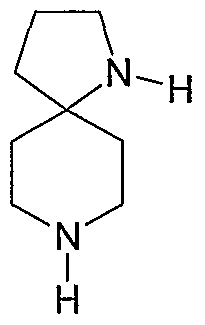
THE USE OF N-ARYL DIAZASPIRACYCLIC COMPOUNDS
IN THE TREATMENT OF ADDICTION
This application claims benefit of U.S. Provisional Patent Application
No. 60/603,479, filed August 20, 2004.
Field of the Invention
The present invention relates to nicotinic antagonists, particularly antagonists
and partial antagonists that have more potent antagonistic activity with respect to
dopamine release than at the α4β2 receptor, pharmaceutical compositions including
these compounds, and the use of these compounds in the treatment of addiction,
including smoking addiction, addiction to narcotics and other drugs, and obesity that
occurs following drug cessation.
Background of the Invention
Smoking addiction is a complex phenomenon believed to involve cognition
enhancement, psychological conditioning, stress adaptation, reinforcing properties and relief from withdrawal. Consequently, providing therapeutic treatment for smoking
addiction is an extremely difficult challenge.
The nicotine in tobacco may be partially responsible for the difficulty some
individuals face in overcoming smoking addiction. Numerous methods have been
developed to assist with smoking cessation, including reducing consumption over
time, and providing alternate delivery vehicles for nicotine, including gums and skin
patches.
Neuronal nicotinic acetylcholine receptors (nAChRs) are widely distributed
throughout the central and peripheral nervous systems including several regions of the
brain. The two most prominent CNS subtypes of nAChRs are α4β2 andα7. However,
the predominance of a particular nicotinic receptor subtype in the brain does not
necessarily reflect its functional importance. For example, although of lesser
prevalence in the brain, the α3β2-containing receptor subtypes are believed to be at
least partially responsible for mediating dopamine release, based on studies in which
antagonists of these receptors (i.e., bungarotoxin and α-conoxin partially inhibited
dopamine release (Dworsin et al, J. Pharm. Ex. Titer. 10(10):1561-1581 (2000)).
Accordingly, it is believed that there are multiple receptor subtypes involved in
nicotine-evoked dopamine release in striatum. Nicotine antagonists active against one
or more of these receptors one are well known in the art, and are described, for
example, in Dwoskin et al., J. Pharm. Ex. Titer. 298(2):395 (2001).
One pharmaceutical approach to causing smoking cessation involves blocking
the nicotine signal from tobacco with another agent, such as Bupropion. At low
micromolar concentrations, Buproprion non-competitively inhibits α3β2, α4β2 and α7 nAChRs, and is now marketed as an aid to smoking cessation. Other non-competitive
nicotinic antagonists have also been considered as an approach to smoking cessation.
One theory is that the nicotine antagonists block the reinforcing signal from nicotine
associated with smoking addiction. Mecamylamine, an antagonist at both α4β2 and α7
receptors, is an example of a nicotine antagonist that has been used, alone and in
combination with nicotine replacement therapy, to promote smoking cessation.
In spite of the known methods for treating smoking addiction, there remains an
interest in new methods and pharmaceutical compositions for treating smoking
addiction.
It is also difficult to overcome addiction to other compounds, including
opiates, cocaine, and other ilicit drugs. Mecamylamine and other nicotinic
compounds have been proposed for use in overcoming addiction to these illicit drugs
(see for example, Reid, Neuropsychopharmacology, 20(3):297-307 (1999);
Campiani et al., J Med Chem, 46:3822-39 (2003) (discussing the role of dopamine
D3/D2 receptors), Chi and de Wit H , Alcoholism: Clinical and Experimental
Research, 27:780-786 (2003); PiUa et al., Nature, 400:371-5 (1999) (discussing the
role of partial dopamine D3 receptor agonists); Reid et al.,
Neuropsychopharmacology, 20:297-307 (1999); Slemmer et al., J. Pharmacol. Exp.
Ther. 295:321-327 (2000); Vorel et al., J. Neurosci., 22:9595-603 (2002) (discussing
how dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-
enhanced brain reward in rats), and Zachariou et al., Neuropsychopharmacology,
24:576-589 (2001), the contents of each of which are hereby incorporated by reference
in their entirety) .
Weight gain is often associated with drug cessation (see, for example, Dwoskin et al., "Recent developments in neuronal nicotinic acetylcholine receptor antagonists," Exp. Opin. Ther. Patents 10:1561-1581 (2000). It would be desirable to provide methods and compositions for inhibiting this weight gain.
Dopamine release is believed to be associated with the physiological "reward" associated with consumption of these substances of addiction. Modulation of
dopamine release has been proposed for use in treating addiction. Modulation of the α4β2 receptor is one way to modulate dopamine release, and may be at least part of the
mechanism by which mecamylamine is effective at treating drug addiction. However, it may be desirable in some instances to modulate dopamine release without
antagonizing α4β2 activity. Thus, the availability of a variety of ligands that bind with
high affinity and selectivity for receptors other than α4β2, and that modulate dopamine
release, are of interest.
Further, a limitation of some nicotinic compounds is that they are associated
with various undesirable side effects, for example, by stimulating muscle and
ganglionic receptors. It would be desirable to have compounds, compositions and
methods for preventing and/or treating drug addiction, promoting smoking cessation,
and inhibiting obesity associated with overcoming addiction, where the compounds
exhibit pharmacology with a beneficial effect (e.g., inhibition of dopamine secretion),
but without significant associated side effects.
The present invention provides such compounds, compositions and methods.
Summary of the Invention
Compounds, pharmaceutical compositions, and methods of treating nicotine
addiction, drug addiction, and/or obesity associated with drug and/or nicotine
cessation are disclosed. The compounds function by decreasing dopamine release,
without significantly affecting the α4β2 receptor. Decreased dopamine release results
in a decreased physiological "reward" associated with administration of nicotine or
illicit drugs, and thus helps overcome addiction.
The compounds are N-aryl diazaspirocyclic compounds, bridged analogs of N-
heteroaryl diazaspirocyclic compounds, or prodrugs or metabolites of these
compounds. The aryl group can be a five- or six-membered heterocyclic ring
(heteroaryl). Examples of the N-aryl diazaspiocyclic compounds include 7-(3-
pyridyl)-l,7-diazaspiro[4.4]nonane and l-(3-pyridyl)-l,7-diazaspiro[4.4]nonane.
Examples of bridged analogs of N-heteroaryl diazaspirocyclic compounds include 1'-
(3 -pyridyl)-spiro [ 1 -azabicyclo [2.2.1 ]heptane-2,3 '-pyrrolidine] .
The compounds and compositions can be used to treat and/or prevent a wide
variety of conditions or disorders, particularly those disorders characterized by
dysfunction of nicotinic cholinergic neurotransmission, including disorders involving
neuromodulation of neurotransmitter release, such as dopamine release. CNS
disorders, which are characterized by an alteration in normal neurotransmitter release,
are another example of disorders that can be treated and/or prevented. The
compounds and compositions can also be used to alleviate pain. The methods involve
administering to a subject an effective amount of an N-aryl diazaspirocyclic compound, bridged analog of an N-heteroaryl diazaspirocyclic compound, or prodrug
or metabolite thereof to alleviate the particular disorder.
The pharmaceutical compositions include an effective amount of the
compounds described herein. When employed in effective amounts, the compounds
can cause a decrease in dopamine release in a subject, without demonstrating
stimulant sensitization properties. The pharmaceutical compositions provide therapeutic benefit to individuals
suffering from such disorders and exhibiting clinical manifestations of such disorders.
The pharmaceutical compositions are believed to be safe and effective with regards to
treating these disorders.
The foregoing and other aspects of the present invention are explained in detail
in the detailed description and examples set forth below.
Detailed Description of the Invention
Compounds, pharmaceutical compositions including the compounds, and
methods of preparation and use thereof are disclosed.
The following definitions will be useful in understanding the metes and
bounds of the invention as described herein.
As used herein, "alkyl" refers to straight chain or branched alkyl radicals
including C1-C8, preferably C1-C5, such as methyl, ethyl, or isopropyl; "substituted
alkyl" refers to alkyl radicals further bearing one or more substituent groups such as
hydroxy, alkoxy, aryloxy, mercapto, aryl, heterocyclo, halo, amino, carboxyl,
carbamyl, cyano, and the like; "alkenyl" refers to straight chain or branched
hydrocarbon radicals including C1-C8, preferably C1-C5 and having at least one
carbon-carbon double bond; "substituted alkenyl" refers to alkenyl radicals further
bearing one or more substituent groups as defined above; "cycloalkyl" refers to
saturated or unsaturated, non-aromatic, cyclic ring-containing radicals containing
three to eight carbon atoms, preferably three to six carbon atoms; "substituted
cycloalkyl" refers to cycloalkyl radicals further bearing one or more substituent groups
as defined above; "aryl" refers to aromatic radicals having six to ten carbon atoms;
"substituted aryl" refers to aryl radicals further bearing one or more substituent groups
as defined above; "alkylaryl" refers to alkyl-substituted aryl radicals; "substituted
alkylaryl" refers to alkylaryl radicals further bearing one or more substituent groups as
defined above; "arylalkyl" refers to aryl-substituted alkyl radicals; "substituted
arylalkyl" refers to arylalkyl radicals further bearing one or more substituent groups as defined above; "heterocyclyl" refers to saturated or unsaturated cyclic radicals
containing one or more heteroatoms (e.g., O, N, S) as part of the ring structure and
having two to seven carbon atoms in the ring; "substituted heterocyclyl" refers to
heterocyclyl radicals further bearing one or more substituent groups as defined above.
As used herein, an "agonist" is a substance that stimulates its binding partner,
typically a receptor. Stimulation is defined in the context of the particular assay, or
may be apparent in the literature from a discussion herein that makes a comparison to
a factor or substance that is accepted as an "agonist" or an "antagonist" of the
particular binding partner under substantially similar circumstances as appreciated by
those of skill in the art. Stimulation may be defined with respect to an increase in a
particular effect or function that is induced by interaction of the agonist or partial
agonist with a binding partner and can include allosteric effects.
As used herein, an "antagonist" is a substance that inhibits its binding partner,
typically a receptor. Inhibition is defined in the context of the particular assay, or may
be apparent in the literature from a discussion herein that makes a comparison to a
factor or substance that is accepted as an "agonist" or an "antagonist" of the particular
binding partner under substantially similar circumstances as appreciated by those of
skill in the art. Inhibition may be defined with respect to a decrease in a particular
effect or function that is induced by interaction of the antagonist with a binding
partner, and can include allosteric effects.
As used herein, a "partial agonist" is a substance that provides a level of
stimulation to its binding partner that is intermediate between that of a full or
complete antagonist and an agonist defined by any accepted standard for agonist
activity.
As used herein, a "partial antagonist" is a substance that provides a level of
inhibition to its binding partner that is intermediate between that of a full or complete
antagonist and an inactive ligand.
It will be recognized that stimulation, and hence, inhibition is defined
intrinsically for any substance or category of substances to be defined as agonists,
antagonists, or partial agonists. As used herein, "intrinsic activity", or "efficacy,"
relates to some measure of biological effectiveness of the binding partner complex.
With regard to receptor pharmacology, the context in which intrinsic activity or
efficacy should be defined will depend on the context of the binding partner (e.g.,
receptor/ligand) complex and the consideration of an activity relevant to a particular
biological outcome. For example, in some circumstances, intrinsic activity may vary
depending on the particular second messenger system involved. See Hoyer, D. and
Boddeke, H., Trends Pharmacol Sd. 14(7):270-5 (1993). Where such contextually
specific evaluations are relevant, and how they might be relevant in the context of the
present invention, will be apparent to one of ordinary skill in the art.
As used herein, neurotransmitters whose release is mediated by the compounds
described herein include, but are not limited to, acetylcholine, dopamine,
norepinephrine, serotonin, and glutamate, and the compounds described herein
function as agonists or partial agonists at one or more of the Central Nervous System
(CNS) nAChRs.
I. Compounds
The compounds are N-aryl diazaspirocyclic compounds, bridged analogs of N-
heteroaryl diazaspirocyclic compounds, prodrugs or metabolites of these compounds,
and pharmaceutically acceptable salts thereof.
The compounds can bind to, and modulate nicotinic acetylcholine receptors in
the patient's brain in the cortex, hippocampus, thalamus, basal ganglia, and spinal cord. When so bound, the compounds express nicotinic pharmacology and, in
particular, can antagonize the release of dopamine at effective concentrations that do
not significantly antagonize the α4β2 receptor.
Receptor binding constants provide a measure of the ability of the compound
to bind to half of the relevant receptor sites of certain brain cells of the patient. See,
for example, Cheng et al., Biochem. Pharmacol. 22:3099 (1973). The receptor
binding constants of the compounds described herein, at one or more receptors other
than the α4β2 receptor that mediate dopamine release, generally exceed about 0.1 nM,
often exceed about 1 nM, and frequently exceed about 10 nM, and are often less than
about 100 μM, often less than about 10 μM and frequently less than about 5 μM.
Preferred compounds generally have receptor binding constants less than about 2.5
μM, sometimes are less than about 1 μM, and can be less than about 100 nM.
Preferably, the compounds can cross the blood-brain barrier, and thus enter the
central nervous system of the patient. Log P values provide a measure of the ability of
a compound to pass across a diffusion barrier, such as a biological membrane,
including the blood brain barrier. See, for example, Hansch et al., J. Med. Chem.llύ
(1968). Typical log P values for the compounds described herein are generally greater
than about -0.5, often are greater than about 0, and frequently are greater than about
0.5, and are typically less than about 3, often are less than about 2, and frequently are
less than about 1.
In one embodiment, the compounds have the structure represented by Formula
1 below:
Formula 1
In the formula, Q1 is (CZ2)U, Qπ is (CZ2)V, Qm is (CZ2)W, and Qw is (CZ2)X
where u, v, w and x are individually 0, 1, 2, 3 or 4, preferably 0, 1, 2 or 3. R is
hydrogen, lower alkyl, acyl, alkoxycarbonyl or aryloxycarbonyl, preferably hydrogen
or lower alkyl. When the value of u is 0, the value of v must be greater than 0, and, in
the case of Formula 1, when the value of w is 0, the value of x must be greater than 0.
In addition, the values of u, v, w and x are selected such that the diazaspirocyclic ring
contains 7, 8, 9, 10 or 11 members, preferably 8, 9 or 10 members.
Formula 2
In another embodiment, the compounds are represented by Formula 2, above.
In Formula 2 Q1 is (CZ2)U, Qπ is (CZ2)V, Qm is (CZ2)W, Qw is (CZ2)X, Qv is(CZ2)y and
QVI is (CZ2)Z where u, v, w, x, y and z are individually 0, 1, 2, 3 or 4, preferably 0, 1
or 2. The values of u, v, w, x, y and z are selected such that the bridged diazaspirocyclic ring contains 8, 9, 10, 11, 12 or 13 members, preferably 9, 10, 11 or
12 members. In the case of Formula 2, the values w and x can be simultaneously 0.
In addition, R is hydrogen, lower alkyl, acyl, alkoxycarbonyl or aryloxycarbonyl,
preferably hydrogen or lower alkyl.
Each individual Z represents either hydrogen or a suitable non-hydrogen substituent species (e.g., alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl,
heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, alkylaryl, substituted
alkylaryl, arylalkyl or substituted arylalkyl; but preferably lower alkyl or aryl).
In either formula, Cy represents a suitable five- or six-membered
heteroaromatic ring. In one embodiment, Cy is a six membered ring of the formula:
Each of X, X', X", X"' and X"" is individually nitrogen, nitrogen bonded to
oxygen (e.g., an N-oxide or N-O functionality) or carbon bonded to a substituent
species. No more than three of X, X', X", X"' and X"" are nitrogen or nitrogen bonded
to oxygen, and it is preferred that only one or two of X, X', X", X'" and X"" be nitrogen
or nitrogen bonded to oxygen. In addition, it is highly preferred that not more than
one of X, X', X", X'" and X"" be nitrogen bonded to oxygen; and it is preferred that if
one of those species is nitrogen bonded to oxygen, that species is X'". Most
preferably, X'" is nitrogen. In certain preferred circumstances, both X' and X'" are
nitrogen. Typically, X, X" and X"" are carbon bonded to a substituent species, and it
is typical that the substituent species at X, X" and X"" are hydrogen. For certain other preferred compounds where X"1 is carbon bonded to a substituent species such as
hydrogen, X and X" are both nitrogen. In certain other preferred compounds where X'
is carbon bonded to a substituent species such as hydrogen, X and X" are both
nitrogen.
In another embodiment, Cy is a five 5-membered heteroaromatic ring, such as pyrrole, furan, thiophene, isoxazole, isothiazole, oxazole, thiazole, pyrazole, 1,2,4-
oxadiazole, 1,3,4-oxadiazole and 1,2,4-triazole. Other examples of such rings are
described in U.S. Patent No. 6,022,868 to Olesen et al., the contents of which are
incorporated herein by reference in their entirety. One way of depicting Cy is as
follows:
where Y and Y" are individually nitrogen, nitrogen bonded to a substituent species,
oxygen, sulfur or carbon bonded to a substituent species, and Y' and Y1" are nitrogen
or carbon bonded to a substituent species. The dashed lines indicate that the bonds
(between Y and Y' and between Y1 and Y") can be either single or double bonds.
However, when the bond between Y and Y' is a single bond, the bond between Y' and
Y" must be a double bond and vice versa. In cases in which Y or Y" is oxygen or
sulfur, only one of Y and Y" is either oxygen or sulfur. At least one of Y, Y', Y" and
Y'" must be oxygen, sulfur, nitrogen or nitrogen bonded to a substituent species. It is
preferred that no more than three of Y, Y', Y" and Y'" be oxygen, sulfur, nitrogen or
nitrogen bonded to a substituent species. It is further preferred that at least one, but no
more than three, of Y, Y', Y" and Y'" be nitrogen.
Substituent species associated with any of X, X, X", X1", Xtm, Y, Y', Y" and
Y'" (when any is carbon bonded to a substituent species or nitrogen bonded to a
substituent species), typically have a sigma m value between about -0.3 and about
0.75, frequently between about -0.25 and about 0.6; and each sigma m value individually can be 0 or not equal to zero; as determined in accordance with Hansch et
al, Chem. Rev. 91:165 (1991).
Examples of suitable substituent species associated with any of X, X, X", X'", X"", Y, Y', Y" and Y'" (when any is carbon bonded to a substituent species or nitrogen
bonded to a substituent species), include hydrogen, alkyl, substituted alkyl, alkenyl,
substituted alkenyl, heterocyclyl, substituted heterocyclyl, cycloalkyl, substituted
cycloalkyl, aryl, substituted aryl, alkylaryl, substituted alkylaryl, arylalkyl, substituted
arylalkyl, halo (e.g., F, Cl, Br, or I), -OR1, -NR1R", -CF3, -CN, -NO2, -C2R1, -SR1, -N3, -
C(=O)NR'R", -NR'C(=O) R", -C(=O)R', -CO=O)OR1, -OC(^O)R1, -O(CR'R")rC(=O)R!,
-O(CR1R")rNR"C(=O)R', -0(CR1R1OrNR11SO2R, -OC(=O)NR'R", -NR'C(=O)O R", -
SO2R1, -SO2NR1R11, and -NR1SO2R", where R and R" are individually hydrogen, lower
alkyl (e.g., straight chain or branched alkyl including C1-C8, preferably C1-C5, such as
methyl, ethyl, or isopropyl), cycloalkyl, heterocyclyl, aryl, or arylalkyl (such as
benzyl), and r is an integer from 1 to 6. R1 and R" can combine to form a cyclic
functionality. The term "substituted" as applied to alkyl, aryl, cycloalkyl and the like
refers to the substituents described above, starting with halo and ending with -
NR1SO2R".
Examples of suitable Cy groups include 3-pyridyl (unsubstituted or substituted
in the 5 and/or 6 position(s) with any of the aforementioned substituents), 5-
pyrimidinyl (unsubstituted or substituted in the 2 position with any of the
aforementioned substituents), 4 and 5-isoxazolyl, 4 and 5-isothiazolyl, 5-oxazolyl, 5-
thiazolyl, 5-(l,2,4-oxadiazolyl), 2-(l,3,4-oxadiazolyl) or 3-(l,2,4-triazolyl).
Representative aryl groups include phenyl, naphthyl, furanyl, thienyl,
pyridinyl, pyrimidinyl, pyrazinyl, pyridazinyl, quinolinyl, and indolyl. Other
representative aromatic ring systems are set forth in Gibson et al., J. Med. Chem.
39:4065 (1996). Any of these aromatic group containing species can be substituted
with at least one substituent group, such as those described above that are associated with x' and the like. Representative substitevely include alkyl, aryl, halo, hydroxy,
alkoxy, aryloxy or amino substituents.
Adjacent substituents of X, X1, X", Xm, X"", Y, Y1, Y" and Y1" (when
substituents are present) can combine to form one or more saturated or unsaturated,
substituted or unsubstituted carbocyclic or heterocyclic rings containing, but not
limited to, ether, acetal, ketal, amine, ketone, lactone, lactam, carbamate, or urea
functionalities.
The compounds can occur in stereoisomeric forms, including both single
enantiomers and racemic mixtures of such compounds, as well as mixtures of varying
degrees of enantiomeric excess.
The compounds can be in a free base form or in a salt form (e.g., as
pharmaceutically acceptable salts). Examples of suitable pharmaceutically acceptable
salts include inorganic acid addition salts such as sulfate, phosphate, and nitrate;
organic acid addition salts such as acetate, galactarate, propionate, succinate, lactate,
glycolate, malate, tartrate, citrate, maleate, fumarate, methanesulfonate, p-
toluenesulfonate, and ascorbate; salts with an acidic amino acid such as aspartate and
glutamate; alkali metal salts such as sodium and potassium; alkaline earth metal salts
such as magnesium and calcium; ammonium salt; organic basic salts such as
trimethylamine, triethylamine, pyridine, picoline, dicyclohexylamine, and N5N'-
dibenzylethylenediamine; and salts with a basic amino acid such as lysine and
arginine. The salts can be in some cases hydrates or ethanol solvates. The
stoichiometry of the salt will vary with the nature of the components. Representative
salts are provided as described in U.S. Patent Nos. 5,597,919 to Dull et al., 5,616,716
to Dull et al. and 5,663,356 to Ruecroft et al., the disclosures of which are incorporated herein by reference in their entirety.
Representative compounds include the following:
7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(5-pyrimidinyl)- 1 ,7-diazaspiro [4.4]nonane
7-(5-isoxazolyl)-l,7-diazaspiro[4.4]nonane
7-(5-isothiazolyl)- 1 ,7-diazaspiro[4.4]nonane
7-(5-(l ,2,4-oxadiazol)yl)- 1 ,7-diazaspiro[4.4]nonane
7-(2-(l ,3,4-oxadiazol)yl)-l ,7-diazaspiro[4.4]nonane
7-(2-pyrazinyl)- 1 ,7-diazaspiro [4.4]nonane
7-(3-pyridazinyl)-l,7-diazaspiro[4.4]nonane
7-(5-methoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(5-cyclopentyloxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(5-(4-hydroxyρhenoxy)-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(5-ethynyl-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(6-chloro-3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(6-methoxy-3-pyridazinyl)-l,7-diazaspiro[4.4]nonane
l-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
1 -(5-pyrimidinyl)-l ,7-diazaspiro[4.4]nonane
1 -(5-isoxazolyl)-l ,7-diazaspiro[4.4]nonane
1 -(5-isothiazolyl)-l ,7-diazaspiro[4.4]nonane
l-(5-(l,2,4-oxadiazol)yl)-l,7-diazaspiro[4.4]nonane
l-(2-(l,3,4-oxadiazol)yl)-l,7-diazaspiro[4.4]nonane
1 -(2-pyrazinyl)- 1 ,7-diazaspiro[4.4]nonane
1 -(3 -pyridazinyl)- 1 ,7-diazaspiro [4.4]nonane
1 -methyl-7-(3-pyridyl)-l ,7-diazaspiro [4.4]nonane
1 -methyl-7-(5-pyrimidinyl)- 1 ,7-diazaspiro [4.4]nonane
1 -methyl-7-(5-isoxazolyl)- 1 ,7-diazaspiro [4.4]nonane
1 -methyl-7-(5-isothiazolyl)- 1 ,7-diazaspiro[4.4]nonane
1 -methyl-7-(5-(l ,2,4-oxadiazol)yl)- 1 ,7-diazaspiro [4.4]nonane
1 -methyl-7-(2-(l ,3,4-oxadiazol)yl)- 1 ,7-diazaspiro[4.4]nonane
1 -methyl-7-(2-pyrazinyl)-l ,7-diazaspiro[4.4]nonane
l-methyl-7-(3-pyridazinyl)-l,7-diazaspiro[4.4]nonane
l-methyl-7-(5-methoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
1 -methyl-7-(5-cyclopentyloxy-3-pyridyl)- 1 ,7-diazaspiro[4.4]nonane
1 -methyl-7-(5-phenoxy-3-ρyridyl)-l ,7-diazasρiro[4.4]nonane
1 -methyl-7-(5 -(4-hydroxyphenoxy)-3 -pyridyl)- 1 ,7-diazaspiro [4.4]nonane
l-methyl-7-(5-ethynyl-3-pyridyl)-l,7-diazaspiro[4.4]nonane
1 -methyl-7-(6-chloro-3 -pyridyl)- 1 ,7-diazaspiro[4.4]nonane
l-methyl-7-(6-methoxy-3-pyridazinyl)-l,7-diazaspiro[4.4]nonane
7-methyl-l-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-methyl- 1 -(5-pyrimidinyl)- 1 ,7-diazaspiro[4.4]nonane
7-methyl-l-(5-isoxazolyl)-l,7-diazaspiro[4.4]nonane
7-methyl-l-(5-isothiazolyl)-l,7-diazaspiro[4.4]nonane
7-methyl- 1 -(5-( 1 ,2,4-oxadiazol)yl)- 1 ,7-diazaspiro[4.4]nonane
7-methyl-l-(2-(l,3,4-oxadiazol)yl)-l,7-diazaspiro[4.4]nonane
7-methyl-l-(2-pyrazinyl)-l,7-diazaspiro[4.4]nonane
7-methyl- 1 -(3 -pyridazinyl)- 1 ,7-diazaspiro[4.4]nonane
2-(3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(5-pyrimidinyl)-2,7-diazaspiro[4.4]nonane
2-(5-isoxazolyl)-2,7-diazaspiro[4.4]nonane
2-(5-isothiazolyl)-2,7-diazaspiro[4.4]nonane
2-(5-(l,2,4-oxadiazol)yl)-2,7-diazaspiro[4.4]nonane 2-(2-(l ,3,4-oxadiazol)yl)-2,7-diazaspiro[4.4]nonane
2-(2-pyrazinyl)-2,7-diazaspiro[4.4]nonane
2-(3-pyridazinyl)-2,7-diazaspiro[4.4]nonane
2-(5-methoxy-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(5-cyclopentyloxy-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(5-phenoxy-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(5-(4-hydroxyphenoxy)-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(5-ethynyl~3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(6-chloro-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-(6-methoxy-3-pyridazinyl)-2,7-diazaspiro[4.4]nonane
2-methyl-7-(3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-methyl-7-(5-methoxy-3-pyridyl)-2,7-diazaspiro[4.4]nonane
2-methyl-7-(5-phenoxy-3-pyridyl)-2,7-diazaspiro[4.4]nonane
6-(3-pyridyl)- 1 ,6-diazaspiro[3.4] octane
1 -methyl-6-(3 -pyridyl)- 1 ,6-diazaspiro[3.4] octane
2-(3-pyridyl)-2,5-diazaspiro[3.4]octane
5-methyl-2-(3-pyridyl)-2,5-diazaspiro[3.4]octane
6-(3-pyridyl)-l,6-diazaspiro[3.5]nonane
1 -methyl-6-(3 -pyridyl)- 1 ,6-diazaspiro[3.5]nonane
2-(3-pyridyl)-2,5-diazaspiro[3.5]nonane
5-methyl-2-(3-pyridyl)-2,5-diazaspiro[3.5]nonane
2-(3-pyridyl)-2,6-diazaspiro[4.5]decane
6-methyl-2-(3-pyridyl)-2,6-diazaspiro[4.5]decane
7-(3-pyridyl)-l,7-diazaspiro[4.5]decane
1 -methyl-7-(3 -pyridyl)- 1 ,7-diazaspiro[4.5] decane 8-(3 -pyridyl)- 1 ,8-diazaspiro[5.5]undecane
l-methyl-8-(3-pyridyl)-l,8-diazaspiro[5.5]undecane
Other representative compounds of the present invention include the
following:
1 '-(3-pyridyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
1 '-(5-ethoxy-3-pyridyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
1 '-(5-cyclopentyloxy-3-pyridyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
r-(5-phenoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
1 '-(5-(4-hydroxyphenoxy)-3-pyridyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '- pyrrolidine]
r-(5-pyrimidinyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
1 '-(5-isoxazolyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
l'-(5-isothiazolyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
1 '-(5-(1 ,2,4-oxadiazol)yl)-spiro[ l-azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
r-(2-(l,3,4-oxadiazol)yl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
1 '-(2-pyrazinyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine]
1 '-(3-pyridazinyl)-spiro[ 1 -azabicyclo[2.2.1 ]heptane-2, 3 '-pyrrolidine]
r-(5-ethynyl-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
r-(6-chloro-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]
r-(6-methoxy-3-pyridazinyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidme]
1 '-(3 -pyridyl)-spiro [ 1 -azabicyclo [2.2.2] octane-2,3 '-pyrrolidine] r-(5-methoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
r-(5-cyclopentyloxy-3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-ρyrrolidine]
r-(5-phenoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3 '-pyrrolidine]
1 '-(5-(4-hydroxyphenoxy)-3-pyridyl)-spiro[ 1 -azabicyclo[2.2.2]octane-2,3 '-pyrrolidine] l'-(5-ethynyl-3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
l'-(6-chloro-3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
l'-(5-pyrimidinyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
1 '-(2-pyrazinyl)-spiro[ 1 -azabicyclo[2.2.2]octane-2,3 '-pyrrolidine]
1 '-(3-pyridazinyl)-spiro[ 1 -azabicyclo[2.2.2]octane-2,3 '-pyrrolidine]
1 '-(6-methoxy-3 -pyridazinyl)-spiro [ 1 -azabicyclo [2.2.2] octane-2,3 '-pyrrolidine]
l'-(5-isoxazolyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
l'-(5-isothiazolyl)-spiro[l-azabicyclo[2.2.2]octane-2,3 '-pyrrolidine]
r-(5-(l,2,4-oxadiazol)yl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
r-(2-(l,3,4-oxadiazol)yl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine]
r-(3-pyridyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
1 '-(5-methoxy-3-pyridyl)-2'H-spiro[ 1 -azabicyclo[2.2.1 ]heptane-7,3 '-pyrrolidine]
1 '-(5-cyclopentyloxy-3-pyridyl)-2'H-spiro[ 1 -azabicyclo[2.2.1 ]heptane-7,3 '-
pyrrolidine]
r-(5-phenoxy-3-pyridyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine] 1 '-(5-(4-hydroxyphenoxy)-3-pyridyl)-2'H-spiro[l -azabicyclo[2.2.1 ]heptane-7,3 '-
pyrrolidine] r-(6-chloro-3-pyridyl)-2Η-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
1 '-(5-pyrimidinyl)-2'H-spiro[ 1 -azabicyclo[2.2.1 ]heptane-7,3 '-pyrrolidine]
r-(2-pyrazinyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
r-(3-pyridazinyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
r-(6-methoxy-3-pyridazinyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine] 1 '-(5-isoxazolyl)-2'H-spiro[ 1 -azabicyclo[2.2.1 ]heptane-7,3 '-pyrrolidine]
r-(5-isothiazolyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3 '-pyrrolidine]
r-(5-(l,2,4-oxadiazol)yl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
r-(2-(l,3,4-oxadiazol)yl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine]
II. Methods of Preparing the Compounds
Scheme 1
Reacrion 1
Reaction 2
Ar
The compounds of Formulas 1 and 2 can be prepared using a general method involving arylation of one amino group of an optionally protected diazaspiroalkane
(Scheme 1). Arylation at N with an appropriate aryl, or preferably heteroaryl, halide
or triflate can be performed according to methods known to those skilled in the art, for
example, employing metal (e.g., copper or palladium compounds) catalysis. The
preferred general method in the present invention utilizes the teachings of Buchwald
or Hartwig (Buchwald et al, J. Org. Chem., 61: 7240 (1996); Hartwig et al., J.
Org..Chem., 64: 5575 (1999); see also Old et al., J. Am. Chem. Soc. 120: 9722
(1998)), wherein an amine is treated with a palladium(O) catalyst, a phosphine ligand
and base. Thus, 1 -benzyl- l,7-diazaspiro[4.4]nonane is reacted with 3-bromopyridine
in the presence of tris(dibenzylideneacetone)dipalladium(0), 2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl and sodium tert-butoxide in toluene, to give
l-benzyl-7-(3-pyridyl)diazaspiro[4.4]nonane. Removal of the benzyl group by
hydrogenation, over 10% palladium on carbon, provides 7-(3-pyridyl)-
diazaspiro[4.4]nonane. Alternatively, one skilled in the art will recognize that various
protecting group strategies can be employed to provide products bearing an aryl group
on nitrogen N', as opposed to N (Reaction 1 , Scheme 1). A particularly useful
combination of protecting groups in the present invention is benzyl and a carbamate,
specifically, tert-butylcarbamate. Thus, 1 -benzyl- l,7-diazaspiro[4.4]nonane is
converted into l-benzyl-7-(tert-butoxycarbonyl)-l,7-diazaspiro[4.4]nonane by
treatment with di-tert-butyl dicarbonate. Subsequent hydrogenation and palladium-
catalyzed arylation, with 3-bromopyridine, gives 7-(tert-butoxycarbonyl)-l-(3- pyridyl)diazaspiro[4.4]nonane. Removal of the tert-butoxycarbonyl group, with
hydrochloric acid, provides l-(3-pyridyl)-diazaspiro[4.4]nonane. Finally, in many
cases where N and N' are sterically dissimilar, and whenever N is tertiary (as in
Reaction 2, Schemel), selective arylation of N can be accomplished without first protecting N'. Thus, reaction of l,7-diazaspiro[4.4]nonane with 3-bromopyridine,
under the palladium-catalyzed conditions reported previously, gives almost
exclusively 7-(3-pyridyl)-diazaspiro[4.4]nonane.
It will be obvious to those skilled in the art that incorporation of substituents
on the heteroaryl ring introduced onto the diazaspiroalkane can be readily realized.
Such substituents can provide useful properties in and of themselves or serve as a
handle for further synthetic elaboration. A suitably protected heteroaryl
diazaspiroalkane can be elaborated to give a number of useful compounds possessing
substituents on the heteroaryl ring. For example, l-benzyl-7-(5-bromo-3-pyridyl)-l,7-
diazaspiro[4.4]nonane can be made by reacting 3,5-dibromopyridine with 1-benzyl-
l,7-diazaspiro[4.4]nonane according to procedures described previously. The
conversion of l-benzyl-7-(5-bromo-3-pyridyl)diazaspiro[4.4]nonane into the
corresponding 5-amino-substituted compound can be accomplished by the general
method of Zwart et al., Recueil Trav. Chim. Pays-Bas 74: 1062 (1955), in which the
bromo compound heated with aqueous ammonia in the presence of a copper catalyst.
5-Alkylamino substituted compounds can be prepared in a similar manner. 5-Ethynyl-
substituted compounds can be prepared from the 5-bromo compound by palladium
catalyzed coupling using 2-methyl-3-butyn-2-ol, followed by base- catalyzed (sodium hydride) removal of the acetone unit, according to the general techniques described in
Cosford et al., J Med. Chem. 39: 3235 (1996). The 5-ethynyl analogs can be
converted into the corresponding 5-ethenyl, and subsequently to the corresponding 5-
ethyl analogs by successive catalytic hydrogenation reactions. The 5-azido-substituted
analogs can be prepared from the 5-bromo compound by reaction with lithium azide in N,N-dimethylformamide. 5-Alkylthio-substituted analogs can be prepared from the
5-bromo compound by reaction with an appropriate sodium alkylmercaptide (sodium
alkanethiolate), using techniques known to those skilled in the art of organic synthesis.
A number of other analogs, bearing substituents in the 5 position of the pyridine ring, can be synthesized from the corresponding amino compounds, vide
supra, via a 5-diazonium salt intermediate. Examples of other 5-substituted analogs
that can be produced from 5-diazonium salt intermediates include, but are not limited
to: 5-hydroxy, 5-alkoxy, 5-fluoro, 5-chloro, 5-iodo, 5-cyano, and 5-mercapto. These
compounds can be synthesized using the general techniques set forth in Zwart et al.,
supra. For example, l-benzyl-7-(5-hydroxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane can
be prepared from the reaction of the corresponding 5-diazonium salt intermediate with
water. Likewise, l-benzyl-7-(5-alkoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonanes can be
made from the reaction of the diazonium salt with alcohols. Appropriate 5-diazonium
salts can be used to synthesize cyano or halo compounds, as will be known to those
skilled in the art. 5-Mercapto substitutions can be obtained using techniques
described in Hoffman et al., J. Med. Chem. 36: 953 (1993). The 5-mercaptan so
generated can, in turn, be converted to a 5-alkylthio substitutuent by reaction with
sodium hydride and an appropriate alkyl bromide. Subsequent oxidation would then
provide a sulfone. 5-Acylamido analogs of the aforementioned compounds can be
prepared by reaction of the corresponding 5-amino compounds with an appropriate
acid anhydride or acid chloride using techniques known to those skilled in the art of
organic synthesis.
5-Hydroxy-substituted analogs of the aforementioned compounds can be used
to prepare corresponding 5-alkanoyloxy-substituted compounds by reaction with the appropriate acid, acid chloride, or acid anhydride. Likewise, the 5-hydroxy
compounds are precursors of both the 5-aryloxy and 5-heteroaryloxy via nucleophilic aromatic substitution at electron deficient aromatic rings (e.g., 4-fluorobenzonitrile
and 2,4-dichloropyrimidine). Such chemistry is well known to those skilled in the art
of organic synthesis. Ether derivatives can also be prepared from the 5-hydroxy
compounds by alkylation with alkyl halides and a suitable base or via Mitsunobu
chemistry, in which a trialkyl- or triarylphosphine and diethyl azodicarboxylate are
typically used. See Hughes, Org. React. (N. Y.) 42: 335 (1992) and Hughes, Org.
Prep. Proced. Int. 28: 127 (1996) for typical Mitsunobu conditions.
5-Cyano-substituted analogs of the aforementioned compounds can be
hydrolyzed to afford the corresponding 5-carboxarnido-substituted compounds.
Further hydrolysis results in formation of the corresponding 5-carboxylic acid-
substituted analogs. Reduction of the 5-cyano-substituted analogs with lithium
aluminum hydride yields the corresponding 5-aminomethyl analogs. 5-Acyl-
substituted analogs can be prepared from corresponding 5-carboxylic acid-substituted
analogs by reaction with an appropriate alkyllithium using techniques known to those
skilled in the art of organic synthesis.
5-Carboxylic acid-substituted analogs of the aforementioned compounds can
be converted to the corresponding esters by reaction with an appropriate alcohol and
acid catalyst. Compounds with an ester group at the 5-pyridyl position can be reduced
with sodium borohydride or lithium aluminum hydride to produce the corresponding
5-hydroxymethyl-substituted analogs. These analogs in turn can be converted to compounds bearing an ether moiety at the 5-pyridyl position by reaction with sodium
hydride and an appropriate alkyl halide, using conventional techniques. Alternatively,
the 5-hydroxymethyl-substituted analogs can be reacted with tosyl chloride to provide the corresponding 5-tosyloxymethyl analogs. The 5-carboxylic acid-substituted
analogs can also be converted to the corresponding 5-alkylaminoacyl analogs by sequential treatment with thionyl chloride and an appropriate alkylamine. Certain of
these amides are known to readily undergo nucleophilic acyl substitution to produce ketones. Thus, the so-called Weinreb amides (N-methoxy-N-methylamides) react
with aryllithium reagents to produce the corresponding diaryl ketones. For example,
see Selnick et al, Tet. Lett. 34: 2043 (1993).
5-Tosyloxymethyl-substituted analogs of the aforementioned compounds can
be converted to the corresponding 5 -methyl-substituted compounds by reduction with
lithium aluminum hydride. 5-Tosyloxymethyl-substituted analogs of the
aforementioned compounds can also be used to produce 5-alkyl-substituted
compounds via reaction with an alkyllithium reagent. 5-Hydroxy-substituted analogs
of the aforementioned compounds can be used to prepare 5-N-alkyl- or 5-N-
arylcarbamoyloxy-substituted compounds by reaction with N-alkyl- or N-
arylisocyanates. 5-Amino-substituted analogs of the aforementioned compounds can
be used to prepare 5-alkoxycarboxamido-substituted compounds and 5-urea
derivatives by reaction with alkyl chloroformate esters and N-alkyl- or N-
arylisocyanates, respectively, using techniques known to those skilled in the art of
organic synthesis.
Chemistries analogous to those described hereinbefore for the preparation of
5-substituted pyridine analogs of diazaspiro compounds can be devised for the synthesis of analogs bearing substituents in the 2, 4, and 6 positions of the pyridine
ring. For example, a number of 2-, 4-, and 6-aminopyridyldiazaspiroalkanes can be
converted to the corresponding diazonium salt intermediates, which can be
transformed to a variety of compounds with substituents at the 2, 4, and 6 positions of
the pyridine ring as was described for the 5-substituted analogs above. The requisite 2-, A-, and 6-aminopyridyl diazaspiroalkanes are available via the Chichibabin
reaction of unsubstituted pyridyl diazaspiroalkanes (e.g., l-benzyl-7-(3-pyridyi)-l,7-
diazaspiro[4.4]nonane, described previously) with sodium amide. Similar reactions
are described in Chemistry of Heterocyclic Compounds, Volume 14, part 3, pp.3-5
(Interscience Publishers, 1962) and by Lahti et al., J. Med. Chem. 42: 2227 (1999).
After the desired heteroaryl ring functional group manipulation has been
accomplished, the optional protecting group can be removed from the diazabicycle
using appropriate conditions. Thus, for example, hydrogenolysis of l-benzyl-7-(5-
alkoxy-3- pyridyl)-l,7-diazaspiro[4.4]nonane will generate 7-(5-alkoxy-3-pyridyl)-
l,7-diazaspiro[4.4]nonane. Those skilled in the art of organic chemistry will
appreciate the necessity of pairing protecting groups with the chemistries required to
generate particular functionalities. In some cases it can be necessary, to retain a
particular functionality, to replace one protecting group with another.
In an alternative approach to the synthesis of pyridine-substituted pyridyl
diazaspiroalkanes, 3,5-dibromopyridine can be converted into the corresponding 5-
alkoxy-3-bromo- and 5-aryloxy-3-bromopyridines by the action of sodium alkoxides
or sodium aryloxides. Procedures such as those described by Comins et al., J. Org.
Chem. 55: 69 (1990) and Hertog et al., Recueil Trav. CUm. Pays-Bas 74: 1171 (1955)
are used. This is exemplified by the preparation 7-(5-(4-methoxyphenoxy)-3-pyridyi)-
l,7-diazaspiro[4.4]nonane. Reaction of 3,5-dibromopyridine with sodium 4-
methoxyphenoxide in N,N-dimethylformamide gives 3-bromo-5-(4-
methoxyphenoxy)pyridine. Coupling of 3-bromo-5-(4-methoxyphenoxy)pyridine with l-benzyl-7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane in the presence of sodium tert-
butoxide, and a catalytic amount of tris(dibenzylideneacetone)dipalladium(0) and 2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl, in toluene, followed by hydrogenolysis of the
benzyl protecting group, will provide 7-(5-(4-methoxyphenoxy)-3-pyridyi)-l,7-
diazaspiro[4.4]nonane.
Other aryl halides undergo the palladium-catalyzed coupling reaction
described previously. Thus 7-(5-pyrimidinyl)-l,7-diazaspiro[4.4]nonane is prepared
in a similar manner from 5-bromopyrimidine and optionally 1 -position protected 1,7-
diazaspiro[4.4]nonane followed by deprotection, if necessary. This technology is
especially applicable in cases, such as 3-bromopyridine, 3,5-dibromopyridine, and 5-
bromopyrimidine, where the aromatic ring is not activated toward nucleophilic
aromatic substitution.
In some cases, coupling of the heteroaromatic ring to the diazaspirocycle can
be accomplished without the use of palladium catalysis. Examples of both five- and
six-membered heteroaromatic ring compounds, which are activated toward
nucleophilic aromatic substitution, are known by those skilled in the art of organic
synthesis. For example, 7-(6-chloro-3-pyridazinyl)-l,7-diazaspiro[4.4]nonane can be synthesized from 3,6-dichloropyridazine and 1 ,7-diazaspiro[4.4]nonane. Likewise,
2,6-dicloropyrazine, and 2-bromothiazole will react with l,7-diazaspiro[4.4]nonane to
give 7-(6-chloro-2-pyrazinyl)-l,7-diazaspiro[4.4]nonane and 7-(2-thiazoyl)-l,7- diazaspiro[4.4]nonane, respectively.
The coupling reactions described in this application, whether palladium catalyzed or not, are amenable to high through-put synthetic techniques. Thus a
library of compounds of the present invention can be produced by coupling, in a 96-
well plate format, for instance, various haloarenes with various diazaspiro
compounds.
Specific Diazaspiro Ring Systems
Optionally protected diazaspiroalkane intermediates used to prepare the
compounds of Formulas I and II can be prepared by numerous methods. Several of
these diazaspiroalkane intermediates are known and can be prepared using prior art
methods. However, the synthesis of the intermediates using palladium chemistry is
new to the art, and the pharmaceutical activity of the intermediates was not
appreciated in the prior art.
The compounds of Formula 1, where U=V=I, w=0 and x=3, possess a 2,5-
diazaspiro [3 ,4] octane core which can be prepared as depicted in Scheme 2.
Alkylation of N-benzyl-L-proline ethyl ester (Aldrich Chemical), using a
strong base such as lithium diisopropylamide (LDA) and the aminomethyl equivalent
cyanomethylbenzylamine, provides a beta-lactam, according to the procedure reported
by Overman, J. Am. Chem. Soc. 107:1698 (1985) and Tet. Lett. 25: 1635 (1985). This
can subsequently be reduced with lithium aluminum hydride to provide the 2,5-
dibenzyl derivative of 2,5-diazaspiro[3,4]octane. Removal of the benzyl protecting
groups, by either hydrogenation or oxidative cleavage with, for example, eerie
ammonium nitrate, will produce 2,5-diazaspiro[3,4]octane. Alternatively, chemistry
similar to that described in EP patent application 90117078.7 (publication number EP
0 417 631) can be used to produce a geminal bis(hydroxymethyl) derivative and
subsequently convert it to the desired 2,5-diazaspiro[3,4]octane (Scheme 2). The
subsequent palladium-catalyzed arylation, as described previously, would be expected
to proceed with selectivity for the less sterically hindered azetidinyl nitrogen,
producing 2-aryl-2,5-diazaspiro[3,4]octanes. The isomeric 5-aryl-2,5-
diazspiro[3,4]octanes can be made by first protecting the azetidinyl nitrogen (with, for
instance, a carbamate) and then performing the arylation, followed by deprotection.
Scheme 2
The compounds of Formula 1, wherein u=2, v=l, w=0 and x=3, possess the
1 ,7-diazaspiro[4.4]nonane system which can be prepared according to numerous
methods, several of which are shown above in Scheme 3. In one embodiment
(Method A), a suitably protected proline ester, for example N-benzyl-L-proline ethyl
ester, can be deprotonated with lithium diisopropylamide and allowed to react by
Michael addition to nitroethylene. This provides methyl 2-(2-nitroethyl)-l-
benzylpyrrolidine-2-carboxylate. Subsequent reduction of the nitro group using
Raney nickel, followed by lactamization by methods known to those skilled in the art
(for example, heating in a suitable solvent with or without an acidic or basic catalyst),
provides 1 -benzyl- l,7-diazaspiro[4.4]nonan-6-one.
The l,7-diazaspiro[4.4]nonane-6-one can alternatively be prepared according
to one of several other methods reported in the literature. Such teachings indicate that
a suitably protected proline ester can be deprotonated with lithium diisopropylamide
and allowed to react with an alkylating agent such as chloroacetonitrile, then subjected
to nitrile reduction and cyclization (Method B, Scheme 3) as reported by Culbertson et
al, J. Med Chem. 33:2270 (1990).
Other teachings indicate that a suitably protected proline ester can be
deprotonated with lithium diisopropylamide and allowed to react with an alkylating agent such as allyl bromide (Method C, Scheme 3). The resulting olefin can then be
oxidatively cleaved to an aldehyde, as reported by Genin et al., J. Org. Chem. 58:2334
(1993); Hinds et al., J. Med. Chem. 34:1777 (1991); Kim et al., J. Org. Chem.
61:3138 (1996); EP 0 360 390 and U.S. Patent No. 5,733,912. The aldehyde can then
be subjected to reductive animation with an ammonium salt or primary aliphatic or
aromatic amine, according to methods known to those skilled in the art. Alternatively,
the aldehyde can be reduced to the corresponding alcohol and the alcohol then
transformed to an amine by conversion to a leaving group, followed by displacement
with the appropriate amine. This can also be achieved by displacing the leaving group
with an azide ion and subsequently reduction to the primary amine using methods
known to those skilled in the art. The alcohol can be converted to an amine using
Mitsunobu conditions, as discussed previously. The alkyl 2-aminoethyl pyrrolidine-2-
carboxylate, obtained according to one of the methods described above, can be
cyclized to a spirolactam by methods known to those skilled in the art, such as heating
in a suitable solvent with or without an acidic or basic catalyst.
The lactam obtained by any one of the above methods (Methods A, B or C)
can be treated with a suitable reducing agent, such as lithium aluminum hydride, to
provide the protected l,7-diazaspiro[4.4]nonane, in this example, 1 -benzyl- 1,7-
diazaspiro[4.4]nonane. The protecting group can be removed using methods known
those skilled in the art to provide the desired l,7-diazaspiro[4.4]nonane. Arylation at
either nitrogen can be accomplished using methods described herein.
Scheme 3
a) (i) LDA; (ii) nitroethylene (Method A) or CICH2CN (Method B); (iii) RaNi; (iv) PhCH3, heat b) (i) LDA; (ii) allyl bromide; (iii) O3 or OsO4, NaIO4; (iv) NH4CI, NaBH(OAc)3 (Method C) c) (i) LiAIH4 or BH3; (ii) [-PG]
Alternatively, the l,7-diazaspiro[4.4]nonane core can also be prepared
according to Scheme 4. The conversion of l,4-dioxaspiro[4.5]decan-8-one to 4-
benzoyloxycyclohexanone can be readily achieved by those skilled in the art.
Subsequent transformation of 4-benzoyloxycyclohexanone to 1,7-
diazaspiro[4.4]nonane (through the intermediacy of 4-oxocaprolactam, as shown) can
be performed according to the teachings of Majer et al., Coll. Czech. Chem. Comm.
47:950 (1982).
Scheme 4
The compounds of Formula 1, wherein u=2, V=I, w=l and x=2, possess the symmetrical 2,7-diazaspiro[4,4]nonane system which can be prepared according to
Scheme 5. This method is reported by Overman et al., J. Org. Chem. 46: 2757 (1981)
and Culbertson et al., J. Med. Chem. 33:2270 (1990).
Scheme 5
The compounds of Formula 1, wherein u=3, v=l, w=0 and x=3, possess the
l,7-diazaspiro[4.5]decane system which can be prepared according to Scheme 6. The
teachings of Kim et al., J. Org. Chem. 61:3138 (1996), patent EP360390 and US
patent 5,733,912 indicate that a suitably protected proline ester (e.g., N-benzyl-L-
proline ethyl ester) can be deprotonated with lithium diisopropylamide and allowed to
react with an alkylating agent such as allyl bromide. US patent 5,733,912 also teaches
that hydroboration/oxidation of the allyl side chain can be performed to provide the 2-
(3-hydroxypropyl) group. Those skilled in the art will appreciate that the hydroxyl
group can then be converted to an amino group by a number of methods, for example
oxidation followed by reductive animation. Alternatively, a suitably protected proline
ester can be deprotonated with lithium diisopropylamide and allowed to react with an
alkylating agent such as diiodopropane. Conversion of the primary iodide to an amine
can then be performed according to known methods, for example treatment with
ammonia in the presence of a copper catalyst. The resulting amino ester can be
cyclized to afford a protected 1 ,7-diazaspiro[4.5]decan-6-one using any number of
known procedures, for example heating in a suitable solvent in the presence or
absence of an acidic or basic catalyst, as discussed previously. Alternatively, the known l,7-diaza-spiro[4.5]decan-6-one can be prepared according to the teachings of
Loefas et al, J. Het. Chem. 21:583 (1984), in which the ring contraction of 2,10-
diazabicyclo[4.4.0]dec-l-ene is used.
The l,7-diazaspiro[4.5]decan-6-one, obtained by any of the above methods,
can then be treated with a reducing agent, such as lithium aluminum hydride, followed
by removal of the protecting group, to provide the desired l,7-diazaspiro[4.5]decane.
Arylation can then be carried out at either nitrogen using methods described herein.
Scheme 6
a) X=OH: (i) LDA, allyl bromide; (ii) BH
3, H
2O
2 X=I: LDA, 1 ,3-diiodopropane b) X=OH: (i) PCC or Swern; (ii) NH
4CI, NaBH(OAc)
3; (iii) heat (÷catalyst?) X=I: (i) NH
3, CuI; (ii) heat (÷catalyst?) c) (i) BH
3 or LiAIH
4; (ii) [-PG]
The compounds of Formula 1, wherein u=2, v=l, w=0, and x=4, possess the
2,6-diazaspiro[4.5]decane core which can be prepared according to the method of
Ciblat, et al., Tet. Lett. 42: 4815 (2001). Thus, commercially available l-benzyl-3-
pyrrolidinone can be reacted with 2-methyl-2-(2-aminoethyl)-l,3-dioxolane (Islam
and Raphael, J. Chem. Soc. 3151 (1955)) in an intramolecular Mannich reaction. The product, the ethylene ketal of 2-benzyl-2,10-diazaspiro[4,5]decan-7-one, can then be
hydrolyzed to the ketone, using aqueous hydrochloric acid. Deoxygenation of the ketone can then be accomplished by standard methods, such as conversion to the
corresponding 1,3-dithiane, followed by treatment with Raney nickel. The 2-benzyl-
2,6-diazaspiro[4,5]decane thus produced can be directly arylated on the 6-position
nitrogen or converted into 6-(tert-butoxycarbonyl)-2,6-diazaspiro[4,5]decane by
treatment with di-tert-butyl dicarbonate, followed by hydrogenation. The latter
derivative can then be arylated at the 2-position nitrogen. Similar chemistry can be
used to convert other azacyclic ketones into the corresponding spirodiaza compounds.
Thus, reaction of any of various N-protected 3-azetidinones (the synthesis of which is
described by LaIl, et al, J. Org. Chem. 67: 1536 (2002) and Marchand, et al,
Heterocycles 49: 149 (1998)) with 2-methyl-2-(2-aminoethyl)-l,3-dioxolane, followed
by deoxygenation (as described above), will produce the corresponding protected 2,5-
diazaspiro[3.5]nonane (Formula 1, wherein u=l, v=l, w=0, and x=4).
The compounds of Formula 1, wherein u=v=2, w=0, and x=3, possess the 1,8-
diazaspiro[4.5]decane core which can be prepared according to Scheme 7. According
to the teachings reported by Wittekind et al., J Het. Chem. 9:11 (1972), a protected A-
piperidone can be converted to the 4-nitropiperidine. Subsequent Michael addition
with ethyl acrylate, for example, followed by reduction of the nitro group with Raney
nickel, provides the l,8-diazaspiro[4.5]decan-2-one. This lactam can be reduced with
an appropriate reducing agent, such as lithium aluminum hydride, followed by removal of the protecting group, to provide the optionally substituted 1,8-
diazaspiro[4.5jdecane. Arylation on either nitrogen can be accomplished using
methods described herein.
Scheme 7
1 ■ ) L -|A™IH4
The compounds of Formula 1, wherein u=2, v=l, and w=x=2, possess the 2,8-
diazaspiro[4.5]decane core which can be prepared according to Scheme 8. According
to various teachings (HeIv. Chun. Acta 60: 1650 (1977); Smith et al., J. Med. Chem.
19:3772 (1995); Elliott et al., Biorg. Med. Chem. Lett. 8:1851 (1998)), a protected 4-
piperidone can be converted to the 4-piperidinylidene acetic acid ester via Wittig
olefination. Subsequent Michael addition with the anion of nitromethane, followed by
reduction of the nitro group and spontaneous cyclization with Raney nickel, provides
the protected 2,8-diazaspiro[4.5]decan-3-one. Treatment of the protected 2,8-
diazaspiro[4.5]decan-3-one with a reducing agent, such as lithium aluminum hydride,
followed by removal of the protecting group, provides the 2,8-diazaspiro[4.5]decane.
Arylation can be accomplished on either nitrogen using the methods described herein.
Scheme 8
The compounds of Formula 1, wherein u=2, v=l, w=4 and x=0, possess the l,8-diazaspiro[5.5]decane core and can be prepared according to the procedures
utilized for the analogous l,7-diazaspiro[4.4]nonanes by substituting pipecolinate
ester for proline ester. Alternatively, the procedure reported in Zhu et al., J. Org.
Chem. 58:6451 (1993) can be employed.
The compounds of Formula 1 wherein u=3, v=l, w=l and x=3, possess the
symmetrical 2,8-diazaspiro[5.5]undecane core and can be prepared according to the
procedures reported in HeIv. Chim. Acta 36:1815 (1953), J. Org. Chem. 28:336 (1963)
or, preferably, Culbertson et al., J. Med. Chem. 33:2270 (1990).
The compounds of Formula 1, wherein u=v=2 and w=x=2, possess the
symmetrical 3,9-diazaspiro[5.5]undecane core and can be prepared according to
procedures reported in Rice et al., J. Het. Chem. 1:125 (1964), US patent 3,282,947,
or J. Med. Chem. 8:62 (1965).
Single enantiomer compounds of the present invention can be made by various
methods. One method, well known to those skilled in the art of organic synthesis,
involves resolution using diastereomeric salts. Compounds of the present invention
contain basic nitrogen atoms and will react with acids to form crystalline salts.
Various acids, carboxylic and sulfonic, are commercially available in enantiomerically
pure form. Examples include tartaric, dibenzoyl- and di-p-toluoyltartaric, and
camphorsulfonic acids. When any one of these or other single enantiomer acids is
reacted with a racemic amine base, diastereomeric salts result. Fractional
crystallization of the salts, and subsequent regeneration of the bases, results in
enantiomeric resolution thereof.
Another means of separation of involves conversion of the enantiomeric mixture into diastereomeric amides or carbamates, using a chiral acid or
chloroformate. Thus, when racemic 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane is
coupled with N-(tert-butoxycarbonyl)-S-proline, using diphenyl chlorophosphate, and the protecting group removed (with trifluoroacetic acid), the resulting diastereomeric
proline amides of 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane are separable by liquid
chromatography. The separated amides are then transformed into (+) and (-) 7-(3- pyridyl)-l,7-diazaspiro[4.4]nonane by the Edman degradation.
Selective synthesis of single enantiomers can also be accomplished by
methods known to those skilled in the art. Such methods will vary as the chemistry
used for construction of the diazaspiro rings varies. For instance, for the syntheses in
which the alkylation of a proline derivative is used to form the diazaspiro system
(such as described for the l,7-diazaspiro[4.4]nonane system), the alkylation of proline
can be carried out in a stereospecific manner. Thus, methods such as those described
by Beck et al., Org. Synth. 72: 62 (1993) or Wang and Germanas, Synlett : 33 (1999)
(and references therein) can be used to control the stereochemistry of the alkylation
step. When enantiomerically pure proline ester (commercially available from Aldrich)
is used as the starting material for such a process, the alkylation product is also a
single enantiomer. A variety of electrophiles can be used in such alkylations,
including allyl halides, which have been useful in assembling spiro systems related to
compounds of the present invention Genin and Johnson, J. Amer. Chem. Soc. 114:
8778 (1992).
Bridged Spiro Ring Systems The compounds of Formula 2, wherein u=l, v=2, w=0, x=0, y=2 and z=2,
possess the spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine] core and can be prepared according to Scheme 9. The anion of ethyl nitroacetate, formed in the
presence of an amine base, can be condensed with tetrahydropyran-4-one using the
procedure reported in Fornicola et al., J. Org. Chem. 63:3528 (1998). Simultaneous reduction of the nitro group and the olefin under catalytic hydrogenation conditions
provides the 2-(4-oxanyl)glycine ester. This compound can be treated with
hydrobromic acid to afford a dibromide, which is cyclized under basic conditions to
the azabicyclo[2.2.1]heptane-7-carboxylic acid. Treatment of the acid with ethanol
and sulfuric acid provides the ethyl azabicyclo[2.2.1]heptane-7-carboxylate. This
compound is then deprotonated with lithium diisopropylamide and reacted by
Michael addition with nitroethylene to give the ethyl aza-7-(2-
nitroethyl)bicyclo[2.2.1]heptane-7-carboxylate. Reduction of the nitro group with
Raney nickel, followed by spontaneous cyclization, affords the spirolactam.
Treatment of the lactam with lithium aluminum hydride affords the spiro[l-
azabicyclo[2.2.1]heptane-7,3'-pyrrolidine], which is subsequently arylated on the
pyrrolidine nitrogen to produce compounds of the present invention.
Scheme 9
The compounds of Formula 2, wherein u=l, v=2, w=l, x=0, y=l and z=2, possess the spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine] ring system and can be prepared according to Scheme 10. Conversion of tetrahydrofuran-3-ylmethanol
(Aldrich) to 3-(bromomethyl)tetrahydrofuran can be achieved through mesylation and subsequent treatment with lithium bromide. The reaction of ethyl glycinate with
benzophenone imine provides ethyl 3-aza-4,4-diphenyl-but-3-enoate which serves to
both protect the amine and activate the methylene carbon toward alkylation.
Alkylation of this imine can be performed, according to the method of Hansen, J. Org.
Chem. 63:775 (1998), by deprotonating with potassium tert-butoxide and reacting
with the 3-(bromomethyl)tetrahydrofuran. Deprotection under acidic conditions
gives the desired 2-amino-3-(tetrahydrofuran-3-yl) propionic ester. Ring opening of
the tetrahydrofuran can be achieved by treatment with hydrobromic acid to afford the
dibromoamino acid intermediate, which, upon heating under basic conditions, cyclizes
to the l-azabicyclo[2.2.1]heptane-2-carboxylic acid. This acid iscan subsequently
converted to the ethyl ester, using ethanol and sulfuric acid. Alkylation iscan then
performed by deprotonation with lithium diisopropylamide and reaction with
nitroethylene. Subsequent reduction of the nitro group using Raney nickel, followed
by lactamization by methods known to those skilled in the art, gives the spiro[l-
azabicyclo[2.2.1]heptane-2,3'-pyrrolidine]-2'-one. Treatment of the lactam with
lithium aluminum hydride, gives the desired spiro[l-azabicyclo[2.2.1]heptane-2,3'- pyrrolidine], which is subsequently arylated on the pyrrolidine nitrogen to give
compounds of the present invention.
Scheme 10
ethyl glycinate
The compounds of Formula 2, wherein u=l ,v=2, w=l, X=O, y=2 and z=2,
possess the spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine] core and can be prepared
in a manner similar to that for the corresponding spiro[l-azabicyclo[2.2.1]heptane-
2,3 '-pyrrolidine], as seen in Scheme 11. Ethyl quinuclidine-2-carboxylate can be
generated from (4-bromomethyl)tetrahydropyran by the procedures discussed
previously for ethyl l-azabicyclo[2.2.1]heptane-2-carboxylate. The requisite 4-
(bromomethyl)tetrahydropyran can be prepared according to procedures found in
Burger, et al, J. Am. Chem. Soc. 72:5512 (1950), Thomas, et al., J. Pharm.
Pharmacol. 15:167 (1963) and J. Am. Chem. Soc. 115:8401 (1993). Ethyl
quinuclidine-2-carboxylate iscan then deprotonated with lithium diisopropylamide and
reacted with nitroethylene. Subsequent treatment with Raney nickel gives directly the
spirolactam, spiro[azabicyclo[2.2.2]octane-2,3'-pyrrolidine]-2'-one, by reduction of
the nitro group followed by spontaneous cyclization. This lactam iscan then reduced
with lithium aluminum hydride to provide the desired spiro[l-
azabicyclo[2.2.2]octane-2,3'-pyrrolidine], which is then arylated on the pyrrolidine nitrogen.
11A"
ethyl glycinate
Alternate Synthetic Methods
The compounds can be produced using varying methods. Alternatives to the
palladium catalyzed coupling protocol described above can be used. For instance,
those skilled in the art of organic synthesis will recognize that one or more of the
nitrogen containing rings can be formed by any one of many common amine
syntheses. Thus, an arylamine can be reacted with a protected cyclic amine derivative
(see scheme 12), which contains two reactive electrophiles, to generate an N-
aryldiazaspiro compound. A variety of electrophiles participate in such chemistry (e.g., halides and sulfonates via nucleophilic displacement, aldehydes via reductive
animation, esters and other acid derivatives via acyl substitution, followed by
reduction).
Scheme 12
Dn PG
Q-N reagents QU-Q IV
Q-1 ]- Qιv + ArNH2 (vary with E)
E = CH2Z (where Z = X or sulfonate), CHO, CO2R
The requisite bis-electophiles can be synthesized by many diverse methods.
Schemes 2, 3 and 6 all incorporate such intermediates (in reaction with benzylamine
or ammonia). Pedersen, et al., J. Org. Chem. 58: 6966 (1993) and Berkowitz, et al., J.
Org. Chem. 60: 1233 (1995) both report the alkylation of dianions of N-acyl α-
aminoesters. These alkylations also can be used for synthesis of N-aryldiazaspiro
compounds. Thus, dianion of commercially available (Acros) ethyl 2-pyrrolidone-5-
carboxylate can be alkylated with ethyl bromoacetate to generate ethyl 5-
(carboethoxymethyl)-2-pyrrolidone-5-carboxylate. The second spiro ring can be
formed by reacting ethyl 5-(carboethoxymethyl)-2-pyrrolidone-5-carboxylate with an
arylamine. The resulting 2-aryl-2,6-diazspiro[4.4]nonane-l ,3,7-trione can be reduced
with diborane to give 7-aryl-l,7-diazaspiro[4.4]nonane. Depending on the nature of
the aryl group, the order of the synthetic steps can be changed. Likewise, it can be
necessary to incorporate protection/deprotection steps into particular methods.
A wide variety or arylamines are available for use in the approach outlined in
Scheme 12. Pn addition to aminopyridines and aminopyrimidines, 3-aminoisoxazole
is commercially available (Aldrich). This provides a means of synthesizing N-
isoxazolyldiazaspiro compounds. The isomeric 4-aminoisoxazole can be made by
reducing the corresponding nitro compound using the method described by Reiter, J.
Org. Chem. 52: 2714 (1987). Examples of other amino derivatives of 5-membered
aromatic rings include 3-aminoisothiazole, made according to Holland, et al., J. Chem. Soc, 7277 (1965), and 4-aminoisothiazole, made according to Avalos, et al.,
An. Quim. 72: 922 (1976). Thus, a variety of N-aryldiazaspiro compounds of the
present invention, in which the aryl group is a five-membered heterocycle, can be
produced.
III. Pharmaceutical Compositions The compounds described herein can be incorporated into pharmaceutical
compositions and used to bring about smoking cessation, treat drug addiction, or treat
or prevent obesity associated with drug cessattion. The pharmaceutical compositions
described herein include one or more compounds of Formulas 1 or 2 and/or
pharmaceutically acceptable salts thereof. Optically active compounds can be
employed as racemic mixtures or as pure enantiomers.
The manner in which the compounds are administered can vary. The
compositions are preferably administered orally (e.g., in liquid form within a solvent
such as an aqueous or non-aqueous liquid, or within a solid carrier). Preferred
compositions for oral administration include pills, tablets, capsules, caplets, syrups,
and solutions, including hard gelatin capsules and time-release capsules.
Compositions may be formulated in unit dose form, or in multiple or subunit doses.
Preferred compositions are in liquid or semisolid form. Compositions including a
liquid pharmaceutically inert carrier such as water or other pharmaceutically compatible liquids or semisolids maybe used. The use of such liquids and semisolids
is well known to those of skill in the art.
The compositions can also be administered via injection, i.e., intraveneously,
intramuscularly, subcutaneously, intraperitoneally, intraarterially, intrathecally; and intracerebroventricularly. Intravenous administration is a preferred method of
injection. Suitable carriers for injection are well known to those of skill in the art, and
include 5% dextrose solutions, saline, and phosphate buffered saline. The compounds
can also be administered as an infusion or injection (e.g., as a suspension or as an
emulsion in a pharmaceutically acceptable liquid or mixture of liquids).
The formulations may also be administered using other means, for example,
transdermally (e.g., using a transdermal patch, using technology that is commercially
available from Novartis and Alza Corporation). Formulations useful for transdermal
administration are well known to those of skill in the art. The compounds can also be
administered by inhalation (e.g., in the form of an aerosol either nasally or using
delivery articles of the type set forth in U.S. Patent No. 4,922,901 to Brooks et al., the
disclosure of which is incorporated herein in its entirety); topically (e.g., in lotion
form); or rectally. Although it is possible to administer the compounds in the form of
a bulk active chemical, it is preferred to present each compound in the form of a
pharmaceutical composition or formulation for efficient and effective administration.
Exemplary methods for administering such compounds will be apparent to the
skilled artisan. The usefulness of these formulations may depend on the particular
composition used and the particular subject receiving the treatment. These
formulations may contain a liquid carrier that may be oily, aqueous, emulsified or
contain certain solvents suitable to the mode of administration.
The compositions can be administered intermittently or at a gradual,
continuous, constant or controlled rate to a warm-blooded animal (e.g., a mammal such as a mouse, rat, cat, rabbit, dog, pig, cow, or monkey), but advantageously are
administered to a human being. In addition, the time of day and the number of times per day that the pharmaceutical formulation is administered can vary.
Preferably, upon administration, the active ingredients interact with receptor sites within the body of the subject, that control dopamine release. The compounds
may be antagonists at both the α4β2 subtype and those NNR subtypes affecting
dopamine release, as long as the effective concentration needed to effectively control
dopamine release is at least an order of magniture less than that necessary to
significantly affect the α4β2 receptor. In one embodiment, the compounds are partial
antagonists, and the partial antagonism permits the compounds to result in a preferred
side effect profile relative to full antagonists.
The ability of these compounds to partially inhibit the release of dopamine is
especially significant, as it indicates that the compounds can be useful in interrupting
the dopamine reward system, and thus in treating disorders that are mediated by it.
Such disorders include substance abuse, tobacco use and weight gain that
accompanies drug cessation.
Thus, the compounds described herein are a useful alternative in treating
dependencies on drugs of abuse including alcohol, amphetamines, barbiturates,
benzodiazepines, caffeine, cannabinoids, cocaine, hallucinogens, opiates,
phencyclidine and tobacco and the treatment of eating disorders such as obesity that
occurs following drug cessation while reducing side effects associated with the use of
psychomotor stimulants (agitation, sleeplessness, addiction, etc.).
The compounds also advantageously affect the functioning of the CNS, in a
manner which is designed to optimize the effect upon those relevant receptor subtypes
that have an effect upon dopamine release, while minimizing the effects upon muscle-
type receptor subtypes.
Preferably, the compositions are administered such that active ingredients
interact with regions where dopamine production is affected or occurs. The
compounds described herein are very potent at affecting doamine production and/or secretion at very low concentrations, and are very efficacious (i.e., they inhibit
dopamine production and/or secretion to an effective degree).
In certain circumstances, the compounds described herein can be employed as
part of a pharmaceutical composition with other compounds intended to prevent or
treat drug addiction, nicotine addiction, and/or obesity. In addition to effective
amounts of the compounds described herein, the pharmaceutical compositions can
also include various other components as additives or adjuncts. Exemplary
pharmaceutically acceptable components or adjuncts which are employed in relevant
circumstances include antidepressants, antioxidants, free-radical scavenging agents,
peptides, growth factors, antibiotics, bacteriostatic agents, immunosuppressives,
anticoagulants, buffering agents, anti-inflammatory agents, anti-pyretics, time-release
binders, anaesthetics, steroids, vitamins, minerals and corticosteroids. Such
components can provide additional therapeutic benefit, act to affect the therapeutic
action of the pharmaceutical composition, or act towards preventing any potential side
effects which can be imposed as a result of administration of the pharmaceutical
composition.
The appropriate dose of the compound is that amount effective to prevent
occurrence of the symptoms of the disorder or to treat some symptoms of the disorder
from which the patient suffers. By "effective amount", "therapeutic amount" or
"effective dose" is meant that amount sufficient to elicit the desired pharmacological
or therapeutic effects, thus resulting in effective prevention or treatment of the
disorder.
An effective amount of compound is an amount sufficient to pass across the blood-brain barrier of the subject, to bind to relevant receptor sites in the brain of the
subject and to activate relevant nicotinic receptor subtypes (e.g., to antagonize or
partially antagonize dopamine production and/or secretion, thus resulting in effective prevention or treatment of the disorder). Prevention of the disorders is manifested by
delaying the onset of the symptoms of the disorder. Treatment of the disorder is
manifested by decreasing the symptoms associated with the disorder or an
amelioration of the recurrence of the symptoms of the disorder. Preferably, the
effective amount is sufficient to obtain the desired result, but insufficient to cause
appreciable side effects.
The effective dose can vary, depending upon factors such as the condition of
the patient, the severity of the symptoms of the disorder, and the manner in which the
pharmaceutical composition is administered. For human patients, the effective dose
of typical compounds generally requires administering the compound in an amount
sufficient to decrease dopamine release, but the amount should be insufficient to
induce effects on skeletal muscles and ganglia to any significant degree. The effective
dose of compounds will of course differ from patient to patient, but in general
includes amounts starting where desired therapeutic effects occur (i.e., where
dopamine production and/or secretion is sufficiently lowered) but below the amount
where muscular effects are observed.
The compounds, when employed in effective amounts in accordance with the
method described herein, are selective to certain relevant nicotinic receptors, but do
not significantly activate receptors associated with undesirable side effects at
concentrations at least greater than those required for suppressing the release of
dopamine or other neurotransmitters. By this is meant that a particular dose of compound effective in preventing and/or treating drug addiction, nicotine addiction
and/or obesity (primarily but not necessarily the obesity associated drug or nicotine
cessation) is essentially ineffective in eliciting activation of certain ganglionic-type
nicotinic receptors at concentration higher than 5 times, preferably higher than 100 times, and more preferably higher than 1 ,000 times than those required for
suppression of dopamine production and/or release. This selectivity of certain
compounds described herein against those ganglionic-type receptors responsible for
cardiovascular side effects is demonstrated by a lack of the ability of those compounds
to activate nicotinic function of adrenal chromaffin tissue at concentrations greater
than those required for suppression of dopamine production and/or release.
For human patients, the effective dose of typical compounds generally requires
administering the compound in an amount of at least about 1, often at least about 10,
and frequently at least about 25 μg/ 24 hr/ patient. The effective dose generally does
not exceed about 500, often does not exceed about 400, and frequently does not
exceed about 300 μg/ 24 hr/ patient. In addition, administration of the effective dose
is such that the concentration of the compound within the plasma of the patient
normally does not exceed 500 ng/mL and frequently does not exceed 100 ng/mL.
The compounds described herein, when employed in effective amounts in
accordance with the methods described herein, can provide some degree of prevention
of the progression of CNS disorders, ameliorate symptoms of CNS disorders, and
ameliorate to some degree of the recurrence of CNS disorders. The effective amounts
of those compounds are typically below the threshold concentration required to elicit
any appreciable side effects, for example those effects relating to skeletal muscle. The compounds can be administered in a therapeutic window in which certain CNS
disorders are treated and certain side effects are avoided. Ideally, the effective dose of
the compounds described herein is sufficient to provide the desired effects upon the
CNS but is insufficient (i.e., is not at a high enough level) to provide undesirable side effects. Preferably, the compounds are administered at a dosage effective for treating
the CNS disorders but less than 1/5, and often less than 1/10, the amount required to
elicit certain side effects to any significant degree.
Most preferably, effective doses are at very low concentrations, where
maximal effects are observed to occur, with a minimum of side effects.
Concentrations, determined as the amount of compound per volume of relevant tissue,
typically provide a measure of the degree to which that compound affects cytokine
production. Typically, the effective dose of such compounds generally requires
administering the compound in an amount of less than 5 mg/kg of patient weight.
Often, the compounds of the present invention are administered in an amount from
less than about 1 mg/kg patent weight and usually less than about 100 μg/kg of patient
weight, but frequently between about 10 μg to less than 100 μg/kg of patient weight.
For compounds that do not induce effects on muscle-type nicotinic receptors at low
concentrations, the effective dose is less than 5 mg/kg of patient weight; and often
such compounds are administered in an amount from 50 μg to less than 5 mg/kg of
patient weight. The foregoing effective doses typically represent that amount
administered as a single dose, or as one or more doses administered over a 24-hour
period.
For human patients, the effective dose of typical compounds generally requires administering the compound in an amount of at least about 1, often at least about 10,
and frequently at least about 25 μg/ 24 hr/ patient. For human patients, the effective
dose of typical compounds requires administering the compound which generally does
not exceed about 500, often does not exceed about 400, and frequently does not
exceed about 300 μg/ 24 hr/ patient. In addition, the compositions are advantageously administered at an effective dose such that the concentration of the compound within
the plasma of the patient normally does not exceed 500 pg/mL, often does not exceed
300 pg/mL, and frequently does not exceed 100 pg/mL. When employed in such a
manner, the compounds are dose dependent, and, as such, inhibit cytokine production
and/or secretion when employed at low concentrations but do not exhibit those
inhibiting effects at higher concentrations. The compounds exhibit inhibitory effects
on dopamine production and/or secretion when employed in amounts less than those amounts necessary to elicit activation to any significant degree of nicotinic receptor
subtypes associated with side effects.
IV. Methods of Using the Compounds and/or Pharmaceutical Compositions
The compounds can be used to treat drug addiction, nicotine addiction and/or
obesity, such as the obesity associated with drug cessation. The compounds can also
be used as adjunct therapy in combination with existing therapies in the management
of the aforementioned types of diseases and disorders. In such situations, it is
preferable to administer the active ingredients to in a manner that optimizes effects
upon dopamine production and/or secretion, while minimizing effects upon receptor
subtypes such as those that are associated with muscle and ganglia. This can be
accomplished by targeted drug delivery and/or by adjusting the dosage such that a
desired effect is obtained without meeting the threshold dosage required to achieve
significant side effects.
The compounds have the ability to bind to, and in most circumstances, antagonize or partially antagonize one or more nicotinic receptors of the brain of the
patient that modulate dopamine release, other than the α4β2 receptor, at concentrations
at which the α4β2 receptor is largely unaffected. As such, such compounds have the
ability to express nicotinic pharmacology, and in particular, to act as dopamine
antagonists. The receptor binding constants of typical compounds useful in carrying out the present invention generally exceed about 0.1 nM, often exceed about 1 nM,
and frequently exceed about 10 nM. The receptor binding constants of such typical
compounds generally are less than about 1 μM, often are less than about 100 nM, and
frequently are less than about 50 nM. Receptor binding constants provide a measure of
the ability of the compound to bind to half of the relevant receptor sites of certain
brain cells of the patient. See, Cheng, et al., Biochem. Pharmacol. 22: 3099 (1973).
The compounds, when employed in effective amounts as described
herein, are selective to certain relevant nicotinic receptors, but do not significantly
activate receptors associated with undesirable side effects. By this is meant that a
particular dose of compound that is effective at suppressing dopamine production
and/or release is essentially ineffective in eliciting activation of certain ganglionic-
type nicotinic receptors. This selectivity of the compounds of the present invention
against those receptors responsible for cardiovascular side effects is demonstrated by a
lack of the ability of those compounds to activate nicotinic function of adrenal
chromaffin tissue.
The compounds demonstrate poor ability to cause isotopic rubidium ion flux
through nicotinic receptors in cell preparations expressing muscle-type nicotinic
acetylcholine receptors. Thus, the compounds exhibit receptor activation constants or
EC50 values (i.e., which provide a measure of the concentration of compound needed
to activate half of the relevant receptor sites of the skeletal muscle of a patient) which
are extremely high (i.e., greater than about 100 μM). Generally, typical preferred
compounds useful in carrying the present invention activate isotopic rubidium ion flux
by less than 10 percent, often by less than 5 percent, of that maximally provided by S(-
) nicotine.
Accordingly, the compounds are effective at suppressing of dopamine
production and/or release, and can be used to treat drug addiction, nicotine addiction, and/or obesity at effective at concentrations that are not sufficient to elicit any
appreciable side effects, as is demonstrated by decreased effects on preparations
believed to reflect effects on the cardiovascular system, or effects to skeletal muscle.
As such, administration of the compounds provides a therapeutic window in which
treatment of drug addiction, nicotine addiction and/or obesity is effected, and side effects are avoided. That is, an effective dose of a compound of the present invention
is sufficient to provide the desired antagonistic effects on dopamine production and/or
secretion, but is insufficient (i.e., is not at a high enough level) to provide undesirable
side effects. Preferably, the compounds results in treatment of drug addiction, nicotine
addiction and/or obesity upon administration of less 1/3, frequently less than 1/5, and
often less than 1/10, that amount sufficient to cause any side effects to a significant
degree.
The following examples are provided to illustrate the present invention, and
should not be construed as limiting thereof. In these examples, all parts and
percentages are by weight, unless otherwise noted. Reaction yields are reported in mole percentages. Several commercially available starting materials are used
throughout the following examples. 3-Bromopyridine, 3,5-dibromopyridine, 5- bromonicotinic acid, 5-bromopyrimidine, and 4-penten-2-ol were obtained from
Aldrich Chemical Company or Lancaster Synthesis Inc. 2-Amino-5-bromo-3- methylpyridine was purchased from Maybridge Chemical Company Ltd. (R)-(+)-
propylene oxide was obtained from Fluka Chemical Company, and (S)-(-)-propylene
oxide was obtained from Aldrich Chemical Company. Column chromatography was
done using either Merck silica gel 60 (70-230 mesh) or aluminum oxide (activated, neutral, Brockmann I, standard grade, about 150 mesh). Pressure reactions were done
in a heavy wall glass pressure tube (185 mL capacity), with Ace-Thread, and plunger
valve available from Ace Glass Inc. Reaction mixtures were typically heated using a
high-temperature silicon oil bath, and temperatures refer to those of the oil bath. The
following abbreviations are used in the following examples: CHCl3 for chloroform,
CH2Cl2 for dichloromethane, CH3OH for methanol, DMF for N5N-
dimethylformamide, and EtOAc for ethyl acetate, THF for tetrahydrofuran, and Et3N for triethylamine.
V. Assays
Binding Assay
The ability of the compounds to bind to relevant receptor sites was determined
in accordance with the techniques described in U.S. Patent No. 5,597,919 to Dull et al.
Inhibition constants (Kj values) were calculated from the IC50 values using the method
of Cheng et al., Biochem. Pharmacol. 22:3099 (1973). For the α4β2 subtype, the Ki
value for each of the examples in this application was less than 1 μM, indicating that
compounds of the present invention bind tightly to the receptor.
Determination of Log P Value
Log P values, which have been used to assess the relative abilities of
compounds to pass across the blood-brain barrier (Hansch, et al., J. Med. Chem. 11: 1
(1968)), were calculated using the Cerius software package Version 3.5 by Molecular
Simulations, hie. Determination of Dopamine Release
Dopamine release was measured using the techniques described in U.S. Pat. No. 5,597,919 to Dull et al. Release is expressed as a percentage of release obtained with a concentration of (S)-(-)-nicotine resulting in maximal effects. Reported EC50 values are expressed in nM, and Emax values represent the amount released relative to (S)-(-)-nicotine on a percentage basis.
Antagonism of dopamine release can also be evaluated using the assays
described in Gradyet al., "Characterization of nicotinic receptor mediated
[3H]dopamine release from synaptosomes prepared from mouse striatum," J.
Neurochem. 59: 848-856 (1992) and Soliakov and Wonnacott, "Voltage-sensitive
Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat
striatal synaptosomes," J. Neurochem. 67:163-170 (1996).
Determination of Rubidium Ion Release
Rubidium release was measured using the techniques described in Bencherif et
al., JPET 279: 1413-1421 (1996). Reported EC50 values are expressed in nM, and
Emax values represent the amount of rubidium ion released relative to 300μM
tetramethylammonium ion, on a percentage basis.
Determination of Interaction with Muscle Receptors
The determination of the interaction of the compounds with muscle receptors
was carried out in accordance with the techniques described in U.S. Pat. No.
5,597,919 to Dull et al. The maximal activation for individual compounds (Emax) was
determined as a percentage of the maximal activation induced by (S)-(-)-nicotine.
Reported Emax values represent the amount released relative to (S)-(-)-nicotine on a percentage basis.
Determination of Interaction with Ganglion Receptors
The determination of the interaction of the compounds with ganglionic
receptors was carried out in accordance with the techniques described in U.S. Pat. No. 5,597,919 to Dull et al. The maximal activation for individual compounds (Emax) was
determined as a percentage of the maximal activation induced by (S)-(-)-nicotine.
Reported Emax values represent the amount released relative to (S)-(-)-nicotine on a percentage basis.
Selectivity
The selectivity of the compounds for a given receptor can be evaluated by
comparing the binding of the compounds to different receptors using known
methodology.
VI. Synthetic Examples
The following synthetic examples are provided to illustrate the present
invention and should not be construed as limiting the scope thereof. In these
examples, all parts and percentages are by weight, unless otherwise noted. Reaction
yields are reported in mole percentage.
Example 1
Sample No. 1 is 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane dihydrochloride,
which was prepared according to the following techniques:
Nitroethylene
Nitroethylene was prepared accordingly to the procedure reported by
Ranganathan, et al., J. Org. Chem. 45: 1185 (1980).
Ethyl 2-(2-nitroethyl)-l-benzylpyrrolidine-2-carboxylate
Under a nitrogen atmosphere, a solution of diisopropylamine (4.34 g, 6.01 mL,
42.9 mmol) in dry THF (50 mL) was cooled in an ice bath as n-butyllithium (17.1 mL
of 2.5 M in hexane, 42.8 mmol) was added by syringe. The ice bath was removed and the solution of lithium diisopropylamide was first warmed to ambient temperature and
then transferred by cannula into a stirred solution of ethyl (S)-N-benzyl pyrrolidine-2-
carboxylate (10.0 g, 42.9 mmol) (Fluka) in dry THF (50 mL), held at -78°C under
nitrogen. The addition took 10 min. After stirring an additional 30 min at -78 "C, the enolate solution was treated (via cannula) with a solution of nitroethylene (3.13 g,
42.9 mmol) in dry THF (20 mL). The mixture was then stirred for 1 h at -78°C.
Saturated aqueous ammonium chloride solution was then added (at -78°C), and the
mixture was warmed to ambient temperature and extracted the ethyl acetate (4 x 30
mL). The extracts were dried (K2CO3) and concentrated by rotary evaporation. The
residue was purified by chromatography on a Merck silica gel 60 (70-230 mesh)
column with 9:1 (v/v) hexane/ethyl acetate. Concentration of selected fractions gave
10.0 g (76.3%) of viscous, tan oil.
6-Benzyl-2,6-diazaspiro[4.4]nonan-l-one
Raney nickel (~2 g) was added to a solution of ethyl 2-(2-nitroethyl)-l-
benzylpyrrolidine-2-carboxylate (6.00 g, 19.6 mmol) in absolute ethanol (200 mL) in
a hydrogenation bottle. The mixture was shaken for 12 h under a hydrogen
atmosphere (50 psi) in a Parr hydrogenation apparatus, filtered through a Celite pad
and concentrated by rotary evaporation. GCMS analysis indicated that the
hydrogenation product was a mixture of the primary amine and the lactam resulting
from cyclization of the amine onto the ester. The mixture was dissolved in toluene
(150 mL). A catalytic amount of p-toluenesulfonic acid (~30 mg) was added and the
mixture was heated at reflux under a nitrogen atmosphere for 24 h. Upon evaporation
of the toluene, the residue (now entirely lactam, by GCMS) crystallized to give 4.20 g (93.1%) of tan solid (mp 152-153°C).
l-Benzyl-l,7-diazaspiro[4.4]nonane
Lithium aluminum hydride (1.98 g, 52.2 mmol) was added in portions, under
argon, to a ice bath cooled solution of 6-benzyl-2,6-diazaspiro[4.4]nonan-l-one (4.00
g, 17.4 mmol) in dry THF (100 mL). The addition funnel was replaced with a reflux condenser, and the mixture was heated at reflux for 24 h. The mixture was cooled to
0°C and treated drop-wise (caution: exothermic reaction) with 10 M aqueous sodium
hydroxide until hydrogen evolution ceased and the aluminate salts were granular. The
mixture was stirred 1 h at 0°C and filtered through Celite. The filtrate was dried
(K2CO3) and concentrated, leaving 3.60 g (95.7%) of viscous, colorless liquid.
l-Benzyl-7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane A mixture of l-benzyl-l,7-diazaspiro[4.4]nonane (2.00 g, 9.26 mmol), 3-
bromopyridine (1.38 g, 8.73 mmol), potassium tert-butoxide (2.50 g, 22.3 mmol),
tris(dibenzylideneacetone)dipalladium(0) (0.318 g, 0.347 mmol), 2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl (0.324 g, 0.520 mmol) and dry toluene (50
mL) was placed in a pressure tube under argon. The mixture was stirred and heated at
90°C (bath temperature) for 24 h and cooled. Water (20 mL) was added and the
mixture was extracted with ethyl acetate (6 x 25 mL). The extracts were dried
(K2CO3) and concentrated. Column chromatography of the residue on Merck silica
gel 60 (70-230 mesh), with 6:4 (v/v) chloroform/acetone, gave 1.80 g (66.2%) of light
brown oil, after concentration of selected fractions.
7-(3-Pyridyl)-l,7-diazaspiro[4.4]nonane dihydrochloride
Aqueous hydrochloric acid (0.5 mL of 12 M) and 10% palladium on carbon
(0.100 g) were added to a solution of l-benzyl-7-(3-pyridyi)-l,7- diazaspiro[4.4]nonane (1.0 g, 3.41 mmol) in methanol (30 mL). The mixture was
shaken under a hydrogen atmosphere (50 psi) in a Parr hydrogenation apparatus for 24
h and filtered through Celite. The filtrate was concentrated by rotary evaporation and
column chromatographed on Merck silica gel 60 (70-230 mesh). Elution with
0.01:1:9 (v/v) aqueous ammonia/methanol/chloroform, and concentration of selected fractions, gave 0.650 g (93.8%) of viscous, brown oil. A portion (300 mg, 1.48
mmol) of this material was treated with aqueous hydrochloric acid (2 mL). The water
was azeotropically removed by repeated treatment with small volumes of ethanol (~ 5
mL) and rotary evaporation. The resulting solid was recrystallized from hot
isopropanol to give 360 mg (88.2%) of fine tan crystals.
Example 2
Sample 2 is l-(3-pyridyl)-l,7-diaza-spiro[4.4]nonane dihydrochloride, which
was prepared according to the following techniques:
tert-Butyl 6-benzyl-2,6-diazaspiro [4.4]nonane-2-carboxylate
Di-t-butyl dicarbonate (1.45 g, 6.64 mmol) was added to a solution of 1-
benzyl-l,7-diazaspiro[4.4]nonane (1.30 g, 6.01 mmol) and triethylamine (1 mL) in
dichloromethane (25 mL), and the mixture was stirred at ambient temperature
overnight. The mixture was poured into saturated aqueous sodium bicarbonate (10
niL) and extracted with chloroform (4 x 25 niL). The extracts were dried (K2CO3)
and concentrated by rotary evaporation. The residue was column chromatographed on
Merck silica gel 60 (70-230 mesh), eluting with, to give 1.85 g (97.4%) of viscous,
colorless oil, after concentration of selected fractions.
tert-Butyl 2,6-diazaspiro[4.4]nonane-2-carboxyIate
A solution of t-butyl 6-benzyl-2,6-diazaspiro[4.4]nonane-2-carboxylate (1.70
g, 5.37 mmol) in methanol (30 mL) was mixed with 10% palladium on carbon (50
mg). The mixture was shaken under a hydrogen atmosphere (50 psi) in a Parr
hydrogenation apparatus for 8 h and filtered through Celite. The filtrate was
concentrated by rotary evaporation and high vacuum treatment, leaving 1.26 g of
viscous, light brown oil (>100%), which was of sufficient purity to be used in the subsequent reaction.
tert-Butyl 6-(3-pyridyl)-2,6-diazaspiro[4.4]nonane-2-carboxylate
A mixture of tert-butyl 2,6-diazaspiro[4.4]nonane-2-carboxylate (1.00 g, ~4.4 mmol), 3-bromopyridine (0.736 g, 4.66 mmol), potassium tert-butoxide (1.22 g, 10.9
mmol), tris(dibenzylideneacetone)dipalladium(0) (0.155 g, 0.169 mmol), 2,2'-
bis(diphenylphosphino)-l,r-bmaphthyl (0.158 g, 0.254 mmol) and dry toluene (25 mL) was placed in a pressure tube under argon. The mixture was stirred and heated at
180° C (bath temperature) for 8 h and cooled. Thin layer analysis indicated that very
little conversion had taken place. A second charge, equal in quantity to the first, of all
reagents except the tert-butyl 2,6-diazaspiro[4.4]nonane-2-carboxylate was added to
pressure tube and the tube was returned to the bath for another 8 h. Again relatively
little reaction seemed to have occurred, so a third charge of reagents was added and
heating (at 180°C) was continued for a third 8 h period. Water (20 mL) was added
and the mixture was extracted with ethyl acetate (6 x 25 mL). The extracts were dried
(K2CO3) and concentrated. Column chromatography of the residue on Merck silica
gel 60 (70-230 mesh), with 6:4 (v/v) chloroform/acetone, gave 150 mg (-11%) of
light brown oil, after concentration of selected fractions.
l-(3-Pyridyl)-l,7-diazaspiro[4.4]nonane dihydrochloride
A solution of tert-butyl 6-(3-pyridyl)-2,6-diazaspiro[4.4]nonane-2-carboxylate
(100 mg, 0.330 mmol) in dichloromethane (5 mL) was rapidly stirred with 1 mL of 12 M hydrochloric acid at ambient temperature for Ih, during which time the biphasic
mixture became monophasic. The dichloromethane was evaporated, and the residue was dissolved in water (3 mL) and made strongly basic (pH 9) with potassium
carbonate. The mixture was saturated with sodium chloride and extracted with
chloroform (4 x 10 mL). The extracts were dried (K2CO3) and concentrated , first by rotary evaporation and then by high vacuum treatment. The viscous brown oil which
resulted was 98% pure by GCMS and weighed 50 mg (73%). A sample of this free base (40 mg, 020 mmol)was dissolved in 10 drops of 12 M hydrochloric acid. The
water was azeotropically removed by repeated treatment with small volumes of
ethanol (~ 5 mL) and rotary evaporation. The resulting solid was recrystallized from
hot isopropanol to give 40 mg (72%) of fine tan crystals (mp 170-175°C).
Example 3
Sample 3 is l-methyl-7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane, which was
prepared according to the following techniques:
l-Methyl-7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
7-(3-Pyridyl)-l,7-diazaspiro[4.4]nonane (30 mg, 0.15 mmol) was dissolved in
98% formic acid (0.5 mL) and formaldehyde (1 mL, 28% aqueous solution). The
reaction mixture was heated to reflux for 8 h. The reaction mixture was cooled to
room temperature, basified with saturated aqueous sodium bicarbonate to pH 9-10 and
extracted with chloroform (4 x 3mL). The combined chloroform extracts were dried
(K2CO3), filtered and concentrated on a rotary evaporator to afford 30 mg of the
desired compound (93.6%) as a light brown liquid.
Example 4
Sample 4 is l-methyl-7-(5-ethoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane, which
was prepared according to the following techniques:
5-bromo-3-ethoxypyridine
Under a nitrogen atmosphere, sodium (4.60 g, 200 mmol) was added to absolute ethanol (100 mL) at 0-50C, and the stirring mixture was allowed to warm to
ambient temperature over 18 h. To the resulting solution was added 3,5-
dibromopyridine (31.5 g, 133 mmol), followed by DMF (100 mL). The mixture was
heated at 70°C for 48 h. The brown mixture was cooled, poured into water (600 mL), and extracted with ether (3 x 500 mL). The combined ether extracts were dried
(Na2SO4), filtered, and concentrated by rotary evaporation. Purification by vacuum
distillation afforded 22.85 g (85.0%) of an oil, bp 89-90°C at 2.8 mm Hg (lit. bp
11 TC at 5 mm Hg, see K. Clarke, et al., J. Chem. Soc. 1885 (I960)).
l-Benzyl-7-(5-ethoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane l-Benzyl-l,7-diazaspiro[4.4]nonane (500.0 mg, 2.4 mmol) was dissolved in
dry toluene (15 mL) in a 50 mL round bottom flask equipped with a magnetic stirring
bar. Nitrogen was bubbled through the solution in a slow stream. To the stirring
solution was added 3-bromo-5-ethoxypyridine (513.8 mg, 2.55 mmol), potassium tert-
butoxide (1039.0 mg, 9.26 mmol), rac-2,2'-bis(diphenylphosphino)-l,l'-binaphthyl (
86.4 mg, 0.14 mmol) and tris(dibenzylideneacetone)dϊpalladium(0) (63.6 mg, 0.06
mmol), while continuing to purge with nitrogen. Nitrogen flow was discontinued and
the flask was sealed and heated at 9O0C for 8 h. The reaction was cooled and the
solvent was removed by rotary evaporation. The resulting residue was suspended in
saturated aqueous sodium bicarbonate (10 mL) and extracted with chloroform (4 x 25
mL). The combined organic extracts were dried (Na2SO4), filtered, and concentrated
by rotary evaporation to a thick dark mass. Purification by column chromatography,
using methanol/chloroform (2:98, v/v) as the eluent, gave 0.54 g of the desired
compound as a light brown viscous liquid (69%).
7-(5-Ethoxy-3-pyridyl)- 1 ,7-diazaspiro [4.4] nonane
To a solution of l-benzyl-7-(5-ethoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
(540 mg, 1.6 mmol) in ethanol (25 mL) in a pressure bottle was added concentrated
HCl (1 mL) and Pearlman's catalyst (Pd(OH)2, 20% on carbon, 50 mg). The solution was shaken under 50 psi of hydrogen gas for 8 h. The catalyst was removed by
filtration through Celite, and the filter cake was washed with ethanol (20 mL). The
solvent was removed by rotary evaporation, and the residue was basified with saturated aqueous sodium bicarbonate to pH 8-9. Solid sodium chloride (2 g) was
added, and the mixture was extracted with chloroform (4 x 20 mL). The combined
chloroform extracts were dried (Na2SO4), filtered and concentrated by rotary
evaporation to afford 360.7 mg of the desired compound as a light brown viscous
liquid (91.1%).
l-Methyl-7-(5-ethoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane To a stirring solution of 7-(5-ethoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
(360.4mg, 1.4 mmol) in 37% aqueous solution of formaldehyde (4 mL) was added
98% formic acid (2 mL) under nitrogen. The reaction mixture was heated to reflux
for 8 h. The reaction mixture was cooled to room temperature, then basified with
saturated aqueous sodium bicarbonate to pH 8-9 and extracted with chloroform (4 x
15mL). The combined chloroform extracts were dried (Na2SO4), filtered and
concentrated by rotary evaporation to afford a viscous brown liquid. This was
distilled using a Kugelrohr apparatus (2mm, 18O0C) to give a very light cream-colored
syrup (340 mg, 89.3%).
Example 5
Sample 5 is l-methyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane,
which was prepared according to the following techniques:
3-Bromo-5-phenoxypyridine
Sodium hydride (1.35 g of 80% in mineral oil, 45.0 mmol) was added to a
stirred solution of phenol (4.26 g, 45.3 mmol) in DMF (30 mL) at 0°C, under
nitrogen. The mixture was stirred at room temperature for 3 h, treated with 3,5-
dibromopyridine (4.0 g, 16.9 mmol) and heated at 100°C for 48 h. The reaction
mixture was cooled to room temperature, poured into a mixture of water (100 mL)
and 5M sodium hydroxide (10 mL), and extracted with ether (3 x 60 mL). The
combined ether extracts were dried (Na2SO4), filtered, and rotary evaporated to a pale
yellow semi-solid (4.9 g). This was chromatographed on a silica gel (200 g) column
with hexane/ethyl acetate/chloroform (8:1:1, v/v) as eluant to give 2.86 g (68% yield) of a colorless oil.
l-Benzyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane l-Benzyl-l,7-diazaspiro[4.4]nonane (500.0 mg, 2.4 mmol) was dissolved in
dry toluene (15 mL) in a 50 mL round bottom flask equipped with a magnetic stirring
bar. Nitrogen was bubbled through the solution in a slow stream. To the stirring
solution was added 3-bromo-5-phenoxypyridine (636.8 mg, 2.55 mmol), potassium
tert-butoxide (1039.0 mg, 9.26 mmol), rac-2,2'-bis(diphenylphosphino)-l,l'-
binaphthyl (86.4 mg, 0.14 mmol) and tris(dibenzylideneacetone)dipalladium(0) (63.6
mg, 0.06 mmol), while continuing to purge with nitrogen. Nitrogen flow was
discontinued and the flask was sealed and heated at 9O0C for 8 h. The reaction was
cooled and the solvent was removed by rotary evaporation. The resulting residue was
suspended in saturated aqueous sodium bicarbonate (10 mL) and extracted with
chloroform (4 x 25 mL). The combined organic extracts were dried (Na2SO4),
filtered, concentrated by rotary evaporation to a thick dark mass. This was purified by
column chromatography, using methanol/chloroform (2:98, v/v) as the eluent, to
afford 0.70 g of the desired compound as a light brown viscous liquid (78.6%).
7-(5-Phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
To a solution of l-benzyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
(690 mg, 1.79 mmol) in ethanol (25 mL) in a pressure bottle was added concentrated
HCl (1 mL) and Pearlman's catalyst (Pd(OH)2, 20% on carbon, 50 mg). The solution was shaken under 50 psi of hydrogen gas for 8 h. The catalysts was removed by
filtration through Celite, and the filter cake was washed with ethanol (20 mL). The
solvent was removed by rotary evaporation, and the residue was basifϊed with
saturated aqueous sodium bicarbonate to pH 8-9. Solid sodium chloride (2 g) was
added, and the solution was extracted with chloroform (4 x 20 mL). The combined
chloroform extracts were dried (Na2SO4), filtered and concentrated by rotary
evaporation to afford 490 mg of the desired compound as a light brown viscous liquid
(92.7 %).
l-Methyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
To a stirring solution of 7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane
(420 mg, 1.42 mmol) in 37% aqueous solution of formaldehyde (5 mL) was added
98% formic acid (3 mL) under nitrogen. The reaction mixture was heated to reflux
for 8 h. The reaction mixture was cooled to room temperature, then basified with
saturated aqueous sodium bicarbonate to pH 8-9 and extracted with chloroform (4 x
15 mL). The combined chloroform extracts were dried (Na2SO4), filtered and
concentrated by rotary evaporation to afford a thick brown viscous liquid. This was
distilled using a Kugelrohr apparatus (2mm, 18O0C) to give a very pale cream-colored syrup (400 mg, 90.9 %).
l-Methyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane dihydrochloride l-Methyl-7-(5-phenoxy-3-pyridyl)-l,7-diazaspiro[4.4]nonane (200 mg, 0.65
mmol) was dissolved in concentrated HCl (1 mL) and sonicated for 5 min. The
excess acid and water were removed by repeated azeotropic evaporation with small portions of ethanol. A pale yellow solid was obtained. The solid was dissolved in the
minimum amount of absolute ethanol (~ 1 mL), and then ether was added drop-wise
until the solution became opaque. Cooling in the refrigerator overnight produced
cream-colored crystals, which were filtered, washed with ether and dried in a vacuum oven to yield 210 mg (85.4 %) of pure dihydrochloride salt, m.p. 180-1910C. Example 6
Sample 6 is r-(3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine] dihydrochloride, which was prepared according to the following techniques:
(3-oxolanyl)methyl methanesulfonate
To a stirring solution of (3-oxolanyl)methan-l-ol (25 g, 245 mmol) and triethylamine (34.37 mL, 245 mmol) in dry dichloromethane (250 mL) at O0C under
N2 atmosphere was added dropwise methanesulfonyl chloride (18.94 mL, 245 mmol).
The reaction mixture was stirred overnight after warming to room temperature, then a
saturated solution OfNaHCO3 (100 mL) was added and the mixture stirred for another
30 min. The biphasic mixture was separated and the organic layer was discarded.
The aqueous layer was extracted with dichloromethane (3 x 25 mL) and the combined
dichloromethane extracts were dried (Na2SO4), filtered and concentrated by rotary
evaporation to give 42.16 g of (3-oxolanyl)methyl methanesulfonate (99 %) as a light
brown liquid.
3-(Bromomethyl)oxolane
To a stirring solution of (3-oxolanyl)methyl methanesulfonate (42.16 g, 239.5
mmol) in dry acetone (600 mL) was added lithium bromide (101.7 g, 1198 mmol).
The reaction mixture was heated to reflux for 3 h, then it was cooled and the solvent
removed by rotary evaporation. The residue was dissolved in water (200 mL) and
extracted with dichloromethane (2 x 100 mL). The combined extracts were dried
(Na2SO4), filtered and concentrated by rotary evaporation to afford a light brown liquid. It was distilled at 70°C and lmm of pressure to give 33.00 g (86.77 %) of 3-
(bromomethyl)oxolane as a colorless liquid.
Methyl 3-aza-4,4-diphenyl-but-3-enoate
To a stirring solution of methyl glycine ester hydrochloride (17.49 g, 139 mmol) in dry dichloromethane (150 mL) under N2 at room temperature was added
diphenylimine (25.00 g, 137 mmol) in one portion. The reaction mixture was stirred
for 24 h, during which time ammonium chloride precipitated. Water (20 mL) was
added and the layers were separated. The organic layer was washed with saturated
Na2CO3 solution (2 x 20 mL) and brine (20 mL). The organic layer was dried
(Na2SO4), filtered and concentrated by rotary evaporation to give ~35 g of a thick light
brown syrup (99% pure) in ~ 100% yield. This was taken on to the next reaction
without further purification.
Methyl 3-(3-oxolanyl)-2-aminopropanoate
To a stirring solution of methyl 3-aza-4,4-diphenyl-but-3-enoate (23.00 g, 90
mmol) under N2 in dry DMF (25 mL) and toluene (25 mL) was added potassium tert-
butoxide (10.20 g, 90 mmol) in one portion. The reaction mixture was stirred for 15
min; it changed color from yellow to dark reddish-brown. Then, a solution of 3-
(bromomethyl)oxolane (15 g, 90 mmol) in DMF (20 mL) and dry toluene (20 mL)
was added via cannula over a period of 30 min. The reaction mixture was stirred for
an additional 16 h at ambient temperature. Then, IN HCl (100 mL) was added to the
reaction mixture and it was stirred for another 30 min. The mixture was extracted with
ethyl acetate (3 x 50 mL). The aqueous layer was basified with solid K2CO3 to pH 8-9,
then saturated with solid NaCl and extracted with ethyl acetate (4 x 50 mL). The
combined ethyl acetate extracts were dried (K2CO3), filtered and concentrated by
rotary evaporation to give methyl 3-(3-oxolanyl)-2-aminopropanoate (10 g, 59.37 %)
as a brown liquid.
Ethyl l-azabicyclo[2.2.1]heptane-2-carboxylate
Methyl 3-(3-oxolanyl)-2-aminopropanoate (6.00 g, 3.46 mmol) was placed in a
sealed pressure tube, then 48% aqueous HBr (20 mL) was added and the solution was saturated with HBr gas. The tube was sealed carefully and heated at 110°- 120° C for 8
h. The reaction was then cooled and the contents transferred to a 250 mL round
bottom flask with 20 mL of water. The excess acid was removed by rotary
evaporation to give a semi solid brown mass. Then 30% aqueous ammonium
hydroxide (150 mL) was added at O0C and the mixture was heated at gentle reflux for
4 h. The solvent was removed by rotary evaporation to give a brown solid, which then
was dissolved in absolute ethanol (50 mL). Concentrated H2SO4 (10 mL) was added
and the solution was refluxed for 8 h. The contents were cooled in an ice bath, and
then basified with concentrated NaHCO3 solution to pH 8-9 and extracted with
chloroform (4 x 4OmL). The combined chloroform extracts were dried (K2CO3),
filtered and concentrated to give a brown-black liquid which was distilled using a
Kugelrohr apparatus (lmm, 14O0C) to afford a colorless liquid (4 g, 68.25%) as a
mixture of the exo and endo isomers of ethyl l-azabicyclo[2.2.1]heptane-2- carboxylate.
Ethyl l-aza-2-(nitroethyl)bicyclo[2.2.1]heptane-2-carboxylate
Lithium diisopropylamide (LDA) was prepared at O0C from diisopropylamine
(2.078g, 20.53mmol) and n-butyllithium (8.2ImL, 20.53 mmol) in dry THF (20 mL)
under an N2 atmosphere. To a stirring solution of a mixture of the exo and endo
isomers of ethyl l-azabicyclo[2.2.1]heptane-2-carboxylate (2.67g, 15.79 mmol) in dry THF (35 mL) at -780C under N2 atmosphere was added via cannula the LDA solution
over a period of 15 min. The reaction mixture was stirred for an additional 40
minutes. Then a solution of nitroethylene (1.45 g, 20.53 mmol) in dry THF (20 mL)
was added dropwise via cannula to the reaction mixture over a period of 15 min.
After stirring for 2 h at -780C, the reaction was quenched by adding a saturated solution of ammonium chloride (20 mL). It was extracted with ethyl acetate (5 x 25
mL), dried (Na2SO4), filtered and concentrated by rotary evaporation to give 3.82 g of
the desired product (86% pure) as a light brown liquid, which was taken on to the next
■ ■ step without further purification.
2 Η-spiro[azabicyclo[2.2.1]heptane-2,3 f-pyrroIidin]-2 '-one
Ethyl l-aza-2-(nitroethyl)bicyclo[2.2.1]heptane-2-carboxylate (3.82 g, 86%
pure, 15.78 mmol) was dissolved in ethanol (50 mL) in a hydrogenolysis bottle. A
catalytic amount of Raney nickel was added and the mixture was subjected to
hydrogenolysis at 50 psi on a Parr apparatus for 16 h. The catalyst was removed by
filtration through a celite plug and washed with ethanol (20 mL). A catalytic amount
(5 mg) of p-toluenesulfonic acid was added and the reaction mixture was refluxed for
12 h. The solvent was removed by rotary evaporation to afford a light brown solid.
This was dissolved in cone. NaHCO3 solution (10 mL), saturated with NaCl and
extracted with chloroform (4 x 40 mL). The combined chloroform extracts were dried
(K2CO3), filtered and concentrated by rotary evaporation to give a light brown solid.
It was purified by column chromatography, using MeOH:CHCl3:NH4OH (9:1:0.01,
v/v) as the eluent, to afford 1.96 g (75%) of 2'H-spiro[azabicyclo[2.2.1]heptane-2,3'-
pyrrolidin]-2'-one as a cream-colored solid (m.p. 980C).
Spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine]
To a solution of 2Η-spiro[azabicyclo[2.2.1]heptane-2,3'-pyrrolidin]-2'-one (1.00 g, 6.02 mmol) in dry THF (20 mL) at O0C under N2 atmosphere was added
lithium aluminum hydride (647 mg, 17.7 mmol) and the mixture was refluxed for 24
h. The reaction mixture was cooled in ice bath and then ether (20 mL) was added.
Excess hydride was quenched by the dropwise addition of 5 M solution of NaOH. The resulting solid aluminate salts were removed by filtration through a celite plug.
The filtrate was dried (Na2SO4), filtered and concentrated by rotary evaporation to
yield 800 mg of spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine] as a colorless
liquid (87.43%).
1 '-(3-Pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine] dihydrochloride A mixture of spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine] (300 mg, 1.98
mmol), 3-bromopyridine (344 mg, 2.18 mmol),
tris(dibenzylideneacetone)dipalladium(0) (54.57mg, 0.0654 mmol), rac-2,2'-
bis(diphenylphosphino)- 1 , 1 '-binaphthyl (74.22 mg, 0.131 mmol) and potassium tert-
butoxide (668.8 mg, 5.96 mmol) in dry toluene (20 mL) was heated in a sealed tube
flushed with argon gas at 9O0C for 8 h. The reaction was cooled to O0C and the
contents transferred to a 10OmL round bottom flask. The solvent was removed by
rotary evaporation and the residue was dissolved in a saturated solution OfNaHCO3
(10 mL) and extracted with chloroform (4 x 15mL). The combined chloroform
extracts were dried (K2CO3), filtered and concentrated by rotary evaporation to give a
dark colored syrup. This was purified by column chromatography, using
MeOH:CHCl3:NH4OH (8:2:0.01, v/v) as the eluent, to afford 350 mg (79.0%) of 1'- (3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine] as a light brown syrup.
A portion of the free base (200 mg) was converted to a hydrochloride salt, which was
crystallized from isopropanol and ethanol to yield 200 mg (76%) of a light brown
solid, (m.p. 232°-236°C).
Example 7
Sample 7 is l'-(5-ethoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'- pyrrolidine], which was prepared according to the following techniques:
l'-(5-Ethoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine]
A mixture of spiro[l-azabicyclo[2.2.1]heptane-2,3'-pyrrolidine] (50 mg, 0.3 mmol) tris(dibenzylideneacetone)dipalladium(0) (9 mg, 0.009 mmol), rac-2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl (12 mg, 0.018 mmol), potassium tert-butoxide
(147 mg, 1.2 mmol), and 5-bromo-3-ethoxypyridine (73 mg, 0.36 mmol) in dry
toluene (5 mL) was placed in a sealed tube under argon and heated at 160° C for 17 h.
The reaction was cooled to O0C and the contents transferred to a 10OmL round bottom
flask. The solvent was removed by rotary evaporation and the residue was dissolved
in a saturated solution OfNaHCO3 (10 mL) and extracted with chloroform (4 x
15mL). The combined chloroform extracts were dried (K2CO3), filtered and
concentrated by rotary evaporation to give a dark colored syrup. This was purified by
column chromatography, using MeOH : CHCl3 :NH4OH (8:2:0.01, v/v) as the eluent, to
give 28 mg (27%) of l'-(5-ethoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-
pyrrolidine] as a viscous brown oil.
Example 8
Sample 8 is l'-(5-phenoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-
pyrrolidine], which was prepared according to the following techniques:
l'-(5-Phenoxy-3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine] A mixture of spiro[ 1 -azabicyclo[2.2.1 ]heptane-2,3 '-pyrrolidine] (50 mg,
0.3mmol), tris(dibenzylideneacetone)dipalladium(0) (9mg, 0.009 mmol), rac-2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl (12 mg, 0.018 mmol), potassium tert-butoxide
(147 mg, 1.3 mmol), and 5-bromo-3-phenoxypyridine (90 mg, 0.36 mmol) in dry
toluene (5 mL) was heated in a sealed tube under argon at 160° C for 17 h. The
reaction was cooled to O0C and the contents transferred to a 10OmL round bottom
flask. The solvent was removed by rotary evaporation and the residue was dissolved in a saturated solution OfNaHCO3 (10 mL) and extracted with chloroform (4 x
15mL). The combined chloroform extracts were dried (K2CO3), filtered and
concentrated by rotary evaporation to give a dark colored syrup. This was purified by column chromatography, using MeOH: CHCl3 :NH4OH (8:2:0.01, v/v) as the eluent, to
afford 55.8 mg of l'-(5-phenoxy- 3-pyridyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-
pyrrolidine] (52%) as a viscous tan oil.
Example 9
Sample 9 is r-(5-pyrimidinyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'-
pyrrolidine], which was prepared according to the following techniques:
l'-(5-Pyrimidinyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine]
A mixture of spiro[l-azabicyclo[2.2.1]heptane-2,3 '-pyrrolidine] (100 mg, 0.06
mmol), tris(dibenzylideneacetone)dipalladium(0) (18mg, 0.0018 mmol), rac-2,2'-
bis(diphenylphosphino)-l,l'-binaphthyl (24 mg, 0.0036 mmol), potassium tert-
butoxide (300 mg, 2.6 mmol), and 5-bromopyrimidine (114 mg, 0.7 mmol) in dry
toluene (10 niL) was placed in a sealed tube under argon and heated at 125° C for 17
h. The reaction was cooled to O0C and the contents transferred to a 10OmL round
bottom flask. The solvent was removed by rotary evaporation and the residue was
dissolved in a saturated solution OfNaHCO3 (10 mL) and extracted with chloroform
(4 x 15mL). The combined chloroform extracts were dried (K2CO3), filtered and
concentrated by rotary evaporation to give a dark colored syrup. This was purified by
column chromatography, using MeOH : CHCl3 :NH4OH (8:2:0.01, v/v) as the eluent, to
afford 49.0 mg of r-(5-pyrimidinyl)-spiro[l-azabicyclo[2.2.1]heptane-2,3'- pyrrolidine] (32%) as a viscous brown oil.
Example 10
Sample 10 is r-(3-pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine], which was prepared according to the following techniques:
Ethyl quinuclidine-2-carboxylate
The ethyl quinuclidine-2-carboxylate for this synthesis was prepared according
to the method described by Ricciardi and Doukas (Heterocycles 24:971 (1986)). We have also prepared ethyl quinuclidine-2-carboxylate using chemistry analogous to that
used for the synthesis of ethyl l-azabicyclo[2.2.1]heptane-2-carboxylate, but using 4- (bromomethyl)oxane in place of 3-(bromomethyl)oxolane.
Ethyl 2-(2-nitroethyl)quinuclidine-2-carboxylate Lithium diisopropylamide was prepared at O0C from lithium diisopropylamine
(193.53mg, 1.91 mmol) and n-butyllithium (0.764mL, 1.91 mmol) under N2. It was
added via cannula to a stirring solution of ethyl quinuclidine-2-carboxylate (320 mg,
1.74 mmol) in dry THF (10 mL) at -780C. After Ih, a solution of nitroethylene
(140.41 mg, 1.91 mmol) in THF (5 mL) was added dropwise to the reaction mixture.
After stirring for 2 h at -780C, the reaction was quenched by adding a saturated
solution of ammonium chloride (20 mL). It was extracted with ethyl acetate (5 x 25
mL), dried (Na2SO4), filtered and concentrated by rotary evaporation to give 325 mg
(70% pure) ethyl 2-(2-nitroethyl)quinuclidine-2-carboxylate as a light brown liquid,
which was taken on to the next step without further purification.
2 Η-spiro[azabicyclo[2.2.2] octane-2,3 '-pyrrolidin]-2 '-one
A solution of ethyl 2-(2-nitroethyl)quinuclidine-2-carboxylate (320 mg, ) in
ethanol (10 mL) was subjected to hydrogenolysis at 50 psi on a Parr apparatus for 16 h
using Raney nickel as a catalyst. The catalyst was removed by filtration through a celite plug and washed with ethanol (20 mL). A catalytic amount (5 mg) of p-
toluenesulfonic acid was added and the reaction mixture was refluxed for 12 h. The
solvent was removed by rotary evaporation to afford a light brown solid. This was
dissolved in cone. NaHCO3 solution (10 mL), saturated with NaCl and extracted with chloroform (4 x 40 mL). The combined chloroform extracts were dried (K2CO3),
filtered and concentrated by rotary evaporation to give a light brown solid. It was
purified by chromatography, using MeOH: CHCl3 :NH4OH (8:2:0.01, v/v) as the eluent, to give 120 mg (38.2%) of desired compound as light cream-colored solid
(m.p. 103°-105°C).
Spiro [1-azabicyclo [2.2.2] octane-2,3 '-pyrrolidine]
To a solution of 2Η-spiro[azabicyclo[2.2.2]octane-2,3'-pyrrolidin]-2'-one (100
mg, 0.55 mmol) in dry THF (10 mL) at O0C under N2 atmosphere was added lithium
aluminum hydride (74 mg, 1.94 mmol) and the mixture was refluxed for 24 h. The
reaction mixture was cooled in ice bath and then ether (20 mL) was added. Excess
hydride was quenched by the dropwise addition of 5 M solution of NaOH. The
resulting solid aluminate salts were removed by filtration through a celite plug. The
filtrate was dried (Na2SO4), filtered and concentrated by rotary evaporation to yield 83
mg of spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine] as a colorless liquid (90 %).
1 '-(3-Pyridyl)-spiro [1-azabicyclo [2.2.2] octane-2,3 '-pyrrolidine]
A stirring solution of spiro[l-azabicyclo[2.2.2]octane-2,3 '-pyrrolidine] (80 mg, 0.48 mmol), tris(dibenzylidineacetone)dipalladium(0) (26.47 mg, 0.024 mmol), rac-
2,2'-bis(diphenylphosphino)-l,r-binaphthyl (30 mg, 0.048 mmol) and potassium tert-
butoxide (215 mg, 1.92 mmol) in dry toluene (15 mL) was placed in a sealed tube
under argon and heated at 9O0C for 16 h. The reaction was cooled to O0C and the
contents transferred to a 10OmL round bottom flask. The solvent was removed by
rotary evaporation and the residue was dissolved in a saturated solution OfNaHCO3
(10 mL) and extracted with chloroform (4 x 15mL). The combined chloroform
extracts were dried (K2CO3), filtered and concentrated by rotary evaporation to give a
dark colored syrup. This was purified by column chromatography, using MeOH:CHCl3:NH4OH (8:2:0.01, v/v) as the eluent, to give 102 mg (85.7 %) of l'-(3-
pyridyl)-spiro[l-azabicyclo[2.2.2]octane-2,3'-pyrrolidine] as a light brown syrup. Example 11
Sample 11 is l'-(3-pyridyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-
pyrrolidine], which was prepared according to the following techniques:
Ethyl 2-(2H,3H,5H-4-oxinyl)-2-nitroacetate
A 2 M solution of titanium tetrachloride in THF was made by slow addition of
the titanium tetrachloride (7.59 g, 40 mmol) to dry THF (20 mL) at O0C under an
nitrogen, atmosphere. Ethyl nitroacetate (2.66 g, 20 mmol) was then added to the
stirring solution, and the mixture was stirred for 5 min. Next, tetrahydro-4-H- pyran-
4-one (2.00 g, 20 mmol) was added in one portion. Then, a 1.0 M solution of N-
methyl morpholine in THF (8.09 g, 80 mmol) was added dropwise over a period of 2
h at O0C. The mixture was then allowed to warm to room temperature and was stirred
for 18 h. It was then poured into water (20 mL) and extracted with ethyl acetate (5 x
4OmL). The combined extracts were dried over Na2SO4, filtered and concentrated by
rotary evaporation. The thick brown syrup was purified by column chromatography,
using ethyl acetate :hexane (1 :9, v/v) as eluent, to afford 3.00 g of pure compound as a
light-brown syrup (70%).
Ethyl 2-(4-oxanyl)-2-aminoacetate
Raney nickel (~2 g) was added to a solution of ethyl 2-(2H,3H,5H-4-oxinyl)-2-
nitroacetate (2.50 g, 11.62 mmol) in ethanol (50 mL) and cone. HCl (1 mL). The
mixture was subjected to hydrogenolysis at 50 psi on a Parr apparatus for 18 h. The
catalyst was removed by careful filtration through a celite plug. The solvent was
removed by rotary evaporation. The residue was basifϊed with saturated aqueous
NaHCO3 to pH 8-9, then saturated with NaCl and extracted with chloroform (4 x 25 mL). The combined extracts were dried over K2CO3, filtered and concentrated to yield
2.40 g (~ 100%) of desired compound as a tan liquid.
l-azabicyclo[2.2.1] heptane-7- carboxylic acid hydrochloride
Ethyl 2-(oxanyi)-2-aminoacetate (1.50 g, 8.02 mmol) was dissolved in 48%
HBr (10 mL) in a pressure tube and saturated with HBr gas. The tube was sealed
carefully and heated for 12 h at 120°- 13 O0C. The reaction was cooled to room
temperature, transferred to a 250 mL round bottom flask, and the acid was removed by
rotary evaporation. The dark colored residue was dissolved in 30% ammonia solution
(50 mL). This mixture was stirred for 5 h at room temperature, until cyclization to the
desired acid was complete. The ammonia solution was removed by rotary evaporation
to afford a light brown solid, which was redissolved in 5 mL of water and purified on
an ion exchange resin using water as the eluent and ammonia (30% aq.). Ammoniacal
fractions containing the desired acid were combined and concentrated to afford pure
acid, which was converted to an HCl salt and crystallized from isopropanol and
diethyl ether to give 1.21 g (85%) of a cream-colored solid (m.p. 232° turns brown,
melts at 253°-254°C).
Ethyl l-azabicycIo[2.2.1]heptane-7-carboxylate
A solution of 1 -azabicyclo [2.2.1] heptane-7- carboxylic acid hydrochloride
(1.20 g, 6.76 mmol) in absolute ethanol (10 mL) and concentrated sulfuric acid (2 mL)
was refluxed for 8 h. The reaction mixture was cooled and then basified with
saturated aqueous NaHCO3 to pH 8-9. The solution was saturated with solid NaCl
and extracted with chloroform (4 x 20 mL). The combined chloroform extracts were
dried over Na2SO4, filtered and concentrated by rotary evaporation to give a light
brown liquid. This was purified by Kugelrohr distillation at 12O0C and 2.5mm pressure to afford 1.00 g (90%) as a colorless liquid.
Ethyl l-aza-7-(2-nitroethyl)bicyclo[2.2.1]heptane-7-carboxylate
Lithium diisopropylamide was prepared by the addition of n-butyllithium (1.70
mL, 6.26 mmol) to diisopropylamine (431.1 mg, 6.26 mmol) in dry THF (5 mL) at 0° under a N2 atmosphere. The reaction was stirred at room temperature for 15 min and
then transferred via cannula to a stirring solution of ethyl l-azabicyclo[2.2.1]heptane-
7-carboxylate (600 mg, 3.55 mmol) in THF (2OmL) at -78° C under a N2 atmosphere.
The reaction mixture was stirred for 30 min at -78°C, then a solution of nitroethylene
(285.3 mg, 3.91 mmol) in THF (10 mL) was added via cannula and the reaction was
stirred for additional 2 h at -78°C. Then the reaction was quenched with saturated
NH4Cl solution (10 mL). The reaction mixture was allowed to warm to room
temperature and then was extracted with ethyl acetate (4 x 20 mL). The combined
fractions were dried (K2CO3), filtered and concentrated by rotary evaporation to give
650 mg of a light-brown liquid. It was purified by column chromatography, using
ethyl acetate:dichloromethane (8:2, v/v), to give 600 mg (85%) of tan liquid.
2Η-Spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidin]-2'-one
Ethyl l-aza-7-(2-nitroethyl)bicyclo[2.2.1]heptane-7-carboxylate (550 mg, 2.27
mmol) was dissolved in ethanol (25 mL) and subjected to hydrogenolysis at 50 psi for
18 h, using Raney nickel as a catalyst. The catalyst was removed by filtration through
a celite plug. The solvent was removed by rotary evaporation. The resultant residue
was dissolved in toluene (50 mL) and a catalytic amount of p-toluenesulfonic acid (10
mg) was added. The solution was refluxed for 12 h and then the solvent was removed
by rotary evaporation. The residue was added to saturated NaHCO3 (10 mL) solution
and extracted with chloroform (5 x 15 mL). The combined chloroform extracts were
dried (K2CO3), filtered, and concentrated. The residue was purified by column chromatography, using CHCl3: MeOH :NH4OH (9:1:0.01, v/v) as the eluent, to afford
320 mg (85%) of pure compound as a cream-colored thick syrup.
2 Η-Spiro[l-azabicyclo[2.2.1]heptane-7,3 '-pyrrolidine] To a stirring solution of 2'H-spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidin]-
2'-one (300 mg, 1.80 mmol) in dry THF (30 mL) at 0° under N2 was added LiAlH4
(274.33 mg, 7.22 mmol). The ice bath was removed and the reaction mixture was
refluxed for 24 h. The reaction mixture was cooled to O0C, diethyl ether (20 mL) was
added and 5M NaOH was added dropwise with constant stirring until all unreacted
LiAlH4 solidified. The reaction mixture was filtered through celite and then the
filtrate was dried (K2CO3), filtered and concentrated by rotary evaporation to yield
250 mg (70 %) of a colorless syrup.
l'-(3-Pyridyl)-2'H-spiro[l-azabicyclo[2.2.1]heptane-7;,3'-pyrrolidine]
2Η-Spiro[l-azabicyclo[2.2.1]heptane-7,3'-pyrrolidine] (100 mg, 0.66 mmol),
tris(dibenzylideneacetone)dipalladium(0) (30mg, 0.020 mmol), rac-2,2'-
bis(diphenylphosphino)-l,r-binaphthyl (45mg, 0.040 mmol), potassium tert-butoxide
(369mg, 3.3mmol) and 3-bromopyridine (114mg, 0.72 mmol) and dry toluene (10
niL) were placed in a pressure tube which was flushed with argon. The tube was
carefully sealed and heated for 8 h at 9O0C. The reaction mixture was cooled,
transferred to a round bottom flask and the solvent removed by rotary evaporation.
The residue was poured into saturated NaHCO3 solution (5 mL) and extracted with
chloroform (4 x 15 mL). The combined chloroform extracts were dried over K2CO3,
filtered and concentrated by rotary evaporation. The residue was purified by column
chromatography, using CHCl3 :MeOH:NH4OH (8:2:0.01, v/v) as eluent, to afford 130
mg (86.7%) of a light brown syrup. The product turns dark brown on exposure to
light and air. Examples 12 and 13
Samples 12 and 13 are (+) and (-) 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
respectively, which were prepared according to the following techniques:
Diastereomeric 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane S-proline amides
Triethylamine (6.0 mL, 43 mmol) and diphenyl chlorophosphate (4.0 mL, 19
mmol) were added, in that order, to a stirred suspension of N-(tert-butoxycarbonyl)-S-
proline (4.67 g, 21.7 mmol) in dichloromethane (100 mL) under a nitrogen
atmosphere. After stirring for 1.5 h at ambient temperature, the reaction mixture was
treated with a solution of 7-(3-pyridyl)-l ,7-diazaspiro[4.4]nonane (4.40 g, 21.6 mmol)
in dichloromethane (10 mL). The mixture was stirred 3 days at ambient temperature.
Sodium hydroxide solution (30 mL of 5 M) was then added. After stirring an
additional hour, the mixture was poured into a separatory funnel with chloroform (30
mL) and water (30 mL). The mixture was shaken vigorously, and the layers were
separated. The organic layer and a 30 mL chloroform extract of the aqueous layer
were combined, dried (MgSO4) and concentrated by rotrary evaporation. The residue
(7.2 g) was dissolved in dichloromethane (100 mL) and conbined with trifluroacetic
acid (50 mL). The mixture was stirred at ambient temperature for 1 h. The volatiles
were evaporated, first by rotary evaporation and then on the vacuum pump. The
residue was purified by preparative HLPC, using 10% acetonitrile, 0.1%
trifluoroacetic acid in water as eluent. Selected fractions were combined and
concentrated, leaving 3.13 g (79% yield) of the diastereomer which elutes at 11.4 min and 2.90 g (74% yield) of the diastereomer that elutes at 13.2 min, both as white
foams (presumably mono trifluoroacetate salts).
(+) and (-) 7-(3-pyridyl)-l,7-diazaspiro[4.4]nonane
Each of the two diastereomeric S-proline amides was dissolved in
dichloromethane (50 mL) and triethylamine (2-3 mL), and then combined with
phenylisothiocyanate (1.73 g, 12.8 mmol for the earlier eluting diastereomer and 1.57 g, 11.6 mmol for the later eluting diastereomer). The two reactions were stirred at
ambient temperature for 16 h, at which point thin layer chromatography indicated that
the reactions were complete. The mixtures were concentrated by rotary evaporation,
and each of the residues was taken up in dichloromethane (10 mL) and treated with
trifluoroacetic acid (10 mL). These reactions were held at 50°C for 16 h and
concentrated to dryness. Column chromatography on silica gel with 80:20:2
chlorform/methanol/ammonia gave 620 mg (derived from the earlier eluting
diastereomer, 40.5% yield) and 720 mg (derived from the later eluting diastereomer,
50.7% yield), as light brown oils. Chiral HPLC analysis was perormed on a Chiralcel
OD ® column, using 7:3 heaxane/ethanol. The isomer derived from the earlier eluting
diastereomer had the longer retention time on the chiral column (10.9 min); that
derived from the later eluting isomer exhibited a retention time of 8.7 min on the
chiral column. The samples were enantiomerically pure within the limits of detection
(-2%).
Example 14
The study of the in vitro pharmacology of 7-(3-pyridyl)-l,7-
diazaspiro[4.4]nonane showed it to be an antagonist at both the α4β2 subtype (IC50 =
193 μM; Imax = 50%) and those NNR subtypes affecting dopamine release (IC50 =
901 nM; Imax = 67%). The ability of this compound to partially inhibit the release of
dopamine is especially significant, as it indicates that this compound (and others in the
N-arylspirodiazaalkane genus) may be useful in interrupting the dopamine reward
system, and thus treating disorders that are mediated by it. Such disorders include
substance abuse, tobacco use and weight gain that accompanies drug cessation.
The in vivo evidence that N-arylspirodiazaalkanes can be useful in this manner
was derived from a fourteen-day preclinical safety pharmacology study, in which 7-(3-
pyridyl)-l,7-diazaspiro[4.4]nonane reduced weight gain in rats, without demonstrating stimulant sensitization properties.
Based on this data, it is anticipated that compounds of the N-
arylspirodiazaalkane genus described herein present a useful alternative in treating
dependencies on drugs of abuse including alcohol, amphetamines, barbiturates,
benzodiazepines, caffeine, cannabinoids, cocaine, hallucinogens, opiates,
phencyclidine and tobacco and in treating eating disorders such as obesity that occurs
following drug cessation while reducing side effects associated with the use of
psychomotor stimulants (agitation, sleeplessness, addiction, etc.).
Having hereby disclosed the subject matter of the present invention, it should
be apparent that many modifications, substitutions, and variations of the present
invention are possible in light thereof. It is to be understood that the present invention
can be practiced other than as specifically described. Such modifications,
substitutions and variations are intended to be within the scope of the present application.