WO2005058888A2 - Oxazolidinone-quinolone hybrid antibiotics - Google Patents
Oxazolidinone-quinolone hybrid antibiotics Download PDFInfo
- Publication number
- WO2005058888A2 WO2005058888A2 PCT/EP2004/014500 EP2004014500W WO2005058888A2 WO 2005058888 A2 WO2005058888 A2 WO 2005058888A2 EP 2004014500 W EP2004014500 W EP 2004014500W WO 2005058888 A2 WO2005058888 A2 WO 2005058888A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- oxo
- fluoro
- oxazolidin
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*(*)NC[C@@](*N1c2cc(F)c(C=CC(N(CC3)CCN3c(c(F)c3)cc(N(C4CC4)C=C4C(O)=O)c3C4=O)=O)cc2)OC1=O Chemical compound C*(*)NC[C@@](*N1c2cc(F)c(C=CC(N(CC3)CCN3c(c(F)c3)cc(N(C4CC4)C=C4C(O)=O)c3C4=O)=O)cc2)OC1=O 0.000 description 1
- LFQCRYBCQONNDC-FNQPIQQGSA-N CC(C)(C)OC(N(CC1)CCN1C(/C=C/c(ccc(N(C[C@H](CNC(C)=O)O1)C1=O)c1)c1F)=O)=O Chemical compound CC(C)(C)OC(N(CC1)CCN1C(/C=C/c(ccc(N(C[C@H](CNC(C)=O)O1)C1=O)c1)c1F)=O)=O LFQCRYBCQONNDC-FNQPIQQGSA-N 0.000 description 1
- DUTSDAAIIVNUAG-XSFVSMFZSA-N CC(C1C(O)=O)N(C2CC2)c(c(OC)c(cc2)N(CC3)CCC3(COc(ccc(N(C/C(/O3)=C\N)C3=O)c3)c3F)O)c2C1=O Chemical compound CC(C1C(O)=O)N(C2CC2)c(c(OC)c(cc2)N(CC3)CCC3(COc(ccc(N(C/C(/O3)=C\N)C3=O)c3)c3F)O)c2C1=O DUTSDAAIIVNUAG-XSFVSMFZSA-N 0.000 description 1
- MNABRWLVTSGIMU-UHFFFAOYSA-N CC(NCC(CN1c(cc2F)ccc2OCC(CC2)(CCN2c(cc(c2c3)N(C4CC4)C=C(C(O)=O)C2=O)c3F)OP(O)(O)=O)OC1=O)=O Chemical compound CC(NCC(CN1c(cc2F)ccc2OCC(CC2)(CCN2c(cc(c2c3)N(C4CC4)C=C(C(O)=O)C2=O)c3F)OP(O)(O)=O)OC1=O)=O MNABRWLVTSGIMU-UHFFFAOYSA-N 0.000 description 1
- BNNCQDUNVMOUSZ-SVQMELKDSA-N CC(NC[C@@H](CN1c(cc2)cc(F)c2OCC(C(N(CC2)CCN2c(c(F)c2)cc(N(C3CC3)C=C3C(O)=O)c2C3=O)=O)O)OC1=O)=O Chemical compound CC(NC[C@@H](CN1c(cc2)cc(F)c2OCC(C(N(CC2)CCN2c(c(F)c2)cc(N(C3CC3)C=C3C(O)=O)c2C3=O)=O)O)OC1=O)=O BNNCQDUNVMOUSZ-SVQMELKDSA-N 0.000 description 1
- LCJJIHFQGUSREF-NRFANRHFSA-N CC(NC[C@@H](CN1c(cc2)cc(F)c2OCC(CC2)(CCN2c(c(F)c2)nc(N(C3CC3)C=C3C(O)=O)c2C3=O)OC(CCC(O)=O)=O)OC1=O)=O Chemical compound CC(NC[C@@H](CN1c(cc2)cc(F)c2OCC(CC2)(CCN2c(c(F)c2)nc(N(C3CC3)C=C3C(O)=O)c2C3=O)OC(CCC(O)=O)=O)OC1=O)=O LCJJIHFQGUSREF-NRFANRHFSA-N 0.000 description 1
- HCTYWQCEGDHIQR-FQEVSTJZSA-N CC(NC[C@@H](CN1c(cc2F)ccc2OCC(N(CC2)CCN2c(c(F)c2)cc(N(C3CC3)C=C3C(O)=O)c2C3=O)=O)OC1=O)=O Chemical compound CC(NC[C@@H](CN1c(cc2F)ccc2OCC(N(CC2)CCN2c(c(F)c2)cc(N(C3CC3)C=C3C(O)=O)c2C3=O)=O)OC1=O)=O HCTYWQCEGDHIQR-FQEVSTJZSA-N 0.000 description 1
- HQAQIPZKPURBJS-CVMIBEPCSA-N CC1N(C=C(C(O)=O)C(c2cc(F)c3N(CC4)CCC4(COc(ccc(N(C[C@H](CNC(C)=O)O4)C4=O)c4)c4F)O)=O)c2c3OC1 Chemical compound CC1N(C=C(C(O)=O)C(c2cc(F)c3N(CC4)CCC4(COc(ccc(N(C[C@H](CNC(C)=O)O4)C4=O)c4)c4F)O)=O)c2c3OC1 HQAQIPZKPURBJS-CVMIBEPCSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/06—Peri-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D263/00—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings
- C07D263/02—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings
- C07D263/08—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D263/16—Heterocyclic compounds containing 1,3-oxazole or hydrogenated 1,3-oxazole rings not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D263/18—Oxygen atoms
- C07D263/20—Oxygen atoms attached in position 2
- C07D263/24—Oxygen atoms attached in position 2 with hydrocarbon radicals, substituted by oxygen atoms, attached to other ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4071—Esters thereof the ester moiety containing a substituent or a structure which is considered as characteristic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4071—Esters thereof the ester moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4087—Esters with arylalkanols
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
Definitions
- the present invention describes new compounds in which the pharmacophores of quinolone and oxazolidinone are linked together through a linker that is stable under physiological conditions and a pharmaceutical antibacterial composition containing these compounds.
- These dual action compounds are useful antimicrobial agents effective against a variety of human and veterinary pathogens including Gram positive aerobic bacteria such as multiply-resistant staphylococci, streptococci and enterococci as well as Gram negative bacteria such as Moraxella catarrhalis and Haemophilus influenzae and anaerobic organisms such as bacteroides spp. and Clostridia spp . species and acid-fast organism such as Mycobacterium tuberculosis, Mycobacterium avium spp .
- Oxazolidinone-quinolone hybrid antibiotics have already been described ( O02059116, O03002560, WO03031443, WO03032962) .
- the major drawback of the compounds known in the state of the art is the poor water solubility, which makes the development of a formulation difficult .
- the present invention provides new compounds of formula (I) , that are useful antimicrobial agents and effective against a variety of multi-drug resistant bacteria
- A is an alkylene group, an alkenylene group, an alkynylene group, a heteroalkylene group, a cycloalkylene group, a heterocycloalkylene group, an arylene group or a heteroarylene group all of which groups may be substituted;
- Q is CR 4 or N (especially CR 4 ) ;
- X is CR' or N
- Y is CR 6 or N
- n 1, 2 or 3;
- n 1, 2 or 3;
- R 1 is H, F, CI, Br, I, OH, NH 2 , an alkyl group or a heteroalkyl group;
- R 2 is H, F or Cl ;
- R 3 is H, an alkyl group, an alkenyl group, an alkynyl group, a heteroalkyl group, a cycloalkyl group, a heterocycloalkyl group, an aryl group, a heteroaryl group, an alkylaryl group or a heteroarylalkyl group; all of which groups may be substituted with one, two or more halogen atoms like F or Cl or amino groups.
- R 4 is hydroxy, a group of formula OP0 3 R 9 2 or OS0 3 R 10 or a heteroalkyl group carrying at least one OH, NH 2 , S0 3 R 10 , P0 3 R 9 2 or COOH group or an ester of a naturally occurring amino acid or a derivative thereof, wherein the groups R 9 independently of each other are H, alkyl, cycloalkyl, aryl or aralkyl and wherein R 10 is H, alkyl, cycloalkyl, aryl or aralkyl;
- R 5 is selected from following groups:
- R 6 is H, F , Cl or OMe ;
- R 7 is H, F, Cl, OH, NH 2 , a substituted or unsubstituted alkyl group or a substituted or unsubstituted heteroalkyl group, or R 3 and R 7 can be linked via an alkylene, an alkenylene or a heteroalkylene group or be a part of a cycloalkylene or heterocycloalkylene group; in case R 3 is no H and R 7 is no H, F, OH, NH 2 or Cl ; and R 8 is a C ⁇ - 6 heteroalkyl, a heteroarylalkyl, a hetero- alkylaryl or a heteroalkylheteroaryl group; or a pharmacologically acceptable salt, solvate, hydrate or formulation thereof.
- alkyl refers to a saturated straight or branched chain alkyl group, preferably containing from one to ten, preferably one to six carbon atoms, for example methyl, ethyl, propyl, isopropyl, butyl, iso-butyl, sec- butyl, tert-butyl, n-pentyl , iso-pentyl, n-hexyl, 2,2- dimethylbutyl , n-octyl or n-pentyl groups.
- Any alkyl group as defined herein may be substituted with one, two or more substituents, for example F, Cl , Br, I, H 2 , OH, SH or N0 2 .
- alkenyl and alkynyl refer to an unsaturated straight or branched chain alkyl group (having one, two or more double and/or triple bonds, an alkenyl preferably having one or two double bonds and an alkynyl preferably having one or two triple bonds) , preferably containing two to ten, preferably two to six carbon atoms for example: ethenyl (vinyl), propenyl (allyl) , iso-propenyl , n- pentenyl, butenyl, isoprenyl or hexa-2-enyl; ethynyl, propynyl or butynyl groups .
- Any alkenyl or alkynyl group as defined herein may be substituted with one, two or more substituents, for example F, Cl, Br, I, NH 2 , OH, SH or N0 2 .
- the term heteroalkyl refers to an alkyl, alkenyl or alkynyl group as defined herein where one or more carbon atoms are replaced by an oxygen, nitrogen, phosphorous or sulphur atom, for example an alkoxy group such as methoxy, ethoxy, propoxy, iso-propoxy, butoxy or tert .
- an alkoxyalkyl group such as methoxymethyl , ethoxymethyl , 1- methoxyethyl, 1-ethoxyethyl, 2-methoxyethyl or 2- ethoxyethyl
- an alkylamino group such as methylamino, ethylamino, propylamino, isopropylamino, dimethylamino or diethylamino
- an alkylthio group such as methylthio, ethylthio or isopropylthio or a cyano group. It may also refer to one of the above groups containing a keto group.
- heteroalkyl furthermore refers to a group derived from a carboxylic acid or carboxylic acid amide such as acetyl, propionyl, acetyloxy, propionyloxy, acetylamino or propionylamino, a carboxyalkyl group such as carboxymethyl , carboxyethyl or carboxypropyl, a carboxyalkyl ester, an alkylthiocarboxyamino group, an alkoxyimino group, an alkylaminothiocarboxyamino group or an alkoxycarbonylamino group.
- Any heteroalkyl group as defined herein may be substituted with one, two or more substituents, for example F, Cl, Br, I, NH 2 , OH, SH or N0 2 .
- cycloalkyl refers to a saturated or partially unsaturated (having one, two or more double and/or triple bonds) cyclic group with one, two or more rings, having three to 14 carbon ring-atoms, preferably from five or six to ten carbon ring-atoms, for example cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, tetralin, cyclopentenyl or cyclohex-2-enyl groups.
- Any cycloalkyl group as defined herein may be substituted with one, two or more substituents, for example F, Cl, Br, I, OH, NH 2 , SH, N 3 , N0 2 , alkyl groups such as methyl or ethyl, heteroalkyl groups such as methoxy, methylamino, dimethylamino or cyanide .
- heterocycloalkyl refers to a cycloalkyl group as defined herein where one, two or more carbon ring-atoms are replaced by one, two or more oxygen, nitrogen, phosphorous or sulphur atoms or S ⁇ 0)1-2 groups for example piperidino, morpholino or piperazino groups.
- aryl refers to an aromatic cyclic group with one, two or more rings, having five to 14 carbon ring-atoms preferably from five or six to ten carbon ring-atoms, for example phenyl or naphthyl groups .
- Any aryl group as defined herein may be substituted with one, two or more substituents, for example F, Cl, Br, I, OH, NH 2 , SH, N 3 , N0 2 , alkyl groups such as methyl or ethyl, heteroalkyl groups such as methoxy, methylamino, dimethylamino or cyanide .
- heteroaryl refers to an aryl group as defined herein where one, two or more ring-carbon atoms are replaced by an oxygen, nitrogen, boron, phosphorous or sulphur atom, for example pyridyl, imidazolyl, pyrazolyl, quinolinyl, isoquinolinyl, pyrrolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, 1, 2 , 3-triazolyl , 1, 2 , 4-triazolyl , oxadiazolyl, thiadiazolyl, indolyl, indazolyl, tetrazolyl, pyrazinyl, pyrimidinyl and pyridazinyl groups.
- aralkyl refers to groups that comprise both aryl as well as alkyl and/or cycloalkyl groups .
- h teroarylalkyl refers to an aralkyl group as defined herein where one, two, three or more carbon atoms are replaced by one, two, three or more oxygen, nitrogen, phosphorous or sulphur atoms or S(0) ⁇ - 2 groups.
- Any alkyl, alkenyl, alkynyl, heteroalkyl, cycloalkyl, heterocycloalkyl, aryl, heteroaryl, aralkyl or heteroaryl- alkyl groups as defined herein may be substituted with one or more halogen atoms, NH 2 , SH, N0 2 or OH groups or unsubstituted alkyl, heteroalkyl, aryl, aralkyl, aralkyl- oxy, heteroaryl, cycloalkyl or heterocycloalkyl groups as defined herein.
- optionally substituted or “substituted” refer to groups wherein one or more hydrogen atoms may be replaced by a halogen atom, a NH 2 , SH, N0 2 or OH group or by an unsubstituted alkyl, heteroalkyl, aryl, aralkyl, aralkyloxy, heteroaryl, cycloalkyl or heterocycloalkyl group as defined herein.
- Preferred and/or advantageous embodiments of the invention are subject-matter of the subclaims. Preferred are compounds of Formula (I) , wherein R 1 is H.
- R 3 is an ethyl, a 2 -propyl, a C 3 -C 6 cycloalkyl (i.e, cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl) , a phenyl or a pyridyl group. All these groups may be substituted with one, two, three or more fluorine atoms or amino groups .
- R 3 is a cyclopropyl group.
- R 4 is hydroxy or a group of formula OS0 3 H, OP0 3 H 2 , OCH 2 OP0 3 H 2 , OCOCH 2 CH 2 COOH or an ester of a naturally occurring amino acid or a derivative thereof (i.e. a group of formula -OCOCHR ⁇ NH 2 or a derivative like an ester, amide or alkylamine thereof, wherein R' is the side chain of a naturally occurring amino acid like aspartic acid, glutaric acid, lysine, etc; e.g. dimethyl aminoglycine 0C0CH 2 N(CH 3 ) 2 ) .
- R 5 has the following structure:
- R 7 is H, F, Cl or a methoxy group that may be substituted by one, two or three fluorine atoms.
- A is C ⁇ _ 6 alkylene, C 2 - 6 alkenylene, C 2 - 6 alkynylene, C ⁇ - 6 heteroalkylene, cyclopropylene, epoxide, aziridine, thioepoxide, lactame or lactone, all of which groups may be subsitituted.
- A is a group of Formula -0-B-, wherein B is a C ⁇ _ 4 alkylene group, a C 2 - 4 alkenylene group, a C 2-4 alkynylene group or a C ⁇ _ 4 heteroalkylene group, all of which groups may be substituted by one, two or more hydroxy or amino groups.
- B is CH 2 or CH 2 CH 2 .
- B is CH 2 or CH 2 CH 2 ;
- X is CH, N or C-OMe and R 3 is cyclopropyl or X is CR 7 and R 7 and R 3 together form a bridge of the formula -0-CH 2 -CH (Me) - , wherein the preferred stereochemistry at the chiral center is the one giving the (S) configuration in the final compound,
- n is 1, 2 or 3, is 1, 2 or 3 and R 4 is hydroxy or a group of formula OS0 3 H, 0P0 3 H 2 , 0CH 2 0P0 3 H 2 , 0C0CH 2 CH 2 C00H or an ester of a naturally occurring amino acid or a derivative thereof .
- the mono, di or tri sodium salts are preferably the mono, di or tri sodium salts (most preferred the mono sodium salts) of compounds of formula (I) or (II) or mixtures thereof.
- the mono, di or tri sodium salts are preferably the mono sodium salts of compounds of formula (I) or (II) , wherein R 4 is OP0 3 H 2 or OS0 3 H or mixtures thereof.
- the present invention also relates to pharmacologically acceptable salts, or solvates and hydrates, respectively, and to compositions and formulations of compounds of Formula (I) or (II) .
- the present invention describes procedures to produce pharmaceutically useful agents, which contain these compounds, as well as the use of these compounds for the production of pharmaceutically useful agents.
- compositions according to the present invention contain at least one compound of Formula (I) or (II) as the active agent and optionally carriers and/or diluents and/or adjuvants.
- the pharmaceutical compositions according to the present invention may also contain additional known antibiotics.
- Examples of pharmacologically acceptable salts of sufficiently basic compounds of Formula (I) or (II) are salts of physiologically acceptable mineral acids like hydrochloric, hydrobromic, sulfuric and phosphoric acid; or salts of organic acids like methanesulfonic, p- toluenesulfonic, lactic, acetic, trifluoroacetic, citric, succinic, fumaric, maleic and salicylic acid.
- a sufficiently acidic compound of Formula (I) or (II) may form alkali or earth alkaline metal salts, for example sodium, potassium, lithium, calcium or magnesium salts; ammonium salts; or organic base salts, for example methylamine, dimethylamine, trimethylamine, triethylamine, ethylenediamine, ethanolamine, choline hydroxide, meglumin, piperidine, morpholine, tris- (2-hydroxyethyl) amine, lysine or arginine salts.
- Compounds of Formula (I) or (II) may be solvated, especially hydrated.
- the hydratisation can occur during the process of production or as a consequence of the hygroscopic nature of the initially water free compounds of Formula (I) or (II) .
- the compounds of Formula (I) or (II) may contain asymmetric C-atoms and may be present either as achiral compounds, mixtures of diastereomers, mixtures of enantiomers or as optically pure compounds.
- the present invention also relates to pro-drugs which are composed of a compound of Formula (I) or (II) and at least one pharmacologically acceptable protective group which will be cleaved off under physiological conditions, such as an alkoxy- , aralkyloxy-, acyl-, S0 3 H, P0 3 H 2 , acyloxymethyl group (e.g. pivaloyloxymethyl) , an 2-alkyl-, 2-aryl- or 2-aralkyl-oxycarbonyl-2-alkylidene ethyl group or an acyloxy group as defined herein, e.g. ethoxy, benzyloxy, acetyl or acetyloxy.
- a pharmacologically acceptable protective group which will be cleaved off under physiological conditions, such as an alkoxy- , aralkyloxy-, acyl-, S0 3 H, P0 3 H 2 , acyloxymethyl group (e.g.
- therapeutically useful agents that contain compounds of Formula (I) or (II) , their solvates, salts or formulations are also comprised in the scope of the present invention.
- compounds of Formula (I) or (II) will be administered by using the known and acceptable modes known in the art, either alone or in combination with any other therapeutic agent.
- Such therapeutically useful agents can be administered by one of the following routes: oral, e.g.
- TDS transder al delivery system
- the therapeutically useful product may be mixed with pharmaceutically inert, inorganic or organic excipients as are e.g. lactose, sucrose, glucose, gelatin, malt, silica gel, starch or derivatives thereof, talc, stearinic acid or their salts, dried skim milk, and the like.
- pharmaceutically inert, inorganic or organic excipients as are e.g. lactose, sucrose, glucose, gelatin, malt, silica gel, starch or derivatives thereof, talc, stearinic acid or their salts, dried skim milk, and the like.
- excipients e.g. vegetable, petroleum, animal or synthetic oils, wax, fat, polyols.
- liquid solutions, emulsions or suspensions or syrups one may use as excipients e.g.
- excipients as are e.g. vegetable, petroleum, animal or synthetic oils, wax, fat and polyols.
- compressed gases suitable for this purpose as are e.g. oxygen, nitrogen and carbon dioxide.
- the pharmaceutically useful agents may also contain additives for conservation, stabilisation, e.g. UV stabilizers, emulsifiers, sweetener, aromatisers, salts to change the osmotic pressure, buffers, coating additives and antioxidants .
- a daily dosage per patient of about l g to about 4000mg especially about 50mg to 3 g is usual with those of ordinary skill in the art appreciating that the dosage will depend also upon the age, conditions of the mammals, and the kind of diseases being treated or prevented.
- the daily dosage can be administrated in a single dose or can be divided over several doses .
- An average single dose of about 50mg, lOOmg, 250mg, 500mg, lOOOmg and 2000mg can be contemplated .
- step 1 CH 2 C1 2 , KOH (50%), 3h, rt ; 97%.
- step 2 H 2 , Pt/C, 20h, rt; followed by Z-Cl (Cbz-Cl), acetone/water, NaHC0 3 , 12h, rt, 98%.
- step 3 n-BuLi, -60°C, 24h, 80%.
- step 4 MsCI, triethylamine, CH 2 C1 2 ; 100%.
- step 5 NaN 3 in DMF,
- step 6 H 2 , Pd(OH) 2 , THF, MeOH, 24h, followed by AcOH, Ac 2 0, rt , 2h, 70%.
- step 7 DMF, NaH, 70°C, 12h, 75%.
- step 8 H 2 , Pd(OH) 2 , MeOH, THF, 24h, RT, 100%.
- step 9 N-Methylpyrrolidinone, l-Cyclopropyl-7- chloro-6-fluoro-1,4-dihydro-4-oxo-1,8-naphthydrin-3 - carboxylic acid (commercially available), TMS-Cl, H ⁇ nig Base or K 2 C0 3 , 80°C, 5h, 80%.
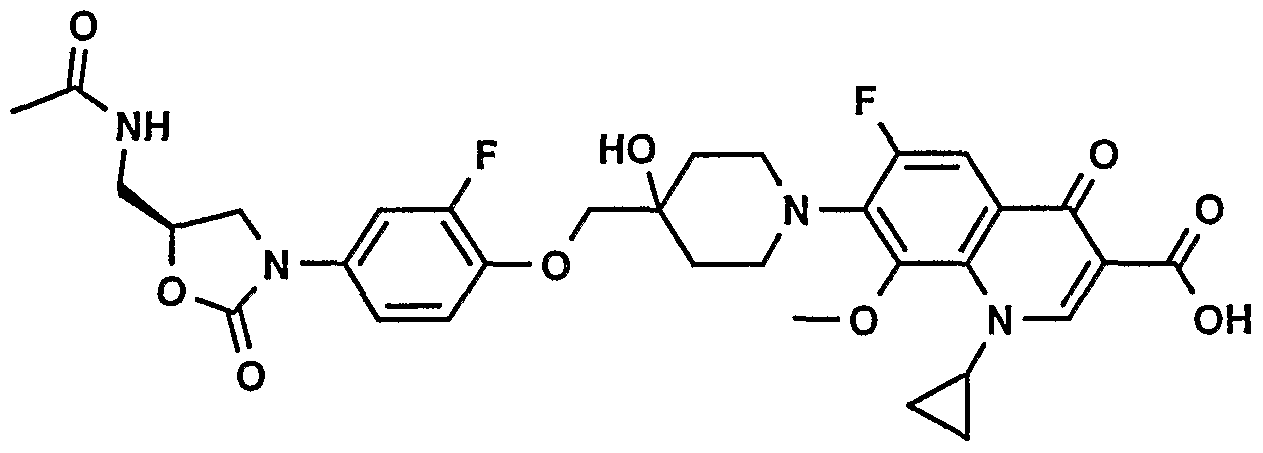
- Example 1 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy- piperidin-1-yl) -1-cyclopropyl-6-fluoro-4 -oxo-1,4-dihydro- [1,8] naphthyridine-3 -carboxylic acid
- Step 1 (4-Benzyloxy-3-fluoro-phenyl) -carbamic acid benzyl ester:
- Step 3 (5S) -5-azid ⁇ methyl-3- (4-benzyloxy-3-fluorophenyl) -oxazolidin-2-one: A solution of lOg (5R) -3- (4-benzyloxy-3-fluoro-phenyl) -5- hydroxymethyl-oxazolidin-2-one (MW: 317.32, 31.51mmol) and 4.78g triethylamine (MW: 101.19, 47.26mmol) in 300ml dichloromethane was treated under stirring at 10°C with 4.32g of methane sulfonyl chloride (MW: 114.55, 37.82mmol).
- the reaction was stirred at room temperature for one hour and monitored by TLC (ethyl acetate: hexane 1:1) .
- the reaction mixture was quenched with 100ml water and the organic layer washed with 100ml brine. The organic layer was dried over magnesium sulfate, filtered and the filtrate evaporated under reduced pressure. The residue was dissolved in 100ml dimethylformamide, 5.12g sodium azide (MW: 65.01, 78.7mmol) and a catalytic amount of tetrabutyl ammonium iodide were added.
- the suspension was stirred at 90 °C over night. The reaction was monitored by HPLC.
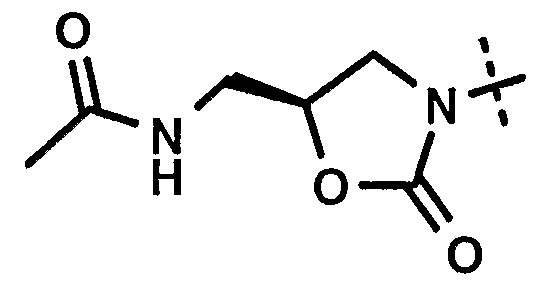
- Step 4 N- [ (5S) - ⁇ 3- (3 -fluoro-4-hydroxy-phenyl) ⁇ -2-oxo- oxazolidin-5-ylmethyl] -acetamide :
- Step 6 N- [ ⁇ (5S) -3 [3-fluoro-4- (4-hydroxy-piperidin-4-yl- methoxy) -phenyl] -2-oxo-oxazolidin-5-ylmethyl ⁇ ] -acetamide : A suspension of 31g 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluorophenoxymethyl ⁇ -4 -hydroxy- piperidine-1-carboxylic acid benzylester (MW: 515,54 60.13mmol) and 2.5 g of palladium 10% on activated carbon in 310ml methanol and 150ml ethyl acetate was stirred under hydrogen for 4 hrs . The reaction was monitored by TLC
- Step7 7- (4- ⁇ [ (5S) -5- (Acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy-piperidin-l-yl) -1- cyclopropyl-6-fluoro-4-oxo-1, 4 -dihydro- [1,8] naphthyridine- 3 -carboxylic acid:
- Step 1 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (bis-benzyloxy- phosphoryloxy) -piperidin-1-yl] -1-cyclopropyl-6-fluoro-4- oxo-1, 4-dihydro- [1, 8] naphthyridine -3-carboxylic acid:
- the original suspension slowly cleared.
- the solution was stirred at room temperature for two hours and monitored by TLC. (dichloromethane/methanol 9:1) .
- the reaction mixture was cooled to 0°C and treated with a 0.6ml of a 0.5M m- chloroperbenzoic acid solution in dichloromethane.
- the mixture was stirred for two hours at room temperature and diluted with 20ml dichloromethane.
- the organic layer was washed successively with 20ml of a saturated aqueous sodium bicarbonate solution and 20ml of brine and dried over magnesium sulfate.
- the slurry was filtered and the filtrate evaporated under reduced pressure.
- Step 2 7- (4- ⁇ 4- [ (5S) - (5-Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4-phosphonooxy- piperidin-1-yl) -l-cyclopropyl-6-fluoro-4-oxo-1, 4-dihydro- [1, 8] naphthyridine -3-carboxylic acid: A suspension of 158mg 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) - 2-oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (bis- benzyloxy-phosphoryloxy) -piperidin-1-yl] -1-cyclopropyl-6- fluoro-4-oxo-l, 4-dihydro- [1, 8] naphthyridine -3-carboxylic acid (MW: 8
- the catalyst was filtered off using a glass fibre filter paper. The solvents were evaporated under reduced pressure and the residue dissolved in 10ml methanol. The solution was diluted with 20ml water while a white solid precipitated. The solid was collected and dried. Yield: 85mg, 68%. MS: 709.0 (M+H) + , 706.5 (M-H) " Method ESI ⁇ ESI " .
- Example 3 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2, 6-diamino- hexanoyloxy) -piperidin-1-yl] -1-cyclopropyl-6-fluoro-4-oxo- 1, -dihydro- [1, 8] naphthyridine-3 -carboxylic acid
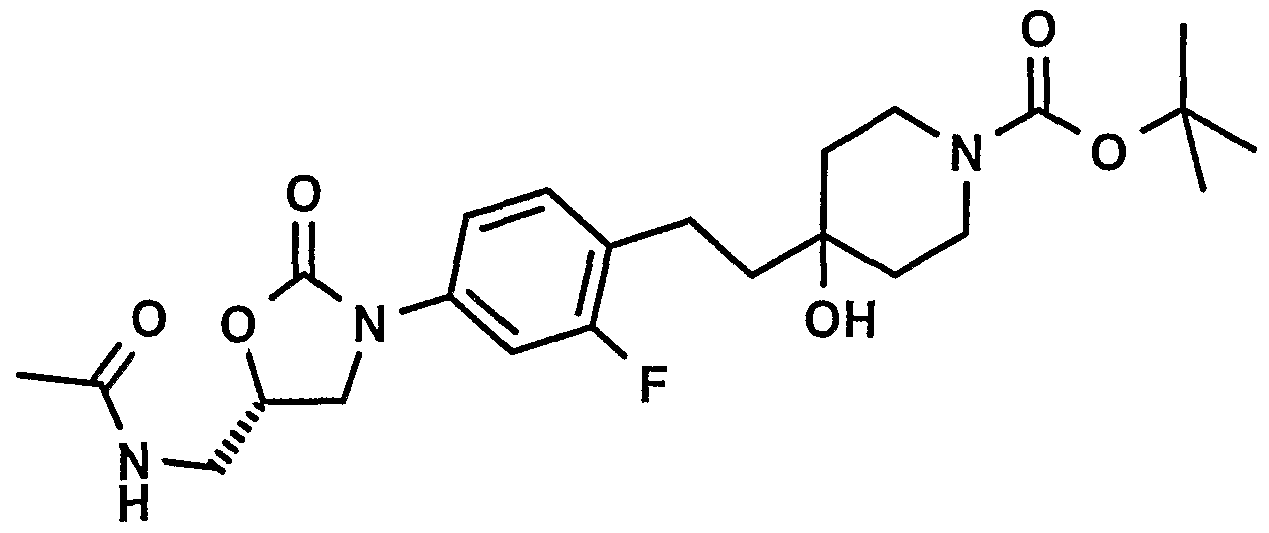
- Step 1 4- ⁇ 4- [ (5S) - (5-Acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy-piperidine-1- carboxylic acid tert-butyl ester:
- step 5 by reacting 3.83g l-oxa-6- aza-spiro [2.5] octane-6-carboxylic acid tert-butyl ester (WO0204462) (MW: 213.28 l ⁇ mmol) , 4.02g N- [ (5S) - ⁇ 3- (3- fluoro-4-hydroxy-phenyl) ⁇ -2-oxo-oxazolidin-5-ylmethyl] - acetamide (MW: 268.246, 15mmol) and 3. lg potassium carbonate (MW: 138.20, 22.5mmol) in 30ml dimethylformamide . Yield: 4.89-g, 67%. MS: 482.6 (M+H) + , Method ESI + .
- Step 2 4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2 , 6-bis- benzyloxycarbonylamino-hexanoyloxy) -piperidine-1-carboxylic acid tert-butyl ester:
- Step 3 2 , 6-Bis-benzyloxycarbonylamino-hexanoic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2- fluoro-phenoxymethyl ⁇ -piperidin-4-yl ester hydrochloride : 200mg of 4- ⁇ 4- [5- (5S) - (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2 , 6-bis- benzyloxycarbonylamino-hexanoyloxy) -piperidine-1-carboxylic acid tert-butyl ester (MW: 977.97, 0.22mmol) were dissolved in 4ml of a 1.25M dry hydrochloric acid in methanol. The reaction was stirred at 40°C for two hours, and the solvent removed by distillation under
- Step 4 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2, 6-bis- benzyloxycarbonylamino-hexanoyloxy) -piperidin-1-yl] -1- cyclopropyl-6-fluoro-4-oxo-l, 4-dihydro- [1, 8] naphthyridine- 3-carboxylic acid:
- Step 5 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2 , 6-diamino- hexanoyloxy) -piperidin-1-yl] -1-cyclopropyl- 6-fluoro-4-oxo- 1, 4-dihydro- [1, 8] naphthyridine-3-carboxylic acid: A suspension of 94mg 7- [4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) - 2-oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (2,6-bis- benzyloxycarbonylamino-hexanoyloxy) -piperidin-1-yl] -1- cyclopropyl-6-fluoro-4-oxo-l,
- Example 4 Succinic acid mono- [4- ⁇ 4- [ (5S) -5- (acetylamino- methyl) -2-oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -1- (6-carboxy-8-cyclopropyl-3-fluoro-5-oxo-5 , 8-dihydro- [1, 8] naphthyridin-2-yl) -piperidin-4-yl] ester
- Step 1 Succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -1-tert-butoxy- carbonyl-piperidin-4-yl ester benzyl ester:
- step 2 with 825mg 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2-fluoro- phenoxymethyl ⁇ -4 -hydroxy-piperidine-1 -carboxylic acid tert- butyl ester (MW: 481.52, 1.71mmol), 1.07g of succinic acid monobenzyl ester (MW: 208.21, 5.14mmol) and 0.63g of 4- dimethylaminopyridine (MW: 122.17, 5.1mmol) in 10ml dichloromethane was treated under stirring at room temperature with 1.3g N- (3-dimethylaminopropyl) -N'-ethyl- carbodiimid HC1 (MW: 191.70, 6.8mmol). Yield: 820mg, 70%. MS: 673.3 (M+H) + , Method ESI + .
- Step 2 Succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -piperidin-4-yl ester benzyl ester: 820mg of succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -1-tert-butoxy- carbonyl-piperidin-4-yl ester benzyl ester (MW: 671.72, 1.23mmol) were dissolved in 4ml of trifluoro acetic acid.
- Step 3 Succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -1- (6-carboxy- 8-cyclopropyl-3-fluoro-5-oxo-5, 8-dihydro- [1, 8] naphthyridin- 2-yl) -piperidin-4-yl ester benzyl ester: In analogy to example 1 step 7, with 113mg 7-chloro-l- cyclopropyl-6-fluoro-l,4-dihydro-4-oxo- [1, 8] naphthyridine- 3-carboxylic acid (MW:282.66, 0.4mmol ), 230mg succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2-oxo-oxazolidin-3- yl] -2-fluor
- Step 4 Succinic acid mono- [4- ⁇ 4- [ (5S) -5- (acetylamino- methyl) -2-oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -1- (6-carboxy-8 -cyclopropyl-3 -fluoro-5 -oxo-5,8-dihydro- [1, 8] naphthyridin-2-yl) -piperidin-4-yl] ester: In analogy to example 3 step 5 with 22mg succinic acid 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2- fluoro-phenoxymethyl ⁇ -1- (6-carboxy-8-cyclopropyl-3-fluoro- 5-OXO-5, 8-dihydro- [1, 8] naphthyridin-2-yl) -piperidin-4-yl ester benz
- Example 5 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy- piperidin-1-yl) -1-cyclopropyl-6-fluoro-4-oxo-1 , 4-dihydro- quinoline-3-carboxylic acid
- N-methyl-pyrrolidin-2-one was treated with 67.81g (7-chloro-1-cyclopropyl -6-fluoro-1, 4 -dihydro-4-oxo- 3-quinolinecarboxylic acid-boron diacetate complex (MW:410.57, 0.165 mole) and the mixture was stirred at 80°C for 5 hours.
- the N-methyl-pyrrolidin-2-one was evaporated under reduced pressure and residue was dissolved in 300ml of methanol.
- Example 6 7- [4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4- (bis-benzyloxy- phosphoryloxy) -piperidin-1-yl] -1-cyclopropyl-6-fluoro-4 - oxo-1 , 4 -dihydro-quinoline-3 -carboxylic acid
- the reaction was monitored by TLC (dichloromethane/methanol 9:1). The reaction was stirred for one hour and the mixture was washed at 0°C with 200ml IN aqueous hydrochloric acid and 100ml of a saturated sodium bicarbonate solution. The water layer were backwashed with 200ml dichloromethane. The combined organic layer were concentrated to 500ml and treated at roomtemperature with 13,2ml of a 70 % ter-butyl hydroperoxid solution in water (MW:90.12, 95mmol) . The reaction was stirred for 30 min, diluted with 500ml dichloromethane and the organic layer washed with 200ml IN aqueous hydrochloric acid and with 300ml brine.
- Example 7 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2-fluoro-phenoxymethyl ⁇ -4 -phosphonooxy- piperidin-1-yl) -l-cyclopropyl-6-fluoro-4-oxo-l, 4-dihydro- quinoline-3 -carboxylic acid
- the yellow suspension was diluted with 150ml of acetic acid and was heated to 45 °C. The reaction was monitored by HPLC/MS and was complete after 3 hours .
- the sticky suspension was added to 1.5 L of water under stirring.
- the off white crystals were collected, washed with 300ml water, 150ml ethanol and 150ml ether.
- the solid was suspended in 1.3L water and treated with 35ml (35mmol) of a 1M aqueous sodium hydroxide solution.
- the solid dissolved, and the brown-yellow solution was treated with 15 g of activated charcoal and filtered.
- the filtrate was extracted with 3 portions of 200ml of a 95/5 dichloromethane/ methanol mixture.
- Example 8 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy- piperidin-1-yl) -1-cyclopropyl-6-fluoro-8-methoxy-4-oxo-l , 4- dihydro-quinoline-3-carboxylic acid
- Example 9 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy- piperidin-1-yl) -1-cyclopropyl-8-methoxy-4-oxo-1 , 4-dihydro- quinoline-3 -carboxylic acid O
- Example 10 9- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy- piperidin-1-yl) -8-fluoro-3 -methyl-6-oxo-2, 3 -dihydro-6H-1- oxa-3a-aza-phenalene-5-carboxylic acid
- Example 11 7- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -3-hydroxy- pyrrolidin-1-yl) -1-cyclopropyl-6-fluoro- -oxo-1, 4-dihydro- [1, 8] naphthyridine-3 -carboxylic acid
- Step 1 l-0xa-5-aza-spiro[2.4] heptane-5-carboxylic acid benzyl ester:
- the reaction mixture was stirred at room temperature for three hours.
- the reaction mixture was diluted with 20ml of a saturated aqueous sodium sulfite solution and 45ml of dichloromethane.
- Step 2 3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo-oxazolidin- 3 -yl] -2 -fluoro-phenoxymethyl ⁇ -3-hydroxy-pyrrolidine- 1- carboxylic acid benzyl ester: A solution of 420mg of N- [ (5S) - ⁇ 3- (3-fluoro-4-hydroxy- phenyl) ⁇ -2-oxo-oxazolidin-5-ylmethyl] -acetamide (MW:
- Step 3 N- ⁇ (5S) -3- [3-Fluoro-4- (3-hydroxy-pyrrolidin-3-yl- methoxy) -phenyl] -2-oxo-oxazolidin-5-ylmethyl ⁇ -acetamide: A suspension of 660mg 3- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -3-hydroxy- pyrrolidine-1-carboxylic acid benzyl ester (MW: 501.51, 1.31mmol) and 20mg palladium 10% on activated carbon in 20ml of a 1/1 ethyl acetate / methanol mixture was stirred for twelve hours under hydrogen. The catalyst was filtered on a glass fiber filter paper and the filtrate evaporated under reduced pressure to afford a colorless oil. Yield: 400mg, 83.2 %. MS: 368.4 (M+H
- Step 4 7- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2 -fluoro-phenoxymethyl ⁇ -3 -hydroxy- pyrrolidin-1-yl) -l-cyclopropyl-6-fluoro-4 -oxo-1, 4 -dihydro- [1, 8] naphthyridine-3 -carboxylic acid: In analogy to example 1, step 7 with 39mg 7-chloro-l-cyclo- propyl-6-fluoro-1, 4-dihydro-4-oxo- [1,8] naphthyridine-3- carboxylic acid (MW: 282.66, 0.24mmol ), 99mg N- ⁇ (5S) -3- [3- fluoro-4- (3-hydroxy-pyrrolidin-3-ylmethoxy) -phenyl] -2-oxo- oxazolidin-5-
- Example 12 7- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2 -fluoro-phenoxymethyl ⁇ -3 -hydroxy- pyrrolidin-1-yl) -1-eyelopropyl-6-fluoro-4-oxo-1,4-dihydro- quinoline-3 -carboxylic acid
- Example 13 7- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2-fluoro-phenoxymethyl ⁇ -3 -hydroxy- pyrrolidin-1-yl) -l-cyclopropyl-6-fluoro-8-methoxy-4-oxo- 1 , 4-dihydro-quinoline-3 -carboxylic acid
- Example 14 7- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -3-hydroxy- pyrrolidin-1-yl) -1-cyclopropyl-8-methoxy-4 -oxo-1,4-dihydro- quinoline-3 -carboxylic acid
- Example 15 9- (3- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -3-hydroxy- pyrrolidin-1-yl) -8-fluoro-3 -methyl -6-oxo-2 , 3 -dihydro-6H-1- oxa-3a-aza-phenalene-5-carboxylic acid
- Example 16 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4 -hydroxy-azepan- 1-yl) -1-cyclopropyl-6-fluoro-4 -oxo-1,4-dihydro-quinoline-3 - carboxylic acid
- Step 1 4-Methylene-azepane-l-carboxylic acid tert-butyl ester:
- the yellow suspension was cooled to -78 °C and treated with a solution of 595mg 4-oxo-azepane-l-carboxylic acid tert-butyl ester (WO 2000044376) (MW: 213.279, 2.78mmol) in 10ml tetrahydrofuran.
- the reaction mixture was stirred at room temperature for one and half hour.
- the reaction mixture was quenched with 30ml of a saturated aqueous solution of ammonium chloride, diluted with 30ml of ethyl acetate.
- the organic layer was successively washed with 30ml water and 30ml brine, dried over magnesium sulfate and filtered.
- Step 2 l-Oxa-6-aza-spiro [2.6] nonane-6-carboxylic acid tert-butyl ester:
- step 1 with 4-methylene-azepane-l- carboxylic acid tert-butyl ester (MW:211.307, 1.73mmol), 1.16g sodium bicarbonate (MW: 84.01 13.8mmol) and 1.36g of 80% m-chloroperbenzoic acid (MW172.57, 6.05mmol) in 5ml of dichloromethane. Yield: 250mg, 63 %.
- Step 3 4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy-azepane-l- carboxylic acid tert-butyl ester:
- step 5 with 247mg of l-oxa-6-aza- spiro [2.6] nonane-6-carboxylic acid tert-butyl ester. (MW: 227.31 1.08mmol), 296mg N- [ (5S) - ⁇ 3- (3-fluoro-4-hydroxy- phenyl) ⁇ -2-oxo-oxazolidin-5-ylmethyl] -acetamide (MW: 268.246, 80mmol) and 228mg potassium carbonate (MW: 138.20, 1.65mmol) in 150ml dimethylformamide . Yield: 334mg, 62 %. MS: 496.8 (M+H) + , 440.8 (M-C (CH 3 ) 3 +H) + , Method ESI + .
- Step 4 N- ⁇ (5S) -3- [3-Fluoro-4- (4-hydroxy-azepan-4- ylmethoxy) -phenyl] -2-oxo-oxazolidin-5-ylmethyl ⁇ -acetamide : A solution of 334mg 4- ⁇ 4- [ (5S) -5- (acetylamino-methyl) -2- oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4 -hydroxy- azepane-1-carboxylic acid tert-butyl ester (MW:495.55, 0.674mmol) in 3ml of a 1.25 M anhydrous hydrogen chloride solution in methanol was stired at 35°C for four hours.
- Step 5 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2 -fluoro-phenoxymethyl ⁇ -4 -hydroxy-azepan- 1-y1) -1-eye1opropyl-6-fluoro-4 -oxo-1,4-dihydro-quino1ine-3 - carboxylic acid: In analogy to example 5 with 150mg N- ⁇ (5S) -3- [3-fluoro-4- ( -hydroxy-azepan-4-ylmethoxy) -phenyl] -2-oxo-oxazolidin-5- ylmethyl ⁇ -acetamide (MW: 395.43) and 98mg of ethyl diisopropylamine (MW: 129.25, 0.758mmol), 163mg (7-chloro- 1-cyclopropyl-6-fluoro-1, 4-dihydro-4-
- Example 17 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino-methyl) -2-oxo- oxazolidin-3 -yl] -2-fluoro-phenoxymethyl ⁇ -4-hydroxy-azepan- 1-yl) -l-cyclopropyl-6-fluoro-4-oxo-l , 4-dihydro- [1, 8] naphthyridine-3 -carboxylic acid
- Example 18 sodium salt of 7- (4- ⁇ 4- [ (5S) -5- (Acetylamino- methyl) -2-oxo-oxazolidin-3-yl] -2-fluoro-phenoxymethyl ⁇ -4 - phosphonooxy-piperidin-1-yl) -1-cyclopropyl -6-fluoro-4-oxo- 1,4-dihydro-quinoline-3-carboxylic acid
- the resulting suspension was stirred for another hour and the resulting crystals were collected by filtration, washing the solid with ethyl acetate (10 x 500 ml) to remove the DMSO and then dried in vacuo. If there is still amounts of DMSO and/or ethyl acetate remaining, then the solid was slurred with acetone / water (99:1) for 24 h. The mixture was then filtered, washed with acetone / water (99:1) (2 x 500 ml) and then allowed to suck dry on the filter for 12 h. The solid was then dried in vacuo. Yield: 90%.
- Example 19 Formation of building blocks via a Sonogashira reaction - 4- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenylethynyl ⁇ -4-hydroxy-piperidine-l- carboxylic acid tert-butyl ester.
- Step 1 (4-Bromo-3-Fluoro-phenyl) -carbamic acid benzyl ester.
- the aqueous layer was then extracted with ethyl acetate (3 x 150 ml) .
- the combined organic layers were then washed with a saturated sodium chloride solution, and dried over MgS0 4 . After filtration, the solvent was removed, and n-hexane added. The mixture was stirred during 30min at room temperature, the crystals were filtrated and washed with hexane to give the first crop of solid.
- the filtrate was evaporated, and the solid mixed with heptane at 0°C and stirred during
- Step 2 (5R) 3- (4-Bromo-3-Fluoro-phenyl) -5-hydroxymethyl oxazolidin-2-one Butyl lithium (2.3M in n-hexanes, 118,3 ml, 0,272 mol, l,06eq) was added at -30°C to anhydrous tert-butanol (25.0 g, 0,53 mol, 2,07eq) in anhydrous THF (170 ml), under nitrogen. The mixture was stirred for 30min at -30°C, and was then allowed to warm slowly to 0°C.
- Step 3 Methanesulfonic acid 3- (4-Bromo-3-Fluoro-phenyl) -2- oxo-oxazolidin- (5R) -ylemthyl ester.
- Methansulfonyl chloride (27.4 ml, 0,354 mol, 1.9 eq) was added to an ice-cold solution of the (5R) 3- (4-Bromo-3- Fluoro-phenyl) -5-hydroxymethyl oxazolidin-2-one (54.0 g, 0,186 mol, leq) and triethylamine (51.8 ml, 0,372 mol, 2eq) in anhydrous DCM (420 ml) at 0°C. The resulting solution was allowed to come to room temperature, and then stirred over 3 hours. The mixture was then washed with 10% sodium hydrogen carbonate solution giving a precipitate.
- the solid was filtered, The washed with DCM, and the filtrate and washings dried over MgS0. After filtering, the solvent was removed, and the resulting solid was slurried with diethyl ether. The solid was then filtered, washed with ice cold diethyl ether and dried to give the product (68,5 g, quantitative) .
- Step 4 (5R) - Azidomethyl-3- (4 -Bromo-3-Fluoro-phenyl) oxazolidin-2-one
- a suspension of the Methanesulfonic acid 3- (4-Bromo-3- Fluoro-phenyl) -2-oxo-oxazolidin- (5R) -ylemthyl ester (68.5 g, 0.186 mol, leq), tetrabutyl ammonium iodide (0.686 g, 0.00186 mol, O.Oleq) and sodium azide (24.57g, 0.378 mol, 2.03eq) in anhydrous DMF (500 ml) was stirred 80°C under nitrogen over night.
- Step 5 (5R) - Aminomethyl-3 - (4-Bromo-3-Fluoro-phenyl) oxazolidin-2-one .
- Step 7 4-oxo-piperidine-l-carboxylic acid tert-butyl ester.
- Step 8 4-Hydroxy-4-trimethylsilanylethynyl piperidine-1- carboxylic acid tert-butyl ester.
- n-Butyl lithium (2.3M solution in n-hexanes, 16.0 ml, 36.8 mmol, 1. leq) was added to a solution of TMS-alkyne (6.03 ml, 42.4 mmol,1.26eq) in THF (124 ml) at - 78°C under nitrogen.
- Step 9 4-Ethynyl-4-Hydroxy-piperidine-l-carboxylic acid tert-butyl ester.
- Step 10 4- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin-3- yl] -2-fluoro-phenylethynyl ⁇ -4-hydroxy-piperidine-1- carboxylic acid tert-butyl ester.
- PdCl 2 (P(C 6 H 5 )3)2 (297 mg, 0.422 mmol, 0.1 eq) and 148mg of copper (I) iodide (160 mg, 0.78 mmol, 0.2 eq) were stirred at RT, under argon.
- Example 20 Formation of building blocks via a Heck reaction - 4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2- fluoro-phenyl ⁇ -acryloyl) -piperazine-1-carboxylic acid tert butyl ester.
- Step 1 Piperazine-1-carboxylic acid tert butyl ester A solution of 24g of di-tert-butyl dicarbonate (24 g, O.llmol, leq) in 200ml dichloromethane (200ml) was added to a stirred solution of 20g piperazine (20 g, 0.23mol, 2eq) in dichloromethane (800ml) at RT. The mixture was stirred overnight at RT and the mixture was then filtered and the filtrate was evaporated. Diethylether was then added to the residue, and the mixture was filtered again, and to the filtrate n- heptane was added. The suspension was filtrated again and the filtrate was evaporated to give the product (19 g, 43,9%) as a white solid.
- Step 2 4-Acryloyl-piperazine-l-carboxylic acid tert butyl ester
- Step 3 4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenyl ⁇ -acryloyl) -piperazine-1-carboxylic acid tert butyl ester (5S) -N- [ (4-bromo-3-Fluoro- henyl) -2-oxo-oxazolidin-5- ylmethyl] -acetamide (1.998 g, 6.0 mmol, 1 eq) , 4-Acryloyl- piperazine-1-carboxylic acid tert butyl ester (1.6 g, 6 .
- Example 21 Formation of building blocks via epoxide ring opening with a phenol- 4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2- fluoro-phenoxy ⁇ -2-hydroxy-propionyl) -piperazine-1- carboxylic acid tert butyl ester
- Step 1 4-Oxiranecarbonyl piperazine-1-carboxylic acid tert butyl ester
- Step 2 4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2-fluoro-phenoxy ⁇ -2-hydroxy-propionyl) -piperazine-1- carboxylic acid tert butyl ester
- the 4-Oxiranecarbonyl piperazine-1-carboxylic acid tert butyl ester (0.1 g, 0,39mmol, leq) was added to a stirred solution of N- [ (5S) - ⁇ 3- (3-fluoro-4-hydroxy-phenyl) ⁇ -2-oxo- oxazolidin-5-ylmethyl] -acetamide (step 4, example 1) (0.104 g, 0,39mmol, leq) and K 2 C0 3 (0.081 g, 0,585mmol, l,5eq) in DMF (2 ml) .
- Example 22 Formation of building blocks via alkylation of a phenol group -
- Step 1 4- (2-bromo acetyl) -piperazine-1-carboxylic acid tert butyl ester Bromo acetyl bromide (4.86 ml, 21.47 mmol, leq) was added dropwise to a stirred ice cold mixture of the Piperazine-1- carboxylic acid tert butyl ester (4.0 g, 21.47 mmol, leq) and diisopropyl ethyl amine (12.05 g, 92.5 mmol, 4,3eq) in dichloromethane (108 ml) .
- Step 2 4- (2- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin- 3-yl] -2 -fluoro-phenoxy ⁇ -acetyl) -piperazine-1-carboxylic acid tert butyl ester
- the 4- (2-bromo acetyl) -piperazine-1-carboxylic acid tert butyl ester (2.0 g, 6.5 mmol, leq) was added to a stirred solution of N- [ (5S) - ⁇ 3- (3-fluoro-4 -hydroxy-phenyl) ⁇ -2-oxo- oxazolidin-5-ylmethyl] -acetamide (step 4, example 1) (1.75 g, 6.5 mmol, leq) and potassium carbonate (1.138 g, 9.75 mmol, l,5eq) in DMF (32 ml) .
- Example 23 Formation of building blocks via triple bond reduction 4- (2- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo-oxazolidin-3-yl] -2- fluoro-phenyl ⁇ -ethyl) -4-hydroxy-piperidine-l-carboxylic acid tert butyl ester
- Example 24 7- (4- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2 -fluoro-phenylethynyl ⁇ -4 -hydrox - piperidin-1-yl) -1-cyclopropyl-6-fluoro-4-oxo-1 , 4 -dihydro- quinoline-3 -carboxylic acid
- Example 26 7- [4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenyl ⁇ -acryloyl) -piperazin-1-yl] -1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydro-quinoline-3- carboxylic acid
- Example 27 7- [4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenyl ⁇ -acryloyl) -piperazin-1-yl] -1-cyclopropyl-6-fluoro-4-oxo-l, 4-dihydro- [1,8]- napthyridine-3 -carboxylic acid
- Example 28 7- [4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxy ⁇ -2-hydroxy-propionyl) -1- cyclopropyl-6-fluoro-4-oxo-1, 4-dihydro-quinoline-3- carboxylic acid
- Example 29 7- [4- (3- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxy ⁇ -2-hydroxy-propionyl) -1- cyclopropyl-6-fluoro-4-oxo-1, 4-dihydro- [1,8] -napthyridine 3-carboxylic acid
- the resulting amine was then coupled to the required quinoline using the method described in example 1 - step 7 This gave the required product in 4 % yield over two steps
- Example 30 7- [4- (2- ⁇ 4- [5S- (acetylamino-methyl) -2-oxo- oxazolidin-3-yl] -2-fluoro-phenoxy ⁇ -acetyl) -piperazin-1-yi; l-cyclopropyl-6-fluoro-4-oxo-1, 4 -dihydro-quinoline-3- carboxylic acid
- Example 32 7- [4 - (2- ⁇ 4- [5S- (acetylamino-methyl) -2 -oxo- oxazolidin-3 -yl] -2 -f luoro-phenyl ⁇ -ethyl) -4 -hydroxy- piperidin- 1 -yl ] - 1 -cyclopropyl - 6 - f luoro-4 -oxo- 1 , 4 -dihydro- quinoline- 3 -carboxylic acid
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
Abstract
Description


































Claims


Priority Applications (19)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MXPA06006769A MXPA06006769A (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics. |
| CA 2549675 CA2549675C (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| JP2006544382A JP2007516263A (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotic |
| ES04804099T ES2310299T5 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone-based hybrid antibiotics |
| KR20067014403A KR101176450B1 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| US10/583,419 US8158797B2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| NZ547382A NZ547382A (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| EP04804099.2A EP1709044B2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| DK04804099T DK1709044T4 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| PL04804099T PL1709044T5 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| SI200430867T SI1709044T2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| BRPI0417193A BRPI0417193B8 (en) | 2003-12-18 | 2004-12-20 | oxazolidinone-quinolone hybrid antibiotics, their monosodium, disodium or trisodium salts and their prodrugs, pharmaceutical compositions, and their use |
| AU2004299278A AU2004299278C9 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| DE200460015158 DE602004015158D1 (en) | 2003-12-18 | 2004-12-20 | OXAZOLIDINONE-CHINOLON HYBRID ANTIBIOTICS |
| HK06112470.8A HK1090647B (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| IL175409A IL175409A (en) | 2003-12-18 | 2006-05-04 | Oxazolidinone-quinolone hybrid derivatives and pharmaceutical compositions containing the same |
| US13/368,667 US8501774B2 (en) | 2003-12-18 | 2012-02-08 | Oxazolidinone-quinolone hybrid antibiotics |
| US13/933,811 US9133213B2 (en) | 2003-12-18 | 2013-07-02 | Oxazolidinone-quinolone hybrid antibiotics |
| US14/816,789 US20150336992A1 (en) | 2003-12-18 | 2015-08-03 | Oxazolidinone-quinolone hybrid antibiotics |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53082203P | 2003-12-18 | 2003-12-18 | |
| US60/530,822 | 2003-12-18 | ||
| EP04001506.7 | 2004-01-23 | ||
| EP04001506A EP1557416A1 (en) | 2004-01-23 | 2004-01-23 | Oxazolidinone-quinolone hybrid antibiotics |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/583,419 A-371-Of-International US8158797B2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| US10/583,419 Continuation US8158797B2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
| US13/368,667 Continuation US8501774B2 (en) | 2003-12-18 | 2012-02-08 | Oxazolidinone-quinolone hybrid antibiotics |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005058888A2 true WO2005058888A2 (en) | 2005-06-30 |
| WO2005058888A3 WO2005058888A3 (en) | 2005-08-18 |
Family
ID=39109376
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/014500 Ceased WO2005058888A2 (en) | 2003-12-18 | 2004-12-20 | Oxazolidinone-quinolone hybrid antibiotics |
Country Status (21)
| Country | Link |
|---|---|
| US (3) | US8501774B2 (en) |
| EP (1) | EP1709044B2 (en) |
| JP (2) | JP2007516263A (en) |
| KR (1) | KR101176450B1 (en) |
| CN (2) | CN106957316A (en) |
| AT (1) | ATE401326T1 (en) |
| AU (1) | AU2004299278C9 (en) |
| BR (1) | BRPI0417193B8 (en) |
| CA (1) | CA2549675C (en) |
| CY (1) | CY1108409T1 (en) |
| DE (1) | DE602004015158D1 (en) |
| DK (1) | DK1709044T4 (en) |
| ES (1) | ES2310299T5 (en) |
| IL (1) | IL175409A (en) |
| MX (1) | MXPA06006769A (en) |
| NZ (1) | NZ547382A (en) |
| PL (1) | PL1709044T5 (en) |
| PT (1) | PT1709044E (en) |
| RU (1) | RU2371443C2 (en) |
| SI (1) | SI1709044T2 (en) |
| WO (1) | WO2005058888A2 (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007017828A2 (en) | 2005-08-08 | 2007-02-15 | Actelion Pharmaceuticals Ltd | Oxazolidinone-quinolone hybrids as antibacterial compounds |
| WO2007114326A1 (en) | 2006-03-31 | 2007-10-11 | Research Foundation Itsuu Laboratory | Novel compound having heterocyclic ring |
| WO2008062379A3 (en) * | 2006-11-24 | 2008-07-17 | Actelion Pharmaceuticals Ltd | 4- (2-oxo-oxazolidin-3yl)-phenoxymethyle derivativeas as antibacterials |
| WO2009044777A1 (en) | 2007-10-02 | 2009-04-09 | Research Foundation Itsuu Laboratory | Oxazolidinone derivative having 7-membered hetero ring |
| JP2010132677A (en) * | 2006-11-10 | 2010-06-17 | Actelion Pharmaceuticals Ltd | 5-hydroxymethyl-oxazolidin-2-one derivative |
| US7820823B2 (en) | 2001-10-04 | 2010-10-26 | Morphochem Aktiengesellschaft Fur Kominatorische Chemi | Dual action antibiotics |
| EP2374806A1 (en) * | 2003-09-03 | 2011-10-12 | Morphochem Aktiengesellschaft Für Kombinatorische Chemie | OXAZOLIDINON-QUINOLONe HYBRIDes and intermediates for their preparation |
| US8124623B2 (en) | 2006-11-10 | 2012-02-28 | Actelion Pharmaceuticals Ltd. | 5-hydroxymethyl-oxazolidin-2-one-derivatives and their uses as antibacterials |
| US8268812B2 (en) | 2003-04-30 | 2012-09-18 | Morphochem Aktiengesellschaft fül Kombinatorische Chemie | Use of oxazolidinone-quinoline hybrid antibiotics for the treatment of anthrax and other infections |
| US8354401B2 (en) | 2008-10-27 | 2013-01-15 | Mitsubishi Tanabe Pharma Corporation | Oxazolidinone amide aromatic compounds for supressing MMP-9 production |
| WO2014191075A1 (en) | 2013-05-28 | 2014-12-04 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| WO2014191109A1 (en) * | 2013-05-28 | 2014-12-04 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Combination therapy comprising oxazolidinone-quinolones for use in treating bacterial infections |
| CN108059624A (en) * | 2016-11-08 | 2018-05-22 | 上海医药工业研究院 | It is used to prepare the preparation method of click azoles amine key intermediate |
| US10087171B2 (en) | 2016-12-19 | 2018-10-02 | Actelion Pharmaceuticals Ltd | Crystalline forms of cadazolid |
| CN114478486A (en) * | 2022-01-28 | 2022-05-13 | 西南大学 | Trimolecular conjugate with p-aminosalicylic acid as parent nucleus, intermediate, preparation method and use |
| WO2024024962A1 (en) | 2022-07-29 | 2024-02-01 | 住友ファーマ株式会社 | Nitrogen-containing saturated heterocyclic ring derivative |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR101592046B1 (en) * | 2008-02-22 | 2016-02-05 | 액테리온 파마슈티칼 리미티드 | Oxazolidinone derivatives |
| JP5613656B2 (en) * | 2008-03-26 | 2014-10-29 | グローバル、アライアンス、フォア、ティービー、ドラッグ、ディベロップメント | Bicyclic nitroimidazoles covalently linked to substituted phenyloxazolidinones |
| SG11201401710PA (en) * | 2011-11-08 | 2014-09-26 | Actelion Pharmaceuticals Ltd | 2-oxo-oxazolidin-3,5-diyl antibiotic derivatives |
| CN103435611B (en) * | 2013-09-07 | 2015-05-27 | 吉首大学 | Multi-target point alpha-pyridoin compounds and preparation method and application thereof |
| CN103483321B (en) * | 2013-09-07 | 2014-08-13 | 吉首大学 | Alkyl-linked pyrrolone-quinolinone type compound and its preparation method and use |
| CN104725330B (en) * | 2013-12-18 | 2017-06-13 | 四川好医生药业集团有限公司 | Oxazolidinone compounds and its production and use |
| CN107286182A (en) * | 2016-04-12 | 2017-10-24 | 李靖 | Novel oxazolidinone fluoro quinolone derivative and purposes |
| CN112898317B (en) * | 2021-01-21 | 2021-12-07 | 河南科技大学第一附属医院 | Oxazole compound for sterilization and disinfection in hospital care and preparation method and application thereof |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4840956A (en) | 1986-02-18 | 1989-06-20 | Warner-Lambert Company | Novel disubstituted-7-pyrrolidinoquinoline antibacterial agents |
| NZ222047A (en) | 1986-10-08 | 1991-01-29 | Bristol Myers Co | Quinoline - or naphthyridine - carboxylic acid anti-bacterial agents |
| JPH0269478A (en) | 1988-09-05 | 1990-03-08 | Sagami Chem Res Center | Quinolonecarboxylic acid derivative |
| US5491139A (en) | 1988-10-24 | 1996-02-13 | The Procter & Gamble Company | Antimicrobial quinolonyl lactams |
| DE69019859T2 (en) | 1989-03-30 | 1995-10-05 | Wakunaga Seiyaku Kk | Quinolone derivatives and their salts, processes for their preparation and antibacterial active ingredients containing them. |
| IL100572A (en) | 1991-01-03 | 1997-01-10 | Lepetit Spa | Amides of antibiotic ge 2270 factors their preparation and pharmaceutical compositions containing them |
| EP0610265B1 (en) | 1991-11-01 | 1996-12-27 | PHARMACIA & UPJOHN COMPANY | Substituted aryl- and heteroarylphenyloxazolidinones useful as antibacterial agents |
| US5221676A (en) | 1992-02-06 | 1993-06-22 | Warner-Lambert Company | 7-substituted quinolones and naphthyridones as antibacterial agents |
| DK0992501T3 (en) | 1995-09-22 | 2002-10-28 | Wakunaga Pharma Co Ltd | Pyridonecarboxylic acid derivatives as antibacterial agents |
| DE19601265A1 (en) | 1996-01-16 | 1997-07-17 | Bayer Ag | 2-oxo and 2-thio-1,2-dihydroquinolinyl oxazolidinones |
| GB9702213D0 (en) | 1996-02-24 | 1997-03-26 | Zeneca Ltd | Chemical compounds |
| DE19633480A1 (en) | 1996-08-20 | 1998-02-26 | Bayer Ag | Orally administrable formulations of quinolone and naphthyridonecarboxylic acids |
| GB9725244D0 (en) | 1997-11-29 | 1998-01-28 | Zeneca Ltd | Chemical compounds |
| BR9907183A (en) | 1998-01-23 | 2003-06-10 | Versicor Inc | Oxazolidinone combinatorial collections, compositions and preparation processes |
| TW572757B (en) | 1998-08-24 | 2004-01-21 | Bristol Myers Squibb Co | Novel isoxazolinone antibacterial agents |
| KR100324621B1 (en) * | 1999-04-27 | 2002-02-27 | 박호군 | Oxazolidinone derivatives having quinolone moiety and their uses for antibacterials |
| WO2001009107A1 (en) | 1999-07-28 | 2001-02-08 | Pharmacia & Upjohn Company | Oxazolidinones and their use as antiinfectives |
| US6518285B2 (en) | 1999-12-21 | 2003-02-11 | Ortho Mcneil Pharmaceutical, Inc. | Piperidinyloxy and pyrrolidinyloxy oxazolidinone antibacterials |
| US6689769B2 (en) * | 2000-12-21 | 2004-02-10 | Pharmacia & Upjohn Company | Antimicrobial quinolone derivatives and use of the same to treat bacterial infections |
| ES2186550B2 (en) | 2001-06-27 | 2003-11-16 | Vita Lab | NEW DERIVATIVES OF OXAZOLIDINONES AS ANTIBACTERIALS. |
| WO2003031441A1 (en) * | 2001-10-04 | 2003-04-17 | Morphochen Aktiengesellschaft Für Kombinatorische Chemie | Multiple action compounds |
| WO2003031443A1 (en) * | 2001-10-04 | 2003-04-17 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Dual actions antibiotics comprising a oxazoldinone and a quinolone or naphthyridinone moiety |
| US6624593B2 (en) | 2001-10-08 | 2003-09-23 | Randall D. Blanchard | Dimmable ballast for electrodeless fluorescent lamps |
| US20040132764A1 (en) | 2002-10-23 | 2004-07-08 | Morphochem Aktiengesellschaft Fuer Kombinatorische Chemie | Antibiotics for the treatment of infections in acidic environments |
| EP1594852A1 (en) | 2003-02-07 | 2005-11-16 | Ranbaxy Laboratories, Ltd. | Oxazolidinone derivatives as antimicrobials |
| JP4805139B2 (en) * | 2003-04-30 | 2011-11-02 | モルホケム アクツィエンゲゼルシャフト フューア コンビナートリッシュ ヒェミー | Use of oxazolidinone-quinoline hybrid antibiotics to treat anthrax and other infections |
| DE10340485B4 (en) * | 2003-09-03 | 2015-05-13 | Morphochem AG Aktiengesellschaft für kombinatorische Chemie | Process for the preparation of oxazolidinone-quinolone hybrids |
| AR054608A1 (en) | 2005-08-08 | 2007-06-27 | Actelion Pharmaceuticals Ltd | OXAZOLIDINONE DERIVATIVES LINKED TO QUINOLONES AS ANTIBACTERIALS |
| KR200430712Y1 (en) | 2006-08-30 | 2006-11-13 | 명 식 신 | Ceiling light |
| HRP20120385T1 (en) † | 2006-11-10 | 2012-06-30 | Actelion Pharmaceuticals Ltd. | 5-hydroxymethyl-oxazolidin-2-one derivatives |
| CL2007003332A1 (en) | 2006-11-24 | 2008-06-20 | Actelion Pharmaceuticals Ltd | COMPOUNDS DERIVED FROM CONDENSED HETEROCICLES; INTERMEDIARY COMPOUNDS; PHARMACEUTICAL COMPOSITION; AND USE IN THE PREVENTION OR TREATMENT OF BACTERIAL INFECTIONS. |
| NZ589734A (en) † | 2008-05-09 | 2012-11-30 | Actelion Pharmaceuticals Ltd | 5-hydroxymethyl-oxazolidin-2-one derivatives for treating bacterial intestinal diseases |
-
2004
- 2004-12-20 BR BRPI0417193A patent/BRPI0417193B8/en not_active IP Right Cessation
- 2004-12-20 WO PCT/EP2004/014500 patent/WO2005058888A2/en not_active Ceased
- 2004-12-20 NZ NZ547382A patent/NZ547382A/en not_active IP Right Cessation
- 2004-12-20 MX MXPA06006769A patent/MXPA06006769A/en active IP Right Grant
- 2004-12-20 CN CN201611120339.0A patent/CN106957316A/en active Pending
- 2004-12-20 SI SI200430867T patent/SI1709044T2/en unknown
- 2004-12-20 CA CA 2549675 patent/CA2549675C/en not_active Expired - Lifetime
- 2004-12-20 CN CNA200480038072XA patent/CN1898238A/en active Pending
- 2004-12-20 PT PT04804099T patent/PT1709044E/en unknown
- 2004-12-20 DK DK04804099T patent/DK1709044T4/en active
- 2004-12-20 KR KR20067014403A patent/KR101176450B1/en not_active Expired - Lifetime
- 2004-12-20 DE DE200460015158 patent/DE602004015158D1/en not_active Expired - Lifetime
- 2004-12-20 AU AU2004299278A patent/AU2004299278C9/en not_active Expired
- 2004-12-20 PL PL04804099T patent/PL1709044T5/en unknown
- 2004-12-20 ES ES04804099T patent/ES2310299T5/en not_active Expired - Lifetime
- 2004-12-20 RU RU2006125510A patent/RU2371443C2/en active
- 2004-12-20 JP JP2006544382A patent/JP2007516263A/en active Pending
- 2004-12-20 EP EP04804099.2A patent/EP1709044B2/en not_active Expired - Lifetime
- 2004-12-20 AT AT04804099T patent/ATE401326T1/en active
-
2006
- 2006-05-04 IL IL175409A patent/IL175409A/en active IP Right Grant
-
2008
- 2008-10-13 CY CY081101128T patent/CY1108409T1/en unknown
-
2011
- 2011-07-20 JP JP2011158523A patent/JP5722149B2/en not_active Expired - Lifetime
-
2012
- 2012-02-08 US US13/368,667 patent/US8501774B2/en not_active Expired - Fee Related
-
2013
- 2013-07-02 US US13/933,811 patent/US9133213B2/en not_active Expired - Lifetime
-
2015
- 2015-08-03 US US14/816,789 patent/US20150336992A1/en not_active Abandoned
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7820823B2 (en) | 2001-10-04 | 2010-10-26 | Morphochem Aktiengesellschaft Fur Kominatorische Chemi | Dual action antibiotics |
| US8329908B2 (en) | 2001-10-04 | 2012-12-11 | Morphochem Aktiengesellschaft Fur Kombinatorische Chemie | Dual action antibiotics |
| US8513231B2 (en) | 2003-04-30 | 2013-08-20 | Morphochem Aktiengesellschaft fü Kombinatorische Chemie | Use of oxazolidinone-quinoline hybrid antibiotics for the treatment of anthrax and other infections |
| US8268812B2 (en) | 2003-04-30 | 2012-09-18 | Morphochem Aktiengesellschaft fül Kombinatorische Chemie | Use of oxazolidinone-quinoline hybrid antibiotics for the treatment of anthrax and other infections |
| EP2374806A1 (en) * | 2003-09-03 | 2011-10-12 | Morphochem Aktiengesellschaft Für Kombinatorische Chemie | OXAZOLIDINON-QUINOLONe HYBRIDes and intermediates for their preparation |
| US8124772B2 (en) | 2003-09-03 | 2012-02-28 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Intermediate products for producing oxazolidinone-quinolone hybrids |
| EP1660489B1 (en) * | 2003-09-03 | 2014-07-02 | Morphochem Aktiengesellschaft Für Kombinatorische Chemie | Intermediate products for producing oxazolidinone-quinolone hybrids |
| EP2256120A1 (en) * | 2005-08-08 | 2010-12-01 | Actelion Pharmaceuticals Ltd. | Oxazolidinone-quinolone hybrids as antibacterial compounds |
| CN101238135B (en) * | 2005-08-08 | 2011-05-11 | 埃科特莱茵药品有限公司 | Oxazolidinone-quinolone hybrids as antibacterial compounds |
| WO2007017828A3 (en) * | 2005-08-08 | 2007-07-19 | Actelion Pharmaceuticals Ltd | Oxazolidinone-quinolone hybrids as antibacterial compounds |
| WO2007017828A2 (en) | 2005-08-08 | 2007-02-15 | Actelion Pharmaceuticals Ltd | Oxazolidinone-quinolone hybrids as antibacterial compounds |
| US8148362B2 (en) | 2006-03-31 | 2012-04-03 | Research Foundation Itsuu Laboratory | Compound having heterocyclic ring |
| WO2007114326A1 (en) | 2006-03-31 | 2007-10-11 | Research Foundation Itsuu Laboratory | Novel compound having heterocyclic ring |
| US8785625B2 (en) | 2006-03-31 | 2014-07-22 | Research Foundation Itsuu Laboratory | Compound having heterocyclic ring |
| EP2181994A1 (en) | 2006-03-31 | 2010-05-05 | Research Foundation Itsuu Laboratory | Antimicrobial compounds |
| US8124623B2 (en) | 2006-11-10 | 2012-02-28 | Actelion Pharmaceuticals Ltd. | 5-hydroxymethyl-oxazolidin-2-one-derivatives and their uses as antibacterials |
| JP2010132677A (en) * | 2006-11-10 | 2010-06-17 | Actelion Pharmaceuticals Ltd | 5-hydroxymethyl-oxazolidin-2-one derivative |
| RU2453546C2 (en) * | 2006-11-10 | 2012-06-20 | Актелион Фармасьютиклз Лтд | 5-hydroxymethyloxazolidin-2-one derivatives |
| WO2008062379A3 (en) * | 2006-11-24 | 2008-07-17 | Actelion Pharmaceuticals Ltd | 4- (2-oxo-oxazolidin-3yl)-phenoxymethyle derivativeas as antibacterials |
| KR101424839B1 (en) * | 2006-11-24 | 2014-08-01 | 액테리온 파마슈티칼 리미티드 | 4-(2-oxo-oxazolidin-3-yl)-phenoxymethyle derivativeas as antibacterials |
| JP2010510977A (en) * | 2006-11-24 | 2010-04-08 | アクテリオン ファーマシューティカルズ リミテッド | 4- (2-Oxo-oxazolidine-3-yl) -phenoxymethyl derivatives as antibacterial agents |
| US8039466B2 (en) | 2006-11-24 | 2011-10-18 | Actelion Pharmaceutical Ltd. | 5-hydroxymethyl-oxazolidin-2-one antibacterials |
| WO2009044777A1 (en) | 2007-10-02 | 2009-04-09 | Research Foundation Itsuu Laboratory | Oxazolidinone derivative having 7-membered hetero ring |
| US8530646B2 (en) | 2007-10-02 | 2013-09-10 | Research Foundation Itsuu Laboratory | Oxazolidinone derivative having 7-membered hetero ring |
| EP2669283A1 (en) | 2007-10-02 | 2013-12-04 | Shionogi&Co., Ltd. | Oxazolidinone derivative having 7-membered hetero ring |
| EP2233484A2 (en) | 2007-10-02 | 2010-09-29 | Research Foundation Itsuu Laboratory | Oxazolidinone derivatives having a 7-membered heterocyclic ring |
| US8354401B2 (en) | 2008-10-27 | 2013-01-15 | Mitsubishi Tanabe Pharma Corporation | Oxazolidinone amide aromatic compounds for supressing MMP-9 production |
| AU2014273510B2 (en) * | 2013-05-28 | 2019-10-10 | Morphochem Aktiengesellschaft Fur Kombinatorische Chemie | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| US10723746B2 (en) | 2013-05-28 | 2020-07-28 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| KR20160013193A (en) * | 2013-05-28 | 2016-02-03 | 모르포켐 악티엥게셀샤프트 퓌르 콤비나토리셰 케미 | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| US11261205B2 (en) | 2013-05-28 | 2022-03-01 | Morphochem Gmbh | Oxazolidinone-quinolone hybrid antibacterial for the parenteral treatment of prophylaxis of bacterial diseases |
| US9993469B2 (en) | 2013-05-28 | 2018-06-12 | Morphochem Aktiengesellschaft Für Kombinatorishe Chemie | Combination therapy comprising oxazolidinone-quinolones for use in treating bacterial infections |
| AU2014273471B2 (en) * | 2013-05-28 | 2019-10-03 | Morphochem Aktiengesellschaft Fur Kombinatorische Chemie | Combination therapy comprising oxazolidinone-quinolones for use in treating bacterial infections |
| WO2014191109A1 (en) * | 2013-05-28 | 2014-12-04 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Combination therapy comprising oxazolidinone-quinolones for use in treating bacterial infections |
| RU2702364C2 (en) * | 2013-05-28 | 2019-10-08 | Морфохем Акциенгезельшафт Фюр Комбинаторише Хеми | Hybrid antibacterial agents oxazolidinone-quinolone intended for parenteral administration for treatment or prevention of bacterial diseases |
| EP3517106A1 (en) | 2013-05-28 | 2019-07-31 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| WO2014191075A1 (en) | 2013-05-28 | 2014-12-04 | Morphochem Aktiengesellschaft für kombinatorische Chemie | Oxazolidinone-quinolone hybrid antibacterials for the parenteral treatment or prophylaxis of bacterial diseases |
| CN108059624A (en) * | 2016-11-08 | 2018-05-22 | 上海医药工业研究院 | It is used to prepare the preparation method of click azoles amine key intermediate |
| US10087171B2 (en) | 2016-12-19 | 2018-10-02 | Actelion Pharmaceuticals Ltd | Crystalline forms of cadazolid |
| CN114478486A (en) * | 2022-01-28 | 2022-05-13 | 西南大学 | Trimolecular conjugate with p-aminosalicylic acid as parent nucleus, intermediate, preparation method and use |
| CN114478486B (en) * | 2022-01-28 | 2023-07-25 | 西南大学 | Three-molecule conjugate taking para-aminosalicylic acid as parent nucleus, intermediate, preparation method and application |
| WO2024024962A1 (en) | 2022-07-29 | 2024-02-01 | 住友ファーマ株式会社 | Nitrogen-containing saturated heterocyclic ring derivative |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1709044B2 (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| EP1620098B1 (en) | Use of oxazolidinone-quinoline hybrid antibiotics for the treatment of anthrax and other infections | |
| CN101381365B (en) | dual action antibiotic | |
| US8158797B2 (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| CN100544719C (en) | Use of oxazolidinone-quinoline hybrid antibiotics in the preparation of medicines for treating anthrax and other infections | |
| HK1090647C (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| HK1090647B (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| AU2011202771A1 (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| ZA200604180B (en) | Oxazolidinone-quinolone hybrid antibiotics | |
| HK1078806B (en) | Use of oxazolidinone-quinoline hybrid antibiotics for the treatment of anthrax and other infections | |
| HK1128469B (en) | Dual action antibiotics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480038072.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 175409 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004804099 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 547382 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/04180 Country of ref document: ZA Ref document number: 200604180 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 693/MUMNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2549675 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/006769 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004299278 Country of ref document: AU Ref document number: 2006544382 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006125510 Country of ref document: RU Ref document number: 1020067014403 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2004299278 Country of ref document: AU Date of ref document: 20041220 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004299278 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004804099 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0417193 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10583419 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10583419 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2004804099 Country of ref document: EP |