WO2006044457A1 - N-benzenesulfonyl substituted anilino-pyrimidine analogs - Google Patents
N-benzenesulfonyl substituted anilino-pyrimidine analogs Download PDFInfo
- Publication number
- WO2006044457A1 WO2006044457A1 PCT/US2005/036674 US2005036674W WO2006044457A1 WO 2006044457 A1 WO2006044457 A1 WO 2006044457A1 US 2005036674 W US2005036674 W US 2005036674W WO 2006044457 A1 WO2006044457 A1 WO 2006044457A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- group
- compound
- formula
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC1(C)OB(C=CC*)OC1(C)C Chemical compound CC1(C)OB(C=CC*)OC1(C)C 0.000 description 7
- WCNPNHJYHJLXHL-UHFFFAOYSA-N CC(C1)C([N+]([O-])=O)=CC=CC1(C)S(Cl)(=O)=O Chemical compound CC(C1)C([N+]([O-])=O)=CC=CC1(C)S(Cl)(=O)=O WCNPNHJYHJLXHL-UHFFFAOYSA-N 0.000 description 1
- YCVDPBWJLBQWJH-UHFFFAOYSA-N CCOC(CCNC)OCC Chemical compound CCOC(CCNC)OCC YCVDPBWJLBQWJH-UHFFFAOYSA-N 0.000 description 1
- GHTDAXTYNRRXEC-UHFFFAOYSA-N CCOC(CNC)OCC Chemical compound CCOC(CNC)OCC GHTDAXTYNRRXEC-UHFFFAOYSA-N 0.000 description 1
- LNJMHEJAYSYZKK-UHFFFAOYSA-N Cc1ncccn1 Chemical compound Cc1ncccn1 LNJMHEJAYSYZKK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/10—Drugs for genital or sexual disorders; Contraceptives for impotence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/12—Drugs for genital or sexual disorders; Contraceptives for climacteric disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/04—Drugs for skeletal disorders for non-specific disorders of the connective tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to anilino-pyrimidine analogs that are useful for inhibiting kinase activity.
- Nuclear factor- ⁇ B is a transcriptional factor that regulates the expression of important genes related to cell survival. Activation of NF- ⁇ B is central to inflammatory response because it regulates the expression of pro-inflammatory cytokines such as tumor necrosis factor- ⁇ (TNF- ⁇ ). TNF- ⁇ not only induces inflammation, but also acts as a survival factor for many cancers and can stimulate the production of angiogenic factors. TNF- ⁇ has been found in ovarian, breast, prostate, bladder, and colorectal cancer as well as in lymphomas and leukemias. The role of NF- ⁇ B in cancer has been further illuminated by research showing that NF -KB promotes tumorigenesis by suppressing apoptosis and stimulating cell proliferation.
- NF- ⁇ B inhibitors may prove useful as anti-cancer and anti-inflammation therapeutic agents.
- the primary form of NF- ⁇ B is retained in the cytoplasm of resting cells by IKB, an inhibitor of NF- ⁇ B.
- NF- ⁇ B is activated by stimulation of a cellular kinase complex known as IKB kinase ("IKK”) complex, comprising subunits IKK ⁇ , ⁇ , and ⁇ .
- IKK IKB kinase
- IKK phosphorylates IKB and triggers ubiquitination-dependent degradation through the proteasome pathway. With IKB destroyed, NF- ⁇ B is free to enter the nucleus and activate transcription.
- IKK Aberrant expression of IKK has been correlated with activation of NF- ⁇ B and, in turn, tumorigenesis and cell proliferation. High IKK levels may also promote tumorigenesis by negatively regulating other transcription factors, such as FOXO factors. Hu, M. (2004) "IKB Kinase Promotes Tumorigenesis through Inhibition of Forkhead FOXC ⁇ a," Cell, 117, 225- 237. Thus, inhibiting IKK may inhibit cell proliferation and tumorigenesis. Other anilino- pyrimidine derivatives have been shown to inhibit inappropriately high kinase activity. See, e.g., U.S. Patent No. 6,048,866. However, there remains a need for agents that selectively inhibit kinase activity, including IKK. The present invention fulfills this need.
- the present invention provides a compound of formula I:
- R 1 is hydrogen
- R 2 is selected from the group consisting OfNR 7 R 8 , guanidinyl, ureido, optionally substituted imidazolyl, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, hydroxy, and alkoxy;
- R 5 is selected from the group consisting of hydrogen, methyl, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, hydroxymethyl, and alkylaminomethyl;
- R 7 and R 8 are independently selected from the group consisting of hydrogen; optionally substituted alkyl; optionally substituted alkenyl; optionally substituted alkynyl; optionally substituted aryl; optionally substituted heteroaryl; hydroxy; alkoxy; alkylamino; arylamino; heteroarylamino; -NCOR 9 ; -COR 9 ; -CONR 7 R 8 ; SO 2 R 10 ; optionally substituted 3 to 10 membered cyclic amines containing O to 3 heteroatoms; optionally, R and R together form an optionally substituted 3 to 12 membered monocyclic or bicyclic ring containing O to 4 heteroatoms;
- R 9 is selected from the group consisting of hydrogen, methyl, trifluoromethyl, optionally substituted alkyl, optionally substituted aryl, and optionally substituted heteroaryl
- R 10 is selected from the group consisting of methyl, trifluoromethyl, optionally substituted alkyl, optionally substituted aryl, optionally substituted heteroaryl, and NR 7 R 8 ; and the salts, solvates, and hydrates thereof.
- the present invention provides preferred substituents and specific compounds of formula I.
- the present invention also provides pharmaceutical compositions comprising a compound of the present invention and a pharmaceutically acceptable carrier.
- the present invention provides a method of inhibiting kinase action, especially IKK, in a cell by providing a compound of the present invention.
- the present invention also provides a method of inhibiting kinase activity, especially IKK, in a mammal, especially a human, by administering a compound or pharmaceutical composition of the present invention.
- the present invention also provides a method of treating a kinase-dependent condition, especially inflammation or cancer, by administering a compound of the present invention.
- the present invention provides methods of treating diseases associated with NF- ⁇ B activation by administering a compound of the present invention.
- the present invention provides methods of treating cancer; inflammatory or autoimmune conditions; cardiovascular, metabolic, or ischemic conditions; infectious diseases, particularly viral infections; as well as pre- or post-menopausal conditions, particularly osteoporosis, by administering a compound of the present invention.
- the present invention also provides methods further comprising administering an additional inhibitor of a protein kinase of the NF- ⁇ B pathway.
- the present invention provides processes for making a compound of formula I as defined above.
- the present invention also encompasses intermediates of these processes.
- Figures 1 -8 depict exemplary guanidine and enaminone reactions.
- Figures 9-14 depict exemplary halogen displacement reactions.
- the present invention relates to anilino-pyrimidine analogs, pharmaceutical compositions, and methods using the same.
- the present invention provides a compound of formula I:
- R 1 is hydrogen
- R is selected from the group consisting of NR R , guanidinyl, ureido, optionally substituted imidazolyl, optionally substituted alkyl, optionally substituted alkenyl, optionally substituted alkynyl, hydroxy, and alkoxy;
- R 4 is hydrogen
- R 5 is selected from the group consisting of hydrogen, methyl, alkyl, alkylcarbonyl, alkoxycarbonyl, alkylsulfonyl, hydroxymethyl, and alkylaminomethyl;
- R and R are independently selected from the group consisting of hydrogen; optionally substituted alkyl; optionally substituted alkenyl; optionally substituted alkynyl; optionally substituted aryl; optionally substituted heteroaryl; hydroxy; alkoxy; alkylamino; arylamino; heteroarylamino; -NCOR 9 ; -COR 9 ; -CONR 7 R 8 ; SO 2 R 10 ; optionally substituted 3 to 10 membered cyclic amines containing O to 3 heteroatoms; optionally, R 7 and R 8 together form an optionally substituted 3 to 12 membered monocyclic or bicyclic ring containing O to 4 heteroatoms;
- R 9 is selected from the group consisting of hydrogen, methyl, trifluoromethyl, optionally substituted alkyl, optionally substituted aryl, and optionally substituted heteroaryl;
- R 10 is selected from the group consisting of methyl, trifluoromethyl, optionally substituted alkyl, optionally substituted aryl, optionally substituted heteroaryl, and NR 7 R 8 ; and the salts, solvates, and hydrates thereof.
- the R groups of the present invention are optionally substituted. Unless otherwise specified, optionally substituted means having zero, one, or more than one substituents. Unless otherwise specified, substituted means having one or more substituents.
- Substituents include hydrogen, halogen, cyano, nitro, alkylamino, hydroxy, alkoxy, alkanoyl, carbonyl, carbamoyl, trifluoromethyl, trifluoromethoxy, aryl, heteroaryl, aralkyl, aryloxy, alkylthio, arylthio, thioyl, -COOR 9 , -CONR 7 R 8 , NR 7 R 8 (including cyclic amines as described below), SR 7 , and -SO 2 R 10 .
- the substituents further include methyl groups and optionally substituted C 2 - I o straight, branched, or cyclic alkyl, alkenyl, or alkynyl groups.
- the substituents on the R groups can also be optionally substituted.
- halogens include, but are not limited to fluorine, chlorine, bromine, and iodine.
- alkyl, alkenyl, and alkynyl groups have 1 to 10 carbon atoms and may be straight, branched, or cyclic.
- Alkyl means a straight chain or branched, cyclic or non-cyclic hydrocarbon.
- Alkenyl means a straight chain or branched, cyclic or non-cyclic hydrocarbon having at least 2 carbon atoms and including at least one carbon-carbon double bond.
- Alkynyl means a straight chain or branched hydrocarbon having at least 2 carbon atoms and including at least one carbon-carbon triple bond.
- Heteroatom means an atom selected from nitrogen, which can be quatemized; oxygen; and sulfur, including sulfoxide and sulfone.
- Alkoxy means a group -OR, wherein R is an alkyl, alkenyl, or alkynyl group which can optionally be substituted with one or more functional groups.
- Hydroxy means -OH.
- Amino means the -NH 2 group.
- Hydrates are solid compounds containing water molecules combined in a definite ratio as an integral part of the crystal.
- Solvates are solid compounds containing solvent molecules combined in a definite ratio as an integral part of the crystal.
- aryl groups include, but are not limited to phenyl and naphthyl groups.
- Heteroaryl means an aromatic heterocycle ring, including both mono- bi- and tricyclic ring systems, wherein at least one carbon atom of ring sytem is replaced with a heteroatom independently selected from nitrogen, oxygen and sulfur.
- heteroaryl groups include, but are not limited to pyridyl, pyrimidyl, thienyl, furanyl, imidazolyl, triazinyl, oxazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, pyrrole, pyrazinyl, and thiazolyl groups.
- heterocyclic groups include, but are not limited to saturated or partially saturated heteroaryls, including but not limited to pyrazoline, oxazolone, thiazolone, thiadiazolone, piperazine, pyrrolidine, piperidine, morpholine, benzoimidazolone, benzoxazolone, benzodioxazol, benzodioxazolone, benzo[l,4]oxazin-3-one, 3,4-dihydroquinoxaline-2-one, benzo[l,4]dioxene-2-one, and 1,2,3,4-tetrahydroquinoxaline.
- heteroaryls including but not limited to pyrazoline, oxazolone, thiazolone, thiadiazolone, piperazine, pyrrolidine, piperidine, morpholine, benzoimidazolone, benzoxazolone, benzodioxazol, benzodioxazolone, benzo[
- Examples of a benzene ring fused to a heterocyclic ring include, but are not limited to benzofuran, isobenzofuran, dihydrobenzofuran, dihydrobenzopyran, benzoxazolidinone, benzimidazolinone, benzooxazinone, indole, isoindole, benzothiophene, quinoline, and isoquinoline.
- the heteroaryl and heterocyclic groups contain one or more heteroatoms selected from the group consisting of sulfur, nitrogen, and oxygen.
- R 2 is selected from the group consisting OfNR 7 R 8 , optionally substituted imidazolyl, and optionally substituted alkyl. In a preferred embodiment, R 2 is
- NR R , and R and R are independently selected from the group consisting of hydrogen, alkyl, amino and alkylamino (including cyclic amines), alkylhydroxy, alkanoyl, alkoxy, alkoxycarbonyl, carbonyl, carboxyl, aralkyl, optionally substituted phenyl, heteroaryl, and COR 9 where R 9 is alkyl or aralkyl.
- R 2 is NH 2 , -(dimethylamino)ethyl, or -(dimethylamino)propyl.
- R 7 and R 8 together form an optionally substituted 3 to 12 membered monocyclic or bicyclic ring containing 0 to 4 heteroatoms.
- R is an optionally substituted 5 to 6 membered heterocyclic group containing at least one nitrogen atom and 0 to 1 additional heteroatoms.
- R 2 can be, for example, an optionally substituted morpholinyl group, an optionally substituted piperazinyl group, or an optionally substituted pyrrolidinyl group.
- R 2 is NR 7 R 8 , and R 2 is selected from the groups listed as Set 2a: Set 2a:
- R 2 is selected from the groups listed as Set 2b:
- the SO 2 R 2 group is at position 3 of the phenyl ring. In another embodiment, the SO 2 R 2 group is at position 4 of the phenyl ring such that the compound is a compound of formula II: ⁇
- R 3 is an optionally substituted phenyl group. Preferred substituents for this embodiment include alkoxy, trifluoromethyl, fluoro, hydroxy, and NR 7 R 8 where R 7 is COR 9 and R 8 is hydrogen. In one embodiment, R 3 is selected from the groups listed as Set 3a:
- R is an optionally substituted thienyl group.
- Preferred substituents for this embodiment include hydrogen (i.e., an unsubstituted thienyl group), bromo, and methyl.
- R 3 is selected from the groups listed as Set 3b:
- R is selected from the groups listed as Set 3c: Set 3c:
- R 5 is hydrogen or methyl. In a preferred embodiment, R 5 is hydrogen.
- R 6 is hydrogen, methyl, ethyl, chloro, methoxy, NH 2 , or trifluoromethyl. In a preferred embodiment, R 6 is hydrogen.
- Exemplary compounds of the present invention include the following compounds and salts, solvates, and hydrates thereof:
- salts of the compounds of formula I may enable salts of the compounds to be formed.
- Suitable salts include pharmaceutically acceptable salts, for example acid addition salts derived from inorganic or organic acids, and salts derived from inorganic and organic bases.
- pharmaceutically acceptable salt is a salt formed from an acid and a basic nitrogen group of a pharmaceutically active agent.
- Illustrative salts include, but are not limited, to sulfate; citrate, acetate; oxalate; chloride; bromide; iodide; nitrate; bisulfate; phosphate; acid phosphate; isonicotinate; lactate; salicylate; acid citrate; tartrate; oleate; tannate; pantothenate; bitartrate; ascorbate; succinate; maleate; gentisinate; fumarate; gluconate; glucaronate; saccharate; formate; benzoate; glutamate; methanesulfonate; ethanesulfonate; benzenesulfonate; p-toluenesulfonate; pamoate (i.e., 1,1 '-methylene-bis-(2-hydroxy-3-naphthoate)); and salts of fatty acids such as caproate, laurate, myristate, palmitate,
- phrases "pharmaceutically acceptable salt” also refers to a salt prepared from a pharmaceutically active agent having an acidic functional group, such as a carboxylic acid functional group, and a pharmaceutically acceptable inorganic or organic base.
- Suitable bases include, but are not limited to, hydroxides of alkali metals such as sodium, potassium, and lithium; hydroxides of alkaline earth metal such as calcium and magnesium; hydroxides of other metals, such as aluminum and zinc; ammonia, and organic amines, such as unsubstituted or hydroxy-substituted mono-, di-, or trialkylarnines; dicyclohexylamine; tributyl amine; pyridine; N-methyl,N-ethylamine; diethylamine; triethylamine; mono-, bis-, or tris-(2-hydroxy-lower alkyl amines), such as mono-, bis-, or tris-(2-hydroxyethyl)amine, 2- hydroxy-tert-but
- Acid addition salts include hydrochlorides, hydrobromides, hydroiodides, alkylsulphonates, e.g. methanesulphonates, ethanesulphonates, or isethionates, arylsulphonates, e.g. p-toluenesulphonates, besylates or napsylates, phosphates, sulphates, hydrogen sulphates, acetates, trifluoroacetates, propionates, citrates, maleates, fumarates, malonates, succinates, lactates, oxalates, tartrates and benzoates.
- Salts derived from inorganic or organic bases include alkali metal salts such as sodium or potassium salts, alkaline earth metal salts such as magnesium or calcium salts, and organic amine salts such as morpholine, piperidine, dimethylamine or diethylamine salts.
- Particularly useful salts of compounds according to the invention include pharmaceutically acceptable salts, especially acid addition pharmaceutically acceptable salts.
- the present invention provides processes for making a compound of formula I as defined above. The present invention also encompasses intermediates of these processes. Throughout the description of the processes, the numbered R groups are defined above with respect to formula I, and generic (not numbered) R groups represent independent substituents as described above.
- the compounds shown in the Figures are numbered by figure number and, where appropriate, a parenthetical note designating the corresponding general structure is also included.
- the term "reacting” includes, but is not limited to, adding, stirring, heating, heating to reflux, dissolving, titurating, and any combination thereof.
- One skilled in the art would appreciate the meaning of reacting given the reaction components and given the examples provided herein.
- the processes preferably include a step of isolating the compound of formula I.
- the present invention provides methods for preparing a compound of formula I by reacting an enaminone and a guanidine (Scheme 1).
- an enaminone of formula G-I is reacted with a guanidine of formula G-2 in the presence of l-methyl-2-pyrrolidinone (NMP).
- Scheme 1 :
- the reaction is conducted in the presence of a base, such as potassium carbonate or potassium hydroxide.
- a base such as potassium carbonate or potassium hydroxide.
- the enaminone G-I can be prepared by any method known in the art, such as the reaction of an acetyl derivative with an acetal, preferably N,N-dimethylformamide dimethyl acetal, or tert-butoxybis(dimethylamino)methane. See Figure 1.
- the guanidine G-2 can be prepared by reacting an amine of formula G-3 with cyanamide or 1-H-pyrazole-l-carboximidine. See also Figure 1.
- the guanidine G-2 can be prepared by reacting a halogenated sulfonamide of formula G-4 with guanidine. See Figure 2. halogen.
- the SO 2 R 2 group is added after the formation of the pyrimidine.
- This method includes the steps of: reacting an enaminone G-I with a guanidine derivative of formula 3-1 and NMP to form a pyrimidine; reacting the pyrimidine with chlorosulfonic acid to form a sulfonyl chloride of formula 3-3; and reacting the sulfonyl chloride 3-3 with an amine having the formula HNR 7 R 8 to form a compound of formula I. See Figure 3.
- the present invention provides methods for preparing a compound of formula I by halogen displacement (Scheme 2).
- the Scheme 2 reactions can be conducted in a solvent, preferably dioxane.
- R 3 is an optionally substituted phenyl or optionally substituted thienyl group.
- an amine G-3 is reacted with a halogenated pyrimidine of formula G-5.
- the halogen of the halogenated pyrimidine is chlorine.
- the reaction is conducted in the presence of p-toluenesulfonic acid.
- a halogenated sulfonamide of formula G-4 is reacted with a pyrimidine of formula G-6.
- the halogen of the halogenated sulfonamide is bromine.
- the reaction includes a step of adding sodium tert- butoxide (NaOtBu).
- the reaction is preferably conducted in the presence of tris(dibenzylideneacetone)dipalladium(0) (Pd 2 dba 3 ) and 2,2'-bis(diphenylphosphino)-l,l'- binaphthyl (BINAP).
- the present invention also provides pharmaceutical compositions comprising a compound of the present invention and a pharmaceutically acceptable carrier.
- Pharmaceutical compositions are prepared in accordance with acceptable pharmaceutical procedures, such as described in Remingtons Pharmaceutical Sciences, 17th edition, ed. Alfonoso R. Gennaro, Mack Publishing Company, Easton, Pa. (1985).
- Pharmaceutically acceptable carriers are those that are compatible with the other ingredients in the formulation and biologically acceptable.
- the present invention provides a method of inhibiting kinase action, especially IKK, by providing one or more compounds or pharmaceutical compositions of the present invention.
- Providing includes, but is not limited to, administration by pharmaceutical acceptable methods and routes of administration known by one of skill in the art.
- Providing also means exposing to or contacting with.
- Compounds of the present invention are useful to inhibit kinase activity, particularly IKK. Inhibiting includes total inhibition as well as decreasing or reducing.
- compounds of the present invention are believed to inhibit the ability of the IKK complex to phosphorylate IKB. AS such, NF- ⁇ B is not released and does not enter the nucleus to activate transcription.
- a binding assay demonstrates that compounds of the present invention affect the association of IKK ⁇ and I ⁇ B ⁇ .
- the binding assay is performed by contacting compounds of the present invention with IKK ⁇ enzyme and I ⁇ B ⁇ substrate and then detecting whether the compound inhibits association of IKK ⁇ and I ⁇ B ⁇ .
- Compounds of the present invention that inhibit the association of IKK ⁇ and I ⁇ B ⁇ may inhibit the ability of IKK to phosphorylate IKB and as such may inhibit the release of NF- ⁇ B and the transcription of NF- ⁇ B controlled genes.
- the present invention also provides a method of inhibiting kinase activity, especially IKK, in a mammal, especially a human, by administering a kinase-inhibiting amount, especially an IKK-inhibiting amount, of a compound or pharmaceutical composition of the present invention.
- Administering includes all pharmaceutical acceptable methods and routes of administration known by one of skill in the art.
- one embodiment provides a method of treating a kinase-dependent condition, such as an IKK dependent condition, comprising administering to a subject a kinase- inhibiting amount, such as an IKK-inhibiting amount, of a compound or pharmaceutical composition of the present invention.
- Kinase-dependent conditions include, but are not limited to autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus, transplant rejection, graft versus host disease, hyperproliferative disorders such as tumors, psoriasis, pannus formation in rheumatoid arthritis, restenosis following angioplasty and atherosclerosis, osteoporosis and in diseases in which cells receive pro-inflammatory signals such as asthma, inflammatory bowel disease, and pancreatitis.
- autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus
- transplant rejection transplant rejection
- graft versus host disease hyperproliferative disorders
- hyperproliferative disorders such as tumors, psoriasis, pannus formation in rheumatoid arthritis, restenosis following angioplasty and atherosclerosis, osteoporosis and
- the pharmaceutical compositions comprising compounds of the present invention may inhibit kinase activity, particularly IKK.
- Kinase inhibition would in turn inhibit the downstream expression of genes responsible for kinase-dependent conditions such as inflammation and cancer.
- IKK inhibits the activation of NF- ⁇ B, which in turn reduces expression of NF- ⁇ B dependent genes.
- pharmaceutical compositions comprising compounds that inhibit IKK may be useful to treat inflammation and cancer.
- the present invention also provides methods of treating diseases associated with NF-
- Treating includes, but is not limited to, complete treatment, where no symptoms are seen, as well as reducing symptoms and ameliorating symptoms.
- Treating includes, but is not limited to, complete treatment, where no symptoms are seen, as well as reducing symptoms and ameliorating symptoms.
- the phrase "treating,” “treatment of,” and the like includes the amelioration or cessation of a specified condition.
- Diseases associated with NF- ⁇ B activation include, but are not limited to inflammatory disorders; particularly rheumatoid arthritis, inflammatory bowel disease, and asthma; dermatosis, including psoriasis and atopic dermatitis; autoimmune diseases; tissue and organ rejection; Alzheimer's disease; stroke; epilepsy; Parkinson's disease, atherosclerosis; restenosis; cancer, including Hodgkins disease; and certain viral infections, including AIDS; osteoarthritis; osteoporosis; and Ataxia Telangiestasia.
- the present invention provides methods of treating cancer by administering a pharmaceutical composition of the present invention.
- Cancer includes an abnormal growth of cells, which tend to proliferate in an uncontrolled way and, in some cases, to metastasize (spread).
- Treating cancer encompasses, but is not limited to inhibiting or reducing tumor cell proliferation, tumor cell growth, and inhibiting tumorigenesis.
- Cancer includes, but is not limited to cancer of the colon, rectum, prostate, liver, lung, bronchus, pancreas, brain, head, neck, stomach, skin, kidney, cervix, blood, larynx, esophagus, testes, urinary bladder, ovary, or uterus.
- the present invention provides methods of treating an inflammatory or autoimmune condition by administering a pharmaceutical composition of the present invention.
- Treating inflammation encompasses, but is not limited to reducing inflammation and treating an inflammatory condition.
- Inflammatory and autoimmune conditions include, but are not limited to rheumatoid arthritis, rheumatoid spondylitis, osteoarthritis, gout, asthma, bronchitis, allergic rhinitis, chronic obstructive pulmonary disease, cystic fibrosis, inflammatory bowel disease, irritable bowel syndrome, mucous colitis, ulcerative colitis, diabrotic colitis, Crohn's disease, gastritis, esophagitis, hepatitis, pancreatitis, nephritis, psoriasis, eczema, dermatitis, hives, multiple sclerosis, Lou Gehrig's disease, sepsis, conjunctivitis, acute respiratory distress syndrome, pur
- the present invention provides methods of treating a cardiovascular, metabolic, or ischemic condition by administering a pharmaceutical composition of the present invention.
- Cardiovascular, metabolic, and ischemic conditions include, but are not limited to atherosclerosis, restenosis following angioplasty, left ventricular hypertrophy, insulin resistance, Type I diabetes, Type II diabetes, hyperglycemia, hyperinsulinemia, dyslipidemia, obesity, polycystic ovarian disease, hypertension, syndrome X, osteoporosis, erectile dysfunction, cachexia, myocardial infraction, ischemic diseases of heart kidney, liver, and brain, organ transplant rejection, graft versus host disease, endotoxin shock, and multiple organ failure.
- the present invention provides methods of treating an infectious disease, particularly a viral infection, by administering a pharmaceutical composition of the present invention.
- Viral infections include, but are not limited to those caused by human immunodeficiency virus (HIV), hepatitis B virus, hepatitis C virus, human papilomavirus, human T-cell leukemia virus, and Epstein-Barr virus.
- the present invention provides methods of treating a pre- or post-menopausal condition by administering a pharmaceutical composition of the present invention.
- a pharmaceutical composition of the present invention can be used to treat osteoporosis. Treating osteoporosis includes preventing osteoporosis as well as combating the existing condition.
- the present invention also provides methods of inhibition and treatment further comprising administering an additional inhibitor of a protein kinase of the NF- ⁇ B pathway.
- Inhibitors of a protein kinase of the NF- ⁇ B pathway include, but are not limited to IKK inhibitors and GSK-3 inhibitors.
- IKK inhibitors include, but are not limited to heterocyclic carboxamides, substituted benzimidazoles, substituted indoles, ⁇ -carbolines such as PS-1145, SPC0023579, SPC839/AS602868 (AS2868), NVPIKK004, and NVPIKK005.
- GSK-3 inhibitors include, but are not limited to maleimides such as SB410111, SB495052, SB517955, SB216763, SB415286, diamino-l,2,4-triazole carboxylic acid derivatives and 2,5-dihydro-lH-pyrrole-2,5,-dione derivatives, diaminothiazoles, bicyclic compounds, pyrazine derivatives, pyrimidine- or pyridine derivatives, and purine derivatives such as CT 98014, CT98023, CT99021, 2-amino-3-(alkyl)-pyrimidone derivatives, lH-imidazol-4-amine derivatives, and 3-indolyl-4-phenyl-lH-pyrrole-2,5-dione derivatives.
- maleimides such as SB410111, SB495052, SB517955, SB216763, SB415286, diamino-l,2,4-triazole carboxylic acid derivatives and 2,5-di
- compositions of the present invention may comprise the compound of the present invention alone or in combination with other kinase-inhibiting compounds or chemotherapeutic agents.
- Chemotherapeutic agents include, but are not limited to exemestane, formestane, anastrozole, letrozole, fadrozole, taxane and derivatives such as paclitaxel or docetaxel, encapsulated taxanes, CPT-11, camptothecin derivatives, anthracycline glycosides, e.g., doxorubicin, idarubicin, epirubicin, etoposide, navelbine, vinblastine, carboplatin, cisplatin, estramustine, celecoxib, tamoxifen, raloxifen, Sugen SU-
- the pharmaceutical compositions of the present invention may contain one or more excipients. Excipients are added to the composition for a variety of purposes. Diluents increase the bulk of a solid pharmaceutical composition, and may make a pharmaceutical dosage form containing the composition easier for the patient and caregiver to handle. Diluents for solid compositions include, for example, microcrystalline cellulose (e.g.
- Avicel ® microfme cellulose, lactose, starch, pregelatinized starch, calcium carbonate, calcium sulfate, sugar, dextrates, dextrin, dextrose, dibasic calcium phosphate dihydrate, tribasic calcium phosphate, kaolin, magnesium carbonate, magnesium oxide, maltodextrin, mannitol, polymethacrylates (e.g. Eudragit ® ), potassium chloride, powdered cellulose, sodium chloride, sorbitol and talc.
- Solid pharmaceutical compositions that are compacted into a dosage form, such as a tablet may include excipients whose functions include helping to bind the active ingredient and other excipients together after compression.
- Binders for solid pharmaceutical compositions include acacia, alginic acid, carbomer (e.g. carbopol), carboxymethylcellulose sodium, dextrin, ethyl cellulose, gelatin, guar gum, hydrogenated vegetable oil, hydroxyethyl cellulose, hydroxypropyl cellulose (e.g. Klucel ® ), hydroxypropyl methyl cellulose (e.g.
- Methocel ® liquid glucose, magnesium aluminum silicate, maltodextrin, methylcellulose, polymethacrylates, povidone (e.g. Kollidon ® , Plasdone ® ), pregelatinized starch, sodium alginate and starch.
- povidone e.g. Kollidon ® , Plasdone ®
- pregelatinized starch sodium alginate and starch.
- the dissolution rate of a compacted solid pharmaceutical composition in the patient's stomach may be increased by the addition of a disintegrant to the composition.
- Disintegrants include alginic acid, carboxymethylcellulose calcium, carboxymethylcellulose sodium (e.g. Ac-Di-Sol ® , Primellose ® ), colloidal silicon dioxide, croscarmellose sodium, crospovidone
- Glidants can be added to improve the flowability of a non-compacted solid composition and to improve the accuracy of dosing.
- Excipients that may function as glidants include colloidal silicon dioxide, magnesium trisilicate, powdered cellulose, starch, talc and tribasic calcium phosphate.
- a dosage form such as a tablet is made by the compaction of a powdered composition
- the composition is subjected to pressure from a punch and dye.
- Some excipients and active ingredients have a tendency to adhere to the surfaces of the punch and dye, which can cause the product to have pitting and other surface irregularities.
- a lubricant can be added to the composition to reduce adhesion and ease the release of the product from the dye.
- Lubricants include magnesium stearate, calcium stearate, glyceryl monostearate, glyceryl palmitostearate, hydrogenated castor oil, hydrogenated vegetable oil, mineral oil, polyethylene glycol, sodium benzoate, sodium lauryl sulfate, sodium stearyl fumarate, stearic acid, talc and zinc stearate. Flavoring agents and flavor enhancers make the dosage form more palatable to the patient. Common flavoring agents and flavor enhancers for pharmaceutical products that may be included in the composition of the present invention include maltol, vanillin, ethyl vanillin, menthol, citric acid, fumaric acid, ethyl maltol and tartaric acid.
- Solid and liquid compositions may also be dyed using any pharmaceutically acceptable colorant to improve their appearance and/or facilitate patient identification of the product and unit dosage level.
- liquid pharmaceutical compositions of the present invention the compound of formula I and any other solid excipients are dissolved or suspended in a liquid carrier such as water, vegetable oil, alcohol, polyethylene glycol, propylene glycol or glycerin.
- Liquid pharmaceutical compositions may contain emulsifying agents to disperse uniformly throughout the composition an active ingredient or other excipient that is not soluble in the liquid carrier.
- Emulsifying agents that may be useful in liquid compositions of the present invention include, for example, gelatin, egg yolk, casein, cholesterol, acacia, tragacanth, chondrus, pectin, methyl cellulose, carbomer, cetostearyl alcohol and cetyl alcohol.
- Liquid pharmaceutical compositions of the present invention may also contain a viscosity enhancing agent to improve the mouth-feel of the product and/or coat the lining of the gastrointestinal tract.
- a viscosity enhancing agent include acacia, alginic acid bentonite, carbomer, carboxymethylcellulose calcium or sodium, cetostearyl alcohol, methyl cellulose, ethylcellulose, gelatin guar gum, hydroxyethyl cellulose, hydroxypropyl cellulose, hydroxypropyl methyl cellulose, maltodextrin, polyvinyl alcohol, povidone, propylene carbonate, propylene glycol alginate, sodium alginate, sodium starch glycolate, starch tragacanth and xanthan gum.
- Sweetening agents such as sorbitol, saccharin, sodium saccharin, sucrose, aspartame, fructose, mannitol and invert sugar may be added to improve the taste.
- Preservatives and chelating agents such as alcohol, sodium benzoate, butylated hydroxy toluene, butylated hydroxyanisole and ethylenediamine tetraacetic acid may be added at levels safe for ingestion to improve storage stability.
- a liquid composition may also contain a buffer such as guconic acid, lactic acid, citric acid or acetic acid, sodium guconate, sodium lactate, sodium citrate or sodium acetate. Selection of excipients and the amounts used may be readily determined by the formulation scientist based upon experience and consideration of standard procedures and reference works in the field.
- a buffer such as guconic acid, lactic acid, citric acid or acetic acid, sodium guconate, sodium lactate, sodium citrate or sodium acetate.
- the solid compositions of the present invention include powders, granulates, aggregates and compacted compositions.
- the dosages include dosages suitable for oral, buccal, rectal, parenteral (including subcutaneous, intramuscular, and intravenous), inhalant and ophthalmic administration. The most suitable administration in any given case will depend on the nature and severity of the condition being treated.
- the dosages may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the pharmaceutical arts.
- Dosage forms include solid dosage forms like tablets, powders, capsules, suppositories, sachets, troches and lozenges, as well as liquid syrups, suspensions and elhcirs.
- the dosage form of the present invention may be a capsule containing the composition, such as a powdered or granulated solid composition of the invention, within either a hard or soft shell.
- the shell may be made from gelatin and optionally contain a plasticizer such as glycerin and sorbitol, and an opacifying agent or colorant.
- compositions and dosage forms may be formulated into compositions and dosage forms according to methods known in the art.
- a composition for tableting or capsule filling may be prepared by wet granulation,- In wet granulation, some or all of the active ingredients and excipients in powder form are blended and then further mixed in the presence of a liquid, typically water, that causes the? powders to clump into granules.
- the granulate is screened and/or milled, dried and then screened and/or milled to the desired particle size.
- the granulate may then be tableted, or- other excipients may be added prior to tableting, such as a glidant and/or a lubricant.
- a tableting composition may be prepared conventionally by dry blending.
- the blended composition of the actives and excipients may be compacted into a slug or a sheet and then comminuted into compacted granules. The compacted granules may subsequently be compressed into a tablet.
- a blended composition may be compressed directly into a compacted dosage form using direct compression, techniques.
- Direct compression produces a more uniform tablet without granules.
- Excipients that are particularly well suited for direct compression tableting include microcrystalline cellulose, spray dried lactose, dicalcium phosphate dihydrate and colloidal silica. The proper use of these and other excipients in direct compression tableting is known to those in the art with experience and skill in particular formulation challenges of direct compression tableting.
- a capsule filling of the present invention may include any of the aforementioned blends and granulates that were described with reference to tableting, however, they are not subjected to a final tableting step.
- Methods of administration of a pharmaceutical composition encompassed by the invention are not specifically restricted, and can be administered in various preparations depending on the age, sex, and symptoms of the patient.
- tablets, pills, solutions, suspensions, emulsions, granules and capsules may be orally administered.
- Injection preparations may be administered individually or mixed with injection transfusions such as glucose solutions and amino acid solutions intravenously. If necessary, the injection preparations are administered singly intramuscularly, intracutaneously, subcutaneously or intraperitoneally. Suppositories may be administered into the rectum.
- the amount of the compound of formula I contained in a pharmaceutical composition according to the present invention is not specifically restricted, however, the dose should be sufficient to treat, ameliorate, or reduce the targeted symptoms.
- the dosage of a pharmaceutical composition according to the present invention will depend on the method of use, the age, sex, and condition of the patient.
- Step 2 A mixture of sulfanilamide (0.86 g, 5 mmol) and 1-H-pyrazole-l- carboxamidine HCl (0.73g, 5 mmol) in 3 mL nitrobenzene is heated to reflux for 2 hrs. The solution is decanted from the solid that is formed. jV-butanol (8 mL), aqueous NaOH solution (0.73 mL 10N), and the crude l-(5-chloro-thiophen-2-yl)-3-dimethylamino-propenone is added to the solid. The reaction is heated to reflux overnight.
- Exemplary compounds 5-34 can also be synthesized according to this method.
- HPLC Conditions (Condition A): Hewlett Packard 1100 MSD with ChemStation Software; Xterra C 1S column, 30 mm x 2.1 mm, 5 ⁇ particle size, at 5O 0 C;
- Solvent A Water (0.02% formic acid buffer);
- Solvent B Acetonitrile (0.02 % formic acid buffer);
- Gradient Time 0: 5% B; 0.3 min: 5% B; 3.0 min: 90% B; Hold 90% B 2 min;
- Flow rate 1.0 niL/min; Detection: 254 run DAD; API-ES Scanning Mode Negative 150-700; Fragmentor 70 mV.
- Example Ib Preparation of 4-[4-(5-Pyridin-2-ylethvnyl-thiophen-2-yl)-pyrimidin-2- ylamino]-benzenesulfonamide (exemlary compound 35) See Figure 1.
- Step 1 4-[4-(5-Bromo-thiophen-2-yl)-pyrimidin-2-ylamino]-benzenesulfonamide is prepared by the procedure described in Example Ia.
- Step 2 A 10 mL glass microwave reaction vessel with stir bar contained palladium acetate (5 mg, 22 ⁇ mol), tri-o-tolylphosphine (13 mg, 44 ⁇ mol), and 4-[4-(5-bromo-thiophen- 2-yl)-pyrimidin-2-ylamino]-benzenesulfonamide (80 mg, 200 ⁇ mol).
- Anhydrous dimethylformamide (DMF) (3.5 mL), 2-ethynylpyridine (46 mg, 450 ⁇ mol), and triethylamine (50 ⁇ L) is added to the reaction vessel.
- DMF dimethylformamide
- 2-ethynylpyridine 46 mg, 450 ⁇ mol
- triethylamine 50 ⁇ L
- the reaction vessel is sealed and heated to 180°C for 660 seconds in a microwave reactor (Emrys Microwave Reactor, personal Chemistry AB, Uppsala, Sweden).
- the reaction is filtered through celite, concentrated, redissolved in dimethylsulfoxide (DMSO), and purified by reverse phase (RP) HPLC to obtain 10 mg of the title compound.
- LC/MS data Condition A; molecular ion and retention time: m/z 434 (M+H); 2.52 min.
- Exemplary compounds 36-38 can also be synthesized according to this method.
- Example 2 Preparation of A r -[ ⁇ 4-(Morpholin-4-ylsulfonyl)phenyl '
- Step 1 Preparation of 4-[(4-fluorophenyl)sulfonyl]morpholine To a solution of 4-fluorobenzenesulfonyl chloride (3.97 g, 20 mmol) in methylene chloride (40 ml), at O 0 C, under nitrogen, with stirring, is added morpholine (4.4 mL, 50 mmol). The mixture is stirred at 0°C for 15 min.
- Step 2A Preparation of iV-[4-(morpholin-4-ylsulfonyl)phenyl]guanidine
- a mixture of 4-[(4-fluorophenyl)sulfonyl]morpholine (0.25 g, 1 mmol), cesium carbonate (1.30 g, 4 mmol), and guanidine carbonate (1.08 g, 6 mmol) in 2 ml of l-methyl-2- pyrrolidinone (NMP) is stirred at 85 to 9O 0 C for 24 hrs. It is then cooled to room temperature and diluted with ether.
- Exemplary compounds 40-67 can also be synthesized according to this method.
- Example 3 Preparation of N-(3-hvdroxypropyl)-4-( ⁇ 4-[4-(trifluoromethyl)phenyl1pyrimidin- 2-vU amino Vbenzenesulfonamide (exemplary compound 68) See Figure 3.
- Step 1 Preparation of iV-phenyl-4-[4-(trifluoromethyl)phenyl]pyrimidin-2 -amine
- Step 2 Preparation of 4-( ⁇ 4-[4-(trifluoromethyl)phenyl]pyrimidin-2- yl ⁇ amino)benzenesulfonyl chloride
- a solution of N-phenyl-4-[4-(trifluoromethyl)phenyl]pyrimidin-2-amine (0.16 g, 0.5 mmol) in 1.5 ml of chlorosulfonic acid is stirred at 65 to 7O 0 C for 1 hr. It is then cooled to room temperature, and added slowly to a stirred mixture of ice and water.
- Step 3 Preparation of iV-(3-hydroxypropyl)-4-( ⁇ 4-[4- (trifluoromethyl)phenyl]pyrimidin-2-yl ⁇ amino)benzenesulfonamide
- 4-( ⁇ 4-[4-(trifiuoromethyl)phenyl]pyrimidin-2- yl ⁇ amino)benzenesulfonyl chloride (0.10 g, 0.25 mmol)
- 3- amino-1-propanol (0.19 g, 2.5 mmol) with stirring, at O 0 C.
- the mixture is stirred at room temperature for 1 hr and then quenched with water (10 ml).
- Exemplary compounds 67 can also be synthesized according to this method.
- Example 4 General experimental for the preparation of 2-anilino-4-aryl/heteroarylpyrimidine primary sulfonamides. See Figure 4.
- Aniline target molecules of structure (I) may also be prepared using the procedure first outlined by Bredereck (Bredereck, H. et al. Ber., Dtsch. Chem. Ges. 1964, 97, 3397).
- Amines (G-3) can be converted to the corresponding aryl guanidines (G-2) using pyrazole-1- carboxamidine according to the procedure of Bernatowicz (Bernatowicz, M.S. et al. J. Org. Chem. 1992, 57, 2497).
- the guanidines can be combined with 3 -dimethylamino- 1- aryl/heteroaryl-propenones (G-I), prepared by heating methyl ketones (4-3) with DMF DMA, in the presence of a base such as KOH, NaOH, or Et 3 N, or an acid such as HOAC in hot EtOH or MeOH to give the desired 2-aminopyrimidines (I).
- G-I 3 -dimethylamino- 1- aryl/heteroaryl-propenones
- a 0.1 M solution of a methyl ketone is heated at 13O 0 C for 12 h. After cooling to 23 0 C, all volatiles are evaporated. The remaining material is dissolved in a minimum of CH 2 Cl 2 and passed through as short SPE SiO 2 gel cartridge eluting with additional CH 2 Cl 2 . The eluant is concentrated to a minimum volume, and an equal amount of hexanes is added. Cooling to 5 0 C produces crystals of the title compound as a yellow or orange solid.
- Step 2 Preparation of 2-anilino-4-aryl/heteroarylpyrimidine primary sulfonamides (I) Aniline (1 equiv.) is combined with 1.5 equiv. of lH-pyrazole-1 -carboxamidine hydrochloride as a 0.1 M nitrobenzene solution and heated to 200°C for 6 h. After cooling to 23 °C, 1 equiv. of 3 -dimethylamino- 1 -aryl/heteroaryl-propenone is added followed by 1.25 equiv. of KOH, EtOH (equal volume to that of nitrobenzene) and H 2 O, (1/lOth the volume of EtOH).
- This mixture is heated at 12O 0 C for 12 h, cooled to 23°C, and evaporated in a Speed- Vac.
- This crude material is dissolved in 0.5 ml DMSO: 1.5 ml MeCN, filtered through a 0.45 ⁇ m GMF, and purified on a Gilson HPLC, using a Phenomenex LUNA C 18 column: 60 mm x 21.20 mm LD. , 5 urn particle size: with ACN/water (containing 0.2% TFA or Et 3 N) gradient elution. The appropriate fractions are analyzed by LC/MS. To isolate the title compound, the pure fractions are combined and the solvent is evaporated in a Speed- Vac.
- Exemplary compounds 1, 4, 9, 10, 12, 13, 15, 16, 18-29, 31, 32, 34, 35, 37, 38, 69, and 70 can be synthesized according to this method.
- HPLC Conditions Instrument - Agilent 1100; Column: Keystone Aquasil Cl 8 (as above); Mobile Phase A: 10 mM NH 4 OAC in 95% water / 5% CAN; Mobile Phase B: 10 niM NH 4 OAC in 5% water / 95% CAN; Flow Rate: 0.800 ml/min; Column Temperature: 4O 0 C; Injection Volume: 5 ul; UV: monitor 215, 230, 254, 280, and 300nm; Purity is reported at 254nm unless otherwise noted.
- Example 5 The enamino is added to a solution of the substituted guanidine in NMP, and the mixture is heated at 105 °C for 48 hours. The reaction is cooled to room temperature. Water is added, and the aqueous layer is extracted with EtOAc. The solvent is removed by evaporation, and the residue is purified by pre-plate with DCM/EtOAc/MeOH (8:8:1).
- Exemplary compounds 45-66 and 89-92 can be synthesized according to this method.
- Exemplary compounds 104, 107, 125, 129, 219, 294, 295, 296, 297 can also be synthesized according to this method.
- Exemplary compounds 243-244 can also be synthesized according to this method.
- Example 6 Preparation of 4-rf4- ⁇ 4-r(lE)-3-hvdroxyprop-l-en-l-yl]phenyl)pyrimidin-2- yl)amino]benzenesulfbnamide (Exemplary compound 272) See Figures 6a and 6b.
- Step 1 Tert-Butyl(dimethyl) ⁇ [(2E)-3 -(4,4,5,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2- yl)prop-2-en- 1 -yl]oxy ⁇ silane
- tert-butyl-dimethyl-prop-2-ynyloxy-silane 3.00 g, 17.6 mmol
- 4,4,5,5-tetramethyl-l,2,3-dioxaborolane (2.80 ml, 2.50 g, 19.4 mmol
- bis(cyclopentadienyl)zirconium (IV) chloride hydride 0.454 g, 1.76 mmol
- triethylamine (0.250 ml, 0.178 g, 1.76 mmol).
- Step 3 4-[(4- ⁇ 4-[(lE)-3-hydroxyprop-l-en-l-yl]phenyl ⁇ pyrimidin-2- yl)amino]benzenesulfonamide
- a vial is charged with the anilino-pyrimidine, N,N-diethyl aniline, and NMP. The mixture is cooled to 0°C, and acyl chloride is added. The reaction is warmed to room temperature and stirred for 4 hours. Water is added, and the precipitate is washed with ether, DCM.
- Exemplary compounds 221-225 can be synthesized according to this method.
- the aldehyde is dissolved in THF and cooled to 0°C.
- the amine is added, followed by Na(OAc)3BH, and the reaction is stirred at O 0 C for 15 minutes.
- HOAc is added dropwise, and the reaction is warmed to room temperature for 3 hours.
- the reaction is quenched with water.
- the product is extracted with ethyl acetate, washed with sodium bicarbonate and brine, and purified with EtOAc/MeOH (10:1).
- Exemplary compounds 132-134, 141, and 143-149 can be synthesized according to this method.
- Example 9 General experimental for the preparation of 2-anilino-4-aryl/heteroarylpyrimidme sulfonamide secondary and tertiary sulfonamides. See Figure 9.
- Amino sulfonamides (G-3) can be purchased commercially or prepared by the process depicted in Figure 9: nitrobenzenesulfonyl chlorides (9-1) can be converted to the corresponding sulfonamides (9-2) via reaction with HNR 7 R 8 in an amine solvent such as pyridine or in a polar aprotic solvent such as CH 2 Cl 2 or THF in the presence of a hindered amine base such as /-Pr 2 NEt or Et 3 N and DMAP.
- an amine solvent such as pyridine
- a polar aprotic solvent such as CH 2 Cl 2 or THF
- a hindered amine base such as /-Pr 2 NEt or Et 3 N and DMAP.
- nitrobenzenesulfonamides (9-2) can be reduced to the corresponding amines using conditions such as 10% Pd/C, NH 4 HCO 2 , MeOH, or SnCl 2 -H 2 O, EtOH, heat or Fe, HCl, EtOH, H 2 O, heat.
- Step 1 Preparation of Substituted-4-nitro-benzenesulfonamides (9-2) 1.25 eq of /-Pr 2 NEt, 0.1 equiv. of DMAP 5 and 1.25 equiv. of amine is added to 1 equiv. of 4-nitrobenzenesulfonyl chloride as a 0.1 M solution in CH 2 Cl 2 . This mixture is stirred at 23 0 C until judged complete by TLC. After quenching with sat. NaHCO 3 solution and separating the organic and aqueous layers, the organic layer is evaporated to yield nearly pure 4-nitrobenzenesulfonamides as off-white to colorless solids (Yield range: 56-100% yields).
- Step 2 Preparation of 4-amino-benzenesulfonamide secondary and tertiary sulfonamides (G-3) 0.1 wt. equiv. of 10% Pd/C and 5 equiv. of ammonium formate is added to 1 eq of a
- Step 3 Preparation of 2-chloro-4-aryl/heteroaryl-pyrimidine (G-5)
- a -30 0 C solution of a Ar/HetLi (10.66 mmol, 1.08 eq, generated via deprotonation of Li for Br exchange) in 20 ml Of Et 2 O is added portion wise a suspension of 2- chloropyrimidine (9.84 mmol, 1 equiv.) in 20 ml Et 2 O in 2 ml portions over 15 min.
- the resulting suspension is stirred for 30 min at -30 0 C and at 0 0 C for 60 rnin.
- Aniline target molecules of structure (I) can be prepared by reacting 2- chloropyrimidine (9-4) with aryl or heteroaryllithiums, prepared by reacting aryl bromides/heteroaryl bromides with a strong base such as «-BuLi, MeLi or PhLi or via deprotonation of aryls/heteroaryls with a strong base such as n-BuLi, IsAeLi, PhLi, LDA, or LiN(TMS) 2 , followed by oxidation with DDQ to give 4-aryl/heteroaryl-2-chloropyrimidines (G-5) according to the procedures of Czarny and Harden.
- aryl or heteroaryllithiums prepared by reacting aryl bromides/heteroaryl bromides with a strong base such as «-BuLi, MeLi or PhLi or via deprotonation of aryls/heteroaryls with a strong base
- UV Detector Agilent 1100 Diode Array Detector
- MS Parameters Mass Range 100 - 1000, Fragmentor 140, Gain EMV 1.0
- Exemplary compounds 2, 3, 71-79, 86, and 87 can be synthesized according to this method.
- HPLC Conditions Instrument - Agilent 1100; Column: Keystone Aquasil Cl 8 (as above); Mobile Phase A: 10 niM NH 4 OAC in 95% water / 5% CAN; Mobile Phase B: IO mM NH 4 OAC in 5% water / 95% CAN; Flow Rate: 0.800 ml/min; Column Temperature : 40 °C; Injection Volume: 5 ul; UV: monitor 215, 230, 254, 280, and 300nm; Purity is reported at 254nm unless otherwise noted.
- Example 10 General experimental for the preparation of 2-NfMe)-anilino-4- aryl/heteroarylpyrimidine sulfonamides. See Figure 10.
- 4-Methylaminobenzene sulfonamides (10-6) are prepared according to the process depicted in Figure 10.
- N-methyl acetamide (10-1) can be converted to sulfonyl chloride (10- 2) according to the procedure of Stojanovic (Stojanovic, O. K. et al. Chem. Abstr. 1973, 3902) using neat ClSO 3 H.
- the sulfonyl chloride is converted to the corresponding sulfonamides (10-3) using amines, NaOAc in EtOH, and NaOH hydrolysis of the acetyl group to produce the desired 4-methylaminobenzene sulfonamides (10-4) according to the procedure of Oinuma (Oinuma, H.
- N-Methylaminosulfonamide analogs can be prepared according to the process depicted in Figure 10. 4-aryl/heteroaryl-2-chloropyrimidines (10-5) are combined with 4- methylaminobenzene sulfonamides (10-4) in hot dioxane in the presence ofp-TsOH ⁇ 2 O to give the desired N-methylaminosulfonamide sulfonamides (10-6).
- Step 1 4-(Acetyl-methyl-arnino)-benzenesulfonyl chloride (10-2) (Based on the procedure of O. K. Stojanovic et al. Chem. Abstr. 1973, 78, 3902s.) N-
- Step 4 The protocol described in Example 9, Step 4 is used except that 4-methylamino- benzenesulfonamides are used in place of primary amino-benzenesulfonamides.
- Exemplary compounds 80-85 can be synthesized according to this method.
- HPLC Conditions Instrument - Agilent 1100; Column: KLeystone Aquasil Cl 8 (as above); Mobile Phase A: 10 mM NH 4 OAC in 95% water / 5% CAN; Mobile Phase B: 10 mM NH 4 OAC in 5% water / 95% CAN; Flow Rate: 0.800 ml/min; Column Temperature: 4O 0 C; Injection Volume: 5 ul; UV: monitor 215, 230, 254, 280, and 300nm; Purity is reported at 254nm unless otherwise noted.
- MS Conditions Instrument: Agilent MSD; Ionization Mode: API-ES; Gas Temperature: 350 C; Drying Gas: 11.0 L/min.; Nebulizer Pressure: 55psig; Polarity: 50% positive, 50% negative; VCap: 3000V (positive), 2500V (negative); Fragmentor: 80 (positive), 120 (negative); Mass Range: 100 - lOOOm/z; ThreshoLd: 150; Step size: 0.15; Gain: 1 ; Peak width: 0.15min
- Example 11 The starting materials are combined in a vial in dioxane and stirred at 90°C overnight, then cooled to the room temperature. 50% NaHCO 3 is added, and the reaction is stirred for 10 minutes. The precipitate is filtered, then dissolved in THF, and purified by pre-plate with THF/MeOH (10:1). See Figure 11. Exemplary compound 184 can be synthesized according to this method.
- a halogenated (Br) sulfonamide (G-4) is reacted with a pyrimidine (G-6) by adding sodium tert-butoxide (NaOtBu) in the presence of tris(dibenzylideneacetone)dipalladium(0) (Pd 2 dba 3 ) and 2,2'-bis(diphenylphosphino)-l,l'-binaphthyl (BINAP).
- Exemplary compounds 115-118, 123, 127, and 128 can be synthesized according to this method.
- Example 13 Sodium t-butoxide is added to a stirred suspension of anilino-pyr ⁇ midines, substituted sulfonamides, tris(dibenzylideneacetone)dipalladium(0), and 2,2'-bis(diphenylphosphino)- 1,1 '-binaphthyl in dioxane. The mixture is heated at 80 0 C for 50 hours. The reaction is cooled to room temperature, and the mixture is filtered and washed with THF and MeOH. The solvent is removed by evaporation, and the residue is purified by pre-plate with EtOAc/MeOH (10:l).
- Exemplary compounds 130, 131, 139, 161-164, 166-169, 171-174, and 299 can be synthesized according to this method.
- Example 14 Sodium t-butoxide is added to a stirred suspension of anilino-pyrimidines, substituted sulfones, tris(dibenzylideneacetone)dipalladium(0), and 2,2'-bis(diphenylphosphino)-l,l '- binaphthyl in dioxane. The mixture is heated at 80°C for 72 hours. The reaction is cooled to room temperature, and the mixture is filtered and washed with THF and MeOH. The solvent is removed by evaporation, and the residue is purified by pre-plate with EtOAc/MeOH (10:1.5).
- Exemplary compounds 98-103 can be synthesized according to this method.
- Human IKK ⁇ cDNA is amplified by reverse transcriptase-polymerase chain reaction from human placental RNA (CLONTECH) using primers that incorporated the FLAG- epitope at the C terminus of IKK ⁇ .
- FLAG- IKK ⁇ is inserted into the baculovirus expression plasmid pF ASTB AC (Life Technologies).
- BAC-TO-BAC Baculovirus Expression System
- recombinant baculoviruses expressing the IKK ⁇ enzyme are made. Briefly, 9 X 10 5 SF9 cells per well of a 6-well plate are transfected with one ⁇ g of miniprep bacmid DNA using the CeIlFECTIN TM reagent. Virus is harvested 72 hours post transfection, and a viral titer is performed, after which a high titer viral stock (2 x 10 8 pfu/ml) is amplified by three to four rounds of infection.
- the pellets are thawed on ice and resuspended in cell lysis buffer (50 mM HEPES, pH 7.5, 100 mM NaCl, 1 % NP-40, 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail from Pharmingen).
- cell lysis buffer 50 mM HEPES, pH 7.5, 100 mM NaCl, 1 % NP-40, 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail from Pharmingen.
- cell lysis buffer 50 mM HEPES, pH 7.5, 100 mM NaCl, 1 % NP-40, 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail from Pharming
- the resulting supernatant is loaded onto an anti-FLAG M2 agarose affinity column (Sigma) at 4°C and the column is washed with 60 mL of wash buffer (50 mM HEPES, pH 7.5, 300 mM NaCl 5 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, and 1 mM PMSF).
- wash buffer 50 mM HEPES, pH 7.5, 300 mM NaCl 5 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, and 1 mM PMSF.
- the FLAG-IKK ⁇ is eluted using 200 ⁇ g/mL Flag peptide (Sigma) in elution buffer (50 mM HEPES, pH 7.5, 100 mMNaCl, 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail from Pharmingen) in 500 ⁇ L aliquots, which are tested for protein levels using SDS-PAGE followed by Coomassie Blue staining (BioRad). After testing for activity as described below, fractions with high IKK enzyme activity are combined, aliquotted, and stored at -80°C.
- elution buffer 50 mM HEPES, pH 7.5, 100 mMNaCl, 10% glycerol, 1 mM Na 3 VO 4 , 1 mM EDTA, 1 mM DTT, and protease inhibitor cocktail from Pharmingen
- LANCE reactions are carried out based upon the suggestions of Wallac/Perkin Elmer.
- Purified Flag-IKK ⁇ enzyme (2 nM final concentration) is typically used in the kinase reaction buffer described above supplemented with 0.0025% Brij solution (Sigma) to help stabilize the enzyme.
- Biotinylated substrate I ⁇ B ⁇ (1-54) is synthesized and purified (> 95% pure) and is used at 500 nM final concentration.
- ATP is used at a final concentration of 2 ⁇ M.
- the total reaction volumes are 25 ⁇ L and the inhibitor compounds are preincubated with enzyme before substrate and ATP are added. Reactions are conducted for 30 minutes at room temperature in black, low binding plates (Dynex).
- HeIa cells are plated at 6- well plate for 24 hours and treated with compounds for 30 min before the addition of TNF ⁇ (10 ng/ml). After one hour, HeIa cells are harvested in MPER reagent (Pierce, Rockford, IL) containing 400 mM NaCl. Protein from all samples is quantified by the Bradford method. Cell lysates containing 30 ⁇ g of protein are electrophoresed on a 12% SDS-PAGE gel and transferred to a PVDF membrane using a Bio Rad liquid transfer apparatus. The PVDF membrane is incubated in TBST (TBS with 0.1% Tween-20) with 3% milk for 15 minutes before the addition of the first antibody, mouse anti- I ⁇ B ⁇ (Santa Cruz).
- the PVDF membrane is washed 3 times with TBST and incubated with secondary antibodies coupled with horseradish peroxidase (Transduction Labs) for one hour.
- the PVDF membrane is then washed 3 times with TBST and protein is detected using an enhanced chemiluminescence detection system (Pierce).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Physical Education & Sports Medicine (AREA)
- Oncology (AREA)
- Virology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Endocrinology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Reproductive Health (AREA)
- Molecular Biology (AREA)
- Obesity (AREA)
- Urology & Nephrology (AREA)
- Biotechnology (AREA)
- Vascular Medicine (AREA)
- Psychiatry (AREA)
Abstract
Description





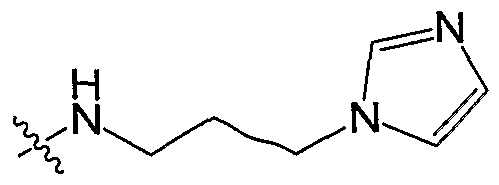








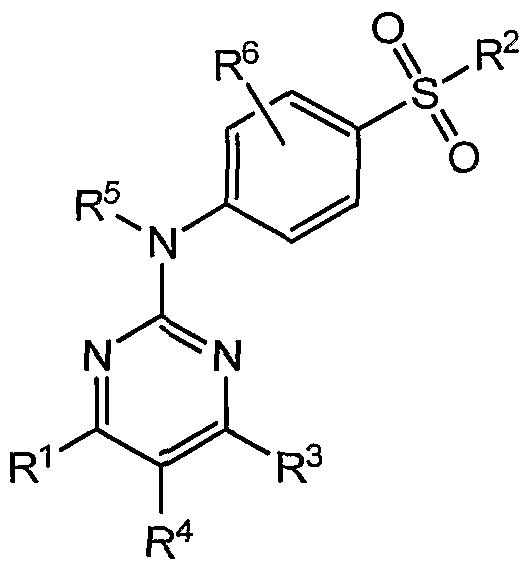





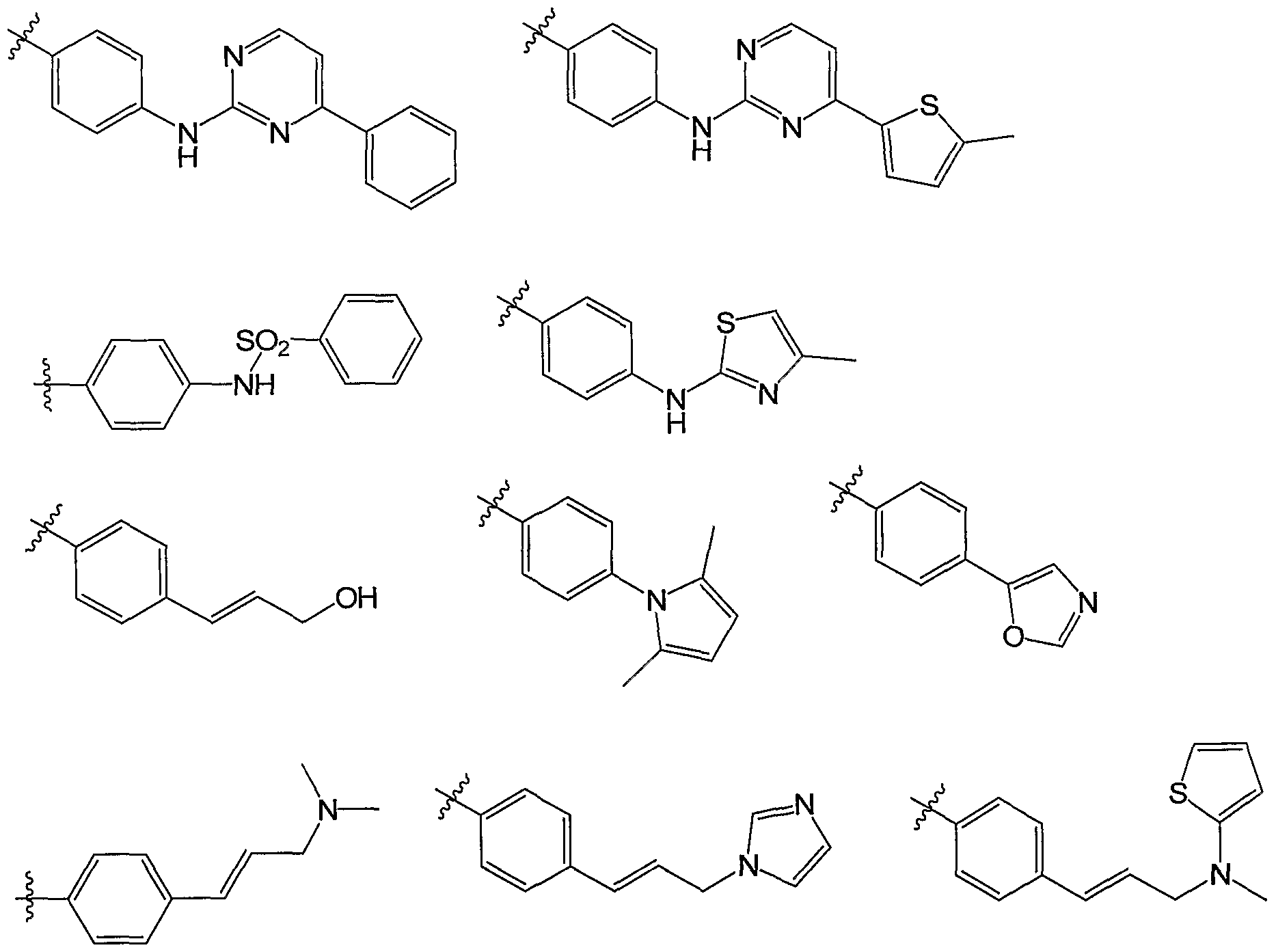

























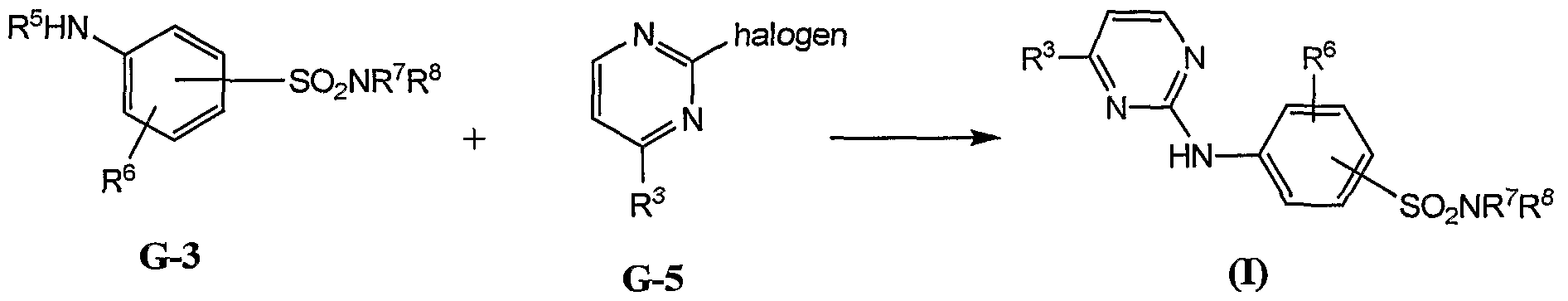

Claims





















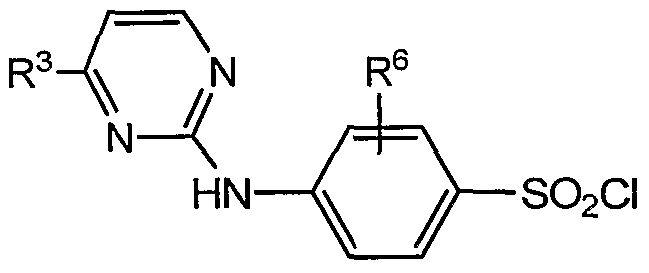










Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0516597-0A BRPI0516597A (en) | 2004-10-13 | 2005-10-13 | compound of the formula |
| MX2007004488A MX2007004488A (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogs. |
| CA002580913A CA2580913A1 (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
| AU2005295788A AU2005295788A1 (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
| JP2007536838A JP2008515986A (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogues |
| EP05812654A EP1799652A1 (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
| NO20071642A NO20071642L (en) | 2004-10-13 | 2007-03-28 | N-Benzenesulfonyl-substituted anilino-pyrimidine analogs |
| IL182316A IL182316A0 (en) | 2004-10-13 | 2007-03-29 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61766804P | 2004-10-13 | 2004-10-13 | |
| US60/617,668 | 2004-10-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006044457A1 true WO2006044457A1 (en) | 2006-04-27 |
Family
ID=35945218
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/036674 Ceased WO2006044457A1 (en) | 2004-10-13 | 2005-10-13 | N-benzenesulfonyl substituted anilino-pyrimidine analogs |
Country Status (20)
| Country | Link |
|---|---|
| US (1) | US7799915B2 (en) |
| EP (1) | EP1799652A1 (en) |
| JP (1) | JP2008515986A (en) |
| KR (1) | KR20070084067A (en) |
| CN (1) | CN101039919A (en) |
| AR (1) | AR051387A1 (en) |
| AU (1) | AU2005295788A1 (en) |
| BR (1) | BRPI0516597A (en) |
| CA (1) | CA2580913A1 (en) |
| CR (1) | CR9048A (en) |
| GT (1) | GT200500286A (en) |
| IL (1) | IL182316A0 (en) |
| MX (1) | MX2007004488A (en) |
| NO (1) | NO20071642L (en) |
| PA (1) | PA8649401A1 (en) |
| PE (1) | PE20060582A1 (en) |
| RU (1) | RU2007114080A (en) |
| TW (1) | TW200626559A (en) |
| WO (1) | WO2006044457A1 (en) |
| ZA (1) | ZA200703022B (en) |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007053776A1 (en) * | 2005-11-03 | 2007-05-10 | Sgx Pharmaceuticals, Inc. | Pyrimidinyl-thiophene kinase modulators |
| WO2007120593A1 (en) * | 2006-04-12 | 2007-10-25 | Wyeth | Anilino-pyrimidine phenyl and benzothiophene analogs |
| WO2008011560A3 (en) * | 2006-07-20 | 2008-03-27 | Mehmet Kahraman | Benzothiophene inhibitors of rho kinase |
| FR2911138A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New N,N'-2,4-dianilino-pyrimidine derivatives, are kinase inhibitors, especially IKK inhibitors, useful for treating inflammatory diseases, diabetes and cancers |
| FR2911140A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New 2-anilino-4-heteroaryl-pyrimidine derivatives, are kinase inhibitors, especially IKK inhibitors, useful for treating inflammatory diseases, diabetes and cancers |
| FR2911139A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New 2,4-diaminopyrimidine derivatives useful for treating inflammatory diseases, diabetes or cancer |
| JP2010533652A (en) * | 2007-07-13 | 2010-10-28 | アイカジェン, インコーポレイテッド | Sodium channel inhibitor |
| JP2010538076A (en) * | 2007-09-04 | 2010-12-09 | ザ スクリプス リサーチ インスティチュート | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| JP2011506281A (en) * | 2007-12-05 | 2011-03-03 | ビーエーエスエフ ソシエタス・ヨーロピア | Pyridylmethyl-sulfonamide compound |
| JP2011512380A (en) * | 2008-02-22 | 2011-04-21 | エフ.ホフマン−ラ ロシュ アーゲー | Modulator of amyloid β |
| US8486941B2 (en) | 2007-03-12 | 2013-07-16 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| WO2014106762A1 (en) * | 2013-01-07 | 2014-07-10 | Vichem Chemie Kutató Kft. | 4-pyrimidinylamino-benzenesulfonamide derivatives and their use for the inhibition of polo-like kinase 1 (plk1) for the treatment of cancer and their use for the treatment of bacterial infections |
| US8809359B2 (en) | 2012-06-29 | 2014-08-19 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine bicyclic compounds and uses thereof |
| USRE48285E1 (en) | 2014-06-12 | 2020-10-27 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| WO2023150197A1 (en) * | 2022-02-03 | 2023-08-10 | Nexys Therapeutics, Inc. | Aryl hydrocarbon receptor agonists and uses thereof |
| USRE50528E1 (en) | 2014-10-13 | 2025-08-12 | Yuhan Corporation | Compounds and compositions for modulating EGFR mutant kinase activities |
Families Citing this family (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7442697B2 (en) * | 2002-03-09 | 2008-10-28 | Astrazeneca Ab | 4-imidazolyl substituted pyrimidine derivatives with CDK inhibitory activity |
| GB0205693D0 (en) | 2002-03-09 | 2002-04-24 | Astrazeneca Ab | Chemical compounds |
| GB0205690D0 (en) * | 2002-03-09 | 2002-04-24 | Astrazeneca Ab | Chemical compounds |
| GB0205688D0 (en) * | 2002-03-09 | 2002-04-24 | Astrazeneca Ab | Chemical compounds |
| GB0311274D0 (en) * | 2003-05-16 | 2003-06-18 | Astrazeneca Ab | Chemical compounds |
| GB0311276D0 (en) * | 2003-05-16 | 2003-06-18 | Astrazeneca Ab | Chemical compounds |
| TW200528101A (en) * | 2004-02-03 | 2005-09-01 | Astrazeneca Ab | Chemical compounds |
| US7279288B2 (en) * | 2004-09-03 | 2007-10-09 | California Institute Of Technology | I kappa B kinase complex as a target for the treatment of Huntington's disease |
| NZ555474A (en) * | 2004-12-17 | 2010-10-29 | Astrazeneca Ab | 4-(4-(imidazol-4-yl) pyrimidin-2-ylamino) benzamides as CDK inhibitors |
| MX2008001428A (en) * | 2005-07-30 | 2008-04-04 | Astrazeneca Ab | Imidazolyl-pyrimidine compounds for use in the treatment of proliferative disorders. |
| WO2007036732A1 (en) * | 2005-09-30 | 2007-04-05 | Astrazeneca Ab | Imidazo [1,2-a] pyridine having anti-cell-proliferation activity |
| GB0520955D0 (en) * | 2005-10-14 | 2005-11-23 | Cyclacel Ltd | Compound |
| TW200811169A (en) * | 2006-05-26 | 2008-03-01 | Astrazeneca Ab | Chemical compounds |
| AR063946A1 (en) * | 2006-09-11 | 2009-03-04 | Cgi Pharmaceuticals Inc | CERTAIN REPLACED PIRIMIDINS, THE USE OF THE SAME FOR THE TREATMENT OF DISEASES MEDIATED BY THE INHIBITION OF THE ACTIVITY OF BTK AND PHARMACEUTICAL COMPOSITIONS THAT UNDERSTAND THEM. |
| US20110117121A1 (en) * | 2006-10-27 | 2011-05-19 | James Dao | Compositions for treatment and inhibition of pain |
| AU2016200866B2 (en) * | 2007-03-12 | 2017-06-22 | Glaxosmithkline Llc | Phenyl amino pyrimidine compounds and uses thereof |
| AU2013201306B2 (en) * | 2007-03-12 | 2015-11-12 | Glaxosmithkline Llc | Phenyl Amino Pyrimidine Compounds and Uses Thereof |
| BRPI0810411B8 (en) * | 2007-04-18 | 2021-05-25 | Pfizer Prod Inc | sulfonyl amide derivatives for the treatment of abnormal cell growth, their sos, as well as pharmaceutical composition |
| EP2148874B1 (en) * | 2007-05-04 | 2012-02-08 | Irm Llc | Pyrimidine derivatives and compositions as c-kit and pdgfr kinase inhibitors |
| US20090048282A1 (en) * | 2007-08-14 | 2009-02-19 | Wyeth | Pyrimidine sulfonamide analogs and their use as agonists of the wnt-beta-catenin cellular messaging system |
| CA2697081C (en) | 2007-08-22 | 2013-04-23 | Irm Llc | 5-(4-(haloalkoxy)phenyl)pyrimidine-2-amine compounds and compositions as kinase inhibitors |
| CA2697077C (en) * | 2007-08-22 | 2012-10-16 | Irm Llc | 2-heteroarylamino-pyrimidine derivatives as kinase inhibitors |
| JP2011518769A (en) * | 2008-03-14 | 2011-06-30 | ビーエーエスエフ ソシエタス・ヨーロピア | Substituted pyrazinylmethylsulfonamides for use as fungicides |
| US9273077B2 (en) | 2008-05-21 | 2016-03-01 | Ariad Pharmaceuticals, Inc. | Phosphorus derivatives as kinase inhibitors |
| PT2300013T (en) | 2008-05-21 | 2017-10-31 | Ariad Pharma Inc | Phosphorous derivatives as kinase inhibitors |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| TWI546290B (en) | 2008-06-27 | 2016-08-21 | 賽基艾維洛米斯研究股份有限公司 | Heteroaryl compound and its use |
| US11351168B1 (en) | 2008-06-27 | 2022-06-07 | Celgene Car Llc | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| US8513242B2 (en) | 2008-12-12 | 2013-08-20 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| KR101705158B1 (en) | 2009-05-05 | 2017-02-09 | 다나-파버 캔서 인스티튜트 인크. | Egfr inhibitors and methods of treating diseases |
| WO2010151747A1 (en) * | 2009-06-26 | 2010-12-29 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimine compounds and methods of making and using same |
| CN101991553B (en) * | 2009-08-21 | 2015-02-25 | 北京以岭生物工程技术有限公司 | Exemestane tablet and preparation method thereof |
| WO2011046970A1 (en) * | 2009-10-12 | 2011-04-21 | Myrexis, Inc. | Amino - pyrimidine compounds as inhibitors of tbkl and/or ikk epsilon |
| FR2954317B1 (en) * | 2009-12-23 | 2012-01-27 | Galderma Res & Dev | NOVEL PHENOLIC DERIVATIVES, AND THEIR PHARMACEUTICAL OR COSMETIC USE |
| AU2010339531A1 (en) * | 2009-12-30 | 2012-08-23 | Arqule, Inc. | Substituted naphthalenyl-pyrimidine compounds |
| US8334292B1 (en) | 2010-06-14 | 2012-12-18 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| GB201012105D0 (en) | 2010-07-19 | 2010-09-01 | Domainex Ltd | Novel pyrimidine compounds |
| CN105566229A (en) | 2010-08-10 | 2016-05-11 | 西建阿维拉米斯研究公司 | Besylate salt of a BTK inhibitor, application and preparation method thereof |
| EP2635285B1 (en) | 2010-11-01 | 2017-05-03 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| MX382354B (en) | 2010-11-01 | 2025-03-13 | Celgene Car Llc | HETEROCYCLIC COMPOUNDS AND THEIR USES. |
| JP5957003B2 (en) | 2010-11-10 | 2016-07-27 | セルジーン アヴィロミクス リサーチ, インコーポレイテッド | Mutant selective EGFR inhibitor and use thereof |
| US9834518B2 (en) | 2011-05-04 | 2017-12-05 | Ariad Pharmaceuticals, Inc. | Compounds for inhibiting cell proliferation in EGFR-driven cancers |
| AR088570A1 (en) | 2011-10-28 | 2014-06-18 | Celgene Avilomics Res Inc | METHODS TO TREAT AN ILLNESS OR DISORDER RELATED TO BRUTON TYROSINE KINASE |
| WO2013096744A1 (en) | 2011-12-21 | 2013-06-27 | Novira Therapeutics, Inc. | Hepatitis b antiviral agents |
| KR102081042B1 (en) | 2012-03-15 | 2020-02-26 | 셀젠 카르 엘엘씨 | Solid forms of an epidermal growth factor receptor kinase inhibitor |
| PL2825042T3 (en) | 2012-03-15 | 2019-02-28 | Celgene Car Llc | Salts of an epidermal growth factor receptor kinase inhibitor |
| US20150166591A1 (en) | 2012-05-05 | 2015-06-18 | Ariad Pharmaceuticals, Inc. | Methods and compositions for raf kinase mediated diseases |
| EA038942B1 (en) | 2012-08-28 | 2021-11-12 | Янссен Сайенсиз Айрлэнд Юси | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| US9126950B2 (en) | 2012-12-21 | 2015-09-08 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| AU2014214846A1 (en) | 2013-02-08 | 2015-07-23 | Celgene Avilomics Research, Inc. | ERK inhibitors and uses thereof |
| HUE034820T2 (en) | 2013-02-28 | 2018-02-28 | Janssen Sciences Ireland Uc | Sulfamoyl-arylamides and the use thereof as medicaments for the treatment of hepatitis b |
| CA2903103C (en) | 2013-03-15 | 2020-06-09 | Discoverybiomed, Inc. | Coumarin derivatives and methods of use in treating cystic fibrosis, chronic obstructive pulmonary disease, and misfolded protein disorders |
| KR20150132483A (en) | 2013-03-15 | 2015-11-25 | 디스커버리바이오메드 인코포레이티드 | Coumarin derivatives and methods of use in treating hyperproliferative diseases |
| EA027068B1 (en) | 2013-04-03 | 2017-06-30 | Янссен Сайенсиз Айрлэнд Юси | N-phenyl-carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| US9611283B1 (en) | 2013-04-10 | 2017-04-04 | Ariad Pharmaceuticals, Inc. | Methods for inhibiting cell proliferation in ALK-driven cancers |
| TWI651300B (en) | 2013-05-17 | 2019-02-21 | 健生科學愛爾蘭無限公司 | Amidoximepyrroleamine derivatives and their use as medicaments for the treatment of hepatitis B |
| EA035500B1 (en) | 2013-05-17 | 2020-06-25 | Янссен Сайенсиз Айрлэнд Юси | Sulphamoylthiophenamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| DK3024819T3 (en) | 2013-07-25 | 2018-06-06 | Janssen Sciences Ireland Uc | GLYOXAMIDE-SUBSTITUTED PYRROLAMIDE DERIVATIVES AND THE USE THEREOF AS MEDICINES FOR TREATING HEPATITIS B |
| US9492471B2 (en) | 2013-08-27 | 2016-11-15 | Celgene Avilomics Research, Inc. | Methods of treating a disease or disorder associated with Bruton'S Tyrosine Kinase |
| KR102290189B1 (en) | 2013-10-23 | 2021-08-17 | 얀센 사이언시즈 아일랜드 언리미티드 컴퍼니 | Carboxamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| US9415049B2 (en) | 2013-12-20 | 2016-08-16 | Celgene Avilomics Research, Inc. | Heteroaryl compounds and uses thereof |
| US10392349B2 (en) | 2014-01-16 | 2019-08-27 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| WO2015118057A1 (en) | 2014-02-06 | 2015-08-13 | Janssen Sciences Ireland Uc | Sulphamoylpyrrolamide derivatives and the use thereof as medicaments for the treatment of hepatitis b |
| US10005760B2 (en) | 2014-08-13 | 2018-06-26 | Celgene Car Llc | Forms and compositions of an ERK inhibitor |
| CN106188029B (en) * | 2015-05-05 | 2018-09-18 | 山东轩竹医药科技有限公司 | Two and ring class anaplastic lymphoma kinase inhibitor |
| US10875876B2 (en) | 2015-07-02 | 2020-12-29 | Janssen Sciences Ireland Uc | Cyclized sulfamoylarylamide derivatives and the use thereof as medicaments for the treatment of hepatitis B |
| KR101767602B1 (en) * | 2016-04-04 | 2017-08-14 | 한국원자력의학원 | Novel N-phenylpyrimidin-2-amine derivatives compound, Manufacturing method thereof, Pharmaceutical composition for preventing and treating cancer containing the same and Methods for preventing and treating cancer using the same |
| SG10202011827YA (en) | 2016-04-15 | 2021-01-28 | Novira Therapeutics Inc | Combinations and methods comprising a capsid assembly inhibitor |
| ES2837157T3 (en) * | 2016-06-30 | 2021-06-29 | Janssen Pharmaceutica Nv | Cyanoindoline derivatives as NIK inhibitors |
| KR102585048B1 (en) * | 2017-01-17 | 2023-10-05 | 아스트라제네카 아베 | JAK1 selective inhibitor |
| CN106946888B (en) * | 2017-04-06 | 2018-06-01 | 福建拓路建设有限公司 | A kind of sulfoamido derivative and its purposes in antitumor drug is prepared |
| EP3749697A4 (en) | 2018-02-05 | 2021-11-03 | Bio-Rad Laboratories, Inc. | CHROMATOGRAPHIC RESIN WITH A LIGAND WITH ANION EXCHANGE-HYDROPHOBIC MIXED MODE |
| CA3090125A1 (en) | 2018-03-14 | 2019-09-19 | Janssen Sciences Ireland Unlimited Company | Capsid assembly modulator dosing regimen |
| CN109438365B (en) * | 2018-12-06 | 2022-04-05 | 华南师范大学 | N- (3- ((4-trifluoromethyl) -2-pyrimidinyl) aminophenyl) -2, 6-difluorobenzenesulfonamide derivative |
| WO2020150668A1 (en) * | 2019-01-17 | 2020-07-23 | Board Of Trustees Of Michigan State University | Compositions and methods for immune modulation and treatment of cancer |
| MX2021010145A (en) | 2019-02-22 | 2021-09-14 | Janssen Sciences Ireland Unlimited Co | Amide derivatives useful in the treatment of hbv infection or hbv-induced diseases. |
| CN109912584B (en) * | 2019-03-22 | 2021-08-13 | 中国药科大学 | A kind of BRD4 protein inhibitor with antitumor activity and its preparation method and application |
| MX2021013531A (en) * | 2019-05-05 | 2022-02-11 | Qilu Regor Therapeutics Inc | CDK INHIBITORS. |
| MA55879A (en) | 2019-05-06 | 2022-03-16 | Janssen Sciences Ireland Unlimited Co | AMIDE DERIVATIVES USEFUL IN THE TREATMENT OF HEPATITIS B VIRUS INFECTION OR DISEASES INDUCED BY HEPATITIS B VIRUS |
| CN114901072B (en) | 2019-12-16 | 2024-12-17 | 埃科莱布美国股份有限公司 | Effect of anionic surfactants on virucidal efficacy |
| CN111574413B (en) * | 2020-06-08 | 2022-04-05 | 杭州尚合生物医药科技有限公司 | A kind of preparation method of sulfonyl amidine with 2-aminomethylpyridine and DMF-DMA as amine source |
| CN114014847B (en) * | 2021-12-08 | 2023-11-03 | 滨州医学院 | Benzothiophene pyrimidine derivative, preparation method thereof and application thereof in preparation of antitumor drugs |
| CN120157658B (en) * | 2025-03-07 | 2026-01-09 | 辽宁大学 | Aminopyrimidine compound containing toluene structure and preparation method and application thereof |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR829926A (en) * | 1936-11-28 | 1938-07-11 | Ig Farbenindustrie Ag | Ortho and para-aminoarylsulfones and their preparation process |
| CH210790A (en) * | 1938-01-31 | 1940-07-31 | Chem Ind Basel | Process for the preparation of a benzene sulfamide derivative. |
| US3775168A (en) * | 1972-02-28 | 1973-11-27 | Cities Service Co | Process for protecting a substrate with an n{11 -pyrimidylamino-arylsulfonamide intumercent agent |
| WO2001012621A1 (en) * | 1999-08-13 | 2001-02-22 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF c-JUN N-TERMINAL KINASES (JNK) AND OTHER PROTEIN KINASES |
| WO2001014375A1 (en) * | 1999-08-21 | 2001-03-01 | Astrazeneca Ab | Imidazo[1,2-a]pyridine and pyrazolo[2,3-a]pyridine derivatives |
| WO2001064654A1 (en) * | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | Pyrimidine compounds |
| WO2002020512A1 (en) * | 2000-09-05 | 2002-03-14 | Astrazeneca Ab | Imidazolo-5-yl-2-anilino-pyrimidines as agents for the inhibition of the cell proliferation |
| WO2002066481A1 (en) * | 2001-02-17 | 2002-08-29 | Astrazeneca Ab | Pyrimidine derivatives for inhibition of cell-proliferation |
| WO2002083668A1 (en) * | 2001-04-10 | 2002-10-24 | Vertex Pharmaceuticals Incorporated | Isoxaxole derivatives as inhibitors of src and other protein kinases |
| WO2002092573A2 (en) * | 2001-05-16 | 2002-11-21 | Vertex Pharmaceuticals Incorporated | Heterocyclic substituted pyrazoles as inhibitors of src and other protein kinases |
| WO2002096905A1 (en) * | 2001-06-01 | 2002-12-05 | Vertex Pharmaceuticals Incorporated | Thiazole compounds useful as inhibitors of protein kinases |
| WO2003074515A1 (en) * | 2002-03-01 | 2003-09-12 | Smithkline Beecham Corporation | Diamino-pyrimidines and their use as angiogenesis inhibitors |
| WO2003076434A1 (en) * | 2002-03-09 | 2003-09-18 | Astrazeneca Ab | 4- imidazolyl substuited pyrimidine derivatives with cdk inhibitiory activity |
| WO2004043953A1 (en) * | 2002-11-14 | 2004-05-27 | Cyclacel Limited | Pyrimidine compounds |
| WO2004043467A1 (en) * | 2002-11-14 | 2004-05-27 | Cyclacel Limited | Anti-viral compounds |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4876252A (en) * | 1986-01-13 | 1989-10-24 | American Cyanamid Company | 4,5,6-substituted-N-(substituted-phenyl)-2-pyrimidinamines |
| US4788195A (en) * | 1986-01-13 | 1988-11-29 | American Cyanamid Company | 4,5,6-substituted-N-(substituted-phenyl)-2-pyrimidinamines |
| JPH0796543B2 (en) * | 1987-08-11 | 1995-10-18 | サントリー株式会社 | Acylaminobenzenesulfonamide derivative and herbicide containing the same as an active ingredient |
| PL313973A1 (en) * | 1993-10-12 | 1996-08-05 | Du Pont Merck Pharma | 1 n-alkyl-n-arylopyrimidin amines and their derivatives |
| US5786354A (en) * | 1994-06-21 | 1998-07-28 | Celltech Therapeutics, Limited | Tri-substituted phenyl derivatives and processes for their preparation |
| GB9526245D0 (en) * | 1995-12-21 | 1996-02-21 | Celltech Therapeutics Ltd | Chemical compounds |
| GB2302021A (en) | 1996-10-16 | 1997-01-08 | Lilly Co Eli | Inhibiting bone loss or resorption |
| GB9622363D0 (en) * | 1996-10-28 | 1997-01-08 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9705361D0 (en) * | 1997-03-14 | 1997-04-30 | Celltech Therapeutics Ltd | Chemical compounds |
| CA2287387C (en) * | 1997-05-14 | 2010-02-16 | Sloan-Kettering Institute For Cancer Research | Methods and compositions for destruction of selected proteins |
| GB9914258D0 (en) * | 1999-06-18 | 1999-08-18 | Celltech Therapeutics Ltd | Chemical compounds |
| DE19942354A1 (en) * | 1999-09-04 | 2001-03-08 | Aventis Pharma Gmbh | Substituted 3-phenyl-5-alkoxi-1,3,4-oxdiazol-2-one, their manufacture and use in medicinal products |
| GB9924862D0 (en) * | 1999-10-20 | 1999-12-22 | Celltech Therapeutics Ltd | Chemical compounds |
| CA2419301C (en) * | 2000-08-21 | 2009-12-08 | Astrazeneca Ab | Quinazoline derivatives |
| US7122544B2 (en) | 2000-12-06 | 2006-10-17 | Signal Pharmaceuticals, Llc | Anilinopyrimidine derivatives as IKK inhibitors and compositions and methods related thereto |
| EP1343782B1 (en) * | 2000-12-21 | 2009-05-06 | SmithKline Beecham Corporation | Pyrimidineamines as angiogenesis modulators |
| US20020132823A1 (en) * | 2001-01-17 | 2002-09-19 | Jiahuai Han | Assay method |
| EP1423388B1 (en) * | 2001-02-20 | 2008-12-03 | AstraZeneca AB | 2-arylamino-pyrimidines for the treatment of gsk3-related disorders |
| EP1373257B9 (en) * | 2001-03-29 | 2008-10-15 | Vertex Pharmaceuticals Incorporated | Inhibitors of c-jun n-terminal kinases (jnk) and other protein kinases |
| MXPA03009378A (en) * | 2001-04-13 | 2004-01-29 | Vertex Pharma | Inhibitors of c-jun n-terminal kinases (jnk) and other protein kinases. |
| KR100874791B1 (en) * | 2001-05-29 | 2008-12-18 | 바이엘 쉐링 파마 악티엔게젤샤프트 | CDV-inhibited pyrimidine, preparation method thereof and use as medicament |
| US6825190B2 (en) * | 2001-06-15 | 2004-11-30 | Vertex Pharmaceuticals Incorporated | Protein kinase inhibitors and uses thereof |
| US6939874B2 (en) * | 2001-08-22 | 2005-09-06 | Amgen Inc. | Substituted pyrimidinyl derivatives and methods of use |
| US6703414B2 (en) * | 2001-09-14 | 2004-03-09 | Arizona Board Of Regents On Behalf Of The University Of Arizona | Device and method for treating restenosis |
| CA2476281A1 (en) * | 2002-02-08 | 2003-08-14 | Smithkline Beecham Corporation | Pyrimidine compounds |
| GB0215844D0 (en) * | 2002-07-09 | 2002-08-14 | Novartis Ag | Organic compounds |
| JP2006512314A (en) * | 2002-11-01 | 2006-04-13 | バーテックス ファーマシューティカルズ インコーポレイテッド | Use of the compositions as JAK inhibitors and other protein kinase inhibitors |
| EP1560824A1 (en) * | 2002-11-05 | 2005-08-10 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of jak and other protein kinases |
-
2005
- 2005-10-13 MX MX2007004488A patent/MX2007004488A/en unknown
- 2005-10-13 BR BRPI0516597-0A patent/BRPI0516597A/en not_active IP Right Cessation
- 2005-10-13 TW TW094135620A patent/TW200626559A/en unknown
- 2005-10-13 EP EP05812654A patent/EP1799652A1/en not_active Withdrawn
- 2005-10-13 GT GT200500286A patent/GT200500286A/en unknown
- 2005-10-13 CN CNA2005800349350A patent/CN101039919A/en not_active Withdrawn
- 2005-10-13 JP JP2007536838A patent/JP2008515986A/en active Pending
- 2005-10-13 PA PA20058649401A patent/PA8649401A1/en unknown
- 2005-10-13 AR ARP050104293A patent/AR051387A1/en not_active Application Discontinuation
- 2005-10-13 RU RU2007114080/04A patent/RU2007114080A/en not_active Application Discontinuation
- 2005-10-13 WO PCT/US2005/036674 patent/WO2006044457A1/en not_active Ceased
- 2005-10-13 AU AU2005295788A patent/AU2005295788A1/en not_active Abandoned
- 2005-10-13 CA CA002580913A patent/CA2580913A1/en not_active Abandoned
- 2005-10-13 US US11/248,495 patent/US7799915B2/en not_active Expired - Fee Related
- 2005-10-13 PE PE2005001207A patent/PE20060582A1/en not_active Application Discontinuation
- 2005-10-13 KR KR1020077010445A patent/KR20070084067A/en not_active Withdrawn
-
2007
- 2007-03-28 NO NO20071642A patent/NO20071642L/en not_active Application Discontinuation
- 2007-03-29 IL IL182316A patent/IL182316A0/en unknown
- 2007-04-11 CR CR9048A patent/CR9048A/en unknown
- 2007-04-12 ZA ZA200703022A patent/ZA200703022B/en unknown
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR829926A (en) * | 1936-11-28 | 1938-07-11 | Ig Farbenindustrie Ag | Ortho and para-aminoarylsulfones and their preparation process |
| CH210790A (en) * | 1938-01-31 | 1940-07-31 | Chem Ind Basel | Process for the preparation of a benzene sulfamide derivative. |
| US3775168A (en) * | 1972-02-28 | 1973-11-27 | Cities Service Co | Process for protecting a substrate with an n{11 -pyrimidylamino-arylsulfonamide intumercent agent |
| WO2001012621A1 (en) * | 1999-08-13 | 2001-02-22 | Vertex Pharmaceuticals Incorporated | INHIBITORS OF c-JUN N-TERMINAL KINASES (JNK) AND OTHER PROTEIN KINASES |
| WO2001014375A1 (en) * | 1999-08-21 | 2001-03-01 | Astrazeneca Ab | Imidazo[1,2-a]pyridine and pyrazolo[2,3-a]pyridine derivatives |
| WO2001064654A1 (en) * | 2000-03-01 | 2001-09-07 | Astrazeneca Ab | Pyrimidine compounds |
| WO2002020512A1 (en) * | 2000-09-05 | 2002-03-14 | Astrazeneca Ab | Imidazolo-5-yl-2-anilino-pyrimidines as agents for the inhibition of the cell proliferation |
| WO2002066481A1 (en) * | 2001-02-17 | 2002-08-29 | Astrazeneca Ab | Pyrimidine derivatives for inhibition of cell-proliferation |
| WO2002083668A1 (en) * | 2001-04-10 | 2002-10-24 | Vertex Pharmaceuticals Incorporated | Isoxaxole derivatives as inhibitors of src and other protein kinases |
| WO2002092573A2 (en) * | 2001-05-16 | 2002-11-21 | Vertex Pharmaceuticals Incorporated | Heterocyclic substituted pyrazoles as inhibitors of src and other protein kinases |
| WO2002096905A1 (en) * | 2001-06-01 | 2002-12-05 | Vertex Pharmaceuticals Incorporated | Thiazole compounds useful as inhibitors of protein kinases |
| WO2003074515A1 (en) * | 2002-03-01 | 2003-09-12 | Smithkline Beecham Corporation | Diamino-pyrimidines and their use as angiogenesis inhibitors |
| WO2003076434A1 (en) * | 2002-03-09 | 2003-09-18 | Astrazeneca Ab | 4- imidazolyl substuited pyrimidine derivatives with cdk inhibitiory activity |
| WO2004043953A1 (en) * | 2002-11-14 | 2004-05-27 | Cyclacel Limited | Pyrimidine compounds |
| WO2004043467A1 (en) * | 2002-11-14 | 2004-05-27 | Cyclacel Limited | Anti-viral compounds |
Non-Patent Citations (2)
| Title |
|---|
| ANDERSON, MALCOLM ET AL: "Imidazo[1,2a]pyridines: A potent and selective class of cyclin-dependent kinase inhibitors identified through structure-based hybridisation", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 13, no. 18, 15 September 2003 (2003-09-15), pages 3021 - 3026, XP002372577 * |
| CROSS, PETER ET AL: "Cerebrovasodilatation through selective inhibition of the enzyme carbonic anhydrase. 1. Substituted benzenedisulfonamides", J.MED.CHEM., vol. 21, no. 9, 1 September 1978 (1978-09-01), pages 845 - 849, XP002372578 * |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007053776A1 (en) * | 2005-11-03 | 2007-05-10 | Sgx Pharmaceuticals, Inc. | Pyrimidinyl-thiophene kinase modulators |
| US7977481B2 (en) | 2005-11-03 | 2011-07-12 | Sgx Pharmaceuticals, Inc. | Pyrimidinyl-thiophene kinase modulators |
| US7803806B2 (en) | 2005-11-03 | 2010-09-28 | Sgx Pharmaceuticals, Inc. | Pyrimidinyl-thiophene kinase modulators |
| WO2007120593A1 (en) * | 2006-04-12 | 2007-10-25 | Wyeth | Anilino-pyrimidine phenyl and benzothiophene analogs |
| WO2008011560A3 (en) * | 2006-07-20 | 2008-03-27 | Mehmet Kahraman | Benzothiophene inhibitors of rho kinase |
| WO2008099075A1 (en) * | 2007-01-05 | 2008-08-21 | Sanofi-Aventis | 2-anilino-4-heteroaryl pyrimidine derivatives, and preparation thereof as medicaments, pharmaceutical compositions, and in particular ikk inhibitors |
| WO2008099074A1 (en) * | 2007-01-05 | 2008-08-21 | Sanofi-Aventis | Phenyl- (4-phenyl-pyrimidin-2-yl) - amine derivatives as ikk inhibitors, preparation thereof, and pharmaceutical compositions thereof |
| FR2911139A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New 2,4-diaminopyrimidine derivatives useful for treating inflammatory diseases, diabetes or cancer |
| WO2008099073A1 (en) * | 2007-01-05 | 2008-08-21 | Sanofi-Aventis | N, n' -2, 4-dianilinopyrimidines preparation and use thereof as ikk inhibitors preparation and teh pharmacuetical compositions thereof |
| FR2911140A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New 2-anilino-4-heteroaryl-pyrimidine derivatives, are kinase inhibitors, especially IKK inhibitors, useful for treating inflammatory diseases, diabetes and cancers |
| FR2911138A1 (en) * | 2007-01-05 | 2008-07-11 | Sanofi Aventis Sa | New N,N'-2,4-dianilino-pyrimidine derivatives, are kinase inhibitors, especially IKK inhibitors, useful for treating inflammatory diseases, diabetes and cancers |
| US12570614B2 (en) | 2007-03-12 | 2026-03-10 | Glaxosmith Kline Llc | Phenyl amino pyrimidine compounds and uses thereof |
| US8486941B2 (en) | 2007-03-12 | 2013-07-16 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| CN101861313B (en) * | 2007-03-12 | 2014-06-04 | Ym生物科学澳大利亚私人有限公司 | Phenylaminopyrimidine compounds and uses thereof |
| CN104030990B (en) * | 2007-03-12 | 2017-01-04 | Ym生物科学澳大利亚私人有限公司 | Phenyl amino pyrimidine compounds and application thereof |
| US9238628B2 (en) | 2007-03-12 | 2016-01-19 | YM Biosicences Australia PTY LTD | Phenyl amino pyrimidine compounds and uses thereof |
| US9233934B2 (en) | 2007-03-12 | 2016-01-12 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine compounds and uses thereof |
| JP2010533652A (en) * | 2007-07-13 | 2010-10-28 | アイカジェン, インコーポレイテッド | Sodium channel inhibitor |
| JP2010538076A (en) * | 2007-09-04 | 2010-12-09 | ザ スクリプス リサーチ インスティチュート | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| US9018205B2 (en) | 2007-09-04 | 2015-04-28 | The Scripps Research Institute | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| JP2015007102A (en) * | 2007-09-04 | 2015-01-15 | ザ スクリプス リサーチ インスティテュート | Substituted pyrimidinyl-amines as protein kinase inhibitors |
| JP2011506281A (en) * | 2007-12-05 | 2011-03-03 | ビーエーエスエフ ソシエタス・ヨーロピア | Pyridylmethyl-sulfonamide compound |
| US8962834B2 (en) | 2008-02-22 | 2015-02-24 | Hoffmann-La Roche Inc. | Modulators of amyloid beta |
| JP2011512380A (en) * | 2008-02-22 | 2011-04-21 | エフ.ホフマン−ラ ロシュ アーゲー | Modulator of amyloid β |
| US8809359B2 (en) | 2012-06-29 | 2014-08-19 | Ym Biosciences Australia Pty Ltd | Phenyl amino pyrimidine bicyclic compounds and uses thereof |
| WO2014106762A1 (en) * | 2013-01-07 | 2014-07-10 | Vichem Chemie Kutató Kft. | 4-pyrimidinylamino-benzenesulfonamide derivatives and their use for the inhibition of polo-like kinase 1 (plk1) for the treatment of cancer and their use for the treatment of bacterial infections |
| USRE48285E1 (en) | 2014-06-12 | 2020-10-27 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| USRE49445E1 (en) | 2014-06-12 | 2023-03-07 | Sierra Oncology, Inc. | N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| USRE50497E1 (en) | 2014-06-12 | 2025-07-22 | Glaxosmithkline Llc | (N-(cyanomethyl)-4-(2-(4-morpholinophenylamino)pyrimidin-4-yl)benzamide |
| USRE50528E1 (en) | 2014-10-13 | 2025-08-12 | Yuhan Corporation | Compounds and compositions for modulating EGFR mutant kinase activities |
| WO2023150197A1 (en) * | 2022-02-03 | 2023-08-10 | Nexys Therapeutics, Inc. | Aryl hydrocarbon receptor agonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0516597A (en) | 2008-09-16 |
| ZA200703022B (en) | 2009-04-29 |
| KR20070084067A (en) | 2007-08-24 |
| NO20071642L (en) | 2007-06-01 |
| TW200626559A (en) | 2006-08-01 |
| GT200500286A (en) | 2006-06-13 |
| EP1799652A1 (en) | 2007-06-27 |
| US20060079543A1 (en) | 2006-04-13 |
| JP2008515986A (en) | 2008-05-15 |
| PA8649401A1 (en) | 2006-09-22 |
| CA2580913A1 (en) | 2006-04-27 |
| MX2007004488A (en) | 2007-09-11 |
| AR051387A1 (en) | 2007-01-10 |
| US7799915B2 (en) | 2010-09-21 |
| RU2007114080A (en) | 2008-11-27 |
| IL182316A0 (en) | 2007-07-24 |
| CN101039919A (en) | 2007-09-19 |
| AU2005295788A1 (en) | 2006-04-27 |
| PE20060582A1 (en) | 2006-08-17 |
| CR9048A (en) | 2007-08-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7799915B2 (en) | Anilino-pyrimidine analogs | |
| US20070244140A1 (en) | Anilino-pyrimidine phenyl and benzothiophene analogs | |
| JP5692747B2 (en) | Novel triazine derivative and pharmaceutical composition containing the same | |
| AU2002220195B2 (en) | Anilinopyrimidine derivatives as IKK inhibitors and compositions and methods related thereto | |
| CN103124730B (en) | Heterocyclic alkyne benzene compounds and their pharmaceutical compositions and applications | |
| JP6075621B2 (en) | Novel heterocyclic derivatives and pharmaceutical compositions containing them | |
| JP5637562B2 (en) | Novel pyrrolinone derivative and pharmaceutical composition containing the same | |
| RU2625791C2 (en) | Derivatives of 1,2,4-triazine-4-amine | |
| US20040106627A1 (en) | Dihydroxypyrimidine carboxylic acids as viral polymerase inhibitors | |
| CN104003940B (en) | 2,4-difluoro-5-( phthalazone-1-methyl)-benzoyl piperazine compound and application thereof | |
| EP1701944B1 (en) | 2-(amino-substituted)-4-aryl pyramidines and related compounds useful for treating inflammatory diseases | |
| CN102099036A (en) | Compounds and methods for treating inflammatory and fibrotic disorders | |
| TW200922932A (en) | Heterocyclic compound and pharmaceutical composition thereof | |
| CN101443318A (en) | Heterobicyclic carboxamides as inhibitors for kinases | |
| CN112313207B (en) | Cyano-substituted pyridine and cyano-substituted pyrimidine compounds, and preparation methods and applications thereof | |
| CN103739550B (en) | 2,3-dimethyl-6-urea-2H-indazole compounds and its preparation method and application | |
| CN114539263B (en) | Nitrogen-containing heterocyclic compound, and pharmaceutical composition and application thereof | |
| RU2771819C2 (en) | 1,2,4-triazine-3-amine derivatives | |
| HK1138280A (en) | 2-(amino-substituted)-4-phenyl pyrimidines useful for treating inflammatory diseases |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2580913 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 182316 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005812654 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007500746 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 554369 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2696/DELNP/2007 Country of ref document: IN Ref document number: CR2007-009048 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007536838 Country of ref document: JP Ref document number: 200580034935.0 Country of ref document: CN Ref document number: 07036277 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/004488 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077010445 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005295788 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007114080 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2005295788 Country of ref document: AU Date of ref document: 20051013 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005295788 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005812654 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0516597 Country of ref document: BR |