WO2004113322A1 - Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives - Google Patents
Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives Download PDFInfo
- Publication number
- WO2004113322A1 WO2004113322A1 PCT/IB2004/001983 IB2004001983W WO2004113322A1 WO 2004113322 A1 WO2004113322 A1 WO 2004113322A1 IB 2004001983 W IB2004001983 W IB 2004001983W WO 2004113322 A1 WO2004113322 A1 WO 2004113322A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- cycloalkyl
- formula
- independently selected
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCCCC*1(CC(C)(C*2)C*2=*)CC1 Chemical compound CCCCC*1(CC(C)(C*2)C*2=*)CC1 0.000 description 3
- HPWDSQAMGYDJJC-UHFFFAOYSA-N CC(C)c1ccc(C2CCC2)cc1C Chemical compound CC(C)c1ccc(C2CCC2)cc1C HPWDSQAMGYDJJC-UHFFFAOYSA-N 0.000 description 1
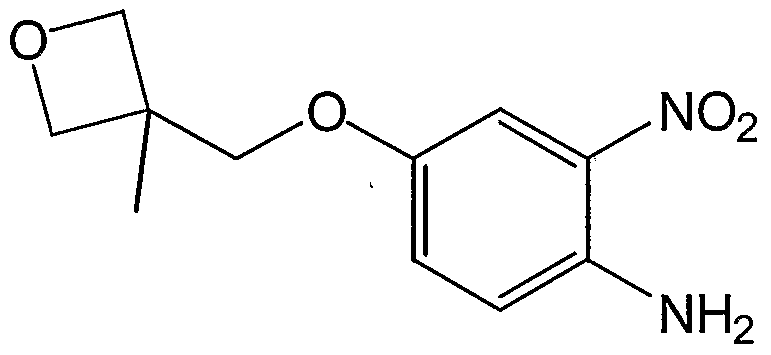
- XQYDJANMTIYABZ-UHFFFAOYSA-N CC1(COc(cc2)cc([N+]([O-])=O)c2N)COC1 Chemical compound CC1(COc(cc2)cc([N+]([O-])=O)c2N)COC1 XQYDJANMTIYABZ-UHFFFAOYSA-N 0.000 description 1
- DZXWNUQNMRNACR-UHFFFAOYSA-N CC1(COc(cc2)cc([N+]([O-])=O)c2Nc(ccc2ccc3)nc2c3OCc2ccccc2)COC1 Chemical compound CC1(COc(cc2)cc([N+]([O-])=O)c2Nc(ccc2ccc3)nc2c3OCc2ccccc2)COC1 DZXWNUQNMRNACR-UHFFFAOYSA-N 0.000 description 1
- NIEHWTGNWJVDEU-UHFFFAOYSA-N Clc1ccc(cccc2OCc3ccccc3)c2n1 Chemical compound Clc1ccc(cccc2OCc3ccccc3)c2n1 NIEHWTGNWJVDEU-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- This invention relates to novel processes for preparing benzimidazole derivatives that are useful in the treatment of abnormal cell growth, such as cancer, in mammals.
- This invention also relates to novel processes for preparing intermediates that may be converted to the aforementioned benzimidazole derivatives.
- Benzimidazole derivatives, intermediates useful in preparing such bezimidazole derivatives and processes for preparing such benzimidazole derivatives and intermediates have been disclosed in International Patent Publication WO 01/40217, published June 7, 2001 , and United States Provisional Patent Application Serial Numbers 60/406,524 and 60/417,047, filed August 28, 2002, and October 28, 2002 respectively.
- each P. 1 , P ⁇ , and R 3 i ⁇ independently selected from ihe group consisting of H, (C Cyal l, (G ⁇ - C 6 )cycloalkyl, halo, cyano, CF 3 , difluoromethoxy, trifluoromethoxy, -0(C C 6 )alkyl, -0(C 3 - C 6 )cycloalkyl, and -NR 12 R 13 ; wherein R 4 is -(CR 5 R 6 ) m H, or -(CR 7 R 8 ) n (4 to 10 memb ⁇ red)-aromatic or nonaromatic heterocyclic containing one or more heteroatoms each selected from O, S and N, wherein m is an integer ranging from 1 to 5, wherein n is an integer ranging from 0 to 5, wherein said 4 to 10 membered heterocyclic when aromatic is optionally substituted by
- BOC is t-butoxycarbonyl
- R 1 , R 2 , R 3 and R 4 are as defined above for the compound of formula I
- a metal alkoxide preferably an alkaline earth metal in the presence of water to give a compound of the formula I
- the water is preseni in an amount of about one equivalent (i.e., one equivalent with respect to the compound of formula II).
- the alkaline earth metal alkoxide is preferably an alkaline earth metal (C ⁇ -C 6 )alkoxide.
- the alkaline earth metal is is preferably sodium or potassium, and the (CrC 6 )alkoxide is preferably t-butoxide.
- the reaction is preferably conducted in the presence of a solvent, such as an ether.
- a solvent such as an ether.
- the ether is preferably a cyclic ether, although acyclic ethers may also be used.
- suitable ethers include dioxane, dimethoxymethane, diethoxymethane, tetrahydrofuran and 2- methyl tetrahydrofuran, or mixtures of at least two thereof. Tetrahydrofuran, 2- methyltetrahydrofuran or mixtures thereof are especially preferred.
- the reaction is conducted at a temperature of about 50°C to about 110°C, and in a more preferable embodiment at a temperature of about 60°C to about 80°C.
- An embodiment of the present invention refers to thoses processes wherein the 4 to 10 membered heterocyclic is a 4 to 8 membered heterocyclic, in another embodiment a 4 to 6 membered heterocyclic, in another embodiment a 6-membered heterocyclic, in another embodiment a 5-membered heterocyclic and in another embodiment a 4-membered heterocyclic.
- Another embodiment of the present invention refers to thoses processes wherein m is an integer from 1 to 5, in another embodiment 1 , and in another embodiment 2.
- Another embodiment of the present invention refers to thoses processes wherein n is an integer from 0 to 5, in another embodiment 1 , and in another embodiment 2.
- Another embodiment of the present invention refers to thoses processes wherein when the 4 to 10 membered heterocyclic is aromatic, it may be optionally substituted by 1 R 9 substituent.
- An embodiment of the present invention refers to thoses processes wherein the 4 to 10 membered heterocyclic group is an aromatic heterocyclic group.
- suitable of such aromatic hetercyclic groups include: pyridinyl, pyrimidinyl, pyrazinyl, quinolinyl, isoquinolinyl, pyrrolyl, pyrazolyl, imidazolyl, thiophenyl, furanyl, indolyl and benzofuranyl.
- Another embodiment of the present invention refers to those processes wherein the the 4 to 10 membered hetercyclic group is a non-aromatic heterocyclic group.
- suitable non-aromatic heterocyclic groups include tetrahydrothiopyranyl, thiomorpholino, dioxanyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydropyranyl, piperidino, morpholino, piperazinyl, homopiperazinyl, azetidinyl, oxetanyl, homopiperidinyl, 3-azabicyclo[3.1.0]hexanyl, 3- azabicyclo[4.1.0]heptanyl, azabicyclo[2.2.2]hexanyl, 3H-indolyl, and 4H-pyranyl.
- Another embodiment of the present invention refers to those processes wherein the 4 to 10 membered aromatic heterocyclic group containing one or more heteroatoms each selected from O, S and N contains one to four heteroatoms each selected from O, S and N, with the proviso that said 4 to 10 membered aromatic heterocylclic does not contain two adjacent O or S atoms.
- the 4 to 10 membered hetercyclic group contains one to two O atoms, and in another embodiment one O atom.
- the -I to 10 membered hetercyclic group contains one to two M atoms, and in a preferred embodiment one N atom.
- Another embodiment of the present invention refers to those processes wherein the compound of formula I is selected from the group consisting of 1- ⁇ 2-[5-(3-Morpholin-4-yl- propoxy)-benzoimidazol-1-yl]-quinolin-8-yl ⁇ -piperidin-4-ylamine;
- the present invention refers to those processes wherein the compound of formula I is the benezenesulfonate salt of 1- ⁇ 2-[5-(3- Methyl-oxetan-3-ylmethoxy)-benzoimidazol-1-yl]-quinolin-8-yl ⁇ -piperidin-4-ylamine.
- the present invention also relates to a process for preparing a compound of formula VI
- R 1 and R 2 are as defined above for formula I, in the presence of 1 ,2- Bis(diphenylphosphino)ethane, a base and a a palladium catalyst, such as a palladium (0) or a palladium (II) catalyst.
- the palladium catalyst is preferably tris(dibenzylidene acetone) dipalladium (0) or palladium acetate, with the latter being most preferred.
- suitable bases include potassium phosphate, sodium t-butoxide and cesium carbonate.
- One especially preferred embodiment of the present invention refers to those processes wherein the palladium catalyst is palladium acetate, and the base is cesium carbonate.
- the reaction is preferably carried out in the presence of an aromatic solvent, such as toluene, an ether, such as dioxane, dimethoxyethane, or tetrahydrofuran, or a polar nitrogen-containing solvent such as dimethylformamide (DMF). Solvent mixtures can also be used.
- the reaction may be carried out at a temperature of of about 90°C to about 120°C.
- An especially preferred embodiment of the present invention refers to those processes wherein R 1 and R 2 in the compound of formula VIII are both hydrogen, and the compound of formula VII is a compound of formula VIIA
- halo as used herein, unless otherwise indicated, includes fluoro, chloro, bromo or iodo. Preferred halo groups are fluoro and chloro.
- alkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight or branched moieties. It is understood that for said alkyl group to include cyclic moieties it must contain at least three carbon atoms.
- cycloalkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having cyclic (including mono- or multi-cyclic) moieties.
- alkenyl as used herein, unless otherwise indicated, includes alkyl groups, as defined above, having at least one carbon-carbon double bond.
- allynyl as used herein, unless otherwise indicated, includes all .yl groups, as defined above, having at least one carbon-carbon triple bond.
- alkoxy as used herein, unless otherwise indicated, includes -O-alkyl groups wherein alkyl is as defined above.
- solvate includes, a compound of the invention or a salt thereof, that further includes a stoichiometric or non-stoichiometric amount of a solvent bound by non-covalent intermolecular forces.
- Preferred solvents are volatile, non-toxic, and/or acceptable for topical administration to humans.
- hydrate refers to a compound of the invention or a salt thereof, that further includes a stoichiometric or non-stoichiometric amount of water bound by non-covalent intermolecular forces.
- 4 to 10 membered heterocyclic includes aromatic and non-aromatic heterocyclic groups containing one or more heteroatoms each selected from O, S and N, wherein each heterocyclic group has from 4 to 10 atoms in its ring system.
- Non-aromatic heterocyclic groups include groups having only 4 atoms in their ring system, but aromatic heterocyclic groups must have at least 5 atoms in their ring system.
- the heterocyclic groups include benzo-fused ring systems and ring systems substituted with one or more oxo moieties.
- An example of a 4 membered heterocyclic group is azetidinyl (derived from azetidine).
- An example of a 5 membered heterocyclic group is thiazolyl and an example of a 10 membered heterocyclic group is quinolinyl.
- non-aromatic heterocyclic groups are pyrrolidinyl, tetrahydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 1 ,2,3,6- tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H-pyranyl, dioxanyl, 1 ,3- dioxolanyl, pyrazolinyl, dithianyl, dithiolanyl,
- aromatic heterocyclic groups are pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinox
- Spiro moieties are also included within the scope of this definition including 1-oxa- 6-a ⁇ a-spiro[2.5]oct-6-yl.
- the foregoing groups may be C-attached or N-attached where such is possible.
- a group derived from pyrrole may be pyrrol-1-yl (N-attached) or pyrrol-3-yl (C-attached).
- a group derived from imidazole may be imidazol-1-yl (N-attached) or imidazol-3-yl (C-attached).
- pharmaceutically acceptable salt(s) includes salts of acidic or basic groups which may be present in the compounds of formula I.
- the compounds of formula I that are basic in nature are capable of forming a wide variety of salts with various inorganic and organic acids.
- acids that may be used to prepare pharmaceutically acceptable acid addition salts of such basic compounds of formula I are those that form non-toxic acid addition salts, Le ⁇ salts containing pharmacologically acceptable anions, such as the acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, calcium edetate, camsylate, carbonate, chloride, davulanate, citrate, dihydrochloride, edetate, edislyate, estolate, esylate, ethylsuccinate, fumarate, gluceptate, gluconate, giutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylsulfate,
- prodrugs of the compounds of the formula I are functional derivativatives of the compounds of formula " ! which are readily convertible convertible in vivo into the required compound of formula I.
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H.
- a prodrug may be a pharmacologically inactive derivative of a biologically active substance (the "parent drug” or “parent molecule”) that requires transformation within the body in order to release the active drug, and that has improved delivery properties over the parent drug molecule.
- the transformation in vivo may be, for example, as the result of some metabolic process, such as chemical or enzymatic hydrolysis of a carboxylic, phosphoric or sulphate ester, or reduction or oxidation of a susceptible functionality.
- Compounds referrred to in the processes of the present invention having free amino, amido, hydroxy or carboxylic groups can be converted into prodrugs.
- Prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more (e.g., two, three or four) amino acid residues is covalently joined through an amide or ester bond to a free amino, hydroxy or carboxylic acid group of compounds of the present invention.
- the amino acid residues include but are not limited lo the 20 naturally occurring amino acids commonly designated by three letter symbols and also includes 4-hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhislidine, norvalin, bela-alanine, gamma- aminobulyric acid, citrulline homocysteine, homoserine, ornithine and melhionine sulfone. Additional types of prodrugs are also encompassed.
- tree carbo 'yl groups can be derivatized as amides or alkyl esters.
- Free hydroxy groups may be derivatized using groups including but not limited to hemisuccinates, phosphate esters, dimelhylaminoacetates, and phosphoryloxymethyloxycarbonyls, as outlined in Advanced Drug Delivery Reviews, 1996, 19, 115.
- Carbamate prodrugs of hydroxy and amino groups are also included, as are carbonate prodrugs, sulfonate esters and sulfate esters of hydroxy groups.
- acyl group may be an alkyl ester, optionally substituted with groups including but not limited to ether, amine and carboxylic acid functionalities, or where the acyl group is an amino acid ester as described above, are also encompassed.
- Prodrugs of this type are described in J. Med. Chem. 1996, 39, 10. Free amines can also be derivatized as amides, sulfonamides or phosphonamides. All of these prodrug moieties may incorporate groups including but not limited to ether, amine and carboxylic acid functionalities.
- the compounds referred to in the processes of the present invention may have one or more asymmetric centres, and may accordingly exist both as enantiomers and as diastereoisomers. It is to be understood that all such isomers and mixtures thereof are encompassed within the scope of such compounds.
- Certain compounds of formula I may have asymmetric centers and therefore exist in different enantiomeric forms. All optical isomers and stereoisomers of the compounds of formula I, and mixtures thereof, are considered to be within the scope of the compounds of formula I.
- the compounds of formula I may include a racemate, one or more enantiomeric forms, one or more diastereomeric forms, or mixtures thereof.
- the compounds of formula I may also exist as tautomers. Reference to the compound of formula l includes reference to the use of all such tautomers and mixtures thereof.
- Me means methyl
- Et means ethyl
- Ac means acetyl
- DMF dimethylformamide
- NMP N- methylpyrrolidinone (also known as 1-Methyl-2-pyrrolidinone).
- DIPHOS 2- Bis(diphenylphosphino)ethane
- Bll lAP (abbreviation for 2,2'-Bi3(diphenylphosphino)-1 , l'-binaphthyl) ; a ⁇ used herein, unless otherwise indicated, is represented by the following formula:
- the compounds referred to in the processes of the present invention also include isotopically-labelled compounds, which are identical to compounds referred to herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 13 C, 14 C, 15 N, 18 0, 17 0, 31 P, 32 P, 35 S, 18 F, and 36 Cl, respectively.
- Isotopically labelled compounds of Formula I of this invention and prodrugs thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples and Preparations below, by substituting a readily available isotopically labelled reagent for a non-isotopically labelled reagent.
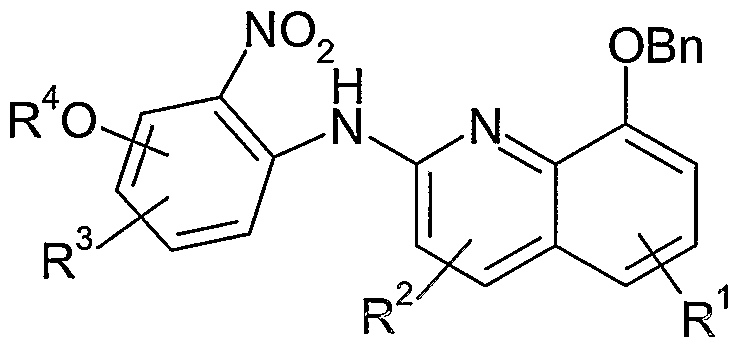
- the compond of formula I may be prepared starting with the palladium amination of reaction of a 2-chloro-8-ben ⁇ yloxyquinoline (VIII) and an appropriate 2-amino-nitrobenzene (VII) to provide the quinoline (VI).
- Reduction of the nitro group and removal of the benzyl group via catalytic hydrogenation, followed by addition of formamidine acetate provides the benzimidazole (V) which can then be transformed into the corresponding triflate (IV).
- a second palladium catalyzed amination with amine (III) provides piperidinyl quinoline (II) and subsequent removal of the t-butyloxycarbonyl group provides (I).
- the reaction of the compound of formula VIII with the compound of formula VII in the presence of palladium acetate and DIPHOS (1 , 2-Bis(diphenylphosphino)ethane) to produce the compound of foi mula VI is particularly and unexpectedly advantageous compared to the same reaction using palladium acetate, BINAP and PhB(OH) 2 .
- the reaction in the presence of DIPHOS results in higher (e.g., 15-25% higher) yields of the product and takes less time to go to completion, particularly in high scale (e.g., 100 grams and higher) synthesis. This process has significant commercial advantages for the production of active ingredients for use in the preparation of a drug.
- pressure is not critical unless otherwise indicated. Pressures from about 0.5 atmospheres to about 5 atmospheres are generally acceptable, and ambient pressure, L , about 1 atmosphere, is preferred as a matter of convenience.
- HPLC chromatography is referred to in the preparations and examples below, the general conditions used, unless otherwise indicated, are as follows.
- the column used is a ZORBAX RXC18 column (manufactured by Hewlett Packard) of 150 mm distance and 4.6 mm interior diameter.
- the samples are run on a Hewlett Packard-1100 system.
- a gradient solvent method is used running 100 percent ammonium acetate / acetic acid buffer (0.2 M) to 100 percent acetonitrile over 10 minutes.
- the system then proceeds on a wash cycle with 100 percent acetonitrile for 1.5 minutes and then 100 percent buffer solution for 3 minutes.
- the flow rate over this period is a constant 3 mL/ minute.
- the flask and the filter cake were rinsed with 2 by 6 volumes of ethyl acetate.
- the filtrate was then washed with 25 volumes of 0.5 N sodium hydroxide solution, followed by 25 volumes of saturated sodium chloride solution.
- the resulting solution was concentrated to low volume and isopropanol (25 mL, 5 volumes) was added.
- the solids were granulated at 20-25°C for at least 10 hours and then collected and dried under vacuum at 40°C with a slight nitrogen bleed to provide 7.7 g of a reddish orange fluffy solid (74% yield).
- MethyI-oxetan-3-ylmethoxy)-2-nitro-phenyIamine 5.3 g, 22.2 mmol, 1.2 equivalents
- cesium carbonate 8.46 g, 26 mmol, 1.4 equivalents
- DIPHOS 1-Bis(diphenylphosphino)ethane; 443 mg, 111 ⁇ mol, 0.06 equivalents
- toluene 75 mL, 15 volumes
- reaction completion the reaction was cooled to 55 °C and dichloroethane ("DCE"; 75 mL, 15 volumes) was charged. The slurry was filtered through a pad of Celite and then the flask and filter were rinsed once with additional DCE (50 mL, 10 volumes). The organics were concentrated to low volume and ethyl acetate (50 mL, 10 volumes) was added. The reaction was heated to reflux and allowed to cool to 20-25 °C.
- DCE dichloroethane
- the slurry was filtered through a pad of Celite and rinsed with dichloroethane (5 volumes). The mother liquor was then concentrated to low volume and ethyl acetate (75 mL, 15 volumes) was charged. The thin slurry was granulated al 20-25X for 8-15 h and then filtered. The mother liquor was collected and washed with a 2.5% NaH 2 P0 4 solution (3 :: 9 volumes). The organics were again concentrated to low volume and aceionilrile (25 mL, 5 volumes) was charged.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Epoxy Compounds (AREA)
Abstract
Description

















Claims





Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2004249511A AU2004249511B2 (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
| DE602004017731T DE602004017731D1 (en) | 2003-06-24 | 2004-06-14 | PROCESS FOR THE PREPARATION OF 1-Ä (BENZIMIDAZOL-1-YL) CHINOLIN-8-YLIPIPERIDIN-4-YLAMINE DERIVATIVES |
| MXPA05014203A MXPA05014203A (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives. |
| HK06109559.8A HK1089170B (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
| CA002529032A CA2529032C (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[2-benzimidazol-1-yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
| DK04736779T DK1641780T3 (en) | 2003-06-24 | 2004-06-14 | Methods for Preparation of 1 - [(benzimidazol-1yl) quinolin-8-yl] -piperidin-4-ylamine derivatives |
| NZ543714A NZ543714A (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[(benzoimidazol-1yl)quinolin-8-yl]piperidin-4-ylamine derivatives |
| EP04736779A EP1641780B1 (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1- [(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
| BRPI0411794-8A BRPI0411794A (en) | 2003-06-24 | 2004-06-14 | processes for the preparation of 1 - [(benzoimidazol-1-yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
| JP2006516552A JP2007516168A (en) | 2003-06-24 | 2004-06-14 | Process for producing 1- [2- (benzimidazol-1-yl) quinolin-8-yl] -piperidin-4-ylamine derivative |
| IL172064A IL172064A (en) | 2003-06-24 | 2005-11-20 | Processes for the preparation of 1-[(benzoimidazole-1-yl) quinolin- 8-yl] piperidin-4-ylamine derivatives |
| NO20060376A NO20060376L (en) | 2003-06-24 | 2006-01-24 | Methods for Preparation of 1 - [(Benzothmidazol-1-yl) quinol-8-yl] piperidin-4-ylamine derivatives |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US48217603P | 2003-06-24 | 2003-06-24 | |
| US60/482,176 | 2003-06-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004113322A1 true WO2004113322A1 (en) | 2004-12-29 |
Family
ID=33539339
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2004/001983 Ceased WO2004113322A1 (en) | 2003-06-24 | 2004-06-14 | Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives |
Country Status (24)
| Country | Link |
|---|---|
| US (2) | US7183414B2 (en) |
| EP (1) | EP1641780B1 (en) |
| JP (1) | JP2007516168A (en) |
| KR (2) | KR100787649B1 (en) |
| CN (1) | CN100404530C (en) |
| AR (1) | AR044849A1 (en) |
| AT (1) | ATE414077T1 (en) |
| AU (1) | AU2004249511B2 (en) |
| BR (1) | BRPI0411794A (en) |
| CA (1) | CA2529032C (en) |
| CL (1) | CL2004001470A1 (en) |
| CO (1) | CO5650165A2 (en) |
| DE (1) | DE602004017731D1 (en) |
| DK (1) | DK1641780T3 (en) |
| ES (1) | ES2315663T3 (en) |
| IL (1) | IL172064A (en) |
| MX (1) | MXPA05014203A (en) |
| NO (1) | NO20060376L (en) |
| NZ (2) | NZ543714A (en) |
| RU (1) | RU2323214C2 (en) |
| SG (1) | SG161745A1 (en) |
| TW (1) | TWI294425B (en) |
| WO (1) | WO2004113322A1 (en) |
| ZA (1) | ZA200509253B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006038111A1 (en) * | 2004-10-07 | 2006-04-13 | Pfizer Products Inc. | Benzoimidazole derivatives useful as antiproliferative agents |
| JP2008534689A (en) * | 2005-04-05 | 2008-08-28 | ファーマコペイア, インコーポレイテッド | Purine and imidazopyridine derivatives for immunosuppression |
| US7504513B2 (en) | 2006-02-27 | 2009-03-17 | Hoffman-La Roche Inc. | Thiazolyl-benzimidazoles |
| US8044213B2 (en) | 2008-12-18 | 2011-10-25 | Hoffmann-La Roche Inc. | Thiazolyl-benzimidazoles |
| US8518952B2 (en) | 2008-08-06 | 2013-08-27 | Pfizer Inc. | 6 substituted 2-heterocyclylamino pyrazine compounds as CHK-1 inhibitors |
| CN107382984A (en) * | 2017-08-24 | 2017-11-24 | 扬州市三药制药有限公司 | A kind of preparation method for treating leukemia medicament |
| CN109678849A (en) * | 2019-01-16 | 2019-04-26 | 北京悦康科创医药科技股份有限公司 | A kind of preparation method of anticancer drug |
| US20220017500A1 (en) * | 2020-07-20 | 2022-01-20 | Arog Pharmaceuticals, Inc. | Crystal forms of crenolanib and methods of use thereof |
| CN116234549A (en) * | 2020-08-05 | 2023-06-06 | 总医院公司 | Salt-Inducible Kinase Inhibitors |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0411421D0 (en) * | 2004-05-21 | 2004-06-23 | Glaxo Group Ltd | Novel compounds |
| CN101993416B (en) * | 2009-08-27 | 2013-09-11 | 中国科学院上海药物研究所 | Quinoline compound and preparation method thereof, medicament combination containing compound and application of compound |
| KR101738063B1 (en) | 2012-09-21 | 2017-05-19 | 아로그 파마슈티칼스, 인코퍼레이티드 | Method of inhibiting constitutively active phosphorylated flt3 kinase |
| US11642340B2 (en) | 2012-09-26 | 2023-05-09 | Arog Pharmaceuticals, Inc. | Method of inhibiting mutant C-KIT |
| TWI599356B (en) | 2012-09-26 | 2017-09-21 | 安羅格製藥有限責任公司 | Method of inhibiting mutant c-kit |
| US10835525B2 (en) | 2012-09-26 | 2020-11-17 | Arog Pharmaceuticals, Inc. | Method of inhibiting mutant C-KIT |
| BR112015016282A2 (en) | 2013-01-07 | 2017-07-11 | Arog Pharmaceuticals, Inc. | crenolanib for treatment of mutated flt3 proliferative disorders |
| US10463658B2 (en) | 2013-10-25 | 2019-11-05 | Videra Pharmaceuticals, Llc | Method of inhibiting FLT3 kinase |
| US12534764B2 (en) | 2016-11-02 | 2026-01-27 | Arog Pharmaceuticals, Inc. | Crenolanib for treating FLT3 mutated proliferative disorders associated mutations |
| EA201991078A1 (en) | 2016-11-02 | 2019-11-29 | CRENOLANIB FOR TREATMENT OF PROLIFERATIVE DISORDERS ASSOCIATED WITH FLT3 MUTATION | |
| CN107382983B (en) * | 2017-08-24 | 2020-08-07 | 扬州市三药制药有限公司 | Synthesis method of medicine for treating leukemia |
| CN110840893A (en) | 2018-12-13 | 2020-02-28 | 安罗格制药有限责任公司 | Claranib-containing pharmaceutical composition and use thereof |
| US11471451B2 (en) | 2019-08-19 | 2022-10-18 | Arog Pharmaceuticals, Inc. | Uses of crenolanib |
| CN117447376A (en) * | 2019-12-09 | 2024-01-26 | 苏州恩华生物医药科技有限公司 | Siama-1 receptor inhibitors of the bicyclic structure |
| CN118767143A (en) | 2019-12-12 | 2024-10-15 | 听治疗有限责任公司 | Compositions and methods for preventing and treating hearing loss |
| CA3186319A1 (en) * | 2020-07-20 | 2022-01-27 | Arog Pharmaceuticals, Inc. | Crystal forms of crenolanib and methods of use thereof |
| US20220079934A1 (en) * | 2020-09-17 | 2022-03-17 | Arog Pharmaceuticals, Inc. | Crenolanib for treating pain |
| US11524006B2 (en) | 2020-09-17 | 2022-12-13 | Arog Pharmaceuticals, Inc. | Crenolanib for treating TRK kinase associated proliferative disorders |
| US11969420B2 (en) | 2020-10-30 | 2024-04-30 | Arog Pharmaceuticals, Inc. | Combination therapy of crenolanib and apoptosis pathway agents for the treatment of proliferative disorders |
| WO2022090547A1 (en) | 2020-10-30 | 2022-05-05 | Dsm Ip Assets B.V. | Production of carotenoids by fermentation |
| CN113683594B (en) * | 2021-09-07 | 2022-12-27 | 曲靖师范学院 | Quinoline-benzimidazole salt compound and synthesis method and application thereof |
| JP2023063189A (en) | 2021-10-22 | 2023-05-09 | アログ・ファーマシューティカルズ・インコーポレイテッド | Crenolanib for Treating FLT3 Mutant Proliferative Disorders Relapsed/Refractory to Previous Therapy |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001040217A1 (en) * | 1999-11-30 | 2001-06-07 | Pfizer Products Inc. | Novel benzoimidazole derivatives useful as antiproliferative agents |
| WO2004020431A2 (en) * | 2002-08-28 | 2004-03-11 | Pfizer Products Inc. | Novel benzoimidazole derivatives useful as antiproliferative agents |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4897401A (en) * | 1987-06-19 | 1990-01-30 | Janssen Pharmaceutical N.V. | N-(4-piperidinyl) bicyclic condensed 2-imidazolamine derivatives useful in treating allergic diseases |
| US5478832A (en) * | 1992-05-08 | 1995-12-26 | The Green Cross Corporation | Quinoline compounds |
| ES2300119T3 (en) * | 1997-02-27 | 2008-06-01 | Takeda Pharmaceutical Company Limited | AMINA COMPOUNDS, ITS PRODUCTION AND ITS USE AS INHIBITORS OF THE PRODUCTION OF BETA-AMILOID. |
| EP1245390A4 (en) * | 1999-12-10 | 2009-04-01 | Fujifilm Corp | Ink-jet head and printer |
| JPWO2002022549A1 (en) * | 2000-09-14 | 2004-01-22 | 鐘淵化学工業株式会社 | Method for removing nitrobenzenesulfonyl group |
-
2004
- 2004-06-14 EP EP04736779A patent/EP1641780B1/en not_active Expired - Lifetime
- 2004-06-14 NZ NZ543714A patent/NZ543714A/en not_active IP Right Cessation
- 2004-06-14 KR KR1020057024593A patent/KR100787649B1/en not_active Expired - Fee Related
- 2004-06-14 DK DK04736779T patent/DK1641780T3/en active
- 2004-06-14 ES ES04736779T patent/ES2315663T3/en not_active Expired - Lifetime
- 2004-06-14 RU RU2005138367/04A patent/RU2323214C2/en active
- 2004-06-14 MX MXPA05014203A patent/MXPA05014203A/en active IP Right Grant
- 2004-06-14 AT AT04736779T patent/ATE414077T1/en not_active IP Right Cessation
- 2004-06-14 DE DE602004017731T patent/DE602004017731D1/en not_active Expired - Lifetime
- 2004-06-14 CL CL200401470A patent/CL2004001470A1/en unknown
- 2004-06-14 JP JP2006516552A patent/JP2007516168A/en active Pending
- 2004-06-14 AU AU2004249511A patent/AU2004249511B2/en not_active Ceased
- 2004-06-14 SG SG200700254-6A patent/SG161745A1/en unknown
- 2004-06-14 WO PCT/IB2004/001983 patent/WO2004113322A1/en not_active Ceased
- 2004-06-14 CA CA002529032A patent/CA2529032C/en not_active Expired - Lifetime
- 2004-06-14 CN CNB2004800175706A patent/CN100404530C/en not_active Expired - Lifetime
- 2004-06-14 KR KR1020077019713A patent/KR20070092333A/en not_active Withdrawn
- 2004-06-14 NZ NZ571898A patent/NZ571898A/en not_active IP Right Cessation
- 2004-06-14 BR BRPI0411794-8A patent/BRPI0411794A/en not_active IP Right Cessation
- 2004-06-22 AR ARP040102160A patent/AR044849A1/en unknown
- 2004-06-23 US US10/875,030 patent/US7183414B2/en not_active Expired - Lifetime
- 2004-06-23 TW TW093118112A patent/TWI294425B/en not_active IP Right Cessation
-
2005
- 2005-11-15 ZA ZA200509253A patent/ZA200509253B/en unknown
- 2005-11-20 IL IL172064A patent/IL172064A/en active IP Right Grant
- 2005-12-20 CO CO05127734A patent/CO5650165A2/en active IP Right Grant
-
2006
- 2006-01-24 NO NO20060376A patent/NO20060376L/en not_active Application Discontinuation
- 2006-12-05 US US11/567,071 patent/US20070088032A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001040217A1 (en) * | 1999-11-30 | 2001-06-07 | Pfizer Products Inc. | Novel benzoimidazole derivatives useful as antiproliferative agents |
| WO2004020431A2 (en) * | 2002-08-28 | 2004-03-11 | Pfizer Products Inc. | Novel benzoimidazole derivatives useful as antiproliferative agents |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006038111A1 (en) * | 2004-10-07 | 2006-04-13 | Pfizer Products Inc. | Benzoimidazole derivatives useful as antiproliferative agents |
| JP2008534689A (en) * | 2005-04-05 | 2008-08-28 | ファーマコペイア, インコーポレイテッド | Purine and imidazopyridine derivatives for immunosuppression |
| US7504513B2 (en) | 2006-02-27 | 2009-03-17 | Hoffman-La Roche Inc. | Thiazolyl-benzimidazoles |
| US8518952B2 (en) | 2008-08-06 | 2013-08-27 | Pfizer Inc. | 6 substituted 2-heterocyclylamino pyrazine compounds as CHK-1 inhibitors |
| US8044213B2 (en) | 2008-12-18 | 2011-10-25 | Hoffmann-La Roche Inc. | Thiazolyl-benzimidazoles |
| CN107382984A (en) * | 2017-08-24 | 2017-11-24 | 扬州市三药制药有限公司 | A kind of preparation method for treating leukemia medicament |
| CN109678849A (en) * | 2019-01-16 | 2019-04-26 | 北京悦康科创医药科技股份有限公司 | A kind of preparation method of anticancer drug |
| US20220017500A1 (en) * | 2020-07-20 | 2022-01-20 | Arog Pharmaceuticals, Inc. | Crystal forms of crenolanib and methods of use thereof |
| US11713310B2 (en) * | 2020-07-20 | 2023-08-01 | Arog Pharmaceuticals, Inc. | Crystal forms of crenolanib and methods of use thereof |
| CN116234549A (en) * | 2020-08-05 | 2023-06-06 | 总医院公司 | Salt-Inducible Kinase Inhibitors |
| EP4192459A4 (en) * | 2020-08-05 | 2024-12-04 | The General Hospital Corporation | Salt inducible kinase inhibitors |
| US12600719B2 (en) | 2020-08-05 | 2026-04-14 | The General Hospital Corporation | Salt inducible kinase inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1641780B1 (en) | Processes for the preparation of 1- [(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives | |
| JP3367945B2 (en) | 2-Substituted-1-piperidylbenzimidazole compounds as ORL1 receptor agonists | |
| EP1044969B1 (en) | Processes and intermediates for preparing anti-cancer compounds | |
| TWI508961B (en) | Heterocyclic compounds | |
| EP1551803B1 (en) | Azabicyclo derivatives as muscarinic receptor antagonists | |
| JP5746821B2 (en) | Pyrazolylbenzimidazole derivatives, compositions containing them and their use | |
| CN116157396B (en) | Pyridazinone derivatives and their use in medicine | |
| WO2022040469A1 (en) | Spiro compounds as kras inhibitors | |
| EP1940823A2 (en) | Substituted 1-amino-phthalzine derivatives, preparation and therapeutic use thereof | |
| CN114206836B (en) | New pyrrole compounds | |
| KR20040032639A (en) | The preparation method for pyrimidinone compound and trihydrate of the salt of the pyrimidinone compound | |
| AU2014252281A1 (en) | Derivatives of 1-(substituted sulfonyl)-2-aminoimidazoline as antitumor and antiproliferative agents | |
| DE60003169T2 (en) | PROCESS FOR PREPARING [S- (R *, S *)] - G (B) - [[1- [1-OXO-3- (4-PIPERIDINYL) PROPYL] -3-PIPERIDINYL] CARBONYL] AMINO] -3 -PYRIDINPROPANSÄUREDERIVATE | |
| KR100617953B1 (en) | Method for preparing pyrimidinone compound and salts thereof | |
| JP3140155B2 (en) | N, N'-disubstituted amide derivatives | |
| KR20240110861A (en) | Compounds used as kinase inhibitors and their uses | |
| HK1089170B (en) | Processes for the preparation of 1-[(benzoimidazole-1yl) quinolin-8-yl] piperidin-4-ylamine derivatives | |
| CN110963991A (en) | PI3K inhibitor and preparation method and application thereof | |
| KR0184340B1 (en) | N-substituted heterocyclic compound and preparation method thereof | |
| CN106800538A (en) | A kind of benzimidizole derivatives and its synthetic method | |
| CN119638695A (en) | Phenanthroline-substituted methyl guanidine compound, and preparation and application thereof | |
| JP2009529575A (en) | Pyridine-containing large heterocyclic compounds as kinase inhibitors | |
| WO1994021641A1 (en) | Process for production of an imidazole derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005/09253 Country of ref document: ZA Ref document number: 200509253 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 172064 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 543714 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5390/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12005502132 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004249511 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006516552 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2529032 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2004249511 Country of ref document: AU Date of ref document: 20040614 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004249511 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 05127734 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/014203 Country of ref document: MX Ref document number: 2004736779 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057024593 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048175706 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200600110 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005138367 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057024593 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004736779 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0411794 Country of ref document: BR |