明 細 書
PLK阻害剤
技 術 分 野
本努明は、 ポロ様キナーゼ (Polo- like kinase、 PLK) P且害剤、 PLK阻害活性また は抗腫瘍活性等を有するピリミジン誘導体等に関する。
背 景 技 術 ,
細胞周期において、 有糸分裂期 (M期) は真核細胞の染色体を分配し、 正確な遺 伝情報を娘細胞に伝えるための重要なステップであり厳密に制御されている。 M期 の進行には様々なプロテインキナーゼが関与していることが知られている。 PLKフ ァミリーは、真核生物において M期への進入から脱出まで様々なステージに関与し ているセリンスレオ ンキ^ "一ゼ群である [ジーンズ'アンド'ディべロプメント (Genes & Dev. )、 12卷、 3777頁 (1998年) ]。 哺乳動物 (マウス)の PLK,遺伝子の 単離は 1994年に Hamanakaらにより初めて報告された [セル ·グロウス ·アンド · ディファレンシエーション (Cell Growth & Diff. )、 5卷、 249頁 (1994年) ;]。 多くの癌組織では、正常細胞に比べ PLKが過剰発現しており、癌化や癌の進行に 関与していると考えられている [US6180380;ィンターナショナル■ジャーナル · ォブ.オンコロジ一 (Int. J. Oncol. )、 15卷、 687頁 (1999年) ]0非小細胞肺癌、 頭頸部癌及び卵巣癌では PLKの高発現と癌の悪性化や予後の再発との相関が認めら れている [オンコジーン (0ncogene)、 14卷、 543頁 (1997年) ;インターナショナ ル .ジャーナル.ォブ .キャンサー (Int. j. Cancer)、 89卷、 535頁 (2000年) ; キャンサー ' レターズ (Cancer Letters)、 169卷、 41頁 (2000年) ]。
PLKの機能阻害変異体の導入実験では、 PLK阻害が正常細胞よりも高い割合で癌 細胞に細胞死を誘導するという報告がある [セル 'グロウス 'アンド■ディファレ ンシエーション (Cell Growth & Diff. )、 11卷、 615頁 (2000年) ;]。 また、 PLK のアンチセンスオリゴヌクレオチドがマゥス Xenograftモデルで抗腫瘍効果を示す ことも報告されている レ ィォケミカル 'アンド 'バイオフィジカル · リサーチ■ コミユエケーシヨンズ (Biochem. Biophys. Res. Comm. ) 260卷、 352 頁 (2000 年) ]。
また、 アルツハイマー病患者の脳では PLKの発現が上昇しており、神経変性疾患
と PLKの関連が明らかになりつつある [ニューロバイオロジー ·ォブ ·エージング (Neurobiol. Aging)、 21卷ヽ 837頁 (2000年) ]。 このことから、 PLK阻害剤は神 経変性疾患、例えばアルツハイマー病、 パーキンソン病等の治療剤としても有用で あると考えられる。 さらに末梢血液由来の静止リンパ球は PLKを発現しないが、 フ ィ トへマグルチニン刺激または IL- 2 刺激により、 PLK の発現が誘導される (US6180380)。 このことから、 PLK阻害剤は免疫系の活性化に関わる疾患、例えばァ レ^/ギーゃリゥマチの治療剤としても有用であると考えられる。
以上のように、 PLKを阻害する薬剤は癌や神経変性疾患、 アレルギー疾患等の細 胞シグナル伝達異常に関わる疾患の治療剤として有望であると考えられる。
PLK 酵素阻害活性を示す化合物としては、 cyanobacteria から単離された scytoneminが報告されている (W0 01/62900)。
5位にシァノまたは複素環基を有するピリミジン誘導体としては、 以下の化合物 が知られている。
後記の式 (I) において Z1及び Z2が Nで、 Xがシァノで、 W - Arに相当する基が芳 香族ァミノであるピリミジン誘導体が KDRキナーゼ及び Zまたは FGFrキナーゼ阻 害剤として (W0 00/78731)、 後記の式 (I) において Z1及び Z2が Nで、 R1が二環式 芳香族複素環基であるピリミジン誘導体が Src キナーゼ阻害剤として (¾Ό· 01/00213)、後記の式(I)において Ζ1及び Ζ2が Νで、 Xがシァノで、 R1がァミノで、 W-Ar に相当する基が芳香族ァミノであるピリミジン誘導体がサイクリン依存性キ ナーゼ (CDK) 阻害剤として (W0 01/727i7)、,後記の式 (I) において Xがシァノで ある誘導体が CD40機能阻害剤として (特開 2001- 89452)、 後記の式 (I) において Z1及び Z2が Nで、 Xがシァノで、 W - Arに相当する基が芳香族ァミノであるピリミジ ン誘導体がグリコーゲンシンターゼキナーゼ 3阻害剤として (W0 02/04429)、 後記 の式 (I) において Xが芳香族複素環基である誘導体がグリコーゲンシンターゼキ ナーゼ 3阻害剤として (W0 99/65897、 W0 02/20495)、 後記の式 (I) において Z1 及び Z2が Nであるピリミジン誘導体が抗ゥィルス剤 (W0 99/41253) または抗腫瘍 剤 (W0 00/39101) としてそれぞれ知られている。
また、 後記の式 (I) において Z1及ぴ Z2が Nで、 R1がァミノで、 Xがテトラゾリ ルである化合物が知られている [ケミカ一 -ツァイ トゥン (Chemiker- Zeitung)、
112卷、 135頁 (1988年) ]。
発 明 の 開 示
本発明の目的は、 PLK阻害剤、 PLK阻害作用または抗腫瘍作用等を有するピリミ ジン誘導体またはその薬理学的に許容される塩等を提供することにある。 ' 本発明は、 以下の (1 ) ~ ( 1 9 ) に関する。
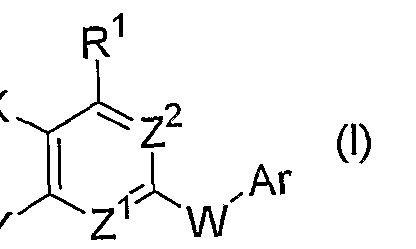
[式中、 Arは置換もしくは非置換のァリールまたは置換もしくは非置換の芳香族複 素環基を表し、
Z1及び Z2は、 同一または異なって窒素原子 (N) または CHを表し、
Xは水素原子、 ニトロ、 カルボキシ、 低級アルコキシカルボ二ル、 .置換もしくは非 置換の低級アルキル、 置換もしくは非置換のシクロアルキル、 - CQNHR7 (式中、 R7 は水素原子、 置換もしくは非置換の低級アルキル、 換もしくは非置換の低級アル ケュル、 置換もしくは非置換の低級アルキニル、 置換もしくは非置換のシクロアル キル、置換もしくは非置換のァラルキルまたは置換もしぐは非置換の複素環アルキ ルを表す)、 シアノまたはテトラゾリルを表し、
1 ) Xがシァノまたはテトラゾリルを表す場合、 ' Υは水素原子を表し、 . R1は- OR2 (式中、 は置換もしくは非置換の低級アルキルを表す) または - NR¾4 (式 中、 R3は置換もしくは非置換の低級アルキル、 置換もしくは非置換のシクロアルキ ル、置換もしくは非置換のァラルキルまたは置換もしくは非置換のァリールを表し、 は水素.原子または置換もしくは非置換の低級アルキルを表すか、 または R3と R4 が隣接する窒素原子と一緒になつて置換もしくは非置換の複素環基を形成する) を 表し、
Wは- NHCR¾6- (式中、 R5及ぴ R6は同一または異なって水素原子または置換もしくは 非置換の低級アルキルを表す) または- NHCR¾6CR5AR6A- (式中、 R5及び R6はそれぞれ 前記と同義であり、 R5A及び RMはそれぞれ前記 R5及び R6と同義である) を表し、
2 ) Xがシァノ及ぴテトラゾリル以外の基を表す場合、
Yは水素原子、 ニトロ、 カルボキシ、 低級アルコキシ力ルポニル、 置換もしくは非 置換の低級アルキル、置換もしくは非置換のシク口アルキルまたは- C0NHR7A (式中、 R7Aは水素原子、置換もしくは非置換の低級アルキル、置換もしくは非置換の低級ァ ルケニル、 置換もしくは非置換の低級アルキニル、 置換もしくは非置換のシクロア ルキル、置換もしくは非置換のァラルキルまたは置換もしくは非置換の複素環アル キルを表す) を表し、
R1は- 0R2A (式中、 R2Aは置換もしくは非置換の低級アルキル、置換もしくは非置換の 低級アルケニル、 置換もしくは非置換の低級アルキニル、 置換もしくは非置換のシ クロアルキル、 置換もしくは非置換のァラルキル、 置換もしくは非置換のァリール または置換もしくは非置換の複素環基を表す)、 ~SR2B (式中、 R2Bは前記 R2Aと同義 である) または- NR3AR4A (式中、 R3Aは置換もしくは非置換の低級アルキル、 置換も しくは非置換の低級ァルケ-ル、 置換もしくは非置換の低級アルキニル、 置換もし ぐは非置換のシクロアルキル、 置換もしくは非置換のァラルキル、 置換もしくは非 置換のァリールまたは置換もしくは非置換の複素環基を表し、 R4Aは水素原子または 置換もしくは非置換の低級アルキルを表す力 \ または R3Aと Aが隣接する窒素原子 と一緒になつて置換もしくは非置換の複素環基を形成する) を表し、
Wは- NHCR5¾6B- (式中、 R5B及び R6Bは同一または異なって水素原子、 ハロゲンまたは 置換もしくは非置換の低級アルキルを表す)、 -NHCR5BR6BCR5CR6C- (式中、 R5B及び R6B はそれぞれ前記と同義であり、 及び R6Gはそれぞれ前記 R5B及び R6Bと同義である) または- NHCR5BR6BCR5¾6¾R5DR6D- (式中、 R5B、 R6B、 R 及び R6eはそれぞれ前記と同義で あり、 R5D及び R6Dはそれぞれ前記 R5B及び R6Bと同義である) を表す] で表される化 合物またはその薬理学的に許容される塩を有効成分として含有する PLK阻害剤。
( 2 ) Xがシァノまたはテトラゾリルであり、
R1カ 0R2a (式中、 R2aは低級アルキル、 シクロアルキルアルキルまたは脂環式複素環 アルキルを表す) または- NR¾4 (式中、 及び R4はそれぞれ前記と同義である) で あり、 ' '
Z1及び Z2が窒素原子 (N) である前記 (1 ) 記載の PLK阻害剤。
( 3 ) 式 (IA)
[式中、 ArAは置換または非置換の芳香族複素環基を表し、
nは 0または 1を表し、
R8は- 0RW (式中、 R1Qは低級アルキル、 シクロアルキルアルキルまたは脂環式複素環 アルキルを表す) または- NRUR12 (式中、 R11は低級アルキル、 シクロアルキルアル キルまたは置換もしくは非置換のァリールを表し、 R12は水素原子または低級アルキ ルを表すか、または R11と R12が隣接する鲎素原子と一緒になつてテトラヒドロイソ キノリン -2-ィル、 3, 5-ジメ,チルピペリジノまたは 2, 6 -ジメチルモルホリノを形成 する) を表し、
R9\ R9b、 R9e及び R9dは、 同一または異なって水素原子または低級アルキルを表す] で表されるピリミジン誘導体またはその薬理学的に許容される塩。
( 4 ) R8が-丽 RUa (式中、 RUaは置換もしくは非置換のァリールを表す) であり、 R9a、 R9b、 R9c及び R9dが水素原子である前記 ( 3 ) 記載のピリミジン誘導体またはそ の薬理学的に許容される塩。
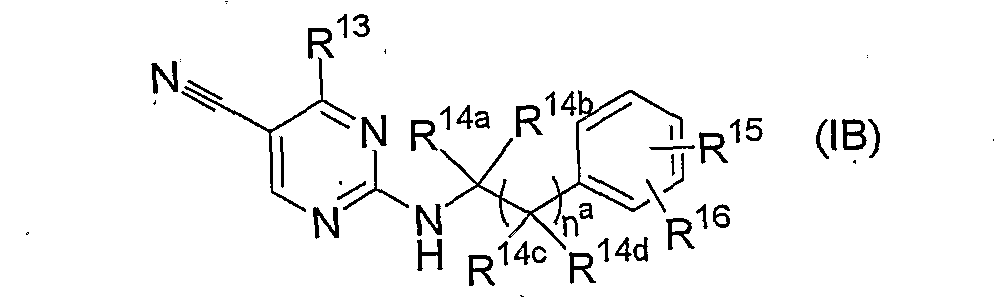
. ( 5 ) 式 (IB)
{式中、 naは 0または 1を表し、'
R13は- OR17 (式中、 R17はシクロアルキルアルキルまたは含酸素脂環式複素環アルキ ルを表す) または- NR18R19 [式中、 R18は低級アルキル、 シクロアルキルアルキル、 置換もしくは非置換のァリールまたは式'(Π)
(式中、 nbは 1〜3の整数を表す) で表される基を表し、 R19は水素原子または低級
アルキルを表すか、または R1Sと R19が隣接する窒素原子と一緒になつてテトラヒド 口イソキノリン- 2-ィル、 3, 5-ジメチルピペリジノまたは 2, 6-ジメチルモルホリノ を形成する] を表し、
Rl4a、 Rl4b、 R14e及び Rl4dは、同一または異なって水素原子または低級アルキルを ¾し、 R15及び R16は同一または異なって水素原子、 ハロゲン、 低級アルキルまたは低級ァ ルコキシを表すか、 または R15と R16が一緒になつて - 0(C¾)m0- (式中、 mは 1また は 2を表す) を形成する. } で表されるピリミジン誘導体またはその薬理学的に許容 される塩。
(6) naが 0であり、 R15及ぴ R16が同一または異なって水素原子または低級アルコ キシであるか、または R15と R16が一緒になつて - 0CH20- である前記(5)記载のピ リミジン誘導体またはその薬理学的に許容される塩。
(7) 前記 (1) または (2) に記載の化合物またはその薬理学的に許容される塩 を有効成分として含有する PLKが関与する疾患の予防及び Zまたは治療剤。
(8) PLK阻害剤の製造のための、 前記 (1) または (2) に記載の化合物または その薬理学的に許容される塩の使用。 '
(9) PLKが関与する疾患の予防及び Zまたは治療剤の製造のための、 前記 (1) または (2) に記載の化合物またはその薬理学的に許容される塩の使用。
(10) 前記 (1) または (2) に記載の化合物またはその薬理学的に許容される 塩の有効量を投与する工程を含む、 PLKが関与する疾患の予防及び/または治療方 法。
(1 1) 前記 (3) 〜 (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩を有効成分として含有する PLK阻害剤。
(12) 前記 (3) 〜 (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩を有効成分として含有する PLKが関与する疾患の予防及び/ま たは治療剤。
(1 3) 前記 (3) 〜 (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩を有効成分として含有する抗腫瘍剤。
(14) 前記 (3) ~ (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩を有効成分として含有する医薬。
(15) PLK阻害剤の製造のための、 前記 (3) 〜 (6) のいずれかに記載のピリ ミジン誘導体またはその薬理学的に許容される塩の使用。
(16) PLKが関与する疾患の予防及ぴ Zまたは治療剤の製造のための、前記(3) 〜 (6) のいずれかに記載のピリミジン誘導体またはその薬理学的に許容される塩 の使用。
'(1 7) 前記 (3) ~ (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩の有効量を投与する工程を含む、 PLKが関与する疾患の予防及 び/または治療方法。
(18) 抗腫瘍剤の製造のための、 前記 (3) 〜 (6) のいずれかに記載のピリミ ジン誘導体またはその薬理学的に許容される塩の使用。
(19) 前記 (3) 〜 (6) のいずれかに記載のピリミジン誘導体またはその薬理 学的に許容される塩の有効量を投与する工程を含む、腫瘍の予防及び/または治療 方法。
以下、 式 (1)、 (IA) 及び (IB) で表される化合物をそれぞれ化合物 (1)、 (IA) 及び (IB) という。 他の式番号の化合物についても同様である。
式 (1)、 (IA) 及び (IB) の各基の定義において、 以下の例示が挙げられる。
(i) ハロゲンは、 フッ素、 塩素、 臭素及ぴヨウ素の各原子を表す。
(ii) 低級アルキル、低級アルコキシ及び低級アルコキシ力ルポニルの低級アルキ ル部分としては、 例えば直鎖または分岐状の炭素数 1〜10のアルキルが挙げられ、 具体的にはメチル、 ェチル、 プロピル、 イソプロピル、 ブチル、 イソプチル、 sec- ' プチノレ、 tert-プチル、 ペンチノレ、 ィソペンチノレ、 ネオペンチノレ、 へキシノレ、 ヘプ チル、 ォクチル、 イソォクチル、' ノエル、 デシル等が挙げられる。
(iii) シク口アルキル及びシク口アルキルアルキルのシク口アルキル部分として は、例えば炭素数 3~8のシク口アルキルが挙げられ、具体的にはシク口プロピル、 シクロプチノレ、 シクロペンチノレ、 シクロへキシノレ、 シクロへプチノレ、 シクロォクチ ル等が挙げられる。
(iv) 低級ァルケ-ルとしては、 例えば直鎖、 分岐または環状の炭素数 2〜8のァ ルケニルが挙げられ、 具体的にはビュル、 ァリル、 1 -プロべエル、 ブテニル、 ペン テニノレ、 へキセニル、 ヘプテニノレ、 ォクテ二ノレ、 シクロへキセニノレ、 2,6-ォクタジ
ェ-ル等が挙げられる。
(V) 低級アルキエルとしては、 例えば直鎖または分岐状の炭素数 2〜8のアルキエ ルが挙げられ、 具体的にはェチニル、 プロビュル、 ブチニノレ、 ペンチニル、 へキシ ニル、 へプチニル、 ォクチニル等が挙げられる。
(vi) ァリールとしては、 例えば炭素数 6〜14の単環式、 二環式または三環式のァ リール及び炭素数 8〜14のべンゾシクロアルキルが挙げられ、具体的にはフエニル、 ナフチル、 ィンデュル、 アントラニノレ、 ィンダニノレ、 1, 2, 3, 4 -テトラヒ ドロナフチ ノレ、 6, 7, 8, 9-5H -ベンゾシク口へプチル等が挙げられる。
(vii) ァラルキル、 複素環アルキル、 脂環式複素環アルキル、 含酸素脂,環式複素 環アルキル及ぴシクロアルキルアルキルのアルキレン部分は、 前記低級アルキル
(ii) の定義から水素を一つ除いたものと同義である。
(viii) ァラルキルのァリー'ル部分としては、 前記ァリール (vi) の定義で挙げた 基が挙げられる。
(ix) 複素環基及び複素環アルキルの複素環基部分としては、 例えば窒素原子、 酸 素原子及び硫黄原子から選ばれる少なくとも 1個の原子を含む 5員、 6員または 7 員の単環式複素環基、 3〜8員の環が縮合した二環または三環式であつて窒素原子、 酸素原子及び硫黄原子から選ばれる少なくとも 1個の原子を含む縮環式複素環基等 が挙げられ、 具体的にはピリジル、 ピラジニル、 ピリミジ -ル、 ピリダジ -ル、 ベ ンゾィミダゾリノレ、 2-ォキソベンゾィミダゾリル、 ベンゾトリァゾリル、 ベンゾフ リノレ、 ベンゾチェ二ノレ、 プリ二ノレ、 ベンゾォキサゾリノレ、 ベンゾチアゾリノレ、 ベン ゾジォキソリル、 ィンダゾリル、 インドリル、 イソインドリル、 キノリル、 イソキ ノリル、フタラジニル、ナフチリジニル、キノキサリニル、 ピロリノレ、 ピラゾリル、 キナゾリニル、 シンノリ -ル、 トリアゾリノレ、 テトラゾリル、 イミダゾリル、 ォキ サゾリル、 イソォキサゾリル、 チアゾリル、 イソチアゾリル、 チェニル、 フリル、 ピロリジニノレ、 '2, 5-ジォキソピロリジニノレ、 チアゾリジニノレ、 ォキサゾリジニノレ、 ピペリジル、 ピペリジノ、 ピペラジニル、 ホモピペラジニル、 ホモピペリジル、 ホ モピぺリジノ、 モルホリ -ル、 モルホリノ、 チオモルホリニル、 チオモルホリノ、 ビラニル、テトラヒドロピリジル、テトラヒドロピラエル、テトラヒドロフラニル、 テトラヒドロキノリル、 テトラヒドロイソキノリル、 ォクタヒドロキノリル、 イン
ドリ-ル等が挙げられる。 脂環式複素環アルキルの脂環式複素環基部分は、 上記の うち芳香族性を有さないものを表し、芳香族複素環基は上記のうち芳香族性を有す るものを表す。 また、含酸素脂環式複素環ァ キ の含酸素脂環式複素環基部分と しては、例えばテトラヒドロフラ -ル、テトラヒドロビラニル、ジヒ ロビラニル、 ピラエル等が挙げられる。
(X) P舞接する窒素原子と一緒になつて形成される複素環基としては、少なくとも 1 個の窒素原子を含む 5員または 6員の単環式複素環基 (該単環式複素環基は、 他の 窒素原子、 酸素原子または硫黄原子を含んでいてもよい)、 3〜8員の環が縮合した 二環または三環式で少なくとも 1個の窒素原子を含む縮環式複素環基(該縮環式複 素環基は、 他の窒素原子、 酸素原子または硫黄原子を含んでいてもよい) 等が挙げ られ、 具体的にはテトラヒドロピリジル、 インドリ-ル、 イソインドリ-ル、 ピロ リジエル、 チアゾリジニル、 ォキサゾリジニル、 ピペリジノ、 ホモピペリジノ、 ピ ペラジ-ル、 ホモピペラジニル、 モノレホリノ、 チオモルホリノ、 ペルヒドロアゼピ ニル、ペルヒドロアゾシニル、テトラヒドロキノリノレ、テトラヒドロイソキノリル、 ォクタヒ ドロキノリル、 ベンゾイミダゾリル、 インダゾリル、 インドリル、 イソィ ンドリル、プリニル、ジヒドロインドリノレ、 ピロリル、 ピラゾリル、 トリァゾリル、 テトラゾリル、 イミダゾリル等が挙げられる。
(xi)置換低級アルキルにおける置換基としては、 同一または異なって、例えば置換 数 1〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アル カノィル、 低級アルコキシ、 シクロアルキル、 ァリールォキシ、置換ァリールォキシ
[該置換ァリールォキシにおける置換基としては、同一または異なって、例えば置換 数 1〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アル キル、低級アルコキシ、 低級アルコキシカルボニル等が挙げられる。 ここでハロゲン は前記ハロゲン ) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義 であり、低級アルコキシ及び低級アルコキシカルボ-ルの低級アルキル部分は前記低 級アルキル (ii) と同義である]、 ァラルキルォキシ、 置換ァラルキル.ォキシ [該置 換ァラルキルォキシにおける置換基としては、 同一または異なって、 例えば置換数 1 〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級ァレキ ル、 低級アルコキシ、低級アルコキシ力ルボニル等が挙げられる。 ここでハロゲンは
前記ハロゲン (i) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義で あり、低級アルコキシ及ぴ低級アルコキシカルボエルの低級アルキル部分は前記低級 アルキル (ii) と同義である]、 複素環基、 置換複素環基 [該置換複素環基における 置換基としては、 同一または異なって、 例えば置換数 1〜3の、 ハロゲン、 ァミノ、 二 トロ、 ヒドロキシ、 シァノ、 力ルポキシ、 低級アルキル、 低級アルコキシ、 低級アル コキシカルボニル等が挙げられる。 ここでハロゲンは前記ハロゲン (i) と同義であ り、 低級アルキルは前記低級アルキル (i i) と同義であり、 低級アルコキシ及び低級 アルコキシカルボ-ルの低級アルキル部分は前記低級アルキル(ii)と同義である]、 低級アルカノィルォキシ、 低級アルコキシカルボニル、低級アルキルチオ、 モノまた はジ低級アルキルァミノ、低級アルキルスルホニル、低級アルキルスルフィエル、低 級アルコキシカルボエルァミノ、低級アル力ノィルァミノ、モノまたはジ低級アルキ ルァミノカルボ-ル、モノまたはジ低級アルキルァミノカルボニルォキシ等が挙げら れる。
ここで示したハロゲン、 ァリールォキシ及ぴァラルキルォキシのァリール部分、 シク口アルキル、 複素環基ならびに低級アル力ノィル、 低級アルコキシ、 低級アル カノィルォキシ、 低級アルコキシカルボ-ル、 低級アルキルチオ、 モノまたはジ低 級アルキルァミノ、 低級アルキルスルホニル、 低級アルキルスルフィエル、 低級ァ ルコキシカルボニルァミノ、 低級アル力ノィルァミノ、 モノまたはジ低級アルキル ァミノカルボニル及びモノまたはジ低級アルキルァミノカルボニルォキシの低級 アルキル部分は、それぞれ前記ハロゲン(i)、ァリール(vi)、シク口アルキル(iii)、 複素環基 (ix) ならびに低級アルキル (H) と同義であり、 ァラルキルォキシのァ ルキレン部分は、前記低級アルキル(ii)から水素を一つ除いたものと同義である。 ジ低級アルキルァミノ、ジ低級アルキルァミノカルボニル及びジ低級アルキルァ ミノカルボ-ルォキシの 2つの低級アルキル部分は、それぞれ同一でも異なってい てもよい。
(xii) 置換ァリール、 置換ァラルキル、 置換シクロアルキル、 置換低級アルケニ ル、 置換低級アルキニル、 置換複素環基、 置換複素環アルキル、 置換芳香族複素環 基及び隣接する窒素原子と一緒になつて形成される置換複素環基における置換基 としては、 前記置換低級アルキルにおける置換基 (xi) の定義で挙げた基に加え、
低級アルキル、 ハロゲン置換低級アルキル [該ハロゲン置換低級アルキルは、 1か ら置换可能な数のハロゲン、 好ましくは 1〜6個のハロゲン、 より好ましくは 1〜3 個のハロゲンで置換された低級アルキルである〕、 ァリール、 置換ァリール [該置 換ァリールにおける置換基としては、同一または異なって、例えば置換数 1〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アルキル、 低 級アルコキシ、 低級アルコキシカルボニル等が挙げられる。 ここでハロゲンは前記 ハロゲン (i) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義であ り、低級アルコキシ及び低級アルコキシ力ルポニルの低級アルキル部分は前記低級 アルキル (ii) と同義である]、 ァラルキル、 置換ァラルキル [該置換ァラルキル における置換基としては、同一または異なって、例えば置換数 1〜3の、ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 力ルポキシ、 低級アルキル、 低級アルコキ シ、低級アルコキシカルポニル等が挙げられる。ここでハ,口ゲンは前記ハロゲン(i) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義であり、 低級アルコ キシ及ぴ低級アルコキシカルボニルの低級アルキル部分は前記低級アルキル (ii) と同義である]、 ァリ一ルカルポニル、 置換ァリ一ルカルポニル [該置換ァリール カルボニルにおける置換基としては、同一または異なって、例えば置換数 1〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アルキル、 低 級アルコキシ、 低級アルコキシカルボ二'ル等が挙げられる。 ここでハロゲンは前記 ハロゲン(i)と同義であり、 低級アルキルは前記低級アルキル(ii)と同義であり、 低級アルコキシ及び低級アルコキシカルボニルの低級アルキル部分は前記低級ァ ルキル (ii)と同義である]、 複素環アルキル、 置換複素環アルキル [該置換複素環 アルキルにおける置換基としては、 同一または異なって、 例えば置換数 1〜3の、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アルキル、 低 級アルコキシ、低級アルコキシカルボニル等が挙げられる。 ここでハロゲンは前記 ハロゲン (i) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義であ り、低級アルコキシ及ぴ低級アルコキシカルボニルの低級アルキル部分は前記低級 アルキル (ii) と同義である]、 N -低級アルキル - N -低級アルカノィルァミノ等が挙 げられる。 さらに、 置換ァリール及び隣接する窒素原子と一緒になつて形成される 置換複素環基における置換基は置換低級アルキル [該低級アルキルは前記低級アル
キル (ii) と同義であり、 該置換低級アルキルにおける置換基としては、 同一また は異なって、 例えば置換数 1〜3の、 ヒ ドロキシ、 カルボキシ、 低級アルコキシ力 ルポエル等が挙げられる。 ここで低級アルコキシカルボニルの低級アルキル部分は 前記低級アルキル (ii) と同義である] であってもよい。
ここで示したハロゲン置換低級アルキルのハロゲン部分、 低級アルキル及び N- 低級アルキル- N -低級アル力ノィルァミノの2つの低級アルキル部分、 ァリール、 ァラルキル及びァリールカルボ二ルのァリール部分、 ァラルキル、複素環アルキル の複素環基部分ならびにァラルキル及び複素環アルキルのアルキレン部分は、それ ぞれ前記ハロゲン (i)、 低級アルキル (ii)、 ァリール (vi)、 複素環基 (ix)、 了 ラルキルのアルキレン部分 (vii) 及びァラルキルのァリール部分 (viii) と同義 である。 またハロゲン置換低級アルキルのアルキレン部分は、 前記低級アルキル (ii) から水素を一つ除いたものと同義である。
また置換ァリール及び置換ァラルキルにおける置換ァリール部分は、以下に示す アルキレンジォキシフエ二ノレであってもよい。
(式中、 n。は 1〜3の整数を表す)
また置換シクロアルキルは、 例えば以下に示す縮合環基のような、 シクロアルキ ルがァリールまたは置換ァリールと縮合して形成される縮合環基であってもよい。
[式中、 ndは 1〜5の整数を表し、 R17、 R18及び R19は、同一または異なつて水素原子、 ハロゲン、 ァミノ、 ニトロ、 ヒドロキシ、 シァノ、 カルボキシ、 低級アルキル、 低 級アルコキシ、 低級アルコキシカルボエル等を表す。 ここでハロゲンは前記ハロゲ ン (i) と同義であり、 低級アルキルは前記低級アルキル (ii) と同義であり'、 低 級アルコキシ及ぴ低級アルコキシカルボニルの低級アルキル部分は前記低級アル キル (ii) と同義である]
化合物 (1)、 (IA) 及び(IB) の薬理学的に許容される塩としては、毒性のない、 水溶性のものが好ましく、 例えば塩酸塩、 臭化水素酸塩、 硝酸塩、 硫酸塩、 リン酸 塩等の無機酸塩、 ベンゼンスルホン酸塩、 安息香酸塩、 クェン酸塩、 フマル酸塩、 ダルコン酸塩、 乳酸塩、 マレイン酸塩、 リンゴ酸塩、 シユウ酸塩、 メタンスルホン 酸塩、酒石酸塩等の有機酸塩等の酸付加塩、 ナトリゥム塩、 力リゥム塩等のアル力 リ金属塩、 マグネシウム塩、 カルシウム塩等のアルカリ土類金属塩、 アルミニウム 塩、 亜鉛塩等の金属塩、 アンモ-ゥム、 テトラメチルアンモニゥム等のアンモニゥ ム塩、モルホリン付加塩、 ピぺリジン付加塩等の有機ァミン付加塩、 グリシン付加 塩、 フエ二ルァラニン付加塩、 リジン付加塩、 ァスパラギン酸付加塩、 グルタミン 酸付加塩等のァミノ酸付加塩等が挙げられる。
次に化合物 (IA)、 (IB) の製造法について説明する。
なお、 以下に示した製造法において、 定義した基が反応条件下変化するか、 また は方法を実施するのに不適切な場合、有機合成化学で常用される方法、例えば官能 基の保護、脱保護等 [例えば、プロテクティブ'グループス 'イン-オーガ二ック · シンセシス 第三版 (Protective Groups in Organic Synthesis, the third edition)、 グリーン (T. W. Greene)、 ワッツ (Peter G. M. Wuts) 著、 ジョン ' ワイリー 'アン ド -サンズ ·インコーポレイテッド (John Wiley & Sons Inc. )■ (1999年)] の手段 に付すことにより容易に製造を実施することができる。 また、必要に応じて置換基 導入等の反応工程の順序を変えることもできる。
化合物 (IA) は、 例えば以下に示す製造法 1によって得ることが出来る b 製造法 1 : '
(A) (C)
(D)
(式中、 ArA、 R8、 R9a、 R9b、 R9\ R9d及び nは、 それぞれ前記と同義であり、 racは 1 または 2を表す)
[工程 1 ]
2 -エトキシメチレン -2-シァノ酢酸ェチル (A) を溶媒中、 アルカリ存在下、 0. 5 当量から大過剩量、 好ましくは 0. 5〜2当量の硫酸メチルイソチォ尿素 (B) と反応 させることにより、 化合物 (C) を得ることができる。
溶媒としては、 例えばメタノール、 エタノール、 2 -プロパノール、 テトラヒドロ フラン、 1, 4 -ジォキサン等を単独でまたはそれらを混合して用いることができ、 中 でもエタノールが好ましい。
アル力リとしては、例えば水酸化リチウム、水酸化力リウム、水酸化ナトリウム、 水酸化マグネシゥムまたは水酸化カルシゥム等のアル力リ水溶液、カリウム tert - ブトキシドの水溶液、 テトラヒ ドロフラン溶液もしくは 2 -メチル- 2 -プロパノール 溶液;ナトリウムメトキシドの水溶液もしくはメタノール溶液等を用いることがで き、 中でも水酸ィ匕ナトリウム水溶液が好ましい。
反応は、 0〜50°Cの間の温度、 好ましくは 0〜15°Cの間の温度で行われ、 通常 1〜 48時間で終了する。
[工程 2 ]
工程 1で得られる化合物(C) を、反応に不活性な溶媒中または無溶媒で、 1当量 から大過剰量の塩素化剤と反応させることにより化合物 Φ)を得ることができる。 塩素化剤としては、 例えばォキシ塩化リン等が用いられる。
反応に不活性な溶媒は、 反応に不活性なものであればいずれでもよく、 特に限定 されないが、例えば 1, 2-ジクロロェタン、 テトラヒドロフラン、 ジォキサン、 クロ 口ホ^/ム、 ベンゼン、 トルエン、 キシレン、 酢酸ェチ /レ、 トリェチルァミン、 ピリ ジン、 N, N -ジメチルァニリン等を単独でまたはそれらを混合して用いることができ' る。
反応は、 0°Cから溶媒の沸点の間の温度、 好ましくは 50°Cから溶媒の沸点の間の 温度で行われ、 通常 1〜48時間で終了する。
なお、 本工程で得られる化合物 (D) は上記の方法以外に、 ジャーナル'オブ' へテ口サイクリック ·ケミストリー(J. Heterocycl. ' Chem. )、 8 (3)卷、 445頁(1971 年)、 W099/61444等に記載の方法またはそれらに準じた方法によっても得ることが できる。
[工程 3 ] .
工程 2で得られる化合物 (D) を反応に不活性な溶媒中、 1当量から大過剩量、好 ましくは 1〜10当量の塩基の存在下に、 1当量から大過剩量、好ましくは 1〜3当量 の化合物 (E) と反応させることにより、 化合物 (F) を得ることができる。
反応に不活性な溶媒は、反応に不活性なものであればいずれでもよく、 特に限定 されないが、 例えばテトラヒドロフラン、 ジォキサン、 ジェチルエーテル、 ジイソ プロピルエーテル、ベンゼン、 トルエン、 キシレン、酢酸ェチル、 ァセトニトリル、
ジクロロ'メタン、 クロ口ホルム、 1, 2 -ジクロロェタン、 ジメチルホルムアミ ド、 ジ メチルァセトアミ ド、 N -メチルピロリ ドン、 ジメチルスルホキシド等を単独でまた はそれらを'混合して用いることができ、 中でもテトラヒドロフラン、 クロ口ホルム またはそれらの混合溶媒が好ましい。 - 反応は、 0~100°Cの間の温度、 好ましぐは 0〜50°Cの間の温度で行われ、 通常 10 分間から 48時間で終了する。
塩基としては、 例えばトリェチルァミン、 ジイソプロピルェチルァミン、 1, 8 -ジ ァザビシク口 [5. 4. 0]ゥンデセ -7-ェン (DBU)、 N, N-ジメチルァ-リン、 ピリジン、 キノリン等の有機塩基、 炭酸カリウム、 炭酸ナトリウム、 炭酸リチウム、 炭酸水素 ナトリゥム、水酸化カリゥム、水酸化ナトリゥム、水酸ィヒリチウム、カリゥム tert - ブトキシド等の無機塩基、ァンバーリスト A_21 (ロームアンドハース社製)、 AG 1-X8
(バイオラッド社製) 等の塩基性ァニオン交換レジン、 ポリビニルピリジン、 モル ホリノメチルポリスチレン等の固相に担持された塩基等が用いられ、中でもモルホ リノメチルポリスチレン、 DBUが好ましい。
化合物 (E) は、 市販品としてまたはコンプリヘンシブ 'オーガニック. ' トラン スフォーメーションズ第二 (Comprehensive Organic Transformations, second edition) , ラ口ック (R. C. Larock) 著、 ジョン - ワイリー 'アンド ·サンズ■ィ ンコーポレイテツド (John Wiley & Sons Inc. ) (1999年) 等に記載の方法に準じ て得ることができる。 - [工程 4 ]'
工程 3で得られる化合物 (F)を、反応に不活性な溶媒中、 1当量から大過剰量、好 ましくは 1~5当量の酸化剤で処理することにより、 化合物 (G) を得ることができ る。 '
反応に不活性な溶媒は、 反応に不活性なものであればいずれでもよく、 特に限定 されないが、 例えばジクロロメタン、 クロロホ ム、 1, 2-ジクロロェタン、 テトラ ヒドロフラン、 ジォキサン、 ジェチノレエーテ /レ、 ジイソプロピノレエーテノレ、 メタノ 一ノレ、 エタノーノレ、 イソプロピノレアノレコーノレ、 ベンゼン、 トノレェン、 キシレン、 酢 酸ェチル、 ァセトェトリノレ、 水等を単独でまたはそれらを混合して用いることがで き、 中でもジクロロメタンが好ましい。
酸化剤としては、 例えばメタクロ口過安息香酸、 過酸化べンゾィル、 過酢酸、 過 矣化水素水、過ヨウ素酸ナトリウム等を用いることができ、 中でもメタクロ口過安 息香酸が好ましい。
反応は 0〜100°Cの間の温度、 好ましくは 0〜50°Cの間の温度で行われ、 通常 10 分間から 24時間で終了する。
なお、 化合物 (G) において、 mcが 1である化合物と mcが 2である化合物は、 例 えば酸化剤の当量数、反応の温度等の条件等を調節することにより一方を選択的に 得ることも可能であり、 それらが混在することもある。 混在する場合、 その比率は 特に限定されず、 いずれの場合もそのまま次工程に使用することができる。
[工程 5 ]
工程 4で得られる化合物(G) を、反応に不活性な溶媒中、 1〜5当量の化合物(H) と反応させることにより、 化合物 ) を得ることができる。
反応に不活性な溶媒は、反応に不活性なものであればいずれでもよく、 特に限定 されないが、 例えばジクロロメタン、 クロロホノレム、 1, 2 -ジクロロェタン、 ジメチ ルホルムアミ ド、 ジメチルァセトアミ ド、 N -メチノレピロリ ドン、 ジメチルスルホキ シド、 テ,トラヒ ドロフラン、 ジォキサン、 ジェチノレエーテ Λ^、 ジィソプロピ /レエ一 テル、 ベンゼン、 トルエン、 キシレン、 酢酸ェチル、 ァセトニトリル等を単独でま たはそれらを混合して用いることができ、 中でもテトラヒドロフランが好ましい。 反応は 0〜100°Cの間の温度、 好ましくは 20〜60°Cの間の温度で行われ、 通常 1 〜72時間で終了する。
化合物 (H) は、 市販品としてまたはコンプリヘンシブ'オーガニック ' トラン スフォーメーションズ第二 |¾¾ (Comprehensive Organic Transformations, second edition) , ラロック (R. C. Larock) 著、 ジョン ' ワイリー 'アンド ·サンズ■ィ ンコーポレイテツド (John Wiley & Sons Inc. ) (1999年) 等に記載の方法に準じ て得ることができる。
[工程 6 ]
工程 5で得られる化合物 ひ) を、反応に不活性な溶媒中、 1〜10当量のアジ化ナ トリウムまたはアジ化アンモ-ゥムと反応させることにより、 化合物 (ΙΑ) を得る ことができる。
反応に不活性な溶媒は、 反応に不活性なものであればいずれでもよく、 特に限定 されないが、 例えばク口口ホ^/ム、 1, 2 -ジク口口エタン、 ジメチ レムァミ ド、 ジメチノレアセトアミ ド、 Ν -メチノレピロリ ドン、 ジメチノレスノレホキシド、 テトラヒ ド 口フラン、ジォキサン、 ジイソプロピ /レエーテノレ、ベンゼン、 トノレェン、 キシレン、 酢酸ェチル、ァセトニトリル等を単独でまたはそれらを混合して用いることができ、 中でもジメチルホルムアミドが好ましい。 また、 反応を加速する目的で、 反応系中 へ塩化アンモニゥム、塩化ェチルアンモニゥム等を 1当量以上添加することもでき る。 ' 反応は 0〜180°Cの間の温度、 好ましくは 50〜120°Cの間の温度で行われ、 通常 1
〜72時間で終了する。
化合物 (IB) は、 例えば以下に示す製造法 2によって得ることができる。
製造法 2 :
(式中、 、 R"a、 R14b、 、 Rl4d、 Rl5、 6、 na及び mcは、 それぞれ前記と同義であ る) '
[工程 7 ]
製造法 1の工程 3に記した方法に準じ、製造法 1の工程 2で得られる化合物 (D) を化合物 (κ) と反応させることにより、 化合物 ) を得ることができる。
工程 7において溶媒、 塩基、 温度、 当量、 反応時間等は、 製造法 1の工程 3に記
載したものに準じて化合物 (D を製造することができる。 .
[工程 8 ] . 製造法 1の工程 4に記した方法に準じ、 工程 7で得られる化合物 (L) から化合 物 (M) を得ることができる。
工程 8において溶媒、 塩基、 温度、 当量、 反応時間等は、 製造法 1の工程 4に記 载したものに準じて化合物 (M) を製造することができる。
[工程 9 ]
製造法 1の工程 5に記した方法に準じ、 工程 8で得られる化合物 (M) を化合物 (N) 反応させることにより、 化合物 (IB) を得ることが出来る。
工程 9において溶媒、 塩基、 温度、 当量、 反応時間等は、 製造法 1の工程 5に記 載したものに準じて化合物 (IB) を製造することができる。
化合物(IA)、 (IB) 及び原料化合物における各官能基の変換ならびに置換基に含 まれる官能基の変換は、.公知の方法 [例えば、 コンプリへンシブ 'オーガニック - トランスフォーメーションズ 第二 (Comprehensive Organic Transformations, the second edition)、 R. C.ラロック (Larock) 著、 ジョン ' 'ワイリー ' アンド - サンズ 'インコーポレーティッド (John Wiley & Sons Inc. ) (1999年) に記載の 方法] 等によって行うことができる。
上記の方法等を適宜組み合わせて実施することにより、所望の位置に所望の官能 基を有する化合物 (IA)、 (IB) を得ることができる。
上記製造法における中間体及ぴ生成物の単離 ·精製は、通常の有機合成で用いら れる方法、 例えば濾過、 抽出、 洗浄、 乾燥、 濃縮、 結晶化、 各種クロマトグラフィ 一等を適宜組み合わせて行うことができる。 さらに一般的な並列合成法で常用され る精製法、例えばスカベンジャーレジン、 イオン交換レジンを用いた精製法によつ て行うことができる。 また、 中間体においては、 特に精製することなく次の反応に 供することもできる。
化合物 (IA)、 (IB) には、 位置異性体、 幾何異性体、 光学異性体または互変異性 体のような異性体が存在し得るものもあるが、これらを含め可能な全ての異性体及 ぴ該異性体のいかなる比率における混合物も本発明に包含される。
化合物 (IA)ヽ (IB) の塩を取得したい場合には、 化合物 (IA)、 (IB) の塩が得ら
れるときはそのまま精製すればよく、 また化合物 (IA)、 (IB) が遊離の形で得られ るときは化合物 (IA)、 (IB) を適当な溶媒に溶解または懸濁し、 酸または塩基を加 えて単離 '精製すればよい。
また、 化合物 (IA)、 (IB) またはその薬理学的に許容される塩は、 水または各種 溶媒との付加物の形で存在することもある力 これらの付加物も本発明に包含され る。
化合物 (I) の中で、 化合物 (IA)、 (IB) 以外の化合物 (I) は、 公知の方法 [例 えば、 シンレツト (Synlett)、 625頁 (2000年) ;ジャーナノレ ·ォブ ·ケミカノレ - ソサェティ一( J. Chem. Soc. )、 800頁 (1952年);テトラへドロン(Tetrahedron)、 43卷、 2557頁 (1987年);ジャーナル■ォブ■ヘテロサイクリック 'ケミストリー (J. Heterocyclic Chem. )ヽ 24卷、 85頁 (1987年) ;ジャーナノレ■ォブ ·オーガ二 ック 'ケミストリー (J. Org. Chem. )、 60卷、 1875頁 (1995年)] 等によって合成 することができる。
以下、 第 1表〜第 2表に本発明によって得られる化合物 (IA)、 (IB) の具体例を 示すが、 本発明の範囲はこれらの化合物に限定されることはない。
TSM0/C00Zdf/X3d 9£6£請 00Z OAV
zz
.(Η+ΙΛ1) 9 SIAl
.(Η+ΙΛΙ) fr ζ/山 SIAl
Η+ΙΛΙ) ZO Ζ/山 SIAl
.(Η+ΙΛΙ) 89ε ζ/山 SIAl
辇 3— L蕖
TSM0/C00Zdf/X3d 9£6£請 OOZ OAV
第 2— 1表
次に、代表的な化合物(I)の薬理作用について試験例により具体的に説明する。 「試験例 1」 PLK酵素に対する阻害活性
以下に示す方法にて PLK酵素に対する阻害活性を測定した。 GST (グルタチオン S -トランスフェラーゼ) を N末端に融合した全長のマウス PLK [セル■グロウス ' アンド 'ディファレンシエーション (Cell Growth & D f. )、 5卷、 249頁 (1994 年)] を発現するバキュロウィルスを、 Spodoptera frugiperda (スポドプテラ フ ルギペルダ) (Sf) 9 昆虫細胞に感染させた。 その後、 Sf9 細胞を ESF921 培地 (EXPRESSION SYSTEMS, カタ口グ番号 96 - 001) を用いて、 28°Cで 48時間振とう培 養した。 培養後、 細胞を遠心して沈殿物を回収し、 一 20°Cで凍結した。 凍結した細 胞沈殿物を融解し、 200 mLの Lysis Buffer 〔20 mmol/L Hepes (pH7. 5)、 20 mmol/L
]3 グ リ セ 口 ホ ス フ ェ ー ト 、 1% CHAPS { 3- [ (3-Cholaraidopropyl) dimethylammonio]propanesulfonic acid} ヽ 1 mmolん DTT (Dithiothreitol)ヽ 10 μ mol/L MG132 (カルビオケム社、カタ口グ番号 474790)、 I X Complete Mix EDTA free (ロシュ .ダイァグノスティックス社、 カタ口グ番号 18"73580)〕 に懸濁した。 水上 で 10分'間静置後、 4°C、 10, 000 gで 15分間遠心し、 上清を回収した。 上清に Lysis Bufferで平衡化した 50%グルタチオンビーズ (アマシャムフアルマシア社、 カタ口 グ番号 27- 4574-01) 8 mLを加えて、 ローテ一ター RT - 1 (タイテック社)で回転させ ながら 4°Cで 5時間反応させた。 反応液を 4°C、 3, 000 g、 1分間遠心後、 上清を取 り除き沈殿したビーズを回収した。 Lysis Buffer 8 mLで 2回洗浄後、 Wash ffer [50 mmol/L Tris/HCl (pH7. 5) ] 16 mLで 2回洗浄した。 洗浄したビーズに 9 raL の Elute Buffer [50 mmol/L Tris/HCl (pH7. 5)、 10 mmol/L グルタチオン、 10 mol/L MG132, 100 nmol/L オカダ酸] を加え、 ローテ一ターで回転させながら 4°C で.5時間反応させた。 反応液を 4°C、 3, 000 g、 1分間遠心後、 上清8 mLを回収し た。 回収した溶出液を最終的に以下の保存状態になるように保存した [最終保存条- 件; GST-PLK 38 μ g/mL、 25 mmol/L Tris/HCl (pH7. 5)、 3. 3 mmol/L グルタチオン、 0. 5 mmol/L EDTA, 5 ramol/L NaCl、 0. 5 mmol/L DTT、 5 μ mol/L MG132、 50 nmol/L オカダ酸、 50% グリセロールで一 20°Cで保存]。
PLK酵素阻害 50%の濃度を求めるために、以下の系を構築した。 試験用薬剤サン プルは DMS0 (ジメチルスルホキシド) に溶解した 1 mmol/Lサンプルを順次希釈し て調製した。 コントロールとプランクには薬剤を溶解していない DMS0を用いた。 40 の容量で最終濃度が GST- PLK 5 ^ g/ ^ L, 脱リン酸化型 α -カゼイン (シグ マアルドリツチ社、カタ口グ番号 C- 8032) 250 μ / μ 20 ramol/L Hepes (pH7. 5)、 1 ramol/L DTT、 DMS0 1%、 試験用薬剤濃度が 0. 12、 0. 37、 1. 11、 3. 33、 10 μ mol/L になるようにそれぞれ溶液を調整した。ブランクには GST- PLKを加えないサンプル を用いた。その後 5 X ATP溶液 { [ γ - 33P] -ATP 1. 6 /i Ci (アマシャムフアルマシア社、 カタ口グ番号 A H9968、 1000-3000Ci/ mmol)、 25 μ mol/L cold ATP, 100 mmol/L MgCl2、 20 mmol/L Hepes (pH7. 5)、 1 mmol/L DTT} を 10 Lカ卩え、 30°Cで 50分間反応さ せた。 反応液 50 のうち 20 x Lを P81フィルター (ワットマン社、 カタログ番 号 3698023) に添加し、 0. 75%リン酸液に入れた。 フィルターは 0. 75%リン酸液で
4回洗浄し、 65°Cで 30分間乾燥させた。 乾燥させたフィルターの 33P放射能の力ゥ ントは TRI- CARB 2700TR液体シンチレーションアナライザ一 (パッカード社) によ つて測定した。 フィルターに添加してから測定までの操作は 2連で行った。 このァ ッセィによって測定した化合物濃度 10 ^u mol/Lにおける PLK阻害率 (%) 'を第 3表 に、また一部の化合物について PLK酵素阻害 50%の濃度(1C5。) を第 4表に示した。 第 3表
' 化合物番号 阻害率 (10 μ mol/L, )
1 86
2 86
3. 83 '
4 72
5 72
6 77
7 88
8 88
9 74
10 76
11 78
12 88
13 74
14 72
15 71
16 76
17 76
18 82
19 76
20 88
21 82
22 74
23 79
24 54
25 87
26 77
27 77
28 85
第 4表
化合物番号 ICgo ^ mol/L)
1 1. 04
2 0. 33
3 1. 01
7 0. 96
8 0. 49
16 0. 58
20 0. 55
22 0. 75
23 1. 60
28 1. 60
「試験例 2」 PKAキナーゼアツセィ
酵素阻害の選択性を調べるため、 PLKと同じセリンスレオニンキナーゼである PKA に対する酵素阻害率を求めるために、 以下の系を構築した。 PKA 酵素は cAMP-dependent Protein Kinase (PKA)、 catalytic subunit (ニューイングランド バイオラブ社、カタログ番号 6000S) を用いた。試験用薬剤サンプルは DMSOに溶解 した 1 議 ol/Lサンプルを順次希釈して調整した。 コントロールとブランクには薬 剤を溶解していない DMS0を用いた。 40 /i Lの容量で最終濃度が PKA 5unit/ mL、 MBP, bovine, purified (アップステート社、 カタログ番号 13- 104) 400 g/ ^ L 50 mmol/L Tris (pH7. 5) s 1 mraol/L DTT、 DMSO 1%、 試験用薬剤濃度が δθまたは 10 mol/Lになるようにそれぞれ溶液を調整した。 ブランクには PKAを加えないサン プルを用いた。 その後 5 XATP溶液 { [ γ - 33P]- ATP 1. 6 Ci (1000 - 3000Ci/ mmol)、 25 μ raol/L cold ATP, 100 mmol/L MgCl2、 50 mmol/L Tris (pH7. 5)、 1 mmol/L DTT} を 10 x L加え、 30°Cで 30分間反応させた 9 10 μ ΚΌ 10% リン酸バッファーを添カロ して、反応を止めた。反応液 60 ;/ Lのうち 20 /i Lを P81フィルターに添加し、 0. 75% リン酸液に入れた。 フィルタ^"は 0. 75%リン酸液で 4回洗浄し、 65°Cで 30分間乾 燥させた。 乾燥させたフィルターの 33P放射能のカウントは TRI- CARB 2700TR液体
シンチレーシヨンアナライザー (パッカード社) によって測定した。 フィルターに 添加してから測定までの操作は 2連で行った。 このアツセィによつて測定した化合 物濃度 50 mol/L及び 10 mol/Lでの PKA酵素阻害率 を第 5表に示した。 いずれの化合物も 50 μ mol/L という高濃度でも阻害活性を示さず、 PKA と PLK の阻害作用に選択性があることが示された。
「試験例 3」 Erk2キナーゼアツ.セィ
酵素阻害の選択性を調べるため、 PLK と同じセリンスレオニンキナーゼである Extracel lular Signal-Regulated Kinase 2 (Erk2) 酵素に対する阻害率を求める ために、 以下の系を構築した。 Erk2酵素は MAP Kinase 2/ Erk2, active (アップ ステート社、 カタログ番号 14- 173) を用いた。 試験用薬剤サンプルは DMS0に溶解' した 1 墮 ol/Lサンプルを順次希釈して調整した。 コントロールとブランクには薬 剤を溶解していない DMS0を用いた。 40 Lの容量で最終濃度が Erk2 400 ng/ mL、 MBP, bovine, purified 400 μも I μ 、 50 mmol/L Tris (pH7, 5)ヽ 1 mmol/L DTTヽ DMS0 1%、 試験用薬剤濃度が 50または 10 μ mol/L になるようにそれぞれ溶液を調 整した。ブランクには Erk2を加えないサンプルを用いた。その後 5 X ATP溶液 { [ γ — 33Ρ] - ATP 1. 6 /i Ci (lOO0-3000Ci/ mmol)、 25 μ mol/L cold ATP、 100 ramol/L MgCl2、 50 mmol/L Tris (pH7. 5)、 1 mmol/L DTT} を 10 ^u Lカ卩え、 30°Cで 30分間反応させ た。 10 z Lの 10%リン酸バッファーを添加して、 反応を止めた。 反応液 60 の うち 20 を P81フィルターに添加し、 0. 75%リン酸液に入れた。 フィルタ一は 0. 75%リン酸液で 4回洗浄し、 65°Cで 30分間乾燥させた。 乾燥させたフィルター の 33P放射能の力ゥントは TRI- CARB 2700TR液体シンチレーシヨンアナライザ一(パ ッカード社) によって測定した。 フィルターに添加してから測定までの操作は 2連 で行った。 このアツセィによって測定した化合物濃度 50 z raol/L及び 10 μ mol/L での Erk2酵素阻害率 (%) を第 5表に示した。
いずれの化合物も 50 μ mol/Lという高濃度でも 50%を超える阻害活性を示さず、 Erk2と PLKの阻害作用に選択性があることが示された。
以上の結果より、本発明で見出された PLK P且害剤は、他のキナーゼとの選択性が 高く、 PLK阻害剤として優れた特徴を有する化合物と結論された。
第 5表
化合物濃度 50 μ mol/L及び 10 μ raol/Lでの各酵素の阻害率 (%)
PKA Erk2 '
50 μ mol/L 10 mol/L 50 mol/L 10 mol/L 化合物 16 -2. 1 ' 3. 4 17. 9 8. 1
化合物 20 4. 7 7. 5 29. 7 19. 9
「試験例 4」 抗細胞試験
ヒ ト子宮頸癌 HeLa細胞 (ATCC番号: CCL-2) に対する増殖阻害を以下の方法で実 施した。 HeLa細胞 の培養には 10%FBS入り RPMI [RPMI1640 (GIBC0 BRL カタログ 番号 11875- 093) ]培地を用いた。 1 X 103個/ ゥエルの HeLa細胞を TC MICROWELL 96F プレート (ナルジェン 'ヌンク社、 カタログ番号 167008) に 100 /z Lずつ播種し、 37°Cで 24時間、 5%炭酸ガスインキュベーター内で培養した。 ブ ンクには培地の みを 100 Ai L添加したサンプルを使用した。 その後、 段階的に希釈した試験化合物 を 50 カロえ、 さらに 37°Cで 72時間、 5%炭酸ガスインキュベーター内で培養し た。 コント口ールとブランクには 0. 3%の DMS0入り RPMI培地を 50 L添加した。 RPMI培地を取り除き、 100 i Lの PBS (Phosphate buffer saline) を加えた後、 取 り 除 い た 。 そ の 後 RPMI 培 地 で 10% に 希 釈 し た WST {4 - [3 - (4-Iodophenyl) -2- (4-nitrophenyl) - 2H - 5 -" etrazolio] - 1, 3- benzene disulfonate} 試薬 (ロシュ 'ダイァグノスティックス社、 カタログ番号 1644807) を 100 /x L加え、 37°Cで 40分間インキュベートした。 マイクロプレート分光光度 計 SPECTRA max 340PC (モレキユラーデバイス社) を用い、 450 nm (対照波長 690 nra) の吸光度を測定した。代表的な化合物の結果を HeLa細胞増殖阻害 50%の濃度として 第 6表に示した。
第 6表
HeLa細胞増殖阻害 50%の濃度 ( μ mol/L) 化合物 1 6 6. 70
化合物 2 0 8. 71
「試験例 5」 M期集積能試験
PLKは細胞周期の M期への進入から脱出までの過程で機能することが知られてお り、 PLKの活性阻害により、細胞周期、特に M期の進行に影響が出ると予想される。 PLK阻害剤による細胞周期への影響を、 M期の細胞数を調べる方法 (M期集積能試験) により測定した。 HeLa細胞 の培養には 10%FBS入り RPMI培地を用いた。 MULTIDISH 6プレート (ナルジェン'ヌンク社、 カタログ番号 152795) にカバーグラスを入れ ておき、 HeLa細胞を 1 X 105cells/raLを 2 mL播種し、 37°Cで 24時間、 5%炭酸ガス インキュベーター内で培養した。 RPMI培地を取り除き、 さらに 2 mLの PBSを加え た後、取り除いた。その後 RPMI培地で希釈した試験化合物を 2 mL加え、さらに 37°C で 15時間、 5%炭酸ガスィンキュベータ一内で培養した。 培地を静かに,取り除き、 3. 7°/。ホルムアルデヒドを含む PBSを 2mLずつ添加し、 10分間固定した。 100 μ mol/L Hoechest 33342 (シグマアルドリッチ社、 カタログ番号 B-2261) を含む PBSを 2 mL ずつ添加し、静かに摇らした後、 10分間室温に放置した。カバーグラスを取り出し、 倒立顕微鏡 ECLIPSE TE300 (ニコン社) で観察した。 染色体が核一様に染まる細胞 を間期の細胞とし、染色体が凝集したパターンを示す細胞を M期の細胞として、各 細胞数を生細胞のみカウントした。 なお核の断片化、細胞膜の湾曲等のアポトーシ スの兆候が認められない細胞を生細胞と定義した。各条件下最低 100個以上の細胞 をカウントし、 各形態の細胞の割合 (%) を算出した。 カウントは 2連で行つた。 M 期集積作用が知られている微小管作用性物質ノコダゾール (Nocodazole) をコント ロールに用いた。 代表的な化合物の結果を第 7表に示す。 PLK阻害活性を有する化 合物 1 6及ぴ 2 0では、化合物未処理の細胞に比べ明らかに M期の細胞数が増大し ており、 これら化合物が HeLa細胞に対し M期集積作用を有することが明らかにな つた。この結果は、本発明で見出された PLK阻害剤が M期集積という作用を介して、 癌細胞の増殖を抑制する効果、 すなわち抗腫瘍作用を有することを示している。
第 7表
HeLa細胞に対する M期集積作用
DMS0 4. 7% 95. 3%
ノコダゾール (Ι μ ιηοΐ/L) 48. 9% , 51. 1%
化合物 1 6 (10 /i raol/L) 28. 1% 71. 9%
化合物 2ひ (10 /^nol/L) 43. 6% 56. 4% 本発明に係る医薬は、 式 (I) で表される化合物及びその薬理学的に許容される 塩、ならびにそれらの水和物及び溶媒和物からなる群から選ばれる物質を有効成分 として含むことを特徴とする。本発明に係る医薬としては、有効成分である上記物 質をそのまま投与してもよいが、一般的には、有効成分である上記の物質と 1また は 2以上の製剤用添加物とを含む医薬糸且成物の形態で投与することが望ましい。 こ のような医薬組成物は、それ自体製剤学の分野で周知または慣用の方法に従って製 造することが可能である。 また、 医薬組成物の形態である本発明に係る医薬には、 他の医薬の有効成分が 1または 2以上含まれていてもよい。なお、本発明の E薬は、 ヒトを含む哺乳類動物に適用可能である。
本発明の医薬の投与経路は特に限定されず、経口投与または静脈内投与等の非経 口投与のいずれかから治療及び/または予防のために最も効果的な投与経路を適 宜選択することができる。 経口投与に適する製剤の例としては、 例えば、 錠剤等を 挙げることができ、 非経口投与に適する製剤の例としては、 例えば、 注射剤等を挙 げることができる。
錠剤等の固形製剤の製造には、例えば、,乳糖、 マンニット等の賦形剤;デンプン 等の崩壊剤;ステアリン酸マグネシゥム等の滑沢剤;ヒドロキシプロピルセルロー ス等の結合剤;脂肪酸エステル等の界面活性剤;グリセリン等の可塑剤を用いるこ とができる。
非経口投与に適する製剤のうち注射剤等の血管内投与用製剤は、好ましくはヒ ト 血液と等張の水性媒体を用いて調製することができる。例えば、注射剤は、塩溶液、 ブドウ糖溶液、塩水とブドウ糖溶液の混合物等から選ばれる水性媒体を用い、 常法
に従って適当な助剤とともに溶液、懸濁液または分散液として調製することができ る。非経口用の製剤の製造には、例えば、希釈剤、香料、防腐剤、賦形剤、崩壊剤、 滑沢剤、 結合剤、 界面活性剤、 可塑剤等から選択される 1または2以上の製剤用添 加物を用いることもできる。
本発明の医薬の投与量及び投与頻度は特に限定されず、有効成分である上記物質 の種類、 投与経路、 治療及び/または予防の目的、 患者の年齢及び体重、 症状の性 質及び重篤度等の種々の条件に応じて適宜選択することが可能である。 例えば、成 人 1日当り ひ. 1〜ίθΟ mg/kgを 3〜4回に分けて投与するのが好ましい。 し力 ^しな がら、 これら投与量及ぴ投与回数は前述の種々の条件等により変動する。
発明を実施するための最良の形態
以下に、本発明を実 ¾例及び参考例によりさらに具体的に説明するが、本発明の 範囲はこれら実施例により限定されることはない。
下記実施例及び参考例中の各化合物の物理化学的データは、以下の機器類によつ て測定した。
¾雇 R: JEOL EX- 270型 (270MHz) または JEOL GX-270型 (270MHz)
IR: HORIBA FT - 200
MS: JEOL J1S-DX303 (FAB法) または Micromass Quattro (APCI法)
参考例 1 : 4 -ク口口- 5 -シァノ -2-メチルチオピリミジンの合成
工程 1 : 5 -シァノ -3, 4-ジヒドロ- 2-メチルチオピリミジン- 4-オンの合成
硫酸メチルイソチォ尿素 (90. 5 g, 0. 325 mmol) を永酸化ナトリウム水溶液 (2 mol/L, 325 mL) に溶解し、 氷冷下、 その水溶液に 2 -エトキシメチレン- 2 -シァノ酢 酸ェチル (100 g, 0. 591 mmol) のエタノール溶液 (350 mL) を、 内温が 15°Cを超 えないように注意しながら少しずつ滴下した。 全てのェタノール溶液を加えた後、 水酸化ナトリウム水溶液 (2 mol/L, 300 mL) を少しずつ加え、 さらにエタノール
(150 mL) を加え、 室温で一晚攪拌した。 反応混合物中に析出した結晶を濾取し、 エタノール (500 raL) で洗浄した。 得られた白色結晶を減圧乾燥し、 目的とする 5_ シァノ -3, 4 -ジヒドロ- 2-メチルチオピリミジン- 4 -オン (113. 3 g, 100%) を得た。 工程 2 : 4-クロ口 -5-シァノ- 2-メチルチオピリミジンの合成
工程 1で得た 5-シァノ -3, 4 -ジヒドロ- 2-メチルチオピリミジン - 4 -オン(30. 0 g,
0: 159 mmol) にォキシ塩ィ匕リン (150 mL) を加え、 7時間加熱還流を行った。 反応 混合物を放冷した後、ォキシ塩化リンを減圧留去した。得られた残渣を氷 (約 1000 mL)にあけ、析出した淡黄色固体を濾取し、水で洗浄した。その固体を減圧乾燥し、 目的とする 4 -クロ口 -5-シァノ -2 -メチルチオピリミジン (11. 9 g, 40%) を得た。 実施例 1 :化合物 1の合成
工程 1
参考例 1で得られた 4 -クロ口- 5 -シァノ -2 -メチルチオピリミジン (0. 25 mol/L クロロホルム溶液, 0. 20 mL, 0. 050 mmol) と 6 -ァミノ- 1, 4 -べンゾジォキサン (1. 0 mol/L クロ口ホルム溶液, 0. 060 mL, 0. 060 膽 ol) の混合液に、 モルホリノメチル ポリスチレン (35 rag, 0. 12 mmol) を加え、 50°Cで 1日間攪拌した。 その後、 反応 混合物にベンゾイルクロリ ドポリマーバウンド (23 mg, 0. 058 瞧 ol) 及びクロ口 ホルム (0. 50 mL) を加え、 室温でさらに 1 3間攪拌した。 反応混合物中の樹脂を 濾別し、 樹月旨をクロ口ホルムとメタノールの混合溶媒 (クロ口ホルム/メタノール = 3/1, 1. 2 mL) で洗浄した。 得られた溶液を全て合わせ、 溶媒留去して対応する 化合物を得た。
工程 2
工程 1で得られた化合物をジクロロメタン (0. 20 mL) に溶解し、 メタクロ口過 安息香酸 (0. 50 mol/L ジクロロメタン溶液, 0. 125 mL, 0. 063 醒 ol) を加え、 室 温で 0. 5〜1時間攪拌.した。 反応混合物に飽和重曹水 (0. 20 mL) を加え、 攪拌した 後、 水層を除去した。 その後、 再度飽和重曹水 (0. 20 mL) を加えて攪 した ¾、 分液した。 有機層を硫酸マグネシウムと珪藻土の混合物 (硫酸マグネシウム/珪藻 土 = 1/1, 140 mg) で乾燥した。 硫酸マグネシウムと珪藻土の混合物をジクロロメ タン (0. 80 mL) で洗浄した。 得られた溶液を全て合わせ、 溶媒を留去して、 対応 する化合物を得た。 ■
工程 3
工程 2で得られた化合物をテトラヒドロフラン (0. 20 mL) に溶解し、 2- (2 -チェ -ル)ェチルァミン (1. 0 mol/L クロ口ホルム溶液, 0. 060 mL, 0. 060 mmol) を加 え、 室温で 1日間攪拌した。 その,後、 反応混合物にクロ口ホルム (0. 40 mL)、 ベン ゾイルクロリ ドポリマーパウンド (23 mg, 0. 058 mmol) s ポリ(4-ビニルピリジン)
(23 rag) を加え、 室温で 1 0間攪拌した。 反応混合物中の樹脂を濾別し、 樹脂を クロ口ホルムとメタノールの混合溶媒 (クロ口ホルム/メタノール =3/1, 1.4 mL) で洗浄した。 得られた溶液を全て合わせ、 溶媒を留去して対応する化合物を得た。 工程 4
工程 3で得られた化合物をジメチルホルムアミド (0. lO raL) に溶解し、 アジ化 アンモニゥムのジメチノレホノレムアミド溶 ί夜 (1.0 mol/L, 0.080 mL, 0.080 mmol) を加え、 100°Cで 1 .日間撹拌した。 その後、 溶媒を留去し、 残渣をクロ口ホルムと 1,1,1, 3, 3, 3 -へキサフルォロ- 2-プロパノールの混液(3:1, 0.40 raL)に溶解し、 5% クヱン酸水溶液で洗浄した。 有機層を乾燥 ·濃縮後、残渣をエタノール (0.300 mL) に溶解し、 イオン交換樹脂 AG1-X8 (バイオラッド社製) を加え、室温で 2時間撹拌 した。 樹脂をメダノール (0.60 mL) で洗浄後、 樹脂にクロ口ホルムとメタノール の混液 (1:1, 0.40 mL) 及び塩酸の酢酸ェチル溶液 (4mol/L, 0.050 mL) を加え、 目的物を溶出した。 榭脂をクロ口ホルムとメタノールの混液 (1:1, 1.2 mL) で洗 浄し、 溶出液と洗浄液を合わせて濃縮し、 目的とする化合物 1を得た。
その他の化合物 (IA) (化合物 2〜 14) も上記と同様の方法において、 工程 1 及び工程 3において対応する試薬を用いることで合成した。なおそれらの通算収率 は約 30%であった。
得られた化合物は質量分析により同定した。各化合物の分析結果は第 1一 1表〜 第 1一 2表に機器データとして記載する。また、以下に代表的化合物のプロトン應 R
(¾ NMR) を記載する。
化合物 1
¾應 R (270 MHz, DMSO-o^) 8 (ppm): 3.07 (t, /= 6.8 Hz, 2 H), 3.49-3.50 (br, 2 H), 4.24 (s, 4 H), 6.84—7.10 (m, 5 H), 7.32-7.47 (m, 2 H), 8.53 (s, 1 H), 8.76 (s, 1 H), 10.77 (s, 0.2 H), 10.58 (s, 0.8 H)
化合物 2
¾丽 R (270 MHz, DMS0- 4) δ (ppm): 1.79 (s, 3 H), 3.11 (t, /= 7.8 Hz, 2 H), 3.16 (s, 3 H), 3.61 (t, / = 7.8 Hz, 2 H), 6.88 (s, 1 H), 6.94 (dd, /= 3.5 Hz, 5.0 Hz, 1 H), 7.25—7.33 (m, 3 H), 7.79 (d, /= 8. Hz, 2 H), 7.86 (s, 1 H), 8.12 (s, 1 H), 8.69 (s, 1 H), 10.50 (s, 0.4 H), 10.77 (s, 0.6 H)
化合物 3
¾ NMR (270 MHz, DMSO-4) δ (ppm): 3.10 (t, J = '7.3 Hz, 2 H), 3.50—3.51 (m, 2 H), 3.66 (s, 6 H), 3.72 (s, 3 H), 6.85-6.95 (m, 2 H), 7.07 (s, 2 H), 7.31 (dd, 1.2 Hz, 5.1 Hz, 1 H), 7.63 (br, 1 H), 7.81 (br, 1 H), 8,64 (s, 1 H), 10.24 (br, 0.4 H) , 10.68 (br, 0.6 H)
化合物 7 (主なピーク)
¾丽 R (270 MHz, DMSO - δ (ppm): 1.30 (s, 9 H), 4.58 (s, 2 H), 7.22-7.76 (m, 5 H), 7.95 (s, 1 H), 8.13-8.25 (m, 1 H), 8.45 (d, /= 3.5 Hz, 1 H), 8.56 (s, 1 H), 8.66 (s, 1 H), 10.28 (s, 0.4 H), 10.55 (s, 0.6 H)
化合物 8 '
¾ NMR (270 MHz, DMSO- 4) 8 (ppm): 4.60 (d, J : 5.6 Hz, 2 H), 7.35 (dd, J : 4.9 Hz, 7.7 Hz, 1 H), 7.51-7.70 (br, 2 H), 7.77 (d, J = 7.4 Hz, 2 H), 8.10 (br, 1 H), 8.25 (br, 1 H), 8.38 (br, 1 H) , 8.45 (d, = 4.9 Hz, 1 H), 8.59 (s, 1 H), 8.72 (s, 1 H), 10.70 (br, 1 H)
実施例 2 :化合物 1 5の合成 ·
工程 1参考例 1 で得られた 4-ク口口- 5-シァノ -2-メチルチオピリミジン (0.25 mol/L クロ口ホルム溶液, 0.20 mL, 0.050 mmol) と 3 -ェチルァニリン (1.0 mol/L クロ口ホルム溶液, 0.060 mL, 0.060 mmol) の混合液に、 モルホリノメチルポリス チレン (35 mg, 0.12 mmol) を加え、 50°Cで 1日間攪拌した。 その後、 反応混合物 にべンゾィノレクロリ ドポリマーバウンド (23 mg, 0.058 mmol) 及ぴクロロホノレム
(0.50 mL) を加え、室温でさらに 1日間攪拌した。反応混合物中の樹脂を濾別し、 樹脂をクロ口ホルムとメタノールの混合溶媒(ク口口ホルム/メタノール =3/1, 1.2 tnL)で洗浄した。得られた溶液を全て合わせ、溶媒留去して対応する化合物を得た。 工程 2
工程 1で得られた化合物をジクロロメタン (0.20 mL) に溶解し、 メタクロ口過 安息香酸 (0.50 raol/L ジクロロメタン溶液, 0.125 mL, 0.063 mmol) を加え、 室 温で 0.5〜1時間攪拌した。 反応混合物に飽和重曹水 (0.20mL) を加え、 攪拌した 後、 水層を除去した。 その後、 再度飽和重曹水 (0.20 mL) を加えて攪拌した後、 分液した。 有機層を硫酸マグネシウムと珪藻土の混合物 (硫酸マグネシウム/珪藻
土 =1/1, 140 mg) で乾燥した。 硫酸マグネシウムと珪藻土の混合物をジクロロメ タン (0.80 mL) で洗浄した。 得られた溶液を全て合わせ、 溶媒を留去して、 対応 する化合物を得た。'
工程 3
工程 2で得られた化合物をテトラヒドロフラン (0.20 mL) に溶解し、 1 -フエ二 ルェチルァミン (1.0 mol/L クロ口ホルム溶液, 0.060 mL, 0.060 mraol) を加え、 室温で 1曰間攪拌した。 その.後、 反応混合物にク口口ホルム (0.40 mL)、 ベンゾィ ルクロリ ドポリマーバウンド (23 rag, 0.058 ramol)、 ポリ (4 -ビュルピリジン) (23 mg) を加え、 室温で 1日間攪拌した。 反応混合物中の樹脂を濾別し、 樹脂をクロ口 ホルムとメタノールの混合溶媒 (クロ口ホルム/メタノール =3/1, 1.4 mL) で洗浄 した。 得られた溶液を全て合わせ、 溶媒を留去して化合物 1 5を得た。
なお化合物 (IB) の中で、 R13が- NR18R19で表される化合物 (IBa) (化合物 1 6〜 2 2及び化合物 2 5〜 2 8 ) は上記と同様の方法に従い、 工程 1及び工程 3で対応 する試薬を用いることにより合成した。
得られた化合物 (IBa) は質量分析により同定した。 各化合物の分析結果は第 2 一 1表〜第 2— 2表に機器データとして記載する。 また、 以下に代表的化合物のプ ロトン丽 R (¾丽 R) を記載する。 ' 化合物 1 6 ,
應 R (270MHz, CDC13) δ (ppm): 1.55 (d, J = 6.9 Hz, 3 H), 3.79 (s, 3 H), 5.02-5.17 (m, 1 H), 5.90-6.04 (br, 1 H), 6.70 (dd, /= 2.3 Hz, 8.2 Hz, 1 H), 6.83-6.90 (m, 1 H), 7.10 (s, 1 H), 7.13-7. 3 (ra, 6 H), 8.17 (s, 1 H), 8.27 (br s, 1 H)
化合物 20
¾ NMR (270MHz, CDC13) δ (ppm): 0.11—0.26 (m, 2 H), 0.39-0.58 (m, 2 H), 0.89 (t, /= 7.3 Hz, 3 H), 0.81 - 1.03 (br, 1 H), 1.53 (d, /= 6.9 Hz, 3 H), 1.48-1.70 (m, 2H), 3.24-3.70 (m, 4H), 4.90-5.08 (br, 1H), 5.53-5.73 (br, 1H), 7.13-7.42 (m, 5 H), 8.11 (s, 1 H)
化合物 2 2
¾雇 R (270MHz, CDC13) δ (ppm): 1.56 (d, = 6.9 Hz, 3 H), 2.80-3.05 (br, 2
H), 3.80-4.09 (br, 2 H), 4.63—4.90 (m, 2 H), 5.00 (br, 1 H), 5.87 (br, 1 H), 7.11-7.41 (m, 9 H), 8.12 (s, 1 H)
化合物 28
¾雇 R (270MHz, CDCI3) δ (ppra): 0.23-0.29 (m, 2 H), 0.51—0.58 (ra, 2 H>, 0.93 (t, / = 7.3 Hz, 3 H), 1.01-1.14 (m, 1 H), 1.55—1.76 (m, 2 H), 3.52 (d, J二 6.6 Hz, 2 H), 3.63 (t, J= 7.9 Hz, 2 H), 4.45 (d, /= 5.3 Hz, 2 H), 5.95 (s, 2 H), 6.38 (br, 1 H), 6.76—6.79 (m, 3 H) , 7.90 (s, 1 H)
実施例 3 :化合物 23の合成
工程 1
参考例 1で得られる 4 -クロ口- 5-シァノ- 2-メチルチオピリミジン (223 mg, 1.20 ramol) をクロ口ホルム (5 mL) に溶解し、 テトラヒドロビラン- 2-ィルメタノール
(1.44 mraol)、 1, 8 -ジァザビシク口 [5.4.0]ゥンデセ _7 -ェン (0.215 mL, 1.44 mmol) を加え、 室温で 1〜2時間攪拌した。 反応液を 5%クェン酸水溶液で希釈し、 ク 口口ホルムにて抽出した。有機層を無水硫酸マグネシゥムにて乾燥し、濃縮するこ とにより得られる残渣をクロ口ホルム (12 mL) に溶解し、 ベンゾイルク口リ ドポ リマーバウンド (552 mg, 1.66 mraol)、 ポリ(4 -ビュルピリジン) (552 mg) を力!]え、 室温にて 12〜24時間攪拌した。 反応溶液を濾過し、 濃縮することによって、 対応す る化合物を得た。 .
工程 2
工程 1により得られた化合物を、実施例 2の工程 2に示した方法と同一の方法に より反応させ、 対応する化合物を得た。
工程 3
工程 2により得られた化合物を、実施例 2の工程 3に示した方法と同一の方法に より 1-フエニルェチルァミンと反応させ、 化合物 23を得た。
化合物 24も上記と同様の方法において、工程 1でシク口へキシルメタノールを 用いることで合成することが可能である。
化合物 23
¾應 R (270MHz, CDC13) δ (ppm): 1.24-1.76 (m,.3 H), 1.57 (d, = 6.9 Hz, 3 H), 77-1.95 (ra, 2 H), 3.30-3.74 (m, 4 H), 3.85-4.07 (m, 2 H), 4.90 - 5.12 (m, 1
H),6.36 (br, 1 H), 7.23-7.40 (m, 5 H), 8.13 (s, 1 H)
化合物 24
¾ 丽 R (270MHz, CDC13) δ (ppm): 0.74-1.08 (m, 2 H), 1.13-1.38 (ra, 2 H), 1.51-1.80 (m, 7 H), 1.58 (d, /= 6.9 Hz, 3 H), 4.05 (d, /= 6.2 Hz, 2 H), 4.90 一 5.15 (m, 1 H), 6.58 (d, 6.5 Hz, 1 H), 7: 24-7.38 (m, 5 H), 8.17 (s, 1 H) 製剤例 1 :錠剤
常法により、 次の組成からなる錠剤を調製する。
処方 化合物 1 20 mg
乳糖 143.4 mg
' 馬鈴薯デンプン 30 mg
ヒ ドロキシプロピノレセノレロース 6 mg
m ra m m
ステアリン酸マグネシゥム 0.6 mg
200 mg
製剤例 2 :注射剤
常法により、 次の組成からなる注射剤を調製する。
処方' 化合物 10 2
精製ダイズ油 200
24
注射用グリセリン. 50
注射用蒸留水 1.72 ml
2.00 ml
産業上の利用可能性
本発明により、 PLK阻害剤、 PLK阻害作用または抗腫瘍活性等を有するピリミジ ン誘導体等が提供される。