DERIVES D ' INDAZθLECARBOXAMIDES , LEUR PREPARATION ET LEUR UTILISATION COMME INHIBITEURS DES CD 1 , CDK2 ET CDK4
L'invention a pour objet des dérivés de 7/-/-indazole-3-carboxamides, leur préparation et leur utilisation en thérapeutique.
Certains dérivés de . H-indazole-3-carboxamides ont été décrits dans l'art antérieur.
Le N-(4-méthylbenzyl)-7H-indazole-3-carboxamide a été décrit dans J. Gen.
Chem. USSR, 32, 78 (1962) et n'a montré aucune activité pharmacologique.
Par ailleurs, le brevet US 3,457,269 décrit des . /-/-indazoIe-3-carboxamides utiles comme agents hypotenseurs.
Il existe toujours une nécessité de trouver et de développer des produits inhibiteurs de cyclin dépendent kinases (cdks) telles que les cdkl , cdk2 et cdk4.
L'invention répond à ce but en proposant des dérivés de 7/-/-indazole-3- carboxamides, qui présentent des effets d'inhibition des cdkl , cdk2 et cdk4.
L'invention a pour premier objet des composés répondant à la formule générale (I) :
(D
dans laquelle, Ri représente un atome d'hydrogène, d'halogène, un NH2, NHR2, NHCOR2, NO2, CN, CH2NH2,
CH2NHR2 ; ou bien Ri représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, Cι-6 alkyle, Ci ,6 alcoxy, NH2, NHR2, NR2R3;
ou bien R^ représente un groupe heteroaromatique éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, un groupe heteroaromatique, Cι-6 alkyle, un NH2) NHR2, NHCOR2, COOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; Ar représente un groupe phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C1-6 alkyle, C-i _6 thioalkyle, Cι-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2) NHR2, NR2R3, NHSO2R2, CN, S02R2, S02NH2, S02NHR2, COOH, COOR2, CONH2, CONHNHs, CONHR2> CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C-i .6 alkyle, Cι-6 thioalkyler C1-6 alcoxy, CHfeOHr-phénoxy, morpholinyle, -CH2-morpholinyle, NH2) NHR2, NR2R3, NHSO2R2, CN, SO2R2, S02NH2, S02NHR2, COOH, COOR2) CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ;
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un groupe CONH2) par un phényle ou par un groupe heteroaromatique ; ou R et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique ; n représente 0, 1 , 2 ou 3.
Les composés suivants ne font pas partie de l'invention :
- N-phényl- . H-indazole-3-carboxamide ;
- N-(2-chlorophényl)- 7H-indazole-3-carboxamide
- N-(3-chlorophényl)- 7 H-indazole-3-carboxamide
- N-(4-chlorophényl)- 7 H-indazole-3-carboxamide
- N-(2,4-dichlorophényl)- . H-indazole-3-car boxamide ;
- N-(3,4-dichlorophényl)- 7 H-indazole-3-carboxamide ;
- N-(2-méthylphényl)-7/-/-indazole-3-carboxamide ;
- N-(2-méthoxyphényl)- 7/-/-indazole-3-carboxamide ;
- N-(4-méthoxyphényl)- 7H-indazole-3-carboxamide ;
- N-(4-thiométhylphényl)-7 -/-indazole-3-carboxamide ;
- N-(3-chloro-4-thiométhylphényl)-5-amino- 7/-/-indazole-3-carboxamide
- N-benzyl- 7H-indazole-3-carboxamide ;
- N-(2-chlorobenzyl)- 7H-indazole-3-carboxamide ;
- N-(4-méthylbenzyl)- 7H-indazole-3-carboxamide ;
- N-(pyridin-2-ylméthyl)- 7H-indazole-3-carboxamide ;
- N-(pyridin-3-ylméthyl)- 7H-indazole-3-carboxamide ;
- N-(pyridin-4-ylméthyl)- 7H-indazole-3-car boxamide ;
- N-(2-phényléthyl)-7H-indazole-3-carboxamide ;
- N-(3,4-diméthoxyphényléthyl)-7H-indazole-3-carboxamide ;
- N-[3-(ρyridin-2-yl)propyl]- 7H-indazole-3-carboxamide ; - N-[3-(2,6-diméthylphényl)propyl]-5-nitro- 7H-indazole-3-carboxamide.
Parmi les composés de formule générale (I), une première famille de composés préférés est constituée des composés pour lesquels : Ri représente un atome d'hydrogène, d'halogène, un NH2) NHR2, NHCOR2) N02, CN, CH2NH2j CH2NHR2 ; ou bien R-i représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, Cι-4 alkyle, Cι-6 alcoxy, NH2) NHR2, NR2R3; ou bien Ri représente un groupe heteroaromatique éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, un groupe heteroaromatique, un Cι-6 alkyle, un NH2, NHR2, NHCOR2> COOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; et/ou Ar représente un groupe phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C-ι-6 alkyle, C1-6 thioalkyle, Cι-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3, NHSO2R2, CN, SO2R2, S02NH2l S02NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un Cι-6 alkyle, C1-6 thioalkyle, Cι-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2) NR2R3, NHS02R2, CN, S02R2, S02NH2) S02NHR2, COOH, COOR≥, CONH2, CONHNH2) CONHR2> CH2NHR2, CH2NR R3 ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un groupe CONH2, par un phényle ou par un groupe heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3 ; avec la condition que
- quand Ri représente un atome d'hydrogène
si n représente 0 et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, les substituants méthyle, méthoxy, thiométhyle, atome de chlore étant exclus ; si n représente 1 , alors et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, les substituants méthyle et atome de chlore étant exclus ; si n représente 1 , alors et Ar est un pyridinyle, alors le pyridinyle est obligatoirement substitué tel que défini ci-dessus ; si n représente 2 et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, le substituant méthoxy étant exclu ; si n représente 3 et Ar est un pyridinyle, alors le pyridinyle est obligatoirement substitué tel que défini ci-dessus ; - — --quand R-i représente un NH2 si n représente 0 et Ar est un phényle, alors le ou les substituants du phényle ne peuvent être choisis parmi un thiométhyle ou un atome de chlore ;
- quand R-i représente un NO2 si n représente 3 et Ar est un phényle, alors le ou les substituants du phényle de Ar ne peuvent être un méthyle .
Parmi les composés de formule générale (I), une seconde famille de composés préférés est constituée des composés pour lesquels :
- quand Ri représente un atome d'hydrogène, alors Ar représente un groupe phényle substitué par un ou deux substituants choisis parmi un atome de brome ou d'iode, un C2-6 alkyle, C2-6 thioalkyle, C2-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3, NHS02R2, CN, S02R2, S02NH2, S02NHR2, COOH, COOR2, CONH2) CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique choisi parmi un pyrrolyle, imidazolyle, pyrazolyle, triazolyle, tetrazolyle, thiazolyle, isothiazolyle, 1 ,3,4- thiadiazolyle, oxazolyle, isoxazolyle, oxadiazolyle, pyridazinyle, pirimidinyle, pyrazinyle, 1 ,3,5-triazinyle, indolyle, isoindolyle, indazolyle, benzimidazolyle, benzothiazolyle, quinolinyle, isoquinolinyle, pyrrolo[2,3-c]pyridinyle, éventuellement substitué par un ou deux substituants ; ou Ar représente un pyridinyle substitué par un ou deux substituants ; les substituants étant choisis parmi un atome d'halogène, un Cι-6 alkyle, Cι-6 thioalkyle, C-|.6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3, NHS02R2, CN,
SO2R2, S02NH2, SO2NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un Ci .6 alkyle éventuellement substitué par un groupe CONH2, par un phényle ou par un groupe heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3 ;
- quand Ri représente un atome d'halogène, un NH2, NHR2, NHCOR2, N02, CN, CH2NH2, CH2NHR2 ; ou bien Ri représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, C1-6 alkyle, Cι-β alcoxy, NH2, NHR2, NR2R3; ou bien Ri représente un groupe hétéroaromatique-éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, un groupe heteroaromatique, un Cι-6 alkyle, un NH2, NHR2, NHCOR2, COOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; alors Ar représente un groupe phényle éventuellement substitué par un ou deux substituants choisis parmi un atome de brome ou d'iode, un C2.6 alkyle, C2.6 thioalkyle, C2-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3, NHS02R2, CN, SO2R2, SO2NH2, S02NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2> CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C1.6 alkyle, Cι-6 thioalkyle, Cι-6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3> NHSO2R2, CN, S02R2, S02NH2, SO2NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; et/ou R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un groupe CONH2, par un phényle ou par un groupe heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3.
Parmi les composés de formule générale (I), une troisième famille de composés préférés est constituée des composés pour lesquels :
- quand Ri représente un atome d'hydrogène, alors Ar représente
un phényle substitué par un ou deux substituants choisis parmi un atome de brome, un CH2OH, phénoxy, NH2, NHR2, NR2R3, CN, S02NH2, COOH, COOR2, CONH2 ; ou bien Ar représente un groupe heteroaromatique choisi parmi un imidazolyle, 1 ,3,4-thiadiazolyle, pyrazinyle, indolyle, indazolyle, quinolinyle, isoquinolinyle, éventuellement substitué par un ou deux substituants ; ou Ar représente un pyridinyle substitué par un ou deux substituants ; les substituants étant choisis parmi un atome d'halogène, plus particulièrement un chlore, un COOH, un Ci .6 alkyle, plus particulièrement un méthyle, un Cι-6 alcoxy, plus particulièrement un méthoxy ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un C -6 alkyle, plus particulièrement un méthyle ou un éthyle ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ; et/ou n représente 0, 2 ou 3 ; - quand Ri représente un atome d'halogène, plus particulièrement un brome ou un iode, un NH2, NHCOR2, N02) CN, CH2NH2 ; ou bien Ri représente un phényle; ou bien Ri représente un groupe heteroaromatique, plus particulièrement un pyrazolyle, tetrazolyle, thiazolyle, oxazolyle, pyridinyle, pyrimidinyle, quinolinyle, isoquinolinyle, pyrrolo[2,3-c]pyridinyle, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, plus particulièrement un chlore ou un fluor, un hydroxy, un groupe heteroaromatique, plus particulièrement un pyridinyle, un Cι-6 alkyle, plus particulièrement un méthyle, un NH2, NHR2, NHCOR2, COOR2, CONH2, CONHR2, CH2OR2 ; alors Ar représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, plus particulièrement un chlore ou un fluor, un morpholinyle, -CH2-morpholinyle, NHSO2R2, CN, S02R2, S02NH2, S02NHR2, COOH, CONHNH2, CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique, plus particulièrement un pyridinyle, éventuellement substitué par un Cι-6 alcoxy, de préférence un méthoxy ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un C1-6 alkyle, plus particulièrement un méthyle, éthyle ou 2-méthylpropyle, éventuellement substitué par un groupe CONH2 ou par un phényle ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique, plus particulièrement un pyridinyle ou pyrimidinyle ; et/ou n représente 0 ou 1.
Parmi les composés de formule générale (I), une quatrième famille de composés particulièrement préférés est constituée des composés pour lesquels : Ri représente un groupe heteroaromatique éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, un groupe heteroaromatique, Cι-6 alkyle, un NH2, NHR2, NHCOR , COOR , CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; et/ou Ar représente un groupe phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C1-6 alkyle, Cι-β thioalkyle, Ci-β alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle, NH2, NHR2, NR2R3, NHSO2R2, CN- SO2R2, S02NH2,— S02NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; ou bien Ar représente un groupe heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C -6 alkyle, Cι-β thioalkyle, Ci .6 alcoxy, CH2OH, phénoxy, morpholinyle, -CH2-morpholinyle NH2, NHR2, NR2R3, NHS02R2, CN, S02R2, S02NH2, S02NHR2, COOH, COOR2, CONH2, CONHNH2, CONHR2, CH2NHR2, CH2NR2R3 ; et/ou R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un groupe CONH2, par un phényle ou par un groupe heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un groupe heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3.
Parmi les composés de formule générale (I), une cinquième famille de composés particulièrement préférés est constituée des composés pour lesquels : Ri représente un groupe heteroaromatique, plus particulièrement un pyrazolyle, thiazolyle, oxazolyle, pyridinyle, isoquinolinyle, pyrrolo[2,3-c]pyridinyle, éventuellement substitué par un ou deux substituants choisis . parmi un atome d'halogène, plus particulièrement un chlore, un groupe heteroaromatique, plus particulièrement un pyridinyle, un C1-6 alkyle, plus particulièrement un méthyle, un NH2, CONHR2 ; et/ou Ar représente un phényle éventuellement substitué par un ou deux substituants choisis parmi morpholinyle, -CH2-morpholinyle, NHS02R2, CN, S02R2, S02NH2, S02NHR2, COOH, CH2NHR2, CH2NR2R3 ;
ou bien Ar représente un groupe heteroaromatique, plus particulièrement un pyridinyle, éventuellement substitué par un C1-6 alcoxy, de préférence un méthoxy ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle, plus particulièrement un méthyle ou un éthyle, éventuellement substitué par un groupe CONH2 ou par un phényle ; ou R2 et R3 représentent indépendamment l'un de l'autre un groupe heteroaromatique, plus particulièrement un pyridinyle ou pyrimidinyle ; et/ou n représente 0 ou 1.
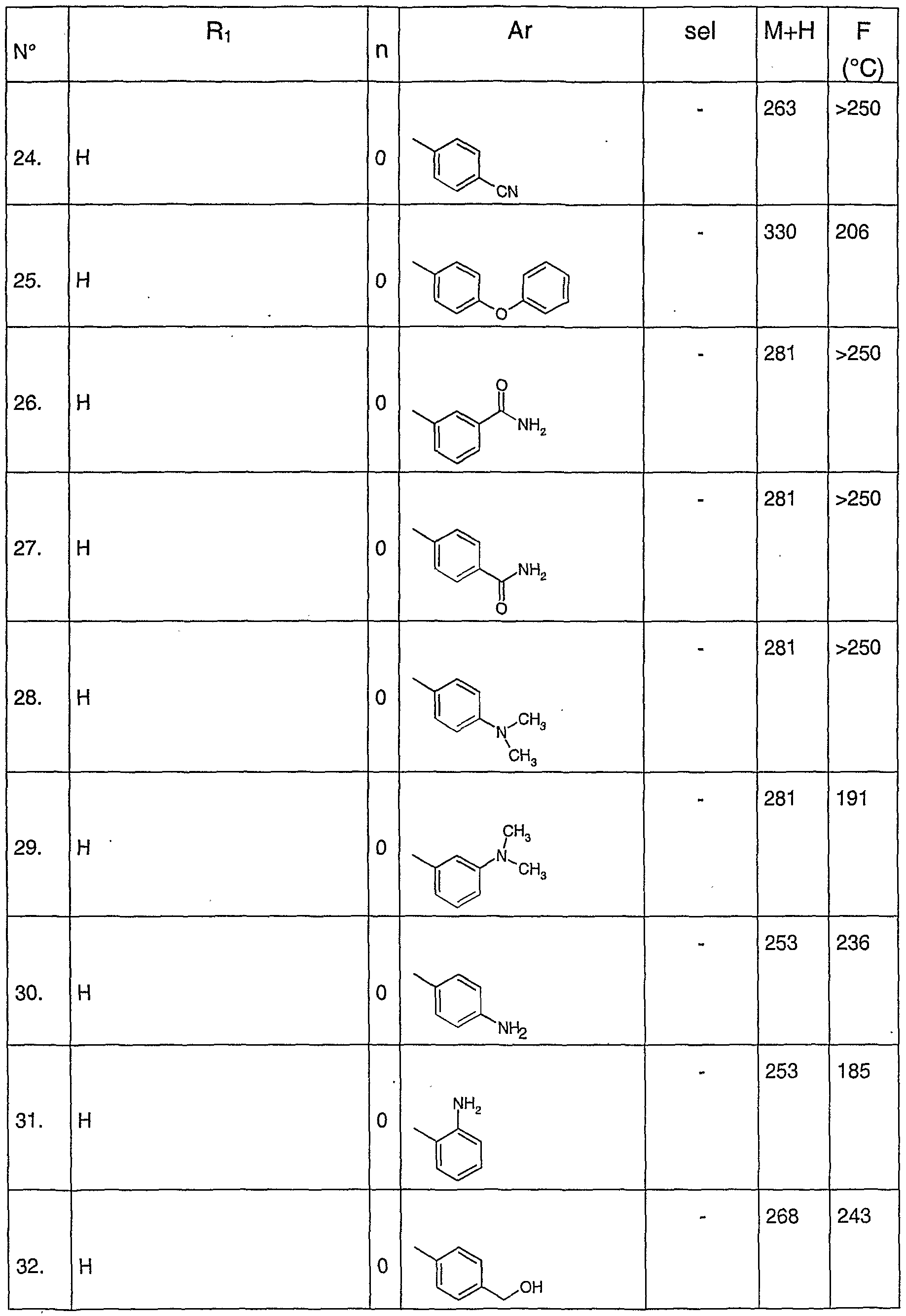
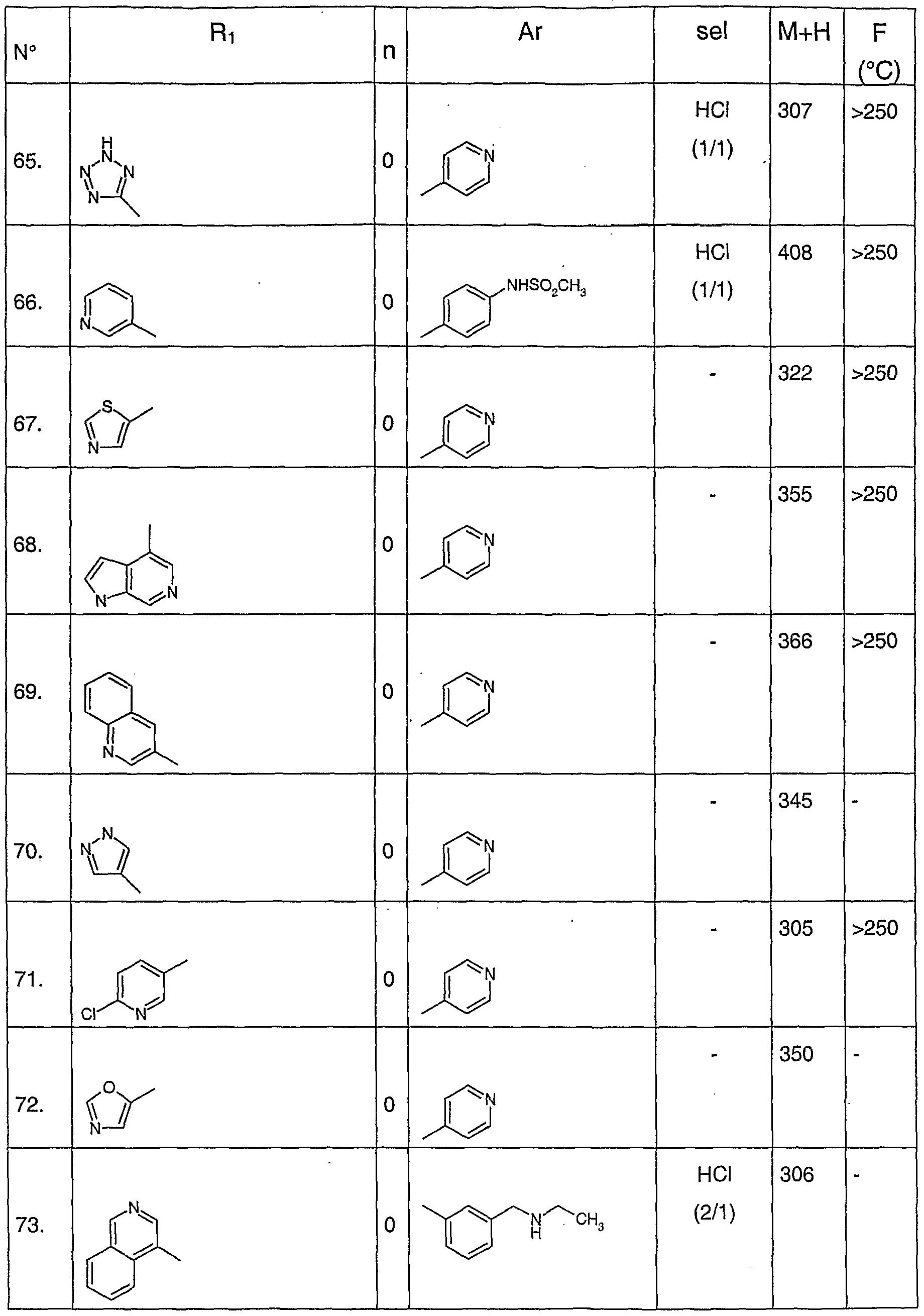
A titre d'exemple de composés préférés de formule générale (I), on peut citer les composés suivants :
- chlorhydrate de N-(pyridin-4-yl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide
- chlorhydrate de N-(pyr idin-4-yl)-5-(4-méthyl-[3,4']-bipyridinyl-5-yl)-1 H-indazole- 3-carboxamide
- N-(pyridin-4-yl)-5-isoquinolin-4-yl-1 --indazole-3-carboxamide
- N-(pyridin-3-ylméthyl)-5-isoquinolin-4-yl-1 H-indazole-3-carboxamide
- N-(3-cyanophényl)-5-isoquinolin-4-yl-1/-/-indazole-3-carboxamide
- N-(4-sulfamoylphényl)-5-isoquinolin-4-yl-1H-indazole-3-carboxamide - acide 4-[5-(isoquinolin-4-yl)-1 -/-indazole-3-carbonylamino]benzoique
- N-(pyridin-4-yl)-5-[4-méthyl-5-(pyridin-3-ylcarbamoyl)-pyridin-3-yl]-1 H-indazole- 3-carboxamide
- N-(pyridin-4-yl)-5-(4-méthyl-[3,3']-bipyridinyl-5-yl)-1 H-indazole-3-carboxamide
- N-{4-[(N-méthylsulfonyl)amino]phényl}-5-pyridin-3-yl-1 H-indazole-3- carboxamide
- N-(pyridin-4-yl)-5-(1 ,3-thiazol-5-yl)-1 H-indazole-3-carboxamide
- N-(pyridin-4-yl)-5-(1 H-pyrrolo[2,3-c]pyridin-4-yl)-1 H-indazole-3-carboxamide
- 5-(1 H-pyrazol-4-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
- N-(pyridin-4-yl)-5-[(2-chloro)pyridin-5-yl]-1 /-/-indazole-3-carboxamide - 5-(1 ,3-oxazol-5-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
- dichlorhydrate de N-{3-[(éthylamino)méthyl]phényl}-5-isoquinolin-4-yl-1H- indazole-3-carboxamide - 5-(5-amino-4-méthylpyridin-3-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
- N-(pyridin-4-yl)-5-(3-(N-aminocétométhyl)carboxamide)pyridin-4-yl-1 -/- indazole-3-carboxamide
- N-(pyridin-3-ylméthyl)-5-(1 ,3-thiazol-5-yl)-1 H-indazole-3-carboxamide
- N-(pyridin-3-ylméthyl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide
- N-(4-sulfamoylphényl)-5-pyridin-3-yl-1 /-/-indazole-3-carboxamide
- N-(3-sulfamoylphényl)-5-pyr idin-3-yl-1 /-/-indazole-3-carboxamide
- N-phényl-5-pyridin-3-yl-1 H-indazole-3-carboxamide
- N-(3-méthylsulfonamidophényl)-5-pyridin-3-yl-1H-indazole-3-carboxamide
- N-(4-morpholin-4-ylphényl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide - 5-pyridin-3-yl-N-{4-[(N-pyrimidin-2-ylamino)sulfonyl]phényl}-1 H-indazole-3- carboxamide
- N-(4-méthylsulfonylphényl)-5-pyridin-3-yl-1r7,-indazole-3-carboxamide - 5-(4-hydroxy-3-méthylpyridin-2-yl)-N-pyridin-4-yl-1 H-indazole-3-carboxamide
- N-(3-cyanophényl)-5-oxazol-5-yl-1 H-indazole-3-carboxamide - N-(2-méthoxypyridin-5-yl)-5-(1 ,3-oxazol-5-yl)-1 H-indazole-3-carboxamide
- N-{3-[(éthylamino)méthyl]phényl}-5-(1 ,3-thiazol-5-yl)-1 H-indazole-3- carboxamide
- N-{3-[(diéthylamino)méthyl]phényl}-5-(1 ,3-oxazob5_yl)-1 H-indazole-3- carboxamide - N-{3-[(diéthylamino)méthyl]phényl}-5-(1 ,3-thiazol-5-yl)-1 H-indazole-3- carboxamide
- N-[3-(morpholin-4-ylméthyl)phényl]-5-pyridin-3-yl-1H-indazole-3-carboxamide
- N-(3,5-difluorobenzyl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide
- N-(3,4-difluorobenzyl)-5-pyridin-3-yl-1H-indazole-3-carboxamide
L'invention a également pour objet, parmi les composés de formule générale (I), des composés répondant à la formule générale (I') :
(!') dans laquelle,
Ri représente un atome d'hydrogène, d'halogène, un NH2, NHR2, NHCOR2, N02, CN, CH2NH2,
CH2NHR2 ; ou bien Ri représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, C1-6 alkyle, Cι-6 alcoxy, NH2, NHR2, NR2R3; ou bien Ri représente un heteroaromatique éventuellement substitué par un ou plusieurs substituants choisis parmi un heteroaromatique, Cι.6 alkyle , un NH2,
NHR2, NHCOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi 0, NH et S ;
Ar représente un groupe phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un C1.6 alkyle, Cι-6 thioalkyle, Cι-6 alcoxy,
CH2OH, phénoxy, NH2, NHR2, NR2R3, CN, SO2NH2, S02NHR2, COOH, COOR2,
CONH2, CONHR2 ; ou bien Ar représente un heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un COOH, Cι-6 alkyle,
C1-6 alcoxy ;
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un phényle ou un heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de — Hautre un phényle ou un heteroaromatique ; n représente 0, 1 , 2 ou 3.
Les composés suivants ne font pas partie des composés répondant à la formule générale (I') :
- N-phényl- 7H-indazole-3-carboxamide ;
- N-(2-chlorophényl)-7H-indazole-3-carboxamide ;
- N-(3-chlorophényl)-7H-indazole-3-carboxamide ;
- N-(4-chlorophényl)-7H-indazole-3-carboxamide ;
- N-(2,4-dichlorophényl)-7H-indazole-3-carboxamide ;
- N-(3,4-dichlorophényl)-7H-indazole-3-carboxamide ;
- N-(2-méthylphényl)-7H-indazole-3-carboxamide ;
- N-(2-méthoxyphényl)-7H-indazole-3-carboxamide ;
- N-(4-méthoxyphényl)- 7/-/-indazole-3-carboxamide ;
- N-(4-thiométhyIphényl)-7H-indazole-3-carboxamide ;
- N-(3-chloro-4-thiométhylphényl)-5-amino- 7H-indazole-3-car boxamide ;
- N-benzyl-7H-indazole-3-carboxamide ;
- N-(2-chlorobenzyl)-7H-indazole-3-carboxamide ;
- N-(4-méthylbenzyl)-7H-indazole-3-carboxamide ;
- N-(pyridin-2-ylméthyl)- 7H-indazole-3-carboxamide ;
- N-(pyridin-3-ylméthyl)-7H-indazole-3-carboxamide ;
- N-(pyridin-4-ylméthyl)-7H-indazole-3-carboxamide ;
- N-(2-phényléthyl)-7/-/-indazole-3-carboxamide ;
- N-(3,4-diméthoxyphényléthyl)-7H-indazole-3-carboxamide ;
- N-[3-(pyridin-2-yl)propyl]-7H-indazole-3-carboxamide ;
- N-[3-(2,6-diméthylphényl)propyl]-5-nitro-7H-indazole-3-carboxamide.
Parmi les composés de formule générale (I'), une première famille de composés préférés est constituée des composés pour lesquels : Ri représente un atome d'hydrogène, d'halogène, un NH2, NHR2, NHCOR2, N02, CN, CH2NH2, CH2NHR2 ; ou bien Ri représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un hydroxy, C alkyle, Cι-6 alcoxy, NH2, NHR2, NR2R3; ou bien Ri représente un heteroaromatique éventuellement substitué par un ou plusieurs substituants choisis parmi un heteroaromatique, Cι-6 alkyle , un NH2, NHR2( NHCOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; et/ou Ar représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un Cι-6 alkyle, Ci .6 thioalkyle, Cι-6 alcoxy, CH2OH, phénoxy, NH2, NHR2, NR2R3, CN, S02NH2, SO2NHR2, COOH, COOR2, ÇONH2, CONHR2 ; ou bien Ar représente un heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un COOH, un Ci .6 alkyle, un d -6 alcoxy ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un phényle ou un heteroaromatique ou R2 représente un phényle ou un heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3 ; avec la condition que
- quand Ri représente un atome d'hydrogène si n représente 0 et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, les substituants méthyle, méthoxy, thiométhyle, atome de chlore étant exclus ; si n représente 1 , alors et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, les substituants méthyle et atome de chlore étant exclus ; si n représente 1 , alors et Ar est un pyridinyle, alors le pyridinyle est obligatoirement substitué tel que défini ci-dessus ; si n représente 2 et Ar est un phényle, alors le phényle est obligatoirement substitué tel que défini ci-dessus, le substituant méthoxy étant exclu ;
si n représente 3 et Ar est un pyridinyle, alors le pyridinyle est obligatoirement substitué tel que défini ci-dessus ;
- quand Ri représente un NH2 si n représente 0 et Ar est un phényle, alors le ou les substituants du phényle ne peuvent être choisis parmi un thiométhyle ou un atome de chlore ;
- quand Ri représente un N02 si n représente 3 et Ar est un phényle, alors le ou les substituants du phényle de Ar ne peuvent être un méthyle .
Parmi les composés de formule générale (I'), une seconde famille de composés préférés est constituée des composés pour lesquels :
- quand Ri représente un atome d'hydrogène, alors Ar -représente un phényle substitué par un ou deux substituants choisis parmi un atome de brome ou d'iode, un C2.6 alkyle, C2-6 thioalkyle, C2.6 alcoxy, CH2OH, phénoxy, NH2, NHR2, NR2R3( CN, S02NH2, S02NHR2, COOH, COOR2, CONH2, CONHR2 ; ou bien Ar représente un heteroaromatique choisi parmi un pyrrolyle, imidazolyle, pyrazolyle, isothiazolyle, 1 ,3,4-thiadiazolyle, pyridazinyle, pirimidinyle, pyrazinyle, indolyle, indazolyle, quinolinyle, isoquinolinyle, éventuellement substitué par un ou deux substituants ; ou Ar représente un pyridinyle substitué par un ou deux substituants ; les substituants étant choisis parmi un atome d'halogène, un COOH, un C|-6 alkyle, un Cι-6 alcoxy ; et/ou R et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un phényle ou un heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3 ;
- quand Ri représente un atome d'halogène, un NH2, NHR2, NHCOR2, N02, CN, CH2NH2, CH2NHR2 ; ou bien Ri représente un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un HYDROXY, Ci .6 alkyle, C -6 alcoxy, NH2, NHR2, NR2R3; ou bien Ri représente un heteroaromatique éventuellement substitué par un ou plusieurs substituants choisis parmi un heteroaromatique, C 6 alkyle , un NH2, NHR2, NHCOR2, CONH2, CONHR2, CH2XR2 où X représente un atome choisi parmi O, NH et S ; alors Ar représente
un phényle éventuellement substitué par un ou deux substituants choisis parmi un atome de brome ou d'iode, un C2-β alkyle, C2-β thioalkyle, C1-6 alcoxy, CH2OH, phénoxy, NH2, NHR2, NR2R3, CN, S02NH2, S02NHR2, COOH, COOR2, CONH2, CONHR2 ; ou bien Ar représente un heteroaromatique, éventuellement substitué par un ou deux substituants choisis parmi un atome d'halogène, un COOH, un C|.6 alkyle, un C1-6 alcoxy ; et/ou
R2 et R3 représentent indépendamment l'un de l'autre un Cι-6 alkyle éventuellement substitué par un phényle ou un heteroaromatique ; ou R2 et R3 représentent indépendamment l'un de l'autre un phényle ou un heteroaromatique ; et/ou n représente 0, 1 , 2 ou 3.
Dans le cadre de l'invention, on entend par : - Ct-z où t et z peuvent prendre les valeurs de 1 à 6, une chaîne carbonée pouvant avoir de t à z atomes de carbone, par exemple Ci .6 une chaîne carbonée qui peut avoir de 1 à 6 atomes de carbone ;
- alkyle, un groupe aliphatique saturé, linéaire ou ramifié; par exemple, un groupe Cι-6 alkyle représente une chaîne carbonée de 1 à 6 atomes de carbone, linéaire ou ramifiée, plus particulièrement un méthyle, éthyle, propyle, isopropyle, butyle, isobutyle, secbutyle, tertbutyle, pentyle...
- alcoxy, un groupe alkyloxy à chaîne aliphatique saturée, linéaire ou ramifiée ;
- thioalkyle, un groupe -S-alkyle à chaîne aliphatique saturée, linéaire ou ramifiée ; - atome d'halogène, un fluor, un chlore, un brome ou un iode ;
- groupe heteroaromatique : un groupe aromatique cyclique comprenant entre 5 et 9 atomes de carbone et comprenant entre 1 et 4 hétéroatomes, tels que l'azote, l'oxygène ou le soufre. A titre d'exemples de groupes hétéroaromatiques, on peut citer les groupes pyrrolyle, imidazolyle, pyrazolyle, triazolyle, tetrazolyle, thiazolyle, isothiazolyle, 1 ,3,4-thiadiazolyle, oxazolyle, isoxazolyle, oxadiazolyle, pyridinyle, pyridazinyle, pirimidinyle, pyrazinyle, 1 ,3,5- triazinyle, indolyle, isoindolyle, indazolyle, benzimidazolyle, benzothiazolyle, quinolinyle, isoquinolinyle, pyrrolo[2,3-c]pyridinyle.
Les composés de formule générale (I) peuvent comporter un ou plusieurs atomes de carbone asymétriques. Ils peuvent donc exister sous forme d'énantiomères ou de diastéréoisomères. Ces énantiomères, diastéréoisomères,
ainsi que leurs mélanges, y compris les mélanges racémiques, font partie de l'invention.
Les composés de formule générale (I) peuvent exister sous forme de tautomères. Ainsi, l'invention a pour objet les composés de l'invention sous toutes leurs formes tautomères.
Les composés de formule générale (I) peuvent exister à l'état de bases ou de sels d'addition à des acides. De tels sels d'addition font partie de l'invention. Ces sels sont avantageusement préparés avec des acides pharmaceutiquement acceptables, mais les sels d'autres acides utiles, par exemple, pour la purification ou l'isolement des composés de formule générale (I) font également partie de l'invention.
Les composés de formule générale (I) peuvent se trouver sous forme d'hydrates ou de solvats, à savoir sous forme d'associations ou de combinaisons avec une ou plusieurs molécules d'eau ou avec un solvant. De tels hydrates et solvats font également partie de l'invention.
La présente invention a également pour objet des procédés de préparation des composés de formule générale (I).
Ainsi, les composés de l'invention peuvent être préparés par les méthodes illustrées dans les schémas qui suivent, dont les conditions opératoires sont classiques pour l'homme du métier.
On entend par groupe protecteur PG, un groupe permettant d'empêcher la réactivité d'une fonction ou position, lors d'une réaction chimique pouvant l'affecter, et qui restitue la molécule après clivage selon des méthodes connues de l'homme du métier. Des exemples de groupes protecteurs ainsi que les méthodes de protection et déprotection sont données, entre autres, dans Protectivθ groups in Organic Synthesis, Green et al., 2nd Ed. (John Wiley & Sons, Inc., New York).
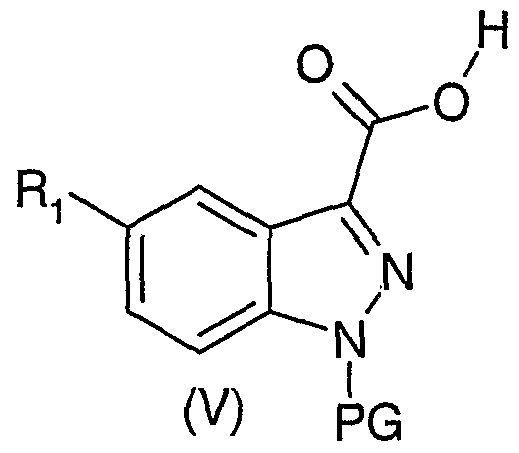
Quand Ri représente un atome d'halogène, un N02 ou un CN, les composés de formule générale (I) peuvent être préparés par la méthode illustrée dans le schéma 1.
Cette méthode consiste à transformer un indole de formule générale (II), où Ri est un N02, un CN ou un atome d'halogène, en indazole-3-carbaldéhyde de formule générale (III) par exemple avec de l'acide nitreux. Le composé de
formule générale (III) est ensuite protégé en milieu basique par un groupe PG, de type triméthylsilyléthoxyméthyle (SEM) ou mésitylènesulfonyle (Mts) pour donner l'indazole-3-carbaldéhyde protégé en position 1 de formule générale (IV). Le composé (IV) est oxydé en acide indazole-3-carboxylique de formule générale (V), par exemple par réaction avec du chlorite de sodium. L'obtention de l'indazole-3-carboxamide protégé en position 1 de formule générale (VII) se fait par couplage du composé de formule générale (V) avec une aminé de formule générale Ar(CH2)nNH2 (VI) dans laquelle Ar et n sont tels que définis dans la formule générale (I). Cette réaction de couplage peut être effectuée par activation d'un composé de formule générale (V) par des réactifs de couplage, tels que du carbonyldiimidazole ou du chloroformiate d'isopropyle ou d'isobutyle. La déprotection du composé de formule générale (VII) peut se faire soit par action d'une base telle que la souder soit en présence de fluorure de tétrabutylammonium (TBAF) et d'éthylènediamine, ou encore en présence d'acide trifluoroacétique puis chauffage avec de l'éthylènediamine. Cette étape de déprotection permet d'obtenir l'indazole-3-carboxamide de formule générale
(!)•
Dans le cas où Ri est un atome d'hydrogène, la méthode de préparation décrite dans le schéma 1 est reprise, en réalisant la réaction de couplage de l'aminé de formule générale Ar(CH2)nNH2 (VI), telle que définie ci-dessus, avec l'acide indazole-3-carboxylique commercial.
Les composés de formule générale (I), où R représente un NH2, sont obtenus par réduction d'un composé de formule générale (I), où Ri est un N02, tel qu'obtenu selon le schéma 1 , par exemple en présence de chlorure d'étain. Les composés de formule générale (I), où Ri représente un NHR2 ou un
NHCOR2, sont obtenus par fonctionnalisation des composés de formule générale (I) correspondants, où Ri est un NH2, selon des techniques connues de l'homme du métier.
Les composés de formule générale (I) où Ri représente un CH2NH2, sont obtenus par hydrogénation à pression atmosphérique d'un composé de formule générale (I), où Ri est un CN, tel qu'obtenu selon le schéma 1 , par exemple en présence de palladium sur charbon.
Les composés de formule générale (I), où R représente un CH2NHR2, sont obtenus par fonctionnalisation des composés de formule générale (I) correspondants, où Ri est un CH2NH2, selon des techniques connues de l'homme du métier.
Schéma 1
Quand Ri représente un groupe phényle ou heteroaromatique éventuellement substitué, les composés de formule générale (I), peuvent être obtenus selon l'une des méthodes illustrées dans les schémas 1 , 2 et 3. Cependant, quand Ri représente un groupe oxazolyle, les composés de formule (I) peuvent être obtenus selon le schéma 4 et quand Ri représente un groupe thiazolyle, les composés de formule (I) peuvent être obtenus selon le schéma 5.
Dans le cas du schéma 1 , le composé de formule générale (II), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I), peut être obtenu par exemple par une réaction de type Suzuki sur le 5-iodoindole selon des techniques connues de l'homme du métier.
Le schéma 2 illustre une méthode de préparation alternative du composé de formule générale (VII) à partir du 5-iodoindole.
Le composé de formule générale (IVa), où SEM est un groupe triméthylsilyléthoxyméthyle, est obtenu en reprenant les deux premières étapes illustrées dans le schéma 1. Une réaction de Suzuki, réalisée par exemple en
présence d'un acide boronique de formule générale R1B(OH)2 (VIII), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I), d'une base minérale, telle que le carbonate de sodium (Na2C03), et de palladium (0), permet d'obtenir le composé de formule générale (IV), dans laquelle PG représente un groupe SEM. Le composé de formule générale (I), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I), est obtenu à partir du composé de formule générale (IV) en reprenant les trois dernières étapes illustrées dans le schéma 1.
Schéma 2
Le schéma 3 illustre une méthode de préparation à partir de la 5-iodo- ou de la 5-bromoisatine.
L'acide 5-iodo- ou 5-bromoindazolecarboxylique peut être obtenu par ouverture du cycle indoledione de la 5-iodo- ou de la 5-bromoisatine, par exemple en présence de soude, puis par diazotation, par exemple grâce à de l'acide nitreux, et enfin par réduction et formation du cycle indazole, par exemple en présence de chlorure d'étain (SnCI2). L'acide 5-iodo- ou 5-bromoindazole-3-carboxylique obtenu est ensuite protégé en milieu basique, par exemple par un groupe SEM, pour donner le composé de formule générale (IX), dans laquelle X représente un atome de brome ou d'iode. L'indazoIe-3-carboxamide de formule générale (X) peut être obtenu par couplage du composé de formule générale (IX) avec une aminé de formule générale Ar(CH2)nNH2 (VI), dans laquelle Ar et n sont tels que définis dans la
formule générale (I). Cette réaction de couplage peut être effectuée par activation d'un composé de formule générale (IX) par des réactifs de couplage, tels que du carbonyidiimidazole ou du chloroformiate d'isopropyle ou d'isobutyle.
Schéma 3
L'obtention du composé de formule générale (VII) à partir du composé de formule générale (X) peut se faire par 2 méthodes :
- soit par une réaction de Suzuki, réalisée en présence d'un acide boronique de formule générale RιB(OH)2 (VIII), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I), d'une base et de palladium (0) ;
- soit par l'intermédiaire d'un dioxaborolane de formule générale (XI) obtenu par réaction de bis(pinacolato)diborane et de 1 ,1'- bis(diphénylphosphino)ferrocènedichloro-palladiumll sur le composé de formule générale (X) ; l'intermédiaire de formule générale (XI) étant ensuite mis en présence d'une base minérale, telle que l'acétate de sodium ou de potassium, de palladium (0) et d'un composé de formule générale R X (XII), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I) et X est un atome de brome ou d'iode. Le composé de formule générale (I), où Ri représente un groupe phényle ou heteroaromatique éventuellement substitué tel que défini dans la formule générale (I), est obtenu par déprotection du composé de formule générale (VII), telle qu'illustrée dans la dernière étape du schéma 1.
Le schéma 4 illustre une méthode de préparation des composés de formule générale (Vlla), c'est à dire les composés de formule générale (VII) pour lesquels Ri représente un groupe oxazolyle et PG représente un groupe SEM. Le composé de formule générale (X), telle que définie ci-dessus et dans laquelle X représente un atome d'iode, est formylé par exemple en présence de monoxyde de carbone et d'un complexe de palladium, tel que le tétrakis(triphénylphosphine)palladium, puis d'un agent réducteur, tel que l'hydrure de tributylétain dans un solvant, tel que le tétrahydrofurane (THF). Le composé de formule générale (XIII) ainsi obtenu est chauffé à reflux dans un solvant, tel que le méthanol, en présence de tosylméthylisocyanate (TosMIC) et d'une base telle que le carbonate de potassium (K2C03), pour obtenir le composé de formule générale (Vlla).
Le composé de formule générale (I), où Ri représente un groupe oxazolyle, est obtenu à partir du composé de formule générale (Vlla) par déprotection telle qu'illustrée dans la dernière étape du schéma 1.
Schéma 4
(Vlla)
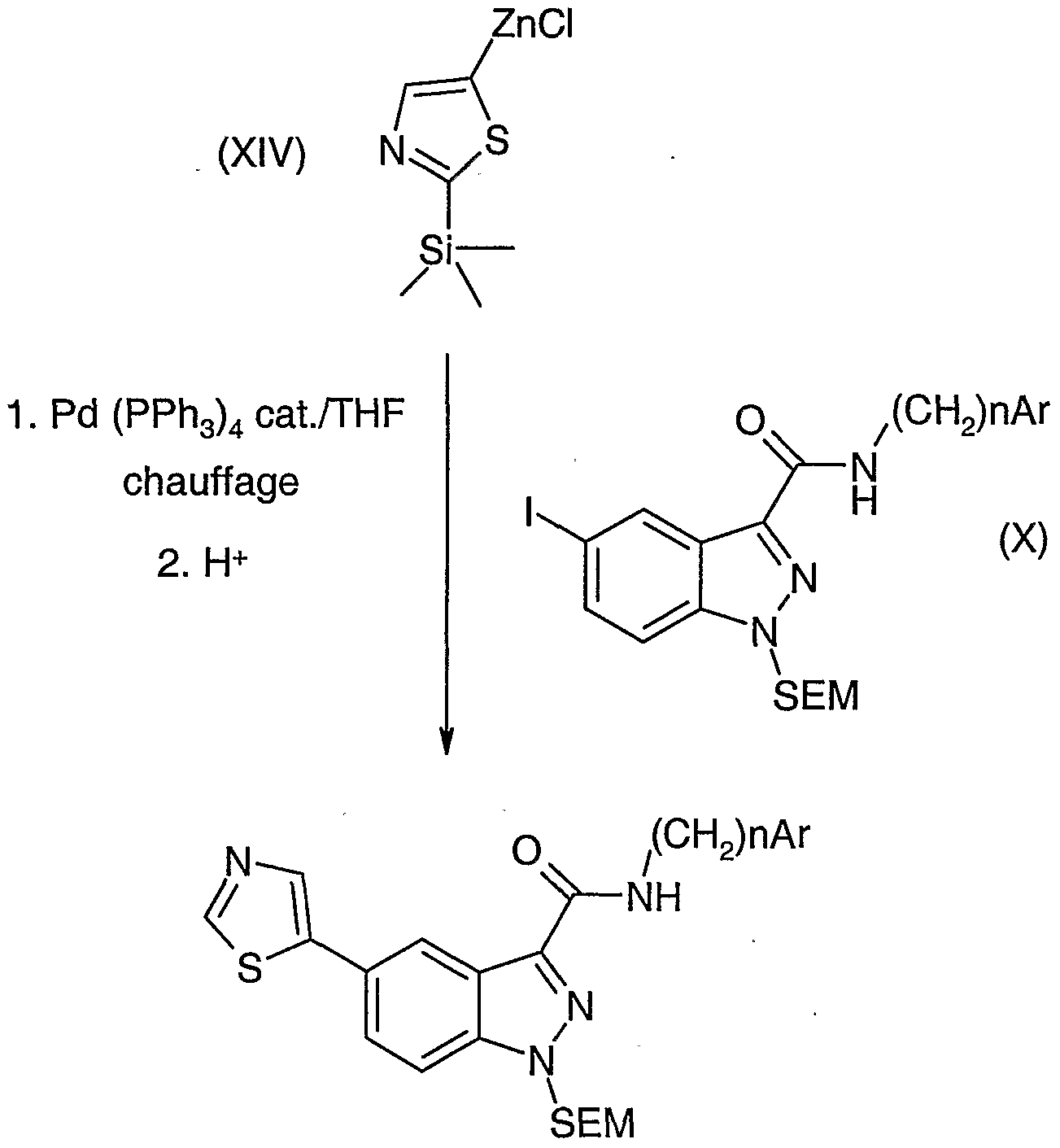
Le schéma 5 illustre une méthode de préparation des composés de formule générale (Vllb), c'est à dire les composés de formule générale (VII) pour lesquels Ri représente un groupe thiazolyle et PG représente un groupe SEM. Le groupe thiazolyle est introduit par chauffage du composé de formule générale (X), telle que définie ci-dessus et dans laquelle X représente un atome d'iode, en présence du dérivé de formule (XIV) illustré dans le schéma 5, de tétrakis(triphénylphosphine)palladium dans du THF anhydre puis par acidification. Le dérivé de formule (XIV) est préparé à partir de 2- triméthylsilyl(thiazole), en présence d'une base forte, telle que le butyllithium, par réaction du chlorure de zinc (ZnCI2) en solution dans de l'éther anhydre. Le composé de formule générale (Vllb) ainsi obtenu est déprotégé selon la dernière étape du schéma 1 afin d'obtenir le composé de formule générale (I), où Ri représente un groupe thiazolyle.
Schéma 5
1- nBuLi/Et20 2. ZnCI,
(Vllb)
Les composés de formule générale (V), (VII) et (X) sont nouveaux et font également partie de l'invention. Ils sont utiles en tant qu'intermédiaires de synthèse pour la préparation des composés de formule générale (I).
Dans les schémas 1 à 5, les composés de départ et les réactifs, quand leur mode de préparation n'est pas décrit, sont disponibles dans le commerce ou
décrits dans la littérature, ou peuvent être préparés par des méthodes qui y sont décrites ou qui sont connues de l'homme du métier.
Les exemples suivants décrivent la préparation de certains composés conformes à l'invention. Ces exemples ne sont pas limitatifs et ne font qu'illustrer l'invention.
Les numéros des composés exemplifiés renvoient à ceux donnés dans le tableau ci-après. Les microanalyses, les spectres I.R. et R.M.N. et/ou la LC/MS/UV (Liquid Chromatography coupled to Mass Spectroscopy and to Ultraviolet analysis) confirment les structures des composés obtenus.
Pour chaque valeur de LC/MS/UV fournie, le pourcentage entre parenthèses représente la pureté UV du composé.
Exemple 1 (composé n°1) N-(3-imidazol-1 -ylpropyl)-1 /-/-indazole-3-carboxamide
De l'acide indazole-3-carboxylique (810 mg, 5 mmol) est chauffé à 60°C en présence de carbonyidiimidazole (891 mg, 5,5 mmol) dans la N,N- diméthylformamide (DMF) (14 ml) sous argon pendant 3 h. Du 1-(3- aminopropyl)imidazole (597 μl, 5 mmol) en solution dans de la DMF (2 ml) est ajouté et le mélange est chauffé pendant 2h et 20 min à 60°C. Après refroidissement, la DMF est évaporée sous vide pour donner une huile jaune qui est chromatographiée sur 54 g de silice. Le composé obtenu est élue par un mélange acétate d'éthyle (AcOEt) / méthanol (MeOH) (9/1). On obtient 690 mg de produit. PF: 154-155°C
LC/MS/UV: MH+ 270 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 1 ,98 (quintuplet, 2H), 3,27 (m, 2H), 4,01 (t, 2H), 6,88 (s, 1 H), 7,20 (s, 1 H), 7,22 (m, 1 H), 7,39 (m, 1 H), 7,58 (m, 1 H), 7,66 (s, 1 H), 8,16 (m, 1 H), 8,52 (m, 1 H), 13,5 (s, 1 H).
Exemple 2 (composé n°3)
N-(4-sulfamoylphenyl)-1 H-indazole-3-carboxamide
Un mélange d'acide indazole-3-carboxylique (324 mg, 2 mmol), d'hydroxy- benzotriazole (297mg, 2,2 mmol) et de disopropylcarbodiimide (344 μl, 2,2 mmol) dans de la DMF (10 ml) est agité 30 min à température ambiante. Dé la sulfanilamide (380 mg, 2,2 mmol) est ajoutée. Le mélange réactionnel est agité pendant la nuit à température ambiante puis filtré. Le filtrat est évaporé puis est extrait avec AcOEt / H20. La phase organique est séchée sur MgS04,
filtrée et évaporée sous vide pour donner un solide jaune (640 mg). Ce composé est recristallisé successivement dans AcOEt / CH2CI2 et AcOEt / MeOH. On obtient 110 mg de produit sous forme d'un solide de couleur crème. PF: >250°C LC/MS/UV: MH+ 317 (96,5 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,26 (s,2H), 7,30 (m, 1 H), 7,46 (m, 1 H), 7,67 (m, 1 H), 7,78 (d, 1 H), 8,07 (d, 1 H), 8,21 (d, 1 H), 10,69 (s,1 H), 13,87 (s,1 H).
Exemple 3 (composé n°4) N-(4-phénylaminophényl)-1 H-indazole-3-carboxamide
Ce composé est synthétisé de manière analogue au mode opératoire décrit à l'exemple 2 par couplage d'acide indazole-3-carboxylique et de N-phenyl-1 ,4- phénylènediamine à l'échelle de Jjrimol, mais en utilisant de la dicyclohexyl- carbodiimide (DCC) comme réactif de couplage. Le produit brut obtenu après extraction est repris dans CHCI3. L'insoluble est filtré et séché. On obtient 190 mg de produit. PF: 218°C
LC/MS/UV: MH+ 329 (100 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 6,76 (t, 1 H), 7,02 (d, 2H), 7,06 (d, 2H), 7,19 (t, 2H), 7,27 (t, 1 H), 7,44 (t, 1 H), 7,64 (d, 1 H), 7,74 (d, 2H), 8,04 (s, 1 H), 8,21 (d, 1 H), 10,14 (s, 1 H), 13,65 (s, 1 H).
Exemple 4 (composé n°6)
N-(1 H-indazol-5-yl)-1 H-indazole-3-car boxamide
Intermédiaire 4.1
Acide 1 -(2,4,6-triméthylbenzènesulfonyl)-1 H-indazole-3-carboxylique
Le NaH (60 % dans l'huile, 40 mmol) est lavé sous argon avec de l'éther de pétrole puis mis en suspension dans 60 ml de DMF anhydre. Une solution d'acide indazole-3-carboxylique (3,35 g, 20 mmol) dans de la DMF anhydre (50 ml) est ajoutée goutte à goutte à cette suspension maintenue à 0°C. Le mélange réactionnel devient limpide. Quand l'addition est terminée, on laisse réagir pendant 30 min à température ambiante puis le mélange réactionnel est à nouveau refroidi à 0°C. Le chlorure de mésitylènesulfonyle (4,82 g, 22 mmol) en solution dans du tétrahydrofurane (THF) anhydre (50 ml) est ajouté. Le bain de glace est enlevé et une suspension apparaît. On laisse réagir 30 min à température ambiante. Les solvants sont évaporés sous vide et le résidu est
repris avec une solution 0,1 N NaOH. La solution obtenue est lavée à l'éther diéthylique puis acidifiée avec HCI 6N. Le composé qui se sépare est filtré, repris avec AcOEt. La solution d'AcOEt est séchée sur Na2S04 puis évaporée. Le résidu (6,18g) est recristallisé dans AcOEt / éther de pétrole. On obtient 4,89 g de produit sous forme d'un solide jaune. PF: 208-209°C.
Intermédiaire 4.2
[1-(2,4,6-triméthylbenzènesulfonyl)-1 H-indazol-3-yl]carbonyIcarbonate d'isopropyle
A une solution d'intermédiaire 4.1 (7,58 g, 22 mmol) dans 300ml de THF anhydre maintenue à 0°C sous argon, on ajoute goutte à goutte une solution de chloroformiate. d'isopropyle (1 M dans le toluène, 22 ml) puis de la N-méthyl- morpholine (2,42 ml, 22 mmol). Un précipité apparaît, l'agitation est maintenue 15 min à 0°C puis on laisse revenir à température ambiante pendant 15 min. Les solvants sont évaporés sous vide puis le résidu est repris avec un mélange éther diéthylique/eau. La phase éthérée est lavée avec HCI 0,5N et NaHC03 10 %, puis séchée sur Na2S0 et évaporée pour donner le produit. On obtient 10,8 g de produit sous forme d'une gomme jaune.
Intermédiaire 4.3
N-(1 H-indazol-5-yl)-1 -(2,4,6-triméthylbenzènesulfonyl)-1 H-indazole-3- carboxamide
Sous argon, un mélange d'intermédiaire 4.2 (431 mg, 1 mmol) et de 5-amino- indazole (137 mg, 1 mmol) dans du THF (10 ml) est chauffé à 60°C pendant 3 jours. Le THF est évaporé pour donner le produit sous forme d'une gomme.
N-(1 H-indazol-5-yl)-1 H-indazole-3-carboxamide
Sous argon, à une suspension d'intermédiaire 4.3 (1 mmol) dans du dioxane (10 ml) est ajoutée une solution de NaOH (0,5 N, 20 ml, dioxane /H20 1/1). Le mélange réactionnel est chauffé pendant 2h à 60°C puis concentré sous vide et repris avec H20. Le précipité est filtré puis recristallisé dans un mélange isopropanol / éther de pétrole.
On obtient 175 mg de produit sous forme d'un solide de couleur crème. PF: > 250°C
LC/MS/UV: MH+ 278 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,28 (t, 1 H), 7,44 (t, 1 H), 7,50 (d, 1 H), 7,65 (d, 1 H), 7,75 (d, 1 H), 8,04 (s, 1 H), 8,23 (d, 1 H), 8,43 (s, 1 H), 10,31 (s, 1 H), 12,99 (s, 1 H), 13, 7 (s, 1 H).
Exemple 5 (composé n°7)
N-(4-bromophényl)-1 /--indazole-3-carboxamide
Sous argon, à une solution d'acide indazole-3-carboxylique (405 mg, 2,5 mmol) dans du THF (50 ml) est ajouté à 0°C de l'isobutylchloroformiate (357 μl, 2,75 mmol) puis de la N-méthylmorpholine (302 μl, 2,75 mmol). Le mélange réactionnel est agité pendant 15 min à 0°C puis de la 4-bromoaniline (860 mg, 5 mmol) est ajoutée. Le bain de glace est enlevé et l'agitation est maintenue pendant la nuit. Le solvant est évaporé sous vide . Le résidu est repris avec un mélange AcOEt / H20. L'évaporation de la solution organique après séchage sur MgS0 donne un composé brut (1 ,27 g) qui est chromatographié sur silice en éluant avec CH2CI2. Le composé ainsi obtenu est recristallisé dans Pisopropanol. On obtient 572 mg de produit. PF: > 250°C LC/MS/UV: MH+ 316 RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,29 (t, 1 H), 7,45 (t, 1 H), 7,52 (d, 2H), 7,65 (d, 1 H), 7,89 (d, 2H), 8,20 (d, 1 H), 10,52 (s, 1 H), 13,81 (s, 1 H).
Exemple 6 (composé n°12)
3-(1 H-indazole-3-carbonylamino)benzoate d'éthyle
Ce composé est synthétisé de manière analogue au mode opératoire décrit à l'exemple 5. La synthèse est réalisée à l'échelle de 5 mmol. Le composé final est recristallisé dans AcOEt.
On obtient 390 mg de produit sous forme d'un solide jaune.
PF: 187-188°C
LC/MS/UV: MH+ 310 (100 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 1 ,33 (t, 3H), 4,32 (q, 2H), 7,30 (t, 1 H),
7,48 (m, 2H), 7,67 (m, 2H), 8,11 (d, 1 H), 8,22 (d, 1 H), 8,63 (s, 1 H), 10,62 (s, 1 H),
13,9 (s, 1 H).
Exemple 7 (composé n°13) Acide 3-(1 H-indazole-3-carbonylamino)benzoique
Une suspension du composé obtenu à l'exemple 6 (310 mg, 1 mmol) dans du dioxane (10 ml) est mise en réaction avec NaOH aqueuse (0,5N, 10 ml, 5 mmol). Le mélange réactionnel est chauffé une nuit à 50°C puis est concentré
sous vide, dilué avec H20 et acidifié avec HCI 6N. La suspension résultante est filtrée. Le solide est lavé avec H20, de l'isopropanol et de l'éther diéthylique puis séché sous vide. On obtient 230 mg de produit. PF: >250°C
LC/MS/UV: MH+ 282 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,29 (t, 1 H), 7,45 (m, 2H), 7,67 (d, 2H),
8,05 (d, 1 H), 8,22 (d, 1 H), 8,60 (s, 1 H), 10,56 (s, 1 H), 12,9 (s, 1 H), 13,82 (s, 1 H)
Exemple 8 (composé n°22)
N-(3-thiométhylphényl)-1 H-indazole-3-carboxamide
Ce composé est préparé de manière analogue au mode opératoire décrit à l'exemple 4 à l'échelle -de -1 ,56 mmol. L'intejπmédiaire 4.2 est mis en réaction avec de la 3-(thiométhyl)aniline pendant une nuit à température ambiante et le produit de la réaction est traité avec NaOH. Le mélange réactionnel est acidifié par HCI 6N puis est évaporé sous vide. Le résidu est extrait par AcOEt/HCI 1 N.
La solution d'AcOEt est lavée successivement avec NaHC03 10 %, H20 et de la saumure. Le composé final est lavé avec MeOH.
On obtient 260 mg de produit sous forme d'une poudre. PF: 183°C
LC/MS/UV: MH+ 284 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,49 (s, 3H), 6,97 (d, 1 H), 7,27 (m, 2H),
7,45 (t, 1 H), 7,66 (d, 1 H), 7,69 (d, 1 H), 7, 88 (s, 1 H), 8,21 (d, 1 H), 10,35 (s, 1 H),
13,79 (s, 1 H).
Exemple 9 (composé n°34)
Chlorhydrate de N-(pyridin-4-yl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide
Intermédiaire 9.1 Acide 5-iodo-1 H-indazole-3-carboxylique
De la 5-iodoisatine (5 g, 18,3 mmol) est chauffée en présence de soude (0,77 g, 19,2 mmol dans 12 ml H20) jusqu'à dissolution puis le mélange réactionnel est refroidi à 0°C. Une solution de nitrite de sodium préalablement refroidie à 0°C (1 ,26g, 18,3 mmol dans 5,5 ml H20) est ajoutée. La pâte obtenue est ajoutée par petites portions, sous agitation vigoureuse à une solution d'acide sulfurique (3,40 g, 34,8 mmol dans 37 ml H20) pré-refroidie à 0°C de sorte que la température n'excède pas 4°C. L'agitation est maintenue 15 min puis une solution de chlorure d'étain (SnCI2.2H20, 9,91 g, 43,9 mmol dans 15 ml HCI
concentré) est additionnée lentement de sorte que la température n'excède pas 4°C. On laisse réagir plusieurs heures. Le mélange réactionnel est filtré. Le solide est lavé avec de l'eau bouillante puis repris avec de l'éthanol à chaud. Les impuretés insolubles sont éliminées par filtration. On obtient 2 g de produit.
Intermédiaire 9.2
5-iodo-1-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3-carboxylate de sodium
Sous argon à de Phydrure de sodium (6,16 g, 55 % dans l'huile, 140 mmol) dans du THF anhydre (200 ml) est ajouté à 0°C de l'intermédiaire 9.1 (20 g, 70 mmol). On laisse remonter la température à l'ambiante et l'agitation est maintenue pendant 20 min. Le mélange réactionnel est refroidi à nouveau à 0°C et du chlorure de 2-(triméthylsilyl)éthoxyméthyle (12,25 g, 73,5 mmol) en solution dans du THF (75 ml) est introduit lentement. La réaction est agitée quelques minutes à 0°C puis 3h à température ambiante. 80 ml d'eau sont ajoutés. Le THF est évaporé sous vide et l'insoluble est filtré. Le solide est lavé avec H20 puis avec un mélange éther diéthylique / éther de pétrole et enfin avec de l'éther diéthylique. Le solide obtenu est séché sous vide sur potasse. On obtient 20,71 g de produit sous forme d'une poudre jaune.
Intermédiaire 9.3
N-(pyridin-4-yl)-5-iodo-1-(2-triméthyIsilyléthoxyméthyl)-1 H-indazole-3- carboxamide
Sous argon, à une solution de l'intermédiaire 9.2 (5,02 g, 11 ,4 mmol) dans du THF anhydre (50 ml), à une température de -10°C, on ajoute lentement une solution de chloroformiate d'isopropyle dans le toluène (1M, 12 ml) puis goutte à goutte de la N-méthylmorpholine (1 ,22 g, 12 mmol). La température est maintenue 5 min à -10°C puis le bain réfrigérant est enlevé. Le mélange est agité pendant 25 min à température ambiante puis refroidi à nouveau et une solution de 4-aminopyridine (1 ,13 g, 12 mmol) dans du THF est ajoutée. Le mélange réactionnel est ensuite agité la nuit à température ambiante, filtré et concentré sous vide. Le produit brut est chromatographié sur gel de silice (500 g) en éluant suivant un gradient, de CH2CI2 à AcOEt. On obtient 4,17 g de produit.
Intermédiaire 9.4
N-(pyridin-4-yl)-5-pyridinyl-3-yl-1-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3- carboxamide Sous argon, à une solution d'intermédiaire 9.3 (685 mg, 1 ,38 mmol) dans du diméthoxyéthane (DME) (5,5 ml), on ajoute de l'acide pyridine-3-boronique (299 mg, 1 ,15 équiv.) puis une solution aqueuse de Na2C03 (734 mg / 2,7 ml H20, 5 équiv.). Le réacteur est dégazé plusieurs fois à l'argon puis, sous argon, du tétrakis(triphénylphosphine)palladium (48 mg, 0,03 équiv.) est additionné. Le mélange réactionnel est chauffé à 85°C pendant une nuit. Les solvants sont évaporés sous vide et le résidu est extrait avec AcOEt / H20. La phase organique est séchée et évaporée. Le produit brut est chromatographié sur gel de silice (200 g). L'élution par AcOEt / MeOH (95 / 5) fournit après évaporation 370 mg de produit.
Chlorhydrate de N-(ρyridin-4-yl)-5-pyridin-3-yl-1 H-indazole-3-carboxamide Sous argon à une solution de l'intermédiaire 9.4 (530 mg, 1 ,19 mmol) dans du THF (15 ml) sont ajoutés une solution de fluorure de tétrabutylammonium (TBAF) dans du THF (1M, 6 ml, 5 équiv.), de l'eau (0,2 ml) et de l'éthylènediamine (0,20 ml, 3 mmol, 2,5 équiv.). Le mélange réactionnel est chauffé à 60°C pendant 3 jours. Une addition supplémentaire de fluorure de tétrabutylammonium (1 N, 3 ml) est effectuée. Le chauffage est maintenu pendant une nuit supplémentaire. Le mélange réactionnel est acidifié par HCI 4N (1 ,2 ml), concentré sous vide puis dilué avec H20. Le précipité est filtré, lavé avec CH3OH et de l'éther diéthylique. Le solide obtenu est recristallisé dans CH2CI2 / MeOH.
On obtient 190 mg de produit sous forme d'une poudre blanche. PF: 196°C LC/MS/UV: MH+ 316 (96,8 %) RMN - H (500 MHz, DMSO-D6) δ (ppm) : 7,53 (dd, 1 H), 7,84 (s, 2H), 7,99 (d, 2H), 8,13 (d, 1 H), 8,46 (s, 1 H), 8,50 (d, 1 H), 8,59 (d, 1 H), 8,92 (s, 1 H), 10,86 (s, 1 H), 14,1 (s, 1 H).
Exemple 10 (composé n°35)
Chlorhydrate de N-(pyridin-4-yl)-5-(4-méthyl-[3,4']-bipyridinyl-5-yl)-1 H-indazole-3- carboxamide
Intermédiaire 10.1
3,5-dibromo-4-méthylpyridine
Sous argon à une solution de diisopropylamine (3,6 ml, 1 ,02 équiv.) dans du
THF anhydre (145 ml) maintenue à -10°C est ajoutée goutte à goutte une solution de n-butyllithium dans de l'hexane (1 ,6 N, 16 ml). Le mélange réactionnel est refroidi à -78°C puis une solution de 3,5-dibromopyridine (5,92 g, 25 mmol) dans le THF (200 ml) refroidie à -78°C est ajoutée goutte à goutte. Le mélange réactionnel est agité pendant 30 min puis de l'iodure de méthyle (2,17 ml, 1 ,4 équiv.) est additionné goutte à goutte. L'agitation esfïriaintenue pendant"" 2 h à -78°C . Une solution aqueuse saturée en NH4CI (120 ml) est ajoutée. Après évaporation des solvants, le mélange réactionnel est extrait avec AcOEt. La phase organique est lavée avec de la saumure, séchée sur MgS04 et évaporée. Le solide jaune obtenu est repris par de AcOEt. La suspension est filtrée. Le filtrat est évaporé puis le résidu est chromatographié sur gel de silice en éluant avec un mélange éther de pétrole / AcOEt (97,5 / 2,5) . On obtient 1 ,67 g de produit sous forme d'un solide blanc.
Intermédiaire 10.2 5-bromo-4-méthyl-[3,4']-bipyridinyle Sous argon, à une solution d'intermédiaire 10.1 (1 ,3 g, 5,18 mmol) dans du DME (18 ml) sont ajoutés de l'ester pinacolique de l'acide pyridine-4-boronique (910 mg, 4,45 mmol), une solution aqueuse de Na2C03 (2,35 g / 9 ml H20) et enfin du tétrakis(triphénylphosphine)palladium (153 mg). Le mélange est chauffé à 85°C pendant 2 jours. Le solvant est évaporé puis le résidu est extrait avec AcOEt / H20. Le composé obtenu après lavage de la solution organique par de la saumure, séchage sur MgS04 et évaporation, est chromatographié sur gel de silice (200 g) en éluant avec un mélange AcOEt/ éther de pétrole (1/1). On obtient 440 mg de produit sous forme d'une huile.
Intermédiaire 10.3
N-(pyridin-4-yl)-5-(4,4,5,5-tétraméthyl-[1 ,3,2]-dioxaborolan-2-yl)-1-(2- triméthylsilyléthoxyméthyl)-1 H-indazole-3-carboxylique
Un mélange d'intermédiaire 9.3 (3,77 g, 7,6 mmol), de bis(pinacolato)diborane
(2,12 g, 8,3 mmol), d'acétate de potassium (2,24 g) dans le diméthylsulfoxyde
(DMSO) (50 ml) est dégazé à l'argon. Du 1 ,1 '- bis(diphénylphosphino)ferrocènedichloropalladiumll (310 mg, 0,38 mmol, 0,05 équiv.) est ajouté sous argon. Le mélange réactionnel est chauffé à 80°C pendant 1 ,5 h. L'extraction par AcOEt / H20 permet d'isoler une huile orange qui est chromatographiée sur gel de silice en éluant avec AcOEt. L'huile jaune obtenue est cristallisée dans l'éther diéthylique. On obtient 2,56 g de produit sous forme d'une poudre blanche.
Intermédiaire 10.4 N-(pyridin-4-yl)-5-(4-méthyl-[3,4']-bipyridinyl-5-yl)-1 -(2-triméthylsilyl- éthoxyméthyl)-1 H-indazole-3-carboxamide
A une solution d'intermédiaire 10.3 (640 mg, 1 ,29 mmol) et d'intermédiaire 10.2
(370 mg, 1 ,15 équiv.) dans du DME (6 ml) est ajoutée une solution aqueuse de
Na2C03 (686 mg / 2,6 ml). Le mélange réactionnel est dégazé à l'argon. Du tétrakis(triphénylphosphine)palladium (46 mg) est ajouté sous argon. Le mélange réactionnel est chauffé à 85°C pendant une nuit. Le solvant est évaporé. L'extraction avec AcOEt / H20 permet d'isoler une huile qui est cristallisée dans un mélange AcOEt / éther de pétrole.
On obtient 530 mg de produit sous forme d'une poudre blanche.
Chlorhydrate de N-(pyridin-4-yl)-5-(4-méthyl-[3,4']-bipyridinyl-5-yl)-1 H-indazole-3- carboxamide
La coupure du groupe protecteur SEM de l'intermédiaire 10.4 est effectuée en présence de fluorure de tétrabutylammonium de manière analogue au mode opératoire décrit à l'exemple 9. Le produit brut est repris dans MeOH / Et20. Le composé obtenu est isolé par filtration et lavé avec MeOH.
On obtient 265 mg de produit sous forme d'une poudre.
PF: 192°C
LC/MS/UV: MH+ 407 (96,1 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,13 (s, 3H), 7,65 (m, 3H), 7,90 (d, J =
8,5, 1 H), 8,01 (d, J = 7,2, 2H), 8,32 (s, 1 H), 8,54 (s, 1 H), 8,55 (d, J = 6,2, 2H),
8,60 (s, 1 H), 8,79 (d, J = 5,7, 2H), 10,90 (s, 1 H), 13 (s, 1 H).
Exemple 11 (composé n°36) N-(pyridin-4-yl)-5-(5-carbamoyl-4-méthylpyridin-3-yl)-1 /-/-indazole-3-carboxamide
Intermédiaire 11.1 5-bromo-Λ-tert-butyl-4-méthylnicotinamide
Sous argon, une solution de n-butyllithium dans l'hexane (1 ,55 N, 6,6 ml) est additionnée goutte à goutte à une solution de 3,5-dibromo-4-méthylpyridine (intermédiaire 10.1 , 2,51 g, 10 mmol) maintenue à -100°C Après 10 min d'agitation, de l'isocyanate de tertiobutyle (2,28 ml, 20 mmol) est ajouté. L'agitation est maintenue pendant 20 min à -100°C puis 1h à -78°C et à température ambiante pendant une nuit. Une solution aqueuse de NH4CI est ajoutée au mélange réactionnel. L'extraction par AcOEt/ H20 fournit un solide brun qui est repris dans un mélange AcOEt / éther de pétrole. Le composé obtenu est isolé par filtration. On obtient 1 ,6 g de produit.
Intermédiaire 11.2 5-bromo-4-méthylnicptjnamide
L'intermédiaire 11.1 (1 ,4 g, 5,18 mmol) est mis en réaction avec H2S04 90 % (25 ml). Le mélange est agité à température ambiante pendant 3 jours. Le mélange réactionnel est neutralisé par une solution aqueuse saturée en Na2C03 puis est extrait avec AcOEt. Le solide obtenu est repris dans AcOEt et filtré. On obtient 880 mg de produit sous forme d'une poudre.
Intermédiaire 11.3
N-(pyridin-4-yl)-5-(5-carbamoyl-4-méthylpyridin-3-yl)-1-(2-triméthylsilanyl- éthoxyméthyl)-1 H-indazole-3-carboxamide
Ce composé est synthétisé d'une manière analogue à l'intermédiaire 10.4, par une réaction de Suzuki entre l'intermédiaire 10.3 et l'intermédiaire 11.2, à l'échelle de 1 ,29 mmol. Il est recristallisé dans un mélange CHCI3 / AcOEt.
On obtient 530 mg de produit sous forme d'un solide blanc.
N-(pyridin-4-yl)-5-(5-carbamoyl-4-méthylpyridin-3-yl)-1 H-indazole-3-carboxamide L'intermédiaire 11.3 (550 mg, 1,09 mmol) est mis en réaction avec de l'acide trifluoroacetique (TFA) pendant 5 min à 0°C puis pendant 1 ,5 h à température ambiante. Le TFA est évaporé sous vide. Les traces de TFA sont éliminés par co-évaporation avec du toluène. Le solide blanc obtenu est mis en réaction avec une solution d'éthylènediamine (366 μl, 5,4 mmol) dans le THF (10 ml). Le mélange réactionnel est chauffé à reflux pendant une nuit. Le mélange réactionnel est filtré puis lavé par un mélange MeOH / H20. On obtient 270 mg de produit sous forme d'un solide blanc. PF: >250°C LC/MS/UV: MH+ 373 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,25 (s, 3H), 7,54 (dd, J = 8,7, J = 1 ,1 , 1 H), 7,73 (s, 1 H), 7,87 (d, J = 8,6, 1 H), 7,98 (d, J = 6,3, 2H), 8,09 (s, 1 H), 8,21 (s, 1 H), 8,49 (d, J = 5,0, 2H), 8,54 (s, 1 H), 8,59 (s, 1 H), 10,87 (s, 1 H), 14,1 (s, 1 H).
Exemple 12 (composé n°37)
Chlorhydrate de N-(pyridin-4-yl)-5-(4-méthyl-5-phénoxyméthylpyridin-3-yl)-1 H- indazole-3-carboxamide
Intermédiaire 12.1
5-bromo-4-méthylpyridine-3-carbaldéhyde
Sous argon, une solution de n-butyllithium dans l'hexane (1 ,55 N, 4,6 ml) est additionnée goutte à goutte à une solution de 3,5-dibromo-4-méthylpyridine (intermédiaire 10.1 , 1 ,76 g, 7 mmol) maintenue à -100°C. Après agitation pendant 5 min, de la DMF (0,84 ml, 1 ,54 équiv) est ajoutée goutte à goutte. L'agitation est maintenue pendant 20 min à -100°C puis 1 h à -78°. Une solution aqueuse de NH4CI est ajoutée et le mélange réactionnel est extrait avec un mélange éther diéthylique/eau. Le solide jaune obtenu est purifié sur gel de silice (100 g) en éluant avec AcOEt / éther de pétrole (1 14). On obtient 610 mg de produit sous forme d'une huile incolore.
Intermédiaire 12.2
(5-bromo-4-phénylpyridin-3-yl)méthanol
A de l'intermédiaire 12.1 (610 mg, 3,05 mmol) en solution dans MeOH (15 ml) est ajouté à 0°C du borohydrure de sodium (358 mg, 9,46 mmol). Le mélange réactionnel est agité pendant 1 ,5 h puis est dilué avec AcOEt / H20. L'extraction par AcOEt fournit un solide blanc qui est chromatographié sur gel de silice (100 g). L'élution avec AcOEt fournit 420 mg de produit.
Intermédiaire 12.3
3-bromo-4-méthyl-5-phénoxyméthylpyridine
A de l'intermédiaire 12.2 (420 mg, 2,08 mmol) en solution dans un mélange de THF (10 ml) et de toluène (3 ml) sont ajoutés du phénol (0,22 ml, 2,50 mmol), de la triphénylphosphine (655 mg, 2,50 mmol) et du diéthylazodicarboxylate (0,393 ml, 2,50 mmol). Le mélange réactionnel est agité 3 jours à température ambiante puis est extrait avec AcOEt / H20. Le composé brut est purifié sur gel de silice (100 g). L'élution par AcOEt / éther de pétrole (1/1) fournit un composé qui repris dans AcOEt et lavé avec NaOH 1 N.
On obtient 380 mg de produit sous forme d'une huile incolore.
Intermédiaire 12.4
N-(pyridin-4-yl)-5-(4-méthyl-5-phénoxyméthylpyridin-3-yl)-1-(2-triméthylsilyl- éthoxyméthyl)-1 H-indazole-3-carboxamide
Ce composé est synthétisé d'une manière analogue à l'intermédiaire 10.4, par une réaction de Suzuki entre les intermédiaires 10.3 et 12.3, à l'échelle de 1 ,37 mmol. Il est chromatographié sur gel de silice en éluant avec AcOEt.
On obtient 567 mg de produit sous forme d'une huile incolore.
Chlorhydrate de N-(pyridin-4-yl)-5-(4-méthyl-5-phénoxyméthylpyridin-3-yl)-1 H- indazole-3-carboxamide
La .coupure du groupe protecteur SEM de l'intermédiaire 12.4 est effectuée par du fluorure de tétrabutylammonium de manière analogue au mode opératoire décrit à l'exemple 9, à l'échelle de 1 mmol. Le composé est repris avec MeOH puis filtré.
On obtient 230 mg de produit sous forme d'une poudre blanche.
PF: 160°C
LC/MS/UV: MH+ 436 (98,8 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,40 (s, 3H), 5,32 (s, 2H), 7,07 (t, J =
4,85, 1H), 7,19 (d, J = 7,9, 2H), 7,42 (t, J = 5,3, 2H), 7,56 (dd, J = 8,4, J = 1 ,5,
1 H), 7,88 (d, J = 8,6, 1 H), 7,99 (d, J = 6,3, 2H), 8,23 (s, 1 H), 8,53 (s, 1 H), 8,54
(d, J = 6,0, 2H), 8,70 (s, 1 H), 10,88 (s, 1 H), 14,05 ,s, 1 H)
Exemple 13 (composé n°41)
N-(pyridin-4-yl)-5-isoquinolin-4-yl-1 H-indazole-3-carboxamide
Intermédiaire 13.1
5-iodo-1 H-indazole-3-carbaldéhyde Sous argon, à une suspension de 5-iodoindole (9,722 g, 40 mmol) dans l'eau est ajouté par portions de 1 g du nitrite de sodium (27,6 g, 400 mmol) puis goutte à goutte une solution d'HCI 6N (59 ml). La température du mélange réactionnel est maintenue en dessous de 15°C puis le mélange réactionnel est laissé sous vive agitation à température ambiante pendant une nuit. Les vapeurs nitreuses sont chassées sous courant d'argon puis le mélange réactionnel est filtré. Le lavage du solide avec H20 suivi d'une purification sur gel de silice (600 g) en éluant suivant un gradient d'élution, de CH2CI2 à un mélange CH2CI2 / AcOEt (9 / 1), permet d'isoler 1 ,37 g de produit sous forme d'un solide brun.
LC/MS/UV : MH+ 273 (88,6 %)
Intermédiaire 13.2
5-iodo-1-(2-triméthylsilyléthoxyméthyl)-1 --indazole-3-carbaldéhyde Sous argon, à une suspension d'hydrure de sodium (50 % dans l'huile, 0,50 g, 10,4 mmol) dans de la DMF anhydre (10 ml) sont ajoutés de l'intermédiaire 13.1 (2,58 g, 9,5 mmol) puis goutte à goutte une solution de chlorure de triméthylsilyléthoxyméthyle (SEMCI, 1 ,60 g,9,6 mmol) dans de la DMF (5 ml). L'agitation est maintenue 1 h à température ambiante. De l'eau est ajoutée puis la DMF est évaporée sous vide. Le résidu est repris dans CH2CI2. La solution organique est lavée avec de la saumure, séchée puis évaporée sous vide. Le produit brut est chromatographié sur gel de silice (500 g). L'élution avec CH2CI2 donne le produit sous forme d'une Jhuile visqueuse brune.
Intermédiaire 13.3
5-isoquinolin-4-yl-1-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3-carbaldéhyde Sous argon à une solution de l'intermédiaire 13.2 (1,07 g, 2,66 mmol) dans du DME (10 ml) sont ajoutés du chlorhydrate d'acide isoquinolin-4-ylboronique (0,56 g, 2,66 mmol), une solution aqueuse de Na2C03 (1 ,42 g, 13,4 mmol dans 5 ml H20) et du tétrakis(triphénylphosphine)palladiumO (0,160 g, 0,14 mmol, 0,05 équiv.). Le mélange réactionnel est chauffé au moyen d'un bain d'huile régulé à 85°C pendant 5 h puis est concentré sous vide. Le résidu est repris dans AcOEt. La solution organique est lavée avec de la saumure, séchée puis évaporée pour donner un produit brut qui est purifié sur gel de silice (150 g). L'élution selon un gradient allant de CH2CI2 à CH2CI2 / AcOEt (9 / 1) fournit 0,84 g de produit.
Intermédiaire 13.4
Acide 5-isoquinolin-4-yl-1 -(2-triméthylsilyléthoxymé thyl)-1 H-indazole-3- carboxylique
A une solution d'intermédiaire 13.3 (0,50 g, 1 ,24 mmol) dans de la DMF (5 ml) maintenue à une température comprise entre 0°C et -5°C refroidie par un bain constitué d'un mélange de glace et de sel sont additionnés du 2-méthyl-2-butène (5 ml), de la DMF (5 ml) puis une solution aqueuse (10 ml) de chlorite de sodium (1 ,12 g) et de dihydrogénophosphate de sodium (1 ,37 g, sous forme d'hydrate). La température du mélange réactionnel est maintenue à 0°C pendant 30 min, puis le mélange réactionnel est agité pendant 4,5 h à température ambiante et, après acidification par HCI 6N (5ml), pendant une nuit. Le mélange réactionnel
est évaporé. Le résidu est repris dans AcOEt. Cette solution est lavée avec H20 et de la saumure, séchée sur Na2S04 et évaporée pour donner 0,50 g de produit sous forme d'un solide blanc.
Intermédiaire 13.5
N-(pyridin-4-yl)-5-isoquinolin-4-yl-1-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3- carboxamide
Sous argon, à une solution d'intermédiaire 13.4 (0,50 g, 1 ,19 mmol) dans du
THF anhydre (10 ml) maintenue à une température entre -5°C et 0°C sont ajoutés de l'isopropylchloroformiate (1 M dans le toluène, 1 ,2 ml) et de la N- méthylmorpholine (0,120 g, 1 ,2 mmol). On laisse réagir 15 min avant d'additionner de la 4-aminopyridine (0,114 g, 1 ,2 mmol). Le mélange réactionnel est agité durant 30 min à 0°C puis une nuit à température ambiante. Il est ensuite évaporé et repris dans AcOEt. La solution organique est lavée avec de la saumure, séchée sur Na2S04 et évaporée. L'huile obtenue est purifiée sur gel de silice. L'élution par un mélange CH2CI2 / MeOH (9/1) permet d'isoler 250 mg de produit sous forme d'une huile jaune.
N-(pyridin-4-yl)-5-isoquinolin-4-yl-1 H-indazole-3-carboxamide Sous argon, un mélange d'intermédiaire 13.5 (0,250 g, 0,5 mmol), de 1 ,2- diaminoéthane (0, 150 g, 2,5 mmol) et de fluorure de tétrabutylammonium dans du THF (1 M, 5 ml) est chauffé à 70°C pendant une nuit. Le mélange réactionnel est évaporé sous vide. Le résidu est repris avec AcOEt. Cette solution est lavée avec une solution aqueuse de NaHC03 saturée et de la saumure, puis séchée et évaporée. Le solide obtenu est lavé avec de l'éther diéthylique puis purifié sur gel de silice (50 g). Le composé obtenu est élue avec un mélange AcOEt / MeOH (9 / 1). Il est repris un mélange éther diéthylique / éther de pétrole puis filtré. On obtient 182 mg de produit sous forme d'un solide blanc. PF: >250°C
LC/MS/UV: MH+ 366 (99,4 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,65 (d, J = 8,5, 1 H), 7,76 (t, J = 7,3, 1 H), 7,81 (t, J = 7,4, 1 H), 7,88 (dd, J = 8,4, J = 3,3, 2H), 7,92 (d, J = 6,1 , 2H), 8,26 (d, J = 8,0, 1 H), 8,34 (s, 1 H), 8,46 (d, J = 5,9, 2H), 8,52 (s, 1 H), 9,38 (s, 1 H), 10,85 (s, 1 H), 14,2 (s, 1 H).
Exemple 14 (composé n°45)
Acide 4-[5-(isoquinolin-4-yl)-1 H-indazole-3-carbonylamino]benzoique
Intermédiaire 14.1
4-[5-(isoquinolin-4-yl)-1-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3- carbonylaminojbenzoate d'éthyle
Ce composé est préparé de manière similaire à l'intermédiaire 13.5 en couplant l'intermédiaire 13.4 à du 4-aminobenzoate d'éthyle, à l'échelle de 2,4 mmol. Le composé brut est purifié sur gel de silice (80 g). Un gradient d'élution, de CH2CI2 à CH2CI2 / AcOEt (713) permet d'isoler 0,62 g de produit sous forme d'une huile.
Intermédiaire 14.2
4-[5-(isoquinolin-4-yl)-1 H-indazole-3-carbonylamino]benzoate d'éthyle Le groupe protecteur SEM de l'intermédiaire 14.1 est coupé par du TBAF de manière analogue au mode opératoire décrit à l'exemple 13, à l'échelle de 1 ,1 mmol. Ce composé est repris dans de l'éther diéthylique puis filtré. On obtient 292 mg d'un mélange de l'ester attendu et de l'acide correspondant.
Acide 4-[5-(isoquinolin-4-yl)-1 --indazole-3-carbonylamino]benzoique
A une suspension de l'intermédiaire 14.2 (290 mg, 0,67 mmol) dans le THF (5 ml) est ajoutée une solution aqueuse d'hydroxyde de lithium (36 mg / 1 ml H2Or 1 ,48 mmol). Le mélange réactionnel est chauffé au reflux pendant une nuit puis acidifié par HCI 1 N (1,5 ml). L'évaporation des solvants donne un solide qui est repris dans l'eau, filtré, lavé avec de l'eau et de l'éther diéthylique (Et20). Le solide obtenu est séché sous vide en présence de P205. On obtient 0,278 g de produit sous forme d'un solide gris. PF: >260°C LC/MS/UV: MH+ 409 (100 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,5- 8,6 (m, 12H), 9,42 (s, 1 H), 10,75 (s, 1 H), 12,67 (s, 1 H), 14,08 (s, 1 H).
Exemple 15 (composé n°46) N-(pyridin-4-yl)-5-amino-1 H-indazole-3-carboxamide
Intermédiaire 15.1 5-nitro-1 H-indazole-3-carbaldéhyde
Cet intermédiaire est préparé de manière analogue à l'intermédiaire 13.1 par réaction de l'acide nitreux sur du 5-nitroindole à l'échelle de 30 mmol. Le mélange réactionnel est extrait avec AcOEt. La phase organique est lavée avec de la saumure puis séchée sur Na2S0 . L'évaporation sous vide donne un solide rougeâtre qui est lavé avec de l'éther de pétrole et séché sous vide. On obtient 5,51 g de produit.
Intermédiaire 15.2 5-nitro-1-(2,4,6-triméthylbenzènesulfonyl)-1r7'-indazole-3-carbaldéhyde Sous argon, à une solution d'intermédiaire 15.1 (5,4 g, 28,25 mmol) dans CH2CI2 anhydre (250 ml) maintenue à 0°C par un bain de glace, sont ajoutés de la 4- diméthylaminopyridine (3,63 g, 29,71 mmol) puis après quelques minutes, goutte à goutte du chlorure de 2-mésitylènesulfonyle (6,50 g, 29,71 mmol) en solution dans CH2CI2 (100 ml). Le mélange réactionnel est agité pendant 1 h à 0°C puis une nuit à température ambiante. De l'eau est ajoutée. Le mélange réactionnel est extrait avec CH2CI2. La phase organique est lavée avec de la saumure, séchée sur Na2S04 et évaporée pour donner un solide brun qui est purifié par chromatographié sur gel de silice (300 g) en éluant avec CH2CI2. Un solide se sépare après concentration des fractions sous vide. Il est filtré puis lavé avec de l'éther diéthylique et de l'éther de pétrole. On obtient 5 g de produit sous forme d'un solide beige.
Intermédiaire 15.3
Acide 5-nitro-1 -(2,4,6-triméthylbenzènesulfonyl)-1 /-/-indazole-3-carboxylique La fonction aldéhyde de l'intermédiaire 15.2 est oxydée en acide carboxylique par du chlorite de sodium de manière similaire à l'intermédiaire 13.4, à l'échelle de 13,4 mmol.
Le produit brut, une huile jaune, est cristallisé dans un mélange éther de pétrole
/ éther diéthylique. On obtient 4,47 g de produit sous forme d'un solide blanc.
Intermédiaire 15.4
N-(pyridin-4-yl)-5-nitro-1-(2,4,6-triméthylbenzènesulfonyl)-1 H-indazole-3- carboxamide L'intermédiaire 15.4 est obtenu de manière similaire à l'intermédiaire 13.5 en couplant l'intermédiaire 15.3 à de la 4-aminopyridine, à l'échelle de 4,78 mmol. Le brut réactionnel est repris dans l'éther diéthylique. Un précipité se forme qui
est filtré puis purifié par chromatographié sur gel de silice (80 g) en éluant par un mélange AcOEt / CH2CI2 (1 / 1).
On obtient 0,69 g de produit sous forme d'un solide blanc.
Intermédiaire 15.5
N-(pyridin-4-yl)-5-amino-1-(2,4,6-triméthylbenzènesulfonyl)-1 -/-indazole-3- carboxamide
L'intermédiaire 15.4 (0,35 g, 0,75 mmol) est mis en suspension dans de l'éthanol
(40 ml). Du chlorure d'étain (SnCI2, 2H20, 0,847 g, 3,75 mmol) est ajouté puis le mélange réactionnel est chauffé à reflux pendant 4 h. Il est évaporé sous vide. Le résidu est repris dans H20 et le pH est ajusté à pH 8 par l'addition de soude 2N. L'extraction par un mélange CH2CI2 / CHCI3 suivi d'un lavage par de la saumure, d'un séchage sur Na2S04 puis d'une évaporation sous vide donne un produit brut qui est repris dans un mélange éther diéthylique / éther de pétrole puis filtré.
On obtient 0,282 g de produit sous forme d'un solide blanc.
N-(pyridin-4-yl)-5-amino-1 H-indazole-3-carboxamide
A de l'intermédiaire 15.5 (0,277 g, 0,636 mmol) en solution dans du 1,4-dioxane est ajoutée une solution de soude 1N (3,18 ml). Le mélange réactionnel est chauffé à 70°C par un bain d'huile, pendant 4 h. Le mélange réactionnel est neutralisé par addition d'une solution HC1 1 N (3,2 ml) puis évaporé. Le résidu est repris avec de l'eau. Le pH est ajusté à pH neutre. Il se forme un précipité qui est filtré, lavé avec de l'eau puis avec de l'éther diéthylique. On obtient 121 mg de produit sous forme d'un solide brun. PF: >260°C
LC/MS/UV: MH+ 254 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 5,13 (s, 2H), 6,88 (d, 1 H), 7,30 (s, 1 H), 7,38 (d, 1 H), 7,78 (d, 2H), 8,44 (d, 2H), 10,49 (s,1H), 13,46 (s, 1 H).
Exemple 16 (composé n°47) N-(pyridin-4-yl)-5-(3-méthylbutyrylamino)-1 /--indazole-3-carboxamide
Intermédiaire 16.1 N-(pyridin-4-yl)-5-(3-méthyl-butyrylamino)-1 -(2,4,6-triméthylbenzènesulfonyl)-1 H- indazole-3-carboxamide
Sous argon, à une solution d'intermédiaire 15.5 (240 mg, 0,55 mmol) dans CH2CI2 anhydre (5 ml) maintenue à 0°C, sont ajoutés de la N,N-
diisopropyléthylamine (72 mg, 0,55 mmol) puis du chlorure d'isovaléryle (68 mg, 0,55 mmol) en solution dans CH2CI2 (2 ml). L'agitation est maintenue pendant 30 min à 0°C puis une nuit à température ambiante. Le mélange réactionnel est évaporé. Le résidu est repris dans AcOEt. La solution organique est lavée avec NaHC03, de la saumure puis séchée sur Na2S0 . Le résidu d'évaporation est repris dans l'éther diéthylique. Le composé obtenu est isolé par filtration puis lavé avec de l'éther diéthylique. On obtient 198 mg de produit.
N-(pyridin-4-yl)-5-(3-méthylbutyrylamino)-1 H-indazole-3-carboxamide
Ce composé est préparé par coupure du groupe protecteur mésitylènesulfonyle de l'intermédiaire 16.1 par de la soude d'une manière analogue au mode opératoire décrit à l'exemple 15.
On obtient 76,5^ mg de produit sous-forme d'un solide brun. PF: >260°C
LC/MS/UV: MH+ 338 (95,3 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 0,95 (d, 6H), 2,12 (m, 1 H), 2,21 (d, 2H),
7,63 (s, 2H), 8,15 (d, 2H), 8,59 (d, 2H), 8,61 (s, 1 H), 10,02 (s, 1 H), 11 ,13 (s, 1 H),
13,96 (s, 1 H).
Exemple 17 (composé n°49)
N-(pyridin-4-yl)-5-nitro-1 H-indazole-3-carboxamide
Le groupe protecteur mésitylènesulfonyle de l'intermédiaire 15.4 est coupé avec de la soude, de manière analogue à l'exemple 15, à l'échelle de 0,7 mmol. On obtient 0,20 g de produit sous forme d'un solide jaune.
PF : >260°C
LC/MS/UV : MH+ 284 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,93 (d, J = 9,0, 1 H), 8,13 (d, J = 6,3,
2H), 8,32 (dd, J = 8,8, J = 1 ,9, 1 H), 8,61 (d, J = 6,9, 2H), 9,08 (d, J = 1 ,9, 1 H), 11 ,35 (s, 1 H), 14,65 (s, 1 H).
Exemple 18 (composé n°50) N-(pyridin-4-yl)-5-iodo-1 H-indazole-3-carboxamide
Le groupe protecteur SEM de l'intermédiaire 9.3 est coupé par du TBAF, de manière analogue au mode opératoire décrit à l'exemple 13, à l'échelle de 0,5 mmol. Le brut de réaction est lavé avec de l'éther diéthylique. On obtient 297 mg de produit. PF : >260°C LC/MS/UV : MH+ 365 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,59 (d, 1 H), 7,76 (d, 1H), 8,09 (d, 2H), 8,54 (d, 2H), 8,61 (s, 1 H), 11 ,01 (s, 1 H), 14,16 (s, 1 H).
Exemple 19 (composé n°51) N-(pyridin-4-yl)-5-cyano-1 H-indazole-3-carboxamide
Intermédiaire 19.1
3-formyl-1 H-indazole-5-carbonitrile
A une solution aqueuse de nitrite de sodium (13,80 g, 200 mmol dans 400 ml H20) sont ajoutés du 5-cyanoindole (2,85 g, 20 mmol) puis goutte à goutte en 30 min une solution d'HCI 6N (30 ml). Le mélange réactionnel est agité pendant 3 h puis filtré. Le solide est repris dans AcOEt. La solution organique est séchée sur Na2S0 puis évaporée. On obtient le produit sous forme d'un solide orange.
Intermédiaire 19.2
3-formyl-1-(2,4,6-triméthylbenzènesulfonyl)-1 --indazole-5-carbonitrile
A une solution de l'intermédiaire 19.1 (20 mmol) dans du THF anhydre (200 ml), maintenue à 0°C sont ajoutées de la 4-dimé thylaminopyridine (2,21 g, 21 mmol) et une solution de chlorure de mésitylènesulfonyle (4,60 g, 21 mmol) dans du THF anhydre (50 ml). Le mélange réactionnel est agité à température ambiante pendant une nuit. Il est filtré puis évaporé. Le résidu est repris dans AcOEt. La solution organique est lavée avec HCI 1 N, H20 et NaHC03 10 % puis séchée et concentrée sous vide. Le résidu est précipité par addition d'éther de pétrole. On obtient 6,11 g de produit sous forme d'un solide orange.
Intermédiaire 19.3
Acide 5-cyano-1 -(2,4,6-triméthylbenzènesul.onyl)-1 --indazole-3-carboxylique
Une solution de l'intermédiaire 19.2 (6,0 g, 17 mmol) dans de la DMF (100 ml) est refroidie à 0°C. Du 2-méthyl-2-butène (70 ml) puis une solution aqueuse (125 ml) de chlorite de sodium (80 %, 15,3 g, 136 mmol) et de dihydrogéno- phosphate de sodium (NaH2P0 .H20, 18,7 g, 136 mmol) sont ajoutés. Le mélange réactionnel est agité pendant 1 h à température ambiante. Une solution d'HCI 4N (100 ml) est additionnée goutte à goutte et le mélange réactionnel est agité pendant 2h. Le mélange est dilué avec de l'eau et extrait avec AcOEt. La phase organique est lavée avec de l'eau, séchée et évaporée. Le résidu est précipité dans un mélange éther diéthylique / éther de pétrole. On obtient 5,32 g de produit sous forme d'une poudre de couleur crème.
Intermédiaire 19.4
[5-cyano-1-(2,4,6-triméthyl-benzènesulfonyl)-1 H-indazol-3-yl]carbonyl- carbonate d'isopropyle Sous argon, à une solution de l'intermédiaire 19.3 (5,32 g, 14,4 mmol) dans du THF anhydre (150 ml) maintenue à 0°C, sont ajoutées une solution d'isopropyl- chloroformiate dans le toluène (1 M, 15,8 ml) et, goutte à goutte de la N- méthylmorpholine (1 ,74 ml, 15,8 mmol). Le mélange réactionnel est agité pendant 15 min à 0°C puis 30 min à température ambiante. Le solvant est évaporé et le résidu est repris par un mélange éther diéthylique / eau. La phase organique est lavée avec de l'eau, HCI 0,5N et NaHC03 5 % puis séchée et évaporée. On obtient 5,61 g de produit sous forme d'un solide de couleur crème.
N-(pyridin-4-yl)-5-cyano-1 H-indazole-3-carboxamide
Sous argon, de la 4-aminopyridine (2,32 g, 24,6 mmol) est ajouté à 5,61 g (12,3 mmol) de l'intermédiaire 19.4 en solution dans du THF anhydre (100 ml). Le mélange réactionnel est chauffé à 60°C pendant 36 h. puis filtré. Le solide ainsi isolé est lavé avec du THF. Il est repris dans un mélange CH2CI2 / MeOH et filtré.
On obtient 3,95 g de produit sous forme d'un solide de couleur crème.
PF : >250°C
LC/MS/UV : MH+254 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,80 (d, 1 H), 7,87 (d, 1 H), 7,91 (d, 2H), 8,47 (d, 2H), 8,65 (s, 1 H),10,95 (s, 1 H), 14,3 (s, 1 H).
Exemple 20 (composé n°52)
Sel d'acide méthanesulfonique de N-(pyridin-4-yl)-5-aminométhyl-1 H-indazole-3- carboxamide Une solution du composé obtenu à l'exemple 19 (263 mg, 1 mmol) dans un mélange AcOH / H20 (25 ml / 5ml) est hydrogénée à pression atmosphérique en présence de 10 % Pd / C (50 mg) pendant 3 h. Le catalyseur est filtré et le mélange est concentré sous vide. Le résidu est cristallisé dans un mélange isopropanol / H20. On obtient 128 mg de produit.
PF : >250°C
LC/MS/UV: MH+268 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : (en accord avec C14H13N50; 1 ,5 CH403S) 2,31 (d, 2H), 7,60 (d, J = 8,8, 1 H), 7,82 (d, J = 8,7, 1H), 8,21 (s, 3H), 8,38 (s, 1 H), 8,43 (d, J = 6,8, 2H), 8,77 (d, J = 7,0, 2H), 11 ,79 (s, 1H), 14,32 (s, 1H).
Exemple 21 (composé n°53) N-(pyridin-4-yl)-5-bromo-1 H-indazole-3-carboxamide
Intermédiaire 21.1 5-bromoindazole-3-carbaldéhyde
A une solution aqueuse de nitrite de sodium (13,80 g, 200 mmol dans 400 ml H20) sont ajoutés du 5-bromoindole (3,93 g, 20 mmol) puis lentement, en 25 min, une solution aqueuse d'HCI 6N (30 rnl) L.e mélange réactionnel est agité vigoureusement pendant 3 h puis filtré. Le solide recueilli est repris par AcOEt, Cette solution est séchée et évaporée.
Intermédiaire 21.2
5-bromo-1-(2,4,6-triméthylbenzènesulfonyl)-1 H-indazole-3-carbaldéhyde
A une solution de l'intermédiaire 21.1 (20 mmol) dans du THF anhydre (200 ml), maintenue à 0°C sont ajoutées de la 4-diméthylaminopyridine (2,21 g, 21 mmol) et une solution de chlorure de mésitylènesulfonyle (4,60 g, 21 mmol) dans du THF anhydre (50 ml). Le mélange réactionnel est agité à température ambiante pendant une nuit, puis est filtré et évaporé. Le résidu est repris dans AcOEt. La solution organique est lavée avec HCI 1 N, H20 et NaHC03 10 % puis séchée et concentrée sous vide. Le résidu est purifié sur gel de silice en éluant suivant un gradient de AcOEt / éther de pétrole (1/9) à AcOEt / éther de pétrole (1/4). Le composé purifié est recristallisé dans AcOEt / éther de pétrole. On obtient 3,60 g de produit sous forme d'un solide brun.
Intermédiaire 21.3
Acide 5-bromo-1 -(2,4,6-triméthylbenzènesulfonyl)-1 H-indazole-3-carboxylique A une solution de l'intermédiaire 21.2 (2,04 g, 5 mmol) dans de la DMF (50 ml) et de 2-méthyl-2-butène (20 ml) refroidie à 0°C est ajoutée en 20 min une solution aqueuse (40 ml) de chlorite de sodium (80 %, 4,50 g, 50 mmol) et de dihydrogénophosphate de sodium (NaH2P0 .H 0, 5,50 g, 40 mmol). Le mélange réactionnel est agité pendant 1 h à température ambiante. Une solution d'HCI 4N (30 ml) est additionnée goutte à goutte et le mélange réactionnel est agité pendant 2h. Il est dilué avec de l'eau (100 ml) et extrait avec de l'éther
diéthylique. La phase organique est lavée avec de l'eau, séchée et évaporée. Le résidu est précipité dans un mélange CH2CI2 / éther de pétrole. On obtient 2,1 g de produit sous forme d'un solide brun.
Intermédiaire 21.4
[5-bromo-1-(2,4,6-triméthylbenzènesulfonyl)-1 --indazol-3-yl]carbonyl- carbonate d'isopropyle
Sous argon, à une solution de l'intermédiaire 21.3 (5 mmol) dans du THF anhydre (50 ml) maintenue à 0°C, sont ajoutées une solution d'isopropyl- chloroformiate dans le toluène (1M, 5,5 ml) et, goutte à goutte, de la N-méthyl- morpholine (605 μl, 5,5 mmol). Le mélange réactionnel est agité pendant 15 min à 0°C puis 30 min à température ambiante. Le solvant est évaporé et le résidu est repris par un mélange éther diéthylique / eau. La phase organique est lavée avec de l'eau, HCI 0,5N et NaHC035 % puis séchée et évaporée. On obtient 2,58 g de produit sous forme d'une gomme rouge-noir.
N-(pyridin-4-yl)-5-bromo-1H-indazole-3-carboxamide
Sous argon, de la 4-aminopyridine (941 mg, 10 mmol) et de l'intermédiaire 21.4
(5 mmol) en solution dans du THF anhydre (35 ml) sont chauffés à 60°C pendant 36 h. Le mélange réactionnel est évaporé. Le résidu est repris avec de l'éther diéthylique. La solution éthérée est évaporée. Le résidu est recristallisé dans MeOH / CH2CI2.
On obtient 186 mg de produit.
LC/MS/UV : MH+317 (99,5 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,59 (d, 1 H), 7,68 (d, 1 H), 7,90 (d, 2H),
8,35 (s, 1 H), 8,46 (d, 2H), 10,80 (s, 1 H), 14,1 (s, 1 H).
Exemple 22 (composé n°54)
N-(pyridin-4-yl)-5-[4-méthyl-5-(pyridin-3-ylcarbamoyl)-pyridin-3-yl]-1 /--indazole-3- carboxamide
Intermédiaire 22.1 Acide 5-bromo-4-méthylnicotinique
Sous argon, une solution de 3,5-dibromo-4-méthylpyridine (intermédiaire 10.1 , 2,51 g, 10 mmol) dans du THF anhydre (100 ml), est mis en réaction à une température de -100°C avec une solution de n-butyllithium dans l'hexane (1,6 N, 6,5 ml). Le mélange réactionnel est agité pendant 15 min puis de la carboglace est ajoutée. La température est maintenue pendant 15 min à -85°C, pendant
1 h30 à -78°C et 2 h à température ambiante. De l'eau est ajoutée. Les solvants sont évaporés sous vide et le résidu est purifié en éluant sur de la silice greffée diol avec du MeOH.
On obtient 1 ,66 g de produit sous forme d'un solide blanc.
Intermédiaire 22.2
5-bromo-4-méthyl-Λ/-pyridin-3-ylnicotinamide
Sous argon, à une solution d'intermédiaire 22.1 (1 ,22 g, 5,65 mmol) dans de la
DMF anhydre (60 ml) est ajouté de l'hexafluorophosphate de 0-(7-azabenzo- triazol-1-yl)-/V,Λ/,/V', Λ/'-tétraméthyluronium (HATU, 2,36 g, 6, 21 mmol). Le mélange réactionnel est agité 10 min avant d'additionner de la 3-aminopyridine
(740 mg, 7,86 mmol). L'agitation est maintenue 3 jours à température ambiante.
La DMF est évaporée. Le résidu est repris par AcOEt / H20. La phase-organique est lavée avec HCI 1 N. Les phase aqueuses sont combinées et portées à pH basique par l'addition de soude 1 N puis extraites avec AcOEt. La phase organique est séchée sur MgS04 puis évaporée.
On obtient 1 ,08 g de produit sous forme d'un solide blanc.
Intermédiaire 22.3 N-(pyridin-4-yl)-5-[4-méthyl-5-(pyridin-3-ylcarbamoyl)-pyridin-3-yl]-1-(2- triméthylsilyléthoxyméthyl)-1 H-indazole-3-carboxamide
A une solution d'intermédiaire 10.3 (640 mg, 1 ,29 mmol) et d'intermédiaire 22.2
(435 mg, 1 ,49 mmol) dans du DME (6 ml) est ajoutée une solution aqueuse de
Na2C03 (686 mg / 2,6 ml). Le mélange réactionnel est dégazé à l'argon. Du tétrakis(tiphénylphosphine)palladium (46 mg) est ajouté sous argon. Le mélange réactionnel est chauffé à 85°C pendant une nuit. Le solvant est évaporé .
L'extraction avec AcOEt / H20 permet d'isoler une huile qui est cristallisée dans un mélange AcOEt / CHCI3.
On obtient 504 mg de produit sous forme d'une poudre.
N-(pyridin-4-yl)-5-[4-méthyl-5-(pyridin-3-ylcarbamoyl)-pyridin-3-yl]-1 H-indazole-3- carboxamide
Le composé est obtenu de manière similaire au mode opératoire décrit à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 22.3 (500 mg, 0,86 mmol). Le produit brut est repris dans MeOH. Le composé obtenu est isolé par filtration.
On obtient 340 mg de produit sous forme d'une poudre blanche.
PF : >250°C
LC/MS/UV: MH+ 450 (97,7 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,29 (s, 3H), 7,48 (dd, J = 8,0, J = 4,7, 1 H), 7,57 (d, J = 8,5), 7,89 (d, J = 8,6, 1 H), 7,98 (d, J = 5,2, 2H), 8,26 (m, 2H), 8,40 (d, J = 4,5, 1 H), 8,52 (d, J = 5,2, 2H), 8,64 (s, 1 H), 8,77 (s, 1 H), 9,86 (s, 1 H), 10,88 (s, 1 H), 13,8 (s, 1 H).
Exemple 23 (composé n°55)
N-(pyridin-4-yl)-5-[8-(3-phénylpropionylamino)isoquinolin-4-yl]-1 H-indazole-3- carboxylique
Intermédiaire 23.1
4-bromo-8-nitroisoquinoléine
Sous argon, à température ambiante, à une solution de 4-bromoisoquinoléine
(100 mmol, 21 ,24 g) dans du H2S04 36N (50 ml) est ajouté du HN03 65 % (200 mmol, 13,85 ml) par petites portions. Le mélange réactionnel est agité 3h à température ambiante. Il est refroidi à 0°C puis est dilué avec de l'eau. Un précipité jaune se forme. Il est filtré. Le pH du filtrat est ajusté à pH 10 par addition lente de NaOH 5N. Le précipité blanc qui apparaît est extrait avec CH2CI2. Cette solution organique est lavée avec de la saumure, séchée sur Na2S04 puis évaporée sous vide. Le résidu est purifié par élution sur gel de silice avec un mélange AcOEt /éther de pétrole. Le produit purifié est recristallisé dans de l'éthanol (EtOH) absolu.
On obtient 2,71 g de produit sous forme de cristaux jaunes. PF: 127-128°C
Intermédiaire 23.2
8-amino-4-bromoisoquinoléine
A une solution de l'intermédiaire 23.1 (2,70 g, 10,7 mmol) dans EtOH (50 ml) est ajouté du chlorure d'étain (SnCI2, 2H20, 12,04 g, 53,3 mmol) en solution dans un mélange d'EtOH (30 ml) et d'HCI 12 N (30 ml). Le mélange réactionnel est chauffé à reflux pendant 1 h. Après refroidissement, le précipité orange est filtré puis repris dans l'eau. Le produit est extrait à pH basique par de l'éther diéthylique. Les sels d'étain sont éliminés par filtration sur célite. La phase organique est séchée sur Na2S04 puis évaporée sous vide. Le résidu est recristallisé dans de l'EtOH absolu. On obtient 2,26 g de produit. PF: 200-201 °C
RMN (DMS0-d6): 6,5 (s, 2H), 6,85 (d,1 H), 7,15 (d, 1 H), 7,6 (t, 1 H), 8,6 (s, 1 H), 9,45 (s, 1 H).
Intemédiaire 23.3 Λ/-(4-bromoisoquinolin-8-yl)-3-phénylpropionamide
A une solution d'intermédiaire 23.2 (265 mg, 1 ,18 mmol) dans du THF anhydre
(20 ml) est ajoutée à 0°C du chlorure de 3-phénylpropionyle (176,5 μl, 1 ,18 mmol). L'agitation est maintenue une nuit à température ambiante. Le mélange réactionnel est évaporé sous vide puis le résidu est extrait avec NaHCÛ3 / AcOEt. La solution organique est lavée avec de la saumure, séchée sur Na2S04 puis évaporée sous vide. Le résidu est purifié sur gel de silice en éluant avec un mélange AcOEt / éther de pétrole (2/3).
On obtient 340 mg de produit—
PF: 174-175°C RMN (DMSO-d6): 2,84 (t, J = 7,9, 2H), 2,99 (t, J = 7,9, 2H), 7,21 (m, 1 H), 7,31
(m, 4H), 7,92 (s, 3H), 8,76 (s, 1 H), 9,30 (s, 1 H), 10,28 (s, 1 H).
Intermédiaire 23.4
N-(pyridin-4-yl)-5-[8-(3-phénylpropionylamino)isoquinolin-4-yl]-1-(2-triméthyl- silyléthoxyméthyl)-1 /-/-indazole-3-carboxamide
L'intermédiaire 23.3 et l'intermédiaire 10.3 sont couplés dans des conditions de
Suzuki de manière analogue au mode opératoire décrit à l'exemple 10, à l'échelle de 0,93 mmol. Le produit brut est repris dans AcOEt / Et20 puis filtré.
On obtient 380 mg de produit sous forme d'une poudre blanche.
N-(pyridin-4-yl)-5-[8-(3-phénylpropionylamino)isoquinolin-4-yl]-1 H-indazole-3- carboxylique
Le composé est obtenu de manière similaire au mode opératoire décrit à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 23.4 (380 mg, 0,59 mmol). Le produit brut est lavé avec du THF puis avec de l'éther diéthylique.
On obtient 150 mg de produit sous forme d'une poudre jaunâtre.
PF: 205°C
LC/MS/UV : MH+513 (100 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,87 (t, J = 6,4, 2H), 3,03 (t, J = 6,4,
2H), 7,28 (m, 1 H), 7,38 (m, 4H), 7,68 (d, J = 8,5, 2H), 7,80 (t, J = 8,0, 1 H), 7,89
(d, J = 7,6, 1 H), 7,93 (d, J = 8,6, 1 H), 7,97 (d, J = 5,1 , 2H), 8,37 (s, 1 H), 8,51 (d,
J = 5,3, 2H), 9,43 (s, 1 H), 10,33 (s, 1 H), 10,89 (s, 1 H), 14,1 (s, 1 H).
Exemple 24 (composé n°56)
Chlorhydrate N-(pyridinyl-4-yl)-5-{8-[(pyridine-3-carbonyl)amino]- isoquinolin-4-yl}-1 H-indazole-3-carboxamide
Intermédiaire 24.1
N-(4-bromoisoquinolin-8-yl)nicotinamide
A une solution de 8-amino-4-bromoisoquinoléine (intermédiaire 23.2, 446 mg,
0,90 mmol) dans du THF anhydre (25 ml) sont ajoutés du chlorhydrate de chlorure de nicotinoyie (356 mg, 2 mmol), de la triéthylamine (558 μl, 2 mmol). Le mélange réactionnel est agité pendant une nuit, dilué avec AcOEt (200 ml) et lavé 2 fois avec NaOH 1 N (100 ml) puis la phase organique est séchée sur Na2S04 et évaporée sous vide. Le résidu est purifié sur gel de silice en éluant— avec AcOEt. On obtient 200 mg de produit. PF: 221-222°C
RMN(DMSO-d6): 7,62 (dd, J = 7,6, J = 4,4, 1 H), 7,96 (dd, J = 6,9, J = 1 ,9, 1 H), 8,00 (dd, J = 8,2, J = 8,2, 1 H), 8,04 (d large, J = 8,8, 1H), 8,44 (ddd, J = 2,2, J = 2,2, J = 7,6, 1 H), 8,80 (s, 1 H), 8,81 (dd, J = 5,7, J = 1 ,3, 1 H), 9,26 (d large, J = 1 ,9, 1 H), 9,48 (s, 1 H), 10,95 (s, 1 H).
Intermédiaire 24.2
N-(pyridinyl-4-yl)-5-{8-[(pyridine-3-carbonyl)amino]isoquinolin-4-yl}-1-(2- triméthylsilyléthoxyméthyl)-1 H-indazole-3-carboxamide L'intermédiaire 24.1 et l'intermédiaire 10.3 sont couplés dans des conditions de
Suzuki de manière analogue au mode opératoire décrit à l'exemple 10, à l'échelle de 0,79 mmol. Le produit brut est repris dans CH3OH/ AcOEt puis filtré.
Le filtrat est concentré. Le résidu est repris dans du AcOEt chaud et le produit obtenu est isolé par filtration. On obtient 360 mg de produit sous forme d'une poudre.
Chlorhydrate N-(pyridinyl-4-yl)-5-{8-[(pyridine-3-carbonyl)amino]-isoquinolin-4-yl}- 1 /-/-indazoIe-3-carboxamide
Le composé du titre est obtenu de manière similaire au mode opératoire décrit à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 24.2 (360 mg, 0,58 mmol). Le produit brut est repris à chaud dans un mélange CH2CI2 / MeOH puis filtré. Le filtrat est acidifié par addition d'HCI 6N. Le précipité qui apparaît est filtré. Le solide est lavé par un mélange CH3OH / Et20.
On obtient 140 mg de produit sous forme d'une poudre. PF : >250°C
LC/MS/UV : MH+486 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7,70 (dd, J = 4,4, J = 6,3, 1 H), 7,74 (d, J = 8,2, 1 H), 7,82 (dd, J = 5,7, J = 3,2, 1 H), 7,99 (m, 3H), 8,40 (s, 1 H), 8,48 (d, J
= 6,3, 2H), 8,55 (d, J = 8,2, 1 H), 8,64 (s, 1 H), 8,77 (d, J = 6,3, 2H), 8,87 (d, J =
3.8, 1 H), 9,34 (s, 1 H), 9,72 (s, 1 H), 11 ,15 (s, 1 H), 11 ,90 (s, 1 H), 14,52 (s, 1 H).
Exemple 25 (composé n°57) N-(pyridin-4-yl)-5-(8-benzylaminoisoquinolin-4-yl)-1 H-indazole-3-carboxamide
Intermédiaire 25.1 benzyl(4-bromoisoquinolin-8-yl)amine
Sous argon, à une solution de 8-amino-4-bromoisoquinoléine (intermédiaire 23.2, 1 ,11g, 5 mmol) dans de l'EtOH absolu (60 ml) est ajouté du benzaldéhyde
(584 μl, 5,75 mmol). Le mélange est chauffé à 90°C pendant 24h. Il est refroidi à
0°C puis du cyanoborohydrure de sodium (NaBH3CN, 3,3 g, 50 mmol) est additionné en petites portions. L'agitation est maintenue 2 h à température ambiante. Le mélange réactionnel est dilué avec AcOEt (600 ml) puis est lavé avec de l'eau. La phase organique est séchée sur Na2S0 puis évaporée. Le résidu est purifié sur gel de silice en éluant avec un mélange AcOEt / éther de pétrole (1/4).
On obtient 640 mg de produit.
PF: 193-194°C RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 4,54 (d, J = 5,7, 2H), 6,60 (d, J = 8,2,
1 H), 7,18 (d, J = 8,2, 1 H), 7,23 (dd, J = 7,2, J = 7,2, 1 H), 7,32 (dd, J = 7,9, J =
7.9, 2H), 7,41 (d, J = 7,9, 2H), 7,56 (dd, J = 8,2, J = 8,2, 1 H), 7,79 (t, J = 6,0, 1 H), 8,64 (s, 1 H), 9,62 (s, 1 H).
Intermédiaire 25.2
N-(pyridin-4-yl)-5-(8-benzylaminoisoquinolin-4-yl)-1-(2-triméthylsilyléthoxy- méthyl)-1 H-indazole-3-carboxamide
L'intermédiaire 25.1 et l'intermédiaire 10.3 sont couplés dans des conditions de
Suzuki de manière analogue au mode opératoire décrit à l'exemple 10, à l'échelle de 0,91 mmol. Le produit brut est cristallisé dans CHCI3 / Et20.
On obtient 510 mg de produit sous forme d'une poudre jaune.
N-(pyridin-4-yl)-5-(8-benzylaminoisoquinolin-4-yl)-1 H-indazole-3-carboxamide
Ce composé est obtenu de manière similaire au mode opératoire décrit à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 25.2 (510 mg, 0,85 mmol). Le produit brut est repris dans MeOH. Le mélange est agité pendant 30 min. Le solide est filtré, lavé avec MeOH puis avec de l'éther diéthylique.
On obtient 60 mg de produit sous forme d'une poudre. PF : >250°C
LC/MS/UV : MH+ 471 (94,8 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 4,47 (d, J = 5,5, 1 H), 6,53 (d, J = 8,2, 1 H), 6,91 (d, J = 8,2, 1 H), 7,23 (d, J = 7,2, 1 H), 7,34 (t, J = 7,2, 2H), 7,39 (t, J = 7,2, 1 H), 7,45 (d, J = 7,2, 2H), 7,56 (d, J = 8,2, 1 H), 7,71 (t, J = 5,3, 1 H), 7,83 (d, J = 8,8, 1 H), 7,91 (d, J = 5,0, 2H), 8,27 (s, 1 H), 8,40 (s, 1 H), 8,45 (d, J = 5,0, 2H), 9,70 (s, 1 H)r 10,80 (s, 1 H), 13,92-(s. 1 H).
Exemple 26 (composé n°59)
N-(pyridin-4-yl)-5-[5-(3-phénylpropionylamino)isoquinolin-4-yl]-1 H-indazole-3- carboxamide
Intermédiaire 26.1 4-bromo-5-nitroisoquinoléine
La nitration de 4-bromoisoquinoléine permet la synthèse de la 4-bromo-8- nitroisoquinoléine (intermédiaire 23.1) et de la 4-bromo-5-nitroisoquinoléine. Le précipité jaune qui s'est formé après addition de H20 (voir synthèse de l'intermédiaire 23.1) est repris dans AcOEt. La solution est lavée avec une solution aqueuse de 10 % NaHC03 puis avec de la saumure. Le produit brut obtenu après séchage sur Na2S04 et évaporation sous vide est recristallisé dans
EtOH absolu.
On obtient 22,35 g de produit sous forme de cristaux.
PF: 174-175°C
Intermédiaire 26.2
5-amino-4-bromoisoquinoléine
Ce composé est obtenu de manière analogue à l'intermédiaire 23.2, à partir de l'intermédiaire 26.1 , à l'échelle de 25 mmol. Le produit brut est recristallisé dans EtOH absolu.
On obtient 5,11 g de produit sous forme de cristaux jaunes.
PF: 156-157°C
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 6,18 (s, 2H), 7,08 (d, 1 H), 7,37 (d, 1 H), 7,48 (dd, 1 H), 8,4 (s, 1 H), 9,0 (s,1 H).
Intermédiaire 26.3 Λ/-(4-bromoisoquinolin-5-yl)-3-phénylpropionamide
Ce composé est préparé à partir de l'intermédiaire 26.2, de manière analogue à l'intermédiaire 23.3, à l'échelle de 1 ,6 mmol. Le produit brut est purifié sur gel de silice en éluant avec un mélange AcOEt / chlorure de méthylène (1/5). On obtient 470 mg de produit. PF: 168-169°C
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 2,75 (t, 2H), 2,95 (t, 2H), 7,3 (m, 5H), 7,55 (d, 1 H), 7,8 (t, 1 H), 8,18 (d, 1 H), 8,67 (s, 1 H), 9,35 (s, 1 H), 10,0 (s, 1 H).
Intermédiaire 26.4 N-(pyridin-4-yl)-5-[5-(3-phénylρropionylamino)isoquinolin-4-yl]-1 -(2-tri- méthylsilyléthoxyméthyl)-1 H-indazole-3-carboxamide
L'intermédiaire 26.3 et l'intermédiaire 10.3 sont couplés dans des conditions de Suzuki de manière analogue au mode opératoire décrit à l'exemple 10, à l'échelle de 1 mmol. La réaction est effectuée dans un mélange de DME (6 ml) et de DMSO (1 ml). Le mélange réactionnel est dilué avec AcOEt / H20 puis filtré. La phase organique est séparée, séchée sur Na2S0 puis évaporée. Le produit brut est purifié sur gel de silice en éluant par un mélange AcOEt / MeOH (98 / 2). On obtient 600 mg de produit sous forme d'un solide de couleur crème.
N-(pyridin-4-yl)-5-[5-(3-phénylpropionylamino)isoquinolin-4-yl]-1 H-indazole-3- carboxamide
Ce composé est obtenu de manière similaire au mode opératoire décrit à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 26.4 (600 mg, 0,93 mmol). Le mélange réactionnel est chauffé pendant 3 jours à 60°C. Le mélange réactionnel est dilué avec de l'eau puis filtré. Le solide obtenu par filtration est recristallisé dans de l'isopropanol, puis séché sous vide à 90°C. On obtient 260 mg de produit. PF : 209-211 °C LC/MS/UV : MH+513 (99,4 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 1 ,41 (m, 1 H), 1 ,50 (m, 1 H), 2,22 (m,1 H), 2,33 (m, 1 H), 6,81 (d, J = 7,6, 1 H), 7,11 (t, J = 7,7, 1 H), 7,18 (t, J = 7,7, 2H), 7,34 (d, J = 8,8, 1 H), 7,59 (d, J = 7,6, 1 H), 7,71 (d, J = 8,8, 1 H), 7,76 (t, J =
8,0, 1 H), 7,93 (d, J = 5,7, 2H), 8,17 (d, J = 8,2, 1 H), 8,22 (s, 1 H), 8,31 (s, 1 H), 8,45 (d, J = 5,7, 2H), 9,00 (s, 1 H), 9,38 (s, 1 H), 10,80(s, 1 H), 14,02 (s, 1 H).
Exemple 27 (composé n°65) 5 Chlorhydrate de N-(pyridin-4-yl)-5-(2H-tétrazol-5-yl)-1 H-indazole-3-carboxamide
Une suspension du chlorhydrate du composé obtenu à l'exemple 19, salifié selon une méthode connue de l'homme du métier, (chlorhydrate de N-(pyridin-4- yl)-5-cyano-1 /-/-indazole-3-carboxamide, 330 mg, 1 ,1 mmol), <_e chlorure d'ammonium (615 mg, 11 ,5 mmol) et d'azide de sodium (715 mg, 11 mmol) dans 0 de la DMF (20 ml) est chauffée sous argon, à 115°C, durant une nuit. Après refroidissement, de l'acide acétique (0,7 ml) est ajouté puis le mélange réactionnel est concentré sous vide. Le résidu est repris par AcOEt/H20, agité ~~~~ pendant 10 min puis filtré. Le solide ainsi séparé, est mis en suspension dans de la DMF et acidifié par HC1 1 N. Le chauffage à 80°C et l'addition d'H20 entraînent 5 la dissolution du solide. Après refroidissement, la suspension est filtrée pour donner le produit attendu.
On obtient 21 g de produit sous forme d'une poudre blanche.
PF : >250°C
LC/MS/UV : MH+307 (100 %) 0 RMN H (500 MHz, DMSO-D6) δ (ppm) : 7.93 (d, J=8.2 Hz, 1 H) 7.94 (s, 1 H)
8.18 (m, 3 H) 8.62 (d, J=6.3 Hz, 2 H) 9.00 (s, 1 H) 11.34 (s, 1 H) 14.39 (s, 1 H).
Exemple 28 (composé n°67)
N-(pyridin-4-yl)-5-(1 ,3-thiazol-5-yl)-1 H-indazoIe-3-carboxamide 5
Intermédiaire 28.1
N-pyridin-4-yl-5-(1 ,3-thiazol-5-yl)-(2-triméthylsilyléthoxyméthyl)-1 H-indazole-3- carboxamide
Sous atmosphère d'argon, à une solution de n-butyllithium (8,25 ml, 1 ,6 N dans 0 l'hexane, 13,2 mmol) dans de l'éther diéthylique anhydre (12 ml) refroidie à -78°C est ajoutée goutte à goutte une solution de 2-triméthylsilyl(thiazole) (1 ,97 ml, 12 mmol) dans de l'éther diéthylique anhydre (12 ml). L'agitation est maintenue 30 min à -78°C puis est additionnée une solution molaire de ZnCI2 (4,91 g, 36 mmol) dans de l'éther diéthylique anhydre (36 ml) fraîchement 5 préparée à partir de ZnCI2 très sec. Le bain réfrigérant est enlevé et le mélange est agité pendant 30 min à température ambiante. Les solvants sont évaporés sous vide. Sous argon, au résidu est ajouté un mélange d'intermédiaire 9.3 (5,37
g, 12 mmol) et de tétrakis(triphénylphosphine)palladium (277 mg) dans du THF anhydre (50 ml) et la suspension est chauffée sous reflux pendant 24 h. Le mélange réactionnel est acidifié à pH 2 par addition d'HC1 1 N puis est concentré sous vide. De la soude 1 N est ajoutée pour atteindre pH 10. Le solide obtenu est agité en présence de CH2CI2 et les sels de zinc sont séparés par filtration. Le filtrat est lavé avec de l'eau. La phase organique est séchée sur MgS0 puis évaporée. L'huile brune obtenue (5,10 g) est filtrée rapidement sur silice. Après cristallisation dans de l'éther diéthylique, un solide jaune (2,40 g) est obtenu; Ce solide est chromatograhié sur silice (300 g) . Un gradient d'élution éther de pétrole / AcOEt (2/3) à éther de pétrole / AcOEt (1/3)] permet de séparer 2 isomères. Le composé attendu est le plus polaire. On obtient 1 ,08 g de produit sous forme d'une poudre blanche.
N-pyridin-4-yl-5-(1 ,3-thiazol-5-yl)-1 H-indazole-3-carboxamide Ce composé est obtenu de manière similaire à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 28.1 (452 mg, 1 mmol). Le produit brut est recristallisé dans un mélange MeOH / AcOEt en présence d'une trace d'eau puis est filtré.
On obtient 22,5 g de produit sous forme d'un solide jaune. PF : >250°C
LC/MS/UV : MH+322 (98,5 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7.77 (d, J=8.8 Hz, 1 H) 7.86 (d, J=8.8
Hz, 1 H) 7.93 (d, J=6.3 Hz, 3 H) 8.35 (s, 1 H) 8.41 (s, 1 H) 8.47 (d, J=6.3 Hz, 2
H) 9.10 (s, 1 H) 10.80 (s, 1 H), 14.0 (s, 1 H)
Exemple 29 (composé n°68)
N-(pyridin-4-yl)-5-(1 H-pyrrolo[2,3-c]pyridin-4-yl)-1 H-indazole-3-carboxamide
Intermédiaire 29.1 3-nitropyridin-4-ol
A une solution d'oléum (H2S04 contenant 20 % S03, d = 1 ,9, 10,5 ml) refroidie à
0°C est ajoutée lentement une solution d'acide nitrique (HNO3>90 %, 13,3 ml).
Du nitrate de 4-pyridol (10 g, 63 mmol) est additionné à 0°C au mélange qui est ensuite chauffé à 90°C pendant 1 h30. Le mélange réactionnel est versé sur 50 g de glace. La suspension est agitée 30 min à 0°C puis est filtrée. Le solide est lavé avec quelques ml d'eau puis séché à l'air.
On obtient 6,59 g de produit sous forme d'une poudre blanche.
Intermédiaire 29.2
3-bromo-5-nitropyridin-4-ol
Du brome (5,07 g, 31 ,7 mmol) est additionné goutte à goutte à une suspension d'intermédiaire 29.1 (4,04 g, 28,4 mmol) dans H20 (40 ml). Le mélange est chauffé avec un bain d'huile à une température de 90°C, pendant 1 h. Après refroidissement, le mélange est filtré. Le solide est lavé avec H20 puis séché au dessiccateur en présence de gel de silice.
On obtient 4,51 g de produit sous forme d'un solide blanc.
Intermédiaire 29.3
3-bromo-4-chloro-5-nitropyridine
De l'intermédiaire 29.2 (5,30 g , 24,0 mmol) est séché à 60°C sous vide (0 ,5 mBar) pendant 2 h dans un ballon bicol. PCI5 (9,12 g, 43,8 mmol) et POCI3 (0,5 ml, 5,4 mmol) sont ajoutés. Le mélange est chauffé à 160°C au bain d'huile. Après 20 min, le solide a été converti en une huile brune limpide. Après refroidissement, le mélange se solidifie. Les substances volatiles sont évaporées sous vide puis le solide mis en suspension dans CHCI3 est traité à 0°C par une solution aqueuse d'acétate de potassium (AcOK 25g / 35 ml H20). La phase organique est séparée puis est évaporée. Le résidu est extrait par un mélange éther diéthylique/NaHC03. la phase organique est lavée avec de la saumure, séchée sur Na2S04 et évaporée. Le résidu est repris dans du CH2CI2. Les insolubles sont éliminés par filtration et le filtrat est évaporé pour donner le produit attendu. On obtient 4,66 g de produit sous forme d'une huile brune.
Intermédiaire 29.4
(3-bromo-5-nitropyridin-4-yl)malonate de diéthyle
Sous azote, à une suspension de NaH (1 ,70g, 55-60 % dans l'huile, 39-46 mmol) dans de la DMF anhydre (30 ml) est ajouté du malonate de diéthyle (7,2 ml, 47,4 mmol) en 10-15 min, de sorte que la température du mélange réactionnel n'excède pas 50°C. L'agitation est maintenue encore pendant 30 min après l'addition. De l'intermédiaire 29.3 (4,46 g, 18,62 mmol) est ajouté sous forme solide par petites portions. L'agitation est maintenue pendant 3h30 à température ambiante. De l'eau (50 ml) et AcOH (5 ml) sont ajoutés. Le mélange est extrait avec de l'éther diéthylique. La phase organique est lavée avec de la saumure, séchée sur Na2S04 et évaporée. Le résidu est élue sur gel de silice (300 g) selon un gradient d'un mélange AcOEt / éther de pétrole (1/9) à un mélange AcOEt / éther de pétrole (3/7).
On obtient 5,15 g de produit sous forme d'une huile brune.
Intermédiaire 29.5
3-bromo-4-méthyl-5-nitropyridine
L'intermédiaire 29.4 (5,15 g, 14,2 mmol) est chauffé à 100°C en présence d'HCI aqueux 18 % (50 ml) pendant 15h. Le mélange est extrait avec de l'éther diéthylique (3 x 80 ml). La solution éthérée est lavée avec de la saumure, séchée sur MgS0 et évaporée sous vide pour donner le produit attendu.
On obtient 2,43 g de produit sous forme d'un solide jaune.
Intermédiaire 29.6
2-(3-bromo-5-nitropyridin-4-yl)-N,N-diméthyléthylènamine Un mélange d'intermédiaire 29.5 (1 ,136 g, 5,19 mmol) et d'acétal de diéthyle de DMF (1 ,46 mi78,52 mmol) dans de a DMF (6 ml) est chauffé à 85°C pendant 1 h15. La DMF est évaporée sous vide pour donner le produit attendu. On obtient 1 ,407 g de produit sous forme d'un solide de couleur pourpre.
Intermédiaire 29.7 4-bromo-1 H-pyrrolo[2,3-c]pyridine L'intermédiaire 29.6 (1 ,407 g, 5,13 mmol) en solution dans de l'acide acétique (15 ml) est chauffé sous azote en présence de fer (poudre 325 mesh, 1 ,85 g, 33,1 mmol) à 120°C pendant 40 min. Le mélange est filtré. Les sels de fer sont lavés avec AcOH (5 ml). Le filtrat est dilué dans 75 ml d'eau pour donner une solution limpide de couleur rouge-orange. Cette solution est neutralisée par addition de K2Cθ3 solide jusqu'à l'obtention de pH 9. La suspension brunâtre est extraite avec CHCI3. La solution organique est lavée avec de la saumure, filtrée sur une membrane de 0,45 μM, séchée sur Na2S04 et enfin est évaporée sous vide.
On obtient 801 mg de produit sous forme d'un solide gris-brun.
Intermédiaire 29.8
N-(pyridin-4-yl)-5-(1 H-pyrrolo[2,3-c]pyridin-4-yl)-1 -(2-triméthylsilyléthoxyméthyl)-
1 H-indazole-3-carboxamide
La synthèse de cet intermédiaire est réalisée de manière analogue à la synthèse de l'intermédiaire 10.4 en effectuant une réaction de Suzuki entre l'intermédiaire 29.7 et l'intermédiaire 10.3 à l'échelle de 2,43 mmol. Le produit brut purifié sur gel de silice (50 g) en éluant avec un mélange AcOEt / MeOH
(95/5).
On obtient 780 mg de produit sous forme d'une poudre blanche.
N-(pyridin-4-yl)-5-(1 H-pyrrolo[2,3-c]pyridin-4-yl)-1 H-indazole-3-carboxamide Ce composé est obtenu de manière similaire à l'exemple 11 par coupure du groupe protecteur SEM de l'intermédiaire 29.8 (780 mg, 1 ,61 mmol) par du TFA suivi d'un chauffage en présence de d'éthylènediamine. Le produit brut est agité 1 h en présence de MeOH puis est filtré. On obtient 480 mg de produit sous forme d'une poudre blanche. PF : >250°C
LÇ/MS/UV : MH+355 (100 %) RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 6.67 (s, 1 H) 7.71 (s, 1 H) 7.84 (s, 2 H) 7.93 (d, J=5.7 Hz, 2 H) 8.27 (s, 1 H) 8.46 (d, J=6.3 Hz, 2 H) 8.55 (s, 1 H) 8.77 (s, 1 H) 10.79 (s, 1 H) 11.80 (s, 1 H) 12-14 (s, 1 H)
Exemple 30 (composé n°74) 5-(5-amino-4-méthylpyridin-3-yl)-N-pyridin-4-yl-1 H-indazole-3-carboxamide
Intermédiaire 30.1 3-bromo-4-méthyl-5-aminopyridine
Ce composé est obtenu par réduction du 3-bromo-4-methyl-5-nitropyridine (intermédiaire 29.5) par du fer d'une manière analogue à la synthèse de l'intermédiaire 29.7, à l'échelle de 10 mmol. On obtient 1 ,20 g de produit sous forme d'un solide de couleur crème.
Intermédiaire 30.2 5-(5-amino-4-méthylpyridin-3-yl)-N-pyridin-4-yl-1-(2-triméthylsilyléthoxyméthyl)-
1 H-indazole-3-carboxamide
La synthèse de cet intermédiaire est réalisée de manière analogue à la synthèse de l'intermédiaire 10.4 en effectuant une réaction de Suzuki entre l'intermédiaire
30.1 et l'intermédiaire 10.3 à l'échelle de 2,48 mmol. Le mélange réactionnel est évaporé. Le résidu est repris par AcOEt / H20. Un insoluble se sépare. Après agitation pendant 1 h, il est filtré.
On obtient 1 ,02 g de produit sous forme d'une poudre de couleur crème.
5-(5-amino-4-méthylpyridin-3-yl)-N-pyridin-4-yl-1 H-indazole-3-carboxamide L'intermédiaire 30.2 (980 mg, 2,06 mmol) en solution dans du THF (30 ml) est mis en réaction avec une solution de fluorure de tétrabutylammonium (solution 1 M dans THF, 10,3 ml), de l'éthylènediamine (0,347 ml, 5,2 mmol) et H20 (0,377 ml). Le mélange est chauffé à 60°C pendant 3 jours puis évaporé. Le résidu est agité dans un mélange AcOEt/ H20 puis filtré. Le solide ainsi récupéré, est agité
en présence de MeOH puis est filtré pour donner le produit attendu (270 mg) sous la forme d'un solide de couleur crème. Le filtrat est évaporé sous vide puis est chromatographié sur gel de silice (50 g) en éluant avec un mélange MeOH/CH2CI2 (1/4) pour donner un 2èmθ lot du produit attendu (1 ,5 g) sous la forme d'un solide de couleur jaune. LC/MS/UV : MH+345 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 1.98 (s, 3 H) 5.19 (s, 2 H) 7.40 (d, J=8.2 Hz, 1 H) 7.67 (s, 1 H) 7.74 (d, J=8.8 Hz, 1 H) 7.91 (d, J=6.3 Hz, 2 H) 7.96 (s, 1 H) 8.08 (s, 1 H) 8.45 (d, J=6.3 Hz, 2 H) 10.76 (s, 1 H) 13.67 (s, 1 H)
Exemple 31 (composé n°70) 5-(1 H-pyrazol-4-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
Intermédiaire 31.1 4-iodo-1-(2-triméthylsilyléthoxyméthyl)-1 H-pyrazole
Sous argon, à une solution de 4-iodopyrazole (1 ,94 g, 10 mmol) dans du CH2CI2 (100 ml) sont ajoutés de la diisopropyléthylamine (DIEA, 1 ,75 ml, 10 mmol) et du chlorure de triméthylsilyléthoxyméthyle (SEMCI, 1,83 ml, 10 mmol). Le mélange est agité pendant une nuit à température ambiante. Le solvant est évaporé sous vide. Le résidu est repris avec un mélange éther diéthylique / eau. La phase organique après séchage et évaporation donne une huile incolore qui est purifiée par élution sur gel de silice avec un mélange AcOEt / CH2CI2 (5/95). On obtient 3,02 g de produit sous forme d'une huile incolore.
Intermédiaire 31.2
N-(pyridin-4-yl)-1-(2-triméthylsilyléthoxyméthyl)-5-(1-(2-triméthylsilyl- éthoxyméthyl)-1 H-pyrazol-4-yl)-1 H-indazole-3-carboxamide La synthèse de cet intermédiaire est réalisée de manière analogue à la synthèse de l'intermédiaire 10.4 en effectuant une réaction de Suzuki entre l'intermédiaire 31.1 et l'intermédiaire 10.3 à l'échelle de 2,0 mmol. Le produit brut est purifié sur gel de silice (50 g) en éluant avec un mélange MeOH/CH2CI2 (2/98). Le composé purifié est cristallisé dans un mélange CHCI3/ E.2O. On obtient 367 mg de produit sous forme d'une poudre blanche. 5-(1 H-pyrazol-4-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
Ce composé est obtenu de manière similaire à l'exemple 11 par coupure des groupements protecteurs SEM de l'intermédiaire 31.2 (550 mg, 0,97 mmol) par du TFA suivi d'un chauffage en présence de d'éthylènediamine. Le mélange réactionnel est filtré. Le solide est agité en présence de CHCI3/H2O. La fraction
insoluble est agitée en présence de MeOH pendant 1 h puis filtrée pour donner le produit attendu.
On obtient 220 mg de produit sous forme d'une poudre blanche. PF: >250°C LC/MS/UV: MH+305 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7.67 (d, J=8.2 Hz, 1 H) 7.75 (d, J=6.9 Hz, 1 H) 7.94 (d, J=6.3 Hz, 2 H), 7.96 (s large, 1H), 8.21 (s large, 1 H) 8.35 (s, 1 H) 8.46 (d, J=5.7 Hz, 2 H) 10.72 (s, 1 H) 13.0 (s, 1 H)
Exemple 32 (composé n°72)
5-(1,3-oxazol-5-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
Intermédiaire 32.1 _-
5-formyl-N-(pyridin-4-yl)-1-(2-triméthylsilyléthoxyméthyl-1H-indazole-3- carboxamide
Un courant de monoxyde de carbone est introduit durant 15 min dans une solution d'intermédiaire 9.3 (4,95 g, 10 mmol) dans du THF (50 ml). Du tétrakis(triphénylphosphine)palladium (578 mg, 0,5 mmol) est ajouté puis on introduit un courant de monoxyde de carbone pendant 10 min. Le mélange est porté à 50°C et une solution d'hydrure de tributylétain (3,05 ml, 11 mmol) dans du THF (20 ml) est ajoutée lentement en 2h30. Après refroidissement, de l'eau (0,5 ml) est ajoutée et le mélange réactionnel est évaporé. Le résidu est purifié sur gel de silice en éluant par MeOH/AcOEt/ CH2CI (2/29/69). On obtient 2,65 g de produit sous forme d'un solide jaune.
Intermédiaire 32.2
5-(1 ,3-oxazol-5-yl)-N-(pyridin-4-yl)-1 -(2-triméthylsilyléthoxyméthyl)-1 H-indazole-
3-carboxamide
A l'intermédiaire 32.1 (2,65 g, 6,68 mmol) en solution dans MeOH (100 ml) sont ajoutés K2C03 (1 ,02 g, 7,35 mmol) et du tosylméthylisocyanate (TosMIC, 1 ,44 g, 7,35 mmol). Le mélange est chauffé à reflux pendant 2h30, puis après refroidissement est concentré sous vide. Le résidu est repris avec AcOEt/H 0. Après séchage et évaporation, la phase organique donne un solide qui est purifié sur gel de silice en éluant avec MeOH/ AcOEt/CH2CI2 (3/28,5/68,5). Le produit purifié est recristallisé dans AcOEt.
On obtient 2,345 g de produit sous forme d'un solide blanc.
5-(1 ,3-oxazol-5-yl)-N-(pyridin-4-yl)-1 H-indazole-3-carboxamide
L'intermédiaire 32.2 (360 mg, 0,82 mmol) est mis en réaction à température ambiante avec du TFA (3 ml) pendant 1 h. Le mélange réactionnel est évaporé sous vide. Le résidu est repris avec du THF provoquant la séparation d'un solide. Le mélange est à nouveau évaporé puis repris dans du THF (10 ml). De l'éthylènediamine (267 μl, 4 mmol) est ajoutée et le mélange est chauffé à 70°C pendant une nuit. Le THF est évaporé. Le résidu est trituré dans MeOH, filtré puis lavé avec de l'éther diéthylique. Il est repris avec 80 ml d'un mélange MeOH/ H20 (9/1). Le mélange est chauffé à reflux. De la DMF est ajoutée jusqu'à dissolution complète. Après refroidissement, apparaît un solide orange qui est filtré et séché sous vide poussé durant 2 jours.
On obtient 206 mg de produit sous forme d'une poudre orange.
LC/MS/UV : MH+306 (94,3 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 7.74 (s, 1 H) 7.77 (d, J=8.8 Hz, 1 H)
7.84 (d, J=8.8 Hz, 1 H) 7.92 (d, J=5.7 Hz, 2 H) 8.45 (d, J=5.0 Hz, 3 H) 8.46 (s, 1 H) 8.51 (s, 1 H) 10.82 (s, 1 H) 13.9 (s, 1 H)
Exemple 33 (composé n°73)
Dichlorhydrate de N-{3-[(éthylamino)méthyl]phényl}-5-isoquinolin-4-yl-1 H- indazole-3-carboxamide
Intermédiaire 33.1
Chlorhydrate de N-(3-nitrobenzyl)éthanamine
Sous argon, à une solution de N-(3-nitrobenzyl)acetamide (3,89 g, 20 mmol) dans du THF anhydre (100 ml) est ajoutée goutte à goutte une solution de BH3- THF (1 M, 60 ml). Le mélange réactionnel est agité à 40°C durant une nuit. MeOH (40 ml) est ajouté. Les solvants sont évaporés sous vide et le résidu est chauffé pendant 4 h à 60°C dans un mélange THF/HCI 4N (80/25). Après refroidissement, un solide se sépare. AcOEt (50 ml) est ajouté en maintenant l'agitation. Le solide est filtré, lavé avec AcOEt et de l'éther diéthylique puis séché.
On obtient 3,95 g de produit sous forme d'une poudre blanche.
Intermédiaire 33.2 (3-aminobenzyl)éthylamine Sous argon, l'intermédiaire 33.1 (3,90 g, 18 mmol) dans de l'acide acétique glacial (100 ml) est agité à 110°C pendant 30 min en présence de fer (6.64 g, 118,8 mmol). Après refroidissement, les sels de fer sont filtrés et lavés avec de l'acide acétique (15 ml). Le filtrat est dilué dans l'eau (200 ml) puis amené à pH 9 par addition de K2C03 solide et à pH 12 par addition de NaOH . La suspension
résultante est extraite avec CH2CI2. La solution organique est séchée puis évaporée pour donner le produit attendu.
On obtient 2,65 g de produit sous forme d'une huile jaune.
Intermédiaire 33.3 tert-butyl (3-aminobenzyl)éthylcarbamate
Sous argon, l'intermédiaire 33.2 (2,60 g, 17,3 mmol) en solution dans du CH2CI2 (150 ml) est mis en réaction avec du di-tert-butyl dicarbonate (3,89 g, 17,3 mmol) pendant 1h30. Le solvant est évaporé. L'huile obtenue est purifiée sur gel de silice en effectuant un gradient d'élution d'un mélange AcOEt/CH2CI2 (1/9) à un mélange AcOEt/ CH2CI2 (1/4). On obtient 1 ,28 g de produit sous forme d'une gomme incolore.
Intermédiaire 33.4 (3-{[(5-bromo-1 -(2-triméthylsilyléthoxyméthyl)-1 H-indazol-3-yl)carbonyl] amino}benzyl)éthylcarbamate de terf-bu tyle
Sous argon, à une solution d'acide 5-bromo-1-(2-triméthylsilyléthoxyméthyl)-1H- indazole-3-carboxylique (1,86 g, 5 mmol), préparé à partir de la 5-bromoisatine de manière analogue à l'intermédiaire 9.2, dans du THF anhydre (45 ml) est ajoutée à 0°C une solution de chloroformiate d'isopropyle dans du toluène (1M, 5,5 ml, 5,5 mmol) puis de la N-méthylmorpholine (600 μl, 5,5 mmol). Le mélange est agité 20 min à 0°C puis 15 min à température ambiante. Après refroidissement à 0°C, est ajouté l'intermédiaire 33.3 (1 ,38 g, 5,5 mmol) en solution dans du THF anhydre (5 ml). Le mélange réactionnel est agité pendant 5 h à température ambiante puis est concentré sous vide. Le résidu est repris avec AcOEt/H20. La phase organique est lavée avec K2C03 10 %, séchée puis évaporée sous vide. On obtient 2,81 g de produit sous forme d'une gomme jaune.
Intermédiaire 33.5 tert-butyl éthyl(3-{[(5-isoquinolin-4-yl-1 -(2-triméthylsilyléthoxymé thyl)-1 H-indazol-
3-yl)carbonyl]amino}benzyl)carbamate
A une solution d'intermédiaire 33.4 (1 ,21 g, 2mmol) dans du DME (12 ml) sont ajoutés de l'acide isoquinolin-4-ylboronique (461 mg, 2 mmol) et une solution aqueuse de Na2C03 (2M, 5 ml, 10 mmol). Le mélange réactionnel est dégazé à l'argon puis du tétrakis(triphénylphosphine)palladium (116 mg, 0,1 mmol) est additionné. Après un nouveau dégazage, le mélange est chauffé à 80°C pendant 20 h. Le mélange réactionnel est évaporé sous vide puis repris dans
AcOEt/ H20 et filtré. La solution d'AcOEt est séchée puis évaporée sous vide. Le résidu est purifié sur gel de silice en éluant avec un mélange AcOEt / DCM (1/3). On obtient 1 ,08 g de produit sous forme d'une gomme jaune.
Dichlorhydrate de N-{3-[(éthylamino)méthyl]phényl}-5-isoquinolin-4-yl-1 H- indazole-3-carboxamide
Sous argon, du TFA (20 ml) refroidi à 4°C est ajouté à l'intermédiaire 33.5 (1 ,0 g, 1 ,53 mmol). Le mélange est agité 2 h à température ambiante. Le TFA est évaporé sous vide. Le résidu est repris avec du toluène puis le mélange est évaporé sous vide. Cette opération est répétée. Le résidu est repris dans du THF (50 ml). De l'éthylènediamine (620 μl, 9,20 mmol) est ajoutée et le mélange est chauffé à 70°C sous argon durant une nuit. Le mélange est évaporé et le résidu est extrait avec AcOEt/H20. La phase organique, après séchage et évaporation donne un composé qui est solubilisé dans un mélange isopropanol/HCL 4N (20 ml 12 ml). Le solide qui se sépare lentement est filtré. On obtient 622 mg de produit sous forme d'un solide jaune. LC/MS/UV : MH+422 (100 %)
RMN 1H (500 MHz, DMSO-D6) δ (ppm) : 1.21 (t, J=6.9 Hz, 3 H) 2.95 (m, 2 H) 4.09 (t, J=6.0 Hz, 2 H) 7.30 (d, J=7.6 Hz, 1 H) 7.40 (t, J=7.9 Hz, 1 H) 7.68 (d, J=10.1 Hz, 1 H) 7.81 (d, J=8.2 Hz, 1 H) 7.92 (d, J=8.2 Hz, 1 H) 7.97 (t, J=7.9 Hz, 1 H) 8.07 (m, 1 H) 8.12 (s, 1 H) 8.12 (s, 1 H) 8.41 (s, 1 H) 8.53 (d, J=7.6 Hz, 1 H) 8.67 (s, 1 H) 9.1 (s large, 2 H) 9.78 (s, 1 H) 10.55 (s, 1 H) 14.2 (s, 1 H).
Le tableau qui suit illustre les structures chimiques et les propriétés physiques de quelques composés de l'invention. Dans ce tableau :
- dans la colonne « sel », « - » représente un composé sous forme de base libre, alors que « HCI » représente un composé sous forme de chlorhydrate et « CH3SO3H » représente un composé sous forme de sel d'acide méthanesulfonique. Le rapport entre parenthèses est le rapport (acide:base) ;
- F(°C) représente le point de fusion du composé en degré Celsius ;
- M+H représente la masse du composé plus 1 ;
- « déc. » signifie décomposition du produit.
Tableau
(0
Les composés de l'invention ont fait l'objet d'essais pharmacologiques qui ont montré leur intérêt comme substances actives en thérapeutique.
Ils ont en particulier été testés quant à leurs effets d'inhibition de certaines cyclin dépendent kinases (cdks) telles que les cdkl (également dénommé cdc2), cdk2 et cdk4.
Les cdks sont des kinases protéiques (sérine/thréonine) qui jouent un rôle décisif dans la progression du cycle de la cellule, fonctionnant uniquement en étant associées à des cyclines. A l'intérieur de ces associations, les cyclines ont une fonction de contrôle, alors que la fonction des cdks est catalytique. A ce jour, neuf cdks (cdkl à cdk9) et onze cyclines ont été identifiées chez l'homme. Les cdks individuelles jouent des rôles distincts dans la progression du cycle cellulaire et peuvent être classées en fonction du stade du cycle cellulaire au cours duquel elles interviennent : transition G1 S (cdk2/cycline E, cdk4/cycline D1-D3, cdk6/cycline D1 ,_çdk8/cycline C), phase.. S (cdk2/cycline A), G2 (cdc2/cycline A), transition G2/M (cdc2/ cycline B, cdk7/ cycline H). Par exemple, les cdk4/cycline D et cdkδ/cycline D actives phosphorylent la protéine du retinoblastome pRb menant à sa dissociation du complexe de facteur de transcription complexe E2F/DP1 et désactivant ainsi son activité de suppresseur de transcription. La libération de E2F/DP1 a pour résultat l'activation de la transcription d'un groupe de gènes nécessaires à l'entrée en phase S, incluant la thymidylate synthetase, la dihydrofolate reductase et la cycline E. A la fin du stade G1 , la cycline E stimule cdk2 qui agit en continuant le processus de phosphorylation de pRb, ayant pour résultat un engagement irréversible dans un processus de division cellulaire et de transition en phase S. Au. cours de la phase S, la cycline A remplace la cycline E en activant cdk2, ceci ayant pour résultat un changement de la spécificité du substrat de cdk2. E2F/DP1 est phosphorylé, ce qui inactive son activité de promoteur de transcription. Ceci résulte en une diminution dans la synthèse de la cycline E et une transition vers G2. L'activité des cdk est inhibée par les inhibiteurs de cdk endogènes (cdkl: p15, p16, p18, p19, p21 , p27 et p57).
Le développement des tumeurs chez l'homme est souvent associé à un dérèglement de l'activité des cdk. p16, un inhibiteur de cdk4 et de cdk6, est détruit ou muté dans 55 % des gliomes et des mésothéliomes ainsi que dans 38 % des cancers du pancréas. Il a été montré que p27, un autre inhibiteur, et les co-activateurs cycline E ou cycline D sont respectivement soit « surexprimés » soit « sous-exprimés », dans notamment les cancers du sein, du côlon, du poumon non à petites cellules, de l'estomac, de la prostate, de la
vessie, du lymphome non hodgkinien, des ovaires. Il a été montré que leur expression altérée est à mettre en corrélation avec un accroissement des activités cdk2 et cdk4.
L'induction de p21 par le supresseur de tumeur activée p53, en réponse au signaux d'altération de l'ADN, inhibe l'activité du cdk2 (le composant clef du contrôle de la phase G1) ; ainsi, la mutation de p53 dans environ 50 % de tous les cancers de l'homme peut indirectement résulter dans le dérèglement de l'activité cdk et la perte du contrôle de la phase G1 , contribuant à la génération de tumeurs (Paulovich A et al., Cell. 88, 315-321 (1997)).
Les composés de l'invention représentent une nouvelle classe d'inhibiteurs de cdk, dont les propriétés incluent la suspension du cycle de la cellule, le blocage de la prolifération des cellules et l'apoptose.
L'activité inhibitrice sur cdk2 a été mise en évidence dans un test où l'activité enzymatique cdk2 / Cycline A est mesurée par quantification de 3P provenant de 33P-ATP qui a été incorporé dans l'histone type lll-S de thymus de veau (Sigma Ref : H-5505). L'essai est réalisé en milieu HEPES 50 mM, pH 7,2, en présence de 1mM DTT (ajouté extemporanément), 1 mM MgCI2,1 mM EGTA et 0,02 % Tween 20.. La concentration d'histone est de 0,4 mg / ml et la concentration d'ATP froid est de 10 μM. La concentration en 33P-ATP est ajustée à une activité d'environ 300 000 cpm. L'inhibiteur 5 μl est ajouté en solution dans 10 % DMSO aqueux, afin d'avoir une concentration finale de 1 % en DMSO. Le mélange réactionnel est incubé pendant 1 heure à température ambiante. La réaction est arrêtée par dépôt sur filtre (Whatman P81 , ion-exchange chromatography paper). Les filtres sont lavés 2 fois par trempage durant 20 min dans une solution d'acide phosphorique 37,5 mM. Les filtres sont placés dans des fioles à scintillation et le scintillant (aquasafe, Zinsser Analytic, 5ml) est ajouté. Ces fioles sont ensuite comptées pendant 1 min sur un compteur Wallac.
Dans ces conditions, les composés les plus actifs de l'invention présentent des CI50 (concentration inhibant 50 % de l'activité enzymatique) inférieures à 20 μM.
L'activité inhibitrice sur cdkl a été mise en évidence dans un test cdkl/ Cycline B. La mesure de l'activité inhibitrice des composés de l'invention sur cdkl se fait suivant le même protocole que pour le test cdk2 / Cycline A
avec les modifications suivantes : la concentration en MgCI2 est de 10 mmol et la concentration d'ATP est de 0,1 μM.
Dans ces conditions, les composés les plus actifs de l'invention présentent des CI5o (concentration inhibant 50 % de l'activité enzymatique) inférieures à 20 μM.
L'activité inhibitrice sur cdk4 a été mise en évidence dans un test cdk4/Cyc!ine D1. La mesure de l'activité inhibitrice des composés de l'invention sur cdk4 se fait suivant le même protocole que pour le test cdkl/Cycline B.
Seul diffère le substrat qui peut être la rétinoblastoma protéine à une concentration de 0,02 mg / ml ou le peptide K10K (K-A-P-L-S-P-K-K-A-K-NH2) à une concentration de 1 mM.
Dans ces conditions, les composés les plus actifs de l'invention présentent des CI50 (concentration inhibant 50 % de l'activité enzymatique) inférieures à 20 μM.
Les résultats des tests biologiques montrent que les composés de l'invention sont des inhibiteurs des cdkl , cdk2 et cdk4.
Ainsi, les composés de l'invention peuvent être employés dans le traitement des pathologies dans lesquelles un inhibiteur des cdkl , cdk2, cdk4 apporte un bénéfice thérapeutique. Notamment de telles pathologies sont les cancers, les maladies auto-immunes et/ou inflammatoires, les maladies cardiovasculaires, les maladies neurodégéneratives, les infections virales et fongiques, les maladies dégénératives du système musculo-squelétique, les maladies hématologiques, les maladies du rein et les maladies du foie dues aux toxines ou à l'alcool.
Les composés de l'invention ont démontré notamment une activité antiproliférative puissante sur certaines lignées cellulaires tumorales.
De plus, étant donné le rôle clef des cdks dans la régulation de la prolifération des cellules en général, les inhibiteurs de l'invention peuvent se comporter comme des agents cytostatiques réversibles, qui peuvent être utiles dans le traitement du processus de maladies présentant des caractéristiques de prolifération cellulaire anormale.
Les composés de formule générale (I) sont également susceptibles de moduler l'apoptose, un processus de mort physiologique de la cellule, crucial pour le développement normal et l'hémostase. Les altérations de l'apoptose contribuent
à la pathogénèse d'une multiplicité de maladies humaines. Les composés de formule générale (I), en tant que modulateur d'apoptose, peuvent être utiles pour le traitement de maladies humaines présentant des aberrations de l'apoptose. Enfin, les inhibiteurs de cdk de l'invention peuvent être utiles dans le traitement de maladies virales et fongiques, puisque les cdk sont activés par la cycline virale (v-cycline) et de ce fait sont impliqués dans la réplication virale et la prolifération de virus comme le virus de l'herpès et HIV-1.
Plus spécifiquement, les composés de formule générale (I) peuvent être utiles dans le traitement :
- de cancers, incluant mais ne se limitant pas au carcinome, incluant le cancer de la vessie, du sein, du côlon, du rein, du foie, du poumon, des ovaires, du pancréas, de l'estomac, du col de l'utérus, de la thyroïde, de la-.pr-os.ate et de la peau ; les tumeurs hématopoïétiques de la famille des lymphoïdes, incluant la leucémie lymphocytique aiguë, les lymphomes folliculaires, le lymphome des cellules B et le lymphome de Burkett ; les tumeurs hématopoïétiques de la famille des myéloïdes, incluant les leucémies myéloïdes aiguës et chroniques et la leucémie promyélocytique ; les tumeurs d'origine mésenchymale incluant le fibrosarcome et le rhabdomyosarcome ; et d'autres tumeurs, incluant le mélanome, le séminome, le teratocarcinome, l'ostéosarcome, le neuroblastome et le gliome, ainsi que les lésions pré-cancéreuses telles que le polypose adénomateux héréditaire ;
- de maladies auto-immunes ou inflammatoires, incluant mais ne se limitant pas à l'arthrite, telle que l'arthrite rhumatoïde, le psoriasis, la glomérulonéphrite immune, l'inflammation de l'intestin, le rejet de transplantation et le choc endotoxique, le lupus systémique, Pérythèmatose, le diabète sucré auto-immun, la rétinite pigmentaire ;
- de maladies cardiovasculaires, incluant mais ne se limitant pas à la neuro- fibromatose, l'athérosclérose, la fibrose pulmonaire, la resténose à la suite d'une angioplastie ou de chirurgie vasculaire, la formation d'une cicatrice hypertrophique, l'angiogénèse, la maladie de la greffe veineuse, la transplantation vasculopathie, les lésions ischémiques faisant suite à un infarctus du myocarde, les lésions dues à une attaque et une reperfusion, l'arythmie cardiaque ; - de maladies neurodégéneratives, incluant mais ne se limitant pas à la maladie d'Alzheimer, aux démences liées au SIDA, à la maladie de Parkinson, à la sclérose latérale amyotrophique, à la retinitis pigmentosa, à l'atrophie spinale musculaire, à la dégénération du cervelet ;
- d'infections virales et fongiques incluant, mais ne se limitant pas au virus de l'herpès, de la variole, d'Eps tein-Barr, de Sindbis et à l'adénovirus, au SIDA ;
-de maladies dégéneratives du système musculo-squelétique incluant, mais ne se limitant pas à l'ostéoporose et l'arthrite, les rhinosinusites réactives à l'aspirine, la fibrose cystique ;
- de maladies hématologiques incluant, mais ne se limitant pas à l'anémie chronique, l'anémie aplasique et les syndromes de myéloplas tie .
De plus, les composés de formule générale (I) peuvent être utilisés pour traiter l'alopécie générée par la chimiothérapie, la thrombocytopenie générée par la chimiothérapie, la leukopénie ou mucite générés par la chimiothérapie.
L'utilisation des composés de formule générale (I), ainsi que des composés suivants, pour la préparation d'un médicament destiné à traiter les pathologies ci-dessus mentionnées fait partie intégrante de l'invention :
- N-phényl- 7 H-indazole-3-carboxamide
- N-(2-chlorophényl)- . H-indazole-3-carboxamide
- N-(3-chlorophényl)- . H-indazole-3-carboxamide
- N-(4-chlorophényl)- . H-indazole-3-carboxamide - N-(2,4-dichlorophényl)-7H-indazole-3-carboxamide
- N-(3,4-dichlor ophényl)- . H-indazole-3-carboxamide
- N-(2-méthylphényl)- 7r.-indazole-3-carboxamide
- N-(2,4-diméthylphényl)- 7H-indazole-3-carboxamide
- N-(2-méthoxyphényl)- 7Η-indazole-3-car boxamide - N-(4-méthoxyphényl)- . H-indazole-3-carboxamide
- N-(4-thiométhylphényl)- . H-indazole-3-carboxamide
- N-(3-chloro-4-thiométhylphényl)-5-amino- 1 H-indazole-3-carboxamide
- N-benzyl- , H-indazole-3-carboxamide
- N-(2-chlorobenzyl)- t H-indazole-3-carboxamide - N-(4-méthylbenzyl)-7H-indazole-3-carboxamide
- N-(pyridin-2-ylméthyl)- 1 H-indazole-3-carboxamide
- N-(pyridin-3-ylméthyl)- . H-indazole-3-carboxamide
- N-(pyridin-4-ylméthyl)- 1 H-indazole-3-carboxamide
- N-(2-phényléthyl)- 7H-indazoIe-3-carboxamide - N-(3,4-diméthoxyphényléthyl)-tH-indazole-3-carboxamide
- N-[3-(pyridin-2-yl)propyl]- . H-indazole-3-carboxamide
- N-[3-(2,6-diméthylphényl)propyl]-5-nitro- . H-indazole-3-carboxamide
L'invention a également pour objet des médicaments qui comprennent un composé de formule générale (I), ou un sel d'addition de ce dernier à un acide pharmaceutiquement acceptable ou encore un hydrate ou un solvat du composé de formule générale (I). Ces médicaments trouvent leur emploi en thérapeutique, notamment dans le traitement des pathologies ci-dessus mentionnées.
Selon un autre de ses aspects, la présente invention concerne des compositions pharmaceutiques comprenant en tant que principe actif, au moins un composé selon l'invention. Ces compositions pharmaceutiques comprennent une dose efficace d'un composé selon l'invention, ou un sel pharmaceutiquement acceptable, un hydrate ou un solvat dudit composé, et éventuellement un —ou plusieurs excipients pharmaceutiquement acceptables. Lesdits excipients sont choisis selon la forme pharmaceutique et le mode d'administration souhaité, parmi les excipients habituels qui sont connus de l'homme du métier.
Dans les compositions pharmaceutiques de la présente invention pour l'administration orale, sublinguale, sous-cutanée, intramusculaire, intraveineuse, topique, locale, intratrachéale, intranasale, transdermique ou rectale, le principe actif de formule générale (I) ci-dessus, ou son sel, solvat ou hydrate éventuel, peut être administré sous forme unitaire d'administration, en mélange avec des excipients pharmaceutiques classiques, aux animaux et aux êtres humains pour la prophylaxie ou le traitement des troubles ou des maladies ci-dessus.
Les formes unitaires d'administration appropriées comprennent les formes par voie orale telles que les comprimés, les gélules molles ou dures, les poudres, les granules, les chewing-gums et les solutions ou suspensions orales, les formes d'administration sublinguale, buccale, intratrachéale, inraoculaire, intranasale, par inhalation, les formes d'administration sous- cutanée, intramusculaire ou intraveineuse et les formes d'administration rectale ou vaginale. Pour l'application topique, on peut utiliser les composés selon l'invention dans des crèmes, pommades ou lotions.
A titre d'exemple, une forme unitaire d'administration d'un composé selon l'invention sous forme de comprimé peut comprendre les composants suivants :
Composé selon l'invention 50,0 mg Mannitol 223,75 mg
Croscaramellose sodique 6,0 mg
Amidon de maïs 15,0 mg
Hydroxypropyl-méthylcellulose . 2,25 mg
Stéarate de magnésium 3,0 mg
Afin d'obtenir l'effet prophylactique ou thérapeutique désiré, la dose de principe actif peut varier entre 0,1 mg et 200 mg par kg de poids du corps et~ par jour. Bien que ces dosages soient des exemples de situation moyenne, il peut y avoir des cas particuliers où des dosages plus élevés ou plus faibles sont appropriés, de tels dosages appartiennent également à l'invention. Selon la pratique habituelle, le dosage approprié à chaque patient est déterminé par le médecin selon le mode d'administration, le poids et la réponse dudit patient.
Chaque dose unitaire peut contenir de 0,1 à 1000 mg, de préférence de 0,1 à 500 mg, de principe actif en combinaison avec un ou plusieurs excipients pharmaceutiques. Cette dose unitaire peut être administrée 1 à 5 fois par jour de façon à administrer un dosage journalier de 0,5 à 5000 mg, de préférence de 0,5 à 2500 mg.
La présente invention selon un autre de ses aspects, concerne également une méthode de traitement des pathologies ci-dessus indiquées qui comprend l'administration d'un composé selon l'invention, d'un sel pharmaceutiquement acceptable, d'un solvat ou d'un hydrate dudit composé.