WO2005092912A1 - Progesterone receptor modulators - Google Patents
Progesterone receptor modulators Download PDFInfo
- Publication number
- WO2005092912A1 WO2005092912A1 PCT/EP2005/051265 EP2005051265W WO2005092912A1 WO 2005092912 A1 WO2005092912 A1 WO 2005092912A1 EP 2005051265 W EP2005051265 W EP 2005051265W WO 2005092912 A1 WO2005092912 A1 WO 2005092912A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optionally substituted
- halogen
- compound according
- cyclopropyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
- C07J43/003—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton not condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J43/00—Normal steroids having a nitrogen-containing hetero ring spiro-condensed or not condensed with the cyclopenta(a)hydrophenanthrene skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J5/00—Normal steroids containing carbon, hydrogen, halogen or oxygen, substituted in position 17 beta by a chain of two carbon atoms, e.g. pregnane and substituted in position 21 by only one singly bound oxygen atom, i.e. only one oxygen bound to position 21 by a single bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J53/00—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class
- C07J53/001—Steroids in which the cyclopenta(a)hydrophenanthrene skeleton has been modified by condensation with a carbocyclic rings or by formation of an additional ring by means of a direct link between two ring carbon atoms, including carboxyclic rings fused to the cyclopenta(a)hydrophenanthrene skeleton are included in this class spiro-linked
Definitions
- the present invention relates to the field of contraception, hormone replacement therapy HRT) and therapy of gynaecological disorders, as well as adjuvant therapy in cancer and other diseases.
- the subject invention provides novel progesterone receptor modulating steroids which have both agonistic and antagonistic modulating activities towards the progesterone receptor, processes for their preparation, and their use in therapy.
- Intracellular receptors are a class of structurally related proteins involved in the regulation of gene transcription.
- Steroid receptors are a subset of these receptors, including the progesterone receptor (PR), androgen receptor (AR), estrogen receptor (ER), glucocorticoid receptor (GR) and mineralocorticoid receptor (MR).
- PR progesterone receptor
- AR androgen receptor
- ER estrogen receptor
- GR glucocorticoid receptor
- MR mineralocorticoid receptor
- Progesterone receptor modulators are known to play an important role in the health of women.
- the natural ligand for PR is the steroid hormone progesterone, but synthetic compounds have been made which may also serve as ligands (see e.g. Jones et al U.S. Patent No. 5,688,810).
- Progestagens are currently widely used for hormonal contraception and in HRT. Other important clinical applications of progestagens are treatment of gynaecological disorders (e.g. endometriosis, dysmenorrhea, dysfunctional uterine bleeding, severe premenstrual syndrome), breast cancer, hot flushes and mood disorders, and luteal support during IVF. In addition, they are applied in combination with other hormones and/or other therapies including, without limitation, chemotherapeutic agents such as cytotoxic and cytostatic agents, immunological modifiers such as interferons and interleukins, growth hormones or other cytokines, hormone therapies, surgery and radiation therapy. The current steroidal progestagens have been proven to be quite safe and are well tolerated.
- Antiprogestagens in combination with progestagens are also useful in contraceptive and hormone replacement regimens as described e.g. in WO 99 25360 and WO 97/49407. It would therefore be useful to find compounds which have both progestagenic and antiprogestagenic properties within one molecule.
- WO 99/45022 describes 20-keto-l l ⁇ -arylsteroids which have either antagonistic or agonistic activity towards the progesterone receptor.
- 20-keto-l l ⁇ -arylsteroids which have either antagonistic or agonistic activity towards the progesterone receptor.
- the compounds described in EP 349481 contain a 4-[(3-pyridyl)phenyl] substituent in position 1 l ⁇ and have no substituent in position 16; none of these compounds possesses a cyclopropylcarbonyl or cyclopropenylcarbonyl substituent in position 17, nor a spirocycloalkanone or spirocycloalkenone substituent in position 17.
- the compounds of EP 349481 have antiprogestagenic properties only.
- novel steroid compounds with an (1 l ⁇ )-[4-(aza-aryl)phenyl] substituent in combination with a variety of substituents in positions 16 and 17 show a mixed profile of PR agonist and PR antagonist activity (hereinafter referred to as mixed P/AP profile) within one compound.
- mixed P/AP profile a mixed profile of PR agonist and PR antagonist activity
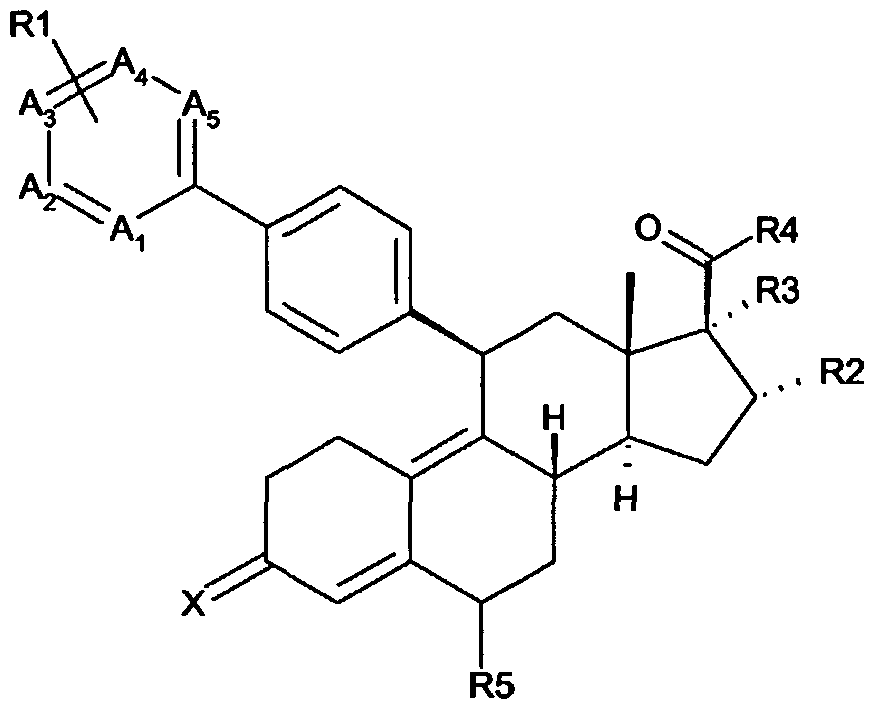
- the subject invention provides a compound of structural Formula I
- X is O, NOH, NO(l-4C)alkyl, NO(l-4C)acyl
- A1-A5 are C, substituted with RI, or N, provided that at least one and not more than three of Al-A5 are N; or one or two of Al, A2 and A5 are N, and the others are C, substituted with RI, and A3 and A4 together represent a fused benzo ring or a fused five- or six-membered nitrogen- containing aromatic ring, both optionally substituted with one or more halogen and/or (l-4C)alkyl; each RI is independently selected from H, halogen, (l-4C)alkyl and (l-4C)alkoxy; R2 is H, (1-4C) alkyl or (1-6C) alkenyl, both optionally substituted with an (6-10C)aryl group, which is optionally substituted with one or more halogen and/or (l-4C)alkyl; and R3 is H or (l-4C)alkyl, optionally substituted with one or more halogen atoms; and R4 is cycloprop
- R2 together with R3 forms a 3-, 4-, 5- or 6-membered carbocyclic ring; and R4 is cyclopropyl or cyclopropenyl, both optionally substituted with one or more halogen and/or (l-4C)alkyl; or R2 is H or (l-4C)alkyl; and
- R5 is H or (l-4C)aIkyl; or a pharmaceutically acceptable salt and/or hydrate form and/or prodrug thereof .
- A1-A5 are C, substituted with RI, or N, provided that at least one and not more than three of A1-A5 are N.
- one or two of Al, A2 and A5 are N, and the others are C, substituted with RI , and A3 and A4 together represent a fused benzo ring or a fused nitrogen-containing ring, both optionally substituted with halogen and/or (l-4C)alkyl.
- R2 is H, (l-4C)alkyl or (l-6C)alkenyl, both optionally substituted with an (6-10C)aryl group, which is optionally substituted with one or more halogen and/or (1 - 4C)alkyl; and R3 is H or (l-4C)alkyl, optionally substituted with one or more halogen; and R4 is cyclopropyl or cyclopropenyl, both optionally substituted with one or more halogen and/or (l-4C)alkyl.
- R2 together with R3 forms a 3-, 4-, 5- or 6-membered carbocyclic ring; and R4 is cyclopropyl or cyclopropenyl, both optionally substituted with one or more halogen and/or (l-4C)alkyl.
- R2 is H or (l-4C)alkyl; and R3 together with R4 forms a 5-, 6- or 7-membered saturated or unsaturated carbocyclic ring.
- X is O.
- R4 is cyclopropyl.
- Al, A3, A4 and ⁇ 5 are C, substituted with RI, and ⁇ 2 is N.
- R2 is H, (l-4C)alkyl or (l-4C)alkenyl.
- X is O, Al, A3, A4 and A5 are C, substituted with RI, and A2 is N;
- R2 is H, (l-4C)alkyl or (l-4C)alkenyl; and
- R3 is H or (l-4C)alkyl, optionally substituted with one or more halogen; and
- R4 is cyclopropyl; or
- R2 together with R3 forms a 3-, 4-, 5- or 6-membered carbocyclic ring; and R4 is cyclopropyl.
- X is O, Al, A3, A4 and A5 are C; A2 is N; RI is H; R2 is methyl; R3 is H; R4 is cyclopropyl; and R5 is H.
- X is O, Al, A3, A4 and A5 are C; A2 is N; RI is H; R2 is ethenyl; R3 is H; R4 is cyclopropyl; and R5 is H .
- the compounds of the subject invention are envisaged for use in therapy.
- the subject invention provides a pharmaceutical composition comprising a compound of the subject invention and a pharmaceutically acceptable carrier.
- a pharmaceutical composition is envisaged for contraceptioa
- a pharmaceutical composition is envisaged for hormone replacement therapy.
- a pharmaceutical composition is envisaged for the treatment of a gynaecological disorder.
- the subject invention further involves a use of a compound of the subject invention for the manufacture of a medicament
- a use of a compound of the subject invention is for the manufacture of a contraceptive.
- a use of a compound of the subject invention is for the manufacture of a medicament for hormone replacement therapy or for the treatment of a gynaecological disorder.
- the subject invention further provides a method of contraception comprising -idmiriistering a pharmaceutically effective amount of a compound of the subject invention to a subject in need thereof.
- the subject invention further provides a method of treating a gynaecological disorder comprising administering a pharmaceutically effective amount of a compound of the subject invention to a subject in need thereof.
- the carbonyl group at position 17 of the steroid was used as such in this reaction sequence, or was masked in the form of a synthetic equivalent such as an hydroxymethyl group (which at a later moment in the synthesis was oxidized back to a carbonyl).
- a protecting group such as ketal.
- Compounds of Formula ⁇ were oxidized to an epoxide of Formula III using various methods known in the art such as treatment with hydrogen peroxide in the presence of trifluoroacetophenone. Treatment of such an epoxide with (4-bromophenyl)magnesium bromide in the presence of a suitable Cu(I) salt such as copper(I) chloride yielded compounds of Formula IV.
- Compounds of Formula IV were transferred into compounds of Formula V using palladium-mediated cross-coupling reactions such as the Suzuki, Stille or Negishi reactions. Removal of the protecting group of compounds of Formula V using methods known in the art such as, in the case of ketals, aqueous acid afforded compounds of Formula I. Such methods of deprotection can be applied to compounds of Formula IV to give compounds of Formula VT. The latter compounds can be transferred into compounds of Formula I using palladium-mediated cross-coupling reactions.
- An example of a suitable combination of reagents is lithium hexamethyldisilazane as base followed by addition of N-phenyl-bis(trifluoromem-mesuIfo--imide) as triflating agent
- the resulting enol triflates were transferred into compounds of Formula V-TJ using a palladium-mediated carbonylation in the presence of N,O-(-imethylhydroxyl--mine.
- Treatment of compounds of Formula VHI with cyclopropyl-Grignard, cyclopropyllithiate, cyclopropenyl-Grignard, or cyclopropenyllithiate yields compounds of Formula DC.
- Compounds of Formula II where R3 together with R4 forms a 5-membered carbocyclic ring can be prepared using the method described in US Patent No. 5,084,450.
- compounds of Formula II where R3 together with R4 forms a 5-, 6-or 7-membered carbocyclic ring can be prepared from compounds of Formula VII using the method described by Mash, E.A. et al. in J. Org. Chem. 55, 2045 (1990). In this publication the method was applied to transform a ketone into a 6-membered spiro compound.
- This method can be extended to 5- or 7-membered spiro compounds by using 4-iodobutyl tert- butyldimethylsilyl ether or 6-iodohexyl t ⁇ rt-butyldimethylsilyl ether instead of the 5- iodopentyl tert-butyldimethylsilyl ether applied in the publication to prepare a 6-membered spiro compound.
- a compound according to the invention is a compound as defined above in Formula I, a salt thereof, a hydrate thereof and/or a prodrug thereof.
- a compound of the invention contains a nitrogen atom of suitable basicity
- the compound may be used as a free base or as a pharmaceutically acceptable salt.
- pharmaceutically acceptable salt represents those salts which are, within the scope of medical judgement, suitable for use in contact with the tissues of humans and/or animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk ratio.
- salts are well known in the art They may be obtained during the final isolation and purification of the compounds of the invention, or separately by reacting the free base function with a suitable mineral acid such as hydrochloric acid, phosphoric acid, or sulfuric acid, or with an organic acid such as for example ascorbic acid, citric acid, tartaric acid, lactic acid, maleic acid, malonic acid, fumaric acid, glycolic acid, succinic acid, propionic acid, acetic acid, methanesulfonic acid, and the like.
- a suitable mineral acid such as hydrochloric acid, phosphoric acid, or sulfuric acid
- organic acid such as for example ascorbic acid, citric acid, tartaric acid, lactic acid, maleic acid, malonic acid, fumaric acid, glycolic acid, succinic acid, propionic acid, acetic acid, methanesulfonic acid, and the like.
- Prodrugs represent compounds which are rapidly transformed in vivo to the parent compound of the above formula, for example by hydrolysis in the stomach and/or in the blood, metabolism in the liver or other processes known to those skilled in the art. For instance, those skilled in the art will recognize that compounds of Formula I where X is H 2 can be expected to be metabolized to the analogous compounds where X is O, which show activity in vitro even if the prodrug where X is H 2 does not.
- - (l-4C)alkyl is a branched or unbranched alkyl group having 1-4 carbon atoms, for example methyl, ethyl, propyL isopropyl, butyl, sec-butyl or tert-butyl;
- - (l-6C)alkenyl is a branched or unbranched alkenyl group having 1-6 carbon atoms, such as ethenyl, 1-methyl-ethenyl, 2-propenyl, 2-butenyl and the like;
- - (6-10)aryl is a carbocyclic aromatic group having 6-10 carbon atoms, such as phenyL 1- naphthyl or 2-naphthyl;
- - (l-4C)acyl is an alkylcarbonyl group having 1-4 carbon atoms, such as formyl, acetyl or propionyl;
- - aza-aryl means a monocyclic or bicyclic aromatic ring system, in which at least one of the rings contains at least one nitrogen ring atom.
- examples include, but are not limited to, pyridyl, pyrimidinyl, quinolinyl, naphthyridyl and the like;
- - carbocyclic when mentioned in the context of a ring, means that all the atoms constituting the ring are carbon atoms;
- - spirocycloalkane is a substituent consisting of an alkanediyl group of which the two terminal atoms are attached to the same (carbon) atom, thus forming a spiro ring system;
- - spirocycloalkene is a substituent consisting of an alkenediyl group of which the two terminal atoms are attached to the same (carbon) atom, thus forming a spiro ring system;
- - the prefixes (1-4C), (2-4C) etc. have the usual meaning to restrict the meaning of the indicated group to those with 1 through 4, 2 through 4 etc. carbon atoms;
- - halogen refers to fluorine, chlorine, bromine and iodine
- - spirocycloalkanone is a spirocycloalkane ring where one of the carbon atoms is forming a carbonyl group
- - spirocycloalkenone is a spirocycloalkene ring where one of the carbon atoms is forming a carbonyl group.
- the progestagen receptor affinity and efficacy of the compounds according to the invention make them suitable for use in control of fertility and reproduction, e.g. in female contraception, and further for female HRT, the treatment of gynaecological disorders, as components of male contraception and in diagnostic methods focussed on the amount and/or location of progesterone receptors in various tissues.
- the treatment of gynaecological disorders focussed on the amount and/or location of progesterone receptors in various tissues.
- isotopically labelled variants of the compounds according to the invention it can be preferred to make isotopically labelled variants of the compounds according to the invention.
- the compounds of the invention may further be useful for the treatment of endometriosis, menorrhagia, menometrorrhagia, dysmenorrhoea, acne, fibroids, osteoporosis as well as other bone disorders, bone fraction repair, sarcopenia, frailty, skin ageing, female sexual dysfunction, postmenopausal symptoms, atherosclerosis, aplastic anaemia, lipodystrophy, side effects of chemotherapy, tumours (located in e.g. breast, ovary or uterus) and others.
- the compounds of the invention may be administered in conjunction with estrogens, androgens, progestagens, antiprogestagens, and other suitable compounds such as folic acid, vitamins, minerals etc.
- isolated DNA encoding the progesterone receptor gene is expressed in suitable host cells.
- a cell might be the Chinese Hamster Ovary (CHO) cell, but other cells are also suitable.
- the cells are of mammalian origin. Methods to construct recombinant progesterone receptor-expressing cell lines are well known in the art (Sambrook et al., Molecular Cloning: a Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, latest edition). Expression of receptor is attained by expression of the DNA encoding the desired protein.
- Portions or all of the DNA encoding the desired protein can be constructed synthetically using standard solid phase techniques, preferably to include restriction sites for ease of ligation.
- Suitable control elements for transcription and translation of the included coding sequence can be provided through the DNA coding sequences.
- expression systems are now available which are compatible with a wide variety of hosts, including prokaryotic hosts such as bacteria and eukaiyotic hosts such as yeast, plant cells, insect cells, mammalian cells, avian cells and the like.
- Cells expressing the receptor are then contacted with a compound of the invention to observe binding, or stimulation or inhibition of a functional response.
- isolated cytosol containing the expressed receptor may be used to measure binding of a compound of the invention.
- radioactive or fluorescence-labelled compounds may be used.
- reference compound the native hormone, or other compounds binding to the receptor, can be used.
- competition binding assays can be performed as well.
- Another assay involves screening for progesterone receptor mixed agonist/antagonist compounds of the invention by determining regulation of receptor-mediated natural target gene mRNA, i.e. genes regulated by the receptor through binding of the receptor in the promoter region of the gene. The levels of target gene mRNA will be reduced or increased, depending on the inhibitory or stimulating effect of a compound of the invention upon binding to the receptor.
- cells can be used which in addition to transfection with receptor encoding DNA have also been transfected with a second DNA encoding a reporter gene, the expression of which responds to binding of the receptor towards responsive elements in the promoter of the particular reporter gene.
- responsive elements might be classical hormone-responsive elements, well known in the art and described e.g. in Beato, M, Chalepakis, G, Schauer, M, Slater, EP J. Steroid Biochem. 5 (1989)737-47 or might be constructed in such a way that they are connected to novel responsive elements.
- reporter gene expression might be controlled by any response element reacting to progesterone receptor binding. Suitable reporter genes are e.g. LacZ, alkaline phosphatase, firefly luciferase and green fluorescence protein.
- testing in the agonistic mode must result in an intrinsic activity of between about 15% and about 85% of the maximal activity when (16 ⁇ )-16-ethyl-21- hydroxy-19-norpregn-4-ene-3,20-dione is used as a reference. Moreover, this maximal agonistic activity should be reached at a concentration of 10 "6 or less, and preferably at a concentration of 10 "8 or less.
- testing must result in an intrinsic activity of between about 85% and about 15% of the maximal activity when (6 ⁇ ,l 1 ⁇ ,17 ⁇ )-l l-[4-(dimethylamino)phenyl]- 4 ⁇ 5'-dihydro-6-methylspiro[estia-4,9-diene-17,2'(3'H)-furan]-3-one is used as a reference.
- IC 5 0 value which must be ⁇ 10 "6 M, preferably ⁇ 10 "8 M. It will be understood by those skilled in the art that for the present invention compounds with a mixed P/AP profile are understood to have a profile ranging from a combination of minimal intrinsic agonistic activity of about 15% and maximal intrinsic antagonistic activity of about 85% to a combination of maximal intrinsic agonistic activity of about 85% and minimal intrinsic antagonistic activity of about 15%. Those skilled in the art will also recognize that, due to the biological variation in the assay, it is not always necessarily the case that the intrinsic agonistic activity and the intrinsic antagonistic activity add up to exactly 100%.
- EC50 and ICso values are dependent on the compound of the invention which is being tested.
- a compound with an EC 50 which is less than 10 "6 M is, generally, considered a candidate for drug selection.
- this value is lower than 10 "8 M.
- a compound which has a higher EC 50 and or IC50,, but has a suitable selectivity (or a combination of agonistic and antagonistic selectivity) for the particular receptor may still be a candidate for drug selection.
- any transactivation assay in mammalian cells that can yield information about the possible receptor activation can be used for the purpose of selecting potent and suitable hgands.
- the added value of using several cell systems, with cells which originate from different organs, will be that information on the potential tissue specificity of the Hgands is obtained.
- examples of cells frequently used to this end are, besides CHO cells, e.g. T47D cells, MCF7 cells, ECC-1 cells, HeLa cells, primary cultures of endometrial cells, and pituitary cells.
- Suitable routes of administration for the compounds of the subject invention are oral, rectal, nasal, topical (including tra-isdermal, buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) administration or administration via an implant.
- the compounds can be administered orally.
- the exact dose and regimen of administration of the active ingredient, or a pharmaceutical composition thereof will necessarily be dependent upon the therapeutic effect to be achieved (e.g. contraception, HRT, endometriosis) and may vary with the particular compound, the route of --dministratio--, and the age and condition of the individual subject to whom the medicament is to be administered.
- parenteral administration requires lower dosages than other methods of administration which are more dependent upon adsorption.
- a dosage for humans is likely to contain 0.0001-25 mg per kg body weight.
- the desired dose may be presented as one dose or as multiple sub-doses -idministered at appropriate intervals throughout the day, or, in case of female recipients, as doses to be administered at appropriate (daily) intervals throughout the menstrual cycle.
- the present invention thus also relates to pharmaceutical compositions comprising a compound according to Formula I in adinixture with pharmaceutically acceptable auxiliaries, and optionally other therapeutic agents.
- the auxiliaries must be "acceptable” in the sense of being compatible with the other ingredients of the composition and not deleterious to the recipients thereof.
- compositions include those suitable for oral, rectal, nasal, topical (including transdermal, buccal and sublingual), vaginal or parenteral (including subcutaneous, intramuscular, intravenous and intradermal) --dministration or --dministration via an implant
- the compositions may be prepared by methods known in the art of pharmacy, for example, using methods such as those described in Gennaro et al., Remington's Pharmaceutical
- auxiliary agent(s) also named accessory ingredients
- auxiliary agent(s) include those conventional in the art (Gennaro, supra), such as carriers, fillers, binders, diluents, disintegrants, lubricants, colorants, flavouring agents, anti-oxidants, and wetting agents.
- compositions suitable for oral administration may be presented as discrete dosage units such as pills, tablets, dragees or capsules, or as a powder or granules, or as a solution or suspension.
- the active ingredient may also be presented as a bolus or paste.
- the compositions can further be processed into a suppository or enema for rectal --dministration.
- compositions as hereinbefore described, in combination with packaging material, including instructions for the use of the composition for the use as hereinbefore described.
- suitable compositions include aqueous and non-aqueous sterile injection.
- the compositions may be presented in unit-dose or multi-dose containers, for example sealed vials and ampoules, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of sterile liquid carrier, for example water, prior to use.
- Compositions or formulations suitable for administration by nasal inhalation include fine dusts or mists which may be generated by means of metered dose pressurized aerosols, nebulisers or insufflators.
- the compounds of the invention can also be administered in the form of devices consisting of a core of active material, encased by a release rate-regulating membrane.
- Such implants are to be applied subcutaneously or locally, and will release the active ingredient at an approximately constant rate over relatively large periods of time, for instance from weeks to years.
- Methods for the preparation of implantable pharmaceutical devices as such are known in the art, for example as described in EP 303,306.
- the compounds of the invention can also be administered in the form of a vaginal ring such as described for example in EP 876815.
- the compounds of the invention can be produced by various methods known in the art of organic chemistry in general. More specifically the routes of synthesis as illustrated in the previous and following schemes and examples can be used. In the schemes and examples the following abbreviations are used:
- SiO 2 silicon dioxide (silica gel)
- Na 2 SO sodium sulfate
- MgSO 4 magnesium sulfate
- Triethylamine (221 mL, 1.59 mol), triphenylphosphine (6.67 g, 25 mmol) and N,O- dimethylhydroxyl--mine.HCl ( 82.2 g, 843 mmol) were added to a solution of 17- [[(trifluoromethyl)sulfonyl]oxy]estra-5(10),9(l l),16-trien-3-one cyclic 1,2-ethanediyl acetal (70.9 g, 159 mmol) in DMF (1.5 L).
- a grain of iodine was added to magnesium (8.4 g, 350 mmol) and heated for 1 minute.
- a solution of 1,4-dibromobenzene (85.1 g, 350 mmol) and a few drops of 1,2-dibromoethane in THF (400 mL) was added dropwise under a nitrogen atmosphere while the temperature was kept at 45 °C.
- reaction mixture was stirred for 2 hours at 100 °C and then cooled to room temperature. Water was added and the mixture was extracted three times with ethyl acetate. The combined organic layers were washed with brine, dried (MgSO ) and evaporated to dryness.
- reaction mixture was cooled to -78 °C and a solution of pyrida-rine (452 ⁇ L, 6.3 mmol) and tributyltin chloride (1.9 mL, 6.9 mmol) were added simultaneously while the temperature was kept below -70 °C.
- the reaction mixture was stirred for 2 hours at -78 °C; subsequently, a saturated aqueous NFLCl solution was added and the reaction mixture was extracted three times with ethyl acetate. The combined organic layers were dried (MgSO 4 ) and evaporated to dryness.
- Example 17 Preparation of (ll ⁇ ,16 ⁇ ,17 ⁇ )-17-cycIopropylcarbonyI-16-cthyl-ll-[4-(6-mcthoxy- pyridin-3-yl)phenyI] -estra-4,9-dien-3-one (16 ⁇ ,17 ⁇ )-17-(Cyclopropylcarbonyl)-16-ethylestra-5(10),9(l l)-dien-3-one cychc 1,2- ethanediyl acetal was transformed into crude title compound using the procedures described in example 1 steps e, f, g and h using 6-methoxy-3-pyridinylboronic acid in the last step. Purification by column chromatography gave the title compound.
- step g (5 ⁇ ,l l ⁇ ,17 ⁇ )-l l-(4- bromophenyl)- 17-(cyclopropylcarbonyl)-5-hydroxyestr-9-en-3-one cyclic 1 ,2-ethanediyl acetal was transformed into the title compound (65% yield).
- J NMR (400 MHz, CDC1 3 ): ⁇ 0.27 (s, 3H), 0*83-2.83 (m, 22H), 4.34 (d, J 7 Hz, IH), 5.79 (s, IH), 7.02-7.07 (m, 2H), 7.37-7.42 (m, 2H).
- the progestagenic activity of a compound of the invention was determined in an in vitro bioassay of Chinese hamster ovary (CHO) cells as described by W.G.E.J. Schoonen et al. (Anal. Biochem. 261 (1998), 222-224).
- the antiprogestagenic activity of a compound of the invention was determined in a setting comparable to the agonistic assay described above, by the inhibition of the transactivation via the progesterone receptor-B of the enzyme luciferase in the presence of 0.1 nM of the inducer (16 ⁇ )-16-ethyl-21-hydroxy-19-norpregn-4-ene- 3,20-dione.
- the efficacy of the antagonistic effect was expressed as the percentage of the effect produced by a standard antagonist, (6 ⁇ ,ll ⁇ ,17 ⁇ )-ll-[4-(dimethylamino)phenylj-4',5'- d-hydro-6-methyl- ⁇ ho[e-rtra-4,9-diene-17,2 3'lT)-fur--n]-3-one.
- Agonistic hgands do not inhibit transactivation of luciferase activity produced by the inducer, whereas strong and weak antiprogestagens as well as compounds with a mixed progestagenic/antiprogestagenic profile can inhibit transactivation dependent on the dose level used of the antiprogestagen or mixed-profile compound in question.
- the EC50 determined is more or less absolute and depends on the intrinsic property of the tested compound itself, however, the IC50 depends on the amount and agonistic EC50 of the inducer as well as on the intrinsic property of the tested compound itself.
- a relatively strong antagonist will be able to produce a measurable IC50 whereas a relatively weak antagonist may fail to produce a detectable result.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Reproductive Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Endocrinology (AREA)
- Gynecology & Obstetrics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Steroid Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description





Claims
Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NZ549862A NZ549862A (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| DE602005003564T DE602005003564T2 (en) | 2004-03-25 | 2005-03-18 | PROGESTERONE RECEPTOR MODULATORS |
| CA2560694A CA2560694C (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| US10/594,103 US7691836B2 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| PL05733590T PL1735330T3 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| AU2005225560A AU2005225560B2 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| HK07104070.8A HK1096405B (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| SI200530144T SI1735330T1 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| JP2007504406A JP4922918B2 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulator |
| DK05733590T DK1735330T3 (en) | 2004-03-25 | 2005-03-18 | progesterone receptor |
| MXPA06010906A MXPA06010906A (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators. |
| EP05733590A EP1735330B1 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
| BRPI0509153-5A BRPI0509153A (en) | 2004-03-25 | 2005-03-18 | compound, pharmaceutical composition, use of a compound, and methods of contraception, and treatment of a gynecological disorder |
| IL178010A IL178010A0 (en) | 2004-03-25 | 2006-09-11 | Progesterone receptor modulators |
| NO20064822A NO20064822L (en) | 2004-03-25 | 2006-10-24 | Progesterone receptor modulators |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US55621004P | 2004-03-25 | 2004-03-25 | |
| EP04101241 | 2004-03-25 | ||
| EP04101241.0 | 2004-03-25 | ||
| US60/556,210 | 2004-03-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005092912A1 true WO2005092912A1 (en) | 2005-10-06 |
Family
ID=34928929
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/051265 Ceased WO2005092912A1 (en) | 2004-03-25 | 2005-03-18 | Progesterone receptor modulators |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US7691836B2 (en) |
| EP (1) | EP1735330B1 (en) |
| JP (1) | JP4922918B2 (en) |
| KR (1) | KR20070028355A (en) |
| CN (1) | CN100549023C (en) |
| AR (1) | AR048330A1 (en) |
| AU (1) | AU2005225560B2 (en) |
| BR (1) | BRPI0509153A (en) |
| CA (1) | CA2560694C (en) |
| CY (1) | CY1107132T1 (en) |
| DE (1) | DE602005003564T2 (en) |
| DK (1) | DK1735330T3 (en) |
| EC (1) | ECSP066881A (en) |
| ES (1) | ES2297699T3 (en) |
| IL (1) | IL178010A0 (en) |
| LV (1) | LV13527B (en) |
| MX (1) | MXPA06010906A (en) |
| MY (1) | MY143074A (en) |
| NO (1) | NO20064822L (en) |
| NZ (1) | NZ549862A (en) |
| PE (1) | PE20060094A1 (en) |
| PL (1) | PL1735330T3 (en) |
| PT (1) | PT1735330E (en) |
| RU (1) | RU2381232C2 (en) |
| SI (1) | SI1735330T1 (en) |
| TW (1) | TW200531977A (en) |
| UA (1) | UA88782C2 (en) |
| WO (1) | WO2005092912A1 (en) |
| ZA (1) | ZA200607801B (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016090139A1 (en) * | 2014-12-03 | 2016-06-09 | Evestra, Inc. | Combination of estrogens plus antiprogestins with significant partial agonistic effect as an effective treatment of menopausal symptoms and for prevention of the occurrence of breast cancer |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD290198A5 (en) * | 1989-09-18 | 1991-05-23 | Zi Fuer Mikrobiologie Und Experimentelle Therapie Jena,De | PROCESS FOR PREPARING 11 BETA-ARYL SUBSITSTATED ESTRA-4,9-DIEN-3-ON-17 (S) -SPIRO-1'-CYCLOHEXAN-2'-ONES AND 11 BETA-ARYL SUBSITSTATED ESTRA-4,9-DIEN-3 -ON-17 (S) -SPIRO-1'-CYCLOHEXAN-2'-OLEN AND THEIR DERIVATIVES |
| WO1999045022A1 (en) * | 1998-03-06 | 1999-09-10 | Research Triangle Institute | 20-KETO-11β-ARYLSTEROIDS AND THEIR DERIVATIVES HAVING AGONIST OR ANTAGONIST HORMONAL PROPERTIES |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE290198C (en) | ||||
| GB8513723D0 (en) | 1985-05-31 | 1985-07-03 | Erba Farmitalia | 11-beta substituted steroids |
| IE60780B1 (en) | 1987-01-23 | 1994-08-10 | Akzo Nv | New 11-aryl steroid derivatives |
| EP0289073B1 (en) | 1987-04-24 | 1991-11-27 | Akzo N.V. | Novel 11-aryloestrane and 11-arylpregnane derivatives |
| EP0303306B1 (en) | 1987-08-08 | 1993-03-10 | Akzo N.V. | Contraceptive implant |
| DE3822770A1 (en) * | 1988-07-01 | 1990-01-04 | Schering Ag | 13-ALKYL-11SS-PHENYLGONANE |
| FR2658516B1 (en) | 1990-02-22 | 1992-06-12 | Roussel Uclaf | NEW STEROUID PRODUCTS COMPRISING A RADICAL SPIRO IN POSITION 17, THEIR PREPARATION METHOD AND THE INTERMEDIATES THEREOF, THEIR APPLICATION AS MEDICAMENTS AND THE PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| ZA929315B (en) | 1991-12-20 | 1993-05-24 | Akzo Nv | 17-spirofuran-3'-ylidene steroids. |
| CA2100514C (en) | 1992-07-29 | 2005-03-29 | Johannes A. M. Hamersma | 17-spiromethylene steroids |
| US5696130A (en) | 1994-12-22 | 1997-12-09 | Ligand Pharmaceuticals Incorporated | Tricyclic steroid receptor modulator compounds and methods |
| NZ332894A (en) | 1996-06-25 | 1999-05-28 | Akzo Nobel Nv | A regimen containing progestogen and/or estrogen with two or more anti-progestogen dosage units |
| IL123813A0 (en) | 1997-04-11 | 1998-10-30 | Akzo Nobel Nv | Drug delivery system for two or more active substances |
| TR200001334T2 (en) | 1997-11-14 | 2000-09-21 | Akzo Nobel N.V. | Progestogen-anti-progestogen regimens. |
| US6172052B1 (en) * | 1998-12-04 | 2001-01-09 | Research Triangle Institute | 17β-acyl-17α-propynyl-11β-arylsteroids and their derivatives having agonist or antagonist hormonal properties |
-
2005
- 2005-03-15 TW TW094107888A patent/TW200531977A/en unknown
- 2005-03-18 US US10/594,103 patent/US7691836B2/en active Active
- 2005-03-18 RU RU2006137578/04A patent/RU2381232C2/en not_active IP Right Cessation
- 2005-03-18 DE DE602005003564T patent/DE602005003564T2/en not_active Expired - Lifetime
- 2005-03-18 SI SI200530144T patent/SI1735330T1/en unknown
- 2005-03-18 CA CA2560694A patent/CA2560694C/en not_active Expired - Fee Related
- 2005-03-18 MX MXPA06010906A patent/MXPA06010906A/en active IP Right Grant
- 2005-03-18 NZ NZ549862A patent/NZ549862A/en unknown
- 2005-03-18 CN CNB2005800096126A patent/CN100549023C/en not_active Expired - Fee Related
- 2005-03-18 DK DK05733590T patent/DK1735330T3/en active
- 2005-03-18 EP EP05733590A patent/EP1735330B1/en not_active Expired - Lifetime
- 2005-03-18 JP JP2007504406A patent/JP4922918B2/en not_active Expired - Fee Related
- 2005-03-18 BR BRPI0509153-5A patent/BRPI0509153A/en not_active IP Right Cessation
- 2005-03-18 WO PCT/EP2005/051265 patent/WO2005092912A1/en not_active Ceased
- 2005-03-18 ES ES05733590T patent/ES2297699T3/en not_active Expired - Lifetime
- 2005-03-18 PT PT05733590T patent/PT1735330E/en unknown
- 2005-03-18 PL PL05733590T patent/PL1735330T3/en unknown
- 2005-03-18 KR KR1020067021801A patent/KR20070028355A/en not_active Ceased
- 2005-03-18 UA UAA200609904A patent/UA88782C2/en unknown
- 2005-03-18 AU AU2005225560A patent/AU2005225560B2/en not_active Ceased
- 2005-03-22 PE PE2005000328A patent/PE20060094A1/en not_active Application Discontinuation
- 2005-03-23 AR ARP050101145A patent/AR048330A1/en unknown
- 2005-03-24 MY MYPI20051276A patent/MY143074A/en unknown
-
2006
- 2006-09-11 IL IL178010A patent/IL178010A0/en unknown
- 2006-09-18 ZA ZA200607801A patent/ZA200607801B/en unknown
- 2006-09-25 EC EC2006006881A patent/ECSP066881A/en unknown
- 2006-10-24 NO NO20064822A patent/NO20064822L/en not_active Application Discontinuation
- 2006-10-24 LV LVP-06-120A patent/LV13527B/en unknown
-
2008
- 2008-01-10 CY CY20081100031T patent/CY1107132T1/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DD290198A5 (en) * | 1989-09-18 | 1991-05-23 | Zi Fuer Mikrobiologie Und Experimentelle Therapie Jena,De | PROCESS FOR PREPARING 11 BETA-ARYL SUBSITSTATED ESTRA-4,9-DIEN-3-ON-17 (S) -SPIRO-1'-CYCLOHEXAN-2'-ONES AND 11 BETA-ARYL SUBSITSTATED ESTRA-4,9-DIEN-3 -ON-17 (S) -SPIRO-1'-CYCLOHEXAN-2'-OLEN AND THEIR DERIVATIVES |
| WO1999045022A1 (en) * | 1998-03-06 | 1999-09-10 | Research Triangle Institute | 20-KETO-11β-ARYLSTEROIDS AND THEIR DERIVATIVES HAVING AGONIST OR ANTAGONIST HORMONAL PROPERTIES |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016090139A1 (en) * | 2014-12-03 | 2016-06-09 | Evestra, Inc. | Combination of estrogens plus antiprogestins with significant partial agonistic effect as an effective treatment of menopausal symptoms and for prevention of the occurrence of breast cancer |
| US9850273B2 (en) | 2014-12-03 | 2017-12-26 | Evestra, Inc. | Combination of estrogens plus antiprogestins with significant partial agonistic effect as an effective treatment of menopausal symptoms and for prevention of the occurrence of breast cancer |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6130087B1 (en) | 17-Hydroxy-17-pentafluoroethyl-estradi-4,9 (10) -dien-11-aryl derivatives, process for producing the same, treatment of various diseases using the derivatives | |
| JP2911553B2 (en) | 11β-Aryl-4-estrene, a process for preparing the same, an intermediate product and a pharmaceutical compound containing an anti-gestagen and anti-glucocorticoid action containing a novel compound | |
| AU736064B2 (en) | 16-hydroxy-11-(substituted phenyl)-estra-4,9-diene derivatives | |
| US9085603B2 (en) | Progesterone receptor antagonists | |
| JP5809172B2 (en) | Progesterone receptor antagonist | |
| CN102596985B (en) | 17-Hydroxy-17-Pentafluoroethyl-Estro-4,9(10)-Diene-11-aryl Derivatives, Preparation Process and Use for Treating Diseases | |
| JP5731498B2 (en) | 17-Hydroxy-17-pentafluoroethyl ester-4,9 (10) -diene 11-ethynylphenyl derivatives, process for their preparation and their use for the treatment of diseases | |
| US7691836B2 (en) | Progesterone receptor modulators | |
| HK1096405B (en) | Progesterone receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005733590 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 178010 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006501786 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 549862 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/07801 Country of ref document: ZA Ref document number: 200607801 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2560694 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 06095222 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/010906 Country of ref document: MX Ref document number: 2007504406 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10594103 Country of ref document: US Ref document number: 2007203105 Country of ref document: US Ref document number: 3513/CHENP/2006 Country of ref document: IN Ref document number: 200580009612.6 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005225560 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200601712 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067021801 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 06120 Country of ref document: LV Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200620060120 Country of ref document: LV |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006137578 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2005225560 Country of ref document: AU Date of ref document: 20050318 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005225560 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005733590 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067021801 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 10594103 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0509153 Country of ref document: BR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2005733590 Country of ref document: EP |