WO2006099459A1 - Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine - Google Patents
Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine Download PDFInfo
- Publication number
- WO2006099459A1 WO2006099459A1 PCT/US2006/009247 US2006009247W WO2006099459A1 WO 2006099459 A1 WO2006099459 A1 WO 2006099459A1 US 2006009247 W US2006009247 W US 2006009247W WO 2006099459 A1 WO2006099459 A1 WO 2006099459A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- epa
- salt
- enriched
- dnt
- dnth
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/16—Radicals substituted by singly bound hetero atoms other than halogen by oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/38—Heterocyclic compounds having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/14—Radicals substituted by singly bound hetero atoms other than halogen
- C07D333/20—Radicals substituted by singly bound hetero atoms other than halogen by nitrogen atoms
Definitions
- the present invention provides processes for synthesis of duloxetine intermediate.
- the present invention also provides processes for converting these duloxetine intermediate into pharmaceutically acceptable salts of duloxetine.
- Duloxetine is a dual reuptake inhibitor of the neurotransmitters serotonin and norepinephrine. It is used for the treatment of stress urinary incontinence (SUI), depression, and pain management.
- Duloxetine hydrochloride CAS Registry No. 136434-34-9, has the chemical structure Formula I.
- An intermediate in the synthesis of duloxetine is (S)-(+)-N,N-dimethyl-3-(l- naphthalenyloxy)-3-(2-thienyl)propanamine.
- the intermediate is also known as (S)-(+)-DNT, has been assigned the CAS Registry No. 132335-46-7, and has the chemical structure Formula II.
- the '269 patent describes the preparation of duloxetine by reacting (S)-(-)-N,N-Dimethyl-3-(2- thienyl)-3-hydroxypropanamine with fluoronaphtalene (Stage a) to produce N,N-Dimethyl-3- (l-naphthalenyloxy)-3-(2-thienyl)propanamine (DNT), followed by demethylation with phenyl chloroformate or trichloroethyl chloroformate (Stage b) and basic hydrolysis (Stage c) in accordance with the following Scheme 1.
- EP 273,658 which corresponds to the '388 and '269 patents, and is incorporated herein by reference in its entirety, discloses 3-aryloxy-3-substituted propanamines capable of inhibiting the uptake of serotonin and norepinephrine.
- the present invention is directed to a method of synthesizing (S)-(+)-DNT of
- the method of the invention comprises: (a) treating a solution of (R 5 S)-DNT with an enantiomerically pure acid (H-EPA); (b) crystallizing a diastereomerically enriched salt of (S)-(+)-DNTH + EPA " ; and, (c) separating the enriched salt.
- H-EPA enantiomerically pure acid
- the present invention also provides a method of synthesizing (R)-(-)-N,N- dimethyl-3-(l-na ⁇ hthalenyloxy)-3-(2-thienyl)propanamine ((R)-(-)-DNT) of formula III.
- the method of the invention comprises: (a) treating a solution of (R 5 S)-DNT with an H-EPA; (b) crystallizing a diastereomerically enriched salt of (R)-(-)-DNTH + EPA " ; and, (c) separating the enriched crystalline diastereomeric salt.
- the invention provides a diasteromerically enriched salt Of(S)-DNTH + EPA " and a diasteromerically enriched salt Of(R)-DNTH + EPA " .
- the invention provides a process for the racemization of DNT.
- the process comprises providing a mixture of DNT, a polar aprotic solvent, and an alkaline metal base, heating the mixture to a temperature of from about room temperature to the reflux temperature of the solvent, and recovering substantially racemic (R 5 S)-DNT.
- the present invention further provides pharmaceutically acceptable salts of duloxetine, prepared by obtaining (S)-(+)-DNT as described above, and converting the (S)-(+)-DNT to pharmaceutically acceptable salts of duloxetine.
- the (S)-(+)-DNT is converted to duloxetine hydrochloride.
- percent refers to percent by weight.
- the present invention provides a method of synthesizing (S)-(+)-DNT of formula II.
- the method comprises: (a) treating a solution of (R 5 S)-DNT with an H-EPA;
- the present invention provides a method of synthesizing (R)-(-)-DNT of formula III.
- the method comprises: (a) treating a solution of racemic DNT with an H-EPA;
- H-EPA refers to an enantiomerically pure acid.
- the H-EPA is greater than about 75 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 75 percent of one enantiomer of the enantiomerically pure acid.
- the H-EPA is greater than about 85 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 85 percent of one enantiomer of the enantiomerically pure acid.
- the H-EPA is greater than about 95 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 95 percent of one enantiomer of the enantiomerically pure acid.
- EPA ' refers to the anion of the corresponding H-EPA.
- a salt of (S)-(+)-DNT and H-EPA is referred to as (S)-(+)-DNTH + EPA " .
- a salt of (R)-(-)-DNT and H-EPA is referred to as (R)-(-)-DNTH + EPA " .
- Preferred enantiomerically pure acids may be selected from the group consisting of di-R-L-sub-tartaric acid, (R)-(-)-mandelic acid and (-)-2,3:4,6-Di-O- isopropylidene-2-keto-L-gulonic acid, and the opposite enantiomerically pure acids.
- Preferred subgroups include, but are not limited to, toluoyl, benzoyl, and pyvaloyl. Most preferably, the subgroup is toluoyl.
- a diasteromerically enriched salt of (S)-DNTH + EPA " is preferably a salt which is greater than about 60 percent enriched (S)-DNTH + EPA " . More preferably, the diasteromerically enriched salt is greater than about 75 percent enriched (S)-DNTH + EPA " . Even more preferably, the diasteromerically enriched salt is greater than about 90 percent enriched (S)-DNTH + EPA " . Most preferably, the diasteromerically enriched salt is greater than about 95 percent enriched (S)-DNTH + EPA " .
- a diasteromerically enriched salt of (R)-DNTH + EPA " is preferably a salt which is greater than about 60 percent enriched (R)-DNTH + EPA " . More preferably, the diasteromerically enriched salt is greater than about 75 percent enriched (R)-DNTH + EPA " . Even more preferably, the diasteromerically enriched salt is greater than about 90 percent enriched (R)-DNTH + EPA " . Most preferably, the diasteromerically enriched salt is greater than about 98 percent enriched (R)-DNTH + EPA " .
- the invention provides a diasteromerically enriched salt Of(S)-DNTH + EPA " and a diasteromerically enriched salt Of(R)-DNTH + EPA " .
- the step of treating a solution of (R 5 S)-DNT with an H-EPA may be performed in an organic solvent or water.
- Preferred organic solvents may be selected from the group consisting of toluene, ethyl acetate, and dichloromethane.
- the step of treating a solution of (R 5 S)-DNT with an H-EPA may also comprise a step of treating a solution of (R 5 S)-DNT with H-EPA at a temperature of from about room temperature to about the reflux temperature of the solvent, preferably at a temperature of from about 50° to about 95 °C. Preferably, the heating is for about 5 minutes to about 48 hours.
- the step of separating the enriched crystalline diastereomeric salt comprises a step of filtration.
- a preferred embodiment embraces a process whereby (R 5 S)-DNT is reacted with an H-EPA to form a diasteromerically enriched salt. Separation of the salt followed by basic hydrolysis results in enantiomerically enriched (S)-DNT and enantiomerically enriched (R)-DNT accordingly. This process is illustrated in Scheme 3.
- the invention provides a process for the racemization of DNT.
- the process comprises providing a mixture of DNT, a polar aprotic solvent, and an alkaline metal base, heating the mixture to a temperature of from about room temperature to about the reflux temperature of the solvent, and recovering substantially racemic (R 5 S)-DNT.
- the DNT used in the process of the invention can be either enantiomerically pure or enantiomerically rich.
- the polar aprotic solvent is selected from the group consisting of dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), dimethylacetamide (DMA), l-methyl-2-pyrolidinone (NMP) and hexamethylphosphoramide (HMPA). More preferably, the polar aprotic solvent is DMSO.
- the alkaline metal base is selected from the group consisting of lithium hydride, lithium N,N-diisopropylamide, sodium hydride, potassium hydride, sodium hydroxide, potassium hydroxide, sodium amide, potassium amide, sodium tert-butoxide, sodium methoxide, sodium ethoxide, potassium tert-butoxide, potassium methoxide, potassium ethoxide. More preferably, the alkaline metal base is potassium hydroxide or potassium tert-butoxide.
- the mixture is heated to a temperature of from about 50° to about
- the mixture is maintained for about fifteen minutes to about 48 hours, and, more preferably, the mixture is maintained for about 18 hours.
- the racemic (R 5 S)-DNT may be recovered by any methods known in the art.
- the present invention further provides pharmaceutically acceptable salts of duloxetine prepared by obtaining (S)-(+)-DNT as described above, and converting the (S)-(+)-DNT to pharmaceutically acceptable salts of duloxetine.
- the (S)-(+)-DNT is converted to duloxetine hydrochloride.
- Example 1 Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00036] A 1.24 g portion of di-p-toluoyl-L-tartaric acid was added to a solution of 2 g
- Example 5 Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00040] A 6.2 g portion of Di-p-toluoyl-L-tartaric acid was added to a solution of 10 g
- Example 6 Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00041] A 0.72 g portion of di-p-toluoyl-L-tartaric acid was added to a solution of 1.16 g (R 5 S)-DNT (ee: 77%) in 11.6 ml of toluene. The resulting mixture was heated to 75°C for 20 minutes, and then cooled to room temperature. The resulting solid was filtered, and dried in a vacuum oven to give 1.1 g of (S)-DNT di-p-toluoyl-L-tartarate (ee: 98%).
- Example 8 Racemization of (S)-DNT with potassium tert-butoxide [00043] A 2.7 g sample of KtBuO was added to a solution of 3.74 g of enantiomerically pure DNT (ee: 99.80%) dissolved in 37 ml of DMSO 5 and the resulting mixture was heated to 60°C. After eighteen hours, the mixture was cooled to room temperature. Water was added to the reaction mixture, followed by the addition of ethyl acetate and AcOH in an amount sufficient to provide a pH of from 8 to 9. After phase separation, the water phase was extracted with ethyl acetate, and the organic extracts were combined and concentrated to dryness to give brownish oil with an ee less than 1%.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pain & Pain Management (AREA)
- Engineering & Computer Science (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Psychiatry (AREA)
- Neurosurgery (AREA)
- Urology & Nephrology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
- Solid-Sorbent Or Filter-Aiding Compositions (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Steroid Compounds (AREA)
Abstract
Diasteromerically enriched salts Of (S)-DNTH+ EPA- and (R)-DNTH+ EPA-, methods of preparing such diasteromerically enriched salts Of (S)-DNTH+ EPA- and (R)-DNTH+ EPA-, and methods of preparing enantiomerically enriched (S)-DNT and enantiomerically enriched (R)-DNT are provided.
Description
PROCESS FOR THE PREPARATION OF OPTICALLY ACTIVE (S)-(+)-iV;N-DIMETHYL-3-(l-NAPHTHALENYLOXY)-3-(2-THIENYL)PROPANAMINE
RELATED APPLICATIONS
[0001] This application claims benefit of U.S. Provisional Application Nos.
60/726,502, filed October 12, 2005, 60/736,746, filed November 14, 2005, 60/661,711, filed March 14, 2005, and 60/773,593, filed February 14, 2006.
Field of the Invention
[0002] The present invention provides processes for synthesis of duloxetine intermediate. The present invention also provides processes for converting these duloxetine intermediate into pharmaceutically acceptable salts of duloxetine.
Background
[0003] Duloxetine is a dual reuptake inhibitor of the neurotransmitters serotonin and norepinephrine. It is used for the treatment of stress urinary incontinence (SUI), depression, and pain management. Duloxetine hydrochloride, CAS Registry No. 136434-34-9, has the chemical structure Formula I.

Formula I
[0004] An intermediate in the synthesis of duloxetine is (S)-(+)-N,N-dimethyl-3-(l- naphthalenyloxy)-3-(2-thienyl)propanamine. The intermediate is also known as (S)-(+)-DNT, has been assigned the CAS Registry No. 132335-46-7, and has the chemical structure Formula II.

Formula II
[0005] U.S. Patents Nos. 4,956,388 ("the '388 patent") and 5,023,269 ("the '269 patent"), incorporated herein by reference in their entirety, disclose 3-aryloxy-3-substituted propanamines capable of inhibiting the uptake of serotonin and norepinephrine. The '269 patent describes the preparation of duloxetine by reacting (S)-(-)-N,N-Dimethyl-3-(2- thienyl)-3-hydroxypropanamine with fluoronaphtalene (Stage a) to produce N,N-Dimethyl-3- (l-naphthalenyloxy)-3-(2-thienyl)propanamine (DNT), followed by demethylation with phenyl chloroformate or trichloroethyl chloroformate (Stage b) and basic hydrolysis (Stage c) in accordance with the following Scheme 1.
Scheme 1 : Synthesis of Duloxetine Hydrochloride

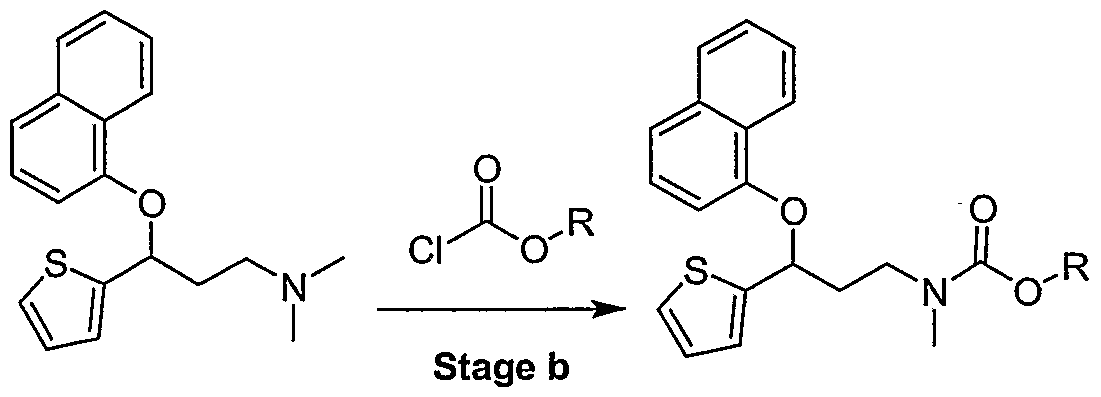
DNT-base Duloxetine alky I carbamate
OH
HCI / solvent


Stage d
DLX-HCI DLX-base
R= Phenyl, trichloroethyl
[0006] U.S. Patent No. 5,491,243, incorporated herein by reference in its entirety, discloses a stereospecific process for the synthesis of (S)-(+)-DNT.
[0007] U.S. Patent No. 6,541,668, incorporated herein by reference in its entirety, discloses the preparation of 3-aryloxy-3-arylpropylamines and intermediates thereof using a nucleophilic aromatic displacement in l,3-dimethyl-2-imidazolidinone or N-methylpyrrolidinone.
[0008] EP 273,658, which corresponds to the '388 and '269 patents, and is incorporated herein by reference in its entirety, discloses 3-aryloxy-3-substituted propanamines capable of inhibiting the uptake of serotonin and norepinephrine.
[0009] Wheeler WJ., et al, J. Label. Cpds. Radiopharm, 1995, 36, 312, incorporated herein by reference in its entirety, discloses the conversion of duloxetine to its hydrochloride salt.
[00010] The prior art discloses that enantiomerically pure duloxetine may be prepared by different routes than those shown in Scheme 1. These prior art routes for preparing enantiomerically pure duloxetine may be summarized as follows: a. Chiral resolution of 3-(Dimethylarnino)-l-(2-thienyl)-l -propanol (alcohol a in Scheme 2), Path A, followed by Path C or D; b. Asymmetric reduction of 3-(Dimethylamino)-l-(2-thienyl)-l-propanone (amino ketone b in Scheme 2), Path B, followed by Path C or D; c. Chiral resolution of 3 -memylamino-l-(2-thienyl)-l -propanol (alcohol c in Scheme 2); and, d. Chiral resolution of the racemic duloxetine.
Scheme 2: Prior art synthetic options for the preparation of enantiomerically pure Duloxetine
Path A Path B / Asymmetric reduction

OH
U T
Vi 1F . . 1. Chloroformate halonaphtalene/ path C Path D \ 2. Basic Hydrolysis
DNT rac
- TjT
1. Chloroformate
2. Basic Hydrolysis


DLX-racemic DLX-base
[00011] The drawback of the processes described in Scheme 2 is the lack of processes for synthesizing an enantiomerically pure DNT and a racemization process for the reprocessing of the undesirable enantiomer.
Summary of the Invention
[00012] The present invention is directed to a method of synthesizing (S)-(+)-DNT of
Formula II.

Formula II
The method of the invention comprises: (a) treating a solution of (R5S)-DNT with an enantiomerically pure acid (H-EPA); (b) crystallizing a diastereomerically enriched salt of (S)-(+)-DNTH+ EPA"; and, (c) separating the enriched salt.
[00013] The present invention also provides a method of synthesizing (R)-(-)-N,N- dimethyl-3-(l-naρhthalenyloxy)-3-(2-thienyl)propanamine ((R)-(-)-DNT) of formula III.

Formula III
The method of the invention comprises: (a) treating a solution of (R5S)-DNT with an H-EPA; (b) crystallizing a diastereomerically enriched salt of (R)-(-)-DNTH+ EPA"; and, (c) separating the enriched crystalline diastereomeric salt.
[00014] In alternative embodiments the invention provides a diasteromerically enriched salt Of(S)-DNTH+ EPA" and a diasteromerically enriched salt Of(R)-DNTH+ EPA".
[00015] In a further embodiment, the invention provides a process for the racemization of DNT. The process comprises providing a mixture of DNT, a polar aprotic solvent, and an alkaline metal base, heating the mixture to a temperature of from about room temperature to the reflux temperature of the solvent, and recovering substantially racemic (R5S)-DNT.
[00016] The present invention further provides pharmaceutically acceptable salts of duloxetine, prepared by obtaining (S)-(+)-DNT as described above, and converting the (S)-(+)-DNT to pharmaceutically acceptable salts of duloxetine. Preferably the (S)-(+)-DNT is converted to duloxetine hydrochloride.
Detailed Description of the Invention
[00017] Unless stated otherwise, as used herein, the term "percent" refers to percent by weight.
[00018] The present invention provides a method of synthesizing (S)-(+)-DNT of formula II.

Formula II
The method comprises: (a) treating a solution of (R5S)-DNT with an H-EPA;
(b) crystallizing a diastereomerically enriched salt of (S)-(+)-DNTH+ EPA"; and,
(c) separating the enriched salt.
[00019] The present invention provides a method of synthesizing (R)-(-)-DNT of formula III.

III
The method comprises: (a) treating a solution of racemic DNT with an H-EPA;
(b) crystallizing a diastereomerically enriched salt of (R)-(^-DNTH+ EPA'; and,
(c) separating the enriched crystalline diastereomeric salt.
[00020] As used herein, "H-EPA" refers to an enantiomerically pure acid. Preferably, the H-EPA is greater than about 75 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 75 percent of one enantiomer of the enantiomerically pure acid. More preferably, the H-EPA is greater than about 85 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 85 percent of one enantiomer of the enantiomerically pure acid. Most preferably, the H-EPA is greater than about 95 percent pure enantiomeric acid; i.e., the H-EPA comprises greater than about 95 percent of one enantiomer of the enantiomerically pure acid.
[00021] As used herein, "EPA'" refers to the anion of the corresponding H-EPA. A salt of (S)-(+)-DNT and H-EPA is referred to as (S)-(+)-DNTH+EPA". A salt of (R)-(-)-DNT and H-EPA is referred to as (R)-(-)-DNTH+ EPA".
[00022] Preferred enantiomerically pure acids may be selected from the group consisting of di-R-L-sub-tartaric acid, (R)-(-)-mandelic acid and (-)-2,3:4,6-Di-O- isopropylidene-2-keto-L-gulonic acid, and the opposite enantiomerically pure acids. Preferred subgroups include, but are not limited to, toluoyl, benzoyl, and pyvaloyl. Most preferably, the subgroup is toluoyl.
[00023] As used herein, a diasteromerically enriched salt of (S)-DNTH+ EPA" is preferably a salt which is greater than about 60 percent enriched (S)-DNTH+ EPA". More preferably, the diasteromerically enriched salt is greater than about 75 percent enriched (S)-DNTH+ EPA". Even more preferably, the diasteromerically enriched salt is greater than
about 90 percent enriched (S)-DNTH+ EPA". Most preferably, the diasteromerically enriched salt is greater than about 95 percent enriched (S)-DNTH+ EPA".
[00024] As used herein, a diasteromerically enriched salt of (R)-DNTH+ EPA" is preferably a salt which is greater than about 60 percent enriched (R)-DNTH+ EPA". More preferably, the diasteromerically enriched salt is greater than about 75 percent enriched (R)-DNTH+ EPA". Even more preferably, the diasteromerically enriched salt is greater than about 90 percent enriched (R)-DNTH+ EPA". Most preferably, the diasteromerically enriched salt is greater than about 98 percent enriched (R)-DNTH+ EPA".
[00025] In further embodiments the invention provides a diasteromerically enriched salt Of(S)-DNTH+ EPA" and a diasteromerically enriched salt Of(R)-DNTH+ EPA".
[00026] The step of treating a solution of (R5S)-DNT with an H-EPA may be performed in an organic solvent or water. Preferred organic solvents may be selected from the group consisting of toluene, ethyl acetate, and dichloromethane.
[00027] The step of treating a solution of (R5S)-DNT with an H-EPA may also comprise a step of treating a solution of (R5S)-DNT with H-EPA at a temperature of from about room temperature to about the reflux temperature of the solvent, preferably at a temperature of from about 50° to about 95 °C. Preferably, the heating is for about 5 minutes to about 48 hours.
[00028] Preferably, the step of separating the enriched crystalline diastereomeric salt comprises a step of filtration.
[00029] A preferred embodiment embraces a process whereby (R5S)-DNT is reacted with an H-EPA to form a diasteromerically enriched salt. Separation of the salt followed by basic hydrolysis results in enantiomerically enriched (S)-DNT and enantiomerically enriched (R)-DNT accordingly. This process is illustrated in Scheme 3.
Scheme 3
H-EPA: Enantiomerically pure acid

(R)-DNT (R)-DNT
[00030] In a further embodiment, the invention provides a process for the racemization of DNT. The process comprises providing a mixture of DNT, a polar aprotic solvent, and an alkaline metal base, heating the mixture to a temperature of from about room temperature to about the reflux temperature of the solvent, and recovering substantially racemic (R5S)-DNT. The DNT used in the process of the invention can be either enantiomerically pure or enantiomerically rich.
[00031] Preferably, the polar aprotic solvent is selected from the group consisting of dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), dimethylacetamide (DMA), l-methyl-2-pyrolidinone (NMP) and hexamethylphosphoramide (HMPA). More preferably, the polar aprotic solvent is DMSO.
[00032] Preferably, the alkaline metal base is selected from the group consisting of lithium hydride, lithium N,N-diisopropylamide, sodium hydride, potassium hydride, sodium hydroxide, potassium hydroxide, sodium amide, potassium amide, sodium tert-butoxide, sodium methoxide, sodium ethoxide, potassium tert-butoxide, potassium methoxide, potassium ethoxide. More preferably, the alkaline metal base is potassium hydroxide or potassium tert-butoxide.
[00033] Preferably, the mixture is heated to a temperature of from about 50° to about
140°C, and, more preferably, to about 80°. Preferably, after heating, the mixture is maintained for about fifteen minutes to about 48 hours, and, more preferably, the mixture is
maintained for about 18 hours. The racemic (R5S)-DNT may be recovered by any methods known in the art.
[00034] The present invention further provides pharmaceutically acceptable salts of duloxetine prepared by obtaining (S)-(+)-DNT as described above, and converting the (S)-(+)-DNT to pharmaceutically acceptable salts of duloxetine. Preferably, the (S)-(+)-DNT is converted to duloxetine hydrochloride.
[00035] The function and advantage of these and other embodiments of the present invention will be more fully understood from the examples below. These examples are intended to illustrate the benefits of the present invention, but are not intended to limit the scope of the invention.
Examples
Example 1: Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00036] A 1.24 g portion of di-p-toluoyl-L-tartaric acid was added to a solution of 2 g
(R3S)-DNT in 10 ml of toluene. The resulting mixture was heated to 75°C for 10 minutes, and then cooled to room temperature. The resulting solid was filtered, and dried in a vacuum oven to give 1.15 g of (S)-DNT di-p-toluoyl-L-tartarate.
Example 2: Preparation of (S)-DNT di-p-toluoyl-L-tartarate in EtO Ac/Ether [00037] A 1.24 g portion of di-p-toluoyl-L-tartaric acid was added to a solution of 2 g
(R5S)-DNT in 10 ml of ethyl acetate, and the resulting mixture was stirred at room temperature for an hour. The addition of 6 ml of ether resulted in a precipitate. The mixture was then heated to reflux, and an additional 20 ml of ethyl acetate were added. The mixture was cooled to room temperature, filtered, washed with 10 ml of ether, and dried in a vacuum oven to give 1.41 g of (S)-DNT di-p-toluoyl-L-tartarate.
Example 3: Preparation of (S)-DNT
[00038] A solution of 10 percent by weight NaOH was added to a mixture of 1 g of
(S)-DNT di-p-toluoyl-L-tartarate in 30 ml of water and 30 ml of dichloromethane to provide a pH of 14, and stirred for an hour. After phase separation, the organic phase was washed with water (30 ml), dried over Na2SO4, filtered, and concentrated to dryness to give 0.4 g of brownish oil (62.17 percent ee).
Example 4: Preparation of (S)-DNT-(R)-mandelate in water
[00039] A 0.49 g portion of (R)-mandelic acid was added to a mixture of 2 g of
(R,S)-DNT in 15 ml of water, and the mixture was heated to 95°C, followed by cooling to room temperature over a period of 2 hours. The solid was filtered out, and the mother liquor was allowed to stand overnight. The resulting solid was filtered out, and the resulting solution analyzed by HPLC giving 45 percent ee of (S)-DNT-(R)-mandelate.
Example 5: Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00040] A 6.2 g portion of Di-p-toluoyl-L-tartaric acid was added to a solution of 10 g
(R5S)-DNT in 100 ml of toluene. The resulting mixture was heated to 75°C for 30 minutes, and then cooled to room temperature. The resulting solid was filtered, and dried in a vacuum oven to give 5.13 g of (S)-DNT di-p-toluoyl-L-tartarate (ee: 77%).
Example 6: Preparation of (S)-DNT di-p-toluoyl-L-tartarate in toluene [00041] A 0.72 g portion of di-p-toluoyl-L-tartaric acid was added to a solution of 1.16 g (R5S)-DNT (ee: 77%) in 11.6 ml of toluene. The resulting mixture was heated to 75°C for 20 minutes, and then cooled to room temperature. The resulting solid was filtered, and dried in a vacuum oven to give 1.1 g of (S)-DNT di-p-toluoyl-L-tartarate (ee: 98%).
Example 7: Racemization of (S)-DNT with KOH
[00042] A 5.3 g sample of KOH was added to a solution of 55 g of enantiomerically pure DNT (ee: 99.80%) dissolved in 50 ml of DMSO5 and the resulting mixture was heated to 80°C. After six hours, the mixture was cooled to room temperature. Water was added to the reaction mixture, followed by the addition of ethyl acetate and AcOH in an amount sufficient to provide a pH of from 8 to 9. After phase separation, the water phase was extracted with ethyl acetate, and the organic extracts were combined and concentrated to dryness to give brownish oil with an ee less than 1%.
Example 8: Racemization of (S)-DNT with potassium tert-butoxide [00043] A 2.7 g sample of KtBuO was added to a solution of 3.74 g of enantiomerically pure DNT (ee: 99.80%) dissolved in 37 ml of DMSO5 and the resulting mixture was heated to 60°C. After eighteen hours, the mixture was cooled to room temperature. Water was added to the reaction mixture, followed by the addition of ethyl acetate and AcOH in an amount sufficient to provide a pH of from 8 to 9. After phase
separation, the water phase was extracted with ethyl acetate, and the organic extracts were combined and concentrated to dryness to give brownish oil with an ee less than 1%.
Example 9: Racemization of (R)-DNT with KOH
[00044] An 8.4 g sample of KOH was added to a solution of 7.5 g of enantiomerically rich DNT dissolved in 75 ml of DMSO, and the resulting mixture was heated to 80°C. After six hours, the mixture was cooled to room temperature. Water was added to the reaction mixture, followed by the addition of ethyl acetate and AcOH in an amount sufficient to provide a pH of from 8 to 9. After phase separation, the water phase was extracted with ethyl acetate, and the organic extracts were combined and concentrated to dryness to give brownish oil with an ee less than 1%.
[00045] While it is apparent that the invention disclosed herein is well calculated to fulfill the objects stated above, it will be appreciated that numerous modifications and embodiments may be devised by those skilled in the art. Therefore, it is intended that the appended claims cover all such modifications and embodiments as falling within the true spirit and scope of the present invention.
Claims
1. A method of synthesizing (S)-(+)-DNT, comprising: a) treating a solution of (R5S)-DNT with an H-EPA; b) crystallizing a diastereomerically enriched salt of (S)-(+)-DNTH+ EPA-; and c) separating the enriched salt.
2. A method of synthesizing (R)-(-)-DNT, comprising: a) treating a solution of (R5S)-DNT with an H-EPA; b) crystallizing a diastereomerically enriched salt of (R)-Q-DNTH+ EPA"; and c) separating the enriched salt.
3. The method of any of claims 1 or 2, wherein the H-EPA is greater than about 75 percent pure enantiomeric acid.
4. The method of claim 3, wherein the H-EPA is greater than about 85 percent pure enantiomeric acid.
5. The method of claim 4, wherein the H-EPA is greater than about 98 percent pure enantiomeric acid.
6. The method of any of claims 1 or 2, wherein the H-EPA is selected from the group consisting of di-R-L-sub-tartaric acid, (R)-(-)-mandelic acid, (-)-2,3:4,6-Di-O- isopropylidene-2-keto-L-gulonic acid, and the opposite enantiomerically pure acids.
7. The method of claim 6, wherein the subgroup is selected from the group consisting of toluoyl, benzoyl, and pyvaloyl.
8. The method of claim 7, wherein the subgroup is toluoyl.
9. The method of any of claims 1 or 2, wherein step a) is performed in an organic solvent or water.
10. The method of claim 9, wherein the organic solvents is selected from the group consisting of toluene, ethyl acetate, and dichloromethane.
11. The method of any of claims 1 or 2, wherein step a) is performed at a temperature of from about room temperature to about the reflux temperature of the solvent.
12. The method of claim 11, wherein step a) is performed at a temperature of from about 50c to about 95°C.
13. The method of claim 11, wherein the heating is for about 5 minutes to about 48 hours.
14. The method of claim 1, wherein step c) is followed by basic hydrolysis, obtaining enantiomerically enriched (S)-DNT.
15. The method of claim 2, wherein step c) is followed by basic hydrolysis, obtaining enantiomerically enriched (R)-DNT.
16. A diasteromerically enriched salt of (S)-DNTH+ EPA".
17. The diasteromerically enriched salt of claim 16, wherein the salt is enriched with greater than about 60 percent Of(S)-DNTH+ EPA".
18. The diasteromerically enriched salt of claim 17, wherein the salt is enriched with greater than about 75 percent Of(S)-DNTH+ EPA".
19. The diasteromerically enriched salt of claim 18, wherein the salt is enriched with greater than about 90 percent of (S)-DNTH+ EPA".
20. The diasteromerically enriched salt of claim 19, wherein the salt is enriched with greater than about 95 percent Of(S)-DNTH+ EPA".
21. A diasteromerically enriched salt of (R)-DNTH+ EPA".
22. The diasteromerically enriched salt of claim 21, wherein the salt is enriched with greater than about 60 percent of (R)-DNTH+ EPA".
23. The diasteromerically enriched salt of claim 22, wherein the salt is enriched with greater than about 75 percent of (R)-DNTH+ EPA".
24. The diasteromerically enriched salt of claim 23, wherein the salt is enriched with greater than about 90 percent Of(R)-DNTH+ EPA".
25. The diasteromerically enriched salt of claim 24, wherein the salt is enriched with greater than about 98 percent Of (R)-DNTH+ EPA".
26. A process for the racemization of DNT, comprising: a) providing a mixture of DNT, a polar aprotic solvent, and an alkaline metal base; b) heating the mixture to a temperature of from about room temperature to about the reflux temperature of the solvent; and c) recovering substantially racemic (R5S)-DNT.
27. The process of claim 26, wherein the DNT is enantiomerically pure or enantiomerically rich.
28. The process of either of claims 26 and 27, wherein the polar aprotic solvent is selected from the group consisting of dimethyl sulfoxide (DMSO), dimethyl formamide (DMF), dimethylacetamide (DMA), l-methyl-2-pyrolidinone (NMP) and hexamethylphosphoramide (HMPA).
29. The process of claim 28, wherein the polar aprotic solvent is DMSO.
30. The process of any of claims 26 to 29, wherein the alkaline metal base is selected from the group consisting of lithium hydride, lithium N,N-diisopropylamide, sodium hydride, potassium hydride, sodium hydroxide, potassium hydroxide, sodium amide, potassium amide, sodium tert-butoxide, sodium methoxide, sodium ethoxide, potassium tert- butoxide, potassium methoxide, potassium ethoxide. More preferably, the alkaline metal base is potassium hydroxide or potassium tert-butoxide.
31. The process of claim 30, wherein the alkaline metal base is potassium hydroxide or potassium tert-butoxide.
32. The process of any of claims 26 to 31, wherein the mixture is heated to a temperature of from about 50° to about 140°C.
33. The process of claim 32, wherein the mixture is heated to a temperature of about 80°.
34. The process of any of claims 26 to 33, wherein, after step b), the mixture is maintained for about fifteen minutes to about 48 hours.
35. The process of claim 34, wherein the mixture is maintained for about 18 hours.
36. A pharmaceutically acceptable salt of duloxetine prepared by obtaining the (S)-(+)-DNT in accordance with the method of any of claims 1 and 3 to 14, and converting the (S)-(+)- DNT to a pharmaceutically acceptable salt of duloxetine.
37. The process of claim 36, wherein the (S)-(+)-DNT is converted to duloxetine hydrochloride.
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2007011254A MX2007011254A (en) | 2005-03-14 | 2006-03-14 | Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propana mine. |
| EP06738321A EP1858859A1 (en) | 2005-03-14 | 2006-03-14 | Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine |
| IL184189A IL184189A0 (en) | 2005-03-14 | 2007-06-25 | Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66171105P | 2005-03-14 | 2005-03-14 | |
| US60/661,711 | 2005-03-14 | ||
| US72650205P | 2005-10-12 | 2005-10-12 | |
| US60/726,502 | 2005-10-12 | ||
| US73674605P | 2005-11-14 | 2005-11-14 | |
| US60/736,746 | 2005-11-14 | ||
| US77359306P | 2006-02-14 | 2006-02-14 | |
| US60/773,593 | 2006-02-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006099459A1 true WO2006099459A1 (en) | 2006-09-21 |
Family
ID=36593962
Family Applications (4)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/009275 Ceased WO2006099468A2 (en) | 2005-03-14 | 2006-03-14 | Process for the purification of duloxetine hydrochloride |
| PCT/US2006/009165 Ceased WO2006099433A1 (en) | 2005-03-14 | 2006-03-14 | Pure duloxetine hydrochloride |
| PCT/US2006/009247 Ceased WO2006099459A1 (en) | 2005-03-14 | 2006-03-14 | Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine |
| PCT/US2006/009241 Ceased WO2006099457A1 (en) | 2005-03-14 | 2006-03-14 | (s)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl) propanamine-di-p-toluoyl-l-tartarate and methods of preparation thereof |
Family Applications Before (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/009275 Ceased WO2006099468A2 (en) | 2005-03-14 | 2006-03-14 | Process for the purification of duloxetine hydrochloride |
| PCT/US2006/009165 Ceased WO2006099433A1 (en) | 2005-03-14 | 2006-03-14 | Pure duloxetine hydrochloride |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/009241 Ceased WO2006099457A1 (en) | 2005-03-14 | 2006-03-14 | (s)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl) propanamine-di-p-toluoyl-l-tartarate and methods of preparation thereof |
Country Status (7)
| Country | Link |
|---|---|
| US (4) | US20060258871A1 (en) |
| EP (4) | EP1858859A1 (en) |
| CA (2) | CA2599478A1 (en) |
| IL (3) | IL184184A0 (en) |
| MX (2) | MX2007011254A (en) |
| TW (2) | TW200639162A (en) |
| WO (4) | WO2006099468A2 (en) |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006071868A2 (en) * | 2004-12-23 | 2006-07-06 | Teva Pharmaceutical Industries Ltd. | Process for preparing pharmaceutically acceptable salts of duloxetine and intermediates thereof |
| EP1856087A1 (en) * | 2005-03-08 | 2007-11-21 | Teva Pharmaceutical Industries Limited | Crystal forms of (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl) propanamine oxalate and the preparation thereof |
| CA2599478A1 (en) * | 2005-03-14 | 2006-09-21 | Teva Pharmaceutical Industries Ltd. | Process for the purification of duloxetine hydrochloride |
| EP1838692A2 (en) * | 2005-09-22 | 2007-10-03 | Teva Pharmaceutical Industries Ltd | Dnt-maleate and methods of preparation thereof |
| US20080207923A1 (en) * | 2005-09-22 | 2008-08-28 | Santiago Ini | Pure DNT-maleate and methods of preparation thereof |
| US7759500B2 (en) | 2005-12-05 | 2010-07-20 | Teva Pharmaceutical Industries Ltd. | 2-(N-methyl-propanamine)-3-(2-naphthol)thiophene, an impurity of duloxetine hydrochloride |
| PL1971592T3 (en) | 2005-12-12 | 2011-09-30 | Medichem Sa | Improved synthesis and preparations of duloxetine salts |
| CZ299270B6 (en) * | 2006-01-04 | 2008-06-04 | Zentiva, A. S. | Process for preparing (S)-N-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride |
| WO2007077580A2 (en) | 2006-01-06 | 2007-07-12 | Msn Laboratories Limited | Improved process for pure duloxetine hydrochloride |
| MX2008001079A (en) * | 2006-05-23 | 2008-03-19 | Teva Pharma | Duloxetine hcl polymorphs. |
| EP1976846A2 (en) * | 2006-05-31 | 2008-10-08 | Teva Pharmaceutical Industries Ltd | Process for preparing duloxetine and intermediates thereof |
| GB0612508D0 (en) | 2006-06-23 | 2006-08-02 | Arrow Int Ltd | Crystalline duloxetine hydrochloride |
| GB0612509D0 (en) | 2006-06-23 | 2006-08-02 | Arrow Int Ltd | Crystalline duloxetine hydrochloride |
| CZ300116B6 (en) * | 2006-12-05 | 2009-02-11 | Zentiva, A. S. | Purification process of (S)-N-methyl-3-(1-naphtyloxy)-3-(2-thienyl) propylamine hydrochloride |
| EP2132192B1 (en) * | 2007-03-05 | 2013-04-24 | Lupin Limited | Novel process for preparation of duloxetine hydrochloride |
| WO2009109992A1 (en) * | 2008-01-23 | 2009-09-11 | Arch Pharmalabs Limited | Novel process for preparation of duloxetine and intermediates for use therein |
| EP2107057A1 (en) * | 2008-04-04 | 2009-10-07 | Ranbaxy Laboratories Limited | Process for the preparation of pure duloxetine hydrochloride |
| WO2009150238A2 (en) * | 2008-06-13 | 2009-12-17 | Krka, D.D. Novo Mesto | Gastro-resistant pharmaceutical oral compositions comprising duloxetine or its pharmaceutically acceptable derivatives |
| WO2010011811A2 (en) | 2008-07-24 | 2010-01-28 | Theravance, Inc. | 3-(phenoxyphenylmethyl)pyrrolidine compounds |
| CZ304602B6 (en) * | 2009-09-02 | 2014-07-30 | Zentiva, K. S. | Crystallization process of (S)-N-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride (duloxetine hydrochloride) |
| EP2575457B1 (en) * | 2010-05-25 | 2016-08-17 | Hetero Research Foundation | Oral pharmaceutical composition of duloxetine |
| CN103626735A (en) * | 2012-08-28 | 2014-03-12 | 石药集团中奇制药技术(石家庄)有限公司 | Duloxetine hydrochloride crystal form and preparation method thereof |
| CN104478849A (en) * | 2014-02-14 | 2015-04-01 | 广东东阳光药业有限公司 | Method for preparing noradrenaline reuptake dual inhibitor |
| JP2016172704A (en) * | 2015-03-17 | 2016-09-29 | 株式会社トクヤマ | Method for producing duloxetine hydrochloride and duloxetine hydrochloride having a novel crystal structure |
| JP2016222628A (en) * | 2015-06-03 | 2016-12-28 | 株式会社トクヤマ | Method for producing duloxetine hydrochloride |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5023269A (en) * | 1986-12-22 | 1991-06-11 | Eli Lilly And Company | 3-aryloxy-3-substituted propanamines |
| US5362886A (en) * | 1993-10-12 | 1994-11-08 | Eli Lilly And Company | Asymmetric synthesis |
| WO2003070720A1 (en) * | 2002-02-22 | 2003-08-28 | Degussa Ag | Preparation of n-methyl-3-hydroxy- 3-(2-thienyl)propylamine via novel thiophene derivatives containing carbamate groups as intermediates |
| WO2004056795A1 (en) * | 2002-12-19 | 2004-07-08 | Cipla Ltd | A process for preparing duloxetine and intermediates for use therein |
| US20040249170A1 (en) * | 2002-01-24 | 2004-12-09 | Alfio Borghese | Process for preparing an intermediate useful for the asymmetric synthesis of duloxetine |
| WO2006027798A2 (en) * | 2004-08-05 | 2006-03-16 | Sun Pharmaceutical Industries Limited | A process for preparation of an antidepressant compound |
| WO2006045255A1 (en) * | 2004-10-26 | 2006-05-04 | Zentiva, A.S. | Method of manufacturing (s)-n-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride (duloxetine) |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH368806A (en) * | 1957-02-28 | 1963-04-30 | Spofa Spojene Farmaceuticke Z | Process for the preparation of 6-azauracil riboside |
| US3105564A (en) * | 1960-10-13 | 1963-10-01 | Alfred N Ormond | Apparatus for measuring static loads |
| GB1022031A (en) * | 1962-05-11 | 1966-03-09 | Egyt Gyogyszervegyeszeti Gyar | Improvements in or relating to basic esters of 3,5-dimethoxy-4-alkoxy-benzoic acids |
| AT255400B (en) * | 1965-03-22 | 1967-07-10 | Chemie Linz Ag | Process for the production of new basic ethers |
| BE786141A (en) * | 1971-07-14 | 1973-01-11 | Pfizer | NEW ALPHA- (ALKYLBENZYL (THENYL)) - BENZYLOXY OF AMINES AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| US4018895A (en) * | 1974-01-10 | 1977-04-19 | Eli Lilly And Company | Aryloxyphenylpropylamines in treating depression |
| US4194009A (en) * | 1974-01-10 | 1980-03-18 | Eli Lilly And Company | Aryloxyphenylpropylamines for obtaining a psychotropic effect |
| US4314081A (en) | 1974-01-10 | 1982-02-02 | Eli Lilly And Company | Arloxyphenylpropylamines |
| US4325953A (en) * | 1979-09-14 | 1982-04-20 | John Wyeth And Brother Limited | 4-Aryl-4-aryloxypiperidines |
| KR880007433A (en) | 1986-12-22 | 1988-08-27 | 메리 앤 터커 | 3-aryloxy-3-substituted propanamine |
| IL98108A0 (en) | 1990-05-17 | 1992-06-21 | Lilly Co Eli | Chiral synthesis of 1-aryl-3-aminopropan-1-ols |
| US5371240A (en) * | 1992-11-30 | 1994-12-06 | Torcan Chemical Ltd. | Process for the preparation of pure thiophene derivatives |
| TW344661B (en) | 1993-11-24 | 1998-11-11 | Lilly Co Eli | Pharmaceutical composition for treatment of incontinence |
| US5508276A (en) | 1994-07-18 | 1996-04-16 | Eli Lilly And Company | Duloxetine enteric pellets |
| GB9812413D0 (en) * | 1998-06-10 | 1998-08-05 | Glaxo Group Ltd | Compound and its use |
| CA2362185C (en) * | 1999-04-09 | 2009-06-02 | Eli Lilly And Company | Methods for preparing 3-aryloxy-3-arylpropylamines and intermediates thereof |
| WO2003097632A1 (en) | 2002-05-20 | 2003-11-27 | Mitsubishi Rayon Co., Ltd. | Propanolamine derivatives, process for preparation of 3-n-methylamino-1-(2-thienyl)-1-propanols and process for preparation of propanolamine derivatives |
| EP1545489A4 (en) * | 2002-07-24 | 2006-08-23 | Cypress Bioscience Inc | Treatment of depression secondary to pain (dsp) |
| US20040235925A1 (en) | 2002-12-17 | 2004-11-25 | Pharmacia Corporation | Method for the treatment, prevention, or inhibition of a CNS disorder and/or pain and inflammation using a combination of duloxetine, venlafaxine or atomoxetine and a cyclooxygenase-2 selective inhibitor and compositions thereof |
| US20040214215A1 (en) | 2003-03-07 | 2004-10-28 | Yu Ruey J. | Bioavailability and improved delivery of alkaline pharmaceutical drugs |
| WO2004105690A2 (en) * | 2003-05-23 | 2004-12-09 | Cypress Bioscience, Inc. | Treatment of chronic pain associated with drug or radiation therapy |
| AU2003263585A1 (en) | 2003-08-25 | 2005-03-10 | Hetero Drugs Limited | Amorphous duloxetine hydrochloride |
| US20050250838A1 (en) * | 2004-05-04 | 2005-11-10 | Challapalli Prasad V | Formulation for sustained delivery |
| GB0410470D0 (en) | 2004-05-11 | 2004-06-16 | Cipla Ltd | Pharmaceutical compound and polymorphs thereof |
| WO2006071868A2 (en) * | 2004-12-23 | 2006-07-06 | Teva Pharmaceutical Industries Ltd. | Process for preparing pharmaceutically acceptable salts of duloxetine and intermediates thereof |
| WO2006081515A2 (en) * | 2005-01-27 | 2006-08-03 | Teva Pharmaceutical Industries Ltd. | Duloxetine hydrochloride polymorphs |
| AT501846B1 (en) | 2005-02-16 | 2007-08-15 | Fronius Int Gmbh | DEVICE AND METHOD FOR IMPLEMENTING SOFTWARE UPDATES IN INVERTERS AND INVERTERS DESIGNED FOR SOFTWARE UPDATES |
| CA2599478A1 (en) | 2005-03-14 | 2006-09-21 | Teva Pharmaceutical Industries Ltd. | Process for the purification of duloxetine hydrochloride |
| WO2006126213A1 (en) | 2005-05-24 | 2006-11-30 | Matrix Laboratories Ltd | An improved process for the preparation of duloxetine |
| PL1971592T3 (en) | 2005-12-12 | 2011-09-30 | Medichem Sa | Improved synthesis and preparations of duloxetine salts |
| WO2007077580A2 (en) | 2006-01-06 | 2007-07-12 | Msn Laboratories Limited | Improved process for pure duloxetine hydrochloride |
| US7538232B2 (en) | 2006-01-19 | 2009-05-26 | Eli Lilly And Company | Process for the asymmetric synthesis of duloxetine |
-
2006
- 2006-03-14 CA CA002599478A patent/CA2599478A1/en not_active Abandoned
- 2006-03-14 EP EP06738321A patent/EP1858859A1/en not_active Withdrawn
- 2006-03-14 EP EP06738348A patent/EP1858873A2/en not_active Withdrawn
- 2006-03-14 US US11/376,574 patent/US20060258871A1/en not_active Abandoned
- 2006-03-14 WO PCT/US2006/009275 patent/WO2006099468A2/en not_active Ceased
- 2006-03-14 MX MX2007011254A patent/MX2007011254A/en unknown
- 2006-03-14 TW TW095108594A patent/TW200639162A/en unknown
- 2006-03-14 WO PCT/US2006/009165 patent/WO2006099433A1/en not_active Ceased
- 2006-03-14 EP EP06738247A patent/EP1874754A1/en not_active Withdrawn
- 2006-03-14 TW TW095108592A patent/TW200639161A/en unknown
- 2006-03-14 CA CA002599475A patent/CA2599475A1/en not_active Abandoned
- 2006-03-14 WO PCT/US2006/009247 patent/WO2006099459A1/en not_active Ceased
- 2006-03-14 US US11/376,573 patent/US20060270861A1/en not_active Abandoned
- 2006-03-14 MX MX2007011255A patent/MX2007011255A/en unknown
- 2006-03-14 US US11/376,552 patent/US7534900B2/en not_active Expired - Fee Related
- 2006-03-14 WO PCT/US2006/009241 patent/WO2006099457A1/en not_active Ceased
- 2006-03-14 EP EP06738315A patent/EP1858874A1/en not_active Withdrawn
- 2006-03-14 US US11/376,754 patent/US20060270731A1/en not_active Abandoned
-
2007
- 2007-06-25 IL IL184184A patent/IL184184A0/en unknown
- 2007-06-25 IL IL184189A patent/IL184189A0/en unknown
- 2007-06-25 IL IL184185A patent/IL184185A0/en unknown
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5023269A (en) * | 1986-12-22 | 1991-06-11 | Eli Lilly And Company | 3-aryloxy-3-substituted propanamines |
| US5362886A (en) * | 1993-10-12 | 1994-11-08 | Eli Lilly And Company | Asymmetric synthesis |
| US20040249170A1 (en) * | 2002-01-24 | 2004-12-09 | Alfio Borghese | Process for preparing an intermediate useful for the asymmetric synthesis of duloxetine |
| WO2003070720A1 (en) * | 2002-02-22 | 2003-08-28 | Degussa Ag | Preparation of n-methyl-3-hydroxy- 3-(2-thienyl)propylamine via novel thiophene derivatives containing carbamate groups as intermediates |
| WO2004056795A1 (en) * | 2002-12-19 | 2004-07-08 | Cipla Ltd | A process for preparing duloxetine and intermediates for use therein |
| WO2006027798A2 (en) * | 2004-08-05 | 2006-03-16 | Sun Pharmaceutical Industries Limited | A process for preparation of an antidepressant compound |
| WO2006045255A1 (en) * | 2004-10-26 | 2006-05-04 | Zentiva, A.S. | Method of manufacturing (s)-n-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride (duloxetine) |
Also Published As
| Publication number | Publication date |
|---|---|
| IL184185A0 (en) | 2007-10-31 |
| US20060258871A1 (en) | 2006-11-16 |
| US20060270861A1 (en) | 2006-11-30 |
| IL184189A0 (en) | 2007-10-31 |
| TW200639161A (en) | 2006-11-16 |
| EP1858874A1 (en) | 2007-11-28 |
| CA2599478A1 (en) | 2006-09-21 |
| TW200639162A (en) | 2006-11-16 |
| WO2006099433A1 (en) | 2006-09-21 |
| US20060270731A1 (en) | 2006-11-30 |
| US7534900B2 (en) | 2009-05-19 |
| EP1858873A2 (en) | 2007-11-28 |
| MX2007011255A (en) | 2007-10-18 |
| WO2006099457A1 (en) | 2006-09-21 |
| IL184184A0 (en) | 2007-10-31 |
| EP1858859A1 (en) | 2007-11-28 |
| EP1874754A1 (en) | 2008-01-09 |
| WO2006099468A2 (en) | 2006-09-21 |
| WO2006099468A3 (en) | 2007-04-05 |
| MX2007011254A (en) | 2007-10-18 |
| US20060276660A1 (en) | 2006-12-07 |
| CA2599475A1 (en) | 2006-09-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006099459A1 (en) | Process for the preparation of optically active (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine | |
| US20060194869A1 (en) | Process for preparing pharmaceutically acceptable salts of duloxetine and intermediates thereof | |
| EP2114912B1 (en) | Process for making duloxetine and related compounds | |
| US20090182156A1 (en) | Synthesis and preparations of duloxetine salts | |
| US7709662B2 (en) | Method of manufacturing (S)-N-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride (duloxetine) | |
| EP1478641A1 (en) | Process for preparing an intermediate useful for the asymmetric synthesis of duloxetine | |
| WO2007095200A2 (en) | A process for the preparation of (s)-(+)-n,n-dimethyl-3-(1-naphthalenyloxy)-3-(2-thienyl)propanamine, a duloxetine intermediate | |
| WO2003070720A1 (en) | Preparation of n-methyl-3-hydroxy- 3-(2-thienyl)propylamine via novel thiophene derivatives containing carbamate groups as intermediates | |
| CN101454306A (en) | Process for preparing duloxetine and intermediates thereof | |
| WO2008107911A2 (en) | Novel process for preparation of duloxetine hydrochloride | |
| WO2007076733A2 (en) | A METHOD FOR THE PREPARATION OF (S)-N-METΗYL-3-(l-NAPHTHYLOXY)-3-(2-THIENYL)PROPYLAMINE HYDROCHLORIDE (DULOXETINE) | |
| US20100280093A1 (en) | Process for the preparation enantiomerically pure salts of n-methyl-3-(1-naphthaleneoxy)-3-(2-thienyl)propanamine | |
| US7560573B2 (en) | Process for the preparation of (S)-(-)-N,N-dimethyl-3-(2-thienyl)-3-hydroxypropananine, a duloxetine intermediate | |
| WO2007123900A2 (en) | Enantiomers of n,n-dimethyl-3-(2-thienyl)-3-hydroxypropanamine borane as intermediates in the synthesis of duloxetine | |
| WO2008093360A2 (en) | A process for preparation of (s)-(+)-n-methyl-3(1-naphthyloxy)-3(2-thienyl)propylamine hydrochloride | |
| EP2125772B1 (en) | A process for the preparation of duloxetin and new key intermediates for use therein | |
| WO2009019719A2 (en) | Process for the preparation of 3-aryloxy-3-arylpropanamines | |
| ES2349047T3 (en) | PREPARATION PROCEDURE FOR A USEFUL INTERMEDIATE FOR ASYMMETRIC SYNTHESIS OF (+) DULOXETIN. | |
| MX2008001519A (en) | Process for preparing duloxetine and intermediates thereof | |
| WO2009109992A1 (en) | Novel process for preparation of duloxetine and intermediates for use therein | |
| CZ297555B6 (en) | Process for preparing (S)-N-methyl-3-(1-naphthyloxy)-3-(2-thienyl)propylamine hydrochloride (duloxetine) | |
| MX2007013043A (en) | Process for the preparation of (s)-(-)-n,n-dimethyl-3-(2-thienyl)-3-hydroxypropanamine, a duloxetine intermediate | |
| WO2007084193A1 (en) | Dnt-succinate and methods of preparation thereof | |
| US20080207923A1 (en) | Pure DNT-maleate and methods of preparation thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006738321 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 184189 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 6797/DELNP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/011254 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |