WO2004005252A1 - Azabicyclo derivatives as muscarinic receptor antagonists - Google Patents
Azabicyclo derivatives as muscarinic receptor antagonists Download PDFInfo
- Publication number
- WO2004005252A1 WO2004005252A1 PCT/IB2003/001367 IB0301367W WO2004005252A1 WO 2004005252 A1 WO2004005252 A1 WO 2004005252A1 IB 0301367 W IB0301367 W IB 0301367W WO 2004005252 A1 WO2004005252 A1 WO 2004005252A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- hydroxy
- formula
- azabicyclo
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(C1C2****C(NC(*)(*)*)=O)NC(*)[C@]12N=C Chemical compound *C(C1C2****C(NC(*)(*)*)=O)NC(*)[C@]12N=C 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention generally relates to muscarinic receptor antagonists which are useful, among other uses, for the treatment of various diseases of the respiratory, urinary and gastrointestinal systems mediated through muscarinic receptors.
- the invention relates to derivatives of azabicyclo compounds, including, for example, 6- substituted azabicyclo[3.1.0] hexanes, 2,6- and 4,6-disubstituted derivatives, and 2,4,6- trisubstituted derivatives, as well as pharmaceutical compositions containing such compounds and methods of treating diseases mediated through muscarinic receptors.
- Muscarinic receptors as members of the G Protein Coupled Receptors are composed of a family of 5 receptor sub-types (Mi, M 2 , M 3 , i and M 5 ) and are activated by the neurotransmitter acetylcholine. These receptors are widely distributed on multiple organs and tissues and are critical to the maintenance of central and peripheral cholinergic neurotransmission. The regional distribution of these receptor sub-types in the brain and other organs has been documented.
- the M] subtype is located primarily in neuronal tissues such as cereberal cortex and autonomic ganglia, the M 2 subtype is present mainly in the heart where it mediates cholinergically induced bradycardia, and the M 3 subtype is located predominantly on smooth muscle and salivary glands (Nature, 323, p.411 (1986); Science, 237, p.527 (1987)).
- Muscarinic agonists such as muscarine and pilocarpine and antagonists such as atropine have been known for over a century, but little progress has been made in the discovery of receptor subtype-selective compounds, making it difficult to assign specific functions to the individual receptors.
- classical muscarinic antagonists such as atropine are potent bronchodilators, their clinical utility is limited due to high incidence of both peripheral and central adverse effects such as tachycardia, blurred vision, dryness of mouth, constipation, dementia, etc.
- azabicyclo derivatives including, for example, 6-substituted azabicyclo[3.1.0]hexanes, 2,6- and 4,6-disubstituted derivatives, and 2,4,6-trisubstituted derivatives, are provided as muscarinic receptor antagonists which can be useful as safe and effective therapeutic or prophylactic agents for the treatment of various diseases of the respiratory, urinary and gastrointestinal systems. Also provided are processes for synthesizing such compounds.
- compositions containing such compounds are provided together with acceptable carriers, excipients or diluents which can be useful for the treatment of various diseases of the respiratory, urinary and gastrointestinal systems.
- the enantiomers, diastereomers, N-oxides, polymorphs, pharmaceutically acceptable salts and pharmaceutically acceptable solvates of these compounds as well as metabolites having the same type of activity are also provided, as well as pharmaceutical compositions comprising the compounds, their metabolites, enantiomers, diastereomers, N-oxides, polymorphs, solvates or pharmaceutically acceptable salts thereof, in combination with a pharmaceutically acceptable carrier and optionally included excipients.
- Ar represents an aryl or a heteroaryl ring having 1-2 hetero atoms selected from the group consisting of oxygen, sulphur and nitrogen atoms, the aryl or heteroaryl rings may be unsubstituted or substituted by one to three substituents independently selected from lower alkyl (C ⁇ -C 4 ), lower perhalo alkyl (C ⁇ -C 4 ), cyano, hydroxy, nitro, lower alkoxy (C ⁇ -C 4 ), lower perhalo alkoxy (C ⁇ -C 4 ), unsubstituted amino, N-lower alkyl (Ci- C 4 ) or -aryl amino, amino carbonyl, or N-lower alkyl (Cj-C 4 ) or -aryl amino carbonyl;
- Ri represents a hydrogen, hydroxy, hydroxy methyl, substituted or unsubstituted amino, alkoxy, carbamoyl or halogen (e.g. fluorine, chlorine, bromine and iodine);
- R 2 represents alkyl, C -C 7 cycloalkyl ring, a C -C 7 cyclo alkenyl ring, an aryl, heterocyclic or a heteroaryl ring having 1 to 2 hetero atoms selected from a group consisting of oxygen, sulphur and nitrogen atoms; the aryl, heteroaryl, heterocyclic or a cycloalkyl ring may be unsubstituted or substituted by one to three substituents independently selected from lower alkyl ( -C 4 ), lower perhalo alkyl (C ⁇ -C 4 ), cyano, hydroxy, nitro, lower alkoxycarbonyl, halogen, lower alkoxy (C ⁇ -C 4 ), lower perhalo alkoxy (C ⁇ -C ), unsubstituted amino, N-lower alkyl (C ⁇ -C 4 ) or -aryl amino, amino carbonyl, or N-lower alkyl (C ⁇ -C 4 ) or -
- W represents (CH 2 ) P , wherein p represents 0 to 1 ;
- X represents an oxygen, sulphur, -NR or no atom, wherein R represents hydrogen or (C ⁇ - 6 ) alkyl;
- Y represents CHR 5 CO or (CH )q wherein R 5 represents hydrogen or methyl and q represents 0 to 4;
- Z represents oxygen, sulphur, or NR 10 , wherein R ⁇ 0 represents hydrogen, or C ⁇ -6 alkyl;
- Q represents (CH 2 ) n (wherein n represents 0 to 4), CHR 8 (wherein R 8 represents H, OH, C ⁇ -6 , alkyl, C ⁇ - 6 alkenyl, C ⁇ - 6 alkoxy) or CH 2 CHR 9 (wherein R represents H, OH, lower alkyl (C C ) or lower alkoxy (Cj-C 4 )); and
- R 6 and R 7 are independently selected from H, CH 3 , COOH, CONH 2 , NH 2 , and CH 2 NH 2 .
- Formula V in accordance with a sixth aspect, there is provided a method for treatment or prophylaxis of an animal or a human suffering from a disease or disorder of the respiratory, urinary and gastrointestinal systems, wherein the disease or disorder is mediated through muscarinic receptors.
- the method includes administration of at least one compound having the structure of Formula I.
- a method for treatment or prophylaxis of an animal or a human suffering from a disease or disorder associated with muscarinic receptors comprising administering to a patient in need thereof, an effective amount of a muscarinic receptor antagonist compound as described above.
- a method for treatment or prophylaxis of an animal or a human suffering from a disease or disorder of the respiratory system such as bronchial asthma, chronic obstructive pulmonary disorders (COPD), pulmonary fibrosis, and the like; urinary system which induce such urinary disorders as urinary incontinence, lower urinary tract symptoms (LUTS), etc.; and gastrointestinal system such as irritable bowel syndrome, obesity, diabetes and gastrointestinal hyperkinesis with compounds as described above, wherein the disease or disorder is associated with muscarinic receptors.
- a disease or disorder of the respiratory system such as bronchial asthma, chronic obstructive pulmonary disorders (COPD), pulmonary fibrosis, and the like
- urinary system which induce such urinary disorders as urinary incontinence, lower urinary tract symptoms (LUTS), etc.
- gastrointestinal system such as irritable bowel syndrome, obesity, diabetes and gastrointestinal hyperkinesis with compounds as described above, wherein the disease or disorder is associated with muscarinic receptors
- the compounds described herein exhibit significant potency in terms of their activity, as determined by in vitro receptor binding and functional assays and in vivo experiments using anaesthetized rabbits.
- the compounds that were found active in vitro were tested in vivo.
- Some of the compounds are potent muscarinic receptor antagonists with high affinity towards M 3 receptors. Therefore, pharmaceutical compositions for the possible treatment for the disease or disorders associated with muscarinic receptors are provided.
- the compounds can be administered orally or parenterally.
- the compounds of Formula I may be prepared, for example, by the reaction sequence as shown in Scheme I.
- the preparation comprises reacting a compound of Formula VII with a compound of Formula VI, wherein
- Ar represents an aryl or a heteroaryl ring having 1-2 hetero atoms (such as oxygen, sulphur or nitrogen atoms), where the aryl or heteroaryl rings may be unsubstituted or substituted by one to three substituents independently selected from lower alkyl (C ⁇ -C 4 ), lower perhalo alkyl (C ⁇ -C 4 ), cyano, hydroxy, nitro, lower alkoxy (Ci- C 4 ), lower perhalo alkoxy (C ⁇ -C 4 ), unsubstituted amino, N-lower alkyl (C ⁇ -C 4 ) or -aryl amino, amino carbonyl, or N-lower alkyl (C ⁇ -C 4 ) or -aryl amino carbonyl; Ri represents a hydrogen, hydroxy, hydroxy methyl, substituted or unsubstituted amino, alkoxy, carbamoyl or halogen (e.g. fluorine, chlorine, bromine and iodine);
- R 2 represents alkyl, C 3 -C 7 cycloalkyl ring, a C 3 -C 7 cyclo alkenyl ring, an aryl, heterocyclic or a heteroaryl ring having 1 to 2 hetero atoms; the aryl, heteroaryl, heterocyclic or a cycloalkyl ring may be unsubstituted or substituted by one to three substituents independently selected from lower alkyl (C ⁇ -C 4 ), lower perhalo alkyl (C ⁇ -C 4 ), cyano, hydroxy, nitro, lower alkoxycarbonyl, halogen, lower alkoxy (CrC 4 ), lower perhalo alkoxy (C ⁇ -C 4 ), unsubstituted amino, N-lower alkyl (C]-C 4 ) or -aryl amino, amino carbonyl, or N-lower alkyl (C ⁇ -C 4 ) or -aryl amino carbonyl;
- W represents (CH 2 ) P , wherein p represents 0 to 1 ;
- X represents an oxygen, sulphur, -NR or no atom, wherein R represents hydrogen or (Ci-6) alkyl;
- Y represents CHR 5 CO or (CH 2 )q wherein R 5 represents hydrogen or methyl and q represents 0 to 4;
- Z represents oxygen, sulphur, or NR ⁇ 0 , wherein R ]0 represents hydrogen, C] -6 or alkyl;
- Q represents -(CH ) n - (wherein n represents 0 to 4), CHR 8 (wherein R 8 represents H, OH, Ci- 6 , alkyl, C ⁇ - 6 alkenyl, C ⁇ - 6 alkoxy) or CH CHR 9 (wherein R 9 represents H, OH, lower alkyl (C ⁇ -C 4 ) or lower alkoxy (C ⁇ -C 4 )); ⁇ and R 7 are independently selected from H, CH 3 , COOH, CONH 2 , NH 2 , and CH 2 NH 2 ; and
- P is any protecting group for an amino group, for example, benzyl or t-butyloxy carbonyl groups.
- the reaction between a compound of Formula VII and a compound of Formula VI can take place in the presence of a condensing agent (for example, l-(3- dimethylaminopropyl)-3 -ethyl carbodiimide hydrochloride (EDC) or 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU)), in a solvent (such as N,N-dimethylformamide, dimethylsulfoxide, toluene, or xylene, at temperatures ranging from about 0 to about 140°C), to give a protected compound of Formula VIII which on deprotection in the presence of a deprotecting agent (for example, palladium on carbon, trifluoroacetic acid (TFA) or hydrochloric acid) in an organic solvent (for example, methanol, ethanol, tetrahydrofuran or acetonitrile, at temperatures ranging from about 10 to about 50°C) gives an unprotected compound of Formula I.
- salts examples include pharmacologically acceptable salts such as inorganic acid salts (for example, hydrochloride, hydrobromide, sulphate, nitrate and phosphate), organic acid salts (for example, acetate, tartarate, citrate, fumarate, maleate, tolounesulphonate and methanesulphonate).
- inorganic acid salts for example, hydrochloride, hydrobromide, sulphate, nitrate and phosphate
- organic acid salts for example, acetate, tartarate, citrate, fumarate, maleate, tolounesulphonate and methanesulphonate.
- carboxyl groups When carboxyl groups are included in the Formula I as substituents, they may be present in the form of an alkaline or alkali metal salt (for example, sodium, potassium, calcium, magnesium, and the like).
- alkaline or alkali metal salt for example, sodium, potassium, calcium, magnesium, and the like.
- the compounds described herein may be administered to an animal for treatment orally, or by a parenteral route.
- the pharmaceutical compositions described herein can be produced and administered in dosage units, each unit containing a certain amount of at least one compound described herein and/or at least one physiologically acceptable addition salt thereof.
- the dosage may be varied over extremely wide limits as the compounds are effective at low dosage levels and relatively free of toxicity.
- the compounds may be administered in the low micromolar concentration, which is therapeutically effective, and the dosage may be increased as desired up to the maximum dosage tolerated by the patient.
- the compounds described herein can be produced and formulated as their enantiomers, diastereomers, N-Oxides, polymorphs, solvates and pharmaceutically acceptable salts, as well as metabolites having the same type of activity.
- Pharmaceutical compositions comprising the molecules of Formulae I, II, III, IV and V or metabolites, enantiomers, diastereomers, N-oxides, polymorphs, solvates or pharmaceutically acceptable salts thereof, in combination with pharmaceutically acceptable carrier and optionally included excipient can also be produced.
- reaction mixture was treated with hydroxy benzotriazole (29.9 mmol, 4.04 gm) followed by N-methyl morpholine (54.4 mmol, 5.2 gm) was stirred at 0°C for 0.5 hours.
- EDC l-[3-(dimethylamino)propyl]-3-ethyl carbodiimide hydrochloride (29.9mmol, 5.7 gms) was added and the reaction mixture was stirred at 0° C for 1 hour and further at room temperature (RT) overnight.
- the reaction mixture was poured into saturated sodium bicarbonate and extracted with ethyl acetate. The organic layers were washed with water and dried over sodium sulphate and concentrated under reduced pressure.
- Step b Preparation of (2R,2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cyclopentyI-2-phenyl acetamide
- the mixture was cooled to room temperature and the reaction mixture was filtered through a bed of hyflo.
- the hyflo bed was washed with methanol (75.0 ml), ethyl acetate (25. 0ml) and water (25.0 ml).
- the filtrate was concentrated under vaccum.
- the residue was diluted with water and pH of the resulting solution was adjusted to pH ⁇ 14 with IN NaOH.
- the solution was extracted with ethyl acetate (2x50 ml) and the ethyl acetate layer was washed with water and brine solution.
- the layer was dried over anhydrous Na 2 SO 4 and concentrated to give the title compound as solid in 96.2% (0.75g, 2.39 mmol) yield with >98 % purity by HPLC.
- Step a Synthesis of (2R)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyi-3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide
- Step a This compound was synthesised following the procedure of Example 1, Step a, using (2R)-2-hydroxy-2-cyclopentyl-2-phenyl acetic acid (synthesised as in Grover et. al, J. Ore. Chem.. 2000; 65:6283-6287), instead of 2-hydroxy-2-cyclopentyl-2-phenyl acetic acid.
- Step b Synthesis of (2R)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6-
- This compound was synthesised following the procedure of Example 1, Step b, using (2R)- (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6(aminomethyl)-yl]-2- hydroxy-2-cyclopentyl-2-phenyl acetamide instead of (2R,2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide.
- the enantiomeric excess (ee) was determined by HPLC (Chinacel OD, mobile phase 90% hexane/10% EtOH/0.1% TFA) by observing the (S) and (R) isomers.
- the (S) isomer elutef at approximately 11.11 min.
- the (R) isomer eluted at approximately 11.81 min.
- the optical purity was > 99%.
- the compound exhibited a melting point of 150.2°C. Infrared spectral data showed ( DCM): 1653.8 cm "1 .
- Example 2 using (2R)-(l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-azabicylo[3.1.0]hexyl-6-(aminomethyl)-yl]-2- hydroxy-2-cyclopentyl-2-phenyl acetamide instead of (2R,2S) (lot, 5 ⁇ , 6 ⁇ )-N-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide.
- Step a Synthesis of (2S)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cycIopentyl-2-phenyI acetamide
- This compound was synthesized following the procedure of Example 1, Step a, using (2S)-2-hydroxy-2-cyclopentyl-2-phenyl acetic acid (synthesised as in Grover et. al, J. Org. Chem., 2000; 65:6283-6287), instead of 2-hydroxy-2-cyclopentyl-2-phenyl acetic acid.
- Step b Synthesis of (2S)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicycIo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyI acetamide
- This compound was synthesized following the procedure of Example 1, Step b, using (2S)-(l ⁇ ,5 ⁇ ,6 )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6(aminomethyl)-yl]-2- hydroxy-2-cyclopentyl-2 -phenyl acetamide instead of (2R,2S) (l ⁇ ,5 ⁇ ,6 )-N-[3-benzyl-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2 -phenyl acetamide.
- the ee was determined by HPLC (Chinacel OD, mobile phase 90% hexane/10% EtOH/0.1% TFA) by observing the (S) and (R) isomers.
- the (S) isomer eluted at approximately 11.11 min.
- the (R) isomer eluted at approximately 11.81 min.
- the optical purity was > 99%.
- Example 2 using (2S)-(l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-azabicylo[3.1.0]hexyl-6-(aminomethyl)-yl]-2- hydroxy-2-cyclopentyl-2-phenyl acetamide instead of (2R,2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide.
- Example 7 Preparation of (2R.
- Step b Synthesis of (2R, 2S) 2-methoxy-2-cyclopentyl-2-phenyI acetic acid
- Step c Preparation of (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0] hexyl-6-(aminomethyI)-yl]-2-methoxy-2-cyciopentyl-2-phenyl acetamide
- Step d Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-methoxy-2-cyclopentyl-2-phenyl acetamide
- Step a Synthesis of (2R, 2S)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-
- Step b Synthesis of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cycloheptyl-2-phenyl acetamide
- This compound was synthesized following the procedure of Example 1, step b, using (2R, 2S)-(l ⁇ -5 ⁇ , 6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethy ⁇ )-y ⁇ ]- 2-hydroxy-2-cycloheptyl-2-phenyl acetamide instead of (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3- benzyl-3-azabicyclo[.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide in 90% yield.
- the compound had
- This compound was synthesized following the procedure of Example 1, step a, using (2R, 2S) 2-hydroxy-2-cyclobutyl-2-phenyl acetic acid (synthesized as per reported procedure of Saul B. Kadin and Joseph G.Cannon., J. Org. Chem., 1962; 27:240-245), instead of 2-hydroxy-2-cyclopentyl-2-phenyl acetic acid.
- Step b Synthesis of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-cyclobutyl-2-phenyl acetamide
- This compound was synthesized following the procedure of Example 1, step b, using (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]- 2-hydroxy-2-cyclobutyl-2-phenyl acetamide instead of (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3- benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide to give the title compound with 90.6% purity by HPLC.
- Example 10 Preparation of (2R. 2S) (l ⁇ .5 ⁇ .6 ⁇ -N-[3-azabicvclo 3.1.01hexyl-6- (aminomethyl -yl ⁇
- Step b Preparation of (2R,5R)-2-tert-butyl-5-[(lRorlS)-3-oxocyclopentyl]-5- phenyl-1 ,3-dioxalan-4-one
- Step d Preparation of (2R)- [(lSorlR)-3,3-difluorocycIopentyl]-2- hydroxy- 2-phenyIaceticacid
- MeOH MeOH
- Step f Preparation of (2R)- (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-
- Step g Synthesis of (2R) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6-
- Step-a Preparation of (2R,2S)-2-tert-butyl-5-[(lR or IS, 3R or 3S)-3-hydroxy cyclopentyl]-5-phenyl-l,3-dioxaIan-4-one.
- Step-b Preparation of (2R, 2S)-2-tert-butyl-5-[lR or IS, 3R or 3S]-3- fluorocyclopentyl]-5-phenyl-l,3-dioxolan-4-one.
- Step-c Preparation of (2R, 2S)-[(1R or IS, 3R or 3S]-3-fluorocycIopentyl]-2- hydroxy-2-phenylacetic acid.
- the compound was synthesised following the procedure of Example 11, step-d using (2R, 2S, 5R)-2-tert-butyl-5-[(lR or IS, 3R or 3S)-3-fluorocyclopentyl]-5-phenyl- l,3-dioxolan-4-one instead of (2R, 5R)-2-tert-butyl-5-[(lR or lS)-3,3- difluorocyclopentyl]-5-phenyl- 1 ,3-dioxolan-4-one.
- Step-d Preparation of (2R, 2S)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-[lR or IS, 3R or 3S]-3- fluorocyclopentyl]-2-hydroxy-2-phenyIacetamide.
- This compound was synthesized following the procedure of Example 1, step a, using the acid synthesized in the above step-c, instead of 2-hydroxy-2-cyclopentyl-2- phenyl acetic acid.
- Step-e Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-(3-fluorocyclopentyl)-2-phenyl acetamide
- This compound was synthesized following the procedure of Example 1, step b, using (2R, 2S)-(l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]- 2-[lR or IS, 3R or 3S]-3-fluorocyclopentyl]-2-hydroxy-2-phenylacetamide instead of (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2- hydroxy-2-cyclopentyl-2-phenyl acetamide.
- the optical purity was 87.27% (HPLC).
- This compound was prepared following the procedure of Example 11, by using (2R, 2S) [(l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-benzyl-3-azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2- hydroxy-2-(3,3-difluorocyclo-pentyl)-2-phenyl acetamidejinstead of (2R) [(l ⁇ , 5 ⁇ , 6 ⁇ )-
- Example 15 Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-r3-azabicvclor3.1.0 "
- Step a Synthesis of (2R, 2S) (l ⁇ , 5 ⁇ , 6 ⁇ )-N-[3-benzyl-3- azabicyclo[3.1.0]hexyl-6-(methyl)-yl]-2-hydroxy-2,2-diphenyl acetate.
- Step b Synthesis of (2R, 2S) (1 ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo [3.1.0] hexyI-6- (methyl)-yl]-2-hydroxy-2,2-diphenyl acetate
- Step a Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-benzyl-3- azabicyclo[3.1.0]hexyl-6-(aminomethyl)-yl]-2-hydroxy-2,2-diphenyl acetamide. It was prepared following the procedure of Example 1, step a by using 2-hydroxy-
- Step b Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo [3.1.0] hexyl-6- (aminomethyl)-yl]-2-hydroxy-2-(3,3-difluorocyclopentyl)-2-phenyl acetamide
- Step a Synthesis of (l ⁇ ,5 ⁇ ,6 ⁇ )-N- [3-benzyl-3-azabicyclo [3.1.0] hexyl-6-
- Step b Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hexyl-6- (aminomethyl)-yl]-2-cyclohexyl-2-phenyl acetamide
- Step a Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-tert-butyloxycarbonyl-3- azabicyclo[3.1.0]-hexyl-6-yl methyl]-2-hydroxy-2-cyclopentyl-2-phenyl acetamide
- Step b Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-tert-butyloxycarbonyl-3- azabicyclo[3.1.0]hexyl-6-yl methyl)-2-(3-silyltrimethyloxy)-2-cyclopentyl-2-phenyl acetamide
- Boc-derivative 2.0 g, 4.8 mmol
- imidazole 1.2 g, 16.9 mmol
- trimethylsilyl chloride (1.54 ml, 12.0 mmol
- Step c Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-tert-butyloxycarbonyl-3- azabicyclo[3.1.0]hex-6-yl methyl)-2-cyclopentyl-2-hydroxy-N-methyl-2-phenyl acetamide
- Step d Preparation of (2R, 2S) (l ⁇ ,5 ⁇ ,6 ⁇ )-N-[3-azabicyclo[3.1.0]hex-6- ylmethyl)-2-cyclopentyl-2-hydroxy-N-methyl-2-phenyl acetamide
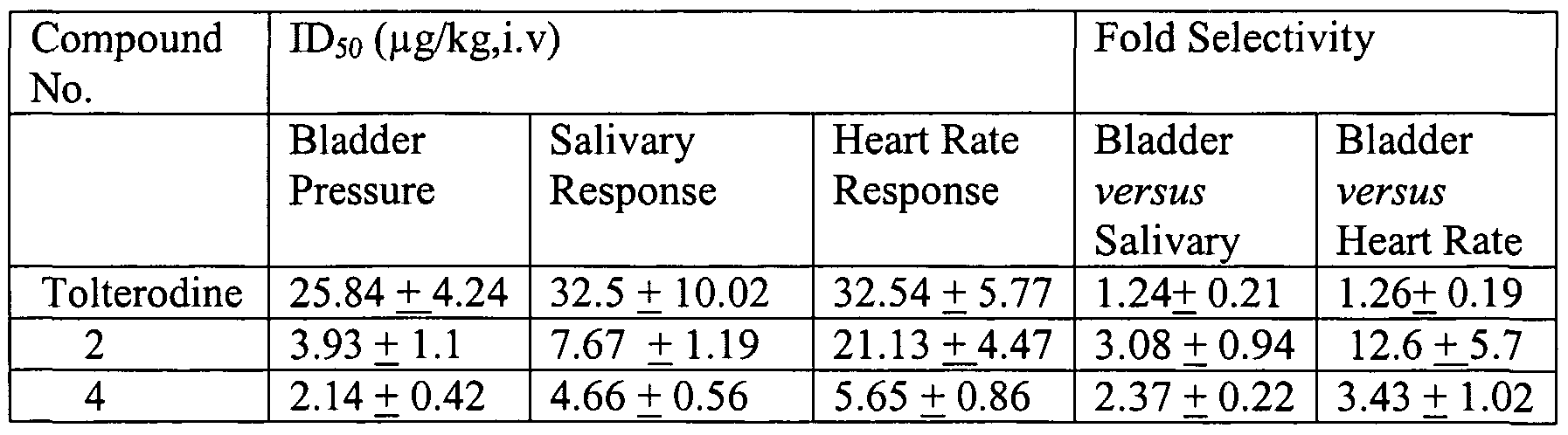
- Biological Activity Radioligand Binding Assays The affinity of test compounds for M 2 and M 3 muscarinic receptor subtypes was determined by [ H]-N-methylscopolamine binding studies, using rat heart and submandibular gland, respectively, as described by Moriya et al., (Life Sci.. 1999; 64(25):2351-2358) with minor modifications as follows. The membrane preparation was done with the following modifications: a low spin step of 500g for 10 minutes at 4°C was used; the buffer was 20 mM HEPES, 10 mM EDTA, at pH 7.4; the high speed spin was done at 40,000g and the homogenate was passed through a filter gauge before any spinning.
- the assay conditions were modified as follows: the assay volume was 250 ⁇ L; the incubation time was 3 hours; the PE concentration was 0.1%; the filtermat used was GF/B from Wallac; the scintillant used was Supermix from Wallac; the amount of scintillant was 500 ⁇ L/well; and the counter used was a 1450 microbeta PLUS, from Wallac.
- Membrane preparation Submandibular glands and heart were isolated and placed in ice cold homogenising buffer (HEPES 20mM, lOmM EDTA, pH 7.4) immediately after sacrifice. The tissues were homogenised in 10 volumes of homogenising buffer and the homogenate was filtered through two layers of wet gauze and filtrate was centrifuged at 500g for lOmin. The supernatant was subsequently centrifuged at 40,000g for 20 min. The pellet thus obtained was resuspended in same volume of assay buffer (HEPES 20 mM, EDTA 5mM, pH 7.4) and were stored at -70°C until the time of assay.
- Ligand binding assay The compounds were dissolved and diluted in DMSO.
- the membrane homogenates (150-250 ⁇ g protein) were incubated in 250 ⁇ l of assay buffer (HEPES 20 mM, pH 7.4) at 24-25°C for 3h. Non-specific binding was determined in the presence of 1 ⁇ M atropine. The incubation was terminated by vacuum filtration over GF/B fiber filters (Wallac). The filters were then washed with ice cold 50mM Tris HCl buffer (pH 7.4). The filter mats were dried and bound radioactivity retained on filters was counted. The IC 5Q and Kd were estimated by using the non-linear curve fitting program using G Pad Prism software.
- Kj IC 50 /(1+L/Ka), where L is the concentration of [ 3 H]NMS used in the particular experiment.
- pK, -[log K,]
- Test compound and saline were infused intravenously via the femoral vein.
- the bladder was exposed through a midline laparotomy and both the ureters were identified, carefully separated and ligated.
- the ureters were incised proximally to allow free flow of urine from the kidney to the exterior.
- Bladder neck was gently held and the urethra was traced and separated from the adjoining tissues.
- PE canula was introduced into the bladder and ligated.
- the bladder was drained and subsequently filled with 15ml of warm saline (37°C).
- the other end of the intravesical catheter was connected to the Grass model 7D polygraph through a Statham P10 EZ pressure transducer to monitor the bladder pressure. Care was taken to keep the exposed area moist and warm. A period of 30-60 min was allowed for stabilization of parameters subsequent to surgery. Salivation response was assessed by placing preweighed absorbent cotton gauze in the buccal cavity for 2 minutes after carbachol administration.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Nutrition Science (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Medicines Containing Plant Substances (AREA)
Abstract
Description











Claims














Priority Applications (34)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SI200330515T SI1551803T1 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| CA002491998A CA2491998A1 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| BR0312572-6A BR0312572A (en) | 2002-07-08 | 2003-04-11 | Azabicyclic derivatives as muscarinic receptor antagonists |
| DE60309057T DE60309057T2 (en) | 2002-07-08 | 2003-04-11 | AZABICYCLODERIVATES AS ANTAGONISTS OF THE MUSCARIN RECEPTOR |
| EA200500172A EA007932B1 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives derivatives as muscarinic receptor antagonists |
| EP03762827A EP1551803B8 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| JP2004519035A JP2005535655A (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| DK03762827T DK1551803T3 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| NZ537585A NZ537585A (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| US10/520,572 US7544708B2 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| AU2003226579A AU2003226579B2 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| MXPA05000435A MXPA05000435A (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists. |
| HK06100635.5A HK1082728B (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
| BRPI0409302-0A BRPI0409302A (en) | 2003-04-11 | 2004-01-06 | azabicyclic derivatives as muscarinic receptor antagonists, method for their preparation and pharmaceutical composition containing them |
| CA002522071A CA2522071A1 (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| CNB2004800144712A CN100436414C (en) | 2003-04-11 | 2004-01-06 | Azabicyclic derivatives as muscarinic receptor antagonists |
| PCT/IB2004/000008 WO2004089900A1 (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| EP04700287A EP1626957A1 (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| US10/552,503 US20070021487A1 (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| NZ542952A NZ542952A (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| JP2006506251A JP2006522787A (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| AU2004228452A AU2004228452A1 (en) | 2003-04-11 | 2004-01-06 | Azabicyclo derivatives as muscarinic receptor antagonists |
| EA200501593A EA009387B1 (en) | 2003-04-11 | 2004-01-06 | Azabicycloderivatives as muscarinic receptor antagonists |
| US10/552,455 US7446123B2 (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| JP2006506252A JP2006522788A (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| CA002521989A CA2521989A1 (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| BRPI0409308-9A BRPI0409308A (en) | 2003-04-11 | 2004-01-07 | azabicyclic derivatives as muscarinic receptor antagonists, pharmaceutical composition and processes for their preparation |
| CNA2004800145024A CN1794985A (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| EA200501595A EA009942B1 (en) | 2002-07-08 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| PCT/IB2004/000012 WO2004089364A1 (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| AU2004228760A AU2004228760A1 (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| NZ542951A NZ542951A (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| EP04700488A EP1620087A1 (en) | 2003-04-11 | 2004-01-07 | Azabicyclo derivatives as muscarinic receptor antagonists |
| CY20061101846T CY1105879T1 (en) | 2002-07-08 | 2006-12-21 | AZADICYCLE DERIVATIVES AS MUSCARINIC RECEPTOR ANTAGONISTS |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2002/002663 WO2004004629A2 (en) | 2002-07-08 | 2002-07-08 | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
| IBPCT/IB02/02663 | 2002-07-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004005252A1 true WO2004005252A1 (en) | 2004-01-15 |
Family
ID=30011691
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2002/002663 Ceased WO2004004629A2 (en) | 2002-07-08 | 2002-07-08 | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
| PCT/IB2003/001367 Ceased WO2004005252A1 (en) | 2002-07-08 | 2003-04-11 | Azabicyclo derivatives as muscarinic receptor antagonists |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2002/002663 Ceased WO2004004629A2 (en) | 2002-07-08 | 2002-07-08 | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
Country Status (19)
| Country | Link |
|---|---|
| US (2) | US7399779B2 (en) |
| EP (2) | EP1546099B1 (en) |
| JP (2) | JP2006502985A (en) |
| CN (2) | CN1668585A (en) |
| AT (2) | ATE419236T1 (en) |
| AU (2) | AU2002345266B2 (en) |
| BR (2) | BR0215801A (en) |
| CA (2) | CA2492121A1 (en) |
| CY (1) | CY1105879T1 (en) |
| DE (2) | DE60230683D1 (en) |
| DK (1) | DK1551803T3 (en) |
| EA (3) | EA200500173A1 (en) |
| ES (1) | ES2274275T3 (en) |
| MX (2) | MXPA05000434A (en) |
| NZ (2) | NZ537584A (en) |
| PT (1) | PT1551803E (en) |
| SI (1) | SI1551803T1 (en) |
| WO (2) | WO2004004629A2 (en) |
| ZA (1) | ZA200500951B (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006032994A3 (en) * | 2004-09-24 | 2006-05-04 | Ranbaxy Lab Ltd | Muscarinic receptor antagonists |
| WO2006035282A3 (en) * | 2004-09-27 | 2006-05-18 | Ranbaxy Lab Ltd | Muscarinic receptor antagonists |
| WO2006064304A1 (en) * | 2004-12-15 | 2006-06-22 | Ranbaxy Laboratories Limited | Acid addition salts of muscarinic receptor antagonists |
| WO2006003587A3 (en) * | 2004-07-01 | 2006-09-14 | Ranbaxy Lab Ltd | Solid oral dosage forms of azabicyclo derivatives |
| JP2006527183A (en) * | 2004-05-31 | 2006-11-30 | アルミラル プロデスファルマ ソシエダッド アノニマ | Combination comprising antimuscarinic agent and beta-adrenergic agonist |
| WO2007039884A1 (en) | 2005-10-05 | 2007-04-12 | Ranbaxy Laboratories Limited | 3 -azabicyclooctane derivatives as muscarinic receptor antagonists |
| WO2007045979A1 (en) * | 2005-10-19 | 2007-04-26 | Ranbaxy Laboratories Limited | Pharmaceutical compositions of muscarinic receptor antagonists |
| US7232835B2 (en) | 2002-12-10 | 2007-06-19 | Ranbaxy Laboratories Limited | 3,6-Disubstituted azabicyclo derivatives as muscarinic receptor antagonists |
| WO2007077510A2 (en) | 2005-12-30 | 2007-07-12 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2007110782A1 (en) | 2005-12-30 | 2007-10-04 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| US7399779B2 (en) | 2002-07-08 | 2008-07-15 | Ranbaxy Laboratories Limited | 3,6-disubstituted azabicyclo [3.1.0] hexane derivatives useful as muscarinic receptor antagonists |
| WO2008075321A3 (en) * | 2006-12-21 | 2008-08-21 | Ranbaxy Lab Ltd | Modified-release formulations of azabicyclo derivatives |
| US7446123B2 (en) | 2003-04-11 | 2008-11-04 | Ranbaxy Laboratories Limited | Azabicyclo derivatives as muscarinic receptor antagonists |
| US7517905B2 (en) | 2003-04-09 | 2009-04-14 | Ranbaxy Laboratories Limited | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| EP2130830A1 (en) | 2008-06-03 | 2009-12-09 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| JP2011157358A (en) * | 2003-10-14 | 2011-08-18 | Pfizer Products Inc | Bicyclic [3.1.0] derivative as glycine transporter inhibitor |
| CN110191709A (en) * | 2017-01-17 | 2019-08-30 | 德州大学系统董事会 | It can be used as the compound of indole amine 2,3-dioxygenase and/or tryptophan dioxygenase inhibitor |
| US11046649B2 (en) | 2018-07-17 | 2021-06-29 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1746998A1 (en) * | 2004-03-22 | 2007-01-31 | Ranbaxy Laboratories, Ltd. | Combination therapy for lower urinary tract symptoms |
| WO2006016245A1 (en) * | 2004-08-05 | 2006-02-16 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2006054162A1 (en) * | 2004-11-19 | 2006-05-26 | Ranbaxy Laboratories Limited | Azabicyclic muscarinic receptor antagonists |
| EP1888525A1 (en) * | 2005-05-03 | 2008-02-20 | Ranbaxy Laboratories Limited | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives as muscarinic receptor antagonists |
| JP2013542929A (en) | 2010-09-28 | 2013-11-28 | パナセア バイオテック リミテッド | New bicyclo ring compounds |
| CN111317754B (en) * | 2018-12-13 | 2022-02-18 | 泰州医药城国科化物生物医药科技有限公司 | Preparation method and application of asiatic moonseed total alkali |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0863141A1 (en) * | 1995-10-13 | 1998-09-09 | Banyu Pharmaceutical Co., Ltd. | Substituted heteroaromatic derivatives |
| WO2002004402A1 (en) * | 2000-07-11 | 2002-01-17 | Banyu Pharmaceutical Co., Ltd. | Ester derivatives |
| WO2002051841A1 (en) * | 2000-12-22 | 2002-07-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
Family Cites Families (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2490714A (en) | 1947-05-13 | 1949-12-06 | Du Pont | Preparation of diazoacetic esters |
| NL267508A (en) | 1960-07-26 | |||
| SE8800207D0 (en) | 1988-01-22 | 1988-01-22 | Kabivitrum Ab | NEW AMINES, THEIR USE AND MANUFACTURING |
| US5001160A (en) | 1988-04-28 | 1991-03-19 | Marion Laboratories, Inc. | 1-aryl-1-hydroxy-1-substituted-3-(4-substituted-1-piperazinyl)-2-propanones and their use in treatment of neurogenic bladder disorders |
| GB8906166D0 (en) | 1989-03-17 | 1989-05-04 | Pfizer Ltd | Therapeutic agents |
| IE66202B1 (en) | 1989-08-16 | 1995-12-13 | Pfizer | Azabicyclo quinolone carboxylic acids |
| US5164402A (en) | 1989-08-16 | 1992-11-17 | Pfizer Inc | Azabicyclo quinolone and naphthyridinone carboxylic acids |
| GB8928042D0 (en) | 1989-12-12 | 1990-02-14 | Pfizer Ltd | Muscarinic receptor antagonists |
| US5281601A (en) | 1989-12-12 | 1994-01-25 | Pfizer Inc. | Muscarinic receptor antagonists |
| FR2659323B1 (en) | 1990-03-07 | 1992-06-12 | Synthelabo | DERIVATIVES OF 4- (AMINOMETHYL) PIPERIDINE, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION. |
| JPH0492921A (en) | 1990-08-03 | 1992-03-25 | Fujitsu Ltd | Exponential function computing element |
| GB9020051D0 (en) | 1990-09-13 | 1990-10-24 | Pfizer Ltd | Muscarinic receptor antagonists |
| GB9202443D0 (en) | 1992-02-05 | 1992-03-18 | Fujisawa Pharmaceutical Co | A novel substituted-acetamide compound and a process for the preparation thereof |
| FI100051B (en) | 1992-02-18 | 1997-09-15 | Favorit Oy | composting |
| JPH0635958A (en) | 1992-07-14 | 1994-02-10 | Hitachi Ltd | Word search method |
| JP3429338B2 (en) | 1992-07-27 | 2003-07-22 | 杏林製薬株式会社 | Novel arylglycinamide derivative and method for producing the same |
| JPH06135958A (en) | 1992-10-28 | 1994-05-17 | Tanabe Seiyaku Co Ltd | Benzocycloheptene derivative and its production |
| SE9203318D0 (en) | 1992-11-06 | 1992-11-06 | Kabi Pharmacia Ab | NOVEL 3,3-DIPHENYL PROPYLAMINES, THEIR USE AND PREPARATION |
| EP0613232B1 (en) | 1993-02-24 | 1997-04-23 | Siemens Aktiengesellschaft | Flyback converter power supply with a semi-conductor switch of low resistance voltage |
| NO2005012I1 (en) | 1994-12-28 | 2005-06-06 | Debio Rech Pharma Sa | Triptorelin and pharmaceutically acceptable salts thereof |
| EP0823423B1 (en) * | 1995-04-28 | 2004-06-16 | Banyu Pharmaceutical Co., Ltd. | 1,4-disubstituted piperidine derivatives |
| CA2179574A1 (en) | 1995-06-26 | 1996-12-27 | Tomomi Okada | Substituted piperidine derivative and medicine comprising the same |
| WO1997045414A1 (en) | 1996-05-31 | 1997-12-04 | Banyu Pharmaceutical Co., Ltd. | 1,4-disubstituted piperidine derivatives |
| WO1998000109A1 (en) | 1996-07-01 | 1998-01-08 | Sepracor, Inc. | Methods and compositions for treating urinary incontinence using enantiomerically enriched (s,r)-glycopyrrolate |
| EP0909172A4 (en) | 1996-07-01 | 1999-06-09 | Sepracor Inc | METHOD AND COMPOSITIONS FOR THE TREATMENT OF URININE CONTINENCE USING ENANTIOMER-ENRICHED (R, S) -GLYCOPYRROLATE |
| WO1998000016A1 (en) | 1996-07-01 | 1998-01-08 | Sepracor, Inc. | Methods and compositions for treating urinary incontinence using enantiomerically enriched (r,r)-glycopyrrolate |
| WO1998000133A1 (en) | 1996-07-01 | 1998-01-08 | Sepracor, Inc. | Methods and compositions for treating urinary incontinence using enantiomerically enriched (s,s)-glycopyrrolate |
| PE92198A1 (en) * | 1996-08-01 | 1999-01-09 | Banyu Pharma Co Ltd | DERIVATIVES OF FLUORINE-CONTAINED 1,4-PIPERIDINE |
| KR20000057548A (en) | 1996-12-13 | 2000-09-25 | 알프레드 엘. 미첼슨 | Optically transmissive material and bond |
| EP1001764A4 (en) | 1997-05-29 | 2005-08-24 | Merck & Co Inc | Heterocyclic amides as cell adhesion inhibitors |
| US6319920B1 (en) | 1998-02-27 | 2001-11-20 | Syntex (U.S.A.) Llc | 2-arylethyl-(piperidin-4-ylmethyl)amine derivatives |
| TWI244481B (en) | 1998-12-23 | 2005-12-01 | Pfizer | 3-azabicyclo[3.1.0]hexane derivatives useful in therapy |
| AU783095B2 (en) | 1999-12-07 | 2005-09-22 | Theravance Biopharma R&D Ip, Llc | Carbamate derivatives having muscarinic receptor antagonist activity |
| UA73543C2 (en) | 1999-12-07 | 2005-08-15 | Тераванс, Інк. | Urea derivatives, a pharmaceutical composition and use of derivative in the preparation of medicament for the treatment of disease being mediated by muscarine receptor |
| SE9904765D0 (en) | 1999-12-23 | 1999-12-23 | Astra Ab | Pharmaceutically useful compounds |
| WO2001090082A1 (en) | 2000-05-25 | 2001-11-29 | F. Hoffmann-La Roche Ag | Substituted 1-aminoalkyl-lactams and their use as muscarinic receptor antagonists |
| PL204753B1 (en) | 2000-05-25 | 2010-02-26 | Hoffmann La Roche | Substituted 1−aminoalkyl−lactams and their use as muscarinic receptor antagonists |
| WO2002006241A1 (en) | 2000-06-14 | 2002-01-24 | Muscagen Limited | 1, 2, 3, 5 -tetrahydrobenzo`c!azepin-4-one derivatives having muscarinic antagonist activity |
| CZ294251B6 (en) | 2000-06-27 | 2004-11-10 | Laboratorios S. A. L. V. A. T., S. A. | Carbamates and their use in the preparation of a pharmaceutical composition |
| IL156558A0 (en) | 2000-12-28 | 2004-01-04 | Almirall Prodesfarma Ag | Novel quinuclidine derivatives and medicinal compositions containing the same |
| US6699866B2 (en) | 2001-04-17 | 2004-03-02 | Sepracor Inc. | Thiazole and other heterocyclic ligands for mammalian dopamine, muscarinic and serotonin receptors and transporters, and methods of use thereof |
| JP2005512974A (en) | 2001-10-17 | 2005-05-12 | ユ セ ベ ソシエテ アノニム | Quinuclidine derivatives, process for their preparation and their use as m2 and / or m3 muscarinic receptor inhibitors |
| WO2003048124A1 (en) | 2001-12-03 | 2003-06-12 | F. Hoffmann-La Roche Ag | 4-piperidinyl alkylamine derivatives as muscarinic receptor antagonists |
| MXPA04005313A (en) | 2001-12-03 | 2004-09-13 | Hoffmann La Roche | Aminotetralin derivatives as muscarinic receptor antagonists. |
| AU2002345266B2 (en) | 2002-07-08 | 2009-07-02 | Ranbaxy Laboratories Limited | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
| HK1079787A1 (en) | 2002-07-31 | 2006-04-13 | Ranbaxy Laboratories Limited | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
| HK1079708A1 (en) | 2002-08-09 | 2006-04-13 | Ranbaxy Laboratories, Ltd. | 3,6-disubstituted azabicyclo [3.1.0] hexane derivatives useful as muscarinic receptor antagonist |
| JP2006501236A (en) * | 2002-08-23 | 2006-01-12 | ランバクシー ラボラトリーズ リミテッド | 3,6-disubstituted azabicyclo (3.1.0) hexane derivatives containing fluoro and sulfonylamino as muscarinic receptor antagonists |
| US7232835B2 (en) | 2002-12-10 | 2007-06-19 | Ranbaxy Laboratories Limited | 3,6-Disubstituted azabicyclo derivatives as muscarinic receptor antagonists |
| WO2004056810A1 (en) | 2002-12-23 | 2004-07-08 | Ranbaxy Laboratories Limited | Xanthine derivatives as muscarinic receptor antagonists |
| EP1581522B1 (en) | 2002-12-23 | 2008-02-20 | Ranbaxy Laboratories Limited | Flavaxate derivatives as muscarinic receptor antagonists |
| AU2002347552A1 (en) | 2002-12-23 | 2004-07-14 | Ranbaxy Laboratories Limited | 1-substituted-3-pyrrolidine derivatives as muscarinic receptor antagonists |
| US7488748B2 (en) | 2003-01-28 | 2009-02-10 | Ranbaxy Laboratories Limited | 3,6-Disubstituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| AU2003205964A1 (en) | 2003-02-07 | 2004-08-30 | Ranbaxy Laboratories Limited | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| WO2004089898A1 (en) | 2003-04-09 | 2004-10-21 | Ranbaxy Laboratories Limited | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| DE60313898T2 (en) | 2003-04-10 | 2008-01-17 | Ranbaxy Laboratories, Ltd. | SUBSTITUTED AZABICYCLO HEXANES DERIVATIVES AS MUSCARIN RECEPTOR ANTAGONISTS |
| AU2003223010A1 (en) | 2003-04-10 | 2004-11-01 | Ranbaxy Laboratories Limited | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| EA009387B1 (en) | 2003-04-11 | 2007-12-28 | Рэнбакси Лабораториз Лимитед | Azabicycloderivatives as muscarinic receptor antagonists |
| FR2855805B1 (en) | 2003-06-06 | 2005-08-05 | Vallourec Vitry | STRUCTURE ELEMENT FOR VEHICLE CAPABLE OF IMPROVED SHOCK BEHAVIOR |
| EP1746998A1 (en) | 2004-03-22 | 2007-01-31 | Ranbaxy Laboratories, Ltd. | Combination therapy for lower urinary tract symptoms |
| EP1796667A2 (en) | 2004-09-27 | 2007-06-20 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2006064304A1 (en) | 2004-12-15 | 2006-06-22 | Ranbaxy Laboratories Limited | Acid addition salts of muscarinic receptor antagonists |
-
2002
- 2002-07-08 AU AU2002345266A patent/AU2002345266B2/en not_active Expired - Fee Related
- 2002-07-08 EA EA200500173A patent/EA200500173A1/en unknown
- 2002-07-08 MX MXPA05000434A patent/MXPA05000434A/en unknown
- 2002-07-08 DE DE60230683T patent/DE60230683D1/en not_active Expired - Fee Related
- 2002-07-08 AT AT02743489T patent/ATE419236T1/en not_active IP Right Cessation
- 2002-07-08 CA CA002492121A patent/CA2492121A1/en not_active Abandoned
- 2002-07-08 WO PCT/IB2002/002663 patent/WO2004004629A2/en not_active Ceased
- 2002-07-08 US US10/520,573 patent/US7399779B2/en not_active Expired - Fee Related
- 2002-07-08 CN CNA028295528A patent/CN1668585A/en active Pending
- 2002-07-08 BR BR0215801-9A patent/BR0215801A/en not_active IP Right Cessation
- 2002-07-08 JP JP2004519029A patent/JP2006502985A/en active Pending
- 2002-07-08 NZ NZ537584A patent/NZ537584A/en unknown
- 2002-07-08 EP EP02743489A patent/EP1546099B1/en not_active Expired - Lifetime
-
2003
- 2003-04-11 EP EP03762827A patent/EP1551803B8/en not_active Expired - Lifetime
- 2003-04-11 EA EA200500172A patent/EA007932B1/en not_active IP Right Cessation
- 2003-04-11 PT PT03762827T patent/PT1551803E/en unknown
- 2003-04-11 MX MXPA05000435A patent/MXPA05000435A/en active IP Right Grant
- 2003-04-11 NZ NZ537585A patent/NZ537585A/en unknown
- 2003-04-11 CN CNB038211300A patent/CN100519526C/en not_active Expired - Fee Related
- 2003-04-11 DE DE60309057T patent/DE60309057T2/en not_active Expired - Fee Related
- 2003-04-11 CA CA002491998A patent/CA2491998A1/en not_active Abandoned
- 2003-04-11 SI SI200330515T patent/SI1551803T1/en unknown
- 2003-04-11 BR BR0312572-6A patent/BR0312572A/en not_active IP Right Cessation
- 2003-04-11 ES ES03762827T patent/ES2274275T3/en not_active Expired - Lifetime
- 2003-04-11 JP JP2004519035A patent/JP2005535655A/en active Pending
- 2003-04-11 DK DK03762827T patent/DK1551803T3/en active
- 2003-04-11 US US10/520,572 patent/US7544708B2/en not_active Expired - Fee Related
- 2003-04-11 AU AU2003226579A patent/AU2003226579B2/en not_active Ceased
- 2003-04-11 WO PCT/IB2003/001367 patent/WO2004005252A1/en not_active Ceased
- 2003-04-11 AT AT03762827T patent/ATE342253T1/en not_active IP Right Cessation
-
2004
- 2004-01-07 EA EA200501595A patent/EA009942B1/en not_active IP Right Cessation
-
2005
- 2005-02-02 ZA ZA200500951A patent/ZA200500951B/en unknown
-
2006
- 2006-12-21 CY CY20061101846T patent/CY1105879T1/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0863141A1 (en) * | 1995-10-13 | 1998-09-09 | Banyu Pharmaceutical Co., Ltd. | Substituted heteroaromatic derivatives |
| WO2002004402A1 (en) * | 2000-07-11 | 2002-01-17 | Banyu Pharmaceutical Co., Ltd. | Ester derivatives |
| WO2002051841A1 (en) * | 2000-12-22 | 2002-07-04 | Almirall Prodesfarma Ag | Quinuclidine carbamate derivatives and their use as m3 antagonists |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7544708B2 (en) | 2002-07-08 | 2009-06-09 | Ranbaxy Laboratories Limited | Azabicyclo derivatives as muscarinic receptor antagonists |
| US7399779B2 (en) | 2002-07-08 | 2008-07-15 | Ranbaxy Laboratories Limited | 3,6-disubstituted azabicyclo [3.1.0] hexane derivatives useful as muscarinic receptor antagonists |
| EP1572648B1 (en) * | 2002-12-10 | 2008-07-09 | Ranbaxy Laboratories, Ltd. | 3,6-disubstituted azabicyclo (3.1.0)-hexane derivatives as muscarinic receptor antagonists |
| US7232835B2 (en) | 2002-12-10 | 2007-06-19 | Ranbaxy Laboratories Limited | 3,6-Disubstituted azabicyclo derivatives as muscarinic receptor antagonists |
| US7517905B2 (en) | 2003-04-09 | 2009-04-14 | Ranbaxy Laboratories Limited | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists |
| US7446123B2 (en) | 2003-04-11 | 2008-11-04 | Ranbaxy Laboratories Limited | Azabicyclo derivatives as muscarinic receptor antagonists |
| JP2011157358A (en) * | 2003-10-14 | 2011-08-18 | Pfizer Products Inc | Bicyclic [3.1.0] derivative as glycine transporter inhibitor |
| JP2006527183A (en) * | 2004-05-31 | 2006-11-30 | アルミラル プロデスファルマ ソシエダッド アノニマ | Combination comprising antimuscarinic agent and beta-adrenergic agonist |
| JP4928258B2 (en) * | 2004-05-31 | 2012-05-09 | アルミラル・ソシエダッド・アノニマ | Combination comprising antimuscarinic agent and beta-adrenergic agonist |
| JP2012012407A (en) * | 2004-05-31 | 2012-01-19 | Almirall Sa | Combination comprising antimuscarinic agent and beta-adrenergic agonist |
| WO2006003587A3 (en) * | 2004-07-01 | 2006-09-14 | Ranbaxy Lab Ltd | Solid oral dosage forms of azabicyclo derivatives |
| WO2006032994A3 (en) * | 2004-09-24 | 2006-05-04 | Ranbaxy Lab Ltd | Muscarinic receptor antagonists |
| WO2006035282A3 (en) * | 2004-09-27 | 2006-05-18 | Ranbaxy Lab Ltd | Muscarinic receptor antagonists |
| WO2006064304A1 (en) * | 2004-12-15 | 2006-06-22 | Ranbaxy Laboratories Limited | Acid addition salts of muscarinic receptor antagonists |
| WO2007039884A1 (en) | 2005-10-05 | 2007-04-12 | Ranbaxy Laboratories Limited | 3 -azabicyclooctane derivatives as muscarinic receptor antagonists |
| WO2007045979A1 (en) * | 2005-10-19 | 2007-04-26 | Ranbaxy Laboratories Limited | Pharmaceutical compositions of muscarinic receptor antagonists |
| WO2007110782A1 (en) | 2005-12-30 | 2007-10-04 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2007077510A2 (en) | 2005-12-30 | 2007-07-12 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| WO2008075321A3 (en) * | 2006-12-21 | 2008-08-21 | Ranbaxy Lab Ltd | Modified-release formulations of azabicyclo derivatives |
| EP2130830A1 (en) | 2008-06-03 | 2009-12-09 | Ranbaxy Laboratories Limited | Muscarinic receptor antagonists |
| CN110191709A (en) * | 2017-01-17 | 2019-08-30 | 德州大学系统董事会 | It can be used as the compound of indole amine 2,3-dioxygenase and/or tryptophan dioxygenase inhibitor |
| EP3570832A4 (en) * | 2017-01-17 | 2020-06-10 | Board Of Regents, The University Of Texas System | NOVEL COMPOUNDS USEFUL AS INHIBITORS OF INDOLEAMINE 2,3-DIOXYGENASE AND / OR TRYPTOPHANE DIOXYGENASE |
| US11173145B2 (en) | 2017-01-17 | 2021-11-16 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
| US11046649B2 (en) | 2018-07-17 | 2021-06-29 | Board Of Regents, The University Of Texas System | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7544708B2 (en) | Azabicyclo derivatives as muscarinic receptor antagonists | |
| US7446123B2 (en) | Azabicyclo derivatives as muscarinic receptor antagonists | |
| AU2003214535B2 (en) | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists | |
| US20070010568A1 (en) | Substituted azabicyclo hexane derivatives as muscarinic receptor antagonists | |
| US7560479B2 (en) | 3,6-Disubstituted azabicyclo hexane derivatives as muscarinic receptor antagonists | |
| HK1082728B (en) | Azabicyclo derivatives as muscarinic receptor antagonists | |
| KR20050023400A (en) | Azabicyclo derivatives as muscarinic receptor antagonists | |
| HK1079783B (en) | 3,6-disubstituted azabicyclo 3.1.0 hexane derivatives useful as muscarinic receptor antagonists | |
| KR20050023401A (en) | 3,6-disubstituted azabicyclo [3.1.0]hexane derivatives useful as muscarinic receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2491998 Country of ref document: CA Ref document number: PA/a/2005/000435 Country of ref document: MX Ref document number: 537585 Country of ref document: NZ Ref document number: 2004519035 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057000402 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003226579 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003762827 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/00951 Country of ref document: ZA Ref document number: 405/DELNP/2005 Country of ref document: IN Ref document number: 200500951 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200500172 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038211300 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057000402 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003762827 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2006111425 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10520572 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10520572 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003762827 Country of ref document: EP |