Thieno -pyridine derivatives as GABA-B allosteric enhancers
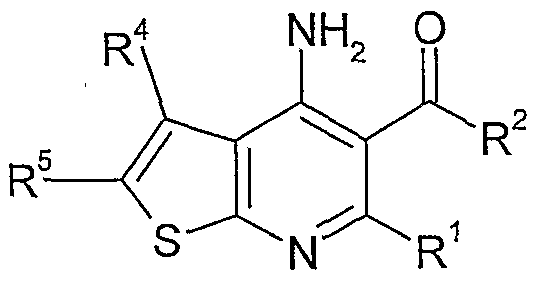
The present invention relates to compounds of formula I
R1 is hydrogen, Ci-C7-alkyl, Ci-C7-haloalkyl, or C3-C8-cycloalkyl; R2 is CrCy-alkyl, Ci-C7-haloaIkyl, C3-C8-cycloalkyl, optionally substituted aryl or optionally substituted heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, Ci-Cy-alkoxy, C1-C7- haloalkyl, CrC7-haloalkoxy, Ci-Cy-alkylsulfonyl, and -C(O)O-Ci-C7-alkyl; R3 is -NRaRb; optionally substituted heterocydoalkyl having 3 to 8 ring atoms; optionally substituted aryl, or optionally substituted heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, Q-Cy-alkoxy, Q-Q-haloalkyl, Ci-Cy-haloalkoxy, Ci-C7-alkylsulfonyl, and -C(O)O-C1-C7-alkyl; R4 is hydrogen, or Ci-C7-alkyl; R5 is hydrogen, halo, CrC7-alkyl, C2-C7-alkenyl, -NRaRb; optionally substituted aryl, or optionally substituted heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, cyano, Ci-C7-alkyl, Ci-C7-haloalkyl, CrCy-alkoxy, Ci-C7-haloalkoxy, Ci-C7-alkylsulfonyl, or - C(O)O-CrC7-alkyl; or R5 together with R may form a ring selected from the group consisting of C5-
C7-cycloalkyl, heterocydoalkyl having 5 to 7 ring atoms, phenyl; pyridinyl, or pyrimidinyl which are optionally substituted or by one or more halo, cyano, Q- C7-alkylsulfonyl, Ci-C7-alkyl, Ci-C7-haloalkyl, Ci-C7-alkoxy, Ci-C7-haloalkoxy, and -C(O)O-Ci-C7-alkyl; Ra, Rb are each independently Ci-C7-alkyl, or Ra and Rb may, together with the nitrogen atom to which they are attached, form an heterocydoalkyl group having 3 to 8 ring atoms
which is optionally substituted by one or more halo, Ci-Cr-alkyl, Ci-Cyalkoxy, hydroxy, phenyl or di(CrC7)alkylamino; as well a pharmaceutically acceptable salts thereof.
The compounds of formula I and their salts are distinguished by valuable therapeutic properties. It has been found that the compounds are active on the GABAB receptor.
γ-Aminobutyric acid (GABA), the most abundant inhibitory neurotransmitter, activates both ionotropic GABAA/C and metabotropic GABAB receptors (Hill and Bowery, Nature, 290, 149-152, 1981). GABAB receptors that are present in most regions of the mammalian brain on presynaptic terminals and postsynaptic neurons are involved in the fine-tuning of inhibitory synaptic transmission. Presynaptic GABAB receptors through modulation of high- voltage activated Ca2+ channels (P/Q- and N-type) inhibit the release of many neurotransmitters. Postsynaptic GABAB receptors activates G-protein coupled inwardly rectifying K+ (GIRK) channel and regulates adenylyl cyclase (Billinton et at, Trends Neuroscu, 24, 277-282, 2001; Bowery et al, Pharmacol. Rev.. 54, 247-264, 2002). Because the GABAB receptors are strategically located to modulate the activity of various neurotransmitter systems, GABAB receptor ligands hence could have potential therapeutics in the treatment of anxiety, depression, epilepsy, schizophrenia and cognitive disorders (Vacher and Bettler, Curr. Drug Target, CNS Neurol. Disord. 2, 248- 259, 2003; Bettler et al, Physiol Rev. 84, 835-867, 2004).
Native GABAB receptors are heteromeric structures composed of two types of subunits, GABABR1 and GABABR2 subunits (Kaupmann etal, Nature, 386, 239-246, 1997 and Nature, 396, 683-687, 1998). The structure of GABABR1 and R2 show that they belong to a family of G-protein coupled receptors (GPCRs) called family 3. Other members of the family 3 GPCRs include the metabotropic glutamate (mGlul-8), Calcium-sensing, vomeronasal, pheromone and putative taste receptors (Pin et al., Pharmaco.. Ther. 98, 325-354, 2003). The family 3 receptors (including GABAB receptors) are characterized by two distinctly separated topological domains: an exceptionally long extracellular amino -terminal domain (ATD, 500-600 amino acids), which contains a venus flytrap module for the agonist binding (orthosteric site) (Galvez et al, J. Biol.
Chem., 275, 41166-41174, 2000) and the 7TM helical segments plus intracellular carboxyl- terminal domain that is involved in receptor activation and G-protein coupling. The mechanism of receptor activation by agonist in GABABR1R2 heterodimer is unique among the GPCRs. In the heteromer, only GABABRI subunit binds to GABA, while the
GABAβR2 is responsible for coupling and activation of G-protein (Havlickova etal, MoI. Pharmacol 62, 343-350, 2002; KniazeffetalJ. NeuroscL, 22, 7352-7361, 2002).
Schuler et al, Neuron, 31, 47-58, 2001 have demonstrated that the GABABR1 knockout (KO) mice exhibit spontaneous seizures and hyperalgesia. These KO mice have lost all the biochemical and electrophysiological GABAB responses. Interestingly, the
GABABRI KO mice were more anxious in two anxiety paradigm, namely the light-dark box (decreased time in light) and staircase tests (decreased rears and steps climbed). They showed a clear impairment of passive avoidance performance model indicating impaired memory processes. The GABABRI KO also displayed increased hyperlocomotion and hyperactivity in new environment. The GABABRI gene is mapped to chromosome
6p21.3, which is within the HLA class I, a region with linkage for schizophrenia, epilepsy and dyslexia (Peters et at, Neurogenetics, 2, 47-54, 1998). Mondabon et al, Am. J. Med. Genet 122B/1, 134, 2003 have reported about a weak association of the Ala20Val polymorphism of GABABRI gene with schizophrenia. Moreover, Gassmann etal, J NeuroscL 24, 6086-6097, 2004 has shown that GABABR2KO mice suffer from spontaneous seizures, hyperalgesia, hyperlocomotor activity and severe memory impairment, comparable to GABAβRlKO mice. Therefore, heteromeric GABAB R1R2 receptors are responsible for these phenotypes.
Baclofen (Lioresalθ, β-chlorophenyl GABA), a selective GABAB receptor agonist with EC50 = 210 nM at native receptor, is the only ligand, which has been used since 1972 in clinical study for the treatment of spasticity and skeletal muscle rigidity in patients following spinal cord injury, multiple sclerosis, amyotrophic lateral sclerosis, cerebral palsy. Most of the preclinical and clinical studies conducted with baclofen and GABAB receptor agonists were for the treatment of neuropathic pain and craving associated with cocaine and nicotine (Misgeld et al, Prog. Neurobiol 46, 423-462, 1995; Enna et al, Life Sd, 62, 1525-1530, 1998; McCarson and Enna, Neuropharmacology, 38, 1767-1773, 1999; Brebner et al, Neuropharmacology, 38, 1797-1804, 1999; Paterson et al, Psychopharmacology, 172, 179-186, 2003). In panic disorder patients, Baclofen was shown to be significantly effective in reducing the number of panic attacks and symptoms of anxiety as assessed with the Hamilton anxiety scale, Zung anxiety scale and Katz-R nervousness subscale (Breslow etal, Am. J. Psychiatry, 146, 353-356, 1989). In a study with a small group of veterans with chronic, combat-related posttraumatic stress disorder (PTSD), baclofen was found to be an effective and well-tolerated treatment. It resulted in significant improvements in the overall symptoms of PTSD, most notably the avoidance, emotional numbing and hyperarousal symptoms and also in reduced accompanying
anxiety and. depression (Drake etal, Ann. Pharmacother. 37., 1177-1181, 2003). In preclinical study, baclofen was able to reverse the reduction in prepulse inhibition (PPI) of the acoustic startle response induced by dizocilpine, but not by apomorphine in rat PPI model of psychosis (Bortolato et al, Psychopharmacology, 171, 322-330, 2004). Therefore, GABAB receptor agonist has a potential in the pharmacological therapy of psychotic disorders. Unfortunately, Baclofen has a number of side-effects including the poor blood-brain-barrier penetration, very short duration of action and narrow therapeutic window (muscle relaxation, sedation and tolerance) that limit its utility.
Urwyler et al, MoI Pharmacol, 60, 963-971, 2001 have reported on a novel class of GABAB receptor ligands, called positive allosteric modulators, CGP7930 [2,6-di-tert- butyl-4-(3-hydroxy-2,2-dimethyl-propyl)-phenoi] and its aldehyde analogue CGP13501. These ligands have no effect on their own at GABAB receptors, but in concert with endogenous GABA, they increase both the potency and maximal efficacy of GABA at the GABABR1R2 (Pin etal, MoI. Pharmacol.,60, 881-884, 2001). Interestingly, recent study with CGP7930 {Binet et al, J Biol Chem., 279, 29085-29091, 2004) has shown that this positive modulator activates directly the seven transmembrane domains (7TMD) of GABABR2 subunit. Mombereau etal, Neuropsychopharmacology, 1-13, 2004 have recently reported on the anxiolytic effects of acute and chronic treatment with the GABAB receptor positive modulator, GS39783 (N5iV_.-dicyclopentyl-2-methylsulfanyl-5-nitro- pyrimidine-4,6-diamine) (Urwyler et al, J. Pharmacol. Exp. Ther., 307, 322-330, 2003) in the light-dark box and elevated zero maze test models of anxiety. No tolerance after chronic treatment (21 days) with GS39783 (10 mg/kg, P.O., once daily) was observed. Because the GABAB enhancers have no effect on receptor activity in the absence of GABA, but do enhance allosterically the affinity of the GABAB receptor for the endogenous GABA, it is expected that these ligands should have an improved side effect profile as compared to baclofen. Indeed, GS39783 at 0.1-200 mg/kg, PO had no effect on spontaneous locomotor activity, rotarod, body temperature and traction test in comparison to baclofen, which showed these side effects at 2.5-15 mg/kg, PO. GS39783 did not have any effect on cognition performance as assessed by passive avoidance behavioral test in mice and rats. Furthermore, GS39783 exhibited anxiolytic-like effects in the elevated plus maze (rat), elevated zero maze (mice and rats), and the stress-induced hyperthermia (mice) test paradigms. Therefore, GS39783 represents a novel anxiolytic without side-effects associated with baclofen or benzodiazepines (Cryan et al, J Pharmacol Exp Ther., 310, 952-963, 2004). The preclinical investigation with the CGP7930 and GS39783 has shown that both compounds were effective at deceasing cocaine self- administration in rats (Smith etal, Psychopharmacology, 173, 105-111, 2004). The positive
modulator, CGP7930 has also been preclinically studied for the treatment of Gastro- Esophageal Reflux Disease (GERD) and was found to be effective (WO 03/090731, Use of GABAB receptor positive modulators in gastro-intestinal disorders).
Positive allosteric modulators have been reported for other family 3 GPCRs including mGlul receptor (Knoflach et al, Proc. Natl Acad. ScI, USA, 98, 13402-13407, 2001; Wichmann etal, Farmaco, 57, 989-992, 2002), Calcium-sensing receptor (NPS R- 467 and NPS R-568) (Hammerland etal, MoI. Pharmacol, 53, 1083-1088, 1998) (US 6,313, 146), mGlu2 receptor [LY487379, N-(4-(2-memoxyphenoxy)-phenyl-N-(2,2,2- trifluoroethylsulfonyl)-pyrid-3-ylmethylamine and its analogs] (WO 01/56990, Potentiators of glutamate receptors) and mGlu5 receptor (CPPHA, N~-{4-chloro-2-[(l,3- dioxo-l,3-dihydro-2H-isoindol-2-yl)methyl] phenyl}-2-hydroxybenzamide) (O'Brien et al, J. Pharmaco. Exp. Ther., 27, Jan. 27, 2004). Interestingly, it has been demonstrated that these positive modulators bind to a novel allosteric site located within the 7TMD region, thereby enhancing the agonist affinity by stabilizing the active state of the 7TMD region (Knoflach et al, Proc. Natl Acad. ScI, USA 98, 13402-13407, 2001; Schaffhauser et al, MoI Pharmacol, 64, 798-810, 2003). Moreover, the NPS R-467, NPS R-568 (Tecalcet) and related compounds represent the first positive allosteric modulators that entered the clinical trails due to their allosteric mode of action.
Objects of the invention are the compounds of formula I and pharmaceutically acceptable acid addition salts thereof, the preparation of the compounds of formula I and salts thereof, medicaments containing a compound of formula I or a pharmaceutically acceptable acid addition salts thereof.
A further object of the invention is the use of the compound of formula I and acceptable acid addition salts thereof for the manufacture of such medicaments useful in the control or prevention of illnesses, especially of illnesses and disorders of the kind referred to earlier, such as anxiety, depression, epilepsy, schizophrenia, cognitive disorders, spasticity and skeletal muscle rigidity, spinal cord injury, multiple sclerosis, amyotrophic lateral sclerosis, cerebral palsy, neuropathic pain and craving associated with cocaine and nicotine, psychosis, panic disorder, posttraumatic stress disorders or gastro- intestinal disorders, and, respectively, for the manufacture of corresponding medicaments.
The following definitions of the general terms used in the present description apply irrespective of whether the terms in question appear alone or in combination.
As used herein, the term "Aryl" means a monovalent cyclic aromatic hydrocarbon moiety containing preferably from 6 to 10 carbon atoms or "C6-Cio-aryl". Preferred aryls include, but are not limited to, optionally substituted phenyl, naphthyl as well as those specifically illustrated by the examples hereinbelow.
"C1-C7-aUcyl" denotes a straight- or branched- carbon chain group containing from
1 to 7 carbon atoms, preferably 1 to 4 carbon atoms. Examples of such groups are are methyl, ethyl, propyl, isopropyl, isobutyl, sec-butyl, tert-butyl, pentyl, n-hexyl as well as those specifically illustrated by the examples herein below.
"C2-C7-alkenyl" denotes a straight- or branched-carbon chain group containing from 2 to 7 carbon atoms and containing 1, 2 or 3 double bond(s), preferably 1 to 4 carbon atoms and 1 double bond. Examples of such groups are are methenyl, 1-ethenyl, 2-ethenyl, 1-propenyl, 2-propenyl, 3-propenyl, isopropenyl, isobutenyl, sec-butenyl, tert- butenyl, pentenyl, n-hexenyl as well as those specifically illustrated by the examples herein below.
"Ci-Cy-haloalkyl" denotes a Ci-Cy-alkyl group as defined above which is substituted by one or more halogen. Examples of Q-Cy-haloalkyl include but are not limited to methyl, ethyl, propyl, isopropyl, isobutyl, sec-butyl, tert-butyl, pentyl or n- hexyl substituted by one or more Cl, F, Br or I atom(s) as well as those groups specifically illustrated by the examples herein below. Prefered Q-Cy-haloalkyl are difluoro- or trifluoro -methyl or ethyl.
"Ci-Cy-alkoxy" denotes a group wherein the alkyl group is as defined above and the alkyl group is connected via an oxygen atom. Prefered alkoxy are MeO- and Et-O as well as those groups specifically illustrated by the examples herein below.
"Ci-C7-haloalkoxy" denotes a Ci-C7-alkoxy group as defined above which is substituted by one or more halogen. Examples of C1-C7 haloalkyl include but are not limited to methoxy, ethoxy, propoxy, isopropoxy, isobutoxy, substituted by one or more Cl, F, Br or I atom(s) as well as those groups specifically illustrated by the examples herein below. Prefered Ci-Cy-haloalkoxy are difluoro- or trifluoro-methoxy or ethoxy groups.
"Halogen" denotes chlorine, iodine, fluorine and bromine.
ccCi-C
7-aIkylsulfonyr' denotes a sulfonyl group which is substituted by a
group as defined herein above. Examples of Ci-C
7-alkylsurfonyl include but are not
limited to methylsulfphonyl and ethylsulfonyl as well as those groups specifically illustrated by the examples herein below.
"di(Ci-C7)aΣkylamino" denotes a -NRcRd group, wherein Rcand Rd are Q-Cy-alkyl groups as defined herein above. Examples of di(Ci-C7)alkylamino groups include but are not limited to di(methyl) amino, di(ethyl)amino, methylethylamino, as well as those groups specifically illustrated by the examples herein below.
"Hydroxy or hydroxy" denotes a -OH group.
KC3-Cs-cycloalkyl" or "Cs-Cy-cycloalkyl" denotes a saturated carbon cyclic ring having 3 to 8 or 5 to 7 carbon atoms as ring members and includes but is not limited to cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, as well as those groups specifically illustrated by the examples herein below.
Ηeterocycloalkyl" or "Heterocycloalkyl having 3 to 8, 4 to 8 or 5 to 7 ring atoms" denote a saturated mono- or bi-cyclic ring comprising of 3 to 8, in particular 4 to 8 and more particularly 5 to 7 ring atoms and furthermore containing one, two, or three ring heteroatoms selected from N, O, or S, the remaining ring atoms being C. Preferred 3 to 8 membered heterocycloalkyl groups are 5 or 6 membered heterocycloalkyl groups. Examples include but are not limited to optionally substituted azetidinyl, piperidinyl, piperazinyl, homopiperazinyl, azepinyl, pyrrolidinyl, pyrazolidinyl, imidazolinyl, imidazolidinyl, pyridinyl, pyridazinyl, pyrimidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiazolidinyl, isothiazolidinyl, quinuclidinyl, quinolinyl, isoquinolinyl, benzimidazolyl, thiadiazolylidinyl, benzothiazolidinyl, benzoazolylidinyl, dihydrofuryl, tetrahydrofuryl, dihydropyranyl, tetrahydropyranyl, thiomorpholinyl, thiomorpholinylsulfoxide, thiomorpholinylsulfone, dihydro quinolinyl, dihydrisoquinolinyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, 1-oxo- thiomorpholin, 1,1-dioxo-thiomorpholin, 1,4-diazepane, 1,4-oxazepane and 8-oxa-3- aza-bicyclo[3.2.1]oct-3-yl, as well as those groups specifically illustrated by the examples herein below.
"Heteroaryl" means a monocyclic, bicyclic or tricyclic radical of 5 to 12 and preferably 5 to 9 ring atoms having at least one aromatic ring and furthermore containing one, two, or three ring heteroatoms selected from N, O, or S, the remaining ring atoms being C. Heteroaryl can optionally be substituted with one, two, three or four substituents, wherein each substituent is independently hydroxy, cyano, alkyl, alkoxy, thioalkyl, halo, haloalkyl, hydroxyalkyl, alkoxycarbonyl, amino, acetyl, -NHCOOC(CH3)3 or halogen as defined above, substituted benzyl, or for the non aromatic part of cyclic
ring also by oxo, unless otherwise specifically indicated. Examples of heteroaryl moieties include, but are not limited to, optionally substituted imidazolyl, optionally substituted oxazolyl, optionally substituted thiazolyl, optionally substituted pyrazinyl, optionally substituted pyrrolyl, optionally substituted pyrazinyl, optionally substituted pyridinyl, optionally substituted pyrimdinyl, optionally substituted indonyl, optionally substituted isoquinolinyl, optionally substituted carbazol-9-yl, optionally substituted furanyl, optionally substituted benzofixranyl, optionally substituted benzo[l,2,3]thiadiazolyl, optionally substituted benzo[b]thiophenyl, optionally substituted 9H-thioxanthenyl, optionally substituted thieno[2,3-c]pyridinyl and the like or those which are specifically exemplified herein.
"R5 together with R4 may form a ring selected from the group consisting of C5-C7- cycloalkyl; heterocycloalkyl having 5 to 7 ring atoms; phenyl; pyridinyl; or pyrimidinyl" denotes a C5-C7-cycloalkyl, heterocycloalkyl having 5 to 7 ring atoms, phenyl, pyridinyl or pyrimidinyl group as defined above which is fused to the thieno-pyridine group via R5 and R . Examples of such group are but are not limited to cyclopentane, cyclohexane, phenyl, pyridine, tetrahydropyrane and 2,2-difiuoro-[l,3]dioxolane. Said groups maybe substituted by -C(O)O-CrC7-alkyl grouρ(s).
The term "pharmaceutically acceptable acid addition salts" embraces salts with inorganic and organic acids, which include but are not limited to hydrochloric acid, nitric acid, sulfuric acid, phosphoric acid, citric acid, formic acid, fumaric acid, maleic acid, acetic acid, succinic acid, tartaric acid, methane-sulfonic acid, p-toluenesulfonic acid.
Encompassed by formula I are the following compounds of formula Ia
wherein R1, R2, R4, R , Ra and R are as defined hereinbefore for the compounds of formula I. In fact, compounds of formula Ia are those compounds of formula I wherein R3 is NRaRb.
Also encompassed by formula I are the following compounds of formula Ib
wherein R1, R2, R3, Ra and Rb are as defined hereinbefore for the compounds of formula I. In fact, compounds of formula Ib are those compounds of formula I wherein R4 is H and R5 is NRaRb.
Also encompassed by formula I are the following compounds of formula Ic
wherein R1, R2, R3 are as defined hereinbefore for the compounds of formula I and R5 is selected from the group consisting of C2-C7-alkenyl and optionally substituted aryl or heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, cyano, Q-Cy-alkyl, Q-Cy-haloalkyL Q-Cy-alkoxy, Q-Cy-haloalkoxy, Ci-Cy-alkylsulfonyL or -C(O)O-C1-C7-aIkyl. In fact, compounds of formula Ic are those compounds of formula I wherein R4 is H and R5 is selected from the group consisting of C2-C7-alkenyl and optionally substituted aryl or heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, cyano, Q-C7- alkyl, Ci-Cz-haloalkyl, Ci-Cy-alkoxy, Q-Cy-haloalkoxy, Q-Cy-aϊkylsulfonyl, and - C(O)O-CrC7-alkyl.
For the compounds of formula I, Ia, Ib and Ic the following groups are preferred: preferred groups for R1 maybe selected from the group consisting of methyl, ethyl, propyl, i-propyl, butyl, i-butyl, and t-butyl.
Preferred groups for R2 may be selected from the group consisting of methyl, ethyl, propyl, i-propyl, butyl, i-butyl, t-butyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, CHF2 and CF3.
Preferred groups for R3 maybe selected from the group consisting of: - phenyl optionally substituted by one or more halo, preferably chloro or fluoro, Ci-C7-alkoxy, preferably methoxy; CrC7-alkylsulfonyl, preferably methylsulfonyl, or -C(O)O-CrC7-alkyl, preferably C(O)-O-CH3,
- thiophenyl, preferably, thiophen-2-yl; furanyl, preferably furan-2-yl; or
- piperidine, morpholine, preferably, piperidin-1-yl.
Preferred groups for R are selected from the group consisting of: - hydrogen, and
- Ci-Cy-alkyl, preferably methyl.
Preferred groups for R5 are selected from the group consisting of: Ci-Cyalkyl, preferably methyl,
- Aryl, preferably phenyl, - Halo, preferably chloro, and
- Hydrogen.
Preferred groups for R5 together with R4 when they form a ring are selected from the group consisting of:
- C5-C7-cycloalkyl, preferably cyclopentyl, cydohexyl, and cycloheptyl, - aryl, preferably phenyl,
- heteroaryl having 5 to 9 ring atoms, preferably pyridinyl or pyrimidinyl, heterocydoalkyl having 5 to 7 ring atoms, preferably tetrahydrofurane, or piperidinyl optionally substituted by -C(O)O-C1-C7-alkyl.
Preferred compounds of the invention are those compounds of formula I, wherein
R1 is Ci -Cy-aϊkyl, preferably methyl;
R2 is Q-Q-alkyl, preferably methyl; Ci-C7-haloalkyl, preferably CF3; or C3-C8- cycloalkyl, preferably cyclopropyl;
R3 is heterocydoalkyl having 3 to 8 ring atoms, preferably piperidin, aryl, preferably phenyl, or heteroaryl having 5 to 9 ring atoms, preferably furanyl or thiophenyl, which are optionally substituted by one or more substituent(s) selected from the group consisting of halo, preferably chloro or fluoro, Ci-C7-alkoxy, preferably methoxy, Ci-C7-alkylsulfonyl, preferably methylsulfonyl, and -C(O)O-Ci-C7- alkyl, preferably C(O)O-Me;
R4 is hydrogen or CrQ-alkyl, preferably methyl; R5 is hydrogen, halo, preferably chloro, Ci-C7-alkyl, preferably methyl, or aryl, preferably phenyl; or R5 together with R4 may form a ring selected from the group consisting of CVC7- cycloalkyl, heterocydoalkyl having 5 to 7 ring atoms, phenyl, pyridinyl, or pyrimidinyl
which are optionally substituted or by one or more halo, cyano, Q-Q-alkylsulfonyl, C1- Cy-aUsyi, Ci-Cy-haloalkyl, Q-Cy-alkoxy, CrC7-haloaϊkoxy, and -C(O)O-C1-C7-alkyi, as well a pharmaceutically acceptable salts thereof.
Also preferred compounds of the invention are those compounds of formula I wherein R2 is C].-C7-alkyl, for example the following compounds: l-(2-Methyl-4-phenyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)- ethanone; l-(2-Methyl-4-thiophen-2-yl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b ] pyridin-3-yl) -ethanone; l-(2-Methyl-4-thiophen-2-yl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3-b]pyridin-
3 -yl)- ethanone; l-[4-(4-Methox7-phenyl)-2-methyl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3- b] pyridin-3-yl] -ethanone; l-(2-methyl-4-phenyl-benzo [4,5 [thieno [2,3-b]pyridin-3-yl)-ethanone; 1- [4-(4-Methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5] thieno [2,3-b] pyridin-3-yl] -ethanone; l-(2,3,6-Trimethyl-4-phenyl-thieno[2>3-b]pyridin-5-yl)-ethanone; l-(4-Furan-2-yl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3- yl) -ethanone; l-(2-Methyl-4-phenyl-9-thia- l,7-diaza-fluoren-3-yl)-ethanone;
3-Ace1yl-2-methyl-4-phenyl-5,8-dihydro-6H-9-thia-l,7-diaza-fluorene-7- carboxylic acid methyl ester; and l-(6-Methyl-2,4-diphenyl-thieno[2,3-b]pyridin-5-yl)-ethanone.
Other preferred are those compounds of formula I wherein, wherein R2 is C1-C7- haloalkyl, for example the following compounds: l-[4-(3,4-Dichloro-phenyl)-2-meth.yl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b] pyridin-3-yl] -2,2,2-trifl.uoro-ethanone;
2,2>2-Trifluoro-l-(2-methyl-4-thiophen-2-yl-6,7-dihydro-5H- cyclopenta[4,5] thieno [2,3-b]pyridin-3-yl)-ethanone; 2,2>2-Trifluoro-l-(2-methyl-4-phenyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b]pyridin-3-yl)-ethanone;
2,2,2-Trifluoro-l-(2-methyl-4-thiophen-2-yl-5,6,7,8-tetrahydro- benzo[4,5]thieno[2,3-b]pyridin-3-yl)-ethanone;
2,2,2-Trifiuoro-l-[4-(4-methoxy-phenyl)-2-methyl-5,6,7,8-tetrahydro- benzo [4,5]thieno [2,3-b]pyridin-3-yl)-ethanone;
2,2,2-Trifluoro-l-[4-(4-methoxy-phenyl)-2-methyl-6>7-diliydro-5H- cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)-ethanone;
2,2,2-Trifluoro-l-(2-methyl-4-phenyl-benzo[4,5[thieno[2>3-b]pyridin-3-yl)- ethanone; " 2,2,2-Trifluoro-l- [4-(4-methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta [4,5 [tbieno [2,3-b] pyridin-3-yl)-ethanone;
2,2,2-Trifluoro-l-(2,3)6-trimethyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-ethanone;
2,2,2-Trifl.uoro-l-[4-(4-fluoro-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno [2,3-b]pyridm-3-yl] -ethanone; 2,2,2-Trifluoro-l-(4-furan-2-yl-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)-ethanone;
2,2,2-Trifluoro-l-(6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-ethanone; l-(2-Chloro-6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-2,2,2-trifluoro- ethanone; 2,2,2-Trifluoro-l- [4-(4-fluoro-phenyl)-6-methyl-thieno [2,3-b] pyridin-5-yl] - ethanone; l-[2-CMoro-4-(4-fluoro-phenyl)-6-methyl-thieno[2,3-b]pyridin-5-yl]-2,2,2- trifluoro-ethanone.
Still other preferred compounds of the invention are those compounds of formula I, wherein R2 is C3-C8-cycloalkyl, for example the following compounds:
Cyclopropyl-(2-methyl-4-phenyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b ] pyridin-3-yl) -methanone;
Cyclopropyl-(6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-methanone; Cyclopropyl-(2-methyl-4-thiophen-2-yl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2)3-b]pyridin-3-yl)-methanone;
Cyclopropyl-(4-furan-2-yl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b ] pyridin- 3 -yl) -methanone;
Cyclopropyl-[(4-(3,4-dichloro-phenyl)-2-methyl-657-dihydro-5H-cyclopenta [4,5]thieno[2,3-b]pyridin-3-yl)-methanone; Cyclopropyl-[(4-(3,4-dichloro-phenyl)-2-methyl-5,6,7,8-tetrahydro- benzo[4,5]thieno [2,3-b]pyridin-3-yl)] -methanone;
Cyclopropyl-[4-(4-methox7-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta [4,5] thieno[2,3-b]pyridin-3-yl)] -methanone;
Cyclopropyl-[4-(4-methoxy-phenyl)-2-methyl-5,6,7,8-tetrahydro- benzo[4,5]thieno[2,3-b]pyridin-3-yl)]-methanone;
4-(3-Cyclopropanecarbonyl-2-methyl-657-dihydro-5H-cyclopenta[4,5]tliieno[2,3- b]pyridin-4-yl)-benzoic acid methyl ester;
4-(3-Cyclopropanecarbonyl-2-methyl-5)6,7,8-tetrahydro-benzo[4,5]thieno[2,3- b ] ρyridin-4-yl) -b enzoic acid methyl ester; 4-(3-Cyclopropanecarbonyl-2-methyl-637,8,9-tetrahydro-5H-10-thia-l-aza- benzo [a] azulen-4-yl) -benzoic acid methyl ester;
Cyclopropyl-[4-(3,4-dichloro-phenyl)-2-methyl-6,7,8,9-tetrahydro-5H-10-thia-l- aza-benzo [a] azulen-3-yl)-methanone;
Cyclopropyl-(2-methyl-4-phenyl-9-thia-l,7-diaza-fluoren-3-yl)-methanone; Cyclopropyl-(2-methyl-4-phenyl-5,8-dihydro-6H-7-oxa-9-thia-l-aza-fluoren-3- yl)-methanone;
Cyclopropyl-(6-methyl-4-piperidin-l-yl-2,3-dihydro-lH-8-thia-7-aza- cyclopenta[a] inden-5-yl)-methanone; and
Cyclopropyl-(6-Methyl-2>4-diphenyl-thieno[2,3-b]pyridin-5-yl)-methanone.
The afore-mentioned compounds of formula I wherein R3 is optionally substituted aryl or heteroaryl having 5 to 9 ring atoms as defined hereinbefore can be manufactured by the following process of the invention comprising the step of reacting a compound of formula IV:
with a compound of formula V:
V in order to obtain the compound of formula I, wherein R1, R2, R3, R4, and R5 are as defined hereinbefore, and if desired, converting the compound of formula I obtained into a pharmaceutically acceptable acid addition salt. This process of manufacture is further detailed in scheme 1 hereinafter.
The afore-mentioned compounds of formula Ia can be manufactured in accordance with the invention by the following process comprising the step of reacting a compound of formula VI with a compound of formula VII:
VII in order to obtain a compound of formula VIII:
VIII and subsequently alkylating the compound of formula VIII in order to obtain the compound of formula Ia, wherein R1, R2, R4, R5, Ra and Rb are as defined hereinbefore for the compounds of formula I; and if desired, converting the compound of formula Ia obtained into a pharmaceutically acceptable acid addition salt. This process of manufacture is further detailed in scheme 2 hereinafter.
The afore-mentioned compounds of formula Ib can be manufactured in accordance with the invention by the following process comprising the step of reacting a compound of formula XI:
XI with a compound of formula V:
in order to obtain the compound of formula Ib, wherein R
1, R
2, R
3, R
a and R
b are as defined hereinbefore for the compounds of formula I; and if desired, converting the compound of formula Ia obtained into a pharmaceutically acceptable acid addition salt. This process of manufacture is further detailed in scheme 3 hereinafter.
The afore-mentioned compounds of formula Ic can be manufactured in accordance with the invention by the following process comprising the step of: a) reacting a compound of formula X:
X with a compound of formula XII:
R5-B(OH)2 (XII) so as to obtain a compound of formula XIII:
XIII b) and reacting the compound of formula XIII with a compound of formula V:
V in order to obtain the compound of formula Ic, wherein R
1, R
2, R
3, are as defined hereinbefore for the compounds of formula I, R
4 is H and R
5 is selected from the group consisting of C
2-C
7-alkenyl and optionally substituted aryl or heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, cyano, Q-Cy-alkyl, Ci-Cyhaloalkyl, Ci-CV-alkoxy, Q-Cy-haloalkoxy, C
1-CV- alkylsulfonyl, or
and if desired, converting the compound of
formula Ic obtained into a pharmaceutically acceptable acid addition salt. This process of manufacture is further detailed in scheme 4 hereinafter.
The invention also encompasses a compound of formulae I, Ia, Ib and Ic whenever it is prepared according to the above-mentioned processes.
The following general schemes 1 to 3 further illustrate certain embodiments of the preparation of the compounds according to the invention. In these schemes, and unless otherwise stated, all starting materials, building blocks and intermediates are commercially available.
In certain embodiments, the compounds of formula I wherein R3 is optionally substituted aryl or heteroaryl having 5 to 9 ring atoms can be prepared according to the general method of schemel:
Scheme 1:
II III IV
a) Morpholine 1 eq., EtOH
1 reflux; b) H
2SO
4 cat., AcOH
The formation of 2-amino-thiophene derivatives of formula IV is achieved from the base- catalyzed (morpholine) condensation of an enolisable carbonyl compounds II with a methylene active nitrile III and sulfur by the Gewald reaction (K. Gewald, E. Schinke, H. Bδettcher, Chem. Ber. 1966, 99, 94-100). Novel thieno pyridine derivatives of formula I were obtained following a Friedlander type reaction (P. Friedlander, Berichte, 1882, 15, 2572) between the 2-amino-thiophene IV and a 1,3-dione V in an acid (e.g. acetic acid) and a catalyst (e.g. a catalytic amount of sulfuric acid) (A. Arcadi, M. Chiarini, S. Giuseppe, F. Marinelli, Synlett, . 2003, 2, 203 and references therein)
In certain embodiments, the compounds of formula I wherein R3 is NRaR are herein designated as compounds Ia and maybe prepared according to the general method described in scheme 2 hereinbelow:
Scheme 2:
c) Morphoϋne, EtOH; d) i.pTsOH cat, PhMe ii. SnCI4; e) NaH, DMF, electrophile
The formation of 2-amino,3-CN-thiophene derivatives VI are either known or maybe prepared using procedures similar to the Gewald reaction described in scheme 1 by reacting malonitrile and sulfur with the appropriate ketone of formula II. (K. Gewald, E. Schinke, H. Bδettcher, Chem. Ber. 1966, 99, 94-100). Novel 4-amino thieno[2,3- bjpyridine derivatives VIII were prepared using a procedure reported in WO93/13104, by a condensation step between compounds of formula VI and VII, followed without isolation of the intermediate by a cydisation step with a Lewis acid. Final derivatives Ia were obtained by conventional procedures for the alkylation of a primary amine.
In certain embodiments, the compounds of formula I wherein R1, R2, R3 are as defined hereinabove for formula I, R4 is H and R5 is -NRaRb , herein designated as compounds Ib, maybe prepared according to the general method described in scheme 3 hereinbelow:
IX x XI Ib f) i.NBS, CH2CI2; ii.Acetic anhydride; g) R3RbNH, Pd2dba3, X-PHOS, CsCO3; b) H2SO4 cat., AcOH
2-Amino thiophenes IX (commercially available or described, in literature) are brominated in 4-position with N-bromo succinimide in CH2CI2 followed by the immediate addition of acetic anhydride to afford derivatives X. Following a methodology developed by S.L. Buchwald et al. (/. Org. Chem. 2000, 65, 1144) novel derivatives XI were obtained, which undergo a Friedlander type reaction with compound of formula V (see scheme l)to yield compounds of formula Ib.
In certain embodiments, the compounds of formula I wherein R1, R2, R3 are as defined hereinabove for formula I, R is H and R5 is selected from the group consisting of Ci-Cy-alkenyl and optionally substituted aryl or heteroaryl having 5 to 9 ring atoms, wherein the substituents are selected from the group consisting of halo, cyano, Q-C7- alkyl, CrCy-haloalkyl, CrCy-alkoxy, CrCy-haloalkoxy, Q-Cγ-aϊkylsulfonyl, or -C(O)O- CrCy-alkyl, herein designated as compounds Ic, maybe prepared according to the general method described in scheme 4 hereinbelow:
Scheme 4:
X XIII Ic
h) i. R5-B(OH)2, Pd(PPh3)4; ii.aq.NaOH, b) dione V, H2SO4 cat., AcOH Following a Suzuki methodology, by coupling a bromothiophene intermediate X (described in scheme 3) with a commercially available boronic acid of formula XII, derivatives XIII were obtained after deprotection of the acetamide under basic conditions. These derivatives XIII undergo a Friedlander type reaction with compound of formula V (see scheme 1) to yield compounds of formula Ic.
The preparation of compounds of formulae I, Ja, Ib and Ic are further described in detail in working examples 1 - 40
As mentioned earlier, the compounds of formula I and their pharmaceutically acceptable addition salts possess valuable pharmacological properties. It has been found that the compounds of the present invention have an affinity to the GABAB receptor.
The compounds were investigated in accordance with the tests given hereinafter.
Intracellular Ca2+ mobilization assay
The Chinese Hamster Ovary (CHO) cells stably expressing human GABA
BRlaR2a and Gαl6 were seeded at 5xlO
4 cells/well in the poly-D-lysine treated, 96-well, black/ clear-bottomed plates (BD Biosciences, Palo Alto, CA). 24 h later, the cells were loaded for 90 min at 37°C with 4 μM Flou-4 acetoxymethyl ester (Catalog No. F- 14202, Molecular Probes, Eugene, OR) in loading buffer (IxHBSS, 20 mM HEPES, 2.5 mM Probenecid). Hanks' Balanced Salt Solution (HBSS) (10X) (catalog No. 14065-049) and HEPES (IM) (catalog No. 15630-056) were purchased from Invitrogen, Carlsbad, CA. Probenecid (250 mM) (catalog No. P8761) was from Sigma, Buchs, Switzerland. The cells were washed five times with loading buffer to remove excess dye and intracellular calcium mobilization, [Ca
2+Ji were measured using a Fluorometric Imaging Plate Reader (FLIPR, Molecular Devices, Menlo Park, CA) as described previously (Porter et al., Br. J. Pharmacol., 128, 13-20, 1999). The enhancers were applied 15 min before the application of the GABA. For GABA shift assay, concentration-response curves of GABA (0.0003-30 μM) were determined in the absence and presence of 10 μM enhancer. The GABA-shift is defined as Log [EC
50 (GABA + 10 μM enhancer) / EC
50 (GABA alone)]. The % maximum enhancing effect (% E
max) and potency (EC
50 value) of each enhancer was determined from concentration-response curve of the enhancer (0.001-30 μM) in the presence of 10 nM GABA (EC
1O). Responses were measured as peak increase in fluorescence minus basal, normalized to the maximal stimulatory effect induced by 10 μM GABA alone (considered 100%) and 10 nM GABA alone (considered 0%). The data were fitted with the equation Y=IOO + (Max- 100)/( 1+(EC
50/ [drug])
11) where Max is the maximum effect, EC
50 the concentration eliciting a half-maximum effect and n the Hill slope.
The compounds of formula I as well as their pharmaceutically usable acid addition salts can be used as medicaments, e.g. in the form of pharmaceutical preparations. The pharmaceutical preparations can be administered orally, e.g. in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions. The administration can, however, also be effected rectally, e.g. in the form of suppositories, or parenterally, e.g. in the form of injection solutions.
The compounds of formula I and their pharmaceutically usable acid addition salts can be processed with pharmaceutically inert, inorganic or organic excipients for the production of tablets, coated tablets, dragees and hard gelatine capsules. Lactose, corn starch or derivatives thereof, talc, stearic acid or its salts etc can be used as such excipients e.g. for tablets, dragees and hard gelatine capsules.
Suitable excipients for soft gelatine capsules are e.g. vegetable oils, waxes, fats, semisolid and liquid polyols.
Suitable excipients for the manufacture of solutions and syrups include but are not limited to water, polyols, saccharose, invert sugar, glucose.
Suitable excipients for injection solutions include but are not limited to water, alcohols, polyols, glycerol, vegetable oils.
Suitable excipients for suppositories include but are not limited to natural or hardened oils, waxes, fats, semi-liquid or liquid polyols.
Moreover, the pharmaceutical preparations can contain preservatives, solubilizers, stabilizers, wetting agents, emulsifiers, sweeteners, colorants, flavorants, salts for varying the osmotic pressure, buffers, masking agents or antioxidants. They can also contain still other therapeutically valuable substances.
The dosage can vary within wide limits and will, of course, be fitted to the individual requirements in each particular case. In general, in the case of oral administration a daily dosage of about 10 to 1000 mg per person of a compound of general formula I should be appropriate, although the above upper limit can also be exceeded when necessary.
Tablet Formulation (Wet Granulation)
Item Ingredients mg/tablet
5 mg 25 mg 100 mg 500
1. Compound of formula I 5 25 100 500 2. Lactose Anhydrous DTG 125 105 30 150
3. Sta-Rx 1500 6 6 6 30
4. Microcrystalline Cellulose e 30 30 30 150
5. Magnesium Stearate 1 1 1 1 Total 167 167 167 831
Manufacturing Procedure
1. Mix items 1, 2, 3 and 4 and granulate with purified water.
2. Dry the granules at 500C.
3. Pass the granules through suitable milling equipment.
4. Add item 5 and mix for three minutes; compress on a suitable press.
Capsule Formulation
Item Ingredients mg/ capsule
5 mg 25 mg 100 mg 500
1. Compound of formula I 5 25 100 500
2. Hydrous Lactose 159 123 148 —
3. Corn Starch 25 35 40 70
4. Talc 10 15 10 25
5. Magnesium Stearate 1 2 2 5
Total 200 200 300 600
Manufacturing Procedure
1. Mix items 1, 2 and 3 in a suitable mixer for 30 minutes.
2. Add items 4 and 5 and mix for 3 minutes.
3. Fill into a suitable capsule.
EXAMPLES
In the following examples, all the starting materials are commercially available.
Example 1 l-[4-(3,4-Dichloro-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b] pyridin-3-yl] -2,2,2- trifluoro-ethanone a.) (2-Amino-5,6-dihydro-4H-cyclopenta[b1thiophen-3-yl)-(3,4-dichloro-phenylV methanone
To a stirred solution of 0.30 g (1.40 mmol) 3-(3,4-dichloro-phenyl)-3-oxo-propionitrile in 10 ml ethanol was added 0.12 ml (1.40 mmol) cyclopentanone, 43 mg (1.40 mmol) sulfur, and 0.12 ml (1.40 mmol) morpholine. The mixture was heated at 400C for 48 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.449 g (89%) (2-amino-5,6-dihydro-4H-cydopenta[b]thiophen-3-yl)- (3,4-dichloro-phenyl)-methanone as a brown solid. ES-MS m/e: 312 (%) (M+ H+, 100). b) l-[4-(3,4-Dichloro-phenyl)-2-methyl-6,7-dihvdro-5H-cyclopentar4,5lthienor2,3- b]pyτidin-3-yll-2,2,2-trifluoro-ethanone To a stirred solution of 50 mg (0.16 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(3,4-dichloro-phenyl)-methanone in 2 ml acetic acid was added 0.020 ml (0.17 mmol) of l,l,l-trifluoro-pentane-2>4-dione and one drop of
sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 10:1) afforded 9 mg (14 %) l-[4-(3,4-dichloro-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl]-2J2,2-trifluoro-ethanone as a yellow solid. ES- MS m/e (%): 430 (M+ H+, 100).
Example 2
2,2,2-Trifluoro-l-(2-methyl-4-thiophen-2-yl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b]pyridin-3-yl)-ethanone a) (2-Amino-5,6-dihydro-4H-cyclopentarblthiophen-3-yl)-thiophen-2-yl-methanone To a stirred solution of 0.30 g (1.98 mmol) 3-oxo-3-thiophenyl-2-yl-propionitrile in 10 ml ethanol was added 0.16 ml (1.98 mmol) cyclopentanone, 63 mg (1.98 mmol) sulfur, and 0.17 ml (1.98 mmol) morpholine. The mixture was heated at 90 °C for 2 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.47 g (96%) (2-amino-5,6-dihydro-4H-cyclopenta[b]thiophen-3-yl)-thiophen- 2-yl-methanone as a brown solid. ES-MS m/e (%): 250 (M+H+, 100). b) 2,212-Trifluoro-l-(2-methyl-4-thiophen-2-yl-6,7-dihydro-5H-cyclopenta[4,5lthieno [2,3-bl pyridin-3-yl)- ethanone
To a stirred solution of 59 mg (0.24 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-thiophen-2-yl-methanone in 2 ml acetic acid was added 0.038 ml (0.24 mmol) of l,l,l-trifiuoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 20:1) afforded 19 mg (22 %) 2,2,2-trifluoro-l-(2-methyl-4-thiophen-2-yl-6,7-dihydro-5H- cyclopenta[4,5]thieno [2,3-b] pyridin-3-yl)- ethanone as a colorless oil. ES-MS m/e (%): 368 (M+ H+, 100).
Example 3
2,2>2-Trifluoro-l-(2-methyl-4-phenyl-6,7-dihydro-5H-cyclopenta[4>5]thieno[2>3- b]pyridin-3-yl)-ethanone a) (2-Amino-5,6-dihydro-4H-cyclopentarblthiophen-3-yl)-phenyl-methanone
To a stirred solution of 0.30 g (2.06 mmol) 3-oxo-3-phenyl-propionitrile in 10 ml ethanol was added 0.18 ml (2.06 mmol) cyclopentanone, 66 mg (2.06 mmol) sulfur, and 0.18 ml (2.06 mmol) morpholine. The mixture was heated at 90 0C for 2 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.50 g
(98%) (2-amino-5,6-dϋiyd.ro-4H-cyclopenta[b]thiophen-3-yl)-phenyl-methanone as a brown solid. ES-MS m/e (%): 244 (M+H+, 100). b) 2^,2-Tri£luoro-l-f2-methyl-4-phenyl-6,7-dihvdro-5H-cvclopenta[4,5ithienor23-bl pyridin-3-yD- ethanone To a stirred solution of 50 mg (0.20 mmol) (2-amino-5,6-dihydro-4H- cydopenta[b]miophen-3-yl)-phenyl-methanone (the preparation of which is described in example 2) in 2 ml acetic acid was added 0.026 ml (0.20 mmol) of 1,1,1-trifluoro- pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 20:1) afforded 20 mg (27 %) 2,2,2-trifluoro-l-(2-methyl-4- phenyl-6,7-dihydro-5H-cyclopenta[4,5]thieno [2,3-b] pyridin-3-yl)- ethanone as an orange oil. ES-MS m/e (%): 362 (M+ H+, 100).
Example 4 l-(2-Methyl-4-phenyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)- ethanone
To a stirred solution of 56 mg (0.23 mmol) (2-amino-5,6-dihydro-4H-cyclopenta[b] thiophen-3-yl)-phenyl-methanone in 2 ml acetic acid was added 0.016 ml (0.24 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 33 mg (47 %) l-(2-methyl-4-phenyl-6,7-dihydro- 5H-cyclopenta[4,5]thieno [2,3-b] pyridin-3-yl)- ethanone as a yellow solid. ES-MS m/e (%): 308 (M+ H+, 100).
Example 5 l-(2-Methyl-4-thiophen-2-yl-6)7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3- yl)-ethanone
To a stirred solution of 55 mg (0.22 mmol) (2-amino-5,6-dihydro-4H-cyclopenta[b] thiophen-3-yl)-thiophen-2-yl-methanone in 2 ml acetic acid was added 0.016 ml (0.24 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 80 °C for 15 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 27 mg (39 %) l-(2-methyl-4- thiophen-2-yl-6,7-dihydro-5H-cyclopenta[4,5]thieno [2,3-b] pyridin-3-yl)- ethanone as a yellow solid. ES-MS m/e (%): 314 (M+ H+, 100).
Example 6
2,2,2-Trifluoro-l-(2-methyl-4-thiophen-2-yl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3- b]pyridin-3-yl)-ethanone
a) (2-Amino-4,5,6J-tetrahydro-benzorb1tMophen-3-ylVtMophen-2-yl-methanone
To a stirred solution of 0.30 g (1.98 mmol) 3-oxo-3-thiophenyl-2-yl-propionitrile in 6 ml ethanol was added 0.21 ml (1.98 mmol) cyclohexanone, 64 mg (1.98 mmol) sulfur, and 0.17 ml (1.98 mmol) morpholine. The mixture was heated at 60 0C for 2 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.52 g (98%) (2-amino-4,5,6,7-tetrahydro-benzo[b]tbiophen-3-yl)-thiophen-2- yl-methanone as an orange solid. ES-MS m/e (%): 264 (M+H+, 100). b) 2,2,2-Trifluoro-l-(2-methyl-4-thiophen-2-yl-5,6J,8-tetrahvdro- benzo [4,5]thieno r2,3-b1pyridin-3-yl)-ethanone
To a stirred solution of 50 mg (0.19 mmol) (2-amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-thiophen-2-yl-methanone in 1.5 ml acetic acid was added 0.024 ml (0.20 mmol) of l,l,l-trifiuoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 17 mg (23 %)
2,2,2-trifiuoro-l-(2-methyl-4-thiophen-2-yl-5,6>7,8-tetrahydro-benzo[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as an orange oil. ES-MS m/e (%): 382 (M+ H+, 100).
Example 7 l-(2-Methyl-4-thiophen-2-yl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3-b]pyridin-3-yl)- ethanone
To a stirred solution of 50 mg (0.19 mmol) (2-amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-tbiophen-2-yl-methanone (the preparation of which is described in example 6) in 1.5 ml acetic acid was added 0.020 ml (0.21 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 46 mg (74 %) l-(2-methyl-4-thioρhen-2-yl-5,6,7,8-tetrahydro- benzo[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a yellow oil. ES-MS m/e (%): 328 (M+ H+, 100).
Example 8 2,2)2-Trifluoro-l-[4-(4-methoxy-phenyl)-2-methyl-5,6,7,8-tetrahydro- benzo [4,5]thieno [2,3-b]pyridin-3-yl)-ethanone a) (2-Amino-4,5 ,6, 7-tetrahydro-benzo [b ) thiophen-3-yl) - ( 4-methoxy-phenyl) - methanone
To a stirred solution of 0.30 g (1.71 mmol) 3-(4-methoxy-phenyl)- 3-oxo-propionitrile in 10 ml ethanol was added 0.17 ml (1.71 mmol) cyclohexanone, 53 mg (1.71 mmol) sulfur,
and 0.14 ml (1.71 mmol) morpholine. The mixture was heated at 40 0C for the week end, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.51 g (85%) (2-amino-4,5,6,7-tetrahydro-benzo[b]thiophen-3-yl)-(4- methoxy-phenyl)-methanone as an orange solid. ES-MS m/e (%): 288 (M+H+, 100). b) 2,2,2-Trifluoro-l-[4-(4-methoχy-ρhenylV2-methyl-5,6,7,8-tetrahydro- benzo[4,5ithieno[2,3-b1pyridine-3-yll-ethanone
To a stirred solution of 50 mg (0.17 mmol) (2-amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone in 1.5 ml acetic acid was added 0.021 ml (0.18 mmol) of l,l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 18 mg (25 %) 2,2,2-trifluoro-l-[4-(4-methoxy-phenyl)-2-methyl-5,6,7,8-tetrahydro- benzo[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a yellow oil. ES-MS m/e (%): 406 (M+ H+, 100).
Example 9 l-[4-(4-Methoxy-phenyl)-2-methyl-5,6)7,8-tetrahydro-benzo[4,5]thieno[2,3-b]pyridin-
3 -yl)- ethanone
To a stirred solution of 50 mg (0.17 mmol) (2-amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone in 1.5 ml acetic acid was added 0.018 ml (0.18 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 38 mg (62 %) l-[4-(4- methoxy-phenyl)-2-methyl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a light yellow solid. ES-MS m/e (%): 352 (M+ H+, 100).
Example 10
2,2,2-Trifluoro-l-[4-(4-methoxy-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)-ethanone a) (2-Amino-5,6-dihydro-4H-cyclopenta[b1thiophen-3-yl)-(4-methoxy-phenyl)- methanone
To a stirred solution of 0.30 g (1.71 mmol) 3-(4-methoxy-phenyl)- 3-oxo-propionitrile in 10 ml ethanol was added 0.15 ml (1.71 mmol) cyclopentanone, 53 mg (1.71 mmol) sulfur, and 0.14 ml (1.71 mmol) morpholine. The mixture was heated at 40 0C for the week end, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.46 g (95%) (2-amino-5,6-dihydro-4H-
cyclopenta[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone as a brown solid. ES-MS m/e (%): 274 (M+H+, 100). b) 2,2,2-Trifluoro-l-r4-f4-methoxy-phenvD-2-meth.yl-6,7-dihvdro-5H- cyclopenta[4,5ltbieno[2,3-b1pyridin-3-yll-ethanone To a stirred solution of 50 mg (0.18 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone in 2 ml acetic acid was added 0.022 ml (0.18 mmol) of l>l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 20 mg (28 %) 2,2,2-trifluoro-l-[4-(4-methoxy-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a white solid. ES-MS m/e (%): 392 (M+ H+, 100).
Example 11 l-(2-methyl-4-phenyl-benzo [4,5 [thieno [2,3-b]pyridin-3-yl)-ethanone a) (2-Amino-4,5,6,7-tetrahydro-benzo[b1thiophen-3-yl)-phenyl-methanone
To a stirred solution of 0.60 g (4.13 mmol) 3-oxo-3-phenyl-propionitrile in 20 ml ethanol was added 0.43 ml (4.13 mmol) cyclohexanone, 133 mg (4.13 mmol) sulfur, and 0.36 ml (4.13 mmol) morpholine. The mixture was heated at 40 0C over the night and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 1.041 g (98%) (2-Amino-4,5,6,7-tetrahydro-benzo[b]thiophen-3-yl)-phenyl- methanone as an orange solid. ES-MS m/e (%): 258 (M+H+, 100). b) N-(3-Benzoyl-4,5,6J-tetrahydro-benzo[b]tHophen-2-yl)-acetamide
To a stirred solution of 1.04 g (4.04 mmol) (2-Amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-phenyl-methanone in 25 ml acetic anhydride was added 0.2 ml pyridine. The mixture was heated at 50 0C for 35 minutes and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo to afford 1.35 g (78%) N-(3-benzoyl- 4,5,6,7-tetrahydro-benzo[b]thiophen-2-yl)-acetamide as an orange oil. ES-MS m/e (%): 300 (M+H+, 100). c) N- ( 3 -B enzoyl-b enzo Tb 1 thiophen-2-yl) -acetamide
To a stirred solution of 0.61 g (2.04 mmol) N-(3-Benzoyl-4,5,6,7-tetrahydro- benzo[b]thiophen-2-yl)-acetamide in 20 ml chloroform was added 1.2 g palladium on charcoal (10%). The mixture was stirred at room temperature for 15 minutes and then the solvent was removed in vacuo. The resulting powder was heated at 100 0C for 48 h
and then suspended, in ethyl acetate and dichloromethane. The suspension was filtered, and the organic phase was concentrated in vacuo to afford 0.26 g (44%) N-(3-benzoyl- benzo[b]thiophen-2-yl)-acetamide as a yellow solid. ES-MS m/e (%): 296 (M+H+, 100). d) (2-Amino-benzo rb1thiophen-3-yl)-phenγl-methanone To a stirred solution of 0.26 g (0.90 mmol) N-(3-benzoyl-benzo[b]thiophen-2-yl)- acetamide in 20 ml ethanol was added 1 ml aqueous sodium hydroxide (1 M). The mixture was heated at 85 °C for 1 hour and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 65 mg (25 %) (2-amino-benzo[b]thiophen-3-yl)-phenyl-methanone as an orange solid. ES-MS m/e (%): 254 (M+H+, 100). e) f2-memyl-4-phenyl-benzor4,5lthieno[2,3-b1pyridin-3-yl)-ethanone
To a stirred solution of 32 mg (0.13 mmol) (2-amino-benzo[b]thiophen-3-yl)-phenyl- methanone in 2 ml acetic acid was added 0.09 ml (0.13 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 21 mg (52 %) (2-methyl-4-phenyl-benzo[4,5]thieno[2,3-b] pyridin- 3-yl)- ethanone as a light yellow solid. ES-MS m/e (%): 318 (M+ H+, 100).
Example 12 2,2,2-Trifluoro-l-(2-methyl-4-phenyl-benzo[4,5[thieno[2,3-b]pyridin-3-yl)-ethanone
To a stirred solution of 32 mg (0.13 mmol) (2-amino-benzo[b]thiophen-3-yl)-phenyl- methanone (the preparation of which is described in example 11) in 2 ml acetic acid was added 0.016 ml (0.13 mmol) of l,l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 20:1) afforded 8 mg (17 %) 2,2,2-trifluoro-l-(2-methyl-4-phenyl-benzo[4,5]thieno[2,3-b] pyridin-3- yl)- ethanone as a light yellow solid. ES-MS m/e (%): 372 (M+ H+, 100).
Example 13
2,2,2-Trifluoro-l-[4-(4-methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5 [thieno [2,3-b]pyridin-3-yl)-ethanone a) 3-(4-Methanesulfonyl-phenyl)-3-oxo-propionitrile
To a stirred suspension of 1.0 g (3.61 mmol) (4-methanesulfonyl-phenyl)-acetyl bromide in 15 ml ethanol was added a solution of 0.47 g (7.22 mmol) potassium cyanide in 5 ml water. The mixture was then stirred at RT for 1 h, then acidified to ph=5-6 with aqueous HCl IM and then extracted with ethyl acetate. The combined organic phases were dried
over sodium sulfate and concentrated in vacuo to afford 0.29 g (37%) 3-(4- methanesulfonyl-phenyl)-3-oxo-propionitrile as a yellow solid. ES-MS m/e (%): 222 (M, 100). b") (2-Amino-5,6-dihydro-4H-cyclopenta[b1thiophen-3-yl)-(4-methanesulfonyl-phenyD- methanone
To a stirred solution of 0.296 g (1.32 mmol) 3-(4-methanesulfonyl-phenyl)- 3-oxo- propionitrile in 20 ml ethanol was added 0.12 ml (1.32 mmol) cyclopentanone, 43 mg (1.32 mmol) sulfur, and 0.12 ml (1.32 mmol) morpholine. The mixture was heated at 40 0C over night, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.37 g (87%) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(4-methanesulfonyl-phenyl)-methanone as an orange solid. ES-MS m/e (%): 322 (M+H+, 100). c) 2,2,2-Trifluoro- 1- r4-(4-methanesulfonyl-phenyl)-2-methyl-6 J-dihvdro-5H- cyclopentar4,5lthieno[2,3-blpyridin-3-yl]-ethanone
To a stirred solution of 50 mg (0.15 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(4-methanesulfonyl-phenyl)-methanone in 2 ml acetic acid was added 0.020 ml (0.15 mmol) of l,l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 20 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 18 mg (27 %) 2,2,2-trifluoro-l-[4-(4-methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a light brown solid. ES-MS m/e (%): 440 (M+ H+, 100).
Example 14 1- [4-(4-Methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta[4,5 [thieno [2,3- b]pyridin-3-yl)-ethanone
To a stirred solution of 50 mg (0.15 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(4-methanesulfonyl-phenyl)-methanone (described above) in 2 ml acetic acid was added 0.011 ml (0.15 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 18 mg (30 %) 1- [4-(4-methanesulfonyl-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a light brown solid. ES-MS m/e (%): 386 (M+ H+, 100). Example 15 l-(2,3,6-Trimethyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-ethanone
a) (2-Amino-4.5-dimethyl-thiophen-3-yl')-phenyl-methanone
To a stirred solution of 0.50 g (3.44 mmol) 3-oxo-3-phenyl-propionitrile in 10 ml ethanol was added 0.31 ml (3.44 mmol) butan-2-one, 110 mg (3.44 mmol) sulfur, and 0.30 ml (3.44 mmol) morpholine. The mixture was heated at 70 0C over the night and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.71 g (89%) (2-Amino-4,5-dimethyl-thiophen-3-yl)-phenyl-methanone as a brown solid. ES-MS m/e (%): 232 (M+H+, 100). b) l-(2,3,6-Trimethyl-4-phenyl-thieno[2,3-b1pyridin-5-yl')-ethanone To a stirred solution of 70 mg (0.30 mmol) (2-Amino-4,5-dimethyl-thiophen-3-yl)- phenyl-methanone in 2 ml acetic acid was added 0.02 ml (0.30 mmol) of pentane-2,4- dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 10 mg (11 %) l-(2,3,6-trimethyl-4-phenyl-thieno[2,3-b]pyridine- 5-yl)-ethanone as an orange solid. ES-MS m/e (%): 296 (M+ H+, 100).
Example 16 2,2,2-Trifluoro-l-(2,3,6-trimethyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-ethanone
To a stirred solution of 50 mg (0.22 mmol) (2-Amino-4,5-dimethyl-thiophen-3-yl)- phenyl-methanone (the preparation of which is described in example 15) in 2 ml acetic acid was added 0.03 ml (0.22 mmol) of l)l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 8 mg (11 %) 2,2,2-trifluoro-l-(2,3,6-trimethyl-4-phenyl-thieno[2,3-b]pyridine-5-yl)-ethanone as an orange solid. ES-MS m/e (%): 350 (M+ H+, 100). Example 17
2,2,2-Trifluoro-l-[4-(4-fluoro-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno
[2,3-b]pyridin-3-yl] -ethanone a) 3-(4-Fluoro-phenyl)-3-oxo-propionitrile
To a stirred suspension of 2.5 g (11.5 mmol) 2-bromo-l-(4-fiuoro-phenyl) -ethanone in 40 ml ethanol was added a solution of 0.90 g (23 mmol) potassium cyanide in 9 ml water. The mixture was then stirred at RT for 2 h, then acidified to ph=5-6 with aqueous HCl IM and then extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 0.11 g (6 %) 3-(4-fluoro-phenyl)-3-oxo-propionitrile as a yellow solid.
h) (2-Ainino-5,6-dihy(lro-4H-cyclopenta[b1thiophen-3-yl)-(4-fluoro-phenvD- methanone
To a stirred solution of 0.11 g (0.67 mmol) 3 -(4-fluoro -phenyl)- 3-oxo-propionitrile in 4 ml ethanol was added 0.06 ml (0.67 mmol) cyclopentanone, 22 mg (0.67 mmol) sulfur, and 0.06 ml (0.67 mmol) morpholine. The mixture was heated at 60 0C for 2 h, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.17 g (98%) (2-amino-5,6-dihydro-4H-cyclopenta[b]thiophen-3-yl)-(4-fluoro- phenyl)-methanone as a red solid. ES-MS m/e (%): 262 (M+H+, 100). c) 2,2,2-Trifluoro-l-[4-f4-fluoro-phenyl)-2-methyl-6J-dihydro-5H- cyclopenta [4,51 thieno [2,3 -bl pyridin-3 -yli -ethanone
To a stirred solution of 50 mg (0.19 mmol) (2-amino~5,6-dihydro-4H-cyclopenta[b] tbJophen-3-yl)-(4-fluoro-phenyl)-methanone in 2 ml acetic acid was added 0.024 ml (0.19 mmol) of l,l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 °C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 10 mg (14 %) 2,2,2-trifluoro-l-[4-(4-fluoro-phenyl)-2-methyl-6,7-dihydro-5H- cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a light brown solid. ES-MS m/e (%): 380 (M+ H+, 100). Example 18 l-(4-Furan-2-yl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)- ethanone a) f2-Amino-5,6-dihydro-4H-cyclopenta[blthiophen-3-yl)-furan-2-yl-methanone
To a stirred solution of 1.0 g (7.40 mmol) 3-furan-2-yl-3-oxo-propionitrile in 20 ml ethanol was added 0.66 ml (7.40 mmol) cyclopentanone, 237 mg (7.40 mmol) sulfur, and 0.64 ml (7.40 mmol) morpholine. The mixture was heated at 70 0C for 2 h, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 1.87 g (90%) (2-amino-5,6-dihydro-4H-cyclopenta[b]thiophen-3-yl)-furan-2- yl-methanone as an orange solid. ES-MS m/e (%): 234 (M+H+, 100). b) l-r4-Furan-2-yl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5ithieno[23-blpyridine-3- yl) -ethanone
To a stirred solution of 50 mg (0.21 mmol) (2-amino-5,6-dihydro-4H-cyclopenta[b] thiophen-3-yl)-furan-2-yl-methanone in 2 ml acetic acid was added 0.016 ml (0.21 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative
HPLC (30% CH3CN/H20) afforded 20 mg (31 %) l-(4-raran-2-yl-2-methyl-6,7-dihydro- 5H-cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)-ethanone as a yellow solid. ES-MS m/e (%): 298 (M+ H+, 100).
Example 19 2,2,2-Tri£luoro-l-(4-furan-2-yl-2-methyl-6>7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b] pyridin-3-yl) -ethanone
To a stirred solution of 50 mg (0.21 mmol) (2-amino-5,6-dihydro-4H-cyclopenta[b] thiophen-3-yl)-furan-2-yl-methanone (the preparation of which is described in example 18) in 2 ml acetic acid was added 0.027 ml (0.21 mmol) of l,l,l-trifluoro-pentane-2,4- dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 19 mg (25 %) 2,2,2-trifluoro-l-(4-furan-2-yl-2-methyl-6,7- dihydro-5H-cyclopenta[4,5]thieno[2,3-b] pyridin-3-yl)- ethanone as a yellow solid. ES- MS m/e (%): 352 (M+ H+, 72). Example 20
2,2,2-Trifluoro-l-(6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-ethanone a) (2-amino-thiophen-3-yl)-phenyl-methanone
Following a procedure described in J.Med.Chem. 2002, 45, 382-389; 1.45 g (10 mmol) 3- oxo-3-phenyl-propionitrile, 0.76 g (5.0 mmol) [l,4]dithiane-2,5-diol, 0.40 ml (10 mmol) diethyl-amine and 5 ml ethanol were charged in a sealed tube. The reaction mixture was heated at 50 0C for 6 h. The tube was then placed in a fridge (about 40C) for the night, and the product was collected by filtration and dried under vacuum to afford 0.42 g (23%) (2-amino-thiophen-3-yl)~phenyl-methanone as a light brown crystals. ES-MS m/e (%): 204 (M+ H+5 100). b) 2,2,2-Trifluoro-l-(6-methyl-4-phenyl-thieno[2,3-b1pyridin-5-yD-ethanone
To a stirred solution of 50 mg (0.24 mmol) (2-amino-thiophen-3-yl)-phenyl-methanone in 2 ml acetic acid was added 0.031 ml (0.24 mmol) of l,l,l-trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H2O) afforded 13 mg (17 %) 2,2,2-trifluoro-l-(6-methyl-4-phenyl-thieno[2,3-b] pyridin-5-yl)- ethanone as a yellow oil. ES-MS m/e (%): 322 (M+ H+, 100).
Example 21 l-(2-Chloro-6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-2,2,2-trirluoro-ethanone a) (*2-amino-5-chloro-thiophen-3-yl)-phenyl-methanone
To a stirred solution of 0.1 g (0.49 mmol) (2-amino-thiophen-3-yl)-phenyl-methanone (the preparation of which is described in example 20) in 10 ml dichloromethane at 00C was added 68 mg (0.49 mmol) N-chloro-succinimide (NCS). The mixture was then stirred from 00C to RT over night, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 3:1) afforded 40 mg (34 %) (2-arnino-5-chloro-thiophen-3-yi)-phenyl-methanone as a yellow solid. b) l-(2-Chloro-6-methyl-4-phenyl-thieno[2,3-blpyridin-5-yl)-2,212-trifluoro-ethanone To a stirred solution of 30 mg (0.13 mmol) (2-amino-5-chloro-thiophen-3-yl)-phenyl- methanone in 2 ml acetic acid was added 0.016 ml (0.13 mmol) of 1,1,1-triftuoro- pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 70 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLG (30% CH3CN/H20) afforded 10 mg (23 %) l-(2-chloro-6-methyl-4-phenyl-thieno[2,3-b] pyridin-5-yl)-2,2,2-trifluoro-ethanone as a light brown solid. ES-MS m/e (%): 356 (M+ H+, 100).
Example 22
2,2,2-Trifluoro-l-[4-(4-fluoro-phenyl)-6-methyl-thieno[2)3-b]pyridin-5-yl]-ethanone a) (2-amino-thiophen-3-yl)-(4-fluoro-phenylVmethanone
Following a procedure described in J.Med.Chem. 2002, 45, 382-389; 0.44 g (2.7 mmol) 3- (4-fluoro-phenyl)-3-oxo-propionitrile (the preparation of which is described in example 17), 0.33 g (2.15 mmol) [l,4]dithiane -2,5-diol, 0.16 ml (2.15 mmol) diethyl-amine and 5 ml ethanol were charged in a sealed tube. The reaction mixture was heated at 50 0C for 6 h. The tube was then placed in a fridge (about 4°C) for the night, and the product was collected by filtration and dried under vacuum to afford 0.41 g (69%) (2-amino- thiophen-3-yl)-(4-fluoro-phenyl)-methanone as a light brown crystals. b) 2,2,2-Trifluoro-l-r4-(4-fluoro-phenyl)-6-methyl-thieno[23-blpyridin-5-yl)-ethanone
To a stirred solution of 30 mg (0.13 mmol) (2-amino-thiophen-3-yl)-(4-fluoro-phenyl)- methanone in 1 ml acetic acid was added 0.017 ml (0.13 mmol) of 1,1,1-trifluoro- pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 15 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 8 mg (17 %) 2,2,2-trifluoro-l-[4-(4-fluoro-phenyl)-6-methyl- thieno[2,3-b] pyridin-5-yl)-ethanone as a yellow oil. ES-MS m/e (%): 340 (M+ H+, 100).
Example 23 l-(2-Methyl-4-phenyl-9-thia-l,7-diaza-fluoren-3-yl)-ethanone
a) 2-Amino-3-benzoyl-4,7-dihydro-5H-thieno[2,3-c1pyridine-6-carboxylic acid tert- butyl ester
To a stirred solution of 2.0 g (13.78 mmol) 3-oxo-3-phenyl-propionitrile in 50 ml ethanol was added 2.74 g (13.78 mmol) 4-oxo-piperidine-l-carboxylic acid tert-butyl ester, 442 mg (13.78 mmol) sulfur, and 1.2 ml (13.78 mmol) morpholine. The mixture was heated at 60 °C for 3 h, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Crystallisation in ethyl acetate afforded 3.60 g (73%) 2-amino-3-benzoyl-4,7-dihydro- 5H-thieno[2,3-c]pyridine-6-carboxyh'c acid tert-butyl ester as yellow crystals. ES-MS m/e (%): 359 (M+H+, 48). b) 3-Acetyl-2-methyl-4-phenyl-5,8-dihγdro-6H-9-thia-l,7-diaza-fluorene-7-carboxylic acid tert-butyl ester
To a stirred solution of 0.50 g (1.39 mmol) 2-amino-3-benzoyl-4,7-dihydro-5H- tbieno[2,3-c]pyridine-6-carboxylic acid tert-butyl ester in 6 ml acetic acid was added 0.19 ml (1.89 mmol) of pentane-2,4-dione and two drops of sulfuric acid. The mixture was then stirred at 1000C for 25 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 4:1) afforded 70 mg (12 %) 3-acetyl-2- methyl-4-phenyl-5,8-diKydro-6H-9-thia- l,7-diaza-fluorene-7-carboxylic acid tert-butyl ester as a yellow oil. ES-MS m/e (%): 423 (M+ H+, 100). c) l-(2-Methyl-4-phenyl-5,6,7,8-tetrahvdro-9-thia-l,7-diaza-fluoren-3-yl)-ethanone
To a stirred solution of 70 mg (0.16 mmol) 3-acetyl-2-methyl-4-phenyl-5,8-dihydro-6H- 9-thia-l,7-diaza-fluorene-7-carboxylic acid tert-butyl ester in 3 ml dicloromethane was added 0.2 ml trifluoroacetic acid. The mixture was stirred over night at RT, and then poured onto an aqueous solution OfNaHCO3, and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo to afford 48 mg (94%) l-(2-methyl-4-phenyl-5,6,7,8-tetrahydro-9-thia-l,7- diaza-fluoren-3-yl)-ethanone as a yellow solid. ES-MS m/e (%): 323 (M+ H+, 100). d) l-(2-methyl-4-phenyl-9-thia-l,7-diaza-fiuoren-3-yl)-ethanone
To a stirred solution of 0.70 g (2.17 mmol) l-(2-methyl-4-phenyl-5,6,7,8-tetrahydro-9- thia-l,7-diaza-fluoren-3-yl)-ethanone in 10 ml diphenylethylene was added 325 mg palladium on charcoal 10%.. The mixture was stirred over night at 1500C, and then poured onto ethyl acetate (about 150 ml), and extracted three times aqueous HCl (IM). The combined aqueous phases were basified with K2CO3 until ρh=8, and then the product was extracted with ethyl acetate. The combined organic phase were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate
1:1) afforded 75 mg (ll%) l-(2-methyl-4-phenyl-9-thia-l,7-diaza-fluoren-3-yl)- ethanone as a light yellow solid. ES-MS m/e (%): 319 (M+ H+, 100).
Example 24 l-[2-Chloro-4-(4-fluoro-phenyl)-6-methyl-thieno[2>3-b]pyridin-5-yl]-2,2,2-trifluoro- ethanone a") (2-ArQino-5-chloro-thiophen-3-yl)-C4-fluoro-phenvD-methanone
To a stirred solution of 0.26 g (1.18 mmol) (2-amino-thiophen-3-yl)-(4-fru.oro-phenyl)- methanone (the preparation of which is described in example 22) in 10 ml dichloromethane at 00C was added 164 mg (1.18 mmol) N-chloro-succinimide (NCS). The mixture was then stirred from O0C to RT over night, and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 4:1) afforded 95 mg (32 %) (2-amino-5-chloro-thiophen-3-yl)-(4-fluoro-phenyl)- methanone as a yellow solid. ES-MS m/e (%): 256 (M+ H+, 100). b) l-l"2-Chloro-4-r4-fluoro-phenyl')-6-methyl-thieno[2,3-b1pyridin-5-vD-2,2,2-trifluoro- ethanone
To a stirred solution of 50 mg (0.19 mmol) (2-amino-5-chloro-thiophen-3-yl)-(4-fluoro- phenyl)-methanone in 2 ml acetic acid was added 0.025 ml (0.19 mmol) of 1,1,1- trifluoro-pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 700C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 9:1) afforded 8 mg (11 %) l-[2-chloro-4-(4- fluoro-phenyl)-6-methyl-thieno[2,3-b]pyridin-5-yl)-2,2)2-trifluoro-ethanone_as an orange oil. ES-MS m/e (%): 374 (M+ H+, 100).
Example 25 Cyclopropyl-(2-methyl-4-phenyl-6>7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-
3-yl)-methanone
To a stirred solution of 60 mg (0.24 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-phenyl-methanone (the preparation of which is described in example 3) in 2 ml acetic acid was added 41 mg (0.32 mmol) of 1-cyclopropyl-butane- 1,3-dione (prep, described in the patent DE 94-4404059) and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 27 mg (33 %) cyclopropyl-(2-methyl-4-phenyl-6>7-dihydro-5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3- yl)-methanone as a light brown oil. ES-MS m/e (%): 334 (M+ H+, 100). Example 26
Cyclopropyl-(6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-methanone
To a stirred solution of 60 mg (0.29 mmol) (2-amino-tHophen-3-yl)-phenyl-methanone (the preparation of which is described in example 20) in 2 ml acetic acid was added 50 mg (0.39 mmol) of l-cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 7 mg (8 %) cyclopropyl-(6-methyl-4-phenyl-thieno[2,3-b]pyridin-5-yl)-methanone as a colorless oil. ES-MS m/e (%): 294 (M+ H+, 100).
Example 27 Cyclopropyl-(2-methyl-4-thiophen-2-yl-6)7-dihydro-5H-cyclopenta[4,5]thieno[2)3- b]pyridin-3~yl)-methanone
To a stirred solution of 60 mg (0.24 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]tHophen-3-yl)-thiophen-2-yl-methanone) (the preparation of which is described in example 2) in 2 ml acetic acid was added 40 mg (0.32 mmol) of 1- cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 15 mg (19 %) cyclopropyl-(2-methyl-4-thiophen-2- yl-6,7-dihydro-5H-cyclopenta[4>5]thieno[2>3-b]pyridin-3-yl)-methanone as a light brown oil. ES-MS m/e (%): 340 (M+ H+, 100). Example 28
Cyclopropyl-(4-furan-2-yl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b]pyridin-3-yl)-methanone
To a stirred solution of 60 mg (0.26 mmol) (2-amino-5,6-dihydro-4H-cyclopenta [b]thiophen-3-yl)-furan-2-yl-methanone (the preparation of which is described in example 18) in 2 ml acetic acid was added 43 mg (0.34 mmol) of 1-cyclopropyl-butane- 1,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 17 mg (21 %) cyclopropyl-(4-furan-2-yl-2-methyl-6>7-dihydro- 5H-cyclopenta[4,5]thieno[2,3-b]pyridin-3-yl)-methanone as a light brown solid. ES-MS m/e (%): 324 (M+ H+, 100).
Example 29
Cyclopropyl- [(4-(3,4-dichloro-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta [4,5]thieno[2)3-b]pyridin-3-yl)-methanone
To a stirred solution of 60 mg (0.19 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(3)4-dichloro-phenyl)-methanone (the preparation of
which is described in example 1) in 2 ml acetic acid was added 32 mg (0.25 mmol) of 1- cydopropyl-butane-l)3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 24 mg (31 %) cyclopropyl-[(4-(3,4-dichloro-phenyl)- 2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno [2,3-b]pyridin~3-yl)-methanone as a light brown solid. ES-MS m/e (%): 402 (M+ H+, 100).
Example 30
Cyclopropyl-[(4-(3,4-dichloro-phenyl)-2-methyl-5,6,7,8-tetrahydro-benzo[4,5]thieno
[2,3-b] pyridin-3-yl) ] -methanone a) f2-Amino-4,5,6,7-tetrahydro-benzo[b1thiophen-3-yl)-f3,4-dichloro-phenyl)- methanone
To a stirred solution of 0.30 g (1.40 mmol) 3-(3,4-dichloro-phenyl)-3-oxo-propionitrile in 10 ml ethanol was added 0.14 ml (1.40 mmol) cydohexanone, 43 mg (1.40 mmol) sulfur, and 0.12 ml (1.40 mmol) morpholine. The mixture was heated at 40 0C for 48 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.478 g (78%) (2-amino-4,5)6,7-tetrahydro-benzo[b]thiophen-3-yl)-(3,4- dichloro-phenyl)-methanone as an orange solid. ES-MS m/e (%): 326 (M+H+, 100). b) Cyclopropyl-[(4-(3,4-dichloro-phenyl')-2-methyl-5,6,7,8-tetrahydro-benzo[4,5lthieno r23-b1pyridin-3-yl)l-methanone
To a stirred solution of 60 mg (0.18 mmol) (2-amino-4,5,6,7-tetrahydro-benzo[b] thiophen-3-yl)-(3,4-dichloro-phenyl)-methanone in 2 ml acetic acid was added 31 mg (0.24 mmol) of l-cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 18 mg (23 %) cydopropyl-[(4- (3,4-dichloro-phenyl)-2-methyl-5,6,7,8-tetrahydro-benzo[4,5]thieno [2,3-b]pyridin-3- yl)] -methanone as a light brown solid. ES-MS m/e (%): 416 (M+ H+, 100).
Example 31
Cyclopropyl-[4-(4-methoxy-phenyl)-2-methyl-6,7-dihydro-5H-cyclopenta [4)5]thieno[2,3-b]pyridin-3-yl)]-methanone
To a stirred solution of 60 mg (0.22 mmol) (2-amino-5,6-dihydro-4H- cyclopenta[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone (the preparation of which is described in example 10) in 2 ml acetic acid was added 37 mg (0.29 mmol) of 1- cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative
HPLC (30% CH3CN/H20) afforded 17 mg (22 %) cyclopropyl-[4-(4-methoxy-phenyl)-2-
methyl-6,7-dihydro-5H-cyclopenta[455]thieno [2,3-b]pyridin-3-yl)-methanone as a light brown solid. ES-MS m/e (%): 364 (M+ H+, 100).
Example 32
Cyclopropyl-^-C^methoxy-phenylj-Z-methyl-Sjo^^-tetrahydro-benzo^Sjthieno^^- b]pyridin-3-yl)]-methanone
To a stirred solution of 60 mg (0.21 mmol) (2-amino-4,5,6,7-tetrahydro- benzo[b]thiophen-3-yl)-(4-methoxy-phenyl)-methanone (the preparation of which is described in example 8) in 2 ml acetic acid was added 35 mg (0.28 mmol) of 1- cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative
HPLC (30% CH3CN/H20) afforded 25 mg (32 %) cyclopropyl-[4-(4-methoxy-ρhenyl)-2- methyl-5)6,7,8-tetrahydro-benzo[4,5]thieno[2)3-b]pyridin-3-vl)-methanone as a light brown oil. ES-MS m/e (%): 378 (M+ H+, 100).
Example 33 4-(3-Cyclopropanecarbonyl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b]pyridin-4-yl)-benzoic acid methyl ester a) 4-(2-Amino-5,6-dihydro-4H-cyclopenta[b1thiophene-3-carbonyl)-benzoic acid methyl ester
To a stirred solution of 0.30 g (1.47 mmol) 4-(2-cyano-acetyl)-benzoic acid methyl ester in 10 ml ethanol was added 0.13 ml (1.47 mmol) cyclopentanone, 46 mg (1.47 mmol) sulfur, and 0.12 ml (1.47 mmol) morpholine. The mixture was heated at 40 0C for 72 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.53 g (98%) 4-(2-Amino-5,6-dihydro-4H-cyclopenta[b]thiophene-3- carbonyl)-benzoic acid methyl ester as a brown solid. ES-MS m/e (%): 302 (M+H+, 100). b) 4-(3-Cyclopropanecarbonyl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5lthieno[2,3- b1pyridin-4-yl)-benzoic acid methyl ester
To a stirred solution of 60 mg (0.20 mmol) 4-(2-amino-5,6-dihydro-4H- cyclopenta[b]thiophene-3-carbonyl)-benzoic acid methyl ester in 2 ml acetic acid was added 33 mg (0.26 mmol) of l-cydopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 22 mg (28 %) 4- (3-cyclopropanecarbonyl-2-methyl-6,7-dihydro-5H-cyclopenta[4,5]thieno[2,3- b]pyridin-4-yl) -benzoic acid methyl ester as a light brown oil. ES-MS m/e (%): 392 (M+ H+, 100).
Example 34
4-(3-Cyclopropanecarbonyl-2-methyl-5,6,7,8-tetrahydro-benzo[4,5]thieno[2,3- b]pyridin-4-yl) -benzoic acid methyl ester a) 4-(2-Ainino-4,5,6,7-tetrahydro-benzorbltl-iophene-3-carbonyl)-benzoic acid methyl ester
To a stirred solution of 0.30 g (1.47 mmol) 4-(2-cyano-acetyl)-benzoic acid methyl ester in 10 ml ethanolwas added 0.15 ml (1.47 mmol) cyclohexanone, 46 mg (1.47 mmol) sulfur, and 0.12 ml (1.47 mmol) morpholine. The mixture was heated at 40 0C for 72 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.45 g (97%) 4-(2-amino-4,5,6,7-tetrahydro-benzo[b]thiophene-3- carbonyO-benzoic acid methyl ester as a brown solid. ES-MS m/e (%): 316 (M+H+, 100). b) 4-(3-Cyclopropanecarbonyl-2-methyl-5,6,7,8-tetrahydro-benzor4>5ithieno[2,3-bl pyridin-4-yl)-benzoic acid methyl ester To a stirred solution of 60 mg (0.19 mmol) 4-(2-Amino-4,5,6,7-tetrahydro-benzo[b] thiophene-3-carbonyl)-benzoic acid methyl ester in 2 ml acetic acid was added 32 mg (0.25 mmol) of l-cyclopropyl-butane-lβ-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CNyH2O) afforded 28 mg (37 %) 4-(3- cyclopropanecarbonyl-2-methyl-5,6,7,8-tetrahydro-benzo [4,5] thieno [2,3-b] pyridin-4- yl) -benzoic acid methyl ester as a light pink solid. ES-MS m/e (%): 406 (M+ H+, 100).
Example 35
4-(3-Cyclopropanecarbonyl-2-methyl-6,7,8>9-tetrahydro-5H-10-thia-l-aza- benzo[a]azulen-4-yl)-benzoic acid methyl ester a) 4-(2-Amino-5,6^7,8-tetrahydro-4H-cycloheptarb1thiophene-3-carbonyl)-benzoic acid methyl ester
To a stirred solution of 0.30 g (1.47 mmol) 4-(2-cyano-acetyl)-benzoic acid methyl ester in 10 ml ethanol was added 0.17 ml (1.47 mmol) cycloheptanone, 46 mg (1.47 mmol) sulfur, and 0.13 ml (1.47 mmol) morpholine. The mixture was heated at 400C for 72 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.10 g (22%) 4-(2-amino-5,657,8-tetrahydro-4H-cyclohepta[b]thiophene- 3-carbonyl)-benzoic acid methyl ester as an orange solid. ES-MS m/e (%): 330 (M+H+, 100).
b) 4-(3-Cydopropanecarbonyl-2-ineth.yl-6,7,819-tetrahydro-5H-lQ-thia-l-aza- benzo[a1azulen-4-yD -benzoic acid methyl ester
To a stirred solution of 20 mg (0.06 mmol) 4-(2-Amino-5,6,7,8-tetrahydro-4H- cyclohepta[b]thiophene-3-carbonyl)-benzoic acid methyl ester in 1 ml acetic acid was added 10 mg (0.06 mmol) of l-cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 11 mg (44 %) 4- (3-cyclopropanecarbonyl-2-methyl-6,7,8,9-tetrahydro-5H-10-thia-l-aza- benzo[a]azulen-4-yl)-benzoic acid methyl ester as a light yellow solid. ES-MS m/e (%): 420 (M+ H+, 100).
Example 36
Cyclopropyl-[4-(3,4-dichloro-phenyl)-2-methyl-6>7,8,9-tetrahydro-5H-10-thia-l-aza- benzo [a] azulen-3-yl)-methanone a) (2-Amino-5,6,7,8-tetrahydro-4H-cycloheptarb1thiophen-3-yl)-(3,4-dichloro-phenyl)- methanone
To a stirred solution of 0.30 g (1.40 mmol) 3-(3,4-dichloro-phenyl)-3-oxo-propionitrile in 10 ml ethanol was added 0.15 ml (1.40 mmol) cycloheptanone, 43 mg (1.40 mmol) sulfur, and 0.12 ml (1.40 mmol) morpholine. The mixture Was heated at 40 0C for 72 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.12 g (26%) (2-amino-5,6,7,8-tetrahydro-4H-cyclohepta[b]thiophen-3- yl)-(3,4-dichloro-phenyl)-methanone as a yellow oil. ES-MS m/e (%): 340 (M+H+, 100). b) Cyclopropyl-[4-(3,4-dichloro-phenyl)-2-meτhyl-6J,8,9-tetrahydro-5H-10-thia-l-aza- benzo [ai azulen-3-yl)-methanone To a stirred solution of 36 mg (0.11 mmol) (2-amino-5,6,7,8-tetrahydro-4H- cyclohepta[b]thiophen-3-yl)-(3,4-dichloro-phenyl)-methanone in 2 ml acetic acid was added 18 mg (0.11 mmol) of l-cyclopropyl-butane-l,3-dione and one drop of sulfuric acid. The mixture was then stirred at 1000C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 10 mg (19 %) cyclopropyl-[4-(3,4-dichloro-phenyl)-2-methyl-6,7,8,9-tetrahydro-5H-10-thia-l-aza- benzo[a]azulen-3-yl)-methanone as a light yellow oil. ES-MS m/e (%): 430 (M+ H+, 100).
Example 37 Cyclopropyl-(2-methyl-4-phenyl-9-thia-l)7-diaza-fluoren-3-yl)-methanone
a) Cyclopropyl-(2-meiiiyl-4-phenyl-5,6J18-tetrahydro-9-thia-l,7-diaza-fluoren-3-yl)- methanone
To a stirred solution of 1.0 g (2.79 mmol) 2-amino-3-benzoyl-4,7-dihydro-5H- thieno[2,3-c]pyridine-β-carboxylic acid tert-butyl ester (the preparation of which is described in example 23) in 15 ml acetic acid was added 0.70 g (5.55 mmol) of 1- cyclopropyl-butane-l,3-dione and three drops of sulfuric acid. The mixture was then stirred at 1000C for 30 minutes and then concentrated in vacuo. Flash chromatography (dichloromethane / methanol 95:5) afforded 0.47 g (48 %) cyclopropyl-(2-methyl-4- phenyl-5,6>7,8-tetrahydro-9-tma-l)7-diaza-fluoren-3-yl)-methanone_as a brown oil. ES- MS m/e (%): 349 (M+ H+, 100). b) Cyclopropyl- (2-methyl-4-phenyl-9-thia- l,7-diaza-fluoren-3-yl)-methanone
To a stirred solution of 0.46 g (1.32 mmol) cyclopropyl-(2-methyl-4-phenyl-5,6,7,8- tetrahydro-9-thia-l>7-diaza-fluoren-3-yl)-methanone in 7 ml diphenylethylene was added 230 mg palladium on charcoal 10%.. The mixture was stirred over night at 1500C, and then poured onto ethyl acetate (about 150 ml), and extracted three times aqueous HCl (IM). The combined aqueous phases were basified with K2CO3 until ph=8, and then the product was extracted with ethyl acetate. The combined organic phase were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 1:1) afforded 35 mg (8%) cyclopropyl- (2-methyl-4-phenyl-9-thia-l,7-diaza- fluoren-3-yl)-methanone as. a light yellow solid. ES-MS m/e (%): 345 (M+ H+, 100).
Example 38
3-Aceτyl-2-methyl-4-phenyl-5,8-dihydro-6H-9-thia-l,7-diaza-fluorene-7-carboxylic acid methyl ester
To a stirred solution of 50 mg (0.15 mmol) l-(2-methyl-4-phenyl-5,6,7,8-tetrahydro-9- thia-l,7-diaza-fluoren-3-yl)-ethanone (the preparation of which is described in example 23) in 5 ml dichloromethane was added 0.02 ml (0.022 mmol) methyl chloroformate and 32 mg (0.022 mmol) potassium carbonate. The mixture was stirred at RT for Ih and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 1:1) afforded 33 mg (56%) 3-Acetyl-2-methyl-4-phenyl-5,8- dihydro-6H-9-thia-l,7-diaza-fluorene-7-carboxylic acid methyl ester as a light yellow solid. ES-MS m/e (%): 381 (M+ H+, 100).
Example 39
Cyclopropyl-(2-methyl-4-phenyl-5,8-dihydro-6H-7-oxa-9-thia-l-aza-fluoren-3-yl)- methanone a) (2-Amino-4,7-dihydro-5H-thieno [2,3-c] pyran-3-yl)-phenyl-methanone
To a stirred solution of 0.725 g (5.00 mmol) 3-oxo-3-phenyl-propionitrile in 5 ml ethanol was added 0.50 g (5.00 mmol) tetrahydro-pyran-4-one, 160 mg (5.00 mmol) sulfur, and 0.44 ml (5.00 mmol) morpholine. The mixture was heated at 550C for 2 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Trituration in ether afforded 0.75 g (58%) (2-amino-4,7-dihydro-5H-tnieno[23-c]pyran-3-yl)-phenyl- methanone as a light yellow solid. ES-MS m/e (%): 260 (M+H+, 100). b) Cyclopropyl-(2-methyl-4-phenyl-5,8-dihydro-6H-7-oxa-9-thia-l-aza-fluoren-3-yl)- methanone To a stirred solution of 100 mg (0.38 mmol) (2-ammo-4,7-dihydro-5H-thieno[2,3- c]pyran-3-yl)-phenγl-methanone in 5 ml acetic acid was added 73 mg (0.57 mmol) of 1- cyclopropyl~butane-l,3-dione (prep, described in the patent DE 94-4404059) and one drop of sulfuric acid. The mixture was then stirred at 70 0C for 2 hours and then concentrated in vacuo. Plash chromatography (heptane / ethyl acetate 3:1) afforded 50 mg (37 %) cyclopropyl-(2-methyl-4-phenyl-5,8-dihydro-6H-7-oxa-9-thia-l-aza-fluoren-3- yl)-methanone_as a yellow solid. ES-MS m/e (%): 350 (M+ H+, 100).
Example 40
C7clopropyl-(6-methyl-4-piperidin-l-yl-2,3-dihydro-lH-8-thia-7-aza-cyclopenta[a] inden-5-yl)-methanone a) 2-Amino-5,6-dmydro-4H-cyclopentafb1thiophene-3-carbonitrile
To a stirred solution of 6.28 g (0.095 mol) malonitrile in 100 ml ethanol was added 8.00 g (0.095 mol) cyclopentanone, 3.04 g (0.095 mol) sulfur, and 8.29 ml (0.095 mol) morpholine. The mixture was heated at 80 0C for 2 h and then poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 3:1) afforded 4.89 g (31%) 2-amino-5)6-dihydro-4H-cyclopenta[bjthiophene-3- carbonitrile as a light brown solid. ES-MS m/e (%): 165 (M+H+, 100). b) (4-Ammo-6-methyl-2,3-dihydro-lH-8-thia-7-aza-cyclopentafa1inden-5-yl')- cyclopropyl-mefhanone To a stirred solution of 0.30 g (1.83 mmol) of 2-ammo-5,6-dihydro-4H-cyclopenta[b] thiophene-3-carbonitrile in 6 ml toluene was added 0.30 g (2.14 mmol) l-cyclopropyl-3- methoxy-but-2-en-l-on.e and 3 mg of p-toluenesulfonic acid. The mixture was heated at reflux for 2 hours, concentrated in vacuo, partitioned between ethyl acetate and water. The combined organic phases were dried over sodium sulfate and evaporated to dryness. The residue was taken up in n-butyl acetate 6 ml and 1.018 g (3.91 mmol) tin (IV) chloride was heated under reflux for 30 minute and allowed to cool. The mixture was
partitioned between aqueous sodium hydroxide and ethyl acetate. Flash chromatography (heptane / ethyl acetate 3:1) afforded 25 mg (5%) (4-amino-6-methyl-2,3-dihydro-lH-8- thia-7-aza-cyclopenta[a]inden-5-yl)-cyclopropyl-methanone as a white solid. ES-MS m/e (%): 273 (M+H+, 100). c) Cyclopropyl-(6-methyl-4-piperidin-l-yl-23-dihydro-lH-8-thia-7-aza-cyclopentaral inden-5-yl)-methanone
To a stirred solution of 12 mg (0.044 mmol) of (4-amino-6-methyl-2,3-dihydro-lH-8- thia-7-aza-cyclopenta[a]inden-5-yl)-cyclopropyl-methanone in 2 ml DMF was added 3.0 mg (0.066 mmol) NaH (55% in oil). After 20 minutes, 10 μl (0.044 mmol) of 1,5- dibromo-pentane and stirring was continued at RT over the night. The mixture was poured onto water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / ethyl acetate 6:1) afforded 8 mg (54%) cyclopropyl-(6-methyl-4-piperidin-l- yl-2,3-dihydro-lH-8-thia-7-aza-cyclopenta[a] inden-5-yl)-methanone as a white solid. ES-MS m/e (%): 341 (M+ H+, 100).
Example 41 l-(6-Methyl-2,4-diphenyl-thieno[2,3-b]pyridin-5-yl)-ethanone a) N-(3-Benzoyl-5-bromo-thiophen-2-yl)-acetamide
To a stirred solution of 1.00 g (4.92 mmol) of (2-amino-thiophen-3-yl)-phenyl- methanone (the preparation of which is described in example 20) in 20 ml CH2Cl2 at 0 0C was added 0.92 g (5.18 mmol) NBS (N-bromo succinimide) in one portion. After 10 minutes, 4.65 ml (49.2 mmol) of acetic anhydride, 0.68 ml (4.92 mmol) OfEt3N and 0.30 g (2.45 mmol) of DMAP were added. Stirring was continued at RT for 2 hours, the mixture was poured onto water and extracted three times with CH2Cl2. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. Flash chromatography (heptane / CH2Cl2 1:1) afforded 0.85 g (53%) N-(3-benzoyl-5-bromo- fhiophen-2-yl)-acetamide as a light brown solid. ES-MS m/e (%): 324, 326 (M+ H+, 100).
b ) N- ( 3 -Benzoyl- 5-phenyl-thiophen-2-yl*) - acetamide
A stirred solution of 0.448 g (1.38 mmol) of N-(3-benzoyl-5-bromo-thiophen-2-yl) acetamide in 20 ml toluene and 10 ml EtOH was degassed with argon. Tetrakis-
(triphenylphosphine) palladium (0) (0.320 g, 0.27 mmol), phenyl boronic acid (0.194 g, 1.59 mmol) and 20 ml of an aqueous solution OfNa2CO3 IN were added. The mixture was heated at 90 °C for 1 hour before cooling down to RT, filtered through Celite and concentrated. The mixture was extracted with ethyl acetate and aq. NaHCO3. The combined organic phases were dried over sodium sulfate and concentrated in vacuo.
Flash chromatography (heptane / ethyl acetate 6:1) afforded 0.393 g (88 %) N-(3- benzoyl-5-phenyl-thiophen-2-yl)-acetamide as a light yellow solid. ES-MS m/e (%): 322 (M+ H+, 100).
c) (2-Amino-5-phenyl-thiophen-3-yl)-phenyl-methanone To a stirred solution of 0.393 g (1.22 mmol) of N-(3-benzoyl-5-phenyl-thiophen~2-yl)- acetamide in 15 ml EtOH was added 2.5 ml of aq. NaOH (4N). The reaction mixture stirred at RT for 1 hour, and then poured onto an aq. solution of NH4CI sat. and the product was extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo to yield 0.45 g (98 %) of (2-amino-5-phenyl- thiophen-3-yl)-phenyl-methanone as a light brown solid. ES-MS m/e (%): 280 (M+ H+, 100).
d) l-(6-Methyl-2,4-diphenγl-thieno['2,3-b1pyridm-5-γl)-ethanone
To a stirred solution of 55 mg (0.197 mmol) (2-amino-5-phenyl-thiophen-3-yl)-phenyl- methanone in 5 ml acetic acid was added 0.016 ml (0.239 mmol) of pentane-2,4-dione and one drop of sulfuric acid. The mixture was then stirred at 100 0C for 10 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H20) afforded 19 mg (28 %) l-(6-methyl-2,4-diphenyl-thieno[2,3-b]pyridin-5-yl)-ethanone as a light grey solid. ES-MS m/e (%): 344 (M+ H+, 100).
Example 42 Cyclopropyl-(6-Methyl-2>4-diphenyl-thieno[2,3-b]pyridin-5-γl)-methanone
To a stirred solution of 0.130 g (0.465 mmol) (2-amino-5-phenyl-thiophen-3-yl)~phenyl- methanone in 7 ml acetic acid was added 62 mg (0.491 mmol) of 1-cyclopropyl-butane- 1,3-dione and one drop of sulfuric acid. The mixture was then stirred at 110 0C for 20 minutes in a microwave and then concentrated in vacuo. Preparative HPLC (30% CH3CN/H2O) afforded 29 mg (17 %) cyclopropyl-(6-methyl-2,4-diphenyl-thieno [2,3- b]pyridin-5-yl)-methanone as a light yellow powder. ES-MS m/e (%): 370 (M+ H+, 100).