WO2006062129A1 - (5α,7α)-3-スピロ-2’-(1’,3’-ジオキソラン)-24-オキソコレスト-22-エン-7-イル ベンゾアートの製造方法 - Google Patents
(5α,7α)-3-スピロ-2’-(1’,3’-ジオキソラン)-24-オキソコレスト-22-エン-7-イル ベンゾアートの製造方法 Download PDFInfo
- Publication number
- WO2006062129A1 WO2006062129A1 PCT/JP2005/022457 JP2005022457W WO2006062129A1 WO 2006062129 A1 WO2006062129 A1 WO 2006062129A1 JP 2005022457 W JP2005022457 W JP 2005022457W WO 2006062129 A1 WO2006062129 A1 WO 2006062129A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- unsaturated ketone
- spiro
- benzoate
- oxocholest
- solution
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J21/00—Normal steroids containing carbon, hydrogen, halogen or oxygen having an oxygen-containing hetero ring spiro-condensed with the cyclopenta(a)hydrophenanthrene skeleton
Definitions
- the present invention relates to a method for producing (5 ⁇ , 70;)-3-spiro2,-(1 ', 3, -dioxolan) 24-oxocholest 22en-7-inole benzoate, which is useful as an intermediate for the synthesis of squalamine. .
- the squalamine shown in [0004] has been reported to have a strong antibacterial activity against gram-positive bacteria, gram-negative bacteria, fungi, etc., and also has anticancer activity, and has attracted attention as a new antibiotic. This compound.
- a solution of potassium bromide was added to a methylene chloride solution of the alcohol compound represented by [0010] (hereinafter sometimes referred to as alcohol (IV)) and 2, 2, 6, 6-tetramethylpiperidine N-oxyl. Then, an aqueous solution adjusted to pH 9.5 by adding sodium hydrogen carbonate to an aqueous sodium hypochlorite solution was added, and the following formula (II)
- step (a ′) A step of obtaining an aldehyde compound represented by the following (hereinafter sometimes referred to as aldehyde ( ⁇ )) (hereinafter also referred to as step (a ′)); and
- step (b ') An unsaturated ketone (I) is reacted with a reaction product of a jetyl (3-methyl-2-oxobutyl) phosphonate (hereinafter sometimes referred to as compound (II ⁇ )) and sodium hydride represented by Step to obtain (hereinafter sometimes referred to as step (b ').)
- Patent Document 1 A powerful method is known (see Patent Document 1 and Non-Patent Document 1).
- Patent Document 1 Pamphlet of International Publication No. 98Z24800
- Non-Patent Document 1 Journal of Organic Chemistry, 1998, 63rd, ⁇ 3786
- unsaturated ketone (I) is usually a compound having gummy properties, and it is difficult to carry out purification by recrystallization, which is one of industrially advantageous purification methods. It is.
- the crude product is dissolved in 5 ml of methanol containing 4 drops of pyridine, and the resulting solution is dissolved.
- the solution was solidified by adding dropwise to 100 ml of water while shaking, and the resulting solid was filtered off and then washed with 20 ml of water three times to obtain 499 mg of unsaturated ketone (I) in a yield of 90%. 85-12 Obtained as a solid with a melting point of 1 ° C.
- the problem to be solved by the present invention is that a large amount of a solvent and an unsaturated ketone
- Unsaturated keton (I) that can be used as an intermediate for the synthesis of squalamine in a simple and industrially advantageous manner, without the need to use a large amount of a poor solvent necessary for precipitating (I) as a solid. Is to provide a method capable of producing a high purity and high recovery rate.
- the unsaturated ketone (I) is insoluble in water.
- the present inventors have disclosed a method for obtaining unsaturated ketone (I) as a solid by the method disclosed in Patent Document 1 and Non-Patent Document 1 in the field of polymer chemistry, which is referred to as so-called reprecipitation operation.
- Unsaturated ketone (I) that can be used as a synthetic intermediate of squalamine has a narrower melting temperature range and higher purity than the prior art, without using a large amount of the poor solvent necessary for precipitating (I) as a solid.
- the present invention was completed by finding that it can be easily obtained at a high recovery rate. That is, the present invention is shown as follows.
- the crude product containing the unsaturated ketone (I) obtained from the reaction step is used in a mass ratio of 1 to the unsaturated ketone (I). Dissolve in methanol in the range of 5 to 3 to obtain a solution, and add the solution to the water in the range of 5 to 30 as a mass ratio of the unsaturated ketone (I) to the unsaturated ketone (I).
- a method for producing an unsaturated ketone (I), comprising solidifying to obtain a suspension, separating a solid from the suspension, and drying the obtained solid.
- unsaturated ketone (I) which is useful as a synthesis intermediate of squalamine having antibacterial activity and anticancer activity, can be produced simply, industrially advantageously and with high purity, which is useful as an intermediate for synthesis. It can be manufactured with a high recovery rate.
- the unsaturated ketone (I) can be preferably produced by a reaction in which an aldehyde ( ⁇ ⁇ ⁇ ⁇ ⁇ ) and a metal phosphonate ( ⁇ ) are condensed.
- the metal phosphonate (III) used in this reaction step has the following general formula ( ⁇ ⁇ ′) [0027] [7]
- the alkyl group represented by R in the general formula ( ⁇ ) and the general formula ( ⁇ ') preferably has 1 to 4 carbon atoms, such as a methyl group, an ethyl group, a propyl group, an isopropyl group, and a butyl group. Can be mentioned.
- the aryl group represented by R preferably has 6 to 10 carbon atoms, and examples thereof include a phenyl group, a tolyl group, and a naphthyl group.
- Examples of the metal atom represented by Met include alkali metal atoms such as lithium, sodium and potassium.
- the basic substance to be reacted with phosphonate ( ⁇ ') to form metal phosphonate ( ⁇ ) is the one that is usually used to produce the corresponding metal salt from ketophosphonate in the field of synthetic organic chemistry. Can be used.
- metal hydrides such as sodium hydride and potassium hydride
- alkali metal alkoxides such as sodium methoxide, sodium ethoxide, sodium t-butoxide, sodium t-amyloxide, potassium t-butoxide
- alkyl metals such as sodium amide, potassium amide, lithium diisopropylamide, lithium jetylamide, lithium hexamethyldisilazide, sodium hexamethyldisilazide, and potassium hexamethyldisilazide.
- Metal alkoxides, especially tertiary alcohol alkali metal alkoxides such as sodium t-butoxide, are preferred from the standpoint of industrial handling and ease of reaction control.
- the amount of basic substance used is usually in the range of 0.8 to 2 moles per mole of phosphonate ( ⁇ ,), but if excess basic substance remains, aldehyde ( ⁇ ) In the reaction with aldehyde ( ⁇ ), side effects such as epimerization of the 20-position methyl group of aldehyde ( ⁇ ) may be adversely affected. so Preferably there is. Specifically, it is preferably in the range of 0.8 to 1.1 mol per mol of phosphonate ( ⁇ ,)! /.
- the reaction of phosphonate ( ⁇ ') and a basic substance is usually carried out in a solvent.
- the solvent used include ethers such as tetrahydrofuran, dioxane, and dimethoxyethane; aromatic hydrocarbons such as toluene; dimethyl sulfoxide, ⁇ , ⁇ ⁇ ⁇ ⁇ -dimethylformamide, ⁇ -methylpyrrolidone, and the like.
- ethers such as tetrahydrofuran, dioxane, and dimethoxyethane
- aromatic hydrocarbons such as toluene
- dimethyl sulfoxide ⁇ , ⁇ ⁇ ⁇ ⁇ -dimethylformamide, ⁇ -methylpyrrolidone, and the like.
- the reaction temperature is usually in the range of ⁇ 20 to 50 ° C.
- the metal phosphonate ( ⁇ ) thus obtained is usually used directly for the reaction with aldehyde ( ⁇ ) without purification.
- the reaction is usually carried out in a solvent.
- the same solvents as those used in the reaction of phosphoner ( ⁇ ′) with a basic substance are the same.
- ethers such as tetrahydrofuran are preferred.
- This reaction is usually carried out by adding a metal phosphate ( ⁇ ) or a solution thereof to a solution obtained by dissolving aldehyde (II) in a solvent, or an aldehyde in a metal phosphonate ( ⁇ ) or a solution thereof. This is done by adding the solution of (ii).
- the amount of aldehyde ( ⁇ ) used is usually in the range of 0.5 to 2 mol per mol of phosphonate ( ⁇ ,,). If aldehyde ( ⁇ ) remains after the reaction, Since separation from the saturated ketone (I) is difficult, it is preferable that the aldehyde (soot) is consumed as much as possible in the reaction. More specifically, the amount of aldehyde ( ⁇ ) used is preferably in the range of 0.5 to 0.95 mol with respect to 1 mol of phosphonate ( ⁇ ,).
- the amount of the solvent for dissolving the aldehyde ( ⁇ ) is usually in the range of 2 to 50 times the mass of the aldehyde ( ⁇ ).
- the reaction temperature is usually in the range of -20 to 50 ° C.
- the reaction time is a force that can vary depending on the type of metal phosphonate ( ⁇ ), the amount ratio used, the reaction temperature, etc. Usually, it is in the range of 0.5 to 48 hours.
- reaction mixture Add water or aqueous sodium hydrogen carbonate solution to the compound, dilute the organic layer with an organic solvent as necessary, separate the organic layer from the aqueous layer, and extract the aqueous layer with an organic solvent such as isopropyl ether or ethyl acetate.
- organic solvent such as isopropyl ether or ethyl acetate.
- the extract is combined with the previously separated organic layer, washed with an aqueous alkaline solution as necessary, and concentrated to obtain a crude product containing an unsaturated ketone (I).
- the crude product containing the unsaturated ketone (I) thus obtained has a mass ratio of 1.5 to 3 with respect to the unsaturated ketone (I) contained in the crude product.
- the resulting methanol solution is added dropwise to the unsaturated ketone (I) contained in the crude product by a method such as dropwise addition to water in the range of 5 to 30 as a mass ratio.
- a suspension in which the unsaturated ketone (I) is dispersed as a fine solid that can be handled industrially can be obtained.
- the ratio of methanol and water used is important.
- the amount of methanol used relative to the unsaturated ketone (I) contained in the crude product is less than 1.5, the crude product of the unsaturated ketone (I) is completely dissolved. If it exceeds 3, the amount of water required to increase the recovery rate of unsaturated ketone (I) will increase, so it is necessary to precipitate a large amount of solvent and unsaturated ketone (I) as a solid. It is necessary to use a large amount of a poor solvent.
- the amount of water used relative to the unsaturated ketone (I) contained in the crude product is less than 5 as a mass ratio, the recovery rate of the unsaturated ketone (I) decreases. As in the comparative example, the entire system is solidified and it becomes extremely difficult to take out. On the other hand, if it exceeds 30, the amount of the poor solvent necessary for precipitating the solvent and product as a solid increases, which is disadvantageous in terms of production cost.
- the power to be added can be selected as appropriate.Since adding too quickly may result in a suspension that is difficult to handle industrially, and if too much time is used, productivity will decrease. It is preferable to add in the range of 5 minutes to 2 hours (for example, dropwise addition). It is also possible to age the suspension after addition. Aging can be carried out usually by stirring at a temperature in the range of 10 to 30 ° C for 5 minutes to 3 hours.
- Industrially available methanol may contain acidic substances such as formic acid.
- acidic substances such as formic acid.
- a basic substance can be added to methanol in which the crude product containing is dissolved.
- powerful basic substances include pyridine and sodium hydrogen carbonate, and the amount used is usually in the range of 1 to 3 times the amount necessary to neutralize acidic substances contained in methanol. It is.
- a basic substance such as sodium hydrogencarbonate can be added to the water side to which the methanol solution of the crude product containing the unsaturated ketone (I) is added.
- the amount used is usually in the range of 1 to 3 times the amount necessary to neutralize the acidic substances contained in methanol.
- Unsaturated ketone (I) can be obtained by separating the solid from the suspension thus obtained.
- a method for separating the solid for example, a method that is usually industrially performed, such as vacuum filtration, pressure filtration, and centrifugal filtration, can be appropriately selected. At this time, for the purpose of smoothly separating the solid, it is preferable to carry out the separation operation while keeping the temperature at the time of addition of the suspension or when it is aged.
- Unsaturated ketone (I) can be obtained with high purity and good recovery rate by washing the obtained solid with water as necessary and then drying.
- the aldehyde ( ⁇ ) can be obtained by oxidizing the hydroxyl group of the alcohol (IV). it can.
- This oxidation can be carried out using various oxidants, for example 2, 2, 6, 6-tetramethylpiperidine N-oxyl, 4-methoxy-2,2,6,6-tetramethylpiperidine N —Oxyl, 4-acetoxy 2, 2, 6, 6—tetramethylpiperidine N-oxyl compound catalysts such as sodium hypochlorite such as sodium hypochlorite Can be mentioned. This method is described in detail below. .
- This oxidation reaction is usually carried out in a solvent.
- the solvent to be used include halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane, and chloroform; esters such as ethyl acetate and butylacetate; aromatic hydrocarbons such as toluene, xylene, and mesitylene; pentane, And hydrocarbons such as hexane, heptane, and octane.
- halogenated hydrocarbons such as dichloromethane, 1,2-dichloroethane, and chloroform
- esters such as ethyl acetate and butylacetate
- aromatic hydrocarbons such as toluene, xylene, and mesitylene
- pentane And hydrocarbons such as hexane, heptane, and octane.
- hypochlorite added as an oxidizing agent is usually added in the form of an a
- the amount of hypochlorite used is usually in the range of 0.8 to 2 times the amount necessary to oxidize the hydroxyl group of alcohol (IV) as the oxidizing agent. However, if the amount used is small, the raw material alcohol (IV) may remain, which may make purification difficult. If the amount used is excessive, the product aldehyde ( ⁇ ) Since the yield may be lowered by further acidification, it is preferable to use only the amount necessary to consume alcohol (IV) within the possible range. Specifically, the conversion of alcohol (IV) is traced during the reaction, and the addition of hypochlorite is terminated when the disappearance of alcohol (IV) is confirmed.
- hypochlorite is preferably added as an aqueous solution of ⁇ 11-12.
- the pH of the aqueous phase of the reaction system becomes too high due to the additive, there is a high risk of side reactions such as epimerization of the 20-position methyl group of the product aldehyde ( ⁇ ).
- the control range of the pH of the aqueous phase is preferably 7 to 11 in consideration of the oxidation reaction rate and the possibility of side reactions.
- Methods for controlling this pH include, for example, a method in which a buffer solution capable of controlling pH to 7 to L 1 is premixed in an aqueous phase, a method in which a substance having a buffering capacity is added in advance, a hypochlorite salt For example, a method of adding an acidic substance in accordance with the addition amount of the borate aqueous solution.
- a buffer solution capable of controlling pH to 7 to L 1 is premixed in an aqueous phase
- a method in which a substance having a buffering capacity is added in advance
- a hypochlorite salt For example, a method of adding an acidic substance in accordance with the addition amount of the borate aqueous solution.
- the substance having a buffer capacity include sodium hydrogen carbonate.
- the amount of the N-oxyl compound used as a catalyst is not particularly limited, but is usually in the range of 0.001 to 0.10 monole with respect to the alcohol (IV), and 0.001 to 0.05 More preferable than monore range power.
- the reaction temperature is preferably in the range of 20 to 70 ° C, more preferably in the range of -10 to 40 ° C.
- the reaction time varies depending on the reaction conditions, and in practice, it can be varied by tracking the conversion rate of alcohol (IV). In general, it is usually in the range of 0.5 to LO time. is there.
- a cocatalyst can be used to facilitate the reaction.
- the cocatalyst include lithium bromide, sodium bromide, potassium bromide, lithium iodide, sodium iodide, potassium iodide, and the like. Among these, sodium bromide and potassium bromide are preferable.
- the amount added is usually in the range of 0.05 to 0.5 mol per 1 mol of alcohol (IV).
- the method for isolating and purifying the aldehyde ( ⁇ ) obtained by vigorous acid-acid reaction is not particularly limited, and the organic compound such as recrystallization or column chromatography is performed after the extraction operation. It is possible to employ a method commonly used for isolation and purification. In addition, without adding a refining operation, the extract can be concentrated and dissolved in a solvent used for the reaction in the next step, and used in the next step.
- Example 1 (5 ⁇ , 7 ⁇ ) — 3-spiro-1, 2- (1, 3, 1, 1-xolane) 1-24-oxocholest 22- 7-yl benzoate (production of unsaturated ketone (I)) )
- the obtained methanol solution was dropped into 1128 g of water in which sodium hydrogen carbonate lg was dissolved (9.98 times by mass with respect to the unsaturated ketone (I)) at 15 to 20 ° C. over 1 hour. After completion of the dropwise addition, the obtained suspension was stirred at 0 to 4 ° C for 1 hour and then filtered at the same temperature. The obtained solid was washed with water and then dried under reduced pressure at 50 ° C. for 10 hours to obtain unsaturated keton (I) having the following physical properties as an off-white powder (117 g, purity 95% (By HPLC analysis), melting point: 87-92 ° C).
- Patent Document 1 a small amount is obtained to obtain 499 mg of unsaturated ketone (I). Dissolve in 5 ml of methanol containing 10 parts of pyridine (10 mass times with respect to unsaturated ketone (I)), and drop the solution into 100 ml of water (200 mass times with respect to unsaturated ketone (I)) to solidify. However, the melting point is 85 to 121 ° C., which is a wide range of low-purity unsaturated ketones (I), and no strength is obtained.
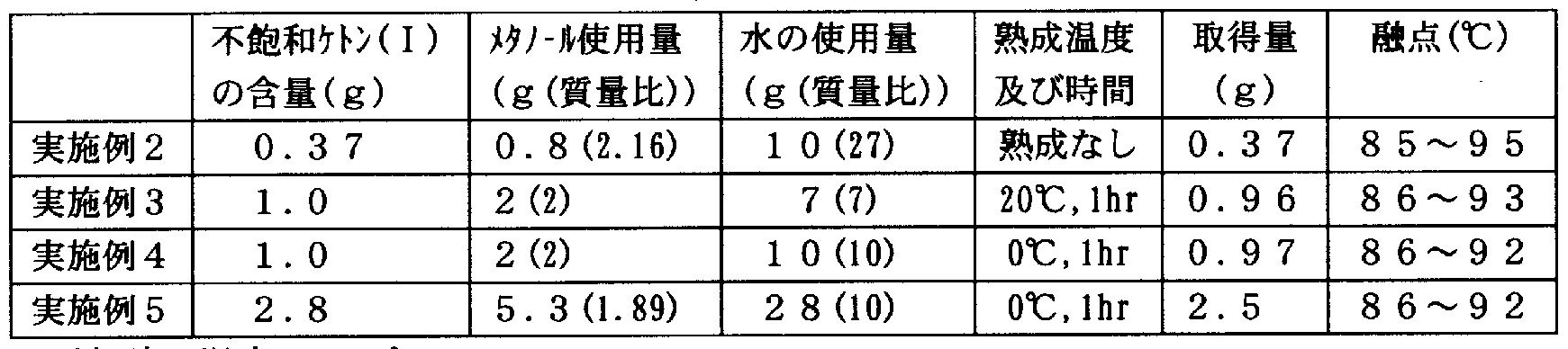
- the crude reaction product obtained in the same manner as in Example 1 was dissolved in various solvents and dropped into water. Summarizes the solvent types used and their usage (usage ratio (mass ratio) to unsaturated ketone (I)) and water (use ratio (mass ratio) to unsaturated ketone (I)). The results are shown in Table 2. In the case of V and shear, the unsaturated ketone (I) was handled! / And could not be obtained as an easy solid.
- Unsaturated ketone (I) can be advantageously produced industrially without using a large amount of solvent and water, which is a poor solvent necessary for solidifying the product.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Steroid Compounds (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description







Claims




Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US11/721,309 US20090259038A1 (en) | 2004-12-08 | 2005-12-07 | Process for producing (5alpha7alpha)-3-spiro-2'-(1',3'-dioxolan)-24-oxocholest-22-en-7-yl benzoate |
| JP2006546733A JPWO2006062129A1 (ja) | 2004-12-08 | 2005-12-07 | (5α,7α)−3−スピロ−2’−(1’,3’−ジオキソラン)−24−オキソコレスト−22−エン−7−イルベンゾアートの製造方法 |
| EP05814172A EP1837343A4 (en) | 2004-12-08 | 2005-12-07 | METHOD OF PREPARING BENZOIC ACID (5 ', 7') - 3-SPIRO-2 '- (1', 3'-DIOXILANE) -24-OXOCHOLEST-22-EN-7-YLESTER |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2004-354787 | 2004-12-08 | ||
| JP2004354787 | 2004-12-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006062129A1 true WO2006062129A1 (ja) | 2006-06-15 |
Family
ID=36577956
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2005/022457 Ceased WO2006062129A1 (ja) | 2004-12-08 | 2005-12-07 | (5α,7α)-3-スピロ-2’-(1’,3’-ジオキソラン)-24-オキソコレスト-22-エン-7-イル ベンゾアートの製造方法 |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20090259038A1 (ja) |
| EP (1) | EP1837343A4 (ja) |
| JP (1) | JPWO2006062129A1 (ja) |
| WO (1) | WO2006062129A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001505207A (ja) * | 1996-12-06 | 2001-04-17 | マガイニン ファーマシューティカルズ インコーポレーテッド | アミノステロール、ビタミンd類似体および他の化合物の製造に有用な24−ヒドロキシル化された化合物の立体選択的合成 |
| WO2003011228A2 (en) * | 2001-08-01 | 2003-02-13 | Biogal Gyogyszergyar Rt | Purification and crystalline forms of zaleplon |
| JP2003512892A (ja) * | 1999-11-03 | 2003-04-08 | グラクソ グループ リミテッド | 結晶性粒子の新規製造装置および製造法 |
-
2005
- 2005-12-07 US US11/721,309 patent/US20090259038A1/en not_active Abandoned
- 2005-12-07 EP EP05814172A patent/EP1837343A4/en not_active Withdrawn
- 2005-12-07 JP JP2006546733A patent/JPWO2006062129A1/ja active Pending
- 2005-12-07 WO PCT/JP2005/022457 patent/WO2006062129A1/ja not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2001505207A (ja) * | 1996-12-06 | 2001-04-17 | マガイニン ファーマシューティカルズ インコーポレーテッド | アミノステロール、ビタミンd類似体および他の化合物の製造に有用な24−ヒドロキシル化された化合物の立体選択的合成 |
| JP2003512892A (ja) * | 1999-11-03 | 2003-04-08 | グラクソ グループ リミテッド | 結晶性粒子の新規製造装置および製造法 |
| WO2003011228A2 (en) * | 2001-08-01 | 2003-02-13 | Biogal Gyogyszergyar Rt | Purification and crystalline forms of zaleplon |
Non-Patent Citations (4)
| Title |
|---|
| BORISSOVA A. ET AL.: "Examination of the Semi-Batch Crystallization of Benzophene from Saturated Methanol Solution via Aqueous Antisolvent Drowing-Out as Monitored In-Process Using ATR FTIR Spectroscopy", CRYST. GROWTH DES., vol. 4, no. 5, 2004, pages 1053 - 1060, XP002996569 * |
| JONES S.R. ET AL.: "Efficient Route to 7alpha-(Benzoyloxy) -3-dioxolane Cholestan-24(R)-ol, a Key Intermediate in the Synthesis of Squalamine", J. ORG. CHEM., vol. 63, no. 11, 1998, pages 3786 - 3789, XP002269175 * |
| KITAMURA M. ET AL.: "Anti-solvent crystallization and transformation of thiazole-derivative Polymorphs- I: effect of addition rate and initial concentrations", J. CRYST. GROWTH, vol. 257, 2003, pages 177 - 184, XP004450900 * |
| See also references of EP1837343A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090259038A1 (en) | 2009-10-15 |
| JPWO2006062129A1 (ja) | 2008-06-12 |
| EP1837343A1 (en) | 2007-09-26 |
| EP1837343A4 (en) | 2009-07-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3904640B2 (ja) | 第1級又は第2級アルコールの酸化方法 | |
| JP4433156B2 (ja) | 光学活性オキソヘプテン酸エステルの製造方法 | |
| JPWO2003042180A6 (ja) | 光学活性オキソヘプテン酸エステルの製造方法 | |
| JP2012121843A (ja) | アミドの脱酸素によるアミンの製造方法 | |
| WO2006062129A1 (ja) | (5α,7α)-3-スピロ-2’-(1’,3’-ジオキソラン)-24-オキソコレスト-22-エン-7-イル ベンゾアートの製造方法 | |
| WO2007034972A1 (ja) | タングステンの回収方法 | |
| JP3282372B2 (ja) | ピペロナールの製法 | |
| JP2014151313A (ja) | 触媒、及び光学活性アンチ−1,2−ニトロアルカノール化合物の製造方法 | |
| WO2005118595A1 (ja) | 3−アルケニルセフェム化合物の製造方法 | |
| JP5215003B2 (ja) | 表面銀固定化ハイドロキシアパタイトを使用したシランの酸化反応 | |
| US20080207936A1 (en) | Method of Producing 3-Hydroxybutyraldehyde Derivative | |
| US9683010B2 (en) | Process for the production of 21-methoxy-11-beta-phenyl-19-nor-pregna-4,9-diene-3,20-dione derivatives | |
| JP5473303B2 (ja) | 2−ブロモ−3−{4−[2−(5−エチル−2−ピリジル)エトキシ]フェニル}プロピオン酸メチルの製造方法 | |
| CN106966942B (zh) | 一种依折麦布的制备方法 | |
| JPH0267250A (ja) | 2,2‐ジフルオロ‐3‐ヒドロキシカルボン酸誘導体の製造法 | |
| CN111233800A (zh) | 一种银催化的叠氮甲基呋喃化合物的方法 | |
| JP3348456B2 (ja) | pーキノン類の製造法 | |
| JP3471067B2 (ja) | 光学活性6―(4―置換インデン―1―イル)―2―メチル―4―ヘプテン―3―オール類の製造法 | |
| TW202539620A (zh) | 大麻二酚之溶劑合物及大麻二酚之精製方法 | |
| JP2003300919A (ja) | 1,3−シクロヘキサンジオール化合物の製造方法 | |
| WO2005095431A1 (ja) | 5α-プレグナン誘導体の製造方法 | |
| JPH0971546A (ja) | トリメチロールアルカンの製造方法 | |
| JP2008143789A (ja) | ビニルナフタレン化合物の精製方法および精製ビニルナフタレン化合物 | |
| JPS6224440B2 (ja) | ||
| JP2003040877A (ja) | 5−[6−(2−フルオロベンジルオキシ)−2−ナフチル]メチル−2,4−チアゾリジンジオンの製造方法及びその精製方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KN KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006546733 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005814172 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11721309 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005814172 Country of ref document: EP |