WO2004018459A1 - Pyrazole derivatives as gnrh inhibitors - Google Patents
Pyrazole derivatives as gnrh inhibitors Download PDFInfo
- Publication number
- WO2004018459A1 WO2004018459A1 PCT/GB2003/003623 GB0303623W WO2004018459A1 WO 2004018459 A1 WO2004018459 A1 WO 2004018459A1 GB 0303623 W GB0303623 W GB 0303623W WO 2004018459 A1 WO2004018459 A1 WO 2004018459A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- alkyl
- formula
- compound
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)(*)C(*)(*)C#N Chemical compound CC(C)(*)C(*)(*)C#N 0.000 description 16
- FYHBVTYDATZUEE-UHFFFAOYSA-N CC(C)(CBr)C(O)=O Chemical compound CC(C)(CBr)C(O)=O FYHBVTYDATZUEE-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N CN1CCNCC1 Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- SJRJJKPEHAURKC-UHFFFAOYSA-N CN1CCOCC1 Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/24—Drugs for disorders of the endocrine system of the sex hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the present invention relates to compounds which are antagonists of gonadotropin releasing hormone (GnRH) activity.
- the invention also relates to pharmaceutical formulations, the use of a compound of the present invention in the manufacture of a medicament, a method of therapeutic treatment using such a compound and processes for producing the compounds.
- Gonadotropin releasing hormone is a decapeptide that is secreted by the hypothalamus into the hypophyseal portal circulation in response to neural and/or chemical stimuli, causing the biosynthesis and release of luteinizing hormone (LH) and follicle- stimulating hormone (FSH) by the pituitary.
- GnRH is also known by other names, including gonadoliberin, LH releasing hormone (LHRH), FSH releasing hormone (FSH RH) and
- LH/FSH releasing factor (LH/FSH RF).
- GnRH plays an important role in regulating the action of LH and FSH (by regulation of their levels), and thus has a role in regulating the levels of gonadal steroids in both sexes, including the sex hormones progesterone, oestrogens and androgens. More discussion of
- GnRH can be found in WO 98/5519 and WO 97/14697, the disclosures of which are incorporated herein by reference.
- sex hormone related conditions such as sex hormone dependent cancer, benign prostatic hypertrophy and myoma of the uterus.
- sex hormone dependent cancers are prostatic cancer, uterine cancer, breast cancer and pituitary gonadotrophe adenoma.
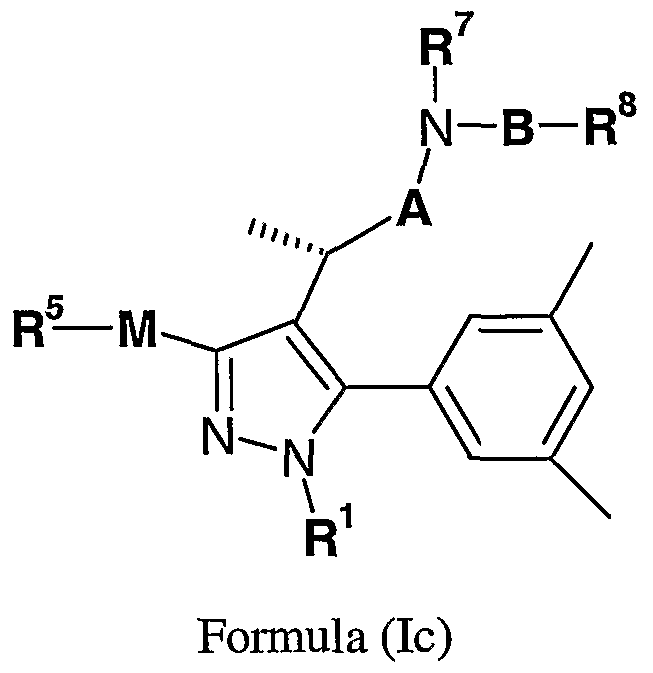
- A represents a direct bond or optionally substituted Q-salkylene
- B is a group of Formula (II):
- Formula (II) wherein at position (a) Formula (II) is attached to the nitrogen atom and the group X is attached to R 8 ; M is -(CH 2 )o- 2 -O-;
- R 1 represents hydrogen; optionally substituted .salkyl; or (CH2)b-R a , wherein
- R a represents C 3 . 8 cycloalkyl and b is zero or an integer from 1 to 6;
- R represents an optionally substituted mono- or bi-cyclic aromatic ring structure wherein the optional substituents are selected from cyano, NR 3 R a , optionally substituted -salkyl, optionally substituted C ⁇ - 8 alkoxy or halo;
- R 3 and R 3a are independently selected from hydrogen; optionally substituted -salkyl and optionally substituted aryl; R 5 is selected from an optionally substituted 3 to 8 membered heterocyclic ring containing from 1 to 4 heteroatoms independently selected from O, N and S; or a group of formula Ili a; III-b; III-c; Ill-d; Ill-e; Ill-f, Ill-g , Ill-h, Ill-i or: III-j, lll-a lll-b lll-c lll-d lll-e
- lll-j wherein het represents an optionally substituted 3 to 8 membered heterocyclic ring containing from 1 to 4 heteroatoms independently selected from O, N and S; R 6 and R 6a , are independently selected from hydrogen and optionally substituted -salkyl; or R 6 and R 6a together represent carbonyl;
- R 7 represents hydrogen or optionally substituted C ⁇ salkyl
- R 6 ⁇ -N-R 7 together from an optionally substituted 3- to 8- membered heterocyclic ring containing from 1 to 3 further heteroatoms independently selected from O, N and S, and R 6a represents hydrogen and optionally substituted Q-salkyl;
- X and R 8 are selected from:
- X represents N and R 8 is selected from: cyano, hydrogen, hydroxy, -O-R b , -NR R c -C(O)O-R b , -CONR b R c or NH-C(O)-R b , where R b and R c are independently selected from hydrogen and Ci- 4 alkyl optionally substituted with hydroxy, amino, N-C ⁇ alkylamino, N,N-di-C ⁇ - 4 alkylamino, HO-C 2 . 4 alkyl-NH- or HO-C 2 . 4 alkyl-N(C 1 . 4 alkyl)-;
- R 11 is a group of the formula: N(R 9 R 10 ) wherein R 9 represents hydrogen, aryl, an optionally substituted 3- to 10 membered heterocyclic ring or optionally-substituted Ci-salkyl and R 10 represents hydrogen or optionally substituted -salkyl; or the structure N(R 9 R 10 ) represents an optionally-substituted 3- to 10 membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S; R 12 and R 12a are independently selected from hydrogen or optionally substituted Ci-salkyl; or R 12 and R 12a together with the carbon to which they are attached form an optionally substituted 3 to 7-membered cycloalkyl ring; R 13 and R 14 are selected from:
- R is selected from hydrogen; optionally substituted Ci-salkyl; optionally substituted aryl; -R d -Ar, where R d represents Ci-salkylene and Ar represents optionally substituted aryl; and optionally substituted 3 to 8 membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S; and R 14 is selected from hydrogen; optionally substituted Ci-salkyl and optionally substituted aryl; (ii) where R 5 represents a group of formula Ill-a , Ill-b or Ill-i, then the group
- NR 13 (-R 14 ) represents an optionally substituted 3 to 8 membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S; or
- R 5 represents structure Ill-e
- group v represents an optionally substituted 3 to 8 membered heterocyclic ring optionally containing from 1 to 4 heteroatoms independently selected from O, N and S; or a salt, pro-drug or solvate thereof.
- a pharmaceutical formulation comprising a compound of Formula (I), or salt, pro-drag or solvate thereof, and a pharmaceutically acceptable diluent or carrier.
- a compound of Formula (I), or salt, pro-drug or solvate thereof (a) the use in the manufacture of a medicament for antagonising gonadotropin releasing hormone activity; (b) the use in the manufacture of a medicament for administration to a patient, for reducing the secretion of luteinizing hormone by the pituitary gland of the patient; and (c) the use in the manufacture of a medicament for administration to a patient, for therapeutically treating and/or preventing a sex hormone related condition in the patient, preferably a sex hormone related condition selected from prostate cancer and pre- menopausal breast cancer.
- a method of antagonising gonadotropin releasing hormone activity in a patient comprising administering a compound of Formula (I), or salt, pro-drug or solvate thereof, to a patient.
- compositions of the invention Whilst pharmaceutically-acceptable salts of compounds of the invention are preferred, other non-pharmaceutically-acceptable salts of compounds of the invention may also be useful, for example in the preparation of pharmaceutically-acceptable salts of compounds of the invention. Whilst the invention comprises compounds of the invention, and salts, pro-drugs or solvates thereof, in a further embodiment of the invention, the invention comprises compounds of the invention and salts thereof.
- alkyl, alkylene or alkenyl moiety may be linear or branched.
- alkylene refers to the group -CH2-.
- C 8 alkylene for example is
- aryl refers to phenyl or naphthyl.
- carbamoyl refers to the group -CONH 2 .
- heterocyclyl or “heterocyclic ring” refers to a 5-10 membered aromatic mono or bicyclic ring or a 5-10 membered saturated or partially saturated mono or bicyclic ring, said aromatic, saturated or partially unsaturated rings containing up to 5 heteroatoms independently selected from nitrogen, oxygen or sulphur, linked via ring carbon atoms or ring nitrogen atoms where a bond from a nitrogen is allowed, for example no bond is possible to the nitrogen of a pyridine ring, but a bond is possible through the 1 -nitrogen of a pyrazole ring.
- 5- or 6-membered aromatic heterocyclic rings examples include pyrrolyl, furanyl, imidazolyl, triazolyl, pyrazinyl, pyrimidinyl, pyridazinyl, pyridinyl, isoxazolyl, oxazolyl, 1,2,4 oxadiazolyl, isothiazolyl, thiazolyl and thienyl.
- a 9 or 10 membered bicyclic aromatic heterocyclic ring is an aromatic bicyclic ring system comprising a 6-membered ring fused to either a 5 membered ring or another 6 membered ring.
- Examples of 5/6 and 6/6 bicyclic ring systems include benzofuranyl, benzimidazolyl, benzthiophenyl, benzthiazolyl, benzisothiazolyl, benzoxazolyl, benzisoxazolyl, indolyl, pyridoimidazolyl, pyrimidoimidazolyl, quinolinyl, isoquinolinyl, quinoxalinyl, quinazolinyl, phthalazinyl, cinnolinyl and naphthyridinyl.
- saturated or partially saturated heterocyclic rings include pyrrolinyl, pyrrolidinyl, morpholinyl, piperidinyl, piperazinyl, dihydropyridinyl and dihydropyrimidinyl.
- This definition further comprises sulphur-containing rings wherein the sulphur atom has been oxidised to an S(O) or S(O2) group.
- aromatic ring refers to a 5-10 membered aromatic mono or bicyclic ring optionally containing up to 5 heteroatoms independently selected from nitrogen, oxygen or sulphur.
- aromatic rings examples include: phenyl, pyrrolyl, furanyl, imidazolyl, triazolyl, pyrazinyl, pyrimidinyl, pyridazinyl, pyridinyl, isoxazolyl, oxazolyl, 1,2,4 oxadiazolyl, isothiazolyl, thiazolyl and thienyl.
- Preferred aromatic rings include 'phenyl, thienyl and pyridyl.
- R 6 ' / ⁇ X" ⁇ -N-R together form an optionally substituted 3- to 8- membered heterocyclic ring containing from 1 to 3 further heteroatoms independently selected from O, N and S, then the groups shown cyclise to form a nitrogen-containing heterocyclic ring, i.e
- Ci-salkyl examples include: methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, tert-butyl and 2-methyl-pentyl;
- example of Ci-salkylene include: methylene, ethylene and 2-methyl-propylene;
- examples of Ci-salkoxy include methoxy, ethoxy and butyloxy;
- examples of N-C ⁇ . 4 alkylamino include N-methylamino and N-ethylamino; examples of N,N-di-C ⁇ . 4 alkylamino, examples of HO-C 2 .
- 4 alkyl-NH include hydroxymethylamino hydroxyethylamino and hydroxypropyamino
- examples of HO-C 2 - 4 alkyl-N(C ⁇ . 4 alkyl) include N-methyl-hydroxymethylamino, N-ethyl-hydroxyethylamino, and N-propyl-hydroxypropyamino.
- the invention includes in its definition any such optically active or racemic form which possesses the property of antagonizing gonadotropin releasing hormone (GnRH) activity.
- GnRH gonadotropin releasing hormone
- the synthesis of optically active forms may be carried out by standard techniques of organic chemistry well known in the art, for example by synthesis from optically active starting materials or by resolution of a racemic form. Similarly, activity of these compounds may be evaluated using the standard laboratory techniques referred to hereinafter.
- the invention also relates to any and all tautomeric forms of the compounds of the different features of the invention that possess the property of antagonizing gonadotropin releasing hormone (GnRH) activity.
- GnRH gonadotropin releasing hormone
- A represents optionally substituted C ⁇ - 5 alkylene. Further preferably C 1 . alkylene. Most preferably methylene or ethylene.
- M is -CH 2 -O-.
- R 1 represents hydrogen or optionally substituted Ci- 6 alkyl. More preferably
- R represents hydrogen, methyl, ethyl or tert-butyl. Most preferably R represents hydrogen.
- R 2 represents an optionally substituted monocyclic aromatic ring structure wherein the optional substituents are selected from cyano, NR e R f , optionally substituted C ⁇ salkyl (preferably, Ci- 4 alkyl, eg, methyl or ethyl), optionally substituted Ci. 8 alkoxy (preferably, Ci_ 6 alkoxy, eg, methoxy, ethoxy or tert-butoxy) or halo (eg, F, Br or CI) wherein R e and R f are independently selected from hydrogen, Ci- 6 alkyl or aryl.
- optional substituents are selected from cyano, NR e R f , optionally substituted C ⁇ salkyl (preferably, Ci- 4 alkyl, eg, methyl or ethyl), optionally substituted Ci. 8 alkoxy (preferably, Ci_ 6 alkoxy, eg, methoxy, ethoxy or tert-butoxy) or halo (eg
- R 2 is optionally substituted phenyl wherein the optional substituents are selected from cyano, NR e R f , optionally substituted Ci- 4 alkyl, optionally substituted Ci- 6 alkoxy, F, Br or CI wherein R e and R f are as defined above. Yet further preferably R 2 is optionally substituted phenyl wherein the optional substituents are selected from methyl, ethyl, methoxy, ethoxy, tert-butoxy, F or CI. Most preferably R 2 represents wherein Me represents methyl. Preferably R bears 1, 2 or 3 substituents.
- R 3 and R 3a are independently selected from hydrogen; optionally substituted Ci- ⁇ alkyl and , optionally substituted aryl . Further preferably R 3 and R 3a are independently selected from methyl, ethyl, tert-butyl and phenyl.
- R 5 is selected from a group of formula Ill-a , Ill-g, Ill-h, or Ill-i or: III-j
- R »5 is selected from one of the following groups:
- R 5 is selected from one of the following groups:
- R 5 is selected from one of the following groups
- R 6 and R 6a each represent hydrogen and A represents Ci_ alkylene (preferably methylene).
- R 6 represents hydrogen
- R 6a represents methyl
- A represents Ci_ 4 alkylene (preferably methylene).
- R 7 is selected from hydrogen or optionally-substituted Ci- 6 alkyl. Further preferably R represents hydrogen, methyl, ethyl or tert-butyl.
- X and R represent either: - (a) X represents N and R 8 represents cyano or -C(O)O-R b ; or (b) X represents N and R 8 represents hydrogen. Further preferably X represents N and R 8 represents cyano or -C(O)O-R b ; wherein R b represents Ci- ⁇ alkyl;
- X represents N and R 8 represents -CONR b R c wherein R b and R c are as defined above.
- R 9 comprise part of the group N(R 9 R 10 ) or is hydrogen, optionally substituted aryl, an optionally substituted 3- to 10 membered heterocyclic ring or optionally substituted Ci- 4 alkyl wherein the optional substituents are selected from: hydroxy, amino, nitro, cyano, optionally-substituted aryl, optionally substituted 3- to 8- membered heterocyclyl containing from 1 to 4 heteroatoms independently selected from O, N and S, -O-R b , C(O)NR b R c , -NR b R c , -NR c C(O)-R b , -C(O)NR b R c , -NR c S(O 0 - 2 )R b , -S(O 0 - 2 )R b , wherein R b and R c are as defined above.
- R 9 is a C ⁇ . 6 a ⁇ kyl group substituted by an optionally-substituted 3 to 10 membered heterocyclic ring containing from 1 to 4 heteroatoms independently selected from O, N and S
- the heterocyclic ring is preferably selected from pyridyl, thienyl, piperidinyl, imidazolyl, triazolyl, thiazolyl, pyrrolidinyl, piperazinyl, morpholinyl, imidazolinyl, benztriazolyl, benzimidazolyl, pyrimidinyl, pyrazinyl, pyridazinyl, oxazolyl, furanyl, pyrrolyl, 1,3-dioxolanyl, 2-azetinyl, each of which is optionally substituted.
- R .16 represents hydrogen, aryl, a 3- to 10 membered heterocyclic ring or optionally substituted Ci- 4 alkyl wherein the optional substituents are selected from: hydroxy, amino, nitro, cyano, optionally-substituted phenyl, optionally substituted 3- to 8- membered heterocyclyl containing from 1 to 4 heteroatoms independently selected from O, N and S, -O-R b ,
- R 10 comprises part of the group N(R 9 R 10 ) or is optionally substituted C ⁇ - 6 alkyl. Further preferably R 10 comprises part of the group N(R 9 R 10 ) or is selected from: methyl, ethyl or tert-butyl.
- N(R 9 R 10 ) represents an optionally substituted 3- to 10- membered heterocyclic ring
- N(R 9 R 10 ) is preferably selected from a 5- or 6-membered monocyclic ring containing between 1 and 3 (preferably 1 or 2) heteroatoms independently selected from O, N and S.
- a 5- or 6-membered monocyclic ring containing between 1 and 3 (preferably 1 or 2) heteroatoms independently selected from O, N and S selected from pyrrolidinyl, thienyl, pyrazolidinyl, piperidinyl, morpholinyl, thiomorpholinyl piperazinyl, imidazole, azetidinyl or azetinyl.
- N(R 9 R 10 ) is a heterocyclic ring selected from an optionally-substituted group of formula, IV-a, IV-b, IV-c, IV-d and IV-e, wherein the optional substituents are preferably selected from the groups listed for R 1S below
- N(R 9 R 10 ) is selected from a group of formula Va, Vb or Vc:
- V-a V-b V-c Most preferably the structure N(R 9 R 10 ) is a group of formula V-c:
- R 15 represents hydrogen, optionally substituted aryl, an optionally substituted 3- to 10 membered heterocyclic ring or optionally substituted C ⁇ _ 4 alkyl wherein the optional substituents on aryl, a heterocyclic ring or Ci- 4 alkyl are selected from: hydroxy, amino, nitro, cyano, halo, optionally-substituted aryl, optionally substituted 3- to 8- membered heterocyclyl containing from 1 to 4 heteroatoms independently selected from O, N and S,
- R 15 is heterocyclyl. Further preferably R 15 is selected from: pyridyl, pyrazinyl, pyridazinyl, pyrimidinyl or thiazolyl. Most preferably R 15 is pyridyl.
- N(R 9 R 10 ) represent an optionally substituted 3- to 10- membered heterocyclic ring, wherein the optional substituents are selected from R 15 as defined above.
- R 12 and R 12a are independently selected from: hydrogen, optionally substituted C ⁇ - 6 alkyl or R 12 and R 12a together with carbon to which they are attached from an optionally substituted 3- to 6-membered cycloalkyl ring. Further preferably R 12 and R 12a are independently selected from: hydrogen, methyl, ethyl or tert-butyl. Most preferably R and R 12a are both methyl.
- R 13 and R 14 are independently selected from hydrogen, optionally substituted Ci_ 6 alkyl, optionally substituted phenyl and -R d -phenyl, where R d represents Ci_ 6 alkylene or and an optionally substituted 3- to 8- membered heterocyclic ring (preferably, a 5- or 6-membered monocyclic ring) containing from 1 to 3 (preferably 1 or 2) further heteroatoms independently selected from O, N and S.
- R 13 and R 14 are independently selected from hydrogen or C ⁇ . 6 alkyl. Where optional substitution is mentioned at various places, this refers to one, two, three or more optional substituents.
- each substituent can be independently selected from C ⁇ - 8 alkyl (eg, C 2 - 6 alkyl, and most preferably methyl, ethyl or tert-butyl); C 3 .
- cycloalkoxy preferably cyclopropoxy, cyclobutoxy or cyclopentoxy
- Ci- 6 alkoxy preferably methoxy or C 2 - 4 alkoxy
- halo preferably CI or F
- R h N- wherein R g and R h independently represent hydrogen or Ci- 8 alkyl (preferably methyl or C 2 . 6 alkyl or C 2 . 4 alkyl), or R g -R h N- represents an optionally substituted C 3 . 8 , preferably C 3 . 6 , heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S; hydrogen; or R k C(O)O- or R k C(O)-, R k representing hydrogen, optionally substituted phenyl or Ci_ 6 alkyl (preferably methyl, ethyl, ,-fo-propyl or tert-butyl).
- At least one (eg, c two or three) substituents may be provided independently selected from Ci- 6 alkyl (eg, C 2- 4 alkyl, more preferably methyl); phenyl; CF 3 O-; F CHO-; Ci-salkoxy, preferably methoxy, ethoxy or C 3 _6alkoxy; C ⁇ - 8 alkoxyC(O), preferably methoxycarbonyl, ethoxycarbonyl, tert-butoxycarbonyl or C 3 _ 6 alkoxyC(O)-; phenoxycarbonyl; phenoxy; C ⁇ _ 8 alkanoyl, preferably acetyl, ethanoyl or C 3 .
- nn is an integer between 0 and 2, preferably methylthio, ethylthio, C 3 . 6 alkylthio, methylsulphinyl, ethylsulphinyl, C 3 .
- Ci- 8 alkoxy preferably methoxy, ethoxy or C 3 . 6 alkoxy
- Ci- 8 alkylS(O) nn wherein nn is an integer between 0 and 2, preferably methylthio, ethylthio, C 3 - 6 alkylthio, methylsulphinyl, ethylsulphinyl, C 3 . 6 alkylsulphinyl, methylsulphonyl, ethylsulphonyl or C 3 . 6 alkylsulphonyl; halo (eg, F, CI or Br); cyano; and NO2.
- halo eg, F, CI or Br
- a preferred group of compounds of the invention comprise compounds of Formula (I) wherein:
- R 11 is a group of the formula: N(R 9 R 10 );
- N(R 9 R 10 ) represents an optionally-substituted 3- to 8- membered heterocyclic ring optionally containing from 1 to 3 further heteroatoms independently selected from O, N and S, preferably substituted by heterocyclyl; or a salt, pro-drug or solvate thereof.
- a preferred group of compounds of the invention comprise compounds of Formula (I) wherein: R 11 is a group of the formula: N(R 9 R 10 );
- R 9 is a Ci-ealkyl group substituted by an optionally-substituted 3 to 8 membered heterocyclic ring containing from 1 to 4 heteroatoms independently selected from O, N and S; and R 10 represents hydrogen or Ci_ 6 alkyl or a salt, pro-drag or solvate thereof.
- a preferred group of compounds of the invention comprises a compound of Formula
- A, B, M, X, R ⁇ R 5 , R 6 , R 6a , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 and R 12a are as defined above; or a salt, pro-drug or solvate thereof.
- a preferred group of compounds of the invention comprises a compound of Formula (lb):
- R 5 3 is selected from: Ilia, Illb, Illg, Illi or IIIj
- R 1 , R s , R 6 , R 6a , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 12a , R 13 , and R 14 are as defined above; or a salt, pro-drug or solvate thereof.
- a further preferred group of compounds of the invention comprises a compound of Formula (Ic):
- R 5 is selected from a Ilia, Illb, Illg, Illi or IIIj:
- R 1 , R 6 , R 6a , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 , R 12a , R 13 , and R 14 are as defined above; or a salt, pro-drug or solvate thereof.
- a yet further preferred group of compounds of the invention comprises a compound of Formula (la), (lb) or (Ic) wherein: R 5 is a group of formula Ilia:
- NR 13 (-R 14 ) represents an optionally substituted 7- to 8- membered bicyclic heterocyclic ring and A, B, X, R 1 , M, R 6 , R 6a , R 7 , R 8 , R 9 , R 10 , R 11 , R 12 and R 12a are as defined above; or a salt, pro-drag or solvate thereof.
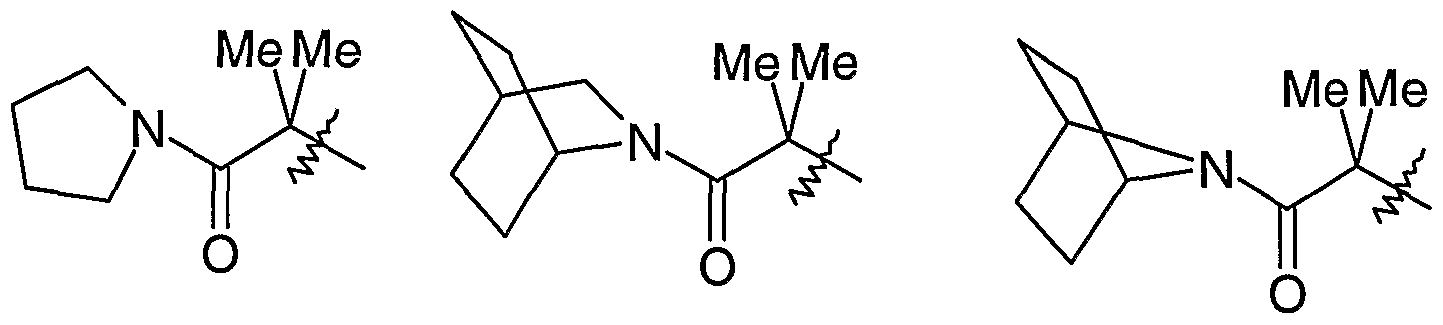
- a preferred compound according to the present invention is: 3-[2,2-dimethyl-3-oxo-3-(azabicyclo[2.2.1]heptan-7-yl)propyl]- 4-[lS-methyl-2-(N'-isopro ⁇ oxycarbonyl-3-pyrid-4-yl-pyrrolidin-l-ylc_-rboximidamido) ethyl]-5-(3,5-dimethylphenyl)-lH-pyrazole; or a salt, pro-drug or solvate thereof.
- particularly preferred compounds according to the present invention are wherein the compound is selected from: isopropyl (lZ)-( ⁇ 2-[3-(2,2-dimethyl-3-oxo-3-pyrrolidin-l-ylpropoxy)-5-(3,5- dimethylphenyl)- lH-pyrazol-4-yl] ethyl ⁇ amino)(3 -pyridin-4-ylpyrrolidin- 1 - yl)methylidenecarbamate; isopropyl (lZ)-( ⁇ 2-[3-(2,2-dimethyl-3-oxo-3-(7-azabicyclo[2.2.
- a pharmaceutical formulation comprising a compound of Formula (la), Formula (lb), Formula (Ic) or preferred compounds of the invention, or salt, pro-drug or solvate thereof, and a pharmaceutically acceptable diluent or carrier.
- a sex hormone related condition in the patient preferably a sex hormone related condition selected from prostate cancer and pre- menopausal breast cancer.
- the compounds of Formula (I) may be administered in the form of a pro-drug which is broken down in the human or animal body to give a compound of the Formula (I).
- pro-drags include in- vivo hydrolysable esters of a compound of the Formula (I). Narious forms of pro-drugs are known in the art. For examples of such pro-drag derivatives, see: a) Design of Prodrags, edited by H.
- Bundgaard (Elsevier, 1985) and Methods in Enzymology, Nol. 42, p. 309-396, edited by K. Widder, et al. (Academic Press, 1985); b) A Textbook of Drug Design and Development, edited by Krogsgaard-Larsen and H. Bundgaard, Chapter 5 "Design and Application of Prodrags", by H. Bundgaard p. 113- 191 (1991); c) H. Bundgaard, Advanced Drug Delivery Reviews, 8, 1-38 (1992); d) H. Bundgaard, et al., Journal of Pharmaceutical Sciences, 77, 285 (1988); and e) ⁇ . Kakeya, et al., Chem Pharm Bull, 32, 692 (1984).
- An in- vivo hydrolysable ester of a compound of the Formula (I) containing a carboxy or a hydroxy group is, for example, a pharmaceutically-acceptable ester which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
- Suitable pharmaceutically-acceptable esters for carboxy include C ⁇ - 6 alkoxymethyl esters for example methoxymethyl, Ci_ 6 alkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters, C 3 . 8 cycloalkoxycarbonyloxyC ⁇ . alkyl esters for example
- An in- vivo hydrolysable ester of a compound of the Formula (I) containing a hydroxy group includes inorganic esters such as phosphate esters (including phosphoramidic cyclic esters) and ⁇ -acyloxyalkyl ethers and related compounds which as a result of the in- vivo hydrolysis of the ester breakdown to give the parent hydroxy group/s.
- Examples of ⁇ -acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxy-methoxy.
- a selection of in- vivo hydrolysable ester forming groups for hydroxy include alkanoyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl, alkoxycarbonyl (to give alkyl carbonate esters), dialkylcarbamoyl and ⁇ -(dialkylaminoethyl)- ⁇ -alkylcarbamoyl (to give carbamates), dialkylaminoacetyl and carboxyacetyl.
- a suitable pharmaceutically-acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifluoroacetic, citric or maleic acid.
- a suitable pharmaceutically-acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, Irimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation
- a salt with methylamine, dimethylamine, Irimethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine for example a salt with methylamine, dimethylamine, Irimethylamine, piperidine, morpholine
- the compounds of Formula (I) can be prepared by a process comprising a step selected from (a) to (f) as follows, these processes are provided as a further feature of the invention:- (a) for compounds wherein X is N and R is CN, reaction of a compound of formula XXXII as follows
- XXXXI XXXX1I and thereafter if necessary: i) converting a compound of the Formula (I) into another compound of the Formula (I); ii) removing any protecting groups; iii) forming a salt, pro-drug or solvate.
- certain functional groups such as hydroxyl or amino groups in the starting reagents or intermediate compounds may need to be protected by protecting groups.
- the preparation of the compounds of Formula (I) may involve, at an appropriate stage, the addition and subsequent removal of one or more protecting groups.
- the protection and de-protection of functional groups is described in 'Protective Groups in Organic Chemistry', edited by J.W.F. McOrnie, Plenum Press (1973) and 'Protective Groups in Organic Synthesis', 2nd edition, T.W. Greene and P.G.M. Wuts, Wiley-Interscience (1991).
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or tert-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the de- protection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a tert-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example dimethylaminopropylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the de-protection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a tert-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a tert-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
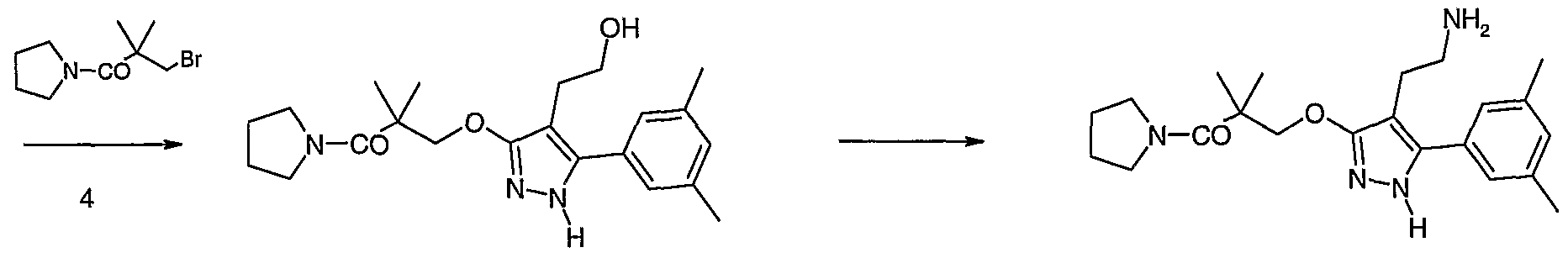
- the amine 6 can be prepared from a compound of formula 5 and phthalimide using a Mitsunobu reaction with an activating agent such as diethyldiazocarboxylate (DEAD), diisopropyldiazocarboxylate or the like with triphenylphosphine, tri-butylphosphine and the like, in an inert solvent such as benzene, toluene, tetrahydrofuran or mixtures thereof, followed by deprotection with hydrazine (Scheme b).
- DEAD diethyldiazocarboxylate
- diisopropyldiazocarboxylate or the like with triphenylphosphine, tri-butylphosphine and the like
- an inert solvent such as benzene, toluene, tetrahydrofuran or mixtures thereof, followed by deprotection with hydrazine (Scheme b).
- a suitable pyrazole 6 can be converted to a urea by either direct treatment with an iso- cyanate in an inert solvent such as methylene chloride, chloroform or THF and the such like, or by a two step procedure of reaction with triphosgene (6— >7) followed by addition of an amine (7— >8), bearing the required substitution to yield 8 (Scheme c).
- a suitable pyrazole (6) can be converted to a guandine or guanidine derivative (12) by reaction with a suitable isothiocyanate (9) to form a compound of formula 10, followed by displacement by a suitable amine (11) (Scheme d).
- R 2 is as defined above; and R x and R y are independently selected from: optionally substituted alkyl, optionally substituted aryl or optionally substituted heteocyclyl.
- the starting material Bb4 was prepared as follows:
- Example 5.1 shows the R group relating to the above structure, the reaction conditions and characteristics for each example, corresponding to the description of the preparation of Example 5 given above:- Example 5.1
- Example 6 A solution of Ce (170 mg ; 0.4 mmol) in CH 2 C1 2 (5 ml) was cooled to 0°C and DIEA (140 ⁇ ; 0.8 mmol) was added. A solution of 4-nitrophenyl chloroformate (85 mg ; 0.42 mmol) in CH 2 C1 2 (1 ml) was added and the mixture was allowed to stir for 30 min. 4-(3-Pyrrolidyl)- pyridine (71 mg ; 0.48 mmol) was added and the mixture allowed to warm to room temperature for 1 h. The mixture was directly purified by flash chromatography eluting with MeOH/CH 2 Cl 2 (0-10% MeOH) to give Example 6 as a pale yellow powder (212 mg). Yield : 88%
- the table shows the R group relating to the above structure, the reaction conditions and characteristics for each example, corresponding to the description of the preparation of
- a compound of Formula (I) is provided as medicaments for antagonising gonadotropin releasing hormone (GnRH) activity in a patient, eg, in men and/or women.
- a compound of Formula (I) can be provided as part of a pharmaceutical formulation which also includes a pharmaceutically acceptable diluent or carrier (eg, water).
- the formulation may be in the form of tablets, capsules, granules, powders, syrups, emulsions (eg, lipid emulsions), suppositories, ointments, creams, drops, suspensions (eg, aqueous or oily suspensions) or solutions (eg, aqueous or oily solutions).
- the formulation may include one or more additional substances independently selected from stabilising agents, wetting agents, emulsifying agents, buffers, lactose, sialic acid, magnesium stearate, terra alba, sucrose, corn starch, talc, gelatin, agar, pectin, peanut oil, olive oil, cacao butter and ethylene glycol.
- stabilising agents wetting agents, emulsifying agents, buffers, lactose, sialic acid, magnesium stearate, terra alba, sucrose, corn starch, talc, gelatin, agar, pectin, peanut oil, olive oil, cacao butter and ethylene glycol.
- the patient may receive a daily dose of O.lmgkg "1 to 30mgkg _1 (preferably, 5mgkg _1 to 20mgkg "1 ) of the compound, the compound being administered 1 to 4 times per day.
- the intravenous, subcutaneous and intramuscular dose may be given by means of a bolus injection.
- the intravenous dose may be given by continuous infusion over a period of time.
- the patient may receive a daily oral dose which is approximately equivalent to the daily parenteral dose, the composition being administered 1 to 4 times per day.
- a suitable pharmaceutical formulation is one suitable for oral administration in unit dosage form, for example as a tablet or capsule, which contains between lOmg and lg (preferably, 100 mg and lg) of the compound of the invention.
- Buffers eg, pharmaceutically acceptable co-solvents (eg, polyethylene glycol, propylene glycol, glycerol or EtOH) or complexing agents such as hydroxy-propyl ⁇ cyclodextrin may be used to aid formulation.
- pharmaceutically acceptable co-solvents eg, polyethylene glycol, propylene glycol, glycerol or EtOH
- complexing agents such as hydroxy-propyl ⁇ cyclodextrin may be used to aid formulation.
- One aspect of the invention relates to the use of compounds according to the invention for reducing the secretion of LH and/or FSH by the pituitary gland of a patient.
- the reduction may be by way of a reduction in biosynthesis of the LH and FSH and/or a reduction in the release of LH and FSH by the pituitary gland.
- compounds according to the invention can be used for therapeutically treating and/or preventing a sex hormone related condition in the patient.
- preventing we mean reducing the patient's risk of contracting the condition.
- treating we mean eradicating the condition or reducing its severity in the patient.
- sex hormone related conditions are: a sex hormone dependent cancer, benign prostatic hypertrophy, myoma of the uterus, endometriosis, polycystic ovarian disease, uterine fibroids, prostatauxe, myoma uteri, hirsutism and precocious puberty.
- sex hormone dependent cancers are: prostatic cancer, uterine cancer, breast cancer and pituitary gonadotrophe adenoma.
- the compounds of the invention may be used in combination with other drugs and therapies used to treat / prevent sex-hormone related conditions. If formulated as a fixed dose such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically-active agent within its approved dosage range. Sequential use is contemplated when a combination formulation is inappropriate.
- anti-angiogenic agents for example linomide, inhibitors of integrin c.v ⁇ 3 function, angiostatin, endostatin, razoxin, thalidomide
- NEGF vascular endothelial growth factor receptor tyrosine kinase inhibitors
- cytostatic agents such as anti-oestrogens (for example tamoxifen, toremifene, raloxifene, droloxifene, iodoxyfene), progestogens (for example megestrol acetate), aromatase inhibitors (for example anastrozole, letrozole, vorazole, exemestane), anti- progestogens, anti-androgens (for example flutamide, nilutamide, bicalutamide, cyproterone acetate), inhibitors of testosterone 5 -dihydroreductase (for example finasteride), anti- invasion agents (for example metalloproteinase inhibitors like marimastat and inhibitors of urokinase plasminogen
- the compounds of the invention may also be used in combination with surgery or radiotherapy.
- the IC50 of the test compound can be determined as the concentration of the compound required to inhibit radio-ligand binding to GnRH receptors by 50%.
- Compounds according to the present invention have activity at a concentration from InM to 5 ⁇ M.
- Binding Assay Using Human GnRH Receptor Crude membranes prepared from CHO cells expressing human GnRH receptors are sources for the GnRH receptor.
- the binding activity of compounds according to the invention can be determined as an IC50 which is the compound concentration required to inhibit the
- the LH release assay can be used to demonstrate antagonist activity of compounds, as demonstrated by a reduction in GnRH-induced LH release.
- Suitable rats are Wistar male rats (150-200g) which have been maintained at a constant temperature (eg, 25°C) on a 12 hour light/12 hour dark cycle. The rats are sacrificed by decapitation before the pituitary glands are aseptically removed to tube containing Hank's Balanced Salt Solution (HBSS).
- HBSS Hank's Balanced Salt Solution
- the glands are further processed by:-
- test compound is dissolved in DMSO to a final concentration of 0.5% in the incubation medium.
- each well is analysed by removing the medium from the well and centrifuging the medium at 2000 x g for 15 minutes to remove any cellular material. The supernatant is removed and assayed for LH content using a double antibody radio-immuno assay. Comparison with a suitable control (no test compound) is used to determine whether the test compound reduces LH release.
- Compounds according to the present invention have activity at a concentration from InM to 5 ⁇ M.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Detergent Compositions (AREA)
- Pyridine Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description



















































Claims











Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE60314158T DE60314158T2 (en) | 2002-08-21 | 2003-08-19 | PYRAZONE DERIVATIVES AS GNRH INHIBITORS |
| AU2003255811A AU2003255811A1 (en) | 2002-08-21 | 2003-08-19 | Pyrazole derivatives as gnrh inhibitors |
| EP03792484A EP1539743B1 (en) | 2002-08-21 | 2003-08-19 | Pyrazole derivatives as gnrh inhibitors |
| JP2004530359A JP2006501231A (en) | 2002-08-21 | 2003-08-19 | Pyrazole derivatives as GNRH inhibitors |
| US10/525,111 US7253290B2 (en) | 2002-08-21 | 2003-08-19 | Pyrazole derivatives as GnRH inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP02292075 | 2002-08-21 | ||
| EP02292075.5 | 2002-08-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004018459A1 true WO2004018459A1 (en) | 2004-03-04 |
Family
ID=31896980
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2003/003623 Ceased WO2004018459A1 (en) | 2002-08-21 | 2003-08-19 | Pyrazole derivatives as gnrh inhibitors |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US7253290B2 (en) |
| EP (1) | EP1539743B1 (en) |
| JP (1) | JP2006501231A (en) |
| AT (1) | ATE363481T1 (en) |
| AU (1) | AU2003255811A1 (en) |
| DE (1) | DE60314158T2 (en) |
| ES (1) | ES2286493T3 (en) |
| WO (1) | WO2004018459A1 (en) |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997044339A1 (en) * | 1996-05-20 | 1997-11-27 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| WO2000004013A1 (en) * | 1998-07-14 | 2000-01-27 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1208412A (en) | 1995-12-14 | 1999-02-17 | 麦克公司 | GnRH antagonists |
| SK77598A3 (en) | 1995-12-14 | 1999-01-11 | Merck & Co Inc | Nonpeptide derivatives, pharmaceutical composition containing them and their use |
| WO1997021435A1 (en) | 1995-12-14 | 1997-06-19 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| JP3092946B2 (en) | 1995-12-14 | 2000-09-25 | メルク エンド カンパニー インコーポレーテッド | Gonadotropin-releasing hormone antagonist |
| EP0986385A4 (en) | 1997-06-05 | 2001-05-16 | Merck & Co Inc | GONADOLIBERINE ANTAGONISTS |
| WO1998055123A1 (en) | 1997-06-05 | 1998-12-10 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| WO1998055479A1 (en) | 1997-06-05 | 1998-12-10 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| AU729752B2 (en) | 1997-06-05 | 2001-02-08 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| US6156772A (en) | 1997-06-05 | 2000-12-05 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| AU7806998A (en) | 1997-06-05 | 1998-12-21 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| JP2001520997A (en) | 1997-10-28 | 2001-11-06 | メルク エンド カムパニー インコーポレーテッド | Gonadotropin-releasing hormone antagonist |
| CA2308454A1 (en) | 1997-10-28 | 1999-05-06 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| CA2317451A1 (en) | 1998-02-11 | 1999-08-19 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| JP2002503661A (en) | 1998-02-11 | 2002-02-05 | メルク エンド カムパニー インコーポレーテッド | Gonadotropin-releasing hormone antagonist |
| JP2002510631A (en) | 1998-04-02 | 2002-04-09 | メルク エンド カムパニー インコーポレーテッド | Gonadotropin-releasing hormone antagonist |
| WO1999051233A1 (en) | 1998-04-02 | 1999-10-14 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| CA2326143A1 (en) | 1998-04-02 | 1999-10-14 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| AU3117899A (en) | 1998-04-02 | 1999-10-25 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| AU3367999A (en) | 1998-04-02 | 1999-10-25 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| WO1999051595A1 (en) | 1998-04-02 | 1999-10-14 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| AU3514200A (en) | 1999-03-10 | 2000-09-28 | Merck & Co., Inc. | 6-azaindole compounds as antagonists of gonadotropin releasing hormone |
| JP2002538204A (en) | 1999-03-10 | 2002-11-12 | メルク エンド カムパニー インコーポレーテッド | 6-azaindole compounds as gonadotropin-releasing hormone antagonists |
| EP1171125A1 (en) | 1999-03-10 | 2002-01-16 | Merck & Co., Inc. | 6-azaindole compounds as antagonists of gonadotropin releasing hormone |
| JP2002538206A (en) | 1999-03-10 | 2002-11-12 | メルク エンド カムパニー インコーポレーテッド | 6-azaindole compounds as gonadotropin-releasing hormone antagonists |
| EP1161431A4 (en) | 1999-03-10 | 2002-04-24 | Merck & Co Inc | 6-AZAINDOLE COMPOUNDS FOR USE AS GONADOTROPIN RELEASING HORMONE ANTAGONISTS |
| CA2367121A1 (en) | 1999-03-10 | 2000-09-14 | Merck & Co., Inc. | 6-azaindole compounds as antagonists of gonadotropin releasing hormone |
| SE0100566D0 (en) | 2001-02-20 | 2001-02-20 | Astrazeneca Ab | Compounds |
| SE0101692D0 (en) | 2001-05-14 | 2001-05-14 | Astrazeneca Ab | Compounds |
-
2003
- 2003-08-19 DE DE60314158T patent/DE60314158T2/en not_active Expired - Fee Related
- 2003-08-19 WO PCT/GB2003/003623 patent/WO2004018459A1/en not_active Ceased
- 2003-08-19 AU AU2003255811A patent/AU2003255811A1/en not_active Abandoned
- 2003-08-19 US US10/525,111 patent/US7253290B2/en not_active Expired - Fee Related
- 2003-08-19 AT AT03792484T patent/ATE363481T1/en not_active IP Right Cessation
- 2003-08-19 JP JP2004530359A patent/JP2006501231A/en active Pending
- 2003-08-19 ES ES03792484T patent/ES2286493T3/en not_active Expired - Lifetime
- 2003-08-19 EP EP03792484A patent/EP1539743B1/en not_active Expired - Lifetime
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997044339A1 (en) * | 1996-05-20 | 1997-11-27 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
| WO2000004013A1 (en) * | 1998-07-14 | 2000-01-27 | Merck & Co., Inc. | Antagonists of gonadotropin releasing hormone |
Also Published As
| Publication number | Publication date |
|---|---|
| DE60314158T2 (en) | 2008-01-31 |
| US7253290B2 (en) | 2007-08-07 |
| AU2003255811A1 (en) | 2004-03-11 |
| ATE363481T1 (en) | 2007-06-15 |
| ES2286493T3 (en) | 2007-12-01 |
| JP2006501231A (en) | 2006-01-12 |
| EP1539743B1 (en) | 2007-05-30 |
| US20050239858A1 (en) | 2005-10-27 |
| DE60314158D1 (en) | 2007-07-12 |
| EP1539743A1 (en) | 2005-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| BRPI0708016A2 (en) | compound or a salt, solvate or n-oxide thereof, pharmaceutical composition, use of a compound or a salt, solvate or n-oxide thereof, and method for treating and / or preventing disorders | |
| EP1531811B1 (en) | Derivatives of 3-hydroxy-4-(cyclyl-alkylaminoalkyl)-5-phenyl-1h-pyrazole as antagonists of the gonadotropin releasing hormone (gnrh) for use in the treatment of sex hormone related conditions, such as prostatic of uterine cancer | |
| EP1532154B1 (en) | Thieno-pyrrole compounds as antagonists of gonadotropin releasing hormone | |
| JP4216608B2 (en) | Compound | |
| EP1543012B1 (en) | 6h-thien 2, 3-b]pyrrole derivatives as antagonists of gonadotropin releasing hormone (gnrh) | |
| EP1539743B1 (en) | Pyrazole derivatives as gnrh inhibitors | |
| US7449489B2 (en) | Indolylalkylamino-methylidenecarbamate derivatives useful as GnRH antagonists | |
| JP2007523145A (en) | Pyrrole derivatives as gonadotropin releasing hormone (GNRH) antagonists | |
| EP1730155A1 (en) | Thienopyrroles as antagonists of gnrh | |
| US20070185185A1 (en) | Derivatives of thienopyrrole as gnrh antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2003792484 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10525111 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004530359 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003792484 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003792484 Country of ref document: EP |