WO2006050501A2 - Novobiocin analogues as anticancer agents - Google Patents
Novobiocin analogues as anticancer agents Download PDFInfo
- Publication number
- WO2006050501A2 WO2006050501A2 PCT/US2005/039990 US2005039990W WO2006050501A2 WO 2006050501 A2 WO2006050501 A2 WO 2006050501A2 US 2005039990 W US2005039990 W US 2005039990W WO 2006050501 A2 WO2006050501 A2 WO 2006050501A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compounds
- methoxy
- alkyl
- hydrogen
- chromen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C1C(*)C(*)(*)OC(*c(ccc2c3)cc2nc(N(*)C2=*)c3N2*#I)C1* Chemical compound *C1C(*)C(*)(*)OC(*c(ccc2c3)cc2nc(N(*)C2=*)c3N2*#I)C1* 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H17/00—Compounds containing heterocyclic radicals directly attached to hetero atoms of saccharide radicals
- C07H17/04—Heterocyclic radicals containing only oxygen as ring hetero atoms
- C07H17/06—Benzopyran radicals
- C07H17/065—Benzo[b]pyrans
- C07H17/075—Benzo[b]pyran-2-ones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- the present invention is directed to the synthesis and identification of novobiocin analogues useful as a class of anticancer agents and/or neuroprotective agents.
- the compounds of the present invention act by inhibition of the Hsp90 protein-folding machinery.
- Hsp90 The 90 kDa heat shock proteins
- the Hsp90 family of chaperones is comprised of four different isoforms.
- Hsp90 ⁇ inducible/major form
- Hsp90 ⁇ constitutive/minor form
- GFP94 94-kDa glucose-regulated protein
- TRAP-I Hsp75/tumour necrosis factor receptor associated protein 1
- Hsp90 is an ATP-dependent protein with an ATP binding site in the N-terminal region of the active homodimer. Disruption of the ATPase activity of Hsp90 results in the destabilization of multiprotein complexes and subsequent ubiquitination of the client protein, which undergoes proteasome-mediated hydrolysis. More specifically, in an ATP-dependent fashion, Hsp70 binds to newly synthesized proteins cotranslationally and/or posttranslationally to stabilize the nascent peptide by preventing aggregation.
- Hsp70-Hsp90 organizing protein contains highly conserved tetratricopeptide repeats ("TPRs") that are recognized by both Hsp70 and Hsp90, promoting the union of Hsp70/HEP and Hsp90, which results in a heteroprotein complex.
- TPRs tetratricopeptide repeats
- the client protein is transferred from the Hsp70 system to the Hsp90 homodimer with concomitant release of Hsp70, HIP, and HOP.
- the ensemble Upon binding of ATP and an immunophilin with cisltrans peptidyl prolyl-isomerase activity (FKBP51, FKBP52, or CyPA), the ensemble folds the client protein into its three-dimensional structure, hi a subsequent event, p23 binds Hsp90 near the TV-terminal region promoting the hydrolysis of ATP and release of the folded protein, Hsp90 partner proteins, and ADP.
- FKBP51, FKBP52, or CyPA an immunophilin with cisltrans peptidyl prolyl-isomerase activity
- proteins dependent upon Hsp90 for conformational maturation include oncogenic and cellular Src kinases (v-Src, Hck, Lck), Raf, pi 85, mutant p53 (not normal p53), telomerase, steroid hormone receptors, polo-like kinase (“PLK”), protein kinase B (“AKT”), death domain kinase (“RIP”), MET kinase, focal adhesion kinase (“FAK”), aryl hydrocarbon receptor, RNA-dependent protein kinase (“PKR”), nitric oxide synthase (“NOS”), centrosomal proteins, PI3 kinases, androgen receptor (“AR”), matrix metalloproteinase-2 (“MMP2”) and others, hi addition, other proteins, such as cyclin dependent kinase 4 (“CDK4"), cyclin dependent kinase 6 (“CDK6”), estrogen receptor, human epidermal growth factor receptor
- Hsp90 client proteins Raf, PLK, RIP, AKT, FAK, telomerase, HER-2, and MET kinase are directly associated with the six hallmarks of cancer: (1) self-sufficiency in growth signals; (2) insensitivity to antigrowth signals; (3) evasion of apoptosis; (4) unlimited replication potential; (5) sustained angiogenesis; and (6) tissue invasion/metastasis. Consequently, Hsp90 is a target for the development of cancer therapeutics because multiple signaling pathways can be simultaneously inhibited by disruption of the Hsp90 protein folding machinery.
- Hsp90 contains two nucleotide-binding sites: the N-terminal ATP binding site is the region to which geldanamycin ("GDA”), 17-(allylamino)-17-demethoxygeldanamycin (“17-AAG”), herbimycin A (“HB”), and radicicol bind (see Roe et al., Structural Basis for Inhibition of the Hsp90 Molecular Chaperone by the Antitumor Antibiotics Radicicol and Geldanamycin, J. Med. Chem.
- Hsp90 The C-terminal portion of Hsp90 is required for dimerization and represents a promising target for inhibitors.
- the ability of novobiocin to cause degradation of Hsp90 clients is relatively weak (about 700 ⁇ M in SKBr3 breast cancer cells).
- these new Hsp90 inhibitors have decreased toxicity, increased solubility, and/or increased selectivity for Hsp90.
- Hsp90 inhibitors of the present invention will be useful as neuroprotective agents.
- the accumulation of protein aggregates within or outside neurons is a common characteristic of the two most common age-related neurodegenerative diseases, Alzheimer's disease, with plaques enriched in ⁇ -amyloid peptides ("A ⁇ ”) and neurofibrillary tangles ("NFTs”) containing hyperphsophorylated Tau protein, and Parkinson's disease (“PD”) with Lewy bodies composed primarily of fibrillar ⁇ -synuclein.
- a ⁇ ⁇ -amyloid peptides
- NFTs neurofibrillary tangles
- PD Parkinson's disease
- Hsp90 and the cochaperones Hsp70 and CHIP carboxy-terminus of the Hsp70-interacting protein
- Hsp70 and CHIP carboxy-terminus of the Hsp70-interacting protein
- the present invention is directed to novel compounds useful as Hsp90 inhibitors, and in particular as anti-cancer an neuroprotective agents.
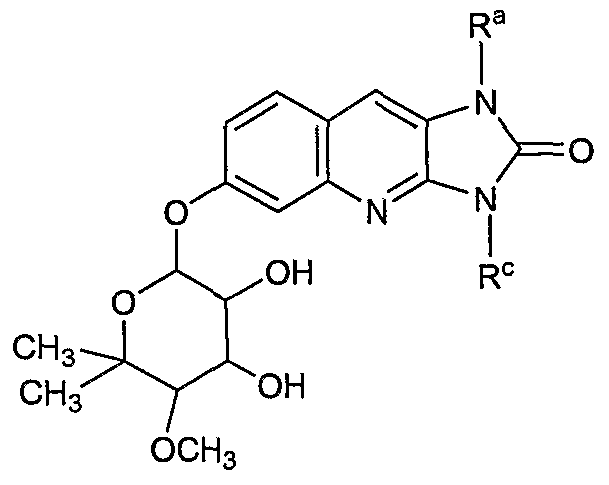
- the invention encompasses compounds according to Formula I
- R 1 is hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, aralkyl, carboxyl, amido, amino, sulfanyl, sulfenyl, sulfonyl, or ether; or R 1 together with X 2 and the atom to which R 1 is attached form a heterocyclic ring having 4 to 8 ring members with at least one heteroatom selected from oxygen or nitrogen; or R 1 together with X 4 and the atom to which R 1 is attached form a heterocyclic ring having 4 to 8 ring members with at least one heteroatom selected from oxygen or nitrogen; wherein R 2 is hydrogen, hydroxy, or -R 8 -OR 9 , wherein R 8 is a covalent bond or alkyl, and R 9 is C-amido or acyl; or R 2 together with R 3 and the atoms to which they are attached form a heterocyclic ring having 4 to 8 ring members with at least one heteroatom
- the present invention is directed to compounds of Formula I that are isocoumarin compounds wherein X 1 is -CO- and X 2 is -O- .
- the present invention is directed to des(dimethyl) derivatives and analogues of novobiocin in which R 4 and R 5 are both hydrogen.
- the present invention is directed to desmethoxy derivatives and analogues of novobiocin in which R 6 is hydrogen.
- the present invention is directed to compounds according to the Formula I(F):
- R a , R b and R c are independently hydrogen, alkyl, alken_yl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl; and wherein R b may also be oxidized to form a carbonyl.
- the invention encompasses compounds according to the Formula I(F)(i):
- R a and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl.
- the present invention is directed to compounds according to the Formula I(F)(ii): wherein X 4 , X 5 , X 6 , and X 8 are defined as set forth above, and wherein R a , R 1* and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, o ⁇ aralkyl; and wherein R may also be oxidized to form a carbonyl.
- the present invention is directed to compounds encompassed by the Formula I(F)(iii):
- the present invention is directed to compounds according to the Formula I(G):
- X 5 , X 6 , n are defined as set forth above, and wherein R a , R b and R Q are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl; and wherein R b may also be oxidized to form a carbonyl.
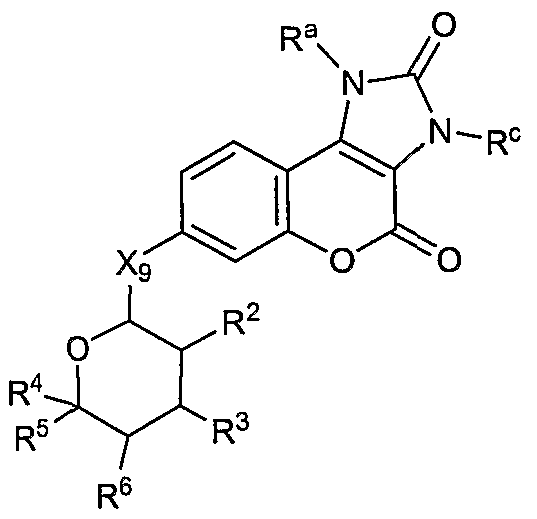
- the present invention is directed to compounds according to the Formula I(G)(i): wherein X 9 , R 2 , R 3 , R 4 , R 5 , R 6 are defined as set forth above, and wherein R a and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl.
- the present invention is directed to compounds according to Formula I(G)(ii):
- X 5 , X 6 , and X 8 are defined as set forth above, and wherein ,R >a , ⁇ R > b and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl; and wherein R b may also be oxidized to form a carbonyl.
- the present invention is directed to compounds according to Formula I(G)(iii):
- the present invention is directed to compounds according to Formula I(H): wherein X 4 , X 6 , X 8 , X 9 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , and n are defined as set forth above.
- the invention comprises compounds according to the Formula I(H)(i):
- the present invention is directed to compounds according to Formula I in which R 2 and R 3 form a cyclic carbonate. In still another aspect, the present invention is directed to compounds according to Formula I in which the sugar ring is modified to include a diol at R 2 and R 3 .
- the present invention is directed to compounds according to Formula I in which sugar is modified to include a 2'-carbamate at R 2 .
- the present invention is directed to the compounds of Formula I in which the coumarin ring is modified to include a lower alkoxy or nitro substitution at the 6-position of the coumarin ring.
- the present invention encompasses compounds according to Formula I(J):
- X 1 , X 2 , X 4 , X 5 , X 8 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , and n are defined as set forth above; and wherein R a and R b are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocyclic, aryl, or aralkyl; and wherein R b may also be oxidized to form a carbonyl.
- the present invention is directed to compounds according Formula I(J)(i):
- X 1 , X 2 , X 4 , X 5 , X 8 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , n, and R a are defined as set forth above.
- the present invention is directed to compounds according to the Formula I( J)(U):
- X 1 , X 2 , X 4 , X 5 , X 8 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , n, and R a are defined as set forth above.
- the present invention is directed to compounds encompassed by Formula I(K) :
- R 18 is hydrogen, alkyl, alkenyl, or alkynyl.
- the present invention is directed to 4-deshydroxy derivatives and analogues of novobiocin in which X 4 is -CR 20 - and R 20 is hydrogen.
- the present invention is directed to 8-desmethyl derivatives and analogues of novobiocin in which X 8 is -CR 22 - and R 22 is hydrogen.
- the present invention encompasses compounds according to the Formula I(L):
- X 9 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , and n are defined as set forth, above.
- the novobiocin derivatives and analogues of the present invention are modified so that the sugar is modified as set forth below:
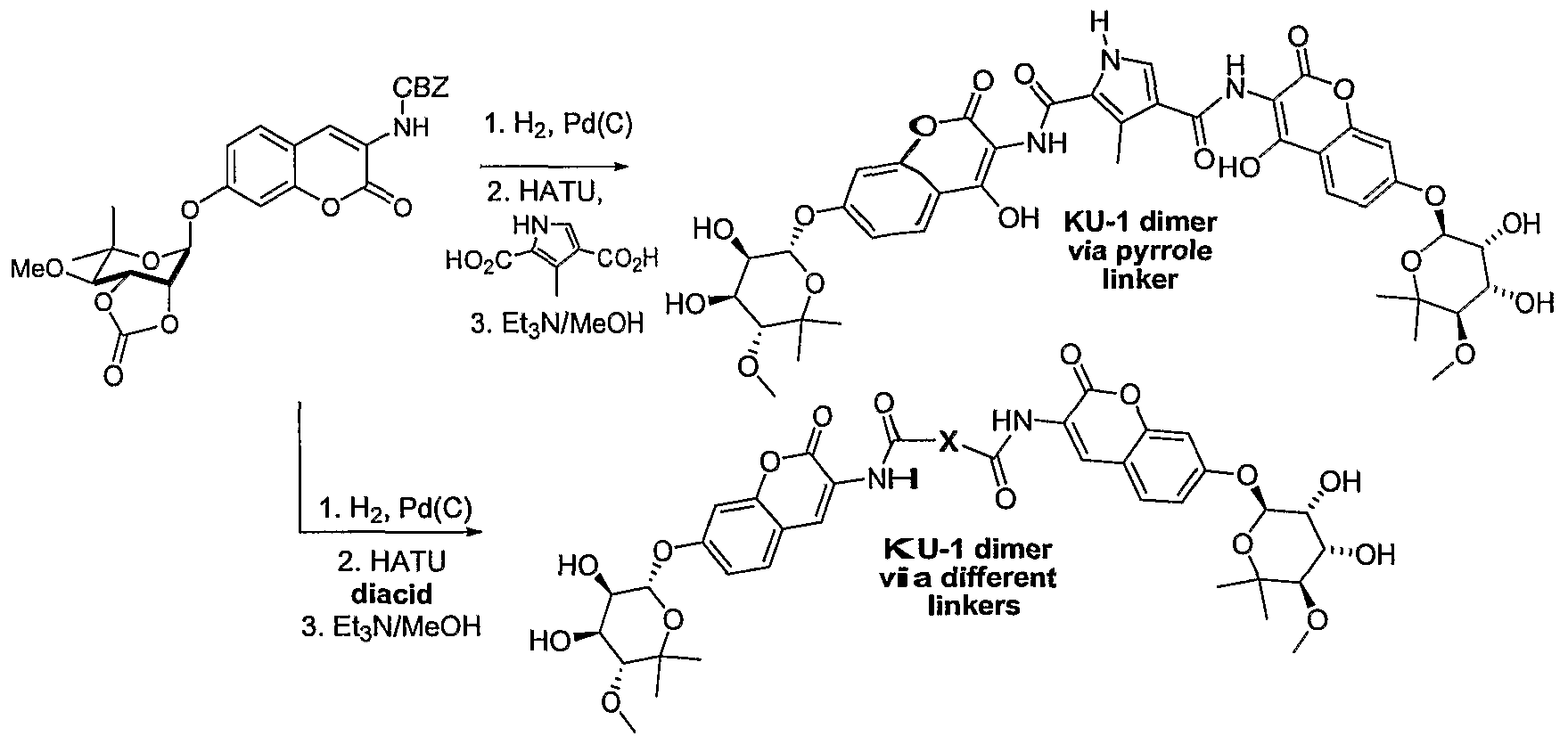
- the present invention is directed to dimers of the foregoing compounds.
- exemplary dimers are provided by the formula:
- X is alkyl, alkenyl, alkynyl, aryl, alkylaryl, carbocyclic or heterocyclic; and wherein R 2 , R 3 , R 4 , R 5 , and R 6 are set forth above.
- the present invention comprises the in wt ⁇ ich the linker is a heterocylic pyrole as shown below:
- the present invention provides a pharmaceutical composition, which comprises a therapeutically-effective amount of one or more compounds of the present invention or a pharmaceutically-acceptable salt, ester or prodrug thereof, together with a pharmaceutically-acceptable diluent or carrier.
- FIG. 1 shows the relative ratios of phospho-AKT by Western, blot analyses when the compounds of Example 1 were tested for their ability to inhibit Hsp90 in Skbr3 breast cancer cells. Total protein concentration of each lysate was determined and equal amounts of protein were run in each lane of the gels. For the graphs shown in FIG. 1 , the O.D.'s (optical density) of the Western bands for phospho-AKT were measured, as were the O.D.'s for actin probed as controls on the same blots. To obtain the graphed values, all specific O.D.'s (for Hsp90 clients) were normalized to the respective actin O.D.
- O.D.'s optical density
- FIG. 2 is a western blot analysis of Skbr3 cells treated with novobiocin analogue denominated herein as KU-3/A2 (2'-carbamate) and KU-1/A4 (diol) for 24 hours. After incubation, the cells were harvested, lysed, and equal amounts of the protein lysates loaded into SDS wells. After electrophoresis, the gel was probed with Her-2 and actin (control) antibodies. The specific decrease in Her-2 levels is a result of Hs ⁇ 90 inhibition that leads to Her-2 degradation.
- FIG. 3 (top panel) is a western blot analysis of prostate cancer LNCaP cells treated with KU-1/A4.
- the bottom panel is a western blot analysis of prostate cancer LAPC-4 cells incubated with KU-1/A4. Actin was used as a control in both assays.
- FIG. 4 shows the dose dependent effects of KU-I /A4 on A ⁇ -indx ⁇ ced cell death in primary neurons.
- the compound was added two hours before the ⁇ A and th.e viability was determined at 4-8 hours.
- the data represents standard error of the means ("S.E.M.") from about 1500 cells from 3 preparations. #, pO.OOOl for control vs. A ⁇ only. **, p ⁇ 0. 001. A ⁇ only vs. A ⁇ + KU-l/A4.
- S.E.M. standard error of the means
- acyl refers to -COR wherein R used in this definition is hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl. Most preferably, R is hydrogen, alkyl, aryl, or aralkyl.
- amido indicates either a C-amido group such as -CONR'R" or an N- amido group such as -NR'COR" wherein R 1 and R" as used in this definition are independently
- a "sulfoamido" group includes the -NR'-SO 2 -R". Most preferably, R' and R" are hydrogen, alkyl, aryl, or aralkyl.
- amino signifies a primary, secondary or tertiary amino group of the formula -NR 1 R" wherein R' and R" as used in this definition are independently hydrogen,
- amino includes unsubstituted, monosubstituted (e.g., monoalkylamino or monoarylamino), and disubstituted (e.g., dialkylamino or aralkylamino) amino groups.
- Amino groups include — NH 2 , methylamino,
- exemplary "amino" groups forming a ring include pyrrolyl, imidazolyl, pyrazolyl, isothiazolyl, isoxazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, indolizinyl, isoindolyl, indolyl, indazolyl, purinyl, quinolizinyl.
- the ring containing the amino group may be optionally substituted with another amino, alkyl, alkenyl, alkynyl, halo, or
- alkyl refers to a branched or unbranched saturated hydrocarbon group of 1 to 24 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, t- butyl, octyl, decyl, tetradecyl, hexadecyl, eicosyl, tetracosyl and the like.
- Preferred “alkyl” groups herein contain 1 to 12 carbon atoms. Most preferred are "lower alkyl” which refer to an alkyl group of one to six, more preferably one to four, carbon atoms.
- the alkyl group may be optionally substituted with an amino, alkyl, halo, or hydroxiyl group.
- alkoxy denotes oxy-containing .groups substituted with an alkyl, or 5 cycloalkyl group. Examples include, without limitation, methoxy, ethoxy, tert-butoxy, and cyclohexyloxy. Most preferred are "lower alkoxy” groups having one to six carbon atoms. Examples of such groups include methoxy, ethoxy, propoxy, butoxy, isopropoxy, and tert- butoxy groups.
- alkenyl and alkynyl refe ⁇ - to unsaturated aliphatic groups 0 analogous in length and possible substitution to the alkyls described above, but that contain at least one double bond or triple bond respectively.
- aryl means a carbocyclic aroimatic system containing one, two or three rings wherein such rings may be attached together in_ a pendant manner or may be fused.
- fused means that a second ring is present (i.e., attached or formed) by having two 5 adjacent atoms in common (Le,, shared) with the first ring.
- fused is equivalent to the term “condensed.”
- aryl embraces aromatic groups such as phenyl, naphthyl, tetrahydronaphthyl, indane, and biphenyl. The aryl group may optionally be substituted with an amino, alkyl, halo, hydroxyl, carbocyclic, heterocyclic, or another aryl group.
- aralkyl embraces aryl-substitutcd alkyl moieties.
- Preferable aralkyl ⁇ 0 groups are "lower aralkyl” groups having aryl groups attached to alkyl groups having one to six carbon atoms. Examples of such groups include benzoyl, diphenylmethyl, triphenylmethyl, phenylethyl, and diphenylethyl.
- benzyl and phexiylmethyl are interchangeable.
- aryloxy embraces aryl groups, as defined above, attached to an oxygen atom.
- the aryloxy groups may optionally be substituted with a halo, hydroxyl, or alkyl
- Examples of such groups include phenoxy, 4-chloro-3-ethylphenoxy, 4-chloro-3- methylphenoxy, 3-chloro-4-ethylphenoxy, 3,4-dichlorophenoxy, 4-methylphenoxy, 3- trifluorornefhoxyphenoxy, 3-trifluoromethylphenoxy, 4-fhuorophenoxy, 3,4-dimethylphenoxy,
- aralkoxy embraces oxy-containing aralkyl groups attached through an oxygen atom to other groups.
- “Lower aralkoxy” groups are those phenyl groups attached to lower alkoxy group as described above. Examples of such groups include benzyloxy, 1- phenylethoxy, 3-trifluoromethoxybenzyloxy, 3-trifluoromethylbenzyloxy, 3,5- difluorobenyloxy, 3-bromobenzyloxy, 4-propylbenzyloxy, 2-fluoro-3- trifluoromethylbenzyloxy, and 2-phenylethoxy.
- Carboxyl includes both carboxylic acids, and carboxylic acid esters.
- carboxylic acid refers to a carboxyl group in which R" is hydrogen. Such acids include formic, acetic, propionic, butryic, valeric acid, 2-methyl propionic acid, oxirane-carboxylic acid, and cyclopropane carboxylic acid.
- carboxylic acid ester or “ester” refers to a carboxyl group in which R" is alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl.
- carbocylic refers to a group that contains one or more covalently closed ring structures, and that the atoms forming the backbone of the ring are all carbon atoms.
- the ring structure may be saturated or unsaturated. The term thus distinguishes carbocyclic from heterocyclic rings in which the ring backbone contains at least one non- carbon atom.
- carbocylic encompasses cycloalkyl ring systems.
- cycloalkane or “cyclic alkane” or “cycloalkyl” refer to a carbocyclic group in which the ring is a cyclic aliphatic hydrocarbon, for example, a cyclic alkyl group preferably with 3 to 12 ring carbons.
- Cycloalkyl inrissas, by way of example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, or cyclooctyl, and the like.
- the cycloalkyl group may be optionally substituted with an amino, alkyl, halo, or hydr ⁇ xyl group.
- ether refers to the group -R'-O-R" wherein R' and R" as used in this definition are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl, and R' can additionally be a covalent bond attached to a carbon.
- R' and R as used in this definition are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl, and R' can additionally be a covalent bond attached to a carbon.
- halo or halogen refer to fluoro, chloro, bromo or iodo, usually regarding halo substitution for a hydrogen atom in an organic compound.
- heterocyclic or heterocycle means an optionally subsituted, saturated or unsaturated, aromatic or non-aromatic cyclic hydrocarbon group with 4 to about 12 carbon atoms, preferably about 5 to about 6, wherein 1 to about 4 carbon atoms are replaced by nitrogen, oxygen or sulfur.
- heterocyclic which are aromatic include groups pyridinyl, furanyl, benzofuranyl, isobenzofuranyl, pyrrolyl, thienyl, 1,2,3-triazolyl, 1,2,4- triazolyl, indolyl, imidazolyl, thiazolyl, thiadiazolyl, pyrimidinyl, oxazolyl, triazinyl, and tetrazolyl.
- heterocycles include benzimidazole, dihydrothiophene, dioxin, dioxane, dioxolane, dithiane, dithiazine, dithiazole, dithiolane, foran, indole, 3-H indazole, 3-H-indole, imidazole, indolizine, isoindole, isothiazole, isoxazole, morpholine, oxazole, oxadiazole, oxathiazole, oxathiazolidine, oxazine, oxadiazine, piperazine, piperidine, purine, pyran, pyrazine, pyrazole, pyridine, pyrimidine, pyrimidine, pyridazine, pyrrole, pyrrolidine, tetrahydrofuran, tetrazine, thiadiazine, thiadiazole, thiatriazole, thi
- the heterocycle may be optionally substituted with an amino, alkyl, alkenyl, alkynyl, halo, hydroxyl, carbocyclic, thio, other heterocyclic, or aryl group.
- exemplary heterocyclic groups include 1-pyrrolyl, 2- pyrrolyl, 3-pyrrolyl, 1-indolyl, 2-indolyl, 3-indolyl, 1-pyridyl, 2-pyridyl, 3-pyridyl, 4-pyridyl, 1-imidazolyl, 2-imidazolyl, 3-imidazolyl, 4-imidazolyl, 1-pyrazolyl, 2 pyrazolyl, 3-pyrazolyl, 4-pyrazolyl, 5-pyrazolyl, 1-pyrazinyl, 2-pyrazinyl, 1-pyrimidinyl, 2-pyrimidinyl, 4- pyrimidinyl, 5-pyrimidinyl, 1 -pyridazinyl, 2-pyridazinyl
- nitro means -NO 2 .
- sulfanyl refers to -SR' where R 1 as used in this definition is hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl.
- sulfenyl refers to -SOR' where R' as used is this definition is hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl.
- sulfonyl refers to -SOR' where R' as used in this definition is hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl.
- the present invention contemplates all such compounds, including cis- and trans-geometric isomers, E- and Z-geometric isomers, R — and S — enantiomers, diastereomers, d-isomers, 1 -isomers, the racemic mixtures thereof and other mixtures thereof, as falling within the scope of the invention.
- pharmaceutically acceptable salts are also included in the family of compounds of the present invention.
- pharmaceutically- acceptable salts embraces salts commonly used to form alkali metal salts and to form addition salts of free acids or free bases. The nature of the salt is not critical, provided that it is pharmaceutically acceptable.
- Suitable pharmaceutically acceptable acid addition salts of compounds of the present invention be prepared from inorganic acid or from an organic acid. Examples of such inorganic acids are hydrochloric, hydrobromic, hydroiodic, nitric, carbonic, sulfuric, and phosphoric acid.
- Appropriate organic acids may be selected from aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclic, carboxylic and sulfonic classes of organic acids, examples of which are formic, acetic, propionic, succinic, glycolic, gluconic, lactic, malic, tartaric, citric, ascorbic, glucuronic, maleic, fumaric, pyruvic, aspartic, glutamic, benzoic, anthranilic, mesylic, salicylic, p-hydroxybenzoic, phenylacetic, mandelic, embonic (pamoic), methanesulfonic, ethylsulfonic, benzenesulfonic, sulfanilic, stearic, cyclohexylaminosulfonic, algenic, galacturonic acid.
- Suitable pharmaceutically-acceptable base addition salts of compounds of the present invention include metallic salts made from aluminum, calcium, lithium, magnesium, potassium, sodium and zinc or organic salts made from N,N'-dibenzylethyleneldiamine, choline, chloroprocaine, diethanolamine, ethylenediamine, meglumine (N-methylglucamine) and procain. All of these salts may be prepared by conventional means from the corresponding compounds of by reacting, for example, the appropriate acid or base with the compounds of the present invention.
- ester refers to esters which hydrolyze in vivo and include, but are not limited to, those that break down readily in the human body to leave the parent compound or a salt thereof.
- Suitable ester groups include, for example, those derived from pharmaceutically acceptable aliphatic carboxylic acids, particularly alkanoic, alkenoic, cycloalkanoic and alkanedioic acids, in which each alkyl or alkenyl moiety advantageously has not more than 6 carbon atoms.
- esters include formates, acetates, propionates, butyrates, acrylates and ethylsuccinates.
- prodrugs refers to those prodrugs of the compounds of the present invention, which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, commensurate with a reasonable risk/benefit ratio, and effective for their intended use, where possible, of the compounds of the invention.
- prodrug refers to compounds that are rapidly transformed in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood.
- the present invention provides a pharmaceutical composition, which comprises a therapeutically-effective amount of one or more compounds of the present invention or a pharmaceutically-acceptable salt, ester or prodrug thereof, together with a pharmaceutically-acceptable diluent or carrier.
- compositions may be formulated for any route of administration, in particular for oral, rectal, transdermal, subcutaneous, intravenous, intramuscular or intranasal administration.
- the compositions may be formulated in any conventional form, for example, as tablets, capsules, caplets, solutions, suspensions, dispersions, syrups, sprays, gels, suppositories, patches and emulsions.
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with trie tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically-acceptable carrier means a pharmaceutically-acceptable material, composition or vehicle, such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting the subject lonidamine analogue or derivative from one organ, or portion of the body, to another organ, or portion of the body.
- a pharmaceutically-acceptable material such as a liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting the subject lonidamine analogue or derivative from one organ, or portion of the body, to another organ, or portion of the body.
- Each carrier must be “acceptable” in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient.
- materials which may serve as pharmaceutically-acceptable carriers include: (1) sugars, such as lactose, glucose and sucrose; (2) starches, such as corn starch and potato starch; (3) cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; (4) powdered tragacanth; (5) malt; (6) gelatin; (7) talc; (8) excipients, such as cocoa butter and suppository waxes; (9) oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; (10) glycols, such as propylene glycol; (11) polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; (12) esters, such as ethyl oleate and ethyl laurate; (13) agar; (14) buffering agents, such as magnesium hydroxide and aluminum hydrox
- inhibitor refers to a statistically significant and measurable reduction in activity, preferably a reduction of at least about 10% versus control, more preferably a reduction of about 50% or more, still more preferably a reduction of about 80% or more.
- a “therapeutically effective amount” is an amount of a compound of the present invention or a combination of two or more such compounds, which inhibits, totally or partially, the progression of the condition or alleviates, at least partially, one or more symptoms of the condition.
- a therapeutically effective amount can also be an amount that is prophylactically effective. The amount that is therapeutically effective will depend upon the patient's size and gender, the condition to be treated, the severity of the condition and the result sought. For a given patient and condition, a therapeutically effective amount can be determined by methods known to those of skill in the art.
- a therapeutically effective amount refers to that amount which has the effect of (1) reducing the size of the tumor, (2) inhibiting (that is, slowing to some extent, preferably stopping) tumor metastasis, (3) inhibiting to some extent (that is, slowing to some extent, preferably stopping) tumor growth, and/or, (4) relieving to some extent (or, preferably, eliminating) one or more symptoms associated with the cancer.
- Several of the compounds of the present invention have been shown to inhibit
- Hsp90 in vitro.
- therapeutically effective amounts of the compounds of the present invention will be useful as anti-cancer agents and/or neuroprotective agents.
- some of the compounds of the present invention may be used with other Hsp90 inhibitors, chemotherapeutic agents, and/or neuroprotective agents.
- novobiocin analogue compounds that contained both modified coumarin and sugar derivatives was prepared.
- the compounds were prepared as set forth in the scheme below along with a procedure recently developed for the synthesis of noviose. See Yu et al., Synthesis of (-)-Noviose from 2,3-0-Isopropylidene-D-erythronolactol, J. Org. Chem, 2004, 69, 7375-7378, which is incorporated by reference.
- the novobiocin analogues prepared according to the scheme included modification of the coumarin ring by shortening of the amide side chain and removal of the 4- hydroxy substituent (A) (see Madhavan et al., Novel Coumarin Derivatives of Heterocyclic Compounds as Lipid Lowering Agents, Bioorg. Med. Chem. Lett. 2003, 13, 2547, which is incorporated by reference), removal of both the 4-hydroxy and amide linker (B), steric replacements of both the 4-hydroxy and benzamide ring (C), and 1,2-positional isomers of the noviosyl linkage (D and E).
- A The novobiocin analogues prepared according to the scheme included modification of the coumarin ring by shortening of the amide side chain and removal of the 4- hydroxy substituent (A) (see Madhavan et al., Novel Coumarin Derivatives of Heterocyclic Compounds as Lipid Lowering Agents, Bioorg. Med. Chem. Let
- Noviosylated coumarin Al (20 mg, 0.047 mmol) was dissolved in methanolic ammonia (7.0 M, 2 mL) at 25 0 C and stirred for 24 h. The solvent was evaporated and the residue purified by preparative HPLC (SiO 2, 20% 2-propanol in hexanes) to afford A2 (4.2 mg, 22%), A3 (8.6 mg, 42%) and A4 (3.5 mg, 20%) as colorless solids.
- Ci 8 H 22 NO 8 requires m/z 380.1345).
- EXAMPLE 2 DEGREDATION OF PHOSPgQ-AKT Inhibition of Hsp90 results in the degradation of Hsp90-dependent clients via ubiquitination of the unfolded client followed by proteasome-mediated hydrolysis. To test whether Hsp90 client proteins were degraded in the presence of these novobiocin analogues,
- A4/KU-1 (diol) and A3/KU-2 (3 '-carbamate) were the most potent novobiocin analogues identified, based on their ability to inhibit Hsp90 and cause the degradation of phosphorylated AKT. As shown in FIG. 1., the most active compound
- the IC 50 for Hsp90 inhibitors is sometimes determined as the concentration of inhibitor required to produce 50% degradation of Her-2, another therapeutically important Hsp90 client protein involved in breast cancer.
- KU-1/A.4 was incubated with Skbr3 breast cancer cells at concentrations of 100 nJVl, l ⁇ M and 10 ⁇ M, a rapid decrease in Her-2 was observed between 100 nM and l ⁇ M, as shown in the Western blot of FIG. 2.
- These data are normalized against actin, a non-Hsp90 client protein, used as a control for non-specific degradation.
- These data suggest the IC 50 of KU-I /A4 is in the Io ⁇ V micromolar range, whereas novobiocin in the same assay produces an IC 50 of 700 ⁇ M.
- EXAMPLE 4 PROSTATE CANCER The steroid hormone receptors are also dependent upon the Hsp90 protein folding machinery for activation and hormone binding.
- LNCaP mutated androgen receptor-dependent prostate cancer cell line
- LAPC-4 ⁇ vild type androgen receptor prostate cancer cell line
- the prostate cancer cells were grown in RPMI with 10% fetal calf serum in a standard fashion. Once the cells had reached near confluence, they were treated with vehicle (DMSO) or varying concentrations of KU-1/A4 ranging from IOnm to lOO ⁇ M for 24 hours.
- DMSO vehicle
- the membranes were blocked for two hours at room temperature in Tris-buffered saline (pH 7.5) containing 0.2% I- block (Tropix, Bedford, MA), 1% milk, and 0.1% Tween-20 (TBS-T). The membranes were subsequently be incubated with a primary antibody to the abo ⁇ ve mentioned proteins (all of which have commercially available antibodies) overnight at 4° C. The next day the membrane was washed three times in TBS-T followed by one hour incubation with an appropriate horseradish peroxidase labeled secondary antibody in blocking buffer (TBS-T).
- TBS-T horseradish peroxidase labeled secondary antibody
- the membranes were again washed in TBS-T and Tris-buffered saline and developed in SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) according to manufacturer's instructions.
- the blots were visualized by exposing the enhanced chemiluminescence-reacted blot to X-ray film.
- KU-1/A4 had a dramatic effect on the concentrations of the mutant androgen receptor, AKT, and HIF- l ⁇ at about 1 ⁇ M in the LNCaP cell line.
- KU-1/A4 drastically reduced levels of the androgen receptor at lower concentrations in the wild type androgen receptor prostate cancer cell line (LAPC-4).
- LAPC-4 wild type androgen receptor prostate cancer cell line
- Hsp90 levels were determined. Under normal conditions, Hsp90 binds heat shock factor 1 (HSFl), but in the presence of Hsp90 inhibitors this interaction is lost and HSFl is able to induce the expression of Hsp90.
- HSFl heat shock factor 1
- Hsp90 levels are significantly increased in a manner dependent on the concentration of KU- 1 /A4 consistent with similar results previously obtained by incubation with geldanamycin and radicicol. Both of these data are in contrast to actin, which is not an Hsp90 client protein and thus remains unaffected by Hsp90 inhibitors.
- PROPHETIC EXAMPLE 4 AMIDE SIDE CHAIN MODIFICATIONS Since KU-1/A4 was shown to be the most potent C- terminal inhibitor of Hsp90 identified in Example 1, additional derivatives of the KU-1/A4 scaffold will be prepared. Modifications of the amide side chain will allow for an in depth study of the hydrophobic cavity that binds to this portion of KU-I /A4 and the analogous benzamide of novobiocin. As such, analogues of KU-1/A4 that have increasingly larger hydrophobic groups by the use of different commercially available or readily synthesized anhydrides, such as those anhydrides shown in the scheme below.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl, (and most preferably R is hydrogen, alkyl, aryl, and aralkyl); and wherein R 1 is hydrogen or CONH 2 .
- the amide linkage will also be reversed to determine the optimal profile of this functionality.
- the 7-hydroxy-3 -ethyl ester coumarin will be hydrolyzed to afford the corresponding acid, which will be coupled with amines that mimic the same side chains used in the KU-1/A4 amide studies for direct comparison of biological activity.
- the free phenols will be noviosylated as described earlier to afford the cyclic carbonate products.
- Treatment of the carbonate with methanolic ammonia will give the diol, 2- and 3-carbamoyl products as shown in the scheme below. See Shen et al., Synthesis of Photolabile Novobiocin Analogues, Bioorg. Med. Chem. Lett. 2004, 14, 5903-5906, which is incorporated by reference.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl; and wherein R' is hydrogen or CONH 2 .
- the R in the amide side chain is hydrogen alkyl, aryl, and alkaryl, and the amines used in the above scheme are NH 3 , methylamine, ethylamine, propylamine, n-butylamine, and phenylamine.
- the amide side chain, coumarin ring, and sugar may be modified in accordance with the other examples shown herein.
- PROPHETIC EXAMPLE 5 ISOCOUMARIN DERIVATIVES
- the isocoumarin derivative of the compounds of the present invention will be prepared.
- the isocoumarin will be prepared from the 4- benzyloxylactone shown in the scheme below.
- Treatment of the lactone with sodium cyanide, followed by HCl/pyridine is known to produce similar isocoumarins. See Wells et al., Facile synthesis of 3-acylaminoisocoumarins, J. Org. Chem. 1971, 36, 1503-1506, which is incorporated by reference.
- isocoumarin derivatives can be prepared in accordance with the above scheme, in addition to the KU-1/A4 analogue shown. That is, the amide side chain, coumarin ring, and sugar may be modified in accordance with the other examples shown herein.
- PROPHETIC EXAMPLE 6 DESfl>IMETHYL) AND DESMETHOXY SUGAR ANALOGUES
- R is lower alkyl; and wherein R' is preferably hydrogen or -CONH 2 .
- demethylated an/or dealkoxylated derivatives can be prepared in accordance with the above scheme, in addition to the modified KU-1/A4 derivative shown above. That is, the amide side chain, coumarin ring, and sugar may be modified in accordance with the other examples shown herein.
- PROPHETIC EXAMPLE 7 MODIFIED NOVOBIOCIN DERIVATIVES
- This example involves the modification to of the compounds of the present invention to complement the hydrogen bonding capabilities of the nucleotide bases (adenine and guanine) with those of the coumarin ring system as shown below.
- these analogues contain conformationally restricted hydrogen bond donors/acceptors of KU-I /A4 (F and G) and strategically placed hydrogen bond acceptors/donors to complement those found in guanine (H-L).
- H-L hydrogen bond acceptors/donors to complement those found in guanine
- the hydrophobic pocket that accommodates the m-substituted benzamide ring of novobiocin will be probed by alteration of the side chain constituents.
- Example 7F Heterocyclic Modifications to Quinolone hi this example, the coumarin ring will be modified to create F analogues that resemble guanine and contain a conformationally biased tiydrogen-bond donor/acceptor.
- the synthesis begins with commercially available 4-hydroxy-2-nitrobenzaldehyde following the procedure of Meanwell, et al., Inhibitors of Blood Platelet cAMP Phosphodiesterase. 2. Structure- Activity Relationships Associated with l,3-DiJhydro-2//-imidazo[4,5-b]quinolin-2- ones Substituted with Functionalized Side Chains, J. Med. Chem. 1992, 35, 2672-2687.
- the phenol will be protected as the benzylether, followed by treatment with hydantoin phosphonate to give the corresponding olefin.
- hydantoin phosphonate See Meanwell et al., Diethyl 2,4-dioxoimidazolidine-5- phosphonate: A Wadsworth-Emmons Reagent for the Mild and Efficient Preparation of C-5 Unsaturated Hydantoins, J. Org. Chem. 1991, 56, 6897-6904.
- Reduction of the benzylether, nitro, and olefin functionalities will provide the appropriate amine for subsequent addition to the carbonyl upon treatment with iodine.
- X 4 , X 5 X 6 X 8 are preferably each -CH-; and wherein R a , R b , and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, h leetteerrooccyylliicc,, aryl, or aralkyl; or wherein R b is oxided to form the carbonyl according to the formula:
- coumarin G will be prepared from 7-benzyloxy-4-hydroxy-3- nitrocoumarin, according to the scheme below.
- Buckle et al. Aryloxyalkyloxy- and aralkyloxy-4-hydroxy-3-nitro coumarins which inhibit histamine release in the rat and also antagonize the effects of a slow reacting substance of anaphylaxis, J. Med. Chem. 1979, 22, 158-168.
- Treatment of the 4-hydroxyl group with phosphorous oxychloride (POCl 3 ) will afford the corresponding 4-amino derivative upon subsequent exposure to ammonia.
- Rassochandran et al. Mild method for the preparation of 4-chloro-3-nitro coumarins, Indian. J.
- X 5 X 6 X 8 are preferably each -CH-; and wherein R a , R b , and R c are independently hydrogen, alkyl, alkenyl, alkynyl, carbocyclic, heterocylic, aryl, or aralkyl; or wherein R b is oxided to form the carbonyl according to the formula:
- the nitrogen-containing H variants of the coumarin ring will be prepared from 2-methyl-3,5-pyridinediol, by bromination of the benzylic methyl group, followed by hydrolysis and oxidation to the corresponding aldehyde as set forth in the scheme below. See Morisawa et al., Anticoccidal agents. IV. Modification at the 5-position of 4-deoxypyridoxol and ⁇ 4-norpyridoxol, Agric. Biol. Chem. 1975, 39, 1275-1281. Using conditions previously employed for the syntheses of other coumarin derivatives by us, the aldehyde will be treated with glycine under basic conditions to yield the azacoumarin ring system.
- the I analogues are directed to other side-chains extending from the coumarin ring.
- the KU-1/A4 coumarin ring will be prepared from 2,4-dihydroxy-5- nitrobenzaldehyde (see Chandrashekhar et al., g-substitution in the resorcinol nucleus, VI. Formylation of 4-nitro and 2-nitro resorcinols, Proc. Ind. Acad. Sd.
- the ⁇ -hydroxybenzaldehyde will be treated with ethyl glycine under acidic conditions to afford the corresponding free amine upon basic workup. Both the amino and hydroxyl functionalities will be acylated with the same anhydrides as shown above. Subsequent hydrolysis of the phenolic ester will provide the coumarin amide, which can be coupled directly with noviose carbonate as described previously.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl; wherein X is alkyl, alkenyl, alkynyl, aryl, aralkyl, alkoxy, halogen or nitro.
- the J analogues will be prepared from 4-chloro-2-hydroxy-5-nitrobenzaldehyde (see Pal et al., New arylsulfonylhydrazones of substituted benzaldehyde as anticancer agents, Neoplasms 1983, 30, 551-556) by treatment with glycine, acetic anhydride, and sodium acetate as mentioned previously for the preparation of other coumarin derivatives as set forth in the following scheme. See Khoo et al., Synthesis of substituted 3-aminocoumarins from ethyl N- 2-Hydroxyarylideneglycinates, Syn. Commun. 1999, 29, 2533-2538.
- the chloro substituent will undergo nucleophilic aromatic displacement with ammonia as a consequence of the electron- withdrawing j?-lactone and o-nitro group.
- the nitro group Upon formation of the 7-amino-6- nitrocoumarin, the nitro group will be reduced and immediately treated with triethyl orthoformate to produce the imidazole ring that resembles guanine. See Buckle et al., Aryloxyalkyloxy- and aralkyloxy-4-hydroxy-3 -nitro coumarins which inhibit hisamine release in the rat and also antagonize the effects of a slow reacting substance of anaphylaxis, J. Med. Chem. 1979, 22, 158-168.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl.
- the K analogues of the KU-1/A4 coumarin moiety will be prepared from 5- methoxy-2-methylbenzonitrile as set forth in the scheme below. See Tomita et al., Schmidt reaction with benzocycloalkenones, J. Chem. Soc. C: Organic 1969, 2, 183-188. Bromination of the benzylic methyl group, followed by displacement with potassium cyanide will furnish the dinitrile product, which is a substrate for acid catalyzed cyclization to form the corresponding 2-bromoisoquinoline. See Johnson et al., The cyclization of dinitriles by anhydrous halogen acids. A new synthesis of isoquinolines, J. Org. Chem. 27, 3953-3958.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl.
- Quinoline derivatives of, L will be prepared from 7-hydroxyquinoline, by first bromination of the quinoline ring, see Zymalkowski et al., Chemistry of 3- quinolinecarboxaldehyde, Ann. Chem., Justis Liebigs 1966, 699, 98-106, followed by a copper-catalyzed animation of the halogenated heterocycle as set forth in the scheme below. See Lang et al., Amination of aryl halides using copper catalysis, Tetrahedron Lett. 2001, 42, 4251-3254. Subsequent treatment with various anhydrides (shown previously), followed by hydrolysis of the phenolic ester and coupling with noviose carbonate will ultimately afford these L analogues.
- R is hydrogen, alkyl, alkenyl, alkynyl, aryl, carbocylic, heterocyclic, aryl, or aralkyl.
- This example involves the modification of the carbohydrate reside. More specifically, analogues similar to that of novobiocin's chlorinated pyrollic ester, chlorobiocin, will be prepared.
- compound KU-1/A4 will be prepared, and then coupled with a variety of acids to selectively afford the equatorial acylated alcohols.
- Selective acylation is based upon previous studies aimed at the preparation of photolabile derivatives of novobiocin. See Shen et al, Synthesis of Photolabile Novobiocin Analogues, JSioorg. Med. Chem. Lett. 2004, 14, 5903-5906, which is incorporated by reference.
- These acids will include the pyrrolic acid found in chlorobiocin as well as several other that are shown in the scheme below.
- Exemplary acids include pyrrolic acids, indolic acids, pyridinic acids, benzoic acids, salicylic acid, para-hydrobenzoic acid, thiobenzoic acid, and pyrazolic acid.
- the sugar will be modified to include a functional group according to the formmla -R-OR", wherein R' is a covalent bond or alkyl, and R" is an acyl group.
- the acyl derivative comprises the group -COR wherein R is alkyl, aryl, aralkyl, or an aromatic heterocyclic group. Alkylated, aralkylated, thiolated, halogenated, and hydroxylated pyroles, indoles, pyridines, and pyrazoles are attached to the sugar ring as shown in the scheme below.
- various substirutents will be added to the amine of the carbamate side chain.
- carbonate KU-9/A1 will be prepared and amines added to provide the 3 '-carbamoyl products as generally set forth in the scheme below.
- the sugar will be modified to include a functional group according to the formula — ROR", wherein R' is a covalent bond or alkyl, and R" is C-amido> .
- the C- amido group is -CONR'R" wherein R 1 is H, and R" is alkyl, ary ⁇ l, aralkyl, or an aromatic heterocyclic group.
- X is alkyl, alkenyl, alkynyl, hydroxyl, halo, and n is an integer, preferably 0, 1, 2, 3, or 4.
- PROPHETIC EXAMPLES 9-11 FURANOSE AND PYRANOSE NOVOBIOCIN DERIVATIVES
- new pyranose and furanose derivatives will be prepared that have affinity with the sugar of GTP and phosphate binding region of Hsp90.
- These selected compounds are shown in below and include ester, amide, sulfonic ester, phosphonic ester, carbamoyl, sulfonamide, and hydroxyl derivatives.
- Initial compounds will be coupled with the coumarin ring present in KU-1/A4, but when a more potent analogue is obtained, the best sugar derivative from these studies will be placed onto the optimized ring system.
- the o-acetyl derivative will be prepared from ribose (9.1, Scheme 9). Treatment of the ribose hemiacetal with benzyl alcohol and hydrochloric gas will provide the benzyloxyacetal, 9.2. See Pigro et al., Readily available carbohydrate-derived imines and amides as chiral ligands for asymmetric catalysis, Tetrahedron 2002, 58, 5459-5466.
- furanose derivatives will be prepared from benzyl-protected ribose carbonate (9.3, Scheme 10). Both the sulfonamide and JV-acetyl analogues will be furnished by conversion of primary alcohol (9.3) to the corresponding azide by a Mitsunobu reaction with bis(azido)zinc pyridine complex. Viaud et al., Zinc azide mediated Mitsunobu substitution. An expedient method for the one-pot azidation of alcohols, Synthesis 1990, 130- 132. The resulting azide (10.1) will be reduced, and the primary amine converted to the sulfonamide and N-acetyl functionalities, 10.2 and 10.3, respectively.
- Both the sulfonic ester and the phosphonic ester will be prepared by conversion of 9.3 to iodide 10.6, followed by generation of the requisite enolate to displace the halide.
- Gallant et al. An efficient preparation and the intramolecular cyclopropanation of Beta-diazo-Beta-ketophosphonates and Beta-diazophosphonoacetates, Syn. Commun. 1984, 14, 155-161.
- Subsequent treatment with palladium (0) and an amine will lead to allyl removal followed by decarboxylation to form 10.10 and 10.8. See Guibe, Allyl esters and their use in complex natural product syntheses, Tetrahedron 1998, 54, 2967-3041.
- the pyranose derivatives which resemble noviose and a ring-expanded ribose ring, will be prepared by our recently reported synthesis of 11.1. See Yu et al., Synthesis of Mono- and dihydroxylated furanoses, pyranoses, and an oxepanose for the Preparation of Natural Product Analogue Libraries, J. Org. Chem. 2005, 70, 5599-56O5, which is incorporated by reference in its entirety.
- the pyranose derivatives will be prepared in a similar manner from the known dihydropyrone ⁇ See Ahmed et al., Total synthesis of the microtubule stabilizing antitumor agent laulimalide and some nonnatural analogues: Tlie power of Sharpless' Asymmetric Epoxidation, J. Org. Chem. 2003, 68, 3026-3042), which is available in four steps from commercially available triacetyl D-glucal (Roth et al.,. Synthesis of a chiral synhton for the lactone portion of compactin and mevinolin, Tetrahedron Lett. 1988, 29, 1255- 12158).
- the pyranose will be furnished by Sharpless asymmetric dihydroxylation (SAD) of the olefin to give the product in high diastereomeric excess (KoIb et al., Catalytic Asymmetric Dihydroxylation, Chem. Rev. 1994, 94, 2483-2547), which can be converted to the cyclic carbonate at a later time.
- SAD Sharpless asymmetric dihydroxylation
- PROPHETIC EXAMPLE 12 PREPARATION QF 3-D YHYDROXY AND 5-DESMETHYOYL ANALOGUES
- the 4-deshydroxy and 8-desmethyl variants of novobiocin will be prepared along with the 8-methyl and 4-hydroxy analogues of KU-2/A3 (3 'carbamate) as shown below. Not only will the 3 '-carbamoyl derivatives of these compounds be prepared, but also the corresponding diols for direct comparison to KU-1/A4 (diol).
- 4-deshydroxynovobiocin will be prepared from 3-N-acetyl-7- hydroxy-8-methyl coumarin and the known carboxylic acid as set forth in the scheme below.
- the C-terminal nucleotide binding sites are in close proximity to the one another along the Hsp90 dimer interface, and therefore dimeric inhibitors of the compounds of the present invention should provide compounds with enhanced inhibitory activity. This is based on the fact that the dimeric compound, coumermycin Al, was shown to be approximately 10 times more active than the monomelic compound, novobiocin.
- the present invention thus includes dimers of the compounds disclosed herein.
- a dimeric inhibitor of KU-1/A4 will be prepared.
- the Cbz group will be removed to furnish the aniline for subsequent coupling with bifunctional linkers to prepare dimeric inhibitors.
- the dimer containing pyrazole linker found in Coumermycin Al will be prepared following the procedure developed by Olson et al., Tetrahedron Letters (2002), Volume Date 2003, 44(1), 61-63.
- the diacid will be coupled with two equivalents of the coumarin amine using C>-(7-azabenzotriazol-l-yl)-N,N,N',iV'- tetramethyluronium hexafluorophosphate (HATU) to furnish the cyclic carbonate precursor to the KU-1/A4 dimer.
- the carbonate will be removed upon treatment with methanolic triethylamine to provide the tetraol product. See Yu et al. Hsp90 Inhibitors Identified from a Library of Novobiocin Analogues. J. Am. Chem. Soc. Ill: 12778-12779 (2005).
- a number of dimeric linkers will be used to perturb the dimeric angle and to extend the dimeric tether in an effort to elucidate structure-activity relationships.
- ortho, meta, and para dibenzoic acids will be used in lieu of the pyrole biscarboxylic acid to determine optimal angles.
- Linker length will be probed by the use of about 3-10 carbon dicarboxylic acids. If the studies support that both angle and linker length are important, then combinations of these linkers will be prepared and coupled to furnisli the conformationally biased, extended compounds such as that shown below.
- PROPHETIC EXAMPLE 14 0 PROSTATE CANCER XENOGRAFT TUMOR MODEL
- This example involves the in vivo effect of the compounds of the present invention using a prostate cancer mouse model. More specifically, four to six week old BALB/c nu/nu nude mice will be obtained commercially and maintained in ventilated cages under Institutional Animal Care and Use Committee approval. Separate male mice will be [5 inoculated subcutaneously with 10 6 LNCaP cells suspended in 0.25 mL of Matrigel (BD, Bioscience, Bedford MA). Stable serum testosterone levels will be maintained in the mice by the implantation of 12.5mg 90-day sustained release testosterone pellets (Innovative Research, Sarasota FL) subcutaneously prior to inoculation with tumor.
- Tumor volume will be measured twice a week with vernier calipers with tumor volumes calculated using the formula [length x width x height x 0.52]. Mice with established tumor volumes of 5 mm will be selected for KU- 1/A4 administration. Utilizing the paradigm for administration of 17- AAG (another Hsp90 inhibitor), animals will be treated with both continuous and intermittent dosing schedules. A control animal will be treated with vehicle alone (DMSO). For the continuous dosing schedule, mice will receive intraperitoneal injections of vehicle or the test compounds (e.g., KU-1/A4) for 5 days per week for 3 weeks. The intermittent group will receive one 5 day cycle and then monitored for progression.
- DMSO vehicle alone
- test compound e.g., KU-1/A4
- KU-1/A4 Differing doses of the test compound (e.g., KU-1/A4) will be utilized based on pharmacokinetic information obtained from toxicity studies. When progression occurs, as defined by an increase in tumor size, the mice will receive a second 5 day cycle of the test compound (e.g., KU-1/A4). Response to the test compound will be assessed by measuring tumor volume and serum PSA levels using the PSA Assay Kit (American Qualex Antibodies, San Clemente, CA).
- PSA Assay Kit American Qualex Antibodies, San Clemente, CA.
- Hsp90's client proteins known to be involved in cancer cell survival mechanisms such as signal transduction (e.g., AKT, Her2, PI3 kinase), angiogenesis (e.g., HIF- l ⁇ ), and metastasis (AR, MMP2).
- signal transduction e.g., AKT, Her2, PI3 kinase
- angiogenesis e.g., HIF- l ⁇
- metastasis e.g., MMP2
- test compound e.g., KU-1/A4
- HPLC high performance liquid chromatography
- EXAMPLE 15 NEUROPROTECTIVE EFFECTS Recently, low concentrations of the Hsp90 inhibitor GDA were reported to induce expression of both Hsp70 and Hsp90, with a concomitant reduction in phosphorylated Tau (Dou et al., 2003).
- KU-1/A4 a novel C-terminal Hsp90 inhibitor, was tested for protective effects against A ⁇ toxicity in primary neurons. See protocols in Michaelis ML, Ansar S, Chen Y, Reiff ER, Seyb KI, Himes RH, Audus KL, Georg GI (2005) B-Amyloidinduced neurodegeneration and protection by structurally diverse microtubule- stabilizing agents. J Pharmacol Exp Ther 312:659-668, which is incorporated by reference.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Pharmacology & Pharmacy (AREA)
- Biochemistry (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description

















































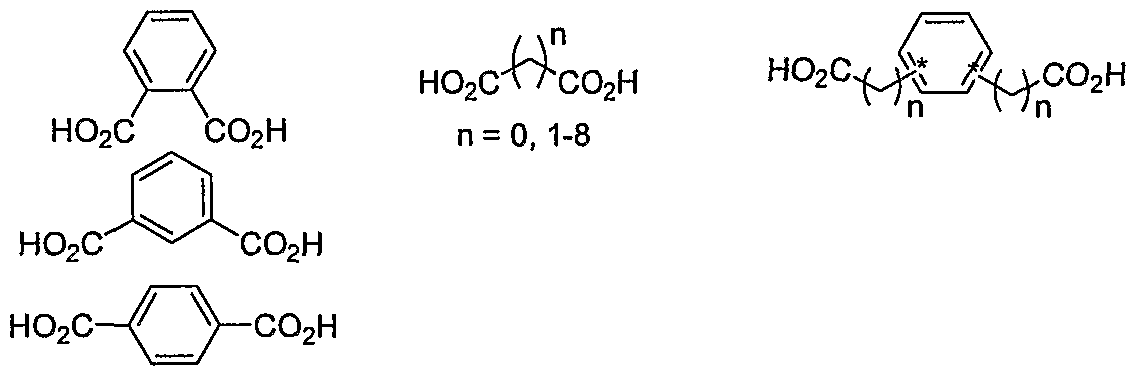
Claims


















Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05824355.1A EP1807440B1 (en) | 2004-11-03 | 2005-11-03 | Novobiocin analogues as anticancer agents |
| CA2585091A CA2585091C (en) | 2004-11-03 | 2005-11-03 | Novobiocin analogues as anticancer agents |
| AU2005301957A AU2005301957B2 (en) | 2004-11-03 | 2005-11-03 | Novobiocin analogues as anticancer agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62456604P | 2004-11-03 | 2004-11-03 | |
| US60/624,566 | 2004-11-03 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006050501A2 true WO2006050501A2 (en) | 2006-05-11 |
| WO2006050501A3 WO2006050501A3 (en) | 2007-05-31 |
Family
ID=36319818
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/039990 Ceased WO2006050501A2 (en) | 2004-11-03 | 2005-11-03 | Novobiocin analogues as anticancer agents |
Country Status (5)
| Country | Link |
|---|---|
| US (2) | US7608594B2 (en) |
| EP (1) | EP1807440B1 (en) |
| AU (1) | AU2005301957B2 (en) |
| CA (1) | CA2585091C (en) |
| WO (1) | WO2006050501A2 (en) |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008049994A1 (en) | 2006-10-24 | 2008-05-02 | Sanofi-Aventis | New fluorene derivatives, compositions containing the same and use thereof as inhibitors of the protein chaperone hsp 90 |
| WO2009059214A1 (en) * | 2007-11-02 | 2009-05-07 | The Regents Of The University Of California | Abeta-binding small molecules |
| US7781577B2 (en) | 2006-09-29 | 2010-08-24 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose co-transporter 2 and methods of their use |
| US7811998B2 (en) | 2004-11-03 | 2010-10-12 | University Of Kansas | Novobiocin analogues as anticancer agents |
| US7846945B2 (en) | 2007-03-08 | 2010-12-07 | Lexicon Pharmaceuticals, Inc. | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use |
| WO2011004132A1 (en) | 2009-07-10 | 2011-01-13 | Sanofi-Aventis | Novel hsp90-inhibiting indole derivatives, compositions containing said derivatives, and use thereof |
| WO2011027081A2 (en) | 2009-09-03 | 2011-03-10 | Sanofi-Aventis | Novel derivatives of 5,6,7,8-tetrahydroindolizine inhibiting hsp90, compositions containing same, and use thereof |
| US7960353B2 (en) | 2007-05-10 | 2011-06-14 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8212011B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues |
| US8212012B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
| JP2012518645A (en) * | 2009-02-20 | 2012-08-16 | ユニバーシティ・オブ・カンザス | Novobiocin analogs having modified sugar moieties |
| US8247408B2 (en) | 2005-10-07 | 2012-08-21 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US8273755B2 (en) | 2006-09-15 | 2012-09-25 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| CN103429254A (en) * | 2010-10-11 | 2013-12-04 | 南加利福尼亚大学 | Secreted Heat Shock Protein-90α (Hsp90α) Fragments as Vaccines or Epitopes or Small Molecule Drug Targets for Monoclonal Antibody Drugs Against a Range of Human Solid Tumors |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| CN103450133A (en) * | 2013-09-16 | 2013-12-18 | 中国药科大学 | Scopoletin derivatives with anti-tumor activity, and preparation method and application thereof |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| WO2014106623A1 (en) * | 2013-01-04 | 2014-07-10 | Max-Planck-Gesellschaft zur Förderung der Wissenschaft e.V. | C-terminal hsp90 inhibitors to treat pituitary adenomas |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN104271587A (en) * | 2012-02-09 | 2015-01-07 | 堪萨斯大学 | Hsp90C terminal inhibitors |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9120774B2 (en) | 2004-11-03 | 2015-09-01 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9994556B2 (en) | 2014-06-13 | 2018-06-12 | University Of Kansas | Triazole modified coumarin and biphenyl amide-based HSP90 inhibitors |
| CN108484495A (en) * | 2018-04-12 | 2018-09-04 | 苏州康润医药有限公司 | A kind of synthetic method of the bromo- 7- oxyquinolines of 3- |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10167309B2 (en) | 2012-07-13 | 2019-01-01 | Wave Life Sciences Ltd. | Asymmetric auxiliary group |
| US10280192B2 (en) | 2011-07-19 | 2019-05-07 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| US10307434B2 (en) | 2009-07-06 | 2019-06-04 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| US10329318B2 (en) | 2008-12-02 | 2019-06-25 | Wave Life Sciences Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| US10485815B2 (en) | 2012-03-21 | 2019-11-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10590065B2 (en) | 2014-06-24 | 2020-03-17 | University Of Kansas | Biphenyl amides with modified ether groups as HSP90 inhibitors and HSP70 inducers |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11224608B2 (en) | 2017-10-16 | 2022-01-18 | Dana-Farber Cancer Institute, Inc. | Compounds and methods for treating cancer |
| US11827664B2 (en) | 2018-05-14 | 2023-11-28 | Reata Pharmaceuticals, Inc | Biaryl amides with modified sugar groups for treatment of diseases associated with heat shock protein pathway |
| US12187721B2 (en) | 2018-10-17 | 2025-01-07 | Array Biopharma Inc. | Protein tyrosine phosphatase inhibitors |
| US12213983B2 (en) | 2012-11-01 | 2025-02-04 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using PI3 kinase isoform modulators |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0957389A1 (en) | 1998-05-13 | 1999-11-17 | Eastman Kodak Company | Precision assembly technique |
| US20070004689A1 (en) * | 2004-03-12 | 2007-01-04 | Agoston Gregory E | Antiangiogenic agents |
| CA2558014A1 (en) * | 2004-03-12 | 2005-09-29 | Entremed, Inc. | Antiangiogenic agents |
| US7622451B2 (en) | 2004-11-03 | 2009-11-24 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| WO2007059111A2 (en) * | 2005-11-14 | 2007-05-24 | Entremed, Inc. | Anti-angiogenic activity of 2-methoxyestradiol in combination with anti-cancer agents |
| KR100802531B1 (en) | 2006-08-28 | 2008-02-13 | 한국화학연구원 | Synthesis of D-erythrose Compound Protected by 4-hydroxy Group |
| WO2008094665A1 (en) * | 2007-01-31 | 2008-08-07 | Entremed, Inc. | Method of treating amyloidosis mediated diseases |
| WO2009036407A2 (en) * | 2007-09-14 | 2009-03-19 | California Institute Of Technology | Compositions and methods for regulating cellular protection |
| WO2010014617A1 (en) * | 2008-07-28 | 2010-02-04 | University Of Kansas | Heat shock protein 90 inhibitor dosing methods |
| MX2011003624A (en) | 2008-10-22 | 2012-01-27 | Graco Minnesota Inc | Portable airless sprayer. |
| WO2011041593A1 (en) * | 2009-09-30 | 2011-04-07 | University Of Kansas | Novobiocin analogues and treatment of polycystic kidney disease |
| WO2012070024A1 (en) * | 2010-11-28 | 2012-05-31 | Metasignal Therapeutics Inc. | Carbonic anhydrase inhibitors with antimetastatic activity |
| MX370138B (en) * | 2014-02-26 | 2019-12-03 | Univ Texas | Nitrobenzaldehyde proton release for manipulation of cellular acidosis. |
| EP3169815B1 (en) | 2014-07-15 | 2020-12-23 | Ontario Institute For Cancer Research | Methods and devices for predicting anthracycline treatment efficacy |
| US12030867B2 (en) | 2018-05-30 | 2024-07-09 | University Of Notre Dame Du Lac | Hsp90β selective inhibitors |
| WO2020132618A1 (en) * | 2018-12-21 | 2020-06-25 | The University Of Kansas | Photocleavable linker for catching and/or releasing of circulating tumor cells or extracellular vesicles |
| CN115739435A (en) | 2019-05-31 | 2023-03-07 | 固瑞克明尼苏达有限公司 | Hand-held fluid sprayer |
| KR20230039665A (en) * | 2020-07-02 | 2023-03-21 | 바이엘 악티엔게젤샤프트 | Heterocycle derivatives as pest control agents |
Family Cites Families (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3105088A (en) * | 1960-04-07 | 1963-09-24 | Upjohn Co | 3-(2-isopentyl) and 3-isoamyl 4-acetoxybenzoyl chloride |
| GB1114470A (en) * | 1965-06-10 | 1968-05-22 | Hoffmann La Roche | Novel coumarin dervatives |
| US3494914A (en) * | 1968-04-29 | 1970-02-10 | Bristol Myers Co | Antibacterial agents |
| US3890297A (en) * | 1973-03-29 | 1975-06-17 | Upjohn Co | Process for n-acylating novenamine |
| CA1294285C (en) * | 1983-03-13 | 1992-01-14 | Robert T. Buckler | Optical indicator chalcogen compounds and methods for their preparation and use |
| JPH06501465A (en) * | 1990-09-07 | 1994-02-17 | シェリング・コーポレーション | antiviral compounds |
| AU1739097A (en) * | 1996-02-23 | 1997-09-10 | Albert Einstein College Of Medicine Of Yeshiva University | Enzyme detection/assay method and substrates |
| EP1161231A2 (en) * | 1999-03-12 | 2001-12-12 | THE UNITED STATES OF AMERICA, represented by THE SECRETARY, DEPT. OF HEALTH AND HUMAN SERVICES NATIONAL INSTITUTES OF HEALTH | Method of inhibiting a chaperone protein |
| FR2792320B1 (en) * | 1999-04-19 | 2003-05-09 | Hoechst Marion Roussel Inc | NOVEL AROMATIC AMIDES SUBSTITUTED BY RIBOSIS, THEIR PREPARATION PROCESS AND THEIR APPLICATION AS MEDICAMENTS |
| EP1287021A2 (en) * | 2000-05-18 | 2003-03-05 | Bayer Aktiengesellschaft | Human galanin receptor-like g protein coupled receptor |
| WO2002094259A1 (en) * | 2001-05-03 | 2002-11-28 | MAX-PLANCK-Gesellschaft zur Förderung der Wissenschaften e.V. | Compounds that inhibit hsp90 and stimulate hsp70 and hsp40, useful in the prevention or treatment of diseases associated with protein aggregation and amyloid formation |
| EP1457499A1 (en) * | 2003-03-12 | 2004-09-15 | Tufts University School Of Medicine | Inhibitors of extracellular Hsp90 |
| US8212011B2 (en) * | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues |
| WO2006050501A2 (en) | 2004-11-03 | 2006-05-11 | University Of Kansas | Novobiocin analogues as anticancer agents |
| US7622451B2 (en) * | 2004-11-03 | 2009-11-24 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| US8212012B2 (en) * | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
-
2005
- 2005-11-03 WO PCT/US2005/039990 patent/WO2006050501A2/en not_active Ceased
- 2005-11-03 EP EP05824355.1A patent/EP1807440B1/en not_active Expired - Lifetime
- 2005-11-03 CA CA2585091A patent/CA2585091C/en not_active Expired - Fee Related
- 2005-11-03 AU AU2005301957A patent/AU2005301957B2/en not_active Ceased
- 2005-11-03 US US11/266,149 patent/US7608594B2/en not_active Expired - Fee Related
-
2009
- 2009-10-01 US US12/571,899 patent/US7811998B2/en not_active Expired - Fee Related
Non-Patent Citations (6)
| Title |
|---|
| MADHAVAN ET AL.: "Novel Coumarin Derivatives of Heterocyclic Compounds as Lipid Lowering Agents", BIOORG. MED. CHERN. LETT., vol. 13, 2003, pages 2547, XP055082572, DOI: doi:10.1016/S0960-894X(03)00490-6 |
| See also references of EP1807440A4 |
| SHEN ET AL.: "Syntheses of Photolabile Novobiocin Analogues", BIOORG. MED. CHEM. LETT., vol. 14, 2004, pages 5903, XP004611145, DOI: doi:10.1016/j.bmcl.2004.09.017 |
| SHEN ET AL.: "Synthesis of Photolabile Novobiocin Analogues", BIOORG. MED. CHERN. LETT., vol. 14, 2004, pages 5903 - 5906, XP004611145, DOI: doi:10.1016/j.bmcl.2004.09.017 |
| YU ET AL.: "Hsp90 Inhibitors Identified from a Library of Novoboicin Analogues", J. AM. CHEM. SOC., vol. 127, 2005, pages 12778 - 12779 |
| YU ET AL.: "Synthesis of (-)-Noviose from 2,3-0-Isopropylidene-D-erythronolactol", J. ORG. CHERN., vol. 69, 2004, pages 7375 - 7378, XP002489260, DOI: doi:10.1021/jo048953t |
Cited By (135)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9120774B2 (en) | 2004-11-03 | 2015-09-01 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
| US8212012B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues having modified sugar moieties |
| US8212011B2 (en) | 2004-11-03 | 2012-07-03 | University Of Kansas | Novobiocin analogues |
| US7811998B2 (en) | 2004-11-03 | 2010-10-12 | University Of Kansas | Novobiocin analogues as anticancer agents |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| US8247408B2 (en) | 2005-10-07 | 2012-08-21 | Exelixis, Inc. | Pyridopyrimidinone inhibitors of PI3Kα for the treatment of cancer |
| US9493467B2 (en) | 2006-04-04 | 2016-11-15 | The Regents Of The University Of California | PI3 kinase antagonists |
| US8642604B2 (en) | 2006-04-04 | 2014-02-04 | The Regents Of The University Of California | Substituted pyrazolo[3,2-d]pyrimidines as anti-cancer agents |
| US8633204B2 (en) | 2006-09-15 | 2014-01-21 | Pfizer Inc. | 4-methylpyridopyrimidinone compounds |
| US8273755B2 (en) | 2006-09-15 | 2012-09-25 | Pfizer Inc | 4-methylpyridopyrimidinone compounds |
| US9365602B2 (en) | 2006-09-29 | 2016-06-14 | Lexicon Pharmaceuticals, Inc. | Sodium glucose co-transporter inhibitors and methods of their use |
| US8476413B2 (en) | 2006-09-29 | 2013-07-02 | Lexicon Pharmaceuticals, Inc. | Sulfanyl-tetrahydropyran-based compounds and methods of their use |
| US7781577B2 (en) | 2006-09-29 | 2010-08-24 | Lexicon Pharmaceuticals, Inc. | Inhibitors of sodium glucose co-transporter 2 and methods of their use |
| WO2008049994A1 (en) | 2006-10-24 | 2008-05-02 | Sanofi-Aventis | New fluorene derivatives, compositions containing the same and use thereof as inhibitors of the protein chaperone hsp 90 |
| US8163750B2 (en) | 2006-10-24 | 2012-04-24 | Sanofi-Aventis | Fluorene derivatives, compositions containing the same and use thereof as inhibitors of the protein chaperone HSP 90 |
| US7846945B2 (en) | 2007-03-08 | 2010-12-07 | Lexicon Pharmaceuticals, Inc. | Piperdine-based inhibitors of sodium glucose co-transporter 2 and methods of their use |
| US7960353B2 (en) | 2007-05-10 | 2011-06-14 | University Of Kansas | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| WO2009059214A1 (en) * | 2007-11-02 | 2009-05-07 | The Regents Of The University Of California | Abeta-binding small molecules |
| US8168800B2 (en) | 2007-11-02 | 2012-05-01 | The Regents Of The University Of California | Aβ-binding small molecules |
| US8703777B2 (en) | 2008-01-04 | 2014-04-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9655892B2 (en) | 2008-01-04 | 2017-05-23 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8785456B2 (en) | 2008-01-04 | 2014-07-22 | Intellikine Llc | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US11433065B2 (en) | 2008-01-04 | 2022-09-06 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9216982B2 (en) | 2008-01-04 | 2015-12-22 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US9822131B2 (en) | 2008-01-04 | 2017-11-21 | Intellikine Llc | Certain chemical entities, compositions and methods |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US9637492B2 (en) | 2008-03-14 | 2017-05-02 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9828378B2 (en) | 2008-07-08 | 2017-11-28 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9629843B2 (en) | 2008-07-08 | 2017-04-25 | The Regents Of The University Of California | MTOR modulators and uses thereof |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US9790228B2 (en) | 2008-09-26 | 2017-10-17 | Intellikine Llc | Heterocyclic kinase inhibitors |
| US8697709B2 (en) | 2008-10-16 | 2014-04-15 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US10329318B2 (en) | 2008-12-02 | 2019-06-25 | Wave Life Sciences Ltd. | Method for the synthesis of phosphorus atom modified nucleic acids |
| EP2438078A4 (en) * | 2009-02-20 | 2012-12-19 | Univ Kansas | NOVOBIOCIN ANALOGUES HAVING MODIFIED GLUCIDIC FUNCTIONAL GROUPS |
| JP2012518645A (en) * | 2009-02-20 | 2012-08-16 | ユニバーシティ・オブ・カンザス | Novobiocin analogs having modified sugar moieties |
| US9315505B2 (en) | 2009-05-07 | 2016-04-19 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US8785454B2 (en) | 2009-05-07 | 2014-07-22 | Intellikine Llc | Heterocyclic compounds and uses thereof |
| US10307434B2 (en) | 2009-07-06 | 2019-06-04 | Wave Life Sciences Ltd. | Nucleic acid prodrugs and methods of use thereof |
| WO2011004132A1 (en) | 2009-07-10 | 2011-01-13 | Sanofi-Aventis | Novel hsp90-inhibiting indole derivatives, compositions containing said derivatives, and use thereof |
| US9206182B2 (en) | 2009-07-15 | 2015-12-08 | Intellikine Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US9522146B2 (en) | 2009-07-15 | 2016-12-20 | Intellikine Llc | Substituted Isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| US8569323B2 (en) | 2009-07-15 | 2013-10-29 | Intellikine, Llc | Substituted isoquinolin-1(2H)-one compounds, compositions, and methods thereof |
| WO2011027081A2 (en) | 2009-09-03 | 2011-03-10 | Sanofi-Aventis | Novel derivatives of 5,6,7,8-tetrahydroindolizine inhibiting hsp90, compositions containing same, and use thereof |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US8604032B2 (en) | 2010-05-21 | 2013-12-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9181221B2 (en) | 2010-05-21 | 2015-11-10 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US9738644B2 (en) | 2010-05-21 | 2017-08-22 | Infinity Pharmaceuticals, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| US10428019B2 (en) | 2010-09-24 | 2019-10-01 | Wave Life Sciences Ltd. | Chiral auxiliaries |
| CN103429254A (en) * | 2010-10-11 | 2013-12-04 | 南加利福尼亚大学 | Secreted Heat Shock Protein-90α (Hsp90α) Fragments as Vaccines or Epitopes or Small Molecule Drug Targets for Monoclonal Antibody Drugs Against a Range of Human Solid Tumors |
| US8901133B2 (en) | 2010-11-10 | 2014-12-02 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9388183B2 (en) | 2010-11-10 | 2016-07-12 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11312718B2 (en) | 2011-01-10 | 2022-04-26 | Infinity Pharmaceuticals, Inc. | Formulations of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one |
| USRE46621E1 (en) | 2011-01-10 | 2017-12-05 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9290497B2 (en) | 2011-01-10 | 2016-03-22 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9840505B2 (en) | 2011-01-10 | 2017-12-12 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1 (2H)-one and methods of use thereof |
| US10550122B2 (en) | 2011-01-10 | 2020-02-04 | Infinity Pharmaceuticals, Inc. | Solid forms of (S)-3-(1-(9H-purin-6-ylamino)ethyl)-8-chloro-2-phenylisoquinolin-1(2H)-one and methods of use thereof |
| US8809349B2 (en) | 2011-01-10 | 2014-08-19 | Infinity Pharmaceuticals, Inc. | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9718815B2 (en) | 2011-07-19 | 2017-08-01 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10280192B2 (en) | 2011-07-19 | 2019-05-07 | Wave Life Sciences Ltd. | Methods for the synthesis of functionalized nucleic acids |
| US9056877B2 (en) | 2011-07-19 | 2015-06-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9605003B2 (en) | 2011-07-19 | 2017-03-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8969363B2 (en) | 2011-07-19 | 2015-03-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9546180B2 (en) | 2011-08-29 | 2017-01-17 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8785470B2 (en) | 2011-08-29 | 2014-07-22 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9115141B2 (en) | 2011-08-29 | 2015-08-25 | Infinity Pharmaceuticals, Inc. | Substituted isoquinolinones and methods of treatment thereof |
| US9321772B2 (en) | 2011-09-02 | 2016-04-26 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US9895373B2 (en) | 2011-09-02 | 2018-02-20 | The Regents Of The University Of California | Substituted pyrazolo[3,4-D]pyrimidines and uses thereof |
| US11021509B2 (en) | 2011-12-22 | 2021-06-01 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10464965B2 (en) | 2011-12-22 | 2019-11-05 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US9073960B2 (en) | 2011-12-22 | 2015-07-07 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10590157B2 (en) | 2012-02-09 | 2020-03-17 | University Of Kansas | C-terminal HSP90 inhibitors |
| CN104271587A (en) * | 2012-02-09 | 2015-01-07 | 堪萨斯大学 | Hsp90C terminal inhibitors |
| US9422320B2 (en) | 2012-02-09 | 2016-08-23 | University Of Kansas | C-terminal HSP90 inhibitors |
| US10882881B2 (en) | 2012-02-09 | 2021-01-05 | University Of Kansas | C-terminal Hsp90 inhibitors |
| US12024536B2 (en) | 2012-02-09 | 2024-07-02 | University Of Kansas | C-terminal HSP90 inhibitors |
| CN107474085B (en) * | 2012-02-09 | 2021-05-11 | 堪萨斯大学 | Hsp90 C-terminal inhibitor and pharmaceutical composition and use thereof |
| CN107474085A (en) * | 2012-02-09 | 2017-12-15 | 堪萨斯大学 | Hsp90 C-terminals inhibitor and its medical composition and purposes |
| US11390640B2 (en) | 2012-02-09 | 2022-07-19 | University Of Kansas | C-terminal Hsp90 inhibitors |
| CN104271587B (en) * | 2012-02-09 | 2017-09-12 | 堪萨斯大学 | Hsp90 C-terminal inhibitor |
| US10030041B2 (en) | 2012-02-09 | 2018-07-24 | University Of Kansas | C-terminal Hsp90 inhibitors |
| USRE48171E1 (en) | 2012-03-21 | 2020-08-25 | Janssen Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US10485815B2 (en) | 2012-03-21 | 2019-11-26 | Alios Biopharma, Inc. | Substituted nucleosides, nucleotides and analogs thereof |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9255108B2 (en) | 2012-04-10 | 2016-02-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US9527847B2 (en) | 2012-06-25 | 2016-12-27 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| US10167309B2 (en) | 2012-07-13 | 2019-01-01 | Wave Life Sciences Ltd. | Asymmetric auxiliary group |
| US10131668B2 (en) | 2012-09-26 | 2018-11-20 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pYRAZINES for modulation of IRE1 |
| US11613544B2 (en) | 2012-09-26 | 2023-03-28 | The Regents Of The University Of California | Substituted imidazo[1,5-a]pyrazines for modulation of IRE1 |
| US10822340B2 (en) | 2012-09-26 | 2020-11-03 | The Regents Of The University Of California | Substituted imidazolopyrazine compounds and methods of using same |
| US12213983B2 (en) | 2012-11-01 | 2025-02-04 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using PI3 kinase isoform modulators |
| WO2014106623A1 (en) * | 2013-01-04 | 2014-07-10 | Max-Planck-Gesellschaft zur Förderung der Wissenschaft e.V. | C-terminal hsp90 inhibitors to treat pituitary adenomas |
| US9877947B2 (en) | 2013-01-04 | 2018-01-30 | Max-Planck-Gesellschaft zur Föderung der Wissenschaften E.V. | C-terminal HSP90 inhibitors to treat pituitary adenomas |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| CN103450133B (en) * | 2013-09-16 | 2015-07-08 | 中国药科大学 | Scopoletin derivatives with anti-tumor activity, and preparation method and application thereof |
| CN103450133A (en) * | 2013-09-16 | 2013-12-18 | 中国药科大学 | Scopoletin derivatives with anti-tumor activity, and preparation method and application thereof |
| US9359365B2 (en) | 2013-10-04 | 2016-06-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9828377B2 (en) | 2013-10-04 | 2017-11-28 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US12152032B2 (en) | 2013-10-04 | 2024-11-26 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9751888B2 (en) | 2013-10-04 | 2017-09-05 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10329299B2 (en) | 2013-10-04 | 2019-06-25 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10160969B2 (en) | 2014-01-16 | 2018-12-25 | Wave Life Sciences Ltd. | Chiral design |
| US10675286B2 (en) | 2014-03-19 | 2020-06-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9775844B2 (en) | 2014-03-19 | 2017-10-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11541059B2 (en) | 2014-03-19 | 2023-01-03 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11110096B2 (en) | 2014-04-16 | 2021-09-07 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11944631B2 (en) | 2014-04-16 | 2024-04-02 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US9994556B2 (en) | 2014-06-13 | 2018-06-12 | University Of Kansas | Triazole modified coumarin and biphenyl amide-based HSP90 inhibitors |
| US10590065B2 (en) | 2014-06-24 | 2020-03-17 | University Of Kansas | Biphenyl amides with modified ether groups as HSP90 inhibitors and HSP70 inducers |
| US11098008B2 (en) | 2014-06-24 | 2021-08-24 | University Of Kansas | Biphenyl amides with modified ether groups as Hsp90 inhibitors and Hsp70 inducers |
| US11708319B2 (en) | 2014-06-24 | 2023-07-25 | University Of Kansas | Biphenyl amides with modified ether groups as HSP90 inhibitors and HSP70 inducers |
| US10941162B2 (en) | 2014-10-03 | 2021-03-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| US10253047B2 (en) | 2014-10-03 | 2019-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11247995B2 (en) | 2015-09-14 | 2022-02-15 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US12384792B2 (en) | 2015-09-14 | 2025-08-12 | Twelve Therapeutics, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US11939333B2 (en) | 2015-09-14 | 2024-03-26 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| US11147818B2 (en) | 2016-06-24 | 2021-10-19 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US11224608B2 (en) | 2017-10-16 | 2022-01-18 | Dana-Farber Cancer Institute, Inc. | Compounds and methods for treating cancer |
| USRE50319E1 (en) | 2017-10-16 | 2025-03-04 | Dana-Farber Cancer Institute, Inc. | Compounds and methods for treating cancer |
| CN108484495B (en) * | 2018-04-12 | 2021-05-04 | 苏州康润医药有限公司 | Synthetic method of 3-bromo-7-hydroxyquinoline |
| CN108484495A (en) * | 2018-04-12 | 2018-09-04 | 苏州康润医药有限公司 | A kind of synthetic method of the bromo- 7- oxyquinolines of 3- |
| US11827664B2 (en) | 2018-05-14 | 2023-11-28 | Reata Pharmaceuticals, Inc | Biaryl amides with modified sugar groups for treatment of diseases associated with heat shock protein pathway |
| US12595278B2 (en) | 2018-05-14 | 2026-04-07 | Reata Pharmaceuticals, Inc | Biaryl amides with modified sugar groups for treatment of diseases associated with heat shock protein pathway |
| US12187721B2 (en) | 2018-10-17 | 2025-01-07 | Array Biopharma Inc. | Protein tyrosine phosphatase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2006050501A3 (en) | 2007-05-31 |
| EP1807440A2 (en) | 2007-07-18 |
| US7608594B2 (en) | 2009-10-27 |
| EP1807440B1 (en) | 2020-02-19 |
| US7811998B2 (en) | 2010-10-12 |
| AU2005301957B2 (en) | 2012-02-23 |
| AU2005301957A1 (en) | 2006-05-11 |
| US20060199776A1 (en) | 2006-09-07 |
| US20100048882A1 (en) | 2010-02-25 |
| CA2585091A1 (en) | 2006-05-11 |
| EP1807440A4 (en) | 2013-12-11 |
| CA2585091C (en) | 2016-07-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1807440B1 (en) | Novobiocin analogues as anticancer agents | |
| US7622451B2 (en) | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders | |
| US7960353B2 (en) | Novobiocin analogues as neuroprotective agents and in the treatment of autoimmune disorders | |
| US8212011B2 (en) | Novobiocin analogues | |
| EP2438078B1 (en) | Novobiocin analogues having modified sugar moieties | |
| US8212012B2 (en) | Novobiocin analogues having modified sugar moieties | |
| US9120774B2 (en) | Novobiocin analogues having modified sugar moieties | |
| Kassem et al. | Synthesis and anticancer activity of new ((Furan-2-yl)-1, 3, 4-thiadiazolyl)-1, 3, 4-oxadiazole acyclic sugar derivatives | |
| US20110082098A1 (en) | Novobiocin analogues and treatment of polycystic kidney disease | |
| US7368434B2 (en) | Compounds useful for the inhibition of ALDH | |
| AU2003234733B2 (en) | Compounds, compositions, and methods | |
| RO120951B1 (en) | COMPOUND FOR TREATMENT OF TUMOR AND INFLAMMATORY DISEASES | |
| JPS5973598A (en) | 20-aminomacrolide derivative | |
| US8916526B2 (en) | Flavanone derivative | |
| EP1846428A1 (en) | Anti-inflammatory conjugates of macrolides and coumarins | |
| CA2135850C (en) | Esculetin derivatives, and method for manufacture thereof, use thereof, and pharmaceutical composition | |
| WO2003069350A2 (en) | Small molecule modulators of apoptosis | |
| JPH0564631B2 (en) |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KN KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005301957 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005824355 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2585091 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005301957 Country of ref document: AU Date of ref document: 20051103 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005824355 Country of ref document: EP |