbon bond such as Li-C or Mg-C. Nucleophiles can have a protecting group. Preferably, R3 is -SH or a protected variant thereof. More preferably, A-R is CH3COSH or C6H5C(O)SH.
A second type of aziridination involves the opening of an epoxide ring with a nucleophilic nitrogen compound. Nucleophilic nitrogen compounds are advantageously generated in situ, whereby the compounds typically have a protecting group that is removed under reaction conditions. Protecting groups typically include those disclosed herein as being suitable for protecting nitrogen atoms (e.g., Boc). Examples of suitable nucleophilic nitrogen compounds include secondary and tertiary, preferably secondary, nitrogen atoms bonded to one or two protecting groups and a second substituent that is preferably not removed under the same conditions as the protecting group (e.g., a substituted phenyl sulfonyl group such as nosyl, tosyl or brosyl). The ring-opened product (an alpha, beta-aminoalcohol) is subsequently reacted with a hydroxyl activating agent and an anhydrous base (e.g., pyridine, dimethylaminopyridine, dimethylmorpholine, another amine disclosed herein or. combination
thereof) in an appropriate solvent in order to form an aziridine. The hydroxyl activating agent converts the hydroxyl group into a better leaving group, such that it can be displaced by the amino moiety. Examples of hydroxyl activating agents include triphenylphosphine and alkyl or aryl sulfonates such as methane sulfonic anhydride, methane sulfonic chloride, toluene sulfonic chloride and trifluoroacetic chloride. A substituted phenyl sulfonyl group can be removed upon formation of the aziridine ring, or a time later in the synthesis. Cleavage of a substituted phenyl sulfonyl group is essentially identical to the cleavage of L--R} and the like from an aziridine, as described below.
Examples of the conversion of epoxides to aziridines can be found, for example, in the following: U.S. Patent No. 5,929,232; PCT Publication No. WOOO/01670; Fuji, K., Kawabata, T., Kiryu, Y., Sugiura, Y., Taga, T., Miwa, Y., "A New Access to Chiral Aziridines by Enzymatic Transesterfication of me-rø-Bis(acetoxymethyl)aziridines," Tetrahedron Lett. 31, 6663-6666 (1990); Ittah, Y., Sasson, Y., Shahak, I., Tsaroom, S., Blum, J., "A New Aziridine Synthesis from 2-Azido Alcohols and Tertiary Phosphines. Preparation of Phenanthrene 9,10-Imine," J. Org. Chem. 43, 4271-4273 (1978); Konsler, R.G., Karl, J., Jacobsen, E.N., "Cooperative Asymmetric Catalysis Using Dimeric Salen Complexes," J. Am. Chem. Soc. 120, 10780-10781 (1998); Lanow, J.F., Roberts, E., Verhoeven, T.R., Ryan, K.M., Senanayake, C.H., Reider, P.J., Jacobsen, E.N., "(lS,2R)-l-Aminoindan-2-ol," Organic Synth. 16, 46-56 (1998), Annis, D. A., Jacobsen, E. N., "Polymer-Supported Chiral Co(Salen) Complexes: Synthetic Applications and Mechanistic Investigations in the
Hydrolytic Kinetic Resolution of Tenninal Epoxides," J. Am. Chem. Soc. 121, 4147-4154 (1999); Senanayake, C. H., Jacobsen, E. N., "Chiral (Salen)Mn(IlI) Complexes in Asymmetric Epoxidations: Practical Synthesis of cis-Aminoindanol and Its Application to Enantiopure Drug Synthesis," in Process Chemistry in the Pharmaceutical Industry, Gadamasetti, K. G., Ed., Deklcer: New York, 1999, pp. 347-368; Myers, J. K., Jacobsen, E.
N., "Asymmetric Synthesis of Amino Acid Derivatives via Catalytic Conjugate Addition of Hydrazoic Acid to Unsaturated hnides," J. Am. Chem. Soc. 121, 8959-8960 (1999); Jacobsen, E. N.,' Wu, M. H., "Ring Opening of Epoxides and Related Reactions," in Comprehensive Asymmetric Catalysis, Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds., Springer: New York, 1999, Chapter 35; Jacobsen, E. N., "Aziridination," in Comprehensive Asymmetric Catalysis,
Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds., Springer: New York, 1999, Chapter 17; Jacobsen, E. N., "Future Perspectives in Asymmetric Catalysis," in Comprehensive
Asymmetric Catalysis, Jacobsen, E. N., Pfaltz, A., Yamamoto, H., Eds., Springer: New York, 1999, Chapter 42; Li, Z., Fernandez, M., Jacobsen, E. N., "Enantioselective Ring-opening of meso Aziridines Catalyzed by Tridentate Schiff-base Cl romium(III) Complexes," Org. Lett. 1, 1611-1613 (1999); Schaus, S. E., Jacobsen, E. N., "Asymmetric Ring-Opening of Meso- Epoxides with TMSCN Catalyzed by (pybox)Lanthanide Complexes," Org. Lett. 2, 1001- 1004 (2000); Brandes, B. D., Jacobsen, E. N., "Regioselective Ring Opening of Enantiomerically Enriched Epoxides via Catalysis with Chiral (Salen)Cr(IH) Complexes," Synlett 1013-1015 (2001); M. K. Gurjar, K. Sadalapure, S. Adhikari, B. V. N. B. S. Sarma and M. S. Chorghade, "Kinetic Resolution of Aryl Glycidyl Ethers: A Practical Synthesis of Optically Pure beta-blocker-S-Metoprolol", Heterocycles 48 (7), 1471 (1998); Mukund K. Gurjar, L. Murali Krishna, Bugga V. N. B. S. Sarma and Mukund S. Chorghade, "A Practical Synthesis of (R)-(-)-Phenylephrine Hydrochloride", Org. Process Res. Dev., 2(6), 422 (1998); M. S. Chorghade, M. K. Gurjar, S. Adhikari, K. Sadalapure, S. V. S. Lalitha, A. M. S. Murugaiah and P. Radha Krishna, "Synthesis of (2S,5S)-trans-5-(4-fluorophenoxymethyl)-2- (l-N-hydiOxyι-reidyl-3-butyn-4-ylH^ Chem. 1071-
74 (1999); Ramesh A. Joshi, Mukund K. Gurjar, Narendra K. Tripathy and Mukund S. Chorghade, "A New and Improved Process for Celiprolol Hydrochloride", Organic Process Research and Development 5(2), 176 (2001); and Mukund K. Gurjar, A. M. S. Murugaiah, P. Radhakrishna, C. V. Ramana and Mukund S. Chorghade, "A Novel and Simple Asymmetric Synthesis of CMI-977 (LDP-977): A potent Anti-Asthmatic Drug Lead", Tetrahedron Asymmetry, In Press, 2003; the contents of each of which are incorporated herein by reference.
Cleavage of L-R4, L-R9 or L-Rι4 can be achieved by, for example, hydrolysis with bases such as potassium hydroxide, sodium hydroxide or methoxides (e.g., sodium methoxide, potassium methoxide); reduction; reaction with compounds such as tetrabutylammonium fluoride and basic thiophenol. Cleavage reactions are further described in Yasuhara, et al, Tet-ahedron Lett. 39: 595-596 (1998); Maligres, et al, Tetrahedron Lett. 38: 5253-5256 (1997); and Vedejs, et al, J. Am. Chem. Soc. 118: 9796-9797 (1996); each of which is incorporated herein by reference. Cleavage of a protecting group is dependent on the nature of the protecting group. For example, an acyl protecting group can be removed by treating the protecting group with acids such as hydrochloric acid, acetic acid, dilute sulfuric acid, and the like; and bases such as
sodiu hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, and ammoma. Other examples of removing protecting groups can be found in "Protective Groups in Organic Synthesis, 3 Edition" by Peter G. M. Wuts and Theodora W. Greene, Wiley-lhterscience, 1999, which is incorporated herein by reference. Protecting group cleavage (e.g., of the carboxylic acid and/or the amino acid sidechain) can occur simultaneously with cleavage of L-R4, L-R9 or L-Rι4.
Ri, R2, R6, Ru, and Rι2 are preferably unsubstituted alkyl groups. Preferably, Ri, R2, R6, Ru, and Ri2 are each methyl. R7 is preferably methyl or benzyl, where the methyl or benzyl group can be substituted or unsubstituted.
R3 and Rι3 include -H, -(CH2)xS(CH2)yH, -(CH2)xO(CH2)yH, -(CH2)xNH(CH2)yH, -(CH2)xC(O)NH2, -(CH2)xC(O)OH, -(CH2)XNHC(NH)NH2, a C1-C6 substituted or unsubstituted alkyl group,
Additional suitable examples of R include -CONH
2, -CH
2CONH
2, -CH
2CH
2CONH
2, -CH
2CH
2CH
2CONH
2, -SH, -CH
2SH, -CH
2CH
2SH, -CH
2CH
2CH
2SH, -CH(CH
3)
2, -CH
2CH(CH
3)
2> -CH
2CH
2CH(CH
3)2, -CH
2CH
2CH
2CH(CH
3)2, -SCH
3, -CH
2SCH
3, -CH
2CH
2SCH
3, -CH
2CH
2CH
2SCH
3, -OH, -CH
2OH, -CH
2CH
2OH, -CH
2CH
2CH
2OH, -COOH, -CH
2COOH, -CH
2CH
2COOH, -CH
2CH
2CH
2COOH, -NHC(NH)NH
2, -CH
2NHC(NH)NH
2, -CH
2CH
2NHC(NH)NH
2, -CH
2CH
2CH
2NHC(NH)NH2, -NH, -CH
2NH
2, -CH
2CH
2NH
2, -CH
2CH
2CH
2NH
2;
Prefened values of x include integers from 0-12, 0-6, 0-4, 0-3, 0-2, and 0-1. Zero is an especially prefened value of x.
Prefened values of y include integers from 0-4, 0-3, 0-2, and 0-1. Zero and one are especially preferable values of y.
Sources of nitrogen also include sulfinimines such as N-benzylidene-p- toluenesulfinimine, 4-methoxybenzyhdene-p-toluenesufinimine, N-isobutylidene-p- toluenesulfinimine, N-(3-phenyl-(E)-2-propylidene)-p-toluenesulfinimine, and N-(2-methyl-
(E)-2-butenylidene)-p-toluenesulfinimine; (N-(p-tolylsulfonyl)imino)phenyliodinane); and tosyl azide.
Methods of Preparing 2-Substituted Amino Acids Via Michael Addition Yet another useful and efficient method of preparing 2-alkyl amino acids involves a
Michael-type addition of a side chain precursor to a dialkyl 2-methylidenylρropan-l,3-dioate, followed by alkylation at the 2-position of the diester. One of the ester groups can be converted into an amino moiety, typically through a reaction with an azide.
Michael-type additions of the present reaction include reacting a nucleophile of the formula A-R3 or A-(R3)2 with a dialkyl 2-methylidenylpropan- 1 ,3-dioate, which forms a 2- substituted propan-l,3-dioate ester. Typically, when the nucleophile does not contain a metal-carbon bond, the Michael-type addition occurs in a protic solvent with either a catalytic amount of abase or a stoichiometric amount of base. When the nucleophile contains a metal- carbon bond such as a lithium-carbon, copper-carbon, or magnesium carbon, the Michael- type addition occurs under conditions where the nucleophile is stable and adds to the dialkyl 2-methylidenylpropan- 1, 3 -dioate at the desired location. Reaction temperature is generally not important, however, the temperature can range from -50°C to 150°C, 0°C to 100°C, or 20°C to 60°C. Michael additions are further described on pages 741-742 and 797-803 of "Advanced Organic Chemistry, Fourth Edition," by Jerry March, Wiley-hiterscience, 1992 and references therein, all of which are incorporated by reference.
Prefened nucleophiles include nucleophiles where R3 is -SH, such as H2S and CH3COSH. Other suitable examples of R3 include -H, -(CH2)xS(CH2)yH, -(CH2)xO(CH2)yH, -(CH2)xNH(CH2)yH, -(CH2)xC(O)NH2, -(CH2)xC(O)OH, -(CH2)XNHC(NH)NH2, a C1-C6 substituted or unsubstituted alkyl group, and aryl and heteroaryl groups such as
and N ; where R
3 optionally comprises a protecting group. The variable x can be an integer of zero or more, such as 0 to about 6, preferably 0-3, or 0 or 1. The variable y is O or l.
Alkylation of a 2-substituted propan-l,3-dioate ester (fonning a 2-alkyl-2-substituted propan-l,3-dioate ester) typically occurs in a protic solvent (e.g., methanol, ethanol, water, propanol, isopropanol, formic acid, acetic acid, DMF, N-ethylacetamide, formaldehyde diethyl acetal), by adding one or more bases, an alkylating agent of the formula R2X or R X, and a phase transfer catalyst. Suitable bases include alkali metal or alkaline earth metal hydroxides, alkoxides, or carbonates such as sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, potassium ethoxide, sodium carbonate, cesium carbonate, calcium carbonate and potassium carbonate, as well as sodium hexamethyl disilazide and potassium hexamethyl disalizide. Prefened alkylating agents include those where R or R is a C1-C4 substituted or unsubstituted alkyl group and X is a halide. Especially prefened alkylating agents include those where R2 or R7 is methyl or benzyl and X is a halide such as iodide. Examples of phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, triethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2-chloroethylamine chloride HCl, bis(2-clιloroethyl)amine HCl, 2- dimethylaminoethyl chloride HCl, 2-ethylaminoethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, methylamine HCl, dimethylamine HCl, trimethylamine HCl, monoethylamine HCl, diethylamine HCl, triethylamine HCl, ethanolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamine HCl, dicyclohexylamine HCl, cyclohexylamine HCl, diisopropylethylamine HCl, ethylenediamine HCl, aniline HCl, methyl salicylate, ethyl salicylate, butyl salicylate amyl salicylate, isoamyl salicylate, 2-ethylsalicylate, and benzyl salicylate.
Converting a 2-alkyl-2-substituted propan-l,3-dioate ester into a 2-alkyl amino acid typically comprises a series of steps where the 2-alkyl-2-substituted propan-l,3-dioate ester is
partially hydrolyzed (at 1- and R8) to give a free carboxylic acid, optionally converting the free carboxylic acid into an acid chloride, and the free carboxylic acid or the acid chloride is reacted with a source of azide and water to give the amino acid. Typically, hydrolysis is achieved by treating the 2-alkyl-2-substituted propan-l,3-dioate ester with acid. This hydrolysis is additionally applicable to other esters. The optional conversion into an acid chloride can be accomplished by reacting the free carboxylic acid with an agent such as SOCl2, PC13, or ClC(O)C(O)Cl. The source of azide for an acid chloride is MN3, where M is H or an alkali metal. The source of azide for a free carboxylic acid is preferably diphenylphosphoryl azide. Following reaction with a source of azide, a carboxy azide is formed, and further reaction with water and heat results in the carboxy azide reananging into isocyanate. The isocyanate can readily be hydrolyzed to an amino moiety.
Alternatively, the above conversion can involve amidatihg a 2-alkyl-2-substituted propan-l,3-dioate ester. Typically, amidation involves hydrolyzing the ester to a free carboxylic acid, converting the free carboxylic acid to an acid chloride (or directly converting the ester to an acid chloride), and reacting the acid chloride with ammonia or a salt thereof. The amide can be converted to an amino moiety by reacting it with 1.) MOR5 and Y2 or 2.) MOY; where M is an alkali metal, R5 is hydrogen, or an alkyl group such as methyl, ethyl, propyl, or isopropyl; and Y is a halogen.
If R3 comprises a protecting group, it can be cleaved. Similarly, a protecting group Z can be cleaved. Cleavage of a protecting group is dependent on the nature of the protecting group. For example, an acyl protecting group can be removed by treating the protecting group with acids such as hydrochloric acid, acetic acid, dilute sulfuric acid, and the like; and bases such as sodium hydroxide, potassium hydroxide, sodium methoxide, potassium methoxide, sodium ethoxide, and potassium ethoxide. Other examples of removing protecting groups can be found in "Protective Groups in Organic Synthesis, 3rd Edition" by
Peter G. M. Wuts and Theodora W. Greene, Wiley-hiterscience, 1999, which is incorporated herein by reference.
Ri, R6, and R9 can optionally be hydrolyzed from the amino acid product. Typically, hydrolysis is achieved by reacting the ester fonn of an amino acid with a sufficient quantity of acid or base to remove Ri, R6, or R . The acid or base used for hydrolysis preferably does not react with or cleave, except to form a salt, other moieties of the amino acid.
Methods of Preparing 2- Alkyl Cysteine Via Oxazolidinone Amide Intermediate
A further useful and efficient method of preparing 2-alkyl cysteine and related compounds involves reacting cysteine (or a related compound, such as a cysteine alkyl ester or the acid chloride of cysteine) and an aryl carboxylic acid, to form a thiazoline intermediate. The thiazoline intermediate can be amidated with an oxazolidinone, and then stereospecifically alkylated. Upon hydrolysis of the thiazoline and the oxazolidinone, a 2- alkyl cysteine (or a related compound) is obtained.
The reaction of cysteine (or a related compound) with an aryl carboxylic acid can occur in a polar solvent, preferably a polar, protic solvent (e.g., methanol, ethanol, water, acetic acid, fonnic acid, isopropanol, propanol, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal) with the addition of a base. Acceptable bases include alkali metal and alkaline earth metal salts, such as sodium hydroxide, sodium methoxide, sodium ethoxide, sodium carbonate, potassium hydroxide, potassium methoxide, potassium ethoxide, cesium carbonate, calcium carbonate, potassium carbonate, sodium hexamethyl disilazide, and potassium hexamethyl disilazide; and trialkylamines such as trimethylamine, triethylamine, and diisopropylethylamine. The aryl carboxylic acid is typically substituted or unsubstituted benzoic acid, but is preferably benzoic acid. The R3 group of cysteine or a related compound can be a poor leaving group, such that peptide bond formation with a ine- containing compounds is minimized. The resultant 2-arylthiazoline-4-carboxylic acid can be amidated with a 4-aryl-2- oxazolidinone or a 4-arylalkyl-2-oxazolidinone. Preferably, the 4-arylalkyl-2-oxazolidinone is 4-benzyl-2-oxazolidinone. The 4-aryl-2-oxazolidone can have either an (R) or (S) configuration at the 4-position of the oxazolidinone ring, which is selected dependent in part upon the desired stereochemistry of the alkylated cysteine product. This amidation reaction is typically conducted in a polar solvent (e.g., acetonitrile, dimethylsulfoxide, dimethylformamide, acetone, hexamethylphosphoramide, methylene chloride, chloroform) in the presence of a catalytic amount of coupling or promoting agent, such as thionyl chloride, dicyclohexylcarbodiimide, diisopropylcarbodiimide, N,N-carbonyldiimidazole, POCl3, TiC , SO2ClF, benzotriazol-1-yl diethyl phosphate, Ti(O-butyl)4, N,N,N',N'- tetramethyl(succinimido)uranium tetrafluoroborate, 1,1' -carbonylbis(3 -methylimidazolium) triflate, Lawesson's reagent, chloro sulfonyl isocyanate, P2L, pyridinium salts with tributyla ine, and a mixture of tributylphosphine and nitrosomethylbenzene.
The 2-aryl-thiazoline-4-carboxamide can be alkylated at the 4-position of the thiazoline ring by reacting it with base and an alkyating agent of the formula R2Y, where R2 and Y are as defined above. The alkylating agent is typically present in excess, such as about a 2-fold, 3-fold, 4-fold, 5-fold, or 10-fold excess. Preferably, R2 is a substituted or unsubstituted C1-C4 alkyl group. Even more preferably, R2 is methyl or benzyl. Y is preferably a halide, such as iodide, chloride, or bromide. Acceptable bases include lithium diisopropylamide (LDA), lithium diisopropylamide, lithium N-isopropyl-N-cyclohexylamide, potassium t-butoxide, sodium t-butoxide, sodium amide, potassium amide, sodium hydride, and potassium hydride. Titanium(IV) chloride or tin(]V) chloride can also be present in the reaction mixture. When titanium chloride is present, the reaction is carried out under nitrogen and in a water-free solvent. The reaction temperature is often about -25°C to about 0°C. Acceptable solvents are polar, aprotic solvents such as acetone, acetonitrile, dimethylformamide, dioxane, ethyl acetate, ethyl ether, hexamethylphosphoramide, tetrahydrofuran, and 1,2-dimethoxyethane. After alkylation, the 4-alkyl-2-arylthiazoline-4-carboxyamide is generally hydrolyzed.
Typically, hydrolysis involves reacting the 4-alkyl-2-arylthiazoline-4-carboxamide with an appropriate amount of base in a polar, protic solvent. Prefened bases include alkali and alkaline earth metal salts such as lithium hydroxide, potassium carbonate, calcium carbonate, and cesium carbonate. Prefened solvents include C1-C4 alcohols such as methanol, ethanol, propanol, isopropanol, butanol, isobutanol, and t-butanol. Methanol is an especially prefened solvent, hi this reaction, the solvent is also a reactant, such that when an alcohol of formula RiOH is the solvent, RiO becomes part of the newly- formed ester. For example, when methanol is the solvent, the product is a 2-alkylcysteine methyl ester. The ester moiety can be hydrolyzed by reacting the cysteine ester with a sufficient quantity of acid or base to remove the methoxy group. The acid or base used for hydrolysis preferably does not react with or cleave, except to form a salt, other moieties of the 2-alkylcysteine.
Methods of Preparing 2- Alkyl Cysteine From Non-Esterified Cysteine
A useful and efficient method of preparing 2-alkylcysteine involves condensing cysteine with an aryl nitrile to form a 2-arylthiazolme-4-carboxylic acid, followed by alkyation at the 4-position of the thiazoline ring. The resulting racemic 2-alkylcysteine
product can be resolved and isolated into a pure or substantially pure enantiomer by a number of methods.
The condensation of an aryl nitrile and cysteine typically occurs in a polar, protic solvent (e.g., water, methanol, ethanol, formamide, formic acid, acetic acid, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal) in the presence of an excess of base. Typically, the aryl nitrile and cysteine are refluxed together for several hours, such as 1-20 hours, 2-15 hours, 4-10 hours, or 6-8 hours. Refluxing preferably occurs in an inert atmosphere, such as nitrogen or argon. An alcohol such as methanol or ethanol is a prefened solvent. Prefened aryl nitriles include aryl nitriles where the aryl group is a substituted or unsubstituted phenyl group. Phenyl is a prefened aryl group. Suitable bases include secondary and tertiary amines such as dimethylamine, diethylamine, trimethylamine, diphenylamine, diisopropylamine, diisopropylethylamine, l,4-diazabicyclo[2.2.2] octane (DABCO), l,5-diazabicyclo[4.3.0]non-5-ene (DBN), and triethylamine. Suitable amounts of base have at least about one equivalent of base, and range from about 1 to about 10, about 1 to about 5, about 1 to about 3, and about 1 to about 2 equivalents, relative to the amount of cysteine.
Alternatively, an aryl imidate (e.g., a benzimidate, where the benzene ring can have one or more substituents, as described below) can be condensed with cysteine. Typically, the aryl imidate is reacted with cysteine under basic conditions. Acceptable bases include those named above. Aryl imidates can be prepared, for example, for aryl nitriles, aryl carboxylic acids, and aryl amides. Examples of aryl imidate preparation can be found, for example, in U.S. Application No. 60/380,909, filed May 15, 2002, the contents of which are incorporated herein by reference. In one example, an aryl carboxylic acid (e.g., benzoic acid) is converted into an acid chloride, then an amide, followed by reaction with a trialkyloxonium hexafluorophosphate or a trialkyloxonium tetrafluoroborate to fonn the aryl imidate. In a second example, an aryl nitrile is converted into an aryl imidate through reaction with an alcohol in the presence of an acid, as is described below.
The 2-arylthiazoline-4-carboxylic acid can be alkylated in the presence of one or more bases, an alkylating agent, and optionally a phase transfer catalyst. Typically, the 2- arylthiazoline-4 carboxylic acid is reacted with one or more equivalents (e.g., about 1 to about
10 equivalents, about 1 to about 5 equivalents, about 1 to about 3 equivalents, or about 1.5 to about 2.5 equivalents) of base and one or more equivalents (e.g., about 1 to about 5
equivalents, about 1 to about 2 equivalents, about 1 to about 1.5 equivalents, about 1 to about 1.1 equivalents) of an alkylating agent in a polar, aprotic solvent (e.g., acetone, acetonitrile, dimethylformamide, dioxane, ethyl acetate, ethyl ether, hexamethylphosphoramide, tetrahydrofuran) at about -80°C to about 40°C, about -50°C to about 25°C, about -20°C to about 10°C, or about -5°C to about 5°C. Alkylating agents are of the formula R2X, where R2 and X are as defined above. Prefened R2 groups include substituted or unsubstituted C1-C4 alkyl groups; methyl and benzyl are prefened R2. The leaving group X is typically a weak base. Suitable leaving groups include halogen, tosyl, triflyl, brosyl, p-nitrophenyl, 2,4- dinitrophenyl, and mesyl groups. Halogens include bromine, chlorine, and iodine. Iodine is a prefened leaving group. Prefened bases include potassium t-butoxide, sodium methoxide, sodium ethoxide, sodium amide, and other alkali and alkaline earth metal alkoxides.
Examples of phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, triethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2- chloroethylamine chloride HCl, bis(2-chloroethyl)amine HCl, 2-dimethylaminoethyl chloride HCl, 2-ethylaminoethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, methylamine HCl, dimethylamine HCl, trimethylamine HCl, monoethylaniine HCl, diethylamine HCl, triethylamine HCl, ethaiiolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamine HCl, dicyclohexylamine HCl, cyclohexylamine HCl, diisopropylethylamine HCl, ethylenediamine HCl, aniline HCl, methyl salicylate, ethyl salicylate, butyl salicylate amyl salicylate, isoamyl salicylate, 2-ethylsalicylate, and benzyl salicylate.
Methods of Preparing 2-Alkyl Cysteine Via Chiral Amide Intermediate
Another useful and efficient method of preparing a 2-alkylcysteine involves condensing cysteine with an aryl nitrile to form a 2-arylthiazoline-4-carboxylic acid, forming a 2-arylthiazoline-4-amide using an amine group comprising at least one substituted or unsubstituted alkyl group that comprises one or more chiral carbon atoms, and alkylating at the 4-ρosition of the thiazoline ring to form a 2-aryl-4-alkyl-thiazoline-4-amide (i.e., an alkylated substituted thiazoline amide). The resulting enantiomers of the product can be
further purified and isolated into pure or substantially pure enantiomer components by a number of methods.
The condensation of an aryl nitrile and cysteine typically occurs in a polar, protic solvent in the presence of an excess of base. Typically, the aryl nitrile and cysteine are refluxed together for several hours, such as 1-20 hours, 2-15 hours, 4-10 hours, or 6-8 hours. Refluxing preferably occurs in an inert atmosphere, such as nitrogen or argon. Prefened aryl nitriles include aryl nitriles where the aryl group is a substituted or unsubstituted phenyl group. Phenyl and heteroaryls, such as pyridines and thiazolines, are prefened aryl groups. Suitable polar, protic solvents include, but are not limited to, water, methanol, ethanol, formic acid, acetic acid, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal, and long chain alcohols (e.g., propanol and isopropanol). An alcohol, such as methanol or ethanol, is a prefened solvent. Suitable bases include secondary and tertiary amines such as dimethylamine, diethylamine, trimethylamine, triethylamine, diisopropylamine, and diisopropylethylamine. The base can be added in excess, such as one or more equivalents relative to the amount of cysteine. Suitable amounts of base have at least about one equivalent of base, and range from about 1 to about 10, about 1 to about 5, about 1 to about 3, and about 1 to about.2 equivalents, relative to the amount of cysteine. hi one example, cysteine, benzonitrile, and 5 equivalents of triethylamine are refluxed in ethanol for about 6-8 hours to obtain a 2-phenylthiazoline-4-carboxylic acid. Alternatively, an aryl imidate (e.g., a benzimidate, where the benzene ring can have one or more substituents, as described below) can be condensed with cysteine to form a substituted thiazoline carboxylic acid. The substituted thiazoline carboxylic acid can be formed by coupling an aryl imidate, such as benzimidate, with a cysteine, such as the cysteine represented by Structural Fonnula (LXXXVI). Typically, coupling of a cysteine or a 2- alkylcysteine with an aryl imidate includes reacting a cysteine (or a related compound) with the aryl imidate under basic conditions. Acceptable bases include trimethylamine; triethylamine; dimethylamine; diethylamine; diphenylamine; diisopropylamine; diisopropylethylamine; l,4-diazabicyclo[2.2.2]octane (DABCO); l,5-diazabicyclo-[4.3.0]- non-5-ene (DB ); and the like. Aryl imidates can be prepared, for example, for aryl nitriles, aryl carboxylic acids, and aryl amides. Methods of fonning aryl imidates are discussed in U.S. Patent Application No. 60/380,909, filed May 15, 2002, the entire contents of which are incorporated herein by
reference. In one example, an aryl carboxylic acid (e.g., benzoic acid) is converted into an acid chloride, then an amide, followed by reaction with a trialkyloxonium hexafluorophosphate or a trialkyloxonium tetrafluoroborate to form the aryl imidate. In a second example, an aryl nitrile is converted into an aryl imidate through reaction with an alcohol in the presence of an acid, as is described below.
The thiazoline amide can be produced, for example, by reacting an amine with a carboxylic acid or with an activated acid (e.g., an ester, anhydride, or acid halide). In one embodiment of the present invention, a substituted thiazoline amide represented by Stractual Formula (LXXXIX) is formed through the reaction of a substituted thiazoline carboxylic acid with an amine represented by Structual Formula (LXXXN-DT). The amine represented by
Structual Formula (LXXXNm) contains at least one substituted or unsubstituted alkyl group comprising one or more chiral carbon atoms. Prefened amines include phenylethylamines and heteroaryl chiral amines such as, for example, cinchonidine and quinidine. In a prefened embodiment, the amine used in this reaction is substantially optically pure. There are several specific pathways by which the substituted thiazoline amide can be fonned. In one pathway, the substituted thiazoline amide is formed by reacting the amine with the substituted thiazolme carboxylic acid with at least stoichiometric quantities of a promoting agent (i.e., with at least one equivalent relative to the substituted thiazoline carboxylic acid). Preferably, the substituted thiazoline amide is fonned by reacting the amine with the substituted thiazoline carboxylic acid with about one equivalent of a promoting agent. The use of a promoting agent allows the formation of the substituted thiazoline amide at temperatures at or near room temperature. Examples of promoting agents include, but are not limited to, dicyclohexylcarbodiimide (DCC); Ν,Ν'-carbonyldiimidazole; POCl3; TiCl4; sulfuryl chloride fluoride; benzotriazol-1-yl diethyl phosphate; Ti(Obu) ; molecular sieves; N,N,N'5N'-tetramethyl(succmimido)uranium tetrafluoroborate; l,l'-carbonylbis(3- methylhnidazolium) triflate; 2,4-bis(4-inethoxyphenyl)-l,3,2,4-dithiadiphosphetane-2,4- disulfide (Lawesson's reagent); chlorosulfonyl isocyanate; P2L; pyridinium salts-Bu3N; Bu3P/PhCNO; SOCl2; imidazoles; and N-hydroxybenzothiazole (HOBt). Without being held to any particular theory, it is believed that the promoting agent DCC acts as a dehydrating agent on the substituted thiazoline carboxylic acid and encourages the formation of carboxylic acid anhydrides (i.e., (RCO)2O, where R is the substituted thiazoline group), which then react with the amine to fonn the substituted thiazoline amide. In a prefened embodiment, the
substituted thiazoline amide is formed by the reaction of a substituted thiazoline carboxylic acid with an amine, along with about one equivalent (relative to the substituted thiazoline carboxylic acid) of dicyclohexylcarbodiimide (DCC).
In another method, a substituted thiazoline amide is produced by pyrolysis of a mixture of the amine and the substituted thiazoline carboxylic acid. Carboxylic acids can also be converted to amides by heating with amides of carboxyhc acids, sulfonic acids, or phosphoric acids. In one example, a substituted thiazoline amide is formed from the reaction of a substituted thiazoline carboxylic acid with an amide of carboxylic acid, sulfonic acid, or phosphoric acid. hi another method, a substituted thiazoline amide is formed from an acyl halide. For example, an acyl halide is formed from the substituted thiazoline carboxylic acid by reaction with a halogenating agent. Halogenating agents include, but are not limited to, thionyl chloride, PC13, PC15, PBr3 and PBr5. hi a prefened embodiment the halogenating agent is thionyl chloride. The acyl halide is then reacted with an amine, such as the amine represented by Structual Formula (LXXXVIII), optionally in the presence of a weak base such as pyridine, to form a substituted thiazoline amide.
The 2-arylthiazoline-4-carboxamide can be alkylated in the presence of one or more bases, an alkylating agent, and optionally a phase transfer catalyst. Typically, the 2-arylthiazoline-4-amide is reacted with one or more equivalents (e.g., about 1 to 10, about 1 to 5, about 1 to 3, or about 1.5 to 2.5 equivalents) of base and one or more equivalents
(e.g., about 1 to 5, about 1 to 2, about 1 to 1.5, or about 1 to 1.1 equivalents) of an alkylating agent h a polar, aprotic solvent at about -80 to 40°C, about -50 to 25°C, about -20 to 10°C, or about -5 to 5°C.
Alkylating agents represented by the formula RiX, where Ri and X are as defined above. Prefened Ri groups include substituted or unsubstituted C1-C4 alkyl groups, for example, methyl or benzyl. The leaving group X is typically a weak base. Suitable leaving groups include halogen, tosyl, and mesyl, triflyl, brosyl, p-nitrophenyl, and 2,4- dinitrophenyl groups. Halogens include bromine, chlorine, and iodine. Iodine is a prefened leaving group. Prefened bases include alkali metal alkoxides, potassium t-butoxide, sodium methoxide, sodium ethoxide, and sodium amide. Suitable polar, aprotic solvents include, but are not limited to, dimethylformamide, dimethyl sulfoxide, acetonitrile, acetone,
tetrahydrofuran (THF), and hexamethylphosphoramide. Tetrahydrofuran (THF) is a prefened solvent.
In one example, a 2-arylthiazoline-4-carboxamide is reacted with about 2 equivalents of base and about 1 equivalent of methyl iodide in tetrahydrofuran (THF) at 0°C to form a 2-aryl-4-methyl-thiazohne-4-carboxamide.
Alternatively, the 2-arylthiazoline-4-carboxamide can be alkylated in the presence of a phase transfer catalyst. Examples of phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, friethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2-chloroethylamine chloride HCl, bis(2-chloroethyl)amine HCl, 2- dimethylaminoethyl chloride HCl, 2-ethylaminoethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, methylamine HCl, dhnethylamine HCl, trimethylamine HCl, monoethylamine HCl, diethylamine HCl, triethylamine HCl, ethanolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamine HCl, dicyclohexylamine HCl, cyclohexylamine HCl, diisopropylethylamine HCl, ethylenediamine HCl, aniline HCl, methyl salicylate, ethyl salicylate, butyl salicylate amyl salicylate, isoamyl salicylate, 2-ethylsalicylate, and benzyl salicylate. hi one embodiment of the invention, the 2-aryl-4-alkyl-thiazoline-4-carboxamide is hydrolyzed to form a 2-aryl-4-alkyl-thiazoline-4-carboxylic acid. Substituted amides (e.g., N- and N,N- substituted amides) can be hydrolyzed with either basic or acid catalysis to form a free carboxylic acid and an amine. For example, 2-aryl-4-alkyl-thiazoline-4-carboxamide can be hydrolyzed with water, heat, and either basic or acid catalyst to form 2-aryl-4-alkyl- thiazoline-4-carboxylic acid. An example of suitable conditions for hydrolysis is heating the amide under reflux in aqueous hydrochloric acid.
Preferably, 2-aryl-4-methyl-thiazoline-4-carboxyHc acid is subsequently reacted with acid to form 2-methylcysteine.
Methods of Alkylating Substituted Tbiazolines
The invention provides a useful and efficient method of preparing an alkylated thiazoline carboxylic acid, or a derivative thereof, represented by Structural Formula (XCDC):
wherein Ri is a substituted or unsubstituted alkyl group; each R2 is, independently, -H or a substituted or unsubstituted alkyl group; R is -H, a substituted or unsubstituted alkyl group or a carboxyl protecting group; and n is an integer from 1 to 5. hidependently, each R2 can be, for example, a substituted or unsubstituted Cl to C4 alkyl group. In a prefened embodiment, each R2 is methyl. Also, each R2 is preferably hydrogen. R3 can be, for example, a substituted or unsubstituted Cl to C4 alkyl group, hi a prefened embodiment, a group protecting an alkylated thiazoline carboxylic acid is removed and R3 is then hydrogen. The method comprises coupling a substituted aryl nitrile such as, for example, 2,4- dimethoxybenzonitrile or 4-methoxybenzonitrile, with a cysteine ester to form a substituted thiazoline carboxylic acid ester; optionally hydrolyzing the substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxylic acid; optionally, protecting the carboxyl group; alkylating the thiazoline ring at the 4-carbon position, as indicated in Structural Formula (XCDC), with a compound of the formula Ri-L, wherein Ri is as defined above and L is a leaving group, in the presence of a phase transfer catalyst; and, optionally, deprotecting the carboxyl group.
For example, the invention provides a method for producing a 2-(2,4- dihydroxyphenyl)-4-alkyl-4,5-dihydro-thiazole-4-caι-boxylic acid, as represented by Structural Formula (CXXVm):

(CXXVm),
wherein Ri is a substituted or unsubstituted alkyl group (e.g., methyl), that comprises coupling a substituted aryl nitrile, such as 2,4-dimethoxybenzonitrile, with a cysteine ester to form a substituted thiazoline carboxylic acid ester; optionally hydrolyzing the substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxyhc acid; optionally protecting the carboxyl group; alkylating the thiazoline ring at the 4-carbon position with a compound of the fonnula Ri-L, wherein Ri is as defined above and L is a leaving group, in the presence of a phase transfer catalyst; and deprotecting the carboxyl group. Also, the ether linkages (methoxy groups) or the phenyl ring are preferably cleaved to give free hydroxyl groups. In one incarnation of the invention, a cinchona-alkaloid derived phase transfer catalyst is used to alkylate a protected thiazoline carboxylic acid. Enantiomeric excesses of either (R)- or (S)-stereoisomers are produced during the synthesis of the alkylated thiazoline carboxylic acid, or derivative thereof, due to the asymmetric alkylation of the protected thiazoline carboxylic acid. For example, one synthesis route will produce an enantiomeric excess of a protected 2-(2,4-dialkyoxyphenyl)-4-alkyl-4,5-dihydro-thiazole-4-(S)-carboxylic acid.
Subsequently, the synthesis product can be purified by further resolving the enantiomers of the alkylated thiazoline carboxylic acid and isolating the desired isomer. hi one aspect of the invention, an aryl nitrile and a cysteine ester are condensed to form a substituted thiazoline carboxylic acid ester. Prefened aryl nitriles include aryl nitriles where the aryl group is a substituted or unsubstituted phenyl group.
An aryl nitrile such as that represented by Structural Formula (C):
wherein each R is, independently, a substituted or unsubstituted alkyl group and n is 1 to 5, can be coupled with a cysteine ester to form a substituted thiazolme carboxylic acid ester. Each R can be, independently, a substituted or unsubstituted Cl to C4 alkyl group. Preferably, each -R is methyl. In one embodiment, the aryl nitrile is a 2,4- dialkoxybenzonitrile, preferably 2,4-dimethoxybenzonitrile. In another embodiment, the aryl nitrile is a 4-alkoxybenzoι itrile, preferably 4-methoxybenzonitrile.
The aryl nitrile can be coupled with a cysteine ester, as represented by Structural Formula (Cl):
wherein A is an anion, preferably a halide such as chloride, bromide or iodide, and R5 is a substituted or unsubstituted alkyl group, to form an ester of a substituted thiazoline carboxylic acid.
For example, a 2,4-dialkoxybenzonitrile, represented by Structual Formula (CXXIX):
wherein R25 and R26 are independently, a substituted or unsubstituted alkyl group, can be coupled with a cysteine ester to form a substituted thiazoline carboxylic acid ester. Rz5 and R26 can be, independently, a substituted or unsubstituted Cl to C4 alkyl group. Preferably, R25 and R26 are each methyl. In one embodiment, a 2,4-dialkoxybenzonitrile is coupled with a cysteine ester, as represented by Structural Formula (CXXX):
wherein A is an anion, preferably a halide such as chloride, bromide or iodide, and R27 is a substituted or unsubstituted alkyl group, to form an ester of a 2-(2,4-dialkoxyphenyl)-4,5- dihydro-thiazole-4-carboxylic acid. R27 can be a Cl to C4 alkyl group. Preferably, R27 is ethyl, hi a prefened embodiment, the cysteine ester is the (R)- stereoisomer, for example,
2,4-dimethoxybenzonitrile is reacted with an (R)-cysteine ethyl ester to form ethyl 2-(2,4- dimethoxyphenyl)-4,5-dihydro-thiazole-4-(R)-carboxylate.
The condensation of an aryl nitrile and a cysteine ester typically occurs in a polar, protic solvent in the presence of an excess of base. Typically, the aryl nitrile and cysteine ester are refluxed together for several hours, such as 1-20 hours, 2-15 hours, 4-10 hours, or 6- 8 hours. Refluxing preferably occurs in an inert atmosphere, such as nitrogen or argon. Suitable polar, protic solvents include, but are not limited to, water, methanol, ethanol, formic acid, acetic acid, dimethylformamide, N-ethylacetamide, formaldehyde diethyl acetal, and long chain alcohols (e.g., propanol and isopropanol). An alcohol, such as methanol or ethanol, is a prefened solvent. Suitable bases mclude secondary and tertiary amines such as dimethylamine, diethylamine, trimethylamine, triethylamine, diisopropylamine, and diisopropylethylamine. The base can be added in excess, such as one or more equivalents relative to the amount of cysteine ester. Suitable amounts of base have at least about one equivalent of base, and range from about 1 to about 10, about 1 to about 5, about 1 to about 3, or about 1 to about 2 equivalents, relative to the amount of cysteine ester. In one example, cysteine ethyl ester, 2,4-dimethoxybenzonitrile, and 5 equivalents of triethylamine are refluxed in ethanol to obtain ethyl 2-(2,4-dimethoxyphenyl)-4,5-dihy<-ho-thiazole-4- carboxylate. In another example, cysteine ethyl ester, 4-methoxybenzonitrile, and 5 equivalents of triethylamine are refluxed in ethanol to obtain ethyl 2-(4-methoxyphenyl)-4,5- dihydro-thiazole-4-carboxylate.
Alternatively, an aryl imidate (e.g., a benzimidate, where the benzene ring can have one or more substituents, as described below) can be condensed with a cysteine ester to fonn a substituted thiazoline carboxylic acid ester. The substituted thiazoline carboxylic acid ester can be formed by coupling a substituted benzimidate such as, for example, 2,4- dihydroxybenzimidate, 2,4-dnnethoxybenzimidate, or a
4-methoxybenzimidate, with a cysteine ester such as the cysteine ester represented by Structural Formula (CXXX). Typically, coupling of a cysteine ester or a 2-alkylcysteine ester with an aryl imidate includes reacting a cysteine ester (or a related compound) with the aryl imidate under basic conditions. Acceptable bases include trimethylamine, triethylamine, dimethylamine, diethylamine, diphenylamine, diisopropylamine; diisopropylethylamine; l,4-diazabicyclo[2.2.2]octane (DABCO); l,5-diazabicyclo-[4.3.0]-non-5-ene (DBN); and the like.
Aryl imidates can be prepared, for example, from aryl nitriles, aryl carboxylic acids, and aryl amides. Methods of forming aryl imidates are discussed in co-pending U.S. Patent Application No. 60/380,909, filed on May 15, 2002, the entire contents of which are incorporated herein by reference, hi one example, an aryl carboxylic acid (e.g., benzoic acid) is converted into an acid chloride, then an amide, followed by reaction with a trialkyloxonium hexafluorophosphate or a trialkyloxonium tetrafluoroborate to form the aryl imidate. In a second example, an aryl nitrile is converted into an aryl imidate through reaction with an alcohol in the presence of an acid, as is described below.
The substituted thiazoline carboxylic acid esters represented by Structural Formulas (CD), (CXH), and (CXXD) can comprise a protected carboxyl group. Alternatively, the substituted thiazoline carboxylic acid ester can be protected by hydrolyzing the substituted thiazoline carboxylic acid ester represented by Structural Formulas (CH), (CXH) and (CXXU) and protecting the resulting substituted thiazoline carboxylic acid. In one aspect, the method of the present invention optionally comprises hydrolyzing a substituted thiazoline carboxylic acid ester to form a substituted thiazoline carboxylic acid as represented by Structural
Formula (CHI). For example, the ester of a 2-(2,4-dialkoxyphenyl)-4,5-dihydro-thiazole-4- carboxylic acid can be hydrolyzed to fonn a substituted thiazoline carboxylic acid, such as a 2-(2,4-dialkoxyphenyl)-4,5-dihydro-thiazole-4-carboxylic acid such as the compound represented by Structural Formula (CXDI). Hydrolysis of carboxylic esters is well known in the art. Hydrolysis of carboxylic esters is typically accomplished through acid or base catalysis. The substituted thiazoline carboxylic acid ester can be reacted with water in the presence of a base to fonn the substituted thiazoline carboxylic acid. Preferably, the substituted thiazoline carboxylic acid ester is refluxed with aqueous sodium hydroxide in methyl tertiary-butyl ether (MTBE) to form a substituted thiazoline carboxyhc acid. A protecting group may be added to the carboxyl group of the substituted thiazolme carboxylic acid to form a protected thiazolme carboxylic acid. Carboxyl groups can be protected by means well known in the art. Carboxyl groups are typically protected as esters (e.g., -COOR' wherein R' can be substituted or unsubstituted Cl to CIO alkyl, up to C30 substituted or unsubstituted aryl or alkyl-aryl wherein the alkyl is substituted or unsubstituted Cl to C5 and the aryl is substituted or unsubstituted and up to C30), or as carboxamide groups (e.g., -CONR"R"' wherein R" and R'" can be, independently, -H, substituted or unsubstituted Cl to CIO alkyl, up to C30 substituted or unsubstituted aryl, or alkyl-aryl
wherein the alkyl is substituted or unsubstituted Cl to C5 and the aryl is substituted or unsubstituted and up to C30). hi one embodiment, the carboxyl group of the substituted thiazoline carboxylic acid is protected as an ester of the fonn
-COOR7 wherein R7 is a carboxyl protecting group. Preferably, R is an alkyl group. Even more prefened, R is isopropyl. The protected thiazoline carboxylic acid ester may be formed through various means. An ester of a carboxylic acid can be produced using, for example, an alcohol, such as through the acid catalyzed reaction of a substituted thiazoline carboxylic acid with an alcohol. In a prefened embodiment, the protected thiazoline carboxylic acid is produced through the acid catalyzed reaction of a substituted thiazoline carboxylic acid with isopropanol. Common acid catalysts include sulfuric acid and p-toluenesulfonic acid. Alternatively, a substituted thiazoline carboxylic acid is treated with an alcohol in the presence of a coupling agent. Coupling agents include, but are not limited to dicyclohexylcarbodiimide (DCC); alkyl chloroformate and triethylamine; pyridinium salts and tributylamine; Amberlyst-15; phenyl dichlorophosphate; diethyl azoicarboxylate and triphenyl phosphine; DCC and an aminopyridine; 2-chloiO-l,3,5-trinitrobenzene and pyridine; l, -carbonylbis(3-methylimidazolium) triflate; di-2-pyridyl carbonate, polystyryl diphenylphosphine; (trimethylsilyl)ethoxyacetylene; chlorosulfonyl isocyanate; chlorosilanes, MeSO2Cl-Et3N; Ph3P-CCl4-Et3N; andN,N'-carbonyldiimidazole. Preferably, dicyclohexylcarbodiimide (DCC) is the coupling agent. As an example, a substituted thiazoline carboxylic acid are treated with an alcohol, such as isopropanol; DCC; and 4- (dimethylamino)pyridine (DMAP) in tetrahydrofuran (THF) at room temperature to form a protected thiazoline carboxylic acid.
Additional protecting groups, methods of adding a protecting group, and methods of removing a protecting group are taught in "Protective Groups in Organic Synthesis, 3 Edition" by Peter G. M. Wuts and Theodora W. Greene, Wiley-Interscience, 1999, the entire contents of which are incorporated herein by reference.
In one embodiment, the protected thiazoline carboxylic acid represented by Structural Fonnula (CV) can be alkylated in the presence of one or more bases, an alkylating agent, and a phase transfer catalyst. For example, isopropyl-2-(2,4-dimethoxyphenyl)-4,5-dihydro- thiazole-4-carboxylate or isopropyl-2-(4-methoxyphenyl)-4,5-dihydro-thiazole-4-carboxylate is reacted with 50% potassium hydroxide and excess methyl iodide in dichloromethane in the presence of a phase transfer catalyst at a temperature of about -80°C to about room
temperature. Preferably, the protected thiazoline carboxylic acid is alkylated using a phase transfer catalyst such that an enantiomeric excess of either the (R)- or (S)-isomer is produced, i.e., the alkylation is stereoselective.
Alkylating agents can have the formula Ri-L, where Ri is a substituted or unsubstituted alkyl group and L is a leaving group. Prefened Ri groups include substituted or unsubstituted C1-C4 alkyl groups, for example, methyl or benzyl. The leaving group L is typically a weak base. Suitable leaving groups include halogen, tosyl, mesyl, triflyl, brosyl, p- nitiOphenyl, and 2,4-dinitrophenyl groups. Halogens include bromine, chlorine, and iodine. Iodine is a prefened leaving group. Suitable amounts of alkylating agent can include about 1 to 20, about 2 to 15, about 3 to 10, or, preferably, about 5 equivalents, relative to the amount of protected thiazoline carboxylic acid.
Prefened bases include alkali or alkaline earth metal hydroxides, alkoxides, amides, or carbonates or their combinations. Available bases include potassium t-butoxide, sodiu methoxide, sodium ethoxide, sodium amide, calcium carbonate, cesium carbonate, and the alkali metal salts of hexamethyl disilazide (HMDS). Prefened bases include potassium hydroxide, sodium hydroxide, and cesium hydroxide monohydate. Suitable amounts of base include about 5 to 25, about 10 to 20, about 10 to 15, or, preferably, about 10 equivalents, relative to the amount of protected thiazolme carboxylic acid. The organic phase of the process can include any organic solvent which is substantially inert to the catalyst, reactants and products. The orgamc phase may comprise a combination of two or more solvents. Solvents generally mclude, but are not limited to, aprotic solvents such as acetonitrile, acetone, dimethylformamide, dimethyl sulfoxide, tetrahydrofuran, and hexamethylphosphoramide. In one embodiment, the organic phase comprises toluene. In a prefened embodiment, the organic phase comprises dichloromethane. The alkylation of the protected thiazoline carboxylic acid can be performed at temperatures ranging from about -80°C to about room temperature such as between about - 80° and 0°C. hi a prefened embodiment, the alkylation is performed at temperatures of between about -80° and -40°C, for example, at about -60°C. hi one aspect of the invention, a cinchona-alkaloid derived phase transfer catalyst is used to alkylate a protected substituted thiazoline carboxyhc acid. In one particular embodiment, a cinchona-alkaloid derived phase transfer catalyst is used to alkylate a
protected 2-(alkoxyphenyl)- 4,5-dihydiO-thiazole-4-carboxylic acid, represented by Structural Formula (CV) (e.g., a protected 2-(2,4-dialkoxyphenyl)- 4,5-dihydro-thiazole-4-carboxylic acid or a protected 2-(4-alkoxyphenyl)- 4,5-dihydro-thiazole-4-carboxylic acid), at the thiazoline 4-position. The phase transfer catalyst can be derived from cinchonine or from cinchonidine. Use of one of these catalysts in the alkylation reaction can yield enantiomeric excesses of either the (R)- or (S)-enantiomer of the alkylated protected thiazoline carboxylic acid, while use of an enantiomer of that catalyst can yield enantiomeric excesses of the other enantiomer of the alkylated cysteine derivative. Thus by selecting the phase transfer catalyst used, one can direct which enantiomer of the alkylated cysteine derivative will form.
In a prefened embodiment, the phase transfer catalyst used is derived from cinchonidine and is represented by Structural Formula (CV-D):
wherein R
8 and Rio are, independently, -H, a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group, or a salt thereof; R
9 is a substituted or unsubstituted alkyl group, a substituted or unsubstituted aromatic group, or a substituted or unsubstituted heterocyclic group, or a salt thereof; and
X is a halogen, hi one prefened embodiment, R
8 is substituted or unsubstituted ethenyl. R can be, for example, substituted or unsubstituted napthyl, anthracenylmethyl, or benzyl. Preferably, R
9 is 9-anthracenylmethyl, represented by Structural Formula (CVHI):
Rio can be, for example, substituted or unsubstituted allyl or benzyl. Rio is preferably substituted or unsubstituted allyl. X is preferably chlorine or bromine. Thus, the phase transfer catalyst can be represented by Structural Formula (CXVI):
Additional examples of phase transfer catalysts suitable for use in these methods are described in U.S. Patent No. 5,554,753 issued to O'Donnell, et al, the entire teachings of which are incorporated herein by reference.
The phase transfer catalyst represented by Structural Formula (CXVI) is preferably prepared using the following method as described by Corey, et al, in "A Rational Approach to Catalytic Enantioselective Enolate Allcylation Using a Structurally Rigidified and Defined Chiral Quaternary Ammonium Salt Under Phase Transfer Conditions" (J. Am. Chem. Soc. 119, 12414-12415 and Corey Supplemental therein 1-25 (1997)), the entire contents of which are incorporated by reference herein by reference. In that method, cinchonidine, represented by Structural Fonnula (CXXXI):
is suspended in toluene and 9-(chloromethyl) anthracene, represented by Structural Formula (CXXXII):
is added. The mixture is stined at reflux for about 2 hours. The product, N-9- anthracenylmethylcinchonidinium chloride represented by Structural Formula (CXXXJH):
is collected as a light yellow solid. The N-9-anthracenylmethylcinchonidinium chloride is then suspended in dichloromethane. To this suspension is added 50% KOH and allyl bromide. The resulting mixture is then stined for about 4 hours at about 23 °C. The product, O(9)-allyl-N-9-anthracenylmethylcinchonidium bromide represented by Structural Fonnula (CXVI), wherein X is bromine, is collected as a light orange solid.
The use of O(9)-allyl-N-9-anthracenylmethylcinchonidium bromide as a phase transfer catalyst is also described in U.S. Patent Application No. 60/381,012, filed on May 15, 2002, the entire contents of which are incorporated herein by reference.
The phase transfer catalyst represented by Structural Formula (CXXXIV):
is also suitable for the purposes of the instant invention.
Examples of other phase transfer catalysts include benzyl triethyl ammonium chloride, benzyl trimethyl ammonium chloride, benzyl tributyl ammonium chloride, tetrabutyl ammonium bromide, tetraethyl ammonium bromide, tetrabutyl ammonium hydrogen sulfate, tetramethyl ammonium iodide, tetramethyl ammonium chloride, triethylbutyl ammonium bromide, tributyl ethyl ammonium bromide, tributyl methyl ammonium chloride, 2- chloroethylamine chloride HCl, bis(2-chloroethyl)amine HCl, 2-dimethylaminoethyl chloride HCl, 2-ethylaminoethyl chloride HCl, 3-dimethylaminopropyl chloride HCl, monoethylamine HCl, diethylamine HCl, triethylamine HCl, ethanolamine HCl, diethanolamine HCl, triethanolamine HCl, cyclohexylamine HCl, dicyclohexylamine HCl, cyclohexylamine HCl, diisopropylethylamine HCl, ethylenediamine HCl, and aniline HCl.
In one aspect of the invention, the phase transfer catalyst, such as O(9)-allyl-N-9- anthracenylmethylcinchonidium bromide, is present in an amount of about 0.05 to 0.4 equivalents relative to the amount of protected thiazoline carboxylic acid. Alternatively, the phase transfer catalyst can be present between about 0.05 and 0.25, between about 0.1 and 0.15, or, preferably, at about 0.1 equivalents (relative to the amount of protected thiazoline carboxylic acid).
In a prefened embodiment, isopropyl 4,5-dihydro-2-(2,4-dimethoxyphenyl)-thiazole- 4-carboxylate is reacted with 50% potassium hydroxide and excess methyl iodide in dichloromethane in the presence of about 10%(mol) of O(9)-allyl-N-9- anthracenylmethylcinchonidium bromide. Subsequent to the alkylation, the group protecting the carboxyl group of the alkylated protected thiazoline carboxylic acid can be removed. Additionally, ether groups present on the alkylated protected thiazoline carboxylic acid can be cleaved. Methods of cleaving ether groups are well known in the art. For example, ether groups can be cleaved by reacting the ether compound with an excess of hydrogen bromide or hydrogen iodide. Ether groups also can be cleaved by reacting the ether compound with boron tribromide (BBr3), methylmagnesium iodide (CH3MgI), or aluminum trichloride (A1C13). In a prefened embodiment of the present invention, an alkylated protected thiazoline carboxylic acid is hydrolyzed, as described above, and its ether groups cleaved to form an alkylated thiazoline carboxylic acid, such as 2-(2,4-dihydroxyphenyl)-4-alkyl-4,5-dihydro-thiazole-4-carboxylic acid.
Common Definitions and Techniques
The section below applies to the invention as a whole, such that these definitions and techniques can generally be applied to the various methods and embodiments of the invention described above. Under circumstances where discussion in the individual sections above duplicates the discussion in this section, the discussion in the individual section should be considered as a prefened embodiment of that method. Unless otherwise indicated, these common definitions and techniques are applicable to the entire invention.
The products, either enantiomers or diastereomers, of the above noted syntheses can be purified or ultrapurified before or after any protecting groups are removed. In a prefened embodiment, the 2-alkylcysteine derivative (e.g., 2-methylcysteine) is purified by resolution into the (R)- and (S)-isomers based on the cysteine 2-carbon position. For example, the 2- alkylcysteine derivative can be purified or ultrapurified using the technique of emulsion crystallization. Emulsion crystallization may be used to purify acids and functionalized derivative of acids such as esters and amides. Optionally, the protective groups are removed after purification to form an unprotected (S)-, or (R)-, 2-alkylcysteine (e.g., 2-methylcysteine) or an (S)-, or (R)— , 2-alkylcysteine derivative.
Altematively, protective groups are removed from the 2-alkylcysteine derivative to form an unprotected 2-alkylcysteine prior to further resolution. For example, protective groups are removed from a protected 2-methylcysteine to form an unprotected 2- methylcysteine, the unprotected 2-methylcysteine is resolved into its (R)- and (S)-isomers, and an (S)-2-alkylcysteine is isolated. The 2-alkylcysteine can be resolved into its (R)- and (S)-isomers using the technique of emulsion crystallization, or the 2-alkylcysteine can be resolved into its enantiomers by forming a diastereomeric salt.
Chiral carboxylic acids and their functionalized derivatives, such as 2-alkylcysteines and their derivatives, can be purified by emulsion crystallization, as described in U.S. Patent Nos. 5,872,259, 6,383,233 and 6,428,583, issued to Reuter, the entire teachings of which are incorporated herein by reference. Briefly, emulsion crystallization is a process for separating a desired substance from an aggregate mixture. The process involves forming a three phase system, the first phase comprising the aggregate mixture, the second phase being liquid and comprising a transport phase, and the third phase comprising a surface upon which the desired substance can crystallize. A chemical potential exists for crystal growth of the desired substance in the third phase of the system, thereby creating a flow of the desired substance from the first phase through the second phase to the third phase, where the desired substance crystallizes and whereby an equilibrium of the activities of the remaining substances in the aggregate mixture is maintained between the first phase and the second phase. In one example of emulsion crystallization, a solution of the racemic mixture is supersaturated (by either cooling, adding a solvent in which one or more components are sparingly soluble or by evaporation of the solution). Ultrasonication eventually helps the process of forming an emulsion. The mixture is then seeded with crystals of the desired, optically active acid along with an additional quantity of surfactant and an anti-foaming agent. The desired product usually crystallizes out and can be separated by filtration.
Chiral carboxylic acids also can be purified through further resolution by forming a diastereomeric salt with the chiral carboxylic acid and a chiral amine. Suitable chiral amines include arylalkylamines such as 1 -alkyl- 1-aminoalkanes and 1-aryl-l-aminoalkanes. Examples include (R)-l-phenylethylamine, (S)-l-phenylethylamine, (R)-l-tolylethylamine, (S)-l-tolylethylamine, (R)-l-phenylpropylamine, (S)-l-propylamine, (R)-l-tolylpropylamine, and (S)-l-tolylpropylamine. Preferably, (R)-l-phenylethylamine is used to further resolve the chiral carboxylic acid mixture. Resolution of chiral compounds using diastereomeric salts is
further described in CRC Handbook of Optical Resolutions via Diastereomeric Salt Formation by David Kozma (CRC Press, 2001), incorporated herein by reference in its entirety.
Once the chiral carboxylic acids or their derivatives have been purified, the desired isomer can be isolated. Typically, the (S)-isomer is isolated. For example, protected or unprotected (S)-2-methylcysteine, (S)-2-alkylcysteines, or (S)-2-alkylcysteine derivatives are isolated. Preferably, a protected (S)-2-methylcysteine is isolated.
In a prefened embodiment, a protected (S)-2-methylcysteine is formed and isolated. (S)-2-methylcysteine can then be formed by removing any protecting groups present, for example, by treating the protected (S)-2-methylcysteine with acid to remove protecting groups. Cysteine or a 2-alkylcysteine such as (S)-2-methylcysteine can be coupled to a substituted or unsubstituted aryl nitrile such as a substituted or unsubstituted benzonitrile. Preferably, the substituents on benzonitrile will not interfere with the coupling reaction. In another prefened embodiment, (S)-2-methylcysteine is coupled to 2,4-dihydroxybenzonitrile to fonn 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4(S)-carboxylic acid (also known as 4'-hydroxydesazadesferrithiocin). In yet another embodiment, (S)-2-methylcysteine is coupled to 2-hydroxybenzonitrile to form 4,5-dihydro-2-(2-hydroxyphenyl)-4- methylthiazole-4(S)-carboxylic acid (also known as desazadesferrithiocin).
Typically, coupling of cysteine or a 2-alkylcysteine and a substituted or unsubstituted benzonitrile includes converting the benzonitrile into a benzimidate. The benzimidate can be formed, for example, by reacting the benzonitrile with an alcohol such as methanol, ethanol, n-propanol, or isopropanol in the presence of an acid such as hydrochloric acid. The benzimidate is then reacted with the cysteine (or related compound) under basic conditions. Acceptable bases include trimethylamine, triethylamine, triphenylamine, and the like. The reaction between the benzimidate and the cysteine results in the thiazoline (or 4,5- dihydrothiazole) containing product. When forming the benzimidate from a hydroxylated benzonitrile (e.g., 2,4-dihydroxybenzonitrile), the hydroxyl groups are advantageously protected (e.g., with a substituted or unsubstituted alkyl or arylalkyl group such as a benzyl group). The protecting groups are subsequently cleaved, typically by catalytic hydrogenation. Suitable benzonitriles and benzimidates for use in the above coupling reaction can be synthesized by methods described in co-pending U.S. Patent Application No. 60/381,013, entitled "Synthesis of Benzonitriles from Substituted Benzoic Acid," filed May 15, 2002,
co-pending U.S. Patent Application No. 60/380,878, entitled "Synthesis of Benzonitriles from Substituted Benzaldehyde," filed May 15, 2002, and co-pending U.S. Patent Application No. 60/380,909, entitled "Synthesis of Benzimidate from Benzoic Acid," filed May 15, 2002. The entire contents of these applications are incorporated herein by reference.
The methods of the claimed invention can be used to manufacture other related desferrithiocin analogs and derivatives. Examples of such analogs include those described in U.S. Patent Nos. 5,840,739, 6,083,966, 6,159,983, 6,521,652 and 6,525,080, all issued to Bergeron, the contents of which are incorporated herein by reference. Additional examples can be found in hitemational Application Nos. PCT/US93/10936, published as WO 94/1137 on May 5, 1994; PCT/US97/04666, published as WO 97/36885 on October 9, 1997; and PCT/US99/19691, published as WO 00/12493 on March 9, 2000, the entire contents of which are incorporated herein by reference.
An alkyl group is a hydrocarbon in a molecule that is bonded to one other group in the molecule through a single covalent bond from one of its carbon atoms. Alkyl groups can be cyclic, branched or unbranched, and/or saturated or unsaturated. Typically, an alkyl group has one to about 24 carbons atoms, or one to about 12 carbon atoms. Lower alkyl groups have one to four carbon atoms and include methyl, ethyl, »-propyl, t-sO-propyl, n-butyl, -sec-butyl and tert-butyl. A cycloahphatic group is cyclic, non-aromatic, consists solely of carbon and hydrogen and may optionally contain one or more units of unsaturation, e.g., double and/or triple bonds. A cycloahphatic group can have one or more rings, wliich can be fused together. Typically, a cycloahphatic group has one to about 24 carbons atoms, or about 1 to about 12 carbon atoms. Examples of cycloahphatic groups include cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl, cyclopentenyl, cyclopenta-l,3-dienyl, cyclohexenyl, cyclohexa-l,3-dienyl, cyclohexa-l,4-dienyl, cycloheptenyl, cyclooctenyl, cycloocta-l,3-dienyl, and cycloocta-1,3,5- trienyl.
A heterocyclic group is a cycloahphatic group where one or more of the carbon atoms is replaced by a heteroatom such as S, O, or N. Examples of heterocyclic groups include oxiryl, oxetyl, oxolyl, oxyl, aziridyl, azetidyl, pynolidyl, piperidyl, tetrahydrothiophyl, and tetrahydrothiopyryl.
Aromatic (or aryl) groups include carbocyclic aromatic groups such as phenyl, p- tolyl, 1-naphthyl, 2-naphthyl, 1-anthracyl and 2-anthracyl. Aromatic groups also include heteroaromatic groups such as N-imidazolyl, 2-imidazole, 2-thienyl, 3-thienyl, 2-furanyl, 3- furanyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 2-pyrimidyl, 4-pyrimidyl, 2-pyranyl, 3-pyranyl, 3- pyrazolyl, 4-pyrazolyl, 5-pyrazolyl, 2-pyrazinyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2- oxazolyl, 4-oxazolyl and 5-oxazolyl.
Aromatic groups also include fused polycyclic aromatic ring systems in which a carbocyclic, alicyclic, aromatic ring or heteroaryl ring is fused to one or more other heteroaryl or aryl rings. Examples include 2-benzothienyl, 3-benzothienyl, 2-benzofuranyl, 3-benzofuranyl, 2-indolyl, 3-indolyl, 2-quinolinyl, 3-quinolinyl, 2-benzothiazole, 2- benzooxazole, 2-benzimidazole, 2-quinolinyl, 3-quinolinyl, 1-isoquinolinyl, 3-quinolinyl, 1- isoindolyl and 3-isoindolyl.
Suitable substituents for alkyl and cycloahphatic groups include -OH, halogen (-Br, -Cl, -I and -F), -O(R'), -O-CO-(R '), -CN, -NO2, -COOH, =O, -NH2, -NH(R'), -N(R')2, -COO(R'), -CONH2, -CONH(R'), -CON(R')2, -SH, -S(R'), and guanidine. Each R' is independently an alkyl group or an aromatic group. Alkyl and cycloahphatic groups can additionally be substituted by a heterocyclic, aromatic, or heteroaromatic group (e.g. an alkyl group can be substituted with an aromatic group to form an arylalkyl group). A substituted alkyl or cycloahphatic group can have more than one substituent. Suitable substituents for heterocyclic, aromatic, and heteroaromatic groups include -
OH, halogen (-Br, -Cl, -I and-F), -O(R'), -O-CO-(R '), -CN, -NO2, -COOH, -O, -NH2, -
NH(R'), -N(R')2, -COO(R'), -CONH2, -CONH(R'), -CON(R')2, -SH, -S(R'), and guanidine. Each R' is independently an alkyl group or an aromatic group. Heterocyclic, aromatic, and heteroaromatic groups can additionally be substituted by an alkyl or cycloahphatic group (e.g. an aryl group can be substituted with an alkyl group to form an alkylaryl group such as tolyl). A substituted heterocyclic, aromatic, or heteroaromatic group can have more than one substituent.
Also included in the present invention are salts of the disclosed carboxylic acids. For example, amino acids can also be present in the anionic, or conjugate base, form, in combination with a cation. Suitable cations include alkali metal ions, such as sodium and potassium ions; alkaline earth ions, such as calcium and magnesium ions; and unsubstituted
and substituted (primary, secondary, tertiary and quaternary) ammonium ions. Suitable cations also include transition metal ions such as manganese, copper, nickel, iron, cobalt, and zinc. Basic groups such as amines can also be protonated with a counter anion, such as hydroxide, halogens (chloride, bromide, and iodide), acetate, formate, citrate, ascorbate, sulfate or phosphate.
Functional groups of the present invention can be protected with a protecting group. As is known in the art, a protecting group reduces or eliminates the ability of a functional group to react with another functional group. For example, a thiol or an alcohol can be protected with an acyl group. Similarly, an alcohol can be protected by a tosyl or a trhnethylsilyl group. An amine can, for example, be protected by an Fmoc group or a Boc group. Additional protecting groups, methods of adding a protecting group, and methods of removing a protecting group are taught in "Protective Groups in Organic Synthesis, 3rd Edition" by Peter G. M. Wuts and Theodora W. Greene, Wiley-Interscience, 1999, which was incorporated by reference above. Protecting groups for basic nitrogen atoms include formyl; 4-toluenesulfonyl; t- butyloxycarbonyl; 2,4-dinitrophenol; benzyloxymethyl; trityl; t-butoxymethyl; 2- chlorobenzyloxy-carbonyl; allyloxycarbonyl; benzyloxycarbonyl (Z); mesitylene-2-sulfonyl; 4-methyloxy-2,3,6-trimethyl- benzyenesulfonyl; 2,2,5,7,8-pentamethyl-chroma n-6-sulfonyl; 9-xanthenyl; and 2,4,6-trimethoxybenzyl. Protecting groups for basic sulfur groups include 4-methylbenzyl, 3-nitro-2- pyridinesulfenyl; trityl; 2,4,6-trimethoxybenzyl; acetamidomethyl; trimethylacetaminomethyl; t-butylsulfonyl; and sulfoxide.
Protecting groups for basic oxide groups include benzyl ether; t-butyl ether; benzyl ether; 2,6-dichlorobenzyl ether; 2-bromobenzyl ether; and 3,5-dibromobenzyl ether. Carboxyl groups can be protected, for example, as ethers or as carboxamides. For example, when a carboxyl group is protected as an ether, it takes the form of-COOR wherein R is a substituted or unsubstituted Cl to CIO alkyl group, a substituted or unsubstituted up to C30 alkyl group, or a substituted or unsubstituted alkyl-aryl group wherein the alkyl group is Cl to C5 and the aryl group is up to C30. When a carboxyl group is protected as a carboxamide, it takes the form of-CONR' wherein R' is -H or as in R above.
EXEMPLIFICATION
Example 1
Preparation of the Phase Transfer Catalyst
A cinchonidine derived phase transfer catalyst is prepared as follows. About 4 grams of cinchonidine is suspended in about 40 mL of toluene. About 3 grams of 9- (choloromethyl)anthracene is then added to the suspension. The mixture is heated to reflux and stined for about 2 hours. Solids are cooled to room temperature, poured onto about 200 mL of diethyl ether and filtered. The product collected is N-9- anthracenylmethylcinchonidinium chloride.
About 5 grams N-9-antliracenyhnethylcinchonidinium chloride is then suspended in about 40 mL dichloromethane. Then about 2.5 mL allyl bromide and about 5 mL of 50% KOH (aq) are added to the suspension. The mixture is stined at about room temperature for about 4 hours. Fifty milliliters of water is then added to the mixture and the mixture is extracted using three aliquots of dichloromethane. The organic extracts are combined and dried over Na2SO4, filtered, and concentrated in vacuo. Recrystalization of the residue from methanol-diethyl ether at -20°C yields the product, O(9)-allyl-N-9- anthracenylmethylcinchonidium bromide.
Example 2
Preparation of (S)-2-Methylcysteine 2-(R)- (9H-Fluoren-9-ylmethoxycarbonylamino)-3 -tritylsulfanyl-propionic acid is reacted with t-butyl alcohol and dicyclohexyl carbodiimide (DCC) in 4-(dimethylamino) pyridine (DMAP) and tetrahydrofuran (THF) at room temperature to form 2-(R)- (9H- fluoren-9-ylmethoxycarbonylamino)-3 -tritylsulfanyl-propionic acid tert-butyl ester.
The Fmoc group is removed from the 2-(R)-(9H-fluoren-9-yhnethoxycarbonylamino)- 3 -tritylsulfanyl-propionic acid tert-butyl ester using diethylamine in dichloromethane to form
2-(R)-amino-3 -tritylsulfanyl-propionic acid tert-butyl ester
The 2-(R)-amino-3 -tritylsulfanyl-propionic acid tert-butyl ester is reacted with benzhydrylideneamine in dichloromethane at room temperature to form 2-(R)- (benzhydrylidene-amino)-3-tritylsulfanyl-propionic acid tert-butyl ester. A mixture of 2-(T<.)-(benzhy(--rylidene-ammo)-3-tritylsulfanyl-propionic acid tert-butyl ester (one equivalent), about 10 equivalents cesium hydroxide monohydrate, about 0.1 equivalents of O(9)-allyl-N-9-anthracenyhnethylcinchonidium bromide, and about 0.5 mL
dichloromethane is prepared. Excess methyl iodide (about 5 equivalents) is then added dropwise at about -80°C to the above formed mixture. The mixture is then stined and allowed to react for about 25-30 hours at about -60°C. The reacted mixture is then diluted with ether, washed with water, washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The product collected is 2-(benzhydrylidene-amino)-2-methyl-3- tritylsulfanyl-propionic acid tert-butyl ester with the (S)-isomer in enantiomeric excess.
The 2-(benzhydrylidene-amino)-2-methyl-3 -tritylsulfanyl-propionic acid tert-butyl ester is resolved using emulsion crystallization. 2(S)-(benzhydrylidene-amino)-2-methyl-3- tritylsulfanyl-propionic acid tert-butyl ester is then isolated and reacted with excess 5M hydrochloric acid to form (S)-2-methylcysteine.
Example 3
All compounds were used without further purification. The surfactants Rhodafac RE 610 and Soprophor FL were obtained from Rhόne-Poulenc, Surfynol 465 from Air Products, Synperonic NP 10 from ICI and sodium lauryl sulfate from Fluka. For agitation a shaking machine was used (Buhler KL Tuttlingen). Purities of the resulting crystals were measured by using a PolarMonitor polarimeter (IBZ Hannover). Ethanol was used as the solvent. The total crystal quantity was dissolved in a 1 mL cell at 20 °C)
45 mg of (R,R) - and (S,S) - amino acid derivatives were dissolved in 1 ml of a mixture of 20% v/v 2-hexanol, 12% v/v Rhodafac RE 610, 6% v/v Soprophor FL and 62% v/v water by heating to 80 °C in a 5 mL vial. After the organic derivative was completely dissolved the microemulsion was cooled down to room temperature and agitated using a shaking machine (420 rpm). During two hours no spontaneous crystallization was observed. The mixture was then seeded with two drops of a dilute, finely ground suspension of pure (S,S) - (-) amino acid or its ester crystals grown under similar conditions. After 2 hours of agitation the resulting crystals were filtered off, washed with water and dried in a gentle nitrogen stream.
Example 4 35 mg of R- and S-4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4- carboxylic acid were dissolved in 1 ml of a mixture of 9% N-methyl-pynolidone, 9% v/v 2- hexanol, 10% v/v Rhodafac RE 610, 5% v/v Soprophor FL and 68% v/v water by heating to
50°C in a 5 mL vial. After the product was completely dissolved, the microemulsion was cooled down to room temperature and agitated with a shaking machine (350 rpm). During two hours, no spontaneous crystallization was observed. The mixture was then seeded with two drops of a dilute, finely ground suspension of pure S-product crystals grown under similar conditions. After two hours of shaking, the resulting crystals were filtered off, washed with water and dried in a gentle nitrogen stream. The procedure yielded 5.4 mg (15.4%) of colorless crystals, with a greater than 90% purity of the S enantiomer.
Example 5 4.00 g (S)-2-methylcysteine hydrochloride (23.3 mmol,1.0 meq) and 3.14 g 2,4- dihydroxy benzonitrile (23.3 mmol, 1.0 meq) were suspended in 40 mL ethanol. After degassing this mixture with nitrogen (30 min) 4.95 g triethylamine (6.8 mL, 48.9 mmol, 2.05 meq) were added. The obtained suspension was heated under reflux in an atmosphere of nitrogen for 20 hours and then cooled to room temperature. From this suspension ethanol was evaporated under reduced pressure until an oil (20 % of the initial volume) was obtained. This oil was dissolved in 50 mL water. The solution was adjusted to pH 7.5 with 1.20 ml 20 % KOH and was extracted two times each with 20 mL methyl t- butyl ether (MTBE). The aqueous layer was separated, adjusted with 20 % KOH to pH 11 and again extracted two times each with 20 mL MTBE. After separating the aqueous layer the pH was set with concentrated HCl to 7.5 and traces of MTBE were distilled off. Then the aqueous solution was acidified with 1.50 ml concentrated HCl to pH 1.5. The product precipitated. This suspension was stined at 4 °C for 1 hour. Then the precipitate was filtered, washed two times each with 10 mL water (5°C) and dried at 45 °C under vacuum. The reaction yielded 5.17 g (87.6 %) of crude 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4- methylthiazole-4(S)-carboxylic acid product. 1H-NMR showed no significant impurity.
Example 6
A single-neck 500 mL round-bottomed flask was flushed with nitrogen. (R)- (+)-L- cysteine hydrochloride monohydrate (12.0g, 68.32 mmol) was fransfened to the flask. Ethanol (240 mL) was added to give a suspension. Anhydrous triethylamine (34.6g, 47.7 mL, 341.6 mmol, 5.0 equiv.) was then added via a syringe over a period of 10 min. at room temperature. A white precipitate of triethylamine hydrochloride formed immediately. After
stirring this thick white turbid solution for 30 min. at room temperature, benzonitrile (7.05g, 68.32 mmol) was added and the reaction mixture was refluxed for 6 hours. TLC (CH2C12 as eluent) indicated that all benzonitrile was consumed. The reaction mixture was cooled to room temperature and the solvent was removed in vacuo. Water (25 mL) was added followed by the addition of solid KOH (5g) with stirring. This reddish clear aqueous solution (pH ~ 11-12) was extracted with ethyl acetate (3 X 100 mL) and the orgamc layer was discarded. The aqueous layer was acidified with dropwise addition of 6M HCl to pH 1.5-2.0 to obtain an off-white to tan colored precipitate. This solid was filtered through a Buchner funnel. After drying under high vacuum, the solid was triturated with ethyl acetate to remove any traces of colored impurities. After filtration and drying, the off-white to white solid was stined over dichloromethane to remove any traces of triethylamine hydrochloride and then filtered. After drying under vacuum, a white powdery solid was obtained (10.49g, 74%).
Example 7
Synthesis of N-p-Toluenesulfonyl -2-carbomethoxy-2-methyI aziridine
Bromamine-T (3 g, 11.028 mmol) was added to a stined mixture of anhydrous CuCl2 (148 mg, 1.1 mmol) and methyl methacrylate (5.14g, 5.88mL, 5.5 mmol) in acetonitrile (30 mL) at room temperature under nitrogen. The reaction mixture was stined at room temperature overnight. Then it was diluted with ethyl acetate (100 mL) and filtered through a pad of silica gel. The filtrate was dried over sodium sulfate and the solvent was concentrated in vacuo. A thick colorless oil obtained was purified through silica gel column chromatography with an eluent mixture of petroleum ether: ethyl acetate (1 :4) to yield 2.1 g (70 %) of the pure aziridine product. The product was characterized by 1H NMR (200 MHz), mass spectrometry, and FT-IR spectral analysis.
Synthesis of methyl (2-N-tosylamino-3-benzoylmercapto) propionate
Thiobenzoic acid (253 mg, 1.85 mmol) was added to a stirred solution of N-p- Toluenesulfonyl-2-carbomethoxy-2-methyl aziridine (250 mg, 0.929 mmol) in anhydrous dichloromethane (5 mL). The reaction mixture was stined at room temperature for 18 h and at 4β°C for 30 h. It was then diluted with ethyl acetate (40 mL) and the combined organic layer was washed with saturated sodium bicarbonate solution and dried. Removal of the
solvent under vacuum yielded an oily residue, which crystallized upon the addition of hexane to yield 150 mg (39 %) of the pure product. The product was characterized by 1H NMR, FT-IR and mass spectrometry.
Synthesis of methyl (2-N-tosylamino-3-mercapto) propionate
Methyl N-tosyl amino-3-benzoyl mercapto propionate was dissolved in 30 mL of 0.2 N NaOH. The solution was kept under nitrogen at room temperature for 15 minutes. The reaction mixture was acidified with dilute sulfuric acid and the solution was extracted with ethyl acetate. The ethyl acetate layer was washed with water until the water extract became neutral and was dried over sodium sulfate and concentrated to a small volume in vacuo. Analysis of the isolated products from the reaction mixture did not show any indication of both tosyl and methyl groups. The product isolated from the ethyl acetate layer contained a cleaved benzoyl compound.
Example 8
Synthesis of N-p-Toluenesulfonyl -2-carbomethoxy-2-methyI aziridine
Bromamine-T (3 g, 11.028 mmol) was added to a stined mixture of anhydrous CuCl2 (148 mg, 1.1 mmol) and methyl methacrylate (5.14g, 5.88mL, 5.5 mmol) in acetonitrile (30 mL) at room temperature under nitrogen. The reaction mixture was then stined at room temperature overnight, then diluted with ethyl acetate (100 mL) and filtered through a pad of silica gel. The filtrate was dried over sodium sulfate and the solvent was concentrated in vacuo. An oil was obtained and purified through silica gel column chromatography with an eluent mixture of petroleum ether: ethyl acetate (1:4) to yield 2.1 g (70 %) of the pure aziridine product.
Synthesis of methyl (2-N-tosylarnino-3-benzoylmercapto) propionate
Thiobenzoic acid (253 mg, 1.85 mmol) was added to a stined solution of N-p- toluenesulfonyl-2-carbomethoxy-2-methyl aziridine (250 mg, 0.929 mmol) in anhydrous dichloromethane (5 mL). The reaction mixture was stined at room temperature for 18 h and at 4β°C for 30 h. It was then diluted with ethyl acetate (40 mL) and the combined organic layer washed with saturated sodium bicarbonate solution and dried. Removal of the solvent under vacuum yielded an oily residue, which crystallized upon the addition of hexane to
yield 150 mg (39 %) of the pure product. The product was characterized by Η NMR, FT- IR and Mass spectrometry.
Synthesis of methyl (2-N-tosylamino-3-mercapto) propionate Methyl N-tosyl amino-3-benzoyl mercapto propionate is dissolved in an aqueous ammonia solution. The mixture is stined for a time, then acidified. The solution is extracted with a suitable solvent, and the product is isolated.
Example 9 Synthesis of N-p-toluenesulphonyl-2-carbomethyoxy-2-methyI aziridine
Anhydrous Q1CI2 (148 mg, 1.1 mmol) in acetonitrile (30 ml) was stined under nitrogen at room temperature. Methyl methacrylate (1.1 g, 1.17 ml, 11 mmol) was then added to this solution followed by addition of Bromamine-T (1.793 g, 5 mmol). The reaction mixture was stined at room temperature for 6-8 hours. Then it was diluted with ethyl acetate (100 ml) and filtered through a pad of silica gel. The clear solution was dried over sodium sulphate and solvent concentrated under vacuum. A thick colorless oil was obtained, which was purified by silica gel column chromatography (eluent of petroleum etheπethyl acetate: 4:1) to obtain N-p-toluenesulfonyl-2-carbomethoxy-2-methyl aziridine. The reaction yielded 1.3 g (40%). 1H NMR (CDC13) 200 MHz: δ 1.88 (s, 3H, CH3), 2.42 (s, 3H, CH3), 2.70 (s, IH), 2.78 (s, IH), 3.73 (s, 3H, COOCH3), 7.30-7.33 (d, 2H, aromatic), 7.80-7.84 (d, 2H, aromatic).
Synthesis of methyl (2-N-tosylamino-3-benzoyImercapto)propionate
N-p-Toluenesulphonyl-2-carbomethoxy-2-methyl aziridine (250 mg, 0.229 mmol) was dissolved in 5 ml of CH2CI2 and thiobenzoic acid (253 mg, 1.85 mmol) was added to this solution. The reaction mixture was stined at room temperature for 18 hours and at 40°C for 30 hours. It was then diluted with ethyl acetate (40 ml); the combined organic layer was washed with saturated sodium bicarbonate solution and dried. Removal of the solvent under vacuum yielded an oily residue, which crystallized upon the addition of
hexane. The reaction yielded 150 mg methyl (2-N-tosylamino-3-benzoylmercapto)proionate (39%), which had a melting point of 137-138°C. Other analytical data are as follows: IR (CHC13): 3279 cm-1 (NH), 1733 cm"1 (ester), 1667 cm"1 (ketone). 1H NMR (CDC13), 200 MHz : δ 1.54 (s, 3H, CH3), 2.38 (s, 3H, CH3), 3.48-3.55 (d, 2H, CH3), 3.67 (s, 3H, COOCH3), 5.67 (s, IH, NH), 7.25 (d, 2H, aromatic), 7.40-7.77 (m, 5H, aromatic), 7.95 (d, 2H, aromatic). Mass (m/e): 408 (M-^1), 348 (M - COOCH3).
CH analysis Calculated for: Cι9H2ιNO5S2 : C=56.0%; H=5.15%; N=3.43%; S=15.72% Found : C=56.56%; H=4.92%; N=3.12%; S=16.40%.
Example 11
A one-necked, lOOmL, round-bottomed flask was fitted with Dean-Stark apparatus attached with a drying tube (CaCl2) and a magnetic stiner. The flask was charged with 5 g (26.6 mmol) of t-butyl ethyl malonate, 2.39 g (78.8 mmol) of formalin solution (35% formaldehyde in water), 3.4 g (40 mmol) of piperidine and 50 mL of toluene. The mixture was heated to reflux with stining in an oil bath at 120-130°C for 8 hours. After cooling to room temperature, toluene was removed under reduced pressure. The crude oily product was purified by column chromatography on silica gel, and was eluted with ethyl acetate/petroleum ether (6:94) to give 3.56 g (67%) of t-butyl ethyl methylene malonate.
1H NMR (CDC13, 200 MHz) δ 1.35 (t, 3H), 1.55 (s, 9H), 4.30 (q, 2H), 6.40 (d, 2H).
To a one necked 50 mL round-bottomed flask fitted with a reflux condenser and an outlet dipped inside aqueous KmnO solution, 1.0 g (5 mmol) of t-butyl ethyl methylenemalonate and 1.4 g thiol acetic acid (18.4 mmol) was heated under reflux for 12 hours. The mixture was allowed to cool, and the product was purified by silica gel column chromatography using ethyl acetate/petroleum ether (7:93) to afford 0.86 g (62%) of t-butyl ethyl acetylthiomethylmalonate as a colorless liquid.
1H NMR (CDC13, 200 MHz) δ 1.35 (t, 3H), 1.50 (s, 9H), 2.35 (m, 3H), 2.52 (m, 2H), 3.30
(m, IH), 4.20 (m, 2H).
Example 12
Methyl t-butyl 2-methylidenyl-l,3-dipropionate is reacted with thioacetic acid to form methyl t-butyl 2-acetyltbiomethyl-l,3-dipropionate. Methyl t-butyl 2-acetylthiomethyl-l,3- dipropionate is alkylated with potassium carbonate and methyl iodide in the presence of a phase transfer catalyst to form methyl t-butyl 2-acetylthiomethyl-2-methyl-l,3-dipropionate. The t-butyl group is hydrolyzed by acidifying the reaction mixture. The free carboxylic acid group produced by hydrolyzing the t-butyl group is converted to an amino group through a reaction with diphenylphosphoryl azide, thereby forming S-acetyl-2-methylcysteine methyl ester. The acetyl group is removed to form 2-methylcysteine methyl ester.
Example 13
(S)-Cysteine is reacted with benzoic acid to form 2-phenylthiazoline-4-carboxylic acid. 2-Phenylthiazoline-4-carboxylic acid is amidated with 4-benzyloxazolidone. The amidated 2-phenylthiazoline-4-carboxylic acid is alkylated with methyl iodide in the presence of TiCl4 and lithium diisopropylamide. The alkylated species is hydrolyzed by lithium hydroxide in methanol to obtain (S)-2-methylcysteine methyl ester.
Example 14
Cysteine, benzonitrile, and 5 equivalents of triethylamine were refluxed in ethanol for 6-8 hours to obtain a 66-70% yield of 2-phenylthiazoline-4-carboxylic acid. The 2- phenylthiazoline-4 carboxylic acid was reacted with 2.05 equivalents of base and 1 equivalent of methyl iodide in tetrahydrofuran at 0°C to fonn 2-phenyl-4-methylthiazoline-4 carboxylic acid. The 2-phenyl-4-methylthiazoline-4 carboxylic acid can be resolved and isolated as the (S)-enantiomer using emulsion crystallization, and subsequently hydrolyzed with hydrochloric acid, thereby obtaining (S)-2-methylcysteine hydrochloride.
Example 15
Cysteine, benzonitrile, and 5 equivalents of triethylamine were refluxed in ethanol for 6-8 hours to obtain a 66-70% yield of 2-phenylthiazoline-4-carboxylic acid. The 2- phenylthiazoline-4 carboxylic acid was reacted with 2.05 equivalents of base and 1 equivalent of methyl iodide in tetrahydrofuran at 0°C to form 2-phenyl-4-methylthiazoline-4 carboxylic acid. The 2-phenyl-4-methylthiazoline-4-carboxylic acid can be hydrolyzed with
hydrochloride acid, thereby obtaining a mixture of (R)- and (S)-2-methylcysteine hydrochloride.
Example 16
The procedure of Example 15 is followed, such that a mixture of (R)- and (S)-2- methylcysteine hydrochloride is obtained. Classical chemical resolution with (R)- phenylethylamine at a suitable pH is able to resolve the (R)- and (S)-enantiomers of 2- methylcysteine. Subsequent isolation of the resolved products yields substantially enantiomerically pure (R)-2-methylcysteine and (S)-2-methylcysteine.
Example 17
Preparation of 4,5-dihydro-2-(2,4-dihydroxyphenyl)-4-methylthiazole-4(S)-carboxylic acid
2,4-Dimethoxybenzonitrile, (R)-cysteine ethyl ester and about 5 equivalents of triethylamine are refluxed in ethanol for about 6-8 hours to obtain ethyl-2-(2,4- dimethoxyphenyl)-4,5-dihydiO-thiazole-4-(R)-carboxylate.
Ethyl-2-(2,4-dimethoxyphenyl)-4,5-dihydro-thiazole-4-(R)-carboxylate is refluxed with aqueous sodium hydroxide in methyl tertiary-butyl ether (MTBE) to form 2-(2,4- dimethoxyphenyl)-4,5-dihydro-thiazole-4-(R)-carboxylic acid. Next, 2-(2,4-dimethoxyphenyl)-4,5-dihydro-thiazole-4-(R)-carboxylic acid is reacted with isopropanol; DCC; and 4-(dimethylamino)pyridine (DMAP) in tetrahydrofuran (THF) at room temperature to fonn isopropyl-2-(2,4-dimethoxyphenyl)-4,5-dihydro-thiazole-4-(R)- carboxylate.
Isopropyl-2-(2,4-dimethoxyphenyl)-4,5-dihydro-thiazole-4-carboxylate is reacted with 50%) potassium hydroxide and excess methyl iodide in dichloromethane in the presence of
O(9)-allyl-N-9-anthracenylmethylcinchonidium bromide to form isopropyl-2-(2,4- dimethoxyphenyl)-4-methyl-4,5-dihydro-thiazole-4-(S)-carboxylate in enantiomeric excess.
The isopropyl-2-(2,4-dimethoxyphenyl)-4-methyl-4,5-dihydro-thiazole-4-(S)- carboxylate is purified by further resolving the enantiomers using emulsion crystallization. Isopropyl-2-(2,4-dimethoxyphenyl)-4-methyl-4,5-dihydro-thiazole-4-(S)-carboxylate is then reacted with excess hydrochloric acid to form 2-(2,4-dihydroxyphenyl)-4-methyl-4,5-dihydro- thiazole-4-(S)-carboxylic acid.
Example 18
Preparation of isopropyl 2-(4-methoxy-phenyl)- 4,5-dihydrothiazole-
4-benzyl-4-carboxylate
The C-4 benzylation of 2-(4-methoxy-phenyl)-4,5-dihydro-thiazole-4-carboxylic acid isopropyl ester (A) was performed as described below.
Room Temperature, KOH (50% aqueous solution)
Toluene, Benzyl bromide, Phase transfer catalyst (B)
(c) whe rein the phase transfer catalyst (B) is represented by the following structural formula:
0.25 g of 2-(4-methoxy-phenyl)-4,5-dihydro-thiazole-4-carboxylic acid isopropyl ester (A) and 23 mg of phase transfer catalyst (B) were added to 25 milliliters of toluene. To this mixture, 1 milliliter of aqueous KOH (50%) was added and the mixture was stined for approximately 15 minutes. 106 microliters of benzyl bromide (Avocado Research Chemicals Limited, Heysham, United Kingdom) was then added dropwise with stirring. The reaction was monitored using thin-layer chromatography (TLC) and left to stir at room temperature (about 32°C). About two days later, another 5 mole percent of catalyst was added to the reaction mixture.
After about 2 more hours, 5 milliliters of water were added to the reaction mixture, and the phases were separated and concentrated using a rotary evaporator. The organic phase was concentrated to approximately 0.5 milliliters. The reaction mixture was purified by column chromatography. 124 grams of product was collected for a yield of 41%. Infrared Spectioscopy, Proton Nuclear Magnetic Resonance (!H NMR), and 13C Attached Proton Test confirmed the structure of the product as isopropyl-2-(4-methoxy-phenyl)-4,5-dihydro- thiazole-4-benzyl-4-carboxylate (C).
Example 19
6 grams of isopropyl-2-(4-methoxy-phenyl)-4,5-dihydro-thiazole-4-benzyl-4- carboxylate (C) was produced as in Example 18 and dissolved in 10 milliliters trichloromethane. The solution was placed in a 10 cm polarimetry tube and the optical activity of the solution was measured using a polarimeter. Optical activity were taken in each run and the average was recorded as , observed rotation. Table 1 shows the average observed rotation for each of three runs.
Table 1 : Observed Rotation
The rotation of trichloromethane was +0.004 degrees. The overall average observed rotation conesponds to a specific rotation, [α]p , of -32.2 (degrees)(mL)/(dm)(g).
Given that the purified compound is optically active and that none of the asymmetric catalyst was observed in the Proton Nuclear Magnetic Resonance (1H NMR), and 13C Attached Proton Test of Example 18, it can be assumed that the benzylation of 2-(4-methoxy- phenyl)-4,5-dihydro-tlιiazole-4-carboxylic acid isopropyl ester (A) occurred in an enantioselective fashion and that the product produced is the (-)-enantiomer.
Example 20 Preparation of 2-methyl cysteine by aziridination
Synthesis of N-p-ToluenesuIphonyI-2-carbomentoxy-2-methylaziridine
Anhydrous CuCl2 (148 mg, 1.1 mmol) in acetonitrile (30 ml) was stined under nitrogen at room temperature. Methyl methacrylate (1.1 g, 1.17 ml, 11 mmol) was then added to this solution, followed by addition of Bromamine-T (1.79g, 5 mmol). The reaction mixture was stined at room temperature for 6-8 hours. It was diluted with ethyl acetate (100 ml) and filtered through a pad of silica gel. The clear solution was dried over sodium sulphate and the solvent was concentrated under vacuum. A thick colorless oil was obtained, which was purified by silica gel column chromatography (eluent: petroleum etheπethyl acetate 4:1). The reaction yielded 628.9 mg of product (40%).
Spectral data IR (Neat): 3020; 1741; 1331; 1215; 1163; 882; 759 cm'1.
1H NMR(CDC13), 200 MHz: δ 7.78 (d, J = 8Hz, 2H); 7.34 (d, J = 8 Hz, 2H); 3.72 (s, 3H); 3.50 (m, J=15 Hz, IH); 2.75 (d, J = 15 Hz, IH); 2.45 (s, 3H); 1.96 (s, 3H) Mass (m/e): 269 (6); 238 (12); 210 (9); 184 (7); 155 (25); 114 (100); 91 (98); 77 (10); 65 (60).
13C NMR(CDC13): δ 166.27; 143.75; 136.40; 129.16; 126.95; 52.26; 45.90; 38.22; 24.91;
20.87; 14.59. CH analysis Calculated for d2Hι5NO4S: C=53.53%; H=5.51%; N=5.20%, S=11.89% Found C=49.65%; H=5.81%; N=4.93%; S=12.52%
Synthesis of Methyl (2-N-tosyIamino-3-benzoylmercapto)propionate
M.W = 269 M.W = 269
C12H15NO4S C12H19NO5S2
N-p-Toluenesulphonyl-2-carbomethoxy-2-methyl aziridine (200 mg, 0.743 mmol) was added to a 5 ml two necked round bottom flask under nitrogen, then thioacetic acid (0.816 ml, 11.14 mmol) was added. The reaction mixture was heated at 80°C for 6 hours. It was then diluted with ethyl acetate (40 ml); the combined organic layer was washed with saturated sodium bicarbonate solution and dried. Removal of the solvent under vacuum yielded an oily residue, which was purified by column chromatography. The reaction yielded 51.30mg of product (39.9%).
Spectral data
IR (Neat): 3280; 1740; 1694; 1597; 1331; 1159 cm"1. 1H NMR(CDC13), 200 MHz: δ 7.72 (d, J = 8Hz, 2H); 7.3 (d, J = 8 Hz, 2H); 5.53 (s, 3H); 3.65 (s, 3H); 3.40 (q, 2H); 2.42 (2, 3H); 2.32 (s, 3H); 1.26 (s, 3H) Mass (m/e): 286 (4); 256 (4); 155 (77); 139 (12); 114 (10); 9 (100); 77 (3); 65 (13). 13C NMR(CDC13): δ 193.82; 171.54; 142.80; 138.50; 128.90; 126.40; 61.16; 52.45; 37.04; 29.69; 21.35; 20.83. CH analysis Calculated for Cι4Hι9NO5S2: C=48.91%; H-5.51%; N=4.06%, S=18.55% Found C=48.65%; H=5.27%; N=3.93%; S=19.25%
Synthesis of Methyl-(2-N-tosylamino-3-mercapto)propionate
M.W = 345 M.W = 303 C14H19N05S2 C12H17N04S2
Diy sodium metal (10 mg, 0.43 mmol) was placed in a two necked round bottom flask under nitrogen, then dry methanol (5 ml) was added at room temperature (25 °C) with stirring. After dissolving all sodium metal, methyl(2-N-tosylamino-3-acetomercapto) propionate (150 mg, 0.43 mmol) was added and the reaction mixture was stined at room temperature (25°C) for 6-7 hrs. Methanol was removed under pressure. A colorless oil was obtained, which was purified by column chromatography. The reaction yielded 65.86 mg of product (50%).
Spectral data
FT IR (CHC13): 3020; 1738; 1215; 1158; 768; 668 cm-1 1H NMR (D2O): δ 7.75 (d, J = 8 Hz, 2H); 7.26 (d, J = 8 Hz); 5.87 (s, 3H); 3.70 (s, 3H); 3.25 (q, 2H); 2.42 (s, 3H); 2.03 (s, 3H); 1.49 (s, 3H). 13C NMR (CDCI3): δ 172.17; 143.35; 139.27; 129.49; 126.92; 66.22; 53.07; 49.39; 22.38; 21.39
Synthesis of 2-MethyI-Cysteine
M.W = 303 M.W = 135 C12H17N04S2 C4H9N02S
Methyl (2-N-tosylamino-3 -mercapto) propionate (100 mg, 0.33 mmol), phenol
(94.05 mg, 0.99 mmol), and 10 ml of 32% hydrogen bromide in acetic acid were charged in a thick walled glass tube. It was sealed and heated in a metallic bomb for 12 hours at 80°C. The reaction mixture was allowed to cool to room temperature and then was poured into 60 ml of ether and stined for 5 min. The ether solution was decanted and the residue was dissolved in 2 ml of water. This aqueous solution was stined with charcoal and filtered. The filtrate was passed through a Dowex (1x4-50) bed and washed with 3 ml of water. The aqueous solution was concentrated under vacuum at room temperature to obtain 2-methyl cysteine as a sticky mass. The reaction yielded 31.18 mg of product (70%).
Spectral data
FT IR (nujol): 3615.75; 2542.40; 1611.33; 1511.84 cm-1
1H NMR (D2O): δ 3.31 (IH, m-CH2); 2.47 (IH, m-CH2); 1.37 (3H, s-CH3); 1.44 (IH, s-
SH). HPLC (Lichrosphere RP- 18) : 97.11 %
Alternative Synthesis of 2-Methyl-Cysteine
M.W = 345 M.W = 135 C14H19N05S2 C4H9N02S
Methyl (2-N-tosylamino-3-mercapto) propionate (100 mg, 0.28 mmol), phenol (81.73 mg, 0.86 mmol), and 10 ml of 32% hydrogen bromide in acetic acid were charged in a thick walled glass tube. It was sealed and heated in a metallic bomb for 12 hours at 80°C. The reaction mixture was allowed to cool to room temperature and then was poured into 60 ml of ether and stined for 5 min. The ether solution was decanted and the residue was dissolved in 2 ml of water. This aqueous solution was stined with charcoal and filtered. The filtrate was passed through a Dowex (1x4-50) bed and washed with 3 ml of water. The aqueous solution was concentrated under vacuum at room temperature to obtain 2-methyl cysteine as a sticky mass. The reaction yielded 28.17 mg of product (72%).
Spectral data
FT IR (nujol): 3615.75; 2542.40; 1611.33; 1511.84 cm-1
1H NMR (D2O): δ 3.31 (IH, m-CH2); 2.47 (IH, m-CH2); 1.37 (3H, s-CH3); 1.44 (IH, s- SH).
HPLC (Lichrosphere RP-18): 97.11%
Example 21
2,4-Dibenzyloxybenzonitrile (0.121 mol) was dissolved in 5.85 g (0.127 mol) ethanol and 19.4 ml 1,2-dimethoxyethane in a double walled reactor. This solution was cooled to -5 °C, stined and saturated with dry HCl gas over 5 hours at 0-3 °C. The reaction mixture was stined overnight at 2-4 °C under nitrogen. During this time, a product crystallized. The white crystals were filtered off, washed with 1,2-dimethoxyethane (5 °C,
three times each with 13 ml) and dried. A total of 30 g of the protected ethyl benzimidate was isolated (Yield 88.4%, purity 98.9%).
The protected ethyl benzimidate described above was dissolved in methanol to generate a 10% solution and was catalytically hydrogenated at room temperature using 5% Pd/C as a catalyst. The reaction was completed after 8 hours. The solution was filtered and the solvent evaporated to yield the deprotected product as an orange-yellow solid. The reaction yielded 19.6 g (94%) of product. hi contrast, the formation of the imidate with 2,4 dihydroxybenzonitrile was a low yielding process, generating the desired product in only 20% yield and with less than desired purity. While this invention has been particularly shown and described with references to prefened embodiments thereof, it will be understood by those skilled in the art that various changes in form and details maybe made therein without departing from the scope of the invention encompassed by the appended claims.