WO2005042473A1 - Improved process for the manufacture of citalopram hydrobromide - Google Patents
Improved process for the manufacture of citalopram hydrobromide Download PDFInfo
- Publication number
- WO2005042473A1 WO2005042473A1 PCT/IB2003/004757 IB0304757W WO2005042473A1 WO 2005042473 A1 WO2005042473 A1 WO 2005042473A1 IB 0304757 W IB0304757 W IB 0304757W WO 2005042473 A1 WO2005042473 A1 WO 2005042473A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- citalopram
- formula
- acid
- reaction
- aliphatic
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- WSEQXVZVJXJVFP-UHFFFAOYSA-N CN(C)CCCC1(c(cc2)ccc2F)OCc2cc(C#N)ccc12 Chemical compound CN(C)CCCC1(c(cc2)ccc2F)OCc2cc(C#N)ccc12 WSEQXVZVJXJVFP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C215/00—Compounds containing amino and hydroxy groups bound to the same carbon skeleton
- C07C215/02—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton
- C07C215/22—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated
- C07C215/28—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing six-membered aromatic rings
- C07C215/34—Compounds containing amino and hydroxy groups bound to the same carbon skeleton having hydroxy groups and amino groups bound to acyclic carbon atoms of the same carbon skeleton the carbon skeleton being unsaturated and containing six-membered aromatic rings containing hydroxy groups and carbon atoms of six-membered aromatic rings bound to the same carbon atom of the carbon skeleton and at least one hydroxy group bound to another carbon atom of the carbon skeleton
Definitions
- the present invention relates to an improved process for the preparation of extremely pure 1 -(4' -Fluorophenyl)- 1 -(3 -dimethylaminopropyl)-5-phthalanecarbonitrile and its bromide salt (citalopram hydrobromide), which is a well known antidepressant.
- Other aspect of the invention are isolation of crystalline (4-Bromo-2-hydroxymethyl)phenyl-(4-fluorophenyl)-3- (dimethylaminopropyl)methanol (Bromodiol) and conversion of desmethylcitalopram to Citalopram generated in trace during the reaction by treatment with formaldehyde and formic acid in chloroform.
- the resulting citalopram product is optionally further worked up, purified and isolated in the form of a base or a pharmaceutically acceptable salts.
- Citalopram is a selective centrally acting serotonin (5-hydroxytryptamine; 5HT) reuptake inhibitor having antidepressant activity.
- 5HT serotonin
- the activity of citalopram is described in J. Hyttel, Prog. Neuro-Psychopharmacol. & Biol. Phychiat, 1982, 6, 277-295. Its effectiveness in the treatment of dementia and cardiovascular disorder has been disclosed in EP-A 474 580.
- the structure of Citalopram is shown in Formula (I):
- Citalopram was first discussed in DE 2,657,013, corresponding to US patent No. 4,136,193. So far several different processes for the preparation and purification of this active drug have been reported. US Patent 4,136,193 describes preparation of Citalopram from 5-Bromophthalide using double Grignard reactions, namely with 4-Fluorobromobenzene and N,N-Dimethyl-aminopropyl chloride. The bromo function of l-(4'-Fluoro ⁇ henyl)-l-(3-dimethylamino-propyl)-5- bromophthalan thus obtained is substituted by cyano group using copper cyanide in a suitable solvent to get the citalopram base.
- WO 2000/011926 and WO/2000 013648 disclose the use of transition metals like Nickel or Palladium as catalyst for the substitution of halide group by a cyano such as KCN, NaCN or (R' 4 N)CN, where R' indicates four groups which may be same or different and are selected from hydrogen and straight chain or branched C 1-6 alkane.
- Halide group discussed are bromo, iodo, and CF 3 -(CF 2 ) n -SO 2 - wherein n is an integer from the range of 0 to 8, preferably CF -SO 2 -
- US Patent 4,650,884 uses 5-Cyanophthalide as the starting material for the preparation of Citalopram. In that process the ring closure of the dihydroxy compound of formula
- X is a halogen, with organometallic dimethylaminopropyl halide.
- Impurity profile of Citalopram is discussed in WO 2001/ 47877 where thin film distillation process is described for purification. It is well known that synthesis of citalopram in desired quality is very difficult.
- the manufacturing processes of citalopram described in the US patent 4,136,193; WO 2000/11926, WO 2000/13648 and DE 2,657,013 comprises the exchange of 5-halogen with cyano group. It has been found that along with citalopram unacceptable amount of desmethylcitalopram is also formed during the substitution of halogen group. The removal of desmethylcitalopram is very difficult by usual work up procedure, which leads to extensive and expensive purification processes.
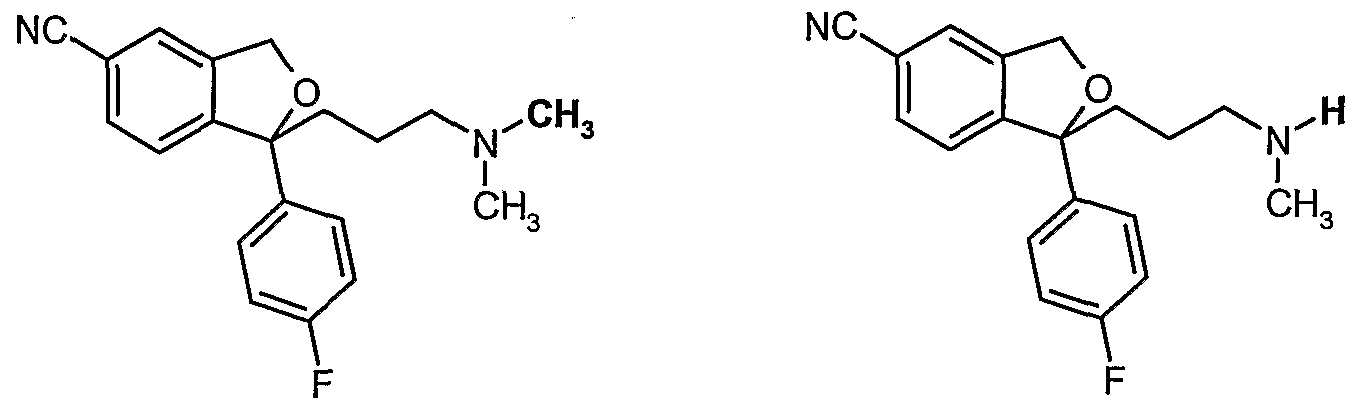
- the chemical structure of citalopram and desmethylcitalopram is shown below:
- WO 2001/045483 discloses the different purification method of citalopram.
- the purification method described in this patent application teaches the removal of desmethylcitalopram formed during the cyanide exchange reaction.
- the crude citalopram obtained in this process after usual purification is subjected to treatment with an amide or an amide like group forming agent from the agents of formulae (a), (b) or (c): o R-co — x Hal — U— W — R" FT— S0 2 — Hal (a) (b) (c) where X is halogen or a group -O-CO-R', Hal is halogen, Y is O or S, W is O, N, or S and R', R" and R'" are each selected from the group consisting of hydrogen, alkyl and optionally substituted aryl or aralkyl.
- a process for the manufacture of highly pure l-(4'-Fluorophenyl)-l-(3-dimethylaminopropyl)-5-phthalanecarbonitrile and its bromide salt (Citalaopram hydrobromide).
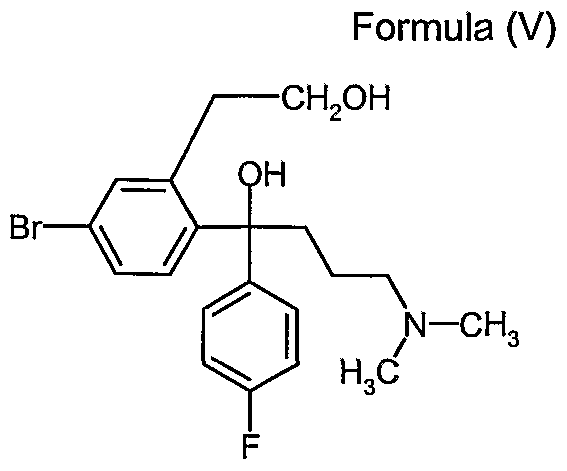
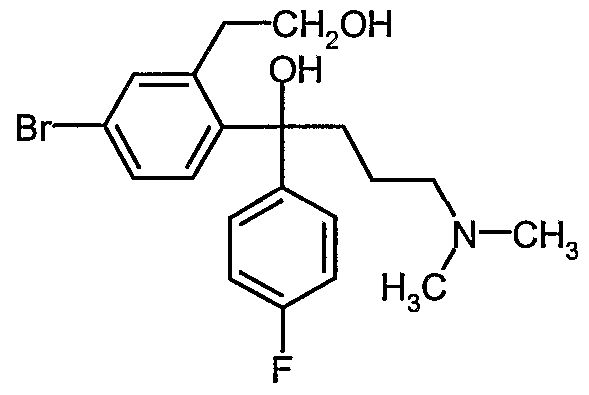
- crystalline (4-Bromo-2-(hydroxymethyl)phenyl)-(4'-fluoro-phenyl)- 3 -dimethylaminopropyl)methanol (Bromodiol) Formula (V)
- the bromodiol is synthesized from 5-Bromophthalide by two successive Grignard reactions, namely with 4-Fluorobromobenzene and N,N-Dimethylaminopropyl chloride.
- the unwanted desmethylcitalopram formed during the cyanide exchange reaction is reconverted to citalopram by refluxing the crude citalopram with formaldehyde and formic acid in chloroform for 8 hours.
- the pictorial process is outlined below:
- Citalopram being an important and active anti-depressant therapeutic agent, a systematic study for its large scale manufacture of very high purity product and having well control over process impurities is under taken. This resulted in a robust manufacturing process, incorporating a step for re-conversion of desmethylcitalopram (the undesired product produced during the manufacture of Citalopram) into Citalopram by treatment with formic acid and formaldehyde.
- desmethylcitalopram the undesired product produced during the manufacture of Citalopram
- the present invention is directed towards the novel manufacturing process of l-(4'- Fluorophenyl)-l-(3-dimethylaminopropyl)-5-phthalanecarbonitrile and its bromide salt (Citalaopram hydrobromide) of formula (I):
- 1 -(4 '-Fluorophenyl)- 1 -(3 -dimethyl- aminopropyl)-5-phthalanecarbonitrile is prepared according to the following synthetic reaction scheme:
- the compound having formula III is added to a cold solution of 5-Bromopthalide (Formula II) slowly over 4-6 hours followed by addition ofcompound having formula IN at -5 to -6 °C.
- the resultant mixture is stirred at -5 to -10 °C for 2 hours and additionally 3 hours at room temperature.
- the molar excess of magnesium halides of 4-Fluorobromo benzene and ⁇ , ⁇ - Dimethylaminopropyl chloride used in this reaction stage is typically between about 1 and about 2 fold preferably about 1.5 fold, relative to the 5-bromophthalide.
- Tetrahydrofuran used in the present reaction is between about 1 to 5 times, more particularly 1 to 2 times of 5- bromophthalide, which provides optimum yield and acceptable purity of Bromodiol.
- Low temperature employed in the present reaction yields the lesser side products.
- organic solvent used in the reaction is distilled under industrial vacuum between 55 to 65 °C. Acetic acid is added to the residue to make it neutral to slightly basic.
- the reaction mass thus obtained is extracted with ethyl acetate and basified to a pH between 8.0 to 9.0 using concentrate ammonia solution. Finally, ethyl acetate extracted bromodiol is crystallized by cooling the solution at 0 °C. It is filtered and dried in the oven at 60 °C for 4 hours.
- Bromodiol is charged in aliphatic halide solvent more particularly in dichloromethane followed by addition of triethylamine.
- the reaction mixture is cool down to -5 °C and to it a solution of methanesulfonyl chloride in dichloromethane is added.
- the reaction mixture is warmed to room temperature and stirred for 1-2 hour till the reactant (Bromodiol) disappears.
- After usual work up crude bromocitalopram is dissolved in petroleum ether and filtered to remove insoluble impurities. This process provides more than 97 % HPLC pure Bromocitalopram.
- Suitable polar solvent for this reaction include dimethylformamide.
- the molar ratio of cyanide source is between about 1 to 5 more preferably about 2 to 3 times of Bromocitalopram. Copper cyanide reacts with Bromocitalopram under rather drastic condition of high temperature preferably around 160 °C. Molar ratio of copper cyanide is critical for this reaction. Copper cyanide about 2-3 times more particularly 2.5 times of Bromocitalopram favors the maximum conversion of bromo function into cyano. It is observed that exchange reaction should be continued until the formation of citalopram occurs, as unreacted bromocitalopram is very difficult to remove from the final product. We have simplified the work-up procedure of this reaction.
- the WO 2001/045483 teaches about the purification of Citalopram, especially removal of desmethylcitalopram. This method is such that it only removes desmethylcitalopram. However, in the present invention desmethylcitalopram is reconverted into Citalopram. This process not only removes the impurity but increases the yield of Citalopram.
- the crude Citalopram is isolated by chloroform extraction from the reaction mixture and purified by treatment with acetic acid and ammonia.
- An interesting and important feature of the present invention is that crude chloroform solution containing citalopram along with desmethylcitalopram is treated with formic acid and formaldehyde as shown below:
- Citalopram hydrobromide is stirred for 8 to 10 hours. It is observed that for getting the right quality Hydrobromide, it is necessary to control the temperature between 30 to 35°C during its formation. Increase of temperature during hydrobromide formation leads to degradation of Citalopram. Finally, Citalopram Hydrobromide thus obtained is re-crystallized from aqueous isopropyl alcohol to get highly pure Citalopram Hydrobromide.
- the following examples illustrate the invention, but is not limiting thereof.
- magnesium turnings (104 gm), 200 ml tetrahydrofuran and a crystal of iodine.
- magnesium turnings (104 gm)
- 200 ml tetrahydrofuran and a crystal of iodine.
- This process raises reaction temperature to 163 to 165 °C and is maintained under stirring for 6 to 8 hours. It is allowed to cool up to 60 °C and is added into a mixture of ethylenediamine (380 ml) and water (1.80 Ltr) under nitrogen stirring. The reaction mass thus obtained is allowed to cool up to 40 °C and chloroform (1.40 Ltr) is added to it. The mixture is stirred for half an hour and filtered to remove metallic impurities. Filtrate is allowed to separate in two layers. Lower organic layer is separated followed by re- extraction of aqueous layer with chloroform (2 X 0.80 Ltr). Combined chloroform layer is washed with water (0.50 Ltr).
- chloroform layer is added formic acid (0.31 Ltr) and formaldehyde (0.29 Ltr) and refluxed for 8 hours. It is then cooled to room temperature and basified to pH 8.0 to 9.0 using ammonia solution. Chloroform layer is separated, washed with water (0.80 Ltr), dried over sodium sulfate and concentrated to thick residue (308 gm). Residue is dissolved in toluene (1.80 Ltr) and toluene solution is extracted using 20% aqueous acetic acid (3 X 1.20 Ltr). Combined aqueous layer is basified to pH 8.0 to 9.0 using Sodium Hydroxide solution (0.75 Ltr).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description











Claims





Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03769715A EP1678122A4 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
| PCT/IB2003/004757 WO2005042473A1 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
| AU2003278409A AU2003278409A1 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
| CA002546422A CA2546422A1 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
| BRPI0318581-8A BR0318581A (en) | 2003-10-28 | 2003-10-28 | Improving process for citalopram hydrobromide manufacturing |
| US11/414,135 US20060293530A1 (en) | 2003-10-28 | 2006-04-28 | Process for the manufacture of citalopram hydrobromide |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/IB2003/004757 WO2005042473A1 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005042473A1 true WO2005042473A1 (en) | 2005-05-12 |
Family
ID=34531835
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2003/004757 Ceased WO2005042473A1 (en) | 2003-10-28 | 2003-10-28 | Improved process for the manufacture of citalopram hydrobromide |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20060293530A1 (en) |
| EP (1) | EP1678122A4 (en) |
| AU (1) | AU2003278409A1 (en) |
| BR (1) | BR0318581A (en) |
| CA (1) | CA2546422A1 (en) |
| WO (1) | WO2005042473A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010527345A (en) * | 2007-05-18 | 2010-08-12 | シプラ・リミテッド | Preparation method of escitalopram |
| CN105439990A (en) * | 2015-12-09 | 2016-03-30 | 山东潍坊润丰化工股份有限公司 | Method for recovering ether solvents from Grignard reaction residues |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4650884A (en) * | 1984-08-06 | 1987-03-17 | H. Lundbeck A/S | Novel intermediate and method for its preparation |
| US6579993B2 (en) * | 2001-01-30 | 2003-06-17 | Orion Corporation, Fermion | Process for the preparation of 1-(3-dimethylaminopropyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile |
| US6660873B2 (en) * | 2000-05-12 | 2003-12-09 | H. Lundbeck A/S | Method for the preparation of citalopram |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1526331A (en) * | 1976-01-14 | 1978-09-27 | Kefalas As | Phthalanes |
| US6310222B1 (en) * | 1999-11-01 | 2001-10-30 | Sumika Fine Chemicals Co., Ltd. | Production method of 5-phthalancarbonitrile compound, intermediate therefor and production method of the intermediate |
| NL1017525C1 (en) * | 2000-12-22 | 2001-04-26 | Lundbeck & Co As H | Process for the preparation of pure Citalopram |
| US6967259B2 (en) * | 2001-09-24 | 2005-11-22 | Pharmachem Technologies Limited | Process for the preparation of Citalopram intermediate |
| AU2003222435A1 (en) * | 2002-01-07 | 2003-07-24 | Sun Pharmaceutical Industries Limited | Process for the preparation of 1-(3-(dimethylamino)propyl)-1-(4-fluorophenyl)- 1,3-dihydro-5-isobenzofuran carbonitrile |
| CA2381341A1 (en) * | 2002-04-09 | 2003-10-09 | Torcan Chemical Ltd. | Process and intermediates for preparing escitalopram |
-
2003
- 2003-10-28 WO PCT/IB2003/004757 patent/WO2005042473A1/en not_active Ceased
- 2003-10-28 CA CA002546422A patent/CA2546422A1/en not_active Abandoned
- 2003-10-28 EP EP03769715A patent/EP1678122A4/en not_active Withdrawn
- 2003-10-28 BR BRPI0318581-8A patent/BR0318581A/en not_active IP Right Cessation
- 2003-10-28 AU AU2003278409A patent/AU2003278409A1/en not_active Abandoned
-
2006
- 2006-04-28 US US11/414,135 patent/US20060293530A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4650884A (en) * | 1984-08-06 | 1987-03-17 | H. Lundbeck A/S | Novel intermediate and method for its preparation |
| US6660873B2 (en) * | 2000-05-12 | 2003-12-09 | H. Lundbeck A/S | Method for the preparation of citalopram |
| US6579993B2 (en) * | 2001-01-30 | 2003-06-17 | Orion Corporation, Fermion | Process for the preparation of 1-(3-dimethylaminopropyl)-1-(4-fluorophenyl)-1,3-dihydroisobenzofuran-5-carbonitrile |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1678122A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2010527345A (en) * | 2007-05-18 | 2010-08-12 | シプラ・リミテッド | Preparation method of escitalopram |
| CN105439990A (en) * | 2015-12-09 | 2016-03-30 | 山东潍坊润丰化工股份有限公司 | Method for recovering ether solvents from Grignard reaction residues |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2546422A1 (en) | 2005-05-12 |
| BR0318581A (en) | 2006-10-10 |
| EP1678122A4 (en) | 2007-05-23 |
| US20060293530A1 (en) | 2006-12-28 |
| EP1678122A1 (en) | 2006-07-12 |
| AU2003278409A1 (en) | 2005-05-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2001100440A4 (en) | Method for the preparation of citalopram | |
| JP3798982B2 (en) | Method for producing pure citalopram | |
| BG64823B1 (en) | Method for the preparation of citalopram | |
| JP2002533450A (en) | Method for producing 5-cyanophthalide | |
| JP3173602B2 (en) | Novel intermediate for the production of enyne derivatives and its production method | |
| EA005491B1 (en) | Method for the preparation of citalopram | |
| US6635773B2 (en) | Process for preparing citalopram | |
| WO2005042473A1 (en) | Improved process for the manufacture of citalopram hydrobromide | |
| US6781003B1 (en) | Preparation of pure citalopram | |
| JP2004529883A (en) | Preparation method of citalopram | |
| HUP0003666A2 (en) | Method for the preparation of citalopram and pharmaceutical compositions containing them | |
| JPH0535136B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 546354 Country of ref document: NZ Ref document number: 2003278409 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003769715 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 461/MUMNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2546422 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005510110 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006113557 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003769715 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0318581 Country of ref document: BR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2003769715 Country of ref document: EP |