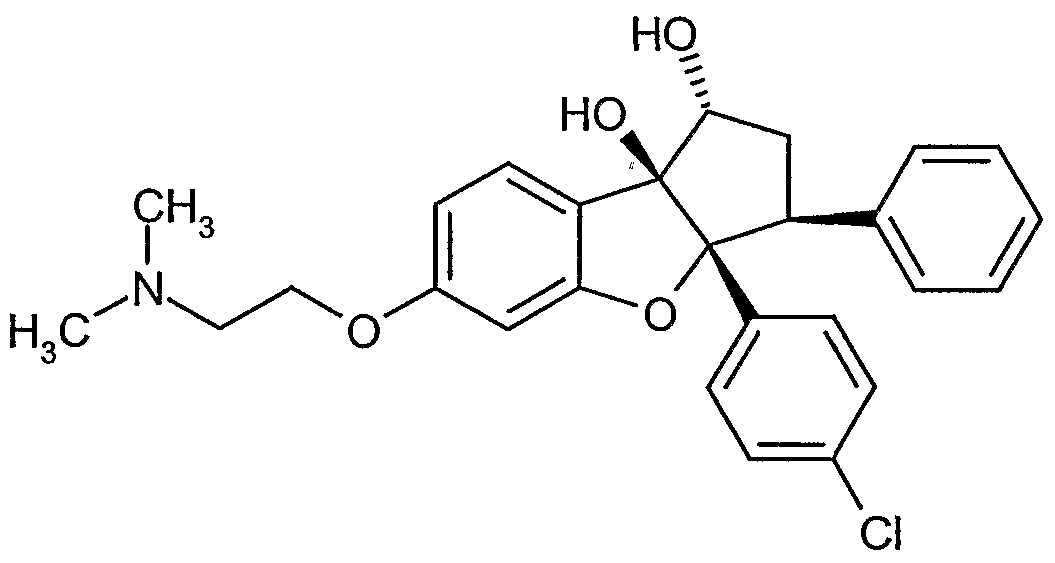
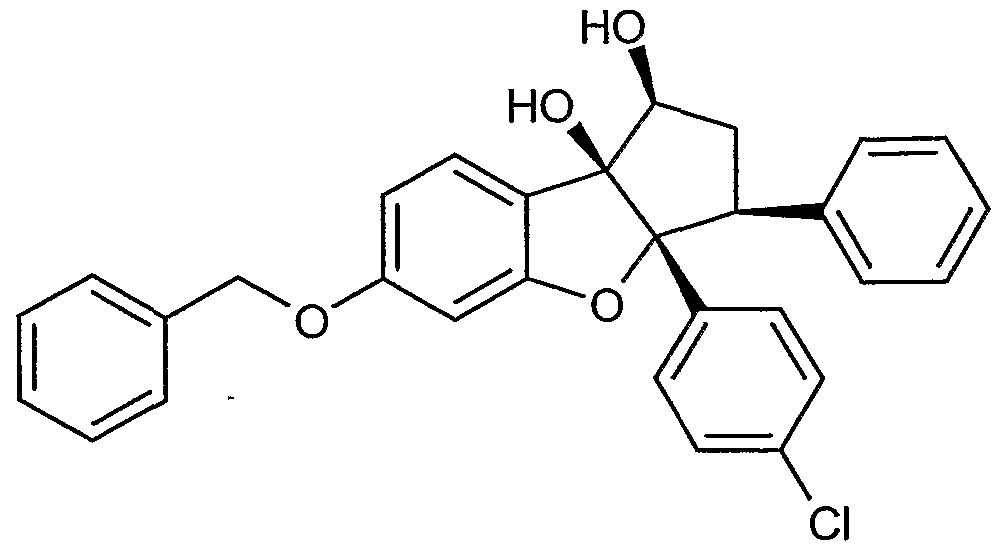
Neue Cylopentaffrlbenzoftiran-Derivate und ihre Verwendung
Die vorliegende Anmeldung betrifft neue Cyclopenta[b]benzofuran-Derivate, Verfahren zu ihrer Herstellung sowie ihre Verwendung zur Herstellung von Arzneimitteln, insbesondere zur Prophylaxe und/oder Therapie akuter oder chronischer Erkrankungen, die gekennzeichnet sind durch erhöhten zellulären Stress, durch lokale oder systemische Entzündungsprozesse oder durch Hyperproliferation.
Die erfindungsgemäßen Verbindungen leiten sich ab von einer als Rocaglaole / Rocaglamide bezeichneten Klasse von Naturstoffen, die aus unterschiedlichen Arten der Pflanze Aglaia extrahiert werden können. Seit der Erst-Isolation eines als Rocaglamid benannten Dihydro- cyclopentabenzofuranol-Derivates (J. Chem. Soc, Chem. Commun. 1982, 1150; US 4,539,414) sind mehrere neue, auch synthetisch hergestellte Derivate und deren biologische Wirkung beschrieben worden (vgl. z.B. J. Chem. Soc, Chem. Commun. 1991, 1137; Phytochemistry 32, 307 (1993); WO 96/04284; Tetrahedron 52, 6931 (1996); Phytochemistry 44, 1455 (1997); Phytochemistry 45, 1579 (1997); Tetrahedron 53, 17625 (1997); JP 11012279; WO 97/08161; WO 00/07579; WO 00/08007; DE 199 34 952-A1).
Die Wirkung von Cyclopentabenzofuran-Derivaten als Inhibitoren der "Nuclear Factor kappa B" (NF-κB)-vermittelten Signalübertragung wurde bereits beschrieben [WO 00/08007; WO 00/07579; J. Biol. Chem. 277, 44791 (2002)]. NF-κB ist ein Transkriptionsfaktor, der eine zentrale Rolle bei Entzündungsprozessen, aber auch bei der Cancerogenese innehat. In seiner aktiven, DNA- bindenden Form setzt er sich aus dimeren Kombinationen unterschiedlicher Mitglieder der NF- κB/Rel-Familie von Proteinen zusammen [Ann. Rev. Immunol. 16, 225 (1998)]. Unter basalen, nicht-stimulierten Bedingungen liegt NF-κB durch Bindung an ein inhibitorisches Protein (I-κB) als zytoplasmatische, inaktive Form vor. Nach Stimulation kommt es zur schnellen Phos- phorylierung von I-κB durch I-κB-Kinasen und infolgedessen zum proteolytischen Abbau von I- KB. Dadurch wird NF-κB in seiner aktiven Form freigesetzt und dessen Translokation in den Zellkern ermöglicht. In seiner Eigenschaft als Transkriptionsfaktor aktiviert oder moduliert NF-κB die Expression verschiedener Gene, im besonderen solcher, deren Produkte für entzündliche Reaktionen und für Zellwachstum und -differenzierung verantwortlich sind [J. Biol. Chem. 274, 27339 (1999)].
Überraschenderweise wurde nun gefunden, dass die erfmdungsgemäßen Verbindungen außerdem die Aktivität eines zweiten Transkriptionsfaktor-Komplexes, des "Activator Protein-1" (AP-1), inhibieren. AP-1 ist ein aus Dimeren der Jun-, Fos-, Maf- und ATF-Familie von Proteinen zusammengesetzter Transkriptionsfaktor, der im Zellkern lokalisiert ist. Die Aktivität von AP-1
wird über eine Reihe unterschiedlichster Stimuli induziert, unter anderem durch Zytokine, bakterielle und virale Infektionen und durch verschiedene Formen von physikalischem oder chemischem Stress. Aktivierende Signale führen einerseits zu einer vermehrten Bildung der Einzelkomponenten des Transkriptionsfaktors, andererseits durch Stimulation bestimmter Kinasen, wie z.B. Jun-Kinasen, zur Phosphorylierung spezifischer Aminosäuren. Beide Prozesse fuhren zu einer verstärkten Wechselwirkung von AP-1 mit dessen Zielgenen und ermöglichen so deren Expression oder Modulation. Zu diesen gehören neben Genen, deren Produkte in Entzündungsprozessen involviert sind, auch solche, die die Zellteilung steuern oder als Regulatoren von Zelltod oder -überleben wirken [Curr. Opin. CellBiol. 9, 240 (1997); Nature CellBiol. 4, E131 (2002)].
Einerseits sind pro-inflammatorische Zytokine, wie z.B. Interleukin-1 (IL-1) oder Tumor Nekrose Faktor (TNF), und oxidativer Stress potente Aktivatoren der NF-κB- und AP- 1 -vermittelten Signalübertragung. Andererseits bewirkt die Aktivierung von NF-κB und/oder AP-1 die Neubildung verschiedener Zytokine (wie z.B. IL-1 und TNF), unterschiedlicher Chemokine (wie z.B. Interleukin-8 (IL-8) und "Monocyte Chemoattractant Protein-1" (MCP-1)) und verschiedener Enzyme (wie z.B. Cyclooxygenase-2 oder "Nitric Oxide Synthase-2" (NOS-2, iNOS)). Die Hauptfunktion der neugebildeten Peptide/Proteine oder der durch die Aktivität der neugebildeten Enzyme entstehenden Endprodukte ist die Rekrutierung und Aktivierung von Entzündungszellen. NF-κB und AP-1 sind damit zentrale Faktoren bei der Induktion und Aufrechterhaltung von Entzündungsprozessen.
Die Pathogenese oder Pathophysiologie einer Vielzahl von Erkrankungen ist charakterisiert durch akute, überschießende oder chronische Entzündungsreaktionen, die lokal auf ein Gewebe beschränkt oder systemischer Natur sein können. Diese Krankheiten zeichnen sich durch lokal oder systemisch erhöhte Zytokin- und/oder Chemokinspiegel sowie durch eine erhöhte Präsenz von verschiedenartigen Entzündungszellen aus, wie z.B. Makrophagen, polymorphkernige Leukozyten, T-Lymphozyten oder B-Zellen. Zu diesen Erkrankungen gehören chronische Entzündungsund Autoimmunkrankheiten (wie z.B. die Crohn'sche Krankheit, ulzerative Colitis, rheumatoide Arthritis, Psoriasis, Multiple Sklerose, Lupus, Asthma, Diabetes), kardiovaskuläre Erkrankungen (wie z.B. koronare Herzerkrankungen, Myokard-Infarkt, Artherosklerose, Restenose, Thrombosen), fibrotische Erkrankungen der Leber und anderer Organe, cerebrovaskuläre Erkrankungen (wie z.B. Schlaganfall, Schädel-Hirn-Trauma, Rückenmarksverletzungen) und chronische neuro- degenerative Erkrankungen (wie z.B. die Alzheimer'sche Krankheit, die Parkinson'sche Krankheit, Chorea Huntington, Amyotrophe Lateralsklerose, periphere Neuropathien und chronischer Schmerz). Eine fehlregulierte oder überschießende Zytokin/Chemokin-Bildung ist ebenfalls ursächlich verknüpft mit der Entstehung oder den Folgen von Strahlenschäden, Transplantat- abstoßung, Sepsis und septischem Schock, sowie bakterieller Meningitis. Die Inhibition oder
Modulation der transkriptioneilen Aktivität von NF-κB und/oder AP-1, wie für die erfindungsgemäßen Verbindungen beschrieben, könnte somit ein erfolgsversprechendes neues Therapieprinzip für die zuvor aufgeführten Erkrankungen darstellen.
Neben ihrer zentralen Funktion in Entzündungsprozessen haben NF-κB und AP-1 wesentliche Bedeutung bei der Regulation von Zellteilung, Zellwachstum und Zelldifferenzierung. Bei der Bildung und beim Wachstum von Tumoren werden zelluläre Signalwege aktiviert, die unter normalen Bedingungen Zellwachstum, Differenzierung und andere biologische Vorgänge steuern. Eine Vielzahl Tumor-induzierender Substanzen und Faktoren (wie z.B. Epidermaler Wachstumsfaktor (EGF), Phorbolester, UV-Strahlung) führen zur Aktivierung von NF-κB und/oder AP-1, und eine Reihe der durch NF-κB und/oder AP-1 gesteuerten Gene gehören zu den Oncogenen (wie z.B. c-myc, c-rel, Melanoma Growth Stimulating Activity (MGSA)). Durch ihre inhibitorische/modulatorische Aktivität auf die NF-κB- und/oder AP- 1 -vermittelte Signalübertragung könnte die Verwendung der erfϊndungsgemäßen Verbindungen damit ein neues Therapieprinzip zur Behandlung hyperproliferativer Erkrankungen wie solider Tumoren (wie z.B. Brustkrebs, Lungenkrebs, Tumore des Gehirns und des Nervensystems, Hautkrebs, Leberkrebs, Tumore der Reproduktionsorgane, Tumore des Verdauungstraktes, Blasenkrebs, Tumore der Harnwegssysteme, Tumore verschiedener Hormondrüsen, Tumore des Auges), Lymphomen (wie z.B. Hodgkin'sche Krankheit, Lymphome des zentralen Nervensystems), Sarkomen (wie z.B. Osteaosarkome, Lympho- sarkome) und Leukämien (wie z.B. akute myeloide Leukämie, lymphoblastische Leukämien, myelogene Leukämien) darstellen.
NF-κB und AP-1 spielen außerdem bei der Replikation lymphotropher Viren wie HIV, HTLV und Epstem-Barr-Virus eine wesentliche Rolle. Die Aktivierung viraler, für die Replikation notwendiger Gene kann über die Virus-mediierte Aktivierung von NF-κB und/oder AP-1 in der Wirtszelle bewirkt werden. Neben der Bedeutung für die Vermehrung lymphotropher Viren wird auch eine positive Beeinflussung der Genexpression beim Zytomegalievirus (CMV) wie auch bei Adenoviren durch NF-κB/AP-1 vermutet. Inhibitoren Modulatoren der NF-κB- und/oder AP-1- Aktivität könnten somit auch anti-virale Effekte ausüben.
Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I)
in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C C6)- Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)- Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (C C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)-Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können,
wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino
und
R Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 oder >C=N-OH bilden,
R5 Mono- oder Di-(C C6)-alkylaminocarbonyl bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (d-C6)-Alkyl, (C,-C6)-Alkoxy, (C6-C10)-Aryl, 5- bis 10- gliedriges Heteroaryl oder eine Gruppe der Formel -NR9R10 bedeutet, wobei Aryl und Heteroaryl ihrerseits jeweils ein- bis zweifach, gleich oder verschieden, durch Halogen, Cyano, (Cι-C4)-Alkylsulfonyl oder eine Gruppe der Formel -NR9R10 substituiert sein können, und worin
R9 und R10 unabhängig voneinander für Wasserstoff, (C C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu Rδ befindet und Wasserstoff, Halogen, (Cι-Cö)-Alkyl, (C C6)-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R
n und R
12 unabhängig voneinander für Wasserstoff,
Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
oder
R
6 und R
7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
Weiterer Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)- Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)- Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C )-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)-Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C )-Alkoxy, Amino, Mono- oder Di-(C C4)-alkylamino substituiert sein können,
wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 oder >C=N-OH bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und (C6-Cι0)-Aryl, 5- bis 10-gliedriges Heteroaryl oder eine Gruppe der Formel -NR9R10 bedeutet, wobei Aryl und Heteroaryl ihrerseits jeweils ein- bis zweifach, gleich oder verschieden, durch Halogen, Cyano, (Cι-C4)-Alkylsulfonyl oder eine Gruppe der Formel -NR9R10 substituiert sein können, und worin
R9 und R10 unabhängig voneinander für Wasserstoff, (Cι-C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C6)-Alkyl, (Cι-C6)-Alkoxy oder eine Gruppe der Formel -NRπR12 bedeutet, worin
Rπ und R12 unabhängig voneinander für Wasserstoff, (Cι-C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
oder
R
6 und R
7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
Weiterer Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)- Alkoxy, Amino, Mono- oder Di-(C C6)-alkylammo, (C3-C8)-Cycloalkylammo, N-(C3-C8)- Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein N-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)-Alkoxy, Amino, Mono- oder Di-(C C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(Cι~C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(C C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können,
wobei R1 oder R2 jedoch nicht beide gleichzeitig für Wasserstoff stehen,
R3 Amino
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=N-OH bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C6)-Alkyl oder (Cι-C6)-Alkoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C6)-Alkyl, (Cι-C6)-Alkoxy oder eine Gruppe der Formel -NRπR12 bedeutet, worin
Rπ und R12 unabhängig voneinander für Wasserstoff, (Cι-C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Weiterer Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Ci- )- Alkoxy, Amino, Mono- oder Di-(C C6)-alkylammo, (C3-C8)-Cycloalkylamino, N-(C3-C8)- Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C C6)-Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkγlammo, (C3-C8)-Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(C1-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C )-alkylamino substituiert sein können,
wobei entweder R1 oder R2 für Wasserstoff steht, jedoch nicht beide gleichzeitig Wasserstoff bedeuten,
R3 Hydroxy
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C6)-Alkyl oder (C C6)-Alkoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C6)-Alkyl, (Cι-C6)-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (C C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Weiterer Gegenstand der vorliegenden Erfindung sind Verbindungen der allgemeinen Formel (I), in welcher
R1 Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C6)-Alkoxy, Amino, Mono- oder Di-(C1-C6)-alkylamino, (C3-C8)-Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(C,-C6)-alkyl- amino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sind, wobei Mono- und Di-(Cι-C6)-alkγlammo ihrerseits durch Hydroxy, ( -G -Alkoxy, Amino, Mono- oder Di-(Cι-C )-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3- Position durch (Cj-C6)-Alkoxy, Amino, Mono- oder Di-(Cι-C6)-alkylamino, (C3-C8)- Cycloalkylamino, N-(C3-C8)-Cycloalkyl-N-(Cι-C6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sind, wobei Mono- und Di-(Cι-C6)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können,
R3 Hydroxy
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C6)-Alkyl oder (Cι-C6)-Alkoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (C C6)-Alkyl, (Cι-C6)-Alkoxy oder eine Gruppe der Formel -ΝRπR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (Cι-C6)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
oder
R
6 und R
7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
bilden,
sowie ihre Salze, Solvate und Solvate der Salze.
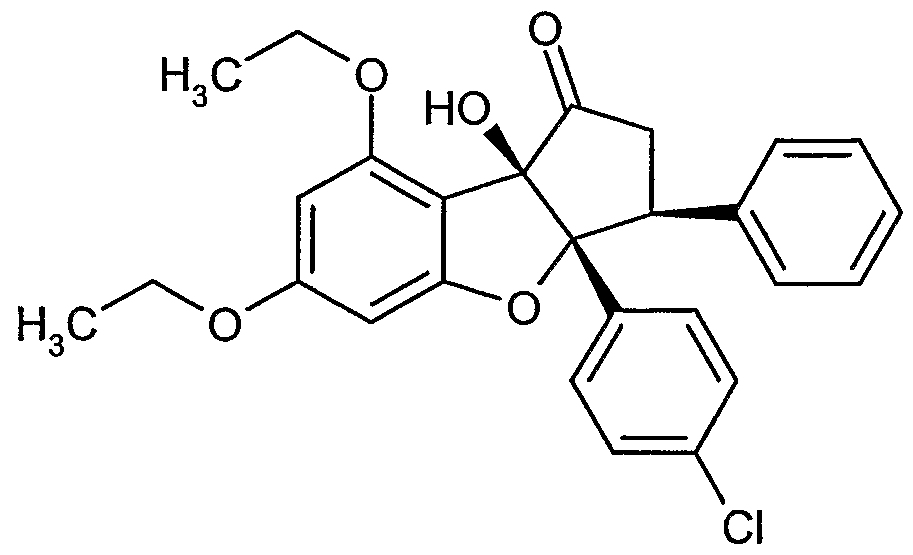
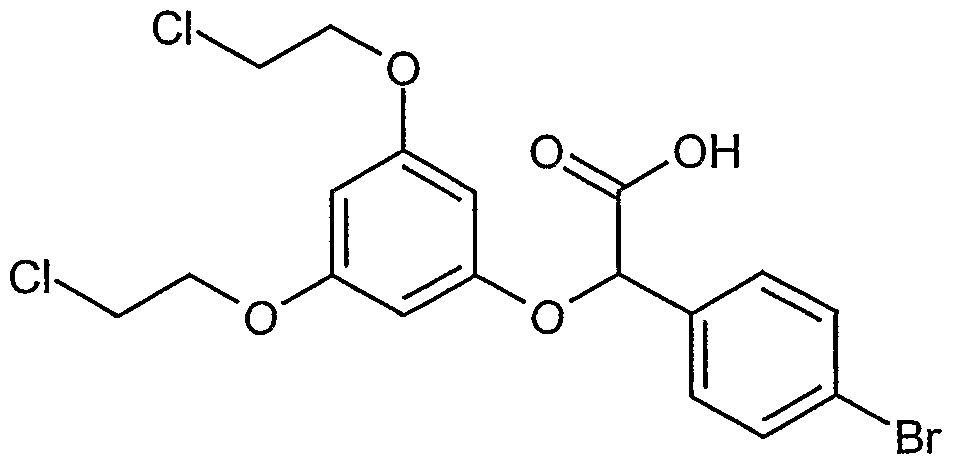
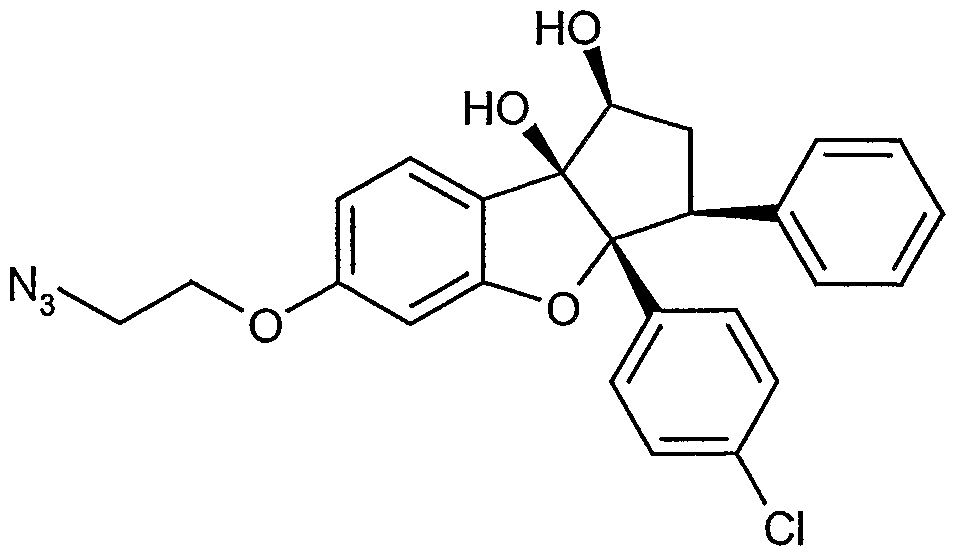
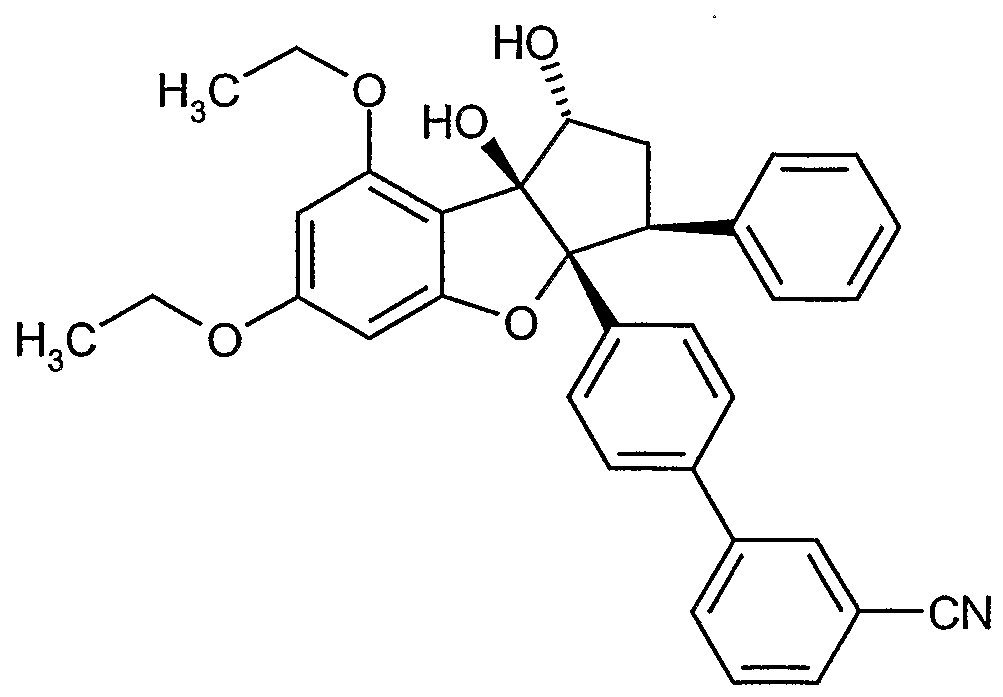
Erfindungsgemäße Verbindungen sind die Verbindungen der Formel (I) und deren Salze, Solvate und Solvate der Salze, die von Formel (I) umfassten Verbindungen der nachfolgend genannten Formeln und deren Salze, Solvate und Solvate der Salze sowie die von Formel (I) umfassten, nachfolgend als Ausführungsbeispiele genannten Verbindungen und deren Salze, Solvate und Solvate der Salze, soweit es sich bei den von Formel (!) umfassten, nachfolgend genannten Verbindungen nicht bereits um Salze, Solvate und Solvate der Salze handelt.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung umfasst deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Sofern die erfindungsgemäßen Verbindungen in tautomeren Formen vorkommen können, umfasst die vorliegende Erfindung sämtliche tautomere Formen.
Als Salze sind im Rahmen der vorliegenden Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt. Umfasst sind auch Salze, die für pharmazeutische Anwendungen selbst nicht geeignet sind, jedoch beispielsweise für die Isolierung oder Reinigung der erfindungsgemäßen Verbindungen verwendet werden können.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoffsäure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure, Ethan- sulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Trifluor- essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumarsäure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen umfassen auch Salze üblicher Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C-Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiisopropylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclohexylamin, Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methyl- morpholin, Arginin, Lysin, Ethylendiamin und N-Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der erfindungsgemäßen Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungsmittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt. Als Solvate sind im Rahmen der vorliegenden Erfindung Hydrate bevorzugt.
Außerdem umfasst die vorliegende Erfindung auch Prodrugs der erfindungsgemäßen Verbindungen. Der Begriff "Prodrugs" umfasst Verbindungen, welche selbst biologisch aktiv oder inaktiv sein können, jedoch während ihrer Verweilzeit im Körper zu erfindungsgemäßen Verbindungen umgesetzt werden (beispielsweise metabolisch oder hydrolytisch).
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
(C Cfi)-Alkyl und (G-G -Alkyl stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkylrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, sec.-Butyl, tert.-Butyl, 1-Ethyl- propyl, n-Pentyl und n-Hexyl.
(C Cs)-Cycloalkyl und (C C≤)-Cvcloalkyl stehen im Rahmen der Erfindung für eine mono- oder gegebenenfalls bicyclische Cycloalkylgruppe mit 3 bis 8 bzw. 3 bis 6 Kohlenstoffatomen. Bevorzugt ist ein monocyclischer Cycloalkylrest mit 3 bis 6 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
( -Cιn)-Aryl steht im Rahmen der Erfindung für einen aromatischen Rest mit vorzugsweise 6 bis 10 Kohlenstoffatomen. Bevorzugte Arylreste sind Phenyl und Naphthyl.
(C C≤)-Alkoxy und ("Cv- VAlkoxy stehen im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkoxyrest mit 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein gerad-
kettiger oder verzweigter Alkoxyrest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methoxy, Ethoxy, n-Propoxy, Isopropoxy und tert.-Butoxy.
Mono-f Cj^-C^-alkylamino und Mono^G-C^-alkylamino stehen im Rahmen der Erfindung für eine Amino-Gruppe mit einem geradkettigen oder verzweigten Alkylsubstituenten, der 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweist. Bevorzugt ist ein geradkettiger oder verzweigter Monoalkyl- amino-Rest mit 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methyl- amino, Ethylamino, n-Propylamino, Isopropylamino und tert.-Butylamino.
D -CfiValkylamino und Di-(C C4. -alkylamino stehen im Rahmen der Erfindung für eine Amino-Gruppe mit zwei gleichen oder verschiedenen geradkettigen oder verzweigten Alkylsubstituenten, die jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatome aufweisen. Bevorzugt sind gerad- kettige oder verzweigte Dialkylamino-Reste mit jeweils 1 bis 4 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: N,N-Dimethylamino, N,N-Diethylamino, N-Ethyl-N-methyl- arnino, N-Methyl-N-n-propylamino, N-Isopropyl-N-n-propylamino, N-tert.-Butyl-N-methylamino, N-Ethyl-N-n-pentylamino und N-n-Hexyl-N-methylamino.
Mono- oder Di-(C
1-Cfi)-alkylaminocarbonyl bzw. Mono- oder DKC^C^-alkylaminocarbonyl stehen im Rahmen der Erfindung für eine Amino-Gruppe, die über eine Carbonylgruppe verknüpft ist und die einen geradkettigen oder verzweigten bzw. zwei gleiche oder verschiedene geradkettige oder verzweigte Alkylsubstituenten mit jeweils 1 bis 6 bzw. 1 bis 4 Kohlenstoffatomen aufweist. Beispielhaft und vorzugsweise seien genannt: Methylaminocarbonyl, Ethylaminocarbonyl, Iso- propylaminocarbonyl, tert.-Butylaminocarbonyl, N,N-Dimethylaminocarbonyl, NN-Diethylamino- carbonyl, N-Ethyl-N-methylaminocarbonyl und N-tert.-Butyl-N-methylaminocarbonyl.

stehen im Rahmen der Erfindung für eine Amino-Gruppe mit einem mono- oder gegebenenfalls bicyclischen Cycloalkyl-Substituenten, der 3 bis 8 bzw. 3 bis 6 Ring-Kohlenstoffatome aufweist. Bevorzugt ist ein monocyclischer Cycloalkyl- Substituent mit 3 bis 6 Ring-Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Cyclopropylamino, Cyclobutylamino, Cyclopentylamino, Cyclohexylamino, Cycloheptylamino und Cyclooctylamino.
(C Cd)-Alkylsulfonyl steht im Rahmen der Erfindung für einen geradkettigen oder verzweigten Alkylsulfonyl-Rest mit 1 bis 4 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkylsulfonyl-Rest mit 1 bis 3 Kohlenstoffatomen. Beispielhaft und vorzugsweise seien genannt: Methylsulfonyl, Ethylsulfonyl, n-Propylsulfonyl, Isopropylsulfonyl, n-Butylsulfonyl und tert.-Butyl- sulfonyl.
Ein 4- bis 7-gliedriger Heterocyclus steht im Rahmen der Erfindung für einen gesättigten oder partiell ungesättigten Heterocyclus mit 4 bis 7 Ringatomen, der ein Ring-Stickstoffatom enthält, über dieses verknüpft ist und ein weiteres Heteroatom aus der Reihe N, O, S, SO oder S02 enthalten kann. Bevorzugt ist ein 4- bis 7-gliedriger gesättigter, N-verknüpfter Heterocyclus, der ein weiteres Heteroatom aus der Reihe N, O oder S enthalten kann. Beispielhaft und vorzugsweise seien genannt: Azetidinyl, Pyrrolidinyl, Pyrrolinyl, Piperidinyl, Piperazinyl, Morpholinyl, Thio- morpholinyl, Azepinyl und 1,4-Diazepinyl.
5- bis 10-gliedriges Heteroaryl steht im Rahmen der Erfindung für einen mono- oder gegebenenfalls bicyclischen aromatischen Heterocyclus (Heteroaromaten) mit bis zu vier gleichen oder verschiedenen Heteroatomen aus der Reihe N, O und/oder S, der über ein Ringkohlenstoffatom oder gegebenenfalls über ein Ringstickstoffatom des Heteroaromaten verknüpft ist. Beispielhaft seien genannt: Furyl, Pyrrolyl, Thienyl, Pyrazolyl, Imidazolyl, Thiazolyl, Oxazolyl, Isoxazolyl, Iso- thiazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Tetrazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl, Benzofuranyl, Benzothienyl, Benzimidazolyl, Benzoxazolyl, Benzothiazolyl, Benzotriazolyl, Indolyl, Indazolyl, Chinolinyl, Isochinolinyl, Naphthyridinyl, Chinazolinyl, Chinoxalinyl. Bevorzugt sind monocyclische 5- oder 6-gliedrige Heteroaryl-Reste mit bis zu drei Heteroatomen aus der Reihe N, O und/oder S wie beispielsweise Furyl, Thienyl, Thiazolyl, Oxazolyl, Isothiazolyl, Isoxazolyl, Pyrazolyl, Imidazolyl, Triazolyl, Oxadiazolyl, Thiadiazolyl, Pyridyl, Pyrimidinyl, Pyridazinyl, Pyrazinyl, Triazinyl.
Halogen schließt im Rahmen der Erfindung Fluor, Chlor, Brom und Iod ein. Bevorzugt sind Fluor, Chlor oder Brom.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach substituiert sein. Im Rahmen der vorliegenden Erfindung gilt, dass für alle Reste, die mehrfach auftreten, deren Bedeutung unabhängig voneinander ist. Eine Substitution mit ein, zwei oder drei gleichen oder verschiedenen Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch ( -C4)- Alkoxy, Amino, Mono- oder Di-(C C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)- Cycloalkyl-N-(C C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können,
wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (C C4)- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (C C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(Cι-C )-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)-Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 6-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (Cι-C )- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino und
R4 Wasserstoff bedeutet oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 oder >C=Ν-OH bilden,
R5 Mono- oder Di-(Cι-C4)-alkylaminocarbonyl bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C4)-Alkyl, (Cι-C4)-Alkoxy, (C6-Cι0)-Aryl, 5- bis 6- gliedriges Heteroaryl oder eine Gruppe der Formel -NR9R10 bedeutet, wobei Aryl und Heteroaryl ihrerseits jeweils ein- bis zweifach, gleich oder verschieden, durch Halogen, Cyano, (Cι-C )-Alkylsulfonyl oder eine Gruppe der Formel -NR9R10 substituiert sein können,
und worin
R9 und R10 unabhängig voneinander für Wasserstoff, (Cι-C )-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C4)-Alkyl, (Cι-C4)-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (Cι-C4)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Ebenfalls bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C1-C4)- Alkoxy, Amino, Mono- oder Di-(Cι-C )-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)- Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können,
wobei Mono- und Di-(Cι-C )-alkylamino ihrerseits durch Hydroxy, ( -C )- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C C4)-Alkoxy, Amino, Mono- oder Di-(C1-C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)-Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (C C4)- Alkoxy, Amino, Mono- oder Di-(C1-C4)-alkylamino substituiert sein können, wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino und
R4 Wasserstoff bedeutet oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 oder >C=Ν-OH bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und (C6-Cι0)-Aryl, 5- bis 6-gliedriges Heteroaryl oder eine Gruppe der Formel -NR9R10 bedeutet, wobei Aryl und Heteroaryl ihrerseits jeweils ein- bis zweifach, gleich oder verschieden, durch Halogen, Cyano, (Cι-C4)-Alkylsulfonyl oder eine Gruppe der Formel -NR9R10 substituiert sein können,
und worin
R9 und R10 unabhängig voneinander für Wasserstoff, (C C )-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (C C )-Alkyl, (Cι-C )-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
Rn und R12 unabhängig voneinander für Wasserstoff, (C C )-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Ebenfalls bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C C4)- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)- Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können,
wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (C C )- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein kömien, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (C C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (C C )-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)-Cycloalkyl-N-(Cι-C4)-alkylammo oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(C C4)-alkylamino ihrerseits durch Hydroxy, (C C )- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, wobei R1 oder R2 jedoch nicht beide gleichzeitig für Wasserstoff stehen,
R3 Amino und
R4 Wasserstoff bedeutet oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=Ν-OH bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C4)-Alkyl oder (Cι-C4)-Alkoxy bedeutet, und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (C C4)-Alkyl, (Cι-C4)-Alkoxy oder eine Gruppe der Formel -NRUR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (C C4)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Ebenfalls bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Benzyloxy, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch ( -Q)- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)- Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7- gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (C C4)- Alkoxy, Amino, Mono- oder Di-(C C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (C)-C4)-Alkyl oder Halogen substituiert sein kann, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (CrC4)-Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino, (C3-C6)-Cycloalkylamino, N-(C3-C6)-Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein N-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sein können, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (Cι-C4)- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, wobei entweder R1 oder R2 für Wasserstoff steht, jedoch nicht beide gleichzeitig Wasserstoffbedeuten,
R3 Hydroxy und
R4 . Wasserstoff bedeutet oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (C C4)-Alkyl oder (Cι-C4)-Alkoxy bedeutet, und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C4)-Alkyl, (Cι-C4)-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (Cι-C4)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Ebenfalls bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (T), in welcher
R1 Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3-Position durch (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(C1-C4)-alkylamino, (C3-C6)-Cycloalkylanιmo, N-(C3-C6)-Cycloalkyl-N-(C,-C4)-alkyl- amino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sind, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, (Cι-C )- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können, und worin
R8 für 5- oder 6-gliedriges Heteroaryl, das durch (Cι-C )-Alkyl oder Halogen substituiert sein kann, steht,
R2 Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 2- oder 3- Position durch (Cι-C4)-Alkoxy, Amino, Mono- oder Di-(C1-C4)-alkylamino, (C3-C6)- Cycloalkylamino, N-(C3-C6)-Cycloalkyl-N-(Cι-C4)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sind, wobei Mono- und Di-(Cι-C4)-alkylamino ihrerseits durch Hydroxy, ( -C4)- Alkoxy, Amino, Mono- oder Di-(Cι-C4)-alkylamino substituiert sein können,
R3 Hydroxy
und
R4 Wasserstoff bedeutet oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0, 1, 2 oder 3 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Halogen, (Cι-C4)-Alkyl oder (Cι-C4)-Alkoxy bedeutet, und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Halogen, (Cι-C4)-Alkyl, (Cι-C )-Alkoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, (Cι-C4)-Alkyl, Phenyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden, oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (ϊ), in welcher
R1 und R2 unabhängig voneinander Wasserstoff, Ethoxy oder n-Propoxy bedeuten, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methyla ino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclo- propyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sind,
wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino bedeutet,
R4 Wasserstoff bedeutet,
R5 Methylaminocarbonyl oder Dimethylaminocarbonyl bedeutet,
n für die Zahl 0 oder 1 steht,
R6 sich in para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy oder Ethoxy bedeutet,
und
R7 Wasserstoff bedeutet,
sowie ihre Salze, Solvate und Solvate der Salze.
Gleichfalls besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-ΝH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können, worin
R8 für Pyridyl, Pyrrolyl, Imidazolyl, Pyrazolyl, Triazolyl oder Thiadiazolyl, die jeweils durch Methyl, Ethyl, Fluor oder Chlor substituiert sein können, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3-Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino,
Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können,
wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Hydroxy oder Amino bedeutet,
R4 Wasserstoff bedeutet,
R5 Wasserstoff bedeutet,
n für die Zahl 0 oder 1 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Phenyl, Thienyl, Indolyl, Chinoxalinyl oder eine Gruppe der Formel -ΝR9R10 bedeutet, wobei Phenyl, Thienyl und Indolyl ihrerseits jeweils ein- bis zweifach, gleich oder verschieden, durch Fluor, Chlor, Brom, Cyano oder Amino substituiert sein können, und worin
R9 und R10 unabhängig voneinander für Wasserstoff, Methyl, Ethyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Pyrrolidino-Ring bilden,
und
R7 Wasserstoff bedeutet,
sowie ihre Salze, Solvate und Solvate der Salze.
Gleichfalls besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3-Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können, worin
R8 für Pyridyl, Pyrrolyl, Imidazolyl, Pyrazolyl, Triazolyl oder Thiadiazolyl, die jeweils durch Methyl, Ethyl, Fluor oder Chlor substituiert sein können, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können,
wobei R1 und R2 jedoch nicht gleichzeitig für Wasserstoff stehen,
R3 Amino bedeutet,
R4 Wasserstoff bedeutet, .
R5 Wasserstoff bedeutet,
n für die Zahl 0 oder 1 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy oder Ethoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy, Ethoxy oder eine Gruppe der Formel -ΝRπR12 bedeutet, worin
Rn und R12 unabhängig voneinander für Wasserstoff, Methyl, Ethyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Pyrrolidino- oder Piperidino-Ring bilden,
oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Gleichfalls besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Wasserstoff, Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropyl- amino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können, worin
R8 für Pyridyl, Pyrrolyl, Imidazolyl, Pyrazolyl, Triazolyl oder Thiadiazolyl, die jeweils durch Methyl, Ethyl, Fluor oder Chlor substituiert sein können, steht,
R2 Wasserstoff, Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sein können,
wobei entweder R1 oder R2 für Wasserstoff steht, jedoch nicht beide gleichzeitig Wasserstoff bedeuten,
R3 Hydroxy
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0 oder 1 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy oder Ethoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy, Ethoxy oder eine Gruppe der Formel -NRπR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, Methyl, Ethyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Pyrrolidino- oder Piperidino-Ring bilden,
oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Gleichfalls besonders bevorzugt im Rahmen der vorliegenden Erfindung sind Verbindungen der Formel (I), in welcher
R1 Ethoxy, n-Propoxy oder eine Gruppe der Formel R8-C(=0)-NH- bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3-Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sind, worin
R8 für Pyridyl, Pyrrolyl, Imidazolyl, Pyrazolyl, Triazolyl oder Thiadiazolyl, die jeweils durch Methyl, Ethyl, Fluor oder Chlor substituiert sein können, steht,
R2 Ethoxy oder n-Propoxy bedeutet, wobei Ethoxy in 2-Position und n-Propoxy in 3 -Position durch Methoxy, Ethoxy, Amino, Methylamino, Ethylamino, Isopropylamino, Dimethylamino, Diethylamino, Cyclopropylamino, N-Cyclopropyl-N-methylamino, Azetidino oder Pyrrolidino substituiert sind,
R3 Hydroxy
und
R4 Wasserstoff bedeutet
oder
R3 und R4 gemeinsam mit dem Kohlenstoffatom, an das sie gebunden sind, eine Gruppe der Formel >C=0 bilden,
R5 Wasserstoff bedeutet,
n für die Zahl 0 oder 1 steht,
R6 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, befindet und Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy oder Ethoxy bedeutet,
und
R7 sich in meta- oder para-Position, relativ zur Verknüpfungsstelle des Phenylrings mit dem Tricyclus, und in ortho-Stellung zu R6 befindet und Wasserstoff, Fluor, Chlor, Brom, Methyl, Ethyl, Methoxy, Ethoxy oder eine Gruppe der Formel -NRnR12 bedeutet, worin
R11 und R12 unabhängig voneinander für Wasserstoff, Methyl, Ethyl, Benzyl oder Pyridylmethyl stehen oder gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Pyrrolidino- oder Piperidino-Ring bilden,
oder
R6 und R7 gemeinsam mit dem Phenylring, an den sie gebunden sind, eine Gruppe der Formel
sowie ihre Salze, Solvate und Solvate der Salze.
Die in den jeweiligen Kombinationen bzw. bevorzugten Kombinationen von Resten im einzelnen angegebenen Reste-Definitionen werden unabhängig von den jeweiligen angegebenen Kombinationen der Reste beliebig auch durch Reste-Definitionen anderer Kombinationen ersetzt.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Die erfindungsgemäßen Verbindungen der Formel (I), in welcher R5 für Wasserstoff steht, können prinzipiell nach den in der WO 00/08007 beschriebenen Verfahren hergestellt werden. Der Inhalt der WO 00/08007, insbesondere die Seiten 14-26, wird hiermit ausdrücklich als Bestandteil der Offenbarung einbezogen. In Abhängigkeit von der spezifischen Bedeutung der Substituenten in (I), insbesondere von R1 und R2, sind jedoch einzelne in der WO 00/08007 beschriebene Verfahrensstufen in manchen Fällen nur mit sehr geringen Ausbeuten verbunden. Weiterer Gegenstand der vorliegenden Erfindung ist daher ein neues Verfahren zur Herstellung der erfindungsgemäßen Verbindungen der Formel (I), in welcher R5 für Wasserstoff steht, dadurch gekennzeichnet, dass man entweder
[A] Verbindungen der Formel (U)
in welcher R
1 und R
2 jeweils die oben angegebene Bedeutung haben, in einem inerten Lösungsmittel in Gegenwart einer Base mit einer Verbindung der Formel (TU)
in welcher R
6 und R
7 jeweils die oben angegebene Bedeutung haben,
X1 für eine geeignete Fluchtgruppe wie beispielsweise Halogen, Mesylat, Tosylat oder Triflat
und für ( -O-Alkyl steht,
zu Verbindungen der Formel (IV)
in welcher R , R , R , R und T jeweils die oben angegebene Bedeutung haben,
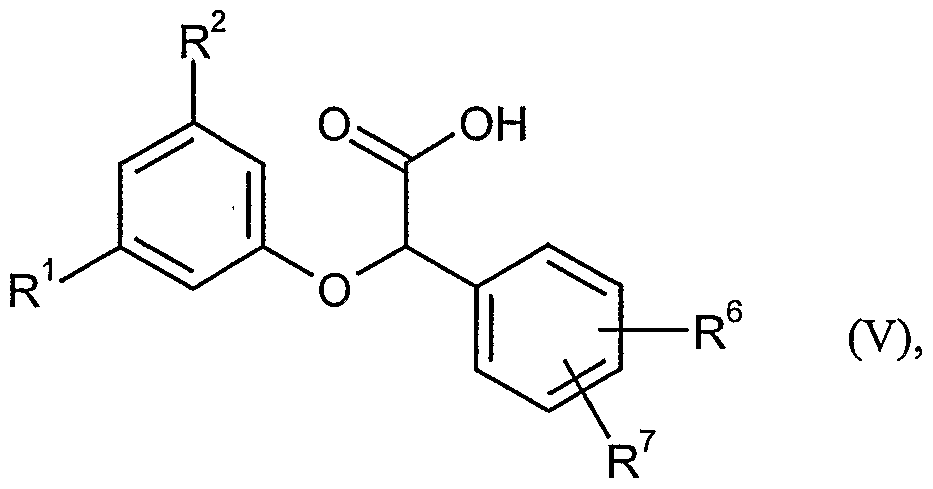
umsetzt, anschließend durch basische oder saure Hydrolyse in Carbonsäuren der Formel (V)
in welcher R1, R2, R6 und R7 jeweils die oben angegebene Bedeutung haben,
überführt, diese dann nach Aktivierung mit Phosphorylchlorid in Gegenwart einer Lewis- Säure zu Verbindungen der Formel (VI)
in welcher R1, R2, R6 und R7 jeweils die oben angegebene Bedeutung haben,
cyclisiert,
oder
[B] Verbindungen der Formel (VH)
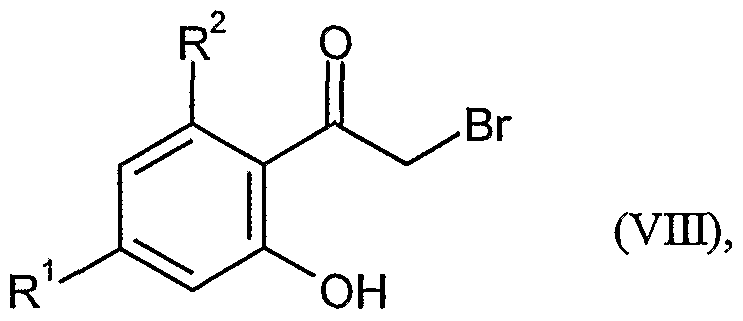
in welcher R und R jeweils die oben angegebene Bedeutung haben, zunächst nach üblichen Methoden in Phenacylbromide der Formel (VIII)
in welcher R
1 und R
2 jeweils die oben angegebene Bedeutung haben, überführt und diese dann in Gegenwart einer Base zu Verbindungen der Formel (IX)
in welcher R
1 und R
2 jeweils die oben angegebene Bedeutung haben, cyclisiert, anschließend in einem inerten Lösungsmittel zu Verbindungen der Formel (X)
in welcher R
1 und R
2 jeweils die oben angegebene Bedeutung haben, bromiert und nach üblichen Methoden in Silylenolether der Formel (XI)
in welcher R und R jeweils die oben angegebene Bedeutung haben
und
T2, T3 und T4 gleich oder verschieden sind und jeweils für (Cι-C4)-Alkyl oder Phenyl stehen,
überführt, nachfolgend in einem inerten Lösungsmittel in Gegenwart eines geeigneten Palladium-Katalysators und einer Base mit einer Verbindung der Formel (XU)
in welcher R und R jeweils die oben angegebene Bedeutung haben
und
Z für Wasserstoff oder Methyl steht oder beide Z-Gruppen zusammen eine CH2CH2- oder C(CH3)2-C(CH3)2-Brücke bilden,
zu Verbindungen der Formel (XIH)
in welcher R1, R2, R6, R7, T2, T3 und T4 jeweils die oben angegebene Bedeutung haben,
umsetzt, die Silylgruppe anschließend nach üblichen Methoden wieder zu Verbindungen der Formel (VI) abspaltet,
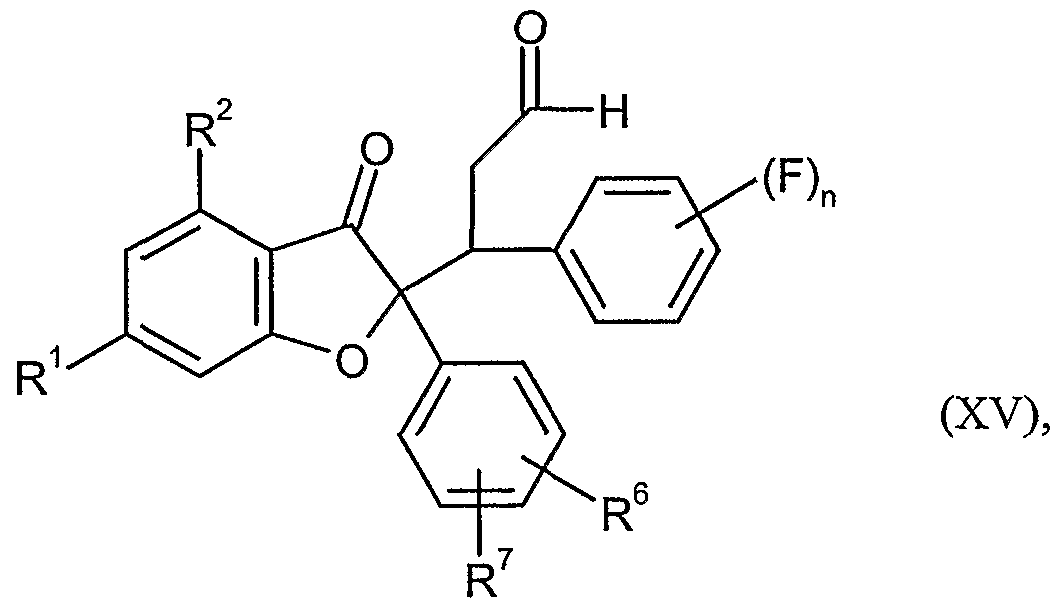
und die jeweils resultierenden Verbindungen der Formel (VI) sodann in einem inerten Lösungsmittel in Gegenwart einer Base gemäß dem in der WO 00/08007 beschriebenen Verfahren mit einem Zimtaldehyd der Formel (XIV)
in welcher n die oben angegebene Bedeutung hat,
in Verbindungen der Formel (XV)
in welcher R1, R2, R6, R7 und n jeweils die oben angegebene Bedeutung haben,
überführt und diese dann nach der in der WO 00/08007 beschriebenen Reaktionssequenz weiter umsetzt,
und die Verbindungen der Formel (I) gegebenenfalls mit den entsprechenden (i) Lösungsmitteln und/oder (ii) Basen oder Säuren zu ihren Solvaten, Salzen und/oder Solvaten der Salze umsetzt.
Der Verfahrensschritt (VH) → (IX) kann auch über ein dreistufiges EintopfVerfahren über den aus (VII) nach üblichen Methoden erhältlichen Silylenolether der Formel (XVI)
in welcher R1 und R2 jeweils die oben angegebene Bedeutung haben,
dessen Bromierung mit N-Bromsuccinimid sowie anschließende Cyclisierung zu (LX) in Gegenwart von Natronlauge durchgeführt werden.
Die Verbindungen der Formeln (II), (DI), (VTf), (XU) und (XTV) sind kommerziell erhältlich, literaturbekannt oder können in Analogie zu literaturbekannten Verfahren hergestellt werden.
Inerte Lösungsmittel für den Verfahrensschritt (II) + (HI) - (TV) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetra- chlorethan, 1 ,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydro- furan, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Ethyl- acetat, Aceton, 2-Butanon, Dimethylformamid, Dimethylsulfoxid, Pyridin oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind 2-Butanon, Diethylether, Dioxan, Tetrahydrofuran, Dichlormethan, Toluol oder Benzol.
Als Basen für den Verfahrensschritt (11) + (DT) -» (IV) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkali- oder Erdalkahcarbonate oder -hydrogencarbonate wie Lithium-, Natrium-, Kalium-, Calcium- oder Cäsiumcarbonat, oder Natrium- oder Kalium- hydrogencarbonat, Alkalihydride wie Natriumhydrid, Amide wie Natriumamid, Lithiumbis(tri- methylsilyl)amid oder Lithiumdiisopropylamid, oder organische Arnine wie Pyridin, Triethylamin, Ethyldiisopropylamin, N-Methylmorpholin oder N-Methylpiperidin. Besonders bevorzugt sind Natrium- oder Kaliumhydroxid, Natrium- oder Kaliumcarbonat oder Natriumhydrid.
Der Verfahrensschritt (D) + (DT) — (TV) wird im Allgemeinen in einem Temperaturbereich von +20°C bis +160°C, bevorzugt von +60°C bis +100°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (IV) -> (V) sind beispielsweise Wasser, Alkohole wie Methanol, Ethanol, n-Propanol, iso-Propanol oder n-Butanol, Kohlenwasserstoffe wie Benzol
oder andere Lösungsmittel wie Aceton, Dimethylformamid, Dimethylsulfoxid oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Methanol, Ethanol, n-Propanol und/oder Wasser.
Als Basen für den Verfahrensschritt (IV) -» (V) eignen sich die üblichen anorganischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkali- oder Erdalkahcarbonate wie Lithium-, Natrium-, Kalium- oder Calciumcarbonat, oder Alkali-Alkoholate wie Natrium- oder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Kalium-tert.-butylat. Besonders bevorzugt sind Natrium- oder Kaliumhydroxid oder Natrium- oder Kaliumcarbonat.
Als Säuren für den Verfahrensschritt (TV) - (V) eignen sich im Allgemeinen Schwefelsäure, Chlorwasserstoff/Salzsäure, Bromwasserstoff/Bromwasserstoffsäure, Phosphorsäure, Essigsäure, Trifluoressigsäure, Toluolsulfonsäure, Methansulfonsäure oder Trifluormethansulfonsäure oder deren Gemische gegebenenfalls unter Zusatz von Wasser. Bevorzugt sind Chlorwasserstoff oder Trifluoressigsäure im Falle der tert.-Butylester und Salzsäure oder Schwefelsäure im Falle der Methylester.
Der Verfahrensschritt (IV) — > (V) wird im Allgemeinen in einem Temperaturbereich von 0°C bis +100°C, bevorzugt von +40°C bis +80°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Der Verfahrensschritt (V) - (VI) wird bevorzugt ohne weiteres Lösungsmittel durchgeführt. Als Lewis-Säuren eignen sich für diesen Verfahrensschritt die üblichen anorganischen Lewis-Säuren wie beispielsweise Aluminiumtrichlorid, Eisentrichlorid, Bortrifluorid, Bortrichlorid, Bortri- bromid, Titantetrachlorid, Titantrichlorid, Zinndichlorid, Zinntetrachlorid oder Zinkdichlorid. Bevorzugt ist Zinkdichlorid.
Der Verfahrensschritt (V) — > (VI) wird im Allgemeinen in einem Temperaturbereich von 0°C bis +100°C, bevorzugt von 0°C bis +40°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für die Verfahrensschritte (Vπ) - (VDT) und (LX) — (X) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetrachlorethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetra- hydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie
Hexan oder Cyclohexan, oder andere Lösungsmittel wie Ethylacetat, Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Diethylether, Dioxan, Tetrahydrofuran, Ethylacetat, Trichlormethan und/oder Tetrachlormethan.
Als Bromierungsmittel für die Verfahrensschritte (VE) → (VTTT) und (LX) -> (X) eignen sich die üblichen anorganischen oder organischen Reagenzien. Hierzu gehören bevorzugt Brom, N-Brom- succinimid, Kupferdibromid, Pyridinhydrotribromid, Dimethylbenzylammoniumtribromid oder Phenyltrimethylammoniumtribromid. Besonders bevorzugt sind Brom und Kupferdibromid.
Die Verfahrensschritte (VII) → (VTTT) und (LX) -» (X) werden im Allgemeinen in einem Temperaturbereich von -20°C bis +150°C, bevorzugt von 0°C bis +80°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (VOT) → (LX) sind beispielsweise Wasser, Alkohole wie Methanol, Ethanol, n-Propanol, iso-Propanol, n-Butanol oder tert.-Butanol, Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetra- chlorethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, Pyridin oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Methanol, Ethanol, Wasser und/oder Tetrahydrofuran.
Als Basen für den Verfahrensschritt (VIII) -» (IX) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkali- oder Erdalkahcarbonate oder -hydrogencarbonate wie Lithium-, Natrium-, Kalium-, Calcium- oder Cäsiumcarbonat, oder Natrium- oder Kalium- hydrogencarbonat, Alkali-Alkoholate wie Natrium- oder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Kalium-tert.-butylat, Alkali-Acetate wie Natrium- oder Kaliumacetat, Alkalihydride wie Natriumhydrid, Amide wie Natriumamid, Lithiumbis(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder organische Amine wie Pyridin, Triethylamin, Ethyldiisopropylamin, N-Methylmorpholin oder N-Methylpiperidin. Besonders bevorzugt sind Natrium- oder Kaliumhydroxid oder Natriumacetat.
Der Verfahrensschritt (VTTT) -» (IX) wird im Allgemeinen in einem Temperaturbereich von 0°C bis +100°C, bevorzugt von +20°C bis +80°C durchgeführt. Die Umsetzung kann bei normalem,
erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (X) -» (XI) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetrachlor- ethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, Pyridin oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Toluol, Hexan, Diethylether oder Tetrahydrofuran.
Als Basen für den Verfahrensschritt (X) -> (XI) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydride wie Natriumhydrid, Amide wie Natriumamid, Lithiumbis(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder organische Amine wie Pyridin, Triethylamin, Ethyldiisopropylamin, N-Methylmorpholin oder N-Methyl- piperidin. Besonders bevorzugt sind Lithiumdiisopropylamid, Triethylamin oder Ethyldiisopropylamin.
Der Verfahrensschritt (X) -» (XI) wird im Allgemeinen in einem Temperaturbereich von -20°C bis +50°C, bevorzugt von 0°C bis +30°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (XI) + (XE) ->■ (XTTT) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetra- chlorethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Alkohole wie Methanol, Ethanol, n-Propanol, iso-Propanol, n-Butanol oder tert.-Butanol, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Ethylacetat, Aceton, Wasser, Dimethylformamid, Dimethylsulfoxid, Pyridin oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Toluol, Tetrahydrofuran, Dioxan oder Dimethylformamid.
Als Basen für den Verfahrensschritt (XI) + (XII) - (XEI) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Lithium-, Natrium- oder Kaliumhydroxid, Alkali-, Erdalkali- oder Schwermetallcarbonate wie Silber-, Thallium-, Lithium-, Natrium-, Kalium-, Cäsium- oder Calciumcarbonat, Alkali- oder Erdalkali- hydrogencarbonate wie Natrium- oder Kaliumhydrogencarbonat, Alkali-Alkoholate wie Natrium-
oder Kaliummethanolat, Natrium- oder Kaliumethanolat oder Lithium-, Natrium- oder Kalium- tert.-butylat, Alkalihydride wie Natriumhydrid, Amide wie Natriumamid, Lithium- oder Natriumbis(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder organische Amine wie Pyridin, Triethylamin, Ethyldiisopropylamin, l,5-Diazabicyclo[5.4.0]undec-5-en (DBU), N-Methylmorpho- lin oder N-Methylpiperidin. Besonders bevorzugt sind Cäsium- oder Νatriumcarbonat, Νatrium- hydrid, Kalium-tert.-butylat, Lithiumdiisopropylamid, DBU, Triethylamin oder Ethyldiisopropylamin.
Als Katalysatoren für den Verfahrensschritt (XI) + (XII) -→- (XDT) eignen sich die für Suzuki- Reaktionsbedingungen üblichen Palladium-Katalysatoren. Bevorzugt sind Katalysatoren wie beispielsweise Dichlorbis(triphenylphosphin)palladium, Tetrakis(triphenylphosphin)palladium(0), Palladium(E)acetat oder Bis(diphenylphosphanferrocenyl)palladium(E)chlorid. Als Katalysatorliganden eignen sich bevorzugt die für Suzuki-Reaktionen üblichen Liganden wie beispielsweise Triphenylphospin, Tri(o-tolyl)phosphin, Tributylphosphin, 2,2'-Bis(diphenylphosphino)-l, -bi- naphthyl (BIΝAP), l,l'-Bis(diphenylphosphino)ferrocen (dppf) oder l,3-Bis(diphenylphosphino)- propan (dppp).
Der Verfahrensschritt (XI) + (XII) -» (XTTT) wird im Allgemeinen in einem Temperaturbereich von +20°C bis +200°C, bevorzugt von +50°C bis +150°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (XIII) - (VI) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetrachlor- ethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Alkohole wie Methanol, Ethanol, n- Propanol, iso-Propanol, n-Butanol oder tert.-Butanol, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan oder Cyclohexan, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid, Acetonitril oder Wasser. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Methanol, Ethanol, Wasser, Tetrahydrofuran oder Dioxan.
Die Abspaltung der Silylgruppe im Verfahrensschritt (X T) - (VT) kann nach den üblichen Methoden alternativ mit Hilfe einer Base oder mit Hilfe einer Säure durchgeführt werden. Als Basen eignen sich bevorzugt Tetrabutylammoniumfluorid, Pyridin oder Triethylamin, als Säuren bevorzugt Fluorwasserstoff, Chlorwasserstoff/Salzsäure, Ameisensäure, Essigsäure, Trifluoressigsäure oder Toluolsulfonsäure.
Der Verfahrensschritt (XTTT) -» (VI) wird im Allgemeinen in einem Temperaturbereich von -80°C bis +100°C, bevorzugt von 0°C bis +80°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die erfmdungsgemäßen Verbindungen der Formel (I), in welcher R5 für Mono- oder Di-(Cι-C6)- alkylaminocarbonyl steht, können hergestellt werden, indem man Verbindungen der Formel (XVII)
in welcher R1, R , R°, R' und n jeweils die oben angegebene Bedeutung haben,
zunächst in einem inerten Lösungsmittel mit Methoxymagnesiummethylcarbonat [M. Stiles, J. Amer. Chem. Soc. 81, 598 (1959)] in Carbonsäuren der Formel (XVEI)
in welcher R1, R2, R6, R7 und n jeweils die oben angegebene Bedeutung haben,
überführt, anschließend in Gegenwart eines Kondensationsmittels und einer Base mit einer Verbindung der Formel (XLX)
HNR13R14 (XLX),
in welcher
R13 für Wasserstoff oder (C C6)-Alkyl
und
RM für (Cι-C6)-Al yl steht,
zu Verbindungen der Formel (XX)
in welcher R1, R2, R6, R7, R13, R14 und n jeweils die oben angegebene Bedeutung haben,
umsetzt und diese dann gegebenenfalls nach den in der WO 00/08007 beschriebenen Reaktionssequenzen weiter umwandelt.
Die Verbindungen der Formel (XVE) sind nach den oben bzw. in WO 00/08007 beschriebenen Verfahren erhältlich. Die Verbindungen der Formel (XIX) sind kommerziell erhältlich oder literaturbekannt.
Inerte Lösungsmittel für den Verfahrensschritt (XVII) -» (XVEI) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetra- chlorethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, oder andere Lösungsmittel wie Dimethylformamid, Dimethylsulfoxid oder Acetonitril. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt ist Dimethylformamid.
Der Verfahrensschritt (XVE) - (XVEI) wird im Allgemeinen in einem Temperaturbereich von 0°C bis +200°C, bevorzugt von +50°C bis +150°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Inerte Lösungsmittel für den Verfahrensschritt (XVEI) + (XLX) -» (XX) sind beispielsweise Halogenkohlenwasserstoffe wie Dichlormethan, Trichlormethan, Tetrachlormethan, Trichlorethan, Tetrachlorethan, 1,2-Dichlorethan oder Trichlorethylen, Ether wie Diethylether, Methyl-tert.-
butylether, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Kohlenwasserstoffe wie Benzol, Toluol, Xylol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Ethylacetat, Aceton, 2-Butanon, Dimethylformamid, Dimethylacetamid, Dimethylsulfoxid, Acetonitril, N-Methylpyrrolidon oder Pyridin. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Dimethylformamid und Tetrahydrofuran.
Als Kondensationsmittel für die Amidbildung im Verfahrensschritt (XVIE) + (XIX) -> (XX) eignen sich beispielsweise Carbodiimide wie N,N'-Diethyl-, N,N'-Dipropyl-, NN'-Diisopropyl-, N,N- Dicyclohexylcarbodiimid (DCC), N-(3-Dimethylaminoisopropyl)-N'-ethylcarbodiimid-Hydro- chlorid (EDC), N-Cyclohexylcarbodiimid-N'-propyloxymethyl-Polystyrol (PS-Carbodiimid) oder Phosgen-Derivate wie N,N'-Carbonyldiimidazol, oder 1,2-Oxazoliumverbindungen wie 2-Ethyl-5- phenyl-l,2-oxazolium-3 -sulfat oder 2-tert.-Butyl-5-methyl-isoxazolium-perchlorat, oder Acyl- aminoverbindungen wie 2-Ethoxy-l-ethoxycarbonyl-l,2-dihydrochinolin, oder Isobutylchlor- formiat, Propanphosphonsäureanhydrid, Cyanophosphonsäurediethylester, Bis-(2-oxo-3-oxazoli- dinyl)-phosphorylchlorid, Benzotriazol-l-yloxy-tris(dimethylamino)phosphonium-hexafluorophos- phat (BOP), Benzotriazol-l-yloxy-tris(pyrrolidino)phosphonium-hexafluorophosphat (PyBOP), O- (Benzotriazol-l-yl)-NN,N',N'-tetramethyluronium-hexafluorophosphat (HBTU), 2-(2-Oxo-l-(2H)- pyridyl)-l,l,3,3-tetramethyluronium-tetrafluoroborat (TPTU) oder <3-(7-Azabenzotriazol-l-yι)- NN,N',N'-tetramethyluronium-hexafluorophosphat (HATU), gegebenenfalls in Kombination mit weiteren Hilfsstoffen wie 1-Hydroxybenzotriazol (HOBt) oder Ν-Hydroxysuccinimid (HOSu), sowie als Basen Alkalicarbonate, z.B. Natrium- oder Kaliumcarbonat oder -hydrogencarbonat, oder organische Amin-Basen wie z.B. Triethylamin, N-Methylmorpholin, N-Methylpiperidin, N,N- Diisopropylethylamin, Pyridin, 4-N,N-Dimethylaminopyridin, l,5-Diazabicyclo[4.3.0]non-5-en (DBΝ) oder l,8-Diazabicyclo[5.4.0]undec-7-en (DBU). Bevorzugt sind BOP, PyBOP oder HATU jeweils in Kombination mit Triethylamin oder NN-Diisopropylethylamin.
Der Verfahrensschritt (XVIE) + (XLX) - ■ (XX) wird im Allgemeinen in einem Temperaturbereich von -20°C bis +100°C, bevorzugt von 0°C bis +50°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die erfindungsgemäßen Verbindungen der Formel (I), in welcher R
1 und R
2 jeweils für Ethoxy oder n-Propoxy stehen, die in 2- bzw. 3-Position durch Amino, Mono- oder Di-(Cι-C
6)-alkylamino, (C
3-C
8)-Cycloalkylamino, N-(C
3-C
8)-Cycloalkyl-N-(Cι-C
6)-alkylamino oder durch einen über ein Ν-Atom gebundenen 4- bis 7-gliedrigen Heterocyclus substituiert sind, können auch hergestellt werden, indem man Verbindungen der Formel (XXI)
in welcher R , R , R , R , R und n jeweils die oben angegebene Bedeutung haben,
X2 für eine geeignete Fluchtgruppe wie beispielsweise Halogen, Mesylat oder Tosylat
und
m für die Zahl 2 oder 3 steht,
in einem inerten Lösungsmittel gegebenenfalls in Gegenwart einer Hilfsbase mit einer Verbindung der Formel (XXII)
HNR^R16 (XXE),
in welcher
R15 für Wasserstoff, (d-C6)-Alkyl oder (C3-C8)-Cycloalkyl
und
R16 für Wasserstoff oder (Cι-C6)-Alkyl steht
oder
R15 und R16 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 4- bis 7-gliedrigen Heterocyclus bilden,
zu Verbindungen der Formel (XXE
in welcher R3, R4, R5, R6, R7, R15, R16, m und n jeweils die oben angegebene Bedeutung haben,
umsetzt und diese dann gegebenenfalls nach den in der WO 00/08007 beschriebenen Reaktionssequenzen weiter modifiziert.
Die Verbindungen der Formel (XXI) sind nach den oben bzw. in WO 00/08007 beschriebenen Verfahren erhältlich. Die Verbindungen der Formel (XXE) sind kommerziell erhältlich oder literaturbekannt.
Inerte Lösungsmittel für den Verfahrensschritt (XXI) + (XXE) -» (XXIE) sind beispielsweise Ether wie Tetrahydrofuran, Dioxan, Glykoldimethylether oder Diethylenglykoldimethylether, Alkohole wie Methanol, Ethanol, n-Propanol, iso-Propanol, n-Butanol oder tert.-Butanol, Kohlenwasserstoffe wie Toluol oder Xylol, oder andere Lösungsmittel wie Aceton, Dimethylformamid oder Dimethylsulfoxid. Ebenso ist es möglich, Gemische der genannten Lösungsmittel einzusetzen. Bevorzugt sind Ethanol, Dimethylformamid, Dimethylsulfoxid oder Xylol.
Als Hilfsbasen für den Verfahrensschritt (XXI) + (XXII) -» (XXEI) eignen sich die üblichen anorganischen oder organischen Basen. Hierzu gehören bevorzugt Alkalihydroxide wie beispielsweise Natrium- oder Kaliumhydroxid, Alkali- oder Erdalkalicarbonate wie Natrium- oder Kalium- carbonat, Alkalihydride wie Natriumhydrid, oder organische Amine wie Triethylamin oder Ethyl- diisopropylamin.
Der Verfahrensschritt (XXI) + (XXE) → (XXEI) wird im Allgemeinen in einem Temperaturbereich von +20°C bis +200°C, bevorzugt von +70°C bis +150°C durchgeführt. Die Umsetzung kann bei normalem, erhöhtem oder erniedrigtem Druck erfolgen (z.B. von 0.5 bis 5 bar). Im Allgemeinen arbeitet man bei Normaldruck.
Die erfindungsgemäßen Verbindungen können, falls zweckmäßig, auch hergestellt werden durch synthetische Umwandlungen von funktionellen Gruppen einzelner Substituenten in Verbindungen der Formel (ϊ), die nach den zuvor beschriebenen Verfahren erhalten werden. Solche Umwand-
lungen funktioneller Gruppen werden nach literaturüblichen Methoden durchgeführt und umfassen beispielsweise Verfahren zur Alkylierung, Acylierung, Aminierung, Veresterung, Esterspaltung, Hydrierung, Oxidation und Reduktion.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch die folgenden Synthese¬ schemata veranschaulicht werden:
Schema 1
[X = Halogen; a): Kaliumcarbonat, 2-Butanon, 80°C; b): Kaliumcarbonat, Methanol/Wasser, 65°C; c): Phosphorylchlorid, Zinkchlorid, 0°C → RT].
Schema 2
[d): 1. Lithiumhexamethyldisilazid, Trimethylsilylchlorid, THF, -78°C -> RT; 2. N-Bromsuccin- imid, 0°C → RT; 3. 1 Ν Natronlauge, RT; oder: 1. Kupfer(E)bromid, Ethylacetat/Chloroform, 65°C; 2. Natriumacetat, Ethanol, 80°C; e): Brom, Dioxan/Diethylether, -5°C → 0°C; oder: Kupfer(E)bromid, Ethylacetat/Chloroform, 65°C; f): tert.-Butyldimethylsilyltrifluormethan- sulfonat, Triethylamin, Diethylether, 0°C — > RT; g): Phenylboronsäure-Derivat, Natriumcarbonat, Tetrakis(triphenylphosphin)palladium(0), Toluol/Wasser, 95°C; h): Chlorwasserstoff in Dioxan, RT; oder: Trifluoressigsäure, RT].
Schema 3
HNR"R"
[m = 2 oder 3, X = Halogen; i): Ethanol, 70°C].
Schema 4
D): Methoxymagnesiummethylcarbonat, DMF, 100°C; k): Benzotriazol-1-yloxy-tripyrrolidino- phosphonium-Hexafluorophosphat (PyBOP), Diisopropylethylamin, Dimethylamin-Hydrochlorid, THF, 0°C].
Die erfmdungsgemäßen Verbindungen besitzen wertvolle pharmakologische Eigenschaften und können zur Vorbeugung und Behandlung von Erkrankungen bei Menschen und Tieren verwendet werden. Sie zeichnen sich zudem durch eine erhöhte metabolische Stabilität im Vergleich zu den in der WO 00/08007 beschriebenen Verbindungen aus.
Die erfindungsgemäßen Verbindungen sind potente Inhibitoren/Modulatoren der NF-κB- und/oder AP- 1 -Aktivität und eignen sich als solche insbesondere zur Behandlung chronischer Entzündungsund Autoimmunkrankheiten (wie z.B. die Crohn'sche Krankheit, ulzerative Colitis, rheumatoide Arthritis, Psoriasis, Multiple Sklerose, Lupus, Asthma, Diabetes), kardiovaskulärer Erkrankungen (wie z.B. koronare Herzerkrankungen, Myokard-Infarkt, Artherosklerose, Restenose, Thrombosen), fibrotischer Erkrankungen der Leber und anderer Organe, cerebrovaskulärer Erkrankungen (wie z.B. Schlaganfall, Schädel-Hirn-Trauma, Rückenmarksverletzungen) und chronischer neuro- degenerativer Erkrankungen (wie z.B. die Alzheimer'sche Krankheit, die Parkinson'sche Krankheit, Chorea Huntington, Amyotrophe Lateralsklerose, periphere Neuropathien und chronischer Schmerz). Darüber hinaus können sie zur Prophylaxe und/oder Therapie von Strahlenschäden, Transplantatabstoßung, Sepsis und septischem Schock sowie der bakteriellen Meningitis verwendet werden.
Weiterhin eignen sich die erfindungsgemäßen Verbindungen aufgrund ihrer inhibitorischen/modulatorischen Aktivität auf die NF-κB- und/oder AP- 1 -vermittelte Signalübertragung zur Behandlung hyperproliferativer Erkrankungen wie solider Tumoren (wie z.B. Brustkrebs, Lungenkrebs, Tumore des Gehirns und des Nervensystems, Hautkrebs, Leberkrebs, Tumore der Reproduktionsorgane, Tumore des Verdauungstraktes, Blasenkrebs, Tumore der Harnwegssysteme, Tumore verschiedener Hormondrüsen, Tumore des Auges), Lymphomen (wie z.B. Hodgkin'sche Krankheit, Lymphome des zentralen Nervensystems), Sarkomen (wie z.B. Osteaosarkome, Lympho- sarkome) und Leukämien (wie z.B. akute myeloide Leukämie, lymphoblastische Leukämien, myelogene Leukämien). Ferner können die erfindungsgemäßen Verbindungen zur Prophylaxe und/oder Therapie viraler Erkrankungen, insbesondere durch HIV, HTLV, Epstein-Barr- Virus, Zytomegalievirus (CMV) und/oder Adenoviren ausgelöster Erkrankungen, eingesetzt werden.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der erfindungsgemäßen Verbindungen zur Herstellung eines Arzneimittels zur Behandlung und/oder Prophylaxe von Erkrankungen, insbesondere der zuvor genannten Erkrankungen.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Behandlung und/oder Prophylaxe von . Erkrankungen, insbesondere der zuvor genannten Erkrankungen, unter Verwendung einer wirksamen Menge von mindestens einer der erfindungsgemäßen Verbindungen.
Die erfindungsgemäßen Verbindungen können allein oder bei Bedarf in Kombination mit anderen Wirkstoffen eingesetzt werden. Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, enthaltend mindestens eine erfindungsgemäße Verbindung und mindestens einen oder mehrere weitere Wirkstoffe, insbesondere zur Behandlung und/oder Prophylaxe der zuvor genannten Erkrankungen. Als geeignete Kombinationswirkstoffe seien beispielhaft und vorzugsweise cytostatisch oder cytotoxisch wirkende Substanzen, entzündungshemmende Substanzen (z.B. Kortikosteroide, NSAXDs) und neuroprotektiv wirkende Substanzen genannt.
Die erfindungsgemäßen Verbindungen können systemisch und/oder lokal wirken. Zu diesem Zweck können sie auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublingual, lingual, buccal, rectal, dermal, transdermal, conjunctival, otisch oder als Implantat bzw. Stent.
Für diese Applikationswege können die erfϊndungsgemäßen Verbindungen in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich nach dem Stand der Technik funktionierende, die erfindungsgemäßen Verbindungen schnell und/oder modifiziert abgebende Applikationsformen, die die erfindungsgemäßen Verbindungen in kristalliner und/oder amorphisierter und/oder gelöster Form enthalten, wie z.B. Tabletten (nicht-überzogene oder überzogene Tabletten, beispielsweise mit magensaftresistenten oder sich verzögert auflösenden oder unlöslichen Überzügen, die die Freisetzung der erfindungsgemäßen Verbindung kontrollieren), in der Mundhöhle schnell zerfallende Tabletten oder Filme/Oblaten, Filme/Lyophylisate, Kapseln (beispielsweise Hart- oder Weichgelatinekapseln), Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Aerosole oder Lösungen.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (z.B. intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (z.B. intramuskulär, subcutan, intracutan, percutan oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikationsformen u.a. Injektions- und Infusions-
zubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten oder sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulver- inhalatoren, Nebulizer), Nasentropfen, -lösungen oder -Sprays, lingual, sublingual oder buccal zu applizierende Tabletten, Filme/Oblaten oder Kapseln, Suppositorien, Ohren- oder Augen- präparationen, Vaginalkapseln, wäßrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, transdermale therapeutische Systeme (z.B. Pflaster), Milch, Pasten, Schäume, Streupuder, Implantate oder Stents.
Bevorzugt sind die orale oder parenterale Applikation, insbesondere die orale Applikation.
Die erfindungsgemäßen Verbindungen können in die angeführten Applikationsformen überführt werden. Dies kann in an sich bekannter Weise durch Mischen mit inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen geschehen. Zu diesen Hilfsstoffen zählen u.a. Trägerstoffe (beispielsweise mikrokristalline Cellulose, Lactose, Mannitol), Lösungsmittel (z.B. flüssige Poly- ethylenglycole), Emulgatoren und Dispergier- oder Netzmittel (beispielsweise Natriumdodecyl- sulfat, Polyoxysorbitanoleat), Bindemittel (beispielsweise Polyvinylpyrrolidon), synthetische und natürliche Polymere (beispielsweise Albumin), Stabilisatoren (z.B. Antioxidantien wie beispielsweise Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie beispielsweise Eisenoxide) und Geschmacks- und/oder Geruchskorrigentien.
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfϊndungsgemäße Verbindung, üblicherweise zusammen mit einem oder mehreren inerten, nichttoxischen, pharmazeutisch geeigneten Hilfsstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Im allgemeinen hat es sich als vorteilhaft erwiesen, bei parenteraler Applikation Mengen von etwa 0.001 bis 1 mg/kg, vorzugsweise etwa 0.01 bis 0.5 mg/kg Körpergewicht zur Erzielung wirksamer Ergebnisse zu verabreichen. Bei oraler Applikation beträgt die Dosierung etwa 0.01 bis 100 mg/kg, vorzugsweise etwa 0.01 bis 20 mg/kg und ganz besonders bevorzugt 0.1 bis 10 mg/kg Körpergewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden
muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die nachfolgenden Ausführungsbeispiele erläutern die Erfindung. Die Erfindung ist nicht auf die Beispiele beschränkt.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
A. Beispiele
Abkürzungen:
DCI direkte chemische Ionisation (bei MS)
DMF N,N-Dimethylformamid
DMSO Dimethylsulfoxid d. Th. der Theorie (bei Ausbeute)
ESI Elektrospray-Ionisation (bei MS)
H Stunde(n)
HPLC Hochdruck- / Hochleistungsflüssigchromatographie
Kp. Siedepunkt
LC-MS Flüssigchromatographie mit gekoppelter Massenspektroskopie min. Minute(n)
MS Massenspektroskopie
NMR Kernresonanzspektroskopie
RF Rückfluss
RT Raumtemperatur (20°C)
Rt Retentionszeit (bei HPLC)
TEA Triethylamin tert. tertiär
THF Tetrahydrofuran
UV Ultraviolett v/v Volumen zu Volumen (bei flüssig/flüssig-Gemischen)
HPLC- und C/MS-Methoden:
Methode 1:
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A → 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A -> 4.5 min 10% A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 nm.
Methode 2:
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A - 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A → 4.5 min 10% A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 nm.
Methode 3:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2 mm, 3.0 μm; Eluent B: Acetonitril + 0.05%» Ameisensäure, Eluent A: Wasser + 0.05% Ameisensäure; Gradient: 0.0 min 5% B → 2.0 min 40% B → 4.5 min 90% B → 5.5 min 90% B; Ofen: 45°C; Fluss: 0.0 min 0.75 ml/min → 4.5 min 0.75 ml/min → 5.5 min 1.25 ml/min; UV-Detektion: 210 nm.
Methode 4:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: TSP P4000, TSP AS300, TSP UV3000; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2 mm, 3.0 μm; Eluent A: Wasser + 250 μl 50%-ige Ameisensäure / 1, Eluent B: Acetonitril + 250 μl 50%-ige Ameisensäure / 1; Gradient: 0.0 min 0% B -» 0.2 min 0% B → 2.9 min 70% B → 3.1 min 90% B → 4.5 min 90% B; Ofen: 50°C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 5:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2 mm, 3.0 μm; Eluent B: Acetonitril + 500 μl 50%-ige Ameisensäure / 1; Eluent A: Wasser + 500 μl 50%-ige Ameisensäure / 1; Gradient: 0.0 min 0% B -> 0.2 min 0% B → 2.9 min 70% B → 3.1 min 90% B → 4.5 min 90% B; Ofen: 50°C; Fluss: 0.8 ml/min; UV- Detektion: 210 nm.
Methode 6:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A, Fluss 1 ml/min -> 2.5 min 30% A, Fluss 2 ml/min → 3.0 min 5% A, Fluss 2 ml/min -» 4.5 min 5% A, Fluss 2 ml/min; Ofen: 50°C; UV-Detektion: 210 nm.
Methode 7:
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A, Fluss lml/min - 2.5 min 30% A, Fluss 2 ml/min → 3.0 min 5% A, Fluss 2 ml/min → 4.5 min 5% A, Fluss 2 ml/min; Ofen: 50°C; UV-Detektion: 210 nm.
Methode 8:
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Grom-Sil 120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A → 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A → 4.5 min 10% A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 210 nm.
Methode 9:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-18e 50 mm x 4.6 mm; Eluent A: Wasser + 500 μl 50%-ige Ameisensäure / 1; Eluent B: Acetonitril + 500 μl 50%-ige Ameisensäure / 1; Gradient: 0.0 min 10% B →- 3.0 min 95% B -> 4.0 min 95% B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min → 3.0 min 3.0 ml/min → 4.0 min 3.0 ml/min; UV-Detektion: 210 nm.
Methode 10:
Gerätetyp MS: Micromass TOF (LCT); Gerätetyp HPLC: 2-Säulen-Schaltung, Waters 2690; Säule: YMC-ODS-AQ, 50 mm x 4.6 mm, 3.0 μm; Eluent A: Wasser + 0.1 % Ameisensäure, Eluent B: Acetonitril + 0.1% Ameisensäure; Gradient: 0.0 min 100% A -> 0.2 min 95% A → 1.8 min 25% A → 1.9 min 10% A → 3.2 min 10% A; Ofen: 40°C; Fluss: 3.0 ml/min; UV-Detektion: 210 nm.
Methode 11:
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule: Uptisphere HDO, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50%-ige Ameisensäure; Gradient: 0.0 min 100% A → 0.2 min 100% A → 2.9 min 30% A → 3.1 min 10% A → 4.5 min 10% A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 nm.
Methode 12:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Uptisphere C 18, 50 mm x 2.0 mm, 3.0 μm; Eluent B: Acetonitril + 0.05% Ameisensäure, Eluent A: Wasser + 0.05% Ameisensäure; Gradient: 0.0 min 5% B → 2.0 min 40% B → 4.5 min 90% B → 5.5 min 90% B; Ofen: 45°C; Fluss:- 0.0 min 0.75 ml/min → 4.5 min 0.75 ml/min → 5.5 min 1.25 ml/min; UV-Detektion: 210 nm.
Methode 13:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: HP 1100 Series; UV DAD; Säule: Phenomenex Synergi 2μ Hydro-RP Mercury 20 mm x 4 mm; Eluent A: 1 1 Wasser + 0.5 ml 50%-ige Ameisensäure, Eluent B: 1 1 Acetonitril + 0.5 ml 50%-ige Ameisensäure; Gradient: 0.0 min 90% A -» 2.5 min 30% A →- 3.0 min 5% A → 4.5 min 5% A; Fluss: 0.0 min 1 ml/min → 2.5 min/3.0 min/4.5 min 2 ml/min; Ofen: 50°C; UV-Detektion: 210 nm.
Methode 14:
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2795; Säule: Merck Chromolith SpeedROD RP-18e 50 mm x 4.6 mm; Eluent A: Wasser + 500 μl 50%-ige Ameisensäure / 1, Eluent B: Acetonitril + 500 μl 50%-ige Ameisensäure / 1; Gradient: 0.0 min 10% B -» 2.0 min 95% B → 4.0 min 95% B; Ofen: 35°C; Fluss: 0.0 min 1.0 ml/min → 2.0 min 3.0 ml/min → 4.0 min 3.0 ml/min; UV-Detektion: 210 nm.
Chirale HPLC-Methoden:
Methode 15A räparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 20 mm; Elutionsmittel: i- Hexan/Ethanol + 0.2% Diethylamin 40:60 (v/v); Fluss: 20 ml/min; UV-Detektion: 220 nm; Temperatur: 30°C; Probenaufgabe in i-Hexan Ethanol 1:1.
Methode 15B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: i- Hexan/Ethanol + 0.2% Diethylamin 40:60 (v/v); Fluss: 0.7 ml/min; UV-Detektion: 220 nm; Temperatur: 30°C; Probenaufgabe im Eluenten.
Methode 16A fpräparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD-H; 5 μm, 250 x 20 mm; Elutionsmittel: Acetonitril/Methanol + 0.2% Diethylamin 90:10 (v/v); Fluss: 20 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe in Acetonitril/Methanol 54:46 (v/v).
Methode 16B (analytisch):
Säule: Daicel Chiralpak AD-H; 5 μm, 250 x 20 mm; Elutionsmittel: Acetonitril/ Methanol + 0.2% Diethylamin 90:10 (v/v); Fluss: 1.0 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 17A fpräparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD-H; 5 μm, 250 x 20 mm; Elutionsmittel: Acetonitril/Methanol + 0.2% Diethylamin 85:15 (v/v); Fluss: 20 ml/min; UV-Detektion: 230 nm; Temperatur: 25°C; Probenaufgabe in Acetonitril.
Methode 17B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD-H; 5 μm, 250 x 4.6 mm; Elutionsmittel: Methanol/Acetonitril + 0.5% Diethylamin 20:80 (v/v); Fluss: 0.5 ml/min; UV-Detektion: 230 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 18A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD-H; 5 μm, 250 x 20 mm; Elutionsmittel: Acetonitril/Methanol + 0.2% Diethylamin 60:40 (v/v); Fluss: 20 ml/min; UV-Detektion: 230 nm; Temperatur: 25°C; Probenaufgabe in Acetonitril/Methanol 58:42 (v/v).
Methode 18B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD-H; 5 μm, 250 x 4.6 mm; Elutionsmittel: Methanol/Acetonitril + 0.5% Diethylamin 20:80 (v/v); Fluss: 1.0 ml/min; UV-Detektion: 230 nm; Temperatur: 30°C; Probenaufgabe im Eluenten.
Methode 19A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 10 mm; Elutionsmittel: Ethanol + 0.2% Diethylamin; Fluss: 10 ml/min; UV-Detektion: 220 nm; Temperatur: 40°C; Probenaufgabe in Ethanol.
Methode 19B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Ethanol + 0.2% Diethylamin; Fluss: 0.7 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 20A (präparativ):
Säule: Chiraler Kieselgelselektor KBD 5326A, basierend auf dem Selektor Poly-(N-methacryloyl- L-leucin-dicyclopropylamid); 10 μm, 250 x 20 mm; Elutionsmittel: iso-Hexan/Methyl-tert.-butyl- ether 1:1 (v/v); Fluss: 25 ml/min; UV-Detektion: 254 nm; Temperatur: 24°C; Probenaufgabe in iso-Hexan/Methyl-tert.-butylether 1:1.
Methode 20B (analytisch):
Säule: Chiraler Kieselgelselektor KBD 5326A; 10 μm, 250 x 4.6 mm; Elutionsmittel: iso- Hexan/Methyl-tert.-butylether 2:3 (v/v); Fluss: 1 ml/min; UV-Detektion: 254 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 21 A (präparativ):
Säule: Chiraler Kieselgelselektor KBD 5326A, basierend auf dem Selektor Poly-(N-methacryloyl- L-leucin-dicyclopropylamid); 10 μm, 250 x 20 mm; Elutionsmittel: iso-Hexan/Methyl-tert.-butyl- ether 2:3 (v/v); Fluss: 25 ml/min; UV-Detektion: 254 nm; Temperatur: 24°C; Probenaufgabe in iso-Hexan/Methyl-tert.butylether 1:1.
Methode 21B (analytisch):
Säule: Chiraler Kieselgelselektor KBD 5326A; 10 μm, 250 x 4.6 mm; Elutionsmittel: iso- Hexan/Methyl-tert.-butylether 2:3 (v/v); Fluss: 1 ml/min; UV-Detektion: 254 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 22A (präparativ):
Säule: Chiraler Kieselgelselektor KBD 8361, basierend auf dem Selektor Poly-(N-methacryloyl-L- leucin-1-menthylamid); 10 μm, 250 x 20 mm; Elutionsmittel: iso-Hexan/Methyl-tert.-butylether 3:2 (v/v); Fluss: 25 ml/min; UV-Detektion: 254 nm; Temperatur: 24°C; Probenaufgabe in iso- Hexan/Methyl-tert.-butylether 3:2.
Methode 22B (analytisch):
Säule: Chiraler Kieselgelselektor KBD 5326A; 10 μm, 250 x 4.6 mm; Elutionsmittel: iso- Hexan Methyl-tert.-butylether 2:3 (v/v); Fluss: 1 ml/min; UV-Detektion: 254 nm; Temperatur: 25 °C; Probenaufgabe im Eluenten.
Methode 23 A (präparativ):
Säule: Kromasil 100 C18; 5 μm, 250 x 20 mm; Fluss: 25 ml/min; Laufzeit: 20 min; Eluent A: Wasser + 0.2% Trifluoressigsäure, Eluent B: Acetonitril; Gradient: 5% B (0 min) - 95% B (15 min) → 5% B (15.1 min) → 5% B (20 min); UV-Detektion: 210 nm; Temperatur: 40°C; Probenaufgabe in Acetonitril/Wasser 2:1 (v/v).
Methode 23B (analytisch):
Säule: Kromasil 100 C18; 5 μm, 250 x 4 mm; Fluss: 1.0 ml/min; Laufzeit: 20 min; Eluent A: Wasser + 0.2% Trifluoressigsäure, Eluent B: Acetonitril; Gradient: 5% B (0 min) → 95% B (10 min) -> 95% B (15.0 min) → 5% B (15.1 min) → 5% B (20 min); UV-Detektion: 210 nm; Temperatur: 40°C; Probenaufgabe in Acetonitril/Wasser 2:1 (v/v).
Methode 24A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 20 mm; Elutionsmittel: i- Hexan Ethanol + 0.2% Diethylamin 90:10 (v/v); Fluss: 25 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe in i-Hexan/Ethanol 5:1 (v/v).
Methode 25 A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 20 μm, 500 x 40 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 85:15 (v/v); Fluss: 100 ml/min; UV-Detektion: 220 nm; Temperatur: 30°C; Probenaufgabe in Isopropanol.
Methode 25B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 85:15 (v/v); Fluss: 1.0 ml min; UV-Detektion: 250 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 26A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 20 μm, 350 x 30 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 75:25 (v/v); Fluss: 50 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe in Isopropanol/Methanol 75:25 (v/v).
Methode 26B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Isopropanol Methanol + 0.2% Diethylamin 85:15 (v/v); Fluss: 1.0 ml/min; UV-Detektion: 250 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 27A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 20 μm, 500 x 40 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 75:25 (v/v); Fluss: 50 ml/min; UV-Detektion: 220 nm; Temperatur: 25 °C; Probenaufgabe in Isopropanol/Methanol 75:25 (v/v).
Methode 27B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Isopropanol/Methanol + 0.2% Diethylamin 85:15 (v/v); Fluss: 1.0 ml/min; UV-Detektion: 250 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 28A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 20 μm, 500 x 40 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 85:15 (v/v); Fluss: 50 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe in Isopropanol/Methanol 85:15 (v/v).
Methode 28B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 5:1 (v/v); Fluss: 1 ml/min; UV-Detektion: 250 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 29 A (präparativ):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 20 mm; Elutionsmittel: i- Hexan Ethanol + 0.2% Diethylamin 85:15 (v/v); Fluss: 25 ml/min; UV-Detektion: 220 nm; Temperatur: 25°C; Probenaufgabe in Ethanol.
Methode 29B (analytisch):
Säule: Chiraler Kieselgelselektor Daicel Chiralpak AD; 10 μm, 250 x 4.6 mm; Elutionsmittel: Isopropanol/Methanol + 0.1% Diethylamin 5:1 (v/v); Fluss: 1 ml/min; UV-Detektion: 250 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Methode 30A (präparativ):
Säule: Chiraler Kieselgelselektor ZWE 803AB, basierend auf dem Selektor Poly-(N- methacryloyl-L-phenylalanin-d-neomenthylamid); 10 μm, 250 x 20 mm; Elutionsmittel: iso- Hexan/Methyl-tert.-butylether 1:4 (v/v); Fluss: 25 ml/min; UV-Detektion: 254 nm; Temperatur: 24°C; Probenaufgabe in Methyl-tert.-butylether.
Methode 30B (analytisch):
Säule: Chiraler Kieselgelselektor ZWE 803 AB; 10 μm, 250 x 4.6 mm; Elutionsmittel: Methyl- tert.-butylether; Fluss: 1 ml/min; UV-Detektion: 254 nm; Temperatur: 25°C; Probenaufgabe im Eluenten.
Ausgangsverbindungen und Intermediate:
Beispiel 1A
3 ,5-Diethoxyphenol
In eine Lösung von 188 g (1.49 mol) Phloroglucinol in 600 ml Ethanol wird unter RF für 5 h Chlorwasserstoff-Gas eingeleitet. Nach dem Abkühlen wird über Nacht bei RT gerührt. Anschließend wird erneut unter RF für 5 h Chlorwasserstoff-Gas eingeleitet. Nach dem Abkühlen wird die Reaktionsmischung eingeengt und der Rückstand in Dichlormethan und Wasser aufgenommen. Die organische Phase wird abgetrennt, die wässrige Phase zweimal mit Dichlormethan extrahiert, und die vereinigten organischen Phasen werden über Natriumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird im Vakuum destilliert (Kp.: 145-150°C / 0.5 mbar). Man löst das Destillat in Dichlormethan, extrahiert zur Entfernung von 5-Ethoxyresorcin fünfmal mit 5%-
iger wässriger Kaliumcarbonat-Lösung, trocknet die organische Phase über Natriumsulfat, filtriert und engt ein. Man erhält 156 g (57% d. Th.) des Produkts.
LC-MS (Methode 1): Rt = 3.32 min.
MS (ESIpos): m/z = 183 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 9.35 (s, 1 H), 5.90 (s, 3 H), 3.91 (q, 4 H), 1.27 (t, 6 H).
Beispiel 2A
(4-Bromphenyl)-(3 ,5 -diethoxyphenoxy)-essigsäuremethylester
20.0 g (110 mmol) 3,5-Diethoxyphenol und 30.7 g (99.8 mmol) Brom-(4-bromphenyl)-essigsäure- methylester werden unter Argon bei RT in 39 ml 2-Butanon gelöst und mit 31.0 g (225 mmol) Kaliumcarbonat versetzt. Man erhitzt die Reaktionsmischung 4 h unter RF. Nach dem Abkühlen wird der Niederschlag abgesaugt, mit 2-Butanon nachgewaschen und das Filtrat eingeengt. Nach Chromatographie an Kieselgel 60 (Laufmittel: Toluol) erhält man 33.1 g (81% d. Th.) des Produkts.
LC-MS (Methode 2): Rt = 4.64 min.
MS (ESIpos): m/z = 409 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.66-7.60 (m, 2 H), 7.53-7.46 (m, 2 H), 6.11 (d, 2 H), 6.09 (t, 1 H), 6.05 (s, 1 H), 3.95 (q, 4 H), 3.66 (s, 3 H), 1.28 (t, 6 H).
Beispiel 3A
(4-Bromphenyl)-(3,5-diethoxyphenoxy)-essigsäure
16.9 g (41.3 mmol) (4-Bromphenyl)-(3,5-diethoxyphenoxy)-essigsäuremethylester werden unter Argon bei RT in 80 ml Methanol und 8 ml Wasser gelöst und mit 7.99 g (57.8 mmol) Kaliumcarbonat versetzt. Man erhitzt 4 h unter RF. Nach dem Abkühlen wird der Niederschlag abfiltriert, das Filtrat eingeengt, der Rückstand in Wasser aufgenommen und die Lösung viermal mit Diethylether extrahiert. Man stellt in der wässrigen Phase durch Zusatz von 10%-iger Salzsäure einen pH- Wert von 2 ein, extrahiert dreimal mit Essigsäureethylester, wäscht die vereinigten Essigsäureethylester-Phasen mit gesättigter Natriumchlorid-Lösung, trocknet über Natriumsulfat, filtriert und engt ein. Man erhält 16.1 g (99% d. Th.) des Produkts.
LC-MS (Methode 4): Rt = 3.85 min.
MS (ESIpos): m/z = 395 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 13.37 (s, 1 H), 7.66-7.58 (m, 2 H), 7.53-7.45 (m, 2 H), 6.11- 6.06 (m, 3 H), 5.83 (s, 1 H), 3.95 (q, 4 H), 1.28 (t, 6 H).
Beispiel 4A
2-(4-Bromphenyl)-4, 6-diethoxybenzofuran-3 (2H)-on
Zu 16.1 g (40.7 mmol) (4-Bromphenyl)-(3,5-diethoxyphenoxy)-essigsäure gibt man unter Argon bei RT 38 ml (407 mmol) Phosphorylchlorid. Man kühlt auf 0°C und setzt 8.32 g (61.0 mmol) Zinkchlorid hinzu. Nach 10 min entfernt man das Eisbad und rührt über Nacht bei RT. Die Reaktionsmischung wird in viel Eiswasser gegossen und 15 min gerührt. Man setzt Dichlormethan hinzu, trennt die Phasen, wäscht die organische Phase mit Wasser, trocknet über Natriumsulfat,
filtriert und engt ein. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester) erhält man 10.2 g (67% d. Th.) des Produkts.
LC-MS (Methode 4): Rt = 4.04 min.
MS (ESIpos): m/z = 377 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.64-7.58 (m, 2 H), 7.28-7.23 ( , 2 H), 6.46 (d, 1 H), 6.19 (d, 1 H), 5.72 (s, 1 H), 4.22-4.05 (m, 4 H), 1.39-1.26 (m, 6 H).
Beispiel 5A
(S*,R*)-3-[2-(4-Bromphenyl)-4,6-diethoxy-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenylpropanal
Unter Argon werden 20.9 g (55.4 mmol) 2-(4-Bromphenyl)-4,6-diethoxybenzofuran-3-on in 422 ml entgastem Methanol und 100 ml entgastem Toluol gelöst. Bei RT setzt man 2.00 g (11.1 mmol) einer 30%-igen methanolischen Natriummethylat-Lösung hinzu. Eine Minute später gibt man in 111 ml entgastem Toluol gelösten Zimtaldehyd (9.52 g, 72.0 mmol) hinzu. Es wird über Nacht bei RT gerührt und dann 48 h stehen gelassen. Anschließend wird gesättigte Ammoniumchlorid- Lösung hinzugesetzt und dreimal mit Dichlormethan extrahiert. Man trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert und engt ein. Nach Säulen-Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester 95:5) erhält man 10.7 g (38% d. Th.) der Titelverbindung als racemisches Gemisch und 6.00 g (21% d. Th.) des Diastereomers (R*,R*)-3- [2-(4-Bromphenyl)-4,6-diethoxy-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenylpropanal als racemisches Gemisch.
LC-MS (Methode 4): Rt = 4.19 min.
MS (ESIpos): m/z = 509 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 9.32 (d, 1 H), 7.69-7.62 (m, 2 H), 7.61-7.54 (m, 2 H), 7.31- 7.10 (m, 5 H), 6.48 (d, 1 H), 5.97 (d, 1 H), 4.24 (dd, 1 H), 4.12 (q, 2 H), 4.01-3.81 (m, 2 H), 3.02 (ddd, 1 H), 2.57-2.44 (m, 1 H), 1.33 (t, 3 H), 1.18 (t, 3 H).
Beispiel 6A
(lS*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta- [b]benzofuran-l ,8b-(lH)-diol
Man legt unter Argon 178 ml (17.9 mmol) einer 0.1 M Lösung von Samariumdiiodid in THF bei 0°C vor und tropft eine Lösung von 2.60 g (5.10 mmol) (S*,R*)-3-[2-(4-Bromphenyl)-4,6- diethoxy-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenylpropanal in 30 ml entgastem THF hinzu. Man rührt 1 h bei 0°C und 1 h bei RT. Man gibt eine eisgekühlte gesättigte Natrium-Kalium- Tartrat-Lösung mit 10% Kaliumcarbonat hinzu, trennt die Phasen, extrahiert die wässrige Phase dreimal mit Dichlormethan, wäscht die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung, trocknet über Natriumsulfat, filtriert und engt ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester 95:5) erhält man 1.55 g (58% d. Th.) des Produkts als racemisches Gemisch.
LC-MS (Methode 3): Rt = 4.62 min.
MS (ESIpos): m/z = 511 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.35-7.28 (m, 2 H), 7.24-7.02 (m, 5 H), 6.91-6.84 (m, 2 H), 6.28 (d, 1 H), 6.14 (d, 1 H), 5.81 (d, 1 H), 4.84 (s, 1 H), 4.71 (ddd, 1 H), 4.13-3.98 (m, 4 H), 3.32- 3.18 (m, 1 H), 2.58-2.41 ( , 1 H), 2.15 (dt, 1 H), 1.35 (t, 3 H), 1.33 (t, 3 H).
Beispiel 7A
(3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l-on
2.60 g (5.08 mmol) (lS*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol werden unter Argon in 19 ml DMSO gelöst und auf 0°C gekühlt. Man tropft 7.80 ml (55.9 mmol) TEA und eine Lösung von 2.43 g (15.3 mmol) Schwefeltrioxid-Pyridin-Komplex in 9.60 ml DMSO hinzu, lässt auf RT erwärmen und rührt über Nacht. Anschließend wird mit eisgekühlter gesättigter Ammoniumchlorid-Lösung versetzt und 30 min gerührt. Der Niederschlag wird abgesaugt und mit wenig Wasser gewaschen. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester) erhält man 2.00 g (77% d. Th.) des Produkts als racemisches Gemisch.
LC-MS (Methode 5): Rt = 4.02 min.
MS (ESIpos): m/z = 509 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.39-6.90 (m, 9 H), 6.43 (d, 1 H), 6.17 (d, 1 H), 5.82 (s, 1 H), 4.20-3.93 (m, 4 H), 3.70 (dd, 1 H), 3.12 (dd, 1 H), 2.93 (dd, 1 H), 1.34 (t, 3 H), 1.30 (t, 3 H).
Beispiel 8A
(lR*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta- [b]benzofuran-l ,8b-(lH)-diol
Man legt unter Argon 814 mg (3.09 mmol) Tetramethylammonium-triacetoxyborhydrid in 1.2 ml Acetonitril vor, gibt 1.2 ml Eisessig hinzu und rührt 0.5 h bei RT. Dann werden 105 mg
(0.21 mmol) (3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-l-on als Lösung in 12 ml Acetonitril hinzugetropft. Man rührt bei RT über Nacht. Bei 0°C gibt man zu der Reaktionsmischung gesättigte Natrium- hydrogencarbonat-Lösung, extrahiert die wässrige Phase zweimal mit Dichlormethan, trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert und engt ein. Nach Gradienten- Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester) erhält man 77 mg (71% d. Th.) des Produkts als racemisches Gemisch.
LC-MS (Methode 1): Rt = 4.20 min.
MS (ESIpos): m/z = 512 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.27-7.18 (m, 2 H), 7.15-6.96 (m, 7 H), 6.24 (d, 1 H), 6.10 (d, 1 H), 4.95 (s, 1 H), 4.54-4.46 (m, 1 H), 4.42 (d, 1 H), 4.13-3.89 (m, 5 H), 2.70 (dt, 1 H), 2.01 (dd, 1 H), 1.33 (t, 3 H), 1.31 (t, 3 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 22A durchgeführt.
Analytische Daten (Methode 22B):
Enantiomer A: Rt = 3.66 min., Enantiomer B: Rt = 4.30 min.
Beispiel 9A
(2R*,3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6,8-diethoxy-8b-hydroxy-l-oxo-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-2 -carbonsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17A ausgehend von Beispiel 7A hergestellt. Die Verbindung wird direkt ohne weitere Charakterisierung in der nächsten Stufe eingesetzt.
Beispiel 10A
(4-Chlo henyl)-(3,5-diethoxyphenoxy)-essigsäuremethylester
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2A ausgehend von Brom-(4- chlorphenyl)-essigsäuremethylester hergestellt.
Ausbeute: 96% d. Th.
LC-MS (Methode 2): Rt = 3.33 min.
MS (ESIpos): m/z = 365 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.58-7.47 (m, 4 H), 6.12-6.08 ( , 4 H), 3.95 (q, 4 H), 3.66 (s, 3 H), 1.28 (t, 3 H).
Beispiel 11A
(4-Chlorphenyl)-(3,5-diethoxyphenoxy)-essigsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 3A ausgehend von Beispiel 10A hergestellt.
Ausbeute: 91% d. Th.
LC-MS (Methode 1): Rt = 3.80 min.
MS (ESIpos): m/z = 351 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 13.26 (br. s, 1 H), 7.57 (d, 2 H), 7.48 (d, 2 H), 6.11-6.09 (m, 3 H), 5.85 (s, 1 H), 3.95 (q, 2 H), 1.28 (t, 3 H).
Beispiel 12A
2-(4-Chlorphenyl)-4,6-diethoxybenzofuran-3( H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel HA hergestellt.
Ausbeute: 85% d. Th.
LC-MS (Methode 1): Rt = 4.00 min.
MS (ESIpos): m/z = 333 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.48 (d, 2 H), 7.31 (d, 2 H), 6.47 (d, 1 H), 6.20 (d, 1 H), 5.75 (s, 1 H), 4.22-4.04 (m, 4 H), 1.39-1.37 (m, 6 H).
Beispiel 13A
(S * ,R*)-3 -[2-(4-Chlorphenyl)-4,6-diethoxy-3 -oxo-2,3 -dihydrobenzofuran-2-yl]-3 -phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 12A hergestellt.
Ausbeute: 35% d. Th. (reines Diastereomer)
LC-MS (Methode 4): Rt = 4.09 min.
MS (ESIpos): m/z = 465 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 9.32 (d, 1 H), 7.64 (d, 2 H), 7.51 (d, 2 H), 7.28-7.25 (m, 2 H), 7.19-7.08 (m, 3 H), 6.46 (d, 1 H), 5.97 (d, 1 H), 4.23 (dd, 1 H), 4.12 (q, 2 H), 3.98-3.85 (m, 2 H), 3.01 (ddd, 1 H), 2.54-2.47 (m, 1 H), 1.33 (t, 3 H), 1.17 (t, 3 H).
Beispiel 14A
(lS*,3S*,3aR*,8bS*)-3a-(4-Chlorphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta- [b]benzofuran-l ,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 13 A hergestellt.
Ausbeute: 55% d. Th.
LC-MS (Methode 3): Rt = 3.79 min.
MS (ESIpos): m/z = 467 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.26-7.05 (m, 7 H), 6.89-6.85 (m, 2 H), 6.28 (d, 1 H), 6.14 (d, 1 H), 5.81 (d, 1 H), 4.84 (s, 1 H), 4.76-4.67 (m, 1 H), 4.12-3.99 (m, 4 H), 3.31-3.19 (m, 1 H), 2.47- 2.42 (m, 1 H), 2.23-2.04 (m, 1 H), 1.39-1.30 (m, 6 H).
Beispiel 15A
(3S*,3aR*,8bR*)-3a-(4-Chlorphenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 14A hergestellt.
Ausbeute: 93% d. Th.
LC-MS (Methode 5): Rt= 1.27 min.
MS (ESIpos): m/z = 465 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.19-6.97 (m, 9 H), 6.43 (d, 1 H), 6.16 (d, 1 H), 5.78 (s, 1 H), 4.12-3.95 (m, 4 H), 3.71 (dd, 1 H), 3.15-2.89 (m, 2 H), 1.34 (t, 3 H), 1.30 (t, 3 H).
Beispiel 16A
(lR*,3S*,3aR*,8bS*)-3a-(4-Chlorphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta- [b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 8A ausgehend von Beispiel 15A hergestellt.
Ausbeute: 83% d. Th.
LC-MS (Methode 2): Rt = 4.59 min.
MS (ESIneg): m/z = 465 (M-TTf
'H-NMR (200 MHz, DMSO-d6): δ = 7.20-6.96 (m, 9 H), 6.24 (d, 1 H), 6.10 (d, 1 H), 4.95 (s, 1 H), 4.54-4.47 (m, 1 H), 4.42 (d, 1 H), 4.13-3.89 (m, 5 H), 2.70 (dt, 1 H), 2.01 (dd, 1 H), 1.33 (t, 3 H), 1.31 (t, 3 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 21 A durchgeführt.
Analytische Daten (Methode 21B):
Enantiomer A: Rt = 4.52 min., Enantiomer B: Rt = 7.56 min.
Beispiel 17A
(2R*,3S*,3aR*,8bR*)-3a-(4-Chlorphenyl)-6,8-diethoxy-8-hydroxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofüran-2-carbonsäure
In einem geschlossenen Gefäß wird eine Mischung aus 500 mg (1.08 mmol) (3S*,3aR*,8bR*)-3a- (4-Chlo henyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tettahyάϊocyclopenta[b]benzofuran- 1-on (Beispiel 15A) und 1.08 ml (2.15 mmol) einer 2 M Lösung von Methoxymagnesiummethyl- carbonat in DMF für 16 h bei 100°C unter Rühren erhitzt. Die entstandene Suspension wird dann auf eine Mischung aus eisgekühlter 5 N Salzsäure und Ethylacetat gegossen. Die organische Phase wird abgetrennt, mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, abgesaugt und eingeengt. 531 mg Rohprodukt werden direkt ohne weitere Charakterisierung in der nächsten Stufe eingesetzt.
Beispiel 18A
3,5-Bis-(2-methoxyethoxy)-phenol
In eine Lösung von 50.0 g (397 mmol) Phloroglucinol in 500 ml Ethylenglykolmonomethylether wird bei 80°C über 4 h Chlorwasserstoff-Gas eingeleitet. Nach Zugabe von weiteren 300 ml Ethylenglykolmonomethylether wird für 2 h bei weiterer Einleitung von Chlorwasserstoff-Gas unter RF erhitzt. Man kühlt ab, engt ein, nimmt den Rückstand in 200 ml DMF auf, gibt 10.9 g (79.2 mmol) Kaliumcarbonat hinzu und erwärmt auf 50°C. Dann werden 10.9 g (78.2 mmol) 2- Bromethylmethylether als Lösung in 100 ml DMF zugetropft. Man rührt 2 h bei 50°C. Anschließend engt man ein, nimmt den Rückstand in einem Gemisch aus Diethylether und Wasser auf, trennt die Phasen und extrahiert die wässrige Phase zweimal mit Diethylether. Die wässrige Phase wird mit konzentrierter Salzsäure auf einen pH-Wert von 5 eingestellt und dreimal mit Diethylether extrahiert. Man wäscht die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung, trocknet über Natriumsulfat, filtriert und engt ein. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester) erhält man 9.20 g (10% d. Th.) des Produkts.
LC-MS (Methode 9): R, = 1.60 min.
MS (ESIpos): m/z = 243 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 9.35 (s, 1 H), 5.97-5.91 (m, 3 H), 4.01-3.96 (m, 4 H), 3.63- 3.58 (m, 4 H), 3.29 (s, 6 H).
Beispiel 19A
[3,5-Bis-(2-methoxyethoxy)-phenoxy]-(4-bromphenyl)-essigsäuremethylester
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2A ausgehend von Beispiel 18A hergestellt.
Ausbeute: 73% d. Th.
LC-MS (Methode 3): Rt = 4.17 min.
MS (ESIpos): m/z = 470 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.66-7.60 (m, 2 H), 7.52-7.46 ( , 2 H), 6.17-6.13 (m, 3 H), 6.07 (s, 1 H), 4.05-4.00 (m, 4 H), 3.66 (s, 3 H), 3.63-3.59 (m, 4 H), 3.29 (s, 6 H).
Beispiel 20A
[3,5-Bis-(2-methoxyethoxy)-phenoxy]-(4-bromphenyl)-essigsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 3A ausgehend von Beispiel 19A hergestellt.
Ausbeute: 86% d. Th.
LC-MS (Methode 3): Rt = 3.68 min.
MS (ESIpos): m z = 456 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 13.3 (s, 1 H), 7.65-7.59 ( , 2 H), 7.53-7.47 (m, 2 H), 6.14 (s, 3 H), 5.86 (s, 1 H), 4.05-4.00 (m, 4 H), 3.64-3.59 (m, 4 H), 3.29 (s, 6 H).
Beispiel 21 A
4,6-Bis-(2-methoxyethoxy)-2-(4-bromphenyl)-benzofuran-3(2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel 20A hergestellt.
Ausbeute: 19% d. Th.
LC-MS (Methode 2): Rt = 4.29 min.
MS (ESIpos): m/z = 438 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.64-7.58 (m, 2 H), 7.29-7.23 (m, 2 H), 6.50 (d, 1 H), 6.26 (d, 1 H), 5.74 (s, 1 H), 4.27-4.15 (m, 4 H), 3.71-3.62 (m, 4 H), 3.32 (s, 3 H), 3.31 (s, 3 H).
Beispiel 22A
(S*,R*)-3-[4,6-Bis-(2-methoxyethoxy)-2-(4-bromphenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3- phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 21 A hergestellt.
Ausbeute: 30% d. Th.
LC-MS (Methode 2): Rt = 4.48 min.
MS (ESIpos): m/z = 570 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 9.32 (d, 1 H), 7.67-7.62 (m, 2 H), 7.61-7.55 (m, 2 H), 7.29- 7.23 (m, 2 H), 7.20-7.09 (m, 3 H), 6.51 (d, 1 H), 6.04 (d, 1 H), 4.24 (dd, 1 H), 4.22-4.17 (m, 2 H), 4.08-3.93 (m, 2 H), 3.68-3.63 (m, 2 H), 3.54-3.49 (m, 2 H), 3.30 (s, 3 H), 3.22 (s, 3 H), 3.01 (ddd, 1 H), 2.56-2.46 (m, 1 H).
Beispiel 23A
3 ,5 -Bis-(2-chlorethoxy)-phenol
In eine Lösung von 150 g (1.19 mol) Phloroglucinol in 1000 ml 2-Chlorethanol wird bei 80°C über 5 h Chlorwasserstoff-Gas eingeleitet. Nach dem Abkühlen wird über Nacht bei RT gerührt. Anschließend erhitzt man unter weiterer Einleitung von Chlorwasserstoff-Gas für 2 h auf 80°C. Man kühlt ab, engt ein, nimmt den Rückstand in einem Gemisch aus Dichlormethan und Wasser auf, trennt die Phasen und extrahiert die wässrige Phase zweimal mit Dichlormethan. Man trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert und engt ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Toluol / Essigsäureethylester 95:5) erhält man 81.2 g (27% d. Th.) des Produkts.
LC-MS (Methode 4): Rt = 3.27 min.
MS (ESIpos): m/z = 252 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 9.51 (s, 1 H), 6.03-5.96 (m, 3 H), 4.20-4.13 (m, 4 H), 3.94- 3.86 (m, 4 H).
Beispiel 24A
[3,5-Bis-(2-chlorethoxy)-phenoxy]-(4-chlorphenyl)-essigsäuremethylester
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2A ausgehend von Beispiel 23A hergestellt.
Ausbeute: 76% d. Th.
LC-MS (Methode 4): Rt = 4.05 min.
MS (ESIpos): m/z = 433 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.59-7.54 (m, 2 H), 7.52-7.46 (m, 2 H), 6.23-6.18 (m, 3 H), 6.13 (s, 1 H), 4.24-4.19 (m, 4 H), 3.93-3.88 (m, 4 H), 3.67 (s, 3 H).
Beispiel 25A
[3,5-Bis-(2-chlorethoxy)-phenoxy]-(4-chlorphenyl)-essigsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 3A ausgehend von Beispiel 24A hergestellt.
Ausbeute: 92% d. Th.
LC-MS (Methode 4): Rt = 3.91 min.
MS (ESIpos): m z = 419 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 13.4 (s, 1 H), 7.61-7.53 (m, 2 H), 7.52-7.45 (m, 2 H), 6.19 (s, 3 H), 5.92 (s, 1 H), 4.25-4.16 (m, 4 H), 3.96-3.88 (m, 4 H).
Beispiel 26A
4,6-Bis-(2-chlorethoxy)-2-(4-chlorphenyl)-benzofuran-3(2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel 25A hergestellt.
Ausbeute: 55% d. Th.
LC-MS (Methode 3): Rt = 3.82 min.
MS (ESIpos): m/z = 401 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.51-7.45 (m, 2 H), 7.36-7.30 (m, 2 H), 6.57 (d, 1 H), 6.32 (d, 1 H), 5.79 (s, 1 H), 4.45-4.35 (m, 4 H), 4.02-3.89 (m, 4 H).
Beispiel 27A
(S*,R*)-3-[4,6-Bis-(2-chlorethoxy)-2-(4-chloφhenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3- phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 26A hergestellt.
Ausbeute: 48% d. Th.
LC-MS (Methode 4): Rt = 3.72 min.
MS (ESIpos): m/z = 533 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 9.33 (d, 1 H), 7.68-7.62 (m, 2 H), 7.54-7.48 (m, 2 H), 7.29- 7.24 (m, 2 H), 7.19-7.09 (m, 3 H), 6.57 (d, 1 H), 6.09 (d, 1 H), 4.40-4.35 (m, 2 H), 4.30-4.09 (m, 3 H), 3.99-3.94 (m, 2 H), 3.81-3.76 (m, 2 H), 3.02 (ddd, 1 H), 2.56-2.46 (m, 1 H).
Beispiel 28A
(lS*,3S*,3aR*,8bS*)-6,8-Bis-(2-chlorethoxy)-3a-(4-chlorphenyl)-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 27A hergestellt.
Ausbeute: 31% d. Th.
LC-MS (Methode 5): Rt = 4.01 min.
MS (ESIpos): m z = 535 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.26-7.15 (m, 4 H), 7.10-7.03 (m, 3 H), 6.92-6.86 (m, 2 H), 6.38 (d, 1 H), 6.25 (d, 1 H), 5.68 (d, 1 H), 4.83 (s, 1 H), 4.82-4.73 (m, 1 H), 4.36-4.28 (m, 4 H), 4.02-3.93 (m, 4 H), 3.29-3.23 (m, 1 H), 2.51-2.43 (m, 1 H), 2.17 (ddd, 1 H).
Beispiel 29A
(3S*,3aR*,8bR*)-6,8-Bis-(2-chlorethoxy)-3a-(4-chlθφhenyl)-8b-hydroxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran- 1 -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 28A hergestellt.
Ausbeute: 71% d. Th.
LC-MS. (Methode 2): Rt = 3.40 min.
MS (ESIpos): m/z = 533 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.21-7.15 (m, 2 H), 7.12-7.05 (m, 3 H), 7.02-6.96 (m, 4 H), 6.54 (d, 1 H), 6.30 (d, 1 H), 5.87 (s, 1 H), 4.36-4.26 (m, 4 H), 3.99-3.94 (m, 2 H), 3.90-3.85 (m, 2 H), 3.73 (dd, 1 H), 3.11 (dd, 1 H), 2.93 (dd, 1 H).
Beispiel 30A
(lR*,3S*,3aR*,8bS*)-6,8-Bis-(2-chlorethoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 8A ausgehend von Beispiel 29A hergestellt.
Ausbeute: 85% d. Th.
LC-MS (Methode 1): Rt = 4.09 min.
MS (ESIpos): m/z = 535 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.11-6.98 (m, 9 H), 6.33 (d, 1 H), 6.21 (d, 1 H), 4.97 (s, 1 H), 4.55-4.50 (m, 1 H), 4.36 (d, 1 H), 4.32-4.26 (m, 4 H), 4.02-3.90 (m, 5 H), 2.72 (ddd, 1 H), 2.03 (ddd, 1 H).
Beispiel 31A
[3,5-Bis-(2-chlorethoxy)-phenoxy]-(4-bromphenyl)-essigsäuremethylester
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2A ausgehend von Beispiel 23A hergestellt.
Ausbeute: 48% d. Th.
LC-MS (Methode 4): Rt = 4.11 min.
MS (ESIpos): m/z = 477 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.68-7.60 (m, 2 H), 7.54-7.45 (m, 2 H), 6.23-6.18 (m, 3 H), 6.13 (s, 1 H), 4.25-4.17 (m, 4 H), 3.95-3.87 (m, 4 H), 3.66 (s, 3 H).
Beispiel 32A
[3,5-Bis-(2-chlorethoxy)-phenoxy]-(4-bromphenyl)-essigsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 3A ausgehend von Beispiel 31A hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 2): Rt = 4.33 min.
MS (ESIpos): m/z = 463 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 13.4 (s, 1 H), 7.64-7.58 (m, 2 H), 7.52-7.46 (m, 2 H), 6.21- 6.16 (m, 3 H), 5.85 (s, 1 H), 4.24-4.18 (m, 4 H), 3.93-3.88 (m, 4 H).
Beispiel 33A
4,6-Bis-(2-chlorethoxy)-2-(4-bromphenyl)-benzofuran-3(2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel 32A hergestellt.
Ausbeute: 16% d. Th.
LC-MS (Methode 4): Rt = 3.96 min.
MS (ESIpos): m/z = 446 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.66-7.57 (m, 2 H), 7.30-7.22 (m, 2 H), 6.58 (d, 1 H), 6.32 (d, 1 H), 5.79 (s, 1 H), 4.46-4.34 (m, 4 H), 4.04-3.88 (m, 4 H).
Beispiel 34A
(S*,R*)-3-[4,6-Bis-(2-chlorethoxy)-2-(4-bromphenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3- phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 33A hergestellt.
Ausbeute: 35% d. Th.
LC-MS (Methode 4): Rt = 4.05 min.
MS (ESIpos): m/z = 578 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 9.33 (d, 1 H), 7.70-7.63 (m, 2 H), 7.62-7.54 (m, 2 H), 7.31- 7.09 (m, 5 H), 6.58 (d, 1 H), 6.10 (d, 1 H), 4.42-4.33 (m, 2 H), 4.31-4.07 (m, 3 H), 4.02-3.93 (m, 2 H), 3.83-3.76 (m, 2 H), 3.03 (ddd, 1 H), 2.58-2.44 (m, 1 H).
Beispiel 35A
(lS*,3S*,3aR*,8bS*)-6,8-Bis-(2-chlorethoxy)-3a-(4-bromρhenyl)-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 34A hergestellt.
Ausbeute: 30% d. Th.
LC-MS (Methode 2): Rt = 4.60 min.
MS (ESIpos): m/z = 579 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.34-7.28 (m, 2 H), 7.18-7.12 (m, 2 H), 7.09-7.03 (m, 3 H), 6.92-6.87 (m, 2 H), 6.37 (d, 1 H), 6.25 (d, 1 H), 5.68 (d, 1 H), 4.83 (s, 1 H), 4.81-4.73 (m, 1 H), 4.35-4.27 (m, 4 H), 4.01-3.93 (m, 4 H), 3.28-3.23 (m, 1 H), 2.54-2.42 ( , 1 H), 2.17 (ddd, 1 H).
Beispiel 36A
(4-Chloφhenyl)-(3-ethoxyphenoxy)-essigsäuremethylester
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2A ausgehend von 3-Ethoxy- phenol hergestellt.
Ausbeute: 97% d. Th.
LC-MS (Methode 5): Rt = 3.86 min.
MS (ESIpos): m/z = 321 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.57 (d, 2 H), 7.49 (d, 2 H), 7.19-7.13 (m, 1 H), 6.54-6.52 (m, 3 H), 6.07 (s, 1 H), 3.98 (q, 2 H), 3.66 (s, 3 H), 1.29 (t, 3 H).
Beispiel 37A
(4-Chlθφhenyl)-(3-ethoxyphenoxy)-essigsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 3A ausgehend von Beispiel 36A hergestellt.
Ausbeute: 96% d. Th.
LC-MS (Methode 4): Rt= 3.18 min.
MS (ESIpos): m/z = 307 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.58 "(d, 2 H), 7.48 (d, 2 H), 7.20-7.12 (m, 1 H), 6.55-6.50 (m, 3 H), 5.88 (s, 1 H), 3.98 (q, 2 H), 1.30 (t, 3 H).
Beispiel 38A
2-(4-Chloφhenyl)-6-ethoxybenzofuran-3 (2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel 37A hergestellt.
Ausbeute: 26% d. Th.
LC-MS (Methode 2): Rt = 3.37 min.
MS (ESIpos): m/z = 289 (M+H)+.
Beispiel 39A
(S * ,R*)-3 -[2-(4-Chloφhenyl)-6-ethoxy-3 -oxo-2,3 -dihydrobenzofuran-2-yl]-3 -phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 38A hergestellt.
Ausbeute: 19% d. Th. (reines Diastereomer)
LC-MS (Methode 12): Rt = 4.53 min.
MS (ESIpos): m/z = 421 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 9.34 (s, 1 H), 7.69 (d, 2 H), 7.52 (d, 2 H), 7.24 (d, 2 H), 7.18- 7.06 (m, 4 H), 6.90 (s, 1 H), 6.50 (dd, 1 H), 4.28 (dd, 1 H), 4.14 (q, 2 H), 3.12-3.04 (m, 1 H), 2.57- 2.52 (m, 1 H), 1.34 (t, 3 H).
Beispiel 40A
(2R*,3S*,3aR*,8bR*)-3a-(4-Chlθφhenyl)-6-ethoxy-8b-hydroxy-l-oxo-3-phenyl-2,3,3a,8b-tetra- hydro-lH-cyclopenta[b]benzofuran-2-carbonsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17A ausgehend von Beispiel 24 hergestellt. Die Verbindung wird direkt ohne weitere Charakterisierung in der nächsten Stufe eingesetzt.
Beispiel 41A
4-(2-Chlorethoxy)-2-hydroxyacetophenon
300 g (1.97 mol) 2,4-Dihydroxyacetophenon, 238 g (2.96 mol) 2-Chlorethanol und 776 g (2.96 mol) Triphenylphosphin werden in 4800 ml THF vorgelegt. Bei RT werden 380 g (2.96 mol) Azodicarbonsäurediisopropylester als Lösung in 1200 ml THF hinzugetropft. Man erhitzt 16 h
unter RF, kühlt dann ab und engt ein. Der Rückstand wird mit 2 N Natronlauge verrührt und abgesaugt. Man verrührt den Feststoff mit Essigsäureethylester, saugt ab, wäscht mit Essigsäureethylester und trocknet den Feststoff. Man nimmt den Feststoff in einem Gemisch aus 4 N Salzsäure und Essigsäureethylester auf, trennt die Phasen und extrahiert die wässrige Phase noch einmal mit Essigsäureethylester. Man wäscht die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung, trocknet über Natriumsulfat, filtriert und engt ein. Nach zweimaliger Säulenchromatographie an Kieselgel 60 (1. Laufmittel: Toluol, 2. Laufmittel: Cyclohexan) erhält man 232 g (55% d. Th.) des Produkts.
LC-MS (Methode 13): Rt = 2.41 min.
MS (ESIpos): m/z = 215 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 12.6 (s, 1 H), 7.86 (d, 1 H), 6.57 (dd, 1 H), 6.50 (d, 1 H), 4.38-4.30 (m, 2 H), 4.00-3.92 (m, 2 H), 2.57 (s, 3 H).
Beispiel 42A
6-(2-Chlorethoxy)-benzofuran-3-on
Man legt 76.0 g (354 mmol) 4-(2-Chlorethoxy)-2-hydroxyacetophenon bei -78°C in 1050 ml THF unter Argon vor und tropft 148 g (885 mmol) Lithiumhexamethyldisilazid als Lösung in 750 ml THF hinzu. Nach Erwärmen auf RT und Zusatz von 96.2 g (885 mmol) Chlortrimethylsilan wird 2 h bei RT gerührt. Anschließend gibt man bei 0°C portionsweise 65.6 g (369 mmol) N-Brom- succinimid hinzu, rührt 0.5 h bei 0°C und 1 h bei RT. Danach gibt man 369 ml 1 Ν Natronlauge hinzu und rührt 0.5 h bei RT. Nach Zugabe von gesättigter Ammoniumchlorid-Lösung extrahiert man die wässrige Phase mehrfach mit Diethylether, trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert und engt ein. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufmittel: Petrolether / Essigsäureethylester) erhält man 59.0 g (78% d. Th.) des Produkts.
LC-MS (Methode 6): Rt= 1.80 min.
MS (ESIpos): m/z = 213 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.54 (d, 1 H), 6.86 (d, 1 H), 6.73 (dd, 1 H), 4.77 (s, 2 H), 4.41-4.36 (m, 2 H), 4.00-3.95 (m, 2 H).
Beispiel 43A
2-Brom-6-(2-chlorethoxy)-benzofuran-3-on
Methode a):
127 g (597 mmol) 6-(2-Chlorethoxy)-benzofuran-3-on werden bei RT unter Argon in 1850 ml Dioxan und 1850 ml Diethylether gelöst und auf -5°C gekühlt. Unter starkem Rühren tropft man langsam 95.6 g (597 mmol) Brom hinzu und rührt 1 h bei 0°C. Anschließend gibt man auf Eiswasser, trennt die Phasen, extrahiert die wässrige Phase zweimal mit Dichlormethan, trocknet die vereinigten organischen Phasen über Natriumsulfat, filtriert und engt ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Toluol) erhält man 150 g (86% d. Th.) des Produkts als Hydrat.
Methode b):
Man gibt zu einer siedenden Suspension von 10.5 g (47.0 mmol) Kupfer(π)bromid in 60 ml Essigsäureethylester 5.00 g (23.5 mmol) 6-(2-Chlorethoxy)-benzofuran-3-on in 30 ml Chloroform. Es wird über Nacht unter RF erhitzt. Nach dem Abkühlen wird der Feststoff abfiltriert und mit Essigsäureethylester gewaschen. Das Filtrat wird mit Wasser und gesättigter wässriger Natriumchlorid-Lösung gewaschen, über Natriumsulfat getrocknet, filtriert und eingeengt. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufmittel: Toluol / Cyclohexan) erhält man 1.50 g (20% d. Th.) des Produkts als Hydrat.
LC-MS (Methode 2): Rt = 4.13 min.
MS (ESIpos): m/z = 291 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.52 (d, 1 H), 7.30 (s, 2 H), 6.75 (d, 1 H), 6.70 (dd, 1 H), 5.57 (s, 1 H), 4.42-4.37 (m, 2 H), 4.00-3.95 (m, 2 H).
Beispiel 44A
2-Brom-3-tert.-butyldimethylsilyloxy-6-(2-chlorethoxy)-benzofuran
1.50 g (5.15 mmol) des Hydrats von 2-Brom-6-(2-chlorethoxy)-benzofuran-3-on (Beispiel 43 A) werden zur azeotropen Entfernung von Wasser dreimal mit Toluol unter Argon eingeengt. Anschließend wird der Rückstand unter Argon in 25 ml Diethylether gelöst und auf 0°C gekühlt. Man tropft nacheinander langsam 0.79 ml (5.66 mmol) TEA und 1.30 ml (5.66 mmol) tert.-Butyl- dimethylsilyltrifluormethansulfonat zu und rührt 10 min bei 0°C und 1 h bei RT. Danach trennt man die Diethylether-Phase ab, extrahiert den Rückstand zweimal mit Diethylether und engt die vereinigten Diethylether-Phasen ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Cyclohexan) erhält man 2.00 g (96% d. Th.) des Produkts.
LC-MS (Methode 2): Rt = 4.92 min.
MS (ESIpos): m/z = 405 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.36 (d, 1 H), 7.21 (d, 1 H), 6.96 (dd, 1 H), 4.32-4.27 (m, 2 H), 3.98-3.93 (m, 2 H), 1.03 (s, 9 H), 0.23 (s, 6 H).
Beispiel 45A
3-tert.-Butyldimethylsilyloxy-6-(2-chlorethoxy)-2-(4-chloφhenyl)-benzofuran
Unter Argon löst man 2.00 g (4.93 mmol) 2-Brom-3-tert.-butyldimethylsilyloxy-6-(2-chlorethoxy)- benzofuran in 41 ml Toluol und gibt 0.92 g (5.91 mmol) 4-Chloφhenylboronsäure und eine Lösung aus 1.15 g (10.8 mmol) Natriumcarbonat in 5.4 ml Wasser hinzu. Man entgast und belüftet zweimal mit Argon, setzt 0.28 g (0.25 mmol) Tetrakis(triphenylphosphin)palladium(0) hinzu und erwärmt für 2 h auf 95°C. Nach dem Abkühlen gießt man die Reaktionsmischung in eine Mischung aus eiskalter, gesättigter Ammoniumchlorid-Lösung und Diethylether, trennt die Phasen, extrahiert die wässrige Phase zweimal mit Diethylether, wäscht die vereinigten organischen Phasen mit Wasser und gesättigter Kochsalz-Lösung, trocknet über Natriumsulfat, filtriert und engt ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Toluol / Cyclohexan 1:1) erhält man 1.69 g (78% d. Th.) des Produkts.
Methode b):
39.4 g (135 mmol) des Hydrats von 2-Brom-6-(2-chlorethoxy)-benzofuran-3-on (Beispiel 43A) werden zur azeotropen Entfernung von Wasser dreimal mit Toluol unter Argon eingeengt. Anschließend wird der Rückstand unter Argon in 1200 ml Toluol gelöst und auf -10°C gekühlt. Man tropft nacheinander langsam 22.7 ml (162 mmol) TEA und 34.2 ml (149 mmol) tert.-Butyl- dimethylsilyltrifluoimethansulfonat zu und rührt 0.5 h bei 0°C und 0.5 h bei RT. Danach trennt man die untere Triethylamin-Trifluormethansulfonsäuresalz-Phase ab und gibt die überstehende Lösung ohne weitere Aufreinigung unter Argon zu 25.4 g (162 mmol) 4-Chloφhenylboronsäure. Nach Zusatz von 31.6 g (298 mmol) Natriumcarbonat als Lösung in 148 ml Wasser wird durch Anlegen eines Vakuums entgast und mit Argon belüftet. Man gibt 7.82 g (6.76 mmol) Tetrakis- (triphenylphosphin)palladium(O) hinzu und entgast und belüftet mit Argon. Unter starkem Rühren wird für 2 h auf 95°C erhitzt. Nach dem Abkühlen trennt man die Phasen, wäscht die organische Phase dreimal mit gesättigter Ammoniumchlorid-Lösung, einmal mit Wasser und einmal mit gesättigter Natriumchlorid-Lösung, trocknet über Magnesiumsulfat, filtriert und engt ein. Nach Säulenchromatographie an Kieselgel 60 (Laufmittel: Toluol / Cyclohexan 1:1) erhält man 53.1 g (90% d. Th.) des Produkts, das noch zu 9% mit 4,4'-Dichlorbiρhenyl verunreinigt ist.
MS (DCI): m/z = 437 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.83-7.77 (m, 2 H), 7.56-7.51 (m, 2 H), 7.43 (d, 1 H), 7.21 (d, 1 H), 6.95 (dd, 1 H), 4.35-4.30 (m, 2 H), 3.99-3.94 (m, 2 H), 1.02 (s, 9 H), 0.12 (s, 6 H).
Beispiel 46A
6-(2-Chlorethoxy)-2-(4-chloφhenyl)-benzofuran-3(2H)-on
66.5 g (152 mmol) 3-tert.-Butyldimethylsilyloxy-6-(2-chlorethoxy)-2-(4-chloφhenyl)-benzofuran werden unter Argon bei RT in 500 ml einer 4 M Chlorwasserstoff-Lösung in Dioxan gelöst und 1.5 h gerührt. Man engt ein und erhält 49.2 g eines Rückstands, den man unter Argon lagert und ohne Aufreinigung weiter umsetzt.
LC-MS (Methode 5): Rt = 3.77 min.
MS (ESIpos): m/z = 323 (M+H)+.
Beispiel 47A
(S*,R*)-3-[6-(2-Chlorethoxy)-2-(4-chlθφhenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenyl- propanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 46A hergestellt.
Ausbeute: 31% d. Th.
LC-MS (Methode 4): Rt = 3.73 min.
MS (ESIpos): m/z = 455 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 9.34 (d, 1 H), 7.75-7.66 (m, 2 H), 7.58-7.49 (m, 2 H), 7.30- 7.05 ( , 6 H), 6.98 (d, 1 H), 6.56 (dd, 1 H), 4.43-4.34 (m, 2 H), 4.30 (dd, 1 H), 4.02-3.94 ( , 2 H), 3.09 (ddd, 1 H), 2.55 (dd, 1 H).
Beispiel 48A
(lS*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofüran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 47A hergestellt.
Ausbeute: 94% d. Th.
LC-MS (Methode 6): Rt = 2.73 min.
MS (ESIpos): m/z = 457 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.32-7.02 (m, 8 H), 6.96-6.88 (m, 2 H), 6.72 (d, 1 H), 6.62 (dd, 1 H), 5.86 (d, 1 H), 5.07 (s, 1 H), 4.45 (q, 1 H), 4.34-4.25 (m, 2 H), 4.00-3.93 (m, 2 H), 3.40- 3.25 (m, 1 H), 2.49-2.36 (m, 1 H), 2.20 (ddd, 1 H).
Beispiel 49A
(lS*,3S*,3aR*,8bS*)-6-(2-Azidoethoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofuran-l,8b-(lH)-diol
Zu 200 mg (0.44 mmol) (lS*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[ό]benzofuran-l,8b-(lH)-diol (Beispiel 48A) in 4 ml DMF unter Argon gibt man 56.9 mg (0.87 mmol) Natriumazid und erhitzt über Nacht auf 100°C. Nach dem Abkühlen engt man ein, nimmt den Rückstand mit Wasser und Dichlormethan auf, trennt die Phasen, extrahiert die wässrige Phase zweimal mit Dichlormethan, wäscht die vereinigten organischen Phasen mit gesättigter Natriumchlorid-Lösung, trocknet über Magnesiumsulfat, filtriert und engt ein. Nach Gradienten-Chromatographie an Kieselgel 60 (Laufinittel: Toluol / Essigsäureethylester) erhält man 190 mg (94% d. Th.) des Produkts.
LC-MS (Methode 9): Rt = 2.66 min.
MS (ESIpos): m/z = 464 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.29 (d, 1 H), 7.26-7.02 (m, 7 H), 6.96-6.90 (m, 2 H), 6.71 (d, 1 H), 6.61 (dd, 1 H), 5.83 (d, 1 H), 5.02 (s, 1 H), 4.49-4.41 (m, 1 H), 4.24-4.19 (m, 2 H), 3.69-3.63 (m, 2 H), 3.34-3.25 (m, 1 H), 2.50-2.40 (m, 1 H), 2.21 (ddd, 1 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 25A durchgeführt.
Analytische Daten (Methode 25B):
Enantiomer A: Rt = 5.52 min., Enantiomer B: Rt = 8.56 min.
Beispiel 50A
(3S*,3aR*,8bR*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-8b-hydroxy-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 48A hergestellt.
Ausbeute: 92% d. Th.
LC-MS (Methode 4): Rt = 3.54 min.
MS (ESIpos): m/z = 455 (M+H)
'H-NMR (200 MHz, DMSO-d6): δ = 7.29-7.00 (m, 10 H), 6.89 (d, 1 H), 6.66 (dd, 1 H), 6.31 (s, 1 H), 4.36-4.28 (m, 2 H), 4.02-3.94 (m, 2 H), 3.65 (dd, 1 H), 3.42-3.20 (m, 1 H), 2.84 (dd, 1 H).
Beispiel 51A
(lR*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofuran-l ,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 8A ausgehend von Beispiel 50A hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 2): Rt = 4.24 min.
MS (ESIneg): m/z = 501 (M-H)-
'H-NMR (200 MHz, DMSO-d6): δ = 7.32 (d, 1 H), 7.14-6.91 (m, 9 H), 6.66 (d, 1 H), 6.55 (dd, 1 H), 5.26 (s, 1 H), 5.17 (d, 1 H), 4.62-4.51 (m, 1 H), 4.32-4.24 (m, 2 H), 4.00-3.92 (m, 2 H), 3.90- 3.77 (m, 1 H), 2.66 (dt, 1 H), 1.98-1.83 (m, 1 H).
Beispiel 52A
(2R*,3S*,3aR:1:,8bR*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-8b-hydroxy-l-oxo-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-2-carbonsäure
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17A ausgehend von Beispiel 50A hergestellt. Die Verbindung wird ohne weitere Aufreinigung in der nächsten Stufe eingesetzt.
Beispiel 53A
(2R*,3S*,3aR*,8bR*)-6-(2-Chlorethoxy)-3a-(4-chlθφhenyl)-2-dimethylcarbamid-8b-hydroxy-3- phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 16 ausgehend von Beispiel 52A hergestellt.
Ausbeute: 14% d. Th. (ausgehend von Beispiel 50A)
LC-MS (Methode 13): Rt = 2.71 min.
MS (ESIpos): m/z = 526 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.23-7.16 (m, 5 H), 7.14-7.05 (m, 3 H), 7.02-6.94 (m, 2 H), 6.89 (d, 1 H), 6.64 (dd, 1 H), 6.47 (s, 1 H), 4.79 (d, 1 H), 4.38-4.29 (m, 2 H), 4.19 (d, 1 H), 4.02- 3.94 (m, 2 H), 3.28 (s, 3 H), 2.77 (s, 3 H).
Beispiel 54A
(lR*,2R*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-2-dimethylcarbamid-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[έ]benzofüran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17 ausgehend von Beispiel 53 A hergestellt.
Ausbeute: 81% d. Th.
LC-MS (Methode 6): Rt = 2.31 min.
MS (ESIpos): m/z = 528 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.36 (d, 1 H), 7.23-7.10 (m, 4 H), 7.06-6.96 (m, 3 H), 6.86- 6.79 (m, 2 H), 6.70 (d, 1 H), 6.55 (dd, 1 H), 5.50 (d, 1 H), 5.32 (s, 1 H), 4.92-4.82 (m, 1 H), 4.34- 4.23 (m, 2 H), 4.20-3.93 ( , 4 H), 3.20 (s, 3 H), 2.72 (s, 3 H).
Beispiel 55A
(3 S*,3 aR*,8bR*)-6-(2-Chlorethoxy)-3 a-(4-chloφhenyl)-8b-hydroxy-3-phenyl-2,3 ,3 a,8b-tetra- hydrocycloρenta[b]benzofuran-l-oxim
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 14 ausgehend von Beispiel 50A hergestellt.
Ausbeute: 82% d. Th.
LC-MS (Methode 6): Rt = 2.55 min.
MS (ESIneg): m/z = 468 (M-HV"
'H-NMR (300 MHz, DMSO-d6): δ = 11.1 (s, 1 H), 7.35 (d, 1 H), 7.23-7.06 (m, 7 H), 7.02-6.97 (m, 2 H), 6.79 (d, 1 H), 6.65 (dd, 1 H), 5.78 (s, 1 H), 4.34-4.28 (m, 2 H), 3.99-3.93 (m, 2 H), 3.45 (t, 1 H), 3.03 (d, 2 H).
Beispiel 56A
(lR*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-8b-hydroxy-3-ρhenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran- 1 -amin
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 15 ausgehend von Beispiel 55A durchgeführt.
Ausbeute: 89% d. Th.
LC-MS (Methode 6): Rt = 1.89 min.
MS (ESIpos): m/z = 456 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.27-6.90 (m, 10 H), 6.69 (d, 1 H), 6.58 (dd, 1 H), 4.32-4.24 (m, 2 H), 3.99-3.92 (m, 2 H), 3.62 (dd, 1 H), 3.43 (dd, 1 H), 2.44-2.31 ( , 1 H), 2.25-2.02 (m, 1 H).
Beispiel 57A
4-Benzyloxy-6-hydroxyacetophenon
Zu einer Lösung von 50.00 g (329 mmol) 2,4-Dihydroxyacetophenon in 500 ml DMF werden 41 ml (345 mmol) Benzylbromid und 47.69 g (345 mmol) Kaliumcarbonat gegeben und die resultierende Suspension über Nacht bei Raumtemperatur gerührt. Über eine Glasfritte wird dann vom Feststoff abgesaugt und das Filtrat auf 200 ml Wasser und 200 ml Ethylacetat gegossen. Die Phasen werden getrennt und die organische Phase mit gesättigter Ammoniumchlorid-Lösung, Wasser, gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Nach Kristallisation aus einem Diethylether/Petrolether-Gemisch resultieren 56.40 g (71% d. Th.) der Titelsubstanz als rosafarbene Kristalle.
LC-MS (Methode 1): Rt = 3.70 min.
MS (ESIpos): m/z = 243 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 12.60 (s, 1 H), 7.85 (d, 1 H), 7.48-7.31 (m, 5 H), 6.60 (dd, 1 H), 6.55 (d, 1 H), 5.19 (s, 2 H), 2.56 (s, 3 H).
Beispiel 58A
6-Benzyloxy-benzofuran-3 -on
Zu einer siedenden Suspension von 18.44 g (82.55 mmol) Kupfer(H)bromid in 70 ml Ethylacetat werden 10 g (41.28 mmol) in 30 ml Chloroform suspendiertes 4-Benzyloxy-6-hydroxyacetophenon
gegeben und die Suspension über Nacht unter Rückfluß erhitzt. Die noch warme Lösung wird dann filtriert und der Filterkuchen mit Ethylacetat nachgewaschen. Die organische Phase wird mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird in 150 ml Ethanol gelöst, 7.72 g (94.06 mmol) Natriumacetat-Trihydrat werden zugegeben und die resultierende Lösung eine Stunde unter Rückfluß erhitzt. Die Reaktionsmischung wird dann auf Eis gegeben und das Ethanol abgezogen. Der wässrige Rückstand wird dreimal mit Ethylacetat extrahiert und die vereinigten organischen Phasen mit 1 N Natronlauge, 1 N Salzsäure und anschließend mit gesättigter Natriumchlorid- Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Nach Säulen- chromatographie an Kieselgel 60 (Laufmittel: Toluol -> Toluol / Ethylacetat 1:1) erhält man 5.52 g (56% d. Th.) des Produkts als beigefarbene Kristalle.
LC-MS (Methode 4): Rt = 3.53 min.
MS (ESIpos): m/z = 241 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.56-7.35 (m, 6 H), 6.91 (d, 1 H), 6.77 (dd, 1 H), 5.23 (s, 2 H), 4.77 (s, 2 H).
Beispiel 59A
2-Brom-6-benzyloxy-benzofuran-3(2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 43 A ausgehend von Beispiel 58A hergestellt.
Ausbeute: 40% d. Th.
LC-MS (Methode 12): Rt = 4.12 min.
MS (ESIpos): m z = 319 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.54-7.37 (m, 5 H), 6.81-6.71 (m, 2 H), 5.56 (s, 1 H), 5.24 (s, 2 H).
Beispiel 60A
3-tert.-Butyldimethylsilyloxy-6-benzyloxy-2-(4-chloφhenyl)-benzofuran
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 44A ausgehend von Beispiel 59A hergestellt.
Ausbeute: 77% d. Th.
LC-MS (Methode 4): Rt = 5.20 min.
MS (ESIpos): m/z = 465 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.85 (d, 2 H), 7.59-7.20 (m, 9 H), 7.04 (dd, 1 H), 5.22 (s, 2 H), 1.07 (s, 9 H), 0.17 (s, 3 H), 0.04 (s, 3 H).
Beispiel 61A
(S*,R*)-3-[6-Benzyloxy-2-(4-chlθφhenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenylpropanal
Die Titelverbindung (als Diastereomerengemisch) wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 60A hergestellt.
Beispiel 62A
2-(2-Chlorethoxy)-6-hydroxyacetophenon
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 41A ausgehend von 2,6- Dihydroxyacetophenon hergestellt.
Ausbeute: 90% d. Th.
LC-MS (Methode 2): Rt = 2.86 min.
MS (ESIpos): m/z = 215 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 12.1 (s, 1 H), 7.34 (t, 1 H), 6.56 (dd, 1 H), 6.52 (dd, 1 H), 4.36-4.29 (m, 2 H), 4.05-3.97 (m, 2 H), 2.61 (s, 3 H).
Beispiel 63A
4-(2-Chlorethoxy)-benzofuran-3-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 42A ausgehend von Beispiel 62A hergestellt.
Ausbeute: 69% d. Th.
LC-MS (Methode 2): Rt = 2.42 min.
MS (ESIpos): m/z = 213 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.61 (t, 1 H), 6.80 (d, 1 H), 6.67 (d, 1 H), 4.70 (s, 2 H), 4.43- 4.35 (m, 2 H), 4.00-3.93 (m, 2 H).
Beispiel 64A
2-Brom-4-(2-chlorethoxy)-benzofüran-3(2i-7)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 43A ausgehend von Beispiel 63A hergestellt.
Ausbeute: 86% d. Th.
LC-MS (Methode 4): Rt = 2.90 min.
MS (ESIpos): m/z = 291 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.62 (t, 1 H), 6.69 (d, 1 H), 6.65 (d, 1 H), 5.47 (s, 1 H), 4.42- 4.37 (m, 2 H), 3.98-3.93 (m, 2 H).
Beispiel 65A
2-Brom-3-tert.-butyldimethylsilyloxy-4-(2-chlorethoxy)-benzofuran
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 44A ausgehend von Beispiel 64A hergestellt.
Ausbeute: 46% d. Th.
LC-MS (Methode 9): Rt = 2.14 min.
MS (ESIpos): m/z = 292 [M+H-Si(CH3)2C(CH3)3]+
'H-NMR (300 MHz, DMSO-d6): δ = 7.22 (t, 1 H), 7.11 (dd, 1 H), 6.84 (dd, 1 H), 4.43-4.38 (m, 2 H), 3.99-3.94 (m, 2 H), 1.03 (s, 9 H), 0.26 (s, 6 H).
Beispiel 66A
3-tert.-Butyldimethylsilyloxy-4-(2-chlorethoxy)-2-(4-chlθφhenyl)-benzofuran
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 45A ausgehend von Beispiel 65A hergestellt.
Ausbeute: 75% d. Th.
LC-MS (Methode 4): Rt = 3.29 min.
MS (ESIpos): m/z = 437 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.82-7.77 (m, 2 H), 7.57-7.52 (m, 2 H), 7.26 (t, 1 H), 7.16 (d, 1 H), 6.83 (d, 1 H), 4.45-4.41 (m, 2 H), 4.02-3.98 (m, 2 H), 1.03 (s, 9 H), -0.02 (s, 6 H).
Beispiel 67A
4-(2-Chlorethoxy)-2-(4-chlθφhenyl)-benzofuran-3-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 46A ausgehend von Beispiel 66A hergestellt. Die Verbindung wird ohne weitere Aufreinigung in der nächsten Stufe eingesetzt.
Beispiel 68A
(S*,R*)-3-[4-(2-Chlorethoxy)-2-(4-chloφhenyl)-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenyl- propanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 67A hergestellt.
Ausbeute: 28% d. Th.
LC-MS (Methode 9): Rt = 2.68 min.
MS (ESIpos): m/z = 455 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 9.33 (dd, 1 H), 7.70-7.65 (m, 2 H), 7.57 (t, 1 H), 7.55-7.49 (m, 2 H), 7.30-7.25 (m, 2 H), 7.17-7.06 (m, 3 H), 6.93 (d, 1 H), 6.51 (d, 1 H), 4.29 (dd, 1 H), 4.28- 4.10 (m, 2 H), 3.83-3.77 (m, 2 H), 3.07 (ddd, 1 H), 2.53 (ddd, 1 H).
Beispiel 69A
(lS*,3S*,3aR*,8bS*)-8-(2-Chlorethoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofüran-l ,8b-(l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 68A hergestellt.
Ausbeute: 55% d. Th.
LC-MS (Methode 2): Rt = 3.47 min.
MS (ESIpos): m/z = 457 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.28 (t, 1 H), 7.25-7.20 (m, 2 H), 7.20-7.15 (m, 2 H), 7.12- 7.03 (m, 3 H), 6.91-6.86 (m, 2 H), 6.72 (d, 1 H), 6.64 (d, 1 H), 5.72 (d, 1 H), 4.91 (s, 1 H), 4.80 (ddd, 1 H), 4.36-4.31 (m, 2 H), 4.03-3.97 (m, 2 H), 3.30-3.24 (m, 1 H), 2.54-2.42 (m, 1 H), 2.19 (ddd, 1 H).
Beispiel 70A
(3S*,3aR*,8bR*)-8-(2-Chlorethoxy)-3a-(4-chlθφhenyl)-8b-hydroxy-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 69A hergestellt.
Ausbeute: 63% d. Th.
LC-MS (Methode 9): Rt = 2.70 min.
MS (ESIpos): m/z = 455 (M+H)+
Η-NMR (400 MHz, DMSO-d6): δ = 7.40 (t, 1 H), 7.22-7.17 (m, 2 H), 7.14-7.05 (m, 3 H), 7.04- 6.96 (m, 4 H), 6.87 (d, 1 H), 6.71 (d, 1 H), 6.03 (s, 1 H), 4.40-4.24 (m, 2 H), 3.93-3.86 (m, 2 H), 3.70 (dd, 1 H), 3.14 (dd, 1 H), 2.95 (dd, 1 H).
Beispiel 71A
(lR*,3S*,3aR*,8bS*)-8-(2-Chlorethoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofuran-l ,8b-(lH)-diol
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 8A ausgehend von Beispiel 70A hergestellt.
Ausbeute: 70% d. Th.
LC-MS (Methode 8): Rt = 3.67 min.
MS (ESIpos): m/z = 457 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.24 (t, 1 H), 7.16-6.98 (m, 9 H), 6.68 (d, 1 H), 6.59 (d, 1 H), 5.11 (s, 1 H), 4.60-4.53 (m, 1 H), 4.50 (d, 1 H), 4.33-4.25 (m, 2 H), 4.03-3.90 (m, 3 H), 2.75 (dt, 1 H), 2.05 (dd, 1 H).
Beispiel 72A
4-Hydroxybenzofuran-3 -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 42A ausgehend von 2,6- Dihydroxyacetophenon hergestellt.
Ausbeute: 54% d. Th.
LC-MS (Methode 4): Rt = 2.04 min.
MS (ESIpos): m/z = 151 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 10.8 (s, 1 H), 7.43 (t, 1 H), 6.56 (dd, 1 H), 6.44 (dd, 1 H), 4.64 (s, 2 H).
Beispiel 73A
4-(2-Methoxyethoxy)-benzofuran-3-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 18A ausgehend von Beispiel 72A hergestellt.
Ausbeute: 68% d. Th.
LC-MS (Methode 4): Rt = 2.23 min.
MS (ESIpos): m/z = 209 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.59 (t, 1 H), 6.75 (d, 1 H), 6.65 (d, 1 H), 4.68 (s, 2 H), 4.25- 4.20 (m, 2 H), 3.71-3.66 (m, 2 H), 3.34 (s, 3 H).
Beispiel 74A
2-Brom-4-(2-methoxyethoxy)-benzofuran-3-on
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 43A ausgehend von Beispiel 73A hergestellt.
Ausbeute: 84% d. Th.
LC-MS (Methode 4): Rt = 2.63 min.
MS (ESIpos): m/z = 287 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 8.24 (s, 2 H), 7.61 (t, 1 H), 6.65 (d, 1 H), 6.63 (d, 1 H), 5.45 (s, 1 H), 4.25-4.20 (m, 2 H), 3.71-3.65 (m, 2 H), 3.34 (s, 3 H).
Beispiel 75A
2-Brom-3-te/-t.-bu1yldimethylsilyloxy-4-(2-methoxyethoxy)-benzofuran
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 44A ausgehend von Beispiel 74A hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 14): Rt = 2.43 min.
MS (ESIpos): m/z = 401 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.21 (t, 1 H), 7.07 (dd, 1 H), 6.81 (dd, 1 H), 4.25-4.20 (m, 2 H), 3.72-3.66 (m, 2 H), 3.29 (s, 3 H), 1.02 (s, 9 H), 0.24 (s, 6 H).
Beispiel 76A
3-tert.-Butyldimethylsilyloxy-2-(4-chloφhenyl)-4-(2-methoxyethoxy)-benzofuran
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 45A ausgehend von Beispiel 75A hergestellt.
Ausbeute: 68% d. Th.
LC-MS (Methode 9): Rt = 3.58 min.
MS (ESIpos): m/z = 433 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.85-7.79 (m, 2 H), 7.58-7.53 (m, 2 H), 7.2"6 (t, 1 H), 7.14 (dd, 1 H), 6.82 (dd, 1 H), 4.27-4.22 (m, 2 H), 3.75-3.70 (m, 2 H), 3.29 (s, 3 H), 1.02 (s, 9 H), -0.02 (s, 6 H).
Beispiel 77A
2-(4-Chloφhenyl)-4-(2-methoxyethoxy)-benzofuran-3-on
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 46A ausgehend von Beispiel 76A hergestellt. Die Verbindung wird ohne weitere Aufreinigung in der nächsten Stufe eingesetzt.
Beispiel 78A
(S *,R*)-3 -[2-(4-Chloφhenyl)-4-(2-methoxyethoxy)-3 -oxo-2,3 -dihydrobenzofuran-2-yl] -3 -phenyl- propanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 77A hergestellt.
Ausbeute: 8% d. Th.
LC-MS (Methode 9): Rt = 2.58 min.
MS (ESIpos): m/z = 451 (M+H)+.
Beispiel 79A
(lS*,3S*,3aR*,8bS*)-6-(3-Chloφropoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[έ]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbmdung wird in Analogie zur Synthese der Beispiele 41A - 48A ausgehend von 2,4- Dihydroxyacetophenon und 3-Chloφropanol hergestellt.
LC-MS (Methode 13) : Rt = 3.02 min.
MS (ESIneg): m/z = 469 (M-H)"
'H-NMR (300 MHz, DMSO-d6): δ = 7.32-7.01 (m, 8 H), 6.94-6.92 (m, 2 H), 6.69 (d, 1 H), 6.60 (dd, 1 H), 5.81 (d, 1 H), 5.01 (s, 1 H), 4.48-4.42 (m, 1 H), 4.13 (t, 2 H), 3.80 (t, 2 H), 3.35-3.27 (m, 1 H), 2.47-2.41 (m, 1 H), 2.30-2.14 (m, 3 H).
Beispiel 80A
N-(3 -Oxo-2,3 -dihydro- 1 -benzofuran-6-yl)-acetamid
Zu einer Lösung von 64.6 g (0.484 mol) Aluminiumchlorid in 100 ml 1,2-Dichlorethan werden bei 0°C unter Argon 32.6 g (0.181 mol) Chloracetylchlorid und anschließend innerhalb von 15 Minuten 20 g (0.121 mol) N-(3-Methoxyphenyl)-acetamid zugegeben. Die Temperatur steigt dabei
auf 10°C an. Der Ansatz wird dann langsam auf Raumtemperatur erwärmt und über Nacht gerührt. Die braune Mischung wird auf Eiswasser gegeben und Ethylacetat hinzugefügt. Nach kräftigem Rühren fällt N-(3-Methoxy-4-chloracetylphenyl)-acetamid aus, welches über eine Νutsche abgesaugt und im Hochvakuum getrocknet wird. Der erhaltene Feststoff wird in 140 ml Ethanol vorgelegt, und nach Zugabe von 13.2 g (0.154 mol) Νatriumacetat wird über Nacht unter Rückfluß erhitzt. Nach dem Abkühlen wird mit Wasser versetzt und das Ethanol am Rotatiorisverdampfer abgezogen. Der ausfallende Feststoff wird über eine Fritte abgesaugt und getrocknet. Es resultieren 9.35 g (40% d. Th.) des Produkts als Feststoff mit leicht rötlicher Färbung.
'H-NMR (200 MHz, DMSO-d6): δ = 10.5 (s, 1 H), 7.7 (s, 1 H), 7.6 (s, 1 H), 7.1 (s, 1 H), 4.8 (s, 2 H), 3.3 (s, 3 H).
Beispiel 81A
6-Amino- 1 -benzofuran-3 (2H)-on
Zu einer Lösung von 500 mg (2.62 mmol) N-(3-Oxo-2,3-dihydro-l-benzofuran-6-yl)-acetamid in 5 ml Methanol werden 5 ml 1 Ν Salzsäure gegeben und eine Stunde unter Rückfluß erhitzt. Nach dem Abkühlen wird der Ansatz auf eine Mischung aus Eiswasser, gesättigter Natriumhydrogen- carbonat-Lösung und Ethylacetat gegeben. Die organische Phase wird abgetrennt, mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Es resultieren 318 mg (61% d. Th.) der Zielverbindung als braunes Pulver mit einer Reinheit von 75%.
LC-MS (Methode 4): Rt = 1.71 min.
MS (ESIpos): m/z = 150 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.24 (s, 1 H), 6.51 (br. s, 2 H), 6.32 (dd, 1 H), 6.12 (d, 1 H), 4.75 (s, 2 H).
Beispiel 82A
Benzyl-(3-oxo-2,3-dihydro-l-benzofuran-6-yl)carbamat
Zu einer Lösung von 23.42 g (157 mmol) 6-Amino-l-benzofuran-3(2H)-on in 400 ml THF werden bei 0°C 54.7 ml (314 mmol) Diisopropylethylamin und 28.3 ml (188 mmol) Chlorameisensäurebenzylester gegeben und dann für 3 Stunden bei Raumtemperatur gerührt. Dann werden erneut 4.7 ml (31 mmol) Chlorameisensäurebenzylester zugegeben und über Nacht gerührt. Der Ansatz wird auf Eiswasser gegeben und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen werden mit Wasser und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Chromatographie an Kieselgel 60 (Laufmittel: Dichlormethan → Dichlormethan / Ethylacetat 5:1) liefert 30.80 g (64% d. Th.) des Produkts als farblose Kristalle.
LC-MS (Methode 4): Rt - 3.43 min.
MS (ESIpos): m/z = 284 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 10.38 (s, 1 H), 7.56-7.31 (m, 7 H), 7.12 (dd, 1 H), 5.20 (s, 2 H), 4.74 (s, 1 H).
Beispiel 83A
Benzyl-[(lS*,3S*,3aR*,8bS*)-3a-(4-chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro- lH-benzo[b]cyclopenta[d]furan-6-yl]-carbamat
Die Titelverbindung wird in Analogie zur Synthese der Beispiele 43A (Methode a), 44A, 45A, 46A, 5A und 6A ausgehend von Beispiel 82A hergestellt.
LC-MS (Methode 7): Rt = 2.90 min.
MS (ESIneg): m/z = 526 (M-HV
Η-NMR (400 MHz, DMSO-d6): δ = 9.88 (s, 1 H), 7.46-6.91 (m, 17 H), 5.87 (d, 1 H), 5.18 (m, 2 H), 5.07 (s, 1 H), 4.45-4.44 (m, 1 H), 3.35-3.28 (m, 1 H), 2.48-2.43 (m, 1 H), 2.25-2.20 ( , 1 H).
Beispiel 84A
(lS*,3S*,3aR*,8bS*)-6-Ammo-3a-(4-chloφhenyl)-3-phenyl-l,2,3,3a-tetrahydrocyclopenta[b]- benzofüran- 1 , 8b-( lH)-diol
Zu einer Lösung von 8.18 g (15.5 mmol) Benzyl-[(lS*,3S*,3aR*,8bS*)-3a-(4-chlθφhenyl)-l,8b- dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-lH-benzo[b]cyclopenta[d]furan-6-yl]-carbamat in 100 ml Methanol wird 1 g 10%-iges Palladium auf Aktivkohle gegeben und für 2 Stunden bei 2 bar hydriert. Nach Abtrennen des Katalysators wird der Rückstand eingeengt, und man erhält 6.06 g (92% d. Th.) der Titelverbindung, welche ca. 7% der entsprechenden dehalogenierten Verbindung als Verunreinigung enthält.
LC-MS (Methode 13): Rt = 2.34 min.
MS (ESIpos): m/z = 394 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.22-6.87 (m, 10 H), 6.24-6.21 (m, 2 H), 5.73 (d, 1 H), 5.19 (s, 2 H), 4.78 (s, 1 H), 4.44-4.37 (m, 1 H), 3.35-3.24 (m, 1 H), 2.48-2.40 (m, 1 H), 2.27-2.11 (m, 1 H).
Beispiel 85A
Brom-(4-chloφhenyl)-essigsäure
80.0 g (469 mmol) 4-Chloφhenylessigsäure werden unter Argon in 200 ml Tetrachlorkohlenstoff gelöst und unter RF erhitzt. In der Siedehitze gibt man 100 g (563 mmol) N-Bromsuccinimid und 7.70 g (46.9 mmol) 2,2'-Azo-bis-2-methylpropannitril hinzu und erhitzt über Nacht unter RF. Anschließend kühlt man auf 0°C, saugt ab, wäscht den Niederschlag mit kaltem Tetrachlorkohlenstoff und engt das Filtrat ein. Der Rückstand wird in Diethylether gelöst und dreimal mit gesättigter wässriger Natriumhydrogencarbonat-Lösung extrahiert. Die vereinigten wässrigen Phasen werden mit konzentrierter Salzsäure auf einen pH-Wert von 1 eingestellt und viermal mit Diethylether extrahiert. Man trocknet die vereinigten organischen Phasen über Magnesiumsulfat, filtriert, engt ein und erhält 83.0 g (71% d. Th.) des Produkts, das ohne weitere Aufreinigung weiter umgesetzt wird.
LC-MS (Methode 11): Rt = 3.42 min.
MS (ESIneg): m/z = 249 (M-H)~
'H-NMR (300 MHz, DMSO-d6): δ = 13.5 (s, 1 H), 7.61-7.55 (m, 2 H), 7.49-7.43 (m, 2 H), 5.80 (s, 1 H).
Beispiel 86A
(4-Chlθφhenyl)-(3,5-dipropoxyphenoxy)-essigsäure
Zu einer Lösung von 10.0 g (47.6 mmol) 3,5-Dipropoxyphenol und 11.9 g (47.6 mmol) Brom-(4- chloφhenyl)-essigsäure in 150 ml THF werden unter Argon portionsweise 4.37 g (109 mmol)
Natriumhydrid als 60%-ige Dispersion in Mineralöl gegeben. Man rührt 0.5 h bei RT und erhitzt über Nacht unter RF. Anschließend gibt man unter Eiskühlung Wasser hinzu, extrahiert dreimal mit Chloroform, wäscht die vereinigten organischen Phasen mit 1 N Natronlauge und gesättigter wässriger Natriumchlorid-Lösung, trocknet über Magnesiumsulfat, filtriert und engt ein (Rückstand 1). Die vereinigten wässrigen Phasen werden mit konzentrierter Salzsäure auf einen pH-Wert von 1 eingestellt und' dreimal mit Diethylether extrahiert. Man wäscht die vereinigten organischen Phasen mit gesättigter wässriger Natriumchlorid-Lösung, trocknet über Magnesiumsulfat, filtriert und engt ein (Rückstand 2). Nach Chromatographie der vereinigten Rückstände 1 und 2 an Kieselgel 60 (1. Laufmittel: Toluol / Essigsäureethylester 9:1, zur Abtrennung von nicht umgesetztem 3,5-Dipropoxy-phenol, 2. Laufmittel: Dichlormethan / Methanol 9:1) erhält man 12.1 g (67% d. Th.) des Produkts.
LC-MS (Methode 1): Rt = 4.21 min.
MS (ESIpos): m/z = 379 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.54 (d, 2 H), 7.38 (d, 2 H), 6.09-6.01 (m, 3 H), 5.41 (s, 1 H), 3.84 (t, 4 H), 1.67 (sext, 4 H), 0.94 (t, 6 H).
Beispiel 87A
2-(4-Chloφhenyl)-4,6-dipropoxybenzofuran-3(2H)-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 4A ausgehend von Beispiel 86A hergestellt.
Ausbeute: 44% d. Th.
LC-MS (Methode 12): Rt = 4.75 min.
MS (ESIpos): m/z = 361 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.50-7.44 (m, 2 H), 7.35-7.30 (m, 2 H), 6.46 (d, 1 H), 6.21 (d, 1 H), 5.74 (s, 1 H), 4.11-3.98 (m, 4 H), 1.82-1.64 (m, 4 H), 0.99 (t, 3 H), 0.96 (t, 3 H).
Beispiel 88A
(S*,R*)-3-[2-(4-Chloφhenyl)-4,6-dipropoxy-3-oxo-2,3-dihydrobenzofuran-2-yl]-3-phenylpropanal
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 5A ausgehend von Beispiel 87A hergestellt.
Ausbeute: 49% d. Th.
LC-MS (Methode 12): Rt = 4.93 min.
MS (ESIpos): m/z = 493 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 9.32 (d, 1 H), 7.69-7.61 (m, 2 H), 7.55-7.47 (m, 2 H), 7.31- 7.09 (m, 5 H), 6.47 (d, 1 H), 5.98 (d, 1 H), 4.24 (dd, 1 H), 4.02 (t, 2 H), 3.90-3.74 (m, 2 H), 3.02 (ddd, 1 H), 2.57-2.43 (m, 1 H), 1.73 (sext, 2 H), 1.57 (sext, 2 H), 0.97 (t, 3 H), 0.84 (t, 3 H).
Ausführungsbeispiele:
Beispiel 1
(3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l-oxim
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 14 ausgehend von Beispiel 7A hergestellt.
Ausbeute: 51% d. Th.
LC-MS (Methode 4): Rt= 3.66 min.
MS (ESIneg): m/z = 522 (M-H)~
Η-NMR (300 MHz, DMSO-d6): δ = 11.0 (s, 1 H), 7.36-7.30 (m, 2 H), 7.14-7.05 (m, 3 H), 7.00- 6.94 (m, 4 H), 6.35 (d, 1 H), 6.14 (d, 1 H), 5.25 (s, 1 H), 4.11-3.95 (m, 4 H), 3.52 (t, 1 H), 3.02- 2.93 (m, 2 H).
Beispiel 2
(lR*,3S*,3aR*,8bS*)-3a-(3'-Aminobiphenyl-4-yl)-6,8-diethoxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l , 8b-( lH)-diol
51.1 mg (0.10 mmol) (lR*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-l,8b-(l/J)-diol (Beispiel 8A), 15.5 mg (0.10 mmol) 3-Amino- phenylboronsäure, 10.6 mg (0.10 mmol) Natriumcarbonat und 5.8 mg (0.005 mmol) Tetrakis- (triphenylphosphin)palladiurn(O) werden unter Argon in einem Gemisch aus 0.1 ml Wasser und 0.5 ml Dioxan über Nacht auf 80°C erhitzt. Anschließend wird mit DMSO verdünnt, filtriert und das Filtrat über präparative HPLC gereinigt. Man erhält 17.4 mg (25% d. Th.) des Produkts.
LC-MS (Methode 10): Rt = 2.07 min.
MS (ESIpos): m/z = 524 (M+H)+.
Beispiel 3
(lR*,3S*,3aR*,8bS*)-6,8-Diethoxy-3-phenyl-2,3,3a,8b-tetrahydro-3a-(4-thiophen-3-ylphenyl)- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.41 min.
MS (ESIpos): m/z = 515 (M+H)+.
Beispiel 4
(lR*,3S*,3aR:1:,8bS*)-3a-(3'-Cyanobiphenyl-4-yl)-6,8-diethoxy-3-ρhenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.38 min.
MS (ESIpos): m/z = 534 (M+H)+
Beispiel 5
(lR*,3S*,3aR*,8bS*)-6,8-Diethoxy-3a-[4-(lH-indol-5-yl)-phenyl]-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.31 min.
MS (ESIpos): m/z = 548 (M+H)+.
Beispiel 6
(lR*,3S*,3aR*,8bS*)-6,8-Diethoxy-3a-(3'-ethylsulfonyl-biphenyl-4-yl)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.26 min.
MS (ESIpos): m/z = 601 (M+H)+.
Beispiel 7
(lR*,3S*,3aR*,8bS*)-3a-(5,-Amino-2'-fluorbiphenyl-4-yl)-6,8-diethoxy-3-ρhenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.05 min.
MS (ESIpos): m/z = 542 (M+H)+
Beispiel 8
(lR5fs,3S*,3aR*,8bS*)-3a-(4-Chinoxalin-6-ylphenyl)-6,8-diethoxy-3-ρhenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 2 hergestellt.
LC-MS (Methode 10): Rt = 2.29 min.
MS (ESIpos): m/z = 561 (M+H)+
Beispiel 9
(lR*,3S*,3aR*,8bS*)-6,8-Diethoxy-3-phenyl-3a-(4-pyrrolidin-l-ylphenyl)-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
51.1 mg (0.10 mmol) (lR*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-l,8b-(17J)-diol (Beispiel 8A), 4.7 mg (0.07 mmol) Pyrrolidin, 7.1 mg (0.07 mmol) Natrium-tert.-butylat, 1.2 mg (0.001 mmol) Tris(dibenzylidenaceton)- dipalladium(O) und 1.7 mg (0.003 mmol) rac-2,2'-Bis-(diphenylphosphino)-l, -binaphthyl werden unter Argon in 0.6 ml Toluol über Nacht auf 80°C erhitzt. Anschließend engt man ein, nimmt mit DMSO auf, filtriert und reinigt das Filtrat über präparative HPLC. Man erhält 14.5 mg (43% d. Th.) des Produkts.
LC-MS (Methode 10): Rt = 2.38 min.
MS (ESIpos): m/z = 502 (M+H)+.
Beispiel 10
(lR*,3S*,3aR*,8bS*)-3a-[4-(Benzyl-methyl-amino)-ρhenyl]-6,8-diethoxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofiιran- 1 , 8b-( 17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 9 hergestellt.
LC-MS (Methode 10): Rt = 2.44 min.
MS (ESIpos): m/z = 552 (M+H)+
Beispiel 11
(lR*,3S*,3aR*,8bS*)-6,8-Diethoxy-3a-[4-(methyl-pyridin-4-ylmethyl-amino)-phenyl]-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 9 hergestellt.
LC-MS (Methode 10): Rt = 1.73 min.
MS (ESIpos): m/z = 553 (M+H)+.
Beispiel 12
(2R*,3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6,8-diethoxy-2-dimethylcarbamid-8b-hydroxy-3- phenyl-2,3 ,3 a, 8b-tetrahydrocyclopenta[&]benzofuran- 1 -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 16 ausgehend von Beispiel 9A hergestellt.
Ausbeute: 29% d. Th.
LC-MS (Methode 4): Rt = 3.53 min.
MS (ESIpos): m z = 581 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.37-7.29 (m, 2 H), 7.15-6.90 (m, 7 H), 6.43 (d, 1 H), 6.16 (d, 1 H), 6.00 (s, 1 H), 4.64 (d, 1 H), 4.28 (d, 1 H), 4.15-3.91 (m, 4 H), 3.30 (s, 3 H), 2.77 (s, 3 H), 1.35 (t, 3 H), 1.22 (t, 3 H).
Beispiel 13
(lR*,2R*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6,8-diethoxy-2-dimethylcarbamid-3-phenyl- 2,3,3 a, 8b-tetrahydrocyclopenta[b]benzofuran- 1 ,8b-( 17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17 ausgehend von Beispiel 12 hergestellt.
Ausbeute: 80% d. Th.
LC-MS (Methode 5): Rt = 3.61 min.
MS (ESIpos): m/z = 583 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.27-7.19 (m, 2 H), 7.12-6.95 (m, 5 H), 6.88-6.80 (m, 2 H), 6.29 (d, 1 H), 6.12 (d, 1 H), 5.20 (s, 1 H), 4.84-4.76 ( , 1 H), 4.60 (d, 1 H), 4.29 (d, 1 H), 4.13- 3.97 (m, 5 H), 3.26 (s, 3 H), 2.75 (s, 3 H), 1.34 (t, 3 H), 1.31 (t, 3 H).
Beispiel 14
(3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran- 1 -oxim
Zu 500 mg (1.08 mmol) in 20 ml Ethanol gelöstem (3S*,3aR*,8bR*)-3a-(4-Chlθφhenyl)-6,8- diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l-on (Beispiel 15A) werden bei Raumtemperatur zunächst 20 ml Pyridin und anschließend 90 mg (1.30 mmol) Hydroxylammoniumchlorid gegeben und die Lösung für einen Tag gerührt. Nach Abzug der flüchtigen Bestandteile am Rotationsverdampfer wird der Rückstand in Ethylacetat gelöst. Die organische Phase wird dann mit 1 N Salzsäure, gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Es resultieren 515 mg (100% d. Th.) des racemischen Produkts als farblose Kristalle.
LC-MS (Methode 9): Rt = 2.66 min.
MS (ESIneg): m/z = 478 (M-H)~
'H-NMR (300 MHz, DMSO-d6): δ = 11.01 (s, 1 H), 7.21-6.94 (m, 9 H), 6.35 (d, 1 H), 6.14 (d, 1 H), 5.25 (s, 1 H), 4.10-3.99 (m, 4 H), 3.55-3.49 (m, 1 H), 3.00-2.94 (m, 2 H), 1.36-1.30 (m, 6 H).
Beispiel 15
(lR*,3S*,3aR*,8bS*)-l-Amino-3a-(4-chloφhenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b- tettahydrocyclopenta[δ]benzofuran
Bei 0°C wird zu einer Lösung von 3.22 ml (3.22 mmol) einer 1 N Lösung von Lithiumaluminiumhydrid in Diethylether in zusätzlichen 6 ml Diethylether portionsweise 115 mg (1.07 mmol) (3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6,8-diethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l-oxim (Beispiel 14) unter Rühren zugegeben. Nachdem die Gasentwicklung beendet ist, wird für 30 Minuten auf Rückfluß erhitzt. Bei 0°C wird dann mit Ethylacetat verdünnt und 1 N Natronlauge zugetropft. Es wird fünf Minuten gerührt und anschließend die Phasen getrennt. Die wäßrige Phase wird zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen werden mit gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand kristallisiert aus einer Mischung aus Dichlormethan, Diethylether und Petrolether. Die erhaltenen Kristalle werden über eine Glasfritte abgesaugt und getrocknet. Es resultieren 155 mg (31% d. Th.) des racemischen Produkts als farblose Kristalle.
LC-MS (Methode 8): Rt = 2.62 min.
MS (ESIneg): m/z = 464 (M-H)"
Η-NMR (400 MHz, DMSO-d6): δ = 7.13-7.03 (m, 7 H), 6.91 (d, 2 H), 6.24 (d, 1 H), 6.11 (d, 1 H), 4.06-4.00 (m, 4 H), 3.75 (dd, 1 H), 3.34 (dd, 1 H), 2.41-2.35 (m, 1 H), 2.15-2.05 (m, 1 H), 1.35- 1.31 (m, l H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 29A durchgeführt.
Analytische Daten (Methode 29B):
Enantiomer A: Rt = 3.89 min., Enantiomer B: Rt = 6.09 min.
Beispiel 16
(2R*,3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6,8-diethoxy-2-dimethylcarbamid-8-hydroxy-3-phenyl- 2,3,3 a, 8b-tetrahydrocyclopenta[b]benzofuran- 1 -on
Zu einer Lösung von 550 mg (1.08 mmol) (2R*,3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6,8-diethoxy- 8-hydroxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-2-carbonsäure (Beispiel 17A) in THF werden bei 0°C 674 mg (1.30 mmol) Benzotriazol-1-yloxy-tripyrrolidinophosphonium-Hexa- fluorophosphat, 106 mg (1.30 mmol) Dimethylamin-Hydrochlorid und 0.47 ml (2.70 mmol) N,N- Diisopropylethylamin zugegeben und die Mischung dann für 4 h bei 0°C gerührt. Die Reaktionsmischung wird dann auf eine Mischung aus 50 ml gesättigter Ammoniumchlorid-Lösung, Eiswasser und Ethylacetat gegossen. Nach Abtrennen der organischen Phase wird die wäßrige Phase zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen werden mit Wasser, gesättigter Natriumhydrogencarbonat-Lösung und gesättigter Natriumchlorid-Lösung gewaschen, über Magnesiumsulfat getrocknet, filtriert und eingeengt. Der Rückstand wird über Kieselgel 60 (Laufmittel: Toluol / Ethylacetat 10:1, 6:1, 4:1) gereinigt. Es resultieren 159 mg (27% d. Th.) racemisches Produkt als farbloser Schaum.
LC-MS (Methode 2): Rt = 3.41 min.
MS (ESIpos): m/z = 536 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.25-6.94 (m, 9 H), 6.43 (d, 1 H), 6.16 (d, 1 H), 6.00 (s, 1 H), 4.63 (d, 1 H), 4.29 (d, 1 H), 4.13-3.94 (m, 4 H), 3.30 (s, 3 H), 2.77 (s, 3 H), 1.35 (t, 3 H), 1.22 (t, 3 H).
Beispiel 17
(lR*,2R*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6,8-diethoxy-2-dimethylcarbamid-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Eine Lösung von 1104 mg (4.20 mmol) Telramethylammoniumtriacetoxyborhydrid in 1.5 ml Acetonitril und 1.5 ml Eisessig wird für 30 Minuten bei Raumtemperatur gerührt. 150 mg (0.28 mmol) (2R*,3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6,8-diethoxy-2-dimethylcarbamid-8-hydroxy-3- phenyl-2,3,3a,8b-tetτahydrocyclopenta[b]benzofuran-l-on werden als Lösung in 13.5 ml Acetonitril zugegeben und für zwei Stunden bei Raumtemperatur gerührt. Bei 0°C wird gesättigte Natriumhydrogencarbonat-Lösung zugegeben und dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Phasen werden über Magnesiumsulfat getrocknet, filtriert und eingeengt. Die aus Diethylether ausfallenden Kristalle werden abgesaugt, mit Diethylether/Petrolether 1:1 gewaschen und getrocknet. Es resultieren 94 mg (62% d. Th.) des Produkts als racemisches Gemisch.
LC-MS (Methode 8): Rt = 3.23 min.
MS (ESIpos): m/z = 538 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.15-6.94 (m, 7 H), 6.85-6.82 (m, 2 H), 6.29 (d, 1 H), 6.12 (d, 1 H), 5.16 (s, 1 H), 4.82-4.79 (m, 1 H), 4.56 (d, 1 H), 4.29 (d, 1 H), 4.08-3.99 (m, 4 H), 3.25 (s, 3 H), 2.76 (s, 1 H), 1.36-1.29 (m, 6 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 26A durchgeführt.
Analytische Daten (Methode 26B):
Enantiomer A: Rt = 3.96 min., Enantiomer B: Rt = 15.29 min.
Beispiel 18
(lR*,3S*,3aR*,8bS*)-6,8-Bis-(2-methoxyethoxy)-3a-(4-bromphenyl)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 22A hergestellt (anders als bei den anderen Reaktionen dieses Typs entsteht hier das trans-Dioi).
Ausbeute: 72% d. Th.
LC-MS (Methode 2): Rt = 4.38 min.
MS (ESIpos): m/z = 572 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.27-7.22 (m, 2 H), 7.13-7.04 (m, 5 H), 7.00-6.95 ( , 2 H), 6.29 (d, 1 H), 6.18 (d, 1 H), 5.01 (s, 1 H), 4.56-4.51 (m, 1 H), 4.41 (d, 1 H), 4.22-4.09 (m, 4 H), 3.86 (dd, 1 H), 3.70-3.64 (m, 4 H), 3.34 (s, 6 H) 2.70 (ddd, 1 H), 1.96 (dd, 1 H).
Beispiel 19
(3S*,3aR*,8bR*)-6,8-Bis-(2-methoxyethoxy)-3a-(4-bromphenyl)-8b-hydroxy-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran- 1 -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 18 hergestellt.
Ausbeute: 33% d. Th.
LC-MS (Methode 3): Rt = 4.30 min.
MS (ESIpos): m/z = 569 (M+H)+.
Beispiel 20
(lS*,3S*,3aR*,8bS*)-6,8-Bis-(2-dimethylaminoethoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b- tettahyά^ocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 28A hergestellt.
Ausbeute: 87% d. Th.
LC-MS (Methode 5): Rt = 2.12 min.
MS (ESIpos): m/z = 553 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.23-7.03 (m, 7 H), 6.93-6.88 (m, 2 H), 6.44 (d, 1 H), 6.30 (d, 1 H), 4.98 (s, 1 H), 4.67 (t, 1 H), 4.38-4.30 (m, 4 H), 3.48-3.23 (m, 5 H), 2.76 (s, 6 H), 2.70 (s, 6 H), 2.53-2.42 (m, 1 H), 2.21 (ddd, 1 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 15A durchgeführt.
Analytische Daten (Methode 15B):
Enantiomer A: Rt = 7.23 min., Enantiomer B: Rt = 9.41 min.
Beispiel 21
(lR*,3S*,3aR*,8bS*)-6,8-Bis-(2-dimethylaminoethoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b- tettahydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 30A hergestellt.
Ausbeute: 82% d. Th.
LC-MS (Methode 2): Rt = 2.03 min.
MS (ESIpos): m/z = 553 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.20-6.95 (m, 9 H), 6.27 (d, 1 H), 6.20 (d, 1 H), 5.16 (s, 1 H), 4.49 (d, 1 H), 4.26-4.11 (m, 1 H), 4.11-4.02 (m, 4 H), 3.84 (dd, 1 H), 2.86-2.55 (m, 5 H), 2.24 (s, 6 H), 2.19 (s, 6 H), 1.95 (dd, 1 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 15A durchgeführt.
Analytische Daten (Methode 15B):
Enantiomer A: Rt = 6.78 min., Enantiomer B: Rt = 8.18 min.
Beispiel 22
(lS*,3S*,3aR*,8bS*)-6,8-Bis-(2-dimethylaminoethoxy)-3a-(4-bromρhenyl)-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 35A hergestellt.
Ausbeute: 59% d. Th.
LC-MS (Methode 4): Rt = 2.19 min.
MS (ESIpos): m/z = 597 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.34-7.29 (m, 2 H), 7.18-7.12 (m, 2 H), 7.11-7.04 (m, 3 H), 6.94-6.88 (m, 2 H), 6.48 (d, 1 H), 6.32 (d, 1 H), 6.10 (s, 1 H), 5.01 (s, 1 H), 4.68 (t, l. H), 4.43-4.37 (m, 4 H), 3.66-3.45 (m, 5 H), 2.87 (s, 6 H), 2.85 (s, 6 H), 2.55-2.43 (m, 1 H), 2.31-2.15 (m, 1 H).
Beispiel 23
(lS*,3S*,3aR:lc,8bS*)-3a-(4-Chloφhenyl)-6-ethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]- benzofuran-l,8b-(lH)-diol
Die Titelverbmdung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 39A hergestellt.
Ausbeute: 73% d. Th.
LC-MS (Methode 3): Rt = 3.57 min.
MS (ESIneg): m/z = 421 (M)'
Η-NMR (200 MHz, DMSO-d6): δ = 7.28-6.90 (m, 10 H), 6.66 (d, 1 H), 6.57 (dd, 1 H), 5.84 (d, 1 H), 5.03 (s, 1 H), 4.49-4.39 (m, 1 H), 4.05 (q, 3 H), 3.31-3.25 (m, 1 H), 2.49-2.38 (m, 1 H), 2.24- 2.15 ( , 1 H), 1.34 (t, 3 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 30A durchgeführt.
Analytische Daten (Methode 30B):
Enantiomer A: Rt = 3.90 min., Enantiomer B: Rt = 5.46 min.
Beispiel 24
(3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6-ethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofuran-l-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 23 hergestellt.
Ausbeute: 81% d. Th.
LC-MS (Methode 9): Rt = 2.66 min.
MS (ESIpos): m/z = 403 [M+H-H20]+
'H-NMR (200 MHz, DMSO-d6): δ = 7.25-7.01 (m, 10 H), 6.82 (d, 1 H), 6.62 (dd, 1 H), 6.28 (s, 1 H), 4.08 (q, 2 H), 3.69-3.58 (m, 1 H), 3.42-3.22 (m, 1 H), 2.93-2.79 (m, 1 H), 1.35 (t, 3 H).
Beispiel 25
(3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6-ethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[&]benzofuran- 1 -oxim
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 14 ausgehend von Beispiel 24 hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 9): Rt = 2.57 min.
MS (ESIneg): m/z = 434 (M-H)~
'H-NMR (300 MHz, DMSO-d6): δ = 11.12 (s, 1 H), 7.32 (d, 1 H), 7.20 (d, 2 H), 7.11-7.09 (m, 5 H), 7.01-6.98 (m, 2 H), 6.73 (d, 1 H), 6.60 (dd, 1 H), 5.74 (s, 1 H), 4.07 (dq, 2 H), 3.40-3.38 (m, 1 H), 3.04-3.01 (m, 2 H), 1.34 (t, 3 H).
Beispiel 26
(lR*,3S*,3aR*,8bR*)-l-Amino-3a-(4-chloφhenyl)-6-ethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 15 ausgehend von Beispiel 25 hergestellt.
Ausbeute: 50% d. Th.
LC-MS (Methode 9): Rt = 1.96 min.
MS (ESIpos): m/z = 422 (M-H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.19 (d, 1 H), 7.12-6.94 (m, 9 H), 6.62 (d, 1 H), 6.52 (dd, 1 H), 5.90 (br. s, 1 H), 4.04 (q, 2 H), 3.61 (dd, 1 H), 3.42 (dd, 1 H), 2.43-2.35 (m, 1 H), 2.19-2.07 (m, 1 H), 1.33 (t, 3 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 27A durchgeführt.
Analytische Daten (Methode 27B):
Enantiomer A: Rt = 4.22 min., Enantiomer B: Rt = 7.38 min.
Beispiel 27
(2R*,3S*,3aR*,8bR*)-3a-(4-Chloφhenyl)-6-ethoxy-2-dimethylcarbamid-8-hydroxy-3-phenyl- 2,3,3a, 8b-tetrahydrocyclopenta[b]benzofuran-l-on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 16 ausgehend von Beispiel 40A hergestellt.
Ausbeute: 26% d. Th.
LC-MS (Methode 9): Rt = 2.49 min.
MS (ESIpos): m/z = 492 (M+H)+
'H-NMR (400 MHz, DMSO-d5): δ = 7.25-6.96 (m, 10 H), 6.82 (d, 1 H), 6.59 (dd, 1 H), 6.40 (s, 1 H), 4.77 (d, J = 13.41 Hz, 1 H), 4.19 (d, J = 13.41 Hz, 1 H), 4.14-4.05 (m, 2 H), 3.27 (s, 3 H), 2.77 (s, 3 H), 1.38-1.33 (m, 3 H).
Beispiel 28
(lR*,2R*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-ethoxy-2-dimethylcarbamid-3-phenyl-2,3,3a,8b- tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 17 ausgehend von Beispiel 27 hergestellt.
Ausbeute: 61% d. Th.
LC-MS (Methode 9): Rt = 2.31 min.
MS (ESIpos): m/z = 494 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.34 (d, 1 H), 7.22-6.80 (m, 9 H), 6.64 (d, 1 H), 6.50 (dd, 1 H), 5.48 (d, 1 H), 5.29 (s, 1 H), 4.91-4.84 (m, 1 H), 4.24-3.95 (m, 4 H), 3.20 (s, 3 H), 2.72 (s, 3 H), 1.34 (t, 3 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 28A durchgeführt.
Analytische Daten (Methode 28B):
Enantiomer A: Rt = 4.48 min., Enantiomer B: Rt = 10.97 min.
Beispiel 29
(lS*,3S*,3aR*,8bS*)-3a-(4-Bromphenyl)-6-ethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]- benzofuran-1 ,8b-(lH)-diol
Die Titelverbmdung wird in Analogie zur Synthese der Beispiele 2A - 6A ausgehend von 3- Ethoxyphenol hergestellt.
LC-MS (Methode 12): Rt = 4.39 min.
MS (ESIpos): m/z = 489 (M+Na)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.32-7.23 (m, 3 H), 7.16-7.06 (m, 5 H), 6.95-6.91 (m, 2 H), 6.66-6.54 (m, 2 H), 5.83 (d, 1 H), 5.03 (s, 1 H), 4.49-4.39 (m, 1 H), 4.05 (q, 2 H), 3.36-3.26 (m, 1 H), 2.51-2.09 (m, 2 H), 1.34 (t, 3 H).
Beispiel 30
(3S*,3aR*,8bR*)-3a-(4-Bromphenyl)-6-ethoxy-8b-hydroxy-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofuran- 1 -on
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 7A ausgehend von Beispiel 29 hergestellt.
Ausbeute: 85% d. Th.
LC-MS (Methode 11): Rt = 4.5 min.
MS (ESIneg): m/z = 463 (M)"
'H-NMR (300 MHz, DMSO-d6): δ = 7.33-7.02 (m, 10 H), 6.81 (d, 1 H), 6.61 (dd, 1 H), 6.23 (s, 1 H), 4.12-4.05 (m, 2 H), 3.68-3.61 (m, 1 H), 3.40-3.23 (m, 1 H), 2.91-2.82 (m, 1 H), 1.35 (t, 3 H).
Beispiel 31
(lR*,3S*,3aR*,8bS*)-3a-(4-Bromρhenyl)-6-ethoxy-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]- benzofuran- 1 , 8b-( l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 8A ausgehend von Beispiel 30 hergestellt.
Ausbeute: 79% d. Th.
LC-MS (Methode 11): Rt = 4.2 min.
MS (ESIneg): m/z = 465 (M-H)"
'H-NMR (200 MHz, DMSO-d6): δ = 7.32-7.23 (m, 3 H), 7.11-6.94 (m, 7 H), 6.60 (d, 1 H), 6.50 (dd, 1 H), 5.22 (s, 1 H), 5.14 (d, 1 H), 4.56 (br. s, 1 H), 4.09-3.98 (m, 2 H), 3.89-3.79 (m, 1 H), 2.74-2.57 (m, 1 H), 1.96-1.84 (m, 1 H), 1.34 (t, 3 H).
Beispiel 32
(lS*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-(2-dimethylaminoethoxy)-3-ρhenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( 17J)-diol
100 mg (0.22 mmol) (lS*,3S*,3aR*,8bS*)-6-(2-Chlorethoxy)-3a-(4-chloφhenyl)-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol (Beispiel 48A) werden unter Argon in einer verschlossenen Apparatur in 10 ml 33%-iger ethanolischer Dimethylamin-Lösung auf 70°C über Nacht erhitzt. Nach dem Abkühlen engt man ein. Nach Säulenchromatographie an Kieselgel 60 (Laufinittel: Dichlormethan / Methanol / Triethylamin 95:5:1) erhält man 89 mg (87% d. Th.) des Produkts als racemisches Gemisch.
LC-MS (Methode 1): Rt = 2.90 min.
MS (ESIpos): m/z = 466 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.30-7.02 (m, 8 H), 6.96-6.88 (m, 2 H), 6.69 (d, 1 H), 6.59 (dd, 1 H), 5.85 (d, 1 H), 5.04 (s, 1 H), 4.51-4.38 (m, 1 H), 4.14-4.04 (m, 2 H), 3.38-3.23 (m, 1 H), 2.73-2.64 (m, 2 H), 2.50-2.37 (m, 1 H), 2.26 (s, 6 H), 2.24-2.10 (m, 1 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 19A durchgeführt.
Analytische Daten (Methode 19B):
Enantiomer A: Rt = 10.52 min., Enantiomer B: Rt = 12.53 min.
Beispiel 33
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-pyrrolidin-l-ylethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Pyrrolidin als Amin- Komponente hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 2): Rt = 2.29 min.
MS (ESIpos): m/z = 492 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.30-7.02 (m, 8 H), 6.96-6.88 (m, 2 H), 6.68 (d, 1 H), 6.59 (dd, 1 H), 5.85 (d, 1 H), 5.04 (s, 1 H), 4.51-4.38 (m, 1 H), 4.09 (t, 2 H), 3.37-3.24 (m, 1 H), 2.79 (t, 2 H), 2.57-2.37 (m, 5 H), 2.20 (ddd, 1 H), 1.73-1.65 (m, 4 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 18A durchgeführt.
Analytische Daten (Methode 18B):
Enantiomer A: Rt = 6.53 min., Enantiomer B: Rt = 8.48 min.
Beispiel 34
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-methylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Methylamin als Amin- Komponente hergestellt.
Ausbeute: 95% d. Th.
LC-MS (Methode 2): Rt = 2.19 min.
MS (ESIpos): m/z = 452 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.79 (s, 1 H), 7.31 (d, 1 H), 7.24-7.13 (m, 4 H), 7.12-7.01 (m, 3 H), 6.95-6.90 (m, 2 H), 6.73 (d, 1 H), 6.64 (dd, 1 H), 5.89 (d, 1 H), 5.03 (s, 1 H), 4.49-4.41 (m, 1 H), 4.22 (t, 2 H), 3.36-3.18 (m, 3 H), 2.57 (s, 3 H), 2.52-2.40 (m, 1 H), 2.22 (ddd, 1 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 16A durchgeführt.
Analytische Daten (Methode 16B):
Enantiomer A: Rt = 9.31 min., Enantiomer B: Rt = 13.90 min.
Beispiel 35
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-diethylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Diethylamin als Amin- Komponente hergestellt.
Ausbeute: 56% d. Th.
LC-MS (Methode 4): Rt = 2.34 min.
MS (ESIpos): m/z = 494 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 7.26 (d, 1 H), 7.23-7.14 (m, 4 H), 7.11-7.01 (m, 3 H), 6.94- 6.90 (m, 2 H), 6.67 (d, 1 H), 6.58 (dd, 1 H), 5.85 (d, 1 H), 5.02 (s, 1 H), 4.48-4.41 (m, 1 H), 4.06 (t, 2 H), 3.37-3.26 (m, 1 H), 2.86-2.73 (m, 2 H), 2.63-2.54 (m, 4 H), 2.49-2.40 (m, 1 H), 2.20 (ddd, 1 H), 1.00 (t, 6 H).
Beispiel 36
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-ethylaminoethoxy)-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Ethylamin als Amin- Komponente hergestellt.
Ausbeute: 31% d. Th.
LC-MS (Methode 13): Rt = 1.97 min.
MS (ESIpos): m/z = 466 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.27 (d, 1 H), 7.24-7.13 (m, 4 H), 7.12-7.03 (m, 3 H), 6.95- 6.90 (m, 2 H), 6.69 (d, 1 H), 6.61 (dd, 1 H), 5.85 (s, 1 H), 5.02 (s, 1 H), 4.45 (t, 1 H), 4.10 (t, 2 H), 3.31 (dd, 1 H), 3.00 (t, 2 H), 2.72 (q, 2 H), 2.48-2.40 (m, 1 H), 2.28-2.14 (m, 1 H), 1.08 (t, 3 H).
Beispiel 37
(lS*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-(2-isopropylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Isopropylamin als Amin- Komponente hergestellt.
Ausbeute: 26% d. Th.
LC-MS (Methode 13): Rt= 1.97 min.
MS (ESIpos): m/z = 480 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.28 (d, 1 H), 7.24-7.13 (m, 4 H), 7.12-7.03 (m, 3 H), 6.95- 6.90 (m, 2 H), 6.69 (d, 1 H), 6.61 (dd, 1 H), 5.85 (s, 1 H), 5.02 (s, 1 H), 4.45 (t, 1 H), 4.10 (t, 2 H), 3.36-3.27 (m, 1 H), 3.00 (t, 2 H), 2.95 (sept, 1 H), 2.48-2.40 (m, 1 H), 2.31-2.14 (m, 1 H), 1.08 (d, 6 H).
Beispiel 38
(lS*,3S*,3aR*,8bS*)-6-(2-Azetidin-l-ylethoxy)-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Azetidin als Amin- Komponente hergestellt.
Ausbeute: 58% d. Th.
LC-MS (Methode 13): R, = 1.96 min.
MS (ESIpos): m/z = 478 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.25 (d, 1 H), 7.23-7.13 (m, 4 H), 7.12-7.02 (m, 3 H), 6.95- 6.90 (m, 2 H), 6.64 (d, 1 H), 6.56 (dd, 1 H), 5.82 (s, 1 H), 5.00 (s, 1 H), 4.44 (t, 1 H), 3.95 (t, 2 H), 3.36-3.27 (m, 1 H), 3.23 (t, 4 H), 2.74 (t, 2 H), 2.48-2.39 (m, 1 H), 2.28-2.14 (m, 1 H), 1.99 (quint, 2 H).
Beispiel 39
(lS*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-(2-moφholin-4-ylethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Moφholin als Amin- Komponente hergestellt.
Ausbeute: 82% d. Th.
LC-MS (Methode 9): Rt = 1.76 min.
MS (ESIpos): m/z = 508 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.29-7.02 (m, 8 H), 6.96-6.87 (m, 2 H), 6.69 (d, 1 H), 6.59 (dd, 1 H), 5.85 (d, 1 H), 5.04 (s, 1 H), 4.50-4.38 (m, 1 H), 4.12 (t, 2 H), 3.63-3.54 (m, 4 H), 3.44- 2.98 (m, 5 H), 2.70 (t, 2 H), 2.48-2.36 (m, 1 H), 2.32-2.07 (m, 1 H).
Beispiel 40
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-cyclopropylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocycloρenta[b]benzofuran-l,8b-(lH)-diol-Hydroformiat
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit Cyclopropylamm als Amin-Komponente hergestellt. Die Aufreinigung des Rohprodukts erfolgt mittels präparativer HPLC (RP18-Säule, Laufmittel: Acetonitril/Wasser-Gradient 5:95 → 95:5 mit 0.1% Ameisensäure).
Ausbeute: 48% d. Th.
LC-MS (Methode 13): Rt = 1.94 min.
MS (ESIpos): m/z = 478 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 8.17 (s, 1 H), 7.30-7.02 ( , 8 H), 6.96-6.88 ( , 2 H), 6.68 (d, 1 H), 6.59 (dd, 1 H), 5.76 (s, 1 H), 5.04 (s, 1 H), 4.50-4.38 (m, 1 H), 4.05 (t, 2 H), 3.48-3.25 (m, 1 H), 2.95 (t, 2 H), 2.47-2.35 (m, 1 H), 2.28-2.08 (m, 2 H), 0.45-0.32 (m, 2 H), 0.30-0.20 (m, 2 H).
Beispiel 41
(lS!,:,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-[2-(2-dimethylamino-ethylamino)-ethoxy]-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(17J)-diol-Dihydroformiat
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 mit 2-Dimethylamino- ethylamin als Amin-Komponente hergestellt. Die Aufreinigung des Rohprodukts erfolgt mittels
präparativer HPLC (RP18-Säule, Laufmittel: Acetonitril/ Wasser-Gradient 5:95 - 95:5 mit 0.1% Ameisensäure).
Ausbeute: 85% d. Th.
LC-MS (Methode 6): Rt = 1.37 min.
MS (ESIpos): m/z = 509 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 8.22 (s, 2 H), 7.30-7.02 (m, 8 H), 6.95-6.90 (m, 2 H), 6.70 (d, 1 H), 6.60 (dd, 1 H), 4.45 (t, 1 H), 4.12 (t, 2 H), 3.31 (dd, 1 H), 3.02 (t, 2 H), 2.81 (t, 2 H), 2.47- 2.36 (m, 1 H), 2.25 (s, 6 H), 2.24-2.14 (m, 3 H).
Beispiel 42
(lS*,3S*,3aR*,8bS*)-6-(2-Aminoethoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclo- penta[b]benzofüran-l ,8b-(lH)-diol
210 mg (0.45 mmol) (lS*,3S*,3aR*,8bS*)-6-(2-Azidoethoxy)-3a-(4-chlθφhenyl)-3-phenyl- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(lH)-diol (Beispiel 49 A) werden in 34 ml Ethanol gelöst. Nach Zusatz von 56 mg 10%-igem Palladium auf Aktivkohle rührt man für 15 Minuten bei RT und Normaldruck unter einer Wasserstoff-Atmosphäre. Man filtriert, engt ein und filtriert den Rückstand über Kieselgel 60. Man wäscht mit Toluol und eluiert mit einem 1:1- Gemisch aus Dichlormethan / Ethanol. Nach dem Einengen erhält man 179 mg (90% d. Th.) des Produkts, das nach Methode 23A feingereinigt wird.
LC-MS (Methode 9): Rt = 1.74 min.
MS (ESIpos): m/z = 438 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.33-6.84 (m, 10 H), 6.70 (d, 1 H), 6.61 (dd, 1 H), 5.89 (s, 1 H), 5.76 (s, 2 H), 5.05 (s, 1 H), 4.52-4.38 (m, 1 H), 4.02 (t, 2 R), 3.39-3.20 (m, 1 H), 2.98 (t, 2 H), 2.50-2.36 (m, 1 H), 2.34-2.10 (m, 1 H).
HPLC (Methode 23B): Rt = 8.76 min.
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 24A durchgeführt (Enantiomer A: Rt = 11.0 min., Enantiomer B: Rt = 23.6 min.).
Beispiel 43
(lR*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-(2-dimethylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 51A hergestellt.
Ausbeute: 94% d. Th.
LC-MS (Methode 5): Rt = 2.41 min.
MS (ESIpos): m z = 466 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.32 (d, 1 H), 7.19-7.02 (m, 7 H), 6.99-6.91 (m, 2 H), 6.60 (d, 1 H), 6.54 (dd, 1 H), 5.26 (s, 1 H), 5.18 (d, 1 H), 4.62-4.52 (m, 1 H), 4.16 (t, 2 H), 3.83 (dd, 1 H), 3.02-2.88 (m, 2 H), 2.75-2.57 (m, 1 H), 2.54 (s, 3 H), 2.46 (s, 3 H), 1.98-1.82 (m, 1 H).
Beispiel 44
(lR*,2R*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-6-(2-dimethylaminoethoxy)-2-dimethylcarbamid-3- phenyl-2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l,8b-(l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 54A hergestellt.
Ausbeute: 64% d. Th.
LC-MS (Methode 2): Rt = 2.10 min.
MS (ESIpos): m/z = 537 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.39 (d, 1 H), 7.25-6.98 (m, 7 H), 6.86-6.78 (m, 2 H), 6.75 (d, 1 H), 6.57 (dd, 1 H), 5.54 (d, 1 H), 5.33 (s, 1 H), 4.96-4.84 (m, 1 H), 4.34-4.12 (m, 3 H), 4.10-3.94 (m, 1 H), 3.48-3.28 (m, 2 H), 3.20 (s, 3 R), 2.72 (s, 3 H), 2.74-2.62 (m, 6 H).
Beispiel 45
(lR*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-8b-hydroxy-3-phenyl-6-(2-pyrrolidin-l-yl-ethoxy)- 2,3,3a,8b-tetrahydrocyclopenta[b]benzofuran-l-amin-Dihydroformiat
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 56A hergestellt. Die Aufreinigung des Rohprodukts erfolgt mittels präparativer HPLC (RP18-Säule, Laufmittel: Acetonitril/Wasser-Gradient 5:95 -» 95:5 mit 0.1% Ameisensäure).
Ausbeute: 67% d. Th.
LC-MS (Methode 6): Rt = 1.20 min.
MS (ESIpos): m/z = 491 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 8.21 (s, 2 H), 7.36 (d, 1 H), 7.21-7.04 (m, 7 H), 6.99-6.94 (m, 2 H), 6.67 (d, 1 H), 6.58 (dd, 1 H), 4.10 (t, 2 H), 3.80-3.70 ( , 1 H), 3.50 (dd, 1 H), 2.82 (t, 2 H), 2.59-2.53 (m, 4 H), 2.48-2.37 (m, 1 H), 2.35-2.18 (m, 1 H), 1.73-1.67 ( , 4 H).
Beispiel 46
(lR*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(2-dimethylaminoethoxy)-8b-hydroxy-3-phenyl- 2,3,3a,8b-tettahydrocyclopenta[b]benzofuran-l-amin-Dihydroformiat
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 56A hergestellt. Die Aufreinigung des Rohprodukts erfolgt mittels präparativer HPLC (RP18-Säule, Laufmittel: Acetonitril/Wasser-Gradient 5:95 — » 95:5 mit 0.1% Ameisensäure).
Ausbeute: 36% d. Th.
LC-MS (Methode 6): Rt = 1.22 min.
MS (ESIpos): m/z = 465 (M+H)+
'H-NMR (400 MHz, DMSO-d6): δ = 8.23 (s, 2 H), 7.35 (d, 1 H), 7.21-7.02 (m, 7 H), 6.99-6.94 (m, 2 H), 6.68 (d, 1 H), 6.58 (dd, 1 H), 4.08 (t, 2 H), 3.75 (dd, 1 H), 3.50 (dd, 1 H), 2.64 (t, 2 H), 2.47- 2.37 (m, 1 H), 2.34-2.23 (m, 1 H), 2.23 (s, 6 H).
Beispiel 47
(lS*,3S*,3aR*,8bS*)-6-Benzyloxy-3a-(4-chloφhenyl)-3-phenyl-2,3,3a,8b-tetrahydrocyclopenta- [b]benzoruran-l ,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 61A hergestellt.
Ausbeute: 54% d. Th.
LC-MS (Methode 3): Rt = 4.85 min.
MS (ESIneg): m/z = 483 (M-H)-
'H-NMR (300 MHz, DMSO-d6): δ = 7.46-7.12 (m, 13 H), 6.88-6.85 (m, 2 H), 6.60-6.55 (m, 2 H), 5.64 (d, 1 H), 5.08-5.06 (m, 3 H), 4.16-4.15 (m, 1 H), 4.07-3.99 (m, 1 H), 1.94-1.91 (m, 2 H).
Beispiel 48
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-3-phenyl-8-(2-pyrrolidin-l-yl-ethoxy)-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 69A hergestellt.
Ausbeute: 70% d. Th.
LC-MS (Methode 7): Rt = 2.10 min.
MS (ESIpos): m/z = 492 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.26 (t, 1 H), 7.21-7.17 (m, 2 H), 7.16-7.11 (m, 2 H), 7.10- 7.02 (m, 3 H), 6.99-6.94 (m, 2 H), 6.68 (d, 1 H), 6.64 (d, 1 H), 6.49 (br. s, 1 H), 4.95 (br. s, 1 H), 4.54 (dd, 1 H), 4.28-4.19 (m, 1 H), 4.16-4.06 (m, 1 H), 3.41-3.25 (m, 1 H), 3.10-2.95 (m, 1 H), 2.75-2.51 (m, 5 H), 2.47-2.37 (m, 1 H), 2.32-2.18 (m, 1 H), 1.79-1.71 (m; 4 H).
Die präparative Trennung des Racemats in die Enantiomeren wird über HPLC an chiraler Phase nach Methode 17A durchgeführt.
Analytische Daten (Methode 17B):
Enantiomer A: Rt = 8.60 min., Enantiomer B: Rt = 9.55 min.
Beispiel 49
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-8-(2-dimethylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran- 1 , 8b-( l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 69A hergestellt.
Ausbeute: 81% d. Th.
LC-MS (Methode 9): Rt = 1.88 min.
MS (ESIpos): m/z = 466 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.26 (t, 1 H), 7.22-7.17 (m, 2 H), 7.16-7.11 (m, 2 H), 7.10- 7.02 (m, 3 H), 6.99-6.93 (m, 2 H), 6.68 (d, 1 H), 6.64 (d, 1 H), 6.52 (br. s, 1 H), 4.92 (br. s, 1 H),
4.55 (dd, 1 H), 4.23-4.15. (m, 1 H), 4.14-4.05 (m, 1 H), 3.38-3.26 (m, 1 H), 2.87-2.76 (m, 1 H), 2.60-2.38 (m, 2 H), 2.32-2.17 (m, 1 H), 2.25 (s, 6 H).
Beispiel 50
(lS*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-8-(2-methylaminoethoxy)-3-phenyl-2,3,3a,8b-tetτa- hydrocyclopenta[b]benzofuran- 1 , 8b-( 17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 69A hergestellt.
Ausbeute: 17% d. Th.
LC-MS (Methode 9): Rt = 1.88 min.
MS (ESIpos): m/z = 452 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.30 (t, 1 H), 7.26-7.14 (m, 4 H), 7.13-7.04 (m, 3 H), 6.95- 6.89 (m, 2 H), 6.74 (d, 1 H), 6.66 (d, 1 H), 4.64 (dd, 1 H), 4.36-4.19 (m, 2 H), 3.41-3.25 (m, 1 H), 2.62 (s, 3 H), 2.48-2.39 (m, 1 H), 2.33-2.21 (m, 1 H).
Beispiel 51
(lR*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-8-(2-dimethylaminoethoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(l/J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 71 A hergestellt.
Ausbeute: 65% d. Th.
LC-MS (Methode 4): Rt = 2.24 min.
MS (ESIpos): m/z = 466 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.23 (t, 1 H), 7.19-7.14 (m, 2 H), 7.13-6.98 (m, 7 H), 6.63 (d, 1 H), 6.61 (d, 1 H), 5.30 (br. s, 1 H), 4.54 (d, 1 H), 4.29-4.12 (m, 3 H), 3.88 (dd, 1 H), 2.74 (dt, 1 H), 2.72-2.58 (m, 2 H), 2.23 (s, 6 H), 1.99 (dd, 1 H).
Beispiel 52
(lR*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-3-phenyl-8-(2-pyrrolidin-l-yl-ethoxy)-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 71 A hergestellt.
Ausbeute: 47% d. Th.
LC-MS (Methode 4): Rt = 2.29 min.
MS (ESIpos): m/z = 492 (M+H)+
'H-NMR (300 MHz, DMSO-d6): δ = 7.22 (t, 1 H), 7.18-7.12 (m, 2 H), 7.11-6.99 (m, 7 H), 6.63 (d, 1 H), 6.61 (d, 1 H), 5.42 (br. s, 1 H), 4.51 (d, 1 H), 4.31-4.13 (m, 3 H), 3.91 (dd, 1 H), 2.90-2.65 (m, 2 H), 2.75 (dt, 1 H), 2.63-2.45 (m, 4 H), 1.99 (dd, 1 H), 1.71-1.60 (m, 4 H).
Beispiel 53
(lR*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-8-(2-methoxyethoxy)-3-phenyl-2,3,3a,8b-tetrahydro- cyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 6A ausgehend von Beispiel 78A hergestellt.
Ausbeute: 12% d. Th.
LC-MS (Methode 4): Rt = 3.50 min.
MS (ESIpos): m/z = 453 (M+H)+
Η-NMR (200 MHz, DMSO-d6): δ = 7.24 (t, 1 H), 7.15-6.94 (m, 9 H), 6.66 (d, 1 H), 6.59 (d, 1 H), 5.14 (s, 1 H), 4.62-4.51 (m, 2 H), 4.22-4.14 (m, 2 H), 3.89 (dd, 1 H), 3.73-3.66 (m, 2 H), 3.35 (s, 3 H) 2.74 (ddd, 1 H), 1.98 (dd, 1 H).
Beispiel 54
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(3-methylaminopropoxy)-3-ρhenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzoftιran- 1 , 8b-( 17J)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 79A hergestellt.
Ausbeute: 94% d. Th.
LC-MS (Methode 7): Rt = 1.83 min.
MS (ESIpos): m/z = 466 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 8.63 (br. s, 1 H), 7.32-6.90 (m, 10 H), 6.70 (d, 1 H), 6.60 (dd, 1 H), 5.92 (d, 1 H), 5.04 (s, 1 H), 4.49-4.40 (m, 1 H), 4.13-4.07 (m, 2 H), 3.35-3.20 (m, 1 H), 3.08- 3.00 (m, 2 H), 2.58 (s, 3 H), 2.55-2.03 (m, 4 H).
Beispiel 55
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-6-(3-dimethylaminopropoxy)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 79A hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 7): Rt = 1.86 min.
MS (ESIpos): m/z = 480 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.30-6.90 (m, 10 H), 6.68 (d, 1 H), 6.58 (dd, 1 H), 5.89 (d, 1 H), 5.04 (s, 1 H), 4.50-4.44 (m, 1 H), 4.08-4.01 (m, 2 H), 3.35-3.25 (m, 1 H), 2.71-2.64 (m, 2 H), 2.39 (s, 6 H), 2.29-2.11 (m, 2 H), 2.02-1.91 (m, 2 H).
Beispiel 56
(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-3-phenyl-6-(3-pyrrolidin-l-yl-propoxy)-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 79A hergestellt.
Ausbeute: 99% d. Th.
LC-MS (Methode 7): Rt = 1.91 min.
MS (ESIpos): m/z = 506 (M+H)+
'H-NMR (200 MHz, DMSO-d6): δ = 7.32-6.90 (m, 10 H), 6.70 (d, 1 H), 6.60 (dd, 1 H), 5.91 (d, 1 H), 5.05 (s, 1 H), 4.50-4.40 (m, 1 H), 4.13-4.07 (m, 2 H), 3.35-3.19 (m, 7 H), 2.50-1.92 (m, 8 H).
Beispiel 57
(lS*,3S*,3aR*,8bS*)-6-(3-Azetidin-l-yl-propoxy)-3a-(4-chlθφhenyl)-3-phenyl-2,3,3a,8b-tetra- hydrocyclopenta[b]benzofuran-l,8b-(lH)-diol
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 32 ausgehend von Beispiel 79A hergestellt.
Ausbeute: 69% d. Th.
LC-MS (Methode 13): Rt = 1.97 min.
MS (ESIpos): m/z = 492 (M+H)+
Η-NMR (300 MHz, DMSO-d6): δ = 7.27-7.03 (m, 8 H), 6.94-6.91 (m, 2 H), 6.64 (d, 1 H), 6.57 (dd, 1 H), 5.82 (d, 1 H), 4.99 (s, 1 H), 4.48-4.41 (m, 1 H), 4.02-3.98 (m, 2 H), 3.12 ( , 4 H), 2.49- 2.40 (m, 2 H), 2.26-2.14 (m, 1 H), 1.96 (t, 2 H), 1.70 (t, 2 H), 0.86-0.81 (m, 2 H).
Beispiel 58
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-17J- benzo[b]cyclopenta[d]furan-6-yl]-2-fluornicotinamid
In 0.5 ml DMSO werden 39.4 mg (0.1 mmol) (lS*,3S*,3aR*,8bS*)-6-Amino-3a-(4-chloφhenyl)- 3-phenyl-l,2,3,3a-tetrahydrocyclopenta[δ]benzofuran-l,8b-(lH)-diol (Beispiel 84A), 14.1 mg (0.1 mmol) 2-Fluoφicolin, 41.7 mg (0.13 mmol) 0-(Benzotriazol-l-yl)-N,N,N,N-tetramethyluronium- Tetrafluoroborat und 25.8 mg (0.2 mmol) Diisopropylethylamin zusammen gegeben und über
Nacht bei Raumtemperatur gerührt. Es wird dann vom Feststoff abfiltriert und das Filtrat mittels präparativer HPLC gereinigt.
Ausbeute: 19.7 mg (38% d. Th.)
LC-MS (Methode 10): Rt = 2.21 min.
MS (ESIpos): m z = 517 (M+H)+.
Beispiel 59
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-lH- benzo[b]cyclopenta[d]furan-6-yl]-6-fluoφyridin-2-carboxamid
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 58 ausgehend von Beispiel 84A hergestellt.
LC-MS (Methode 10): Rt = 2.35 min.
MS (ESIneg): m/z = 515 (M-H)\
Beispiel 60
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-17J- benzo[b]cycloρenta[d]furan-6-yl]-l-ethyl-l/J-pyrazol-3-carboxamid
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 58 ausgehend von Beispiel 84A hergestellt.
LC-MS (Methode 10): Rt = 2.25 min.
MS (ESIpos): m/z = 516 (M+H)+.
Beispiel 61
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-l/J- benzo[b]cyclopenta[d]furan-6-yl]-l,2,3-thiadiazol-4-carboxamid
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 58 ausgehend von Beispiel 84A hergestellt.
LC-MS (Methode 10): Rt = 2.23 min.
MS (ESIneg): m/z = 504 (M-H)".
Beispiel 62
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chlθφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-lH- benzo[b]cyclopenta[d]furan-6-yl]-pyridin-2-carboxamid
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 58 ausgehend von Beispiel 84A hergestellt.
LC-MS (Methode 10): Rt = 2.38 min.
MS (ESIpos): m/z = 499 (M+H)+.
Beispiel 63
N-[(lS*,3S*,3aR*,8bS*)-3a-(4-Chloφhenyl)-l,8b-dihydroxy-3-phenyl-2,3,3a,8b-tetrahydro-lH- benzo [b] cyclopenta[d]furan-6-yl]- \H- 1 ,2,4-triazol-5 -carboxamid
Die Titelverbindung wird in Analogie zur Synthese von Beispiel 58 ausgehend von Beispiel 84A hergestellt.
LC-MS (Methode 10): Rt = 2.00 min.
MS (ESIpos): m z = 489 (M+H)+.
B. Bewertung der pharmakologischen Wirksamkeit
Die pharmakologische Wirkung der erfindungsgemäßen Verbindungen kann in folgenden Assays gezeigt werden:
1. In vftrtf-Bestimmung der NF- B- und AP-1-inhibitorischen Wirkung
l.a) Hemmung der Lnterleukin-8 (IL-8VFreisetzung aus humanen Endothelzellen
Die Expression des humanen IL-8-Gens wird durch eine Vielfalt von regulatorischen und Verstärker-Elementen kontrolliert, die oberhalb der Promoterregion des Gens lokalisiert sind. Diese Elemente enthalten Bindungsstellen für Transkriptionsfaktoren und erst deren Interaktion mit ihren DNA-Bindungsstellen ermöglicht die effiziente Transkription des IL-8-Gens. Die Aktivität der entsprechenden Transkriptionsfaktoren ist durch unterschiedliche Stimuli (z.B. Inter- leukin-lß, Tumor Nekrose Faktor-α) induzierbar und über verschiedene Signaltransduktionswege modulierbar [H. Holtmann et al., Mol. Cell. Biol. 19, 6742-6753 (1999)].
Für die maximale Expression von IL-8 ist die Aktivität zweier unterschiedlicher Transkriptionsfaktoren essentiell: NF-κB und AP-1 [Y.-H. Chang et al, Exp. CellRes. 278, 166-174 (2000)]. Die Zytokin-induzierte IL-8-Synthese lässt sich deshalb als Testsystem zur Identifikation und Charakterisierung von Testsubstanzen einsetzen, die direkt oder indirekt die Bindung von NF-κB und AP- 1 an ihre DNA-Bindungsstellen hemmen.
Versuchsdurchführung:
Humane Nabelschnur-Endothelzellen (HUVEC) wurden von der Firma CellSystems (St. Katharinen, Deutschland) bezogen und entsprechend der Lieferantenempfehlung in EGM-2- Medium inklusive Wachstumszusätzen (CellSystems, St. Katharinen, Deutschland) in 165 cm2- Gewebekulturflaschen kultiviert. Nach Erreichen einer 60-80%-igen Konfluenz des Zellrasens werden die Zellen mittels Trypsin-Behandlung vom Flaschenboden abgelöst, mechanisch dissoziiert und in einer Zelldichte von 5000 Zellen/Vertiefung in 96-Loch-Mikrotiteφlatten (Corning, Wiesbaden, Deutschland) ausgesät. Nach 3 Tagen wird das Kulturmedium komplett durch 170 μl frisches Medium pro Vertiefung ersetzt. Die Zellen werden am nächsten Tag für die Experimente eingesetzt.
Zur Bestimmung der hemmenden Wirkung der erfindungsgemäßen Substanzen auf die E -8-Frei- setzung werden diese zunächst in einer zehnfach höheren Konzentration, verglichen mit der gewünschten Endkonzentration im Test, in Medium, das 1% DMSO enthält, gelöst. Anschließend werden 20 μl der Substanzlösung pro Vertiefung zugesetzt. Die Bildung und Freisetzung von IL-8
wird danach durch Zugabe von Interleukin-lß (IL-lß, Endkonzentration 10 ng/ml; Biosource GmbH, Solingen, Deutschland) induziert und die Zellen für 6 h bei 37°C im C02-Inkubator inkubiert. Im Anschluss werden 150 μl des Zeilüberstandes abgenommen und bis zur Messung des IL-8-Gehaltes mittels ELISA (Biosource GmbH, Solingen, Deutschland) bei -20°C eingefroren. Zur Durchführung des ELISA werden die Proben aufgetaut, 1:10 verdünnt und der Test entsprechend den Herstellerangaben durchgeführt. Die Hemmwirkung der erfindungsgemäßen Verbindungen auf die IL-8-Freisetzung wird durch den Vergleich zu Vehikel-behandelten Zellen ermittelt.
Repräsentative Wirkdaten zu den erfindungsgemäßen Verbindungen sind in der folgenden Tabelle 1 aufgeführt:
Tabelle 1
[IC50 = Wirkstoffkonzentration, die eine 50%-ige Hemmung der IL-8-Freisetzung bezogen auf deren Maximaleffekt bewirkt]. l.b) Hemmung der AP-1 -Aktivität in Astrozyten-Kulturen
AP-1 ist ein aus Homo- oder Heterodimeren der Jun-, Fos- und ATF-Familie gebildeter, im Zellkern lokalisierter Transkriptionsfaktor. Aktivierende Signale führen einerseits zu einer vermehrten Synthese der Einzelkomponenten, andererseits zu einer spezifischen Phosphorylierung der Jun- oder ATF-Untereinheiten. Beide Prozesse führen zu einer verstärkten Wechselwirkung des Proteinkomplexes mit dessen Zielgenen und ermöglicht so deren Expression. Die Stimulusinduzierte Phosphorylierung von z.B. c-Jun kann somit als Indikator für die AP- 1 -Aktivierung dienen. Mithilfe der nachfolgend beschriebenen immunozytochemischen Detektion der c-Jun-
Phosphorylierung lässt sich der Einfluß der erfindungsgemäßen Verbindungen auf die AP-1- Aktivierung untersuchen.
Versuchsdurchführung:
Gemischte Gliazellkulturen wurden aus Gehirnen von 1 Tag-alten Ratten (Wistar) hergestellt. Dazu werden die Tiere durch Enthauptung getötet, die Gehirne entnommen und in kalter Hank's Salzlösung (HBSS, Gibco, Karlsruhe, Deutschland) gesammelt. Hirnstamm und Kleinhirn werden entfernt, die zerebralen Hemisphären von Hirnhäuten befreit und die Gewebestücke mechanisch in Gegenwart von Papain (Papain dissociation kit, CellSystems, St. Katharinen, Deuschland) dissoziiert. Die Zellen werden durch Zentrifugation bei 450 x g gesammelt und in 175 cm2-Gewebe- kulturflaschen ausplattiert. Die Zellen werden für 12-14 Tage in DMEM/Ham's F12-Medium, 10% fötalem Kälberserum, 100 μg/ml Penicillin Streptomycin (Sigma, Taufkirchen, Deutschland) kultiviert. Anschließend werden Zellbruchstücke und auf der Oberfläche des Zellrasens wachsende mikrogliale und oligodendrogliale Zellen für 2 h mithilfe eines Schüttlers abgeschüttelt und die verbleibenden Astrozyten durch Trypsinierung vom Flaschenboden abgelöst. Die Zellen werden mechanisch dissoziiert, durch Zentrifugation bei 450 x g gesammelt, erneut mechanisch dissoziiert und in einer Dichte von 100000 Zellen/Vertiefung auf Poly-D-Lysin-beschichtete 8-Kammer- Objektträger (NUNC, Dänemark) verteilt. Die so angereicherten Astrozyten werden in Phenolrotfreiem DMEM/Ham's F12-Medium, 10% fötalem Kälberserum, 100 μg/ml Penicillin/Streptomycin (Sigma, Taufkirchen, Deutschland) kultiviert. Einen Tag vor ihrer Verwendung im Versuch wird der Anteil von fötalem Kälberserum im Medium auf 1% reduziert.
Zur Untersuchung der Wirkung der erfindungsgemäßen Verbindungen auf die AP- 1 -Aktivierung werden diese in der gewünschten Konzentration dem Kulturmedium zugegeben (in der Regel finale Konzentration von 1 μM im Test). Anschließend wird der AP- 1 -Signalweg (c-Jun-Phos- phorylierung) durch Zugabe von Lipopolysaccherid (LPS, Sigma, Taufkirchen, Deutschland) in einer Endkonzentration von 100 ng/ml oder durch Zugabe von Interleukin-lß (IL-lß, Biosource GmbH, Solingen, Deutschland) in einer Endkonzentration von 30 ng/ml stimuliert. Nach 90 min werden die Zellen kurz mit Phosphat-gepufferter Salzlösung (PBS) gewaschen und dann für 10 min in 4%-iger Paraformaldehyd-Lösung in PBS fixiert. Danach werden die Zellen für 5 min mit -20°C kaltem Methanol permeabilisiert, in PBS, 5% Sucrose, 0.3% TritonX-100 gewaschen und für 30 min bei Raumtemperatur in Blockieφuffer (PBS, 1% Ziegenserum, 2% BSA) inkubiert. Die Inkubation mit den gegen das Astrozyten-spezifische Protein GFAP (glial fibrillary acidic protein; monoklonaler Antiköφer aus Maus, Sigma, Taufkirchen, Deutschland) und gegen die Serin-63- phosphorylierte Form von c-Jun (polyklonaler Antikörper aus Kaninchen, Calbiochem, Bad Soden, Deutschland) gerichteten, primären Antiköφern erfolgt im selben Ansatz bei 4°C über Nacht. Die
Antiköφer gegen GFAP werden 1:400 und die gegen phospho-c-Jun 1:50 in Blockieφuffer verdünnt. Überschüssiger Antiköφer wird anschließend durch dreifaches Waschen in PBS, 5% Sucrose, 0.3% TritonX-100 entfernt und die Zellen bei Raumtemperatur für je 1 h nacheinander mit den jeweiligen sekundären, Spezies-spezifischen und in Blockieφuffer verdünnten Anti- köφern inkubiert (anti-Maus Cy2-konjugierte Antiköφer, 1:500, Amersham Biosciences, Freiburg, Deutschland; anti-Kaninchen Cy3-konjugierte Antiköφer, 1:800, Sigma, Taufkirchen, Deutschland). Überschüssiger Antiköφer wird anschließend durch dreifaches Waschen in PBS, 5% Sucrose, 0.3% TritonX-100 entfernt und die Zellkerne durch 10 min Inkubation der Zellen mit Hoechst 33258 (4 μg/ml in PBS) angefärbt. Abschließend werden die Zellen erneut zweimal für 5 min mit PBS gewaschen, der Dichtring der 8-Kammer-Objekttτäger entfernt und die Zellen in Einbett-Medium (Sigma, Taufkirchen, Deutschland) unter Zuhilfenahme eines Deckgläschens eingebettet. Das Versuchsergebnis läßt sich mithilfe eines Fluoreszenz-Mikroskops (Objektiv- Vergrößerung 25-fach) beurteilen.
Phosphoryliertes c-Jun-Protein ist aufgrund der Bindung des Cy3 -konjugierten sekundären Anti- köφers an den detektierenden, gegen phospho-c-Jun gerichteten, primären Antiköφer durch eine rote, im Zellkern lokalisierte Fluoreszenz erkennbar. Der Zellkern erscheint blau-fluoreszent. Astrozyten sind durch eine Cy-2-vermittelte, grün-fluoreszierende GFAP-Färbung identifizierbar.
Unter basalen Bedingungen enthalten nur wenige Zellkerne phosphoryliertes c-Jun. Durch Zugabe von LPS wird die AP- 1 -vermittelte Signaltransduktion induziert, so dass mehr als 80% der Zellkerne rötlich fluoreszieren, also phospho-c-Jun enthalten. Die Rotfärbung wird durch Zugabe der erfindungsgemäßen Verbindungen deutlich erniedrigt, was zum einen durch eine Reduktion der Farbintensität, zum anderen durch eine Verminderung der gefärbten Kerne zum Ausdruck kommt. Die Kolokalisation der Prozesse in Astrozyten wird durch eine Überlagerung der grünen, roten und blauen Fluoreszenzbilder ermöglicht.
2. Bestimmung der metabolischen Stabilität
In v/tro-Inkubation mit Lebermikrosomen:
Zur Bestimmung der hepatischen mikrosomalen Stabilität werden die erfindungsgemäßen Substanzen mit Lebermikrosomen unterschiedlicher Species inkubiert. Die Inkubationskonzentration der Substanzen wird möglichst niedrig gehalten (bevorzugt < 1 μM), ebenso die Konzentration der mikrosomalen Proteine im Testansatz (bevorzugt 0.2 mg mikrosomales Protein pro ml Inkubationsansatz). Diese Vorgehensweise ermöglicht für die meisten Verbindungen ein Arbeiten im linearen Bereich der Michaelis-Menten-Kinetik. Aus dem Inkubationsansatz wird zu verschiedenen Zeitpunkten (insgesamt 7 Zeitpunkte) eine Probe für die Bestimmung der verbliebenen Substanz-
konzentration entnommen. Aus der Halbwertszeit der Substanz im Inkubationsansatz werden die Clearance (CL) und die maximale Bioverfügbarkeit (Fmax) der Testsubstanz für die jeweilige Species berechnet. Der Gehalt an organischen Lösungsvermittlern beträgt maximal 1% Acetonitril bzw. maximal 0.2% DMSO, um den Einfluss auf die mikrosomalen Enzyme zu minimieren.
Versuchsdurchführung:
Die Versuche werden wie im Detail von J.B. Houston und DJ. Carlile [Drug Metab. Rev. 29, 891- 922 (1997)] beschrieben durchgeführt.
Wie für die in der Tabelle 2 aufgeführten Verbindungen beispielhaft gezeigt, weisen die erfindungsgemäßen Substanzen in Lebeπnikrosomen-Präparationen eine erhöhte Stabilität im Vergleich zu den in WO 00/08007 beschriebenen Verbindungen auf:
Tabelle 2
3. In tro-Proliferation von Tumorzellen
Verschiedene Formen von Krebs-Erkrankungen sind gekennzeichnet durch die unkontrollierte, übermäßige Proliferation unterschiedlicher Zelltypen, was zur Ausbildung von Metastasen und Tumoren führt. Die Verwendungsmöglichkeit der erfindungsgemäßen Substanzen zur Behandlung hypeφroliferativer Erkrankungen lässt sich durch ihre Aktivität auf die Zellteilungsrate von Tumorzellen in vitro untersuchen. Die Verknüpfung zwischen anti-proliferativer Wirkung in vitro und klinischer anti-Tumor- Wirkung ist gut etabliert. Die therapeutische Verwendbarkeit von z.B. Taxol [Silvestrini et al., Stern Cells 11, 528-535 (1993)], Taxotere [Bissery et al., Ana Cancer Drugs 6, 339 (1995)] oder Topoisomerase-Inhibitoren [Edelman et al., Cancer Chemother.
Pharmacol. 37, 385-393 (1996)] wurde durch deren Aktivität in in vz'tro-Tumorzellen- Proliferationstests belegt.
Versuchsdurchführung:
Die Tumorzelllinien, wie z.B. MDA-MB-231 -Zellen (humane Brust-Adenokarzinomzellen), H460- Zellen (humane Lungen-Karzinomzellen) oder HCT-15-Zellen (humane Darm-Adenokarzinom- zellen), werden in entsprechenden, vom Lieferanten (z.B. LGC Promochem, Wesel, Deutschland) empfohlenen Wachstumsmedien (Sigma, Taufkirchen, Deutschland) expandiert. Einen Tag vor Zugabe der erfindungsgemäßen Verbindungen werden die Zellen in einer Dichte von 3000 Zellen/Vertiefung in 100 μl Wachstumsmedium auf schwarze 96-Loch-Mikrotiteφlatten mit klarem Boden verteilt. Am Tag der Testsubstanzzugabe wird je eine Mikrotiteφlatte pro Tumorzelllinie zur Bestimmung der in den Vertiefungen befindlichen Anfangszellzahl verwendet. Den Geschwisteφlatten werden die in Wachstumsmedium und DMSO verdünnten erfindungsgemäßen Substanzen zugegeben. Die Testsubstanzen werden in der Regel in unterschiedlichen Konzentrationen, beginnend mit 10 μM Endkonzentration im Test, zugesetzt. Die DMSO-Konzentration im Test beträgt 0.1%. Vierundzwanzig Stunden nach Substanzzugabe werden die Zellzahlen pro Vertiefung mithilfe des CellTiter-Glo® Luminescent Cell Viability-Tests (Promega GmbH, Mannheim, Deutschland) bestimmt. Der Test wird entsprechend den Herstellerangaben durchgeführt und mithilfe eines Luminometers ausgewertet. Zur Ermittlung der anti-proliferativen Aktivität der erfindungsgemäßen Verbindungen werden die vor Substanzzugabe in Geschwisteφlatten bestimmten Zellzahlen abgezogen und der prozentuale Unterschied der Zellzahländerungen zwischen Substanz-behandelten und Vehikel-behandelten Zellen ermittelt. Die relative Wirksamkeit der unterschiedlichen Testsubstanzen wird über einen Vergleich der Konzentrationen, die 50% ihres Maximaleffektes bewirken (IC50), bestimmt.
Repräsentative Wirkdaten zu den erfindungsgemäßen Verbindungen sind in der folgenden Tabelle 3 aufgeführt:
Tabelle 3
[IC
50 = Wirkstoffkonzentration, die eine 50%-ige Hemmung der Tumorzellproliferation bezogen auf deren Maximaleffekt bewirkt].
4. Maus MPTP-Tiermodell der Parkinson'schen Krankheit
Die Parkinson'sche Krankheit ist histopathologisch durch den selektiven Verlust einer Gruppe von Nervenzellen der Substantia nigra, die den Neurotransmitter Dopamin synthetisieren, gekennzeichnet. l-Methyl-4-phenyl-l,2,3,6-tetrahydropyridin (MPTP) ist eine zu Beginn der 80er Jahre identifizierte Verunreinigung synthetischer Drogen, die beim Menschen Symptome des Parkinsonismus und die charakteristische Degeneration dopaminerger Neurone induziert. Die Applikation von MPTP führt bei verschiedenen Tierarten, wie z.B. Maus oder Affe, zu Verhaltens- und histopatho- logischen Änderungen, die denen der menschlichen Erkrankung vergleichbar sind. Daher gilt das Maus MPTP-Tiermodell als valides Modell für die Parkinson'sche Krankheit und als geeignet, um neuroprotektive Wirkeigenschaften von Prüfsubstanzen zu untersuchen.
Versuchsdurchführung:
Die Versuche werden im wesentlichen wie von E. Bezard et al. [Neurosci. Lett. 234, 47-50 (1997)] beschrieben durchgeführt. Männlichen 8-Wochen-alten C57/BL6-Mäusen wird an drei aufeinander folgenden Tagen je 4 mg/kg MPTP i.p. appliziert. Die erfindungsgemäßen Substanzen werden beginnend unmittelbar vor der ersten MPTP-Applikation täglich oder zweimal täglich oral verabreicht. An Tag 11 nach der ersten MPTP-Gabe werden die Tiere anästhesiert, intrakardial zunächst mit 25 ml 0.9%-iger Salzlösung, dann mit 75 ml 4%-iger Paraformaldehyd-Lösung perfundiert, und die Gehirne zur histologischen Bearbeitung entnommen. Wie von E. Bezard et al. [Neurosci. Lett. 234, 47-50 (1997)] beschrieben, werden dazu die dopaminergen Neurone immuno- histochemisch sichtbar gemacht als Zellen, die das im Dopamin-Stoffwechsel essentielle Enzym Tyrosin-Hydroxylase enthalten. Die Anzahl der verbliebenen dopaminergen Neurone wird mithilfe eines Computeφrogramms in drei unterschiedlichen Schnitten pro Tier, die repräsentativ für eine mittlere Schnittebene durch die Substantia nigra sind, quantitativ erfasst [Nelson et al., J. Comp. Neurol. 369, 361-371 (1996)]. Die neuroprotektive Wirksamkeit der Testsubstanzen wird durch den Vergleich zu Vehikel-behandelten MPTP-Tieren und komplett unbehandelten Tieren ermittelt.
5. Subdurales Hämatom an Ratten als Tiermodell für traumatische Schädel-Hirn- Verletzungen
Schwere traumatische Schädelverletzungen sind oft begleitet durch Einblutungen unterhalb der Hirnhäute. Die subdurale Blutansammlung führt zu einer lokalen Verminderung des zerebralen
Blutflusses der angrenzenden kortikalen Gehirnregion bei gleichzeitigem Anstieg des Glukoseverbrauchs sowie der extrazellulären exzitatorischen Aminosäuren (z.B. Glutamat). Der dem Hämatom benachbarte Gehirnbereich wird ischämisch, was zur Schädigung und zum Absterben von Nervenzellen führt. Die subdurale Injektion von autologem Blut in kortikale Gehirnregionen der Ratte dient als Tiermodell zur Simulation traumatischer Schädel-Hirn- Verletzungen des Menschen. Das Tiermodell eignet sich, um die neuroprotektive Wirksamkeit der erfindungsgemäßen Substanzen zu untersuchen.
Versuchsdurchführung:
Der operative Eingriff an männlichen Wistar-Ratten und die unilaterale Applikation von autologem Blut wird wie im Detail von M. Eijkenboom et al. [Neuropharm. 39, 817-834 (2000)] beschrieben durchgeführt. Direkt nach Verschluss der Wunde und jeweils nach 2 h und 4 h werden die erfindungsgemäßen Substanzen intravenös in den gewünschten Dosen injiziert. Sieben Tage nach der Operation werden die Tiere getötet, die Gehirne entnommen und das Infarktvolumen wie von M. Eijkenboom et al. [Neuropharm. 39, 817-834 (2000)] beschrieben bestimmt. Die neuroprotektive Wirksamkeit der erfindungsgemäßen Substanzen wird durch den Vergleich zu Vehikel- behandelten Tieren ermittelt.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische Zubereitungen überfuhrt werden:
Tablette:
Zusammensetzung:
100 mg der erfindungsgemäßen Verbindung, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrrolidon (PVP 25) (Fa. BASF, Ludwigshafen, Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus erfindungsgemäßer Verbindung, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat 5 Minuten gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse veφresst (Format der Tablette siehe oben). Als Richtwert für die Veφressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der erfindungsgemäßen Verbindung, 1000 mg Ethanol (96%), 400 mg Rhodigel® (Xanthan gum der Firma FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, die erfindungsgemäße Verbindung wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluß der Quellung des Rhodigels wird ca. 6 h gerührt.
Oral applizierbare Lösung:
Zusammensetzung:
500 mg der erfindungsgemäßen Verbindung, 2.5 g Polysorbat und 97 g Polyethylenglycol 400. Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 20 g orale Lösung.
Herstellung:
Die erfindungsgemäße Verbindung wird in der Mischung aus Polyethylenglycol und Polysorbat unter Rühren suspendiert. Der Rührvorgang wird bis zur vollständigen Auflösung der erfindungsgemäßen Verbindung fortgesetzt. v.-Lösung:
Die erfindungsgemäße Verbindung wird in einer Konzentration unterhalb der Sättigungslöslichkeit in einem physiologisch verträglichen Lösungsmittel (z.B. isotonische Kochsalzlösung, Glucose- lösung 5% und/oder PEG 400-Lösung 30%) gelöst. Die Lösung wird steril filtriert und in sterile und pyrogenfreie Injektionsbehältnisse abgefüllt.