WO2005063764A2 - Fused pyrrolocarbazoles and methods for the preparation thereof - Google Patents
Fused pyrrolocarbazoles and methods for the preparation thereof Download PDFInfo
- Publication number
- WO2005063764A2 WO2005063764A2 PCT/US2004/043164 US2004043164W WO2005063764A2 WO 2005063764 A2 WO2005063764 A2 WO 2005063764A2 US 2004043164 W US2004043164 W US 2004043164W WO 2005063764 A2 WO2005063764 A2 WO 2005063764A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- compound
- groups
- alkyl
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- IFKADBQSTWFEOQ-UHFFFAOYSA-N CC(CC1)OC1S Chemical compound CC(CC1)OC1S IFKADBQSTWFEOQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains three hetero rings
- C07D513/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09B—ORGANIC DYES OR CLOSELY-RELATED COMPOUNDS FOR PRODUCING DYES, e.g. PIGMENTS; MORDANTS; LAKES
- C09B57/00—Other synthetic dyes of known constitution
- C09B57/08—Naphthalimide dyes; Phthalimide dyes
Definitions
- the present invention relates generally to fused pyrrolocarbazoles, including pharmaceutical compositions, diagnostic kits, assay standards or reagents containing the same, and methods of using the same as therapeutics.
- the invention is also directed to intermediates and processes for making these novel compounds. BACKGROUND OF THE INVENTION
- Patent Nos. 5,475,110; 5,591,855; 5,594,009; 5,616,724; and 6,630,500 discloses fused pyrrolocarbazole compounds which possess a variety of functional pharmacological activities.
- the fused pyrrolocarbazoles were disclosed to be used in a variety of ways, including: enhancing the function and/or survival of cells of neuronal lineage, either singularly or in combination with neurotrophic factor(s) and/or indolocarbazoles; enhancing trophic factor-induced activity; inhibition of protein kinase C ("PKC") activity; inhibition of trk tyrosine kinase activity; inhibition of proliferation of a prostate cancer cell-line; inhibition of the cellular pathways involved in the inflammation process; and enhancement of the survival of neuronal cells at risk of dying.
- PLC protein kinase C
- trk tyrosine kinase activity inhibition of proliferation of a prostate cancer cell-line
- inhibition of the cellular pathways involved in the inflammation process and enhancement of the survival of neuronal cells at risk of dying.
- novel pyrrolocarbazole derivatives that possess beneficial properties. This invention is directed to this, as well as other important ends.
- the fused pyrrolocarbazoles of the present invention may be used in a variety of ways, including: for inhibition of angiogenesis; as antitumor agents; for enhancing the function and/or survival of cells of neuronal lineage, either singularly or in combination with neurotrophic factor(s) and/or indolocarbazoles; for enhancing trophic factor-induced activity; inhibition of kinase activity, such as trk tyrosine kinase ("trk”), vascular endothelial growth factor receptor (“VEGFR”) kinase, preferably VEGFRl and VEGFR2, mixed lineage kinase (“MLK”), dual leucine zipper bearing kinase (“DLK”), platelet derived growth factor receptor kinase (“PDGFR”), protein kinase C (“PKC”),
- kinase activity such as trk tyrosine kinase (“trk”), vascular endotheli
- the fused pyrrolocarbazoles may useful for inhibition of c-met, c-kit, and mutated FIt-3 containing internal tandem duplications in the juxtamembrane domain. Because of these varied activities, the disclosed compounds find utility in a variety of settings, including research and therapeutic environments. In other embodiments, the compounds of the present invention are useful for treating or preventing angiogenesis and angiogenic disorders such as cancer of solid tumors, endometriosis, retinopathy, diabetic retinopathy, psoriasis, hemangioblastoma, ocular disorders or macular degeneration.
- angiogenesis and angiogenic disorders such as cancer of solid tumors, endometriosis, retinopathy, diabetic retinopathy, psoriasis, hemangioblastoma, ocular disorders or macular degeneration.
- the compounds of the present invention are useful for treating or preventing neoplasia, rheumatoid arthritis, chronic arthritis, pulmonary fibrosis, myelofibrosis, abnormal wound healing, atherosclerosis, or restenosis.
- the compounds of the present invention are useful for treating or preventing neurodegenerative diseases and disorders, such as Alzheimer's disease, a yotrophic lateral sclerosis, Parkinson's disease, stroke, ischemia, Huntington's disease, AIDS dementia, epilepsy, multiple sclerosis, peripheral neuropathy, chemotherapy induced peripheral neuropathy, AIDS related peripheral neuropathy, or injuries of the brain or spinal chord.
- the compounds of the present invention are useful for treating or preventing prostate disorders such as prostate cancer or benign prostate hyperplasia.
- the compounds of the present invention are useful for treating or preventing multiple myeloma and leukemias including, but not limited to, acute myelogenous leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, and chronic lymphocytic leukemia.
- the present invention is directed to pharmaceutical compositions which comprises one or more pharmaceutically acceptable excipients and a therapeutically effective amount of a compound of the present invention.
- the present invention provides a novel compound of Formula I:
- ring A together with the carbon atoms to which it is attached is selected from: (a) a phenylene ring in which from 1 to 3 carbon atoms may be replaced by nitrogen atoms; and (b) a 5-membered aromatic ring in which from 1 to 2 carbon atoms may be replaced by nitrogen atoms;
- R 1 is H or optionally substituted alkyl, wherein said optional substituents are one to three R 10 groups;
- R 19 is selected from optionally substituted cycloalkyl, optionally substituted heterocycloalkyl, and optionally substituted heteroaryl, wherein said optional substituents are one to three R 10 groups;
- R 20 is selected from optionally substituted aryl, optionally substituted heteroaryl, optionally substituted cycloalkyl, and optionally substituted heterocycloalkyl, wherein said optional substituents are one to three R 10 groups;
- R 21 is selected from H, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, optionally substituted aryl, optionally substituted arylalkyl, optionally substituted heteroaryl, optionally substituted cycloalkyl, and optionally substituted heterocycloalkyl, wherein said optional substituents are one to three R 10 groups;
- R" is selected from optionally substituted aryl, and optionally substituted heteroaryl, wherein said optional substituents are one to three R groups;
- the compounds of Formula I as defined herein are not intended to include any compounds disclosed in PCT Publ. No. WO 98/07433.
- a 1 , A 2 O
- rings A and B are each phenylene
- Q is CH-R a
- one of R 2 or R is H and the other is optionally substituted wherein W is optionally substituted alkyl, or NR 27A R 27B ; then any of R 3 , R 4 , R 5 , and R 6 cannot include OR 14 or O-(alkylene)-R 24 .
- Other aspects of the present invention include the compounds of Formula I as defined herein wherein ring A is phenylene; or a 5-membered aromatic ring containing one nitrogen atom, preferably pyrazolylene, and more preferably
- R 1 is H or optionally substituted alkyl.
- Additional aspects include those compounds wherein groups A 1 A 2 are H, H; and B !
- the invention includes compounds wherein Q is selected from an optionally substituted C 1- alkylene, or preferably Q is CH 2 , or CH 2 CH 2 .
- Q is selected from an optionally substituted C 1- alkylene, or preferably Q is CH 2 , or CH 2 CH 2 .
- R 14 is benzoxazole, benzothiazole, pyrimidine, pyrazine or triazine;
- R is a 5-membered heteroaryl group;
- R 20 is heterocycloalkyl or heteroaryl;
- R 23 is heteroaryl or heterocycloalkyl;
- R 24 is heteroaryl; and
- R 26 is heterocycloalkyl.
- Additional aspects of the present invention include any combination of the above preferred substituents, such as, for example, a compound of Formula I with the preferred moieties of groups R 1 and R 2 ; or R 1 and Q; or R 1 , R 2 ; or Q; etc.
- a compound of Formula I with the preferred moieties of groups R 1 and R 2 ; or R 1 and Q; or R 1 , R 2 ; or Q; etc.
- R 1 is H or optionally substituted alkyl.
- Additional aspects include compounds wherein Q is selected from an optionally substituted C 1-2 alkylene, or preferably Q is CH 2 or CH 2 CH 2 . Additional aspects of the present invention include any combination of the above preferred substituents, such as, for example, a compound of Formula II with the preferred moieties of groups R 1 and R 2 ; or R 1 and Q; or R 1 , R 2 ; or Q; etc. In yet another embodiment of the present invention, there are included compounds having a structure of Formula III:
- ring A is phenylene or pyrazolylene, preferably
- R 3 , R 4 , R 5 , and R 6 for compounds of Formulas I- VI and their respective preferred embodiments described heretofore.
- OR 14 paticularly those wherein R 14 is optionally substituted benzoxazole, optionally substituted benzothiazole, optionally substituted pyrimidine, optionally substituted pyrazine or optionally substituted triazine.
- C CH NR 26 ; paticularly those wherein R 26 is optionally substituted heterocycloalkyl 44..
- NR ⁇ C( O)OR 15 00 0( ⁇ 6.
- 7. OC( O)NR 1 l R M ; paticularly those wherein R 20 is optionally substituted cycloalkyl or optionally substituted heterocycloalkyl.
- O-(alkylene)-R 24 paticularly those wherein R 24 is optionally substituted heterocycloalkyl 9.
- the following terms and expressions used herein have the indicated meanings.
- Preferred embodiments include each individual integer in the range, as well as any subcombination of integers.
- preferred integers for "1-6” can include 1, 2, 3, 4, 5, 6, 1-2, 1-3, 1-4, 1-5, 2-3, 2-4, 2-5, 2-6, etc.
- stable compound or “stable structure” refers to a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and preferably capable of formulation into an efficacious therapeutic agent. The present invention is directed only to stable compounds.
- alkyl refers to a straight-chain, or branched alkyl group having 1 to 8 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec- butyl, tert-butyl, pentyl, isoamyl, neopentyl, 1-ethylpropyl, 3-methylpentyl, 2,2- dimethylbutyl, 2,3-dimethylbutyl, hexyl, octyl, etc.
- alkyl moiety of alkyl-containing groups such as alkoxy, alkoxycarbonyl, and alkylaminocarbonyl groups, has the same meaning as alkyl defined above.
- Lower alkyl groups which are preferred, are alkyl groups as defined above which contain 1 to 4 carbons.
- a designation such as “C1-C4 alkyl” refers to an alkyl radical containing from 1 to 4 carbon atoms.
- alkenyl refers to a straight chain, or branched hydrocarbon chains of 2 to 8 carbon atoms having at least one carbon-carbon double bond.
- a designation "C -C 8 alkenyl” refers to an alkenyl radical containing from 2 to 8 carbon atoms.
- alkenyl groups include ethenyl, propenyl, isopropenyl, 2,4- pentadienyl, etc.
- alkynyl refers to a straight chain, or branched hydrocarbon chains of 2 to 8 carbon atoms having at least one carbon-carbon triple bond.
- a designation "C 2 -C 8 alkynyl” refers to an alkynyl radical containing from 2 to 8 carbon atoms. Examples include ethynyl, propynyl, isopropynyl, 3,5-hexadiynyl, etc.
- alkylene refers to a branched or straight chained hydrocarbon of 1 to 8 carbon atoms, which is formed by the removal of two hydrogen atoms.
- phenylene refers to a phenyl group with an additional hydrogen atom removed, ie.
- cycloalkyl refers to a saturated or partially saturated mono- or bicyclic alkyl ring system containing 3 to 10 carbon atoms.
- a designation such as “C 5 -C 7 cycloalkyl” refers to a cycloalkyl radical containing from 5 to 7 ring carbon atoms.
- Preferred cycloalkyl groups include those containing 5 or 6 ring carbon atoms. Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, cycloheptyl, cyclooctyl, etc.
- spirocycloalkyl refers to a cycloalkyl group bonded to a carbon chain or carbon ring moiety by a carbon atom common to the cycloalkyl group and the carbon chain or carbon ring moiety.
- a C 3 alkyl group substituted with an R group wherein the R group is spirocycloalkyl containing 5 carbon atoms refers to:
- aryl refers to a mono- or bicyclic hydrocarbon aromatic ring system having 6 to 12 ring carbon atoms. Examples include phenyl and naphthyl. Preferred aryl groups include phenyl or naphthyl groups. Included within the definition of "aryl” are fused ring systems, including, for example, ring systems in which an aromatic ring is fused to a cycloalkyl ring. Examples of such fused ring systems include, for example, indane and indene.
- heterocycle refers to a mono- di-, tri- or other multicyclic aliphatic ring system that includes at least one heteroatom such as O, N, or S.
- the nitrogen and sulfur heteroatoms may be optionally oxidized, and the nitrogen may be optionally substituted in non-aromatic rings.
- Heterocycles are intended to include heteroaryl and heterocycloalkyl groups.
- heterocyclic groups containing one or more nitrogen atoms include pyrrolidine, pyrroline, pyrazoline, piperidine, morpholine, thiomorpholine, N- methylpiperazine, indole, isoindole, imidazole, imidazoline, oxazoline, oxazole, triazole, thiazoline, thiazole, isothiazole, thiadiazole, triazine, isoxazole, oxindole, pyrazole, pyrazolone, pyrimidine, pyrazine, quinoline, iosquinoline, and tetrazole groups.
- heterocyclic groups formed containing one or more oxygen atoms include furan, tetrahydrofuran, pyran, benzofurans, isobenzofurans, and tetrahydropyran groups.
- Some heterocyclic groups containing one or more sulfur atoms include thiophene, thianaphthene, tetrahydrothiophene, tetrahydrothiapyran, and benzothiophenes.
- heterocycloalkyl refers to a cycloalkyl group in which one or more ring carbon atoms are replaced by at least one hetero atom such as -O-, -N-, or -S-, and includes ring systems which contain a saturated ring group bridged or fused to one or more aromatic groups.
- heterocycloalkyl groups containing both saturated and aromatic rings include phthalamide, phthalic anhydride, indoline, isoindoline, tetrahydroisoquinoline, chroman, isochroman, and chromene.
- heteroaryl refers to an aryl group containing 5 to 10 ring carbon atoms in which one or more ring carbon atoms are replaced by at least one hetero atom such as -O-, -N-, or -S-.
- heteroaryl groups of the present invention include pyridyl, pyrimidyl, purinyl, pyrrolyl, pyridazinyl, pyrazinyl, triazinyl, imidazolyl, triazolyl, tetrazolyl, indolyl, isoindolyl, quinolyl, isoquinolyl, qunioxalinyl, quinazolinyl, cinnolinyl, phthalazinyl, benzoimidazolyl, pyrazolyl, thiazolyl, thiadiazolyl, isothiazolyl, oxazolyl, isoxazolyl, naphthyridinyl, oxindolyl, and benzothiazolyl groups.
- arylalkyl refers to an alkyl group that is substituted with an aryl group.
- arylalkyl groups include, but are not limited to, benzyl, phenethyl, benzhydryl, diphenylmethyl, triphenylmethyl, diphenylethyl, naphthylmethyl, etc.
- arylalkoxy refers to an aryl-substituted alkoxy group, such as benzyloxy, diphenylmethoxy, triphenylmethoxy, phenylethoxy, diphenylethoxy, etc.
- the term “monosaccharide” refers to a simple sugar of the formula
- Examples of monosaccharides include erythrose, threose, ribose, arabinose, xylose, lyxose, allose, altrose, glucose, mannose, gulose, idose, galactose, talose, erythulose, ribulose, xyulose, psicose, fructose, sorbose, tagatose, erythropentulose, threopentulose, glycerotetrulose, glucopyranose, fructofuranose, etc.
- amino acid refers to a group containing both an amino group and a carboxyl group.
- Embodiments of amino acids include ⁇ -amino, ⁇ -amino, ⁇ - amino acids.
- the ⁇ -amino acids have a general formula HOOC-CH(side chain)-NH 2 .
- the amino acids can be in their D, L or racemic configurations.
- Amino acids include naturally-occurring and non-naturally occurring moieties.
- the naturally-occurring amino acids include the standard 20 ⁇ -amino acids found in proteins, such as glycine, serine, tyrosine, proline, histidine, glutamine, etc.
- Naturally-occurring amino acids can also include non- ⁇ -amino acids (such as ⁇ -alanine, ⁇ -aminobutyric acid, homocysteine, etc.), rare amino acids (such as 4-hydroxyproline, 5-hydroxylysine, 3-methylhistidine, etc.) and non-protein amino acids (such as citrulline, ornithine, canavanine, etc.).
- Non-naturally occurring amino acids are well-known in the art, and include analogs of natural amino acids. See Lehninger, A. L. Biochemistry, 2 nd ed.; Worth Publishers: New York, 1975; 71- 77, the disclosure of which is incorporated herein by reference.
- Non-naturally occurring amino acids also include ⁇ -amino acids wherein the side chains are replaced with synthetic derivatives.
- Representative side chains of naturally occurring and non-naturally occurring ⁇ -amino acids include are shown below in Table A. Table A
- trk refers to the family of high affinity neurotrophin receptors presently comprising trk A, trk B, and trk C, and other membrane associated proteins to which a neurotrophin can bind.
- VEGFR refers to the family of high affinity vascular endothelial growth factor receptors presently comprising VEGFRl, VEGFR2, VEGFR3, and other membrane associated proteins to which a VEGF can bind.
- the term "MLK” refers to the family of high affinity mixed lineage kinases presently comprising MLKl, MLK2, MLK3, MLK4 ⁇ & ⁇ , DLK, LZK, ZAK ⁇ & ⁇ , and other serine/threonine kinases classified within this family.
- the terms “enhance” or “enhancing” when used to modify the terms “function” or “survival” means that the presence of a compound of the present invention has a positive effect on the function and/or survival of a trophic factor responsive cell compared with a cell in the absence of the compound.
- a compound of the present invention would evidence enhancement of survival of a cholinergic neuronal population at risk of dying (due to, e.g., injury, a disease condition, a degenerative condition or natural progression) when compared to a cholinergic neuronal population not presented with such a compound, if the treated population has a comparatively greater period of functionality than the non-treated population.
- a compound of the present invention would evidence enhancement of the function (e.g.
- the terms “inhibit” or “inhibition” refer to a specified response of a designated material (e.g., enzymatic activity) is comparatively decreased in the presence of a compound of the present invention.
- the terms “cancer” or “cancerous” refer to any malignant proliferation of cells in a mammal.
- neuronal linesage and neuronal cell refer to a heterogeneous population of neuronal types having singular or multiple transmitters and/or singular or multiple functions; preferably, these are cholinergic and sensory neurons.
- cholinergic neuron means neurons of the Central Nervous System (CNS) and Peripheral Nervous System (PNS) whose neurotransmitter is acetylcholine; exemplary are basal forebrain and spinal cord neurons.
- sensor neuron includes neurons responsive to environmental cues (e.g., temperature, movement) from, e.g., skin, muscle and joints; exemplary is a neuron from the DRG.
- trophic factor refers to a molecule that directly or indirectly affects the survival or function of a trophic factor responsive cell.
- trophic factors include Ciliary Neurotrophic Factor (CNTF), basic Fibroblast Growth Factor (bFGF), insulin and insulin-like growth factors (e.g., IGF-I, IGF-II, IGF-III), interferons, interleukins, cytokines, and the neurotrophins, including Nerve Growth Factor (NGF), Neurotro ⁇ hin-3 (NT-3), Neurotrophin-4/5 (NT-4/5) and Brain Derived Neurotrophic Factor (BDNF).
- CNTF Ciliary Neurotrophic Factor
- bFGF basic Fibroblast Growth Factor
- IGF-I, IGF-II, IGF-III insulin-like growth factors
- interferons interleukins
- cytokines interleukins
- cytokines interferons
- the neurotrophins including Nerve Growth Factor (NGF), Neurotro ⁇ hin-3 (NT-3), Neurotrophin-4/5 (NT-4/5) and Brain Derived Neurotrophic Factor (BDNF
- trophic actor-responsive cell refers to a cell which includes a receptor to which a trophic factor can specifically bind; examples include neurons (e.g., cholinergic and sensory neurons) and non-neuronal cells (e.g., monocytes and neoplastic cells).
- neurotrophic factor activity and “trophic factor-induced activity”, refer to both endogenous and exogenous trophic factors, where “endogenous” refers to a trophic factor normally present and “exogenous” refers to a trophic factor added to a system.
- trophic factor induced activity includes activity induced by (1) endogenous trophic factors; (2) exogenous trophic factors; and (3) a combination of endogenous and exogenous trophic factors.
- a biological material e.g., a cell such as a neuron
- at risk of dying in conjunction with a biological material, e.g., a cell such as a neuron, refers to a state or condition which negatively impacts the biological material such that the material has an increased likelihood of dying due to such state or condition.
- compounds disclosed herein can "rescue” or enhance the survival of motoneurons which are naturally at risk of dying in an in ovo model of programmed cell death.
- a neuron may be at risk of dying due to the natural aging process which occasions the death of a neuron, or due to an injury, such as a trauma to the head, which may be such that neurons and/or glia, for example, impacted by such trauma may be at risk of dying.
- a neuron may be at risk of dying due to a disease state or condition, as in the case of neurons at risk of dying as occasioned by the disease ALS.
- enhancing the survival of a cell at risk of dying by use of a compound of the claimed invention is meant that such compound decreases or prevents the risk of the death of the cell.
- the term "contacting” refers to directly or indirectly causing placement together of moieties, such that the moieties directly or indirectly come into physical association with each other, whereby a desired outcome is achieved.
- a target cell with a compound as disclosed herein even though the compound and cell do not necessarily physically join together (as, for example, is the case where a ligand and a receptor physically join together), as long as the desired outcome is achieved (e.g., enhancement of the survival of the cell).
- Contacting thus includes acts such as placing moieties together in a container (e.g., adding a compound as disclosed herein to a container comprising cells for in vitro studies) as well as administration of the compound to a target entity (e.g., injecting a compound as disclosed herein into a laboratory animal for in vivo testing, or into a human for therapy or treatment purposes).
- a "therapeutically effective amount” refers to an amount of a compound of the present invention effective to prevent or treat the symptoms of a particular disorder.
- the term “subject” refers to a warm blooded animal such as a mammal, preferably a human, or a human child, which is afflicted with, or has the potential to be afflicted with one or more diseases and conditions described herein.
- pharmaceutically acceptable refers to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem complications commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, and the like.
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like
- organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic,
- unit dose refers to a single dose which is capable of being administered to a patient, and which can be readily handled and packaged, remaining as a physically and chemically stable unit dose comprising either the active compound itself, or as a pharmaceutically acceptable composition, as described hereinafter.
- prodrug is intended to include any covalently bonded carriers which release the active parent compound as defined in the present invention in vivo when such prodrug is administered to a mammalian subject. Since prodrugs are known to enhance numerous desirable qualities of pharmaceuticals (e.g., solubility, bioavailability, manufacturing, etc.) the compounds of the present invention may be delivered in prodrug form.
- prodrugs of the claimed compounds may be prepared by modifying functional groups present in the compound in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound.
- prodrugs include, for example, compounds of the present invention wherein a hydroxy, amino, or carboxy group is bonded to any group that, when the prodrug is administered to a mammalian subject, cleaves to form a free hydroxyl, free amino, or carboxylic acid, respectively.
- Examples include, but are not limited to, acetate, formate and benzoate derivatives of alcohol and amine functional groups; and alkyl, carbocyclic, aryl, and alkylaryl esters such as methyl, ethyl, propyl, iso-propyl, butyl, isobutyl, sec-butyl, tert-butyl, cyclopropyl, phenyl, benzyl, and phenethyl esters, and the like. It is recognized that compounds of the present invention may exist in various stereoisomeric forms. As such, the compounds of the present invention include their respective diastereomers or enantiomers.
- the compounds are normally prepared as racemates and can conveniently be used as such, but individual diastereomers or enantiomers can be isolated or synthesized by conventional techniques if so desired. Such racemates and individual diastereomers or enantiomers and mixtures thereof form part of the present invention. It is well known in the art how to prepare and isolate such optically active forms.
- Specific stereoisomers can be prepared by stereospecific synthesis using enantiomerically pure or enantiomerically enriched starting materials.
- the specific stereoisomers of either starting materials or products can be resolved and recovered by techniques known in the art, such as resolution of racemic forms, normal, reverse-phase, and chiral chromatography, recrystallization, enzymatic resolution, or fractional recrystallization of addition salts formed by reagents used for that purpose.
- functional groups present on the compounds of the present invention may contain protecting groups.
- the amino acid side chain substituents of the compounds of the present invention can be substituted with protecting groups such as benzyloxycarbonyl or t-butoxycarbonyl groups.
- protecting groups are known per se as chemical functional groups that can be selectively appended to and removed from functionalities, such as hydroxyl groups and carboxyl groups. These groups are present in a chemical compound to render such functionality inert to chemical reaction conditions to which the compound is exposed. Any of a variety of protecting groups may be employed with the present invention.
- Preferred groups for protecting lactams include silyl groups such as t-butyldimethylsilyl ("TBDMS”), dimethoxybenzhydryl (“DMB”), acyl, benzyl, and methoxybenzyl groups.
- Preferred groups for protecting hydroxy groups include TBS, acyl, benzyl ("Bn”), benzyloxycarbonyl (“CBZ”), t-butyloxycarbonyl (“Boc”), and methoxymethyl.
- Scheme 2 outlines the route to prepare carbamate-type derivatives, such as Examples 1-2, and 70-72.
- An alternate method to preparing N, N-di-substituted carbamates utilized a nitrophenyl carbonate intermediate which may be treated with various primary or secondary amines.
- urea, O-carbamate, and N-carbamate derivatives may be prepared from reaction of the appropriate amine or phenol intermediate with an isocyanate or chloroformate or from the appropriate nitrophenyl carbonate, nitrophenyl carbamate, or trichloromethylcarbonyl (see J. Org. Chem. 2003, 68, 3733- 3735).
- Scheme 3 outlines a route to prepare heteroaryl ethers from the corresponding phenol using a base such as sodium hydride and a heteroaryl bromide or chloride.
- Scheme 4 shows a route for the preparation of N-carbamates (examples 50-69) or amides (examples 74-82) from the corresponding aniline intermediates 1-29.
- Amino intermediates 1-29 were prepared by alkylation of the appropriate cyano-esters with the appropriate alkyl iodide or bromide followed by nitration, and subsequent RaNi reduction to provide the amino-lactam.
- the desired compounds were readily prepared from the amine.
- Heteroaryl ketones may be prepared using standard Friedel-Crafts type acylation reactions.
- Tables 1-5 show activity in the targets described herein at concentrations ranging from 0.1 nM to 10 ⁇ M. These examples are given for illustration of the invention and are not intended to be limiting thereof. Table 1
- Example 3 A suspension of sodium hydride (2.44 mg, 1.22 eq.) in 0.5 mL of THF was stirred under N 2 as phenol intermediate 1-14 (20.6 mg, 0.05 mmol) in 2.0 mL of THF-.DMF (1:1) was added dropwise. After 10 minutes of stirring, 2-bromopyrimidine (8.9 mg, 1.12 equivalents) in 0.5 mL of THF was added. The mixture was stirred at 60 °C for 14 hours. The mixture was cooled to room temperature, diluted with CH 2 Cl 2 /MeOH, filtered through celite and concentrated. Purification was achieved by preparative TLC with CH 2 Cl 2 /MeOH (9:1) to afford the product (4.0 mg, 17%) (MS: 477 m e (M+H) + ).
- Example 4 The compound was prepared according to the procedure of Example 3 using phenol intermediate 1-14 and 2-chlorobenzoxazole; 40 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 28%; MS: 516 m/e (M+l) +
- Example 5 The compound was prepared according to the procedure of Example 3 using intermediate 1-14 and 2-chlorobenzothiazole; 40 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 13%; MS: 531 m/e (M+l) +
- Example 6 To a mixture of Example 3 (25.0 mg, 0.052 mmol) and cesium carbonate (81 mg, 5.0 eq) in 2.0 mL of CH 3 CN was added n-propyl bromide (47 ul, 10.0 eq.) under N 2 . After stirring at 90 °C for 14 hours, the mixture was diluted with CH 2 CI 2 , filtered through celite and concentrated. Purification by preparative TLC with 95% of CH 2 Cl 2 /MeOH afforded the product (15.0 mg, 56%); MS: m e 519 (M+l) + .
- Example 7 The compound was prepared using to the procedure of Example 3 using intermediate 1-14 and 2-bromopyrazine; preparative TLC (10% MeOH in CH 2 CI 2 ); MS 499 m e (M+l) + .
- Example 8 The compound was prepared according to the procedure of Example 6 using Example 7 as starting material. MS m e 519 (M + 1).
- Example 11 The compound was prepared according to the procedure of Example 10 using phenol intermediate 1-18 and 2-chlorobenzoxazole; 36 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 11%; MS: 502 m/e (M+l) +
- Example 12 The compound was prepared according to the procedure of Example 10 using intermediate 1-19 and 2-bromopyrimidine; 36 hr; preparative TLC (10% MeOH in CH 2 CI 2 ); yield 25%; MS: 419 m/e (M+l) + .
- Example 13 The compound was prepared according to the procedure for Example 3 using phenol intermediate 1-22 and 2-bromopyrimidine; 30 hr; preparative TLC (10% MeOH in CH 2 CI 2 ); yield 53%; MS: 423 m/e (M+l) +
- Example 14 The compound was prepared according to the procedure for Example 6 using Example 13 and iodoethane; 14 hr; preparative TLC (10% MeOH in CH 2 C1 ); yield 19%; MS: 451 m/e (M+l) +
- Example 15 The compound was prepared according to the procedure for Example 6 using Example 13 and iodomethane; 14 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 28%; MS: 459 m/e (M+23) + Example 16. The compound was prepared according to the procedure for Example 6 using Example 13 and cyclopentyl bromide; 14 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 38%; MS: 513 m/e (M+23) +
- Example 17 A mixture of phenol intermediate 1-22 (17.2 mg, 0.05 mmol), potassium t- butoxide (33.7 mg, 6 eq.) and t-butylammonium bromide (0.97 mg, 0.06eq) was mixed and stirred for 5 minutes, then 1.0 mL of chloropyrazine was added, then stirred at room temperature for 5 minutes and at 90 °C for 1 hour. The mixture was cooled to room temperature, excess of the chloropyrazine was evaporated off and the resulting residue was diluted with CH 2 Cl 2 MeOH. Purification by preparative TLC with (9:1) CH 2 Cl 2 /MeOH afforded the mono product (11.0 mg, yield 52%) MS: 423 m/e (M+l) + .
- Example 18 The compound was prepared according to the procedure for Example 6 using Example 13 and butyl bromide; 14 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 38%; MS: 479 m/e (M+l) + .
- Example 19 The compound was prepared according to the procedure for Example 10 using Example 13 and 2-propyl bromide; 60 hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 10%; MS: 465 m/e (M+l) +
- Example 20 The compound was prepared according to the procedure for Example 6 using Example 13 and 2-cyclopropylmethyl bromide; 14hr; preparative TLC (10% MeOH in CH 2 C1 2 ); yield 5%; MS: 477 m/e (M+l) + .
- Example 21 The compound was prepared according to the procedure for Example 6 using Example 13 and 2-cyclopropylmethyl bromide; 14 hr; preparative TLC (10% MeOH in CH2CI2); MS: 507 m/e (M+l) +
- Example 22 The compound as prepared according to the procedure for Example 6 using Example 13 and isobutyl bromide; preparative TLC (10% MeOH in CH 2 C1 2 ); MS: 493 m e (M+l) +
- Example 23 The compound was prepared according to the procedure for Example 6 using Example 17 and ethyl iodide; preparative TLC (10% MeOH in CH 2 C1 2 ); MS: 451 m e (M+l) +
- Example 24 The compound was prepared according to the procedure for Example 6 using Example 13 and l-bromo-3,5-dimethoxytriazine; preparative TLC (10% MeOH in CH 2 C1 2 ); MS: 540 m/e (M+l) + .
- Example 25 To 25mg (0.07 mmol) of the N-ethyl intermediate 1-23-1 in methylene chloride/nitromethane (3 mL/2 mL) was slowly added 2-furoyl chloride (69 ⁇ l, 0.7 mmol, 10 eq) followed by aluminum chloride (93 mg, 0.7 mmol, 10 eq). The reaction was stirred at room temperature overnight. The reaction mixture was concentrated, water and a few drops of IN HCl were added to the residue and the mixture was extracted with methylene chloride. The combined organic extracts were dried with sodium sulfate, the drying agents removed by filtration, and the solvent was removed by evaporation.
- Example 26 The compound was prepared by the method described for Example 25. MS m e 438 (M+l).
- Example 27 To the N-ethyl intermediate 1-23-1 (25mg, 0.07 mmol) in nitromethane (5 mL) was added 2-thiophene carbonyl chloride (75 ⁇ l 0.7 mmol, 10 eq) followed by addition of aluminum chloride (94 mg, 0.7 mmol, 10 eq) in small portions. The reaction mixture was stirred at room temperature overnight. The reaction was then concentrated, stirred with water and a few drops of 1 N HCl were added. The product was collected by filtration, dissolved in methylene chloride/methanol and purified by preparative TLC eluting with 10% methanol/methylene chloride. The desired band was collected, stirred with methylene chloride/methanol, filtered, and concentrated. The sample was dried at 80 °C under vacuum overnight. MS m/e 467 (M+l).
- Examples 28-49 were prepared using the general method described for Example 27 using the appropriate N-alkyl intermediate 1-23, and heteroaryl acid chloride with A1C1 3 or FeCl 3 as catalyst.
- Example 28 MS m/e 423 (M+l Example 29. MS m/e 465 (M+l Example 30. MS m/e 479 (M+l Example 31. MS m/e 495 (M+l Example 32. MS m/e 463 (M+l Example 33. MS m/e 509 (M+l Example 34. MS m e 481 (M+l Example 35. MS m e 530 (M+l Example 36. MS m/e 495 (M+l Example 37. MS m/e 479 (M+l Example 38. MS m/e 574 (M+l Example 39. MS m e 481 (M+l Example 40. MS m/e 481 (M+l Example 41.
- MS m/e 608 (M+l Example 42.
- MS m/e 588 (M+l Example 43.
- MS m/e 536 (M+l Example 44.
- MS m/e 520 (M+l Example 45.
- MS m/e 509 (M+l Example 46.
- MS m/e 592 (M+l Example 47.
- MS m/e 550 (M+l Example 48.
- MS m/e 570 (M+l)
- Example 50 To a stirred solution of 3-amino intermediate 1-29-2 (25 mg, 0.0649 mmol) in CH 2 C1 2 (5 mL) was added isopropyl chloroformate (1.0 M in toluene, 125 ⁇ L, 0.125 mmol) and pyridine (20 ⁇ L, 0.247 mmol). After stirring 3 h at room temperature, the resulting precipitate was filtered and dried to give 28 mg (91%) of the desired product.
- isopropyl chloroformate 1.0 M in toluene, 125 ⁇ L, 0.125 mmol
- pyridine 20 ⁇ L, 0.247 mmol
- Example 54 MS m/e 476 (M + H).
- Example 55 MS m/e 492 (M + H).
- Example 59 MS m/e 486 (M + H).
- Example 60 MS m e 472 (M + H).
- Example 64 MS m e 506 (M + H).
- Example 65 MS m/e 550 (M + H).
- Example 68 To 25 mg (0.045 mmol) of the N-p-nitrophenyl intermediate was added 500 ⁇ l N-piperidinylethanol. The reaction was stirred at room temperature for approximately 5 hours, diluted with methylene chloride, washed with water/brine and dried over sodium sulfate. The crude product was purified by preparative TLC eluting with 8-10% MeOH/ CH 2 C1 2 . The pure product was collected, stirred with solvent, filtered, and concentrated. The sample was dried at 80 °C under high vacuum.
- Example 67 The compound was prepared by the method described for Example 68 using the N-p-nitrophenyl intermediate and N-pyrrolidinylethanol. MS m e 527 (M+l).
- Example 69 The compound was prepared by the method described for Example 68 using the N-p-nitrophenyl intermediate and N-pyrrolidinylethanol. MS m/e 538 (M+l).
- Example 70 Step 1 : O-Nitrophenylcarbonate intermediate: A mixture of the phenol intermediate 1-33-1 (192 mg, 0.525 mmol) and p-nitrophenyl carbonate (314 mg, 1.03 mmol) in DMF (4 mL) was heated to 100 °C for 20 h. Solvent was removed by rotary evaporation and the residue was extracted into CH2CI 2 and washed with aqueous NaHCO 3 . The organic layer was dried over MgSO , filtered, and evaporated. The resulting residue was purified by column chromatography (silica gel, 3% MeOH in CH 2 ⁇ 2 ) to afford the carbonate intermediate (156 mg, 56%). 1H NMR (CDC1 3 ) ⁇ 8.86 (s, IH), 8.34 (d, 2H,
- Example 71 MS m e 498 (M + H).
- Example 72 MS m/e 484 (M + H).
- Example 74 To 20 mg (0.052 mmol) of the amine intermediate 1-29-1 in 2 mL CH 2 G 2 /I2.6 ⁇ l pyridine was added 28 mg (0.156 mmol, 3 eq.) nicotinoyl chloride. The reaction was heated to 49 °C for 1 hr, cooled to room temperature, concentrated, stirred with ether, and the solid was filtered off. The solid was taken up in CH 2 Cl 2 /MeOH and purified on preparative TLC eluting with 10% MeOH/CH 2 Cl 2 . The pure product was collected and dried at 80 °C under high vacuum.
- Examples 75-82 were prepared by the method described for Example 74 using the appropriate amine intermediate 1-29 and acid chloride.
- Example 79 MS m/e 510 (M + H).
- Example 80 MS m e 494 (M + H).
- Example 83 The compound was prepared using the N-sec-butyl intermediate 1-36 and 2- thiophene carbonyl chloride by the general procedure described for Example 25. MS m/e 495 (M + H).
- Example 84 The compound was prepared using the N-sec-butyl indazole intermediate I- 36 and 2-furoyl chloride by the general procedure described for Example 25. MS m/e 479 (M + H).
- Example 85 The compound was prepared using the intermediate 1-39 by the general procedure described for Example 13. MS m/e 479 (M + H).
- Example 136 Tan solid (58% yield).
- Example 137 (71% yield).
- Example 138 (19% yield). 1H NMR (CDC1 3 , 300 MHz): ⁇ 1.66 (m, IH), 2.01-2.22 (m, 3H), 2.67 (m, IH), 3.51 (m, 2H), 3.74 (m, 4H), 3.88 (m, 2H), 4.38 (s, 2H), 4.71 (s, 2H), 4.72 (m, 2H), 4.90 (s, 2H), 6.07 (s, IH), 7.36 (t, IH), 7.44-7.68 (m, 5H), 7.80 (s, IH), 9.53 (d, IH); MS (ESI): m/e 483 (M+l) + ; Example 139.
- Example 140 off-white solid (25% yield).
- Example 141 Light yellow solid (17% yield).
- Example 142 Pale yellow solid (26% yield).
- Example 143 Orange residue (21% yield).
- Example 144 Light orange solid (9% yield).
- Example 145 (12.6 mg, 44% yield).
- Example 147 Pale yellow solid (31 mg, 54% yield).
- Example 148 Pale orange solid (12.4 mg, 24% yield).
- Example 149 Pale yellow solid (42.6 mg, 57% yield).
- Example 150 Yellow-tan solid (77% yield).
- Example 151 Tan solid (32% yield). 1H NMR (DMSO-d6, 300 MHz): ⁇ 1.97 (m, 4H), 3.51 (s, 2H), 3.73 (t, 2H), 4.56 (s, 2H), 4.71 (s, 2H), 4.77 (m, 2H), 4.91 (s, 2H), 6.98 (m, 2H), 7.35-7.43 (m, 3H), 7.52 (d, IH), 7.70 (m, 2H), 7.96 (s, IH), 8.60 (s, IH), 9.51 (d, IH); MS (ESI): m/e 509 (M+l) + .
- Example 152 Yellow solid (69%).
- Example 153 (80%) 1H NMR (DMSO-d6, 300 MHz): ⁇ 1.55 (m, 3H), 3.4-3.8 (m, 6H), 4.14 (m, 2H), 4.66 (s, 2H), 4.91 (s, 2H), 7.29-7.73 (m, 5H), 7.98 (s, IH), 8.55 (s, IH), 9.39 (d, IH), 11.94 (s, lH).
- Example 154 (150 mg, 89% yield).1H NMR (DMSO-d6, 300 MHz): ⁇ 1.80 (m, 4H), 3.58-3.78 (m, 4H), 4.02 (m, IH), 4.18 (s, 2H), 4.69 (s, 2H), 4.93 (s, 2H), 7.34-7.49 ( , 2H), 7.56 (t, 2H), 7.70 (d, IH), 7.94 (s, IH), 8.53 (s, IH), 9.39 (d, IH), 11.92 (s, IH).
- Example 155 1H NMR (DMSO-d6, 300 MHz): ⁇ 3.24 (s, 3H), 3.47 (m, 2H), 3.58 (m, 2H), 4.13 (m, 2H), 4.62 (s, 2H), 4.89 (s, 2H), 7.30-7.42 (m, 3H), 7.56 (d, IH), 7.64 (d, IH), 7.91 (s, IH), 8.51 (s, IH), 9.35 (d, IH), 11.89 (s, IH); MS (ESI): m/e 421 (M+Na) + .
- Example 157 A stirred solution of Example 155 (370 mg, 0.93 mmol) in DMF (20 mL) was placed under vacuum and DMF (10 mL) was removed by distillation. The mixture was cooled to room temperature and sodium hydride (45 mg, 0.93 mmol) was added and stirred for 30 min. Glycidol mesylate (170 mg, 1.1 mmol) was added and the reaction mixture heated to 60 °C. After 18 h, the mixture was cooled to room temperature, filtered, and concentrated in vacuo .
- Example 158 To a stirred solution of Example 157 (80mg, 0.18 mmol) in THF (10 mL) was added super hydride (724 uL, 0.72 mmol) dropwise at 0 °C. The reaction mixture was warmed to room temperature and stirred for 2 h. The reaction solvent was removed in vacuo and IN HCl was added.
- Example 159 Using the general procedure for Example 158, a suspension of ester (1.45 g, 2.27 mmol) in methylene chloride (30 mL) was cooled to 0 °C and DIBAL-H (5.7 mL, 5.7 mmol) was added dropwise. The reaction mixture was warmed to room temperature for 2 h then quenched with methanol (20 mL). HCl (IN, 20 mL) was added and the reaction solvent removed in vacuo to give the product as a yellow solid (1.2 g, 78% yield).
- the reaction mixture was stirred at room temperature for 20 min. and acetaldehyde was added and the mixture was stirred for 18 h. Additional cesium carbonate and acetaldehyde was added and the mixture stirred for 3 h.
- the mixture was diluted with methylene chloride, washed with water and brine, and purified by flash chromatography on silica gel using ethyl acetate/ methylene chloride (10%) to give product (45 mg, 43% yield).
- the product (45 mg) was dissolved in methylene chloride (4 mL) and ethanethiol followed by trifluoroacetic anhydride was added at 0 °C.
- Example 162 This compound was prepared using the general procedure as Example 161 starting with 3-aminomethyl-N-ethanol intermediate XIII MS m/e 540 (m+l) + .
- Example 163 This compound was prepared from XII intermediate and ethyl isocyanato acetate. MS m/e 513 (m+l) + .
- Example 165 A mixture of intermediate X (16.5 mg, 0.041 mmol) and cesium carbonate (88 mg, 1.1 eq) in 2.0 mL of CH 3 CN was added cyclopentyl bromide (8.0 ul, 2.0 eq.) under N 2 . After stirred at 70 °C for 24 hours, the mixture was diluted with CH2CI2 and filtered through celite and concentrated. Purification by preparation TLC plate with CH 2 Cl 2 /MeOH afforded the product. MS m/e 533 (M+l).
- Example 166 Prepared by hydrogenation of Example IC in DMF using Pd(OH) 2 and a drop of HCl. MS m/e 443 (M+l) Example IC. A suspension of sodium hydride (2.44 mg, 1.22 eq.) in 0.5 mL of
- THF was stirred under N 2 as phenol intermediate X (3-hydroxy-10-isopropoxy-12,13- dihydro-6H,7H,14H-nephthyl(3,4-a)pyrrolo(3,3-a)pyrrolo(3,4-c)carbazole-7(7H)one) (20.6 mg, 0.05 mmol) in 2.0 mL of THF.DMF (1:1) was added dropwise. After 10 minutes of stirring, 2-bromopyrimidine (8.9 mg, 1.12eq.) in 0.5 mL of THF was added.
- the mixture was stirred at 60 °C for 14 hours. Then, the mixture was cooled to room temperature, diluted with CH 2 ⁇ 2 /MeOH, filtered through celite and concentrated.
- Method A A mixture of hydroxyl intermediate (0.2 mmol), potassium iodide (3.3 mg, 0.1 eq.), N-tetrabutylammonium bromide (0.1 eq), cesium hydroxide hydrate (3 eq) and 20 mg of 4 sieves in 2.0 mL of CH 3 C ⁇ was added the appropriate alkyl bromide or iodide under N 2 . After the mixture was stirred at 50 °C for 14-72 hours, the reaction mixture was diluted with CH3CN and filtered through celite and concentrated. The residue was diluted with CH 2 C1 2 and washed with water and dried over magnesium sulfate.
- Method B A mixture of hydroxy intermediate (0.2 mmol) and cesium carbonate (3 eq) in 2.0 mL of CH 3 CN was added the appropriate alkyl bromide or iodide under N 2 . After the mixture was stirred at 50-80 °C for 14-72 hours, the reaction mixture was diluted with CH 3 CN and filtered through celite and concentrated. The residue was diluted with CH 2 C1 2 and washed with water and dried over magnesium sulfate. Purification by preparation TLC plate or crystallization with CH 2 Cl2/MeOH afforded the desired product.
- Method C A mixture of hydroxyl intermediate (0.1 mmol), sodium hydroxide (1.5 eq.) and N-tetrabutylammonium bromide (0.1 eq) in 0.5 mL of CH2CI2 and 0.5 mL of water was added the appropriate alkyl bromide under ⁇ 2. After the mixture was stirred at room temperature for 14-72 hours, the reaction mixture was concentrated and the residue was washed with water and dried over magnesium sulfate. Purification by preparation TLC plate with CH 2 Cl 2 /MeOH or crystallization afforded the desired product.
- Example 167 A mixture of intermediate phenol XV (19.5 mg, 0.05 mmol), potassium carbonate (34.6 mg, 5 eq.) and potassium iodide (8.7 mg, 1.05 eq) in 1.5 mL of acetone and 0.25 mL of DMF was added the benzyl 2-bromoethyl ether (8.3 uL, 1.05 eq.) under N 2 . After the mixture was stirred at reflux for 24 hours, the reaction mixture was diluted with EtOAc and washed with water, saturated NaCl solution and dried over magnesium sulfate. Purification by preparation TLC plate with 5% of MeOH CH 2 Cl 2 afforded the desired product (10 mg, 39%). MS m/e 519 m/z (M+l) + .
- Example 168 The product was obtained by first forming compound 1681 by Method A, using phenol XV and cyclopentyl bromide; 14 hr; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 10%; MS: m/e 453 m/z (M+l) + .
- a mixture of compound 1681 110 (5 mg, 0.01 mmol), 10% Pd(OH) 2 /C and 0.1 mL of cone.
- HCl in 1.0 mL of EtOH was hydrogenated under 42 psi H 2 on a Parr apparatus for 24 hours at room temperature. Filtration and concentration afforded 2.2 mg (27%) of the title compound.
- Example 169 Method C from phenol XV and epibromohydrin; 22 hour, preparative TLC (10% MeOH in CH 2 C1 2 ); yield 30%; MS: m e 463 m/z (M+Na) + .
- Example 170 Method C; phenol XV and l-bromo-2-(2-methoxyethoxy)ethane, 14 hr; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 11%; MS: 509 m z (M+Na) +
- Example 17 1.
- Method B phenol XV and 2-(2-bromoethyl)-l,3-dioxane, 14 hr reflux; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 54%; MS: 521 m/z (M+l) + .
- Example 172 Method A; phenol XV and (bromomethyl)cyclopropane, 14 hr; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 17%; MS: m e 439 m/z (M+l) + .
- Example 173 Method A; phenol XV and 2-bromomethyl-l,3-dioxolane; 64 hr; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 15%; MS: 471 m/z (M+l) + .
- Example 174 Method B; phenol XV and N-(3-bromopropyl)phthalimide; 48 hr at 80 °C; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 17%; MS: m/e 494 m/z (M+ ⁇ a) +
- Example 176 Method A; phenol XV and methyl 4-chloro-3-methoxy-(E)-2-butenoate; 40 hr at 80 °C; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 21%; MS: m/e 535 m/z (M+Na) +
- Example 177 Method A; phenol XV and 1-bromopinacolone; 14 hr at 60 °C; prep. TLC (10% MeOH in CH 2 C1 2 ); yield 29%; MS: m/e 505 m/z (M+Na) + .
- Example 178 Method A; 20 hr at 50 °C; prep. TLC (10% MeOH in CH 2 C1 2 ); yield (5%); MS: 449 m/z (M+Na) +
- Example 180 Method B (19%) MS m/e 476 (M+l); 1H-NMR (DMSO-d 6 ) 8.56 (s, IH), 8.36 (s, IH), 7.92 (d, IH), 7.85 (m, 2H), 7.66 (d, IH), 7.51 (d, 2H), 7.48 (t, IH), 7.33 (m, IH), 7.27 (t, IH), 6.97 (s, IH), 6.85 (d, IH), 5.20 (s, IH), 4.97 (m, IH), 4.75 (s, 2H), 4.62 (m, 2H).
- Example 185 Method B (88%) M S m e 466 (M+l); 1H-NMR (DMSO-d ⁇ ) 8.35 (s, IH), 7.91 (d, IH), 7.83 (d, IH), 7.64 (d, IH), 7.46 (t, IH), 7.24 (t, IH), 6.86 (s, IH), 6.77 (d, IH), 4.96 (t, IH), 4.75 (s, 2H), 4.61 (m, 2H), 4.03 (t, 2H), 3.78 (m, 2H), 2.74 (m, 2H), 2.54 ), (t, 2H), 1.73 (m, 6H).
- Example 186 Method B.
- Example 188 This compound was formed from Example 185B, ethanol and gaseous hydrogen chloride (85%) MS m/e 512 (M+1);1H-NMR (DMSO-d 6 ) 8.35 (s, IH), 7.91 (s, IH), 7.83 (d, IH), 7.65 (d, 1H),7.46 (t, IH), 7.26 (t, IH), 6.85 (s, IH), 6.76 (d, IH), 4.75 (s, IH), 4.61 (m, 2H), 4.35 (m, 2H), 4.00 (m, 2H), 3.79 (m, 2H), 2.73 (m, 2H), 2.66 (m, 2H), 1.77 (m, 6H), 1.33 (t, 3H).
- Example 189 Example 188 was refluxed in ethanol and concentrated hydrochloric acid for 18 hr. The solution was made basic with sodium hydroxide to pH 10 and refluxed 4 hours. The solution was acidified to precipitate the product. MS m/e 485 (M+l); 1H-NMR (DMSO-de) 12.00 (s, IH), 7.91 (d, IH), 7.82 (d, IH), 7.65 (d, IH), 7.45 (t, IH), 7.24 (m, 2H), 6.85 (s, IH), 6.76 (d, 2H), 4.96 (t, IH), 4.75(s, 2H), 4.61 (m, 2H), 3.98 (t, IH), 3.77 (m, 2H), 2.73(m, 2H), 2.23 (m, 4H), 1.71(m, 8H).
- Example 190 The product was obtained from a reaction of Example 186 with ethanol and gaseous hydrogen chloride (45%) MS m/e 512 (M+l); 1H-NMR (DMSO-d ⁇ ) 8.37 (s, IH), 7.91 (d, IH), 7.82 (d, IH), 7.65 (d, IH), 7.47 (t, IH), 7.23 (m, 2H), 6.86 (s, IH), 6.77 (d, 2H), 6.67 (s, IH), 4.99 (t, IH), 4.76 (s, 2H), 4.61 (m, 2H), 3.98 (t, IH), 3.80 (m, 2H), 2.74 (m, 2H), 2.02 (t, 2H), 1.71 (m, 2H), 1.38 (m, 8H).
- Example 191 and Example 192 Phenol intermediate XVII (25 mg, 79 ⁇ mole), potassium carbonate (17 mg, 123 ⁇ mole), and ethyl bromoacetate (17 ⁇ L, 155 ⁇ mole) were combined in 10 mL dry acetone. A drop of N,N-dimethylformamide was and the mixture was heated at 50°C for three days. HPLC analysis revealed the presence of two products. The two products were separated employing reverse phase C8 high performance liquid chromatography (1:1 acetonitrile:water with 0.1% trifluoroacetic acid). The first product eluted was identified as the mono adduct Example 191B. 2 mg.
- Example 193 Prepared by the method described for Example 192 from bromoacetonitrile: NMR ( ⁇ VDMSO): 11.85 (s, IH), 9.3 (d, IH), 8.48 (s, IH), 7.95 (d, IH), 7.58 (d, IH), 7.4 (m, 2H), 7.2 (dd, IH), 7.1 (d, IH), 5.2 (s, 2H), 4.85 (s, 2H), 4.18 (s, 2H). MS (ES +): 366 (M+l).
- Example 194 Example 192 (10 mg, 24 ⁇ mol) in 10 mL dry tetrahydrofuran was treated with lithium borohydride (0.5 mL of a 2.0 M solution in tetrahydrofuran, 1.0 mmol) and heated at 40°C for 72 h. 1 mL water was then added and the solution was concentrated. The crude solid was taken up into 1 mL DMF and concentrated onto 600 mg silica. The silica was applied to the top of a bed of silica and medium pressure liquid chromatography was effected eluting with 4% methanol: dichloromethane to afford 3.0 mg of a tan solid.
- lithium borohydride 0.5 mL of a 2.0 M solution in tetrahydrofuran, 1.0 mmol
- Example 195 This compound was prepared by the method described for Example 194 from Example 193: NMR (d 6 -DMSO): 9.3 (d, IH), 8.48 (s, IH), 7.95 (d, IH), 7.70 (d, IH), 7.45 (dd, IH), 7.28 (m, IH), 7.22 (s, IH), 6.95 (d, IH), 4.9 (s, 2H), 4.7 (br s, 2H), 4.46 (s, 2H), 4.06 (br s, 2H), 3.80 (br s, 2H), 3.70 (br s, 2H), 3.52 (overlapping s, 2H). MS (ES+): 415 (M+l).
- Example 196 The O-allyl intermediate was prepared using allyl bromide as described for Example 194: NMR (d 6 -DMSO): 11.8 (s, IH), 9.27 (d, IH), 8.48 (s, IH), 7.98 (d, IH), 7.60 (d, IH), 7.45 (dd, IH), 7.30 (s, IH), 7.25 (m, IH), 7.05 (dd, IH), 6.10 (m, IH), 5.4 (dd, IH), 5.3 (dd, IH), 4.95 (s, 2H), 4.7 (d, 2H), 4.18 (s, 2H). MS (ES+): 367 (M+l).
- Example 197 Example 194 (63 mg, 153 ⁇ mol), dimethylamine (3 mL of a 40% solution in water), and ammonium chloride (100 mg) were combined in N,N-dimethylformamide and stirred at ambient temperature in a sealed tube for 5 d. The solution was concentrated onto 0.6 g silica and applied to a bed of silica. Medium pressure liquid chromatography employing a gradient from 5-10% methanol: dichloromethane afforded 60 mg of an orange solid.
- Example 198 The epoxide (42 mg, 0.11 mmol), dimethylamine (3 mL of a 40%solution in water), and ammonium chloride (100 mg) were combined in 10 mL N,N- dimethylformamide and stirred in a sealed tube for 16 h. The mixture was concentrated onto 700 mg silica and applied to a bed of silica. Medium pressure liquid chromatography employing a gradient of 15-25% methanokdichloromethane afforded approximately 5 mg of the polar desired.
- Example 199 This compound was prepared by the same procedure as Example 198 using morpholine: MS (ES+): m/e 470 (M+l).
- the compounds of the present invention are useful, inter alia, as therapeutic agents. Particularly, the compounds are useful for kinase inhibition, such as, for example, trk, VEGFR, PDGFR, PKC, MLK, DLK, Tie-2, FLT-3, and CDK1-6.
- kinase inhibition such as, for example, trk, VEGFR, PDGFR, PKC, MLK, DLK, Tie-2, FLT-3, and CDK1-6.
- Various compounds of the present invention show enhanced pharmaceutical properties over those disclosed in the art and improved pharmacokinetic properties in mammals.
- the compounds of the present invention show enhanced pharmaceutical properties over those disclosed in the art, including increased MLK and DLK dual inhibition activity, or increased VEGFR and Tie- 2 dual inhibition activity, along with improved pharmacokinetic properties in mammals.
- the present invention provides a method for treating or preventing diseases and disorders, such as those disclosed herein, which comprises administering to a subject in need of such treatment or prevention a therapeutically effective amount of a compound of the present invention.
- the present invention provides a method for inhibiting trk kinase activity comprising providing a compound of the present invention in an amount sufficient to result in effective inhibition.
- inhibition of trk implies utility in, for example, diseases of the prostate such as prostate cancer and benign prostate hyperplasia, as well as for the treatment of inflammation, such as neurological inflammation and chronic arthritis inflammation.
- the trk kinase receptor is trk A.
- VEGF vascular endothelial growth factor
- VGTFR VEGF/VEGF receptor
- RTK Receptor tyrosine kinase
- AML vascular endothelial growth factor
- FGF fibroblast growth factor
- the present invention provides a method for treating or preventing angiogenic disorders where VEGFR kinase activity contributes to pathological conditions, the method comprising providing a compound of the present invention in an amount sufficient to result in the vascular endothelial growth factor receptor being contacted with an effective inhibitory amount of the compound.
- Inhibition of VEGFR implies utility in, for example, angiogenic disorders such as cancer of solid tumors, endometriosis, macular degeneration, retinopathy, diabetic retinopathy, psoriasis, hemangioblastoma, as well as other ocular diseases and cancers.
- FLT3, a member of the receptor tyrosine kinase (RTK) class III, is preferentially expressed on the surface of a high proportion of acute myeloid leukemia (AML) and B- lineage acute lymphocytic leukemia (ALL) cells in addition to hematopoietic stem cells, brain, placenta and liver.
- AML acute myeloid leukemia
- ALL B- lineage acute lymphocytic leukemia
- the present invention provides a method for treating disorders characterized by responsiveness to FLT3 inhibition, the method comprising providing a compound of the present invention in an amount sufficient to result in the inhibition of FLT3.
- Platelet-derived growth factor (PDGF) was one of the first polypeptide growth factors identified that signals through a cell surface tyrosine kinase receptor (PDGF-R) to stimulate various cellular functions including growth, proliferation, and differentiation.
- PDGF A and B PDGF A and B
- PDGF-R alpha and beta cognate receptors
- PDGF expression has been shown in a number of different solid tumors, from glioblastomas to prostate carcinomas. In these various tumor types, the biologic role of PDGF signaling can vary from autocrine stimulation of cancer cell growth to more subtle paracrine interactions involving adjacent stroma and even angiogenesis.
- the present invention provides a method for treating or preventing disorders where PDGFR activity contributes to pathological conditions, the method comprising providing a compound of the present invention in an amount sufficient to result in the platelet derived growth factor receptor being contacted with an effective inhibitory amount of the compound.
- Inhibition of PDGFR implies utility in, for example, various forms of neoplasia, rheumatoid arthritis, chronic arthritis, pulmonary fibrosis, myelofibrosis, abnormal wound healing, diseases with cardiovascular end points, such as atherosclerosis, restenosis, post-angioplasty restenosis, and the like.
- the present invention provides a method for treating or preventing disorders where MLK activity contributes to pathological conditions, such as those listed above, wherein the method comprises providing a compound of the present invention in an amount sufficient to result in the MLK receptor being contacted with an effective inhibitory amount of the compound.
- Inhibition of MLK implies utility in, for example, forms of cancer where MLKs play a pathological role as well as in neurological disorders.
- the present invention provides a method for treating disorders characterized by the aberrant activity of trophic factor responsive cells, the method comprising providing a compound of the present invention in an amount sufficient to result in the trophic factor cell receptor being contacted with an effective activity inducing amount of the compound.
- the activity of the trophic factor responsive cells is ChAT activity.
- Fibroblast growth factor receptors are members of a family of polypeptides synthesized by a variety of cell types during the processes of embryonic development and in adult tissues. FGFR have been detected in normal and malignant cells and are involved in biological events that include mitogenic and angiogenic activity with a consequent crucial role in cell differentiation and development.
- FGF fibroblast growth factors
- HS heparan sulfate
- the present invention provides a method for treating disorders characterized by the aberrant activity of FGF responsive cells, the method comprising providing a compound of the present invention in an amount sufficient to result in the FGFR being contacted with an effective activity inducing amount of the compound.
- the compounds of the present invention can also have positive effects on the function and survival of trophic factor responsive cells by promoting the survival of neurons.
- the compound may preserve the survival of a cholinergic neuronal population at risk of dying (due to, e.g., injury, a disease condition, a degenerative condition or natural progression) when compared to a cholinergic neuronal population not presented with such compound, if the treated population has a comparatively greater period of functionality than the non-treated population.
- a variety of neurological disorders are characterized by neuronal cells which are dying, injured, functionally compromised, undergoing axonal degeneration, at risk of dying, etc. These neurodegenerative diseases and disorders include, but are not limited to, Alzheimer's disease; motor neuron disorders (e.g.
- amyotrophic lateral sclerosis Parkinson's disease; cerebrovascular disorders (e.g., stroke, ischemia); Huntington's disease; AIDS dementia; epilepsy; multiple sclerosis; peripheral neuropathies including diabetic neuropathy and chemotherapy induced peripheral neuropathy, AID related peripheral neuropathy; disorders induced by excitatory amino acids; and disorders associated with concussive or penetrating injuries of the brain or spinal cord.
- the compounds of the present invention are useful for treating or preventing multiple myeloma and leukemias including, but not limited to, acute myelogenous leukemia, chronic myelogenous leukemia, acute lymphocytic leukemia, and chronic lymphocytic leukemia.
- the present compounds are also useful in the treatment of disorders associated with decreased ChAT activity or the death, injury to spinal cord motoneurons, and also have utility in, for example, diseases associated with apoptotic cell death of the central and peripheral nervous system, immune system and in inflammatory diseases.
- ChAT catalyzes the synthesis of the neurotransmitter acetylcholine, and it is considered an enzymatic marker for a functional cholinergic neuron.
- a functional neuron is also capable of survival. Neuron survival is assayed by quantification of the specific uptake and enzymatic conversion of a dye (e.g., calcein AM) by living neurons.
- a dye e.g., calcein AM
- the compounds described herein may also find utility in the treatment of disease states involving malignant cell proliferation, such as many cancers. Additional embodiments of the invention are directed to the use of any compound described herein, and stereoisomers or pharmaceutically acceptable salts thereof, in the treatment and/or prevention of any of the conditions, diseases and disorders described above. Further embodiments are directed to the use of the compounds described herein, and stereoisomers or pharmaceutically acceptable salts thereof, in the manufacture of a medicament for treating and/or preventing said conditions, disorders and diseases.
- the compounds of the present invention have important functional pharmacological activities which find utility in a variety of settings, including both research and therapeutic arenas. For ease of presentation, and in order not to limit the range of utilities for which these compounds can be characterized, the activities of the compounds of the present invention can be generally described as follows: A. Inhibition of enzymatic activity
- Inhibition of developmentally programmed motoneuron death Inhibition of enzymatic activity can be determined using, for example, VEGFR inhibition (e.g., VEGFR2 inhibition), MLK inhibition (e.g., MLKl, MLK2 or MLK3 inhibition), PDGFR kinase inhibition, NGF-stimulated trk phosphorylation, PKC inhibition, or trk tyrosine kinase inhibition assays.
- VEGFR inhibition e.g., VEGFR2 inhibition
- MLK inhibition e.g., MLKl, MLK2 or MLK3 inhibition
- PDGFR kinase inhibition e.g., NGF-stimulated trk phosphorylation
- PKC inhibition e.g., trk tyrosine kinase inhibition assays.
- Inhibition of developmentally programmed motoneuron death can be assessed in ovo using embryonic chick somatic motoneurons, which cells undergo naturally occurring death between embryonic days 6 and 10, and analyzing inhibition of such naturally occurring cell death as mediated by the compounds disclosed herein.
- the inhibition of enzymatic activity by the compounds of the present invention can be determined using, for example, the following assays: VEGFR Inhibition Assay MLK Inhibition Assay PKC Activity Inhibition Assay trkA Tyrosine Kinase Activity Inhibition Assay Tie-2 Inhibition Assay CDK1-6 Inhibition Assay Inhibition of NGF-stimulated trk phosphorylation in a whole cell preparation Platelet Derived Growth Factor Receptor (PDGFR) inhibition assay
- PKC Activity Inhibition Assay trkA Tyrosine Kinase Activity Inhibition Assay Tie-2 Inhibition Assay
- CDK1-6 Inhibition Assay Inhibition of NGF-stimulated trk phosphorylation in a whole cell preparation
- PDGFR Platelet Derived Growth Factor Receptor
- VEGF receptor human flk-1, KDR, VEGFR2
- trkA kinase ELIS A assay described above.
- the kinase reaction mixture consisting of 50 mM Hepes, pH 7.4, 40 ⁇ M ATP, 10 mM MnCl 2 , 0.1% BSA, 2% DMSO, and various concentrations of inhibitor, was transferred to PLC- ⁇ /GST-coated plates.
- VEGFR kinase was added and the reaction was allowed to proceed for 15 min. at 37 °C. Detection of phosphorylated product was accomplished by addition of anti-phosphotyrosine antibody (UBI). A secondary enzyme-conjugated antibody was delivered to capture the antibody- phosphorylated PLC- ⁇ /GST complex. The activity of the bound enzyme was measured via an amplified detection system (Gibco-BRL). Inhibition data were analyzed using the sigmoidal dose-response (variable slope) equation in GraphPad Prism.
- each 50- ⁇ l assay mixture contained 20 mM Hepes, pH 7.0, 1 mM EGTA, 10 mM MgCl 2 , 1 mM DTT, 25 mM ⁇ -glycerophosphate, 60 ⁇ M ATP, 0.25 ⁇ Ci [ ⁇ - 32 P] ATP, 0.1% BSA, 500 ⁇ g/ml myelin basic protein (UBI #13-104), 2% DMSO, 1 ⁇ M of test compound, and 1 ⁇ g/ml of baculoviral GST-MLKI KD - Samples were incubated for 15 min at 37°C. The reaction was stopped by adding ice cold 50% TCA and the proteins were allowed to precipitate for 30 min at 4°C. The plates were then washed with ice cold 25% TCA. Supermix scintillation cocktail was added, and the plates were allowed to equilibrate for 1-2 hours prior to counting using the Wallace MicroBeta 1450 PLUS scintillation counter.
- Dual Leucine Zipper Bearing Kinase Assay Compounds were tested for their ability to inhibit the kinase activity of recombinant baculoviral human DLK, containing the kinase domain and leucine zipper. Activity was measured in 384-well FiuoroNunc plates (Cat#460372) using a time-resolved fluorescence readout (PerkinElmer Application Note 1234-968). Plates were coated with 30 ⁇ l of the protein substrate MKK7 (Merritt et al. 1999) at a concentration of 20 ⁇ g/ml in Tris buffered saline (TBS).
- TBS Tris buffered saline
- Each 30 ⁇ l assay contained 20 mM MOPS (pH 7.2), 15 mM MgCl 2 , 0.1 mM Na 3 VO 4 , 1 mM DTT, 5 mM EGTA, 25 mM ⁇ -glycerophosphate, 0.1% BSA, 100 ⁇ M ATP, and 2.5% DMSO. Reactions were started by the addition of 10 ng/ml GST-ITDLK KD/LZ . For IC 5 o determinations, a 10-point dose response curve was generated for each compound. Plates were incubated at 37°C for 30 minutes, and the reactions stopped by the addition of 100 mM EDTA.
- Tie-2 Tyrosine Kinase Assay Compounds were tested for their ability to inhibit the kinase activity of recombinant baculoviral human His 6 -Tie2 cytoplasmic domain using a modification of the ELISA described for trkA (Angeles et al, 1996). A 384-well plate format was used for single-point screening while IC 50 S were performed on 96-well plates.
- each barcoded 384-well Costar High Binding plate (Cat # 3703) was coated with 50 ⁇ l/well of 10 ⁇ g/ml substrate solution (recombinant human GST- PLC- ⁇ ; Rotin et al., 1992) in Tris-buffered saline (TBS).
- the Tie2 activity was measured in 50- ⁇ l assay mixtures containing 50 mM HEPES (pH 7.2), 40 ⁇ M ATP, 10 mM MnCl 2 , 2.5% DMSO, 0.05% BSA, and 200 ng/ml His 6 -Tie2 ⁇ >
- the assays were run as described above but in 96-well Costar High Binding plates (Cat # 3703) and with the volumes doubled. A 10-point dose response curve was generated for each compound. The kinase reaction was allowed to proceed at 37°C for 20 minutes.
- Nl-Eu anti-phosphotyrosine (PT66) antibody (Wallac #AD0041) was added at 1:2000 diluted in block buffer [3% BSA in TBS with 0.05% Tween-20 (TBST)]. After one-hour incubation at 37°C, 50 ⁇ l of enhancement solution (Wallac #1244-105) was added and the plate was gently agitated. The fluorescence of the resulting solution was then measured using the time-resolved fluorescence (TRF) mode in the Multilabel Reader (Nictor2 Model # 1420-018 or Envision Model # 2100). Inhibition data were analyzed using ActivityBase and IC 50 curves were generated using XLFit. The cited references are as follows: 1. Angeles, T.
- the compounds of the present invention can be administered by any means that results in the contact of the active agent with the agent's site of action in the body of the subject.
- the compounds may be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in combination with other therapeutic agents, such as, for example, analgesics.
- the compounds of the present invention are preferably administered in therapeutically effective amounts for the treatment of the diseases and disorders described herein to a subject in need thereof.
- a therapeutically effective amount can be readily determined by the attending diagnostician, as one skilled in the art, by the use of conventional techniques.
- the effective dose will vary depending upon a number of factors, including the type and extent of progression of the disease or disorder, the overall health status of the particular patient, the relative biological efficacy of the compound selected, the formulation of the active agent with appropriate excipients, and the route of administration.
- the compounds are administered at lower dosage levels, with a gradual increase until the desired effect is achieved.
- Typical dose ranges are from about 0.01 mg/kg to about 100 mg/kg of body weight per day, with a preferred dose from about 0.01 mg/kg to 10 mg/kg of body weight per day.
- a preferred daily dose for adult humans includes about 25, 50, 100 and 200 mg, and an equivalent dose in a human child.
- the compounds may be administered in one or more unit dose forms.
- the unit dose ranges from about 1 to about 500 mg administered one to four times a day, preferably from about 10 mg to about 300 mg, two times a day.
- an oral unit dose is one that is necessary to achieve a blood serum level of about 0.05 to 20 ⁇ g/ml in a subject, and preferably about 1 to 20 ⁇ g/ml.
- the compounds of the present invention may be formulated into pharmaceutical compositions by admixture with one or more pharmaceutically acceptable excipients. The excipients are selected on the basis of the chosen route of administration and standard pharmaceutical practice, as described, for example, in Remington: The Science and Practice of Pharmacy, 20 th ed.; Gennaro, A.
- compositions may be formulated to control and/or delay the release of the active agent(s), as in fast-dissolve, modified-release, or sustained-release formulations.
- controlled-release, or extended-release compositions may utilize, for example biocompatible, biodegradable lactide polymers, lactide/glycolide copolymers, polyoxyethylene-polyoxypropylene copolymers, or other solid or semisolid polymeric matrices known in the art.
- compositions can be prepared for administration by oral means; parenteral means, including intravenous, intramuscular, and subcutaneous routes; topical or transdermal means; transmucosal means, including rectal, vaginal, sublingual and buccal routes; ophthalmic means; or inhalation means.
- parenteral means including intravenous, intramuscular, and subcutaneous routes
- transmucosal means including rectal, vaginal, sublingual and buccal routes
- ophthalmic means or inhalation means.
- the compositions are prepared for oral administration, particularly in the form of tablets, capsules or syrups; for parenteral administration, particularly in the form of liquid solutions, suspensions or emulsions; for intranasal administration, particularly in the form of powders, nasal drops, or aerosols; or for topical administration, such as creams, ointments, solutions, suspensions aerosols, powders and the like.
- the tablets, pills, powders, capsules, troches and the like can contain one or more of the following: diluents or fillers such as starch, or cellulose; binders such as microcrystalline cellulose, gelatins, or polyvinylpyrrolidones; disintegrants such as starch or cellulose derivatives; lubricants such as talc or magnesium stearate; glidants such as colloidal silicon dioxide; sweetening agents such as sucrose or saccharin; or flavoring agents such as peppermint or cherry flavoring.
- Capsules may contain any of the afore listed excipients, and may additionally contain a semi-solid or liquid carrier, such as a polyethylene glycol.
- the solid oral dosage forms may have coatings of sugar, shellac, or enteric agents.
- Liquid preparations may be in the form of aqueous or oily suspensions, solutions, emulsions, syrups, elixirs, etc., or may be presented as a dry product for reconstitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as surfactants, suspending agents, emulsifying agents, diluents, sweetening and flavoring agents, dyes and preservatives.
- the compositions may also be administered parenterally.
- the pharmaceutical forms acceptable for injectable use include, for example, sterile aqueous solutions, or suspensions.
- Aqueous carriers include mixtures of alcohols and water, buffered media, and the like.
- Nonaqueous solvents include alcohols and glycols, such as ethanol, and polyethylene glycols; oils, such as vegetable oils; fatty acids and fatty acid esters, and the like.
- Other components can be added including surfactants; such as hydroxypropylcellulose; isotonic agents, such as sodium chloride; fluid and nutrient replenishers; electrolyte replenishers; agents which control the release of the active compounds, such as aluminum monostearate, and various co-polymers; antibacterial agents, such as chlorobutanol, or phenol; buffers, and the like.
- the parenteral preparations can be enclosed in ampules, disposable syringes or multiple dose vials.
- parenteral delivery systems for the active compounds include ethylene-vinyl acetate copolymer particles, osmotic pumps, implantable infusion systems, and liposomes.
- Other possible modes of administration include formulations for inhalation, which include such means as dry powder, aerosol, or drops. They may be aqueous solutions containing, for example, polyoxyethylene-9-lauryl ether, glycocholate and deoxycholate, or oily solutions for administration in the form of nasal drops, or as a gel to be applied intranasally.
- Formulations for topical use are in the form of an ointment, cream, or gel.
- these forms include a carrier, such as petrolatum, lanolin, stearyl alcohol, polyethylene glycols, or their combinations, and either an emulsifying agent, such as sodium lauryl sulfate, or a gelling agent, such as tragacanth.
- a carrier such as petrolatum, lanolin, stearyl alcohol, polyethylene glycols, or their combinations
- an emulsifying agent such as sodium lauryl sulfate
- a gelling agent such as tragacanth.
- Formulations suitable for transdermal administration can be presented as discrete patches, as in a reservoir or microreservoir system, adhesive diffusion-controlled system or a matrix dispersion-type system.
- Formulations for buccal administration include, for example lozenges or pastilles and may also include a flavored base, such as sucrose or acacia, and other excipients such as glycocholate.
- Formulations suitable for rectal administration are preferably presented as unit-dose suppositories, with a solid based carrier, such as cocoa butter, and may include a salicylate.
- a solid based carrier such as cocoa butter
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Diabetes (AREA)
- Endocrinology (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Epidemiology (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Psychiatry (AREA)
- Ophthalmology & Optometry (AREA)
- Pulmonology (AREA)
- Hospice & Palliative Care (AREA)
- Obesity (AREA)
- Reproductive Health (AREA)
- Vascular Medicine (AREA)
- Oncology (AREA)
Abstract
Description

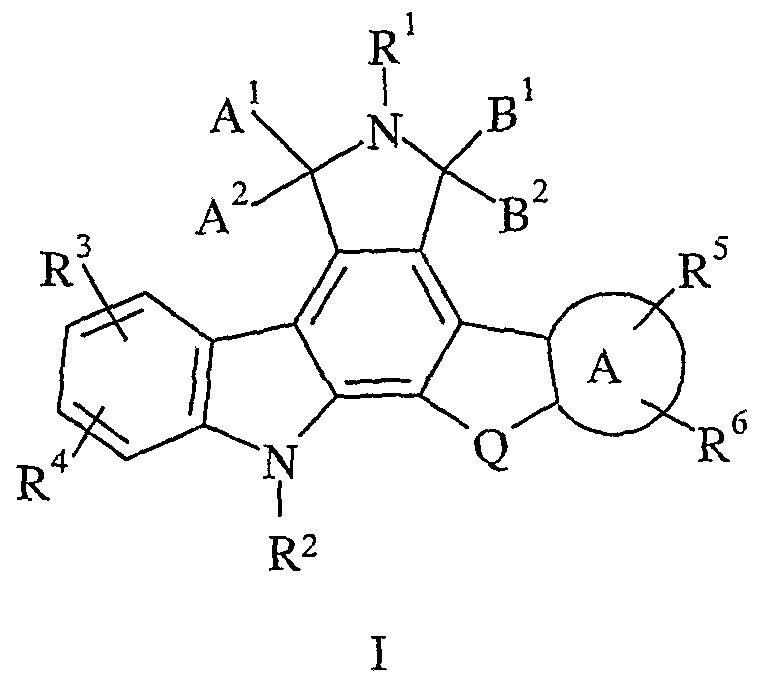




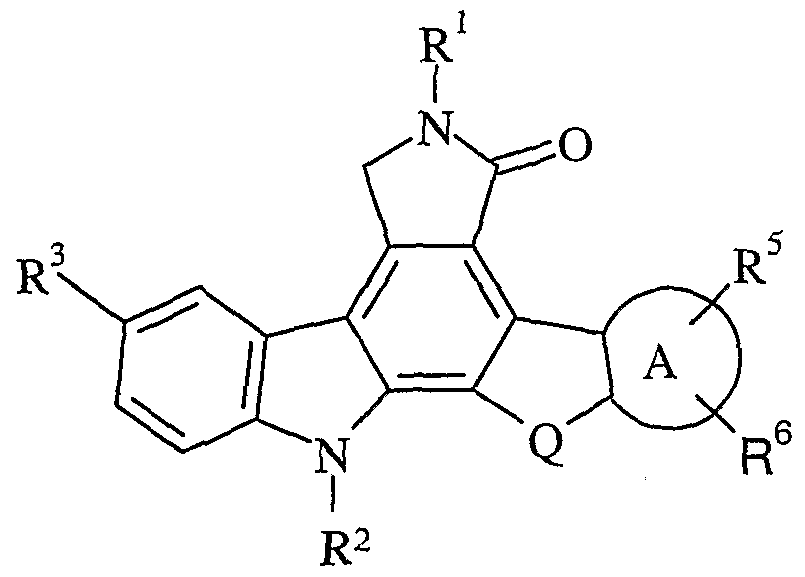


















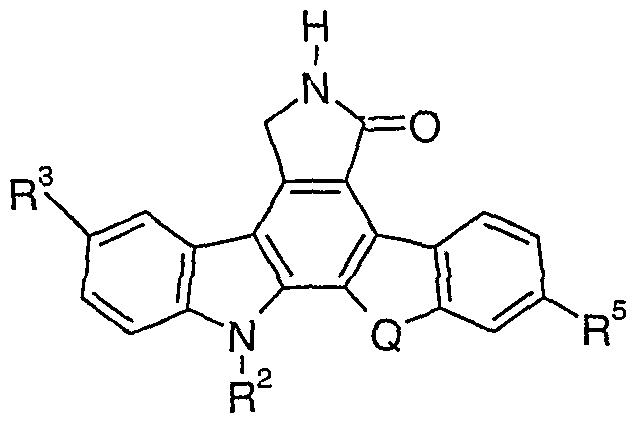





























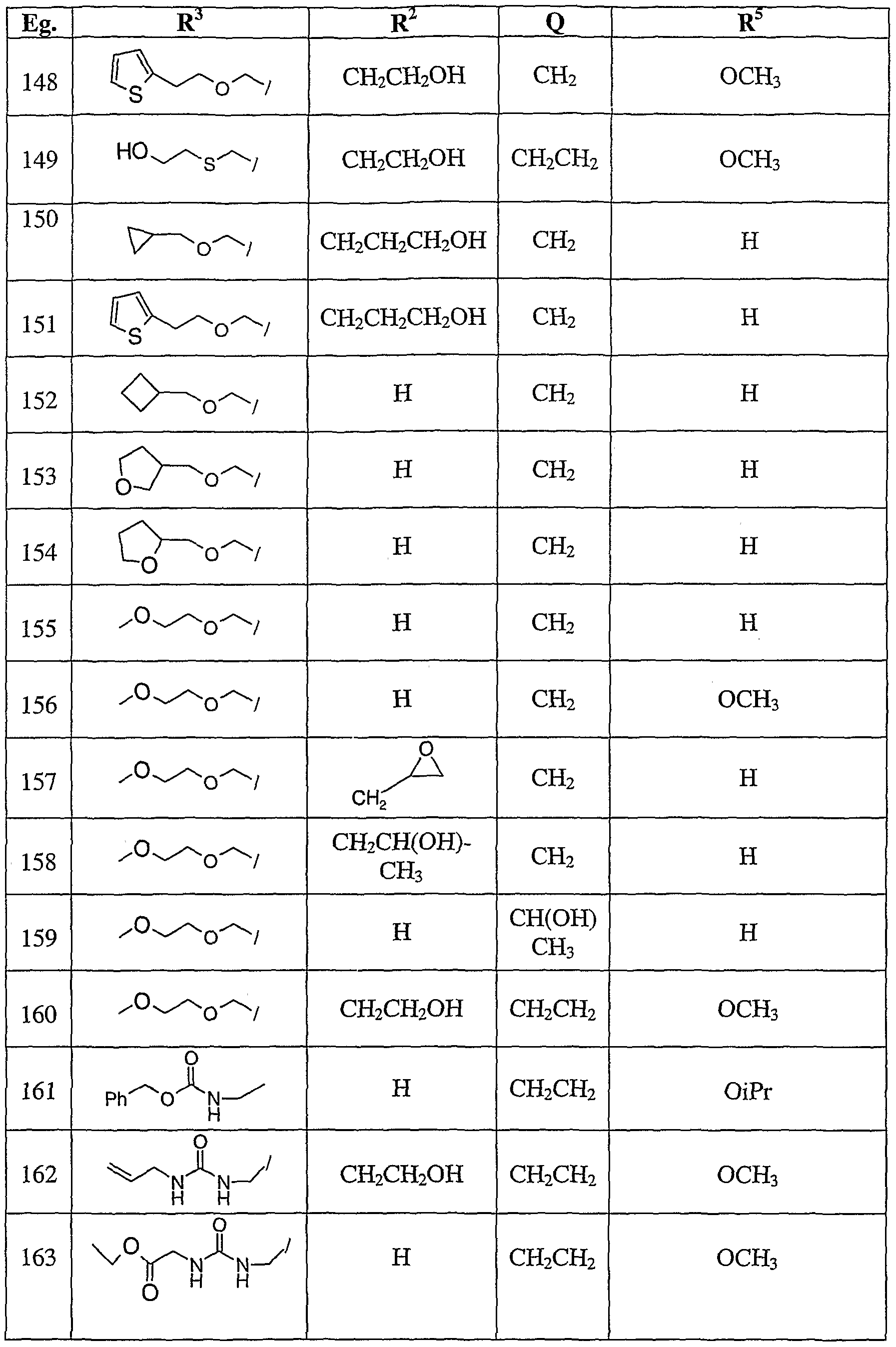



Claims











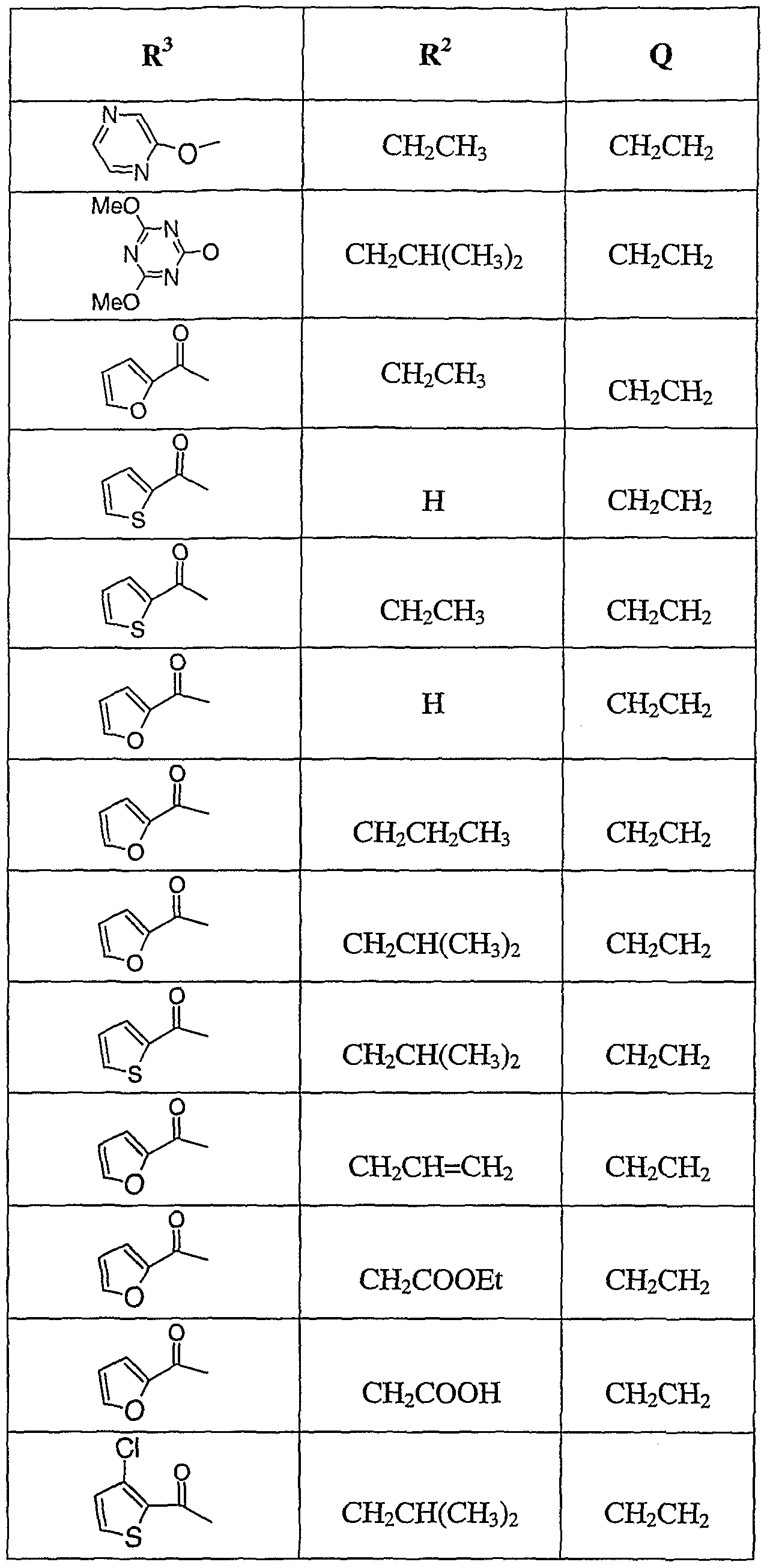


Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2004309397A AU2004309397B2 (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
| CA2549628A CA2549628C (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
| AT04820922T ATE433453T1 (en) | 2003-12-23 | 2004-12-23 | CONDENSED PYRROLOCARBAZOLES AND METHOD FOR THE PRODUCTION THEREOF |
| DE602004021508T DE602004021508D1 (en) | 2003-12-23 | 2004-12-23 | Condensed pyrrolocarbazole and process for its preparation |
| JP2006547325A JP5006047B2 (en) | 2003-12-23 | 2004-12-23 | Novel condensed pyrrolocarbazole |
| EA200601219A EA013182B1 (en) | 2003-12-23 | 2004-12-23 | Novel fused pyrrolocarbazoles |
| EP04820922A EP1711499B1 (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
| BRPI0418120-4A BRPI0418120A (en) | 2003-12-23 | 2004-12-23 | compound, pharmaceutical composition, and methods for treating a disorder, disease or disorder, and for treating multiple myeloma or leukemia |
| HK07103508.2A HK1096394B (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
| KR1020127011222A KR101268704B1 (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
| IL176385A IL176385A (en) | 2003-12-23 | 2006-06-18 | Fused derivatives of pyrrolo[3,4-c]carbazole-1-one and pyrrolo[3,4-c]carbazole-1,3-dione and pharmaceutical compositions comprising same |
| IS8520A IS2775B (en) | 2003-12-23 | 2006-06-22 | Synthesized pyrrolocarbazole and their production methods |
| NO20063193A NO336892B1 (en) | 2003-12-23 | 2006-07-10 | Fused pyrrolocarbazoles, their use, as well as pharmaceutical preparations comprising the same |
| NO20151246A NO338804B1 (en) | 2003-12-23 | 2015-09-23 | Condensed pyrrolocarbazole compounds, their use and pharmaceutical composition. |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53225203P | 2003-12-23 | 2003-12-23 | |
| US60/532,252 | 2003-12-23 | ||
| US11/017,915 | 2004-12-22 | ||
| US11/017,915 US7241779B2 (en) | 2003-12-23 | 2004-12-22 | Fused pyrrolocarbazoles |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005063764A2 true WO2005063764A2 (en) | 2005-07-14 |
| WO2005063764A3 WO2005063764A3 (en) | 2005-08-25 |
Family
ID=34680997
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/043164 Ceased WO2005063764A2 (en) | 2003-12-23 | 2004-12-23 | Fused pyrrolocarbazoles and methods for the preparation thereof |
Country Status (21)
| Country | Link |
|---|---|
| US (3) | US7241779B2 (en) |
| EP (2) | EP1711499B1 (en) |
| JP (1) | JP5006047B2 (en) |
| KR (2) | KR20060126738A (en) |
| CN (2) | CN105294702A (en) |
| AR (1) | AR048808A1 (en) |
| AT (1) | ATE433453T1 (en) |
| AU (1) | AU2004309397B2 (en) |
| BR (1) | BRPI0418120A (en) |
| CA (1) | CA2549628C (en) |
| DE (1) | DE602004021508D1 (en) |
| EA (1) | EA013182B1 (en) |
| ES (2) | ES2387596T3 (en) |
| IL (1) | IL176385A (en) |
| IS (1) | IS2775B (en) |
| MY (2) | MY142713A (en) |
| NO (2) | NO336892B1 (en) |
| NZ (1) | NZ586306A (en) |
| SG (1) | SG149042A1 (en) |
| TW (1) | TWI342309B (en) |
| WO (1) | WO2005063764A2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006031772A1 (en) * | 2004-09-10 | 2006-03-23 | Cephalon, Inc. | Methods of treating proliferative skin diseases using carbazole derivatives |
| WO2007149451A3 (en) * | 2006-06-19 | 2008-02-21 | Cephalon Inc | Cycloalkanopyrrolocarbazole derivatives and the use thereof as parp, vegfr2 and mlk3 inhibitors |
| US9101645B2 (en) | 2008-10-22 | 2015-08-11 | Genentech, Inc. | Modulation of axon degeneration |
| US9777059B2 (en) | 2007-11-30 | 2017-10-03 | Genentech, Inc. | Anti-VEGF antibodies |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7241779B2 (en) * | 2003-12-23 | 2007-07-10 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US7169802B2 (en) | 2003-12-23 | 2007-01-30 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| EP1869141B1 (en) * | 2005-04-14 | 2020-03-11 | Merck Patent GmbH | Compounds for organic electronic devices |
| US20060293378A1 (en) * | 2005-06-28 | 2006-12-28 | Mcintire Gregory | Method of lowering intraocular pressure |
| EP2167075A4 (en) * | 2007-06-08 | 2012-07-11 | Univ Massachusetts | MIXED LINEAR KINASES AND METABOLIC DISORDERS |
| EP2242367A4 (en) * | 2008-01-08 | 2012-07-04 | Univ Pennsylvania | REL INHIBITORS AND METHODS OF USE |
| EP2307456B1 (en) | 2008-06-27 | 2014-10-15 | Amgen Inc. | Ang-2 inhibition to treat multiple sclerosis |
| AU2015202365B2 (en) * | 2008-10-22 | 2016-11-24 | Genentech, Inc. | Modulation of axon degeneration |
| CN102216306B (en) * | 2008-11-19 | 2014-12-24 | 赛福伦公司 | New Forms of Indazolo[5,4-A]pyrrolo[3,4-C]carbazole Compounds |
| CA2778483A1 (en) * | 2009-10-22 | 2011-04-28 | Genentech, Inc. | Modulation of axon degeneration |
| EP2697226B1 (en) * | 2011-04-13 | 2017-01-18 | Merck Patent GmbH | Compounds for electronic devices |
| EP2758374B1 (en) | 2011-09-21 | 2016-08-31 | Givaudan SA | Pyridine derivatives and their use as umami tastants |
| RU2015136387A (en) * | 2013-02-28 | 2017-03-30 | Дженентек, Инк. | DLK STABILITY MODULATION METHODS |
| US9029565B1 (en) * | 2013-11-20 | 2015-05-12 | Transitions Optical, Inc. | Fused ring indeno compounds for preparation of photochromic compounds |
| JP6801159B2 (en) * | 2015-04-21 | 2020-12-16 | ジエンス ヘンルイ メデイシンカンパニー リミテッドJiangsu Hengrui Medicine Co.,Ltd. | Imidazoisoindole derivative, its manufacturing method and its pharmaceutical use |
| JP6501993B1 (en) | 2018-06-06 | 2019-04-17 | 三菱電機株式会社 | Process bus application protection system and intelligent electronic device |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ245203A (en) * | 1991-11-29 | 1997-07-27 | Banyu Pharma Co Ltd | 5h-indolo[2,3-a]pyrrolo[3,4-c]carbazole-5,7(6h)-dione derivatives substituted in position-13 by a pentose or hexose group; corresponding indolo-furano(anhydride)intermediates |
| US5594009A (en) * | 1994-10-14 | 1997-01-14 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US5591855A (en) * | 1994-10-14 | 1997-01-07 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US5705511A (en) * | 1994-10-14 | 1998-01-06 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US5475110A (en) * | 1994-10-14 | 1995-12-12 | Cephalon, Inc. | Fused Pyrrolocarbazoles |
| US5616724A (en) * | 1996-02-21 | 1997-04-01 | Cephalon, Inc. | Fused pyrrolo[2,3-c]carbazole-6-ones |
| HUP9903931A3 (en) | 1996-08-22 | 2000-09-28 | Bristol Myers Squibb Co Wallin | Cytotoxic amino sugar and related sugar derivatives of indolopyrrolocarbazoles |
| US6127401A (en) * | 1998-06-05 | 2000-10-03 | Cephalon, Inc. | Bridged indenopyrrolocarbazoles |
| US6200968B1 (en) * | 1998-08-06 | 2001-03-13 | Cephalon, Inc. | Particle-forming compositions containing fused pyrrolocarbazoles |
| US6841567B1 (en) * | 1999-02-12 | 2005-01-11 | Cephalon, Inc. | Cyclic substituted fused pyrrolocarbazoles and isoindolones |
| US6630500B2 (en) | 2000-08-25 | 2003-10-07 | Cephalon, Inc. | Selected fused pyrrolocarbazoles |
| TR200401316T4 (en) | 2000-09-29 | 2004-07-21 | Eli Lilly And Company | Compounds and methods for treating proliferative diseases |
| US6653290B2 (en) | 2000-10-06 | 2003-11-25 | Bristol-Myers Squibb Company | Tumor proliferation inhibitors |
| US6610727B2 (en) | 2000-10-06 | 2003-08-26 | Bristol-Myers Squibb Company | Anhydro sugar derivatives of indolocarbazoles |
| US6558420B2 (en) | 2000-12-12 | 2003-05-06 | Bausch & Lomb Incorporated | Durable flexible attachment components for accommodating intraocular lens |
| US7018999B2 (en) | 2001-05-16 | 2006-03-28 | Cephalon, Inc. | Methods for the treatment and prevention of pain |
| AU2004288174B2 (en) | 2003-10-28 | 2007-08-16 | Warren Environmental & Coating, Llc | Method for preparing in-ground tunnel structures |
| US7169802B2 (en) * | 2003-12-23 | 2007-01-30 | Cephalon, Inc. | Fused pyrrolocarbazoles |
| US7241779B2 (en) * | 2003-12-23 | 2007-07-10 | Cephalon, Inc. | Fused pyrrolocarbazoles |
-
2004
- 2004-12-22 US US11/017,915 patent/US7241779B2/en not_active Expired - Lifetime
- 2004-12-23 CN CN201510655573.2A patent/CN105294702A/en active Pending
- 2004-12-23 SG SG200809397-3A patent/SG149042A1/en unknown
- 2004-12-23 WO PCT/US2004/043164 patent/WO2005063764A2/en not_active Ceased
- 2004-12-23 KR KR1020067014797A patent/KR20060126738A/en not_active Ceased
- 2004-12-23 JP JP2006547325A patent/JP5006047B2/en not_active Expired - Lifetime
- 2004-12-23 MY MYPI20045330A patent/MY142713A/en unknown
- 2004-12-23 EP EP04820922A patent/EP1711499B1/en not_active Expired - Lifetime
- 2004-12-23 TW TW093140223A patent/TWI342309B/en not_active IP Right Cessation
- 2004-12-23 NZ NZ586306A patent/NZ586306A/en not_active IP Right Cessation
- 2004-12-23 AU AU2004309397A patent/AU2004309397B2/en not_active Ceased
- 2004-12-23 CA CA2549628A patent/CA2549628C/en not_active Expired - Fee Related
- 2004-12-23 DE DE602004021508T patent/DE602004021508D1/en not_active Expired - Lifetime
- 2004-12-23 AT AT04820922T patent/ATE433453T1/en not_active IP Right Cessation
- 2004-12-23 ES ES09075214T patent/ES2387596T3/en not_active Expired - Lifetime
- 2004-12-23 CN CN201510129906.8A patent/CN104788459A/en active Pending
- 2004-12-23 KR KR1020127011222A patent/KR101268704B1/en not_active Expired - Fee Related
- 2004-12-23 ES ES04820922T patent/ES2326225T3/en not_active Expired - Lifetime
- 2004-12-23 EP EP09075214A patent/EP2088150B1/en not_active Expired - Lifetime
- 2004-12-23 BR BRPI0418120-4A patent/BRPI0418120A/en not_active Application Discontinuation
- 2004-12-23 EA EA200601219A patent/EA013182B1/en not_active IP Right Cessation
- 2004-12-23 MY MYPI20085251A patent/MY149041A/en unknown
- 2004-12-27 AR ARP040104891A patent/AR048808A1/en not_active Application Discontinuation
-
2006
- 2006-06-18 IL IL176385A patent/IL176385A/en active IP Right Grant
- 2006-06-22 IS IS8520A patent/IS2775B/en unknown
- 2006-07-10 NO NO20063193A patent/NO336892B1/en not_active IP Right Cessation
-
2007
- 2007-05-03 US US11/744,030 patent/US7514440B2/en not_active Expired - Lifetime
-
2009
- 2009-03-31 US US12/415,525 patent/US8084472B2/en not_active Expired - Lifetime
-
2015
- 2015-09-23 NO NO20151246A patent/NO338804B1/en not_active IP Right Cessation
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006031772A1 (en) * | 2004-09-10 | 2006-03-23 | Cephalon, Inc. | Methods of treating proliferative skin diseases using carbazole derivatives |
| WO2007149451A3 (en) * | 2006-06-19 | 2008-02-21 | Cephalon Inc | Cycloalkanopyrrolocarbazole derivatives and the use thereof as parp, vegfr2 and mlk3 inhibitors |
| US9777059B2 (en) | 2007-11-30 | 2017-10-03 | Genentech, Inc. | Anti-VEGF antibodies |
| US9101645B2 (en) | 2008-10-22 | 2015-08-11 | Genentech, Inc. | Modulation of axon degeneration |
| US9937224B2 (en) | 2008-10-22 | 2018-04-10 | Genentech, Inc. | Modulation of axon degeneration |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8084472B2 (en) | Fused pyrrolocarbazoles | |
| US8044064B2 (en) | Fused pyrrolocarbazoles | |
| HK1096394B (en) | Fused pyrrolocarbazoles and methods for the preparation thereof | |
| MXPA06007302A (en) | Fused pyrrolocarbazoles and methods for the preparation thereof | |
| HK1137439B (en) | Fused pyrrolocarbazoles and methods for the preparation thereof | |
| HK1096096B (en) | Novel fused pyrrolocarbazoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2549628 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 176385 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006501224 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006547325 Country of ref document: JP Ref document number: 548100 Country of ref document: NZ Ref document number: PA/a/2006/007302 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004309397 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200605668 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2004309397 Country of ref document: AU Date of ref document: 20041223 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004820922 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004309397 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067014797 Country of ref document: KR Ref document number: 2052/KOLNP/2006 Country of ref document: IN Ref document number: 200601219 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480041772.4 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004820922 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067014797 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0418120 Country of ref document: BR |