WO2006087140A1 - Novell processes for the preparation of a benzofuran - Google Patents
Novell processes for the preparation of a benzofuran Download PDFInfo
- Publication number
- WO2006087140A1 WO2006087140A1 PCT/EP2006/001179 EP2006001179W WO2006087140A1 WO 2006087140 A1 WO2006087140 A1 WO 2006087140A1 EP 2006001179 W EP2006001179 W EP 2006001179W WO 2006087140 A1 WO2006087140 A1 WO 2006087140A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- pyrimidin
- dimethyl
- chloro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*C(c1c(C(c2cc(c(Cc3cnc(NC(*)=O)nc3NC(*)=O)cc(OC)c3OC)c3[o]2)=O)c2cc(Cl)ccc2[n]1S(c1ccc(C)cc1)(=O)=O)=O Chemical compound C*C(c1c(C(c2cc(c(Cc3cnc(NC(*)=O)nc3NC(*)=O)cc(OC)c3OC)c3[o]2)=O)c2cc(Cl)ccc2[n]1S(c1ccc(C)cc1)(=O)=O)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/06—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
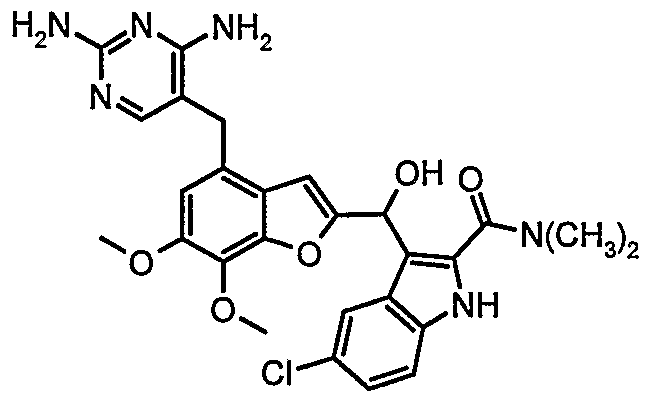
- the present invention relates to novel processes for the preparation of a compound of formula I 1 which compound is related to dihydrofolate reductase inhibitors
- the compound of formula I has valuable antibiotic properties.
- the compound can be used in the control or prevention of infectious diseases in mammals, both humans and non-humans. In particular, it exhibits pronounced antibacterial activity, even against multiresistant Gram-positive strains and against opportunistic pathogens such as e.g. Pneumocystis carinii.
- the compound can also be administered in combination with known substances of antibacterial activity and exhibits synergistic effects with some of them.
- Typical combination partners are e.g. sulfonamides or other inhibitors of enzymes, which are involved in folic acid biosynthesis such as, for example, pteridine derivatives.
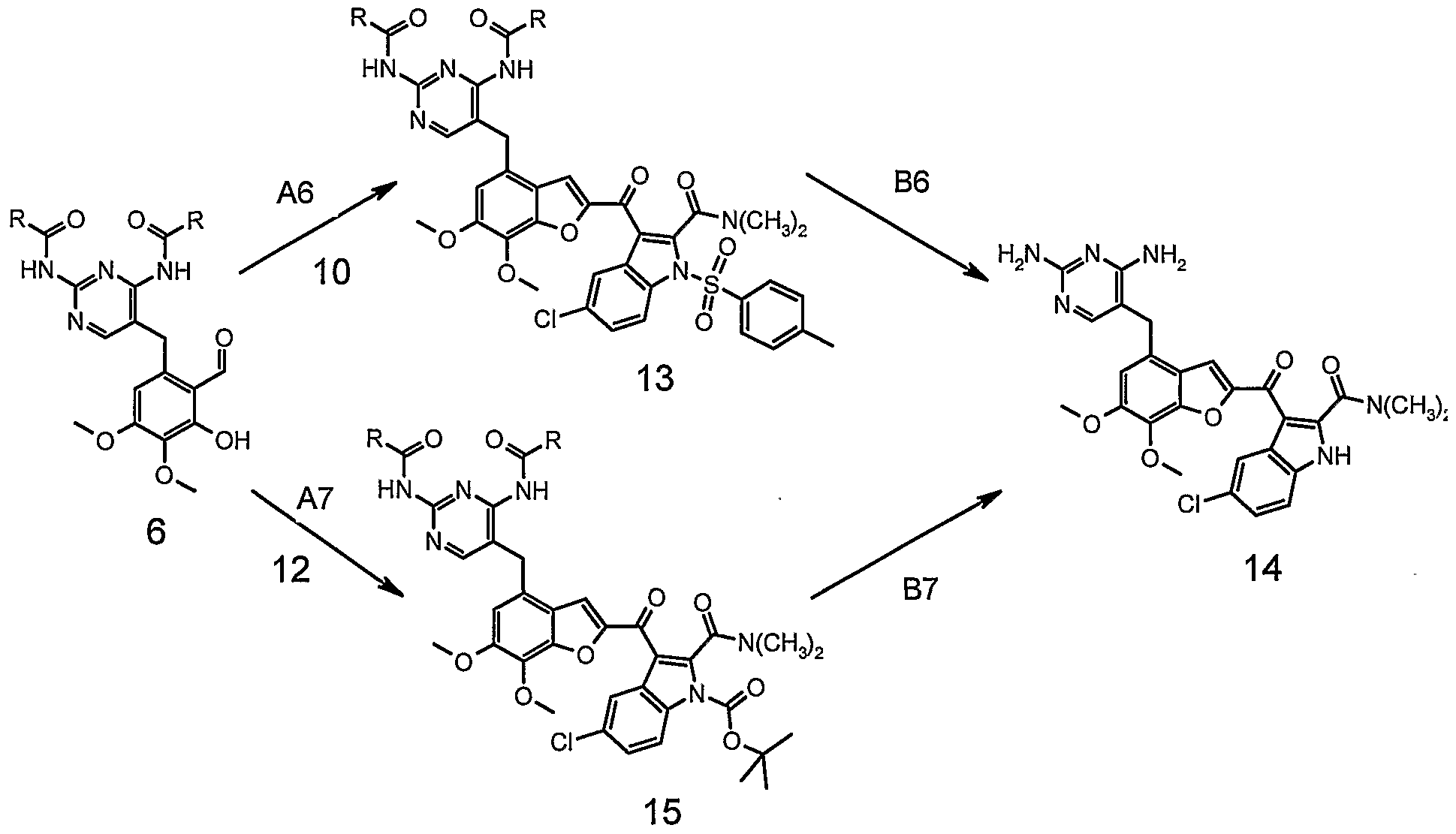
- the present invention provides a process for preparing the compound of the formula I from the intermediate of formula 6.
- the intermediate of formula 3 is synthesized in 3 steps from a readily available starting material 1 (Scheme 1).
- the diamino pyrimidine substituent of 1 is selectively protected according to R.J. Griffin et al., J.Chem.Soc. Perkin Trans I, 1811 (1992) leading to compound of formula 2, which in turn is formylated to a compound of formula 3 (Scheme 1).
- Scheme 1 :
- the compound of formula I is basic in nature and can be, if desired, transformed with an acid into pharmaceutically acceptable salts.
- Suitable acids are, e.g. hydrochloric acid, maleic acid, succinic acid, L(+)-lactic acid, DL-lactic acid, glycolic acid, i-hydroxy-naphthalene-2-carboxylic acid, tartaric acid, citric acid, methane sulfonic acid. Most preferred are carboxylic acids.
- the central intermediate of formula 6 to prepare the compound of formula I may be prepared following the reaction sequences depicted in Schemes 1 to 3.
- the protection A1 of trimethoprim 1 can be done by heating compound of formula 1 with acid anhydrides, e.g. acetic anhydride, isobutyric acid anhydride or pivaloyl acid anhydride in an inert, high boiling solvent like toluene, p-xylene or in plain acid anhydride up to about 120 0 C to 160 0 C.
- the formulation B1 of the protected trimethoprim 2 can be achieved in an inert solvent, e.g.
- dichloromethane dichloroethane, preferably dichloromethane with dichloromethyl-methyl ether and a Lewis acid, e.g. tin tetrachloride at 0 0 C to -30 0 C, preferably at -10 0 C.
- compound of formula 3 can also be synthesized via protection A2 of compound 4 with acid anhydrides, e.g. acetic anhydride, methyl-propionic acid anhydride or pivaloyl acid anhydride in an inert, high boiling solvent like toluene, p- xylene or in plain acid anhydride, preferably methyl-propionic acid anhydride up to about 120 0 C to 160 0 C.
- acid anhydrides e.g. acetic anhydride, methyl-propionic acid anhydride or pivaloyl acid anhydride in an inert, high boiling solvent like toluene, p- xylene or in plain acid anhydride,
- Carbonylation B2 of compound of formula 5 can be effected in an inert atmosphere and solvent, e.g. tetrahydrofuran, with palladium tetrakis as catalyst, carbon monoxide and tri-butyl tin-hydride at 60 0 C to 80 0 C.
- the selective demethylation A3 can be done in an inert solvent, e.g. dichloromethane, acetonitrile, in combination with a Lewis acid like aluminium trichloride, boron trichloride, boron tribromide, manganese dichloride, manganese diiodide, preferably aluminium trichloride and a nucleophile, e.g. sodium iodide, dimethyl sulfide, diethyl sulfide, tetrahydrothiophene, preferably sodium iodide at room temperature up to 40 0 C.
- solvent e.g. tetrahydro
- the convergent synthesis strategy of compound of formula I deserves an additional intermediate of formulae 10 or 12.
- the starting material of formula 7 is acetylated (A4) with acetyl chloride and a Lewis acid like aluminium trichloride or tin tetra-chloride at ambient temperature to the intermediate of formula 8.
- Compound of formula 8 can be converted by protecting first (B4) the nitrogen with a sulfonyl chloride, e.g. benzyl- or p-toluene-sulfonyl chloride with a base like triethylamine, pyridine in an inert solvent at room temperature to the compound of formula 9 followed by bromination (C4) with e.g.
- bromine, N-bromosuccinimid, copper (II) bromide preferably bromine of the acetyl group in dioxane to compound of formula 10, or bromination first with e.g. bromine, N- bromosuccinimid, preferably bromine of 8 (B5) in an inert solvent like dioxane at room temperature to compound of formula 11 and subsequent protection (C5) with di-te/f-butyl dicarbonate and a pyridine base, e.g. 2,6-dimethyl-pyridine with 4- dimethylamino-pyridine as catalyst to compound of formula 12 at ambient temperature.
- a pyridine base e.g. 2,6-dimethyl-pyridine with 4- dimethylamino-pyridine as catalyst to compound of formula 12 at ambient temperature.
- tetrahydrofuran methyl alcohol, preferably tetrahydrofuran and water with a strong base like sodium or potassium hydroxide, preferably sodium hydroxide at 40 0 C to 80 0 C preferably at 50 0 C to the compound of formula 14.
- Reduction A10 of the keto function of compound of formula 14 can be done with a reducing agent, e.g. sodium borohydride, sodium cyanoborohydride, zinc borohydride, sodium acetoxyborohydride, preferably sodium cyanoborohydride, sodium borohydride or zinc borohydride in an organic solvent like methanol, isopropanol, tetrahydrofuran, dimethoxyethane or a mixture thereof, preferably isopropanol or tetrahydrofuran at temperature in the range of -20 0 C up to 70 0 C depending on the reducing agent leading to the target compound I.
- a reducing agent e.g. sodium borohydride, sodium cyanoborohydride, zinc borohydride, sodium acetoxyborohydride, preferably sodium cyanoborohydride, sodium borohydride or zinc borohydride in an organic solvent like methanol, isopropanol, tetrahydrofuran, dimethoxy
- the compounds of formulae 2, 3, 5, 6, 8 to 17 are novel and are also objects of the invention. They can be prepared according to the reaction sequences elucidated in Schemes 1 to 8. The preparation of compounds outlined in Schemes 1 to 8 are, moreover, described in more detail in the examples.
- the compound of formula I or their pharmaceutically acceptable salts have valuable antibacterial properties. These compounds are active against a large number of pathogenic microorganisms such as e.g. S. aureus, P. carinii etc. by virtue of their activity in inhibiting bacterial dihydrofolate reductase (DHFR).
- DHFR bacterial dihydrofolate reductase
- Examples 1 to 11 describe the preparation of compound 6, while examples 12 to 16 describe the preparation of the compound of formulae 10 and 12, and examples 17 to 32 describe the condensation of the compound of formula 6 with those of formulae 10 or 12 to the end product of formula I.
- Compound of formula 4 can be prepared e.g. according to M. Calas et al., Eur.J.Med.Chem.Chim.Ther., 17 (6), 497 (1982).
- Compound 7 can be prepared in analogy to e.g. W.B. Wright et al., J. Med.Chem., 11(6), 1164 (1968). All other reagents and solvents are readily commercially available, for example from Fluka or equivalent commercial suppliers. The temperatures are given in degrees Celsius.
- Solvent A 10 mM Formic acid (Formic acid 377 ⁇ l) was added to HPLC grade water (1 L, Millipore filtered)
- Solvent B Acetonitrile HPLC grade (Biosolve Ltd)
- Wavelength 210 nm to 400 nm.
- HPLC Apparatus Type Finnigan StartSystem SCN 1000, Finnigan Photodiode array detector(PDA) UV6000LP
- a solution of trimethoprim (5 g, 17.24 mmol) in pivalic anhydride (8.74 ml_, 43.10 mmol, 2.5 eq.) was heated during 2 h at 150 0 C under argon. Hot AcOEt was added, and the organic layers were washed with aqueous NaHCO 3 10%, water and brine. The org. layers were then dried over MgSO 4 , filtered and evaporated.
- trimethoprim 50 g, 172.4 mmol
- isobutyric anhydride 100 g, 105 ml_, 632 mmol, 3.6 eq.
- the warm solution was poured into 1 L of cyclohexane from where it slowly crystallized.
- a solution of trimethoprim (50 g, 172.4 mmol) in isobutyric anhydride (62 g, 65.5 ml_, 392 mmol, 2.3 eq.) was heated during 2 h at 150 0 C under Ar and stirred with a mechanical stirrer.
- the reaction mixture is poured into a solution of 300 mL 1 N K 3 PO 4 and 200 mL 1M Na/K-tartrate while cooling with an ice bath.
- the mixture pH was adjusted with 4N NaOH solution to 7-8) was then stirred for 15 minutes until complete hydrolysis, and then extracted with DCM (300 mL) together with AcOEt (500 mL).
- the organic layer was washed with 0.1 N HCI solution (2x200 mL) and brine (2x300 mL), dried over MgSO 4 , filtered and evaporated.
- the slurry was stirred at -15 0 C for two hours, at -10 0 C for one hour and 30 minutes at -5 0 C. Then 40 mL DCM was added at -5 0 C and the separated crystals at the top of the solvent layer were removed with vigorous stirring for 15 minutes. The thin slurry was transferred into a well-stirred mixture of 35 g Na 2 CO 3 (with one crystal water) dissolved in 100 mL water and 35 mL DCM at 10 0 C. The mixture was stirred for 15 minutes at RT and then transferred back to the reaction vessel to finish the workup continuing the stirring at RT.
- Sodium iodide (616 mg, 4.11 mmol) was added, and after 30 minutes, 0.5 mL of acetonitrile was added. The reaction was checked by LCMS 1 and 0.5 mL acetonitrile was added to complete the reaction. The reaction mixture was then poured into 1 N K 3 PO 4 /DCM biphasic solution. The two phases were separated. The aqueous layers were extracted twice with AcOEt and the organic layers were washed with water and brine, then dried on MgSO 4 , filtered and evaporated.
- the mixture was cooled to RT, diluted with 75 mL DCM and quenched by adding the reaction mixture to 30 mL ice-water, then 2.5 mL of concentrated HCI was slowly added, which helped to dissolve the yellow precipitate.
- the organic layer was separated and the aqueous layer extracted once more with DCM (75 mL).
- the combined organic layers were washed with brine (50 mL), twice with sodium bicarbonate solution made from 50 mL saturated sodium bi-carbonate (NaHCO 3 ) + 150 mL water (2x100 mL), 0.1 N HCI solution (50 mL) and again brine (1x50 mL).
- the resulting yellowish solution was dried over MgSO 4 and concentrated.
- step A4 This example illustrates the preparation of 3-Acetyl-5-chloro-1H-indole-2- carboxylic acid dimethylamide 8 (step A4).
- Aluminium trichloride 36 g, 270 mmol was added slowly to a suspension of 7 (30 g, 135mmol) in DCM (675 mL) at 0 0 C under Ar.
- the reaction mixture was stirred for 30 minutes and acetyl chloride (9.6 mL, 135 mmol) was added dropwise at 0 0 C.
- the gold yellow reaction mixture was stirred for an additional 1 hour, until the reaction was completed (verification by LC-MS).
- the reaction mixture is then poured on ice (250 mL).
- the pH was adjusted to pH 4.5 by addition of 4 N NaOH solution (80 mL).
- the phases were separated and the aqueous layer was extracted with DCM (2 x 200 mL). All collected organic layers were then washed with water and brine, then dried over MgSO 4 , filtered and evaporated.
- the compound 8 was obtained as a beige solid and used for the next reaction step without further purification.
- This example illustrates the preparation of 3-(2-Bromo-acetyl)-5-chloro-1-(toluene- 4-sulfonyl)-1H-indole-2-carboxylicacid-dimethyl-amide 10 (step C4).
- Example 15 This example illustrates the preparation of 3-(2-Bromo-acetyl)-5-chloro-1H-indole- 2-carboxylic acid dimethylamide 11 (step B5).
- This example illustrates the preparation of 3-(2-Bromo-acetyl)-5-chloro-2- dimethylcarbamoyl-indole-1-carboxylic acid te/f-butyl ester 12 (step C5).
- This example illustrates the preparation of 5-Chloro-3-[4-(2,4-diamino-pyrimidin-5- yl-methyl)-6,7-dimethoxy-benzofuran-2-carbonyl]-1/-/-indole-2-carboxylic acid dimethyl-amide, mesylate salt 14 (step B6).
- the mixture was cooled to RT and the pH was lowered to approximately 6.5 by adding aqueous HCI solution (75 mL, 300 mmol, 4 M). NaHCO3 saturated solution (700 mL) was added carefully, a minor formation of CO 2 was observed.
- the mixture was extracted with ethyl acetate/isopropanol 85/15 (2x1600 mL). The organic layers were washed with water/brine 90/10 (2x200 mL) and brine (1x200 mL), filtered through a plug of Celite, combined and evaporated to dryness. The resulting yellow foam was kept at high vacuum and RT for 16 hours.
- Example 26 This example illustrates the preparation of 5-Chloro-3- [4-(2,4-diamino-pyrimidin-5- yl-methyl)-6,7-dimethoxy-benzofuran-2-ylmethyl]-1/-/-indole-2-carboxylic acid dimethyl-amide I (step A10).
- the mesylate salt 14 (4.88 g, 6.932 mmol) was dissolved in water (50 ml_). AcOEt (50 ml_) was added, and then the mixture was quenched with NaHCO 3 10 % (50 ml_) and stirred vigorously. The organic layers were separated and the aqueous layers were extracted with AcOEt (50 mL). The combined organic layers were washed with water (100 mL), brine (50 mL) and dried over MgSO 4 , filtered and evaporated to dryness. iPrOH (30 mL) was added to the yellowish compound, then NaBH 4 (352 mg, 9.317 mmol) was added and the mixture heated at 50 0 C during 3 h.
- Example 27 This example illustrates the preparation of 5-Chloro-3- [4-(2,4-diamino-pyrimidin-5- yl-methyl)-6,7-dimethoxy-benzofuran-2-yImethyl] ⁇ 1/-/-indole-2-carboxylic acid dimethyl-amide I (step A10).
- This example illustrates the preparation of 5-Chloro-3- [4-(2,4-diamino-pyrimidin-5- yl-methyO- ⁇ J-dimethoxy-benzofuran ⁇ -ylmethyll-IH-indole ⁇ -carboxylic acid dimethyl-amide I (step A10).
- the mesylate salt 14 (6% isopropanol) (20.0 g, 29.8 mmol) was suspended in tetrahydrofuran (THF) (200 mL) at RT.
- This slurry was cooled with an ethanol bath equipped with a Cryocool (-25 ° C, 1 h), then Zn(BH 4 ) 2 (1 ,5 mol eq, 1.5 M solution in THF, 30 mL) was slowly added dropwise (some H 2 evolution) in 3 portions (3 x 10 mL every 15 minutes). After additional 15 minutes of stirring the slurry was warmed to 0 0 C. HCI solution was added continuously (1 eq, 4 M solution in dioxane, 7.45 mL, 124 ⁇ L/min) over a 1 h period. The solution was stirred for additional 15 minutes at 0 0 C and then allowed to warm up to 20 0 C during a period of 2 h.
- This example illustrates the preparation of 5-Chloro-3- ⁇ [4-(2,4-diamino-pyrimidin-5- ylmethyO-ej-dimethoxy-benzofuran ⁇ -yll-hydroxy-methylJ-IH-indole ⁇ -carboxylic acid dimethylamide 14A (step B10).
- the mesylate salt of the compound 14 (100 mg, 0.155 mmol) was dissolved in a mixture of iPrOH/MeOH (2/0.5 mL). The reaction mixture was cooled to -20 °C, before addition of sodium borohydride (17.6 mg, 0.466 mmol).
- This example illustrates the preparation of 5-Chloro ⁇ 3-[4-(2,4-diamino-pyrimidin-5- yl-methyl)-6,7-dimethoxy-benzofuran-2-ylmethyl]-1 H-indole-2-carboxylic acid dimethyl-amide I (step C10).
- the secondary alcohol 14A (50 mg, 90.9 ⁇ mol) was dissolved in THF (2 mL). The reaction mixture was cooled to -20 0 C, before addition of sodium borohydride (10.3 mg, 0.273 mmol). After 5 minutes stirring at -20 0 C, BF 3 -OEt 2 (34 ⁇ L, 50 %) was added slowly. After each drop of BF 3 OEt 2 , the color of the mixture was turning to violet, then the violet color disappeared again. After complete addition of BF 3 OEt 2 , the violet color was persistent during 3 minutes before returning to a pale yellow solution. The reaction was complete after 5 minutes and then NaOH 0.1 N (10 mL) was added. The mixture was extracted 2 times with EtOAc (15 mL each). The organic layers were washed with brine (50 mL), dried over MgSO 4 , filtered and evaporated to dryness to give the final compound I having the same LCMS signals given in Example 26.
- Example 20 This example illustrates the preparation of 5-Chloro-3-[4-(2,4-diamino-pyrimidin-5- yl-methyO- ⁇ J-dimethoxy-benzofuran ⁇ -ylmethyll-IH-indole ⁇ -carboxylic acid dimethyl-amide 1 (step A11).
- Example 21 illustrates the preparation of 5-Chloro-3-[4-(2,4-diamino-pyrimidin-5- yl-methyl)-6,7-dimethoxy-benzofuran-2-ylmethyl]-1H-indole-2-carboxylic acid dimethyl-amide I (step A12).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Indole Compounds (AREA)
Abstract
Description















Claims






Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ROA200700590A RO122853B1 (en) | 2005-02-18 | 2006-02-10 | Process for preparing a benzofurane derivative |
| US11/816,157 US20080161561A1 (en) | 2005-02-18 | 2006-02-10 | Novel Processing for the Preparation of a Benzofuran |
| MX2007009283A MX2007009283A (en) | 2005-02-18 | 2006-02-10 | Novell processes for the preparation of a benzofuran. |
| EEP200700050A EE200700050A (en) | 2005-02-18 | 2006-02-10 | Method for the preparation of benzofuran and intermediates |
| AU2006215785A AU2006215785A1 (en) | 2005-02-18 | 2006-02-10 | Novel processes for the preparation of a benzofuran |
| CA002596668A CA2596668A1 (en) | 2005-02-18 | 2006-02-10 | Novel processes for the preparation of a benzofuran |
| CNA2006800039630A CN101115746A (en) | 2005-02-18 | 2006-02-10 | A new method for the preparation of benzofurans |
| EP06706809A EP1856109A1 (en) | 2005-02-18 | 2006-02-10 | Novell processes for the preparation of a benzofuran |
| BRPI0607758-7A BRPI0607758A2 (en) | 2005-02-18 | 2006-02-10 | processes for compound manufacturing and compound reduction, and, |
| JP2007555504A JP2008530156A (en) | 2005-02-18 | 2006-02-10 | A new method for the production of benzofuran |
| HU0700605A HUP0700605A3 (en) | 2005-02-18 | 2006-02-10 | Novel processes for the preparation of a benzofuran |
| IL184405A IL184405A0 (en) | 2005-02-18 | 2007-07-04 | Novel processes for the preparation of a benzofuran |
| NO20073678A NO20073678L (en) | 2005-02-18 | 2007-07-17 | New processes for the preparation of benzofuran |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EPEP/2005/001695 | 2005-02-18 | ||
| EP2005001695 | 2005-02-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006087140A1 true WO2006087140A1 (en) | 2006-08-24 |
Family
ID=36283919
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/001185 Ceased WO2006087143A1 (en) | 2005-02-18 | 2006-02-10 | Novel processes for the preparation of a 2h-chromene |
| PCT/EP2006/001179 Ceased WO2006087140A1 (en) | 2005-02-18 | 2006-02-10 | Novell processes for the preparation of a benzofuran |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/001185 Ceased WO2006087143A1 (en) | 2005-02-18 | 2006-02-10 | Novel processes for the preparation of a 2h-chromene |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US20080221324A1 (en) |
| EP (3) | EP1856109A1 (en) |
| JP (2) | JP2008530156A (en) |
| KR (2) | KR20070106635A (en) |
| CN (4) | CN101115746A (en) |
| AU (2) | AU2006215788B2 (en) |
| BG (2) | BG109938A (en) |
| BR (2) | BRPI0607797A2 (en) |
| CA (2) | CA2596668A1 (en) |
| CZ (2) | CZ2007536A3 (en) |
| EE (2) | EE200700050A (en) |
| HU (2) | HUP0700604A3 (en) |
| IL (2) | IL184405A0 (en) |
| MX (2) | MX2007009283A (en) |
| NO (2) | NO20073678L (en) |
| NZ (1) | NZ556800A (en) |
| RO (2) | RO122912B8 (en) |
| RU (2) | RU2397980C2 (en) |
| TR (1) | TR200705187T1 (en) |
| TW (2) | TW200640914A (en) |
| WO (2) | WO2006087143A1 (en) |
| ZA (2) | ZA200706422B (en) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20070106635A (en) * | 2005-02-18 | 2007-11-02 | 아르피다 아게 | Novel methods for the preparation of 2H-chromen |
| CN102112108A (en) * | 2008-04-08 | 2011-06-29 | 阿西诺药品有限公司 | Aqueous pharmaceutical formulation |
| US7947293B2 (en) | 2008-04-08 | 2011-05-24 | Arpida Ag | Aqueous pharmaceutical formulation |
| FR2949465B1 (en) * | 2009-09-01 | 2011-08-12 | Pf Medicament | CHROMIUM DERIVATIVES, PROCESS FOR PREPARING THEM AND THERAPEUTIC APPLICATIONS THEREOF |
| CN110606831A (en) * | 2018-06-14 | 2019-12-24 | 上海度德医药科技有限公司 | Novel intermediate of Icalaprim and preparation method and application thereof |
| CN110818693B (en) * | 2018-08-07 | 2023-06-02 | 上海度德医药科技有限公司 | Crystal form B of ilaypu Lin Jia sulfonate and preparation method thereof |
| CN109988156B (en) * | 2019-03-12 | 2021-12-28 | 广东中科药物研究有限公司 | Aminopyrimidine compound |
| CN110372746A (en) * | 2019-07-11 | 2019-10-25 | 辽宁石油化工大学 | A method for synthesizing β-aminophosphine oxides |
| CN110642792B (en) * | 2019-11-18 | 2023-04-21 | 上海医药工业研究院有限公司 | Preparation method of ilaprine intermediate |
| CN110724135B (en) * | 2019-11-18 | 2023-04-28 | 上海医药工业研究院有限公司 | Esalapril Lin Zhongjian body and preparation method thereof |
| CN110724108B (en) * | 2019-11-18 | 2023-04-28 | 上海医药工业研究院有限公司 | Esalapril Lin Zhongjian body and preparation method thereof |
| CN110746361B (en) * | 2019-11-18 | 2023-04-21 | 上海医药工业研究院有限公司 | Esalapril Lin Zhongjian body and preparation method thereof |
| CN110713483B (en) * | 2019-11-18 | 2023-04-07 | 上海医药工业研究院 | Elaprepilin intermediate and preparation method of elaprilin |
| CN110790753B (en) * | 2019-11-18 | 2023-04-07 | 上海医药工业研究院 | Ealaprilin p-toluenesulfonate, and preparation method and application thereof |
| CN110818694B (en) * | 2019-11-18 | 2023-04-21 | 上海医药工业研究院有限公司 | Esalapu Lin Zhongjian body and application thereof |
| CN113493461A (en) * | 2020-04-01 | 2021-10-12 | 上海医药工业研究院 | Seven-membered heterocyclic compound or salt thereof, and preparation method and application thereof |
| CN117700410B (en) * | 2023-05-20 | 2025-07-04 | 山东康诺生物工程有限公司 | Preparation method of 3- (2-chloroethyl) -2-methyl-9-hydroxy-4H-pyrido [1,2-a ] pyrimidine-4-ketone |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002010157A1 (en) * | 2000-07-29 | 2002-02-07 | Arpida Ag | Benzofuran derivatives and their use as antibacterial agents |
| WO2005005418A1 (en) * | 2003-07-11 | 2005-01-20 | Arpida Ag | Benzofuran derivatives and their use in the treatment of microbial infections |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2709634A1 (en) * | 1977-03-05 | 1978-09-07 | Basf Ag | BENZYLPYRIMIDINE, METHOD FOR THE PRODUCTION THEREOF, AND MEDICINAL PRODUCTS CONTAINING THE SAME |
| US4438267A (en) * | 1980-11-11 | 1984-03-20 | Daluge Susan M | Monoheteroring compounds and their use |
| DE69618986T2 (en) * | 1995-12-04 | 2002-06-20 | Arpida Ag, Muenchenstein | DIAMINOPYRIMIDINE, PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME AND THEIR USE AS AN ANTIBACTERIAL AGENT |
| US7365205B2 (en) * | 2001-06-20 | 2008-04-29 | Daiichi Sankyo Company, Limited | Diamine derivatives |
| WO2005014586A1 (en) * | 2003-08-08 | 2005-02-17 | Arpida Ag | Novel process for the preparation of 2h-chromenes |
| KR20070106635A (en) * | 2005-02-18 | 2007-11-02 | 아르피다 아게 | Novel methods for the preparation of 2H-chromen |
-
2006
- 2006-02-10 KR KR1020077021394A patent/KR20070106635A/en not_active Ceased
- 2006-02-10 RO ROA200700589A patent/RO122912B8/en unknown
- 2006-02-10 RO ROA200700590A patent/RO122853B1/en unknown
- 2006-02-10 KR KR1020077021395A patent/KR20070106636A/en not_active Withdrawn
- 2006-02-10 JP JP2007555504A patent/JP2008530156A/en active Pending
- 2006-02-10 JP JP2007555507A patent/JP2009505943A/en active Pending
- 2006-02-10 WO PCT/EP2006/001185 patent/WO2006087143A1/en not_active Ceased
- 2006-02-10 CZ CZ20070536A patent/CZ2007536A3/en unknown
- 2006-02-10 US US11/816,150 patent/US20080221324A1/en not_active Abandoned
- 2006-02-10 CN CNA2006800039630A patent/CN101115746A/en active Pending
- 2006-02-10 AU AU2006215788A patent/AU2006215788B2/en not_active Ceased
- 2006-02-10 CA CA002596668A patent/CA2596668A1/en not_active Abandoned
- 2006-02-10 TR TR2007/05187T patent/TR200705187T1/en unknown
- 2006-02-10 RU RU2007134584/04A patent/RU2397980C2/en not_active IP Right Cessation
- 2006-02-10 EP EP06706809A patent/EP1856109A1/en not_active Withdrawn
- 2006-02-10 RU RU2007134583/04A patent/RU2007134583A/en not_active Application Discontinuation
- 2006-02-10 BR BRPI0607797-8A patent/BRPI0607797A2/en not_active IP Right Cessation
- 2006-02-10 CN CN2011100324952A patent/CN102079727A/en active Pending
- 2006-02-10 AU AU2006215785A patent/AU2006215785A1/en not_active Abandoned
- 2006-02-10 CA CA002596669A patent/CA2596669A1/en not_active Abandoned
- 2006-02-10 EE EEP200700050A patent/EE200700050A/en unknown
- 2006-02-10 CZ CZ20070537A patent/CZ2007537A3/en unknown
- 2006-02-10 HU HU0700604A patent/HUP0700604A3/en unknown
- 2006-02-10 EP EP10184131A patent/EP2270003A1/en not_active Withdrawn
- 2006-02-10 CN CN2006800039626A patent/CN101115743B/en not_active Expired - Fee Related
- 2006-02-10 EP EP06706815A patent/EP1856106A1/en not_active Withdrawn
- 2006-02-10 US US11/816,157 patent/US20080161561A1/en not_active Abandoned
- 2006-02-10 WO PCT/EP2006/001179 patent/WO2006087140A1/en not_active Ceased
- 2006-02-10 BR BRPI0607758-7A patent/BRPI0607758A2/en not_active IP Right Cessation
- 2006-02-10 HU HU0700605A patent/HUP0700605A3/en unknown
- 2006-02-10 NZ NZ556800A patent/NZ556800A/en not_active IP Right Cessation
- 2006-02-10 MX MX2007009283A patent/MX2007009283A/en unknown
- 2006-02-10 CN CN2011100324933A patent/CN102140094B/en not_active Expired - Fee Related
- 2006-02-10 EE EEP200700051A patent/EE200700051A/en unknown
- 2006-02-10 MX MX2007009282A patent/MX2007009282A/en not_active Application Discontinuation
- 2006-02-17 TW TW095105425A patent/TW200640914A/en unknown
- 2006-02-17 TW TW095105426A patent/TW200640912A/en unknown
-
2007
- 2007-07-04 IL IL184405A patent/IL184405A0/en unknown
- 2007-07-04 IL IL184404A patent/IL184404A0/en unknown
- 2007-07-17 NO NO20073678A patent/NO20073678L/en not_active Application Discontinuation
- 2007-07-18 NO NO20073701A patent/NO20073701L/en not_active Application Discontinuation
- 2007-08-01 ZA ZA200706422A patent/ZA200706422B/en unknown
- 2007-08-01 ZA ZA200706421A patent/ZA200706421B/en unknown
- 2007-08-10 BG BG109938A patent/BG109938A/en unknown
- 2007-08-10 BG BG109937A patent/BG109937A/en unknown
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002010157A1 (en) * | 2000-07-29 | 2002-02-07 | Arpida Ag | Benzofuran derivatives and their use as antibacterial agents |
| WO2005005418A1 (en) * | 2003-07-11 | 2005-01-20 | Arpida Ag | Benzofuran derivatives and their use in the treatment of microbial infections |
Non-Patent Citations (2)
| Title |
|---|
| ROTH B ET AL: "2,4-DIAMINO-5-BENZYLPYRIMIDINES AS ANTIBACTERIAL AGENTS. 13 SOME ALKENYL DERIVATIVES WITH HIGH IN VITRO ACTIVITY AGAINST ANAEROBIC ORGANISMS", JOURNAL OF MEDICINAL CHEMISTRY, AMERICAN CHEMICAL SOCIETY. WASHINGTON, US, vol. 32, no. 8, 1 August 1989 (1989-08-01), pages 1949 - 1958, XP000567729, ISSN: 0022-2623 * |
| TANI M ET AL: "Synthetic studies on indoles and related compounds. XXV. The Friedel-Crafts acylation of ethyl 1H-indole-2-carboxylate. (2)", CHEMICAL AND PHARMACEUTICAL BULLETIN, PHARMACEUTICAL SOCIETY OF JAPAN, TOKYO, JP, vol. 38, no. 12, December 1990 (1990-12-01), pages 3261 - 3267, XP002079475, ISSN: 0009-2363 * |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1856109A1 (en) | Novell processes for the preparation of a benzofuran | |
| AU2003209130B2 (en) | Synthesis of indole thiazole compounds as ligands for the AH receptor | |
| HUT68895A (en) | Process for producing physostigmine carbamate derivatives | |
| Caballero et al. | Stereochemical issues related to the synthesis and reactivity of pyrazino [2′, 1′-5, 1] pyrrolo [2, 3-b] indole-1, 4-diones | |
| ES2204303B2 (en) | PROCEDURE FOR OBTAINING A PHARMACEUTICALLY ACTIVE COMPOUND. | |
| KR100753353B1 (en) | Process for preparing zolmitriptan compounds | |
| KR19980702346A (en) | Amino Tetraron Derivatives and Preparation Methods Thereof | |
| KR100755625B1 (en) | Acyl derivatives of 5-2-4-1,2 benzisothiazole-3-yl-1-piperazinylethyl-6-chloro-1,3-dihydro-2h-indol-2-one having neuroleptic activity | |
| HK1111996A (en) | Novell processes for the preparation of a benzofuran | |
| Bakavoli et al. | Convenient synthesis of some optically active 1, 4-benzodiazepin-2, 5-diones | |
| AU2021371592A1 (en) | Method of producing 3-methyl-4-halo-indole derivative | |
| EP2379549A1 (en) | 7-azaindirubins, 7'azaindirubins, 7,7'-diazaindirubins and the corresponding 3'-oxime ether derivatives thereof, their production and use as a medicament | |
| Tatsugi et al. | Halogenation of 1-alkyl-7-azaisatins using N-halosuccinimides: regioselective synthesis of 1-alkyl-5-halo-7-azaisatins | |
| HK1111997A (en) | Novel processes for the preparation of a 2h-chromene |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006706809 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 184405 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 556799 Country of ref document: NZ Ref document number: 2006215785 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/009283 Country of ref document: MX Ref document number: 2596668 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680003963.0 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV2007-537 Country of ref document: CZ Ref document number: 10993806 Country of ref document: BG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007555504 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007200700590 Country of ref document: RO |
|
| ENP | Entry into the national phase |
Ref document number: 2006215785 Country of ref document: AU Date of ref document: 20060210 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006215785 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3618/CHENP/2007 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P0700605 Country of ref document: HU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007134583 Country of ref document: RU Ref document number: 1020077021395 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006706809 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11816157 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: PV2007-537 Country of ref document: CZ |
|
| ENP | Entry into the national phase |
Ref document number: PI0607758 Country of ref document: BR Kind code of ref document: A2 |