WO2004083199A1 - Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators - Google Patents
Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators Download PDFInfo
- Publication number
- WO2004083199A1 WO2004083199A1 PCT/EP2004/002908 EP2004002908W WO2004083199A1 WO 2004083199 A1 WO2004083199 A1 WO 2004083199A1 EP 2004002908 W EP2004002908 W EP 2004002908W WO 2004083199 A1 WO2004083199 A1 WO 2004083199A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- hydrogen
- white solid
- independently
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCCN(CCc1ccccc1N1CCNCC1)*=O Chemical compound CCCN(CCc1ccccc1N1CCNCC1)*=O 0.000 description 5
- CCGJOCANJCSAPA-XIFFEERXSA-N C=CCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound C=CCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O CCGJOCANJCSAPA-XIFFEERXSA-N 0.000 description 1
- VTASJSIBIOVQPG-PMERELPUSA-N C=CCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1c(CN(CCC3)C3=O)cccc1)=O)=O)=CC2=O Chemical compound C=CCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1c(CN(CCC3)C3=O)cccc1)=O)=O)=CC2=O VTASJSIBIOVQPG-PMERELPUSA-N 0.000 description 1
- HMYNWNYYCMHNRI-WJOKGBTCSA-N C=CCOc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound C=CCOc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O HMYNWNYYCMHNRI-WJOKGBTCSA-N 0.000 description 1
- BLSBQYUJENVWFW-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC=C1S1OC(C)(C)C(C)(C)O1)=O Chemical compound CC(C)(C)OC(N(CC1)CC=C1S1OC(C)(C)C(C)(C)O1)=O BLSBQYUJENVWFW-UHFFFAOYSA-N 0.000 description 1
- QNACALQPXSOODW-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CC=C1c1ccccc1CN(CCC1)C1=O)=O Chemical compound CC(C)(C)OC(N(CC1)CC=C1c1ccccc1CN(CCC1)C1=O)=O QNACALQPXSOODW-UHFFFAOYSA-N 0.000 description 1
- PWBKAWVRUDWXDT-UHFFFAOYSA-N CC(C)(C)OC(N(CC1)CCN1c1c(C)cccc1)=O Chemical compound CC(C)(C)OC(N(CC1)CCN1c1c(C)cccc1)=O PWBKAWVRUDWXDT-UHFFFAOYSA-N 0.000 description 1
- STJJOKVEHLMHJE-DEOSSOPVSA-N CC(C)(C)OC(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O Chemical compound CC(C)(C)OC(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O STJJOKVEHLMHJE-DEOSSOPVSA-N 0.000 description 1
- TZTGUUIDUREVNK-MGBGTMOVSA-N CC(C)(C)c(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound CC(C)(C)c(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O TZTGUUIDUREVNK-MGBGTMOVSA-N 0.000 description 1
- WOMNLXSIQBYHGM-UUWRZZSWSA-N CC(C)(C)c(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound CC(C)(C)c(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O WOMNLXSIQBYHGM-UUWRZZSWSA-N 0.000 description 1
- SHHWWIYNTFUJAR-UUWRZZSWSA-N CC(C)c(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound CC(C)c(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O SHHWWIYNTFUJAR-UUWRZZSWSA-N 0.000 description 1
- UBGCDRGSSZBZNO-YTTGMZPUSA-N CC(N(C)c(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O)=O Chemical compound CC(N(C)c(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O)=O UBGCDRGSSZBZNO-YTTGMZPUSA-N 0.000 description 1
- OJYOSTYARZWPHR-KJZSJKLJSA-N CC1(CN(CCC2)C2=O)C=CC=CC1C(CC1)CCN1C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1O)c2cc1O)=CC2=O)=O)=O Chemical compound CC1(CN(CCC2)C2=O)C=CC=CC1C(CC1)CCN1C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1O)c2cc1O)=CC2=O)=O)=O OJYOSTYARZWPHR-KJZSJKLJSA-N 0.000 description 1
- KVLVWNWFEKEUCW-LDCRUHPQSA-N CC1C(Cl)=CC=C(C[C@H](C(N(CC2)CCN2c2ccccc2CN(CCC2)C2=O)=O)NC(C(Oc(cc2)c3cc2OCC=C)=CC3=O)=O)C1 Chemical compound CC1C(Cl)=CC=C(C[C@H](C(N(CC2)CCN2c2ccccc2CN(CCC2)C2=O)=O)NC(C(Oc(cc2)c3cc2OCC=C)=CC3=O)=O)C1 KVLVWNWFEKEUCW-LDCRUHPQSA-N 0.000 description 1
- DZOJASFMIXADTM-NXDCZNQUSA-N CC1C=CC(N(CC2)CCN2C([C@@H](Cc2cc(cccc3)c3cc2)NC(C(Oc2c3ccc(OC)c2)=CC3=O)=O)=O)=C(CN(CCC2)C2=O)C1 Chemical compound CC1C=CC(N(CC2)CCN2C([C@@H](Cc2cc(cccc3)c3cc2)NC(C(Oc2c3ccc(OC)c2)=CC3=O)=O)=O)=C(CN(CCC2)C2=O)C1 DZOJASFMIXADTM-NXDCZNQUSA-N 0.000 description 1
- YAYHHCRFAVSYDD-OMFPCOHASA-N CC1C=Cc2cc(C[C@H](C(N(CC3)CCN3c3ccccc3CN(CCC3)C3=O)=O)NC(C(Oc(cc3)c4cc3NC(C)=O)=CC4=O)=O)ccc2C1 Chemical compound CC1C=Cc2cc(C[C@H](C(N(CC3)CCN3c3ccccc3CN(CCC3)C3=O)=O)NC(C(Oc(cc3)c4cc3NC(C)=O)=CC4=O)=O)ccc2C1 YAYHHCRFAVSYDD-OMFPCOHASA-N 0.000 description 1
- YJUWMNUCPVHNEV-YTTGMZPUSA-N CCCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound CCCOc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O YJUWMNUCPVHNEV-YTTGMZPUSA-N 0.000 description 1
- ZFPGARUNNKGOBB-UHFFFAOYSA-N CCN(CCC1)C1=O Chemical compound CCN(CCC1)C1=O ZFPGARUNNKGOBB-UHFFFAOYSA-N 0.000 description 1
- JUCWTUQQQBZKJG-MGBGTMOVSA-N CCOc(cc1)cc2c1OC(C(N[C@H](Cc1cc(cccc3)c3cc1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound CCOc(cc1)cc2c1OC(C(N[C@H](Cc1cc(cccc3)c3cc1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O JUCWTUQQQBZKJG-MGBGTMOVSA-N 0.000 description 1
- KCZMLCZLVXAVIP-JGCGQSQUSA-N CCc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound CCc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O KCZMLCZLVXAVIP-JGCGQSQUSA-N 0.000 description 1
- IYKAPLSTFWJRMA-SSEXGKCCSA-N CCc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound CCc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O IYKAPLSTFWJRMA-SSEXGKCCSA-N 0.000 description 1
- FQKNPSFLRCUKPI-PMERELPUSA-N COc(cc1)cc(OC(C(N[C@@H](Cc(cc2)ccc2Cl)C(N(CC2)CCC2c2ccccc2CN(CCC2)C2=O)=O)=O)=C2)c1C2=O Chemical compound COc(cc1)cc(OC(C(N[C@@H](Cc(cc2)ccc2Cl)C(N(CC2)CCC2c2ccccc2CN(CCC2)C2=O)=O)=O)=C2)c1C2=O FQKNPSFLRCUKPI-PMERELPUSA-N 0.000 description 1
- VLHQYFLWKLHLCD-GDLZYMKVSA-N COc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound COc(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O VLHQYFLWKLHLCD-GDLZYMKVSA-N 0.000 description 1
- FQXNWBOAAQFIPD-SSEXGKCCSA-N COc1cccc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O Chemical compound COc1cccc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O FQXNWBOAAQFIPD-SSEXGKCCSA-N 0.000 description 1
- YOFGQHKXANABFX-HKBQPEDESA-N CS(Nc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O)(=O)=O Chemical compound CS(Nc(cc1)cc2c1OC(C(N[C@@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O)(=O)=O YOFGQHKXANABFX-HKBQPEDESA-N 0.000 description 1
- CJGVFHLDBYBBDA-WJOKGBTCSA-N CS(c(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O)(=O)=O Chemical compound CS(c(cc1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O)=O)=O)=CC2=O)(=O)=O CJGVFHLDBYBBDA-WJOKGBTCSA-N 0.000 description 1
- MEKMRCFPMTZCEP-WJOKGBTCSA-N Cc(c(C)c1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound Cc(c(C)c1)cc2c1OC(C(N[C@H](Cc(cc1)ccc1Cl)C(N(CC1)CCC1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O MEKMRCFPMTZCEP-WJOKGBTCSA-N 0.000 description 1
- IRYCLNQPPXHGDK-MIXAMLLLSA-N O=C(/C(/NC(C(Oc1c2ccc(Cl)c1)=CC2=O)=O)=C/c(cc1)ccc1Cl)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O Chemical compound O=C(/C(/NC(C(Oc1c2ccc(Cl)c1)=CC2=O)=O)=C/c(cc1)ccc1Cl)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O IRYCLNQPPXHGDK-MIXAMLLLSA-N 0.000 description 1
- MWBCBFXXQALOJE-UHFFFAOYSA-N O=C(C(Cc(cc1)ccc1Cl)NC(C(Oc1c2ccc(Cl)c1)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C(C(Cc(cc1)ccc1Cl)NC(C(Oc1c2ccc(Cl)c1)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O MWBCBFXXQALOJE-UHFFFAOYSA-N 0.000 description 1
- DHDQDGXEAFLDQS-HHHXNRCGSA-N O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(c(F)c1)c2cc1F)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(c(F)c1)c2cc1F)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O DHDQDGXEAFLDQS-HHHXNRCGSA-N 0.000 description 1
- PKUZYJVJZXWOQC-GDLZYMKVSA-N O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1OC(F)(F)F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1OC(F)(F)F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O PKUZYJVJZXWOQC-GDLZYMKVSA-N 0.000 description 1
- VSYJENPDNZKJFQ-MUUNZHRXSA-N O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2cccc1Cl)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O Chemical compound O=C([C@@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2cccc1Cl)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCCC1)C1=O VSYJENPDNZKJFQ-MUUNZHRXSA-N 0.000 description 1
- OOLUWIDKKFABDU-HHHXNRCGSA-N O=C([C@@H](Cc(cc1Cl)ccc1Cl)NC(C(Oc1c2cccc1)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc(cc1Cl)ccc1Cl)NC(C(Oc1c2cccc1)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O OOLUWIDKKFABDU-HHHXNRCGSA-N 0.000 description 1
- DWWFFCJWFUEQTL-WJOKGBTCSA-N O=C([C@@H](Cc1cc(cccc2)c2cc1)NC(C(Oc(c1c2)ccc2F)=CC1=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc1cc(cccc2)c2cc1)NC(C(Oc(c1c2)ccc2F)=CC1=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O DWWFFCJWFUEQTL-WJOKGBTCSA-N 0.000 description 1
- GIFVRSQNQZPEQX-WJOKGBTCSA-N O=C([C@@H](Cc1cc2ccccc2cc1)NC(C(Oc(cc1)c2cc1SC(F)(F)F)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc1cc2ccccc2cc1)NC(C(Oc(cc1)c2cc1SC(F)(F)F)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O GIFVRSQNQZPEQX-WJOKGBTCSA-N 0.000 description 1
- DFJALWXIGJAOAL-WJOKGBTCSA-N O=C([C@@H](Cc1ccc(cccc2)c2c1)NC(C(Oc(cc1)c2cc1Br)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc1ccc(cccc2)c2c1)NC(C(Oc(cc1)c2cc1Br)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O DFJALWXIGJAOAL-WJOKGBTCSA-N 0.000 description 1
- MUQOYXFUIQJHNR-WJOKGBTCSA-N O=C([C@@H](Cc1ccc(cccc2)c2c1)NC(C(Oc(cc1)c2cc1Cl)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@@H](Cc1ccc(cccc2)c2c1)NC(C(Oc(cc1)c2cc1Cl)=CC2=O)=O)N(CC1)CCN1c1ccccc1CN(CCC1)C1=O MUQOYXFUIQJHNR-WJOKGBTCSA-N 0.000 description 1
- TZIIDUHMMJQKLL-LJAQVGFWSA-N O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1Br)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1Br)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O TZIIDUHMMJQKLL-LJAQVGFWSA-N 0.000 description 1
- FKNBUMGPFLYHSG-LJAQVGFWSA-N O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1OC(C(F)(F)F)(F)F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc(cc1)c2cc1OC(C(F)(F)F)(F)F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O FKNBUMGPFLYHSG-LJAQVGFWSA-N 0.000 description 1
- LHAONUBUCQTMML-PMERELPUSA-N O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2ccc(F)c1)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O Chemical compound O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2ccc(F)c1)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCCC1)C1=O LHAONUBUCQTMML-PMERELPUSA-N 0.000 description 1
- MFUOCORWFXOPIH-LJAQVGFWSA-N O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2cccc1F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O Chemical compound O=C([C@H](Cc(cc1)ccc1Cl)NC(C(Oc1c2cccc1F)=CC2=O)=O)N(CC1)CCC1c1ccccc1CN(CCC1)C1=O MFUOCORWFXOPIH-LJAQVGFWSA-N 0.000 description 1
- DQPPCXKPUWBWOR-UHFFFAOYSA-N O=C1N(Cc(cccc2)c2N2CCNCC2)CCC1 Chemical compound O=C1N(Cc(cccc2)c2N2CCNCC2)CCC1 DQPPCXKPUWBWOR-UHFFFAOYSA-N 0.000 description 1
- SYJUPXPOLHFEIT-SSEXGKCCSA-N Oc(cc1)cc(OC(C(N[C@H](Cc(cc2)ccc2Cl)C(N(CC2)CCC2c2ccccc2CN(CCCC2)C2=O)=O)=O)=C2)c1C2=O Chemical compound Oc(cc1)cc(OC(C(N[C@H](Cc(cc2)ccc2Cl)C(N(CC2)CCC2c2ccccc2CN(CCCC2)C2=O)=O)=O)=C2)c1C2=O SYJUPXPOLHFEIT-SSEXGKCCSA-N 0.000 description 1
- NIDWGKWQUDLPNM-WJOKGBTCSA-N Oc(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound Oc(cc1)cc2c1OC(C(N[C@H](Cc1ccc(cccc3)c3c1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O NIDWGKWQUDLPNM-WJOKGBTCSA-N 0.000 description 1
- UXTCXOZQSCVMQO-SSEXGKCCSA-N Oc1cccc2c1OC(C(N[C@H](Cc1cc3ccccc3cc1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O Chemical compound Oc1cccc2c1OC(C(N[C@H](Cc1cc3ccccc3cc1)C(N(CC1)CCN1c1ccccc1CN(CCC1)C1=O)=O)=O)=CC2=O UXTCXOZQSCVMQO-SSEXGKCCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to novel substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators.
- the compounds of the invention are either selective agonists or selective antagonists of the human melanocortin-4 receptor (MC-4R).
- the agonists can be used for the treatment of disorders and diseases such as obesity, diabetes and sexual dysfunction, whereas the antagonists are useful for the treatment of disorders and diseases such as cancer cachexla, muscle wasting, anorexia, anxiety and depression.
- disorders and diseases such as cancer cachexla, muscle wasting, anorexia, anxiety and depression.
- all diseases and disorders where the regulation of the MC-4R is involve can be treated with the compounds of the invention.
- MCs Melanocortins stem from pro-opiomelanocortin (POMG) via proteolytic cleavage. These peptides, adrenocorticotropic hormone (ACTH), ⁇ -melanocyte-stimufati ⁇ g. hormone ( ⁇ -MSH), ⁇ -MSH and ⁇ -MSH, range i size from 12 to 39 amino acids. The most important endogenous agonist for central MC-4R activation appears to be the tridecapeptide ⁇ -MSH. Among MCs, it was reported that ⁇ -MSH acts as a neurotransmitter or neuromodiflator ir> the brain.
- MC peptides particularly ⁇ -MSH
- ⁇ -MSH have a wide range of effects on biofog ⁇ cal functions including feeding behavior, pigmentation, and exocrine function.
- the biological effects of ⁇ -MSH are mediated by a sub-family of 7-transmembrane G-protefn-coupled receptors, termed melanocortin receptors (MC-Rs). Activation of any of these MC-Rs results in stimulation of cAMP formation.
- MC-Rs melanocortin receptors
- MC-1 R was first found in melanocytes. Naturally occurring inactive variants of MC-1 R in animals were shown to lead to alterations in pigmentation and a subsequent lighter coat color by controlling the conversion of phaeomelanin to eumelanin through the control of tyrosinase. From these, and other studies, it is evident that MC-1 R is an important regulator of melanin production and coat color in animals and skin color in humans.
- the MC-2R is expressed in the adrenal gland representing the ACTH receptor.
- the MC-2R is not a receptor for ⁇ -MSH but is the receptor for the adrenocorticotropic hormone I (ACTH I).
- the MC-3R is expressed in the brain (predominately located in the hypothalamus) and peripheral tissues like gut and placenta, and knock-out studies have revealed that the MC-3R may be responsible for alterations in feeding behavior, body weight and thermogenesis.
- the MC-4R is primarily expressed in the brain. Overwhelming data support the role of MC-4R in energy homeostasis. Genetic knock-outs and pharmacologic manipulation of MC-4R in animals have shown that agonizing the MC-4R causes weight loss and antagonizing the MC-4R produces weight gain. (A. Kask, et al., "Selective antagonist for the melanocortin-4 receptor (HS014) increases food intake in free-feeding rats," Biochem. Biophys. Res. Commun., 245: 90-93 (1998)).
- MC-5R is ubiquitously expressed in many peripheral tissues including white fat, placenta and a low level of expression is also observed in the brain. However its expression is greatest in exocrine glands. Genetic knock-out of this receptor in mice results in altered regulation of exocrine gland function, leading to changes in water repulsion and thermoregulation. MC-5R knockout mice also reveal reduced sebaceous gland lipid production (Chen et al., Cell, 91: 789-798 (1997)).
- MC-3R and MC-4R modulators have potent physiological effects besides their role in regulating pigmentation, feeding behavior and exocrine function.
- ⁇ -MSH recently has been shown to induce a potent anti-inflammatory effect in both acute and chronic models of inflammation including inflammatory bowel-disease, renal ischemia/reperfusion injury and endotoxin-induced hepatitis.
- Administration of ⁇ -MSH in these models results in substantial reduction of inflammation-mediated tissue damage, a significant decrease in leukocyte infiltration, and a dramatic reduction in elevated levels of cytokines and other mediators to near baseline levels.
- ⁇ -MSH anti-inflammatory actions of ⁇ -MSH are mediated by MC-1R.
- the mechanism by which agonism of MC-1 R results in an anti-inflammatory response is likely through inhibition of the pro-inflammatory transcription activator, NF- ⁇ B.
- NF- ⁇ B is a pivotal component of the pro-inflammatory cascade, and its activation is a central event in initiating many inflammatory diseases.
- anti-inflammatory actions of ⁇ -MSH may be in part mediated by agonism of MC-3R and/or MC-5R.
- MC-4R signaling is important in mediating feeding behavior (S.Q. Giraudo et al., "Feeding effects of hypothalamic injection of melanocortin-4 receptor ligands", Brain Research, 80: 302-306 (1998)).
- Further evidence for the involvement of MC-Rs in obesity includes: a) the agouti (A ⁇ ) mouse which ectopically expresses an antagonist of the MC-1 R, MC-3R and MC-4R is obese, indicating that blocking the action of these three MC-Rs can lead to hyperphagia and metabolic disorders; 2) MC-4R knockout mice (D.
- MC-4R appears to play a role in other physiological functions as well, namely controlling grooming behavior, erection and blood pressure.
- Erectile dysfunction denotes the medical condition of inability to achieve penile erection sufficient for successful intercourse.
- the term "impotence" is often employed to describe this prevalent condition.
- Synthetic melanocortin receptor agonists have been found to initiate erections in men with psychogenic erectile dysfunction (H. Wessells et al., "Synthetic Melanotropic Peptide Initiates Erections in Men With Psychogenic Erectile Dysfunction: Double-Blind, Placebo Controlled Crossover Study", J. Urol., 160: 389-393, 1998).
- Activation of melanocortin receptors of the brain appears to cause normal stimulation of sexual arousal.
- Evidence for the involvement of MC-R in male and/or female sexual dysfunction is detailed in WO/0074679.
- Diabetes is a disease in which a mammal's ability to regulate glucose levels in the blood is impaired because the mammal has a reduced ability to convert glucose to glycogen for storage in muscle and liver cells. In Type I diabetes, this reduced ability to store glucose is caused by reduced insulin production.
- Type II diabetes or “Non-Insulin Dependent Diabetes Mellitus” (NIDDM) is the form of diabetes, which is due to a profound resistance to insulin stimulating or regulatory effect on glucose and lipid metabolism in the main insulin-sensitive tissues, muscle, liver and adipose tissue. This resistance to insulin responsiveness results in insufficient insulin activation of glucose uptake, oxidation and storage in muscle and inadequate insulin repression of lipolysis in adipose tissue and of glucose production and secretion in liver.
- NIDDM Non-Insulin Dependent Diabetes Mellitus
- Hyperinsulemia is associated with hypertension and elevated body weight. Since insulin is involved in promoting the cellular uptake of glucose, amino acids and triglycerides from the blood by insulin sensitive cells, insulin insensitivity can result in elevated levels of triglycerides and LDL which are risk factors in cardiovascular diseases.
- the constellation of symptoms which includes hyperinsulemia combined with hypertension, elevated body weight, elevated triglycerides and elevated LDL is known as Syndrome X.
- MC-4R agonists might be useful in the treatment of NIDDM and Syndrome X.
- the MC4 receptor is also of interest in terms of the relationship to stress and the regulation of emotional behavior, as based on the following findings. Stress initiates a complex cascade of responses that include endocrine, biochemical and behavioral events. Many of these responses are initiated by release of corticotropin-releasing factor (CRF), (Owen MJ and Nemeroff CB (1991). Physiology and pharmacology of corticotrophin releasing factor. Pharmacol Rev 43: 425 - 473).
- CCF corticotropin-releasing factor
- MCL0129 (1-[(S)-2-(4-Fluorophenyl)-2-(4-isopropylpiperadin-1-yl)ethyl]-4- [4-(2-methoxynaphthalen-1- yl)butyl]piperazine), a Novel and Potent Nonpeptide Antagonist of the Melanocortin-4 Receptor; Shigeyuki Chaki et al, J. Pharm. Exp. Ther. (2003)304(2), 818-26).
- Chronic diseases such as malignant tumors or infections are frequently associated with cachexia resulting from a combination of a decrease in appetite and a loss of lean body mass.
- Extensive loss of lean body mass is often triggered by an inflammatory process and is usually associated with increased plasma levels of cytokines (e.g. TNF- ⁇ ), which increase the production of ⁇ -MSH in the brain.
- cytokines e.g. TNF- ⁇
- Activation of MC4 receptors in the hypothalamus by ⁇ -MSH reduces appetite and increases energy expenditure.
- Experimental evidence in tumor bearing mice suggests that cachexia can be prevented or reversed by genetic MC4 receptor knockout or MC4 receptor blockade.
- the increased body weight in the treated mice is attributable to a larger amount of lean body mass, which mainly consists of skeletal muscle (Marks D.L. et al. Role of the central melanocortin system in cachexia. Cancer Res. (2001) 61 : 1432-1438).
- WO03009847A1 describes phenylpiperidinyl-phenylalanine derivatives and WO03009850A1 describes phenylpiperazinyl-phenylalanine derivatives for the treatment of obesity.
- Most of the compounds in both patents contain a N-(2-piperidin-4-yl-phenyl)-alkyl, benzyl or aryl sulfonamide group and N-(2-piperazin-4-yl-phenyl)-alkyl, benzyl or aryl sulfonamide group, respectively.
- WO02070511A1 describes phenylpiperazinyl-phenylalanine amides, phenylpiperidinyl- phenylalanine amides and cyclohexyl-phenylalanine amides as modulators of melanocortin receptors 1 and 4.
- the phenylalanine amino group is in the most cases acylated with a second amino acid. For amino acids with a basic side chain the amino group can be acylated. Biological data for the compounds are not given.
- the present invention relates to novel substituted piperidine and piperazine derivatives of the following general structural formula.
- piperidine and piperazine derivatives are effective as melanocortin receptor modulators and are particularly effective as selective melanocortin-4 receptor (MC-4R) modulators. They are therefore useful for the treatment of disorders where the activation or inactivation of the MC-4R are involved.
- Agonists can be used for the treatment of disorders and diseases such as obesity, diabetes, and sexual dysfunction, whereas the antagonists are useful for the treatment of disorders and diseases such as cancer cachexia, muscle wasting, anorexia, anxiety and depression.
- the present invention also relates to pharmaceutical compositions comprising the compounds of the present invention and a pharmaceutically acceptable carrier.
- the present invention relates to novel substituted piperidine and piperazine derivatives useful as melanocortin receptor modulators, in particular, selective MC-4R agonists and MC-4R antagonists.
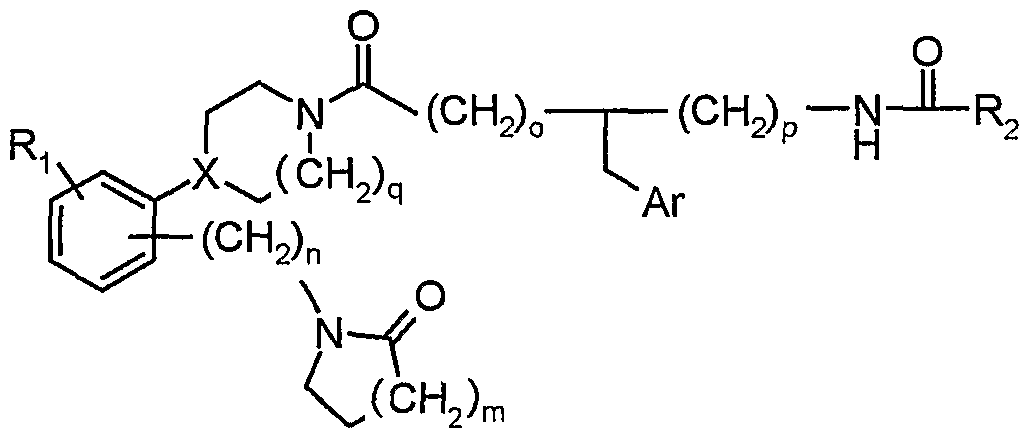
- the compounds of the present invention are represented by structural formula (I).
- Ar is: aryl or heteroaryl which may both be substituted or unsubstituted;
- Ri is independently: hydrogen, hydroxy, cyano, nitro, halo, alkyl, alkoxy or haloalkyl;
- R 2 is:
- each R 3 is independently: hydrogen, halo, alkyl, haloalkyl, hydroxy, alkoxy, S-alkyl, SO 2 -alkyl, O-alkenyl, S-alkenyl, NR 7 C(O)R 7 , NR 7 SO 2 R 7 , N(R 7 ) 2
- heterocyclyl excludes a heterocyclyl containing a single nitrogen, wherein aryl, heteroaryl, heterocyclyl, alkyl and/or cycloalkyl may be substituted or unsubstituted, and two adjacent R 3 may form a 4- to 7-membered ring;
- R 7 and R 8 are each independently: hydrogen, alkyl or cycloalkyl, or
- R 7 and R 8 together with the nitrogen to which they are attached form a 5- to 8-membered ring, wherein alkyl and cycloalkyl are both unsubstituted or substituted;.
- D is a bond or alkyl; X is CH or N; Y is O or NR 7 ; n is 1 - 4; m is 0 - 3; o is 0 - 2; p is 0 - 2; q is 1 or 2; s is 0 - 4.
- variants of formula (I) have the following meanings:
- Ar is as defined above, and is preferably aryl, more preferably phenyl or naphthyl. If aryl or heteroaryl are substituted, it is preferably substituted with one to three, more preferably one or two, most preferably one, substituents.
- the substituents are preferably independently selected from the group consisting of: cyano, nitro, perfluoroalkoxy, halo, alkyl, (D)-cycloalkyl, alkoxy and haloalkyl, more preferably perfluoroalkoxy, halo, alkyl, alkoxy or haloalkyl, even more preferably halo, alkyl, alkoxy and haloalkyl, in particular halo.
- Ar is phenyl or naphthyl which both, preferably phenyl, may be substituted with one to three, in particular one, halo, e.g. CI.
- the substitution can be in any position, preferably in the 4-position.
- R-i is as defined above, preferably hydrogen, hydroxy, halo, alkyl, alkoxy or haloalkyl, more preferably hydrogen, alkoxy, halo or alkyl, most preferably hydrogen.
- R 2 is each of the rings as defined above.
- R 2 is most preferably
- R 3 is as defined above. If aryl, heteroaryl, heterocyclyl, alkyl and/or cycloalkyl are substituted, they are independently preferably substituted with one to three, more preferably one substituent selected from the group consisting of oxo, halo, alkyl, N(R 4 ) 2 , OR 4 , SR 4 and CO 2 R .
- R 3 is preferably hydrogen, halo, unsubstituted alkyl, substituted alkyl, haloalkyl, hydroxyl, alkoxy, S-alkyl, SO 2 -alkyl, O-alkenyl, S-alkenyl, more preferably hydrogen, isopropyl, hydroxyl, alkoxy, S-alkyl, and SO 2 -alkyl.
- R 3 is hydrogen, halo, alkyl, haloalkyl, alkoxy, (D)-cycloalkyl, (D)-aryl, (D)-heteroaryl or (D)-heterocyclyl, wherein heterocyclyl excludes a heterocyclyl containing a single nitrogen, wherein aryl, heteroaryl, heterocyclyl, alkyl and/or cycloalkyl may be substituted or unsubstituted; preferably hydrogen, halo, unsubstituted alkyl, substituted alkyl, haloalkyl, alkoxy, unsubstituted (D)- cycloalkyl or substituted (D)-cycloalkyl; more preferably hydrogen.
- R 4 is independently hydrogen, alkyl, C(O)alkyl, SO 2 alkyl, SO 2 aryl, (D)-aryl or cycloalkyl.
- R 4 is hydrogen or alkyl, more preferably hydrogen
- R 7 and R 8 are each independently as defined above.
- said ring may contain an additional heteroatom, preferably selected from O, S and NR 4 in the ring.
- alkyl and cycloalkyl are substituted, they are preferably substituted with one to three, more preferably one or two groups independently selected from R 9 and oxo.
- R 7 and R 8 are each independently preferably selected from the group consisting of hydrogen, alkyl and cycloalkyl; or R and R 8 together with the nitrogen to which they are attached form a 5- to 7-membered ring. More preferably R 7 and R 8 are each independently selected from the group consisting of hydrogen and alkyl; or R 7 and R 8 together with the nitrogen to which they are attached form a 5- to 6-membered ring optionally containing an additional oxygen atom.
- R 9 is alkyl, (D)-aryl, (D)-cycloalkyl, (D)-heteroaryl, halo, OR ⁇ o, NHSO 2 R ⁇ o, N(R 10 ) 2 , C ⁇ N, CO 2 R 7 , C(R ⁇ o)(R ⁇ o)N(R ⁇ o) 2 , nitro, SO 2 N(R 10 ) 2 , S(O) u R ⁇ o, CF 3 or OCF 3 , and preferably selected from the group consisting of alkyl, OR10, (D)-aryl, (D)-cycloalkyl, (D)-heteroaryl and halo.
- R 10 is independently hydrogen, alkyl, (D)-aryl or cycloalkyl, preferably hydrogen or alkyl, more preferably alkyl.
- D is as defined above, preferably a bond or CH 2 , most preferably a bond.
- X is as defined above. In one embodiment, X is CH.
- Y is as defined above, preferably O. In one embodiment Y is NR 7 , more preferably N-alkyl. Alkyl is as defined below, preferably C C 4 alkyl. In one embodiment, Y is N-propyl.
- n is as defined above, preferably 1 or 2, more preferably 1.
- m is as defined above, preferably 1 , 2 or 3, most preferably 1 or 2.
- 0 is as defined above, preferably 0 or 1 most preferably 0 is 0.
- p is as defined above, preferably 0 or 1 most preferably p is 0.
- q is as defined above, preferably 1.
- s is as defined above, i.e. 0, 1 , 2, 3 or 4, preferably 1 , 2 or 3, most preferably 1 or 2.
- u is O, 1 or 2.
- any of the preferred definitions for each variant can be combined with the preferred definition of the other variants.
- Aryl is an aromatic mono- or polycyclic moiety with 6 to 20 carbon atoms which is preferably selected from phenyl, biphenyl, naphthyl, tetrahydronaphthyl, fluorenyl, indenyl and phenanthrenyl, more preferably from phenyl and naphthyl.
- Heteroaryl is an aromatic moiety having 6 to 20 carbon atoms with at least one heterocycle and is preferably selected from thienyl, benzothienyl, naphthothienyl, furanyl, benzofuranyl, chromenyl, indolyl, isoindolyl, indazolyl, quinolyl, isoquinolyl, phthalazinyl, quinoxalinyl, cinnolinyl and quinazolinyl, more preferably from thienyl, furanyl, benzothienyl, benzofuranyl and indolyl.
- Heterocyclyl is a saturated, unsaturated or aromatic ring containing at least one heteroatom selected from O, N and/or S and 1 to 6 carbon atoms and is preferably selected from thienyl, furyl, piperidinyl, pyranyl, pyrrolyl, imidazolyl, pyrazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, isothiazolyl and isoxazyl, more preferably from pyridyl, piperidinyl, imidazolyl and pyrazinyl.
- Carbocyclyl is a monocyclic or polycyclic ring system of 3 to 20 carbon atoms which may be saturated, unsaturated or aromatic.
- Alkyl is straight chain or branched alkyl having preferably 1 to 8 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, n-pentyl, isopentyl, neopentyl, hexyl or heptyl, more preferably 1 to 4 carbon atoms.
- Cycloalkyl is an alkyl ring having preferably 3 to 8 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl, more preferably 3 to 6 carbon atoms.
- Alkenyi is straight chain or branched alkenyi having preferably 2 to 8 carbon atoms such as vinyl, allyl, methallyl, buten-2-yl, buten-3-yl, penten-2-yl, penten-3-yl, penten-4-yl, 3-methyl- but-3-enyl, 2-methyl-but-3-enyl, 1-methyl-but-3-enyl, hexenyl or heptenyl, more preferably 2 to 4 atoms.
- Alkoxy is O-alkyl wherein alkyl is as defined above and has preferably 1 to 4 carbon atoms, more preferably 1 or 3 carbon atoms.
- Halo or halogen is a halogen atom preferably selected from F, CI, Br and I, more preferably from F, CI and Br.
- Haloalkyl is an alkyl moiety as defined above having preferably 1 to 4 carbon atoms, more preferably 1 or 2 carbon atoms, wherein at least one, preferably 1 , 2 or 3 hydrogen atoms have been replaced by a halogen atom.
- Preferred examples are -CF 3 -CH 2 CF 3 and
- the compounds of structural formula (I) are effective as melanocortin receptor modulators and are particularly effective as selective modulators of MC-4R. They are therefore useful for the treatment and/or prevention of disorders responsive to the activation and inactivation of MC-4R, such as cancer cachexia, muscle wasting, anorexia, anxiety, depression, obesity, diabetes, sexual dysfunction and other diseases with MC-4R involvement.
- the compounds of structural formula (I) contain one or more asymmetric centers and can occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers.
- the present invention is meant to comprehend all such isomeric forms of the compounds of structural formula (I).
- Some of the compounds described herein may exist as tautomers such as keto-enol tautomers.
- the individual tautomers, as well as mixtures thereof, are encompassed within the compounds of structural formula (I).
- the compounds of structural formula (I) may be separated into their individual diastereoisomers by, for example, fractional crystallization from a suitable solvent, for example, methanol or ethyl acetate or a mixture thereof, or via chiral chromatography using an optically active stationary phase.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration.
- any stereoisomer of a compound of the general formula (I) may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known absolute configuration.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, manganous, potassium, sodium, zinc, salts and the like. Particularly preferred are the ammonium, calcium, lithium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically acceptable organic nontoxic bases include salts of primary, secondary and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins such as arginine, betaine, caffeine, choline, N.N'-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyarnine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine and the like.
- basic ion exchange resins such as arginine
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, formic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, malonic, mucic, nitric, parnoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, ptoluenesulfonic, trifluoroacetic acid and the like.
- Particularly preferred are citric, fumaric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric and tartaric acids.
- the compounds of formula (I) are melanocortin receptor modulators and, as such, are useful in the treatment, contro or prevention of diseases, disorders or conditions responsive to the activation or inactivation of one or more of the melanocortin receptors including, but not limited to, MC-1 R, MC-2R, MC-3R, MC-4R and MC-5R.
- Such diseases, disorders or conditions include, but are not limited to, cancer cachexia, muscle wasting, anorexia, anxiety, depression, obesity (by reducing appetite, increasing metabolic rate, reducing fat intake or reducing carbohydrate craving), diabetes mellitus (by enhancing glucose tolerance, decreasing insulin resistance), hypertension, hyperlipidemia, osteoarthritis, cancer, gall bladder disease, sleep apnea, depression, anxiety, compulsion, neuroses, insomnia/sleep disorder, substance abuse, pain, male and female sexual dysfunction (including impotence, loss of libido and erectile dysfunction), fever, inflammation, immune-modulation, rheumatoid arthritis, skin tanning, acne and other skin disorders, neuroprotective and cognitive and memory enhancement, including the treatment of Alzheimer's disease.
- Some compounds encompassed by formula (I) show highly selective affinity for the melanocortin-4 receptor relative to MC-1 R, MC-2R, MC-3R and MC-5R, which makes them especially useful in the prevention and treatment of cancer cachexia, muscle wasting, anorexia, anxiety, depression, obesity, as well as male and/or female sexual dysfunction, including erectile dysfunction.
- “Male sexual dysfunction” includes impotence, loss of libido and erectile dysfunction.
- Female sexual dysfunction can be seen as resulting from multiple components, including dysfunction in desire, sexual arousal, sexual receptivity and orgasm.
- Any suitable route of administration may be employed for providing a mammal, especially a human with an effective dosage of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols and the like.
- the compounds of formula (I) are administered orally or topically.
- the effective dosage of active ingredient employed may vary depending on the particular compound employed, the mode of administration, the condition being treated and the severity of the condition being treated. Such dosage may be ascertained readily by a person skilled in the art.
- the compounds of the present invention are administered at a daily dosage of from about 0.001 milligram to about 100 milligrams per kilogram of body weight, preferably given in a single dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dose will generally be from about 0.07 milligrams to about 3500 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of the present invention are administered at a daily dosage of from about 0.001 milligram to about 100 milligrams per kilogram of body weight, preferably given in a single dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dose will generally be from about 0.07 milligrams to about 3500 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of the present invention are administered at a daily dosage of from about 0.001 milligram to about 100 milligram per kilogram of animal body weight, preferably given in a single dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dose will generally be from about 0.07 milligrams to about 3500 milligrams. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds of the present invention are given in a dose range of 0.001 milligram to about 100 milligram per kilogram of body weight, preferably as a single dose orally, or as a nasal spray.
- the compound of formula (I) is preferably formulated into a dosage form prior to administration. Accordingly, the present invention also includes a pharmaceutical composition comprising a compound of formula (I) and a suitable pharmaceutical carrier.
- the active ingredient (a compound of formula (I)) is usually mixed with a carrier or diluted by a carrier or enclosed within a carrier, which may be in the form of a capsule, sachet, paper or other container.
- a carrier which may be in the form of a capsule, sachet, paper or other container.
- the carrier serves as a diluent, it may be a solid, semisolid, or liquid material which acts as a vehicle, excipient or medium for the active ingredient.
- compositions can be in the form of tablets, pills, powders, lozenges, sachets, cachets, elixirs, suspensions, emulsions, solutions, syrups, aerosol (as a solid or in a liquid medium), soft and hard gelatin capsules, suppositories, sterile injectable solutions or sterile packaged powders.
- Suitable carriers, excipients and diluents include lactose, dextrose, sucrose, sorbitol, mannitol, starches, gum acacia, calcium phosphate, alginates, tragacanth, gelatin, calcium silicate, microcrystalline cellulose, polyvinylpyrrolidone, cellulose, water syrup, methyl cellulose, methyl and propylhydroxybenzoates, talc, magnesium stearate and mineral oil.
- the formulations can additionally include lubricating agents, wetting agents, emulsifying and suspending agents, preserving agents, sweetening agents or flavoring agents.
- the compositions of the invention may be formulated so as to provide quick, sustained or delayed release of the active ingredient after administration to the patient.
- the preparation of the compounds of the present invention may be carried out via sequential or convergent synthetic routes.
- the skilled artisan will recognize that, in general, the three moieties of a compound of formula (I) are connected via amide bonds. The skilled artisan can, therefore, readily envision numerous routes and methods of connecting the three moieties via standard peptide coupling reaction conditions.
- standard peptide coupling reaction conditions means coupling a carboxylic acid with an amine using an acid activating agent such as EDC, dicyclohexylcarbodiimide or benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate, in an inert solvent such as DCM, in the presence of a catalyst such as HOBt.
- an acid activating agent such as EDC, dicyclohexylcarbodiimide or benzotriazol-1-yloxytris(dimethylamino)-phosphonium hexafluorophosphate
- DCM inert solvent
- HOBt a catalyst
- protective groups for amine and carboxylic acids to facilitate the desired reaction and minimize undesired reactions are well documented. Conditions required to remove protecting groups which may be present can be found in Greene et al., Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, NY 1991.
- Protecting groups like Z, Boc or Fmoc are used extensively in the synthesis, and their removal conditions are well known to those skilled in the art.
- removal of Z groups can be achieved by catalytic hydrogenation with hydrogen in the presence of a noble metal or its oxide, such as palladium on activated carbon in a protic solvent such as ethanol.
- a protic solvent such as ethanol.
- removal of Z can also be achieved by treatment with a solution of hydrogen bromide in acetic acid or by treatment with a mixture of TFA and dimethylsulfide.
- Removal of Boc protecting groups is carried out in a solvent, such as methylene chloride, methanol or ethyl acetate, with a strong acid, such as TFA, HCl or hydrogen chloride gas.
- the compounds of formula (I), when existing as a diastereomeric mixture, may be separated into diastereomeric pairs of enantiomers by fractional crystallization from a suitable solvent such as methanol, ethyl acetate or a mixture thereof.
- a suitable solvent such as methanol, ethyl acetate or a mixture thereof.
- the pair of enantiomers, thus obtained, may be separated into individual stereoisomers by conventional means using an optically active acid as a resolving agent.
- any enantiomer of a compound of formula (I) may be obtained by stereospecific synthesis using optically pure starting materials or reagents of known configuration.
- the compounds of formula (I) of the present invention can be prepared according to the procedures of the following schemes and examples using appropriate materials and are further exemplified by the following specific examples. Moreover, by utilizing the procedures described herein, in conjunction with ordinary skills in the art, additional compounds of the present invention can be readily prepared.
- the compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention.
- the examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the instant compounds are generally isolated in the form of their pharmaceutically acceptable salts, such as those described previously.
- the free amine bases corresponding to the isolated salts can be generated by neutralization with a suitable base, such as aqueous sodium hydrogencarbonate, sodium carbonate, sodium hydroxide or potassium hydroxide, and extraction of the liberated amine free base into an organic solvent, followed by evaporation.
- a suitable base such as aqueous sodium hydrogencarbonate, sodium carbonate, sodium hydroxide or potassium hydroxide
- the amine free base, isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent, followed by addition of the appropriate acid and subsequent evaporation, precipitation or crystallization. All temperatures are degrees Celsius.
- Mass spectra (MS) were measured by electron-spray ion-mass spectroscopy.
- an appropriate "A moiety” e.g., 1-(2-piperazin-1-yl-benzyl)- pyrrolidin-2-one
- B moiety e.g., L-Boc-p-CI-Phe-OH
- the coupled AB compound is then coupled to an appropriate "C moiety", followed by deprotection of Boc group and salt formation.
- C moiety is not protected with Boc group, the final compound can be obtained without the deprotection step.
- EDC/HOAt for coupling of A with Boc-B-OH, EDC/HOAt, EDC/HOBt or DCC/HOBt can be used.
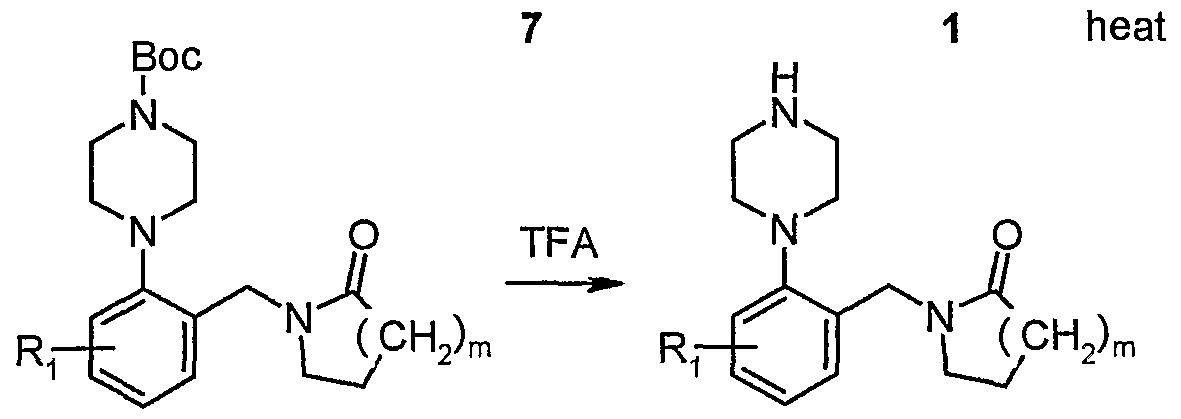
- the starting material of Boc-protected piperazine or piperidine (A moiety) can be deprotected in the presence of TFA/CH 2 CI 2 , HCI/EtOAc, HCl/dioxane or HCl in MeOH/Et 2 O, with or without a cation scavenger, such as dimethyl sulfide (DMS), before being subjected to the coupling procedure. It can be free-based before being subjected to the coupling procedure or, in some cases, used as the salt.
- a cation scavenger such as dimethyl sulfide (DMS)
- a suitable solvent such as CH 2 CI 2 , DMF, THF or a mixture of the above solvents, can be used for the coupling procedure.
- a suitable base includes triethylamine (TEA), diisopropylethylamine (DIPEA), N-methylmorpholine (NMM), collidine and 2,6-lutidine.
- TAA triethylamine
- DIPEA diisopropylethylamine
- NMM N-methylmorpholine
- collidine 2,6-lutidine.
- a base may not be needed when EDC/HOBt is used.
- the reaction mixture can be diluted with an appropriate organic solvent, such as EtOAc, CH 2 CI or Et 2 O, which is then washed with aqueous solutions, such as water, HCl, NaHSO 4 , bicarbonate, NaH 2 PO 4 , phosphate buffer (pH 7), brine or any combination thereof.
- an appropriate organic solvent such as EtOAc, CH 2 CI or Et 2 O
- aqueous solutions such as water, HCl, NaHSO 4 , bicarbonate, NaH 2 PO 4 , phosphate buffer (pH 7), brine or any combination thereof.
- the reaction mixture can be concentrated and then be partitioned between an appropriate organic solvent and an aqueous solution.
- the reaction mixture can be concentrated and subjected to chromatography without aqueous workup.
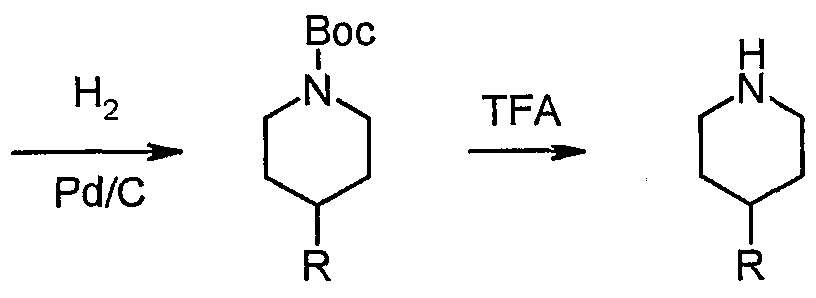
- Protecting groups such as Boc, Z, Fmoc or CF 3 CO, can be deprotected in the presence of H 2 /Pd-C, TFA/DCM, HCI/EtOAc, HCl/dioxane, HCl in MeOH/Et 2 O, NH 3 /MeOH or TBAF with or without a cation scavenger, such as thioanisole, ethane thiol or dimethyl sulfide (DMS).
- the deprotected amines can be used as the resulting salt or are free-based by dissolving in DCM and washing with aqueous bicarbonate or aqueous NaOH.
- the deprotected amines can also be free-based by ion exchange chromatography.
- the "A moieties” of the present invention may be prepared from commercially available starting materials via known chemical transformations.
- the preparation of "A moiety” of the compound of the present invention is illustrated in the reaction scheme below.
- the "A moiety" of the compounds of the present invention can be prepared by coupling halo-substituted aryl 2 (X-R) with 1 -Boc-piperazine 1 in the presence of tri(dibenzylideneacetone) dipalladium (Pd 2 (dba) 3 ), 2,2'-Bis(diphenylphosphino)- 1,1'-binaphtyl (BINAP), and sodium-tert-butoxide (NaOtBu) or cesium carbonate (Cs 2 CO 3 ) in an organic solvent, such as toluene, at a suitable temperature. More detailed examples of "A moiety" preparation are described below.
- the "A moiety" of the compounds ' of the present invention can be prepared by reacting various methyl benzenes 4 with NBS in the presence of a radical starter, such as Bz 2 O 2 , followed by reaction with diethyl phosphite in the presence of a base, such as DIPEA, to give benzylbromides 5, which can then be used to alkylate lactames like 6, in the presence of an appropriate base, such as KF/alumina.
- a radical starter such as Bz 2 O 2
- DIPEA diethyl phosphite
- the substituted bromobenzenes can then be subjected to Buchwald conditions, followed by deprotection using an appropriate reactant, such as TFA.
- carboxylic acids 10 can be reduced to the corresponding alcohols 11 using an appropriate reagent such as BH 3 -THF, which are subsequently transferred to the corresponding alkyl bromides 12 with reagents such as CBr 4 or PPh 3 .
- the alkyl bromides can then be used to alkylate lactames like 6 in the presence of an appropriate base such as KF/alumina.
- the substituted bromobenzenes can then be subjected to Buchwald conditions followed by deprotection using an appropriate reactant such as TFA.
- Reaction Scheme 5 Suzuki Coupling
- Br-R is compound 7 or 13
- 1-(2(H)-pyridine-carboxylic acid-3,6-dihydro-4-(4,4,5,5- tetramethyl-1,3,2-dioxaborolan-2-yl)-1,1 -dimethyl ethyl ester 16 can be reacted with haloaromates such as 7 or 13 in the presence of a base such as K 2 CO 3 and a catalyst such as dichloro(1 ,1'-bis(diphenylphosphino)- ferrocene)palladium(ll) DCM adduct in an organic solvent such as DMF at a suitable temperature.
- the tetrahydropyridines can be hydrogenated in the presence of a catalyst such as Pd/C to yield the protected piperidines 19 which can subsequently be deprotected with a reagent such as TFA to yield piperidines 20.
- ethyl 3-bromo-4-oxochromene-2-carboxylate 21 can be reacted with amines with or without a base such as K 2 CO 3 in an appropriate solvent such as MeCN to form products 22 which are subsequently treated with a reagent such as HBr/HOAc to form carboxylic acids 23.
- R 8 is hydrogen
- the free ⁇ amine can be protected with a reagent such as Boc 2 O in the presence of TEA and DMAP in an appropriate solvent.
- substituted phenols 27 can be reacted with triethylamine followed by dimethyl acetylendicarboxylate in diethyl ether to yield compounds 28 (Aust. J. Chem. 1995, 48, 677-686). Saponification of the latter with aqueous sodium hydroxide leads to acids 29 which are subsequently cyclized to the chromone-2-carboxylic acids -30 using concentrated sulfuric acid in acetyl chloride.
- 2'-hydroxyacetophenones 31 can be reacted with diethyl oxalate 32 in the presence of a base such as sodium methoxide in an appropriate solvent such as methanol or benzene followed by treatment with an acid such as hydrochloric acid to yield chromone-2-carboxylic acid esters 33 (J. Indian Chem. Soc. 1986, 63, 600-602).
- the esters can be cleaved using basic conditions such as sodium bicarbonate in water or acidic conditions such as polyphosphoric acid at an appropriate temperature to the corresponding acids 30.
- methoxy-substituted chromone-2-carboxylic acids can be demethylated with reagents such as hydroiodic acid in an appropriate solvent such as glacial acetic acid to yield the corresponding hydroxy-substituted chromone-2-carboxylic acids.
- reagents such as hydroiodic acid in an appropriate solvent such as glacial acetic acid
- 5,7-Dihydroxychromone-2-carboxylic acid was prepared as described in the literature (OPPI Briefs 1991 , 23, 390-392).
- Example 1 is provided to illustrate the invention and are not limiting the scope of the invention in any manner.
- Example 1 is provided to illustrate the invention and are not limiting the scope of the invention in any manner.
- Example 2 The following examples can be prepared in a similar way:
- R f 0.21 (ethyl acetate); Mp. 121-127 °C.
- R f 0.25 (ethyl acetate); Mp. 115-130.
- R f 0.25 (ethyl acetate); Mp. 115-130.
- R f 0.10 (ethyl acetate); Mp. 125-140 °C.
- R f 0.10 (ethyl acetate); Mp. 125-140 °C.
- R f 0.09 (ethyl acetate); Mp. 120-125.
- R f 0.09 (ethyl acetate); Mp. 120-125.
- R f 0.05 (ethyl acetate); Mp. 165-170 °C.
- R f 0.05 (ethyl acetate); Mp. 165-170 °C.
- R f 0.16 (ethyl acetate).
- R f 0.16 (ethyl acetate).
- R f 0.10 (ethyl acetate).
- R f 0.12 (ethyl acetate).
- R f 0.12 (ethyl acetate).
- Boc-piperazine (895 mg), intermediate 1a) (1004 mg), Pd 2 (dba) 3 (235 mg), BINAP (442 mg) and cesium carbonate (3 g) were mixed together in toluene (20 ml). The mixture was degassed and heated to 100°C for 3 d. The mixture was diluted with ether (100 ml) and filtered over Celite. The filtrate was concentrated and then subjected to chromatography on silica gel to yield the title compound.
- R f 0.70 (ethyl acetate); Mp.125-130 °C.
- R f 0.10 (ethyl acetate); Mp.150-155 °C.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Reproductive Health (AREA)
- Endocrinology (AREA)
- Neurology (AREA)
- Gynecology & Obstetrics (AREA)
- Obesity (AREA)
- Neurosurgery (AREA)
- Pain & Pain Management (AREA)
- Biomedical Technology (AREA)
- Hematology (AREA)
- Psychiatry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Nutrition Science (AREA)
- Pregnancy & Childbirth (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description












































































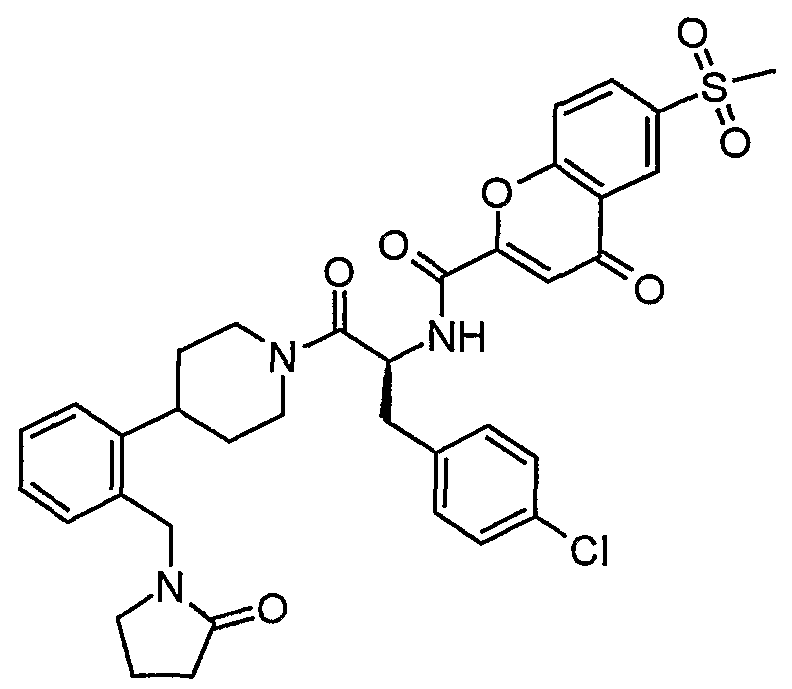

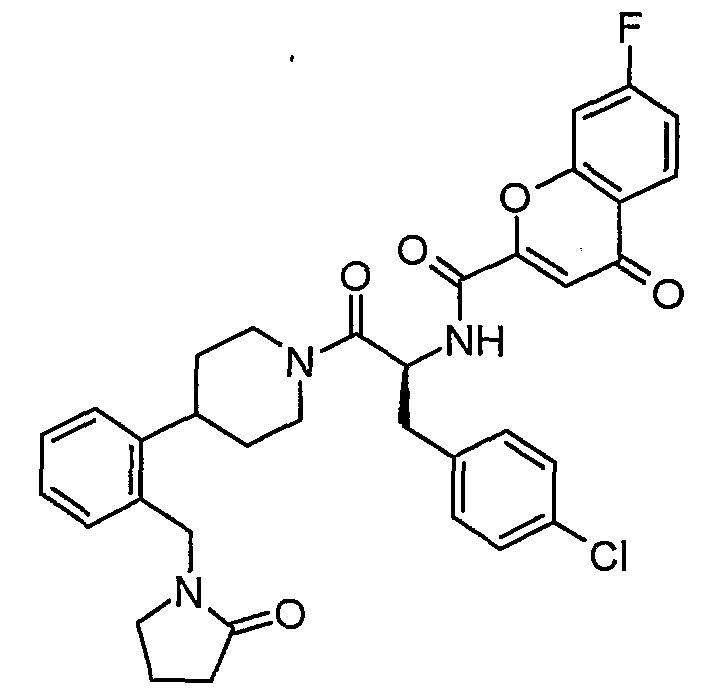









































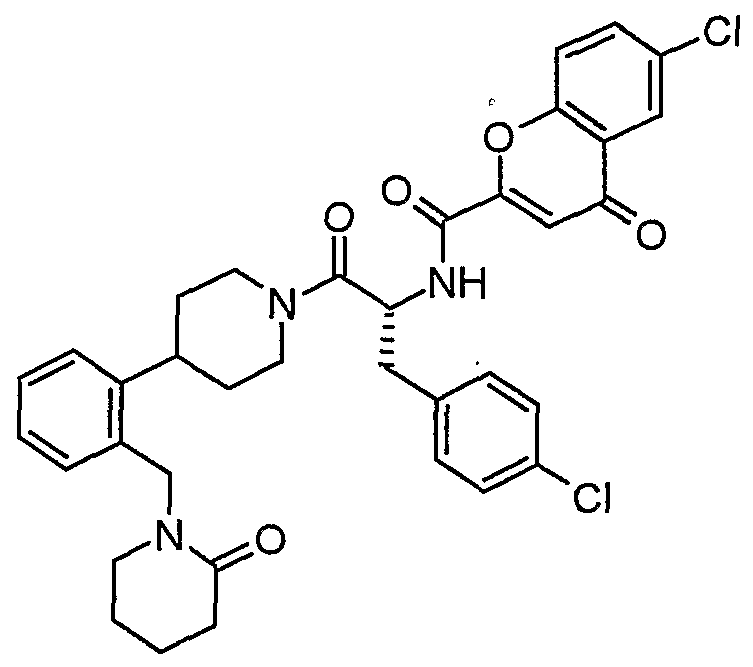































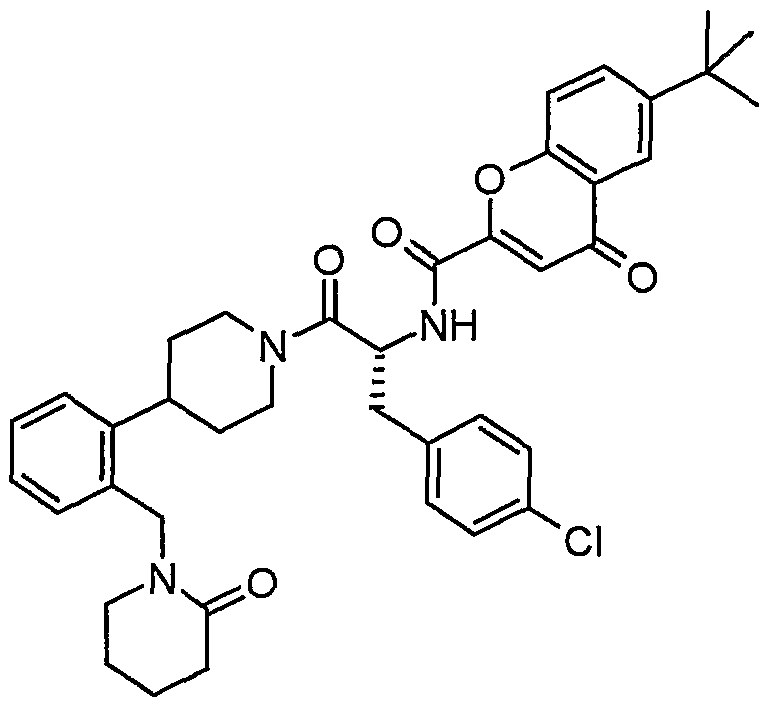

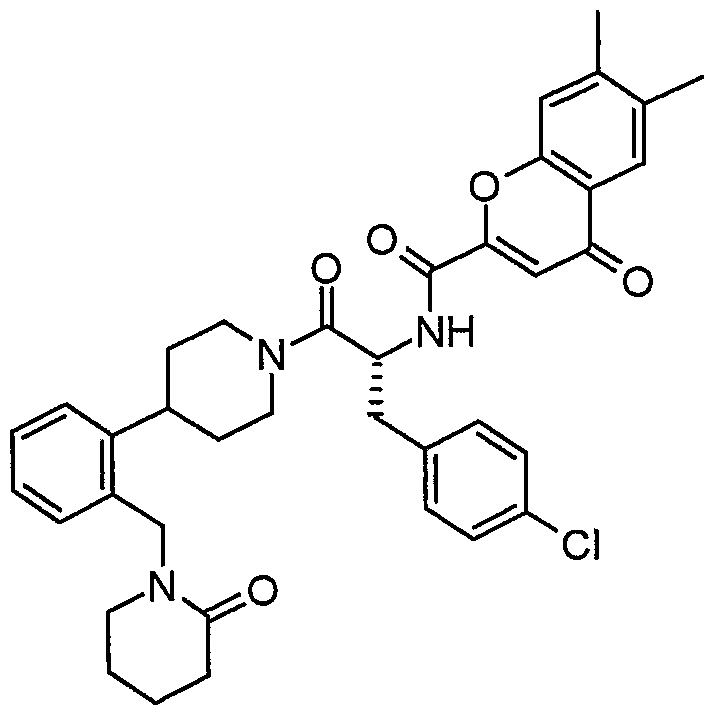























































































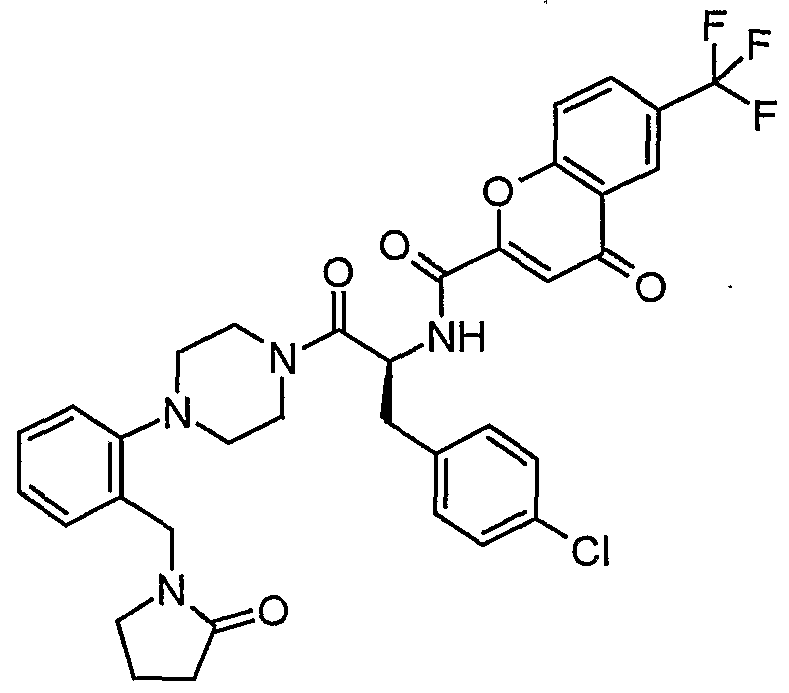









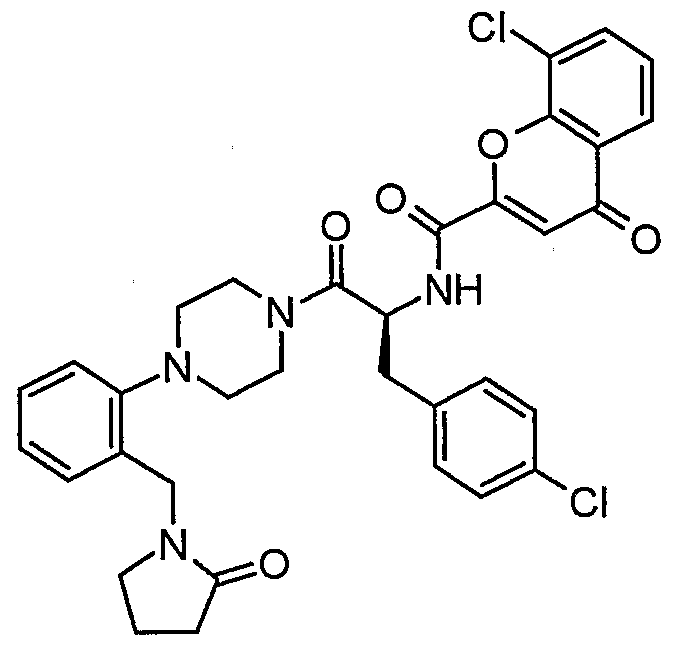









































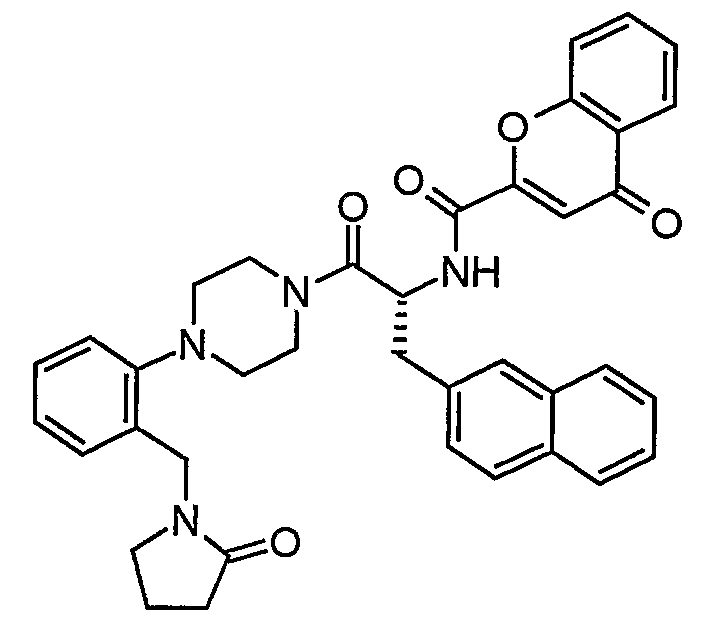

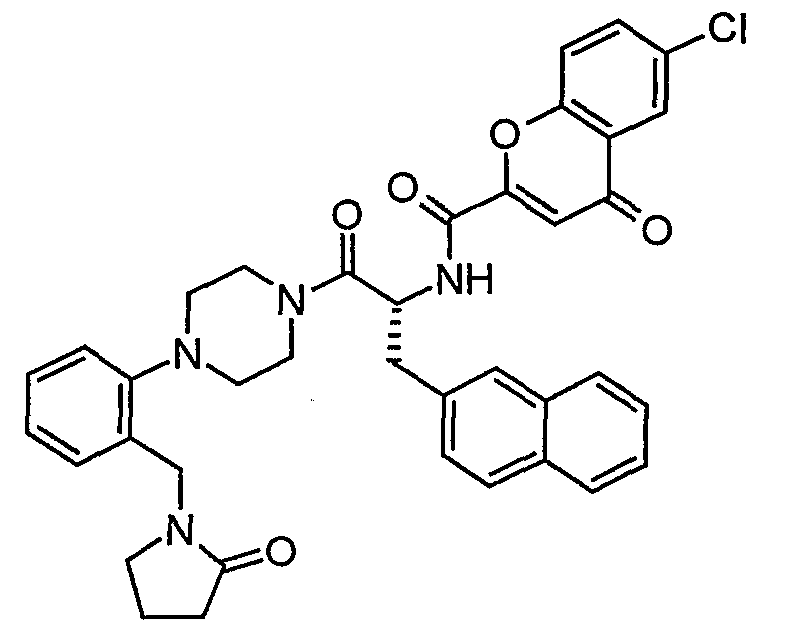





















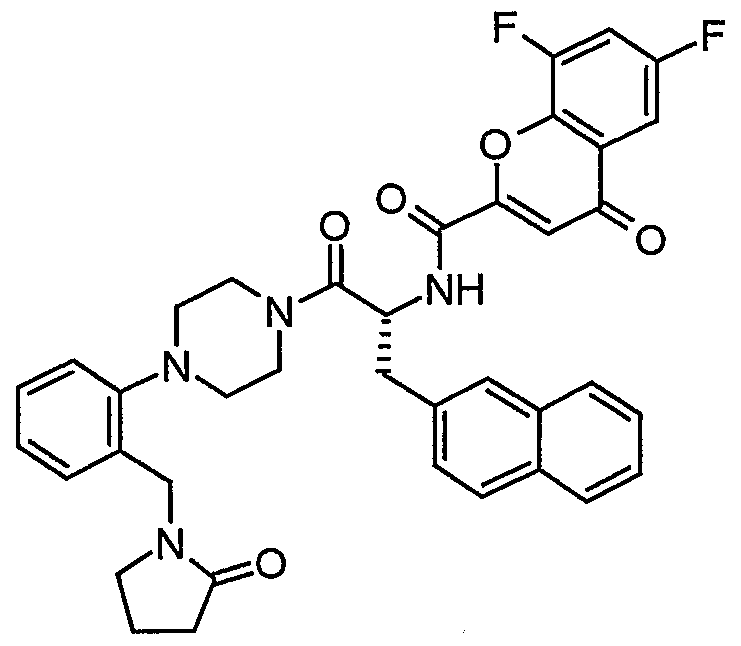












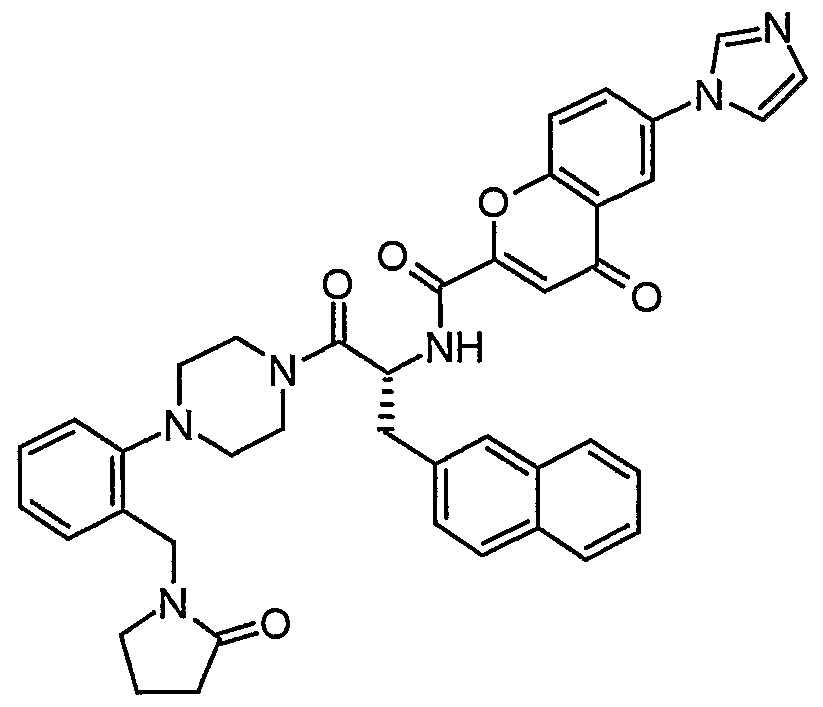































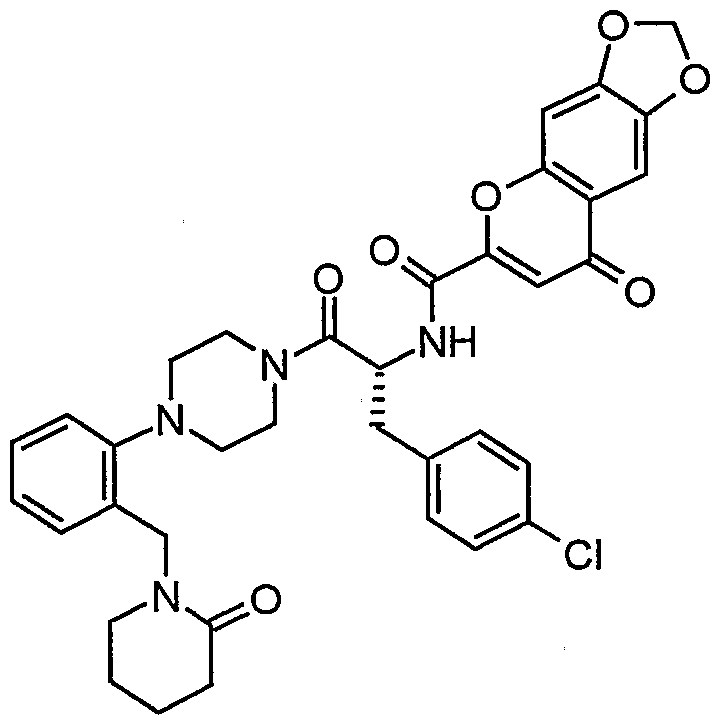

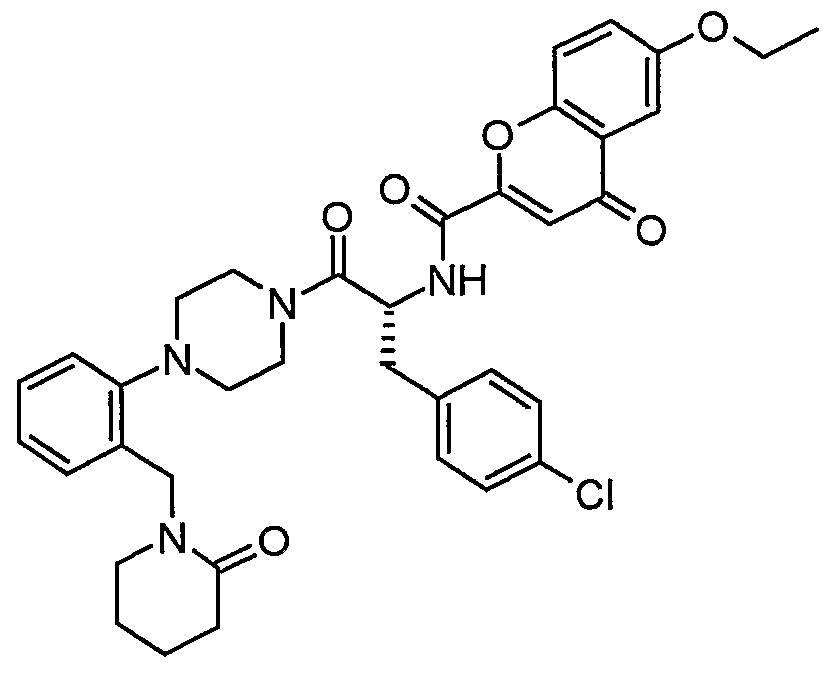










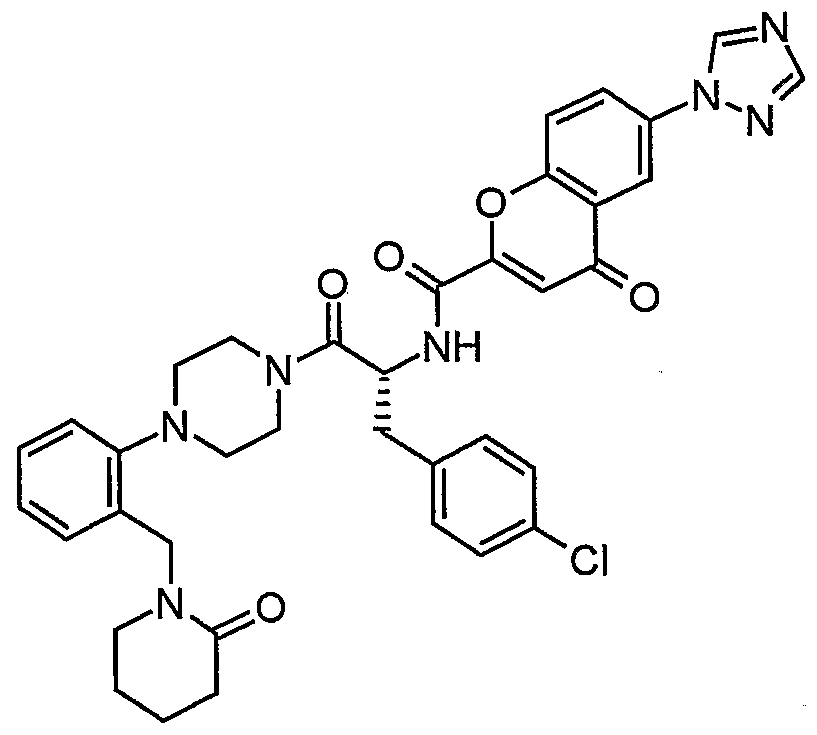
















Claims





Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/550,222 US20060276485A1 (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| CA002519444A CA2519444A1 (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| JP2006504755A JP2006520363A (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| AU2004222096A AU2004222096B2 (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
| EP04721877A EP1606281A1 (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03006253.3 | 2003-03-20 | ||
| EP03006253A EP1460070B1 (en) | 2003-03-20 | 2003-03-20 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004083199A1 true WO2004083199A1 (en) | 2004-09-30 |
Family
ID=32798839
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/002908 Ceased WO2004083199A1 (en) | 2003-03-20 | 2004-03-19 | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20060276485A1 (en) |
| EP (2) | EP1460070B1 (en) |
| JP (1) | JP2006520363A (en) |
| AT (1) | ATE362368T1 (en) |
| AU (1) | AU2004222096B2 (en) |
| CA (1) | CA2519444A1 (en) |
| DE (1) | DE60313844T2 (en) |
| ES (1) | ES2286345T3 (en) |
| WO (1) | WO2004083199A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009510073A (en) * | 2005-09-27 | 2009-03-12 | ノバルティス アクチエンゲゼルシャフト | Carboxyamine compounds and methods of use thereof |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050119252A1 (en) * | 2003-10-22 | 2005-06-02 | Neurocrine Biosciences, Inc. | Ligands of melanocortin receptors and compositions and methods related thereto |
| US8084477B2 (en) * | 2007-10-31 | 2011-12-27 | Bristol-Myers Squibb Company | Alpha-(N-sulfonamido)acetamide compound as an inhibitor of beta amyloid peptide production |
| NZ585131A (en) | 2007-11-05 | 2012-10-26 | Ipsen Pharma Sas | Use melanocortins to treat insulin sensitivity |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003009847A1 (en) * | 2001-07-25 | 2003-02-06 | Amgem, Inc. | Substituted piperidines as modulators of the melanocortin receptor |
| WO2003009850A1 (en) * | 2001-07-25 | 2003-02-06 | Amgen Inc. | Substituted piperazines as modulators of the melanocortin receptor |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2676053B1 (en) * | 1991-05-03 | 1993-08-27 | Sanofi Elf | NOVEL DIALKYLENEPIPERIDINO COMPOUNDS AND THEIR ENANTIOMERS, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| ATE216580T1 (en) * | 1993-07-16 | 2002-05-15 | Merck & Co Inc | BENZOXAZINONE AND BENZOPYRIMIDINONE PIPERIDINYL COMPOUNDS AS TOCOLYTIC OXYTOCIN RECEPTOR ANTAGONISTS |
| US5968929A (en) * | 1996-10-30 | 1999-10-19 | Schering Corporation | Piperazino derivatives as neurokinin antagonists |
| JP2004532838A (en) * | 2001-03-02 | 2004-10-28 | ブリストル−マイヤーズ スクイブ カンパニー | Compounds useful as melanocortin receptor modulators and pharmaceutical compositions containing them |
-
2003
- 2003-03-20 EP EP03006253A patent/EP1460070B1/en not_active Expired - Lifetime
- 2003-03-20 ES ES03006253T patent/ES2286345T3/en not_active Expired - Lifetime
- 2003-03-20 DE DE60313844T patent/DE60313844T2/en not_active Expired - Fee Related
- 2003-03-20 AT AT03006253T patent/ATE362368T1/en not_active IP Right Cessation
-
2004
- 2004-03-19 EP EP04721877A patent/EP1606281A1/en not_active Withdrawn
- 2004-03-19 JP JP2006504755A patent/JP2006520363A/en not_active Withdrawn
- 2004-03-19 US US10/550,222 patent/US20060276485A1/en not_active Abandoned
- 2004-03-19 CA CA002519444A patent/CA2519444A1/en not_active Abandoned
- 2004-03-19 WO PCT/EP2004/002908 patent/WO2004083199A1/en not_active Ceased
- 2004-03-19 AU AU2004222096A patent/AU2004222096B2/en not_active Expired - Fee Related
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003009847A1 (en) * | 2001-07-25 | 2003-02-06 | Amgem, Inc. | Substituted piperidines as modulators of the melanocortin receptor |
| WO2003009850A1 (en) * | 2001-07-25 | 2003-02-06 | Amgen Inc. | Substituted piperazines as modulators of the melanocortin receptor |
Non-Patent Citations (1)
| Title |
|---|
| RICHARDSON, T. I. ET AL.: "Synthesis and Structure-Activity Relationships of Novel Arylpiperazines as Potent and Selective Agonists of the Melanocortin Subtype-4 Receptor", J. MED. CHEM., vol. 47, 2004, pages 744 - 755, XP002282648 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2009510073A (en) * | 2005-09-27 | 2009-03-12 | ノバルティス アクチエンゲゼルシャフト | Carboxyamine compounds and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1460070A1 (en) | 2004-09-22 |
| DE60313844T2 (en) | 2008-01-31 |
| CA2519444A1 (en) | 2004-09-30 |
| AU2004222096B2 (en) | 2007-07-12 |
| JP2006520363A (en) | 2006-09-07 |
| EP1606281A1 (en) | 2005-12-21 |
| AU2004222096A1 (en) | 2004-09-30 |
| DE60313844D1 (en) | 2007-06-28 |
| ES2286345T3 (en) | 2007-12-01 |
| EP1460070B1 (en) | 2007-05-16 |
| US20060276485A1 (en) | 2006-12-07 |
| ATE362368T1 (en) | 2007-06-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2004222097B2 (en) | Substituted cyclohexyl and piperidinyl derivatives as melanocortin-4 receptor modulators | |
| EP2176250B1 (en) | Substituted heteroarylpiperidine derivatives as melanocortin-4 receptor modulators | |
| AU2004222090B2 (en) | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators | |
| WO2007115798A1 (en) | Substituted phenylpiperidine derivatives as melanocortin-4 receptor modulators | |
| EP1538159A1 (en) | Substituted N-benzyl-lactam derivatives as melanocortin-4 receptor agonists | |
| WO2010081666A1 (en) | Substituted heteroarylpiperidine derivatives as melanocortin-4 receptor modulators | |
| EP1603912B1 (en) | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators | |
| AU2004222096B2 (en) | Substituted piperidine and piperazine derivatives as melanocortin-4 receptor modulators | |
| WO2009015867A1 (en) | Substituted aryl or heteroarylpiperdine derivatives as melanocortin-4 receptor modulators | |
| EP1826201A1 (en) | Substituted phenylpiperidine derivatives as melanocortin-4 receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004222096 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2519444 Country of ref document: CA Ref document number: 2004721877 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006504755 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2004222096 Country of ref document: AU Date of ref document: 20040319 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004222096 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004721877 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006276485 Country of ref document: US Ref document number: 10550222 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10550222 Country of ref document: US |