明細書
2 , 2—ジ置換— 1 «, 2 5—ジヒドロキシー 1 9一ノルビタミン D誘導体 技術分野
本発明は、 新規なビタミン D誘導体に関するものであり、 詳しくは、 新規な 2, 2-ジ置換 _19-ノルビ夕ミン D誘導体および新規な 20 - Epi - 19 -ノルビタミン D 誘導体に関するものである。 背景技術
活性型ビタミン D3 ( 1ひ,25-ジヒドロキシビタミン! )3、 l , 25- (OH) 2D3) はカル シゥムおよびリンの代謝調節ホルモンとして知られている他に、 細胞の分化誘 導や増殖抑制作用、 免疫調節作用など多彩な生物作用を発揮する。 これらの作 用は、 核内に存在するビタミン D受容体 (VDR) を介する標的遺伝子の転写制御 により発現する。 1,25- (0H) 2D3は、 腎性骨異栄養症、 D-抵抗性くる病、 副甲状 腺機能低下症、 骨粗鬆症や乾癬症の治療薬に適用されている。 癌や免疫治療薬 としても期待されているが、 癌や免疫治療薬としての有効量で高カルシウム血 症を併発することから、 血清カルシウム上昇作用と細胞の分化誘導作用を分離 したビタミン D誘導体の開発が望まれた。 選択的に細胞の分化誘導作用を持つ 誘導体の合成研究の多くは側鎖の修飾に向けられてきたが、 デル一力等により 1 , 25- (OH) 2D3から 19 -ェキソメチレン基を除去した A環修飾体、 19-ノル- 1 , 25-ジ ヒドロキシビタミン D3 (以下 19- nor_l,25_ (OH) 2D3とも記載する) が合成された (Per lman K. L. , Swenson R. E. , Paaren H. E., Schnoes H. K. , DeLuca H. F. , Tetrahedron Let t. , 1991 , 32, 7663-7666. ) 。 19-Nor-l, 25- (OH) 2D3の生 物作用を活性型 1, 25- (OH) 2D3と比較すると、 VDR結合活性は約 3分の 1、 そして 骨吸収作用は 10分の 1以下と弱くなつたが、 腫瘍細胞に対する分化誘導作用は 同等の活性を保持していることが示され、 19 - nor_l, 25- (OH) 2D3は血清カルシゥ ム上昇作用が弱いビ夕ミン D誘導体の骨格として有用であることが示唆された 。 さらに、 2位に種々の置換基を含む 19-ノルビタミン D誘導体が合成され、 い
くつかの興味深い活性プロフィールが報告されている (Sicinski R. R. , Perlman K. L. , DeLuca H. F., J. Med. Chem. , 1994, 37, 3730-3738 ; Sicinski R. R., Prahl J. M., Smith C. M., DeLuca H. F. , J. Med. Chem. , 1998, 41, 662-4674 ; Sicinski R. R. , Rotkiewicz P., Kolinski A., Sicinska W., Prahl J. M., Smith C. M. , DeLuca H. F. , J. Med. Chem. , 2002, 45, 3366-3380;岩崎由紀子ら、 日本薬学会第 121年会要旨集 3、 p.17, 29[PB]II-01U 2001年 (札幌) ; 吉田彰宏ら、 日本薬学会第 121年会要旨集 3 、 p.17, 29 [PB] 11-014, 2001年 (札幌) ;岩崎由紀子ら、 日本薬学会第 122年 会要旨集 2、 p.180, 28[P]I- 183、 2002年 (千葉) ;吉田彰宏ら、 日本薬学会 第 122年会要旨集 2、 p.180, 28 [P] 1-184, 2002年 (千葉) ;清水正人ら、 日本 ビタミン学会第 54回大会、 要旨、 ビタミン、 76,155-156、 025、 2002年 (東京 ) ;清水正人ら、 第 2 8回反応と合成の進歩シンポジウム講演要旨集、 pp 234-235, 2002年 (東京) ) 。 例えば、 19 - nor- 1, 25 -(0H)2D3の 2位に 1つのァ ルキル基またはアルキリデン基を導入すると (Sicinski R. R. , Prahl J. M. , Smith C. M. , DeLuca H. F., J. Med. Chem. , 1998, 41, 4662-4674 ; Sicinski . R. , Rotkiewicz P. , Kolinski A. , Sicinska W. , Prahl J. . , Smith M., DeLuca H. F. , J. Med. Chem. , 2002, 45, 3366-3380) 、 細胞 の分化誘導作用および血清カルシウム上昇作用の上昇が認められた。
しかしながら、 従来の 2位置換- 19-ノルビタミン D誘導体の研究は、 2位に 1つの置換基を持つものに限定されていた。 また、 20-Epi_19-ノルビタミン D 誘導体については、 現在までに、 2位にメチル、 ェチル、 ヒドロキシメチルな どを持つ誘導体が報告されているが (Sicinski R. R., Prahl J. M. , Smith C. M., DeLuca H. F. , J. Med. Chem. , 1998, 41, 662-4674; Sicinski R. R. , Rotkiewicz P., Kolinski A., Sicinska W. , Prahl J. M. , Smith C. M., DeLuca H. F. , J. Med. Chem. , 2002, 45, 3366-3380)、 さらに優れた生物学 的活性を有する誘導体の開発が望まれていた。 発明の開示
本発明は、 新規な 2, 2-ジ置換- 19-ノルビ夕ミン D誘導体と新規な 20 - Epi- 19-
ノルビタミン D誘導体を合成し、 提供することを目的とするものである。 本発 明はまた、 合成した 2, 2-ジ置換- 19-ノルビタミン D誘導体と新規な 20- Epi - 19- ノルビ夕ミン D誘導体の生物活性を評価することを目的とするものである。 本発明者等は、 上記課題を解決するために鋭意研究した結果、 新規な 2, 2 -ジ 置換- 19-ノルビタミン D誘導体と新規な 20-Epi - 19-ノルビタミン D誘導体を合成 することに成功し、 本発明を完成するに至った。
すなわち、 本発明は一般式 ( I ) :
(式中、
1 1ぉょび11 2は、 同一または異なって、 ハロゲン原子、 または水酸基、 ま たは無置換の炭素数 1一 1 0の直鎖もしくは分岐鎖状アルキル基、 または置換 された炭素数 1一 1 0の直鎖もしくは分岐鎖状アルキル基、 または無置換の炭 素数 2— 1 5の直鎖もしくは分岐鎖状アルケニル基、 または置換された炭素数 2— 1 5の直鎖もしくは分岐鎖状アルケニル基を表すか、 あるいは、 R 1およ び R 2がー緖になって、 無置換の炭素数 3— 6のスピロ環状アルキル基、 また は置換された炭素数 3— 6のスピロ環状アルキル基、 またはへテロ原子として 酸素原子を含む無置換の炭素数 3— 6のスピロへテロ環、 またはへテロ原子と して酸素原子を含む置換された炭素数 3— 6のスピロへテロ環を形成し;
Aは、 水素、 または無置換の炭素数 1一 1 2の直鎖もしくは分岐鎖状アルキ
ル基、 または置換された炭素数 1一 1 2の直鎖もしくは分岐鎖状アルキル基、 または無置換の炭素数 1一 1 2の直鎖もしくは分岐鎖状アルキルォキシ基、 ま たは置換された炭素数 1一 1 2の直鎖もしくは分岐鎖状アルキルォキシ基、 ま たは無置換の炭素数 2— 1 4の直鎖もしくは分岐鎖状アルケニル基、 または置 換された炭素数 2— 1 4の直鎖もしくは分岐鎖状アルケニル基を表す) で表される化合物を提供するものである。
一般式 (I ) において、 尺1ぉょび1 2は、 同一または異なって、 ハロゲン 原子、 または水酸基、 または無置換の炭素数 1一 8の直鎖もしくは分岐鎖状ァ ルキル基、 またはハロゲン原子および無置換の炭素数 1一 4の直鎖もしくは分 岐鎖状のアルキルォキシ基およびァリ一ル基およびアミノ基およびァジド基か らなる群から選択される少なくとも 1個の置換基を有する炭素数 1一 8の直鎖 もしくは分岐鎖状アルキル基、 または無置換の炭素数 2— 8の直鎖もしくは分 岐鎖状アルケニル基、 またはハロゲン原子および無置換の炭素数 1一 4の直鎖 もしくは分岐鎖状のアルキルォキシ基およびァリール基およびアミノ基および アジド基からなる群から選択される少なくとも 1個の置換基を有する炭素数 2 _ 8の直鎖もしくは分岐鎖状アルケニル基を表すか、 あるいは、 R 1および R 2がー緒になって、 無置換のスピロシクロプロピル基、 または少なくとも 1個 の炭素数 1一 4の直鎖もしくは分岐鎖状の無置換のヒドロキシアルキル基で置 換されたスピロシクロプロピル基、 または無置換のスピロォキシラン、 または 炭素数 1一 4の直鎖もしくは分岐鎖状の無置換のヒドロキシアルキル基で置換 されたスピロォキシランを形成し; Aは、 少なくとも 1個のヒドロキシ基で置 換された炭素数 1一 1 2の直鎖もしくは分岐鎖状アルキル基、 または少なくと も 1個のヒドロキシ基で置換された炭素数 1 _ 1 2の直鎖もしくは分岐鎖状ァ ルキルォキシ基、 または少なくとも 1個のヒドロキシ基で置換された炭素数 2 一 1 4の直鎖もしくは分岐鎖状アルケニル基を表すことが好ましい。
さらに、 一般式 (I ) において、 1ぉょび 2は、 同一または異なって、 ハロゲン原子、 または水酸基、 または無置換の炭素数 1一 6の直鎖もしくは分 岐鎖状アルキル基、 またはハロゲン原子および炭素数 1一 3の直鎖もしくは分 岐鎖状の無置換のアルキルォキシ基およびフエニル基およびアミノ基およびァ
ジド基からなる群から選択される少なくとも 1個の置換基を有する炭素数 1一 6の直鎖もしくは分岐鎖状アルキル基、 または無置換の炭素数 2— 4の直鎖も しくは分岐鎖状アルケニル基を表すか、 あるいは、 R 1および R 2が一緒にな つて、 無置換のスピロシクロプロピル基、 または少なくとも 1個の炭素数 1一 3の直鎖もしくは分岐鎖状の無置換のヒドロキシアルキル基で置換されたスピ ロシクロプロピル基、 または無置換のスピロォキシランを形成し; Aは、 水素 、 または少なくとも 1個のヒドロキシ基で置換された炭素数 3— 1 0の直鎖も しくは分岐鎖状アルキル基、 または少なくとも 1個のヒドロキシ基で置換され た炭素数 3 _ 8の直鎖もしくは分岐鎖状アルキルォキシ基、 または少なくとも 1個のヒドロキシ基で置換された炭素数 4一 1 2の直鎖もしくは分岐鎖状アル ケニル基を表すことが好ましい。
一般式 (I ) において、 尺 1ぉょび尺2は、 同一または異なって、 水酸基、 または無置換の炭素数 1一 6の直鎖もしくは分岐鎖状アルキル基、 またはフッ 素原子および無置換の炭素数 1一 3の直鎖もしくは分岐鎖状のアルキルォキシ 基からなる群から選択される少なくとも 1個の置換基を有する炭素数 1— 6の 直鎖もしくは分岐鎖状アルキル基を表すか、 あるいは、 R 1および R 2がー緒 になって無置換のスピロォキシランを形成し; Aは、 水素、 または少なくとも 1個のヒドロキシ基で置換された炭素数 5— 7の直鎖もしくは分岐鎖状アルキ ル基を表すことが好ましい。
一般式 (1 ) において、 2 0位の立体配置は S配置であってもよく、 R配置 であってもよい。
本発明のさらに別の側面によれば、 一般式 ( I V) :
式 (IV)
(式中、
R 4および R 5は、 一方が水素原子を表し、 かつ他方が水酸基で置換された炭 素数 1一 4の直鎖もしくは分岐鎖状アルキル基または— OR 6 (ここで、 R6 は水酸基で置換された炭素数 1一 4の直鎖もしくは分岐鎖状アルキル基を表す )を表す、 あるいは R4および R 5が一緒になつて =CR 7 (ここで、 R7は 水酸基で置換された炭素数 1一 4の直鎖もしくは分岐鎖状アルキル基を表す) )
である化合物が提供される。
本発明のさらに別の側面によれば、 上記一般式 (I) または一般式 (I V) で表される化合物と、 薬学的に許容可能な担体または希釈剤を含む医薬組成物 が提供される。
本発明のさらに別の側面によれば、 細胞の分化に異常を伴う疾患を治療また は予防する方法であって、 そのような治療または予防を必要とする対象に、 上 記一般式 (I) または一般式 (I V) で表される化合物の治療的有効量を投与 することを含む方法が提供される。
本発明のさらに別の側面によれば、 上記一般式 (I) または一般式 (I V) で表される化合物の細胞の分化に異常を伴う疾患治療用の医薬組成物製造への 使用が提供される。
本発明のさらに別の側面によれば、 上記一般式 (I) で表される化合物を製
造する方法であって、 一般:
〇z 式 U I)
(式中、 zは、 同一または異なって、 水素または保護基を表す)
で表される化合物から一般式 (I I I) :
式 (I I I)
(式中、 Zは、 同一または異なって、 水素または保護基を表し、 Phはフエ二 ル基を表す)
の化合物を得る工程を含む方法が提供される。 発明を実施するための好適な形態
なお、 本出願が主張する優先権の基礎となる出願である特願 2002-297366号お よび特願 2003-024183号の開示は、 全て引用により本明細書の中に取り込まれる。 以下に、 本発明の一般式 (I) および (IV) で表されるビタミン D誘導体 の実施態様および実施方法についてより詳細に説明する。
本発明において、 「ビタミン D誘導体」 とは、 9, 10-セコ -5, 7, 10(19)-コレ
スタトリエン構造を有する化合物を指す。 本発明において、 「19-ノル- 1, 25 - ジヒドロキシビタミン D誘導体」 とは、 9, 10-セコ- 5, 7, 10 (19) -コレスタトリエ ン構造を有する化合物から 10 (19) -ェキソメチレン基が除去された化合物を意 味する。
一般式 (I ) の R 1および R 2における、 「ハロゲン原子」 としては、 フッ 素、 塩素、 シユウ素、 ヨウ素原子などが挙げられるが、 フッ素原子が特に好ま しい。
R 1および R 2における、 「無置換の直鎖もしくは分岐鎖状のアルキル基」 としては、 炭素数 1— 1 0の無置換の直鎖もしくは分岐鎖状アルキル基が好ま しい。 炭素数は好ましくは 1— 8、 いっそう好ましくは 1 _ 6、 さらにいっそ う好ましくは 1—4である。 例えば、 メチル基、 ェチル基、 n—プロピル基、 i一プロピル基、 n—ブチル基、 s—ブチル基、 i一ブチル基、 t一ブチル基 、 並びに直鎖及び分岐鎖状のペンチル基、 へキシル基、 ヘプチル基、 ォクチル 基、 ノニル基、 デカニル基等が挙げられるが、 これらに限定されるものではな い。
R 1および R 2における、 「置換された直鎖もしくは分岐鎖状のアルキル基 」 としては、 上記の 「無置換の直鎖もしくは分岐鎖状のアルキル基」 の 1以上 の水素原子が置換されている基を意味する。 置換基としては、 例えば、 ハロゲ ン原子 (例えば、 フッ素原子) 、 置換された直鎖もしくは分岐鎖状のアルキル ォキシ基 (炭素数 1—4、 特に 1一 3であることが好ましい) 、 無置換の直鎖 もしくは分岐鎖状のアルキルォキシ基 (炭素数 1—4、 特に 1一 3であること が好ましい) 、 無置換のァリール基 (たとえば、 フエニル基) 、 ハロゲン原子 もしくは無置換の炭素数 1― 4の直鎖もしくは分岐鎖状のアルキル基で置換さ れたァリール基 (たとえば、 トリル基) 、 アミノ基、 アジド基等が挙げられ、 特に、 フッ素原子、 メトキシ基、 エトキシ基、 フエニル基、 アミノ基、 アジド 基等が好ましい。
R 1および R 2における、 「無置換の直鎖もしくは分岐鎖状アルケニル基」 としては、 少なくとも 1つの二重結合を有する炭素数 2— 1 5の直鎖もしくは 分岐鎖状アルケニル基が好ましい。 炭素数は好ましくは 2— 8、 いっそう好ま
しくは 2— 6、 さらにいつそう好ましくは 2— 4である。 二重結合の数は 1一 3であることが好ましく、 1または 2であることがさらに好ましく、 1である ことがいっそう好ましい。 「置換された直鎖もしくは分岐鎖状アルケニル基」 としては、 上記の 「無置換の直鎖もしくは分岐鎖状アルケニル基」 の 1以上の 水素原子が置換されている基を意味する。 置換基としては、 例えばハロゲン原 子および無置換の炭素数 1一 4の直鎖もしくは分岐鎖状のアルキル基などが挙 げられる。
2位の立体配置は、 R配置であっても S配置であってもよい。
R 1および R 2がー緖になって形成する 「無置換のスピロ環状アルキル基」 は、 炭素数 3— 6、 さらに 3— 4であることが好ましく、 特に、 スピロシクロ プロピル基等が好ましい。
置換されたスピロ環状アルキル基とは、 上記の 「無置換のスピロ環状アルキ ル基」 の 1以上の水素原子が置換されている基を意味する。 置換基としては、 無置換の直鎖もしくは分岐鎖状のヒドロキシアルキル基が好ましく、 炭素数 1 一 4であることがさらに好ましく、 炭素数 1— 3であることがいっそう好まし い。 例えばヒドロキシメチル基、 ヒドロキシェチル基、 ヒドロキシプロピル基 等が挙げられる。
R 1および R 2が一緒になつて形成する 「ヘテロ原子として酸素原子を含む 無置換のスピロへテロ環」 としては、 炭素数 3— 6、 さらに 3— 4であること が好ましい。 ヘテロ原子と'して 1個の酸素原子を含むことが好ましい。 特にス ピロォキシラン等が好ましい。
「ヘテロ原子として酸素原子を含む置換されたスピロへテロ環」 とは、 上記 の 「ヘテロ原子として酸素原子を含む無置換のスピロへテロ環」 の 1以上の水 素原子が置換されている基を意味する。
一般式 (I ) の Aにおける、 「無置換の直鎖もしくは分岐鎖状のアルキル基 」 としては、 炭素数 1一 1 2の無置換の直鎖もしくは分岐鎖状アルキル基が好 ましい。 炭素数は好ましくは 3—1 0、 いっそう好ましくは 5— 7、 もっとも 好ましくは 6である。 例えば、 メチル基、 ェチル基、 n—プロピル基、 iープ 口ピル基、 n—ブチル基、 s—ブチル基、 i 一ブチル基、 t一ブチル基、 並び
に直鎖及び分岐鎖状のペンチル基、 へキシル基、 ヘプチル基、 ォクチル基、 ノ ニル基、 デカニル基等が挙げられるが、 これらに限定されるものではない。
Aにおける 「置換された直鎖もしくは分岐鎖状のアルキル基」 とは、 上記の 「無置換の直鎖もしくは分岐鎖状のアルキル基」 の 1以上の水素原子が置換さ れている基を意味する。 置換基としては、 ヒドロキシ基が好ましい。 置換基の 数は特に制限はないが、 1 _ 3であることが好ましく、 1または 2であること がさらに好ましい。 例えば、 ヒドロキシメチル基、 ヒドロキシェチル基、 ヒド ロキシブチル基、 ヒドロキシプロピル基、 ヒドロキシペンチル基、 ヒドロキシ へキシル基、 ヒドロキシヘプチル基、 ヒドロキシォクチル基、 ヒドロキシノ二 ル基、 ヒドロキシデカニル基、 4ーヒドロキシ— 4—メチルペンチル基、 1, 4—ジヒドロキシ— 4ーメチルペンチル基、 4—ェチル—4—ヒドロキシへキ シル基、 6—ヒドロキシー 6—メチル— 2—ヘプチル基、 7—ヒドロキシ一 7 ーメチルー 2—ォクチル基、 5, 6—ジヒドロキシ _ 6 _メチル _ 2—へプチ ル基等が挙げられる。 '
一般式 ( I ) の Aにおける、 「無置換の直鎖もしくは分岐鎖状アルキルォキ シ基」 としては、 炭素数 1一 1 2の無置換の直鎖もしくは分岐鎖状アルキルォ キシ基が好ましい。 炭素数は好ましくは 3— 8、 さらに好ましくは 4— 6、 も つとも好ましくは 5である。 例えば、 メトシキ基、 エトキシ基、 プロポキシ基 、 ブトキシ基、 ペンチルォキシ基、 へキシルォキシ基等が挙げられるが、 これ らに限定されるものではない。
Aにおける 「置換された直鎖もしくは分岐鎖状のアルキルォキシ基」 とは、 上記の 「無置換の直鎖もしくは分岐鎖状のアルキルォキシ基」 の 1以上の水素 原子が置換されている基を意味する。 置換基としては、 ヒドロキシ基が好まし い。 置換基の数は特に制限はないが、 1—3であることが好ましく、 1または 2であることがさらに好ましい。 例えば、 _0C2 C (C ) 20H、 -0CH2CH0HC (CH3) 20H などが挙げられる。
一般式 (I ) の Aにおける、 「無置換の直鎖もしくは分岐鎖状アルケニル基 」 としては、 炭素数 2 _ 1 4の無置換の直鎖もしくは分岐鎖状アルケニル基が 好ましい。 炭素数は好ましくは 4 _ 1 2、 さらに好ましくは 5— 1 0、 もっと
も好ましくは 6— 9である。 二重結合に関してはシスまたはトランス何れでも よい。 二重結合の数は 1一 3であることが好ましく、 1または 2であることが さらに好ましい。 例えば、 ビニル基、 プロぺニル基、 ブテニル基、 へキセニル 基、 ヘプテニル基、 ォクテニル基、 ノネニル基、 デカネ二ル基、 4—メチルー ペン夕一 1一ェンー 1一^ Γル基、 5—ェチル— 1 , 3—ヘプ夕— 1 , 3—ジェ ン一 1一ィル基等が挙げられるが、 これらに限定されるものではない。
Aにおける 「置換された直鎖もしくは分岐鎖状アルケニル基」 とは、 上記の 「無置換の直鎖もしくは分岐鎖状のアルケニル基」 の 1以上の水素原子が置換 されている基を意味する。 置換基としては、 ヒドロキシ基が好ましい。 置換基 の数は特に制限はないが、 1一 3であることが好ましく、 1または 2であるこ とがさらに好ましい。 例えば、 4—ヒドロキシー 4—メチルーペン夕一 1—ェ ン— 1一ィル基 (- C2 CH2C (C ) 2OH) 、 5—ヒドロキシ— 5—ェチル—ヘプ夕 - 1 , 3 _ジェン—1—ィル基 (_C4H4C (C2H5) 20H) などが挙げられる。
本発明の一般式 (I ) の化合物において、 1位及び 3位の水酸基の立体配置 はひ、 ]3の何れの化合物も本発明に含まれる。 さらに一般式 (I ) 中の Aがァ ルケ二ル基を示す場合、 二重結合により生じるシス、 トランスの幾何異性体も また全て本発明の範囲内に含まれ、 その他、 考えられる光学異性体、 幾何異性 体も全て本発明の範囲に含まれる。
本発明の一般式 (I ) の化合物のうち具体的化合物としては、 下記が挙げら れる。
CS0CT0/C00Zdf/X3d oひ εεο請 OA
εχ
CS0CT0/C00Zdf/X3d οひ εεο請 OA
fl
^ λ'≠≡^^QZ ! c¾-oz
CS0CT0/C00Zdf/X3d oひ εεο請 OA
ST
CS0CT0/C00Zdf/X3d oひ εεο請 OA
CS0CT0/C00Zdf/X3d oひ εεο請 OA
LI
CS0CT0/C00Zdf/X3d oひ εεο請 OA
115a 115b 一般式 (I V) の R 4または R 5の定義における、 「水酸基で置換された炭 素数 1一 4の直鎖もしくは分岐鎖状アルキル基」 の 「炭素数 1一 4の直鎖もし くは分岐鎖状アルキル基」 としては、 メチル基、 ェチル基、 n—プロピル基、 i—プロピル基、 n—ブチル基、 s —ブチル基、 i 一ブチル基、 t一ブチル基 等が挙げられ、 特に炭素数 2— 3の直鎖もしくは分岐鎖状アルキル基、 さらに 特にはェチル基、 n—プロピル基が挙げられる。 置換する水酸基の数は 1また は 2であることが好ましく、 1であることが更に好ましい。
一般式 (I V) の R 6の定義における、 「水酸基で置換された炭素数 1一 4 の直鎖もしくは分岐鎖状アルキル基」 の 「炭素数 1 _ 4の直鎖もしくは分岐鎖 状アルキル基」 としては、 メチル基、 ェチル基、 n—プロピル基、 i 一プロピ ル基、 n _ブチル基、 s—ブチル基、 i 一ブチル基、 t 一ブチル基等が挙げら
れ、 特に炭素数 1 _ 3の直鎖もしくは分岐鎖状アルキル基、 さらに特にはメチ ル基、 ェチル基、 n—プロピル基が挙げられ、 いっそう特にはェチル基が挙げ られる。 置換する水酸基の数は 1または 2であることが好ましく、 1であるこ とが更に好ましい。
一般式 (I V) の R 7の定義における、 「水酸基で置換された炭素数 1—4 の直鎖もしくは分岐鎖状アルキル基」 の 「炭素数 1一 4の直鎖もしくは分岐鎖 状アルキル基」 としては、 メチル基、 ェチル基、 n—プロピル基、 i一プロピ ル基、 n—ブチル基、 s—ブチル基、 i—ブチル基、 t一ブチル基等が挙げら れ、 特に炭素数 1一 3の直鎖もしくは分岐鎖状アルキル基、 さらに特にはメチ ル基、 ェチル基、 いっそう特にはメチル基が挙げられる。 置換する水酸基の数 は 1または 2であることが好ましく、 1であることが更に好ましい。
本発明の一般式 ( I V) の化合物において、 R 4および R 5の一方が水素原 子を表し、 他方が水酸基で置換された炭素数 1一 4の直鎖もしくは分岐鎖状ァ ルキル基または一 O R 6を表す場合には、 水酸基で置換された炭素数 1一 4の 直鎖もしくは分岐鎖状アルキル基や— O R 6は、 2 位にあっても 2 jS位にあ つてもよい。
本発明の一般式 (I V) の化合物において、 R 4および R 5が一緒になつて = C R 7を表す場合には、 この二重結合により生じる (Z ) 体、 (E) 体のい ずれであってもよい。
本発明の一般式 (I V) の化合物のうち具体的化合物としては、 下記が挙げ られる。
OS
CSOCTO/COOZdf/X3d oひ εεο請 OA
CS0CT0/C00Zdf/X3d oひ εεο請 OA
CS0CT0/C00Zdf/X3d oひ εεο請 OA
本発明の一般式 (I) または (IV) で表されるビタミン D誘導体は、 医薬 組成物 (例えば細胞の分化調節剤等) の有効成分として使用することもできる。 本発明の化合物は、 製薬上許容しうる担体、 賦型剤、 崩壊剤、 滑沢剤、 結合 剤、 香料、 着色剤等とともに、 適当な剤型に製剤化して用いるのが好ましく、 そのような剤型としては、 錠剤、 顆粒剤、 細粒剤、 カプセル剤、 散剤、 注射剤 、 溶液剤、 懸濁剤、 乳剤、 経皮吸収剤、 坐剤等が挙げられる。
本発明の化合物の投与経路は特に限定されず、 経口投与でも非経口投与 (静 脈内投与、 筋肉内投与、 腹腔内投与、 経皮投与など) でもよい。
本発明の化合物の投与量は、 対象疾患、 患者の状態、 体重、 体質、 年齢、 性 別、 また投与経路、 剤型等により適宜選択することができるが、 一般に投与量 の下限として、 成人 1日当たり 0. 00 l g— 0. l i gの範囲、 好ましく は 0. 01 g前後で、 投与量の上限としては成人 1日当たり 100 g— 1 0000 の範囲、 好ましくは 200 xg— l 000 gの範囲内で選択で き、 1日 1一 3回に分けて投与することができる。
本発明の一般式 (I) で表されるビタミン D誘導体は新規化合物であり、 そ の合成法は何ら限定されないが、 例えば、
一般式 (I I) :
(式中、 Zは、 同一または異なって、 水素または保護基を表す)
で表される化合物から一般式 (I I I) :
式 (I I I )
(式中、 Zは、 同一または異なって、 水素または保護基を表し、 P hはフエ二 ル基を表す)
の化合物を得る工程を含む方法で合成することができる。
Zにおける保護基としては、 それぞれ同一でも異なっていてもよく、 置換さ れていてもよいアルキル基、 置換シリル基、 ァシル基等があげられるが、 中で も、 ベンジル基、 トリメチルシリル基、 t一プチルジメチルシリル基等が好ま しい。
保護基は、 化学分野でよく知られている従来技術により、 合成の適当な段階 で除去され得る。
本発明の一般式 ( I ) で表されるビタミン D誘導体は、 例えば、 以下に記載 の方法によっても合成することができる。
本発明の A環ホスフィンォキシドの一般的合成スキームを下記に示す。
M
出発化合物である、 シリル基により保護された水酸基を有するシクロへキサ ノン誘導体 (化合物 B ) は既知の方法により、 (一) ーキナ酸から合成するこ とができる (Per lman, K. L., Sewnson, R. E. , Paaren, H. E. , Schnoes, H. K., DeLuca, H. F., Tetrahedron Let t . , 1991 , 32, 7663-7666) 。 シクロへ キサノン誘導体 Bを水素化ホウ素ナトリゥム等の還元剤により一旦アルコール 体 Cに変換する。 ついでアルコール体 Cの水酸基を保護基で保護し化合物 Dと する。 保護基としてベンジル基等を使用してエーテル結合を形成することが好 ましい。 次いで、 酢酸等の酸処理により化合物 Dの 4位のトリメチルシリル基 のみを脱保護し、 さらにジメチルスルホキシドおよび二塩化ォキサリル等によ る酸化反応によりケトン基に変換し化合物 Fを得る。 化合物 Fのケトン基をビ ッティヒ (Wi t t ig) 試薬によりメチレンとし、 得られた化合物 Gを m-クロ口過 安息香酸等の過酸化剤によりスピロエポキシ化合物 Hに変換する。 パラジウム 触媒による水素添加反応により 6位のベンジル保護基を脱保護し化合物 Iとし
、 生成した水酸基を酸化してケトン基とし化合物 Jとした後、 (トリメチルシ リル) 酢酸エステルによる炭素付加、 還元反応によりアルコール体 とする。 アルコール体 Lをジフエニルホスフィン化し、 続いて過酸化水素により酸化を 行うことで、 目的とする A環ホスフィンォキシド化合物 Mを取得することがで きる。
CD環 25—ヒドロキシグランドマンケトン体は、 所望の CD環を有する文献公知 のビタミン D誘導体をオゾン分解することで合成することができる (Sandina, F. J. , Mour ino, S., Cas tedo, L., J. Org. Chem. , 1986, 51, 1264-1269. : Ki egiel , J. , Wovkul ich, P. M. , Uskokovi c, M. R. , Tetrahedron Let t. , 1991 , 32, 6057-6060. : Fernadez, B. , Perez, J. A. Granj a, J. R. , Cas tefo, L., Mour ino, A. , J. Org. Chem. , 1992, 57, 3173-3178. : Fuj i shima, T., Konno, K. , Nakagawa, K. , Kurobe, M., Okano, T. , Takayama, H. , Bi oogr. Med. Chem. , 2000, 8, 123-134. )
所望のビタミン D誘導体の合成は、 上記方法にて合成した A環ホスフィンォ キシド体と CD環 25—ヒドロキシグランドマンケトン体を結合することで行える 。 一般に、 25位のヒドロキシ基を保護することなくカップリング反応を行うと 、 収率が低下するため、 25—ヒドロキシグランドマンケトン体の 25位のヒドロ キシ基を、 ァシル基、 置換シリル基、 置換アルキル基などの適当な保護基 (例 えば、 トリェチルシリル基、 メトキシメチル基等) で保護しておくことが好ま しい。 A環ホスフィンォキシド体をブチルリチウム等の強塩基で処理し、 ホス フイノキシカルバニオンを生成させ、 CD環グランドマンケトン体のケトン基と 反応させる。
2位のスピロォキシランは所望により開環し、 2, 2—ジ置換体とすること ができる。 例えば、 テトラプチルアンモニゥムフロリド等のフッ素化剤を用い れば、 2位にフルォロメチル基と水酸基を有する誘導体を合成することができ る。 また、 リチウムアルミニウムヒドリド等の金属水素化剤を用いれば 2位に メチル基と水酸基を有する誘導体を合成することができる。 メチル化剤ゃメ卜 キシ化剤を用いれば、 それぞれェチル基と水酸基、 メトキシメチル基と水酸基 を有する誘導体を取得することができる。
本発明の一般式 (I V) で表されるビタミン D誘導体は、 新規化合物であり その合成方法は何ら限定されないが、 例えば、 以下の一般的合成スキームに記 載の方法によっても合成することができる。
上記スキームに示すように、 CD環 20—ェピー 25—ヒドロキシグランド マンケトン体は、 例えば、 所望の CD環を有する公知のビタミン D誘導体をォ ゾン分解後、 DBUにより 20—アルデヒド体の 20位をェピメリ化し、 直ち に NaBH
4により還元することで、 主生成物 (非天然型 22—アルコール体 として) として得ることができる (Sandina, F. J., Mourino, S., Castedo, L., J. Org. Chem. , 1986, 51, 1264-1269. : Kiegiel, J., Wovkulich, P. M. Uskokovic, M. R., Tetrahedron Lett., 1991, 32, 6057-6060. : Fernadez, B., Perez, J. A. Granja, J. R. , Castefo, L. , Mourino, A. , J. Org. Chem. , 1992, 57, 3173-3178. : Fujishima, T. , Konno, K. , Nakagawa, K. ,
Kurobe, M., Okano, T. , Takayama, H. , Bioogr. Med. Chem. , 2000, 8, 123- 134.) 。 実施例
以下の実施例により本発明をさらに具体的に説明するが、 本発明は実施例によ つて限定されることはない。
(機器分析条件)
¾ NMRおよび19 F NMRはブルカー製 ARX-400型により測定し、 テトラメチルシラ ン (tetramethylsilane、 TMS) を内部標準として、 また、 19F NMRの場合にはト リフルォロトルエン(trifluorotoluene)を外部標準 (<5=-63ppm) として化学 シフ トを <5値で示した。 NMRの記載は次の略号によった。 s=singlet: d=doublet; t^triplet; m^multiplet; arom=aroiatic; br=broad signal。
MSスぺクトルは日本電子製 JMS-AX505HA型にて電子衝撃法 (EI)により測定し た。 本明細書中、 「no M+」 は M+が観測されないことを意味する。 「HR_MS」 は 高分解能 MSスぺクトルを意味する。
UVスぺクトルはべックマン製 DU- 7500型により測定した。
一部の異性体の混合物は、 日本分光製 MD-910型マルチ検出器を装備した HPLC システムを用いて分離、 精製した。
特別に記載しない限り、 反応はアルゴン気流下で行った。
シリカゲルはヮコーゲル C- 200 (和光純薬工業) を用いた。
以下の実施例において、 1 9一ノルビ夕ミン Dの A環部に相当する化合物 2〜 13、 30〜38、 1 18、 1 1 9、 125〜 135の位置番号は、 I UPA C命名法に基づいて表記する。
た後の 1 9—ノルビタミン D骨格を有する化合物についてはステロイドの位置 番号に基づいて表記する。
(実施例 1 ) (1, 4-c i s) -および(1 , -trans) -3, 5-ビス -[(卜プチルジメチル シリル)ォキシ ]-4- シ]-シクロへキサノール (化合物
0 °Cに冷却した (3R, 5R) -3, 5 -ビス- [(卜プチルジメチルシリル)才キシ] -4 - [ ( トリメチルシリル)ォキシ] -シクロへキサノン (化合物 2) (5.13 g, 11.5 匪 ol) のエタノール (50 mL) 溶液に、 約 10分をかけて水素化ホウ素ナトリウ ム (217.5 mg, 5.75 匪 ol) 加えた後、 1.5時間撹拌した。 反応混合物に氷水を 加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マ グネシゥムで乾燥後、 溶媒を留去した。 残渣はシリカゲルカラムクロマトダラ フィ一 (60 g、 5%酢酸ェチル /へキサン) にて精製し、 化合物 3を 1,4- cis体 と 1,4-trans体の混合物として得た (5.15 g, 99¾) 。 この混合物を構成する立 体異性体の比率は約 2:1であった。 1,4- cis体と 1, 4- trans体のいずれが主生成 物かは判断できなかった。 尚、 C-1位は擬似不斉であり、 1,4- cis体と 1,4 - trans体は互レ こ achiral d i as tereo isomerになる。
2: Ή NMR (CDClj) δ: 0.05 (6 H, Si -Me x 2), 0.06, 0.07 (each 3 H, s, Si -Me x 2), 0.16 (9 H, s, SiMe3), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 2.17 (1 H, m), 2.36 (1 H, dd, J=13.7, 4.5 Hz), 2.73 (2 H, m), 3.80 (1 H, m, H-4), 4.03 (1 H, dd, J=8.3, 2.3 Hz, H-5), 4.24 (1 H, ddd, J=10.6, 4.5, 2.3 Hz, H-3).
MS m/z (¾): no M+, 431 (3), 389 (68), 299 (69), 257 (44), 73 (100).
3: Ή NMR (CDCI3) δ: 0.06〜0.11 (12 H, Si -Me x 4), 0.11, 0.12 (9 H, s, SiMe3), 0.89〜0.91 (18 H, s, Si-tBu x 2), 1.52〜1.92 (4 H, m), 3.43, 3.68 (ca. 2:1) (1 H, m), 3.94〜4.22 (3 H, m).
MS m/z (¾): no M+, 391 (3), 373 (15), 301 (19), 259 (23), 73 (100). (実施例 2) (1,4- cis)-および(l,4-trans)-3,5-ビス- [(t -プチルジメチル シリル)ォキシ] -4- [(トリメチルシリル)ォキシ] -シクロへキサノ一ル ベンジ ルエーテル (化合物 4)
0 °Cに冷却した化合物 3 (主生成物とマイナー生成物の約 2 : 1の混合物、 4.57 g, 10.2 腿 ol) の無水ジメチルホルムアミド (30 mL) 溶液に、 水素化ナ トリウム (1.22 g, 30.5 匪 ol, 60%パラフィンリキッド) および臭化べンジル (3.483 g, 20.4腿 ol) を加え、 8時間撹拌した。 反応混合物に氷水を加え、 酢酸ェチル /へキサン (1:1) にて抽出した。 有機層を飽和食塩水にて洗浄し 、 無水硫酸マグネシウムで乾燥後、 溶媒留去した。 残渣をシリカゲルカラムク 口マトグラフィー (150 g、 3% 酢酸ェチル Zへキサン) にて精製し、 化合物 4 を 1, 4- cis体と 1,4 - trans体の混合物として得た (4.66 g, 85%) 。 この混合物 を構成する立体異性体の比率は約 2:1であった。 1, 4- cis体と 1,4- trans体のい ずれが主生成物かは判断できなかった。 尚、 C-1位は擬似不斉であり、 1,4- cis 体と 1, 4- trans体は互いに achiral diastereoisomerになる。
4a (主生成物) : 'Η NMR (CDC13) δ: 0.03, 0.06 (each 6 H, Si -Me x 4), 0.10 (9 H, s, SiMe3), 0.85, 0.90 (each 9 H, s, Si-tBu x 2), 1.70〜1.93 (4 H, m), 3.57 (1 H, m), 3.64 (1 H, tt, J=11.0, 5.0 Hz), 3.80 (1 H, m), 3.91 (1 H, ddd, J=9.5, 4.3, 2.4 Hz), 4.51, 4.55 (each 1 H, d, J-11.7 Hz, PhCH2) , 7.30〜7.37 (5 H, m, arom-H).
4b (マイナー生成物) : ΐ NMR (CDC13) δ: 0.01, 0.046 (each 3 H, Si -Me x 2), 0.055 (6 H, s, Si -Me x 2), 0.11 (9 H, s, SiMe3), 0.84, 0.88 (each 9 H, s, Si-tBu x 2), 1.37 (2 H, m), 2.07 (1 H, m), 2.19 (1 H, m), 3.28 (1 H, dd, J-8.5, 2.3 Hz), 3.80 (2 H, m), 3.93 (1 H, m), 4.50, 4.52 (each 1 H, d, J=12.1 Hz, PhCH2) , 7.30〜7.37 (5 H, m, arom-H).
混合物の MS m/z (¾): no M+, 481 (5), 391 (9), 373 (20), 349 (6), 259 (6), 91 (100).
(実施例 3) (1 , 4- c i s) -および(1 , 4-trans) _2, 6-ビス- [ (t -プチルジメチル
シリル)才キシ]—4一 [ (ベンジル)ォキシ]-シクロへキサノール (化合物 5 )
化合物 4 (化合物 4aと 4bの約 2:1の混合物、 269 mg, 0.499匪 ol) を、 テトラ ヒドロフラン、 酢酸および水の混合物 (8.5 mL, 8:8:1, v/v/v) に溶解し、 室 温にて 20時間撹拌した。 反応混合液は酢酸ェチルにて希釈し、 5%炭酸水素ナト リウム水溶液、 続いて飽和食塩水にて洗浄した。 有機層は無水硫酸ナトリウム にて乾燥後、 溶媒留去した。 残渣はシリカゲルカラムクロマトグラフィー (15 g、 4% 酢酸ェチル Zへキサン) にて精製し、 化合物 5を 1,4 - cis体と 1,4-trans 体の混合物として得た (187 mg, 80%) 。 この混合物を構成する立体異性体の 比率は約 2:1であった。 1,4-cis体と 1,4-trans体のいずれが主生成物かは判断 できなかった。 尚、 C-1位は擬似不斉であり、 1,4- cis体と 1, 4- trans体は互い に achiral diastereoisome こなる。
5a (主生成物、 高極性) : ¾腿 (CDC13) δ: 0.04 (6 Η, s, Si -Me x 2), 0.07, 0.08 (each 3 H, Si -Me x 2), 0.84, 0.90 (each 9 H, s, Si-tBu x 2), 1.60〜1.73 (2 H, m), 1.89 (1 H, m), 1.98 (1 H, m), 2.43 (1 H, s, OH), 3.57 (1 H, t, J=3.2 Hz, H-4), 3.69(1 H, tt, J=11.4, 4.1 Hz, H-l), 3.96 (1 H, ddd, J=11.6, 4.8, 3.2 Hz), 4.10 (1 H, m), 4.54 (2 H, s, PhCH2) , 7·30〜7·35 (5 H, m, arom-H).
5b (マイナ一生成物、 低極性) : ¾ NMR (CDC13) δ 0.057, 0.076, 0.077, 0.091 (each 3 H, Si -Me x 4), 0.86, 0.90 (each 9 H, s, Si-tBu x 2), 1·36〜1·47 (2 H, m), 2.01 (1 H, d, J=5.7 Hz, OH), 2.13 (1 H, m), 2.22 (1 H, m), 3.28 (1 H, ddd, J=8.7, 5.7, 2.9 Hz, H-4), 3.75 (2 H, m, H-l, 5), 4.13 (1 H, m, H-3), 4.50, 4.54 (each 1 H, d, J=11.8 Hz, PhCH2) , 7.30〜7.35 (5 H, m, arom-H) .
混合物の MS m/z (%): no M+, 409 (6), 319 (2), 301 (17), 277 (6), 259 (4), 211 (9), 169 (31), 91 (100).
(実施例 4) (2R, 6R) -2, 6-ビス- [ (t -プチルジメチルシリル)才キシ] - 4 - [ ( ベンジル)ォキシ] -シクロへキサノン (化合物 6)
-78 °Cに冷却した二塩ィ匕ォキサリル (384 L, 4.40 匪 ol) の無水塩化メチ レン (5 mL) 溶液に、 無水塩化メチレン (2.5 mL) に溶解したジメチルスルホ キシド (621 L, 8.75 mmol) を加え、 5分間撹拌した。 この冷却撹拌溶液に、 化合物 5 (1.71 g, 3.66 mmol, 5a:5b=約 2: 1の異性体の混合物) の無水塩化メ チレン (10 mL) 溶液を加え、 15分間撹拌した。 反応混合物にトリェチルアミ ン (2.55 mL, 18.3 腿 ol) を加え、 反応温度を - 78 °Cから室温に約 1.5時間か けて徐々に上昇させながら撹拌した。 反応液を氷水に投入し、 塩化メチレンに て抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥後 、 溶媒留去した。 残渣はシリカゲルカラムクロマトグラフィー (30 g、 5¾酢 酸ェチル Zへキサン) にて精製し、 化合物 6 (1.69 g, 99%) を単一化合物とし て得た。
Ή NMR (CDC13) δ: 0.02, 0.03, 0.06, 0.12 (each 3 H, Si -Me x 4), 0.86, 0.90 (each 9 H, s, Si-tBu x 2), 1.74 (2 H, m), 2.31 (1 H, m), 2.51 (1 H, m), 4.12 (2 H, m), 4.55, 4.60 (each 1 H, d, J=11.7 Hz, PhCH2) , 4.73 (1 H, dd, J=12.1, 6.4 Hz), 7.27〜7.35 (5 H, m, arom-H).
MS m/z (¾): no M+, 449 (2), 407 (27), 299 (21), 275 (5), 91 (100).
(実施例 5) (3R,5R)- 3, 5-ビス- [(t -プチルジメチルシリル)ォキシ ]-4 -メ チレン-シクロへキサノール ベンジルエーテル (化合物 7)
6
0 °Cに冷却したメチルトリフエニルホスホニゥムブロミド (1.55 g, 4.34 mmol) の無水テトラヒドロフラン (10 mL) 懸濁液に、 n-ブチルリチウム ( 2.71 mL, 4.34 mmol, 1.6 Mへキサン溶液) を加え 10分撹拌し、 更に室温にて 1 時間撹拌した。 得られた黄赤色混合物に、 化合物 6 (1.0 g, 2.15 匪 ol) の無 水テトラヒドロフラン (10 mL) 溶液を約 20分かけて加えた。 反応混合物は 0 °Cで 1時間、 室温で 17時間撹拌した後、 氷水中に投入し酢酸ェチルにて抽出し た。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシウムにて乾燥後、 溶媒 留去した。 残渣はシリカゲルカラムクロマトグラフィー (5 g、 2%酢酸ェチル Zへキサン) にて精製し、 化合物 7 (978.5 mg, 98%, 単一化合物) を得た。
Ή NMR (CDC13) δ: 一 0.01, 0.04, 0.06, 0.07 (each 3 H, Si -Me x 4), 0.84, 0.92 (each 9 H, s, Si-tBu x 2), 1.35〜1.46 (2 H, m), 2.18 (1 H, m), 2.35 (1 H, m), 3.94 (1 H, tt, 1=11.2, 4.2 Hz, H - 1), 4·40〜4.46 (2 H, m, H - 1, 3), 4.54, 4.56 (each 1 H, d, J=11.9 Hz, PhC¾) , 4.84 (1 H, m, OCH), 5.03 (1 H, t, J=2.0 Hz, C=CH) , 7.27〜7.35 (5 H, m, arom-H) .
MS m/z (%) no M+, 405 (84), 355 (2), 313 (35), 297 (15), 273 (20), 223 (18), 165 (22), 91 (100).
(実施例 6) (3, 6- c is) _および(3, 6- trans)- 6-ベンジルォキシ- 4, 8-ビス - [(t-プチルジメチルシリル)ォキシ] -卜ォキサ -スピロ [2.5]オクタン (化合物
0 °Cに冷却した化合物 7 (191.4 mg, 0.414匪 ol) の塩化メチレン (2 mL)
溶液に、 m-クロ口過安息香酸 (106.8 mg, 0.619 mmol) を加えた。 反応混合物 を 0 °Cにて 2時間、 室温にて 16時間撹拌した後、 塩化メチレンを加えた。 塩化 メチレン層は 5% NaHC03および飽和食塩水にて洗浄し、 無水硫酸マグネシウムで 乾燥後、 溶媒留去した。 残渣はシリカゲルカラムクロマトグラフィー (8 g、 2¾→5¾酢酸ェチルノへキサン) にて精製し、 酢酸ェチル含有へキサン溶出 部より化合物 8b (19.0 mg) および 5%酢酸ェチル含有へキサン溶出部より化 合物 8a (178.0 mg) を得た。 合計収率は 99%であった。 3, 6- cis体と 3.6- trans 体のいずれが主生成物かは判断できなかった。
8a (高極性異性体) :¾ NMR (CDC13) δ: 0.01, 0.02, 0.05, 0.06 (each 3 H, s, Si -Me x 4), 0.85, 0.88 (each 9 H, s, Si-tBu x 2), 1.66〜1.75 (2 H, m), 2.05 ( 1 H, m), 2. 4 (1 H, m), 2.62, 3.03 (each 1 H, d, J=5.3 Hz, CH20) , 3.53 (1 H, t, J=3.0 Hz), 3.84 (1 H, tt, J=ll.1, 4.1 Hz, H - 1), 4.21 (1 H, dd, J-11.6, 4.4 Hz), 4.56 (2 H, s, PhCH2) , 7.27〜7.35 (5
H, m, arom-H) .
MS m/z (¾): no M+, 421 (3), 391 (2), 313 (18), 91 (100), 75 (74).
8b (低極性異性体) : ¾ NMR (CDC13) δ: 0.02 (6 Η, s, Si -Me x 2), 0.04, 0.07 (each 3 H, s, Si -Me x 4), 0.856, 0.863 (each 9 H, s, Si-tBu x 2),
I.44〜1.55 (2 H, m), 2.18 ( 1 H, m), 2.35〜2.41 (1 H, m), 2.38, 2.96 (each 1 H, d, J=5.6 Hz, CH20) , 3.55 (1 H, m), 3.93 (1 H, tt, J=11.2, 4.2 Hz, H-l), 4.18 (1 H, dd, J=11.5, 4.8 Hz), 4.54, 4.57 (each 1 H, d, J=11.8 Hz, PhCH2) , 7.27〜7.35 (5 H, m, arom-H).
MS m/z (%): no M+, 421 (7), 391 (3), 313 (11), 91 (96), 75 (100).
(実施例 7) (3,6- 3)_ぉょび(3,6_ &)13)-4,8-ビス-[(卜ブチルジメチル シリル)ォキシ] -卜ォキサ -スピロ [2.5]オクタン- 6-オール (化合物 9 )
エポキシ体である化合物 8a (216.8 mg, 0.453腿 ol) の酢酸ェチル (2 mL) 溶液に、 10%パラジウム付活性炭 (43.4 mg)を加え、 常温常圧下で水素ガスと激 しく 1時間撹拌した。 反応混合物をセライト濾過し、 エタノールおよび酢酸ェ チルにて洗浄した後、 ろ液をまとめて溶媒留去した。 残渣をシリカゲルカラム クロマトグラフィー(8 g、 8% 酢酸ェチル /へキサン)にて精製し、 化合物 9a (176.0 mg, 定量的)を得た。
エポキシ体である化合物 8b (24.6 mg, 0.0514讓 ol) を用いて前述と同様に 水素接触還元反応およびカラム精製を行ない、 脱べンジル体である化合物 9b ( 17.2 mg, 86 %) を得た。
9a: ¾ NMR (CDC13) δ: 0.05, 0.07, 0.10, 0.15 (each 3 H, s, Si -Me x 4), 0.87, 0.91 (each 9 H, s, Si-tBu x 2), 1.62 (1 H, m), 1.80 (1 H, t, J=14.1, 2.9 Hz), 2.08, 2.27 (each 1 H, m), 2.50, 2.95 (each 1 H, d, J=5.5 Hz, CH20) , 3.80 (1 H, m), 4.14 (1 H, m), 4.40 (1 H, m).
MS m/z (%): no M+, 331 (25), 313 (52), 301 (16), 199 (63), 181 (46), 75 (100).
9b : ¾ NMR (CDC13) δ: 0.05, 0.07, 0.08, 0.09 (each 3 H, s, Si -Me x 4), 0.88, 0.89 (each 9 H, s, Si-tBu x 2), 1.68, 1.82, 1.98, 2.14 (each 1 H, m, H-2, 6), 2.71 (1 H, m, CH20) , 2.82 (1 H, d, J=5.4 Hz, CH20) , 4.00 (2 H, m), 4.21 (1 H, m).
MS m/z (%): no M+, 331 (20), 313 (42), 301 (14), 199 (72), 181 (39), 73 (100).
(実施例 8) (4R, 8R) -4, 8-ビス- [ (t-ブチルジメチルシリル)才キシ] -トォ キサ-スピロ [2.5]オクタン- 6-オン (化合物 10 )
-78 °Cに冷却した二塩化ォキサリル(51 L, 0.585 匪 ol)の無水塩化メチレ
ン(0.5 mL)溶液に、 ジメチルスルホキシド(83 L, 1.170 mmol)の無水塩化メ チレン(0.2 mL)溶液を加え 5分間撹拌した後、 化合物 9 (化合物 9a: 9b==約 10: 1の混合物) (190.6 mg, 0.490 醒 ol)の無水塩化メチレン α.3 mL)溶液を加え た。
-78 °Cで 15分間撹拌した後、 トリェチルァミン(341 L, 2.445 mmol)を加え た。 反応温度が- 78 °Cから室温になるまで約 1.5時間撹拌した。 反応混合物に 氷水を加え、 塩化メチレンにて抽出した。 有機層を飽和食塩水にて洗浄、 無水 硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトダラ フィー(8 g、 5¾酢酸ェチル Zへキサン)にて精製し、 化合物 10を単一化合物と して得た(188.5 mg, 99%) 。
¾ NMR (CDC13) δ: 0.047, 0.055, 0.061, 0.086 (each 3 H, s, Si -Me x 4), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 2.45 (1 H, ddd, J=14.4, 7.9, 1.1 Hz), 2.57 (1 H, ddd, J=14.2, 6.2, 1.9 Hz), 2.67 (1 H, ddd, J=14.2, 3.9, 1.1 Hz), 2.79 (1 H, ddd, J=14.4, 4.9, 1.1 Hz), 2.80, 3.02 (each 1 H, d, J-5.3 Hz, CH20), 4.03 (1 H, d, J=6.2, 3.9 Hz), 4.21 (1 H, dd, J=7.9, 4.9 Hz), 4.40 (1 H, m).
MS m/z (%): no M+, 329 (31), 313 (10), 299 (9), 197 (20), 75 (100).
(実施例 9) [(aS*,4R,8R)-および [(aR*, 4R, 8R)- [4, 8-ビス _[(t -プチルジ メチルシリル)ォキシ] - 1-ォキサ -スピロ [2.5]ォクチ- 6 -ィリデン] -酢酸 メチ ルエステル (化合物 1 1)
-78 °Cに冷却したジイソプロピルアミン(115 M L, 0.82 mmol)の無水テトラ ヒドロフラン(1 mL)溶液に、 n-ブチルリチウム (513 zL, 0.82 腿 ol, 1.6 Mへ キサン溶液)を加え 15分撹拌した後、 (トリメチルシリル) 酢酸メチル(135/x
L, 0.82 匪 ol)を加えた。 10分間撹拌後、 化合物 10 (158.8 mg, 0.41 mmol)の 無水テトラヒドロフラン α·2 mL)溶液をゆっくり加え、 -78 °Cで 1時間撹拌し た。 反応混合物に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出し た。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した 。 残渣をシリカゲルカラムクロマトグラフィー(8 g、 2%酢酸ェチル Zへキサ ン)にて精製し、 化合物 11を二種の立体異性体の混合物として得た(172.1 mg, 95¾)。 この混合物を構成する異性体の比率は約 3: 1であったが、 aS*, 4R, 8R体 と aR*, 4R, 8R体のいずれが主生成物かは判断できなかった。
混合物の NMRデータ
11a (主生成物) : ¾ NMR (CDC13) δ: 0.03〜0.08 (12 Η, Si -Me x 4), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 2.41 (1 H, d, J=13.2, 6.7 Hz), 2.47 (1 H, m), 2.74 (1 H, d, J=5.4 Hz, CH20), 2.80 (1 H, dd, J=13.7, 7.6 Hz), 2.90 (1 H, d, J=5.4 Hz, CH20) , 3.40 (1 H, dd, J=13.7, 4.0 Hz), 3.70 (3 H, s, OMe), 3.91 (2 H, m, H-3, 5), 5.76 (1 H, s, OCHC0).
lib (マイナ一生成物) : lH NMR (CDC13) 6: 0.03〜0.08 (12 H, Si -Me x 4), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 2.28 (1 H, m), 2.60 (1 H, dd, J=13.2, 4.6 Hz), 2.67 (1 H, d, J=5.4 Hz, CH20) , 2.72 (1 H, m), 2.92 (1 H, d, J=5.4 Hz, C¾0), 3.48 (1 H, m), 3.69 (3 H, s, OMe), 3.80 (1 H, dd, J=5.9, 3.2 Hz), 4.04 (1 H, dd, J=8.6, 4.6 Hz), 5.81 (1 H, s, OCHC0).
混合物の MS m/z (%) : no M+, 411 (3), 385 (86), 355 (14), 353 (47), 325 (8), 293 (30), 280 (24), 253 (65), 223 (13), 221 (20), 73 (100).
(実施例 10) [(aS*,4R,8R)-および [(aR*, 4R, 8R)-2-[4, 8-ビス - [(t_プチ ルジメチルシリル)ォキシ] -卜ォキサ -スピロ [2.5]ォクチ- 6 -ィリデン]-ェタノ ール (化合物 12 )
- 78 °Cに冷却したァリルエステル体である化合物 11 (190.3 mg, 0.43 mmol, lla:llb=約 3 : 1の混合物)の無水トルエン(2 mL)溶液に、 水素化ジ- iso -プチ ルアルミニウム(1.07 mL, 1.07匪 ol, 1.0 Mトルエン溶液)を加え、 1時間撹拌 した。 還元剤を酒石酸ナトリウムカリウム水溶液により分解した後、 反応混合 物を氷水中に移し、 酢酸ェチルにて抽出した。 有機層は水洗、 飽和食塩水にて 洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムク 口マトグラフィ一(8 g、 10%→15%酢酸ェチル Zへキサン)にて精製し、 10%酢 酸ェチル含有へキサン溶出部より化合物 12を二種の立体異性体の混合物として 得た a67.6 mg, 94%) この混合物を構成する異性体の比率は約 3 :1であった が、 aS*,4R, 8R体と aR*, 4R, 8R体のいずれが主生成物かは判断できなかった。 15% 酢酸ェチル含有へキサン溶出部より開環体 12' (11 mg, 6%,—つの立体異 性体のみ単離された) を得た。
混合物の NMRデータ
12a (主生成物) : ^ NMR (CDC13) δ: 0.034 (3Η, s, Si -Me), 0.056 (6 H, s, Si -Me x 2), 0.068 (3 H, s, Si -Me ), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 1.15 (1 H, t, J=5.6 Hz, OH), 2. 2 (1 H, dd, J=13.6, 6.9 Hz), 2.32 (1 H, dd, J=13.1, 6.9 Hz), 2.39 (1 H, dd, J-13.1, 3.8 Hz), 2.61 (1 H, dd, J=13.6, 4.1 Hz), 2.74, 2.84 (each 1 H, d, J=5.4 Hz, CH20), 3.83〜3.86 (2 H, m, H-3, 5), 4.13〜4.20 (2 H, m, CH20H) , 5.59 (1 H, t, J=7.0 Hz, C=CHC0).
12b (マイナ一生成物) : ¾ NMR (CDC13) δ: 0.026, 0.056, 0.063, 0.09 (each 3 H, s, Si -Me x 4), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 1.22 (1 H, dd, J=6.5, 4.7 Hz, OH), 2.16, 2.21, .53 (each 1 H, m), 2.57 (1 H, d, J=5.5 Hz, CH20) , 2.60 (1 H, m), 2.93 (1 H, d, J=5.5 Hz, CH20) , 3.71 (1 H, dd, J-5.1, 3.1 Hz), 4.00 (1 H, dd, J=9.5, 4.6 Hz), 4.08-
4.20 (2 H, m, CH20H) , 5.67 (1 H, t, J=6.9 Hz, OCHCO).
混合物の MS m/z (%) : no M+, 357 (13), 339 (100), 327 (4), 309 (14), 265 (20), 235 (26), 225 (20), 207 (38), 195 (15), 177 (37), 75 (100).
12' : Ή N R (CDC13) δ: 0.07 (6H, s, Si -Me x 2), 0.08, 0.10 (each 3 H, Si -Me x 2), 0.87, 0.91 (each 9 H, s, Si-tBu x 2), 1.18 (3 H, s, Me), 2.19〜2.38 (4 H, i), 2.46 (1 H, d, J=13.6 Hz), 3.71 (1 H, dd, J=9.6, 4.9 Hz), 3.78 (1 H, dd, J=4.3, 3.4 Hz), 4.09〜 4.16 (2 H, m, CH20H), 5.50 (1 H, t, J=7.0 Hz, C=CHC0) .
(実施例 11) [(aS*,4R,8R)- および [(aR*, 4R, 8R)_4, 8-ビス- [(t-ブチル ジメチルシリル)ォキシ] -6- [2- (ジフエニル-ホスフイノィル) -ェチリデン] -1 - ォキサ -スピロ [2.5]オクタン (化合物 13 )
0 °Cに冷却したァリルアルコール体である化合物 1 2 (167.6 mg, 0.404 腿 ol、 12a:12b=約 3 : 1の混合物)の無水テトラヒドロフラン(2 mL)溶液に、 n-ブチルリチウム(278 L, 0.445 讓 ol, 1.6 Mへキサン溶液)および塩化 p -ト ルエンスルホニル(84.7 mg, 0.445 腿 ol)の無水テトラヒドロフラン(0.3 mL) 溶液を順次加え、 5分間撹拌した。 別の容器にジフエニルホスフィン(141 xL, 0.810 匪 ol)の無水テトラヒドロフラン(1 mL)溶液を作り、 0 撹拌下で n -ブ チルリチウム(505 L, 0.808 匪 ol, 1.6 Mへキサン溶液)を加えたところ濃赤 色になった。 0 °cに冷却した本濃赤色溶液を、 上記のトシル体溶液にゆっくり 滴下し、 反応混合物が赤色を呈するまで加えた。 更に 0 :にて 30分間撹拌し、 水(IOO L)を加え反応を止めた。 反応混合物から溶媒留去後、 残渣を塩化メチ レン(4 mL)に溶解し、 10% 過酸化水素水(6 mL)を加え 0 °Cにて 1時間撹拌した 。 反応液に 2N亜硫酸ナトリウムを加え、 塩化メチレンにて抽出した。 有機層は
水、 飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣を シリカゲルカラムクロマトグラフィー(8 g、 30% 酢酸ェチル Zへキサン)にて 精製し、 化合物 13を二種の立体異性体の混合物として得た(187.0 mg, 77%) 0 この混合物を構成する異性体の比率は約 3 :1であったが、 aS*,4R,8R体と aR* , R, 8R体のいずれが主生成物かは判断できなかった。
混合物の NMRデータ
13a (主生成物) : ¾ NMR (CDC13) δ: -0.01- 0.06 (12 H, Si -Me x 4), 0.83, 0.84 (each 9 H, s, Si-tBu x 2), 1.83 (1 H, m), 2.25〜2.40 (3 H, m), 2.60, 2.82 (each 1 H, d, J=5.5 Hz, CH20) , 3.05〜 3.24 (2 H, m, CH2P0) , 3.70 (1 H, dd, J=5.8, 3.6 Hz), 3.83 (1 H, dd, J=8.5, 4.4 Hz), 5.36 (1 H, i, C=CHC0) , 7.44〜7.77 (10 H, m, arom H) .
13b (マイナ一生成物) : ¾ NMR (CDC13) δ: _0.01〜 0.06 (12 Η, Si -Me x 4), 0.82, 0.86 (each 9 H, s, Si-tBu x 2), 1.93, 2.10, 2.25, 2.46 (each 1 H, m, H-2, 6), 2.55, 2.84 (each 1 H, d, J=5.5 Hz, CH20) , 3.05〜3.24 (2 H, m, CH2P0) , 3.65 (1 H, dd, J=5.6, 3.2 Hz), 3.89 (1 H, dd, J=8.7, 4.6 Hz), 5.36 (1 H, m, OCHC0), 7.44〜7.77 (10 H, m, arom H).
混合物の MS m/z (¾) : no M+, 541 (100), 511 (8), 449 (39), 409 (86), 201 (26), 75 (44). (実施例 12) 1 a-[(t-プチルジメチルシリル)ォキシ ]-2 β, 2' -エポキシ-
25 - [ (トリエチルシリル)ォキシ ]-19 -ノルビタミン D3 t-プチルジメチルシリル エーテルおよび 1 ブチルジメチルシリル)ォキシ ]-2 ,2'-エポキシ- 25- [ (トリエチルシリル) ォキシ ]-19-ノルビタミン D3 t -プチルジメチルシリルェ 一テル (化合物 15 a, b)
-78 °Cに冷却した化合物 13 (100.1 mg, 0.167匪 ol, 13a:13b=約 3 : 1の混 合物)の無水テトラヒドロフラン(1.5 mL)溶液に、 n-ブチルリチウム(104 L, 0.167匪 ol, 1.6 Mへキサン溶液)をゆっくり加えた。 深いオレンジ色の溶液の 溶液が得られた。 15分間撹拌後、 グランドマンケトン体である化合物 14a (44.0 mg, 0.114 mmol)の無水テトラヒドロフラン(0.5 mL)溶液を加えた。 - 78 で 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェ チルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥 、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ一(7 g、 2¾→10¾ →40%酢酸ェチル /へキサン)にて精製し、 2%酢酸ェチル含有へキサン溶出部 より化合物 15を二種の立体異性体の混合物として得た(58.4 mg, 68%)。 この混 合物を構成する異性体の比率は約 3 :,1であった。 実施例 13の結果から 2 ;3, 2'- エポキシ体(15a)が主生成物と考えられた。 10% 酢酸ェチル含有へキサン溶出 部より未反応の化合物 14a (14.0 mg, 32%)および 40%酢酸ェチル含有へキサン 溶出部より化合物 13 (34.0 mg, 34»を回収した。
混合物の NMRデータ
15a (主生成物) : ΐ NMR (CDC13) δ: 0.02 (3 Η, s, Si -Me), 0.055 (3 H, s, Si -Me x 2), 0.065 (6 H, s Si -Me), 0.55 (3 H, s, H-18), 0.56 (6 H, q, SiCH2 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.95 (12 H, t, J=7.9 Hz, SiCH2CH3 x 3, overlapped with H - 21), 1.19 (6 H, s, H-26, 27), 2.27〜2.38 (2 H, m), 2.42 (1 H, dd, J=13.1, 3.6 Hz), 2.66 (1 H, dd, J=13.3, 3.4 Hz), 2.74, 2.82 (each 1 H, d, J=5.5 Hz, CH20) , 3.81 (1 H, dd, J=7.7, 3.9 Hz), 3.88 (1 H, dd, J=7.0, 3.8 Hz), 5.82 ( 1 H, d,
J=ll.1 Hz, H-7), 6.21 (1 H, d, J=ll.l Hz, H- 6) ·
15b (マイナー生成物) : ¾ NMR (CDC13) δ: 0.02 (3 H, s, Si -Me), 0.064 (6 H, s, Si -Me x 2), 0.08 (3 H, Si -Me), 0.55 (3 H, s, H— 18), 0.56 (6 H, q, SiCH2 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.95 (12 H, t, J=7.9 Hz, SiCH2CH3 x 3, overlapped with H-21), 1.19 (6 H, s, H-26, 27), 2.57, 2.92 (each 1 H, d, J=5.5 Hz, CH20) , 3.68 (1 H, 1) , .04 (1 H, dd, J-9.5, 4.5 Hz, H-l), 5.82 ( 1 H, d, J=12.1 Hz, H-7), 6.27 (1 H, d, J-12.1 Hz, H-6).
(実施例 13) 1 a,25 -ジヒドロキシ -2)3, 2' _エポキシ - 19 -ノルビタミン D3 ( 化合物 Y I— 1 a) 、 および 1 ,25-ジヒドロキシ - 2θ!,2' -エポキシ- 19-ノル ビタミン D3 (化合物 Y 1— 1 b) 、 ぉょび1ひ,2)3,25-トリヒドロキシ-20!-フ ルォロメチル- 19 -ノルビ夕ミン D3 (化合物 Y I - 2 a) 、 および 1 α, 2 α, 25-ト リヒドロキシ -2|3-フルォロメチル -19-ノルビタミン D3 (化合物 Y I - 2 b)
トリシリルエーテル体である化合物 15 (58.4 mg, 0.075 讓 ol, 15a: 15b = 約 3 : 1の混合物)の無水テトラヒドロフラン(1 mL)溶液に、 フッ化テトラプチ ルアンモニゥム(301 L, 0.301 mmol, 1· 0 Mテトラヒドロフラン溶液)を加え 、 0 にて 30分間、 次いで室温にて 7時間撹拌した。 反応液に氷水を加え、 酢
酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄、 無水硫酸マグネシウム 乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー(5 g、 60¾ →70%酢酸ェチル Zへキサン)にて精製し、 60 酢酸ェチル含有へキサン溶出部 より化合物 YI- 2aと YI - 2bの混合物 (2.6 mg, 8%)、 70% 酢酸ェチル含有へキサ ン溶出部より化合物 YI- laと YI- lb の混合物 mg, 60%)を得た。
化合物 YI- laと YI_lbを含む混合物を HPLC (LiChrosorb Si 60, 250 x 10 匪, へキサン:塩化メチレン: 2—プロパノール =50 : 50 : 8)にて分離精製 し、 化合物 YI - la (11.0 mg) および化合物 YI_lb (2.6 mg) を得た。 化合物 YI- 2aと YI-2bを含む混合物は HPLC (Y C-Pack ODS-AM SH-342-5, 150 x 20 匪, 20%水/メタノール) にて分離精製し、 化合物 YI- 2a (0.9 mg) および化合物 YI-2b (0.3 mg) を得た。
ΥΙ-la (主生成物) : Ή NMR (CDC13) δ: 0.55 (3 Η, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.30 (1 H, dd, J=13.5, 8.7 Hz, H-10), 2.40 (1 H, dd, J=13.7, 6.1 Hz, H-4), 2.60 (1 H, dd, J=13.7, 3.5 Hz, H-4), 2.81 (1 H, m, H-9), 2.84 (1 H, d, J=4.7 Hz, CH20) , 2.96 (1 H, dd, J=13.5, 4.3 Hz, H-10), 3.08 (1 H, d, J=4.7 Hz, CH20) , 3.80 (1 H, m, H— 3), 3.99 (1 H, m, H-l), 5.86 ( 1 H, d, J=11.2 Hz, H-7), 6.39 (1 H, d, J=11.2 Hz, H-6).
YI - lb (マイナ一生成物) : ¾ NMR (CDC13) <5: 0.56 (3 H, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.30 (1 H, dd, J=13.7, 6.2 Hz, H-4), 2.36 (1 H, dd, J=13.3, 8.6 Hz, H-10), 2.71 (1 H, dd, J=13.7, 3.6 Hz, H-4), 2.81 (1 H, m, H-9), 2.86 (1 H, dd, J=13.3, 4.3 Hz, H-10), 2.94, 2.99 (each 1 H, d, J=4.7 Hz, CH20), 3.81 (1 H, m, H-3, W/2 = 12 Hz), 3.91 (1 H, m, H-l, W/2 = 20 Hz), 5.87 ( 1 H, d, J=11.2 Hz, H-7), 6.37 (1 H, d, J=11.2 Hz, H-6).
MS m/z (¾) : 432 (23, M+), 414 (20), 396 (23), 378 (52), 303 (12), 267 (68), 135 (100).
YI - 2a (主生成物) : ¾ NMR (CDC13) δ 0.53 (3 H, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.45 (1 H, dd, J=13.5,
8.9 Hz, H-4), 2.49 (1 H, dd, J=13.5, 5.4 Hz, H— 4), 2.55 (1 H, dd, J=14.1, 5.8 Hz, H-10), 2.63 (1 H, br. s, OH), 2.69 (1 H, dd, J=14.1, 2.9 Hz, H-10), 2.80 (1 H, m, H-9), 3.86 (1 H, m, H-3), 3.97 (1 H, m, H-l), 4.71, 4.77 (each 1 H, dd, J=47.6, 9.7 Hz, CH2F) , 5.80 ( 1 H, d, J-11.2 Hz, H-7), 6.41 (1 H, d, J-11.2 Hz, H-6) .
19F NMR (CDC13) δ -240.6 (t, J=47.6 Hz).
MS m/z (¾) : 452 (75, M+), 434 (100), 414 (34), 396 (19), 378 (38), 323 (54), 305 (19), 303 (21), 287 (17), 285 (18), 267 (22), 228 (24), 133 (82).
YI-2b (マイナー生成物) : ¾ NMR (CDCI3) δ 0.55 (3 Η, s, H-l 8), 0.94 (3 H, d, J=6.4 Hz, H - 21), 1.22 (6 H, s, H—26, 27), 2.16 (1 H, dd, J=14.1, 4.0 Hz, H-4), 2.27 (1 H, br. t, J=〜12 Hz, H-10), 2.57 (1 H, br. s, OH), 2.79 (1 H, m, H-9), 2.84 (1 H, m, H-4), 2.88 (1 H, dd, J=13.2, 4.9 Hz, H-10), 3.77 (1 H, m, H-l, W/2 = 20 Hz), 3.95 (1 H, m, H-3, W/2=12 Hz), 4.69, 4.77 (each 1 H, dd, J=47.6, 9.6 Hz, CH2F) , 5.86 ( 1 H, d, J=11.3 Hz, H-7), 6.29 (1 H, d, J=11.2 Hz, H-6).
1F NMR (CDCI3) δ: -240.2 (t, J=47.6 Hz).
MS m/z (¾) : 452 (74, M+), 434 (100), 414 (35), 396 (16), 378 (33), 323 (50), 305 (17), 303 (20), 287 (15), 285 (16), 267 (18), 228 (24), 133 (74).
(実施例 14) 1 一 [ (t -プチルジメチルシリル)ォキシ ]-23, 2 '-エポキシ - 25- [(メトキシメチル)ォキシ ]-19-ノルビタミン D
3 t -プチルジメチルシリルェ 一テルおよび 1 - [(t -プチルジメチルシリル)ォキシ ]- 2ひ,2' -エポキシ - 25- [( メトキシメチル)ォキシ ]-19_ノルビタミン D
3 t-プチルジメチルシリルエーテル (化合物 1 6 a, b)
13
-78 °Cに冷却した化合物 13 (185.1 mg, 0.31 匪 ol、 13a: 13b=約 3 : 1の混 合物)の無水テトラヒドロフラン(1 mL)溶液に、 n_ブチルリチウム Q93 L, 0.31 mmol, 1.6 Mへキサン溶液)を加えた。 生じた黄赤色の混合物を 30分間撹 拌した後、 グランドマンケトン体である化合物 14b (66.8 mg, 0.206 mmol)の 無水テトラヒドロフラン(1.2 mL)溶液を加えた。 -78 °Cで 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルで抽出した。 有機層 は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ リカゲルカラムクロマトグラフィー(9 g、 3¾→12¾→40¾酢酸ェチルノへキサ ン)にて精製し、 3%酢酸ェチル含有へキサン溶出部より化合物 16を二種の立体 異性体の混合物(96.6 mg, 67%)として得た。 この混合物を構成する異性体の比 率は約 5 : 1であった。 また、 12% 酢酸ェチル含有へキサン溶出部より未反応の 化合物 14b (22.0 mg, 33%)、 40%酢酸ェチル含有へキサン溶出部より化合物 13 (43.4 mg、 23%)を、 回収した。
混合物の NMRデータ
16a (主生成物) : ¾ NMR (CDC13) δ: 0.02, 0.055 (each 3 H, s, Si -Me), 0.065 (6 H, Si -Me x 2) , 0.55 (3 H, s, H - 18), 0.86, 0.87 (each 9 H, s, Si-tBu x 2), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.28〜2.37 (2 H, m), 2.42 (1 H, dd, J=13.1, 3.7 Hz), 2.65 (1 H, dd, J=13.7, 3.7 Hz), 2.74 (1 H, d, J=5.5 Hz, CH20) , 2.80 (1 H, m, H-9), 2.82 (1 H, d, J=5.5 Hz, CH20) , 3.37 (3 H, s, OMe), 3.81 (1 H, dd, J=7.1, 3.9 Hz, H-3), 3.88 (1 H, dd, J=7.0, 3.9 Hz, H-l), 4.71 (2 H, s, 0CH20), 5.82 (1 H, d, J=11.0 Hz, H-7), 6.21 (1 H, d, J=11.0 Hz, H-6).
16b (マイナ一生成物) : ¾ NMR (CDC13) δ: 0.02〜0.08 (12 Η, Si -Me x 4) , 0.55 (3 Η, s, Η - 18), 0.86, 0.87 (each 9 Η, s, Si-tBu x 2), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.57 (1 H, d, J=5.5 Hz, CH20) , 2.80 (1 H, m, H-9), 2.92 (1 H, d, J=5.5 Hz, CH20) , 3.37 (3 H, s, OMe), 3.68 (1 H, m, H - 3), 4.04 (1 H, dd, J=9.1, 4.9 Hz, H-l), 4.71 (2 H, s, OCH20) , 5.82 (1 H, d, J=11.2 Hz, H_7), 6.28 (1 H, d, J=11.0 Hz, H-6).
(実施例 15) 1ひ-ヒドロキシ- 2 ]3, 2' -エポキシ- 25- [(メトキシメチル)ォ キシ] - 19-ノルビタミン D3および 1ひ-ヒドロキシ- 2 α, 2'-エポキシ- 25- [(メトキ シメチル)ォキシ ]_19-ノルビタミン D3 (化合物 17 a, b)
化合物 16 (34.4 mg, 0.049皿 ol, 16a:16b =約 5 : 1の混合物)の無水テトラ ヒドロフラン(1 mL)溶液に、 フッ化テトラプチルアンモニゥム(122 L, 0.122 腿 ol, 1.0 Mテトラヒドロフラン溶液)を加え、 0 にて 30分間、 室温にて 5時 間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食 塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲル カラムクロマトグラフィー(5 g、 50¾ 酢酸ェチル /へキサン)にて精製し、 化 合物 17を二種の立体異性体の混合物(21.5 mg, 92»として得た。 この混合物を 構成する異性体の比率は約 4 : 1であった。
混合物の匪 Rデータ
17a (主生成物) : ¾ NMR (CDC13) δ 0.55 (3 Η, s, H - 18), 0.93 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.32 (1 H, dd, J=13.5, 8.9
Hz), 2.40 (1 H, dd, J=13.8, 6.1 Hz), 2.62 (1 H, dd, J=13.8, 3.5 Hz), 2.80 (1 H, m, H-9), 2.85 (1 H, d, J=4.7 Hz, CH20) , 2.95 (1 H, dd, J=13.5, 4.5 Hz), 3.07 (1 H, d, J=4.7 Hz, CH20), 3.37 (3 H, s, OMe), 3.82 (1 H, m), 3.98 (1 H, m), 4.71 (2 H, s, OCH20) , 5.86 (1 H, d, J=ll.l Hz, H-7), 6.40 (1 H, d, J=ll.l Hz, H-6).
17b (マイナ一生成物) : ¾ NMR (CDC13) δ: 0.55 (3 H, s, H-18), 0.93 (3 H, d, J=6.4 Hz, H - 21), 1.22 (6 H, s, H-26, 27), 2.72 (1 H,m), 2.94, 2.99 (each 1 H, d, J=4.7 Hz, CH20) , 3.37 (3 H, s, OMe), 3.82 (1 H, m), 3.91 (1 H, m), 4.71 (2 H, s, OCH20) , 5.86 (1 H, d, J=ll.l Hz, H-7), 6.37 (1 H, d, J=ll.1 Hz, H-6).
(実施例 16) 1ひ,2)3-ジヒドロキシ - 2ひ-メチル -および 10;,2 _ジヒド 口キシ- 2/3-メチル -25- [(メトキシメチル)ォキシ ]-19-ノルビタミン D3 (化合物 18 a, b)
17 18 リチウムアルミニウムヒドリド(0.5 mg, 0.014 mmol)の無水ジェチルエーテ ル(0.25 mL)懸濁液中に、 化合物 17 (6.8 mg, 0.014 mmol, 17a: 17b=約 4 : 1 の混合物)の無水ジェチルエーテル(0.25 mL)溶液を加え室温にて 1時間撹拌し た。 反応混合物に酒石酸ナトリウムカリウム水を加え、 酢酸ェチルにて抽出し た。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した 。 残渣をシリカゲルカラムクロマトグラフィー(3 g、 60% 酢酸ェチル /へキサ ン)にて精製し、 化合物 18を二種の立体異性体の混合物 (4.7 mg, 69%)として 得た。 この混合物を構成する異性体の比率は約 4 : 1であった。
混合物の NMRデータ
18a (主生成物) : Ή NMR (CDC13) 6: 0.54 (3 H, s, H-18), 0.93 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 1.27 (3 H, s, 2 - Me); 2.37 (1 H, dd, J=14.4, 4.6 Hz), 2.53 (1 H, m), 2.79 (1 H, m), 2.94 (1 H, dd, J=13.6, 4.3 Hz), 3.37 (3 H, s, OMe), 3.74 (2 H, m, H-l, 3), 4.71 (2 H, s, 0CH20), 5.84 (1 H, d, J=11.2 Hz, H-7), 6.30 (1 H, d, J=11.2 Hz, H— 6).
18b (マイナー生成物) : ¾ NMR (CDC13) δ: 0.54 (3 Η, s, H-18), 0.93 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 1.30 (3 H, s, 2— Me), 2.17 (1 H, m), 2.67 (1 H, m), 3.37 (3 H, s, OMe), 3.74 (2 H, m, H-l, 3), 4.71 (2 H, s, 0CH20) , 5.82 (1 H, d, J=〜ll Hz, H-7), 6.34 (1 H, d, J二〜 11 Hz, H-6).
(実施例 17) 1 , 2 )3, 25-トリヒドロキシ- 2 α -メチル- 19 -ノルビ夕ミン D3 (化合物 Y I - 3 a) および 1 α, 2 α, 25 -トリヒドロキシ- 1 β -メチル -19-ノル ビタミン D3 (化合物 Y I一 3 b)
18 YI-3a Yl-3b
化合物 18 (10.5 mg, 0.022 mmoK 18a : 18b=約 4 : 1の混合物)の無水メタノ —ル(0.5 mL)溶液に、 カンファースルホン酸(10.1 mg, 0.044 mmol)を加え、 0 °Cにて 30分次いで室温にて 2時間撹拌した。 反応混合物を酢酸ェチルで希釈し た後、 有機層は 5%炭酸水素ナトリウム水溶液、 飽和食塩水にて洗浄、 無水硫酸 マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ —(3 g、 70% 酢酸ェチル Zへキサン)にて精製し、 化合物 YI-3aと YI- 3bを含む
混合物(8.8 mg, 93%)を得た。
化合物 YI - 3aと YI- 3bを含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20腿, 20%水/メタノール)にて分離精製し、 化合物 YI- 3a (5.2 mg) および 化合物 YI- 3b (0.7 mg) を得た。
YI-3a: ¾ NMR (CDC13) δ: 0.54 (3 H, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H - 21), 1.22 (6 H, s, H-26, 27), 1.27 (3 H, s, 2-Me) , 2.05 (1 H, m, H- 10), 2.36 (1 H, dd, J=14.4, 4.5 Hz, H-4), 2.55 (2 H, m, H-4, OH), 2.79 (1 H, m, H-9), 2.94 (1 H, dd, J=13.5, 4.4 Hz, H-10), 3.74 (2 H, m, H- 1, 3), 5.84 (1 H, d, J=11.2 Hz, H— 7), 6.30 (1 H, d, J=11.2 Hz, H - 6). YI - 3b: ¾ NMR (CDC13) δ: 0.55 (3 H, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 1.30 (3 H, s, 2-Me) , 2.17 (1 H, dd, J=13.6, 8.4 Hz, H-4), 2.30 (1 H, br s, OH), 2.49 (1 H, dd, J=14.2, 3.3 Hz, H-10), 2.62 (1 H, dd, J=U.2, 6.5 Hz, H-10), 2.67 (1 H, dd, J=13.6, 4.1 Hz, H-4), 2.79 (1 H, m, H-9), 3.73 (1 H, m, H-l, /2 = 12 Hz), 3,78 (1 H, m, H-3, W/2 = 18 Hz), 5.82 (1 H, d, J=11.2 Hz, H-7), 6.34 (1 H, d, J=11.2 Hz, H-6).
MS m/z (¾): 434 (75, M+), 416 (100), 401 (16), 398 (31), 380 (21), 362 (20), 305 (29), 287 (27), 269 (29), 251 (25), 135 (74). (実施例 18) 1 α - [ ( t-プチルジメチルシリル)ォキシ] -2/3-hドロキシ- 2 α-ェチル-および 1 α - [ (卜ブチルジメチルシリル)ォキシ] - 2 α -ヒドロキシ -2 3-ェチル -25- [(メトキシメチル)ォキシ] - 19-ノルビ夕ミン D3 t-ブチルジメチ ルシリルエーテル (化合物 19 a, b)
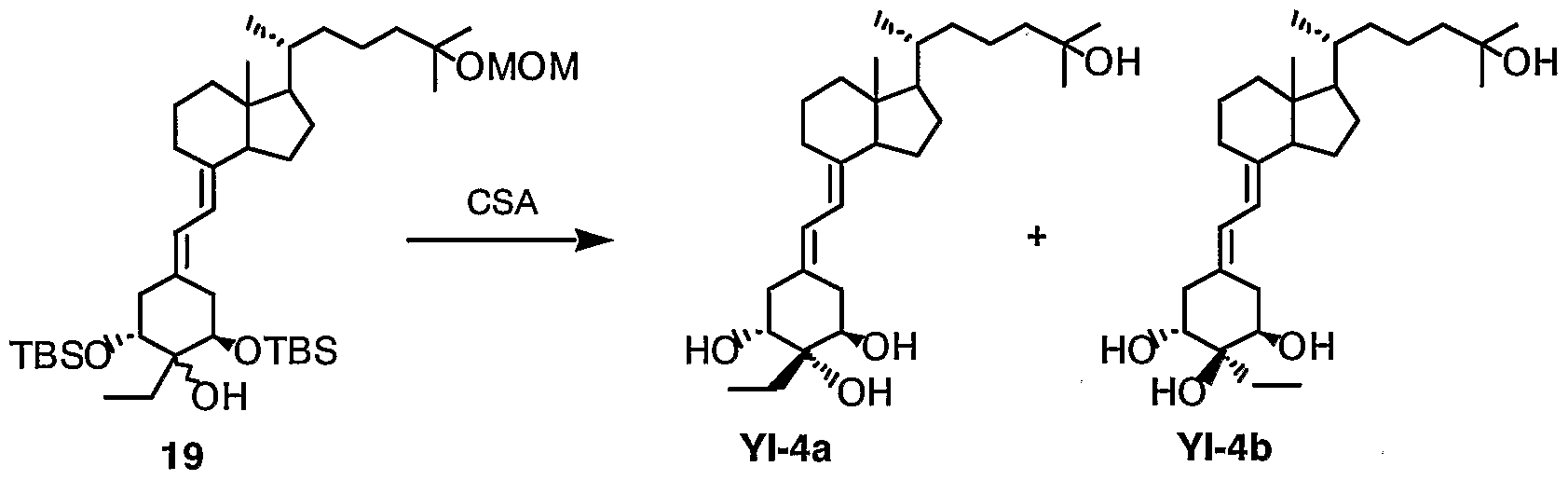
-40 °Cに冷却したシアン化銅 (I) (114.6 mg, 1.280 腿 ol) の無水ェ一テ ル (1.5 mL) 懸濁液に、 メチルリチウム溶液 (2.25 mL, 2.565 腿 ol, 1.14 M エーテル溶液) を加え 30分間撹拌した後、 無水エーテル (3 mL) に溶かした化 合物 16 (113.1 mg, 0.160 醒 ol、 本実施例では 16a: 16b =約 3 : 1の混合物を 使用) を添加した。 反応混合物を- 40 °Cにて 1時間撹拌した後、 0 °Cまで徐々 に温度を上昇させ、 更に 2時間撹拌した。 反応混合物に飽和塩化アンモニゥム 溶液を加え反応を停止後、 氷水に投入し、 酢酸ェチルにて抽出した。 有機層は 飽和食塩水にて洗浄、 無水硫酸マグネシウムにて乾燥、 溶媒を留去した。 残渣 をシリカゲルカラムクロマトグラフィー(ヮコ一ゲル C- 300、 10 g、 2%酢酸ェチ ル Zへキサン溶出部)にて精製し、 2 βヒドロキシ体と 2ひヒドロキシ体との 混合物である化合物 19 (90.3 mg, 78%) と、 未反応の化合物 16 (18.5 mg, 16¾ ) を得た。 化合物 19a, bを構成する異性体の比率は約 3 : 1であった。 クロマ トグラフィ一による分離の過程においては、 化合物 19aのみを含むフラクショ ンゃ化合物 19bのみを含むフラクションも得られたため、 これらを分取した。
19a (主生成物) : ¾ NMR (CDC13) δ: 0.06, 0.07, 0.09, 0.11 (each 3 H, s, Si -Me), 0.55 (3 H, s, H-18), 0.83 (9 H, s, Si-tBu ), 0.90 (9 H, s, Si-tBu, overlapped with CH2CH3) , 0.93 (3 H, d, J=6.5 Hz, H - 21), 1.21 (6 H, s, H - 26, 27), 1.62 (2 H, m, CH2CH3) , 2.26 (1 H, dd. J=13.6, 5.0 Hz), 2.43 (2 H, m), 2.52 (1 H, dd, J=14.4, 4.0 Hz), 2.80 (1 H, m, H-9), 3.37 (3 H, s, OMe), 3.78 (1 H, dd, J-9.7, 5.0 Hz), 3.96 (1 H, t, J=3.0 Hz), 4.71 (2 H, s, 0CH20) , 5.79 (1 H, d, J=11.3 Hz, H-7), 6.16 (1 H, d, J=11.3 Hz, H-6).
19b (マイナー生成物) : ¾ NMR (CDCI3) 6: 0.06, 0.07, 0.09, 0.11 (each 3 H, s, Si -Me), 0.55 (3 H, s, H-18), 0.85 (9 H, s, Si-tBu ), 0.91 (9 H, s, Si-tBu, overlapped with CH2CH3) , 0.93 (3 H, d, J=6.5 Hz, H - 21), 1.21 (6 H, s, H-26, 27), 1.62 (m, CH2CH3) , 2.09 (1 H, dd. J=13.8, 5.0 Hz), 2.30 (1 H, dd, J=12.8, 9.6 Hz), 2.59 (2 H, m), 2.79 (1 H, m, H- 9), 3.37 (3 H, s, OMe), 3.70 (1 H, dd, J=9.2, 4.4 Hz), 3.92 (1 H, dd, J=4.8, 3.2 Hz), 4.71 (2 H, s, 0CH20) , 5.80 (1 H, d, J=11.0 Hz, H-7), 6.10 (1 H, d, J=11.0 Hz, H-6) .
(実施例 19) 1 o;,2j3,25_トリヒドロキシ -2ひ-ェチル -19-ノルビ夕ミン D3 (化合物 Y I -4 a) ぉょび10;,20;,25-トリヒドロキシ-2]3-ェチル-19-ノル ビ夕ミン1)3 (化合物 Y I -4 b)
化合物 19b (17.8 mg, 0.025匪 ol)の無水メタノール(1 mL)溶液に、 カンファ ースルホン酸(48.1 mg, 0.207 mmol)を加え、 室温にて 8時間撹拌した。 反応混 合物に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層 は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ
(3 g、 50% 酢酸ェチル Zへキサン)にて精 製し、 化合物 YI_4b (11.0 mg, 99%)を得た。
主生成物 19a (6.2 mg, 0.009 腿 ol) を用いて上記と同様に反応および後処 理を行ない、 化合物 YI- 4a (2.3 mg, 60%)を得た。
YI-4a (主生成物) : ¾ NMR (CDC13) δ 0.54 (3 Η, s, H - 18), 0.94 (3 H, d, J=6.4 Hz, H-21), 0.98 (3 H, t, J=7.5 Hz, CH2CH3) , 1.22 (6 H, s, H- 26, 27), 1.68, 1.85 (each 1 H, m, CH2CH3) , 2.26 (1 H, dd, J=13.5, 9.2
Hz), 2.38 (1 H, dd, J=13.8, 6.4 Hz), 2.47 (1 H, dd, J=13.8, 3.5 Hz), 2.80 (2 H, m, H-9, 10), 3.83 (2 H, m, H-l, 3), 5.84 (1 H, d, J=11.0 Hz, H-7), 6.33 (1 H, d, J=11.0 Hz, H-6) .
UV Amax (EtOH): 244, 252, 261 nm.
YI-4b (マイナー生成物) : ¾ NMR (CDC13) δ: 0.55 (3 Η, s, H-l 8), 0.94 (3 H, d, J=6.4 Hz, H - 21), 0.99 (3 H, t, J=7.5 Hz, CH2C ), 1.22 (6 H, s, H - 26, 27), 1.72, 1.81 (each 1 H, m, CH2CH3) , 2.18 (1 H, dd, J=13.8, 6.5 Hz, H-4), 2.44 (1 H, dd, J-13.5, 8.5 Hz, H-10), 2.63 (1 H, dd, J=13.5, 4.2 Hz, H-10), 2.72 (1 H, dd, J=13.8, 3.3 Hz, H-4), 2.79 (1 H, m, H-9), 3.73 (1 H, m, W/2 〜17 Hz, H-l), 3.88 (1 H, m, W/2 〜 13 Hz, H-3), 5.84 (1 H, d, J=11.2 Hz, H-7), 6.31 (1 H, d, J=11.2 Hz, H-6).
(実施例 20) 1ひ,2)3, 25 -トリヒドロキシ- 2ひ-メトキシメチル- 19-ノルビ タミン (化合物 Y I— 5 a) および 1ひ,2ひ,25 -トリヒドロキシ- 2j8-メトキ シメチル -19-ノルビタミン D3 (化合物 Y 1— 5 b)
化合物 16 (49.7 mg, 0.070匪 ol, 16a: 16b =約 5 : 1の混合物)の無水メタ ノール(1 mL)溶液に、 カンファースルホン酸(98.2 mg, 0.423 mmol)を加え、 0 °Cで 1時間次いで室温にて 4時間撹拌した。
反応混合物に 5%炭酸水素ナトリゥム水溶液を加え、 酢酸ェチルにて抽出した 。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー(5 g、 70% 酢酸ェチル Zへキサン )にて精製し、 化合物 ΫΙ_5&と YI-5b の混合物(28.5 mg, 87%)を得た。 YI- 5aと YI - 5bの混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20匪, 15%水/メタ
ノール)にて分離精製し、 化合物 YI_5a (8.2 mg) および化合物 YI - 5b (1.6 mg ) を得た。
YI-5a: ¾ NMR (CDC13) δ: 0.53 (3 H, s, H-18), 0.93 (3 H, d, J=6.4 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.41 (2 H, m, H-4), 2.53 (1 H, dd, J=14.1, 5.7 Hz, H - 10), 2.68 (2 H, m, H-10, OH), 2.79 (1 H, m, H-9), 2.97 (1 H, br s, OH), 3.43 (3 H, s, OMe), 3.69, 3.73 (each 1 H, d, J=9.5 Hz, 0CH20), 3.88 (2 H, m, H-l, 3), 5.81 (1 H, d, J=11.2 Hz, H-7), 6.39 (1 H, d, J=11.2 Hz, H-6).
YI-5b: ¾ NMR (CDC13) δ: 0.55 (3 H, s, H-18), 0.94 (3 H, d, J=6.4 Hz, H - 21), 1.22 (6 H, s, H-26, 27), 2.13 (1 H, dd, J=13.8, 3.6 Hz, H-4), 2.21 (1 H, br t, J=〜12 Hz, H-10), 2.77〜2.89 (4 H, HI, H-4, 9, 10, OH), 3.43 (3 H, s, OMe), 3.65, 3.77 (each 1 H, d, J=9.4 Hz, 0CH20) , 3.80 (1 H, m, H-l) 3.85 ( 1H, m, H-3), 5.88 (1 H, d, J=11.2 Hz, H-7), 6.27 (1 H, d, J=ll.2 Hz, H-6).
MS m/z (%) : 464 (52, M+), 446 (74), 428 (23), 410 (10), 401 (65), 383 (100), 335 (13), 317 (10), 299 (12), 281 (13), 222 (12).
2j3,2'-ェポ
エポキシ- 1-19-ノルビ夕ミン^ t -プチルジメチルシリル エーテル (化合物 2 Oa, b)
20
-78 に冷却した化合物 13 (351.0 mg, 0.586 mmol,本実施例では 13a: 13b ==約 2 : 1の混合物を使用)の無水テトラヒドロフラン(3 mL)溶液に、 n-ブチル
リチウム(371 L, 0.586 腿 ol, 1· 58 Mへキサン溶液)をゆっくり加えた。 深い オレンジ色の溶液の溶液が得られた。 15分間撹拌後、 グランドマンケトン体で ある化合物 14c (154.2 mg, 0.391 匪 ol)の無水テトラヒドロフラン(2· 3 mL)溶 液を加えた。 -78 °Cで 2時間撹拌した後、 反応液に飽和塩ィ匕アンモニゥム水溶 液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸 マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ — (15 g、 2%→\Q%→m酢酸ェチル /へキサン)にて精製し、 2%酢酸ェチル含 有へキサン溶出部より化合物 20を二種の立体異性体の混合物として得た(227.0 mg, 75¾) 。 この混合物を構成する異性体の比率は約 3 : 1であった。 また、 10¾ 酢酸ェチル含有へキサン溶出部より未反応の化合物 14c (35.8 mg)および 60¾酢酸ェチル含有へキサン溶出部より化合物 13 (99.0 mg)を回収した。
混合物の NMRデータ
20a (主生成物) : ¾ NMR (CDC13) δ: 0.02 (3 Η, s, Si -Me), 0.05 (3 H, s, Si -Me x 2), 0.06(6 H, s, Si -Me), 0.55 (3 H, s, H-18), 0.56 (6 H, q, SiCH2 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2, overlapped with H-21), 0.95 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3), 1.19 (6 H, s, H - 26, 27), 2.42 (1 H, dd, J=13.2, 3.6 Hz), 2.68 (1 H, dd, J=13.5, 3.8 Hz), 2.79, 2.82 (each 1 H, d, J=5.5 Hz, CH20), 3.81 (1 H, dd, J=7.2, 3.9 Hz, H-3), 3.87 (1 H, dd, J=7.0, 3.9 Hz, H-l), 5.82 ( 1 H, d, J=ll.l Hz, H-7), 6.21 (1 H, d, J^ll.l Hz, H-6).
20b (マイナ一生成物) : ¾ NMR (CDC13) δ: 0.02〜0.07 (12 Η, Si -Me x 4), 0.55 (3 H, s, H-18), 0.56 (9 H, m, SiCH2 x 3, overlapped with H- 18), 0.86, 0.88 (each 9 H, s, Si-tBu x 2, overlapped with H-21), 0.95 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3), 1.19 (6 H, s, H-26, 27), 2.57, 2.92 (each 1 H, d, J=5.5 Hz, CH20), 3.69 (1 H, m, H-3), 4.03 (1 H, d, J=9.3, 4.7 Hz, H-l), 5.82 ( 1 H, d, J=0.7 Hz, H-7), 6.28 (1 H, d, J=10.7 Hz, H-6).
(実施例 22) (20S) - 1 ,25-ジヒドロキシ- 2i3,2'_エポキシ-および(20S) -
1α, 25-ジヒドロキシ -2 α,2'-エポキシ- 19-ノルビタミン D3 (20— Ep i— Y 1— 1 a、 1 b) , ぉょび(203)-1ひ,2|8,25-トリヒドロキシ-2ひ-フルォロメ チル- 19-ノルビタミン D3 (化合物 20— Ep i _Y I—2 a) 、 および (20S) - 1 ひ, 2ひ, 25 -トリヒドロキシ- 2 /3 -フルォロメチル- 19-ノルビタミン D3 (化合物 2 0— Ep i— Y I— 2 b)
卜リシリルエーテル体である化合物 20 (50.0 mg, 0.0645 醒 ol, 20a:20b = 約 3 : 1の混合物)の無水テトラヒドロフラン(1 mL)溶液に、 フッ化テトラプチ ルアンモニゥム(387 L, 0.387 讓 ol, 1.0 Mテトラヒドロフラン溶液)を加え 、 室温にて 7時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残 渣をシリカゲルカラムクロマトグラフィー(5 g、 50¾→70¾ 酢酸ェチル へキ サン)にて精製し、 50%酢酸ェチル含有へキサン溶出部より化合物 20-Epi- YI - 2a と 20- Epi- YI- 2bの混合物 (3.5 mg, 12%, 20-Epi- YI - 2a: 20-Epi- YI- 2b =約 4:1)、 70% 酢酸ェチル含有へキサン溶出部より化合物 20_Epi-YI- laと 20- Epi- YI - lbの混合物 (23.0 mg, 82%, 20- Epi- YI- la: 20- Epi-YI- lb =約 3:1)を得た 化合物 20- Epi- YI- 2aと 20- Epi- YI- 2bの混合物を HPLC (YMC-Pack ODS-AM SH- 342-5, 150 x 20 匪, 20%水/メタノール)にて分離精製し、 化合物 20 - Epi- YI- 2a (2.1 mg) および化合物 20 - Epi - YI- 2b (0.5 mg) を得た。
化合物 ΥΙ-20-Epi-la, lbを含む混合物を HPLC (LiChrosorb Si 60, Hibar, 250 x 4 mm,へキサン:塩化メチレン:メタノール = 50: 50: 8) にて分離精製し、 化合 物 ΥΙ-20-Epi-la (l5.8mg) および化合物 ΥΙ-20-Epi-lb (6.1 mg, Z-異性体) を得た。
ΥΙ-20-Epi-la: Ή NMR (CDC13) δ: 0.55 (3 H, s, H-18), 0.86 (3 H, d,J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.30 (1 H, dd, J=13.4, 8.8 Hz, H-10), 2.40 (1 H, dd, J=13.7, 6.1 Hz, H-4), 2.61 (1 H, dd, j=13.7, 3.5 Hz, H-4), 2.80 (1 H, m, Η-9β), 2.84 (1 H, d, J=4.7 Hz, C¾0), 2.95 (1 H, dd, J=13.4, 4.3 Hz, H-10), 3.08 (1 H, d, J=4.7 Hz, C¾0), 3.81 (1 H, m, H-3), 3.98 (1 H, m, H-1), 5.86 ( 1 H, d, J=11.2 Hz, H-7), 6.39 (1 H, d, J=11.2 Hz, H-6).
MS mlz {%): 432 (M+, 29), 414 (29), 396 (18), 378 (56), 303 (18), 267 (48), 138 (100).
ΥΙ-20-Epi-lb: 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 0.86 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.30 (1 H, dd, J=13.7, 6.2 Hz, H-4), 2.36 (1 H, dd, J=13.5, 8.7 Hz, H-10), 2.72 (1 H, dd, J=13.7, 3.7 Hz, H-4), 2.81 (1 H, m, Η-9β), 2.86 (1 H, dd, J=13.5, 4.4 Hz, H-10), 2.94, 2.99 (each 1 H, d, 7=4.7 Hz, CH20), 3.82 (1 H, m, H-3), 3.90 (1 H, m, H-1), 5.88 ( 1 H, d, J=11.2 Hz, H-7), 6.38 (1 H, d, J=11.2 Hz, H-6).
MS mlz (%): 432 (M+, 68), 414 (77), 396 (35), 378 (50), 303 (35), 267 (42), 133 (100).
20-Epi - YI- 2a (主生成物) : ¾ NMR (CDC13) δ: 0.53 (3 H, s, H-18), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.45 (1 H, dd, j=13.3, 8.7 Hz, H-4), 2.49 (1 H, dd, J=13.3, 5.5 Hz, H-4), 2.56 (1 H, dd, J=14. , 5.8 Hz, H-10), 2.62 (1 H, d, J=1.5 Hz, OH), 2.69 (1 H, dd, j=14.2, 2.9 Hz, H-10), 2.79 (1 H, m, H_9), 3.87 (1 H, m, H-3), 3.97 (1 H, m, H-1), 4.71, 4.76 (each 1 H, dd, J=47.8, 9.7 Hz, C¾F), 5.81 ( 1 H, d, J=11.2 Hz, H-7), 6.40 (1 H, d, J=11.2 Hz, H-6).
19F NMR (CDC13) δ -240.3 (t, J=47.8 Hz).
20-Epi-YI-2b (マイナー生成物) : Ή NMR (CDC13) δ: 0.55 (3 H, s, H- 18), 0.86 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.17 (1 H, dd, J=14.0, 4.0 Hz, H-4), 2.27 (1 H, br. t, J=〜ll Hz, H-10), 2.57 (1 H, d, J=1.8 Hz, OH), 2.79 (1 H, m, H-9), 2.84 (1 H, m, H-4), 2.89 (1 H, dd, J=13.2, 5.0 Hz, H-10), 3.76 (1 H, m, H-1), 3.95 (1 H, m, H_ 3), 4.70, 4.76 (each 1 H, dd, J=47.6, 9.5 Hz, CH2F) , 5.86 ( 1 H, d, j=11.2 Hz, H-7), 6.29 (1 H, d, 1=11.2 Hz, H-6).
19F NMR (CDCI3) δ -240.2 (t, J=47.6 Hz).
(実施例 23) (20S) - 1 Q!,2j3,25-トリヒドロキシ -2 -メチル- 19 -ノルビ夕 ミン D3 (化合物 20 -Ep i -Y I - 3 a) 、 および、 (20S)-1 α, 2ひ, 25 -トリ ヒドロキシ- 2 ]3-メチル -19-ノルビタミン D3 (化合物 20— Ep i _YI— 3 b )
20-Epi-YI-1a, b 20-Epi-YI-3a 20-Epi-YI-3b
(not isolated) エポキシ体である化合物 20- Epi- YI-laと 20- Epi-YI- lb の混合物(23.0 mg, 0.053 mraol, 20_Epi- YI- la : 20-Epi- YI- lb =約 3 : 1)の無水テトラヒドロフラ ン(0.25 mL)溶液に、 リチウムアルミニウムヒドリド(2 mg, 0.053 mmol)を加 え室温にて 7時間撹拌した。 反応 2時間後および 6時間後に、 それぞれ 2 mgのリ チウムアルミニウムヒドリドを追加した。 反応混合物に酒石酸ナトリウムカリ ゥム水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水 硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトダラ フィー(3 g、 60¾ 酢酸ェチル Zへキサン)にて精製し、 化合物 20-Epi-YI-3aと 20-Epi-YI-3b の混合物(15.5 mg, 67%、 比率は不明) を得た。
20- Epi-YI- 3aと 20_Epi-YI_3bを含む混合物を HPLC (YMC-Pack 0DS-AM SH- 342-5, 150 X 20 mm, 20%水/メタノール)にて分離精製し、 化合物 20- Epi- YI- 3a (6.3 mg) を得た。 化合物 20- Epi-YI- 3bは純粋な化合物として単離すること ができなかった。
20-Epi-YI-3a (主生成物) : ¾ NMR (CDC13) δ 0.54 (3 Η, s, H-18), 0.86 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 1.27 (3 H, s, 2 - Me), 2.07 (1 H, m, H - 10), 2.36 (1 H, dd, J=14.4, 4.6 Hz, H-4), 2.54 (2 H, m, H-4, OH), 2.79 (1 H, m, H— 9), 2.94 (1 H, dd, J=13.5, 4.4 Hz),
3.74 (2 H, m, H-l, 3), 5.85 (1 H, d, J=11.3 Hz, H-7), 6.30 (1 H, d, J=11.3 Hz, H-6).
(実施例 24) (20S) - 1 a -[(卜プチルジメチルシリル)ォキシ ]-2i3-ヒドロ キシ- 2 a -ェチル-および(20S) - 1ひ - [ (t -プチルジメチルシリル)ォキシ] - 2ひ - ヒドロキシ- 2 β -ェチル- 25- [(トリェチルシリル)ォキシ] - 19-ノルビタミン D3 t -プチルジメチルシリルエーテル (化合物 21a, 21b)
-40°Cに冷却したシアン化銅 (I) (101.9 mg, 1.138 匪 ol) の無水ジェチル エーテル(1 ml) 懸濁液に、 メチルリチウム(2.0 ml, 2.280 mmol, 1.14 Mへキ サン溶液)をゆっくり加えた。 - 40°Cにて 30分撹拌した後、 エポキシ体である化 合物 20 (110.3 mg, 0.142 匪 ol, 本実施例では約 3 : 1の混合物を使用)の無水 ジェチルェ一テル(1.5 ml)溶液をゆっくり加えた。 反応混合物は- 40 にて 1時 間撹拌した後、 0°Cまで徐々に温度を上昇させ、 更に 2時間撹拌した。 反応混合 物に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は 飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリ 力ゲルカラムクロマトグラフィー ao g、 2%酢酸ェチル /へキサン)にて精製し
、 化合物 21を二種の立体異性体の混合物 (98.5 mg, 87%) として得た。 この混 合物を構成する異性体の比率は 21a : 21b =約 3 : 1であった。
21: ¾ NMR (CDC13) δ: 0.06, 0.07, 0.09, 0.10 (each 3 H, s, Si -Me x 4), 0.54 (3 H, s, H-l 8), 0.56 (6 H, q, J=7.9 Hz, Si— CH2 x 3), 0.84, 0.91 (each 9 H, s, Si-tBo x 2, overlapped with H - 21), 0.94 (9 H, t, J=7.9 Hz, Si-CH2CH3 x 3, overlapped with CH2CH3) , 1.19 (6 H, s, H-26, 27), 2.80 (1 H, m, H— 9), 3.78, 3.92 (ca. 3 : 1) (1 H, m, H-3), 3.70,
3.95 (ca. 1 : 3) (1 H, m, H-l), 5.79 (1 H, d, J=11.3 Hz, H-7), 6.10 6.16 (ca. 1 : 3) (1 H, d, J=11.3 Hz, H-6).
(実施例 25) (20S)-lQ!,2/3,25-トリヒドロキシ- 2ひ-ェチル -19-ノルビタ ミン!) 3 (化合物 20 - epi - YI - 4a) および(20S)_1ひ, 2 α , 25-トリヒドロキシ- 2 ]3 - ェチル- 19 -ノルビタミン D3 (化合物 20- epi- YI-4b)
化合物 21 (98.5 mg, 0.124腿 ol, 21a : 21b = 約 3 : 1の混合物)の無水メ 夕ノール (2 ml ) 溶液に、 カンファースルホン酸(173.5 mg, 0.747 匪 ol)を 加え室温にて 6時間撹拌した。 反応混合物に 5%炭酸水素ナトリウム水溶液を加 え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄、 無水硫酸マグネ シゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー(6 g, 50%酢酸ェチル /へキサン)にて精製し、 化合物 20- epi- YI- 4aおよび 20- epi - YI-4bを約 3 : 1の割合で含む混合物 (45.0 mg, 81%) を得た。
化合物 20- epi- YI- 4aおよび 20- epi- YI- 4bを約 3 : 1の割合で含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 20%水/メタノール, 8ml I min)にて分離精 製し、 化合物 20-epi-YI- 4a (27.4 mg)および化合物 20_epi- YI- 4b (7.3 mg) を 得た。
20-epi-YI-4a: 'Η NMR (CDC13) δ: 0.53 (3 H, s, H - 18), 0.85 (3 H, d, J=6.5 Hz, H - 21), 0.98 (3 H, t, J=7.5 Hz, CH2CH3) , 1.21 (6 H, s, H-26, 27), 2.25 (1 H, dd, J=13.7, 8.9 Hz, H - 10), 2.37 (1 H, dd, J=14.0, 6.2 Hz, H-4), 2.45 (2 H, m, H— 4, OH), 2.80 (2 H, m, H— 9, 10), 3.85 (2 H, m, H-l, 3), 5.83 (1 H, d, J=11.2 Hz, H-7), 6.31 (1 H, d, J=11.2 Hz, H-
6).
UV Amax (EtOH): 244, 252, 261 nm.
20-epi-YI-4b: ¾ NMR (CDC13) δ ·· 0.55 (3 H, s, H-18), 0.85 (3 H, d, J-6.5 Hz, H - 21), 0.99 (3 H, t, J=7.5 Hz, CH2CH3) , 1.22 (6 H, s, H-26, 27), 2.18 (1 H, dd, J=13.8, 6.6 Hz, H - 4), 2.45 (1 H, dd, J=13.6, 8.5 Hz, H-10), 2.62 (1 H, dd, J=13.6, 4.3 Hz, H - 10), 2.71 (1 H, dd, J=13.8, 3.4 Hz, H-4), 2.80 (1 H, m, H-9), 3.74 (1 H, dd, J=8.5, 4.3 Hz, H-l), 3.88 (1 H, dd, J=6.6, 3.4 Hz, H-3), 5.84 (1 H, d, J=11.2 Hz, H-7), 6.31 (1 H, d, J=11.2 Hz, H-6).
UV Amax (EtOH): 244, 252, 261 nm.
(実施例 26) (20S)-la,2i3,25- Uヒドロキシ- 2 -メトキシメチル- 19 - ノルビタミン D3 (化合物 20_epi-YI - 5a) および(20S)_1 α, 2 α, 25-トリヒドロキ シ- 2)3-メトキシメチル- 19-ノルビタミン D3 (化合物 20- epi - YI - 5b)
化合物 20 ( 10.4 nig, 0.013腿 ol, 20a:20b=約 3 : 1の混合物) の無水メタ ノール (0.5 ml) 溶液にカンファースルホン酸 (18.7 mg, 0.080 mmol) を加 え、 0°Cで 1時間、 室温にて 4時間撹拌した。 反応混合物に 5%炭酸水素ナトリウ ム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無 水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマ卜グ ラフィー (5 g, 70%酢酸ェチル /へキサン) にて精製し、 化合物 20- epi- YI- 5a および 20 - epi-YI_5bを約 3 : 1の割合で含む混合物 (5.0 mg, 81¾) を得た。 化 合物 20-epi- YI-5aおよび 20- epi- YI_5bを約 3 : 1の割合で含む混合物を HPLC (
YMC-Pack ODS-AM SH-342-5, 150 x 20 匪, 15%水/メタノール) にて分離精製 し、 化合物 20-epi-YI_5a (2.0 mg) および 20- epi_YI- 5b (0.8 mg) を得た。
20-epi-YI-5a: ¾ N R (CDC13) δ: 0.53 (3 H, s, H - 18), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.41 (2 H, m, H-4, OH), 2.53 (1 H, dd, J=14.1, 5.6 Hz, H-10), 2.68 (2 H, m, H-10, OH), 2.80 (1 H, m, H-9), 2.94 (1 H, s, OH), 3.44 (3 H, s, OMe), 3.69, 3.74 (each 1 H, d, J=9.5 Hz, 0CH20) , 3.89 (2 H, m, H-l, 3), 5.82 (1 H, d, J=11.2 Hz, H- 7), 6.39 (1 H, d, J=11.2 Hz, H-6) .
UV Amax (EtOH): 244, 252, 261 nm.
20-epi-YI-5b: Ή NMR (CDC13) 6: 0.55 (3 H, s, H-l 8), 0.85 (3 H, d, J=6.4 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.12 (1 H, dd, J=13.9, 3.4 Hz, H-4), 2.21 (1 H, br. t, J=〜12 Hz, H-10), 2.77〜2.89 (4 H, m, H-4, 9, 10), 2.89 (1 H, s, OH), 3.43 (3 H, s, OMe), 3.65, 3.77 (each 1 H, d, J=9.5 Hz, 0CH20), 3.79 (1 H, m, H-l), 3.84(1 H, m, H-3), 5.88 (1 H, d, J=11.2 Hz, H-7), 6.27 (1 H, d, J-11.2 Hz, H-6).
UVAmax (EtOH): 244, 252, 261 nm.
(実施例 27) (20S)-1ひ -[(卜プチルジメチルシリル)ォキシ ]-2ひ- [(トリ メチルシリル)才キシ] -および(20S) - 1 - [(t-プチルジメチルシリル)ォキシ]― 2 /3 - [(トリメチルシリル)ォキシ ] -25 - [(トリェチルシリル)ォキシ] -19-ノルビ 夕ミン D3 t-ブチルジメチルシリルエーテル (化合物 23a,
- 78 に冷却した化合物 22 (435.2 mg, 0.660 匪 ol, 22a : 22b = 約 2 : 1の
混合物)の無水テトラヒドロフラン(5 ml)溶液に、 n-ブチルリチウム(412 U, 0.660 mmol, 1.6 Mへキサン溶液)を加え 15分撹拌後、 グランドマンケトン体で ある化合物 14c (173.8 ig, 0.440 mmol) の無水テトラヒドロフラン (3 ml)溶 液を加えた。 -78でで 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液 を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸 マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ 一(20 g, 2%酢酸ェチル /へキサン)にて精製し、 化合物 23 (243.4 mg, 66%) を二種の立体異性体の混合物として得た。 この混合物を構成する異性体 23aお よび 23bの比率は約 3 : 2であった。 5%酢酸ェチル /へキサン溶出部より未反応 の化合物 14c (30.0 mg, 17¾) および 5%酢酸ェチルノへキサン溶出部より化合 物 22 (157.6 mg) を回収した。
混合物の NMRデータ
23a (主生成物) : ¾ NMR (CDC13) δ 0.04, 0.055, 0.058, 0.063 (each 3 H, s, Si -Me x 4), 0.12 (9 H, Si -Me x 3), 0.54 (3 H, s, H-18), 0.56 (6 H, q, J=7.9 Hz, SiC¾ x 3), 0.85 (3 H, d, J=6.5 Hz, H-21), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3), 1.19 (6 H, s, H - 26, 27), 2.30 (1 H, m), 2.50 (2 H, m), 2.79 (1 H, m, H-9), 3.54 (1 H, m, H— 2), 3.80 (1 H, m, H-3), 3.88 (1 H, m, H- 1), 5.81 (1 H, d, J=ll.1 Hz, H-7), 6.10 (1 H, d, J=ll.1 Hz, H - 6).
23b (マイナー生成物) : 'Η NMR (CDC13) δ: 0.04, 0.06 (each 3 H, s, Si -Me x 2), 0.07 (6 H, s, Si -Me x 2), 0.12 (9 H, Si -Me x 3), 0.53 (3 H, s, H-18), 0.56 (6 H, q, J=7.8 Hz, SiCH2 x 3), 0.84 (3 H, d, J=6.6 Hz, H-21), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.8 Hz, SiCH2CH3 x 3), 1.19 (6 H, s, H-26, 27), 2.10 (1 H, m), 2.44 (2 H, m), 2.79 (1 H, m, H-9), 3.60 (1 H, m, H-2), 3.80 (1 H, dd, J-8.7, 4.5 Hz, H-l), 3.94 (1 H, m, H-3), 5.79 (1 H, d, J=11.2 Hz, H-7), 6.13 (1 H, d, J=11.2 Hz, H-6).
(実施例 28) (203)-10;-(卜ブチルジメチルシリルォキシ)-20;,25-ジヒド
口キシ-および(2 OS) -1 - (t-ブチルジメチルシリルォキシ) - 2 i3 , 25-ジヒドロ キシ- 19-ノルビタミン D3 t -プチルジメチルシリルエーテル (化合物 24a, 24b)
化合物 23 (182.5 mg, 0.218 醒 ol、 23a : 23b =約 3: 2の混合物) をテトラ ヒドロフランと酢酸と水との混合物 (9.5 ml, 8 : 8 : 1, v/v/v) に溶解し、 O :で 2時間、 室温で 2.5時間撹拌した。 反応混合液は酢酸ェチルにて希釈し、 5%炭酸水素ナトリウム水溶液、 続いて飽和食塩水にて洗浄した。 有機層は無水 硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカゲルカラムクロマト グラフィー (10 g、 2%酢酸ェチル Zへキサン) にて精製し、 化合物 24a (39.1 mg, 28%) および化合物 24b (26.0 mg, 18%) を得た。 合計収率は 46¾であった
24a: Ή NMR (CDC13) δ 0.067, 0.077, 0.083, 0.10 (each 3 H, s, Si -Me x 4), 0.54 (3 H, s, H-18), 0.86 (3 H, d, J=6.6 Hz, H-21), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 1.22 (6 H, s, H-26, 27), 2.27 (1 H, d, J=3.2 Hz, OH), 2.31 (1 H, dd, J=12.6, 3.7 Hz), 2.48 (2 H, m), 2.79 (1H, m, H-9), 3.51(1 H, m, H-2), 3.91, (1 H, m, H-3), 4.00 (1 H, m, H- 1), 5.80 (1 H, d, J=ll.l Hz, H-7), 6.15 (1 H, d, J=ll.l Hz, H-6).
24b: Ή NMR (CDC13) δ 0.06, 0.07, 0.08, 0.10 (each 3 H, s, Si -Me x 4), 0.53 (3 H, s, H-18), 0.86, 0.90 (each 9 H, s, Si-tBu x 2, overlapped with H-21), 1.21 (6 H, s, H-26, 27), 2.18 (1 H, dd, J=13.0, 4.5 Hz), 2.39 (3 H, m), 2.80 (1H, m, H-9), 3.59 (1 H, m, H-2), 4.00 (2 H, m, H— 1, 3), 5.80 (1 H, d, J=11.2 Hz, H-7), 6.18 (1 H, d, J=11.2 Hz, H-6).
(実施例 29) (20S) -1 α- [ (t-プチルジメチルシリル)ォキシ] -2 -{2- [ (t- プチルジメチルシリル)ォキシ] -ェトキシ} -25-ヒドロキシ- 19-ノルビ夕ミン1)3 t-プチルジメチルシリルエーテル (化合物 25a)
24a 25a
0°Cに冷却した化合物 24a (17.0 mg, 0.026匪 ol) の無水ジメチルホムアミド およびテトラヒドロフランの混合溶液 (9 : 1, 1 ml, v/v) に、 水素化ナトリ ゥム (31.4 mg, 0.786 匪 ol, 60%パラフィンリキッド) および(2-ブロモエト キシ) _t-ブチルジメチルシシラン (2Ί μΛ, 0.126 删 ol) を加え激しく撹拌し た。 22時間後、 反応液に氷水を加え、 酢酸ェチルおよびへキサン混合液 (1:1, v/v) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム 乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 1% 酢酸ェチル /へキサン) にて精製し、 化合物 25a (15.4 mg, 73¾) を得、 20%酢 酸ェチル Zへキサン溶出部より未反応原料である化合物 24a (2.8 mg, 16¾) を 回収した。
25a: Ή NMR (CDC13) (5: 0.04〜0.08 (18 H, Si -Me x 6), 0.54 (3 H, s, H - 18), 0.87, 0.89, 0.91 (each 9 H, s, Si-tBu x 3 overlapped with H- 21), 1.21 (6 H, s, H-26, 27), 2.79 (1 H, m, H - 9), 3.21(1 H, m, H-2), 3.7〜3.9 (4 H, m, OCH2CH20) , 3.96 (1 H, m, H-3), 4.14 (1 H, m, H-l), 5.81 (1 H, d, J=11.2 Hz, H-7), 6.15 (1 H, d, J=11.2 Hz, H-6).
(実施例 30) (20S) -l a- [ (t -プチルジメチルシリリレ)ォキシ] -2 β-{2- [ (t- ブチルジメチルシリル)ォキシ]-エトキシ } -25-ヒドロキシ- 19-ノルビ夕ミン D3
t -プチルジメチルシリルエーテル (化合物 25b)
24b 25b
0。Cに冷却した化合物 24b (15.3 rag, 0.024 mmol) の無水ジメチルホムアミド (1 ml) に、 水素化ナトリウム (18.9 mg, 0.471 mmol, 60%パラフィンリキ ッド) および(2-ブロモエトキシ) -卜ブチルジメチルシシラン (20 ^1, 0.093 匪 ol) を加え激しく撹拌した。 22時間後、 反応液に氷水を加え、 酢酸ェチルお よびへキサン混合液 (1:1, v/v) にて抽出した。 有機層を飽和食塩水にて洗浄 し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロ マトグラフィ一 (5 g、 7%酢酸ェチル Zへキサン) にて精製し、 化合物 25b ( 12.0 mg, 63%) を得、 20%酢酸ェチル Zへキサン溶出部より未反応原料である 化合物 24b (3.4 mg, 22¾) を回収した。
25b: ¾ NMR (CDC13) δ: 0.05〜0.07 (18 Η, Si -Me x 6), 0.53 (3 H, s, H-18), 0.86, 0.88, 0.89 (each 9 H, s, Si-tBu x 3, overlapped with H- 21), 1.21 (6 H, s, H-26, 27), 2.13 (1 H, dd, J=12.8, 4.0 Hz, H-10), 2.35 (2 H, m, H-4), 2.46 (1 H, m, H - 10), 2.80 (1 H, m, H-9), 3.28 (1 H, m, H-2), 3.61(1 H, ) , 3.73 (2 H, m), 3.83 (1 H, m), 3.96 (1 H, dd, J=8.8, 4.3 Hz, H-l), 4.04 (1 H, m, H - 3), 5.79 (1 H, d, J=ll.1 Hz, H- 7), 6.14 (1 H, d, J=ll.l Hz, H— 6). (実施例 31) (20S)- 1 α,25-ジヒドロキシ- 2α- (2-ヒドロキシ -エトキシ) -
19 -ノルビタミン!) 3 (化合物 20- epi- YI- 6a)
20-epi-YI-6a
化合物 25a (15.4 mg, 0.019 mmol) の無水メタノール (0.5 ml) 溶液にカン ファースルホン酸 (26.6 mg, 0.114醒 ol) を加え、 室温にて 2時間撹拌した。 反応混合物に 5%炭酸水素ナトリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。
ル) にて精製し、 続いて HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 腿, 20 水 /メタノール, 8ml/min) にて精製し、 化合物 20- epi- YI-6a (6.4 mg, in) を得た。
20-epi-YI-6a: Ή NMR (CDC13) <5: 0.55 (3 H, s, H - 18), 0.85 (3 H, d,
J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.19 (2 H, m, H - 4, 10),
2.33, 2.41, 2.56 (each 1 H, br. s, OH x 3), 2.63 (1 H, dd, J=13.2, 4.3 Hz, H - 4), 2.80 (1 H, m, H_9), 2.84 (1 H, dd, J=14.4, 5.4 Hz, H— 10), 3.37 (1 H, dd, J=7.8, 2.8 Hz, H-2), 3.72〜3.83 (4 H, m, 0CH2CH20) , 3.96 (1 H, m, H-3), 4.14 (1 H, m, H-l), 5.83 (1 H, d, J=11.2 Hz, H-7), 6.34 (1 H, d, J=11.2 Hz, H-6).
UV Amax (EtOH): 243, 252, 261 nm.
(実施例 32) (20S) - 1ひ, 25 -ジヒドロキシ -2 ]3 - (2 -ヒドロキシ-ェトキシ) - 19 -ノルビタミン!) 3 (化合物 20-epi-Y卜 6b)
25b 20-epi-Yl-6b 化合物 25b (12.0 mg, 0.015 匪 ol) の無水メタノール (0.5 ml) 溶液に、 力 ンファースルホン酸 (20.7 mg, 0.089腿 ol) を加え、 室温にて 2時間撹拌した 。 反応混合物に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した 。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した 。 残渣をシリカゲルカラムクロマトグラフィー (3 g、 2%メタノール Z酢酸ェ チル) にて精製し、 続いて HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 匪, 20%水/メタノール, 8ml / min) にて精製し、 化合物 20- epi - YI - 6b (5.6 ig, 81%) を得た。
20-epi-YI-6b: ¾ NMR (CDC13) δ: 0.54 (3 H, s, H-18), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.35 (1 H, br. d, J=14.2 Hz, H-4), 2.48 (1 H, dm, J=14.2 Hz, H-4), 2.79 (1 H, m, H— 9), 3.09 (1 H, dd, J=13.5, 3.7 Hz, H-10), 3.29 (1 H, dd, J=8.7, 2.7 Hz, H-2), 3.67 (1 H, m), 3.76〜 3.89 (4 H, m, H-l, OCH2CH20) , 4.17 (1 H, m, H-3), 5.84 (1 H, d, J=11.2 Hz, H-7), 6.28 (1 H, d, J=11.2 Hz, H-6).
UVAmax (EtOH): 243, 252, 261 nm.
(実施例 33) (20S)-1ひ- [(t -プチルジメチルシリル)ォキシ ォキソ - 25 -ヒドロキシ -19-ノルビ夕ミン D3 t -プチルジメチルシリルエーテル (化合物 26)
- 78°Cに冷却した二塩化ォキサリル (18 1, 0.206 腿 ol) の無水塩化メチレ ン (1 ml) 溶液に、 ジメチルスルホキシド (29 1, 0.414顧 ol) の無水塩化 メチレン (0.2 ml) 溶液を加え、 5分間撹拌した。 この冷却撹拌溶液に化合物 24 (61.0 mg, 0.094 mmol, 24a : 24b =約 3 : 2の異性体の混合物) の無水塩 化メチレン (1.2 ml) 溶液を加えた。 - 78°Cで 15分間撹拌した後、 トリェチル ァミン (131 l, 0.940 腿 ol) を加え、 - 78°Cで 30分、 Ot:で 10分撹拌した。 反 応液を氷水に投入し、 塩化メチレンにて抽出した。 有機層を飽和食塩水にて洗 浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムク 口マトグラフィー (5 g, 20%酢酸ェチル Zへキサン) にて精製し、 化合物 26 ( 52.0 mg, 86%) を単一化合物として得た。
26: ¾ NMR (CDC13) δ: 0.055, 0.065, 0.069, 0.10 (each 3 H, s, Si -Me x 4), 0.55 (3 H, s, H-18), 0.87, 0.89 (each 9 H, s, Si-tBu x 2, overlapped with H - 21), 1.22 (6 H, s, H-26, 27), 2.45 (1 H, dd, J=13.5, 8.7 Hz), 2.52 (1 H, dd, J=14.2, 4.1 Hz), 2.66 (1 H, dd, J=13.5, 5.5 Hz), 2.72 (1 H, dd, J=14.2, 6.3 Hz), 2.83 (1 H, m, H - 9), 4.35 (1 H, dd, J=6.3, 4.1 Hz), 4.55 (1 H, dd, J=8.7, 5.5 Hz), 5.81 (1 H, d, J=11.2 Hz, H-7), 6.35 (1 H, d, J=11.2 Hz, H- 6). (実施例 34) (20S)-1 a- [(t-プチルジメチルシリル)ォキシ ]-2-シァノメ チレン- 25-ヒドロキシ- 19-ノルビタミン D3 t-ブチルジメチルシリルェ一テルの E -異性体および Z-異性体 (化合物 27a, 27b)
- 40°Cに冷却したジェチルシアノメチルホスホナート (24 l, 0.148 mmol) の無水テトラヒドロフラン (1 ml) 溶液に、 n-ブチルリチウム (95 1, 0.151 腿 ol, 1.58 Mへキサン溶液) を加え 15分撹拌した後、 化合物 26 (48.7 mg, 0.075 腿 ol) の無水テトラヒドロフラン (1.2 ml) 溶液をゆっくり加えた。 - 40°Cで 1.5時間撹拌した後、 反応混合物に飽和塩化アンモニゥム水溶液を加え 、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシ ゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 10%酢酸ェチル Zへキサン) にて精製し、 化合物 27 (50.0 mg, 99¾) を二種の 立体異性体の混合物として得た。 この混合物を構成する異性体 27a (E-異性体 ) および異性体 27b (Z-異性体) の比率は約 1 : 1であった。
混合物の NMRデ一夕
27a: ¾ NMR (CDC13) δ 0.054, 0.067, 0.099, 0.121 (each 3 H, s, Si— Me x 4), 0.55 (3 H, s, H-18), 0.83, 0.92 (each 9 H, s, Si-tBu x 2), 0.86 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.12 (1 H, m, H-10), 4.46 (1 H, m, H-l), 4.99 (1 H, t, J=2.8 Hz, H-3), 5.47 (1 H, d, J=1.8 Hz, OCHCN), 5.82 (1 H, d, J=ll.l Hz, H-7), 6.19 (1 H, d, J=ll.1 Hz, H-6) .
27b: ¾ NMR (CDC13) δ : 0.063, 0.075, 0.112, 0.132 (each 3 H, s, Si— Me x 4), 0.54 (3 H, s, H-18), 0.83, 0.92 (each 9 H, s, Si-tBu x 2), 0.86 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 2.99 (1 H, m, H-10), 4.57 (1 H, m, H-3), 5.04 (1 H, t, J=2.8 Hz, H-l), 5.47 (1 H, d, J=1.8 Hz, C=CHCN) , 5.79 (1 H, d, J=ll.l Hz, H-7),
6.32 (1 H, d, J=11.2 Hz, H-6, 7).
(実施例 35) (20S)-1 -プチルジメチルシリル)ォキシ ]- 2- [2- (ヒド 口キシ) -ェチリデン] -25-ヒドロキシ- 19-ノルビタミン t -プチルジメチルシ リルエーテルの E-異性体および Z-異性体 (化合物 29a, 29b)
_78°Cに冷却した化合物 27 (20.0 mg, 0.030 mmol, 27a : 27b = 約 1 : 1の 混合物) の無水トルエン (1 ml) 溶液に、 水素化- iso-ブチルアルミニウム ( 60 1, 0.060腿 ol, 1.0 Μへキサン溶液) を加えた。 3時間後に- 20°Cに反応温 度を上げた。 1時間撹拌後、 水素化 -iso -ブチルアルミニウム (30 1, 0.030 mmol, 1.0 Mへキサン溶液) を追加し、 更に、 5.5時間撹拌した。 還元剤を酒石 酸ナトリウムカリウム水溶液により分解した後、 反応混合物を氷水に移し、 酢 酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム 乾燥、 溶媒留去した。 残渣をエタノール (1 ml) で溶解し、 水素化ホウ素ナト リウム (1.1 mg, 0.030 mmol) を加え 0°Cにて 1時間撹拌した。 反応液に氷水を 加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マ グネシゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (4 g, 15%酢酸ェチル /へキサン) にて精製し、 化合物 29a (5.5 mg) および 化合物 29b (5.0 mg) を得た。 合計収率は 52%であった。 尚、 未反応原料である 化合物 27 (7.5 mg, 38%) を回収した。
29a: Ή NMR (CDC13) δ 0.02, 0.07, 0.08 (3 Η, 3 Η, 6 Η, s, Si -Me x 4), 0.55 (3 H, s, H-18), 0.85, 0.92 (each 9 H, s, Si-tBu x 2, overlapped with H - 21), 1.22 (6 H, s, H-26, 27), 2.29 (2 H, m, H-4), 2.79 (1 H, m, H-9), 2.88 (1 H, dd, J=12.7, 4.3 Hz, H— 10), 4.19 (1 H,
dd, J=12.7, 6.8 Hz, CH20H) , 4.31 (1 H, dd, J=12.7, 6.7 Hz, CH2OH) , 4.37 (1 H, dd, J=9.7, 4.3 Hz, H-l), 4.81 (1 H, t, J=3.8 Hz, H— 3), 5.72 (1 H, t, J=6.8 Hz, OCH), 5.85 (1 H, d, J=11.2 Hz, H-7), 6.15 (1 H, d, J=11.2 Hz, H - 6).
29b: ¾ NMR (CDC13) δ : 0.01, 0.07, 0.08, 0.09 (each 3 H, s, Si -Me x 4), 0.54 (3 H, s, H-l 8), 0.83, 0.93 (each 9 H, s, Si-tBu x 2), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.22 (6 H, s, H-26, 27), 2.14 (1 H, br. t, J=〜 11.5 Hz, H— 4), 2.55 (1 H, dd, J=12.3, 5.0 Hz, H-4), 2.82 (2 H, m, H - 9, 10), 4.22 (1 H, dd, J=12.3, 7.1 Hz, CH2OH) , 4.30 (each 1 H, dd, J=12.7, 7.0 Hz, CH2OH) , 4.47 (1 H, m, H - 3), 4.86 (1 H, t, J=3.1 Hz, H-l), 5.72 (1 H, m, C=CH) , 5.81 (1 H, d, J=ll.l Hz, H-7), 6.25 (1 H, d, J=ll.1 Hz, H-6).
(実施例 36) (20S) -1 a, 25-ジヒドロキシ -2_ [2- (ヒドロキシ) -ェチリデン ]- 19-ノルビタミン D3 (E -異性体) (化合物 20- epi_YI-8a)
29a 20-epi-YI-8a 化合物 29a (11.0 mg, 0.016 匪 ol) の無水メタノール (0.5 ml) 溶液に、 力 ンファースルホン酸 (11.4 mg, 0.049 匪 ol) を加え室温にて 2時間撹拌した。 反応混合物に 5%炭酸水素ナトリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (3 g、 3%メタノール 酢酸ェチ ル) にて精製し、 化合物 20- epi- YI- 8a (6.4 mg, 88¾) を得た。
20- epi-YI- 8a: [E NMR (CDC13) δ: 0.54 (3 H, s, H-l 8), 0.86 (3 H, d,
J=6.5 Hz, H - 21), 1.21 (6 H, s, H-26, 27), 2.42 (2 H, m, H-4), 2.81 (1
H, m, H-9), 3.15 (1 H, d, J=12.8, 4.9 Hz, H - 10), 4.15 (1 H, dd,
J=12.4, 5.9 Hz, CH20H) , 4.39 (2 H, m, H-l, CH2OH) , 4.84 (1 H, m, H-3),
5.80 (1 H, m, C=CH), 5.88 (1 H, d, J=ll.1 Hz, H-7), 6.29 (1 H, d, J=ll.1 Hz, H-6).
UVAmax (EtOH): 246, 254, 263 nm.
(実施例 37) (20S) - 1 α , 25 -ジヒドロキシ -2- [2- (ヒドロキシ) -ェチリデン ] -19-ノルビタミン!) 3 (Z-異性体) (化合物 20-epi- YI- 8b)
化合物 29b (11.0 mg, 0.016腿 ol) の無水メタノール (0.5 ml) 溶液に、 力 ンファースルホン酸 (11.4 mg, 0.049 mmol) を加え、 室温にて 2時間撹拌した 。 反応混合物に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した 。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した 。 残渣をシリカゲルカラムクロマトグラフィー (3 g、 3%メタノール Z酢酸ェ チル) にて精製し、 化合物 20- epi- YI- 8b (4.8 mg, 66%) を得た。
20-epi-YI-8b: Ή NMR (CDC13) δ 0.55 (3 H, s, H-l 8), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.21 (1 H, br. t. J=〜13 Hz, H-4), 2.33 (1 H, dm, H-10), 2.70 (1 H, d, J=12.8, 4.7 Hz, H-4), 2.82 (2 H, m, H-9, 10), 4.24 (1 H, dd, J=12.6, 6.4 Hz, CH20H) , 4.38 (1 H, dd, J=12.6, 7.4 Hz, CH20H) , 4.46 (1 H, m, H-3), 4.87 (1 H, t, J=4.2 Hz, H-l), 5.83 (2 H, m, H-7, C=CH), 6.40 (1 H, d, J=ll.l Hz, H-6).
UVAmax (EtOH): 246, 254, 263 nm.
(実施例 38) (aS*, 2R, 6R)-および(aR*, 2R, 6R)- {[4-ベンジルォキシ- 2, 6 -ビ ス- [ (t -プチルジメチルシリル)ォキシ]-シクロへキシリデン卜ァセトニトリル (化合物 30)
- 78°Cに冷却したジェチルシアノメチルホスホナート (112 1, 0.69 mmol) の無水テトラヒドロフラン (1 ml) に n -ブチルリチウム (493 1, 0.69 mmol, 1.4 Mへキサン溶液) を加え 15分間撹拌した。 本溶液に実施例 4で得られた化合 物 6 (160 mg, 0.345 匪 ol) の無水テトラヒドロフラン (1.2 ml) 溶液をゆつ くり加えた。 反応温度を 0でに上昇させた後、 1.5時間撹拌した。 飽和塩化アン モニゥム水を加えて反応を停止させた後、 反応混合物に氷水を加え酢酸ェチル にて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥 後、 溶媒留去した。 残渣はシリカゲルカラムクロマトグラフィー (5 g, 2%酢 酸ェチル /へキサン) にて精製し、 化合物 30 (162 mg, 96¾) を二種の立体異 性体の混合物として得た。 この混合物を構成する異性体の比率は約 1 : 2であ つたが、 aS*, 2R, 6R体と aR*, 2R, 6R体のいずれが主生成物かは判断できなかった 混合物の NMRデータ
30a (マイナー生成物、 低極性異性体) : Ή NMR (CDC13) δ: 0.05, 0.06, 0.08, 0.11 (each 3 H, Si -Me x 4), 0.85, 0.91 (each 9 H, s, Si-iBu x 2), 1·32〜1·48 (2 H, m), 2.28 (1 H, m), 2.39 (1 H, m), 3.96 (1 H, m, H-l), 4.54 (2 H, s, PhCH2) , 4.56 (1 H, m), 4.98 (1 H, m), 5.41 (1 H, d, J=2.0 Hz, C=CH), 7.25〜7.35 (5 H, m, arom. H).
30b (主生成物、 髙極性異性体) : ¾ NMR (CDC13) δ 0.07, 0.08, 0.09, 0.11 (each 3 H, Si -Me x 4), 0.84, 0.91 (each 9 H, s, Si-tBu x 2), 1.60 -1.73 (2 H, m), 2.22 (2 H, m), 3.85 (1 H, m, H-l), 4.57, 4.61 (each 1
H, d, 1=12.1 Hz, PhCH2) , 4.94〜 5.00 (2 H, m, H-3, 5), 5.43 (1 H, d, J=1.5 Hz, OCH), 7.25〜7.33 (5 H, m, arom. H).
混合物の MS m/z (¾): no M+, 403 (47), 91 (100). (実施例 39) (&3*,21,61 -ぉょび(&1^2 61 -{[4-べンジルォキシ-2,6-ビ ス- [(t -プチルジメチルシリル)ォキシ]-シクロへキシリデン } -ァセタルデヒド
(化合物 31)
- 78°Cに冷却した化合物 30 (310 ig, 0.635 蘭 ol, 30a : 30b =約 1:2の混合 物) の無水トルエン (3 ml) 溶液に、 水素化ジ- iso-ブチルアルミニウム (763 0.763 mmol, 1.0 Mトルエン溶液) をゆっくりと加え、 1.5時間撹拌した 。 反応液に飽和酒石酸ナトリゥムカリゥム水溶液を加え反応を終結させた後、 混合物に氷水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄 し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣はシリカゲルカラムクロ マトグラフィ一 (13 g, 5%酢酸ェチル /へキサン) にて精製し、 化合物 31 ( 288.5 mg, 93¾) を二種の立体異性体の混合物として得た。 この混合物を構成 する異性体の比率は約 1 : 2であったが、 aS*, 2R,6R体と aR*,2R,6R体のいずれ が主生成物かは判断できなかった。
混合物の NMRデータ
31a (マイナー生成物、 低極性異性体) : ¾ NMR (CDC13) δ: 0·03〜0.07 (12 H, Si -Me x 4), 0.85, 0.92 (each 9 H, s, Si-tBu x 2), 1.47 (2 H, m), 2.33, 2.44 (eachl H, m), 3.97 (1 H, m, H-l), 4.56 (2 H, s, PhCH2) , 4.69 (1 H, ddd, J=11.8, 5.3, 1.7 Hz), 5.53 (1 H, m), 6.16 (1 H, dd, J=7.9, 1.6 Hz, C=CH) , 7.26〜 7.36 (5 H, m, arom. H), 10.09 (1 H, d, J=7.8 Hz, CH0) .
31b (主生成物、 高極性異性体) : ¾ NMR (CDC13) 6: 0·03〜0.07 (12 Η,
Si -Me x 4), 0.88 (18 H, s, Si-tBu x 2), 1.78, 1.90, 2.04, 2.17 (each 1 H, m), 3.93 (1 H, m, H-l), 4.53, 4.58 (each 1 H, d, J=11.8 Hz, Ph¾) , 4.61 (1 H, m), 5.12 (1 H, dd, J=8.9, 4.3 Hz), 5.88 (1 H, d, J=7.7 Hz, OCH), 7.25〜7.36 (5 H, m, arom. H), 10.49 (1 H, d, J=7.7 Hz, CHO). 混合物の MS m/z (%): no M+, 449 (32), 433 (3), 358 (4), 341 (13), 325 (7), 317 (5), 209 (10), 91 (100).
(実施例 40) (aS*, 2R, 6R)-および(aR*, 2R, 6R)- { [4 -ベンジルォキシ- 2, 6 -ビ ス- [ (t-プチルジメチルシリル)ォキシ]-シクロへキシリデン} -エタノール (化 合物 32)
0 に冷却したアルデヒド体である化合物 31 (288 mg, 0.587 mmol, 31a : 31b -約 1 : 2の異性体の混合物) のエタノール (1 ml) 溶液に、 水素化ホウ素 ナトリウム (26.6 mg, 0.704匪 ol) を加え、 1時間撹拌した。 反応混合物に氷 水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫 酸ナトリウム乾燥、 溶媒を留去した。 残渣はシリカゲルカラムクロマトグラフ ィー (10 g, 15%酢酸ェチル へキサン) にて精製し、 化合物 32 (283.3 g, 98%) を二種の立体異性体の混合物として得た。 この混合物を構成する異性体 の比率は約 2 : 1であったが、 aS*,2R,6R体と aR*,2R,6R体のいずれが主生成物 かは判断できなかった。
混合物の NMRデータ
32a (主生成物、 低極性異性体) : 'Η NMR (CDC13) δ: 0.008, 0.04, 0.10, 0.11 (each 3 H, s, Si -Me x 4), 0.85, 0.92 (each 9 H, s, Si-tBu x 2), 1.55 (1 H, m), 1.68 (1 H, q, J=10.8 Hz), 2.08 (1 H, m), 2.20 (1 H, m), 2.94 (1 H, dd, J=10.0, 4.3 Hz, H-l), 3.90 (1 H, tt, J=10.0, 4.3 Hz, H— 1), 4.02 (1 H, ddd, J=13.5, 9.2, 7.0 Hz, CH20H) , 4.34 (1 H, m, H— 3, 5),
4.40 (1H, m, CH20H) , 4.54 (2 H, s, PhCH2) , 4.69 (1 H, dd, J=10.8, 4.0 Hz, H-3, 5), 5.62 (1 H, t, J=5.8 Hz, C=CH) , 7.24〜7.35 (5 H, m, arom. H).
32b (マイナ一生成物、 高極性異性体) : Ή NMR (CDC13) δ -0.01, 0.05 5 (each 3 H, s, Si -Me x 2), 0.07 (6 H, s, Si -Me x 2), 0.83, 0.92 (each 9 H, s, Si-tBu x 2), 1.30〜1.41 (2 H, m), 2.21 (1 H, m), 2.38 (1 H, m), 3.94 (1 H, tt, J=11.3, 4.3 Hz, H-l), 4.23 (2 H, m, CH20H) , 4.48 (1 H, m, H-3, 5), 4.53, 4.57 (each 1 H, d, J=11.8 Hz, PhCH2) , 4.86 (1 H, m, H-3, 5), 5.70 (1 H, dt, J=7.1, 1.8 Hz, OCH), 7.24〜 7.35 (5 H, m, 10 arom. H) .
混合物の MS m/z (¾): no M+, 474 (8), 435 (3), 360 (1), 327 (25), 303 (8), 91 (100).
(実施例 41) (aS*, 3R, 5R)-および(aR*, 3R, 5R)-{3, 5-ビス- [(トプチルジメ 15 チルシリル)ォキシ] -4- [2 -(トプチルジメチルシリル)ォキシ] -ェチリデントシ クロへキシロキシメチル }-ベンゼン (化合物 33)
00°°CCにに冷冷却却ししたた化化合合物物 3322 ((5555 mmgg,, 00..111122讓讓 ooll,, 3322aa :: 3322bb ==約約 22::11のの異異性性体体のの 混混合合物物)) のの無無水水ジジメメチチルルホホルルムムアアミミドド ((11 mmll)) 溶溶液液にに、、 イイミミダダゾゾ一一ルル ((1188..33 2200 mmgg,, 00..226699 mmmmooll)) おおよよびび卜卜ブブチチルルジジメメチチルルシシリリルルククロロリリドド ((2200..22 mmgg,, 00..113344 匪匪 ooll)) をを加加ええ、、 11..55時時間間撹撹拌拌ししたた。。 反反応応混混合合物物にに氷氷水水をを加加ええ、、 酢酢酸酸ェェチチルルーーへへ キキササンン ((11 :: 11)) ににてて抽抽出出ししたた。。 有有機機層層はは飽飽和和食食塩塩水水ににてて洗洗浄浄、、 無無水水硫硫酸酸ママググ ネネシシゥゥムム乾乾燥燥、、 溶溶媒媒をを留留去去ししたた。。
一一
(5 g, 2%酢酸ェチル Zへキサン) にて精製し、 化合物 33 (62 mg, 91%) を二 種の立体異性体の混合物として得た。 この混合物を構成する異性体の比率は約 2 : 1であったが、 aS*,2R,6R体と aR*,2R,6R体のいずれが主生成物かは判断で
きなかった。
混合物の. N MRデータ
33a (主生成物、 低極性異性体) : NMR (CDC13) δ: 0.02, 0.04, 0.051, 0.06 (each 3 H, s, Si -Me x 4), 0.048 (6H, s, Si -Me x 2), 0.86, 0.88, 0.91 (each 9 H, s, Si-tBu x 3), 1.57〜1.68 (2 H, m), 2.02 (1 H, m), 2.10 (1 H, m), 3.86 (1 H, m, H-l), 4.36 (1 H, m), 4.38 (1 H, m, CH20TBS) , 4.52, 4.55 (each 1H, d, J=11.9 Hz, PhCH2) , 4.63 (1 H, m), 4.65 (1 H, m, CH20TBS) , 5.35 (1 H, m, C=CH) , 7.24〜7.35 (5 H, m, arom. H).
33b (マイナー生成物、 高極性異性体) : ¾ NMR (CDC13) δ: -0.01, 0.04, 0.053, 0.054 (each 3 H, s, Si -Me x 4), 0.06 (6H, s, Si -Me x 2), 0.83, 0.88, 0.92 (each 9 H, s, Si-tBu x 3), 1.30〜1.40 (2 H, m), 2.19 (1 H, m), 2.36 (1 H, m), 3.93 (1 H, tt, J=6.9, 4.3 Hz, H-l), 4.24 (2 H, m, CH20TBS) , 4.45 (1 H, m, H-3 or 5), 4.53, 4.56 (each 1H, d, J=11.8 Hz, PhCH2) , 4.83 (1 H, m), 5.60 (1 H, td, J-6.5, 1.8 Hz, C=CH) , 7.24〜7.35 (5 H, m, arom. H) .
混合物の MS m/z (%): no M+, 591 (1), 549 (23), 474 (8), 441 (27), 417 (29), 285 (13), 91 (100).
(実施例 42) 3, 5-ビス- [(t_プチルジメチルシリル)ォキシ ]-4-{ [2- (卜ブ チルジメチルシリル)ォキシ]-ェチル } -シクロへキサノン (化合物 35)
化合物 33 (116.5 mg, 0.192 腿 ol, 33a : 33b = 約 2 : 1の混合物) のエタ ノール (5 ml) 溶液に、 10%パラジウム付活性炭(11.7 mg)を加え、 常温常圧下 水素ガスと激しく撹拌した。 21時間後、 反応混合物をセライト濾過し、 酢酸ェ チルにて洗浄、 ろ液をまとめて溶媒留去した。 残渣をシリカゲルカラムクロマ
トグラフィ一(15 g, 5%酢酸ェチル /へキサン)にて精製し、 溶出部より化合物 34 (42.7 mg, 43°)を得た。 - 78°Cに冷却した二塩化ォキサリル(9 1, 0.099 腿 ol)の無水塩化メチレン(0.3 ml)溶液に、 ジメチルスルホキシド(14 1, 0.197匪 ol)の無水塩ィ匕メチレン(0.3 ml)溶液を加え 5分撹拌した後、 化合物 34 (42.7 mg, 0.082 匪 ol)の無水塩化メチレン(0.5 ml)溶液を加えた。 -78 。(:で 15分撹拌した後、 ト リェチルァミン(57 1, 0.358腿 ol)を加え、 _78°Cで 30分 、 更に室温で 1時間撹拌した。 反応混合物に氷水を加え、 塩化メチレンにて抽 出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去 した。 残渣をシリカゲルカラムクロマトグラフィ一(4 g, 2%酢酸ェチル /へキ サン)にて精製し、 化合物 35 (38.0 mg, 89%, 化合物 34からの収率)を単一化合 物として得た。
35: ¾ NMR (CDC13) δ: 0· 04〜0.06 (18 Η, Si -Me x 6), 0.86, 0.87, 0.90 (each 9 H, s, Si-tBu x 3), 1.69, 1.78 (each 1 H, m, CH2C¾0TBS) , 1.96 (1 H, ddd, J=13.1, 6.9, 3.1 Hz, H-4), 2.30 (1 H, dd, J=14.5, 6.9 Hz), 2.45 (2 H, m), 2.62 (1 H, dd, J=14.5, 4.0 Hz), 3.67〜 3.80 (2 H, m, CH20TBS) , 4.14, 4.38 (each 1 H, i, H-3, 5).
MS m/z (¾): no M+, 459 (31), 327 (41), 195 (100).
(実施例 43) (aS*, 3R, 5R)-および(aR*, 3R, 5R)-{3, 5-ビス- [(t-ブチルジメ チルシリル)ォキシ ]-4- [2- -プチルジメチルシリル)ォキシ]-ェチル卜シクロ へキシリデン } -酢酸 メチルエステル (化合物 36)
- 78Τ:に冷却したジイソプロピルアミン(90 1, 0.641 mmol)の無水テトラヒ ドロフラン(1 ml)溶液に、 n-ブチルリチウム (458 I, 0.641 匪 ol, 1.4 Mへ キサン溶液)を加え 15分撹拌した後、 (トリメチルシリル) 酢酸メチル(105 /
1, 0.641 腿 ol)を加えた。 10分間撹拌後、 化合物 35 (165.7 mg, 0.321 mmol) の無水テトラヒドロフラン(1.3 ml)溶液をゆっくり加え、 - 78 で 2時間撹拌し た。 反応混合物に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出し た。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した 。 残渣をシリカゲルカラムクロマトグラフィー(6 g, 2%酢酸ェチル Zへキサン )にて精製し、 化合物 36 (163.0 mg, 89%, 1 : 1の異性体の混合物)を二種の立 体異性体の混合物として得た。 この混合物を構成する異性体の比率は約 1 : 1 であった。
36 (異性体の混合物): ¾ NMR (CDC13) δ 0.03〜0.08 (18 Η, s, Si -Me x 6), 0.84〜 0.89 (27 H, s, Si-tBu x 3), 1.58〜 1.77 (3 H, m, H-4, CH2CH2OTBS) , 2.14 (1 H, m), 2.26 (1 H, m), 2.47 (1 H, dd, J=13.4, 3.8 Hz), 2.62, 2.70 (1 : 1) (1 H, m), 3.22 (1 H, m), 3.62〜3.73 (2 H, m, CH20TBS) , 3.668, 3.674 (1 : 1) (3 H, s, COOMe) , 3.90, 4.16 (each 1 H, H-3, 5), 5.65, 5.69 (1 : 1) (1 H, s, OCH).
MS m/z (%): no M+, 557 (2), 515 (49). 483 (5), 425 (3), 383 (52), 351 (22), 309 (33), 277 (23), 251 (20), 177 (82), 73 (100).
(実施例 44) (aS*, 3R, 5R)-および(aR*, 3R, 5R)-{3, 5_ビス- [(t-ブチルジメチ ルシリル)才キシ] -4- [2- (t-プチルジメチルシリル)ォキシ] -ェチル]-シク口へ キシリデン } -エタノール (化合物 37)
- 78 に冷却した化合物 36 (163.0 mg, 0.284腿 ol, 36a : 36b =約 1 : 1の 混合物)の無水トルエン(1.5 ml)溶液に、 水素化ジ -iso-ブチルアルミニウム (853 \, 0.853匪 ol, 1.0 Μトルエン溶液)を加えた。 同温にて 1時間撹拌後 、 反応液に酒石酸ナトリウムカリウム水溶液を加え、 酢酸ェチルにて抽出した
。 有機層は水洗、 飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去 した。 残渣をシリカゲルカラムクロマトグラフィー(6 g, 5%酢酸ェチル Zへキ サン)にて精製し、 化合物 37 (143.0 mg, 92%, 1 : 1の混合物)を二種の立体異 性体の混合物として得た。 この混合物を構成する異性体の比率は約 1 : 1であ つた。
37 (異性体の混合物): ¾ NMR (CDC13) δ: 0.03〜0.06 (18 Η, s, Si -Me x 6), 0·86〜 0.89 (27 H, s, Si-tBu x 3), 1.6〜 1.8 (3 H, m, H-4, CH2CH2OTBS) , 2.00〜2.24 (4 H, m, H-2, 6), 3.60〜3.74 (2 H, m, CH20TBS) , 3.78〜3.91 (1 H, m), 4.02〜4.18 (3 H, m, CH20H) , 5.47, 5.51 (1 : 1) (1 H, t, J=7.1 Hz, C=CH) .
MS m/z (¾): no M+, 487 (3), 469 (9), 459 (9), 394 (11), 355 (17), 337 (19), 263 (57), 211 (74), 171 (86), 131 (100), 73 (100).
(実施例 45) (aS*, 3R, 5R)-および(aR*, 3R, 5R)-{3, 5-ビス- [(t -プチルジメ チルシリル)ォキシ] -4 - [2- (卜プチルジメチルシリル)ォキシ] -ェチル卜シク口 へキシリデン }ェチルジフエニルホスフィン ォキシド (化合物 38)
Ot:に冷却した化合物 37 (97.8 mg, 0.179 mmol, 37a : 37b =約 1 : 1の混合 物)の無水テトラヒドロフラン(1 ml)溶液に、 n-ブチルリチウム(141 1, 0.197 匪 ol, 1.4 Mへキサン溶液)と塩化 P-トルエンスルホニル(37.6 mg, 0.197 mmol)の無水テトラヒドロフラン(0.3 ml)溶液とを順次加え、 5分間撹拌 した。 別の容器にジフエ二ルホスフィン(62/ l, 0.358 mmol)の無水テトラヒ ドロフラン(0.5 ml)溶液を作り、 0°C撹拌下で、 n_ブチルリチウム(255 1, 0.358腿 oI, 1.4 Mへキサン溶液)を加えたところ濃赤色になった。 0 に冷却 した本濃赤色溶液を上記のトシル体溶液にゆつくり滴下し、 反応混合物が赤色
を呈するまで加えた。 更に 30分間撹拌し、 水(50/21)を加え反応を止めた。 反 応混合物の溶媒留去後、 残渣を塩化メチレン(3 ml)で溶解し、 10%過酸化水素 水 (4.5 ml)を加え 0°Cにて 1時間撹拌した。 反応液に 2N亜硫酸ナトリウムを加え 、 塩化メチレンにて抽出した。 有機層は水洗、 飽和食塩水にて洗浄、 無水硫酸 マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ 一(6 g, 50%酢酸ェチル Zへキサン)にて精製し、 化合物 38 (110.5 mg, 84.0 %)を二種の立体異性体の混合物として得た。 この混合物を構成する異性体の比 率は約 1 : 1であった。
38 (異性体の混合物): ¾ N R (CDC13) δ - 0.01〜0.02 (18 Η, s, Si -Me x 6), 0.82〜(! .86 (27 H, s, Si-tBu x 3), 3.00〜3.20 (2 H, m, CH2P0) , 3.56 〜3.75, 3.99 (3 H, m, CH20TBS, H-3 or 5), 3.77, 3.99 (ca. 1 : 1) (1 H, m, H-3 or 5), 5.24 (1 H, m, C=CH) , 7.43〜7.75 (10 H, m, arom H).
MS m/z (%): no M+, 671 (100), 539 (63), 464 (15), 407 (21), 202 (53). (実施例 46) (20S)- la- [(t-プチルジメチルシリル)ォキシ ]-2 a- {[2- (ト プチルジメチルシリル)ォキシ] -ェチル} -および(2 OS) -1 a -(t -プチルジメチル シリル)ォキシ] -2β-{ [2- (t -プチルジメチルシリル)ォキシ] -ェチル} -25- [(ト リェチルシリル)ォキシ ] -19-ノルビ夕ミン! )3 t -プチルジメチルシリルエーテル (化合物 39a, 39b)
- 78。Cに冷却した化合物 38 (100.3 mg, 0.138 匪 ol, 38a : 38b=約 1 : 1の 混合物) の無水テトラヒドロフラン (1 ml) 溶液に、 n-ブチルリチウム (87 1, 0.138 匪 ol, 1.58 Mへキサン溶液) を加え、 15分後に、 グランドマンケト
ン体である化合物 14c (36.2 mg, 0.092 匪 ol) の無水テトラヒドロフラン ( 1.2 ml) 溶液をゆっくり加えた。 - 78°Cで 2時間撹拌した後、 反応液に飽和塩化 アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水に て洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラム クロマトグラフィー (10 g, 2%酢酸ェチル /へキサン) にて精製し、 化合物 39 (53.5 mg, 64 %) を二種の立体異性体の混合物として得た。 この混合物を構 成する 2 α -置換体である化合物 39aと 2 β -置換体である化合物 39bとの比率は約 1 : 2であった。 尚、 未反応原料である化合物 14c (10.3 mg, 28%) と化合物 38 (23.4 mg) とを回収した。
39: Ή NMR (CDC13) δ: 0· 03〜0· 07 (18 H, s, Si -Me x 6), 0.54 (3 H, s, H-18), 0.56 (6 H, q, J=7.9 Hz, SiCH2 x 3), 0.84〜0.90 (30 H, Si-tBu x 3, overlapped with H-21), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3), 1.19 (6 H, s, H-26, 27), 2.45, 2.58 (ca. 1 : 2) (1 H, m, H-4), 2.79 (1 H, m, H-9), 3.60〜 3.73 (2 H, m, CH2CH20) , 3.79, 4.09 (each 1 H, m, H-l, 3), 5.81 (1 H, m, H-7), 6.12 (1 H, m, H-6).
(実施例 47) (203)-1 ,25-ジヒドロキシ- 2ひ _ (2 -(ヒドロキシェチル) -お よび(20S)-1 α,25 -ジヒドロキシ- 2 j3- (2- (ヒドロキシェチル) - 19-ノルビタミ ン D3 (化合物 20- epi- YI- 7a, 20-epi-YI-7b)
化合物 39 (53.5 mg, 0.059丽 ol, 39a : 39b =約 1 : 2の混合物) の無水メ 夕ノール (1 ml) 溶液に、 カンファ一スルホン酸 (109.8 mg, 0.437皿 ol) を 加え、 室温にて 2時間撹拌した。 反応混合物に 5%炭酸水素ナトリウム水溶液を
加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マ グネシゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー
(5 g、 3%メタノール Z酢酸ェチル) にて精製し、 化合物 20- epi-YI- 7aおよび 20 - epi- YI-7bの混合物 (21.2 mg, 80 %) を得た。
化合物 20-epi_YI- 7aおよび 20- epi- YI- 7bを含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 20¾ 水/メタノール, 8ml I min) にて分離精製し、 化合物 20-epi-YI-7a (6.3 mg) および化合物 20- epi_YI_7b (11.5 mg) を得た。
20-epi-YI-7a: 'Η NMR (CDC13) δ: 0.53 (3 H, s, H-18), 0.85 (3 H, d, J=6.5 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.17 (2 H, m, H-4, 10), 2.62 (1 H, dd, J=12.8, 4.4 Hz, H-4), 2.80 (1 H, m, H-9), 2.85 (1 H, dd, J=14.2, 4.2 Hz, H-10), 3.70〜3.80 (2 H, m, H-3, CH20H) , 3.83 (1 H, m, CH20H) , 4.06 (1 H, m, H— 1), 5.82 (1 H, d, J=11.2 Hz, H-7), 6.39 (1 H, d, J=11.2 Hz, H-6).
UVAmax (EtOH): 245, 253, 262 nm.
20-epi-YI-7b: ¾ NMR (CDC13) δ: 0.55 (3 H, s, H-18), 0.85 (3 H, d, J=6.4 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.33 (1 H, dm, J=13.5 Hz, H- 4), 2.44 (1 H, br. d, J=13.5 Hz, H-4), 2.79 (1 H, m, H-9), 3.12 (1 H, dd, J=13.0, 4.0 Hz, H-10), 3.63 (1 H, m, H-l), 3.74〜 3.84 (2 H, m, CH20H) , 4.00 (1 H, m, H-3), 5.88 (1 H, d, J=11.2 Hz, H-7), 6.26 (1 H, d, J=ll.2 Hz, H-6).
UV Amax (EtOH): 244, 252, 262 nm.
(実施例 48) (1,4-0;15)-ぉょび(1,4- 3)-3,5-ビス-[(1:-ブチルジ メチルシリル)ォキシ] - 4-ヒドロキシ -4-メチル -シク口へキサノール ベンジ ルエーテル (化合物 125)
リチウムアルミニウムヒドリド (237 mg, 6.24 腿 ol) の無水テトラヒド 口フラン (5 ml) 懸濁液中に化合物 8 (2.99 g, 6.24 mmol, 約 9 : 1 の 混合物) の無水テトラヒドロフラン (15 ml) 溶液を加え、 室温にて 2.5 時間 撹拌した。 リチウムアルミニウムヒドリド (96 mg, 2.53 mmol) を追加し、 更に 3.5 時間撹拌した。 反応混合液に酒石酸ナトリゥムカリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム 乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (80 g, 50%酢酸ェチル Zへキサン) にて精製し、 二種の立体異性体のうち主生成物で ある化合物 125 (1.81 g, 60%) を得た。 尚、 マイナー生成物は単離できなか つた。 1,4-cis体と 1,4- trans体のいずれかが主生成物かは判断できなかつ た。
125 ¾ MR (CDC13) δ: 0.042 (6 H, s. Si -Me x 2), 0.062, 0.082 (each 3 H, s. Si -Me x 2) , 0.84, 0.90 (each 9 H, s, Si-tBu x 2) , 1.17 (3 H, s. Me) , 1.66 (1 H, in) , 1.88 (2 H, m) , 2.00 (1 H, m) , 2.19 (1 H, s, OH), 3.70 (2 H, m) , 3.81 (1 H, m) , 4.51, 4.54 (each 1 H, d, J=12.0 Hz, CH2P ) , 7.26-7.35 (5 H, m, arom H) .
((実実施施例例 4499)) ((11,,44--cciiss))--ままたたはは((11,,44--ttiirraannss))-- 33,,55--ビビスス--[[((11::--ブブチチルル
-- 44 -- [[((トトリリメメチチルルシシリリルル))ォォキキシシ ]] -- 44 --メメチチルル--シシククロロへへ キキササノノーールル ベベンンジジルルェェ一一テテルル ((化化合合物物 112266))
126
5
-78 C に冷却した化合物 125 (62.3 mg, 0.130 ol、 単一主異性体) の無水トルエン (2 ml) 溶液に、 トリェチルァミン (72 μΐ, 0.516 nimol) およびトリフルォロメ夕ンスルホン酸トリメチルシリル (52 μΐ, 0.260 mmol) を加え撹拌した。 約 2.5 時間をかけて反応温度を- 78 Cから- 20 C まで上げた。 5¾炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣 をシリカゲルカラムクロマトグラフィー (5 g, 5る酢酸ェチル Zへキサン) に て精製し、 化合物 126 (66.7 mg, 93%) を得た。
126 ¾ 腿 R (CDC13) δ: 0.018, 0.023, 0.04, 0.05 (each 3 H, s. Si -Me x 4) , 0.11 (9 H, s, Si-Me3) , 0.83, 0.91 (each 9 H, s, Si-tBu x 2) , 1.20 (3 H, s Me) , 1.77 (2 H, m) , 1.89 (2 H, m) , 3.56-3.66 (3 H, m) , 4.52 (2 H, s, CH2P ) , 7.26-7.35 (5 H m, arom H) .
MS m/z (%): no M+, 537 (1) , 495 (9) , 461 (3) , 387 (87) , 91 (100) -
(実施例 50) (1,4-cis)-または(l,4-trans)-3,5-ビス- [(t-ブチル ジメチルシリル)ォキシ ]-4- [(トリメチルシリル)ォキシ ]-4-メチル-シクロへ キサノール (化合物 127)
126 127
化合物 126 (66.7 mg, 0.121 mmol、 単一主異性体) の酢酸ェチル (2 ml) およびエタノール (1 ml) 混合溶液に 10%パラジウム付活性炭 (6.7 mg) を加え、 常温常圧下で水素ガスと激しく 22時間撹拌した。 反応混合物をセ ライト濾過し、 エタノールおよび酢酸ェチルにて洗浄した後、 濾液をまとめて溶 媒留去した。 残渣をシリカゲルカラムクロマトグラフィ一 (6 g, 10 酢酸ェ チル Zへキサン) にて精製し、 化合物 127 (54.0 mg, 97%) を得た。
127 1H R (CDC13) δ: 0.044, 0.055, 0.063, 0.070 (each 3 H, s. Si -Me x 4) , 0.12 (9 H, s, Si-Me3) , 0.88, 0.91 (each 9 H, s, Si-tBu x 2) , 1.22 (3 H, s. Me) , 1.70-1.90 (4 H, m) , 3.58 (1 H, t, J=2.8 Hz) , 3,68 (1 H, dd, J=11.5, 4.2 Hz) , 3.92 (1 H, m) .
MS m/z (%): no M+, 405 (3) , 387 (100) , 273 (25) . (実施例 51) (3J?,5J?)-3,5-ビス- [ (t-ブチルジメチルシリル)ォキシ] -
4 - [ (トリメチルシリル)ォキシ ] - 4 -メチル-シクロへキサノン (化合物 128)
127 128
-78 。C に冷却した二塩化ォキサリル (339 μΐ, 3.89 mmol, 単一主異性 体) の無水塩化メチレン (3 ml) 溶液に、 ジメチルスルホキシド (552 μΐ, 7.78 mmol) の無水塩化メチレン (3 ml) 溶液を加え 5分間撹拌した後、 化合 物 127 (1.50 g, 3.24 mmol) の無水塩化メチレン (6 ml) 溶液を加え た。 -78 。C で 15 分間撹拌した後、 トリェチルァミン (2.26 ml, 16.2 mmol) を加えた。 反応温度が- 78 。Cから室温になるまで約 1.5時間撹拌した。 反応混合物に氷水を加え、 塩化メチレンにて抽出した。 有機層は飽和食塩水にて
洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロ マトグラフィー (30 g, 5る 酢酸ェチル Zへキサン) にて精製し、 化合物 128
(1.36 g, 91%) を単一化合物として得た。
1281H NMR (CDC13) δ: 0.045-0.058 (12 H, Si-Me x 4), 0.15 (9 H, s, Si-Me3), 0.84, 0.90 (each 9 H, s, Si-tBu x 2), 1.35 (3 H, s, Me), 2.16 (1 H, dt, J=14.6, 2.5 Hz), 2.37 (1 H, ddd, J=14.0, 5.0, 2.1 Hz) 2.68 (1 H, dd,J=14.0, 11.3 Hz), 2.93 (1 H, dd,J=14.6, 3.1 Hz), 3.80 (1 H, t, J=3.1 Hz), 3.98 (1 H, dd, J=11.3, 5.0 Hz).
MS m/z (る): no M+, 445 (5), 403 (87), 313 (19) , 271 (56) , 143 (100) -
(実施例 52) (35*,3 ,51?)-ぉょび(32¾*,32?,5 )-[3,5-ビス-[(1:-ブチ ルジメチルシリル)ォキシ ] - 4 - [(トリメチルシリル)ォキシ ] - 4 -メチル-シクロ へキシリデン] -酢酸 メチルエステル (化合物 129)
-78 。C に冷却したジイソプロピルアミン (0.827 ml, 5.90 mmol) の無 水テトラヒドロフラン (5 ml) 溶液に、 n-ブチルリチウム (3.73 ml, 5.90 mmol, 1.58 へキサン溶液) を加え 15分間撹拌した後、 (トリメチルシリ ル) 酢酸メチル (0.969 ml, 5.90 mmol) を加えた。 10分間撹拌した後、 化 合物 128 (1.36 g, 2.95 mmol) の無水テトラヒドロフラン (6 ml) 溶液を ゆっくり加え、 -78 。C にて 1 時間撹拌した。 反応混合物に飽和塩化アンモニ ゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無 水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトダラ フィー (30 g, 2る酢酸ェチル Zへキサン) にて精製し、 化合物 129 を二種の
立体異性体の混合物 (1.30 g, 85%) として得た。 この混合物を構成する異性 体の比率は約 1:1であった。
1291H NMR (CDC13) δ: 0.02- 0.073 (12 H, Si-Me x 4), 0.126, 0.130 (ca.1:1) (9 H, s, Si- Me3), 0.81, 0.84 (ca.1:1) (9 H, s, Si-tBu), 0.91, 0.93 (ca.1:1) (9 H, s, Si-tBu), 1.25 (3 H, s: Me), 1.90-2.85 (4 H), 3.60-3.84 (2 H, m), 3.65, 3.68 (ca.1:1) (3 H, s, C02Me), 5.56, 5.71 (ca.1:1) (1 H, s, C=CH).
MS m/z (%): 516 (M+, 1), 501 (4), 459 (100) , 327 (46), 295 (61).
(実施例 53) (&5*,31?,5 )-ぉょび( *,3 ,5 )-[3,5-ビス-[(1:-ブチ ルジメチルシリル)ォキシ ] - 4 - [(トリメチルシリル)ォキシ ] - 4 -メチル-シクロ へキシリデン] -エタノール (化合物 130)
129 130
-78 。C に冷却した化合物 129 (1.30 g, 2.51 mmol) の無水トルエン (15 ml) 溶液に、 水素化ジ -iso-ブチルアルミニウム (7.53 ml, 7.53 mmol, 1 Mトルエン溶液) を加え 1.5時間撹拌した。 還元剤を酒石酸ナトリウ ムカリウム水溶液により分解した後、 反応混合物を氷水中に移し、 酢酸ェチルに て抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留 去した。 残渣をシリカゲルカラムクロマトグラフィー (30 g, 10%酢酸ェチル /"へキサン) にて精製し、 化合物 130 を二種の立体異性体の混合物 (1.20 g, 98%) として得た。 この混合物を構成する異性体の比率は約 1: 1であった。
130: ¾ NMR (CDC13) δ: 0.03-0.07 (12 H, Si-Me X 4), 0.12 (9 H, s, Si-Me3) , 0.85 (9 H, s, Si-tBu) , 0.91, 0.92 (ca. 1:1)
(9 H, s, Si-tBu) , 1.23 (3 H, s Me) , 1.85-2.75 (4 H) , 3.56-3.67 (2 H, m) , 4.08-4.14 (2 H, m) , 5.36, 5.49 (ca. 1:1) (1 H, m, C=CH) .
MS m/z (%): 488 (M+, 3) , 470 (5) , 455 (4) , 431 (10) , 413 (54) , 380 (9) , 341 (17) , 299 (23) , 73 (100) .
(実施例 54) (35*,3 ,5 )-ぉょび(31?*,3 ,51?)-[3,5-ビス-[ -ブチ シクロ
130 118
0 。Cに冷却した化合物 130 (712 mg, 1.46 匪 ol, 約 1:1の混合物) の 無水テトラヒドロフラン (10 ml) 溶液に、 n-ブチルリチウム (1.16 ml, 1.83 匪 ol, 1.58 Mへキサン溶液) および塩化 p-トルエンスルホニル (349 mg, 1.83 mmol) の無水テトラヒドロフラン (1.5 ml) 溶液を順次加え、 5 分間撹拌した。 別の容器にジフエ二ルホスフィン (0.506 ml, 2.91 mmol) の無水テトラヒドロフラン (3 ml) 溶液を用意し、 0 。Cで撹拌しながら n-ブ チルリチウム (1.84 ml, 2.91 mmol, 1.58 Mへキサン溶液) を加えたとこ ろ濃赤色になった。 0 。C に冷却した本濃赤色溶液を、 上記のトシル体溶液にゆ つくり滴下し、 反応混合物が赤色を呈するまで加えた。 更に 0 。Cにて 30分間 撹拌し、 水 (0.3 ml) を加えて反応を止めた。 反応混合物中の溶媒を留去した 後、 残渣を塩化メチレン (8 ml) に溶解し、 10 過酸化水素水 (12 ml) を加 え 0 。 Cにて 1時間撹拌した。 反応液に 2N亜硫酸ナトリゥム水溶液を加え、 塩 化メチレンにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム
乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (20 g, 30-60%酢酸ェチル へキサン) にて精製し、 化合物 118 を二種の立体異性体 の混合物 (717 mg, 73%) として得た。 この混合物を構成する異性体の比率は 約 1:1であった。
118 MR (CDC13) δ: -0.04-0.02 (12 H, Si -Me x 4) , 0.07, 0.08 (ca. 1:1) (9 H, s, Si-Me3) , 0.80, 0.83 (ca. 1:1) (9 H, s, Si-tBu) , 0.88, 0.89 (ca. 1:1) (9 H, s, Si-tBu) , 1.17, 1.18 (ca. 1:1) (3 H, s Me) , 1.6-2.6 (4 H, m) , 2.9-3.2 (2 H m) , 3.45-3.64 (2 H, m) , 5.17, 5.27 (ca. 1:1) (1 H, m, CH=C) 7.4-7.8 (10 H, m, arom H) .
(実施例 55) ( aS* , 3J? , ) -および( aJ?* , 3i? , 5J? ) - [ 3 , 5 -ビス- [ ( t -ブチ メチルシリル)ォキシ] -ェチリデ
化合物 33b (384 mg, 0.633 mmol,高極性異性体) のエタノール (5 ml) 溶液に 10 パラジウム付活性炭 (40 mg) を加え、 常温常圧下水素ガスと激し く撹拌した。 2.5 時間後、 反応混合物をセライト濾過し、 エタノールおよび酢 酸ェチルにて洗浄、 濾液を溶媒留去した。 有機層は飽和食塩水にて洗浄、 無水硫 酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ 一 (15 g, 5%酢酸ェチル Zへキサン) にて精製し、 化合物 132b (298 mg, 91%) を得た。 化合物 132bは aS*,3J?,5J?体と aR*,3i?,5J?体のいずれかであ るか判断できなかった。
同様に、 化合物 33a (800 mg, 1.32 mmol、 低極性異性体) の水素接触還 元反応により化合物 132aおよび 132bの約 2:1の混合物 (536 mg, 79%) を 得た。 化合物 132bは aS*,3J?,5i?体と a?*, 3 , 5J?体のいずれかであるか判断 できなかった。 化合物 132aは aS*,3i?,5J?体と aJ?* , 32? , 5i?体のいずれかであ るか判断できなかった。 (低極性異性体を用いた接触還元条件下において、 1位 で異性化が起こった) 。
132a (低極性異性体) ½ NMR (CDC13) δ: 0.06 (6 H, s, Si-Me x 2) , 0.07, 0.087, 0.094, 0.12 (each 3 H, s, Si-Me x 4) , 0.89 (18 H, s, Si-tBu x 2) , 0.93 (9 H, s, Si-tBu) , 1.46-1.61 (2 H, m) , 2.12 (1 H, m) , 2.32 (1 H, m) , 4.11 (1 H, m, H-l) , 4.20 (1 H, ddd, J=12.8, 6.2, 1.1 Hz, CH2OTBS) , 4.28 (1 H, ddd, J=12.8, 8.0, 1.1 Hz, CH2OTBS) , 4.82 (1 H, m) , 5.05 (1 H, m) , 5.66 (1 H, m, C=CH) .
MS m/z (%): no M+, 501 (1) , 459 (36) , 441 (18) , 384 (5) , 367 (24) , 327 (83) , 309 (4) , 73 (100) .
132b (高極性異性体) XH NMR (CDC13) δ: 0.02, 0.058, 0.064, 0.067 0.073, 0.075 (each 3 H, s, Si-Me x 6) , 0.87 0.89, 0.92 (each 9 H, s, Si-tBu x 3) , 1.43-1.51 (2 H, m) , 2.01-2.06 (1 H, m) , 2.12-2.18 (1 H, m) , 4.19 (1 H, m, H-l) , 4.27 (1 H, ddd, J=13.1, 6.5, 0.9 Hz, CH2OTBS ) , 4.37 (1 H, ddd, J=13.1, 5.7, 0.9 Hz, CH2OTBS ) , 4.48, 4.86 (each 1 H, m, H-3, 5) , 5.56 (1 H, m, C=CH) .
MS m/z (%): no +, 459 (37) , 441 (22) , 384 (3) , 367 (8) , 327 (100) , 309 (5) , 73 (94) .
-78 。C に冷却した二塩化ォキサリル (7.5 μΐ, 0.086 nunol、 高極性異性 体) の無水塩化メチレン (0.2 ml) 溶液に、 ジメチルスルホキシド (12.2 μΐ 0.172 mmol) の無水塩化メチレン (100 μΐ) 溶液を加え 5分間撹拌した後、 化合物 132b (37 mg, 0.072 mol) の無水塩化メチレン (0.4 ml) 溶液を加 えた。 -78 。C で 15 分間撹拌した後、 トリェチルァミン (50 μΐ, 0.358 mmol) を加え、 反応温度を- 78 。Cから室温まで徐々に上昇させながら 1時間 撹拌した。 反応混合物を氷水中に移し、 塩化メチレンにて抽出した。 有機層は飽 和食塩水にて洗浄、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲ
(3 g, 3%酢酸ェチル /へキサン) にて精製し、 化化合合物物 113333 ( (3366..66 mmgg,, 9999%%)) をを単単一一化化合合物物ととししてて得得たた。。
同同様様にに、、 化化合合物物 113322aa ((7722..55 mmgg,, 00..114400匪匪 ooll、、 低低極極性性異異性性体体)) ののススワワンン酸酸 化化にによよりり化化合合物物 113333 ((6699 raragg,, 9966%%)) をを得得たた。。
1155 113333 ΗΗ NNMMRR ((CCDDCC1133)) δδ:: 00..0022,, 00..0066,, 00..0088,, 00..0099 ((eeaacchh 33 HΗ,, ss., SSii --MMee xx 44)) ,, 00..0077 ((66 HH,, ss,, SSii--Mee xx 22)) ,, 00..8844,, 00..9900,, 00..9911 ((eeaacchh 99 HH,, ss,, SSii--ttBBuu xx 33)) ,, 22..3366 ((11 HH,, dddd,, JJ==1144..22,, 1100..22 HHzz)) ,, 22..4466 ((11 HH,, dddd,, JJ==1144..44,, 33..33 HHzz)) ,, 22..5511 ((11 HH,, dddddd,, JJ==1144..44,, 33..66 11..99 HHzz)) ,, 22..7755 ((11 HH,, dddddd,, JJ==1144..22,, 55..66,, 11..88 HHzz)) ,, 44..3344 ((22 HH,, mm
2200 CCHH22OOTTBBSS)) ,, 44..7766 ((11 HH,, mm)) ,, 55..0044 ((11 HH,, tt ,, JJ==33..44 HHzz)) ,, 55..8811 ((11 HH,, mm,, CC==CCHH)) ..
MMSS mm//zz ((%%)):: nnoo MM++,, 445577 ((110000)) ,, 332255 ((3388)) ,, 119933 ((1133)) ..
((実実施施例例 5577)) ((3355**,,3311??,,55 ))--ぉぉょょびび(( **,,33 ,,55 ))--[[33,,55--ビビスス--[[ ((11::--ブブチチ 2255 ルルジジメメチチルルシシリリルル))ォォキキシシ ]]--44--[[22 --((tt-- * "" 才才キキシシ]] --ェェチチ
ル-シクロへキシリデン]—酢酸 メチルエステル (化合物 134)
-78 。C に冷却したジイソプロピルアミン (0.385 ml, 2.64 mmol) の無 水テトラヒドロフラン (4 ml) 溶液に、 n-ブチルリチウム (1.67 ml, 2.64 mmol, 1.58 へキサン溶液) を加え 15分間撹拌した後、 (トリメチルシリ ル) 酢酸メチル (0.433 ml, 2.64 mmol) を加えた。 10分間撹拌した後、 化 合物 133 (680 mg, 1.32 mmol) の無水テトラヒドロフラン (8 ml) 溶液を ゆっくり加え、 -78 。C にて 1 時間撹拌した。 反応混合物に飽和塩化アンモニ ゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無 水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトダラ フィ一 (15 g, 2%酢酸ェチル Zへキサン) にて精製し、 化合物 134 を二種の 立体異性体の混合物 (699 mg, 93%) として得た。 この混合物を構成する異性 体の比率は約 3: 1であったが、 aS* , 3i?, 51?体と aJ?* , 3R,5R体のいずれが主生 成物であるかは判断できなかった。
1341H NMR (CDC13) δ: 0.04-0.12 (18 H, s, Si-Me x 6), 0.80-0.93 (27 H, s, Si-tBu x 3), 3.67, 3.70 (ca.1:3) (3 H, s, C02Me), 3.90, 4.02 (ca.1:3) (1 H, m), 4.25-4.55 (4 H, m), 4.87 (1 H, m), 5.6-5.8 (2 H, m, C=CH x 2).
MS ri/z (る): no M+, 555 (3) , 513 (79) , 438 (33) , 381 (100) , 349 (8) , 249 (6) .
(実施例 58) (aS*,3J?,5i?)-および(ai?*,3i?,5 ) -[3,5-ビス- [ (1:-ブチ
ォキシ] -ェチ ル-シクロへキシリデン] -エタノール (化合物 135)
-78 。Cに冷却した化合物 134 (699 mg, 1.22 mmol, 約 3:1の混合物) の無水トルエン (6 ml) 溶液に、 水素化ジ -iso-ブチルアルミニウム (3.66 ml, 3.66 mmol, 1 トルエン溶液) を加え 1 時間撹拌した。 還元剤を酒石 酸ナトリウムカリウム水溶液により分解した後、 反応混合物を氷水中に移し、 酢 酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグネシウム乾 燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (10 g, 5% 酢酸ェチル /へキサン) にて精製し、 化合物 135 を二種の立体異性体の混合物 (567 mg, 85%) として得た。 この混合物を構成する異性体の比率は約 3: 1で あつたが、 aS*,3i?,5i?体と ai?*,3i?,5i?体のいずれが主生成物であるかは判断 できなかった。
135: XH MR (CDC13) δ: 0.06-0.07 (18 H, Si -Me x 6), 0.85-0.92 (27 H, Si-tBu x 3), 1.94-2.77 (4 H, m) , 4.0-4.4 (5 H, m) , 4.77, 4.90 (ca. 3:1) (1 H, m) , 5.50, 5.71 (ca. 3:1) (1 H, m, C=CH) , 5.60, 5.66 (ca. 3:1) (1 H, m, C-CH) .
(実施例 59) (aS*,3i?,5i?)-および(3^*,3 ,5 )-[3,5-ビス-[(1:-ブチ ルジメチルシリル)ォキシ ] - 4 - [ 2 - ( t -ブチルシリル)ォキシ〗 -ェチル -シクロ へキシリデン]ェチルジフエニルホスフィン ォキシド (化合物 119)
0 。Cに冷却した化合物 135 (164 mg, 0.302 mmol, 約 3 : 1の混合物) の 無水テトラヒドロフラン (1.5 ml) 溶液に、 n -プチルリチウム (228 μΐ, 0.360 mmol, 1.58 へキサン溶液) および塩化 p-トルエンスルホニル (68.6 mg, 0.360 mmol) の無水テトラヒドロフラン (0.5 ml) 溶液を頁 次加え、 5分間撹拌した。 別の容器にジフエニルホスフィン (105 μΐ, 0.604 mmol) の無水テトラヒドロフラン (0.5 ml) 溶液を用意し、 0 。Cで撹拌しな がら n-ブチルリチウム (382 μΐ. 0.604 mmol, 1.58 Mへキサン溶液) を 加えたところ濃赤色になった。 0 。C に冷却した本濃赤色溶液を、 上記のトシル 体溶液にゆっくり滴下し、 反応混合物が赤色を呈するまで加えた。 更に 0 。cに て 30分間撹拌し、 水 (0.1 ml) を加えて反応を止めた。 反応混合物中の溶媒 を留去した後、 残渣を塩化メチレン (3 ml) に溶解し、 10 過酸化水素水 (4 ml) を加え 0 。 Cにて 1時間撹拌した。 反応液に 2N亜硫酸ナトリゥム水溶液を 加え、 塩化メチレンにて抽出した。 有機層は飽和食塩水にて洗浄、 無水硫酸マグ ネシゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (7 g, 50%酢酸ェチル Zへキサン) にて精製し、 化合物 119 を二種の立体異性体 の混合物 (139 mg, 63%) として得た。 この混合物の ¾ MRスペクトルでは、 各異性体由来の大部分のシグナルが重なり、 正確に異性体の比率を求めることは できなかった。
119: ¾ NMR (CDC13) δ: -0.02-0.05 (18 H, Si -Me x 6), 0.80-0.90 (27 H, Si-tBu x 3), 1.90-2.60 (4 H, m) , 3.15 (2 H, m, CH2PO), 4.20-4.38 (3 H, m) , 4.71 (1 H, m) , 5.29 (1 H, m, C=CH) , 5.56 (1 H, m, CH=C) , 7.40-7,80 (10 H, m, arom H) . (実施例 60) 22-ェン -25-ヒドロキシグランドマンケトン体 (化合物
120)
下記の工程に従って、 化合物 120a、 bを製造した。
120a: R= O
120b: R=TES
5炭素ユニット(v)の合成
文献記載の方法 (Fernandez, B. , Perez, J. A. M. , Granja, J. R. , Castedo, L. , Mourino , A. , J. Org. Chem . , 1992, 57, 3173-3178 、 お よ び Fall, Y. , Vitale, C. , Mourino, A., Tetrahedron Lett. , 2000, 41, 7337 - 7340) に従レ、 ビタミン D
2力、ら 化合物 (VI) を合成した。
化合物 VI (1.17 g, 2.964 mmol) のメタノール (15 ml) 溶液を 0° C に冷却し、 p-トルエンスルホン酸一水和物 (1.69 g, 8.893 mmol) を加え、 0。 Cで 16 時間、 室温で 8 時間撹拌した。 反応液を酢酸ェチルで希釈し、 5る 炭酸水素ナトリゥム水溶液、 飽和食塩水にて洗浄し、 有機層を無水硫酸マグネシ ゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (30 g 30%酢酸ェチル Zへキサン) にて精製し、 化合物 VII (790.7 mg, 95 )を得 た。
VII XH 丽 R (CDC13) δ: 0.93 (3 H, s, H-18) , 1.01 (3 H, d, J=6.6 Hz, H-21) , 1.20 (6 H, s, H-26, 27) , 4.08 (1 H, m, H- 8), 5.37 (2 H, m, H-22, 23) .
-78 。C に冷却した二塩化ォキザリル (127 μΐ, 1.459 mmol) の無水塩化 メチレン (1 ml) 溶液に、 ジメチルスルホキシド (206 μΐ, 2.917 mmol) の無水塩化メチレン (0.5 ml) 溶液を加え 10 分撹拌した後、 化合物 VII (186 mg, 0.663 mmol) の無水塩化メチレン (2 ml) 溶液を加えた。 - 78 °Cで 15分撹拌した後、 トリェチルァミン (924 μΐ, 6.63 mmol) を加 え、 徐々に温度を上げ 0 。(:にした。 反応混合物に氷水を加え、 塩化メチレンに て抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒 留去した。 残渣をシリカゲルカラムクロマトグラフィー (7 g, 25%酢酸ェチ ル含 Zへキサン) にて精製し、 化合物 VIII (178.0 mg, 96 る)を得た。
VIII XH R (CDC13) δ: 0.67 (3 H, s, H-18) , 1.07 (3 H, d, J=6.6 Hz, H - 21), 1.20 (6 H, s, H-26, 27), 5.37 (2 H, m, H - 22, 23) .
0。C に冷却した化合物 VIII (178.0 mg, 0.639 mmol) の無水塩化メチ レン (2 ml) 溶液に、 ジイソプロピルェチルァミン (557 μΐ, 3.196 mmol) およびクロロメチルメチルェ一テル (121 μΐ, 1.596 mmol) を加え 3時間、 室温にて 3時間撹拌した。 更にジイソプロピルェチルァミン (111 μΐ 0.639 mmol) およびクロロメチリレメチレエ一テレ (24 μΐ, 0.320 iranol) を追加し、 室温にて 1.5 時間撹拌した。 反応混合物を氷水中に移し、 塩化メチ レンにて抽出した。 有機層は、 5%炭酸水素ナトリウム水溶液、 飽和食塩水にて 洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムク 口マトグラフィー (7 g, 10% 酢酸ェチル Zへキサン) にて精製し、 化合物 120a (154.2 mg, 75%) を得た。
120a
XH R ( CDC1
3 ) δ: 0.65 (3 H, s, H-18) , 1.05 (3 H, d, «7=6.7 Hz, H-21) , 1.19 (6 H, s, H-26, 27), 2.45 (1 H, dd, J=ll.l, 7.8 Hz, H-9) , 3.37 (3 H, s, OMe) , 5.28, 5.38 (each 1 H, m, H-22, 23) . 化合物 VIII (710.4 mg, 2.55 mmol) の無水ジメチルホルムアミド (10 ml) 溶液を 0。C に冷却し、 イミダゾール (520.8 mg, 7.65 mmol) および クロロトリエチルシラン (868 μΐ, 5.10 mmol) を加え、 2.5 時間撹拌した。 反応液に氷水を加え、 酢酸ェチル Zへキサン (1 : 1)にて抽出した。 有機層を飽 和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカ
(30 g, 4%酢酸ェチル Zへキサン) にて精製 し、 ィ匕合'!? ί 120b 、887.5 rag, 89 %) を得た。
120b ΧΗ NMR (CDC13) δ: 0.57 (6 H, q, J=7.9 Hz, SiCH2 x 3) 0.66 (3 H, s, H-18) , 0.95 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3) 1.05 (3 H, d, J=6.7 Hz, H-21) , 1.16 (6 H, s, H-26, 27) 2.45 (1 H, dd, «7=11.0, 7.5 Hz, H-9) , 5.24, 5.40 (each 1 H m, H-22, 23).
(実施例 61) 22-ォキサ -25-ヒ
(化合物
121)
下記の工程に従って、 化合物 121を製造した。
5炭素ユニット (XI) の合成
文南 記載の方法 (Posner, G. H. , Lee, J. K. , White, . C. , Hutchings , R. Η· , Dai, Η. , Kachinski , J. L. , Dolan, Ρ . , Kensler, Τ. W. , J. Org. Chem . , 1997, 62, 3299-3314) こ従レ 、 ビタミン D2から化合物 (X) を合成した。 ィ匕合物 X (800 mg, 2.56 mmol) ¾よびィ匕合物 XI (4.77 g, 12.80 mmol) の無水ジメチルホルムアミド (30 ml) 溶液に、 水素化ナトリウム
(3.07 g, 76.77 mmol, 60 パラフィンリキッド) を加え、 室温で 16時間 撹拌した。 反応液に氷水を加え、 酢酸ェチル へキサン (1 : 1) にて抽出し た。 有機層を飽和食塩水にて洗浄し、 硫酸マグネシウム乾燥、 溶媒留去した。 残 渣をシリカゲルカラムクロマトグラフィー (85 g、 1% 酢酸ェチル Zへキサ ン) にて精製し、 化合物 XII (1.135 g, 86 %) を得た。
XII 1H NMR (CDC13) δ: -0.01, 0.01, 0.06, 0.07 (each 3 H, s. Si -Me x 4) , 0.85, 0.89 (each 9 H, s, Si-tBu x 2) , 0.93 (3 H s, H-18) , 1.05 (3 H, d, J=6.0 Hz, H-21) , 1.21, 1.22 (each 3 H, s, H-26, 27) , 3.27 (1 H, m, H-20) , 3.31, 3.68 (each 1 H, m, H-23) , 4.00 (1 H, m, H-8) .
MS m/z (%): 512 (no M+) , 455 (1), 497 (1) , 380 (1) , 323 (3) , 295 (20) , 237 (100) , 163 (89) , 75 (81) . 化合物 XII (1.13 g, 2.20 mmol) のメタノール (10 ml) 溶液に、 p-ト ルエンスルホン酸一水和物 (2.10 g, 11.01 mmol) を加え、 室温で 8時間撹 拌した。 反応液を酢酸ェチルで希釈し、 5る炭酸水素ナトリウム水溶液、 飽和食 塩水にて洗浄し、 有機層を無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ リカゲルカラムクロマトグラフィー (30 g、 30%酢酸ェチル /へキサン) にて 精製し、 化合物 XIII (610.2 mg, 97%) を得た。
XIII XH NMR (CDC13) δ: 0.95 (3 H, s, H-18) , 1.11 (3 H, d, J=6.0 Hz, H-21) , 1.23, 1.24 (each 3 H, s, H-26, 27) , 3.30 (1 H, m, H-20) , 3.46 (1 H, m, H-23) , 3.59 (1 H, s, OH) , 3.85 (1 H, dt, J=9.5, 4.1 Hz, H-23) , 4.09 (1 H, m, H-8) . MS m/z (%): 248 (M+, 2) , 226 (1) , 197 (6) , 181 (21) , 163 (84) , 113 (45) , 69 (100) . ィ匕合物 XIII (742.7 mg, 2.611 mmol) 、 4-メチフレモレホリン N-才キシ ド (2.14 g, 18.28 mmol) 、 モレキュラーシ一ブ 4A (450 mg) の無水ジ クロロメタン (15 ml) 溶液に、 テトラプロピルアンモニゥムパ一ルテネー卜
(Pr4NRu04) (45.9 mg, 0.131 mmol) を加え室温で 1時間撹拌した。 反応 液をシリカゲルカラムクロマトグラフィー (30 g、 50% 酢酸ェチル Zへキサ ン) にて精製し、 化合物 XIV (722.8 mg, 98%) を得た。
XIV XH MR (CDC13) δ: 0.65 (3 H, s, H - 18), 1.15 (3 H, d, J=5.9 Hz, H-21) , 1.24, 1.25 (each 3 H, s, H-26, 27), 2.47 (1 H, m, H-9) , 3.25 (1 H, m, H-20) , 3.45 (1 H, m, H-23), 3.44 (1 H, s, OH), 3.88 (1 H, dt , J=9.6, 4.2 Hz, H-23) .
MS m/z (%): 282 (M+, 1), 264 (2), 195 (23), 179 (69) , 161 (41) , 113 (29) , 69 (100) . 化合物 XIV (720.2 mg, 2.550 mmol) の無水ジメチルホルムアミド (10 ml) 溶液を 0 °Cに冷却し、 イミダゾ一ル (1.04 g, 15.30 mmol) およびク 口ロトリエチルシラン (1.3 ml, 7.65 mmol) を加え、 室温で 2時間撹拌し た。 反応液に氷水を加え、 酢酸ェチル Zへキサン (1 : 1) にて抽出した。 有 機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣 をシリカゲルカラムクロマトグラフィー (35 g、 2¾酢酸ェチル Zへキサン) にて精製し、 化合物 121 (1.005 g, 99 %) を得た。
121 XH NMR (CDCI3) δ: 0.57 (6 H, q, «7=7.9 Hz, SiCH2 x 3), 0.65 (3 H, s, H-18) , 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3) , 1.10 (3 H, d, J=5.9 Hz, H-21) , 1.22, 1.24 (each 3 H, s, H- 26, 27) , 2.45 (1 H, dd, J=11.0, 7.5 Hz, H-14), 3.24 (1 H, m H-20) , 3.31, 3.73 (each 1 H, m, H-23) .
(実施例 62) 24a, 26a, 27a-トリホモ- 22, 24-ジェン -25-ヒドロキシグ ランドマンケトン (化合物 122)
文献記載の方法 (Posner, G. H. , Lee, J. K. , White, Μ. C. , Hutchings , R. Η. , Dai, Η. , Kac inski , J. L. , Dolan, P. , Kensler, Τ. W. , J. Org. Chem . , 1997, 62, 3299-3314) に準じて、 下記の工程に従って、 化合物 122を合成した。
Wittig-Horner試薬の合成
Br^\^C02Et + (nBuO)3P (nBuO)2P^"^^C02Et
(XV)
(実施例 63) loc-[(t-プチルジメチルシリル)ォキシ ]_2α-[(トリメチルシリル)ォ キシ-および la-[(t-プチルジメチルシリル)ォキシ ]- 2β- [(トリメチルシリル)ォキ シ] -22-ェン -25- [(メトキシメチル)ォキシ ]-19-ノルビタミン D3 t-プチルジメチルシ リルエーテル (化合物 136a, 136b)
-78 °Cに冷却した A環ホスフィンォキシド体 22 (268.2 mg, 0.407 mmol,約 2: 1 の混合物) の無水テトラヒドロフラン (2 ml) 溶液に、 n-ブチルリチウム (261 μΐ, 0.407 mmol, 1.56 Mへキサン溶液) を加えた。 15分撹拌後、 C/D環ケ トン体 120a (87.5 mg, 0.271 mmol) の無水テトラヒドロフラン (1 ml) 溶液を ゆっくり加えた。 -78 °Cで 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水 溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫 酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィ 一 (10 g, 2%酢酸ェチル /へキサン) にて精製し、 化合物 136 を二種の立体異 性体の混合物 (116.1 mg, 56%) として得た。 この混合物を構成する異性体 136a および 136bの比率は約 5: 4であった。 また、 10%酢酸ェチル Zへキサン溶出部 より未反応の化合物 120a (11.6 mg) を回収した。
混合物の NM Rデータ
136a (主生成物) 1H NMR (CDC13) δ: 0.04-0.06 (12 H, s, Si-Me x 4), 0.12 (9 H, s, Si-Me x 3), 0.55 (3 H, s, H-18), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H- 21), 1.20 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.37 (3 H, s, OMe), 3.54 (1 H, m, H-2), 3.80 (1 H, m, H-3), 3.87 (1 H, m, H-1), 4.73 (2 H, s, OCH20), 5.33 (2 H, m, H-22, 23), 5.81 (1 H, d, J=ll.l Hz, H-7), 6.10 (1 H, d, J=ll.l Hz, H-6).
136b (マイナ一生成物) 1H NMR (CDC13) δ: 0.04-0.06 (12 H, s, Si-Me x 4), 0.12 (9 H, s, Si-Me x 3), 0.54 (3 H, s, H-18), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.02 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.37 (3 H, s, OMe), 3.60 (1 H, m,
H-2), 3.80 (1 H, m, H-3), 3.93 (1 H, m, H-1), 4.73 (2 H, s, OC¾0), 5.33 (2 H, m, H-22; 23), 5.79 (1 H, d, J=11.2 Hz, H-7), 6.13 (1 H, d, J=11.2 Hz, H-6).
混合物の MS mlz ( ): 762 (M+, 18), 700 (28), 630 (39), 568 (57), 511 (18), 465 (25): 309 (36), 147 (35), 109 (56), 75 (100).
(実施例 64) lct-[(t-プチルジメチルシリル)ォキシ ]- 2cc-ヒドロキシ-および 1α- [(t-プチルジメチルシリル)ォキシ ]- 2β-ヒドロキシ -22-ェン -25- [(メトキシメチル) ォキシ ]-19-ノルビタミン D3 t-ブチルジメチルシリルエーテル (化合物 137a, 137b)
化合物 136 (60.0 mg, 0.0786 mmol,約 5 : 4の混合物) をテトラヒドロフラン Z 酢酸 Z水 (8 : 8 : 1, 4.25 ml) に溶解し、 室温で 18時間撹拌した。 反応液は酢酸 ェチルにて希釈し、 5%炭酸水素ナトリウム水溶液、 続いて飽和食塩水にて洗浄 した。 有機層は無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカゲ ルカラムクロマトグラフィー (6 g、 5%酢酸ェチル Zへキサン) にて精製し、 化合物 137を二種の立体異性体の混合物 (53.4 mg, 98%) として得た。 この混合 物を構成する異性体 137aおよび 137bの比率は約 5: 4であった。
混合物の N M Rデータ
137a (主生成物) 1H NMR (CDC13) δ: 0.059-0.096 (12 H, Si-Me x 4), 0.56 (3 H, s, H-18), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 21),
2.80 (1 H, m, H-9), 3.37 (3 H, s, OMe), 3.51 (1 H, m, H-2), 3.90, (1 H, m, H-3), 3.99 (1 H; m, H-1), 4.73 (2 H, s, OCH20), 5.33 (2 H, m, H-22, 23), 5.79 (1 H, d, J=ll.l Hz, H-7), 6.15 (1 H, d, J=ll.l Hz, H-6).
137b (マイナ一生成物) 1H NMR (CDC13) δ: 0.06-0.10 (12 Η, Si-Me χ 4), 0.54 (3 Η, s, Η-18), 0.86, 0.90 (each 9 Η, s, Si-tBu χ 2), 1.02 (3 Η, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H- 26, 27), 2.80 (1 H, m, H-9), 3.37 (3 H, s, OMe), 3.59 (1 H, m, H-2), 3.99 (2 H, m, H-1, 3), 4.73 (2 H, s, OCH20), 5.33 (2 H, m, H-22, 23), 5.80 (1 H, d, J=11.2 Hz, H-7), 6.18 (1 H, d, J=11.2 Hz, H-6).
混合物の MS mlz (%): 690 (M+, 6), 628 (9), 571 (7), 439 (29), 309 (11), 237 (11), 109 (63) 75 (100).
(実施例 65) lo -[(t-プチルジメチルシリル)ォキシ ]- 2a-[2-(t-プチルジメチル シリル)ォキシ] -ェトキシ] -ぉよび la-[(t-プチルジメチルシリル)ォキシ]- 2β-[2-(ί- プチルジメチルシリル)ォキシ] -ェトキシ] -22-ェン -25- [(メトキシメチル)ォキシ] - 19-ノルビ夕ミン D3 t-ブチルジメチルシリルエーテル (化合物 138a, 138b)
0 °Cに冷却した化合物 137 (44.3 mg, 0.064 mmol,約 5: 4の混合物) の無水ジ メチルホルムアミド (1 ml) 溶液に、 水素化ナトリウム (77.0 mg, 1.925 mmol, 60%パラフィンリキッド) および (2-ブロモェトキシ)小ブチルジメチルシラン
(69 μΐ, 0.320 mmol) を加え激しく撹拌した。 20時間後、 反応液に氷水を加え、 酢酸ェチルズへキサン (1 : 1) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグ
ラフィー (10 g, 2%酢酸ェチル /へキサン) にて精製し、 化合物 138 を二種の 立体異性体の混合物 (45.0 mg, 83%) として得た。 この混合物を構成する異性体 138aおよび 138bの比率は約 1: 1であった。
138 1H NMR (CDC13) δ: 0.05-0.09 (18 H, Si-Me x 6), 0.54, 0.56 (ca. 1 : 1) (3 H, s, H-18): 0.86-0.91 (27 H, Si-tBu x 3), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.24 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.19, 3.28 (ca. 1 : 1) (1 H, m, H-2), 3.37 (3 H, s, OMe), 3.5-4.1 (7 H, m, OCH2CH20, H-1, 3), 4.73 (2 H, s, OCH20), 5.32 (2 H, m, H-22, 23), 5.79 (1 H, H-7), 6.13 (1 H, H-6).
MS mlz ( ): no M+, 786 (1), 716 (4), 654 (8), 610 (8), 553 (5), 522 (10), 465 (12), 233 (60), 109 (28), 75 (100).
(実施例 66) 1α,25-ジヒドロキシ -2α-(2-ヒドロキシェトキシ) -ぉよび 1α,25-
138 101 a 101 b 化合物 138 (45.0 mg, 0.053 mmol,約 1:1の混合物) の無水メタノール (1 ml) 溶液にカンファースルホン酸 (73.8 mg, 0.318 mmol) を加え、 室温にて 2時間撹 拌した。 反応液に 5 %炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出 した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去し た。 残渣をシリカゲルカラムクロマトグラフィー (4 g,2% メタノール Z酢酸ェ チル) にて精製し、 化合物 101aと 101bの混合物 (20.3 mg, 83%,約 1: 1の混 合物) を得た。 化合物 101aおよび 101bを含む混合物を HPLC (YMC-Pack ODS- AM SH-342-5, 150 x 20 mm, 25%水/メ夕ノ一ル) にて精製し、 化合物 101a (8.3
mg) および化合物 101b (7.9 mg) を得た。
101a 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.67, 3.03, 3.33 (each 1 H, br. s, OH x 3), 2.62 (1 H, dd, J=13.5, 4.5 Hz, H-4), 2.79 (1 H, m, H-9), 2.86 (1 H, dd, J=14.4, 4.9 Hz, H-10), 3.33 (1 H, dd, J=8.0, 2.8 Hz, H- 2), 3.68-3.83 (4 H, m, OCH2CH20), 3.94 (1 H, m, H-3), 4.15 (1 H, m, H-1), 5.38 (2 H, m: H-22, 23), 5.82 (1 H, d, J=11.2 Hz, H-7), 6.33 (1 H, d, J=11.2 Hz, H-6).
UV max (EtOH): 244 (ε 27400), 252 (ε 32000), 261 (ε 21700) nm.
MS mlz (%): 462 (M+, 55), 444 (58), 426 (43), 408 (22), 346 (32), 317 (68), 299 (39), 255 (69), 237 (76), 133 (100). HR-MS mlz: 462.3348 (Calcd for C28 605: 462.3345).
101b 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.34 (1 H, br. d, J=14.2 Hz, H-4), 2.48 (1 H, dm, J=14.2 Hz, H-4), 2.62 (1 H, br. s, OH), 2.79 (1 H, m, H-9), 3.07 (1 H, dd, J=13.2, 3.8 Hz, H-10), 3.28 (1 H, dd, J=8.7, 2.7 Hz, H-2), 3.28, 3.42 (each 1 H, br. s, OH x 2), 3.64-3.87 (5 H, m, OC¾CH20, H-1), 4.17 (IH, m, H-3), 5.39 (2 H, m, H-22, 23), 5.84 (1H, d, 7=11.2 Hz, H-7), 6.27 (IH, d, J=11.2 Hz, H-6).
UV ληΐ3χ (EtOH): 243 (ε 27700), 251 (ε 32300), 261 (ε 21600) nm.
MS mlz ( ): 462 (M+, 41), 444 (44), 426 (37), 408 (17), 346 (39), 317 (55), 299 (29), 255
(59), 237 (75), 133 (100). HR-MS mlz: 462.3362 (Calcd for 8 605: 462.3345). (実施例 67) la-[(t-プチルジメチルシリル)ォキシ ]-2-ォキソ -22-ェン -25- [(メ トキシメチル)ォキシ ]-19-ノルビタミン D3 t-プチルジメチルシリルエーテル (化 合物 139)
137 139
-78 °Cに冷却した二塩化ォキサリル (7.8 μΐ, 0.088 mmol) の無水塩化メチレン (l ml) 溶液にジメチルスルホキシド (12.4 μΐ, 0.175 mmol) の無水塩化メチレン (0.2 ml) 溶液を加え、 5分撹拌した。 この冷却撹拌溶液に化合物 137 (50.4 mg, 0.073 mmol,約 5 : 4の混合物) の無水塩化メチレン (1.2 ml) 溶液を加えた。 - 78°Cで 15分撹拌した後、 トリェチルァミン (51 μΐ, 0.365 mmol) を加え、 -78°C で 40分、 0 °Cで 20分撹拌した。 反応混合物に氷水を加え、 塩化メチレンにて抽 出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去 した。 残渣をシリカゲルカラムクロマトグラフィー (5 g、 5% .酢酸ェチルズへ キサン) にて精製し、 化合物 139 (49.0 mg, 97%) を単一化合物として得た。
139 1H NMR (CDC13) δ: 0.055, 0.065, 0.069, 0.094 (each 3 H, s, Si-Me x 4), 0.56 (3 H, s, H-18), 0.88, 0.89 (each 9 H, s, Si-tBu x 2), 1.03 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H- 26, 27), 2.44 (1 H, dd, J=13.3, 8.9 Hz), 2.52 (1 H, dd, J=14.2, 3.8 Hz), 2.69 (2 H, m), 2.81 (1 H, m, H-9), 3.37 (3 H, s, OMe), 4.35 (1 H, dd, J=6.4, 4.2 Hz), 4.55 (1 H, dd, J=8.7, 5.5 Hz), 4.73 (2 H, s, OCH20), 5.34 (2 H, m, H-22, 23), 5.80 (1H, d, J=11.2 Hz, H-7), 6.34 (lH, d, J=11.2 Hz, H-6).
MS mlz (%): no M+, 631 (5), 569 (100), 437 (22), 325 (17), 109 (81), 75 (52).
(実施例 68) la-[(t-プチルジメチルシリル)ォキシ ]-2-シァノメチレン -22-ェ ン -25- [(メトキシメチル)ォキシ] -19-ノルビタミン D3 t-プチルジメチルシリルエー テルの E-異性体および Z-異性体 (化合物 140a, 140b)
139 140a 140b
-40 °Cに冷却したジェチルシアノメチルホスホナート (32 μΐ, 0.197 mmol) の 無水テトラヒドロフラン (1 ml) 溶液に、 n-ブチルリチウム (126 μ1, 0.197 mmol, 1.56 Mへキサン溶液) を加え 15分撹拌した後、 化合物 139 (68.0 mg, 0.099 mmol) の無水テトラヒドロフラン (1.2 ml) 溶液をゆっくり加えた。 -40 °Cで 2 時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて 抽出した。 有機層は飽和食塩水にて洗浄後、 無水硫酸マグネシウム乾燥、 溶媒留 去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 3%酢酸ェチルズへ キサン) にて精製し、 化合物 140を二種の立体異性体の混合物 (59.6 mg, 85%) として得た。 この混合物を構成する異性体 140a (E-異性体) および異性体 140b
(Z-異性体) の比率は約 1 : 1であった。
混合物の NM Rデータ
140a 1H NMR (CDC13) δ: 0.054, 0.065, 0.094, 0.120 (each 3 Η, s, Si-Me x 4), 0.56 (3 H, s, H-18), 0.84, 0.92 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.12 (1 H, m, H-10), 3.37 (3 H, s, OMe), 4.46 (1 H, m, H- 1), 4.73 (2 H, s, OCH20), 4.99 (1 H, t, J=2.8 Hz, H-3), 5.33 (2 H, m, H-22, 23), 5.47 (1 H d, J=1.8 Hz, C=CHCN), 5.82 (1 H, d, J=ll.l Hz, H-7), 6.18 (1 H, d, J=ll.l Hz, H-6). 140b 1H NMR (CDCI3) 5: 0.065, 0.075, 0.111, 0.133 (each 3 H, s, Si-Me x 4), 0.55 (3 H, s, H-18), 0.84, 0.92 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.80 (1 H, m, H-9), 2.99 (1 H, m, H-10), 3.37 (3 H, s, OMe), 4.57 (1 H, m, H- 3), 4.73 (2 H, s, OCH20), 5.04 (1 H, t, J=2.8 Hz, H-1), 5.33 (2 H, m, H-22, 23), 5.47 (1 H, d, J=1.8 Hz, C=CHCN), 5.78 (1 H, d, J=ll.l Hz, H-7), 6.31 (1 H, d, J=ll'.l Hz, H-6).
塞 O ϋ ZAV
じさ Θζ 9))O寸 Ι) 6寸 9 S19 ΐ%)ョ 92 Sτ, .
)()) ¾OS ε 666ζ, .
S
οτ
s
01
一 ()) SO90∞ 9 t0§3 H q ε6Ό Ή ¾ £寸is 0Γw ¾7 :。 I-.
(each 9 H, s, Si-tBu x 2), 1.03 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.65 (1 H: m, H-4), 2.80 (1 H, m, H-9), 3.00 (1 H, m, H-10), 3.37 (3 H, s, OMe), 4.70 (1 H, m, H-3), 4.73 (2 H, s, OCH20), 5.35 (2 H, m, H-22, 23), 5.53 (1 H, m, H-1), 5.84 (1 H, d, J=11.3 Hz, H-7), 6.17 (1 H, m, C=CH), 6.31 (1 H, d, J=11.3 Hz, H-6), 10.16 (1 H, d, J=7.9 Hz, CHO).
混合物の MS mlz ( ): 714 (M+, 9), 652 (13), 595 (20), 582 (13), 520 (34), 491 (23), 463 (14), 411 (17), 109 (33), 75 (100).
(実施例 70) 1α- デン] -22-ェン -25- [(メトキシメチル)ォキシ ]-19-ノルビ夕ミン D3 t-プチルジメチル シリルエーテルの E-異性体および Z-異性体 (化合物 141a, 141b)
141 142a 142b
0 °Cに冷却した化合物 141 (56.0 mg, 0.078 mmol,約 1: 1の混合物) のメタノ
(2: 1, 1.5 ml) 溶液に、 水素化ホウ素ナトリウム
(3.6 mg, 0.094 mmol) を加え、 1時間撹拌した。 反応液に氷水を加え、 酢酸ェチ ルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (6 g, 8%酢酸ェチ ル Zへキサン) にて精製し、 化合物 142a (26.3 mg, E-異性体) および化合物 142b (22.7 mg, Z-異性体) を得た。 合計収率は 87%であった。
混合物の N M Rデータ
142a 1H NMR (CDC13) δ: 0.02, 0.06, 0.08 (3 Η, 3 Η, 6 Η, s, Si-Me χ 4), 0.56 (3 Η, s, Η-
18), 0.85, 0.92 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.30 (2 H, m, H-4), 2.80 (1 H, m, H-9), 2.88 (1 H, dd, J=12.7, 4.6 Hz, H-10), 3.37 (3 H, s, OMe), 4.20, 4.30 (eachl H, m, CH2OH), 4.37 (1 H, dd, J=9.5, 4.0 Hz, H-1), 4.73 (2 H, s, OCH20), 4.81 (1 H, t, J=3.8 Hz, H-3), 5.33 (2 H, m, H-22, 23), 5.72 (1 H, m, C=CH), 5.85 (1 H, d, J=ll.l Hz, H-7), 6.14 (1 H, d, J=ll.l Hz, H-6).
142b 1H NMR (CDC1
3) δ: 0.01, 0.08, 0.08, 0.09 (each 3 H, s, Si-Me x 4), 0.55 (3 H, s, H- 18), 0.84, 0.93 (each 9 H, s, Si-tBu x 2), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.55 (1 H, dd, J=12.5, 4.9 Hz, H-4), 2.83 (2 H, m, H-9, 10), 3.37 (3 H, s, OMe), 4.22 (1 H, dd, J=12.4, 7.0 Hz, CH
2OH), 4.30 (1 H, dd, J=12.4, 7.0 Hz, C¾OH), 4.48 (1 H, m, H-3), 4.73 (2 H, s, OCH
20), 4.86 (1 H, t, J=3.2 Hz, H-1), 5.33 (2 H, m, H-22, 23), 5.72 (1 H, dt, J=7.0, 1.3 Hz, C=CH), 5.80 (1 H, d, J=ll.l Hz, H-7), 6.25 (1 H, d, J=ll.l Hz, H-6). 混合物の MS mlz (%): 716 (M
+, 1), 584 (39), 522 (14), 491 (9), 147 (8), 109 (19), 75 (100). ((実実施施例例 7711)) 11αα,,2255--ジジヒヒ
ノノルルビビタタミミンン DD33 ((EE--異異性性体体)) ((化化合合物物 110022aa))
化合物 142a (26.3 mg, 0.037 mmol) の無水メ夕ノ一ル (1 ml) 溶液にカンファ —スルホン酸 (51.2 mg, 0.220 mmol) を加え、 室温にて 2時間撹拌した。 反応液 に 5 %炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を 飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリ 力ゲルカラムクロマトグラフィー (5 g, 2% メタノール Z酢酸ェチル) にて精製
し、 化合物 102a (15.6 mg, 96%) を得、 更にこの化合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 20%水/メタノール) にて精製し、 純粋な化合 物 102a (12.5 mg) を得た。
102a 1H NMR (CD3OD) δ: 0.59 (3 Η, s, H-18), 1.05 (3 Η, d, J=6.6 Hz, H-21), 1.16 (6 H, s, H-26, 27), 2.35 (1 H, br, d, J=13.9 Hz, H-4), 2.43 (1 H, dd, J=13.9, 2.9 Hz, H-4), 2.86 (1 H, m, H-9), 3.11 (1 H, d, J=12.8, 5.0 Hz, H-10), 4.24 (2 H, m, CH2OH), 4.30 (1 H, m, H-1), 4.83 (1 H, m, H-3), 5.31, 5.42 (each 1 H, m, H-22, 23), 5.79 (1 H, dt, J=6.9, 1.8 Hz, C=CH), 5.91 (1 H, d, J=ll.l Hz, H-7), 6.23 (1 H, d, J=ll.l Hz, H-6).
UV max (EtOH): 246 (ε 37000), 254 (ε 42000), 263 (ε 27800) nm.
MS mlz (%): 444 (M+, 7), 426 (5), 408 (22), 390 (9), 372 (14), 281 (4), 263 (11), 252 (100), 147 (9), 109 (12). HR-MS mlz: 444.3246 (Calcd for C28H4404: 444.3240).
(実施例 72) 1α,25-ジヒドロキシ -2-[2- (ヒド口キシ) -ェチリデン] -22-ェン -19- ノルビタミン D3 (Z-異性体) (化合物 102b)
化合物 142b (22.7 mg, 0.032 mmol) の無水メタノール (1 ml) 溶液にカンファ ースルホン酸 (44.1 mg, 0.190 mmol) を加え、 室温にて 2.5時間撹拌した。 反応 液に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層 を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ
2% メタノール Z酢酸ェチル) にて精 製し、 化合物 102b (13.5 mg, 96%) を得、 更にこの化合物を HPLC (YMC-Pack
ODS-AM SH-342-5, 150 x 20 mm, 20%水 Zメタノール) にて精製し、 純粋な化合 物 102b (12.6 mg) を得た。
102b 1H NMR (CD3OD) δ: 0.61 (3 H, s, H-18), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.16 (6 H, s, H-26, 27), 2.65 (1 H, dd, J=12.4, 5.0 Hz, H-4), 2.85 (1 H, m, H-9), 2.93 (1 H, d, J=14.
3.0 Hz, H-10), 4.25 (2 H, m, CH2OH), 4.39 (1 H, m, H-3), 4.87 (1 H, t, J=3.0 Hz, H-1),
5.31, 5.42 (each 1 H, m, H-22, 23), 5.77 (1 H, dt, J=6.9, 1.7 Hz, C=CH), 5.89 (1 H, d,
J=ll.l Hz, H-7), 6.32 (1 H, d, J=ll.l Hz, H-6).
UV max (EtOH): 246 (ε 32500), 254 (ε 37200), 263 (ε 24500) nm.
MS mlz (%): 444 (M+, 10), 426 (5), 408 (23), 390 (27), 372 (91), 281 (54), 263 (79), 252
(57), 147 (86), 109 (100). HR-MS mlz: 444.3227 (Calcd for C28H4404: 444.3240).
(実施例 73) lot-[(t-プチルジメチルシリル)ォキシ ]-2β,2,-エポキシ-およぴ la-[(t-ブチルジメチルシリル)ォキシ ]-2α,2,-エポキシ- 22-ェン -25-[(トリェチルシ リル)ォキシ ]-19-ノルビ夕ミン D3 t-プチルジメチルシリルェ-テル (化合物 143a, 143b)
-78 °Cに冷却した A環ホスフィンォキシド体 13 (106.0 mg, 0.177 mmoL 約 3 : 1 の混合物) の無水テトラヒドロフラン (1 ml) 溶液に、 n-ブチルリチウム
(112.0 μΐ, 0.177 mmol, 1.58 Mへキサン溶液) を加え 15分撹拌後、 C/D環ケト ン体 120b (46.4 mg, 0.118 mmol) の無水テトラヒドロフラン (1.3 ml) 溶液をゆ つくり加えた。 -78 から 0 °Cまで徐々に昇温させながら 3時間撹拌した。 反
応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は 飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリ 力ゲルカラムクロマトグラフィー (10 g, 2%酢酸ェチル /へキサン) にて精製 し、 化合物 143 を二種の立体異性体の混合物 (44.4 mg, 49%) として得た。 この 混合物を構成する異性体 143aおよび 143b の比率は約 3: 1であった。 また、 15%酢酸ェチル /へキサン溶出部より未反応の化合物 120b (22.6 mg) を回収し た。
混合物の NMRデータ
143a (主生成物) 1H NMR (CDC13) δ: 0.02-0.06 (12 H, Si-Me x 4), 0.56 (3 H, s, H-18), 0.57 (6 H, q, J=7.9 Hz, Si-CH2CH3 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.95 (9 H, t, J=7.9 Hz, Si-CH2CH3 x 3), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.17 (6 H, s, H-26, 27), 2.73, 2.82 (each 1 H, d, J=5.5 Hz, CH20), 3.81, 3.87 (each 1 H, m), 5.23-5.42 (2 H, m), 5.82 (1 H, d, J=11.0 Hz, H-7), 6.21 (1 H, d,J=11.0 Hz, H-6).
143b (マイナ一生成物) 1H NMR (CDC13) δ: 0.02-0.06 (12 H, Si-Me x 4), 0.56 (3 H, s, H-18), 0.57 (6 H, q, J=7.9 Hz, Si-C¾CH3 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.95 (9 H, t, J=7.9 Hz, S1-CH2CH3 x 3), 1.02 (3 H, d, J=6.6 Hz, H-21), 1.17 (6 H, s, H-26, 27), 2.57, 2.92 (each 1 H, d, 7=5.5 Hz, CH2-0), 3.68, 4.03 (each 1 H, m), 5.23-5.42 (2 H, m), 5.82 (1 H, d, J=11.0 Hz, H-7), 6.27 (1 H, d, J=11.0 Hz, H-6).
混合物の MS mlz ( ): 772 (M+, 4), 715 (10), 583 (6), 451 (3), 173 (100).
(実施例 74) 1α,25-ジヒドロキシ -2β,2,-エポキシ- 22-ェン -19-ノルビタミン D3 (化合物 103a) 、 1α,25-ジヒドロキシ -2α,2,-エポキシ- 22-ェン -19-ノルビタミ ン D3 (化合物 103b) 、 1α,2β,25-トリヒドロキシ -2α-フルォロメチル -22-ェン -19- ノルビタミン D3 (化合物 104a) および 1α,2α,25-トリヒドロキシ -2β-フルォロメ チル -22-ェン -19-ノルビタミン D3 (化合物 104b)
化合物 143 (75.1 mg, 0.097 mmol,約 3:1の混合物) の無水テトラヒドロフラン (1 ml) 溶液に、 フッ化テトラブチルアンモニゥム (0.583 ml, 0.583 mmol, 1 M テトラヒドロフラン溶液) を加え、 室温にて 4時間撹拌した。 反応液に氷水を加 え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄、 無水硫酸マグネシ ゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 70%酢酸ェチル Zへキサン) にて精製し、 化合物 103 を二種の立体異性体の混 合物 (31.1 mg, 74%) として得た。 これらの混合物を構成する異性体 103aおよ び 103bの比率はそれぞれ約 3 : 1であった。 化合物 104は単離されなかった。 化合物 103aおよび 103bを含む混合物 (11 mg) を HPLC (liChrosorb Si 60, Hibar, 250 x 4 mm,へキサン:塩化メチレン:メタノール = 50: 50: 6) にて分離 精製し、 化合物 103a (4.8mg) および化合物 103b (807 g) を得た。
103a 1H NMR (CDC13) δ: 0.58 (3 H, s, Η-18), 1.04 (3 Η, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 2.31 (1 H, dd, J=13.5, 8.6 Hz, H-10), 2.40 (1 H, dd,J=13.6, 6.2 Hz, H-4), 2.61 (1 H, dd, J=13.6, 3.3 Hz, H-4), 2.81 (m, H-9), 2.84 (1 H, d, J=4.7 Hz, CH20), 2.94 (1 H, dd, J=13.5, 4.0 Hz, H-10), 3.07 (1 H, d, J=4.7 Hz, CH20), 3.81 (1 H, m, H-3), 3.98 (1 H,
m, H-l), 5.39 (2 H, m, H-22, 23), 5.85 (1 H, d, J=ll.l Hz, H-7), 6.39 (1 H, d, J=ll.l Hz, H-6).
103b 1H NMR (CDC13) δ: 0.58 (3 H, s, H-18), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.21 (6 H, s, H-26, 27), 2.31 (1 H, dd, /=13.7, 6.2 Hz, H-4), 2.36 (1 H, dd, J=13.7, 8.4 Hz, H-10), 2.71 (1 H, dd, J=13.7, 3.6 Hz, H-4), 2.81 (m, H-9), 2.86 (1 H, d, J=13.7, 4.4 Hz, H-10), 2.94, 2.99 (each 1 H, d, J=4.7 Hz, C¾0), 3.82 (1 H, m, H-3), 3.90 (1 H, m, H-l), 5.40 (2 H, m, H-22, 23), 5.87 (1 H, d, 7=11.2 Hz, H-7), 6.37 (1 H, d, J=11.2 Hz, H-6).
(実施例 75) 1α,2β,25-トリヒドロキシ -2α-メチル -22-ェン -19-ノルビタミン D3 (化合物 105a) および 1α, 2α,25-トリヒドロキシ -2β-メチル -22-ェン -19-ノルビ 夕ミン D3 (化合物 105b)
103 105a 105b 化合物 103 (11.7 mg, 0.027 mmol,約 3:1の混合物) の無水テトラヒドロフラン (1 ml) 溶液に、 リチウムアルミニゥムヒドリド (1.0 mg, 0.027 mmol) を加え、 室温にて 3時間撹拌した。 反応混合物に酒石酸ナトリゥムカリゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄、 無水硫酸マグネシウム 乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 75% 酢酸ェチル Zへキサン) にて精製し、 化合物 105 を二種の立体異性体の混合物 (3.9 mg, 33%, 105a: 105b=約 3:1) として得た。 化合物 105aおよび 105bを含む 混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 25%水 Zメ夕ノー ル) にて精製し、 化合物 105a (1.58 mg) および化合物 105b (261 g) を得た。 105a 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s,
H-26, 27), 1.24 (3 H, s, 2-Me), 2.36 (1 H, dd, J=14.5, 4.6 Hz, H-4), 2.54 (1 H, d, J=14.5 Hz, H-4), 2.79 (1 H, m, H-9), 2.93 (1 H, dd, J=12.6, 4.3 Hz, H-10), 3.73 (2 H, m, H-1, 3), 5.39 (2 H, m, H-22, 23), 5.83 (1 H, d, J=11.2 Hz, H-7), 6.29 (1 H, d, J=11.2 Hz, H-6). MS mlz (%): 432 (M+, 37), 414 (34), 396 (11), 378 (12), 360 (14), 305 (21), 287 (37), 269 (37), 251 (21), 135 (100). HR-MS mlz: 432.3221 (Calcd for C27H4404: 432.3240).
UV max (EtOH): 244, 252, 261 nm.
105b 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.20 (6 H, s, H-26, 27), 1.30 (3 H, s, 2-Me), 2.49 (1 H, dd, 7=14.2, 3.4 Hz, H-4), 2.62 (1 H, dd, J=14.2, 6.5 Hz, H-4), 2.67 (1 H, dd, J=13.6, 4.0 Hz, H-10), 2.80 (1 H, m, H-9), 3.74, 3.77 (each 1 H, m, H-1, 3), 5.39 (2 H, m, H-22, 23), 5.81 (1 H, d, J=11.2 Hz, H-7), 6.33 (1 H, d, J=11.2 Hz, H-6).
MS mlz (%): 432 (M+, 66), 414 (31), 396 (17), 378 (20), 360 (35), 305 (29), 287 (49), 269 (48), 251 (43), 135 (100). HR-MS mlz: 432.3246 (Calcd for C27H4404: 432.3240).
UV max (EtOH): 244, 252, 261 nm.
(実施例 76) 20-ェピ -loc-[(t-プチルジメチルシリル)ォキシ ]-2α-[(トリメチル シリル)ォキシ]-および 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]-2β-[(トリメ
D3 1-ブチルジメチルシリルェ一テル (化合物 144a. 144b)
-78でに冷却した A環ホスフィンォキシド体 22 (212.0 mg, 0.321 mmol,約 2:
1の混合物) の無水テトラヒドロフラン (3 ml) 溶液に、 n-ブチルリチウム (206 μΐ, 0.321 mmol, 1.56 Mへキサン溶液) を加え 15分撹拌した後、 C/D環ケトン体 121 (85.0 mg, 0.214 mmol) の無水テトラヒドロフラン (1 ml) 溶液をゆつくり加 えた。 -78 °Cで 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシゥ ム乾燥、 溶媒留去した。 残'渣をシリカゲルカラムクロマトグラフィー (10 g) に て精製し、 2%酢酸ェチル Zへキサン溶出部より化合物 144を二種の立体異性体 の混合物 (158.8 mg, 88%) として得た。 この混合物を構成する異性体 144aおよ び 144bの比率は約 3:2であった。 また、 5%酢酸ェチル Zへキサン溶出部より 未反応の化合物 121 (5.1 mg) を回収した。
混合物の NMRデータ
144a (主生成物) 1H NMR (CDC13) δ: 0.04-0.06 (12 H, Si-Me x 4), 0.13 (9 H, s, Si-Me x 3), 0.56 (3 H, s, H-18), 0.57 (6 H, q, J=7.9 Hz, SiCH2x 3), 0.87, 0.88 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiC¾CH3 x 3), 1.08 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.26 (1 H, m, H-20), 3.32, 3.69 (each 1 H, m, H-23), 3.53 (1 H, m, H-2), 3.80 (1 H, m, H-3), 3.89 (1 H, m, H-l), 5.79 (1 H, d, J=ll.l Hz, H-7), 6.11 (1 H, d, J=ll.l Hz, H-6).
144b (マイナ一生成物) 1H NMR (CDC13) δ: 0.04-0.06 (12 Η, Si-Me χ 4), 0.12 (9 Η, s, Si-Me χ 3), 0.54 (3 Η, s, H-18), 0.57 (6 Η, q, J=7.9 Hz, SiCH2 3), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiC¾C¾ x 3), 1.08 (3 H, d, 7=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.26 (1 H, m, H-20), 3.32, 3.69 (each 1 H, m, H-23), 3.59 (1 H, m, H-2), 3.80 (1 H, m, H-3), 3.93 (1 H, m, H-l), 5.77 (1 H, d, J=11.2 Hz, H-7), 6.14 (1 H, d, J=11.2 Hz, H-6).
混合物の MS mlz ( ): no M+, 704 (10), 647 (3), 618 (7), 572 (19), 486 (20), 469 (13), 383 (17), 309 (19), 75 (100).
(実施例 77) 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]- 2α,25-ジヒドロ キシ-および 20-ェピ -la-[(t-ブチルジメチルシリル)ォキシ ]-2β,25-ジヒドロキシ - 22-ォキサ -19-ノルビタミン D3 t-プチルジメチルシリルエーテル (化合物 145a,
145b) および 20-ェピ -lcx-[(t-ブチルジメチルシリル)ォキシ ]-2cx-ヒドロキシ-およ び 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]- 2β-ヒドロキシ -22-ォキサ -25- [(トリエチルシリル)ォキシ ]-19-ノルビタミン D3 t-プチルジメチルシリルェ一テ ル (化合物 146a, 146b)
146a 146b
化合物 144 (153.0 mg, 0.183 mmol,約 3 : 2の混合物) をテトラヒドロフラン 酢酸 Z水 (8: 8: 1; 4.25 ml) に溶解し、 室温にて 16時間撹拌した。 反応液は酢 酸ェチルにて希釈し、 5%炭酸水素ナトリウム水溶液、 続いて飽和食塩水にて洗 浄した。 有機層は無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカ ゲルカラムクロマトグラフィー (6 g) にて精製し、 5%酢酸ェチル へキサン 溶出部より化合物 146を二種の立体異性体の混合物 (37.0 mg,27%) として得た。 更に 10%酢酸ェチル Zへキサン溶出部より化合物 145を二種の立体異性体の混 合物 (77.1 mg, 65%) として得た。 これらの混合物を構成する異性体 145aおよ び 145bまたは 146aおよび 146bの比率は、 それぞれ約 3: 2であった。
混合物の NMRデータ
145a (主生成物) 1H NMR (CDC13) δ: 0.06-0.10 (12 Η, Si-Me χ 4), 0.56 (3 Η, s, Η-18), 0.87, 0.88 (each 9 Η, s, Si-tBu χ 2), 1.14 (3 Η, d, J=5.9 Hz, H-21), 1.22, 1.24 (each 3 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.28 (1 H, m, H-20), 3.46, 3.85 (each 1 H, m, H-23), 3.51 (1 H, m, H-2), 3.59 (1 H, s, OH), 3.92 (1 H, m, H-3), 4.00 (1 H, m, H-1), 5.78 (1 H, d, J=ll.l Hz, H-7), 6.16 (1 H, d, J=ll.l Hz, H-6).
145b (マイナー生成物) 1H NMR (CDCI3) δ: 0.06-0.10 (12 Η, Si-Me χ 4), 0.54 (3 Η, s, Η-18), 0.86, 0.89 (each 9 Η, s, Si-tBu χ 2), 1.14 (3 Η, d, J=5.9 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.28 (1 H, m, H-20), 3.46, 3.85 (each 1 H, m, H-23): 3.59 (1 H, m, H-2), 3.59 (1 H, s, OH), 4.00 (2 H, m, H-3, 1), 5.78 (1 H, d, J=11.2 Hz, H- 7), 6.19 (1 H, d, J=11.2 Hz, H-6).
混合物の MS mlz (%): 650 (M+, 2), 632 (8), 546 (6), 489 (8), 443 (10), 357 (8), 265 (22), 113(30), 75 (100).
混合物の N M Rデータ
146a (主生成物) 1H NMR (CDC13) δ: 0.06-0.10 (12 H, Si-Me x 4), 0.56 (3 H, s, H-18), 0.56 (6 H, q, J=7.9 Hz, SiCH2 x 3), 0.87, 0,88 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, S1CH2CH3 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.81 (1 H, m, H-9), 3.26 (1 H, m, H-20), 3.32, 3.70 (each 1 H, m, H-23), 3.51 (1 H, m, H- 2), 3.91 (1 H, m, H-3), 4.01 (1 H, m, H-1), 5.78 (1 H, d, J=ll.l Hz, H-7), 6.17 (1 H, d, J=ll.l Hz, H-6).
146b (マイナ一生成物) 1H NMR (CDC13) δ: 0.06-0.10 (12 Η, Si-Me χ 4), 0.55 (3 Η, s, Η-18), 0.56 (6 Η, q, J=7.9 Hz, SiCH2 x 3), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3 x 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.81 (1 H, m, H-9), 3.26 (1 H, m, H-20), 3.32, 3.70 (each 1 H, m, H-23), 3.59 (1 H, m, H-2), 4.01 (2 H, m, H-1, 3), 5.78 (1 H, d, J=11.2 Hz, H-7), 6.20 (1 H, d, J=11.2 Hz, H- 6).
混合物の MS mlz (%): 764 (M+, 1), 707 (1), 632 (4), 575 (2), 546 (3), 489 (4), 443 (5), 357 (20), 265 (11), 103 (31), 75 (100).
(実施例 78) 20-ェピ -loc-[(t-プチルジメチルシリル)ォキシ ]- 2a-[2-(t-ブチル
ジメチルシリル)ォキシ] -ェトキシ] -および 20-ェピ -la-[(t-プチルジメチルシリル) ォキシ]- 2β-[2-(ί-ブチルジメチルシリル)ォキシ] -ェトキシ] -22-ォキサ -25-ヒドロキ シ -19-ノルビ夕ミン D3 1-プチルジメチルシリルェ一テル (化合物 147a, 147b)
0 °Cに冷却した化合物 145 (73.5 mg, 0.113 mmol,約 3: 2の混合物) の無水ジ メチルホルムアミド (2 ml) 溶液に、 水素化ナトリウム (135.0 mg, 3.375 mmol, 60% パラフィンリキッド) および (2-プロモエトキシ) -t-プチルジメチルシラン (118 μΐ, 0.550 mmol) を加え激しく撹拌した。 19時間後、 反応液に氷水を加え、 酢酸ェチル /へキサン (1 : 1) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグ ラフィ一 (10 g、 10 ~15%酢酸ェチルノへキサン) にて精製し、 化合物 147を 二種の立体異性体の混合物 (65.0 mg, 71%) として得た。 この混合物を構成する 異性体 147aおよび 147bの比率は約 3: 2であった。
147 1H NMR (CDC13) δ: 0.05-0.07 (18 H, Si-Me x 6), 0.54, 0.55 (ca. 2 : 3) (3 H, s, H-18), 0.86-0.89 (27 H, Si-tBu x 3), 1.13 (3 H, d, J=5.5 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.80 (1 H, m, H-9), 3.2-4.1 (10 H, m, OCH2CH20, H-l, 2, 3, 20, 23), 5.77 (1 H, H- 7), 6.14 (1 H, H-6).
MS mlz ( ): no M+, 790 (1), 676 (4), 658 (5), 572 (6), 526 (5), 397 (18), 233 (74), 75 (100).
(実施例 79) 20-ェピ -1α,25-ジヒドロキシ -2α-(2-ヒドロキシエトキシ) -およ
び 20-ェピ -1α,25-ジヒドロキシ- 2β-(2-ヒドロキシェトキシ) -22-ォキサ -19-ノルビ 夕ミン D3 (化合物 106a, 106b)
147 106a 106b
化合物 147 (63.0 mg, 0.0778 mmol) の無水メタノール (1.5 ml) 溶液にカンフ ァ一スルホン酸 (108.5 mg, 0.467 mmol) を加え、 室温にて 2時間撹拌した。 反 応液に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機 層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣を シリカゲルカラムクロマトグラフィー (5 g, 2% メタノール 酢酸ェチル) にて 精製し、 化合物 106aと 106bの混合物 (33.0 mg, 91%,約 3: 2の混合物) を得た。 化合物 106aと 106bを含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 25%水 Zメタノール) にて分離精製し、 化合物 106a (13.9 mg) および 化合物 106b (10.3 mg) を得た。
106a 1H NMR (CDC13) δ: 0.55 (3 H, s, H-18), 1.12 (3 H, d, J=5.9 Hz, H-21), 1.22, 1.23 (each 3 H, s, H-26, 27), 2.60 (1 H, dd, J=13.4, 4.5 Hz, H-4), 2.79 (1 H, m, H-9), 2.86 (1 H, dd, J=14.5, 4.8 Hz, H-10), 3.26 (1 H, m, H-20), 3.31 (1 H, dd, J=8.1, 2.7 Hz, H-2), 3.45 (1H, m, H-23), 3.57 (1 H, s, OH), 3.66 ~ 3.86 (5 H, m, OCH2CH20, H-23), 3.92 (1 H, m, H-3), 4.14 (1 H, m, H-l), 5.79 (1 H, d, J=11.2 Hz, H-7), 6.33 (1 H, d, J=11.2 Hz, H-6). UV max (EtOH): 243 (ε 29600), 251 (ε 34500), 261 (ε 23200) nm.
MS mlz (%): 466 (M+, 39), 448 (30), 430 (13), 362 (14), 345 (12), 317 (13), 237 (9), 133
(20), 113 (50), 69 (100). HR-MS mlz: 466.3267 (Calcd for 7¾606: 466.3294).
106b 1H NMR (CDCI3) δ: 0.55 (3 H, s, H-18), 1.13 (3 H, d, J=5.9 Hz, H-21), 1.22, 1.24
(each 3 H, s, H-26, 27), 2.34 (1 H, br. d, J=14.1 Hz, H-4), 2.48 (1 H, dm, J=14.1 Hz, H-4): 2.67 (1 H, br. s, OH), 2.79 (1 H, m, H-9), 3.07 (1 H, dd, J=13.4, 3.8 Hz, H-10), 3.27 (2 H, m, H-2, 20), 3.45 (1 H, m, H-23), 3.56 (1 H, s, OH), 3.64-3.87 (6 H, m, OC¾CH20, H-l; 23), 4.17 (1H, m, H-3), 5.82 (1H, d, J=11.2 Hz, H-7), 6.27 (1H, d, J=11.2 Hz, H-6).
UV max (EtOH): 243 (ε 32500), 251 (ε 37900), 261 (ε 25100) nm.
MS mlz (%): 466 (M+, 28), 448 (22), 430 (11), 362 (9), 345 (9), 317 (9), 237 (11), 133 (19), 113 (43), 69 (100). HR-MS mlz: 466.3300 (Calcd for C27H4606: 466.3294).
(実施例 80) 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]-2-ォキソ -22-ォ キサ -25-[(トリェチルシリル)ォキシ ]-19-ノルビ夕ミン D3 t-ブチルジメチルシリル エーテル (化合物 148)
-78 °Cに冷却した二塩ィ匕ォキサリル (8.3 μ1, 0.095 mmol) の無水塩化メチレン (l ml) 溶液にジメチルスルホキシド (13.5 μΐ, 0.190 mmol) の無水塩化メチレン (0.2 ml) 溶液を加え 5分撹拌した後、 化合物 146 (60.7 mg, 0.079 mmol,約 3 : 2 の混合物) の無水塩化メチレン (1.2 ml) 溶液を加えた。 -78 °Cで 15分撹拌した 後、 トリェチルァミン (55 μΐ, 0.397 mmol) を加え、 -78 °Cで 30分、 0でで 10 分撹拌した。 反応混合物に氷水を加え、 塩化メチレンにて抽出した。 有機層を飽 和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカ ゲルカラムクロマトグラフィ一 (5 g, 2%酢酸ェチル /へキサン) にて精製し、 化合物 148 (59.5 mg, 98%) を単一化合物として得た。
148 Ή NMR (CDCI3) δ: 0.057, 0.066, 0.070, 0.097 (each 3 H, s, Si-Me x 4), 0.56 (3 H, s, H-18), 0.57 (6 H, q, J=7.9 Hz, SiCH2x 3), 0.87, 0.89 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, S1CH2CH3X 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.45 (1 H, dd, J=13.2, 8.7 Hz), 2.52 (1 H, dd, J=14.0, 4.0 Hz), 2.70 (2 H, m), 2.81 (1 H, m, H-9), 3.26 (1 H, m, H-20), 3.32, 3.70 (each 1 H, m, H-23), 4.36 (1 H, dd, J=6.3, 4.2 Hz), 4.55 (1 H, dd, J=8.7, 5.5 Hz), 5.79 (IH, d, J=11.0 Hz, H-7), 6.37 (1H, d, J=11.0 Hz, H-6).
MS mlz ( ): no M+, 705 (8), 573 (12), 487 (22), 355 (12), 103 (51), 75 (100).
(実施例 81 ) 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]-2-シァノメチレ ン -22-ォキサ -25-[(トリエヂルシリル)ォキシ ]-19-ノルビ夕ミン D3 t-ブチルジメチ ルシリルエーテルの E-異性体および Z-異性体 (化合物 149a, 149b)
-40 °Cに冷却したジェチルシアノメチルホスホナート (51 μ1, 0.315 mmol) の 無水テトラヒドロフラン (1 ml) 溶液に n-ブチルリチウム (202 μΐ, 0.315 mmol, 1.56 Mへキサン溶液) を加え 15分撹拌した後、 化合物 148 (120.3 mg, 0.157 mmol) の無水テトラヒドロフラン (1.2 ml) 溶液をゆっくり加えた。 -40。 で 1.5時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチル にて抽出した。 有機層は飽和食塩水にて洗浄後、 無水硫酸マグネシウム乾燥、 溶 媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (8 g, 1%酢酸ェチル /へキサン) にて精製し、 化合物 149 を二種の立体異性体の混合物 (120.6 mg,
97%) として得た。 この混合物を構成する異性体 149a (E-異性体) および異性体 149b (Z-異性体) の比率は約 1:1であった。
混合物の NMRデータ
149a 1H NMR (CDC13) δ: 0.05, 0.07, 0.10, 0.12 (each 3 Η, s, Si-Me x 4), 0.56 (3 H, s, H- 18), 0.56 (6 H, q, J=7.9 Hz, SiCH2 x 3), 0.84, 0.92 (each 9 H, s, 2 x Si-tBu x 2), 0.93 (9 H, t, J=7.9 Hz, SiCH2CH3 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.31, 2.37 (each 1 H, m, H-4), 2.80 (1 H, m, H-9), 3.12 (1 H, m, H-10), 3.26 (1 H, m, H-20), 3.32, 3.69 (each 1 H, m, H-23), 4.46 (1 H, m, H-1), 4.99 (1 H, t, J=2.8 Hz, H-3), 5.47 (1 H, d, J=1.8 Hz, C=CHCN), 5.80 (1 H, d, J-ll.l Hz, H-7), 6.20 (1 H, d, J=ll.l Hz; H-6).
149b 1H NMR (CDCls) δ: 0.06, 0.08, 0.11, 0.13 (each 3 H, s, Si-Me x 4), 0.56 (3 H, s, H- 18), 0.56 (6 H, q, J=7.9 Hz, SiCH2 3), 0.84, 0.92 (each 9 H, s, 2 x Si-tBu), 0.94 (9 H, t, 7=7.9 Hz, SiCH2CH3x 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.61 (1 H, m, H-4), 2.82 (1 H, m, H-9), 2.99 (1 H, m, H-10), 3.26 (1 H, m, H-20), 3.32, 3.70 (each 1 H, m, H-23), 4.58 (1 H, ddd, j=11.0, 5.9, 1.9 Hz, H-3), 5.04 (1 H, t, J=2.7 Hz, H-1), 5.47 (1 H, d, J=1.9 Hz, C=CHCN), 5.77 (1 H, d,J=11.2 Hz, H-7), 6.33 (1 H, d, J=11.2 Hz, H-6).
混合物の MS mlz (%): 785 (M+, 2), 728 (8), 701 (12), 653 (6), 596 (9), 569 (16), 510 (17), 483 (11), 103 (66), 75 (100).
(実施例 82) 20-ェピ -la-[(t-プチルジメチルシリル)ォキシ ]-2-[2- (ホルミル) - ェチリデン] -22-ォキサ -25-[(トリェチルシリル)ォキシ ]-19-ノルビ夕ミン D3 1-プチ ルジメチルシリルェ一テルの E-異性体および Z-異性体 (化合物 150a, 150b)
-78 °Cに冷却した化合物 149 (77.0 mg, 0.098 mmol,約 1: 1の混合物) の無水 トルエン (1 ml) 溶液に水素化ジ -iso-ブチルアルミニウム (147 μΐ, 0.147 mmol, 1.0 M トルエン溶液) を加え、 2時間撹拌した。 反応液をへキサンにて希釈し、 直接シリカゲルカラムクロマトグラフィー (8 g、 5%酢酸ェチル Zへキサン) にて精製し、 化合物 150を二種の立体異性体の混合物 (66.9 mg, 87%) として得 た。 この混合物を構成する異性体 150a (E-異性体) および異性体 150b (Z-異性 体) の比率は約 1:1であった。
混合物の NMRデータ
150a 1H NMR (CDC13) δ: 0.01, 0.07, 0.09, 0.10 (each 3 Η, s, Si-Me x 4), 0.57 (3 H, s, H- 18), 0.56 (6 H, q, J=7.9 Hz, SiC¾x 3), 0.84, 0.92 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3x 3), 1.09 (3 H, d, 7=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.42 (2 H, m, H-4), 2.80 (1 H, m, H-9), 3.05 (1 H, dd, J=12.8, 5.3 Hz, H-10), 3.26 (1 H, m, H-20), 3.32, 3.69 (each 1 H, m, H-23), 4.57 (1 H, m, H-1), 5.46 (1 H, t, 7=3.3 Hz, H-3), 5.83 (1 H, d, J=ll.l Hz, H-7), 6.15 (1 H, dd, J=7.9, 1.1 Hz, C=CH), 6.19 (1 H, d, J=ll.l Hz, H-6), 10.18 (1 H, d, J=7.9 Hz, CHO).
150b 1H NMR (CDCI3) δ: 0.02, 0.08, 0.10, 0.11 (each 3 H, s, Si-Me x 4), 0.56 (3 H, s, H- 18), 0.57 (6 H, q, J=1.9 Hz, SiCH2x 3), 0.84, 0.93 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3 3), 1.09 (3 H, d, j=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.65 (1 H, m, H-4), 2.80 (1 H, m, H-9), 3.00 (1 H, m, H-10), 3.26 (1 H, m, H-20), 3.32, 3.71 (each 1 H, m, H-23), 4.69 (1 H, m, H-3), 5.54 (1 H, m, H-1), 5.78 (1 H, d, J=11.2 Hz, H-7), 6.16 (1 H, dd, 7=7.9, 1.1 Hz, C=CH), 6.32 (1 H, d, J=ll.l Hz, H-6),
10.16 (1 H, d, J=7.9 Hz, CHO).
混合物の MS mlz (%): 788 (M+, 5), 731 (5), 656 (8), 627 (7), 599 (4), 524 (5), 495 (3), 409 (5), 103 (42), 75 (100). (実施例 83) 20-ェピ -lcc-[(t-ブチルジメチルシリル)ォキシ ]-2-[2- (ヒドロキ シ) -ェチリデン] -22-ォキサ -25-[(トリェチルシリル)ォキシ ]-19-ノルビ夕ミン D3 t- プチルジメチルシリルェ一テルの E-異性体および Z-異性体 (化合物 151a, 151b)
0 °Cに冷却した化合物 150 (97.0 mg, 0.123 mmol,約 1: 1の混合物) のメ夕ノ —ル/テトラヒドロフラン (2: 1, 1.5 ml) 溶液に水素化ホウ素ナトリゥム (5.6 mg, 0.148 mmol) を加え、 0.5 時間撹拌した。 反応液に氷水を加え、 酢酸ェチル にて抽出した。 有機層を飽和食塩水にて洗、净し、 無水硫酸マグネシウム乾燥、 溶 媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (10 g、 7%酢酸ェチ ル Zへキサン) にて精製し、 化合物 151a (43.6 mg, E-異性体) および化合物 151b (35.5 mg, 異性体) を得た。 全体収率は 81%であった。
混合物の NM Rデータ
151a 1H NMR (CDC13) δ: 0.01, 0.07, 0.08 (3 Η, 3 Η, 6 Η, s, Si-Me χ 4), 0.56 (3 Η, s, Η- 18), 0.56 (6 Η, q, j=7.9 Hz, SiCH2x 3), 0.85, 0.92 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3x 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.31 (2 H, m, H-4), 2.80 (1 H, m, H-9), 2.88 (1 H, dd, J=12.6, 4.6 Hz, H-10), 3.26 (1
H, m, H-20), 3.32, 3.70 (each 1 H, m, H-23), 4.18, 4.31 (eachl H, m, CH2OH), 4.37 (1 H, m, H-1), 4.82 (1 H, t, J=3.8 Hz, H-3), 5.72 (1 H, m, C=CH), 5.83 (1 H, d, J=11.0 Hz, H- 7), 6.15 (1 H, d, J=11.0 Hz, H-6).
151b 1H NMR (CDC13) δ: 0.01, 0.07, 0.08, 0.10 (each 3 H, s, Si-Me x 4), 0.56 (3 H, s, H- 18), 0.57 (6 H, q, J=7.9 Hz, SiCH2x 3), 0.84, 0.93 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, S1CH2CH3 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.55 (1 H, dd, J=12.5, 5.0 Hz, H-4), 2.83 (2 H, m, H-9, 10), 3.26 (1 H, m, H-20), 3.32: 3.70 (each 1 H, m, H-23), 4.22, 4.27 (each 1 H, m, CH2OH), 4.48 (1 H, m, H-3), 4.86 (1 H, t, J=3.1 Hz, H-1), 5.72 (1 H, dt, J=7.0, 1.4 Hz, C=CH), 5.79 (1 H, d, J=ll.l Hz, H-7), 6.26 (1 H, d, J=ll.l Hz, H-6).
混合物の MS mlz (%): 790 (M+, 1), 772 (1), 733 (1), 658 (45), 627 (11), 526 (7), 508 (7), 376 (5), 103 (33), 75 (100).
(実施例 84) 20-ェピ -1α,25-ジヒドロキシ -2-[2- (ヒドロキシ) -ェチリデント 22-ォキサ -19-ノルビ夕ミン D3 (E-異性体) (化合物 107a)
151a 107a
化合物 151a (43.6 mg, 0.055 mmol) の無水メ夕ノ一ル (1 ml) 溶液にカンファ ースルホン酸 (76.8 mg, 0.331 mmol) を加え、 室温にて 2時間撹拌した。 反応液 に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を 飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリ
一 (5 g, 2% メタノール Z酢酸ェチル) にて精製
し、 化合物 107a (23.7 mg, 96%) を得た。 更にこの化合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 20%水 Zメ夕ノール) にて精製し、 純粋な化合 物 107a (20.1 mg) を得た。
107a XH NMR (CDC13) δ: 0.54 (3 H, s, H-18), 1.13 (3 H, d, J=5.9 Hz, H-21), 1.22, 1.23 (each 3 H, s, H-26, 27), 2.33, 2.43 (each 1 H, m, H-4), 2.79 (1 H, m, H-9), 3.12 (1 H, d, J=12.5, 4.4 Hz, H-10), 3.26 (1 H, m, H-20), 3.44 (1 H, m, H-23), 3.51, 3.58, 3.90 (each 1 H, br. s, OH x 3), 3.84 (1 H, dt, J=9.4, 4.3 Hz, H-23), 4.08 (1 H, dd, J=12.4, 5.2 Hz, CH2OH), 4.33 (2 H, m, H-1, CH2OH), 4.79 (1 H, m, H-3), 5.74 (1 H, m, C=CH), 5.84 (1 H, d, J=ll.l Hz, H-7), 6.26 (1 H, d,J=ll.l Hz, H-6).
UV λπΐ3χ (EtOH): 246 (ε 34600), 254 (ε 39700), 263 (ε 26500) nm.
MS mlz (%): 448 (M+, 9), 430 (8), 412 (14), 394 (26), 376 (12), 308 (13), 263 (12), 131 (20), 113 (39), 69 (100). HR-MS mlz: 448.3188 (Calcd forじ27¾405: 448.3189).
(実施例 85) 20-ェピ -1α,25-ジヒドロキシ -2-[2- (ヒドロキシ) -ェチリデント 22-ォキサ -19-ノルビ夕ミン D3 (Z-異性体) (化合物 107b)
化合物 151b (35.5 mg, 0.045 mmol) の無水メタノール (1 ml) 溶液にカンファ —スルホン酸 (62.5 mg, 0.269 mmol) を加え、 室温にて 2時間撹拌した。 反応液 に 5%炭酸水素ナトリウム水溶液を加え、 酢酸ェチルにて抽出した。 有機層を 飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリ 力ゲルカラムクロマトグラフィー (5 g, 2% メタノール Z酢酸ェチル) にて精製
し、 化合物 107b (19.3 mg, 96%) を得た。 更にこの化合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 20%水 Zメ夕ノ一ル) にて精製し、 純粋な化合 物 107b (17.6 mg) を得た。
107b 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 1.13 (3 H, d, J=5.9 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.68 (1 H, dd, J=12.6, 4.5 Hz, H-4), 2.81 (1 H, m, H-9), 2.88 (1 H: d, J=14.2, 3.5 Hz, H-10), 3.28 (1 H, m, H-20), 3.45, 3.84 (each 1 H, m, H-23), 3.61 (1 H, s, OH), 4.14 (1 H, dd, J=12.5, 5.6 Hz, CH2OH), 4.34 (1 H, dd, J=12.5, 8.4 Hz, CH2OH), 4.44 (1 H, m, H-3), 4.84 (1 H, m, H-l), 5.75 (1 H, m, C=CH), 5.82 (1 H, d, J=ll.l Hz, H- 7), 6.38 (1 H, d,J=ll.l Hz, H-6).
UV max (EtOH): 246 (ε 32300), 254 (ε 37100), 263 (ε 24600) nm.
MS mlz (%): 448 (M+, 7), 430 (7), 412 (14), 394 (25), 376 (12), 308 (13), 263 (12), 131 (21), 113 (39), 69 (100). HR-MS mlz: 448.3214 (Calcd forじ27¾405: 448.3189).
(実施例 86) 20-ェピ -lcc-[(t-プチルジメチルシリル)ォキシ ]-2β,2,-エポキシ- および la-[(t-ブチルジメチルシリル)ォキシ ]-2α,2'-エポキシ- 22-ォキサ -25-[(トリ ェチルシリル)ォキシ ]-19-ノルビタミン D3 t-プチルジメチルシリルエ-テル (化 合物 152a. 152b)
(110.1 μ1, 0.174 mmol, 1.58 Mへキサン溶液) を加え 15分撹拌した後、 22-ォキ
サグランドマンケトン体 121 (43.6 mg, 0.110 mmol) の無水テトラヒドロフラン (1.3 ml) 溶液をゆっくり加えた。 -78 。 ら 0 °Cまで徐々に昇温させながら 2 時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて 抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留 去した。 残渣をシリカゲルカラムクロマトグラフィー (10 g, 2%酢酸ェチル Z へキサン) にて精製し、 化合物 152 を二種の立体異性体の混合物 (49.8 mg, 58%) として得た。 この混合物を構成する異性体 152aおよび 152b の比率は約 5 : 1であった。 15% 酢酸ェチル Zへキサン溶出部より未反応の化合物 121 (9.4 mg) を回収した。
混合物の NM Rデータ
152a (主生成物) 1H NMR (CDC13) δ: 0.03-0.08 (12 H, Si-Me x 4), 0.56 (3 H, s, H- 18), 0.57 (6 H, q, J=7.9 Hz, SiCH2 x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2C¾ x 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H-26, 27), 2.73, 2.82 (each 1 H, d, j=5.6 Hz, OCH2), 3.25, 3.32 (each 1 H, m, H-20, 23), 3.71 (1 H, m, H-23), 3.82, 3.86 (each 1 H, m, H-1, 3), 5.80 (1 H, d, J=11.2 Hz, H-7), 6.22 (1 H, d, J=11.2 Hz, H-6).
152b (マイナー生成物) 1H NMR (CDC13) δ: 0.03-0.08 (12 Η, Si-Me χ 4), 0.56 (3 Η, s, Η-18), 0.57 (6 Η, q, J=7.9 Hz, SiCH2x 3), 0.86, 0.88 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, S1CH2CH3.X 3), 1.09 (3 H, d, J=5.9 Hz, H-21), 1.21, 1.23 (each 3 H, s, H- 26, 27), 2.57, 2.92 (each 1 H, d, J=5.5 Hz, OCH2), 3.25, 3.32 (each 1 H, m, H-20, 23), 3.71 (1 H, m), 3.82 (1 H, m), 4.04 (1 H, m), 5.82 (1 H, d, J=11.0 Hz, H-7), 6.28 (1 H, d, J=11.0 Hz, H-6).
(実施例 87) 20-ェピ -1α,25-ジヒドロキシ -2β,2,-エポキシ- 22-ォキサ -19-ノル ビタミン D3 (化合物 108a) 、 20-ェピ -1α,25-ジヒドロキシ -2α,2,-エポキシ- 22-ォ キサ -19-ノルビタミン D3 (化合物 108b) 、 20-ェピ -1α,2β,25-トリヒドロキシ -2α- フルォロメチル -22-ォキサ -19-ノルビ夕ミン D3 (化合物 109a) および 20-ェピ- 1α,2α,25-トリヒドロキシ -2β-フルォロメチル -22-ォキサ -19-ノルビタミン D3 (化 合物 109b)
109a 109b
化合物 152 (49.8 mg, 0.064 mmol,約 5:1の混合物) の無水テトラヒドロフラン (1 ml) 溶液にフッ化テトラブチルアンモニゥム (0.385 ml, 0.385 mmol, 1 Mテ トラヒドロフラン溶液) を加え、 室温にて 4時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシゥ ム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g) に て精製し、 60%酢酸ェチル Zへキサン溶出部より化合物 109 を二種の立体異性 体の混合物 (1.6 mg, 5%, 109a:109b=約 5:1) として得た。 70%酢酸ェチル Zへキ サン溶出部より化合物 108 を二種の立体異性体の混合物 (23.0 mg, 83%, 108a:108b=約 5:1) として得た。 化合物 108a と 108b を含む混合物を HPLC (LiChrosorb Si60, RT 250-4, 250 10 mm,へキサン:塩化メチレン: 2-プロパノ ール =50:50:6) にて分離精製し、 化合物 108a (9.46 mg ) および化合物 108b (995 μg) を得た。 化合物 109aと 109bを含む混合物を HPLC (YMC-Pack ODS- AM SH-342-5, 150 x 20 mm, 25%水 Zメ夕ノール) にて分離精製し、 化合物 109a
(813 μδ) および化合物 109b (170 g) を得た。
108a 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 1.13 (3 H, d, J=5.9 Hz, H-21), 1.22, 1.24
(each 3 H, s, H-26, 27), 2.29 (1 H, dd, J=13.5, 8.6 Hz, H-10), 2.40 (1 H, dd, J=13.7, 6.2 Hz, H-4), 2.60 (1 H, dd, J=13.7, 3.5 Hz, H-4), 2.81 (1 H, m, H-9), 2.83 (1 H, d, J=4.8 Hz, CH20), 2.94 (1 H, dd, J=13.5, 4.3 Hz, H-10), 3.07 (1 H, d, J=4.8 Hz, CH20), 3.27 (1 H, m), 3.45 (1 H, m), 3.82 (2 H, m), 3.98 (1 H, dd, J=8.6, 4.2 Hz), 5.83 (1 H, d, J=ll.l Hz, H-7), 6.39 (1 H, d, J=ll.l Hz, H-6).
UV max (EtOH): 243, 251, 261 nm.
108b 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 1.14 (3 H, d, J=5.9 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.31 (1 H, dd, J=13.7, 6.0 Hz, H-4), 2.36 (1 H, dd, J=13.6, 8.7 Hz; H-10), 2.72 (1 H, dd, J=13.7, 3.6 Hz, H-4), 2.81 (1 H, m, H-9), 2.85 (1 H, dd, J=13.6, 4.2 Hz, H-10), 2.94, 2.98 (each 1 H, d, J=4.7 Hz, CH20), 3.27 (1 H, m), 3.46 (1 H, m), 3.80-3.95 (3 H, m), 5.85 (1 H, d, J=11.2 Hz, H-7), 6.38 (1 H, d, J=11.2 Hz, H-6).
UV max (EtOH): 243, 251, 261 nm.
109a 1H NMR (CDC13) δ: 0.54 (3 H, s, H-18), 1.13 (3 H, d, J=5.9 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.48 (2 H, m, H-4), 2.55 (1 H, dd, J=14.4, 6.0 Hz, H-10), 2.67 (1 H, br. d, J=UA Hz, H-10), 2.78 (1 H, m, H-9), 3.27 (1 H, m, H-20), 3.45 (1 H, m, H-23), 3.48 (1 H, s, OH), 3.85 (2 H, m, H-3, 23), 3.97 (1 H, m, H-1), 4.72, 4.75 (each 1 H, dd, J=47.5, 9.7 Hz, C¾F), 5.78 (1 H, d, J=ll.l Hz, H-7), 6.40 (1 H, d, J=ll.l Hz, H-6). 19F NMR (CDC13) δ: -240.3 (t,J=47.5 Hz).
MS mlz (%): 454 (M+, 24), 436 (9), 416 (3), 380 (1), 323 (16), 303 (4), 287 (3), 69 (100). UV max (EtOH): 243, 251, 261 nm.
HR-MS mlz: 454.3087 (Calcd for C26H43F05: 454.3095).
109b 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 1.14 (3 H, d, J=6.0 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 2.75-2.90 (3 H, m, H-4, 9, 10), 3.28 (1 H, m, H-20), 3.45 (1 H, m, H-23), 3.48 (1 H, s, OH), 3.77 (1 H, m, H-3), 3.84 (1 H, m, H-23), 3.94 (1 H, m, H-1), 4.70, 4.76 (each 1 H, dd, J=48.0, 9.6 Hz, CH2F), 5.83 (1 H, d, J=11.3 Hz, H-7), 6.29 (1 H, d, J=11.3 Hz, H-6). 19F NMR (CDCI3) δ: -240.4 (t, J=48.0 Hz).
MS mlz (%): 454 (M+, 30), 436 (9), 434 (10), 416 (3), 323 (16), 303 (6), 69 (100).
UV max (EtOH): 243, 251, 261 nm.
HR-MS mlz: 454.3109 (Calcd for C26¾3F05: 454.3095).
(実施例 88) 20-ェピ -laイ
シリル)ォキシ ]-2α-メチル-および 20-ェピ -loc-[(t-プチルジメチルシリル)ォキシ] - 2α-[(トリメチルシリル)ォキシ ]-2β-メチル -22-ォキサ -25-[(トリェチルシリル)ォキ シ] -19-ノルビ夕ミン D3 1-プチルジメチルシリルエ-テル (化合物 153a, 153b)
-78 °Cに冷却した A環ホスフィンォキシド体 118 (214.7 mg, 0.32 mmol,約 1: 1 の混合物) の無水テトラヒドロフラン (3 ml) 溶液に、 n-ブチルリチウム (202.9 μ1, 0.32 mmol, 1.58 Mへキサン溶液) を加え 15分撹拌した後、 22-ォキサ グランドマンケトン体 121 (54.6 mg, 0.14 mmol) の無水テトラヒドロフラン (2 ml) 溶液をゆつくり加えた。 -78 °Cから- 30 °Cまで徐々に昇温させながら 3時間 撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出 した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去し た。 残渣をシリカゲルカラムクロマトグラフィー (7 g, 3%酢酸ェチル Zへキサ ン) にて精製し、 化合物 153を二種の立体異性体の混合物 (52.1 mg, 44%) とし て得た。 この混合物を構成する異性体 153aおよび 153bの比率は約 3 : 1であつ た。 また 5%酢酸ェチルズへキサン溶出部より未反応の化合物 121 (25.6 mg) を回収した。
153 1H NMR (CDC13) δ: 0.02-0.12 (21 H, Si-Me x 4, Si-Me3), 0.5588 (3 H, s, H-18), 0.5598 (6 H, q, J=7.8 Hz, Si-CH2CH3 x 3), 0.83, 0.92 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.8 Hz, Si-CH2CH3 x 3), 1.08 (3 H, d, J=5.9 Hz, H-21), 3.20-3.30 (2 H, m),
3.55-3.80 (3 H, m), 5.76, 5.82 (ca. 1:3) (1 H, d, J=ll.l Hz, H-7), 6.04, 6.15 (ca. 3:1) (1 H: d, J=ll.l Hz, H-6).
MS mlz (%): 850 (M+, 3), 718 (74), 661 (5), 586 (100), 454 (3). (実施例 89) 1α,2β,25-トリヒドロキシ -2α-メチル -22-ォキサ -19-ノルビタミ ン D3 (化合物 110a) および 1α,2α,25-トリヒドロキシ -2β-メチル -22-ォキサ -19-ノ ルビ夕ミン ¾ (化合物 110b)
153 110a 110b 化合物 153 (52.1 mg, 0.061 mmol,約 3:1の混合物) の無水テトラヒドロフラン (1 ml) 溶液にフッ化テトラブチルアンモニゥム (0.490 ml, 0.490 mmol, 1 Mテ トラヒドロフラン溶液) を加え、 室温にて 18時間撹拌した。 反応液に氷水を加 え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネ シゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (10 g) にて精製し、 70%酢酸ェチル Zへキサン溶出部より化合物 110を二種の立体 異性体の混合物 (25.7 mg,96%) として得た。 この混合物を構成する異性体 110a および 110b の比率は約 3 : 1であった。 化合物 110a と 110b を含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 30%水 /メタノール) にて 分離精製し、 化合物 110a (14.7 mg) および化合物 110b (4.43 mg) を得た。
110a XH NMR (CDC13) δ: 0.55 (3 H, s, H-18), 1.13 (3 H, d, J=6.0 Hz, H-21), 1.22, 1.24 (each 3 H, s, H-26, 27), 1.29 (3 H, s, Me), 2.47 (1 H, dd, J=14.1, 3.3 Hz, H-10), 2.64 (2 H; m, H-4, 10), 2.78 (1 H, m, H-9), 3.27 (1 H, m, H-20), 3.45 (1 H, m, H-23), 3.58 (1 H, s, OH), 3.72 (2 H, m, H-1, OH), 3.78 (1 H, m, H-3), 3.84 (1 H, m, H-23), 5.79 (1 H, d,
J=ll.l Hz, H-7), 6.33 (1 H, d, J=ll.l Hz, H-6).
MS mlz (%): 436 (M+, 54), 418 (13), 400 (8), 305 (15), 69 (100).
UV Xmax (EtOH): 243, 251, 261 nm.
110b 1H NMR (CDC13) δ: 0.54 (3 H, s, H-18), 1.13 (3 H, d, J=6.0 Hz, H-21), 1.23, 1.24 (each 3 H, s, H-26, 27), 1.26 (3 H, s, Me), 2.36 (1 H, dd, J=14.4, 4.6 Hz), 2.54 (1 H, br. d, J=13.8 Hz), 2.78 (1 H, m, H-9), 2.92 (1 H, dd, 7=14.5, 4.5 Hz), 3.26 (1 H, m, H-20), 3.45 (1 H, m, H-23), 3.57 (1 H, br. s, OH), 3.73 (3 H, m, H-1, 3, OH), 3.84 (1 H, m, H-23), 5.82 (1 H, d, j=ll.l Hz, H-7), 6.29 (1 H, d, j=ll.l Hz, H-6).
UV max (EtOH): 243, 251, 261 m.
(実施例 90) 24a,26a,27a-トリホモ- lct-[(t-プチルジメチルシリル)ォキシ] - 2α-[(トリメチルシリル)ォキシ] -および 24a,26a,27a-トリホモ- lct-[(t-ブチルジメチ シリル)ォキシ ]-19-ノルビ夕ミン D3 t-プチルジメチルシリルエーテル (化合物 154a, 154b)
-78 に冷却した A環ホスフィンォキシド体 22 (260.0 mg, 0.394 mmol,約 2: 1の混合物) の無水テトラヒドロフラン (3 ml) 溶液に、 n-ブチルリチウム (253 μΐ, 0.394 mmol, 1.56 Mへキサン溶液) を加え 15分撹拌した後、 C/D環ケトン体 122 (101.8 mg, 0.235 mmol) の無水テトラヒドロフラン (1 ml) 溶液をゆつくり 加えた。 -78 °Cで 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加
え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネ シゥム乾燥、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (10 g) にて精製し、 1%酢酸ェチル /へキサン溶出部より化合物 154を二種の立体異 性体の混合物 (106.4 mg, 52%) として得た。 この混合物を構成する異性体 154a および 154bの比率は約 3:2であった。 また、 5%齚酸ェチルズへキサン溶出部 より未反応の化合物 122 (36.1 mg) を回収した。
混合物の NMRデータ
154a (主生成物) 1H NMR (CDC13) δ: 0.039, 0.051, 0.059, 0.064 (each 3 H, s, Si-Me x 4): 0.12 (9 H, s, Si-Me x 3), 0.56 (3 H, s, H-18), 0.56 (6 H, q, J=7,9 Hz, SiCH2x 3), 0.82 (6 H, t, J=7.5 Hz, H-26a, 27a), 0.868, 0.874 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 2.80 (1 H, m, H-9), 3.53 (1 H, m, H-2), 3.80 (1 H, m, H-3), 3.88 (1 H, m, H-l), 5.52 (1H, d, J=15.2 Hz, H-24a, overlapped with H-22), 5.81 (1 H, d, j=ll.l Hz, H-7), 5.94 (1 H, dd, J=14.9, 10.4 Hz, H-23), 6.05 (1 H, dd: J=15.2, 10.4 Hz, H-24), 6.10 (1 H, d, J=ll.l Hz, H-6).
154b (マイナ一生成物) 1H NMR (CDC13) δ: 0.039, 0.051, 0.059, 0.064 (each 3 H, s, Si- Me x 4), 0.12 (9 H, s, Si-Me x 3), 0.54 (3 H, s, H-18), 0.56 (6 H, q, 7=7.9 Hz, SiCH2x 3), 0.82 (6 H, t, J=7.5 Hz, H-26a, 27a), 0.86, 0.89 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, j=7.9 Hz, SiCH2CH3x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 2.80 (1 H, m, H-9), 3.59 (1 H, m, H-2), 3.80 (1 H, m, H-3), 3.94 (1 H, m, H-l), 5.52 (1H, d, J=15.2 Hz, H-24a, overlapped with H-22), 5.78 (1 H, d, J=ll.l Hz, H-7), 5.94 (1 H, dd, J=14.9, 10.4 Hz, H-23), 6.05 (1 H, dd, J=15.2, 10.4 Hz, H-24), 6.13 (1 H, d, J=ll.l Hz, H-6).
混合物の MS mlz (%): no M+, 740 (33), 683 (7), 608 (65), 551 (17), 505 (43), 459 (18), 324 (31), 149 (100), 75 (99).
(実施例 91 ) 24a,26a,27a-トリホモ -1α,2α,25-トリヒドロキシ-および 24a,26a,27a-トリホモ- 1α,2β,25-トリヒドロキシ -22,24-ジェン -19-ノルビ夕ミン D3 (化合物 155a, 155b)
154 155a 155b
化合物 154 (55 mg, 0.063 mmol,約 3 : 2の混合物) の無水テ卜ラヒドロフラン (l ml) 溶液に、 トリェチルァミン (20 μΐ) およびフッ化テトラブチルアンモニ ゥム (504 μΐ, 0.504 mmol, 1.0 Mテトラヒドロフラン溶液) を加え、 室温で 4時 間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩 水にて洗浄し、 無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残潦をシリカゲ ルカラムクロマトグラフィー (5 g, 2% メタノール /酢酸ェチル) にて精製し、 化合物 155を二種の立体異性体の混合物 (28.0 mg, 97%) として得た。 この混合 物を構成する異性体 155aおよび 155bの比率は約 3:2であった。
混合物の NMRデータ
155a 1H NMR (CDC13) δ: 0.57 (3 Η, s, Η-18), 0.87 (6 Η, t, J=7.4 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.55, 1.56 (each 2 H, d, J=7.4 Hz, H-26, 27), 2.62 (1 H, dd, J=12.8, 4.1 Hz, H-4), 2.80 (1 H, m, H-9), 2.89 (1 H, dd, J=14.7, 4.3 Hz, H-10), 3.53 (1 H, dd, J=8.2, 2.9 Hz, H-2), 3.79 (1 H, m, H-3), 4.09 (1 H, m, H-l), 5.53 (1H, d, J=15.2 Hz, H- 24a, overlapped with H-22), 5.80 (1 H, d, J=ll.l Hz, H-7), 5.98 (1 H, dd, J=15.0, 10.3 Hz; H-23), 6.15 (1 H, dd, =15.2, 10.3 Hz, H-24), 6.37 (1 H, d, J=ll.l Hz, H-6).
155b 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 0.87 (6 H, t, J=7.4 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.55, 1.56 (each 2 H, d, J=7.4 Hz, H-26, 27), 2.43 (2 H, m, H-4), 2.80 (1 H, m, H-9), 3.07 (1 H, dd, 7=13.2, 4.9 Hz, H-10), 3.48 (1 H, dd, J=8.8, 3.0 Hz, H- 2), 3.67 (1 H, m, H-3), 4.09 (1 H, m, H-l), 5.53 (1H, d, J=15.2 Hz, H-24a, overlapped with H-22), 5.83 (1 H, d, J=ll.l Hz, H-7), 5.98 (1 H, dd, J=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.2, 10.3 Hz, H-24), 6.29 (1 H, d, J=ll.l Hz, H-6).
混合物の MS mlz (%): 458 (M+, 33), 440 (95), 422 (14), 404 (16), 386 (52), 318 (40),
289 (90), 237 (44), 149 (100).
(実施例 92) 24a,26a,27a-トリホモ- lot-[(t-プチルジメチルシリル)ォキシ] - 2α-ヒドロキシ-および 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキ シ] _2β-ヒドロキシ -22,24-ジェン -25-[(t-ブチルジメチルシリル)ォキシ] -19-ノルビ 夕ミン D3 1-プチルジメチルシリルエーテル (化合物 156a, 156b)
155 156a 156b 化合物 155 (48.0 mg, 0.105 mmol,約 3 : 2の混合物) の無水ジメチルホルムァ ミド (1 ml) 溶液に、 トリエチルァミン (117 μ1, 0.840 mmol) 、 t-ブチルジメチ ルシリルクロリド (63.9 mg, 0.424 mmol) および 4,4-ジメチルァミノピリジン
(6.4 mg, 0.052mmol) を加え、 室温にて 2時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸ナトリウム 乾燥後、 溶媒留去した。 残渣をシリカゲルカラムクロマトグラフィー (5 g, 5% 酢酸ェチル Zへキサン) にて精製し、 化合物 156 を二種の立体異性体の混合物 (55.3 mg, 66%) として得た。 この混合物を構成する異性体 156aおよび 156bの 比率は約 3:2であった。
混合物の NMRデータ
156a (主生成物) 1H NMR (CDC13) δ: 0.06-0.10 (18 H, Si-Me x 6), 0.57 (3 H, s, H-18), 0.87 (6 H, t, /=7.5 Hz, H-26a, 27a), 0.86-0.92 (27 H, Si-tBu x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 1.54, 1.55 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.80 (1 H, m, H-9), 3.51 (1 H, m, H- 2), 3.92 (1 H, m, H-3), 4.00 (1 H, m, H-l), 5.53 (1H, d, J=15.3 Hz, H-24a), 5.55 (1 H, dd,
J=15.2, 8.5 Hz, H-22), 5.79 (1 H, d, J=ll.l Hz, H-7), 5.98 (1 H, dd, J=15.2, 10.4 Hz, H- 23), 6.14 (2 H, m, H-6, 24).
156b (マイナ一生成物) 1H NMR (CDC13) δ: 0.06-0.10 (18 Η, Si-Me χ 6), 0.56 (3 Η, s, Η-18), 0.87 (6 Η, t, j=7.5 Hz, H-26a, 27a), 0.86-0.92 (27 H, Si-tBu x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 1.54, 1.55 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.80 (1 H, m, H-9), 3.59 (1 H, m, H-2), 4.00 (2 H, m, H-l, 3), 5.53 (1 H, d, J=15.3 Hz, H-24a), 5.55 (1 H, dd, 7=15.2, 8.5 Hz, H-22), 5.79 (1 H, d, J=ll.l Hz, H-7), 5.98 (1 H, dd, J=15.2, 10.4 Hz, H-23), 6.14 (2 H, m, H-6, 24).
混合物の MS m/z ( ): no M+, 668 (6), 611 (2), 536 (3), 479 (12), 386 (6), 149 (100), 75 (79).
(実施例 93) 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキシ] - 2a-[2-(t-ブチルジメチルシリル)ォキシ] -ェトキシ] -および 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキシ ]-2β-[2-0プチルジメチルシリル)ォキシ] -ェ プチルジメチルシリルエーテル (化合物 157a, 157b)
0 °Cに冷却した化合物 156 (52.4 mg, 0.065 mmol,約 3 : 2の混合物) の無水ジ メチルホルムアミド (1 ml) 溶液に、 水素化ナトリウム (78.5 mg, 1.962 mmo, 60%パラフィンリキッド) および (2-ブロモェトキシ)小ブチルジメチルシラン (68 μΐ, 0.317 mmol) を加え激しく撹拌した。 18時間後、 反応液に氷水を加え、 酢酸ェチルノへキサン (1 : 1) にて抽出した。 有機層を飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ
ラフィ一 (10 g, 1%~10 酢酸ェチル Zへキサン) にて精製し、 化合物 157を二 種の立体異性体の混合物 (44.8 mg, 71%) として得た。 この混合物を構成する異 性体 157aおよび 157bの比率は約 3:2であった。
157 1H NMR (CDC13) δ: 0.05-0.10 (24 H, Si-Me x 8), 0.55, 0.57 (ca. 2 : 3) (3 H, s, H-18): 0.85 ~ 0.92 (42 H, 4 x Si-tBu, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.54, 1.55 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.80 (1 H, m, H-9), 3.18-4.45 (7 H, m, OCH2CH20, H-1, 3), 5.52 (1 H, d, J=15.1 Hz, H-24a), 5.54 (1 H, dd, J=15.0, 8.6 Hz, H-22), 5.79 (1 H, H-7), 5.97 (1 H, dd,J=15.0, 10.4 Hz, H-23), 6.15 (2 H, m, H-6, 24).
MS mlz (%): no M+, 649 (14), 651 (11), 562 (12), 519 (24), 233 (100).
(実施例 94) 24a,26a,27a-トリホモ- 1α,25-ジヒドロキシ -2α-(2-ヒドロキシェ トキシ) -および 24a,26a,27a-トリホモ- 1α,25- シ) -22,24-ジェン -19-ノルビ夕ミン D3 (化合物 111a, 111b)
化合物 157 (42.0 mg, 0.044 mmol,約 3 : 2の混合物) の無水テトラヒドロフラ ン (1 ml) 溶液に、 トリェチルァミン (30 μΐ) およびフッ化テトラプチルアンモ ニゥム (350 μ1,0.350 πιπιο1, 1.0 Mテトラヒドロフラン溶液) を加え、 室温にて 5 時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食 塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカ ゲルカラムクロマトグラフィー (5 g、 2% メタノール/酢酸ェチル) にて精製 し、 化合物 111aと 111bの混合物 (20.0 mg, 91%) を得た。 化合物 111aと 111b を含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 20%水 Zメ
タノ一ル) にて分離精製し、 化合物 Ula (9.4 mg) および化合物 111b (8.3 mg) を得た。
111a 1H NMR (CDC13) δ: 0.57 (3 Η, s, H-18), 0.86 (6 Η, t, /=7.4 Hz, H-26a, 27a), 1.04 (3 H, d, J=6.6 Hz, H-21), 1.54, 1.56 (each 2 H, d, J=7.4 Hz, H-26, 27), 2.61 (1 H, dd, J=13.4: 4.4 Hz, H-4), 2.80 (1 H, m, H-9), 2.86 (1 H, dd, J=14.4, 4.8 Hz, H-10), 3.32 (1 H, dd, J=8.0, 2.5 Hz, H-2), 2.72, 3.12, 3.48 (each 1 H, br. s, OH x 3), 3.67 ~ 3.81 (4 H, m, OCH2CH20), 3.93 (1 H, m, H-3), 4.15 (1 H, m, H-1), 5.53 (IH, d, J=15.3 Hz, H-24a, overlapped with H-22), 5.81 (1 H, d, J=ll.l Hz, H-7), 5.97 (1 H, dd, J=15.0, 10.3 Hz, H- 23), 6.14 (1 H, dd, J=15.3, 10.3 Hz, H-24), 6.33 (1 H, d, J=ll.l Hz, H-6).
UV max (EtOH): 235 (ε 44000), 243 (ε 44200), 251 (ε 38000), 261 (ε 23800) nm.
MS mlz (%): 502 (M+, 11), 484 (62), 466 (33), 448 (7), 386 (17), 333 (40), 237 (29), 149 (100), 133 (43), 93 (49). HR-MS mlz: 502.3658 (Calcd for C31H50O5: 502.3658).
111b 1H NMR (CDC13) δ: 0.56 (3 H, s, H-18), 0.86 (6 H, t, J=7.5 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.55, 1.56 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.34, 2.47 (each 1 H, m, H-4), 2.64 (1 H, br. s, OH), 2.80 (1 H, m, H-9), 3.07 (1 H, dd, J=13.2, 4.0 Hz, H-10), 3.27 (1 H, dd, J=8.7, 2.6 Hz, H-2), 3.64 ~ 3.86 (5 H, m, OCH2CH20, H-1), 4.16 (1 H, m, H-3), 5.53 (IH, d, J=15.3 Hz, H-24a, overlapped with H-22), 5.83 (1 H, d, j=ll.l Hz, H- 7), 5.97 (1 H, dd, J=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.3, 10.3 Hz, H-24), 6.26 (1 H, d, J=ll.l Hz, H-6).
UV rnax (EtOH): 235 (ε 44000), 243 (ε 44500), 251 (ε 38900), 261 (ε 24000) nm.
MS mlz (%): 502 (Μ+, 13), 484 (78), 466 (39), 448 (7), 386 (13), 333 (48), 237 (27), 149 (100), 133 (46), 93 (49). HR-MS m/z: 502.3664 (Calcd for C31H50O5: 502.3658).
(実施例 95) 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキシト2- [2-(t-プチルジメチルシリル)ォキシ] -ェチリデン] -22,24-ジェン -25-[(トリェチルシ リル)ォキシ ]-19-ノルビ夕ミン D3 t-プチルジメチリレシリルエーテルの E-異性体お よび 異性体 (化合物 158a, 158b)
119
-78 °Cに冷却した A環ホスフィンォキシド体 119 (119.5 mg, 0.164 mmol,約 4: 1の混合物) の無水テトラヒドロフラン (2 ml) 溶液に、 n-ブチルリチウム (105 μΐ, 0.164 mmol, 1.56 Mへキサン溶液) を加え 15分撹拌した後、 C/D環ケトン体 122 (47.4 mg, 0.109 mmol) の無水テトラヒドロフラン (1 ml) 溶液をゆつくり加 えた。 -78 で 2時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和食塩水にて洗浄し、 無水硫酸マグネシゥ ム乾燥、 溶媒留去した。 残澄をシリカゲルカラムクロマトグラフィー (10 g) に て精製し、 1%酢酸ェチル /へキサン溶出部より、 化合物 158を二種の立体異性 体の混合物 (27.7 mg, 27%) として得た。 この混合物を構成する異性体 158aお よび 158bの比率は約 6:1であった。 また、 5%酢酸ェチル /へキサン溶出部よ り未反応の化合物 122 (27 mg, 57%) を回収した。
158 1H NMR (CDC13) δ: 0.01-0.08 (18 H, Si-Me x 6), 0.571 (3 H, s, H-18), 0.569 (6 H, q, J=7.9 Hz, SiCH2x 3), 0.83 (6 H, t, J=7.5 Hz, H-26a, 27a), 0.84, 0,90, 0.92 (each 9 H, s, Si-tBu x 3), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 2.80 (1 H, m, H-9), 2.97, 3.28 (ca. 6 : 1) (1 H, dd, J=12.5, 4.7 Hz, H-10), 4.24 ~ 4.47 (3 H, m, H- 1 or 3, CH2OH), 4.79, 4.84 (ca. 6 : 1) (1 H, m, H-l or 3), 5.52 (1 H, dd, J=14.9, 8.3 Hz, H-22), 5.53 (1H, d, J=15.2 Hz, H-24a), 5.61 (1 H, m, C=CH), 5.87 (1 H, d, J=ll.l Hz, H- 7), 5.95 (1 H, dd, j=14.9, 10.4 Hz, H-23), 6.06 (1 H, dd, J=15.2, 10.4 Hz, H-24), 6.13 (1 H, d, J=ll.l Hz, H-6).
(実施例 96)
化合物 158 (56 mg, 0.0595 mmol,約 6: 1の混合物) の無水テトラヒドロフラ ン (1 ml) 溶液に、 トリェチルァミン (40 μΐ) およびフッ化テトラプチルアンモ ニゥム (476 μΐ, 0.476 mmol, 1.0 Mテトラヒドロフラン溶液) を加え、 室温で 20 時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食 塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカ ゲルカラムクロマトグラフィー (5 g、 2% メタノール 酉乍酸ェチル) にて精製 し、 化合物 112aと 112bの混合物 (23.0 mg, 80%,約 10: 1の混合物) を得た。 ィ匕 合物 112aと 112bを含む混合物を HPLC (LiChrosorb Si 60, ffibar RT 250-10, 250 x 10 mm,へキサン:塩化メチレン:メタノール = 50 : 50 : 4) にて分離精製し、 化合物 112a (17.1 mg, E-異性体) および化合物 112b (1.9 mg, Z-異性体) を得た。 112a 1H NMR (CDC1
3) δ: 0.56 (3 H, s, Η-18), 0.87 (6 Η, t,J=7.5, H-26a, 27a), 1.05 (3 H, d, J=6.5 Hz, H-21), 1.55, 1.56 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.34, 2.44 (eac 1 H, m, H-4), 2.81 (1 H, m, H-9), 3.14 (1 H, d, J=12.5, 4.3 Hz, H-10), 3.38, 3.74 (each 1 H, br. s, OH x 2), 4.09 (1 H, dd, J=12.3, 5.2 Hz, CH
2OH), 4.34 (2 H, m, H-l, CH
2OH), 4.80 (1 H, m, H-3), 5.53 (1H, d, J=15A Hz, H-24a, overlapped with H-22), 5.75 (1 H, m, C=CH), 5.88 (1 H, d, J=ll.l Hz, H-7), 5.97 (1 H, dd, 7=15.0, 10.4 Hz, H-23), 6.15 (1 H, dd, J=15.4, 10.4 Hz, H-24), 6.27 (1 H, d,J=ll.l Hz, H-6).
UV max (EtOH): 236 (ε 47300), 245 (ε 48000), 254 (ε 43200), 264 (ε 27200) nm.
MS mlz (%): 484 (M+, 7), 466 (15), 448 (17), 430 (44), 412 (34), 279 (33), 263 (25), 149 (100), 133 (35), 93 (38). HR-MS mlz: 484.3526 (Calcd for C3iH4804: 484.3553).
112b 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 0.87 (6 H, t, J=7.5 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 1.55, 1.56 (each 2 H, d, J=7.5 Hz, H-26, 27), 2.22 (1 H, m, H-4), 2.33 (1 H, m, H-10), 2.70 (1 H, dd, j=13.0, 4.7 Hz, H-4), 2.82 (2 H, m, H-9, 10), 4.25 (1 H, dd, J=12.6, 6.4 Hz, CH2OH), 4.38 (1 H, dd, J=12.6, 7.3 Hz, CH2OH), 4.46 (1 H, m, H- 3), 4.87 (1 H, t, J=4.2 Hz, H-1), 5.54 (1H, d, J=15.3 Hz, H-24a, overlapped with H-22), 5.84 (2 H, m, H-7, C=CH), 5.98 (1 H, dd, J=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.3, 10.3 Hz, H-24), 6.40 (1 H, d, J=ll.l Hz, H-6).
MS mlz (%): 484 (M+, 4), 466 (12), 448 (16), 430 (44), 412 (38), 279 (35), 263 (32), 149 (100), 133 (39), 93 (42). HR-MS mlz: 484.3561 (Calcd for C31H4s04: 484.3553).
(実施例 97) 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキシ] - 2β,2,-エポキシ-および 24a,26a,27a-トリホモ- la-[(t-ブチルジメチルシリル)ォキ
13 -78 °Cに冷却した A環ホスフィンォキシド体 13 (260 mg, 0.434 mmol,約 3: 1 の混合物) の無水テトラヒドロフラン (2 ml) 溶液に、 n-ブチルリチウム (276 μ1, 0.436 mmol. 1.58 Mへキサン溶液) を加え 15分撹拌した後、 C/D環ケトン体
122 (100 mg, 0.231 mmol) の無水テトラヒドロフラン (1.5 ml) 溶液をゆつくり 加えた。 -78 °Cから- 20 °Cまで徐々に昇温させながら 3時間撹拌した後、 反応液 に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層は飽和 食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシリカゲ ルカラムクロマトグラフィー (10 g, 2%酢酸ェチル /へキサン) にて精製し、 化合物 159を二種の立体異性体の混合物 (31.1 mg, 17%) として得た。 この混合 物を構成する異性体 159aおよび 159bの比率は約 10: 1であった。 5%酢酸ェ チルズへキサン溶出部より未反応の化合物 122 (48.0 mg) を回収した。
159 XH NMR (CDC13) δ: 0.02, 0.05 (each 3 H, s, Si-Me x 2), 0.06 (6 H, Si-Me x 2), 0.567 (6 H, q, J=7.9 Hz, SiCH2 x 3), 0.570 (3 H, s, H-18), 0.82 (6 H, t, j=7.5 Hz, H-26a, 27a), 0.86, 0.87 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.9 Hz, SiCH2CH3x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 3.80 (1 H, m), 3.87 (1 H, m), 5.52 (1H, d,J=15.0 Hz, H-24a, overlapped with H-22), 5.81 (1 H, d, J=ll.l, H-7), 5.80-6.08 (3 H, m), 6.21, 6.27 (ca. 10:1) (1 H, d, J=ll.l Hz, H-7).
(実施例 98) 24a,26a,27a-トリホモ- 1α,25-ジヒドロキシ -2β,2,-エポキシ-およ び 24a,26a,27a-トリホモ- 1α,25-ジヒドロキシ -2α,2,-エポキシ- 22,24-ジェン -19-ノル ビタミン D3 (化合物 113a, 113b) ならびに 24a,26a,27a-トリホモ- 1α,2β,25-トリヒ ドロキシ -2α-メチル-および 24a,26a,27a-トリホモ- 1α,2α,25-トリヒドロキシ -22,24- ジェン -19-ノルビ夕ミン D3 (化合物 114a, 114b)
114a 114b
化合物 159 (34.0 mg, 0.042 mmol,約 10 : 1の混合物) の無水テトラヒドロフラ ン (1 ml) 溶液に、 トリェチルァミン (75 μΐ) およびフッ化テトラプチルアンモ ニゥム (251 μΐ, 0.24 mmol, 1.0 Μテトラヒドロフラン溶液) を加え、 室温で 7.5 時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽和食 塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシリカ ゲルカラムクロマトグラフィー (5 g) にて精製し、 50%酢酸ェチルズへキサン 溶出部より化合物 114を二種の立体異性体の混合物 (1.5 mg,7%) として得た。 また、 70%酢酸ェチル /へキサン溶出部より化合物 113 を二種の立体異性体の 混合物 (14.7 mg, 75%) として得た。 これらの混合物の NMRスぺクトルでは、 各異性体由来のシグナルが互いに重なり、 正確な異性体の比率を求めることはで きなかった。
化合物 113aおよび 113bを含む混合物を HPLC (LiChrosorb Si 60, Hibar RT 250- 10, 250 x 10 mm, hexane: CH2C12: MeOH = 40 : 60 : 5)にて分離精製し、 化合物 113a (4.73 mg) および化合物 113b (628 g) を得た。
113a 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 0.87 (6 H, t, J=7.4 Hz, H-26a, 27a), 1.06 (3 H, d, J=6.5 Hz, H-21), 2.31 (1 H, dd, J=13.4, 8.6 Hz, H-10), 2.40 (1 H, dd,J=13.6, 6.2 Hz, H-4), 2.61 (1 H, dd, J=13.6, 3.3 Hz, H-4), 2.80 (1 H, m, H-9), 2.84 (1 H, d, J=4.7 Hz, OCH), 2.95 (1 H, dd, J=13.4, 4.1 Hz, H-10), 3.08 (1 H, d, J=4.7 Hz, OCH), 5.53 (1 H, d, j=15.5 Hz, H-24a, overlapped with H-22), 5.68 (1 H, d, J=ll.l Hz, H-7), 5.97 (1 H, dd, j=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.5, 10.3 Hz, H-24), 6.39 (1 H, d, J=ll.l Hz, H-6).
UV Xmax (EtOH): 235, 243, 251, 261 nm.
113b 1H NMR (CDC13) δ: 0.58 (3 H, s, H-18), 0.87 (6 H, t, J=7.6 Hz, H-26a, 27a), 1.06 (3 H, d, J=6.6 Hz, H-21), 2.28-2.38 (2 H, m, H-4, 10), 2.71 (1 H, dd, J=13.8, 3.5 Hz, H-4), 2.80 (1 H, m, H-9), 2.86 (1 H, dd, J=13.2, 4.2 Hz, H-10), 2.94, 2.99 (each 1 H, d, J=4.7 Hz, OC¾), 5.53 (1 H, d, J=15.2 Hz, H-24a, overlapped with H-22), 5.87 (1 H, d, J=11.0 Hz, H-7), 5.98 (1 H, dd, 7=15.0, 10.3 Hz, H-23), 6.16 (1 H, dd, 7=15.2, 10.3 Hz, H-24), 6.38 (1 H, d, j=11.0 Hz, H-6).
UV ax (EtOH): 234, 243, 251, 261 nm.
114 1H NMR (CDCI3) δ: 0.55 (3 H, s, H-18), 0.88 (6 H, t, J=7.5 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 3.87, 3.97 (each 1 H, m, H-1, 3), 4.72, 4.76 (each 1 H, dd, J=48.0, 9.7 Hz, C¾F), 5.53 (1 H, d, J=15.1 Hz, H-24a, overlapped with H-22), 5.80 (1 H, d, J=11.2 Hz, H-7), 5.97 (1 H, dd, j=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.1, 10.3 Hz, H-24), 6.40 (1 H, d, J=11.2 Hz, H-6). 19F NMR (CDC13) δ: -240.5 (t, J=48.0 Hz).
(実施例 99) 24a,26a,27a-トリホモ- la-[(t-プチルジメチルシリル)ォキシ ]-2β- [(トリメチルシリル)ォキシ ]-2α-メチル-および 24a,26a,27a-トリホモ- la-[(t-ブチル ジメチルシリル)ォキシ ]-2α-[(トリメチルシリル)ォキシ ]-2β-メチル -22,24-ジェン- 25- [(トリェチルシリル)ォキシ ]-19-ノルビタミン D3 t-プチルジメチルシリルエ- テル (化合物 160a, 160b)
118
-78 °Cに冷却した A環ホスフィンォキシド体 118 (253.9 mg, 0.377 mmol,約 1: 1 の混合物) の無水テトラヒドロフラン (2.5 ml) 溶液に、 n-ブチルリチウム (238.6 μΐ, 0.377 mmol, 1.58 Mへキサン溶液) を加え 15分撹拌した後、 C/D環ケ トン体 122 (84.0 mg, 0.194 mmol) の無水テトラヒドロフラン (1.5 ml) 溶液をゆ つくり加えた。 -78 から- 30 °Cまで徐々に昇温させながら 3時間撹拌した後、 反応液に飽和塩化アンモニゥム水溶液を加え、 酢酸ェチルにて抽出した。 有機層 は飽和食塩水にて洗浄し、 無水硫酸マグネシウム乾燥、 溶媒留去した。 残渣をシ リカゲルカラムクロマトグラフィー (6 g, 1%酢酸ェチル /へキサン) にて精製 し、 化合物 160を二種の立体異性体の混合物 (44.6 mg, 26%) として得た。 この 混合物を構成する異性体 160aおよび 160bの比率は約 1 : 2であった。 5%酢酸 ェチルズへキサン溶出部より未反応の化合物 122 (47.0 mg) を回収した。
160 1H NMR (CDC13) δ: 0.02-0.09 (12 H, Si-Me x 4), 0.11, 0.12 (ca. 1:2) (9 H, s, Si-Me x 3), 0.569 (3 H, s, H-18), 0.570 (6 H, q, J=7.6 Hz, Si-CH2CH3 x 3), 0.82 (6 H, t, J=7.5 Hz, H-26a, 27a), 0.83, 0.92 (each 9 H, s, Si-tBu x 2), 0.94 (9 H, t, J=7.6 Hz, Si-CH2CH3 x 3), 1.06 (3 H, d, J=6.6 Hz, H-21), 3.56 (1 H, m), 3.60, 3.70 (ca. 2:1) (1 H, m), 5.52 (1 H, d, J=15.2 Hz, H-24a, overlapped with H-22), 5.78, 5.84 (ca. 1:2) (1 H, d, J=11.2 Hz, H-7): 5.90-6.15 (3 H, m). (実施例 100) 24a,26a,27a-トリホモ- 1α,2β,25-トリヒドロキシ -2α-メチル -お よび 24a,26a,27a-トリホモ- 1α,2α,25-トリヒドロキシ -2β-メチル -22,24-ジェン -19- ノルビ夕ミン D3 (化合物 115a, 115b)
160 115a 115b 化合物 160 (56.0 mg, 0.063 mmol,約 2 : 1の混合物) の無水テトラヒドロフラ ン (1.5 ml) 溶液に、 トリェチルァミン (75 μΐ) およびフッ化テトラプチルアン モニゥム (505 μΐ, 0.505 mmol, 1.0 Mテトラヒドロフラン溶液) を加え、 室温で 24時間撹拌した。 反応液に氷水を加え、 酢酸ェチルにて抽出した。 有機層を飽 和食塩水にて洗浄し、 無水硫酸ナトリウムにて乾燥後、 溶媒留去した。 残渣をシ リカゲルカラムクロマトグラフィー (5 g, 60%酢酸ェチル Zへキサン) にて精 製し、 化合物 115 を二種の立体異性体の混合物 (25.1 mg, 84%,約 5:4 の混合 物) として得た。
化合物 115aおよび 115bを含む混合物を HPLC (YMC-Pack ODS-AM SH-342-5, 150 x 20 mm, 20%水/メ夕ノール)にて精製し、 化合物 115a (5.25 mg) および化 合物 115b (6.68 mg) を得た
115a 1H NMR (CDC13) δ: 0.56 (3 H, s, Η-18), 0.88 (6 Η, t, J=7.5 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 2.37 (1 H, dd, J=14.4, 4.4 Hz, H-4), 2.54 (1 H, br. d, J=14.4 Hz, H-4), 2.80 (1 H, m, H-9), 2.94 (1 H, dd, J=13.5, 4.4 Hz, H-10), 3.73 (2 H, m, H-l, 3), 5.53 (1 H, d, J=15.4 Hz, H-24a, overlapped with H-22), 5.84 (1 H, d, J=11.2 Hz, H-7), 5.97 (1 H, dd, J=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd, J=15.4, 10.3 Hz, H-24), 6.29 (1 H, d, J=11.2 Hz, H-6).
MS m/z (%): 472 (M+, 13), 454 (100), 436 (38), 418 (29), 400 (46).
UV max (EtOH): 235, 243, 252, 261 nm.
115b 1H NMR (CDC13) δ: 0.57 (3 H, s, H-18), 0.87 (6 H, t, J=7.5 Hz, H-26a, 27a), 1.05 (3 H, d, J=6.6 Hz, H-21), 2.48 (1 H, dd, 7=14.2, 3.2 Hz, H-10), 2.63 (1 H, dd, J=14.2, 6.4 Hz,
H-10), 2.66 ( 1 H, dd, J=14.0, 4.0 Hz, H-4), 2.80 (1 H, m, H-9), 3.72, 2.78 (each 1 H, m, H-1, 3), 5.53 (1 H, d, J=15.4 Hz, H-24a, overlapped with H-22), 5.82 (1 H, d, J=ll.l Hz, H-7), 5.98 (1 H, dd, J=15.0, 10.3 Hz, H-23), 6.15 (1 H, dd,J=15.4, 10.3 Hz, H-24), 6.33 (1 H, d,J=ll.lHz, H-6).
MS m/z ( ): 472 (M+, 2), 454 (100), 436 (18), 418 (10), 400 (12).
UV max (EtOH): 235, 243, 251, 261 nm.
(試験例) ゥシ胸腺ビタミン D受容体 (VDR) への結合実験
上記の実施例で合成された本発明の化合物について、 ゥシ胸腺由来のビタミ ン D受容体 (VDR) に対する結合能を試験した。
結合試験はャマサ醤油株式会社の操作マニュアルに従い、 下記のように行な つた。
1 α, 25-ジヒドロキシビタミン D3 (標準物質として使用) および化合物 YI - la、 ΥΙ-lb, YI-2a, YI-3a, YI - 3b、 YI - 4a、 YI_4b、 YI - 5a、 YI-5b, 20-Epi-YI-la, 20-Epi- Yl-lb, 20- Epi-YI- 2a、 20- Epi - YI- 3a、 20-Epi-YI-4a, 20- Epi- YI-4b、 20-Epi- YI- 5a 、 20_Epi- YI-5b、 20- Epi- YI- 6a、 20- Epi- YI_6b、 20- Epi- YI_7a、 20 - Epi-YI - 7b、 20- Epi- YI_8a、 20- Epi- YI- 8bのそれぞれについて、 各種濃度のエタノール溶液 (サ ンプル) を調製した。 すなわち、 後の工程で調製される、 受容体溶液とサンプ ルと [¾] 1 α, 25-ジヒドロキシビタミン D3溶液との混合物における各化合物の最 終濃度が、 100 nM、 30 nM、 10 nM、 1 nM、 300 pM、 100 pM、 30 pM、 10 pM、 3 pM、 1 pMとなるような希釈系列を作製した。
凍結乾燥ゥシ胸腺ビタミン D受容体 (Lot. No. 111931) をャマサ醤油株式会 社 (Choshi, Chiba, Japan) より購入し、 使用直前に 45 mLのリン酸緩衝液 ( 0.3 M KC1, 0.05 M K2HP04-KH2P04, pH 7.4) に溶解し、 受容体溶液とした。
化合物 YI - la、 Yl-lb, YI-2a、 YI - 3a、 YI-3b、 YI— 4a、 YI— 4b、 YI- 5a、 YI - 5b、 20- Epi- YI- la、 20 - Epi— YI_lb、 20-E i-YI-2a> 20 - Epi- YI - 3a、 20- Epi - YI- 4a、 20 - Epi-YI - 4b 、 20- Epi- YI- 5a、 20- Epi - YI- 5b、 20- Epi - YI- 6a、 20-Epi-YI-6b, 20-Epi-YI-7a, 20- Epi-YI-7b, 20- Epi- YI- 8a、 20- Epi - YI- 8bまたは 1 , 25-ジヒドロキシビタミン D3 のサンプル 50^L と受容体溶液 500 L とをカルチヤ一チューブに入れ、 2—
3回ポルテックスにかけ、 室温にて 1時間プレインキュベーションした。 各チュ ーブに、 50 Lに溶解した約 10000 dpmの [¾] 1ひ, 25 -ジヒドロキシビタミン D3 を加え、 2— 3回ポルテックスにかけた後、 4 °C (冷蔵庫内) にて 18時間インキ ュべ一シヨンした。 各チューブに 200 Lの DCC (デキストラン被覆チヤコール 、 ャマサ醤油株式会社より購入) を加えた後、 4 にて 30分間放置し、 4 °C、 3000 rpmで 15分間遠心分離を行なうことにより、 受容体に結合した [¾] 1 α, 25- ジヒドロキシビタミン D3と、 遊離の [¾] 1 α, 25-ジヒドロキシビタミン D3とを分 離した。 各チューブより 500 Lの上清を取りバイアル瓶に入れ、 10 mLの液体 シンチレ一夕一 ACS- II (Amersham, Buckinghamshire, U.K.) と混合し、 放 射活性を測定した。
1 α , 25-ジヒドロキシビタミン1)3の VDRへの結合性を 1としたときの本発明の ビタミン D誘導体の VDRへの相対結合活性を算出した。 計算式は以下の通りであ る。
X=y/x
X:本発明の化合物の VDRへの相対的結合性
y: 1 α,25-ジヒドロキシビタミン D3が、 [¾] 1 α, 25-ジヒドロキシビタミン D3と VDRとの結合を 5 0%阻害する濃度
X :本発明の化合物が、 [¾] 1 ,25-ジヒドロキシビタミン D3と VDRとの 結合を 50%阻害する濃度
表 1
化合物番号 相対結合活性
YI-la 1/23
YI-lb 1/290
YI-2a 1/43
YI-3a 1/3
YI-3b 1/730
YI - 4a 1/50
YI-4b 1/2000
YI- 5a 1/26
YI - 5b 1/820
20-Epi-YI-la 1/2
20-Epi-YI-lb 1/5
20-Epi-YI-2a 2
20-Epi-YI-3a 1
20-Epi-YI-4a 1/20
20-Epi-YI-4b 1/1000
20-Epi-YI-5a 1/5
20-Epi-YI-5b 1/10
20-Epi-YI-6a 1
20-Epi-YI-6b 5
20-Epi-YI-7a 1
20-Epi-YI-7b 1/5
20-Epi-YI-8a 1. 6
20-Epi- YI-8b 1/50 産業上の利用の可能性
本発明の一般式 ( I ) および (I V) で表される化合物は新規化合物であり
、 細胞の分化に異常を伴う疾患等に使用する医薬として有用である可能性があ る。 また、 本発明の化合物は、 活性型ビタミン D3 (即ち、 1 , 2 5 —ジヒド ロキシビタミン D3) の代謝の研究において有用な試薬となる可能性がある。