WO2004016614A2 - Pyrrolopyrazines as kinase inhibitors - Google Patents
Pyrrolopyrazines as kinase inhibitors Download PDFInfo
- Publication number
- WO2004016614A2 WO2004016614A2 PCT/EP2003/009515 EP0309515W WO2004016614A2 WO 2004016614 A2 WO2004016614 A2 WO 2004016614A2 EP 0309515 W EP0309515 W EP 0309515W WO 2004016614 A2 WO2004016614 A2 WO 2004016614A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- alkoxy
- pyrrolo
- hal
- derivatives
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- FJCDNHZPKAXZGS-UHFFFAOYSA-N C1Nc(cc(-c2c[s]cc2)[nH]2)c2NC1 Chemical compound C1Nc(cc(-c2c[s]cc2)[nH]2)c2NC1 FJCDNHZPKAXZGS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
Definitions
- the invention relates to derivatives of pyrrolo- pyrazines having a kinase inhibitory activity and their biological applications.
- Protein kinases catalyse the phosphorylation of serine, threonine and tyrosine residues of proteins, using ATP or GTP as the phosphate donor. Protein phosphorylation is considered as one of the main post- translational mechanisms used by cells to finely tune their metabolic and regulatory pathways.
- Protein kinases (an estimated 800 in the human genome) , and their counterparts the protein phosphatases, appear to be involved in most human diseases. This is the reason why screening for potent and selective inhibitors of protein kinases has intensified over the last few years.
- CDKs cyclin-dependent kinases
- GSK-3 glycogen synthase kinase-3
- CDKs are involved in controlling the cell cycle, apoptosis, neuronal functions and neurodegeneration, transcription and exocytosis.
- GSK-3 an essential element of the NT signaling pathway, is involved in multiple physiological processes including cell cycle regulation by controlling the levels of cyclin Dl and ⁇ -catenin, dorso-ventral patterning during development, insulin action on glycogen synthesis, axonal outgrowth, HIV-1 Tat-mediated neurotoxicity, and phosphorylation of tau, a characteristic of Alzheimer's disease.
- Applications of CDK/GSK-3 inhibitors are being evaluated against cancers, neurodegenerative disorders such as Alzheimer's disease, diabetes, proliferation of protozoan parasites and viral infections (HIV, cytomegalovirus and herpes virus) (1) .
- CDK inhibitors include the purines olomoucine, roscovitine, purvalanols, CVT-313, C2-alkylynated purines, H717 and NU2058, piperidine-substituted purines, toyocamycin, flavopiridol, indirubins, paullones, ⁇ -butyrolactone, hymenialdisine, mdenopyrazoles, the pyrimidmes NU6027 and CGP60474, pyridopyri idine, the aminopyrimidine PNU 112455A, oxindoles, PD0183812, cinnamaldehydes, quinazolines, fasclaplysin, SU9516 and benzocarbazoles (reviewed in ref. 1, 2-8).
- GSK-3 inhibitors include indirubins, paullones, aleimides and lithium.
- the inventors have now identified a new family of kinase inhibitors selective for CDKl/2/5 and GSK-3 ⁇ / ⁇ , acting in the sub-micromolar range by competing with ATP for binding to the kinase active site, as revealed by enzymological studies and crystal structure studies.
- Said family has a therapeutical value in pathological situations involving CDKs and/or GSK-3 / ⁇ deregulations.
- the invention thus relates to novel derivatives of pyrrolo-pyrazines . It also relates to a method for preparing said derivatives .
- the invention relates to the use of said derivatives as active principle of drugs.
- pyrrolo [2, 3b]-pyrazine derivatives of the invention have the general formula (I) :
- R2 and R3 are identical or different and represent
- C1-C6 alkyl said alkyl being a straight or branched - chain alkyl, which can be substituted
- - R6 is an optionally substituted aromatic cycle Ar or a cycloalkyl, said cycloalkyl being optionally substituted by an aryl group which can also be substituted
- CH 2 -cycloalkyl, CH 2 -Ar, - Z is H or CH 3 .
- R2 and R3, and/or Z and/or R7 are different from H.
- Ar is preferably phenyl, naphtyl, furyl, thienyl, pyridyl, cyclopropyl phenyl, phenyl dioxolyl.
- Cycloalkyl is a C3-C6 cycloalkyl.
- Substitutions of the alkyl group, aromatic cycle or cycloalkyl are selected in the group comprising one or more halogen (F,C1, Br, I, CF 3 ) , OH, NH 2 , N (H, alkyl), N (alkyl) 2 , O-alkyl, COOH, COO-alkyl, CONH 2 , CON (H, alkyl) , CON (alkyl) 2 , NHCONH 2/ NHCON (H, alkyl) , NHCON (alkyl) 2 , N (alkyl) CONH 2 , N (alkyl) CO (H, alkyl) , N (alkyl) CO (alkyl) 2 , alkoxy, CN, 0-S0 2 - NH 2 0-S0 2 -N (H, alkyl) ,-0-S0 2 -N (alkyl) 2 , SH,S-alkyl.
- One or more substituents can be present.
- Alk is a C1-C6 alkylene group, n is 1-6, and "hal.” is F, CI, Br, I or CF 3 .
- Said pyrrolo [2,3-b] pyrazines are potent kinase inhibitory scaffold, and selective for CDKs and GSK-3 ⁇ / ⁇ , acting for most of them in the sub- icromolar range.
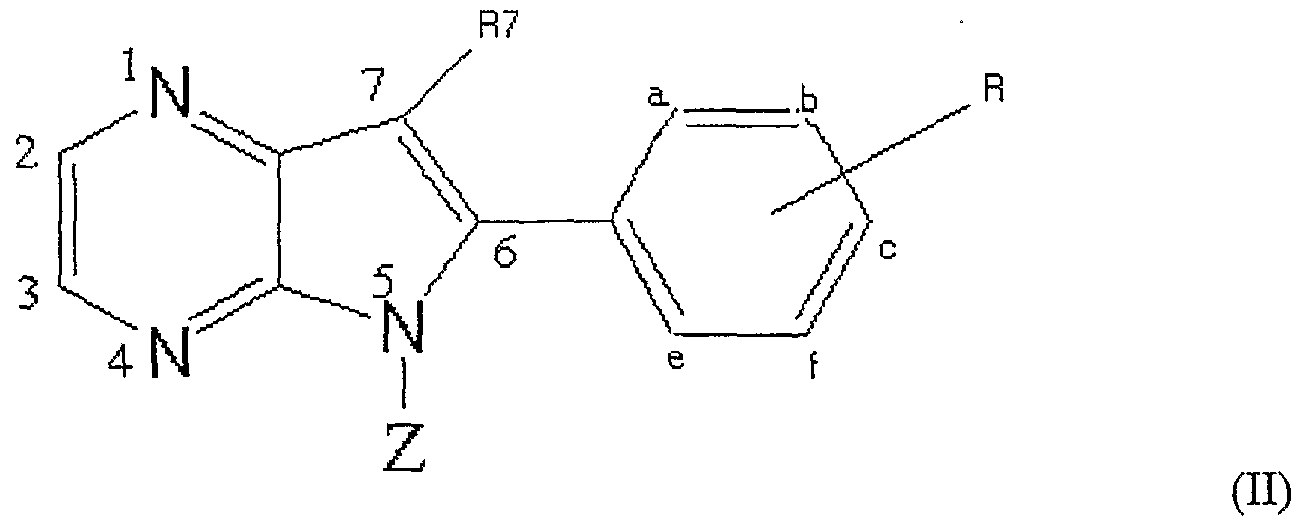
- Preferred derivatives of pyrrolo-pyrazines have formula (ID :
- Z is H or CH 3 .
- Z and/or R7 are different from H.
- a preferred group of said family has an IC 5 o value ⁇ lO ⁇ M with respect to correspond to CDKl/cyclin B, CDK5/p25 and GSK-3. They correspond to the derivatives of formula (II) wherein
- a more preferred group of said family of formula (II) has an IC 5 0 value ⁇ 5 ⁇ M with respect to CDKl/cyclin B, CDK5/p25 and GSK-3.
- Ra, Rb and Rd H
- Ra, Rb and Rd H
- a still more preferred group of said family of formula (II) has an IC 50 ⁇ l ⁇ M with respect to CDKl/cyclin B, CDK5/p25 and GSK-3.
- the derivatives have an IC50 ⁇ 0.5 ⁇ M with respect to CDKl/cyclin B, CDK5/p25 and GSK-3.
- Another preferred group of said family of formula (II) has an IC 50 value ⁇ lO ⁇ M with respect to CDKl/cyclin B and CDK5/p25 or GSK-3, or to CDK5/p25 and GSK-3.
- the invention particularly relates to the group with derivatives having an IC 50 ⁇ lO ⁇ M with respect to CDK5/p25 and GSK-3.
- Preferred derivatives of said group have an IC 50 value 5 ⁇ M with respect to CDK5/p25 and GSK-3.
- Still more preferred derivatives have an IC 50 ⁇ 0.5 ⁇ M with respect to CDK5/p25 and GSK-3.
- the derivatives have an IC 50 ⁇ lO ⁇ M with respect to CDK1 and GSK3.
- the derivatives have an IC 50 ⁇ 5 ⁇ M with respect to CDK1 and GSK-3.
- Ra, Rb and Rd H
- Re alkoxy, OH, hal., alkyl
- CN and R7 H
- Ra, Rb, Rd H
- the derivatives have an IC 5 o value ⁇ I ⁇ M with respect to CDKl and GSK-3.
- the derivatives have an IC 50 value ⁇ 0.5 ⁇ M with respect to CDKl/cyclin B and GSK-3.
- the invention also relates to the group with derivatives having an IC 50 ⁇ lO ⁇ M with respect to CDKl/cyclin B and CDK5/p25.
- Preferred derivatives have an IC 5 0 ⁇ 5 ⁇ M with respect to CDKl/cyclin B and GSK-3.
- the derivatives have an IC 50 ⁇ l ⁇ M with respect to CDKl/cyclin B and GSK-3.
- the derivatives have an IC 50 ⁇ 0.5 ⁇ M with respect to CDKl/cyclin B and GSK-3.
- Ra, Rb, and Rd H
- Re alkoxy or OH
- R7 alkyl
- Another preferred family with an IC 50 ⁇ lO ⁇ M with respect to CDKl/cylein B, CDK5 and GSK-3 has formula (III), and even ⁇ 5 ⁇ M with respect to CDK5/p25 and GSK-3.
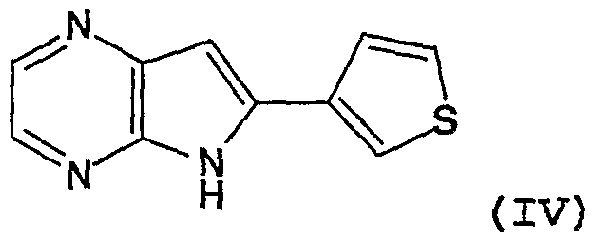
- Still another preferred family has formula IV with an IC5 0 ⁇ 5 ⁇ M with respect to CDKl/cyclin B, CDK5/p25 and
- Said derivatives have interestingly an IC 50 value ⁇ l ⁇ M with respect to CDK5/p25 and GSK-3.
- the invention also relates to a method for preparing said aloisines comprising reacting alkyl-pyrazines of formula (V) :
- Rl and R3 are as above defined, and Alkyl is a C1-C6 alkyl, ith aromatic nitriles, R ⁇ CN, wherein R ⁇ is as above defined.
- the alkylpyrazine derivatives of formula (V) are added to an organic solvent containing butyllithium or analog, at a temperature not exceeding 0°C and preferably of about -40°C.
- the resulting solution is stirred during 30 in to about lh.
- the nitrile derivative is then added and the solution is stirred during 30 min to about lh, and further at the ambient (around 20°C) for about 1 to 20h.
- Alkylpyrazines can be obtained by reaction of pyrazinylmethyllithium with bromoalcanes, and benzonitriles. Demethylation of methoxy compounds can be achieved by refluxing in acidic conditions. The time required for demethylation varied from 3-20 h. As above mentioned, and as illustrated in the Examples hereinafter, said compounds strongly inhibit CDKl and/or CDK5 and/or GSK-3.
- the invention thus relates to pharmaceutical compositions comprising an effective amount of at least one derivative as above defined as active principle in association with a pharmaceutically acceptable carrier.
- Said carrier may be solid, or liquid, depending on the administration form.
- Said pharmaceutical compositions are useful for treating or preventing, neurodegenerative disorders such as Alzheimer's disease or Parkison's diseases. They are also useful for treating invention also relates to the use of said pharmaceutical compositions for treating proliferative disorders such as cancers, or the proliferation of unicellular or pluricellular parasites. Other applications comprise the use of said pharmaceutical compositions against cardiovascular disorders linked to proliferation. They also comprise their use for treating viral injections (HIV, cytomegalovirus and herpes virus. The invention also relates to the use of said derivatives as herbicides.
- Said pharmaceutical compositions can be administered in various forms e.g. orally, topically, by injection (intravenously, subcutaneously, intraperitoneally, or rectally) . They are more particularly administered by the oral route.
- compositions For administration by the oral route, lozenges, compressed tablets, pills, tablets, capsules, drops, syrups, suspensions or emulsions, may be used. These compositions advantageously comprise 100 to 1000 g of active principle per dose unit, preferably 300 to 600 mg.
- injectable solutions for the intravenous, subcutaneous or intramuscular route formulated from sterile or sterilizable solutions. They can also be suspensions or emulsions.
- injectable forms comprise 100 to 1000 mg of active principle preferably 300 to 600 mg, per dose unit.
- the dosage which can be used in a patient in need thereof corresponds to the following doses : for example, 100 to 1000 mg/day are thus administered to the patient 1 to 4 times per day for the treatment of neurodegenerative disorders.
- the invention also relates to biological reagents, the 5. active principles of which consist of the compounds of formula (I) as above-defined.
- FIG. 1 CDKl/cyclin B, CDK5/p25 and GSK-3 ⁇ inhibition results with aloisines of the invention
- figures 2A to 2C enzyme activities for aloisine A 5 in the presence of ATP (CDl/cyclin B : figure 2A ; CDK5/p25 : figure 2B, and GSK-3 ⁇ : figure 2C)
- figure 3 stereo view of the interactions between aloisine B and the CDK2 ATP binding site
- figures 4A to 4E reversible inhibition of 0 exponential cell growth by aloisine A, and figure 5, the comparison of the effects of aloisine A on G0/G1 (A, B, C) and G2/M (D, E, F)
- the organic layer was dried over Na 2 S0 4 , and concentrated under vacuum.
- the crude product was chromatographied on silicagel, eluted with ethylene chloride, then ethyl acetate. If necessary the product was crystallized from ethanol or methylene chloride-ethanol mixture .
- hydrobromic acid was redistilled over a trace of 50% hypophosphorus acid: 1 g for each 100 g of 48% hydrobromic acid.
- Methoxy compound (0.003 mol) was heated with hydrobromic acid (20 ml) . After removal of the aqueous forerun, the temperature reaches 126 °C. The time required for demethylation varies from 3-10 h. The excess of hydrobromic acid was removed under reduced pressure, and the crude product was crystallised from ethanol .
- [ ⁇ - 32 P]-ATP (PB 168) was obtained from Amersham.
- the GS-1 peptide has sequence SEQ ID N°l YRRAAVPPSPSLSRHSSPHQSpEDEEE.
- Ho ogenization Buffer 60 mM ⁇ -glycerophosphate, 15 mM p-nitrophenylphosphate, 25 mM Mops (pH 7.2), 15 mM EGTA, 15 mM MgCl 2 , 1 mM DTT, 1 mM sodium vanadate, 1 mM NaF, 1 mM phenylphosphate, 10 ⁇ g leupeptin/ml, 10 ⁇ g aprotinin/ml, 10 ⁇ g soybean trypsin inhibitor/ml and 100 ⁇ M benzamidine.
- Buffer A 10 mM MgCl 2 , 1 mM EGTA, 1 mM DTT, 25 mM Tris- HC1 pH 7.5, 50 ⁇ g heparin/ l.
- Buffer C homogenization buffer but 5 mM EGTA, no NaF and no protease inhibitors.
- Tris-Buffered Saline -Tween-20 50 mM Tris pH 7.4, 150 mM NaCl, 0.1% Tween-20.
- Hypotonic Lysis Buffer 50 mM Tris-HCl pH 7.4, 120 mM NaCl, 10 % glycerol, 1 % Nonidet-P40, 5 mM DTT, 1 mM EGTA, 20 mM NaF, 1 mM orthovanadate, 5 ⁇ M microcystin, 100 ⁇ g/ml each of leupeptin, aprotinin and pepstatin.
- kinases activities were assayed in Buffer A or C (unless otherwise stated), at 30 °C, at a final ATP concentration of 15 ⁇ M. Blank values were subtracted and activities calculated as pmoles of phosphate incorporated for a 10 min. incubation. The activities are usually expressed in % of the maximal activity, i.e. in the absence of inhibitors. Controls were performed with appropriate dilutions of dimethylsulfoxide. In a few cases phosphorylation of the substrate was assessed by autoradiography after SDS-PAGE.
- GSK-3 ⁇ / ⁇ was either purified from porcine brain or expressed in and purified from insect Sf9 cells. It was assayed, following a 1/100 dilution in 1 mg BSA/ml 10 mM DTT, with 5 ⁇ l 40 ⁇ M GS-1 peptide as a substrate, in buffer A, in the presence of 15 ⁇ M [ ⁇ - 32 P] ATP (3,000 Ci/mmol; 1 Ci/ml) in a final volume of 30 ⁇ l. After 30 min. incubation at 30 °C, 25 ⁇ l aliquots of supernatant were spotted onto 2.5 x 3 cm pieces of Whatman P81 phosphocellulose paper, and, 20 sec. later, the filters were washed five times (for at least 5 min. each time) in a solution of 10 ml phosphoric acid/liter of water. The wet filters were counted in the presence of 1 ml ACS (Amersham) scintillation fluid.
- CDKl/cyclin B was extracted in homogenisation buffer from M phase starfish (2Vfart asterias glacialis) oocytes and purified by affinity chromatography on p9 CKShsl -sepharose beads, from which it was eluted by free p9 CKShsl .
- the kinase activity was assayed in buffer C, with 1 mg histone HI /ml,
- CDK5/p25 was reconstituted by mixing equal amounts of recombinant mammalian CDK5 and p25 expressed in E. coli as
- GST Glutathione-S-transferase fusion proteins and purified by affinity chromatography on glutathione-agarose (p25 is a truncated version of p35, the 35 kDa CDK5 activator) . Its activity was assayed in buffer C as described for CDKl/cyclin B.
- IC 50 ' s were calculated from the dose-response curves and are given in Table 1 hereinafter in ⁇ M.
- Table 1 (continued) . Structure activity relationship of aloisine-related compounds .
- Figure 1 gives the results obtained in the presence of increasing concentrations of aloisines A and B. Activity is presented as % of maximal activity, i.e. measured in the absence of inhibitors.
- Aloisine is a competitive inhibitor of ATP binding
- Human CDK2 was expressed from a recombinant baculo-virus in Sf9 insect cells and purified. Monomeric unphosphorylated CDK2 crystals were grown as previously described in (11) .
- the CDK2-aloisine B dataset was collected from a monomeric CDK2 crystal soaked for 60 h in ImM aloisine B in 1 x mother liquor solution (50 mM ammonium acetate, 10% PEG3350, 15 mM NaCl, 100 mM HEPES, pH7.4) plus 5% DMSO. Data was collected on beamline X-RAY DIFFRACTION at the Elettra Light Source at 100K after the crystal had been transferred briefly to cryo-protectant (mother liquor adjusted to contain 20% glycerol) . The images were integrated with the MOSFLM package (12) and reflections were subsequently scaled and merged using SCALA (13) . Subsequent data reduction and structure refinement were pursued through programs of the CCP4 suite.
- Aloisine B occupies the CDK2 ATP binding site and makes two hydrogen bonds to the CDK2 backbone within the hinge sequence that links the two lobes of the kinase.
- CDK2 is drawn in ribbon representation and colour-ramped from blue through to red starting at the N-terminus.
- the N-terminal lobe is dominated by a 5-stranded anti-parallel ⁇ -sheet and the C-terminal lobe is predominantly -helical.
- Aloisine B is drawn in ball and stick mode bound at the ATP-binding site which lies in the cleft between the two domains.
- Aloisine B carbon atoms are coloured cyan, nitrogen atoms blue and the chlorine atom is drawn in yellow.
- aloisine B does not interact with the backbone oxygen of Glu ⁇ l, but instead accepts and donates a hydrogen bond respectively from the backbone nitrogen and oxygen atoms of Leu 83 (Fig. 3) : residues that lie within 4A of the bound aloisine B molecule are drawn in ball and stick mode.
- Aloisine B carbon atoms are drawn in cyan and those of CDK2 in green.
- Oxygen atoms are coloured red, nitrogen atoms are blue and the chlorine atom is drawn in yellow.
- Dotted lines represent hydrogen bonds (d 0->N or dN-O ⁇ 3.4 A) between aloisine B and the backbone nitrogen and oxygen atoms of Leu 83.
- the Figure also includes (2Fo-Fc) -calc electron density for aloisine B calculated at the end of refinement using map coefficients output from REFMAC with resolution between 20 and 1.9 A.
- the map is contoured at a level of 0.19 e- A "3 corresponding to 1.0 times the r.m.s. deviation of the map from its mean value.
- This hydrogen-bonding pattern has previously been observed in the structures of monomeric CDK2 in complex with olomoucine, roscovitine, purvalanol B, OL567 and H717.
- the CDK2 ATP-binding site is tolerant of a number of positions for the planar heterocyclic ring systems which are a characteristic of the CDK inhibitors identified to date.
- the position of the aloisine B fused ring system within the CDK2 ATP binding site most closely resembles that of indirubin-5- sulphonate and oxindole-3.
- aloisine B does not fill the back of the ATP- binding cleft and form an equivalent edge-to-ring stacking interaction with the side-chain of Phe 80.
- Aloisine A the most active aloisine so far, was tested for selectivity on 26 highly purified kinases.
- Kinase activities were assayed with appropriate substrates (for example histone HI, casein, myelin basic protein and peptides), with 15 ⁇ M ATP and in the presence of increasing concentrations of aloisine A.
- IC 50 values were estimated from the dose-response curves and are presented in Table 3. Table 3.
- Table 3 Kinase inhibition selectivity of aloisine A.
- CDKs Most kinases tested were poorly or not inhibited (IC 50 > lO ⁇ M) .
- GSK-3 ⁇ and CDKs were strongly sensitive to aloisine A (IC 50 's of 0.65 and 0.15 ⁇ M, respectively) (Fig. 1; Table 3).
- CDKs CDKl, CDK2 and CDK5
- CDK4 were inhibited by aloisine A. This is pronounced of other CDK inhibitors, such as purines, hymenialdisine, paullones, and indirubins, which inhibit CDKl/2/5 but have much less effect on or not CDK4/6.
- Clonal human NT2 teraterocarcinoma cells were obtained from Stratagene (La Jolla, CA) and grown in Dulbecco' s Modified Eagle Medium: Nutrient Mixture F-12 with 2mM L- Glutamine (BIO WHITTAKER) supplemented with 5% FCS and containing penicillin (20 Ul/ml) and streptomycin (20 ⁇ g/ l) at 37 °C, in a humidified atmosphere containing 5% C0 2 in air.
- Dulbecco' s Modified Eagle Medium Nutrient Mixture F-12 with 2mM L- Glutamine (BIO WHITTAKER) supplemented with 5% FCS and containing penicillin (20 Ul/ml) and streptomycin (20 ⁇ g/ l) at 37 °C, in a humidified atmosphere containing 5% C0 2 in air.
- Exponentially growing cells were incubated for 24 h with aloisine A (stock solution dissolved in dimethylsulfoxide) .
- Nocodazole treatment of cells was performed at a concentration of 0.04 ⁇ g nocodazole/ml of medium for 24 h. Following the nocodazole treatment, cells were washed twice with fresh medium and cultured with or without aloisine A for 24 h. To perform serum deprivation, cells were maintained in serum-free medium for 40 h. Following serum deprivation, cells were washed twice and cultured in fresh serum- containing medium with or without aloisine A for 40 h.
- RNAse A/ml were washed in PBS, incubated with 10 ⁇ g RNAse A/ml and stained with 25 ⁇ g propidiu iodide/ml for 1 h at 37 °C. The stained cells were then analysed for cell cycle distribution on a FACSort flow cytometer (Becton Dickinson) . Cell cycle analyses were performed multiCYCLE (18) . The effect of aloisine A on the cell cycle distribution was investigated for NT2 cells by flow cytometry. Unsynchronised cells (Fig. 4E) were exposed to 20 ⁇ M aloisine A for 40 h (Fig. 4F) . The proliferation arrest induced by aloisine A in exponentially growing cells was clearly accompanied by an accumulation in G2/M phase. No signs of apoptosis were detectable, confirming the lack of apparent toxicity observed before.
- NT2 cells were synchronized by nocodazole treatment (0.2 ⁇ g/ l) for 24 h (D) , then cultured for 40 h in fresh medium without (E) or with 20 ⁇ M aloisine A (F) . Serum deprivation for 24 h lead to a significant increase of cells in G0/G1 (Fig. 5A) . Cells were then re- exposed to a serum-enriched media for 40 h in the absence (Fig. 5B) or presence (Fig. 5C) of 20 ⁇ M aloisine A. Aloisine A-treated cells remained essentially in G0/G1, with a small additional accumulation of G2/M cells, most probably derived from the initial S phase sub-population (Fig. 5C) .
- Control cells redistributed in a classical cell cycle pattern (Fig. 5B) .
- Nocodazole treatment for 24 h lead to a massive accumulation of cells in G2/M (Fig. 5D) .
- Cells were then washed to remove nocodazole and incubated for 40 h in the absence (Fig. 5E) or presence (Fig. 5F) of 20 ⁇ M aloisine A.
- 40 h after nocodazole withdrawal control cells redistributed in the various cell cycle phases (Fig. 8E) .
- the majority of cells exposed to aloisine A after nocodazole treatment remained in G2/M (Fig. 5F) , precluding the increase in G1/G0 seen in control cells.
- a small sub-G2-peak may indicate a minor onset of apoptotic cell death.
- a Gl arrest correlates with aloisine A' s high potency against CDK2/cyclin E.
- the inability to enter S phase might also be result from inhibition of GSK-3, a kinase known to be involved in cyclin Dl degradation.
- the G2/M arrest correlates well with the potency of aloisine A against CDKl/cyclin B.
- CCP4 Cold Processing Computational Project Number 4
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Hospice & Palliative Care (AREA)
- Tropical Medicine & Parasitology (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description









Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2495060A CA2495060C (en) | 2002-08-09 | 2003-08-08 | Derivatives of pyrrolo-pyrazines having a kinase inhibitory activity and their biological applications |
| AU2003271566A AU2003271566A1 (en) | 2002-08-09 | 2003-08-08 | Pyrrolopyrazines as kinase inhibitors |
| EP03753362A EP1527077A2 (en) | 2002-08-09 | 2003-08-08 | Pyrrolopyrazines as kinase inhibitors |
| JP2004528508A JP4780549B2 (en) | 2002-08-09 | 2003-08-08 | Pyrrolopyrazine derivatives having kinase inhibitory activity and biological uses thereof |
| US10/524,044 US8106050B2 (en) | 2002-08-09 | 2003-08-08 | Derivatives of pyrrolo-pyrazines having a kinase inhibitory activity and their biological applications |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP20020292019 EP1388541A1 (en) | 2002-08-09 | 2002-08-09 | Pyrrolopyrazines as kinase inhibitors |
| EP02292019.3 | 2002-08-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004016614A2 true WO2004016614A2 (en) | 2004-02-26 |
| WO2004016614A3 WO2004016614A3 (en) | 2004-05-06 |
Family
ID=30129256
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/009515 Ceased WO2004016614A2 (en) | 2002-08-09 | 2003-08-08 | Pyrrolopyrazines as kinase inhibitors |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8106050B2 (en) |
| EP (2) | EP1388541A1 (en) |
| JP (1) | JP4780549B2 (en) |
| AU (1) | AU2003271566A1 (en) |
| CA (1) | CA2495060C (en) |
| WO (1) | WO2004016614A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2876377A1 (en) * | 2004-10-11 | 2006-04-14 | Univ Claude Bernard Lyon | NOVEL 9H-PYRIDO [2,3-B] INDOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION, AND PHARMACEUTICAL COMPOSITIONS CONTAINING SUCH COMPOUNDS |
| FR2918061A1 (en) * | 2007-06-28 | 2009-01-02 | Sanofi Aventis Sa | 6-CYCLOAMINO-3- (PYRIDIN-4-YL) IMIDAZO [1,2-B] -PYRIDAZINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE. |
| JP2010500365A (en) * | 2006-08-07 | 2010-01-07 | インサイト・コーポレイション | Triazolotriazines as kinase inhibitors |
| US8470310B2 (en) | 2008-03-04 | 2013-06-25 | The Trustees Of The University Of Pennsylvania | Simian adenoviruses SAdV-36, -42.1, -42.2, and -44 and uses thereof |
| US8658649B2 (en) | 2006-09-11 | 2014-02-25 | Sanofi | Kinase inhibitor |
| US9217159B2 (en) | 2012-05-18 | 2015-12-22 | The Trustees Of The University Of Pennsylvania | Subfamily E simian adenoviruses A1302, A1320, A1331 and A1337 and uses thereof |
Families Citing this family (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1388541A1 (en) | 2002-08-09 | 2004-02-11 | Centre National De La Recherche Scientifique (Cnrs) | Pyrrolopyrazines as kinase inhibitors |
| FR2876582B1 (en) * | 2004-10-15 | 2007-01-05 | Centre Nat Rech Scient Cnrse | USE OF PYRROLO-PYRAZINE DERIVATIVES FOR THE MANUFACTURE OF MEDICAMENTS FOR THE TREATMENT OF MUCOVISCIDOSIS AND DISEASES ASSOCIATED WITH A DEFECT OF ADDRESSING PROTEINS IN CELLS |
| FR2899107B1 (en) * | 2006-03-30 | 2008-06-13 | Neurokin Entpr Unipersonnelle | USE OF (S) -ROSCOVITINE FOR THE MANUFACTURE OF A MEDICINAL PRODUCT |
| EP2012822B1 (en) * | 2006-04-28 | 2010-01-20 | The Trustees of the University of Pennsylvania | Modified adenovirus hexon protein and uses thereof |
| WO2008063888A2 (en) | 2006-11-22 | 2008-05-29 | Plexxikon, Inc. | Compounds modulating c-fms and/or c-kit activity and uses therefor |
| US8110576B2 (en) | 2008-06-10 | 2012-02-07 | Plexxikon Inc. | Substituted pyrrolo[2,3b]pyrazines and methods for treatment of raf protein kinase-mediated indications |
| MY172424A (en) | 2009-04-03 | 2019-11-25 | Hoffmann La Roche | Propane- i-sulfonic acid {3- (4-chloro-phenyl)-1h-pyrrolo [2, 3-b] pyridine-3-carconyl] -2, 4-difluoro-phenyl} -amide compositions and uses thereof |
| CN106220623A (en) | 2009-11-06 | 2016-12-14 | 普莱希科公司 | Compounds and methods for and indication thereof for kinases regulation |
| US8754114B2 (en) | 2010-12-22 | 2014-06-17 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9624213B2 (en) | 2011-02-07 | 2017-04-18 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2013028952A2 (en) * | 2011-08-25 | 2013-02-28 | Rutgers, The State University Of New Jersey | Compositions and methods for enhancing lipid production in microalgae via induction of cell cycle arrest |
| US9150570B2 (en) | 2012-05-31 | 2015-10-06 | Plexxikon Inc. | Synthesis of heterocyclic compounds |
| RS58514B1 (en) | 2012-06-13 | 2019-04-30 | Incyte Holdings Corp | Substituted tricyclic compounds as fgfr inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| PH12015502383B1 (en) | 2013-04-19 | 2023-02-03 | Incyte Holdings Corp | Bicyclic heterocycles as fgfr inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| MA41551A (en) | 2015-02-20 | 2017-12-26 | Incyte Corp | BICYCLIC HETEROCYCLES USED AS FGFR4 INHIBITORS |
| TWI712601B (en) | 2015-02-20 | 2020-12-11 | 美商英塞特公司 | Bicyclic heterocycles as fgfr inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| WO2016190847A1 (en) * | 2015-05-26 | 2016-12-01 | Calitor Sciences, Llc | Substituted heteroaryl compounds and methods of use |
| AR111960A1 (en) | 2017-05-26 | 2019-09-04 | Incyte Corp | CRYSTALLINE FORMS OF A FGFR INHIBITOR AND PROCESSES FOR ITS PREPARATION |
| HRP20241288T1 (en) | 2018-05-04 | 2024-12-06 | Incyte Corporation | Solid forms of an fgfr inhibitor and processes for preparing the same |
| AU2019262579B2 (en) | 2018-05-04 | 2024-09-12 | Incyte Corporation | Salts of an FGFR inhibitor |
| MX2021003904A (en) * | 2018-10-05 | 2021-10-26 | Annapurna Bio Inc | COMPOUNDS AND COMPOSITIONS FOR THE TREATMENT OF CONDITIONS ASSOCIATED WITH APJ RECEPTOR ACTIVITY. |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2021067374A1 (en) | 2019-10-01 | 2021-04-08 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| CA3157361A1 (en) | 2019-10-14 | 2021-04-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| WO2021076728A1 (en) | 2019-10-16 | 2021-04-22 | Incyte Corporation | Bicyclic heterocycles as fgfr inhibitors |
| CA3163875A1 (en) | 2019-12-04 | 2021-06-10 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| JP7832891B2 (en) | 2019-12-04 | 2026-03-18 | インサイト・コーポレイション | Derivatives of FGFR inhibitors |
| US12012409B2 (en) | 2020-01-15 | 2024-06-18 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| WO2022221170A1 (en) | 2021-04-12 | 2022-10-20 | Incyte Corporation | Combination therapy comprising an fgfr inhibitor and a nectin-4 targeting agent |
| CA3220155A1 (en) | 2021-06-09 | 2022-12-15 | Incyte Corporation | Tricyclic heterocycles as fgfr inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5286860A (en) * | 1992-11-12 | 1994-02-15 | Neurogen Corporation | Certain aryl substituted pyrrolopyrazines; a new class of GABA brain receptor ligands |
| US5834469A (en) * | 1994-06-09 | 1998-11-10 | Smithkline Beecham Corporation | Endothelin receptor antagonists |
| KR20000057137A (en) * | 1996-11-19 | 2000-09-15 | 스티븐 엠. 오드레 | Aryl and heteroaryl substituted fused pyrrole antiinflammatory agents |
| BR9812944A (en) * | 1997-10-20 | 2000-08-08 | Hoffmann La Roche | Bicyclic kinase inhibitors |
| SK286640B6 (en) * | 1998-06-19 | 2009-03-05 | Pfizer Products Inc. | Pyrrolo[2,3-d]pyrimidine compounds, the use thereof for producing a medicament, pharmaceutical composition containing the same, the use of its combination with additional agents and kits containing the same for producing a medicament |
| PA8474101A1 (en) * | 1998-06-19 | 2000-09-29 | Pfizer Prod Inc | PYROLEUM [2,3-D] PIRIMIDINE COMPOUNDS |
| ES2253930T3 (en) * | 1998-09-18 | 2006-06-01 | ABBOTT GMBH & CO. KG | 4-AMINOPIRROLOPIRIMIDINAS AS QUINASA INHIBITORS. |
| JP4649046B2 (en) * | 1999-05-21 | 2011-03-09 | ブリストル−マイヤーズ スクイブ カンパニー | Pyrrotriazine inhibitor of kinase |
| DE60037345T2 (en) * | 1999-12-10 | 2008-11-13 | Pfizer Products Inc., Groton | -Pyrrolo (2,3-d) pyrimidin-compounds |
| WO2001047922A2 (en) * | 1999-12-24 | 2001-07-05 | Aventis Pharma Limited | Azaindoles |
| KR20010111298A (en) | 2000-02-05 | 2001-12-17 | 버텍스 파마슈티칼스 인코포레이티드 | Pyrazole compositions useful as inhibitors of erk |
| JP2004503553A (en) * | 2000-06-14 | 2004-02-05 | ワーナー−ランバート・カンパニー、リミテッド、ライアビリティ、カンパニー | 6,5-fused bicyclic heterocycle |
| ATE423120T1 (en) * | 2000-06-26 | 2009-03-15 | Pfizer Prod Inc | PYRROLOÄ2,3-DÜPYRIMIDINE COMPOUNDS AS IMMUNOSUPPRESSIVE ACTIVES |
| KR20050008691A (en) | 2002-04-19 | 2005-01-21 | 셀룰러 지노믹스 아이엔씨 | Imidazo[1,2-a]Pyrazin-8-ylamines Method Of Making And Method Of Use Thereof |
| EP1388541A1 (en) | 2002-08-09 | 2004-02-11 | Centre National De La Recherche Scientifique (Cnrs) | Pyrrolopyrazines as kinase inhibitors |
| DE60313872T2 (en) | 2002-09-04 | 2008-01-17 | Schering Corp. | PYRAZOLOE1,5-ATPYRIMIDINES AS INHIBITORS CYCLINE-DEPENDENT KINASES |
| AU2003279230A1 (en) | 2002-10-09 | 2004-05-04 | Scios Inc. | AZAINDOLE DERIVATIVES AS INHIBITORS OF p38 KINASE |
| FR2876582B1 (en) | 2004-10-15 | 2007-01-05 | Centre Nat Rech Scient Cnrse | USE OF PYRROLO-PYRAZINE DERIVATIVES FOR THE MANUFACTURE OF MEDICAMENTS FOR THE TREATMENT OF MUCOVISCIDOSIS AND DISEASES ASSOCIATED WITH A DEFECT OF ADDRESSING PROTEINS IN CELLS |
-
2002
- 2002-08-09 EP EP20020292019 patent/EP1388541A1/en not_active Withdrawn
-
2003
- 2003-08-08 JP JP2004528508A patent/JP4780549B2/en not_active Expired - Fee Related
- 2003-08-08 CA CA2495060A patent/CA2495060C/en not_active Expired - Fee Related
- 2003-08-08 US US10/524,044 patent/US8106050B2/en not_active Expired - Fee Related
- 2003-08-08 WO PCT/EP2003/009515 patent/WO2004016614A2/en not_active Ceased
- 2003-08-08 AU AU2003271566A patent/AU2003271566A1/en not_active Abandoned
- 2003-08-08 EP EP03753362A patent/EP1527077A2/en not_active Withdrawn
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2876377A1 (en) * | 2004-10-11 | 2006-04-14 | Univ Claude Bernard Lyon | NOVEL 9H-PYRIDO [2,3-B] INDOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION, AND PHARMACEUTICAL COMPOSITIONS CONTAINING SUCH COMPOUNDS |
| WO2006040451A3 (en) * | 2004-10-11 | 2006-06-08 | Univ Claude Bernard Lyon | Novel 9h-pyrido[2,3-b]indole derivatives serving as cdk and gsk3 inhibitors, method for the preparation thereof, and the pharmaceutical compositions containing such compounds |
| JP2010500365A (en) * | 2006-08-07 | 2010-01-07 | インサイト・コーポレイション | Triazolotriazines as kinase inhibitors |
| US8658649B2 (en) | 2006-09-11 | 2014-02-25 | Sanofi | Kinase inhibitor |
| FR2918061A1 (en) * | 2007-06-28 | 2009-01-02 | Sanofi Aventis Sa | 6-CYCLOAMINO-3- (PYRIDIN-4-YL) IMIDAZO [1,2-B] -PYRIDAZINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE. |
| WO2009016286A3 (en) * | 2007-06-28 | 2009-04-09 | Sanofi Aventis | 6-cycloamino-3-(pyridin-4-yl)imidazo[1,2-b]pyridazine derivatives, preparation thereof and therapeutic use thereof |
| US8354405B2 (en) | 2007-06-28 | 2013-01-15 | Sanofi | 6-cycloamino-3-(pyrid-4-yl)imidazo[1,2-b]pyridazine derivatives, preparation thereof and therapeutic use thereof |
| US8846676B2 (en) | 2007-06-28 | 2014-09-30 | Sanofi | 6-cycloamino-3-(pyrid-4-yl)imidazo[1,2-b]pyridazine derivatives, preparation thereof and therapeutic use thereof |
| US8470310B2 (en) | 2008-03-04 | 2013-06-25 | The Trustees Of The University Of Pennsylvania | Simian adenoviruses SAdV-36, -42.1, -42.2, and -44 and uses thereof |
| US9597363B2 (en) | 2008-03-04 | 2017-03-21 | The Trustees Of The University Of Pennsylvania | Simian adenoviruses SAdV-36, -42.1, -42.2, and -44 and uses thereof |
| US9217159B2 (en) | 2012-05-18 | 2015-12-22 | The Trustees Of The University Of Pennsylvania | Subfamily E simian adenoviruses A1302, A1320, A1331 and A1337 and uses thereof |
| US10113182B2 (en) | 2012-05-18 | 2018-10-30 | The Trustees Of The University Of Pennsylvania | Subfamily E simian adenoviruses A1302, A1320, A1331 and A1337 and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP4780549B2 (en) | 2011-09-28 |
| EP1527077A2 (en) | 2005-05-04 |
| WO2004016614A3 (en) | 2004-05-06 |
| US20080161312A1 (en) | 2008-07-03 |
| AU2003271566A1 (en) | 2004-03-03 |
| AU2003271566A8 (en) | 2004-03-03 |
| CA2495060C (en) | 2012-01-03 |
| US8106050B2 (en) | 2012-01-31 |
| JP2006502137A (en) | 2006-01-19 |
| EP1388541A1 (en) | 2004-02-11 |
| CA2495060A1 (en) | 2004-02-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8106050B2 (en) | Derivatives of pyrrolo-pyrazines having a kinase inhibitory activity and their biological applications | |
| Mettey et al. | Aloisines, a new family of CDK/GSK-3 inhibitors. SAR study, crystal structure in complex with CDK2, enzyme selectivity, and cellular effects | |
| JP5542196B2 (en) | 1-Heterocyclic-1,5-dihydro-pyrazolo [3,4-D] pyrimidin-4-one derivatives and their use as PDE9A modulators | |
| EP2387315B1 (en) | IMIDAZO[1,2-a]PYRIDINES AND IMIDAZO[1,2-b]PYRIDAZINES AS MARK INHIBITORS | |
| Legraverend et al. | Cyclin-dependent kinase inhibition by new C-2 alkynylated purine derivatives and molecular structure of a CDK2− inhibitor complex | |
| EP3444254B1 (en) | Heterocyclic-substituted pyridinopyrimidinone derivative as cdk inhibitor and use thereof | |
| AU2002225096B8 (en) | Cyclin-dependent kinase (CDK) and glycogen synthase kinase-3 (GSK-3) inhibitors | |
| ES2338234T3 (en) | USEFUL COMPOSITIONS AS INHIBITORS OF KINASE PROTEINS. | |
| AU2013366513B2 (en) | Novel benzimidazole derivatives as kinase inhibitors | |
| RU2753520C2 (en) | Derivatives of n-(substituted phenyl)-sulfonamide as kinase inhibitors | |
| JP2005526765A (en) | CDK-inhibiting 2-heteroaryl-pyrimidines, their production and use as pharmaceuticals | |
| JP2004535414A (en) | CDK-inhibiting pyrimidines, their production and use as medicaments | |
| US20130109693A1 (en) | Derivatives of pyrido [3,2-d] pyrimidine, methods for preparation thereof and therapeutic uses thereof | |
| EP2351751A1 (en) | Substituted pyrazolopyrimidines | |
| SK8982002A3 (en) | Benzazole derivatives, process for their preparation, pharmaceutical composition comprising the same and their use | |
| JP2004532248A (en) | 3,5-Diamino-1,2,4, -triazole as kinase inhibitor | |
| WO2013060098A1 (en) | Kinase inhibitor and method for treatment of related diseases | |
| EP1295885A1 (en) | Substituted 2-pyridinyl-6,7,8,9-tetrahydropyrimido(1,2-a)pyrimidin-4-one and 7-pyridinyl-2,3-dihydroimidazo(1,2-a)pyrimidin-5(1H)one derivatives | |
| EP3077383B1 (en) | Sulfoximine substituted quinazolines for pharmaceutical compositions | |
| HK1064084A (en) | Pyrrolopyrazines as kinase inhibitors | |
| Demange et al. | Synthesis and evaluation of new potent inhibitors of CK1 and CDK5, two kinases involved in Alzheimer’s disease | |
| CN109476666B (en) | A compound that acts as a kinase inhibitor | |
| CN108822110B (en) | Aromatic heterocycle-containing a, β -unsaturated ketone compound and preparation method and application thereof | |
| US20080261974A1 (en) | Novel Chemical Compounds | |
| CN106336412A (en) | 2-(N-Oxyaryl-2-ylamino)-pyrrolopyrimidines and purines as CDK4/6 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2495060 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003753362 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004528508 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003753362 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10524044 Country of ref document: US |