WO2004096870A1 - リビングラジカルポリマーの製造方法及びポリマー - Google Patents
リビングラジカルポリマーの製造方法及びポリマー Download PDFInfo
- Publication number
- WO2004096870A1 WO2004096870A1 PCT/JP2004/005989 JP2004005989W WO2004096870A1 WO 2004096870 A1 WO2004096870 A1 WO 2004096870A1 JP 2004005989 W JP2004005989 W JP 2004005989W WO 2004096870 A1 WO2004096870 A1 WO 2004096870A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- group
- reaction
- formula
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/44—Polymerisation in the presence of compounding ingredients, e.g. plasticisers, dyestuffs, fillers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2/00—Processes of polymerisation
- C08F2/38—Polymerisation using regulators, e.g. chain terminating agents, e.g. telomerisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F10/00—Homopolymers and copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F12/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an aromatic carbocyclic ring
- C08F12/02—Monomers containing only one unsaturated aliphatic radical
- C08F12/04—Monomers containing only one unsaturated aliphatic radical containing one ring
- C08F12/06—Hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1804—C4-(meth)acrylate, e.g. butyl (meth)acrylate, isobutyl (meth)acrylate or tert-butyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1806—C6-(meth)acrylate, e.g. (cyclo)hexyl (meth)acrylate or phenyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
- C08F220/12—Esters of monohydric alcohols or phenols
- C08F220/16—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms
- C08F220/18—Esters of monohydric alcohols or phenols of phenols or of alcohols containing two or more carbon atoms with acrylic or methacrylic acids
- C08F220/1811—C10or C11-(Meth)acrylate, e.g. isodecyl (meth)acrylate, isobornyl (meth)acrylate or 2-naphthyl (meth)acrylate
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
Definitions
- the present invention relates to a method for producing a living radical polymer and a method for producing the same.
- Non-Patent Document 1 a method of obtaining polystyrene by polymerizing styrene using AIBN and diphenylditelluride (DPDTe) is known (for example, see Non-Patent Document 1).
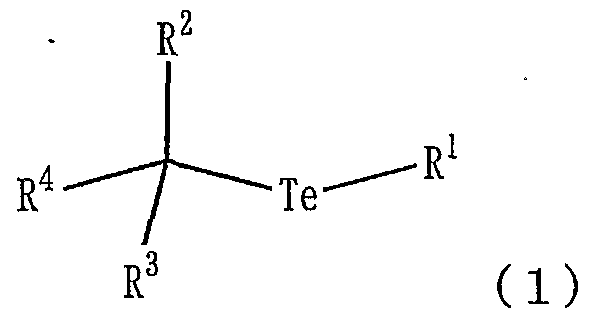
- An object of the present invention is to use an organic tellurium compound represented by the formula (1), an azo-based polymerization initiator, and a ditelluride compound represented by the formula (2) to produce not only styrene but also other (meth) compounds.
- R 1 represents a C ⁇ Cs alkyl group, an aryl group, a substituted aryl group or an aromatic heterocyclic group.
- R 2 and R 3 represent a hydrogen atom or an alkyl group of ⁇ C s.
- R 4 represents an aryl group, a substituted aryl group, an aromatic heterocyclic group, an acyl group, an oxycarbonyl group or a cyano group.
- the living radical polymer of the present invention is obtained by polymerizing a vinyl monomer using an azo-based polymerization initiator in the presence of an organic tellurium compound represented by the formula (1) and a compound represented by the formula (2). It is manufactured by
- R 1 represents a C i Cs alkyl group, an aryl group, a substituted aryl group or an aromatic heterocyclic group.
- R 2 and R 3 represent a hydrogen atom or a C i C s alkyl group.
- R 4 represents an aryl group, a substituted aryl group, an aromatic heterocyclic group, an acyl group, an oxycarbonyl group or a cyano group.
- the organic tellurium compound represented by the formula (1) used in the present invention is as follows.
- R 1 represents an alkyl group of ( ⁇ to ( 8) , an aryl group, a substituted aryl group or an aromatic heterocyclic group.
- R 2 and R 3 each represent a hydrogen atom or a CCS alkyl group.
- R 4 represents an aryl group, a substituted aryl group, an aromatic heterocyclic group, an acyl group, an oxycarbonyl group or a cyano group.
- R 1 The group represented by R 1 is specifically as follows.
- the alkyl group of C E ⁇ C 8, a methyl group, Echiru group, n- propyl Isopropyl, cyclopropyl, n-butyl, sec-butyl, tert-butyl, cyclobutyl, n-pentyl, n-hexyl, n-heptyl, n-octyl
- Examples thereof include a linear, branched or cyclic alkyl group having 1 to 8 carbon atoms such as a group.
- the preferred alkyl group is a linear or branched alkyl group having 1 to 4 carbon atoms, more preferably a methyl group, an ethyl group or an n-butyl group.
- Aryl groups such as phenyl and naphthyl groups; substituted aryl groups such as phenyl groups having a substituent; naphthyl groups having a substituent; and pyridyl groups as aromatic heterocyclic groups.
- substituent of Ariru group having the above substituent for example, a halogen atom, a hydroxyl group, an alkoxy group, ⁇ amino group, a nitro group, Shiano group, Karuponiru containing group represented by _ COR a
- aryl groups are a phenyl group and a trifluoromethyl-substituted phenyl group. These substituents may be substituted with one or two substituents, preferably at the para or ortho position.
- R 2 and R 3 are specifically as follows.
- the alkyl group of C ⁇ 8 may be mentioned the same alkyl groups as indicated above R 1.
- Each group represented by R 4 is specifically as follows.
- the Ariru group, a substituted Ariru group, an aromatic heterocyclic group can be exemplified the same groups as those indicated above R 1.
- the Ashiru group include a formyl group, Asechiru group, Puchiriru group, Benzoi group, a Ashiru group of C ⁇ C 8, such as toluoyl.
- Preferable oxycarbonyl groups are a methoxycarbonyl group and an X-methoxycarbonyl group.
- Ariru group a substituted Ariru group, it is Okishi force Ruponiru group or Shiano group.
- a preferred aryl group is a phenyl group.
- Preferred substituted aryl groups include halogen-substituted phenyl groups and trifluoromethyl-substituted phenyl groups.
- substituents are preferably substituted.
- substituents are preferably substituted.
- substituents are preferably substituted.
- the para position or the ortho position is preferable, and in the case of two substitution, the meta position is preferable.
- Preferred oxycarbonyl groups are methoxycarbonyl and ethoxycarbonyl.
- R 1 represents a C 4 alkyl group
- R 2 and R 3 represent a hydrogen atom or an alkyl group of CC
- R 4 represents an aryl group
- C is R 2 and R 3 each represent a hydrogen atom or a C alkyl group
- R 4 is preferably a phenyl group, a substituted phenyl group, a methoxycarbonyl group, or a ethoxycarbonyl group.
- the organic tellurium compound represented by the formula (1) is specifically as follows.
- Examples of organic tellurium compounds include (methyl teraniline methyl) benzene, (1-methyl teraniline methyl) benzene, (2-methyl teraniline methyl) benzene, and 1-chloro-4- (methyl teraniline methyl) benzene.
- Benzene 1-hydroxy-41- (methyl-teranyl-methyl) benzene, 1-methoxy-141- (methyl-teranyl-methyl) benzene, 1-amino-41- (methyl-teranyl-methyl) benzene, 1-212-low 41- (methylteranyl-methyl) Benzene, 1-Cyanol 4- (Methyl Teranilomethyl) Benzene, 1-Methylcarbonyl 4- (Methylteranilomethyl) Benzene, 1-Fenylcarponyl-4-1 (Methylteranilomethyl) Benzene, 1-Methoxycarbonyl — 4 1 (Methyl Teranirumethyl) Benzene, 1 1 Phenoxycarbone 4 1 (Methyl Teranilumethyl) Benzene, 1 1 Sulfonyl-4 1 (Methyl Teranirumethyl) Benzene, 1 _ Trifluoromethyl-4 One (Methyl Ter
- methylteranyl, 1-methylteranyl and It also includes all compounds changed to luteranyl, 1-ethylterranyl, 2-ethyltilanyl, butylteranyl, 1-butylteranyl, and 2-butylteranyl.
- benzene (1 methyl terrenyl methyl) benzene, (1 methyl terraryl methyl) benzene, (2 1 methyl terraryl methyl) benzene, 1 chloro-41 (1 1 methyl terraryl methyl) benzene, 1-trifluoromethyl — 4-(1 _Methylteranirubetyl) benzene, 2 Methylterarinyl 2 Methyl methylpropionate, 2 Methylteranyl 1 2 Methylethyl propionate, [Ethyl-2-methyl-2-methyl teraniline loop pionate], 2- (n-butylteranyl) -1-ethyl methyl propionate [ethyl-2-methyl-2-n-butylteranyl-propionate], 1- (1-methylteranyl-ethyl) -3,5-bis-trifluoro Methylbenzene, 1,2,3,4,5_Penyu Full Oro
- the compound represented by the above formula (3) is specifically as follows.
- R 2 , R 3 and R 4 are as described above.
- Examples of the group represented by X include a halogen atom such as fluorine, chlorine, bromine or iodine. Preferably, chlorine and bromine are good.
- Specific compounds include benzyl chloride and benzyl bromide.
- the compound represented by the above formula (4) is specifically as follows.
- M represents an alkali metal, an alkaline earth metal or a copper atom.
- m is 1, when M is an alkaline earth metal, m is 2, when M is a copper atom, m is 1 or 2.
- R 1 The group represented by R 1 is as described above.
- M alkali metals such as lithium, sodium and potassium, alkaline earth metals such as magnesium and calcium, and copper.
- lithium is good.
- compound (4) may be Mg (R 1 ) 2 or a compound represented by R 1 Mg X (X is a halogen atom) (Grignard reagent). X is preferably a chlorine atom or a bromo atom.
- Specific compounds include methyllithium, ethyllithium, n-butyllithium, phenyllithium, p-chlorophenylphenyl, p-methoxyphenyllithium, p-nitrophenyllithium, and the like. .
- methyl lithium, ethyl lithium, n-butyl lithium and phenyl lithium are good.
- the above manufacturing method is specifically as follows.
- the tellurium metal is suspended in the solvent.
- Solvents that can be used include polar solvents such as N, N-dimethylformamide (DMF) and tetrahydrofuran (THF), aromatic solvents such as toluene and xylene, and aliphatic carbons such as hexane. And ethers such as hydrogen chloride and dialkyl ether.
- THF is good.
- the amount of the solvent used may be appropriately adjusted, but is usually from 1 to L: 0 m 1, preferably from 5 to 20 m 1 per 1 g of metal tellurium.
- Compound (4) is slowly dropped into the above suspension, and the mixture is stirred.
- the reaction time varies depending on the reaction temperature and pressure, but is usually 5 minutes to 24 hours, preferably 10 minutes to 2 hours.
- the reaction temperature is from ⁇ 20 ° C. to 80 ° C., preferably from 110 ° C. to 40 ° C., and more preferably from ⁇ 5 ° C. to 4.0 ° C.
- the pressure is usually normal pressure, but may be increased or decreased.
- the reaction time varies depending on the reaction temperature and pressure, but is usually 5 minutes to 24 hours, preferably 10 minutes to 2 hours.
- the reaction temperature is preferably from 20 ° C to 80 ° C, more preferably from —10 ° C to 40 ° C, and even more preferably from 15 ° C to 40 ° C.
- the pressure is usually at normal pressure, but may be increased or decreased.
- the proportions of the metal tellurium, the compound (3) and the compound (4) were as follows: 1 mol of metal tellurium, 0.5 to 1.5] 1101 of the compound (3) and compound (1). 4) should be 0.5 to 1.51111, preferably the compound (3) should be 0.8 to 1.2 mol, and the compound (4) should be 0.8 to 1.2 mol.
- the solvent is concentrated and the target compound is isolated and purified.
- the purification method can be appropriately selected depending on the compound, but usually, vacuum distillation, recrystallization purification, or the like is preferable.
- the azo polymerization initiator used in the present invention is not particularly limited as long as it is an azo polymerization initiator used in ordinary radical polymerization.
- AI BN 2,2′-azobis (isobutyronitrile)
- AMBN 2,2'-azobis (2-methylbutyronitrile)
- ADVN 2,2'-azobis (2,4-dimethyl)
- ACVA 4, 4'- Azobis (4-cyanovaleric acid)
- AC VA 1,1'-azobis (1-acetoxy-1-phenylenyl
- 2, 2'-azobis (2-methylbutylamide 1,1'-azobis ( Methyl 1-cyclohexanecarboxylate), 2,2'-azobis (4-methoxy2,4-dimethylvaleronitrile), 2,2'-azobis (2,4,
- the compound represented by the formula (2) used in the present invention is as follows.
- R 1 The group represented by R 1 is as described above.
- 1 ⁇ 1 is preferably an alkyl group of 1 to 4 or a phenyl group.
- the compound represented by the formula (2) is, specifically, dimethyl ditelluride, diethyl ditelluride, di-n-propyl ditelluride, diisopropyl ditelluride, dicyclopropyl ditelluride, di-n-butyl ditelluride , Di-sec-butyl ditelluride, di-tert-butyl ditelluride, dicyclopropyl ditelluride, diphenyl ditelluride, bis- (p-methoxyphenyl) ditelluride, bis- (p-aminophenyl) ditelluride, bi- Sue (p-ditrophenyl) ditelluride, bis- (p-cyanophenyl) ditelluride, bis- (p-sulfonylphenyl) ditelluride, dinaphthyl ditelluride, dipyridyl ditelluride and the like can be mentioned.
- dimethyl ditelluride Preferred are dimethyl ditelluride, getyl ditelluride, di-n-propyl ditelluride, di-n-butyl ditelluride, and diphenylditelluride. Especially good More preferably, dimethyl ditelluride, getyl ditelluride, di-n-propyl ditelluride, and g-n-butyl ditelluride are good.
- Specific examples of the production method include a method in which metal tellurium is reacted with a compound represented by the formula (4).
- the tellurium metal is suspended in the solvent.
- solvents include dimethylformamide (DMF), a polar solvent such as tetrahydrofuran (THF), an aromatic solvent such as toluene and xylene, an aliphatic hydrocarbon such as hexane, and a dialkyl ether. Ethers and the like.
- THF is good.
- the amount of the organic solvent used may be appropriately adjusted, but is usually 1 to 100 m 1, preferably 5 to 20 m 1 per 1 g of metal tellurium.
- the compound represented by the formula (4) is slowly dropped into the above suspension solution, and then stirred.
- the reaction time varies depending on the reaction temperature and pressure, but is usually 5 minutes to 24 hours, preferably 10 minutes to 2 hours.
- the reaction temperature is from 120 to 80 ° C, preferably from 110 to 40 ° C, and more preferably from 15 to 40.
- the pressure is usually set at normal pressure, but may be increased or decreased.
- water neutral water such as saline, alkaline water such as ammonium chloride solution, or acidic water such as hydrochloric acid
- the reaction time depends on the reaction temperature and pressure. Although different, usually 5 minutes to 24 hours, preferably 10 minutes to 2 hours.
- the reaction temperature is preferably 20 ° C. (: up to 80 ° C., preferably 0 ° (up to 40 ° C., more preferably 15 ° C. to 40 ° C.). Pressure is used, but pressure may be increased or reduced.
- the ratio of the metal tellurium and the compound of the formula (4) is 0.5 to 1.5 mol of the compound of the formula (4) per 1 mo 1 of metal tellurium, and preferably 0.8 to 1.2 mol. 1 is good.
- the solvent is concentrated and the target compound is isolated and purified.
- the purification method can be appropriately selected depending on the compound, but usually, vacuum distillation, reprecipitation purification, or the like is preferable.
- the vinyl monomer used in the present invention is not particularly limited as long as it is capable of radical polymerization. Examples thereof include methyl (meth) acrylate, (meth) ethyl acrylate, propyl (meth) acrylate, and (meth) acrylate. ) Butyl acrylate, octyl (meth) acrylate, lauryl (meth) acrylate,
- (Meth) acrylic acid (meth) acrylates such as 2-hydroxyhexyl, (meth) cyclohexyl (meth) acrylate, cyclohexyl (meth) methyl acrylate, isopolnyl (meth) acrylate, (meth) acrylic Unsaturated monomers containing cycloalkyl groups such as cyclododecyl acid, and unsaturated methoxy groups such as methyl such as (meth) acrylic acid, maleic acid, fumaric acid, itaconic acid, citraconic acid, crotonic acid, and maleic anhydride.
- Unsaturated monomers containing quaternary ammonium bases such as acryloyloxypropyl-N, N, N-trimethylammonium chloride, N-methacryloylaminoethyl-N, N, N-dimethylbenzylammonium chloride
- Epoxy group-containing unsaturated monomers such as glycidyl (meth) acrylate, styrene, -methylstyrene, 4-methylstyrene (p-methylstyrene), 2-methylstyrene (0
- (meth) acrylates unsaturated monomers containing a cycloalkyl group, aromatic unsaturated monomers (styrene-based monomers), (meth) acrylamide-based monomers, (meth) acrylonitrile, and methylvinylketone. Is good.
- Preferred (meth) acrylate monomers include methyl (meth) acrylate, ethyl (meth) acrylate, propyl (meth) acrylate, butyl (meth) acrylate, and 12- (meth) acrylic acid. Droxityl [2-hydroxyxyl (meth) acrylate]. Particularly preferred are methyl methacrylate, ethyl methacrylate, propyl methacrylate, butyl methacrylate, and 2-methacrylic acid. Loxoshetyl [2-hydroxyethyl methacrylate] is preferred.
- Preferred examples of the unsaturated monomer having an alkyl group having a mouth opening are cyclohexyl (meth) acrylate and isopornyl (meth) acrylate. Particularly preferred are hexyl methacrylate and isopropyl methacrylate.
- Preferred styrene monomers include styrene, «-methylstyrene, o-methylstyrene, p-methylstyrene, p-methoxystyrene Pt-butylstyrene, p_n-butylstyrene, p-tert-butylstyrene, p-chlorostyrene, p-styrenesulfonic acid or an alkali metal salt thereof (sodium salt, potassium salt, etc.). Particularly preferred are styrene and p-chlorostyrene.
- Preferred (meth) acrylamide monomers include N-isopropyl (meth) acrylamide. Particularly preferred is N-isopropyl methacrylamide.
- (meth) acrylic acid is a general term for “acrylic acid” and “methacrylic acid”.
- the method for producing the living radical polymer of the present invention is specifically as follows.
- a vinyl monomer, an organic tellurium compound represented by the formula (1), an azo-based polymerization initiator, and a compound represented by the formula (2) are mixed.
- the mixture is stirred.
- the reaction temperature and reaction time may be adjusted as appropriate, but usually, the mixture is stirred at 20 to 150 ° C for 1 minute to 100 hours.
- the mixture is stirred at 40 to 100 for 0.1 to 30 hours.
- the pressure is usually at normal pressure, but may be increased or decreased.
- examples of the inert gas include nitrogen, argon, and helium.
- argon and nitrogen are good. Particularly preferred The nitrogen is good.
- the amount of the vinyl monomer and the amount of the organic tellurium compound represented by the formula (1) may be appropriately adjusted according to the molecular weight or molecular weight distribution of the resulting radical radical polymer.
- the vinyl monomer is preferably set to 5 to 100,000 O mo 1, and more preferably, to 50 to 500,000 O mo 1.
- the amount of the organic tellurium compound represented by the formula (1) and the azo-based polymerization initiator to be used is usually 1 mol of the organic tellurium compound represented by the formula (1).
- the amounts of the organic tellurium compound represented by the formula (1) and the compound represented by the formula (2) are usually expressed by the formula (2) based on 1 mo 1 of the organic tellurium compound represented by the formula (1).
- the compound is preferably 0.1 to 100 mol, preferably 0.1 to 1 Omo 1, and particularly preferably 0.1 to 5 mol.
- the t reaction is usually carried out without a solvent.
- a commonly used organic solvent or aqueous solvent may be used.
- Organic solvents that can be used include, for example, benzene, toluene, N, N-dimethylformamide.
- the aqueous solvent include, for example, water, methanol, ethanol, isopropanol, n-butanol, ethyl sorb, butyl sorb, 1-methoxy-2-propanol, and the like.
- the amount of the solvent used may be appropriately adjusted.
- the solvent is used in an amount of 0.01 to 50 m 1, preferably 0.05 to 10 m 1 per 1 g of the vinyl monomer.
- L m 1 is good.
- the reaction temperature and reaction time It may be appropriately adjusted depending on the molecular weight or molecular weight distribution of the radical polymer. Generally, stirring is carried out at 20 to 150 ° C. for 1 minute to 100 hours. Preferably, stirring is carried out at 40 to 100 ° C. for 0.1 to 30 hours. More preferably, the mixture is stirred at 40 to 80 for 0.1 to 15 hours. It is a feature of the present invention that a high yield and precise PD can be obtained even at such a low polymerization temperature and short polymerization time. At this time, the pressure is usually set at normal pressure, but may be increased or decreased.
- the solvent used and the remaining monomer are removed under reduced pressure by a conventional method to take out the target polymer, or the target product is isolated by reprecipitation using a solvent insoluble in the target polymer.
- any treatment method can be used as long as the object is not hindered.
- a plurality of Bier monomers can be used.
- a random copolymer can be obtained by simultaneously reacting two or more types of vinyl monomers.
- the random copolymer can obtain a polymer having the same ratio (mol ratio) of the monomers to be reacted irrespective of the type of the monomer.
- vinyl monomer A and vinyl monomer B are simultaneously reacted to obtain a random copolymer, almost the same ratio of raw materials (mol ratio) can be obtained.
- a block copolymer can be obtained by sequentially reacting two kinds of pinyl monomers.
- a polymer can be obtained according to the order of the monomers to be reacted, regardless of the type of the monomer.
- vinyl monomer A and vinyl monomer B are reacted in order to obtain a block copolymer
- A-B products and B-A products can be obtained depending on the reaction order.
- the living radical polymerization initiator of the present invention can perform excellent control of molecular weight and control of molecular weight distribution under extremely mild conditions.
- the reaction time can be reduced compared to conventional living radical polymerization. Wear.
- the molecular weight of the living radical polymer obtained in the present invention can be adjusted by the reaction time and the amount of the organic tellurium compound, a living radical polymer having a number average molecular weight of 500 to 1,000,000 Obtainable. Particularly, it is suitable for obtaining a living radical polymer having a number average molecular weight of 1,000 to 500,000.
- the terminal group of the living radical polymer obtained in the present invention is an alkyl group, an aryl group, a substituted aryl group, an aromatic heterocyclic group, an acyl group, an oxycarbonyl group or a cyano group derived from an organic tellurium compound. It has been confirmed that the terminal is tellurium with high reactivity. Therefore, by using an organic tellurium compound for living radical polymerization, it is easier to convert a terminal group into another functional group than a living radical polymer obtained by conventional living radical polymerization. With these, the living radical polymer obtained in the present invention can be used as a macro living radical polymerization initiator (macro initiator).
- macro initiator macro living radical polymerization initiator
- an AB diblock copolymer such as methyl methacrylate-styrene
- a BA diblock copolymer of styrene-methyl methyl acrylate are used.
- an A—B—A triblock copolymer such as methyl methacrylate-styrene-methyl methacrylate
- an A—B—C triblock copolymer such as methyl methacrylate-styrene-butyl acrylate Can be. This is the formula of the present invention.
- the organic tellurium compound represented by (1), the azo-based polymerization initiator and the ditelluride compound represented by the formula (2) can control various types of vinyl-based monomers. This is due to the presence of highly reactive tellurium at the growth terminal of the V-bing radical polymer obtained by the initiator.
- the method for producing the block copolymer is specifically as follows.
- the A—B diblock copolymer for example, in the case of the methyl styrene methacrylate copolymer, first, as in the above-described method for producing a living radical polymer, methyl methacrylate and an organic compound represented by the formula (1) are first used.
- a tellurium compound, an azo-based polymerization initiator and a ditelluride compound represented by the formula (2) are mixed to produce polymethyl methacrylate, and then styrene is mixed to obtain a methyl methacrylate-styrene copolymer. There is a method of obtaining.
- the vinyl monomer (A) is prepared after the A—B diblock copolymer is produced by the above method.
- a bimer monomer (C) is mixed to obtain an A—B—A triblock copolymer or an A—B—C triblock copolymer.
- At least one of the production of the first monomer homopolymer and the subsequent production of the diblock copolymer is represented by the formula (1).
- An organic tellurium compound, an azo-based polymerization initiator and a ditelluride compound represented by the formula (2) can be used.
- an organic tellurium compound represented by the formula (1) an organic tellurium compound represented by the formula (1), an azo polymerization initiator and a compound represented by the formula
- the ditelluride compound represented by (2) can be used.
- the reaction of the next block may be started as it is, or once the reaction is completed, the reaction of the next block may be started after purification.
- the block copolymer can be isolated by a usual method. BEST MODE FOR CARRYING OUT THE INVENTION
- Organic tellurium compounds and ditelluride compounds were identified from — NMR and MS measurement results.
- the molecular weight and molecular weight distribution of the living radical polymer were determined using GPC (gel permeation chromatography). The measuring instruments used are as follows.
- Metallurgical tellurium [A 1drich, trade name ⁇ Telluri um (—40 mesh)] 3.19 g (25 mm o 1) is suspended in 5 ml of THF, and methyllithium (Kanto Chemical Co., Ltd.) 25 ml (28. 5 mmo1) (manufactured by company, getyl ether solution) was slowly added at 0 ° C (10 minutes). The reaction solution was stirred until the metal tellurium completely disappeared (10 minutes). To this reaction solution, 20 ml of an ammonium chloride solution was added at room temperature, and the mixture was stirred for 1 hour. The organic layer was separated, and the aqueous layer was extracted three times with getyl ether. The collected organic layer was dried over sodium sulfate and concentrated under reduced pressure to obtain 2.69 g (9.4 mMol: yield: 75%) of a black-purple oil.
- Metallurgical tellurium (same as above) 3.
- 19 g (25 mmo 1) in 25 ml of THF and add n-butyllithium (Aldrich, 1.6 M hexane solution). 2 ml (27.5 mm o 1) was slowly added at 0 (10 minutes). The reaction solution was stirred until the metal tellurium completely disappeared (10 minutes).
- 20 ml of an ammonium chloride solution was added at room temperature, and the mixture was stirred for 1 hour. Separate the organic layer and getyl ether the aqueous layer And extracted three times. The collected organic layer was dried over sodium sulfate and concentrated under reduced pressure to obtain 4.41 g (1.193 mmo1: yield: 95%) of a black-purple oily substance.
- methyl methacrylate [stabi 1ized with Hydroquinone (HQ)] 1.0 g (1 O mmol) and the compound 21.1 mg (0.10 mmo) produced in Synthesis Example 1 1) and AI BN (manufactured by Otsuka Chemical Co., Ltd., trade name: AI BN) 16.4 mg (0.10 mmo 1) and the compound produced in Synthesis Example 4 28.5 mg (0.10 mm o 1) was stirred at 60 ° C. for 2 hours. After the completion of the reaction, the reaction mixture was dissolved in chloroform (5 ml), and the solution was poured into hexane (200 ml) with stirring. Suction filtration of the precipitated polymer at room temperature and drying 0.977 g of polymethyl methacrylate was obtained.
- HQ Hydroquinone
- Table 1 shows the results of the GPC analysis (based on the molecular weight of a polymethylmethacrylate standard sample).
- Example 1 As is clear from comparison between Example 1 and Comparative Example 1, when the compound represented by the formula (1) is used, a living radical polymer having a narrow molecular weight distribution (PD value is closer to 1) can be obtained. You can see that.
- Mn 1200 and PD was 1.09.
- Mn 1200 and PD was 1.10.
- Example 7 In a glove box purged with nitrogen, 1.0 g (10 mm o 1) of methyl methacrylate (same as above) and 0.18 mg (0.10 mm o 1) of the compound prepared in Synthesis Example 3 And AIBN (same as above) 8.2 mg (0.05 mmo 1) and the compound 14.3 mg (0.05 mmo 1) prepared in Synthesis Example 4 at 60 ° C for 2 hours Stirred. After the completion of the reaction, the mixture was dissolved in 5 ml of chloroform and the solution was poured into 200 ml of hexane under stirring. The precipitated polymer was subjected to suction filtration at room temperature and dried to obtain polymethylmethacrylate (yield: 64.3).
- Mn1480 and Pd were 1.17.
- n-butyl methyl acrylate [stabi 1 ized with Hydroquinone (HQ) J Nero manufactured by Kojunyaku Kogyo Co., Ltd.) Compound 30.18 mg (0.10 mm o 1) and MA IB (Otsuka Chemical A product of Shikisha Co., Ltd., trade name: MA IB) 1 1.52 mg (0.05 mmo 1) and the compound 18.5 mg (0.05 mmo 1) produced in Synthesis Example 5
- the mixture was stirred at 0 ° C for 2 hours. After the completion of the reaction, the solution was dissolved in 5 ml of chloroform, and the solution was poured into 200 ml of hexane while stirring. The precipitated polymer was subjected to suction filtration at room temperature and dried to obtain poly n-butyl methacrylate (yield: 87.0%).
- Isobornyl methacrylate [stabilized with hydroquinone mo nomethylether (ME HQ)] (manufactured by Mitsubishi Rayon Co., Ltd.) 2. 22 g (10 mm o 1) and the compound prepared in Synthesis Example 3 0.18 mg (0.10 mm o 1) and 1,1'-azobis-l-cyclohexanecarbonitrile (Otsuka Chemical Co., Ltd., trade name: AC HN) 60 mg of 12.2 mg (0.05 mm o 1) and 18.5 mg (0.05 mm o 1) of the compound produced in Synthesis Example 5: For 5 hours.
- ME HQ hydroquinone mo nomethylether
- Mn 13,000 and PD was 1.34.
- reaction mixture was dissolved in 5 ml of chloroform, and the solution was poured into 200 ml of hexane with stirring.
- the precipitated polymer was subjected to suction filtration at room temperature and dried to obtain 0.845 g of polymethyl methacrylate.
- reaction mixture was dissolved in 5 ml of chloroform, and the solution was poured into 200 ml of stirring hexane.
- the precipitated polymer was subjected to suction filtration at room temperature and dried to obtain 0.887 g of polymethyl methacrylate.
- the mixture was dissolved in 5 ml of chloroform and then the solution was poured into 200 ml of hexane under stirring.
- the precipitated polymer was subjected to suction filtration at room temperature and dried to obtain 0.639 g of polymethyl methacrylate.
- Example 17 In a nitrogen-purged glove box, methyl methyl acrylate 10.0 1 g (100 mmo 1) and ethyl 2-methyl-2 monomethylteraniloop loop pionate 26.0 mg (0 10 mm o 1), AIBN (manufactured by Otsuka Chemical Co., Ltd., trade name: AIBN) 16.4 mg (0.1 l Ommol) and dimethyl ditelluride 28.5 mg produced in Synthesis Example 4 0.1 mmO 1) was stirred at room temperature to obtain a homogeneous solution. From this, 1.0 g of the solution was withdrawn, added to another reaction vessel, and stirred at 60 ° C for 3 hours.
- AIBN manufactured by Otsuka Chemical Co., Ltd., trade name: AIBN
- the mixture was dissolved in 5 ml of chloroform, and the solution was poured into 200 ml of stirring hexane.
- the precipitated polymer was subjected to suction filtration at room temperature and dried to obtain 0.740 g of polymethyl methacrylate.
- AIBN manufactured by Otsuka Chemical Co., Ltd., trade name: AI BN
- the solution was dissolved in 5 ml of chloroform, and the solution was poured into 200 ml of stirring hexane.
- the precipitated polymer was subjected to suction filtration at room temperature and dried to obtain 0.359 g of polymethacrylonitrile.
- Mn 170 and PDI was 1.44.
- N-isopropyl methyl Mid (manufactured by Aldrich) 1.27 g (10 mmo1) and the ethyl 2-ethyl-2-methyltera 2 loop mouth ionate prepared in Synthesis Example 3 0.18 mg (0.1 0 mm o 1) and AIBN (Otsuka Chemical Co., Ltd., trade name: AI BN) 16.4 mg (0.10 mm o 1) and dimethyl ditelluride 28.5 prepared in Synthesis Example 4 mg (0.10 mmo 1) was dissolved in 2 ml of DMF, and the mixture was stirred at 60 ⁇ for 20 hours.
- Mn 14,000 and Pd was 1.27.
- Mn 1500 and PD was 1.36.
- the solution during the reaction was sampled, and the residual monomer ratio was determined using an NMR spectrum.
- the ratio of N-isopropylacrylamide was N-isopropylpropylmethacrylamide at about 65:55. It was found that the polymerization proceeded at almost the same ratio as the charge ratio, although it slightly remained.
- the residue was dissolved in 5 ml of a black form.
- the solution was poured into 200 ml of stirring hexane heated to 55 ° C.
- the precipitated polymer was filtered by suction at room temperature and dried to obtain 0.373 g of poly (N-isopropylacrylamide) -mono (N-isopropylmethacrylamide) copolymer. Obtained.
- Mn4300PD 1.13.
- Example 2 Each of the various random copolymers obtained in 429 was subjected to elemental analysis using an elemental analyzer (organic element analyzer MI CRO CORD ERJM 10 manufactured by J-Science Lab Co., Ltd.). . Table 2 shows the results.
- elemental analyzer MI CRO CORD ERJM 10 manufactured by J-Science Lab Co., Ltd.
- a method for producing a living radical polymer that enables precise control of molecular weight and molecular weight distribution under mild conditions.
- Living radio Calpolymers can be produced.
- the living radical polymer obtained by the polymerization method of the present invention can easily convert a terminal group into another functional group, furthermore, synthesize a macromonomer, use as a crosslinking point, a compatibilizer, It can be used as a raw material for block polymers.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Graft Or Block Polymers (AREA)
- Polymerization Catalysts (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Polymerisation Methods In General (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description





Claims

Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/554,242 US20060199927A1 (en) | 2003-04-25 | 2004-04-26 | Process for production of living-radical polymers and polymers |
| EP04729496A EP1619211B1 (en) | 2003-04-25 | 2004-04-26 | Process for production of living-radical polymers and polymers |
| JP2005505899A JP3845109B2 (ja) | 2003-04-25 | 2004-04-26 | リビングラジカルポリマーの製造方法及びポリマー |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2003121223 | 2003-04-25 | ||
| JP2003-121223 | 2003-04-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004096870A1 true WO2004096870A1 (ja) | 2004-11-11 |
Family
ID=33410035
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2004/005989 Ceased WO2004096870A1 (ja) | 2003-04-25 | 2004-04-26 | リビングラジカルポリマーの製造方法及びポリマー |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20060199927A1 (ja) |
| EP (1) | EP1619211B1 (ja) |
| JP (1) | JP3845109B2 (ja) |
| KR (1) | KR100721224B1 (ja) |
| CN (1) | CN100347202C (ja) |
| TW (1) | TWI288139B (ja) |
| WO (1) | WO2004096870A1 (ja) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004323693A (ja) * | 2003-04-25 | 2004-11-18 | Otsuka Chemical Co Ltd | リビングラジカルポリマーの製造方法及びポリマー |
| JP2005126459A (ja) * | 2003-10-21 | 2005-05-19 | Jsr Corp | 酸解離性基含有樹脂およびその製造方法 |
| JP2005344009A (ja) * | 2004-06-03 | 2005-12-15 | Shin Etsu Chem Co Ltd | レジスト材料用高分子化合物及びその製造方法並びに化学増幅ポジ型レジスト材料 |
| WO2006062255A1 (ja) | 2004-12-10 | 2006-06-15 | Otsuka Chemical Co., Ltd. | 有機ビスマス化合物、その製造方法、リビングラジカル重合開始剤、それを用いるポリマーの製造方法及びポリマー |
| WO2007034965A1 (ja) * | 2005-09-26 | 2007-03-29 | Nippon Shokubai Co., Ltd. | 重合体、その重合体の製造方法およびその重合体を用いたセメント混和剤 |
| WO2007119884A1 (ja) * | 2006-04-14 | 2007-10-25 | Otsuka Chemical Co., Ltd. | 樹脂組成物および耐熱性粘着剤 |
| JP2009191209A (ja) * | 2008-02-15 | 2009-08-27 | Otsuka Chemical Co Ltd | 樹脂組成物および分散剤 |
| JP2009215472A (ja) * | 2008-03-11 | 2009-09-24 | Kyoto Univ | 末端に官能基を有するリビングラジカルポリマーの製造方法 |
| JP2009249606A (ja) * | 2008-04-11 | 2009-10-29 | Otsuka Chem Co Ltd | ポリ乳酸用改質剤およびポリ乳酸樹脂組成物。 |
| JP2009270105A (ja) * | 2008-04-11 | 2009-11-19 | Otsuka Chem Co Ltd | ポリマーアロイ用相溶化剤およびポリマーアロイ調製用マスターバッチ |
| WO2018164147A1 (ja) * | 2017-03-09 | 2018-09-13 | Agc株式会社 | ポリマーの製造方法 |
| WO2018199000A1 (ja) | 2017-04-28 | 2018-11-01 | 国立大学法人京都大学 | 有機テルル化合物及びその製造方法、リビングラジカル重合開始剤、ビニル重合体の製造方法、並びにビニル重合体 |
| JPWO2018117038A1 (ja) * | 2016-12-22 | 2019-10-24 | 日本ゼオン株式会社 | 末端変性アクリルゴム、アクリルゴム組成物、アクリルゴム架橋物、及び末端変性アクリルゴムの製造方法 |
| JP2020180214A (ja) * | 2019-04-25 | 2020-11-05 | 大塚化学株式会社 | N−アルケニルラクタム系ブロック共重合体を含有する重合生成物および重合生成物の製造方法 |
| WO2022025166A1 (ja) | 2020-07-30 | 2022-02-03 | 日本化薬株式会社 | インクジェット用着色分散液、インクジェット記録用インク、及びインクジェット記録方法 |
| WO2022054851A1 (ja) | 2020-09-09 | 2022-03-17 | 日本ペイント・オートモーティブコーティングス株式会社 | 塗料用組成物 |
| WO2023153189A1 (ja) | 2022-02-10 | 2023-08-17 | 日本ペイント・オートモーティブコーティングス株式会社 | 塗料用組成物 |
| WO2024018770A1 (ja) | 2022-07-21 | 2024-01-25 | 住友化学株式会社 | 組成物、膜及び表示装置 |
| WO2025004549A1 (ja) | 2023-06-30 | 2025-01-02 | 大塚化学株式会社 | 黄色アゾ系顔料含有着色組成物、および、その製造方法 |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN103113278A (zh) * | 2013-02-03 | 2013-05-22 | 北京化工大学 | 有机碲化合物链转移剂的合成方法及其在活性自由基聚合的应用 |
| JP6829726B2 (ja) * | 2016-09-30 | 2021-02-10 | 日東電工株式会社 | 偏光フィルム用粘着剤組成物、偏光フィルム用粘着剤層の製造方法、粘着剤層付偏光フィルム、及び、画像表示装置 |
| US10676575B2 (en) | 2016-10-06 | 2020-06-09 | Johnson & Johnson Vision Care, Inc. | Tri-block prepolymers and their use in silicone hydrogels |
| US10996491B2 (en) | 2018-03-23 | 2021-05-04 | Johnson & Johnson Vision Care, Inc. | Ink composition for cosmetic contact lenses |
| US11891526B2 (en) | 2019-09-12 | 2024-02-06 | Johnson & Johnson Vision Care, Inc. | Ink composition for cosmetic contact lenses |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014962A1 (ja) * | 2002-08-08 | 2004-02-19 | Otsuka Chemical Co., Ltd. | リビングラジカルポリマーの製造方法及びポリマー |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4124633A (en) * | 1977-08-01 | 1978-11-07 | Atlantic Richfield Company | Tellurium catalyzed decomposition of peroxide intermediates resulting from the autoxidation of unsaturated aldehydes |
| US6518364B2 (en) * | 2000-09-28 | 2003-02-11 | Symyx Technologies, Inc. | Emulsion living-type free radical polymerization, methods and products of same |
| US20060135711A1 (en) * | 2003-02-17 | 2006-06-22 | Shigeru Yamago | Process for the production of living radical polymers and polymers |
-
2004
- 2004-04-23 TW TW093111574A patent/TWI288139B/zh not_active IP Right Cessation
- 2004-04-26 EP EP04729496A patent/EP1619211B1/en not_active Expired - Lifetime
- 2004-04-26 CN CNB2004800111973A patent/CN100347202C/zh not_active Expired - Lifetime
- 2004-04-26 WO PCT/JP2004/005989 patent/WO2004096870A1/ja not_active Ceased
- 2004-04-26 KR KR1020057020171A patent/KR100721224B1/ko not_active Expired - Lifetime
- 2004-04-26 US US10/554,242 patent/US20060199927A1/en not_active Abandoned
- 2004-04-26 JP JP2005505899A patent/JP3845109B2/ja not_active Expired - Lifetime
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004014962A1 (ja) * | 2002-08-08 | 2004-02-19 | Otsuka Chemical Co., Ltd. | リビングラジカルポリマーの製造方法及びポリマー |
Non-Patent Citations (4)
| Title |
|---|
| GOTO A. ET AL: "Mechanism-based invention of high-speed living radical polymerization using organotellurium compounds and azo-initiators", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 125, no. 29, 23 July 2003 (2003-07-23), pages 8720 - 8721, XP002979332 * |
| POLYMER BULLETIN, vol. 43, 1999, pages 143 - 150 |
| See also references of EP1619211A4 |
| YAMAGO S. ET AL: "Tailored synthesis of structurally defined polymers by organotellurium-mediated living radical polymerization (TERP)", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 124, no. 46, 20 November 2002 (2002-11-20), pages 13666 - 13667, XP002964725 * |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2004323693A (ja) * | 2003-04-25 | 2004-11-18 | Otsuka Chemical Co Ltd | リビングラジカルポリマーの製造方法及びポリマー |
| JP2005126459A (ja) * | 2003-10-21 | 2005-05-19 | Jsr Corp | 酸解離性基含有樹脂およびその製造方法 |
| JP2005344009A (ja) * | 2004-06-03 | 2005-12-15 | Shin Etsu Chem Co Ltd | レジスト材料用高分子化合物及びその製造方法並びに化学増幅ポジ型レジスト材料 |
| WO2006062255A1 (ja) | 2004-12-10 | 2006-06-15 | Otsuka Chemical Co., Ltd. | 有機ビスマス化合物、その製造方法、リビングラジカル重合開始剤、それを用いるポリマーの製造方法及びポリマー |
| WO2007034965A1 (ja) * | 2005-09-26 | 2007-03-29 | Nippon Shokubai Co., Ltd. | 重合体、その重合体の製造方法およびその重合体を用いたセメント混和剤 |
| WO2007119884A1 (ja) * | 2006-04-14 | 2007-10-25 | Otsuka Chemical Co., Ltd. | 樹脂組成物および耐熱性粘着剤 |
| JP5256515B2 (ja) * | 2006-04-14 | 2013-08-07 | 大塚化学株式会社 | 樹脂組成物および耐熱性粘着剤 |
| JP2009191209A (ja) * | 2008-02-15 | 2009-08-27 | Otsuka Chemical Co Ltd | 樹脂組成物および分散剤 |
| JP2009215472A (ja) * | 2008-03-11 | 2009-09-24 | Kyoto Univ | 末端に官能基を有するリビングラジカルポリマーの製造方法 |
| JP2009249606A (ja) * | 2008-04-11 | 2009-10-29 | Otsuka Chem Co Ltd | ポリ乳酸用改質剤およびポリ乳酸樹脂組成物。 |
| JP2009270105A (ja) * | 2008-04-11 | 2009-11-19 | Otsuka Chem Co Ltd | ポリマーアロイ用相溶化剤およびポリマーアロイ調製用マスターバッチ |
| JP7010242B2 (ja) | 2016-12-22 | 2022-01-26 | 日本ゼオン株式会社 | 末端変性アクリルゴム、アクリルゴム組成物、アクリルゴム架橋物、及び末端変性アクリルゴムの製造方法 |
| JPWO2018117038A1 (ja) * | 2016-12-22 | 2019-10-24 | 日本ゼオン株式会社 | 末端変性アクリルゴム、アクリルゴム組成物、アクリルゴム架橋物、及び末端変性アクリルゴムの製造方法 |
| US10961332B2 (en) | 2017-03-09 | 2021-03-30 | AGC Inc. | Method for producing polymer |
| JPWO2018164147A1 (ja) * | 2017-03-09 | 2019-11-07 | Agc株式会社 | ポリマーの製造方法 |
| WO2018164147A1 (ja) * | 2017-03-09 | 2018-09-13 | Agc株式会社 | ポリマーの製造方法 |
| KR20190138796A (ko) | 2017-04-28 | 2019-12-16 | 고쿠리츠 다이가쿠 호진 교토 다이가쿠 | 유기 텔루륨 화합물 및 그의 제조 방법, 리빙 라디칼 중합 개시제, 비닐 중합체의 제조 방법, 그리고 비닐 중합체 |
| WO2018199000A1 (ja) | 2017-04-28 | 2018-11-01 | 国立大学法人京都大学 | 有機テルル化合物及びその製造方法、リビングラジカル重合開始剤、ビニル重合体の製造方法、並びにビニル重合体 |
| US11427538B2 (en) | 2017-04-28 | 2022-08-30 | Kyoto University | Organic tellurium compound, method for producing same, living radical polymerization initiator, method for producing vinyl polymer, and vinyl polymer |
| JP2020180214A (ja) * | 2019-04-25 | 2020-11-05 | 大塚化学株式会社 | N−アルケニルラクタム系ブロック共重合体を含有する重合生成物および重合生成物の製造方法 |
| JP7329956B2 (ja) | 2019-04-25 | 2023-08-21 | 大塚化学株式会社 | N-アルケニルラクタム系ブロック共重合体を含有する重合生成物および重合生成物の製造方法 |
| WO2022025166A1 (ja) | 2020-07-30 | 2022-02-03 | 日本化薬株式会社 | インクジェット用着色分散液、インクジェット記録用インク、及びインクジェット記録方法 |
| WO2022054851A1 (ja) | 2020-09-09 | 2022-03-17 | 日本ペイント・オートモーティブコーティングス株式会社 | 塗料用組成物 |
| WO2023153189A1 (ja) | 2022-02-10 | 2023-08-17 | 日本ペイント・オートモーティブコーティングス株式会社 | 塗料用組成物 |
| WO2024018770A1 (ja) | 2022-07-21 | 2024-01-25 | 住友化学株式会社 | 組成物、膜及び表示装置 |
| WO2025004549A1 (ja) | 2023-06-30 | 2025-01-02 | 大塚化学株式会社 | 黄色アゾ系顔料含有着色組成物、および、その製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1619211B1 (en) | 2012-10-17 |
| US20060199927A1 (en) | 2006-09-07 |
| TWI288139B (en) | 2007-10-11 |
| CN100347202C (zh) | 2007-11-07 |
| JP3845109B2 (ja) | 2006-11-15 |
| EP1619211A4 (en) | 2007-09-26 |
| KR100721224B1 (ko) | 2007-05-23 |
| JPWO2004096870A1 (ja) | 2006-07-13 |
| KR20060006936A (ko) | 2006-01-20 |
| TW200426159A (en) | 2004-12-01 |
| CN1780860A (zh) | 2006-05-31 |
| EP1619211A1 (en) | 2006-01-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004096870A1 (ja) | リビングラジカルポリマーの製造方法及びポリマー | |
| KR100633200B1 (ko) | 리빙 라디칼 폴리머의 제조방법 및 폴리머 | |
| JP3839829B2 (ja) | リビングラジカルポリマーの製造方法及びポリマー | |
| JP4107996B2 (ja) | リビングラジカルポリマーの製造方法及びポリマー | |
| JP5193480B2 (ja) | リビングラジカルポリマーの製造方法およびポリマー | |
| EP3594250B1 (en) | Method for producing polymer | |
| JP2006299278A (ja) | リビングラジカルポリマーの製造方法 | |
| WO2022130919A1 (ja) | テルル含有化合物、重合体、及び重合体の製造方法 | |
| JP5380709B2 (ja) | リビングラジカル重合反応助触媒 | |
| KR100885109B1 (ko) | 유기 비스무트화합물, 그의 제조방법, 리빙 라디칼중합개시제, 그것을 사용하는 폴리머의 제조방법 및 폴리머 | |
| WO2006001496A1 (ja) | 有機アンチモン化合物、その製造方法、リビングラジカル重合開始剤、それを用いるポリマーの製造方法及びポリマー | |
| JP2010126583A (ja) | ブロック共重合体およびその製造方法 | |
| KR100589035B1 (ko) | 리빙 라디칼 폴리머의 제조방법 및 폴리머 | |
| Serhatli et al. | Synthesis of liquid crystalline–amorphous block copolymers by combination of CFRP and ATRP mechanisms | |
| KR100708959B1 (ko) | 유기 텔루르화합물, 그의 제조방법, 리빙 라디칼중합개시제, 그것을 사용하는 폴리머의 제조방법 및 폴리머 | |
| KR100852030B1 (ko) | 유기 안티몬화합물, 그의 제조방법, 리빙 라디칼중합개시제, 그것을 사용하는 폴리머의 제조방법 및 폴리머 | |
| TW202323319A (zh) | 化合物及該化合物之製造方法 | |
| ÇOLAKOĞLU | ISTANBUL TECHNICAL UNIVERSITY★ INSTITUTE OF SCIENCE AND TECHNOLOGY | |
| KR20050047087A (ko) | 유기 텔루르화합물, 그의 제조방법, 리빙 라디칼중합개시제, 그것을 사용하는 폴리머의 제조방법 및 폴리머 | |
| JP2015034241A (ja) | マレイミド系共重合体 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005505899 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004729496 Country of ref document: EP Ref document number: 1020057020171 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10554242 Country of ref document: US Ref document number: 20048111973 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057020171 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004729496 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10554242 Country of ref document: US |