WO2004110453A1 - Pyrimidine derivatives as cannabinoid receptor ligands - Google Patents
Pyrimidine derivatives as cannabinoid receptor ligands Download PDFInfo
- Publication number
- WO2004110453A1 WO2004110453A1 PCT/IB2004/001971 IB2004001971W WO2004110453A1 WO 2004110453 A1 WO2004110453 A1 WO 2004110453A1 IB 2004001971 W IB2004001971 W IB 2004001971W WO 2004110453 A1 WO2004110453 A1 WO 2004110453A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- aryl
- heteroaryl
- substituents
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*1C(*)(*)*INC1(C)* Chemical compound C*1C(*)(*)*INC1(C)* 0.000 description 1
- FPMYFZGKXYTUJR-UHFFFAOYSA-N CC(C)NC1(CNC1)C(N)=O Chemical compound CC(C)NC1(CNC1)C(N)=O FPMYFZGKXYTUJR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/26—Psychostimulants, e.g. nicotine, cocaine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/32—Alcohol-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to substituted pyrimidine-2-carboxamide compounds as cannabinoid receptor ligands, in particular CB1 receptor antagonists, and uses thereof for treating diseases, conditions and/or disorders modulated by cannabinoid receptor antagonists.
- BMI body mass index
- Overweight is typically defined as a BMI of 25-29.9 kg/m 2
- obesity is typically defined as a BMI of 30 kg/m 2 .
- Obesity is now recognized as a chronic disease that requires treatment to reduce its associated health risks.
- weight loss is an important treatment outcome
- one of the main goals of obesity management is to improve cardiovascular and metabolic values to reduce obesity-related morbidity and mortality. It has been shown that 5-10% loss of body weight can substantially improve metabolic values, such as blood glucose, blood pressure, and lipid concentrations. Hence, it is believed that a 5-10% intentional reduction in body weight may reduce morbidity and mortality.
- Currently available prescription drugs for managing obesity generally reduce weight by inducing satiety or decreasing dietary fat absorption. Satiety is achieved by increasing synaptic levels of norepinephrine, serotonin, or both.
- stimulation of serotonin receptor subtypes 1 B, 1 D, and 2C and 1 - and 2-adrenergic receptors decreases food, intake by regulating satiety. See, Bray GA, "The New Era of Drug Treatment. Pharmacologic Treatment of Obesity: Symposium Overview," Obes_Res., 3(suppl 4), 415s-7s (1995).
- Adrenergic agents e.g., diethylpropion, , benzphetamine, phendimetrazine, mazindol, and phentermine
- Older adrenergic weight-loss drugs e.g., amphetamine, methamphetamine, and phenmetrazine
- amphetamine e.g., amphetamine, methamphetamine, and phenmetrazine
- Fenfluramine and dexfenfluramine both serotonergic agents used to regulate appetite, are no longer available for use. , .
- CB1 cannabinoid receptor antagonists/inverse agonists have been suggested as potential appetite suppressants. See, e.g., Arnone, M., et al., "Selective Inhibition of Sucrose and ( ⁇ thanol Intake by SR141716, an Antagonist of Central Cannabinoid (CB1) Receptors.” Psvchopharmacol. 132, 104-106 (1997); Colombo, G., et al., "Appetite Suppression and Weight Loss after the Cannabinoid Antagonist SR141716.” Life ScL.
- Alcoholism affects approximately 10.9 million men and 4.4 million women in the United States. Approximately 100,000 deaths per year have been attributed to alcohol abuse or dependence. Health risks associated with alcoholism include impaired motor control and decision making, cancer, liver disease, birth defects, heart disease, drug/drug interactions, pancreatitis and interpersonal problems. Studies have suggested that endogenous cannabinoid tone plays a critical role in the control of ethanol intake.
- the endogenous CB1 receptor antagonist SR-141716A has been shown to block voluntary ethanol intake in rats and mice.
- the present inventio'n provides compounds of Formula (I) that act as cannabinoid receptor ligands (in particular, CB1 receptor antagonists)
- R 1 are R 2 are each independently aryl or heteroaryl, where the aryl and the heteroaryl moieties are optionally substituted with one or more substituents, provided that R 1 and R 2 are not both a mono-substituted (C r C 4 )alkoxyphenyl;
- R 3 is hydrogen, (C r C 4 )alkyl, or halo-substituted (C r C 4 )alkyl;
- R 4 is -(NH) n -N(R 4a )(R 4a' ), where n is 0 or 1 , R 4a is hydrogen or an optionally substituted (C r C 8 )alkyl and R 4a' is a chemical moiety selected from the group consisting of (C r C 8 )alkyl, aryl, heteroaryl, aryl(C r C 4 )alkyl, a partially or fully saturated (C 3 -C 10 )cycloalkyl, heteroaryl
- R 1 is phenyl substituted with one or more substituents, 2-pyridyl optionally substituted with one or more substituents, or 4-pyridyl optionally substituted with one or more substituents; more preferably, R 1 is a phenyl substituted with one to three substituents independently selected from the group consisting of halo (preferably, chloro or fluoro), (C 1 - C 4 )alkoxy, (C 1 -C 4 )alkyl, halo-substituted (CrC 4 )alkyl (preferably fluo ' ro-substituted alkyl), and cyano; most preferably, R 1 is 2-chlorophenyl, 2-fluorophenyl, 2,4- dichlorqphenyl, 2-fluoro-4-chlorophenyl, 2-chloro-4-fluorophenyl, or 2,4- difluorophenyl: and
- R 2 is a phenyl substituted with one or more substituents or a 2-pyridyl substituted with one or more substituents; more preferably, R 2 is a phenyl substituted with one to three substituents independently selected from the group consisting of halo (preferably, chloro or fluoro), (Ci-C 4 )alkoxy, (C r C 4 )alkyl, halo-substituted (C 1 - C 4 )alkyl (preferably fluoro-substituted alkyl), and cyano; most preferably, R 2 is 4- chlorophenyl or 4-fluorpphenyl.
- a compound of Formula (I) where R 4 is -'(NH) n -N(R 4a )(R 4a' ), where n is 0 or 1 , R 4a is hydrogen and R 4a' , is a chemical moiety selected from the group consisting of (CrC 8 )alkyl, aryl, heteroaryl, aryl(CrC 4 )alkyl, a partially or fully saturated (C 3 -C 10 )cycloalkyl, heteroaryl(Ci-C 3 )alkyl, 5-6 membered lactone, 5- to 6-membered lactam, and a 3- to 6-membered partially or fully saturated heterocycle, where said chemical moiety is optionally substituted with one or more substituents; a pharmaceutically acceptable salt thereof or a solvate or hydrate of the compound or the salt.
- R 4a is preferably a chemical moiety selected from (C r C 8 )alkyl, phenyl(C r C 4 )alkyl, or a partially or fully saturated (C 3 - C 10 )cycloalkyl, where the chemical moiety is optionally substituted with one or more substituents (preferably, 1 to 3 substituents).
- Preferred compounds where n is 0 include: 5-(4-chloro-phenyl)-4-(2,4-dichloro ⁇ phenyl)-pyrimidine-2-carboxylic acid benzylamide;
- R 4 is -(NH) n -N(R 4a )(R 4a ), where n is 0, R 4a is an optionally substituted ' (CrC 8 )alkyl, and R 4a ' is a, chemical moiety selected from the group consisting of (C 1 - C 8 )alkyl, aryl, heteroaryl, 317!(C 1 -C 4 )BlRyI, a partially or fully saturated (C 3 - C 10 )cycloalkyl, heteroaryl(C r C 3 )alkyl, 5-6 membered lactone, 5- to 6-membered , lactam, and a 3- to 6-membered partially or fully saturated heterocycle, where the chemical moiety is optionally substituted with one or more substituents; a pharmaceutically acceptable salt thereof or a solvate or hydrate of the compound or the salt.
- R 4a is preferably (C r C 6 )alkyl, and R 4a' is preferably a chemical moiety selected from (C ⁇ -C 8 )alkyl, aryl, heteroaryl, aryl(CrC 4 )alkyl, a partially or fully
- Representative compounds of this embodiment include: 5-(4-chloro-phenyl)-4-(2,4-dichloro-phenyl)-pyrimidine-2-carboxylic acid cyclohexyl-methyl-amide;
- a compound of Formula (I) where R 4 is -(NH) n -N(R 4a )(R 4a '), where n is 0, and R 4a and R 4a' are taken together to form a heterocycle having Formula (IA)
- R 4b and R 4b' are each independently hydrogen, cyano, hydroxy, amino, H 2 NC(O)-, or a chemical moiety selected from the group consisting of (Ci-C 6 )alkyl, (CrC ⁇ )alkoxy, acyloxy, acyl, (C r C 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-NH-C(O)-, (C 1 - C 4 )alkyl) 2 N-C(O)-, (C r C 6 )alkylamino-, ((C r C 4 )alkyl) 2 amino-, (C 3 -C 6 )cycloalkylamino-, acylamino-, aryl(C r C 4 )alkylamino-, heteroaryKCrC ⁇ Jalkylamino 1 , aryl, heteroaryl, a 3- 6 membered partially or fully saturated heterocycle, ,and
- X is a bond, -CH 2 CH 2 - Or -C(R 4c )(R 4c' )-, where R 4c and R 4c' are each independently hydrogen, cyano, hydroxy, amino, H 2 NC(O)-, or a chemical moiety , selected from the group consisting of (C r C 6 )alkyl, (Ci-C 6 )alkoxy, acyloxy, acyl, (C 1 - C 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-NH-C(O)-, ((CrC 4 )alkylj 2 N-C(O)-, (C r C 6 )alkylamino-, di(C- ⁇ -C 4 )alkylamino-, (Cs-Cejcycloalkylamino-, acylamino-, aryl(C 1 -C 4 )alkylamino-, heteroaryl(C
- Y is oxygen, sulfur, -C(O)-, or -C(R 4d )(R 4d' )-, where R 4d and R 4d' are each independently hydrogen, cyano, hydroxy, amino, H 2 NC(O)-, or a chemical moiety selected from the group consisting of (CrC 6 )alkyl, (C 1 -C 6 JaIkOXy, acyloxy, acyl, (C 1 - C 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-NH-C(O)-, ((C r C 4 )alkyl) 2 N-C(O)-, (C r C 6 )alkylamino-, di(CrC 4 )alkylamino-, (C 3 -C 6 )cycloalkylamino-, acylamino-, aryl(CrC 4 )alkylamino-, heteroaryl(Ci-
- R 4d' or R 4d" taken together with R 4c , R 4c' , R 4e , or R 4e' forms a fused aromatic ring
- Y is -NR 4d " -, where R 4d" is a hydrogen or a chemical moiety selected from the group consisting of (Ci-C 6 )alkyl, (C 3 -C 6 )cycloalkyl, (C r C 3 )alkylsulfonyl-, (C r C 3 )alkylaminosulfonyl-, di(C 1 -C 3 )alkylaminosulfonyl-, acyl, (C r C 6 )alkyl-O-C(O)-, aryl, and heteroaryl, where said moiety is optionally substituted with one or more substituents;
- Z is a bond, -CH 2 CH 2 -, or -C(R 4e )(R 4e' )-, where R 4
- R 4f and R 4f are each independently hydrogen, cyano, hydroxy, amino, H 2 NC(O)-, or a chemical moiety selected from the group consisting of (CrC 6 )alkyl, (CrC ⁇ )alkoxy, acyloxy, acyl, (C r C 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-N H-C(O)-, ((C 1 - C 4 )alkyl) 2 N-C(O)-, (C r C 6 )alkylamino-, di(Ci-C 4 )alkylamino-, (C 3 -C 6 )cycloalkylamino-, acylamino-, aryl(CrC 4 )alkylamino-, heteroaryl(C r C 4 )alkylamino-, aryl, heteroaryl, a 3- 6 membered partially or fully saturated heterocycle, and a 3-6
- R 4b is hydrogen, an optionally substituted (C r C 3 )alkyl, or taken , together with R 4e , R 4e' , R 4f , or R 4f forms a bond, a methylene bridge, or an ethylene bridge;
- R 4b is hydrogen, an optionally substituted (C r C 3 )alkyl, or taken together with R 4 ⁇ , R 4e ', R 4f , or R 4f forms a bond, a methylene bridge, or an ethylene bridge;
- R 4f is hydrogen, an optionally substituted (C r C 3 )alkyl, or taken together with R 4b , R 4b' , R 4c , or R 4c' forms a bond, a methylene, bridge, or an ethylene bridge;
- R 4f is hydrogen, an optionally substituted (C r C 3 )alkyl, or taken together with 'R 4b , R 4b> , R 4c , or R 4c
- R 4d is preferably a hydrogen, heteroary, or an optionally substituted (C r C 6 )alkyl
- X is -CH 2 CH 2 - or -C(R 4c )(R 4c' )-, where R 4 ° and, R 4c' are each independently hydrogen, or an optionally substituted (Ci-C 6 )alkyl, or either , ' R 40 Or R 4c' taken together with R 4e , R 4e' , R 4f , or R 4f forms a bond, a methylene bridge or an ethylene bridge; and Z is -CH 2 CH 2 - or -C(R 4e )(R 4 ⁇ ' )-, where R 4e and R 4e' are each independently hydrogen, or an optionally substituted (CrC 6 )alkyl, or either R 4e or R 4e' taken together with R 4b , R 4b' , R 4c ,
- Preferred compounds include:
- R 4d is preferably hydrogen, cyano, hydroxy, amino, H 2 NC(O)-, or a chemical moiety selected from the group consisting of (C 1 - C 6 )alkyl, (C r C 6 )alkoxy, acyloxy, acyl, (CrC 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-NH-C(O)-, ((C r C 4 )alkyl) 2 N-C(O)-, (C r C 6 )alkylamino-, ((C r C 4 )alkyl) 2 amino-, (C 3 - C 6 )cycloalkylamino-, acylamino-, aryl(CrC 4 )alkylamino-, heteroaryl(CrC 4 )alkylarnino- , (C r C 6
- R 4d' is hydrogen, H 2 NC(O)-, or a chemical moiety selected from the group consisting of (CrC 6 )alkyl, acyl, (C r C 3 )alkyl-O-C(O)-, (C r C 4 )alkyl-NH-C(O)-, (C 1 - C 4 )alkyl) 2 NrC(O)-, aryl, heteroaryl, a 3-6 membered partially or fully saturated heterocycle, and a 3-6 membered partially or fully saturated carbocyclic ring, where the moiety is optionally substituted with one or more substituents; or either R 4d or R 4d" taken together with R 4c , R 4c> , R 4e , or R 4e' forms a fused aromatic ring; X is a bond or -C(R 4c )(R 4c' )-, where R 4c and R 4c' are hydrogen or either R 4c or R 4c' is hydroxy
- Preferred compounds include:
- X is preferably -C(R 4c )(R 4c' )-, where R 40 and R 4c' are each independently hydrogen or (CrC 6 )alkyl; and Z is-C(R 4e )(R 4e' )-, where R 4e and R 4e' are each independently hydrogen or (CrC 6 )alkyl; a pharmaceutically acceptable salt thereof or a solvate or hydrate of said compound or said salt.
- Representative compounds of this embodiment include: [5-(4-chloro-phenyl)-4-(2,4-dichloro-phe'nyl)-pyrimidin-2-yl]-morpholin-4-yl- methanone; and
- R 4 is -(NH) n -N(R 4a )(R 4a ), where n is 1 ; a pharmaceutically acceptable salt thereof or a solvate or hydrate of the compound or the salt.
- Preferred embodiments include R 4a and R 4a as defined above for those compounds where n is 0.
- a preferred compound of this embodiment is 5-(4-Chloro-phenyl)-4-(2- chloro-phenyl)-pyrimidine-2-carboxylic acid piperidin-1-ylamide; a pharmaceutically acceptable salt thereof, or a solvate or hydrate of the compound or the salt.
- a pharmaceutical composition comprising (1) a compound of the present invention; and (2) a pharmaceutically acceptable excipi ⁇ nt, diluent; or carrier.
- the composition comprises a thereapeutically effective amount of a compound of the present invention.
- the composition may also contain at least one additional pharmaceutical agent (described herein).
- Preferred agents include nicotine receptor partial agonists, opioid antagonists (e.g., naltrexone' and nalmefene), dopaminergic agents (e.g., apomorphine), attention deficit activity disorder (ADHD) agents (e.g.,
- a method for treating a disease, condition or disorder modulated by a cannabinoid receptor (in particular, a CB1 receptor) antagonist in animals that includes the step of administering to an animal in need of such treatment a therapeutically effective amount of a compound of the present invention including compounds where I-? 1 and R 2 are both a mono- substituted (C r C 4 )alkoxyphenyl,(or a pharmaceutical composition thereof).
- Diseases, conditions, and/or disorders modulated by cannabinoid , receptor antagonists include eating disorders (e.g., binge eating disorder, anorexia, and bulimia), weight loss or control (e.g., reduction in calorie or food intake, and/or appetite suppression), obesity, depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e.g., conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse, addictive disorders, impulsivity, alcoholism (e.g., alcohol abuse, addiction and/or dependence including treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e.g., smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e.g.
- Compounds of the present invention may be administered in combination with other pharmaceutical agents.
- Preferred pharmaceutical agents include nicotine . receptor partial agonists, opioid antagonists (e.g., naltrexone (including naltrexone depot), antabuse, and nalmefene), dopaminergic agents (e.g., apomorphine), ADD/ADHD agents (e.g., methylphenidate hydrochloride (e.g., RitalinTM and ConcertaTM), atomoxetine (e.g., StratteraTM), and amphetamines (e.g., AdderallTM)) and anti-obesity agents, such as apo-B/MTP inhibitors, 11 ⁇ -hydroxy steroid dehydrogenase- 1 (11 ⁇ -HSD type 1) inhibitors, peptide YY 3-36 or analogs thereof, MCR-4 agonists, CCK-A agonists, monoamine reuptake inhibitors, sympathomimetic agents, ⁇ 3 a
- bombesin receptor agonists e.g., NPY-5 receptor antagonists such as those described herein below
- neuropeptide-Y receptor antagonists e.g., NPY-5 receptor antagonists such as those described herein below
- .thyromimetic agents e.g., dehydroepiandrosterone or analogs thereof, glucocorticoid receptor antagonists, orexin receptor antagonists, glucagon- 1 like peptide-1 receptor agonists, ciliary neurotrophic, factors, human agouti-related protein antagonists, ghrelin receptor antagonists, histamine 3 receptor antagonists or inverse agonists, and neuromedin U receptor agonists, and the like.
- the combination therapy may be administered as (a) a single pharmaceutical composition which comprises a compound of the present invention, at least one additional pharmaceutical agent described herein and a pharmaceutically acceptable excipient, diluent, or carrier; or (b) two separate pharmaceutical compositions comprising (i) a first composition comprising a compound of the present invention and a pharmaceutically acceptable excipient, diluent, or carrier, and (ii) a second composition comprising at least one additional pharmaceutical agent described herein and a pharmaceutically acceptable excipient, diluent, or carrier.
- the pharmaceutical compositions may be administered simultaneously or sequentially and in any order.
- a pharmaceutical kit for use by a consumer to treat diseases, conditions or disorders modulated by cannabinoid receptor antagonists in an animal.
- the kit comprises a) a suitable , dosage form comprising a compound of the present invention; and b) instructions describing a method of using the dosage form to tr ⁇ at diseases, conditions or disorders that are modulated by cannabinoid receptor (in particular, the CB 1 receptor) antagonists.
- a pharmaceutical kit comprising: a) a first dosage form comprising (i) a compound of the present invention and (ii) a pharmaceutically acceptable carrier, excipient or diluent; b) a second dosage form comprising (i) an additional pharmaceutical agent described herein, and (ii) a pharmaceutically acceptable carrier, excipient or diluent; and c) a container.
- alkyl refers to a hydrocarbon radical of the general , formula C n H 2n +i-
- the alkane 1 radical may be straight or branched.
- (CrC 6 )alkyl refers to a monovalent, straight, or branched aliphatic group containing 1 to 6 carbon atoms (e.g., methyl, ethyl, n-propyl, /-propyl, n-butyl, /-butyl, s-butyl, f-butyl, n-pentyl, 1-methylbutyl, 2-methylbutyl, 3-methylbutyl, neopentyl, 3,3- dimethylpropyl, hexyl, 2-methylpentyl, and the Ijke),
- the alkyl portion i.e., alkyl moiety
- acyl e.g., alkanoyl
- alkane radical or alkyl moiety may be unsubstituted or substituted with one or more substituents (generally, one to three substituents except in the case of halogen substituents such as perchloro or perfluoroalkyls) independently selected from the group of substituents listed below in the definition for "substituted.”
- Halo- substituted alkyl refers to an alkyl group substituted with one or more halogen atoms (e.g., fluoromethyl, difluoromethyl, trifluoromethyl, perfluoroethyl, and the like).
- the alkane radicals or alkyl moieties are preferably substituted with 1 to 3 fluoro substituents, or 1 or 2 substituents independently selected from (CrC 3 )alkyl, (C 3 -C 6 )cycloalkyl, (C 2 -C 3 )alkenyl, aryl, heteroaryl, 3- to 6-membered heterocycle, chloro, cyano, hydroxy, (CrC 3 )alkoxy, aryloxy, amino, (C r C 6 )alkyl amino, (Ji-(C 1 - C 4 )alkyl amino, aminocarboxylate (i.e., (C r C 3 )alkyl-O-C(O)-NH-), hydroxy(C 2 - C 3 )alkylamino, or keto (oxy), and more preferably, 1 to 3 fluoro groups, or 1 substituent selected from (CrC 3 )alkyl, (C 3 -C 6 )cyclo
- partially or fully saturated cycloalkyl refers to nonaromatic rings that are either partially or fully hydrogenated and may exist as a single ring, bicyclic ring or a spiral ring. Unless specified otherwise, the carbocyclic ring is generally a 3- to 8-membered ring.
- partially or fully saturated carbocyclic rings include groups such as cyclopropyl, cyclopropenyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclpentenyl, cyclopentadienyl, cyclohexyl, cyclohexenyl, cyclohexadienyl, norbornyl (bicyclo[2.2.1]heptyl), norbornenyl, bicyclo[2.2.2]octyl, and the like.
- the partially saturated or fully saturated cycloalkyl group may be unsubstituted or substituted with one or more substituents (typically, one to three substituents) independently selected from the group of substituents listed below in the definition for "substituted.”
- a substituted carbocyclic ring also includes groups wherein the carbocyclic ring is fused to a phenyl ring (e.g., indanyl).
- the carbocyclic group may be attached to the chemical entity or moiety by any one of the carbon atoms within the carbocyclic ring system.
- the carbocyclic group is preferably substituted with 1 or 2 substituents independently selected from (C r C 3 )alkyl, (C 2 -C 3 )alkenyl, (Ci-C 6 )alkylidenyl, aryl, heteroaryl, 3- to 6- membered heterocycle, chloro, fluoro, cyario, hydroxy, (C r C 3 )alkoxy, aryloxy, amino, (C r C 6 )alkyl amino, di-(C-
- any cycloalkyl portion of a group (e.g., cycloalkylalkyl, cycloalkylamino, etc.) has the same definition as above.
- the term "partially saturated or fully saturated heterocyclic ring” (also referred to as “partially saturated or fully saturated heterocycle”) refers to nonaromatic rings that are either partially or fully hydrogenated and may exist as a single ring, bicyclic ring or a spiral ring.
- the heterocyclic ring is generally a 3- to 6-membered ring containing 1 to 3 heteroatoms (preferably 1 or 2 heteroatoms) independently selected from sulfur, oxygen or nitrogen.
- Partially saturated or fully saturated heterocyclic rings include groups such as epoxy, aziridinyl, tetrahydrofuranyl, dihydrofuranyl, dihydropyridinyl, pyrrolidinyl, N-methylpyrrolidinyl, imidazolidinyl, imidazolinyl, piperidinyl, piperazinyl, pyrazolidinyl, 2H-pyranyl, 4H- pyranyl, 2H-chromenyl, oxazinyl, morpholino, thiomorpholino, tetrahydrothienyl, tetrahydrothienyl 1 ,1 -dioxide, and the like.
- the partially saturated or fully saturated heterocycle group may be unsubstiuted or substituted with one or more substijuents 1 (typically, one to three substituents) independently selected from the group of substituents listed below in the definition for "substituted.”
- a substituted heterocyclic ring includes groups wherein the heterocyclic ring is fused to an aryl or heteroaryl ring (e.g., 2,3- dihydrobenzofuranyl, 2,3-dihydroindolyl, 2,3-dihydrobenzothiophenyl, 2,3- dihydrobenzothiazolyl, etc.).
- the heterocycle group is preferably substituted with 1 or 2 substituents independently selected from (C r C 3 )alkyl, (C 3 - C 6 )cycloalkyl, (C 2 -C 4 )alkenyl, aryl, heteroaryl, 3- to 6-membered heterocycle, chloro, fluoro, cyano, hydroxy, (C r C 3 )alkoxy, aryloxy, amino, (C r C 6 )alkyl amino, di-(Cr , C 3 )alkyl amino, aminocarboxylate (i.e., (C r C 3 )alkyl-O-C(O)-NH-), or keto (oxy), and , more preferably with 1 or 2 substituents independently selected from (C r C 3 )alkyl, (C 3 -C 6 )cycloalkyl, (C 6 )aryl, 6-membered-heteroaryl, 3- to 6-membered heterocycle, or fluor
- heterocyclic group may be attached to the chemical entity or moiety by any one of the ring atoms within the heterocyclic ring system.
- any heterocycle portion of a group e.g., heterocycle-si ⁇ stituted alkyl, heterocycle carbo ⁇ yl, etc. has the same definition as above.
- aryl or "aromatic carbocyclic ring” refers to aromatic r ⁇ oieties having a single (e.g., phenyl) or a fused ring system (e.g., naphthalene, anthracene, phenanthrene, etc.).
- a typical aryl group is a 6- to 10-membered aromatic carbocyclic ring(s).
- the aryl groups When indicated as being “optionally substituted,” the aryl groups may be unsubstituted or substituted with one or more substituents (preferably no more than three substituents) independently selected from the group of substituents listed below in the definition for "substituted.”
- substituents preferably no more than three substituents
- Substituted aryl groups include a chain of aromatic moieties (e.g., biphenyl, terphenyl, phenylnaphthalyl, etc.).
- the aromatic moieties are preferably substituted with 1 or 2 substituents independently selected from (C r C 4 )alkyl, (C 2 -C 3 )alkenyl, aryl, heteroaryl, 3- to 6- membered heterocycle, bromo, chloro, fluoro, iodo, cyano, hydroxy, (C r C 4 )alkoxy, aryloxy, amino, (C-
- 1 or 2 substituents selected independently selected from (C r C 4 )alkyl, chloro, fluoro,
- the aryl group may be attached to the chemical entity or moiety by any one of the carbon atoms within the aromatic ring system.
- the aryl portion (i.e., aromatic moiety) of an aroyl, aroyloxy (i.e., (aryl)-C(O)-O-), aryl substituted alkyl, and til ' so on has th ⁇ same. definition as above.
- heteroaryl or “heteroaromatic ring” refers to aromatic moieties containing at least one heteratom (e.g., oxygen, sulfur, nitrogen or combinations thereof) within a 5- to 10-membered aromatic ring -system (e.g., pyrrolyl, pyridyl, .
- heteroaromatic moiety may consist of a single pr fused ring system.
- a typical single heteroaryl ring is a 5- to 6- membered ring containing one to three heteroatoms independently selected from oxygen, sulfur and nitrogen and a typical fused heteroaryl ring system is a 9- to 10- membered ring system containing one to four heteroatoms independently selected from oxygen, sulfur and nitrogen.
- the heteroaryl groups may be unsubstituted or substituted with one or more substituents (preferably no more than three substituents) independently selected from the group of substituents listed below in the definition for "substituted.”
- the heteroaromatic moieties are preferably substituted with 1 or 2 substituents independently selected from (C r C 4 )alkyl, (C 2 -C 3 )alkenyl, aryl, heteroaryl, 3- to 6-membered heterocycle, bromoj chlord, fludro, iodo, cyano, hydroxy, (Cr C 4 )alkoxy, aryloxy, amino, (C r C 6 )alkyl amino, di-(C r C 3 )alkyl amino, or aminocarboxylate (i.e., (C r C 3 )alkyl-O-C(O)-NH-); and more preferably, 1 or 2 substituents independently selected from (C r C 4 )alkyl,
- the heteroaryl group may be attached to the chemical entity or moiety by any one of the atoms within the aromatic ring system (e.g., imidazol-1-yl, imidazol-2-yl, imidazol-4-yl, imidazol-5-yl, pyrid-2-yl, pyrid-3-yl, pyrid-4-yl, pyrid-5-yl, or pyrid-6-yl).
- the heteroaryl portion i.e., heteroaromatic moiety
- a heteroaroyl i.e., (heteroaryl)-C(O)-O-
- heteroaryl substituted alkyl i.e., (heteroaryl)-C(O)-O-
- acyl refers to alkyl, partially saturated or fully saturated cycloalkyl, partially saturated or fully saturated heterocycle, aryl, and heteroaryl substituted carbonyl groups.
- acyl includes groups such as (C r C 6 )alkanoyl (e.g., formyl, acetyl, propionyl, butyryl, valeryl, caproyl, f-butylacetyl, etc.), (C 3 - C 6 )cycloalkylcarbonyl (e.g., cyclopropylcarbonyl, cyclobutylcarbonyl, cyclopentylcarbonyl, cyclohexylcarbonyl, etc.), heterocyclic carbonyl (e.g., - -
- pyrrolidinylcarbonyl pyrrolid-2-one-5-carbonyl, piperidinylcarbonyl, piperazinylcarbonyl, tetrahydrofuranylcarbonyl, etc.
- aroyl e.g., benzoyl
- heteroaroyl e.g., thiophenyl-2-carbonyl, thiophenyl-3-carbonyl, furanyl-2-carbonyl, furanyl-3-carbonyl, 1 H-pyrroyl-2-carbonyl, 1 H-pyrroyl-3-carbonyl, benzo[b]thiophenyl- 2-carbonyl, etc.
- the alkyl, cycloalkyl, heterocycle, aryl and heteroaryl i . . • > ⁇ • portion Df the acyl group may be any one of the groups described in the respective definitions above.
- the acyl group may be unsubstituted or optionally substituted with.
- one or more substituents typically, one to three substituents independently selected from the group of substituents listed below in the definition for "substituted” or the alkyl, cycloalkyl, heterocycle, aryl and heteroaryl portion of the acyl group may be substituted as , described above in the preferred and more preferred list of substituents, respectively.
- substituents specifically envisions and allows for one or more substitutions that are common in the art. However, it is generally understood by those skilled in the art that the substituents should be selected so as to not adversely affect the pharmacological characteristics of the compound or adversely interfere with the use of the medicament.
- Suitable substitue ⁇ ts fpr any of the groups defined above include (C r C 6 )alkyl, (C 3 -C 7 )cycloalkyl, (C 2 -C 6 )alkenyl, (C r C 6 )alkylidenyl, aryl, .
- heteroaryl 3- to 6-membered heterocycle, halo (e.g 1 ., chloro, bromo, iodq and fluoro), cyano, hydroxy, (C r C 6 )alkoxy, aryloxy, sulfhydryl (mercapto), (C r C 6 )alkylthio, arylthio, amino, mono- or di-(Ci-C 6 )alkyl amino, quaternary ammonium salts, amino(Ci-C 6 )alkoxy, aminocarboxylate (i.e., (Ci-C 6 )alkyl-O-C(O)-NH-), hydroxy(C 2 - C 6 )alkylamino, amino(CrC 6 )alkylthio, cyanoamino, nitro, (Ci-C 6 )carbamyl, keto (oxy), acyl, (CrC 6 )alkyl-CO 2 -, glycolyl,
- substituted combinations such as "substituted aryl(Ci-C 6 )alkyl"
- either the aryl or the alkyl group may be substituted, or both the aryl and the alkyl groups may be substituted with one or more substituents (typically, one to three substituents except in the case of perhalo substitutions).
- An aryl or heteroaryl substituted carbocyclic or heterocyclic group may be a fused ring (e.g., indanyl, dihydrobenzofuranyl, dihydroindolyl, etc.).
- solvate refers to a molecular complex of a compound represented by Formula (I) (including prodrugs and pharmaceutically acceptable salts thereof) with one or more solvent molecules.
- solvent molecules are those commonly used in the pharmaceutical art, which are known to be innocuous to the recipient, e,g.> water, ethanol, and the like.
- hydrate refers to the complex where the solvent molecule is water.
- protecting group refers to a substituent that is commonly employed to block or protect a particular functionality while reacting other functional groups on the compound.
- an “amino-protecting group” is a substituent attached to an amino group that blocks or protects the amino functionality in the compound. Suitable amino-protecting groups include acetyl, trifluor ⁇ acetyl, t- butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethylenoxycarbonyl (Fmoc).
- a "hydroxy-protecting group” refers to a substituent of a hydroxy group that blocks or protects the hydroxy functionality.
- Suitable protecting groups include acetyl and silyl.
- a "carboxy-protecting group” refers to a substituent of the carboxy group that blocks or protects the carboxy functionality.
- Common carboxy- protecting groups include -CH 2 CH 2 SO 2 Ph, cyanbethyl, 2-(trimethylsilyl)ethyl, 2- (trimethylsilyl)ethoxymethyl, 2-(p-toluenesulfonyl)ethyl, 2-(p-nitrophenylsulfenyl)ethyl, 2-(diphenylphosphino)-ethyl', nitroethyl and the like.
- protecting groups and fheir use see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
- terapéuticaally effective amount means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition, or disorder described herein.
- animal refers to humans (male or female), companion animals
- Edible animals refers to food-source animals such as cows, pigs, sheep and poultry.
- phrases "pharmaceutically acceptable” indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- the terms “treating”, “treat”, or “treatment” embrace both preventative, i.e., prophylactic, and palliative treatment.
- the terms “modulated by a cannabinoid receptor” or “modulation of a cannabinoid receptor” refers to the activation or deactivation of a cannabinoid receptor.
- a ligand may act as an agonist, partial agonist, inverse agonist, antagonist, or partial antagonist.
- the term “antagonist” includes both full antagonists and partial antagonists, as well as inverse agonists.
- CB-1 receptor refers to the G-protein coupled type 1 cannabinoid receptor.
- compounds of the present invention refer to compounds of Formula (I), prodrugs thereof, pharmaceutically acceptable salts of the compounds, and/or prodrugs, and hydrates or solvates of t,he compounds, salts, and/or prodrugs, as well as, all stereoisomers (including diastereoisomers and enantiomers), tautomers and isotopically labeled compounds.
- the present invention provides compounds and pharmaceutical formulations thereof that are useful in the treatment of diseases, conditions and/or disorders modulated by cannabinoid receptor antagonists,. ,
- Compounds of the present invention may be synthesized by synthetic' routes that include processes analogous to those well-known in the chemical arjs, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources such as Aldrich Chemicals (Milwaukee, Wl) or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1 -19, Wiley, New York (1967-1999 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, including supplements (also available via the Beilstein online database)).
- reaction schemes depicted below provide potential routes for synthesizing the compounds of the present invention as well as key intermediates.
- Examples section below For a more detailed description of the individual reaction steps, see the Examples section below.
- Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds.
- specific starting materials and reagents are depicted in the schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions.
- many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
- Suitable amino- protecting groups include acetyl, trifluoroa ' cetyl, f-butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethyleneoxycarbonyl (Fmoc).
- NH-Pg amino- protecting groups
- BOC f-butoxycarbonyl
- CBz benzyloxycarbonyl
- Fmoc 9-fluorenylmethyleneoxycarbonyl
- Intermediate alcohol (1a) can be prepared by condensing the desired halide (e.g., chloride or bromide) with the desired aldehyde (R 1 CHO) using a conventional Grignard reaction.
- the chloride is first reacted with Magnesium to form the Grignard reagent (R 2 -CH 2 -Mg-CI) which is then condensed with the aldehyde (R 1 - CHO) to form the desired alcohol (1a).
- the intermediate alcohol (1a) can then be oxidized to the corresponding ketone (1b) using procedures well-known to those skilled in the art.
- the ketone (1 b) is formed by reacting aldehyde (1a) with a Jones reagent (Chromic acid).
- the enamine (1c) can be produced by condensing the ketone (1b) with N,N-dimethylformamide dimethyl acetal.
- the condensation is generally performed by heating the reactants in a polar solvent (e.g., tetrahydrofuran (THF)).
- THF tetrahydrofuran
- the pyrimidine ring of intermediate (1d) may then be formed by condensing the enamine (1c) with acetamidine hydrochloride.
- the reaction is - -
- an inorganic base e.g., sodium or potassium hydroxide, carbonate, alkoxide, etc.
- organic base triethylamine, pyridine, M- methylmorpholine, dimethylbenzylamine, etc.
- the methyl group at the 2-position of the pyrimidine ring of intermediate (1d) can then be oxidized to the corresponding carboxylic acid (1e) using procedures well-known to those skilled in the art.
- pyrimidine intermediate (Id) is treated with selenium dioxide in refluxing pyridine.
- the final amide compound (I) can then be formed by first activating the carboxylic acid (1e).
- the acid chloride may be formed by treating the carboxylic acid (1e) with thionyl chloride.
- the activated carboxylic acid can then be reacted with the desired amine compound (R 4 -H) to form a compound of the presence invention (I).
- Suitable amino compounds can be either purchased commercially or easily prepared using standard procedures well-known to those skilled in the art. Preparations for various piperidine and azetidine starting materials (R 4 -H, where R 4 is an amino group of Formula (IA)) may be found in U.S. Provisional Application Nos. 60/421874, filed on October 28, 2002, and 60/445728 filed' on February 6, 2003, 'both of which are incorporated herein by reference.
- R 4 -H, where R 4 is an amino group of Formula (IA) see Scheme III (below) and the "Preparation of Key Intermediates" in the Example section (below).
- Scheme III (below)
- the keto alkylene intermediate (2a) can be easily sythnesjzed using a
- the methoxy group of intermediate (2b) can then be converted to the corresponding hydroxy group by treating intermediate (2b) with boron tribromide at a temperature from about -78°C to about 0°C followed by quenching with a protic solvent (e.g., methanol) at about - 78 0 C.
- a protic solvent e.g., methanol
- the pendant hydroxy-methylene group can then be oxidized to the corresponding carboxylic acid by first treating the hydroxy-intermediate (2c) using the Swern reaction (treatment with oxalyl chloride in the presence of dimethylsulfoxide) followed by oxidation of the resultant aldehyde using procedures well-known to those skilled in the art.
- the aldehyde is treated with sodium chlorite and sodium dihydrogen phosphate at ambient temperature.
- the amide compound (I) may then produced from the carboxylic acid intermediate (2d) using procedures described above in Scheme I for the conversion of intermediate carboxylic acid (1e) via (1f).
- the amide linkage may be formed by treating the carboxylic acid (2d) in the presence of the desired amine (R 4 -H) and triethylamine withi- propanephosphonic acid cyclic anhydride.
- R 4 -H desired amine
- triethylamine i- propanephosphonic acid cyclic anhydride
- the amino group of 4-piperidinone is first protected to provide intermediate (3a).
- a useful protecting group is benzyl.
- 4-Piperidinone and derivatives thereof may be purchased commercially from a variety of sources (e.g., lnterchem Corporation,, Paramus, NJ and Sigma-Aldrich Co., St. Louis, MO 1 ).
- Piperidinone (3a) is then reacted with the desired alkylamine and potassium pyanide in an aqueous HCI/ethanol solvent mixture at about 0 0 C to about 30 0 C.
- the cyano group is converted to the corresponding amide w,ith acid and water.
- the protecting group is then removed using conventional .methods for the particular protecting group employed. For example, a benzyl-protecting group may be removed by hydrogenation in the presence of Pd/C.
- a detailed description of some representative amines having Formula (3c) above may be found in the "Preparation of Key Intermediates" section of the Examples below.
- the compounds of the present invention may be isolated and used perse or in the form of its pharmaceutically acceptable salt, solvate and/or hydrate.
- salts refers to inorganic and organic salts of a compound of the present invention. These salts can be prepared in situ during the final isolation and purification of a compound, or by separately reacting the compound, or prodrug with a suitable organic or inorganic acid or base and isolating the salt thus formed.
- Representative salts include the hydrobromide, hydrochloride, hydro'iodide, sulfate, bisulfate, nitrate, acetate, trifluoroacetate, oxalate, besylate, palmitiate, pamoate, malonate, stearate, laurate, malate, borate, benzoate, lactate, phosphate, hexafluorophosphate, benzene sulfonate, tosylate, formate, citrate, maleate, fumarate, succinate, tartrate, naphthylate, mesylate, glucoheptonate, lactobionate, and laurylsulfonate salts, and the like.
- a preferred salt of the compounds of the present invention is the hydrochloride salt.
- the salts may include cations based on the alkali and alkaline earth metals, such as sodium, lithium, potassium, calcium, magnesium, and the like, as well as non-toxic ammonium, quaternary ammonium, and amine cations including, but not limited to, ammonium, tetramethylammonium, tetraethylammonium, - -
- methylamine dimethylamine, trimethylamine, triethylamine, ethylamine, and the like. See, e.g., B ⁇ rge, et al., J. Pharm. Sd., 66, 1-19 (197,7).
- prodrug means a compound that is transformed in vivo to yield a compound of Formula (I) or a pharmaceutically acceptable salt, hydrate or solvate of the compound. The transformation may occur by various mechanisms, such as . through hydrolysis in blood.
- a discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A.C.S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
- a prodrug can comprise an ester formed by the replacement of the hydrogen atom of the acid group with a group, such as (CrC 8 )alkyl, (C 2 - Ci 2 )alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1-(alkanoyloxy)-ethyl having from 5 to 10' carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-
- a group such as (CrC 8 )alkyl, (C 2 - Ci 2 )alkanoyloxymethyl, 1-(alkanoyloxy)ethyl having from 4 to 9 carbon atoms, 1- methyl-1-(alkanoyloxy)-ethyl having from 5 to 10' carbon atoms, alkoxycarbonyloxymethyl having from 3 to 6 carbon atoms, 1-
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as (Ci-C 6 )alkanoyloxymethyl, 1-((C 1 - C 6 )alkanoyloxy)ethyl, 1-methyl-1-((Ci-C 6 )alkanoyloxy)ethyl, (C 1 - C 6 )alkoxycarbonyloxymethyl, N-(CrC 6 )alkoxycarbonylaminomethyl, succinoyl, (C 1 - C 6 )alkanoyl, ⁇ -amino(CrC 4 )alkanoyl, arylacyl and ⁇ -aminoacyl, or ⁇ -aminoacyl- ⁇ - aminoacyl, where each ⁇ -aminoacyl group is selected from the naturally occurring L- amino acids, P(O)(OH) 2 , P(O)(O(C
- a prodrug can be formed by the replacement of a hydrogen atom in the amine group with a group such as R-carbonyl, RO-carbonyl, NRR'-carbonyl where R and R' are each independently (C r C 10 )alkyl, (C 3 -C 7 )cycloalkyl, benzyl, or R-carbonyl is a , natural ⁇ -aminoacyl or natural ⁇ -aminoacyi-natural cc ⁇ aminoacyl, -C(OH)C(O)OY' wherein Y' is H, (C r C 6 )alkyl or benzyl, -C(OY 0 )Yi wfiereirt Y 0 is (C 1 -C 4 ) alkyl and Y 1 is (C r C 6 )alkyl, carboxy(C r C 6 )alkyl, amino(C r
- the compounds of the present invention may contain asymmetric or chiral centers, and, therefore, exist in different stereoisomer ⁇ forms. It is intended that all stereoisomeric forms of the compounds of the present invention as well as mixtures thereof, including racemic mixtures, form part of the present invention.
- the present invention embraces all geometric and positional isomers. For exampl ⁇ , if a compound of the present invention incorporates a double bond or a fused ring, both , the , c/s- and trans- forms, as well as mixtures, are embraced within the scope of the invention. Both the single positional isomers and mixture of positional isomers resulting from the N-oxidation of the nitrogen containing heterocyclic rings are also within the scope of the present invention.
- Diastereomeric mixtures can be separated into their individual diaster.eoisomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereoisomers and converting (e.g., hydrolyzing) the individual diastereoisomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride
- converting e.g., hydrolyzing
- some of the compounds of the present invention may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention.
- the compounds of the present invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- the compounds of the present invention may exist in different tautomeric forms, and all such forms are embraced within the scope of the invention.
- all of the tautomeric forms of the triazinone moiety are included in the invention.
- all keto-enol and imine-enamine forms of the compounds are included in the invention.
- the present invention also embraces isotopically-labeled compounds of the present invention which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number , different from the atomic mass or m.ass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, iodine, and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 13 N, 15 N, 15 0, 17 0, 18 0, 31 P, 32 P, 35 S, 18 F, 123 1, 125 I and 36 CI, respectively.
- Certain isotopically-labeled compounds of the present invention are useful in compound and/or substrate tissue distribution assays.
- Tritiated (i.e., 3 H) and carbon-14 (i.e., 14 C) isotopes are particularly preferred for their ease of preparation and detectability.
- substitution with heavier isotopes such as deuterium (i.e., 2 H) ,may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) ,and hence may be preferred in some circumstances.
- Positron emitting isotopes such as 15 0, 13 N, 11 C, and 18 F are useful for positron er ⁇ ission tomography (PET) studies to examine substrate receptor occupancy
- lsotopically i labeled compounds of the present invention can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herein below, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
- Another aspect of the present invention is a method of treating diseases, conditions and/or disorders modulated by cannabinoid receptor antagonists in an animal that includes administering to an animal in need of such treatment a therapeutically effective amount of a compound of the present invention or a pharmaceutical composition comprising an effective amount of a compound of the present invention and a pharmaceutically acceptable excipient, diluent, or carrier.
- the method is particularly useful for treating diseases, conditions and/or disorders modulated by cannabinoid receptor (in particular, CB1 receptor) antagonists.
- cannabinoid receptor in particular, CB1 receptor
- Preliminary investigations have indicated that the following diseases, conditions, and/or disorders are modulated by cannabinoid receptor antagonists: eating disorders (e.g., binge eating disorder, anorexia, and bulimia), weight loss or . -3 -
- control e.g., reduction in calorie or food intake, and/or appetite suppression
- obesity depression, atypical depression, bipolar disorders, psychoses, schizophrenia, behavioral addictions, suppression of reward-related behaviors (e.g., conditioned place avoidance, such as suppression of cocaine- and morphine-induced conditioned place preference), substance abuse; addictive disorders, impuls ' ivity, alcoholism (e.g., alcohol 'abuse, addiction and/or dependence includipg treatment for abstinence, craving reduction and relapse prevention of alcohol intake), tobacco abuse (e.g., smoking addiction, cessation and/or dependence including treatment for craving reduction and relapse prevention of tobacco smoking), dementia (including memory loss, Alzheimer's disease, dementia of aging, vascular dementia, mild cognitive impairment, age-related cognitive decline, and mild neurocognitive disorder), sexual dysfunction in males (e.g., erectile difficulty), seizure disorders, epilepsy, /inflammation, gastrointestinal disorders (e.g., dysfunction of gastrointestinal . motility or intestinal
- the compounds of the present invention are useful in treating diseases, conditions, or disorders that are modulated by cannabinoid receptor antagonists. Consequently, the compounds of the present invention . (including the compositions and processes used therein) may be used in, the manufacture of a medicament for the therapeutic applications described herein.
- disorders associated with impulsive behaviours such as, disruptive behaviour disorders (e.g., anxiety/depression, executive function improvement, tic disorders, conduct disorder and/or oppositional defiant disorder), adult personality disorders (e.g., borderline personality disorder and antisocial personality disorder), diseases associated with impulsive behaviours (e.g., substance abuse, paraphilias and self-mutilation), and impulse control disorders (e.g., intermittene explosive disorder, kleptomania, pyromania, pathological gambling, and trichotillomania)), obsessive compulsive disorder, chronic fatigue syndrome, sexual dysfunction in males (e.g., premature ejaculation), sexual dysfunction in females, disorders of sleep (
- the compounds of the present invention described herein are useful in treating diseases, conditions, or disorders that are modulated by cannabinoid receptor antagonists. Consequently, the compounds of the present invention (including the compositions and processes used therein) may be used in the manufacture of a medicament for the therapeutic applications described herein.
- the compounds of the present invention can be administered to a patient at dosage levels in the range of from about 0.7 mg to about 7,000 mg per day.
- dosage levels in the range of from about 0.7 mg to about 7,000 mg per day.
- a dosage in the range of from about 0.01 mg to about 100 mg per kilogram body weight is typically sufficient.
- some variability in the general dosage range may be required depending on
- sustained release, controlled release, and delayed release formulations which forms are also well known to one of ordinary skill in the art.
- the compounds of this invention may also be used in conjunction with other pharmaceutical agents for the treatment of the diseases, conditions and/or disorders described herein. Therefore, methods of treatment that include administering
- Suitable pharmaceutical agents that may be used in combination with the compounds of the present invention include anti-obesity agents such as apolipoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, 11 ⁇ -hydroxy steroid dehydrogenase-1 (11 ⁇ -HSD type 1) inhibitors, peptide
- anti-obesity agents such as apolipoprotein-B secretion/microsomal triglyceride transfer protein (apo-B/MTP) inhibitors, 11 ⁇ -hydroxy steroid dehydrogenase-1 (11 ⁇ -HSD type 1) inhibitors, peptide
- MCR-4 agonists cholecystokinin-A (CCK-A) agonists, monoamine reuptake inhibitors (such as sibutramine), sympathomimetic agents, ⁇ 3 adrenergic receptor agonists, dopamine agonists (such as bromocriptine), melanocyte-stimulating hormone receptor analogs, 5HT2c agonists, melanin concentrating hormone antagonists, leptin (the OB protein), leptin analogs, leptin - -
- Neuropeptide-Y receptor antagonists e.g., NPY Y5 receptor antagonists, such as the spiro compounds described in US Patent Nos. 6,566,367; 6,649,624; 6,638,942; 6,605,720; 6,495,559; 6,462,053; 6,388,077; 6,335,345; and 6,326,375; US Publication Nos. 2002/0151456 and 2003/036652; and PCT Publication Nos. WO 03/010175.
- thyromimetic agents dehydroepia ⁇ droster ⁇ ne or an analog thereof, glucocorticoid receptor agonists or antagonists, orexin receptor antagonists, glucagon-like peptide-1 receptor agonists, ciliary neurotrophic factors (such as AxokineTM available from Regeneron Pharmaceuticals, Inc., Tarrytown, NY and
- anti-obesity agents selected from the group consisting of orlistat, sibutramine, bromocriptine, ephedrine, leptin, pseudoephedrine; peptide YY 3-36 Or an analog thereof; and 2-oxo-N-(5-phenylpyrazinyl)spiro- [is X Beingzofuran-1 (3H),4'-piperidine]-1 '-carboxamide.
- compo ⁇ nds of the present invention and combination therapies are administered in conjunction with exercise and a sensible diet.
- anti-obesity agents for use in the combinations, pharmaceutical compositions, and methods of the invention can be prepared using methods known to one of ordinary skill in the art, for example, sibutramine can be prepared as described in U.S. Pat. No. 4,929,629; bromocriptine can be prepared as described in U.S. Pat. Nos. 3,752,814 and 3,752,888; orlistat can be prepared as described in U.S. Pat. Nos. 5,274,143; 5,420,305; 5,540,917; and 5,643,874; PYY 3 - 36 (including analogs) can be prepared as described in US Publication No.
- NPY Y5 receptor antagonist 2-o ⁇ o-N- (5-phenylpyrazinyl)spiro[isobenzofuran-1(3H),4'-piperidine]-1 '-carboxamide can be prepared as described in US Publication No. 2002/0151456.
- Other useful NPY Y5 receptor antagonists include those described in PCT Publication No.
- 03/082190 such as 3-oxo-N-(5-phenyl-2-pyrazinyl)-spiro[isobenzofuran-1(3H), 4'-piperidine]-1'- carboxamide; 3-oxo-N-(7-trifluoromethylpyrido[3,2-b]pyridin-2-yl)-spiro- - -
- tobacco abuse e.g., nicotine receptor partial agonists, bupropion hypochloride (also known under the tradename ZybanTM) and nicotine replacement therapies
- agents to treat erectile dysfunction
- agents for reducing alcohol withdrawal symptoms may also be co-administered, such as benzodiazepines, beta-blockers, clonidine, carbamazepine, pregabalin, and gabapentin (NeurontinTM).
- Treatment for alcoholism is preferably administered in combination with behavioral therapy including such components as motivational enhancement therapy, cognitive - -
- antihypertensive agents include antihypertensive agents; anti-inflammatory agents (e.g., COX-2 inhibitors); antidepressants (e.g., fluoxetine hydrochloride (ProzacTM)); 1 cognitive improvement agents (e.g., donepezil hydrochloride (AirceptTM) and other acetylcholinesterase inhibitors); neuroprotective -agents (e.g., memantine); antipsychotic medications (e.g., zi'prasidone (GeodonTM), risperidone (RisperdalTM), and olanzapine (ZyprexaTM)); insulin and insulin analogs (e.g., LysPro insulin); GLP-1 (7-37) (insulinotropin) and GLP-1 (7-36)-NH 2 ; sulfonylureas and analogs thereof: chlorpropamide, glibenclamide, tolbutamide, tolazamide, acetohexamide, Glypizide
- the compounds of the present invention may also be administered in combination with a naturally occurring compound that acts to lower plasma cholesterol levels.
- a naturally occurring compound that acts to lower plasma cholesterol levels.
- Such naturally occurring compounds are commonly called nutraceuticals and include, for example, garlic extract, Hoodia plant extracts, and niacin.
- the dosage of the additional pharmaceutical agent e.g., anti-obesity agent
- the dosage range of an anti-obesity agent is in the range of from about 0.001 mg to about 100 mg per kilogram body weight of the individual per day, preferably from about 0.1 mg to about 10 mg per kilogram body weight of the individual per day.
- some variability in the general dosage ' range may also be required depending upon the age and weight of the subject being treated, the intended route of administration, the particular anti-obesity agent being administered and the like.
- the determination of dosage ranges arid optimal dosages for a particular patient is also well within the ability of one of ordinary skill in the art having the benefit of the instant disclosure.
- the compounds of tihe present invention are useful for treating diseases, conditions and/or disorders modulated by cannabinoid receptor antagonists; therefore, another embodiment of the present invention is a pharmaceutical comp ⁇ sition comprising a therapeutically effective amount of a compound of the present invention and a pharmaceutically acceptable excipient, diluent or carrier.
- a compound of the present invention may be , administered in combination with at least one additional pharmaceutical agent
- a compound of the present invention or a combination can be administered in any conventional oral, rectal, transdermal, parenteral, (for example, intravenous, intramuscular, or subcutaneous) intracisternal, intravaginal, intraperitoneal, intravesical, local (for example, powder, ointment or drop), or buccal, or nasal, dosage form.
- the compound of the present invention and at least one other pharmaceutical agent e.g., anti-obesity agent described above
- a combination When a combination is administered, such administration can be sequential in time or simultaneous with the simultaneous method being generally preferred.
- the combination can be administered in any order. It is , generally preferred that such administration be oral., Jt is especially preferred that ⁇ such administration be oral and simultaneous.
- the administration of the compound of the present invention and the additional pharmaceutical agent can'be by the same or by different methods.
- a typical formulation is prepared by mixing a, compound of the present invention and a excipient, diluent or carrier.
- Suitable excipiehts, diluents and carriers are well known to those skilled in the art and include materials such as carbohydrates, waxes, water soluble and/or swellable polymers, hydrophilic or hydrophobic materials, gelatin, oils, solvents, water, and the like.
- the particular excipient, diluent or carrier used will depend upon the means and purpose for whiph the compound of the present invention is being applied.
- Solvents are generally selected based on solvents 'recognized by persons skilled in the art as safe (GRAS) to be administered to a mammal.
- safe solvents are non-toxic aqueous solvents such as water and other non-toxic solvents that are soluble or miscible in water.
- Suitable aqueous solvents include water, ethanol, propylene glycol,
- the formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents; emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- the formulations may be prepared using conventional dissolution and mixing procedures.
- compositions suitable for parenteral injection generally include pharmaceutically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions, or emulsions.
- compositions generally include sterile excipients, diluents or carriers for reconstitution into sterile injectable solutions or dispersions.
- aqueous and nonaqueous excipients, diluents or carriers examples include water, ethan'ol, polyols (propylene glycol, polyethylene glycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate.
- a coating such as lecithin
- surfactants for example
- compositions may also contain adjuvants such as preserving, wetting, emulsifying, and dispersing agents. Prevention of microorganism contamination of the compositions can be accomplished with various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example, sugars, sodium chloride, and the like. Prolonged absorption of injectable pharmaceutical compositions can be brought about by the use of agents capable of delaying absorption, for example, aluminum monostearate and gelatin.
- Solid dosage forms for oral administration include capsules, tablets, powders, and granules.
- a compound of the present invention or a combination is admixed with' at least one pharmaceutically acceptable excipient, diluent or carrier.
- Suitable excipients, diluents, or carriers include sodium citrate or dicalcium phosphate, or (a) fillers or extenders (e.g., starches, lactose, sucrose, mannitol, silicic acid and the like); (b) binders (e.g., carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose, acacia and the like); (c) humectants (e.g., glycerol and the like); (d) disintegrating agents (e.g., agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain complex silicates, sodium carbonate and the like); (e) solution retarders (e.g., paraffin and the like); (f) absorption accelerators (e.g., quaternary ammonium compounds and the like); (g) wetting agents (e.g., cetyl alcohol, glycerol mono
- compositions of a similar type may also be used as fillers in soft or hard filled gelatin capsules using such excipients as lactose or milk sugar, as well as high molecular weight polyethylene glycols, and the like.
- Solid dosage forms such as tablets, dragees, capsules, and granules can be prepared with coatings and shells, such as enteric coatings and others well known in the art. They may also contain opacifying agents, and can also be of such -3 -
- compositions that they release the compound of the present invention and/or the , additional pharmaceutical agent in a delayed manner.
- embedding compositions that can be used are polymeric substances and waxes.
- the drug can also be in micro-encapsulated form, if appropriate, with one or more of the above- mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs.
- the liquid dosage form may contain excipients, diluents or carriers.
- Suitable excipients, diluents or carriers include additives such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, bepzyl alcohol, benzyl benzoate, propylene glycol, 1 ,3-butylene glycol, dimethylformamide, , oils (e.g., cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil, sesame seed oil and the like), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, or mixtures of these substances, and the like.
- additives such as water or other solvents, solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate,
- suitable additives include wetting agents, emulsifying and suspending agents, sweetening, flavoring, and perfuming agents.
- Suspensions in addition, to the compound of the present invention or the ' combination, may further comprise suspending agents, e.g., ethoxylated,isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar, and tragacanth, or mixtures of these substances, and the like.
- compositions for rectal or vaginal administration preferably comprise suppositories, which can be prepared by mixing a compound of the present invention or a combination with suitable non-irritating excipients, diluents, or carriers, such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ordinary room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity thereby releasing the active component(s).
- suitable non-irritating excipients, diluents, or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at ordinary room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity thereby releasing the active component(s).
- Dosage forms for topical administration of the compounds of the present invention and combinations of the compounds of the present invention with anti- obesity agents may comprise ointments, powders, sprays and inhalants.
- the drugs are admixed under sterile condition with a pharmaceutically acceptable excipient, diluent or carrier, and any preservatives, buffers, or propellants that may be required.
- Ophthalmic formulations, eye ointments, powders, and solutions are also intended to be included .within the scope of the present invention.
- an article for distribution includes a container having deposited therein the pharmaceutical formulation in an appropriate form.
- suitable containers are well-known to those skilled in the art and include materials such as bottles (plastic and glass), sachets, ampoules, plastic bags, metal cylinders, and the like.
- the container may also include a tamper-proof assemblage to prevent indiscreet access to the contents of the package.
- the container has deposited thereon a label that describes the contents of the container. The label may also include appropriate warnings.
- a compound of the present invention or combination i.e., a compound of the present invention with at least one additional pharmaceutical agent
- administration of a compound of the present invention or combination can be effected orally or non-orally (e.g., by injection).
- a daily dose that is administered orally to an animal is between about 0.01 and about 1 ,000 mg/kg of body weight, preferably between about 0.01 and about 300 mg/kg of body weight.
- a compound of the present invention (or combination) can be carried in the drinking water so that a therapeutic dosage of the compound is ingested with the daily water supply.
- the compound can be directly metered into drinking water, preferably in the form of a liquid, water-soluble concentrate (such as an aqueous solution of a water-soluble salt).
- a compound of the present invention can also be added directly to the feed, as such, or in the form of an animal feed supplement, also referred to as a premix or concentrate.
- a premix or concentrate of the compound with an excipient, diluent or carrier is more commonly employed for the inclusion of the agent in the feed.
- Suitable carriers are liquid or solid, as desired, - -
- a particularly effective carrier is the respective animal feed itself; that is, a small portion of such feed.
- the carrier facilitates uniform distribution of the compound in the finished feed with which the premix is blended.
- the compound is thoroughly blended into the premix and, subsequently, the feed.
- the compound may be dispersed or dissolved in a suitable oily vehicle such as soybean oil, corn oil, cottonseed oil, and the like, or in a volatile organic solvent and then blended with the carrier.
- a suitable oily vehicle such as soybean oil, corn oil, cottonseed oil, and the like, or in a volatile organic solvent and then blended with the carrier.
- High potency concentrates may be blended by the feed manufacturer with proteinaceous carrier such as soybean oil meal and other meals, as described above, to produce concentrated supplements, which are suitable for direct feeding to animals.
- the animals are pe,rmit,ted to consume the usual diet.
- concentrated supplements may be added directly to the feed to produce a nutritionally balanced, finished feed containing a therapeutically effective level of a compound of the present invention.
- the mixtures are thoroughly blended by standard procedures, such as in a twin shell blender, to ensure homogeneity.
- Drinking water and feed effective for increasing lean meat deposition and for improving lean meat to fat ratio are generally prepared by mixing a compound of the present invention with a sufficient amount of animal feed to provide from about 1CT 3 to about 500 ppm of the compound in the feed or water.
- the preferred medicated swine, cattle, sheep and goat feed generally contain from about 1 to about 400 grams of a compound of the present invention (or combination) per ton of feed, the optimum amount for these animals usually being about 50 to about 300 grams per ton of feed.
- the preferred poultry and domestic pet feeds usually contain about 1 to about 400 grams and preferably about 10 to about 400 grams of a compound of the present invention (or combination) per ton of feed. - -
- the compounds of the present hi invention may be prepared in the form of a paste or a pellet and administered as an implant, usually under the skin of the head or ear of the animal in which increase in lean meat deposition and improvement in lean meat to fat ratio is sought.
- parenteral administration involves injection of a sufficient amount of a compound of the present invention (or combination) to provide the animal with about 0.01 to about 20 mg/kg/day of body weight of the drug.
- the preferred dosage for poultry, swine, cattle, sheep, goats and domestic pets is in the range of from about 0.05 to about 10 mg/kg/day of body weight of drug.
- Paste formulations can be prepared by dispersing the drug in a pharmaceutically acceptable oil such as peanut oil, sesame oil, corn oil or the like.
- Pellets containing an effective amount of a compound of the present invention, pharmaceutical composition, or combination can be prepared by admixing a compound of the present invention, or combination with a diluent such as carbowax, carnuba wax, and the like, and a lubricant, such as magnesium or calcium stearate, can be added to imprqve the pelleting process.
- a diluent such as carbowax, carnuba wax, and the like
- a lubricant such as magnesium or calcium stearate
- pellets may be administered to an animal to achieve the desired dose level which will provide the increase in lean , meat deposition and improvement in lean meat to fat ratio desired.
- implants may also be made periodically during the animal treatment period in order to maintain the proper drug level in the animal's body.
- the present invention has several advantageous veterinary features.
- the instant invention provides the means by which this may be accomplished.
- utilization of the method of the present invention yields leaner animals that command higher sale prices from the meat industry.
- starting materials are generally available from commercial sources such as Aldrich Chemicals Co. (Milwaukee, Wl), Lancaster Synthesis, Inc. (Windham, NH), Acros Organics (Fairlawn, NJ), Maybridge Chemical Compafiy, Ltd. (Cornwall, England), TygerScientific ' (Priricet ⁇ n, ' NJ), and AstraZeneca Pharmaceuticals (London, England).
- NMR spectra were recorded on a Varian UnityTM 400 or 500 (available from Varian Inc., Palo Alto, CA) at room temperature at 400 and 500 MHz 1 H, respectively. Chemical shifts are expressed in parts per million ( ⁇ ) relative to residual solvent as an internal reference.
- the peak shapes are denoted as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br s, broad singlet; v br s, very broad singlet; br m, broad multiplet; 2s, two singlets. In some cases only representative 1 H NMR peaks are given. '
- Mass spectra were recorded by direct flow analysis using positive and negative atmospheric pressure chemical ionization" (APcI) scan modes.
- a Waters ' ' APcl/MS model ZMD mass spectrometer equipped with Gilson 215 liquid handling system was used to carry out the experiments Mass spectrometry analysis was also obtained by RP-HPLC gradient method for chromatographic separation. Molecular weight identification was recorded by positive and negative electrospray ionization (ESI) scan modes.
- ESI electrospray ionization
- a Waters/Micromass ESI/MS model ZMD or LCZ mass spectrometer equipped with Gilson 215 liquid handling system and HP 1100 DAD was used to carry out the experiments.
- reaction mixture wgs concentrated in vacuo,' azeotroped with methanol, and concentrated on high vacuum.
- the solid was stirred in methanol/ 6N HCI (30 ml/ 2.5 ml) and concentrated in vacuo.
- the residue was dissolved in ethyl acetate, washed with water, brine, dried (Na 2 SO 4 ) and concentrated in vacuo to afford the title compound I -2c as a brown oil (1.0 g).
- the mixture was diluted with methanol (350 ml) filtered through Celite®, rinsing with additional methanol.
- the methanol fractions were filtered through a 0.45 Dm filter disk, and, then concentrated undenreduced pressure to give a solid residue that was triturated ifrom diethyl ether, collected by vacuum filtration, washed with ether and then dried, in vacuo, to afford l-5c (6.3 g, 91%) as a tan solid.
- a protein assay was performed and 200 ⁇ l of tissue totaling 20 ⁇ g was added to the assay.
- the test compounds were diluted in drug buffer (0.5% BSA, 10% DMSO and TME) and then 25 ⁇ l were added to a deep well polypropylene plate.
- [ 3 H] SR141716A was diluted in a ligand buffer (0.5% 'BSA plus TME) and 25 ⁇ l were added to the plate,
- a BCA protein assay was used to determine the appropriate tissue concentration and then 200 ⁇ l of rat brain tissue at the appropriate concentration was added to the plate.
- the plates were covered and placed in an incubator at 20 0 C for 60 minutes.
- a protein assay was performed and 200 ⁇ l of tissue totaling 20 ⁇ g was added to the assay. /6
- test compounds were diluted in drug buffer (0.5% BSA, 10% DMSO , and TME) and then 25 ⁇ l were added to a deep welj polypropylene plate.
- [3H] SR141716A was diluted in a ligand buffer (0.5% BSA plus TME) and 25 ⁇ l were added to the plate.
- the plates were covered and placed in an incubator at 3O 0 C for 60 minutes. At the end of the incubation period 250 ⁇ l of stop buffer (5% BSA plus TME) was added to the reaction plate.
- the plates were then harvested by Skatron onto GF/B filtermats presoaked in BSA (5 mg/ml) plus TME. Each filter was washed twice. The filters were dried overnight. In the morning the filters were counted on a Wallac BetaplateTM counter (available from PerkinElmer Life SciencesTM, Boston, MA). . .
- the homogenate was then .spun. at ,1 ,0Q0X g for 5 minut ⁇ s at 4 0 C.
- the supernatant was recovered and centrifuged at 100,000X G for 1 hour at 4°C.
- a protein assay was performed and 200 ⁇ l of tissue totaling 10 ⁇ g was added to the assay.
- test compounds were diluted in drug buffer (0.5% BSA, 10% DMSO, and 80.5% TME) and then 25 ⁇ l were added to the deep well polypropylene plate.
- [3H] CP-55940 was diluted a ligand buffer (0.5% BSA and 99.5% TME) and then 25 ⁇ l were added to each well at a concentration of 1 nM.
- a BCA protein assay was used to determine the appropriate tissue concentration and 200 ⁇ l of the tissue at the appropriate concentration was added to the plate.
- the plates were covered and placed in an incubator at 30 0 C for 60 minutes. At the end of the incubation period 250 ⁇ l of stop buffer (5% BSA plus TME) was added to the reaction plate.
- Membranes were. prepared from CHO-K1 cells stably transfected with the human CB-1 receptor cDNA. Membranes were prepared from cells as described by Bass et al, in "Identification and characterization of novel somatostatin antagonists," Molecular Pharmacology. 50, 709-715 (1996).
- GTPy [ 35 S] binding assays were performed in a 96 well FlashPlate TM format in duplicate using 100 pM GTPy[ 35 S] and 10 ⁇ g membrane per well in assay buffer composed of 50 mM Tris HCI, pH 7.4, 3 mM MgCI 2 , pH 7.4; 10 mM MgCI 2 , 20 mM EGTA, 100 mM NaCI, 30 ⁇ M GDP, 0.1 % bovine serum albumin and the following protease inhibitors: 100 ⁇ g/ml bacitracin, 100 ⁇ g/ml benzamidine, 5 ⁇ g/ml aprotinin, 5 ⁇ g/ml leupeptin.
- cyclic-AMP assay protocol using intact cells was used to determine inverse agonist activity.
- ⁇ Cells were plated into a 96-well plate at a plating density of 10,000-14,000 cells pe'r well at a concentration of 100 ⁇ l per well. , The plates were incubated for
- Serum free medium containing 1 mM IBMX was added to each well followed by 10 ⁇ l of test compound (1 :10 stock solution (25 mM compound in DMSO) into 50% DMSO/PBS) diluted 10X in PBS with 0.1% BSA. After incubating for 20 minutes at 37°C, 2 ⁇ M of Forskolin was added and then incubated for an additional 20 minutes 1 at 37°C. The media was removed, 100 ⁇ l of 0.01 N HCI was added and then incubated for 20 minutes at room temperature. Cell lysate (75 ⁇ l) along with 25 ⁇ l of assay buffer (supplied in FlashPlateTM cAMP assay kit available from NEN Life ⁇ . Science Products Boston, MA) into a. Flashplate.
- Cannabinoid agoinists such as ⁇ 9 -tetrahydrocannabinol ( ⁇ 9 -THC) and CP-55940 have been shown to affect four characteristic behaviors in mice, collectively known as the Tetrad.
- Tetrad ⁇ 9 -tetrahydrocannabinol
- the rats were individually housed and fed powdered chow. They were maintained on a 12 hour light/dark cycle and received food and water ad , libitum. The animals were acclimated to the vivarium for a period of one week before testing was conducted. Testing was completed during the light portion of the cycle.
- rats were transferred to individual test cages without food the aftemoop prjor to testing, and the rats were fasted overnight. After the overnight fast, rats- were dosed the following morning wtth vehicle or test compounds.
- a known antagonist was dosed (3 mg/,kg) as a positive control, and a control group received vehicle alone (no compound).
- the test compounds were dosed at ranges between 0.1 and 100 mg/kg depending upon the compound.
- the standard vehicle was 0.5% (w/v) methylcellulose in water and the standard route of administration was oral. However, different vehicles and routes of administration were used to accommodate various compounds when required.
- Food was provided to the rats 30 minutes after dosing and the Oxymax automated food intake system (Columbus Instruments, Columbus, Ohio) was started.
- mice were individually housed and, given unlimited access to powdered rat chow, water and a 10 % (w/v) alcohol solution. After 2-3 weeks of unlimited access, wate.r was restricted for 20 hours and alcohol was restricted to only 2 hours access daily. This was done in a manner that the access period was the last 2 hours qf the dark part of the light cycle.
- mice were considered stable when the average alcohol consumption for 3 days was ⁇ 20% of the average for all 3 days.
- day 2 and 3 mice were injected with vehicle or test compound and the same protocol as the , previous day was followed. ' Day 4 was wash out and no injections were given. Data was analyzed using repeated measures ANOVA. Change in water or alcohol consumption was compared back to vehicle for each day of the test. Positive results would be interpreted as a compound that was able to significantly reduce alcohol consumption while having no effect on water , ,
- Oxygen Consumption Methods Whole body oxygen consumption is measured using an indirect calorimeter
- Rats (Oxymax from Columbus Instruments, Columbus, OH) in male Sprague Dawley rats (if another rat strain or female rats are used, it will be specified). Rats (300-38Og body weight) are placed in the calorimeter chambers and the chambers are placed in activity monitors. These studies are done during the light cycle. Prior to the measurement of oxygen consumption, the rats are fed standard chow ad libitum. During the measurement of oxygen consumption, food is not available. Basal pre- dose oxygen consumption and ambulatory activity are measured every 10 minutes for 2.5 to 3 hours.
- the chambers are opened and the animals are administered a single dose of compound (the usual dose range is 0.001 to 10 mg/kg) by oral gavage (or other route of administration as specified, i.e. s.c, i.p., Lv.).
- Drugs are prepared in methylcellulose, water or other specified vehicle (examples include PEG400, 30% beta-cyclodextran and propylene - -
- Oxygen consumption and ambulatory activity are measured every 10 minutes for an additional 1-6 hours post-dosing.
- the Oxymax calorimeter software calculates the oxygen consumption (ml/kg/h) based on the flow rate of air through the chambers and difference in oxygen content at inlet and output ports.
- the activity monitors have 15 infrared' light beams spaced one inch apart on each axis, ambulatory activity is recorded when two consecutive beams are broken and the results are recorded as counts.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Psychiatry (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Addiction (AREA)
- Nutrition Science (AREA)
- Hospice & Palliative Care (AREA)
- Psychology (AREA)
- Child & Adolescent Psychology (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Reproductive Health (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description












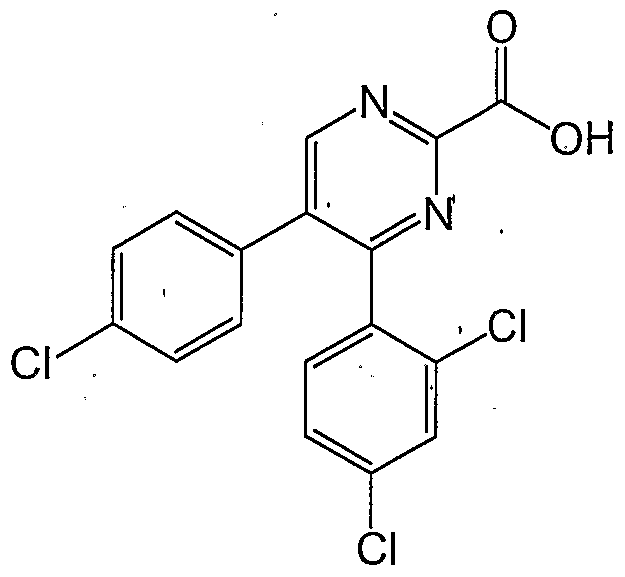




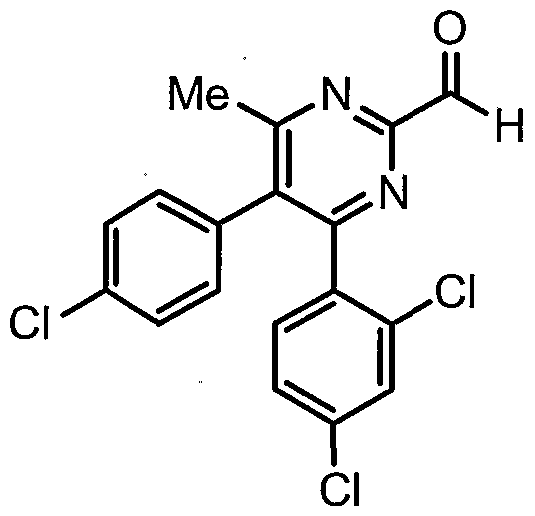













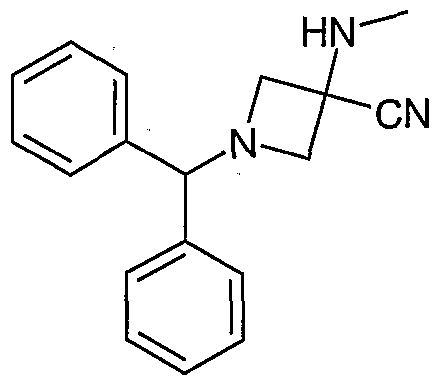

















Claims


Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006516548A JP2006527759A (en) | 2003-06-18 | 2004-06-09 | Pyrimidine derivatives as cannabinoid receptor ligands |
| BRPI0411617-8A BRPI0411617A (en) | 2003-06-18 | 2004-06-09 | pyrimidine derivatives as cannabinoid receptor ligands |
| CA002529068A CA2529068A1 (en) | 2003-06-18 | 2004-06-09 | Pyrimidine derivatives as cannabinoid receptor ligands |
| MXPA05013282A MXPA05013282A (en) | 2003-06-18 | 2004-06-09 | Pyrimidine derivatives as cannabinoid receptor ligands. |
| EP04736431A EP1638570A1 (en) | 2003-06-18 | 2004-06-09 | Pyrimidine derivatives as cannabinoid receptor ligands |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47974603P | 2003-06-18 | 2003-06-18 | |
| US60/479,746 | 2003-06-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004110453A1 true WO2004110453A1 (en) | 2004-12-23 |
Family
ID=33551899
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2004/001971 Ceased WO2004110453A1 (en) | 2003-06-18 | 2004-06-09 | Pyrimidine derivatives as cannabinoid receptor ligands |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20040259887A1 (en) |
| EP (1) | EP1638570A1 (en) |
| JP (1) | JP2006527759A (en) |
| BR (1) | BRPI0411617A (en) |
| CA (1) | CA2529068A1 (en) |
| MX (1) | MXPA05013282A (en) |
| WO (1) | WO2004110453A1 (en) |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7094572B2 (en) | 2003-03-14 | 2006-08-22 | Bristol-Myers Squibb | Polynucleotide encoding a novel human G-protein coupled receptor variant of HM74, HGPRBMY74 |
| FR2888237A1 (en) * | 2005-07-08 | 2007-01-12 | Sanofi Aventis Sa | N - [(4,5-DIPHENYLPYRIMIDIN-2-YL) METHYL] AMINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE |
| US7229999B2 (en) | 2005-04-06 | 2007-06-12 | Hoffmann-La Roche Inc. | Pyridine-3-carboxamide derivatives as CB1 inverse agonists |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US7405221B2 (en) | 2002-09-27 | 2008-07-29 | Merck & Co., Inc. | Substituted pyrimidines |
| WO2008084081A3 (en) * | 2007-01-11 | 2008-10-09 | Basf Se | 2-substituted 5-phenylpyrimidines for the treatment of proliferative disorders |
| WO2008130953A3 (en) * | 2007-04-17 | 2008-12-11 | Abbott Lab | 2-imin0is0thiaz0le derivatives as cannabinoid receptor ligands |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US7553861B2 (en) | 2005-04-22 | 2009-06-30 | Alantos Pharmaceuticals Holding, Inc. | Dipeptidyl peptidase-IV inhibitors |
| US7629346B2 (en) | 2006-06-19 | 2009-12-08 | Hoffmann-La Roche Inc. | Pyrazinecarboxamide derivatives as CB1 antagonists |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US7781593B2 (en) | 2006-09-14 | 2010-08-24 | Hoffmann-La Roche Inc. | 5-phenyl-nicotinamide derivatives |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| US8410107B2 (en) | 2010-10-15 | 2013-04-02 | Hoffmann-La Roche Inc. | N-pyridin-3-yl or N-pyrazin-2-yl carboxamides |
| US8501794B2 (en) | 2007-04-17 | 2013-08-06 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8569282B2 (en) | 2007-12-11 | 2013-10-29 | Cytopathfinder, Inc. | Carboxamide compounds and their use |
| US8669254B2 (en) | 2010-12-15 | 2014-03-11 | Hoffman-La Roche Inc. | Pyridine, pyridazine, pyrimidine or pyrazine carboxamides as HDL-cholesterol raising agents |
| US8735434B2 (en) | 2007-05-18 | 2014-05-27 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8841334B2 (en) | 2006-05-31 | 2014-09-23 | Abbvie Inc. | Compounds as cannabinoid receptor ligands and uses thereof |
| US8846730B2 (en) | 2008-09-08 | 2014-09-30 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8859596B2 (en) | 2008-09-16 | 2014-10-14 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8865753B2 (en) | 2007-03-28 | 2014-10-21 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8895592B2 (en) | 2008-12-16 | 2014-11-25 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US9006275B2 (en) | 2006-05-31 | 2015-04-14 | Abbvie Inc. | Compounds as cannabinoid receptor ligands and uses thereof |
| US9193713B2 (en) | 2007-10-12 | 2015-11-24 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| EP4392418A4 (en) * | 2021-08-24 | 2025-06-18 | The Johns Hopkins University | COMPOSITIONS AND METHODS FOR TREATING DISEASES ASSOCIATED WITH PROTEOTOXICITY |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN100445269C (en) * | 2003-04-04 | 2008-12-24 | 巴斯福股份公司 | 2-substituted pyrimidines |
| US7151097B2 (en) | 2003-11-07 | 2006-12-19 | Pfizer Inc. | Bicyclic pyrazolyl and imidazolyl compounds and uses thereof |
| CA2606288A1 (en) * | 2005-04-18 | 2006-10-26 | Neurogen Corporation | Subtituted heteroaryl cb1 antagonists |
| KR100970055B1 (en) * | 2005-06-22 | 2010-07-16 | 에프. 호프만-라 로슈 아게 | 6- (Fluoro-benzo [1,3] dioxylyl) -morpholin-4-yl-methanone and its use as CVII ligands |
| DE102006021874B4 (en) * | 2006-05-11 | 2008-03-27 | Sanofi-Aventis | 4,5-diphenyl-pyrimidinyl-amino substituted carboxylic acids, process for their preparation and their use as medicaments |
| DE102006021872B4 (en) * | 2006-05-11 | 2008-04-17 | Sanofi-Aventis | 4,5-Diphenyl-pyrimidinyl-oxy or -mercapto substituted carboxylic acids, process for their preparation and their use as medicaments |
| WO2008156607A1 (en) * | 2007-06-12 | 2008-12-24 | Neurogen Corporation | Substituted pyrimidinones |
| FR2948120A1 (en) * | 2009-07-16 | 2011-01-21 | Sanofi Aventis | 5,6-BISARYL-2-PYRIMIDINE CARBOXAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE AS ANTAGONISTS OF UROTENSIN II RECEPTORS |
| CN107531721B (en) * | 2015-04-30 | 2020-07-17 | 诺华股份有限公司 | Fused tricyclic pyrazole derivatives for modulating farnesoid X receptors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003027076A2 (en) * | 2001-09-21 | 2003-04-03 | Solvay Pharmaceuticals B.V. | 1h-imidazole derivatives having cb1 agonistic, cb1 partial agonistic or cb1- antagonistic activity |
| WO2004018433A1 (en) * | 2002-08-21 | 2004-03-04 | Glaxo Group Limited | Pyrimidine derivatives and their use as cb2 modulators |
| WO2004029204A2 (en) * | 2002-09-27 | 2004-04-08 | Merck & Co., Inc. | Substituted pyrimidines |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8613591D0 (en) * | 1986-06-04 | 1986-07-09 | Roussel Lab Ltd | Chemical compounds |
| ES2037739T3 (en) * | 1986-10-22 | 1993-07-01 | Ciba-Geigy Ag | DERIVATIVES OF THE 1,5-DIPHENYLPIRAZOLE-3-CARBOXYLIC ACID FOR THE PROTECTION OF CROP PLANTS. |
| PH27357A (en) * | 1989-09-22 | 1993-06-21 | Fujisawa Pharmaceutical Co | Pyrazole derivatives and pharmaceutical compositions comprising the same |
| FR2665898B1 (en) * | 1990-08-20 | 1994-03-11 | Sanofi | DERIVATIVES OF AMIDO-3 PYRAZOLE, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2692575B1 (en) * | 1992-06-23 | 1995-06-30 | Sanofi Elf | NOVEL PYRAZOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2714057B1 (en) * | 1993-12-17 | 1996-03-08 | Sanofi Elf | New derivatives of 3-pyrazolecarboxamide, process for their preparation and pharmaceutical compositions containing them. |
| US5596106A (en) * | 1994-07-15 | 1997-01-21 | Eli Lilly And Company | Cannabinoid receptor antagonists |
| FR2741621B1 (en) * | 1995-11-23 | 1998-02-13 | Sanofi Sa | NOVEL PYRAZOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME |
| FR2742148B1 (en) * | 1995-12-08 | 1999-10-22 | Sanofi Sa | NOVEL PYRAZOLE-3-CARBOXAMIDE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| DE69822449T2 (en) * | 1997-01-21 | 2005-01-27 | Smithkline Beecham Corp. | NEW CANNABINOID RECEPTOR MODULATORS |
| FR2758723B1 (en) * | 1997-01-28 | 1999-04-23 | Sanofi Sa | USE OF CENTRAL CANNABINOID RECEPTOR ANTAGONISTS FOR THE PREPARATION OF DRUGS |
| FR2761266B1 (en) * | 1997-03-28 | 1999-07-02 | Sanofi Sa | PHARMACEUTICAL COMPOSITION FORMED BY WET GRANULATION FOR THE ORAL ADMINISTRATION OF A DERIVATIVE OF N-PIPERIDINO-3- PYRAZOLECARBOXAMIDE, ITS SALTS AND THEIR SOLVATES |
| IL141769A0 (en) * | 1998-09-11 | 2002-03-10 | Aventis Pharma Sa | Azetidine derivatives, preparation and medicines containing them |
| FR2789079B3 (en) * | 1999-02-01 | 2001-03-02 | Sanofi Synthelabo | PYRAZOLECARBOXYLIC ACID DERIVATIVE, ITS PREPARATION, PHARMACEUTICAL COMPOSITIONS CONTAINING SAME |
| WO2001058869A2 (en) * | 2000-02-11 | 2001-08-16 | Bristol-Myers Squibb Company | Cannabinoid receptor modulators, their processes of preparation, and use of cannabinoid receptor modulators in treating respiratory and non-respiratory diseases |
| US6479479B2 (en) * | 2000-03-03 | 2002-11-12 | Aventis Pharma S.A. | Azetidine derivatives, their preparation and pharmaceutical compositions containing them |
| US6566356B2 (en) * | 2000-03-03 | 2003-05-20 | Aventis Pharma S.A. | Pharmaceutical compositions containing 3-aminoazetidine derivatives, novel derivatives and their preparation |
| US6355631B1 (en) * | 2000-03-03 | 2002-03-12 | Aventis Pharma S.A. | Pharmaceutical compositions containing azetidine derivatives, novel azetidine derivatives and their preparation |
| UA74367C2 (en) * | 2000-03-23 | 2005-12-15 | Сольве Фармас'Ютікалз Б.В. | DERIVATIVES OF 4,5-DIHYDRO-1Н-PYRAZOLE, REVEALING ANTAGONISTIC ACTIVITY RELATIVE TO СВ<sub>1, A METHOD FOR PRODUCING THEREOF, PHARMACEUTICAL COMPOSITION AND A METHOD FOR PRODUCING THEREOF, A METHOD FOR THE TREATMENT OF DISEASES (VARIANTS) |
| US20020019421A1 (en) * | 2000-07-05 | 2002-02-14 | Roni Biberman | Compositions and therapy for substance addiction |
| US20020091114A1 (en) * | 2000-10-04 | 2002-07-11 | Odile Piot-Grosjean | Combination of a CB1 receptor antagonist and of sibutramine, the pharmaceutical compositions comprising them and their use in the treatment of obesity |
| WO2003007887A2 (en) * | 2001-07-20 | 2003-01-30 | Merck & Co., Inc. | Substituted imidazoles as cannabinoid receptor modulators |
| US6509367B1 (en) * | 2001-09-22 | 2003-01-21 | Virginia Commonwealth University | Pyrazole cannabinoid agonist and antagonists |
| US20030139386A1 (en) * | 2001-12-21 | 2003-07-24 | Sophie Cote | Pharmaceutical compositions based on azetidine derivatives |
| US6825209B2 (en) * | 2002-04-15 | 2004-11-30 | Research Triangle Institute | Compounds having unique CB1 receptor binding selectivity and methods for their production and use |
| AU2003267728A1 (en) * | 2002-10-18 | 2004-05-04 | Pfizer Products Inc. | Cannabinoid receptor ligands and uses thereof |
| US7129239B2 (en) * | 2002-10-28 | 2006-10-31 | Pfizer Inc. | Purine compounds and uses thereof |
-
2004
- 2004-05-13 US US10/846,963 patent/US20040259887A1/en not_active Abandoned
- 2004-06-09 JP JP2006516548A patent/JP2006527759A/en active Pending
- 2004-06-09 MX MXPA05013282A patent/MXPA05013282A/en unknown
- 2004-06-09 EP EP04736431A patent/EP1638570A1/en not_active Withdrawn
- 2004-06-09 BR BRPI0411617-8A patent/BRPI0411617A/en not_active Application Discontinuation
- 2004-06-09 WO PCT/IB2004/001971 patent/WO2004110453A1/en not_active Ceased
- 2004-06-09 CA CA002529068A patent/CA2529068A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003027076A2 (en) * | 2001-09-21 | 2003-04-03 | Solvay Pharmaceuticals B.V. | 1h-imidazole derivatives having cb1 agonistic, cb1 partial agonistic or cb1- antagonistic activity |
| WO2004018433A1 (en) * | 2002-08-21 | 2004-03-04 | Glaxo Group Limited | Pyrimidine derivatives and their use as cb2 modulators |
| WO2004029204A2 (en) * | 2002-09-27 | 2004-04-08 | Merck & Co., Inc. | Substituted pyrimidines |
Non-Patent Citations (1)
| Title |
|---|
| HETZHEIM A. ET AL.: "Ringtransformation von O,N-Heterocyclen", PHARMAZIE, vol. 40, no. 1, 1985, pages 17 - 20, XP002295697 * |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7405221B2 (en) | 2002-09-27 | 2008-07-29 | Merck & Co., Inc. | Substituted pyrimidines |
| US7094572B2 (en) | 2003-03-14 | 2006-08-22 | Bristol-Myers Squibb | Polynucleotide encoding a novel human G-protein coupled receptor variant of HM74, HGPRBMY74 |
| US7371822B2 (en) | 2003-03-14 | 2008-05-13 | Bristol-Myers Squibb Company | Human G-protein coupled receptor variant of HM74, HGPRBMY74 |
| US7229999B2 (en) | 2005-04-06 | 2007-06-12 | Hoffmann-La Roche Inc. | Pyridine-3-carboxamide derivatives as CB1 inverse agonists |
| US7553861B2 (en) | 2005-04-22 | 2009-06-30 | Alantos Pharmaceuticals Holding, Inc. | Dipeptidyl peptidase-IV inhibitors |
| WO2007006928A1 (en) * | 2005-07-08 | 2007-01-18 | Sanofi-Aventis | N-[(4,5-diphenylpyrimidin-2-yl)methyl]amine derivatives, the preparation thereof and their therapeutic use |
| FR2888237A1 (en) * | 2005-07-08 | 2007-01-12 | Sanofi Aventis Sa | N - [(4,5-DIPHENYLPYRIMIDIN-2-YL) METHYL] AMINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE |
| JP2009500381A (en) * | 2005-07-08 | 2009-01-08 | サノフイ−アベンテイス | N-[(4,5-diphenylpyrimidin-2-yl) methyl] amine derivatives, their preparation and their therapeutic use |
| US7541361B2 (en) | 2005-07-08 | 2009-06-02 | Sanofi-Aventis | N-[4,5-diphenylpyrimidin-2-yl)methyl]amine derivatives, the preparation thereof and their therapeutic use |
| US8841334B2 (en) | 2006-05-31 | 2014-09-23 | Abbvie Inc. | Compounds as cannabinoid receptor ligands and uses thereof |
| US9006275B2 (en) | 2006-05-31 | 2015-04-14 | Abbvie Inc. | Compounds as cannabinoid receptor ligands and uses thereof |
| US7629346B2 (en) | 2006-06-19 | 2009-12-08 | Hoffmann-La Roche Inc. | Pyrazinecarboxamide derivatives as CB1 antagonists |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| US7781593B2 (en) | 2006-09-14 | 2010-08-24 | Hoffmann-La Roche Inc. | 5-phenyl-nicotinamide derivatives |
| WO2008084081A3 (en) * | 2007-01-11 | 2008-10-09 | Basf Se | 2-substituted 5-phenylpyrimidines for the treatment of proliferative disorders |
| US8865753B2 (en) | 2007-03-28 | 2014-10-21 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8501794B2 (en) | 2007-04-17 | 2013-08-06 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US7872033B2 (en) | 2007-04-17 | 2011-01-18 | Abbott Laboratories | Compounds as cannabinoid receptor ligands |
| WO2008130953A3 (en) * | 2007-04-17 | 2008-12-11 | Abbott Lab | 2-imin0is0thiaz0le derivatives as cannabinoid receptor ligands |
| US8835475B2 (en) | 2007-04-17 | 2014-09-16 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8735434B2 (en) | 2007-05-18 | 2014-05-27 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US9193713B2 (en) | 2007-10-12 | 2015-11-24 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8569282B2 (en) | 2007-12-11 | 2013-10-29 | Cytopathfinder, Inc. | Carboxamide compounds and their use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US8846730B2 (en) | 2008-09-08 | 2014-09-30 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| US8859596B2 (en) | 2008-09-16 | 2014-10-14 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US8895592B2 (en) | 2008-12-16 | 2014-11-25 | Abbvie Inc. | Compounds as cannabinoid receptor ligands |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| US8410107B2 (en) | 2010-10-15 | 2013-04-02 | Hoffmann-La Roche Inc. | N-pyridin-3-yl or N-pyrazin-2-yl carboxamides |
| US8669254B2 (en) | 2010-12-15 | 2014-03-11 | Hoffman-La Roche Inc. | Pyridine, pyridazine, pyrimidine or pyrazine carboxamides as HDL-cholesterol raising agents |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| EP4392418A4 (en) * | 2021-08-24 | 2025-06-18 | The Johns Hopkins University | COMPOSITIONS AND METHODS FOR TREATING DISEASES ASSOCIATED WITH PROTEOTOXICITY |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2529068A1 (en) | 2004-12-23 |
| MXPA05013282A (en) | 2006-03-09 |
| JP2006527759A (en) | 2006-12-07 |
| EP1638570A1 (en) | 2006-03-29 |
| BRPI0411617A (en) | 2006-08-08 |
| US20040259887A1 (en) | 2004-12-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004110453A1 (en) | Pyrimidine derivatives as cannabinoid receptor ligands | |
| US7232823B2 (en) | Cannabinoid receptor ligands and uses thereof | |
| EP1622903B1 (en) | Cannabinoid receptor ligands and uses thereof | |
| US7145012B2 (en) | Cannabinoid receptor ligands and uses thereof | |
| US7176210B2 (en) | Cannabinoid receptor ligands and uses thereof | |
| US7329658B2 (en) | Cannabinoid receptor ligands and uses thereof | |
| US7247628B2 (en) | Cannabinoid receptor ligands and uses thereof | |
| US20040235926A1 (en) | Cannabinoid receptor ligands and uses thereof | |
| EP1622902A1 (en) | Cannabinoid receptor ligands and uses thereof | |
| WO2007020502A2 (en) | Cannabinoid receptor ligands and uses thereof | |
| US20050026983A1 (en) | Imidazole compounds and uses thereof | |
| US20060241100A1 (en) | Acylaminobicyclic heteroaromatic compounds and uses thereof | |
| WO2005103052A1 (en) | Pyrazolo [1,5-a] pyrimidin-7-one compounds and uses thereof | |
| WO2005061507A1 (en) | Bicyclic pyrazol-4-one cannabinoid receptor ligands and uses thereof | |
| WO2005061504A1 (en) | Bicyclic pyridazinone cannabinoid receptor ligands and uses thereof | |
| WO2005061506A1 (en) | Bicyclic pyrazolyl and imidazolyl compounds as cannabinoid receptor ligands and uses thereof | |
| WO2005061505A1 (en) | Bicyclic imidazolyl pyrimidin-4-one cannabinoid receptor ligands and uses thereof | |
| US20070213334A1 (en) | Cannabinoid receptor ligands and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004736431 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/013282 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2529068 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006516548 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004736431 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0411617 Country of ref document: BR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2004736431 Country of ref document: EP |