WO2004039822A2 - 4’’ - modified erythromycin derivatives - Google Patents
4’’ - modified erythromycin derivatives Download PDFInfo
- Publication number
- WO2004039822A2 WO2004039822A2 PCT/EP2003/012068 EP0312068W WO2004039822A2 WO 2004039822 A2 WO2004039822 A2 WO 2004039822A2 EP 0312068 W EP0312068 W EP 0312068W WO 2004039822 A2 WO2004039822 A2 WO 2004039822A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- hydrogen
- optionally substituted
- group
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC(*1C=C=*)=C(*)C=C(**)C1=O Chemical compound CCC(*1C=C=*)=C(*)C=C(**)C1=O 0.000 description 6
- PVOAHINGSUIXLS-UHFFFAOYSA-N CN1CCNCC1 Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/08—Hetero rings containing eight or more ring members, e.g. erythromycins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
Definitions
- Macrolide antibacterial agents are known to be useful in the treatment or prevention of bacterial infections
- the emergence of macrohde-resistant bacte ⁇ al strains has resulted in the need to develop new macrolide compounds
- EP 0 895 999 describes derivatives modified at the 4" position of the macrolide ⁇ ng having antibacte ⁇ al activity
- R. IS hydrogen or a hydroxyl protecting group
- R is hydrogen, C ⁇ alkyl or C3_6alkenyl optionally substituted by 9 to 10 membered fused bicyclic heteroaryl,
- R5 IS hydroxy, C .galkenyloxy optionally substituted by 9 to 10 membered fused bicyclic heteroaryl, or 0(CH2)pO(CH 2 ) q R 10 ,
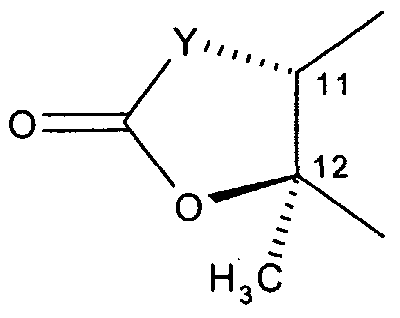
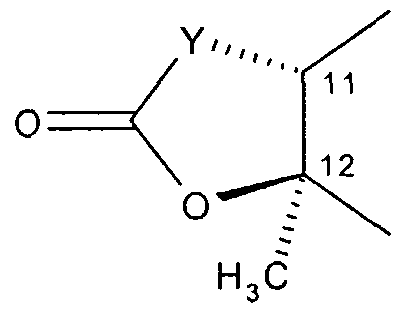
- R" is hydroxy, or R5 and R ⁇ taken together with the intervening atoms form a cyclic group having the following structure:
- Y is a bivalent radical selected from -CH2-, -CH(CN)-, -0-, -N(R ⁇ 1 )- and
- R' is a heterocychc group having the following structure:
- R 8 and R 9 are each independently selected from hydrogen and C j ⁇ alkyl
- R 10 is hydrogen or NR 8 R 9 ;
- R' hydrogen or C ⁇ alkyl substituted by a group selected from optionally substituted phenyl, optionally substituted 5 or 6 membered heteroaryl and optionally substituted 9 to 10 membered fused bicyclic heteroaryl;
- R 12 is hydrogen, C(0)OR 15 , C(0)NHR 15 or C(0)CH 2 N0 2 ;
- Rl3 is hydrogen, C ⁇ alkyl optionally substituted by hydroxy or C1 ⁇ alkoxy, C3.
- R 14 is halogen, C ⁇ alkyl, C ⁇ thioalkyl, C alkoxy, NH 2 , NH ⁇ alkyl) or N(C ⁇ _ 4alkyl)2;
- R ⁇ is hydrogen or C ⁇ alkyl optionally substituted by up to three groups independently selected from halogen, C ⁇ alkoxy, OC(0)C j ⁇ alkyl and OC(0)OC j ⁇ alkyl;
- R i is hydrogen, C ⁇ alkyl, C . cycloalkyl, optionally substituted phenyl or benzyl, acetyl or benzoyl; Rl is hydrogen or R ⁇ , or R ⁇ and R ⁇ are linked to form the bivalent radical -0(CH2)2- or
- X is -U(CH2) S Z- or X is a group selected from:
- U and Z independently are a divalent radical selected from -N(R*")- -0-, -S(0) t - ,
- W is CRl or a nitrogen atom; m is 0 or an integer from 1 to 5; n, r and t are each independently selected from 0, 1 and 2, p and q are each independently selected from 1 to 6 ; s is an integer from 2 to 8, and v is 2 or 3; and pharmaceutically acceptable derivatives thereof
- the present mvention provides compounds of general formula (IA)
- R3 IS hydrogen, C ⁇ _4alkyl or C3_6alkenyl optionally substituted by 9 to 10 membered fused bicyclic heteroaryl;
- R4 is hydrogen, C ⁇ alkyl, C3_7cycloalkyl, C3_.galkenyl or a 5 or 6 membered heterocychc group, wherein the alkyl, cycloalkyl, alkenyl and heterocychc groups are optionally substituted by up to three substituents independently selected from optionally substituted 5 or
- R5 is hydroxy, C3_galkenyloxy optionally substituted by 9 to 10 membered fused bicyclic heteroaryl or O(CH 2 )pO(CH2)qR 10 ; R° is hydroxy, or
- Y is a bivalent radical selected from -CH2-, -CH(CN)-, -O-, -N(Rl )- and ⁇
- R ' is a heterocychc group having the following structure:
- R 8 and R 9 are each independently selected from hydrogen and C ⁇ alkyl
- R 10 is hydrogen or NR 8 R 9 ;
- Ri MS hydrogen or C ⁇ ⁇ alkyl substituted by a group selected from optionally substituted phenyl, optionally substituted 5 or 6 membered heteroaryl and optionally substituted 9 to 10 membered fused bicyclic heteroaryl;
- R 12 is hydrogen, C(0)OR 15 , C(0)NHR 1 5 or C(0)CH 2 N0 2 ;
- R! 5 IS hydrogen or C ⁇ alkyl
- R!6 IS hydrogen, C ⁇ alkyl, C _7cycloalkyl, optionally substituted phenyl or benzyl, acetyl or benzoyl;
- X is -U(CH2) S Z- or X is a group selected from:
- U and Z independently are a divalent radical selected from -N(R ⁇ ). ) -0-, -S(0) t - , - N(R 16 )C(0)-, -C(0)N(R!6)- and -N[C(0)R 16 ]-, W is a carbon or a nitrogen atom; m is 0 or an integer from 1 to 5; n, r and t are each independently selected from 0, 1 and 2; p and q are each independently selected from 1 and 2; and s is an integer from 2 to 8; and pharmaceutically acceptable salts and solvates thereof.
- salts and solvates of compounds of the mvention which are suitable for use in medicine are those wherein the counte ⁇ on or associated solvent is pharmaceutically acceptable.
- salts and solvates having non-pharmaceutically acceptable countenons or associated solvents are within the scope of the present invention, for example, for use as intermediates m the preparation of other compounds of the invention and their pharmaceutically acceptable salts and solvates
- prodrug as used herein means a compound which is converted within the body, e.g. by hydrolysis in the blood, into its active form that has medical effects.
- pharmaceutically acceptable prodrugs are described in T. Higuchi and N. Stella, "Prodrugs as Novel Delivery Systems", Nol. 14 of the A.C.S. Symposium Series, Edward B. Roche, ed., "Bioreversible Carriers in Drug Design", American Pharmaceutical Association and Pergamon Press, 1987, and in D. Fleisher, S. Ramon and H. Barbra "Improved oral drug delivery: solubility limitations overcome by the use of prodrugs", Advanced Drug Delivery Reviews (1996) 19(2) 115-130, each of which are incorporated herein by reference.
- Prodrugs are any covalently bonded carriers that release a compound of structure (I) in vivo when such prodrug is administered to a patient.
- Prodrugs are generally prepared by modifying functional groups in a way such that the modification is cleaved, either by routine manipulation or in vivo, yielding the parent compound
- Prodrugs include, for example, compounds of this invention wherein hydroxy, amme or sulfhydryl groups are bonded to any group that, when administered to a patient, cleaves to form the hydroxy, amme or sulfhydryl groups.
- prodrugs include (but are not limited to) acetate, formate and benzoate de ⁇ vatives of alcohol, sulfhydryl and amme functional groups of the compounds of structure (I).
- esters may be employed, such as methyl esters, ethyl esters, and the like. Esters may be active in their own ⁇ ght and/or be hydrolysable under in vivo conditions m the human body Suitable pharmaceutically acceptable in vivo hydrolysable ester groups include those which break down readily in the human body to leave the parent acid or its salt.
- the compound of formula (I) and salts thereof may form solvates (e.g. hydrates) and the invention includes all such solvates.
- the solid wedge shaped bond indicates that the bond is above the plane of the paper.
- the broken bond indicates that the bond is below the plane of the paper.
- ether groups include those in which R 2 is a t ⁇ alkylsilyl (i.e. t ⁇ methylsilyl).
- R 2 represents an acyloxy group
- suitable groups R 2 include acetyl or benzoyl.
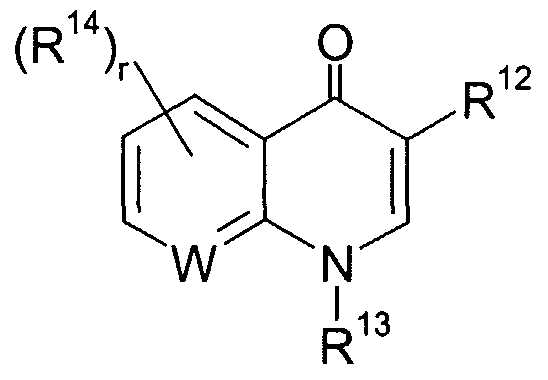
- R' is a heterocychc group having the following structure:
- W is CR* 7 or nitrogen, where R ⁇ is hydrogen or R ⁇ 4 , said heterocychc is linked in the 7 or 6 position to the Z group as above defined or to one of the nitrogen atoms contained in the following structures:
- R' is a heterocychc group having the following structure:
- R' is a heterocychc group having the following structure:
- W is CR 17 , where R 17 is hydrogen or R 14 , said heterocychc is linked in the 8 or 7 position to the Z group as above defined or to one of the nitrogen atoms contained in the following structures:
- R 7 is a heterocychc group having the following structure:
- W is CR* 7 , where R. ⁇ and R ⁇ are linked to form the bivalent radical -0(CH2)2" or -(CH2) V -, said heterocychc is linked in the (i) or (ii) position to the Z group as above defined or to one of the nitrogen atoms contained in the following structures:
- heterocychc is linked in the 2 or 3 position to the Z group as above defined or to one of the nitrogen atoms contained in the following structures:
- Ci ⁇ alkyl refers to a straight or branched alkyl group containing from 1 to 4 carbon atoms; examples of such groups include methyl, ethyl, propyl, isopropyl, n-butyl, isobutyl, tert-butyl.
- C2_6alkenyl group refers to a straight or branched alkenyl group containing from 2 to 6 carbon atoms; examples of such groups include 2-propenyl, 1-propenyl, isopropenyl, 2-butenyl, 2-pentenyl, 2-hexenyl and the like. It will be appreciated that in groups of the form -0-C2-6alkenyl, the double bond is preferably not adjacent to the oxygen.
- C3_7Cycloalkyl group means a non-aromatic monocyclic hydrocarbon ring of 3 to 7 carbon atoms such as, for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl.
- 5 or 6 membered heteroaryl as used herein as a group or a part of the group refers to furanyl, thiophenyl, imidazolyl, oxazolyl, thiazolyl, pyrazolyl, pyridyl, pyridazinyl or pyrimidinyl.
- heterocyclyl groups include, but are not limited to, pyrrolidinyl, imidazolidinyl, pyrazolidinyl, piperidyl, piperazinyl, morpholino, tetrahydropyranyl, tetrahydrofuranyl, and thiomorpholino.
- halogen refers to a fluorine, chlorine, bromine or iodine atom.
- C ⁇ alkoxy group may be a straight chain or a branched chain alkoxy group, for example methoxy, ethoxy, propoxy, prop-2-oxy, butoxy, but-2-oxy or 2-methylprop-2-oxy.
- 9 to 10 membered fused bicyclic heteroaryl refers to quinolinyl, isoquinolinyl, 1,2,3,4-tetrahydroisoquinolinyl, benzofuranyl, benzimidazolyl, benzothienyl, benzoxazolyl, 1,3-benzodioxazolyl, indolyl, benzothiazolyl, furylpyridine, oxazolopyridyl or benzothiophenyl.
- optionally substituted phenyl, optionally substituted 5 or 6 membered heteroaryl, optionally substituted 9 to 10 membered fused bicyclic heteroaryl or optionally substituted 5 or 6 membered heterocychc group refer to a group which is substituted by 1 to 3 groups selected from halogen, Cj ⁇ alkyl, C ⁇ alkoxy, hydroxy, nitro, cyano, amino, Cj ⁇ alkylammo or d ⁇ C ⁇ _4 alkylamino, phenyl and 5 or 6 membered heteroaryl.
- Preferred compounds of formula (I) are those wherein R 2 is hydrogen
- R ⁇ is hydrogen
- R 4 is hydrogen or C j ⁇ alkyl optionally substituted by up to three substituents independently selected from optionally substituted 5 or 6 membered heteroaryl, OR 8 , S(0) n R 8 , NR R 9 , halogen and cyano In another embodiment, R 4 is hydrogen or Ci .
- R 4 is C ⁇ alkyl optionally substituted by up to three substituents independently selected from optionally substituted 5 or 6 membered heteroaryl, OR 8 , S(0) n R 8 , NR 8 R 9 , halogen and cyano
- R 4 is Ci ⁇ alkyl optionally substituted by up to two substituents independently selected from optionally substituted 5 or 6 membered heteroaryl, OR 8 , S(0) n R 8 , NR 8 R 9 , halogen and cyano
- Representative examples of R 4 include include hydrogen and C ⁇ alkyl, for example methyl, ethyl, propyl or isopropyl, optionally substituted by optionally substituted 5 or 6 membered heteroaryl such as pyridyl,
- R 5 is 0(CH2) p O(CH2) q R 10 , wherein R 10 is preferably NR 8 R 9
- R ⁇ is hydroxy
- R ⁇ is hydroxy
- R5 and R° taken together with the intervening atoms form a cyclic group having the following structure
- R ' include heterocychc groups having the following structure
- W is preferably CR ⁇ where R ⁇ is hydrogen
- R 8 and R 9 include C ⁇ alkyl, for example methyl and ethyl
- R ⁇ 2 is C(0)OR ⁇ , wherein R ⁇ is preferably hydrogen.
- R ⁇ is hydrogen, Ci _4alkyl, C3_7cycloalkyl, or optionally substituted phenyl or benzyl.
- a representative example of R ⁇ is C3_7cycloalkyl, for example C3.
- gcycloalkyl such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, in particular cyclopropyl.
- R* 4 include halogen, in particular fluorine and chlonne.
- R ⁇ is hydrogen or C ⁇ alkyl. Preferably, R ⁇ is hydrogen.
- X is -U(CH2) S Z-.
- X is -U(CH2) S Z- wherein U and Z are preferably -NH-.
- X is -U(CH2) S Z- wherein U and Z are preferably independently -NH- or -O-.
- W is carbon 1 e. CR ⁇ ' where R* ' is hydrogen or R* 4 , m particular where R ⁇ 7 is hydrogen, or nitrogen
- m is 1 to 3, in particular 2.
- n 0.
- p and q are each independently selected from 1 and 2.
- p is 1, q is preferably 2.
- s is 2 to 4, in particular 2.
- Particularly preferred compounds of the invention are- 4"-6>-[3 -[[2-[(3 -carboxy-7-chloro- 1 -cyclopropyl- 1 ,4-dihydro-4-oxo-6- quinolinyl ) amino]ethyl]amino]propionyl]- 11 - ⁇ 9-(2-dimethylaminoethoxymethyl)-(9E)- methoximino erythromycin A,
- the compounds of the invention may also be active against resistant strains, for example erythromycin resistant strains.
- the compounds of the invention may be active against erythromycin resistant strains of Streptococcus pneumoniae and Streptococcus pyogenes.
- the compounds of the mvention may therefore be used for treating a variety of diseases caused by pathogenic microorganisms, in particular bacteria, in human beings and animals.
- a compound of formula (I) or a physiologically acceptable derivative, for example salt, thereof for use in the therapy or prophylaxis of systemic or topical microbial infections, in particular bacterial infections, in a human or animal subject.
- a compound of formula (I) or a physiologically acceptable derivative, for example salt, thereof for the manufacture of a therapeutic agent for the treatment or prophylaxis of systemic or topical microbial infections, in particular bacterial infections, in a human or animal body.
- a method of treatment of the human or non-human animal body to combat microbial infections, in particular bacterial infections comprises administering to the body an effective amount of a compound of formula (I) or a physiologically acceptable derivative, for example salt, thereof. While it is possible that, for use m therapy, a compound of the invention may be administered as the raw chemical it is preferable to present the active ingredient as a pharmaceutical formulation
- compositions comp ⁇ smg a compound of the invention adapted for use in human or vete ⁇ nary medicine.
- Such compositions may be presented for use in conventional manner with the aid of one or more suitable carriers or excipients.
- the compositions of the invention include those in a form especially formulated for parenteral, oral, buccal, rectal, topical, implant, ophthalmic, nasal or genito-unnary use.
- the compounds of the invention may also, for example, be formulated as suppositories e g containing conventional suppository bases for use in human or veterinary medicine or as pessaries e g containing conventional pessary bases
- the compounds according to the mvention may be formulated for topical administration, for use in human and veterinary medicine, in the form of ointments, creams, gels, lotions, shampoos, powders, (including spray powders), pessaries, tampons, sprays, dips, aerosols, drops (e.g eye ear or nose drops) or pour-ons.
- Aerosol sprays are conveniently delivered from pressurised packs, with the use of a suitable propellant, eg dichlorodifluoromethane, t ⁇ chlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the compounds according to the invention may be delivered for use in human or veterinary medicine via a nebuhser.
- compositions for topical administration may also contain other active ingredients such as corticosteroids or antifungals as appropriate.
- compositions may contain from 0 01 -99% of the active material
- the composition will generally contain from 0 01-10%, more preferably 0.01-1% of the active material.
- the daily dose as employed for adult human treatment it will range from 2-100mg/kg body weight, preferably 5-60mg/kg body weight, which may be administered in 1 to 4 daily doses, for example, depending on the route of administration and the condition of the patient.
- each unit will preferably contain 200mg to lg of active ingredient.
- the duration of treatment will be dictated by the rate of response rather than by arbitrary numbers of days.
- the compounds of general formula (I) and de ⁇ vatives thereof may be pu ⁇ fied by conventional methods known in the art.
- the compounds may be purified by HPLC using an aqueous solution of an acid such as formic acid or t ⁇ fluoroacetic acid.
- Suitable activated derivatives of the carboxyl group include the corresponding acyl hahde, mixed anhydride or activated ester such as a thioester.
- the reaction is preferably carried out m a suitable aprotic solvent such as a halohydrocarbon (e.g. dichloromethane) or N,N- dimethylformamide optionally in the presence of a tertiary organic base such as dimethylaminopy ⁇ dine or tnethylamme or in the presence of inorganic base (i.e sodium hydride) and at a temperature within the range of 0° to 120°C.
- a suitable aprotic solvent such as a halohydrocarbon (e.g. dichloromethane) or N,N- dimethylformamide
- a tertiary organic base such as dimethylaminopy ⁇ dine or tnethylamme or in the presence of inorganic base (i.e sodium hydride) and at
- Compounds of formula (I) wherein m is 0 and U is selected from -N(R*")- , -O- and -S(O) ⁇ - wherem t is 0, may be prepared by reaction of compounds of formula (II), in which the 4" hydroxy is suitably activated, with a compound of formula X a R a (IN), followed where necessary by subsequent removal of the hydroxyl protecting group R 2 and conversion of the X a R a group to XR 7 .
- Suitable activated de ⁇ vatives of the 4" hydroxy group include for example carbonyl lmidazole.
- the reaction is preferably earned out m a suitable aprotic solvent such as a halohydrocarbon (e.g.
- dichloromethane or ⁇ , ⁇ -d ⁇ methylformam ⁇ de optionally in the presence of a tertiary base such as dimethylaminopy ⁇ dme or tnethylamme and at a temperature within the range of 0° to 120°C.
- a tertiary base such as dimethylaminopy ⁇ dme or tnethylamme and at a temperature within the range of 0° to 120°C.
- bases examples include organic bases such as diisopropylethylamine, triethylamine and l,8-diazabicyclo[5.4.0]undec-7-ene, and inorganic bases such as potassium hydroxide, cesium hydroxide, tetraalkylammonium hydroxyde, sodium hydride, potassium hydride and the like.
- Suitable leaving groups for this reaction include halide (e.g. chloride, bromide or iodide) or a sulfonyloxy group (e.g. tosyloxy or methansulfonyloxy).
- compounds of formula (I) wherein m is 2, U is a group selected from - ⁇ (Rl")-, -O- and -S-, may be prepared by Michael reaction of a compound of formula (IX), wherein R 2 is optionally a hydroxyl protecting group
- compounds of formula (I) may be converted into other compounds of formula (I).
- compounds of formula (I) wherein U is -S(0)f and t is 1 or 2 may be prepared by oxidation of the corresponding compound of formula (I) wherein t is 0.
- the oxidation is preferably carried out using a peracid, e.g. peroxybenzoic acid, followed by treatment with a phosphine, such as t ⁇ phenylphosphine
- the reaction is suitably earned out in an organic solvent such as methylene chloride.
- R 7a R 7a (IN), wherein X a has the meaning defined above with R ⁇ 7 OC(0)(CH2) m L (X) wherein R* 7 is carboxyl protecting group and L is a suitable leaving group, followed by removal of R ⁇ .
- L is a suitable leaving group such as chlorine, fluorine or bromine
- a compound of formula -U(CH2) S Z- (XII) in which Z is -N(R1 ")-, -O- or -S- with piperazine or with lH-pyrrolo[3,4-b]pyridine, octahydro.
- Suitable hydroxy protecting reagents are those described by T.W. Greene and P.G.M Wuts in Protective Groups in Organic Synthesis 2 nd ed., John Wiley & Son, Inc 1991, which is incorporated by reference.
- suitable hydroxy protecting reagents include acetic anhydride, benzoic anhydride or a trialkylsilyl chloride in a protic solvent.
- aprotic solvents are dichloromethane, NN-dimethylformamide, dimethylsulfoxide, tetrahydrofuran and the like.
- Suitable R 7 carboxyl protecting group include t-butyl, allyl or benzyl.
- the crude product was treated with HC1 (6% aqueous solution) heating for 1 h in the presence of charcoal. After filtration, the solution was cooled to 35-40°C and a first precipitation happened. The precipitate was filtered, washed with water and dried at 110°C for 1 h. The title compound (6.4 g) was obtained as the hydrochloride salt. The hydrochloride salt was then converted to the free base using standard conditions.
- Example 4 4"-0-[3-[[2-[(3-Carboxy-l-cycIopropyI-l,4-dihydro-6-fluoro-4-oxo-7- quinolinyl) amino]ethyl]amino]propionyl]-ll,12-carbonate-(9E)-methoximino erythromycin A
- Example 8 4"-0-[3-[[2-[(3-Carboxy-7-chloro-l-cyclopropyl-l,4-dihydro-4-oxo-6- quinolinyl) amino]ethyl]amino]propionyl]-ll,12-carbonate-(9E)-0- (ethoxymethyl)oximino erythromycin A
- Example 14 4"-0-[3-[[2-[(3-Carboxy-7-chloro-l-cycIopropyl-l,4-dihydro-4-oxo-6-quinolinyl)- amino]ethyI]amino]propionyl]-(9E)-0-propyloximino erythromycin A
- MIC ⁇ g/ml
- test compounds against various organisms were determined including: S. aureus Smith ATCC 13709, S. pneumoniae SP030, S. pyogenes 3565, E. faecalis ATCC 29212, H. influenzae ATCC 49247, M. catarrhalis ATCC 23246.
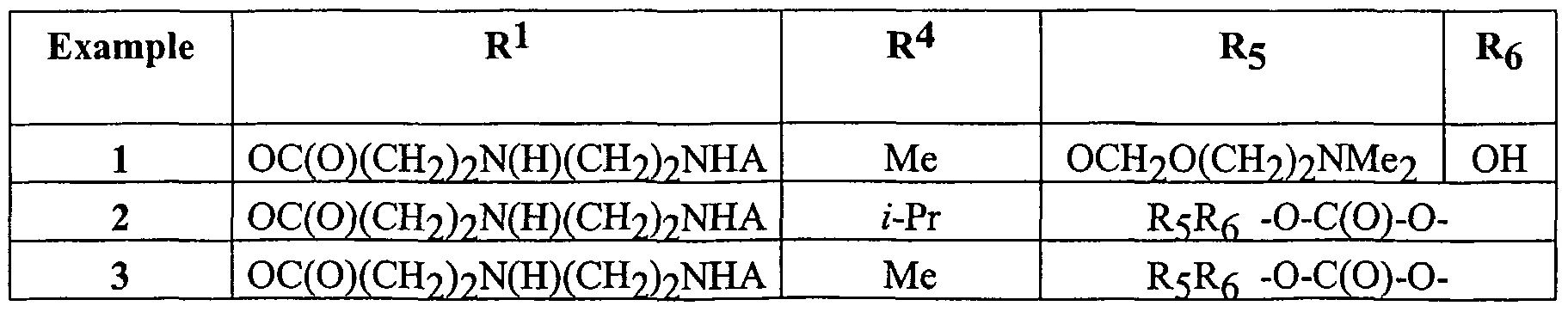
- Examples 1-3 and 6-8 have an MIC ⁇ 1 ⁇ g/ml against S. aureus Smith ATCC 13709, S. pneumoniae SP030, S. pyogenes 3565 and E. faecalis ATCC 29212.
- Examples 1 and 3 have an MIC ⁇ 2 ⁇ g/ml against H. influenzae ATCC 49247 and M. catarrhalis ATCC 23246.
- Examples 1-3 and 6-8 have an MIC ⁇ 0.25 ⁇ g/ml against erythromycin resistant strains of Streptococcus pneumoniae and Streptococcus pyogenes.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Oncology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Description




























Claims













Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2003293635A AU2003293635A1 (en) | 2002-10-31 | 2003-10-29 | 4'' - modified erythromycin derivatives |
| EP03788981A EP1558625A2 (en) | 2002-10-31 | 2003-10-29 | 4''-modified erythromycin derivatives |
| US10/533,461 US7351696B2 (en) | 2002-10-31 | 2003-10-29 | Compounds |
| JP2004547617A JP2006506389A (en) | 2002-10-31 | 2003-10-29 | 4 "modified erythromycin derivative |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0225384.7A GB0225384D0 (en) | 2002-10-31 | 2002-10-31 | Novel compounds |
| GB0225384.7 | 2002-10-31 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004039822A2 true WO2004039822A2 (en) | 2004-05-13 |
| WO2004039822A3 WO2004039822A3 (en) | 2004-07-08 |
Family
ID=9946942
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/012068 Ceased WO2004039822A2 (en) | 2002-10-31 | 2003-10-29 | 4’’ - modified erythromycin derivatives |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7351696B2 (en) |
| EP (1) | EP1558625A2 (en) |
| JP (1) | JP2006506389A (en) |
| AR (1) | AR041860A1 (en) |
| AU (1) | AU2003293635A1 (en) |
| GB (1) | GB0225384D0 (en) |
| WO (1) | WO2004039822A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004101589A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 membered-ring compounds |
| WO2004101585A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Macrolides substituded at the 4'-position |
| WO2004101587A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 memberred-ring compounds |
| WO2004101588A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 membered ring compounds |
| WO2005108412A1 (en) * | 2004-05-06 | 2005-11-17 | Glaxosmithkline Istrazivacki Centar Zagreb D.O.O. | Ester linked macrolides useful for the treatment of microbial infections |
| WO2006050941A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| WO2006050942A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolones - amino substituted quinolones |
| WO2006050943A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| WO2006050940A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| US7547679B2 (en) | 2005-05-10 | 2009-06-16 | Glaxosmithkline Istrazivacki Center Zagreb D.O.O | Ether linked macrolides useful for the treatment of microbial infections |
| WO2012038372A1 (en) | 2010-09-20 | 2012-03-29 | Novartis Ag | Novel process for the preparation of 9-deoxo-9a-aza- 9a-homoerythromycin a modified in the c-4'' of the cladinose ring by an epoxide group |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU1204288A (en) | 1987-02-24 | 1988-08-25 | Beecham Group Plc | Erythromycins |
| WO1996034007A1 (en) * | 1995-04-27 | 1996-10-31 | Laboratorios Aranda, S.A. De C.V. | Quinolonylcarboxyerythromycin derivatives and pharmaceutical compositions containing them |
| AU710532B2 (en) * | 1996-05-07 | 1999-09-23 | Abbott Laboratories | 3-descladinose-2,3-anhydroerythromycin derivatives |
| EP0895999A1 (en) * | 1997-08-06 | 1999-02-10 | Pfizer Products Inc. | C-4" substituted macrolide antibiotics |
| HUP0201516A3 (en) | 1999-05-24 | 2003-03-28 | Pfizer Prod Inc | 13-methyl-erythromycin derivatives, process for their preparation and pharmaceutical compositions containing them |
| US6946446B2 (en) | 2000-02-24 | 2005-09-20 | Abbott Laboratories | Anti-infective agents useful against multidrug-resistant strains of bacteria |
| ATE312839T1 (en) | 2000-02-24 | 2005-12-15 | Abbott Lab | ANTI-INFECTIVE AGENTS USEFUL AGAINST MULTIPLE DRUG-RESISTANT STRAINS OF BACTERIA |
| GB0127349D0 (en) * | 2001-11-14 | 2002-01-02 | Glaxo Group Ltd | Macrolides |
-
2002
- 2002-10-31 GB GBGB0225384.7A patent/GB0225384D0/en not_active Ceased
-
2003
- 2003-10-29 WO PCT/EP2003/012068 patent/WO2004039822A2/en not_active Ceased
- 2003-10-29 AU AU2003293635A patent/AU2003293635A1/en not_active Abandoned
- 2003-10-29 JP JP2004547617A patent/JP2006506389A/en active Pending
- 2003-10-29 EP EP03788981A patent/EP1558625A2/en not_active Withdrawn
- 2003-10-29 US US10/533,461 patent/US7351696B2/en not_active Expired - Fee Related
- 2003-10-29 AR ARP030103954A patent/AR041860A1/en unknown
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004101589A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 membered-ring compounds |
| WO2004101585A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Macrolides substituded at the 4'-position |
| WO2004101587A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 memberred-ring compounds |
| WO2004101588A1 (en) * | 2003-05-13 | 2004-11-25 | Glaxo Group Limited | Novel 14 and 15 membered ring compounds |
| WO2005108412A1 (en) * | 2004-05-06 | 2005-11-17 | Glaxosmithkline Istrazivacki Centar Zagreb D.O.O. | Ester linked macrolides useful for the treatment of microbial infections |
| WO2006050941A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| WO2006050942A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolones - amino substituted quinolones |
| WO2006050943A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| WO2006050940A1 (en) * | 2004-11-11 | 2006-05-18 | Glaxo Group Limited | Macrolone compounds |
| US7718621B2 (en) | 2004-11-11 | 2010-05-18 | Glaxo Group Ltd. | Macrolones—amino substituted quinolones |
| US7547679B2 (en) | 2005-05-10 | 2009-06-16 | Glaxosmithkline Istrazivacki Center Zagreb D.O.O | Ether linked macrolides useful for the treatment of microbial infections |
| WO2012038372A1 (en) | 2010-09-20 | 2012-03-29 | Novartis Ag | Novel process for the preparation of 9-deoxo-9a-aza- 9a-homoerythromycin a modified in the c-4'' of the cladinose ring by an epoxide group |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2003293635A1 (en) | 2004-05-25 |
| AU2003293635A8 (en) | 2004-05-25 |
| GB0225384D0 (en) | 2002-12-11 |
| US7351696B2 (en) | 2008-04-01 |
| JP2006506389A (en) | 2006-02-23 |
| US20070037757A1 (en) | 2007-02-15 |
| WO2004039822A3 (en) | 2004-07-08 |
| AR041860A1 (en) | 2005-06-01 |
| EP1558625A2 (en) | 2005-08-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1453846B1 (en) | 14 or 15 membered macrolides with antibacterial activity | |
| US6900183B2 (en) | Macrolide antibiotics | |
| ZA200301047B (en) | New macrolides with antibacterial activity. | |
| WO2004039822A2 (en) | 4’’ - modified erythromycin derivatives | |
| KR20070085919A (en) | Macroron-Amino Substituted Quinolone | |
| US20070293472A1 (en) | Novel 14 and 15 Membered Ring Compounds | |
| AU2005240849B2 (en) | Ester linked macrolides useful for the treatment of microbial infections | |
| JP2008519787A (en) | Macrolone compounds | |
| EP1328535B1 (en) | Macrolides | |
| MXPA05012163A (en) | Novel 14 and 15 membered-ring compounds. | |
| WO2004101589A1 (en) | Novel 14 and 15 membered-ring compounds | |
| JP2006528947A (en) | 4 "-substituted macrolide | |
| EP1633765B1 (en) | Novel 14 and 15 membered-ring compounds | |
| CA2525457A1 (en) | Novel 14 and 15 membered ring compounds | |
| EP1756134A1 (en) | Carbamate linked macrolides useful for the treatment of microbial infections | |
| US20080312167A1 (en) | 4'' Amino Linked Macrolides Useful for the Treatment of Microbial Infections | |
| JP2008519789A (en) | Macrolone compounds | |
| KR20070031900A (en) | Ester linked macrolides useful in the treatment of microbial infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004547617 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003788981 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003788981 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007037757 Country of ref document: US Ref document number: 10533461 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10533461 Country of ref document: US |