WO2006136857A1 - Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors - Google Patents
Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors Download PDFInfo
- Publication number
- WO2006136857A1 WO2006136857A1 PCT/GB2006/002337 GB2006002337W WO2006136857A1 WO 2006136857 A1 WO2006136857 A1 WO 2006136857A1 GB 2006002337 W GB2006002337 W GB 2006002337W WO 2006136857 A1 WO2006136857 A1 WO 2006136857A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- compound according
- alkoxy
- heteroaryl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)*(C(C(C)=C(C)C)N(*)C(C)=*)=* Chemical compound CC(C)*(C(C(C)=C(C)C)N(*)C(C)=*)=* 0.000 description 1
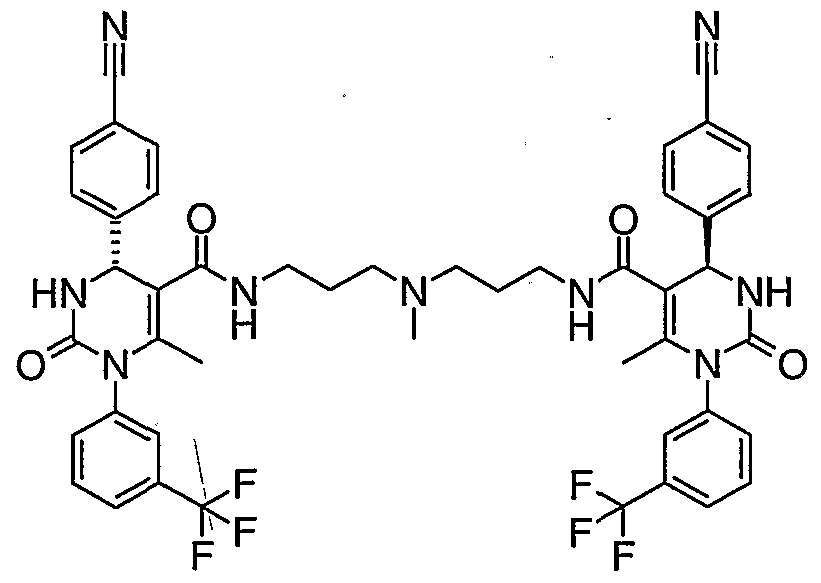
- LYLAOTRMFTXPIX-LJEWAXOPSA-N CC(N(c1cc(C(F)(F)F)ccc1)C(N[C@@H]1c(cc2)ccc2C#N)=O)=C1C(NCCN(C)CCNC(C([C@@H](c(cc1)ccc1C#N)N1)=C(C)N(c2cc(C(F)(F)F)ccc2)C1=O)=O)=O Chemical compound CC(N(c1cc(C(F)(F)F)ccc1)C(N[C@@H]1c(cc2)ccc2C#N)=O)=C1C(NCCN(C)CCNC(C([C@@H](c(cc1)ccc1C#N)N1)=C(C)N(c2cc(C(F)(F)F)ccc2)C1=O)=O)=O LYLAOTRMFTXPIX-LJEWAXOPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/20—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D239/22—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/513—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim having oxo groups directly attached to the heterocyclic ring, e.g. cytosine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
Definitions
- This invention relates to heterocyclic compounds and their use in therapy.
- Human neutrophil elastase is a 32 kDa serine proteinase found in the azurophilic granules of neutrophils. It has a role in the degradation of a wide range of extracellular matrix proteins, including fibronectin, laminin, proteoglycans, Type III and Type IV collagens as well as elastin (Bieth, G. In Regulation of Matrix accumulation, Mecham, R. P. (Eds), Academic Press, NY, USA 1986, 217-306). HNE has long been considered to play an important role in homeostasis through repair and disposal of damaged tissues via degradation of the tissue structural proteins.
- HNE has been implicated in the upregulation of IL-8 gene expression and also induces IL-8 release from the epithelial cells of the lung.
- IL-8 In animal models of Chronic Obstructive Pulmonary Disease induced by tobacco smoke exposure both small molecule inhibitors and protein inhibitors of HNE inhibit the inflammatory response and the development of emphysema (Wright, J. L. et al. Am. J. Respir. Crit. Care Med.2002, 166, 954-960; Churg, A. et al. Am. J. Respir. Crit. Care Med.2003, 168, 199-207).
- HNE may play a role both in matrix destruction and in amplifying inflammatory responses in chronic respiratory diseases where neutrophil influx is a characteristic feature.
- HNE is believed to play a role in several pulmonary diseases, including chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory distress syndrome (ARDS), pulmonary emphysema, pneumonia, severe asthma, sarcoidosis, bronchiectasis and lung fibrosis. It is also implicated in several cardiovascular diseases in which tissue remodelling is involved, for example, in heart failure and the generation of ischaemic tissue injury following acute myocardial infarction.
- COPD chronic obstructive pulmonary disease
- CF cystic fibrosis
- ARDS acute respiratory distress syndrome
- Elevated HNE levels are also correlated with the severity of inflammation in inflammatory bowel disease (Silberer H et al, Clin Lab.2005;51 (3-4):117-26) and may play a role in impaired mucosal repair in patients with ulcerative colitis.
- COPD is an umbrella term encompassing three different pathological conditions, all of which contribute to limitation of airflow: chronic bronchitis, emphysema and small- airway disease. Generally all three will exist to varying extents in patients presenting with COPD, and all three may be due to neutrophil-mediated inflammation, as supported by the increased number of neutrophils observed in bronchoalveolar leakage (BAL) fluids of COPD patients (Thompson, A.
- BAL bronchoalveolar leakage
- the major pathogenic determinant in COPD has long been considered to be the protease-anti-protease balance (also known as the 'elastase:anti-elastase hypothesis'), in which an imbalance of HNE and endogenous antiproteases such as ⁇ 1 - antitrypsin (Ci 1 -AT) 1 Secretory leukocyte protease inhibitor (SLPI) and pre-elafin leads to the various inflammatory disorders of COPD.
- protease-anti-protease balance also known as the 'elastase:anti-elastase hypothesis'
- an imbalance of HNE and endogenous antiproteases such as ⁇ 1 - antitrypsin (Ci 1 -AT) 1 Secretory leukocyte protease inhibitor (SLPI) and pre-elafin leads to the various inflammatory disorders of COPD.
- Multimeric ligands consist of multiple binding domains which are tethered together through a suitable scaffold.
- Multimeric compounds are unlikely to be orally bioavailable (as predicted by Lipinski's "Rule of 5") which may be advantageous where an inhaled route of administration to the lungs is targeted, since even after inhaled administration, a large proportion of drug is likely to enter the Gl tract. Thus such compounds may be expected to show reduced systemic exposure after inhalation administration and hence an improved toxicity profile over orally administered therapies.
- Monomers of formula (II) are described as inhibitors of human neutrophil elastase in WO2004/024700, WO2004/024701 , GB2392910, WO2005/082863 and WO2005/082864. Summary of the Invention
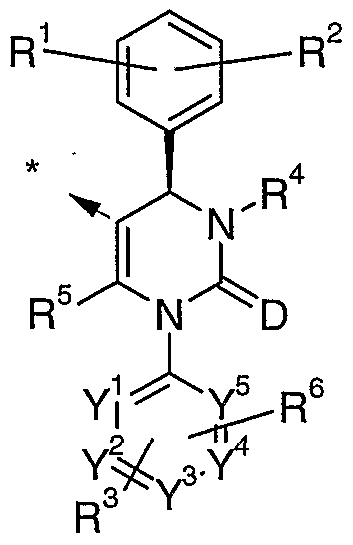
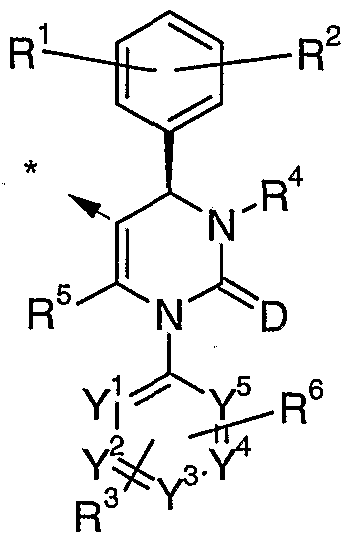
- a first aspect of the invention is a compound of formula (I) or formula (IV):
- each M is independently an inhibitor of HNE; and each L is independently a linker group; t is 2 to 20; and G is aryl, heteroaryl, alkyl, cycloalkyl, nitrogen, a dendrimer or a group of any of formulae (V) to (VII):
- Ar is aryl or heteroaryl; and u is 2-20;
- M is a group of Formula (II)
- A is aryl or heteroaryl; D is oxygen or sulphur;
- R 1 , R 2 and R 3 are independently each hydrogen, halogen, nitro, cyano, alkyl, hydroxy or alkoxy, wherein alkyl and alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and alkoxy;
- R 4 is hydrogen, alkyl, alkylcarbonyl, alkoxycarbonyl, alkenoxycarbonyl, hydroxycarbonyl, aminocarbonyl, arylcarbonyl, heteroarylcarbonyl, heterocycloalkylcarbonyl, heteroaryl, heterocycloalkyl or cyano, wherein alkylcarbonyl, alkoxycarbonyl, and aminocarbonyl can be further substituted with one to three identical or different radicals selected from the group consisting of cycloalkyl, hydroxy, alkoxy, alkoxycarbonyl, hydroxycarbonyl, aminocarbonyl, cyano, amino, heteroaryl, heterocycloalkyl and tri-(alkyl)-silyl, and wherein heteroarylcarbonyl, heterocycloalkylcarbonyl, heteroaryl and heterocycloalkyl can be further substituted with alkyl; or
- R 4 represents a group of Formula (VIII)
- R 4A , R 4B , R 4D , R 4E , R 4G , R 4H , R 41 and R 4J are independently hydrogen or alkyl, or R 4H and R 41 may may be joined together with the nitrogen atom to which they are attached to form a ring;
- R 4F is a lone pair or R 4F is alkyl and the nitrogen atom to which it is attached is quaternary and carries a positive charge;
- R 4G is a lone pair or R 4C is alkyl and the nitrogen atom to which it is attached is quaternary and carries a positive charge; or any two of R 4C , R 4D or R 4E may be joined together with the nitrogen atom to which they are attached to form a ring, optionally containing a further heteroatom selected from oxygen or nitrogen; v1 is 1-3; v2 is 1-6;
- R 5 is alkyl, which can be substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy, alkoxy, alkenoxy, alkylthio, amino, hydroxycarbonyl, alkoxycarbonyl and the radical -O-(alkyl)-O-(alkyl); or R 5 is amino;
- R 6 is halogen, nitro, cyano, alkyl, hydroxy or alkoxy, wherein alkyl and alkoxy can be further substituted with one to three identical or different radicals selected from the group consisting of halogen, hydroxy and alkoxy; and ⁇ i ⁇ ⁇ 2 ⁇ ⁇ 3 ⁇ ⁇ 4 and ⁇ 5 are j nc j e p enc
- L a is a bond or group -C(O)-;
- L b is a bond or group -C(O)-;
- R 7 is a bond or an alkylene or cycloalkylene group;
- W is a bond or is selected from the following divalent radicals
- R 8A is an alkylene or cycloalkylene group
- R 8B is an alkylene or cycloalkylene group, or a group of Formula A 2 ;
- R 9A is hydrogen or alkyl; one of R 9B or R 9C is a lone pair and the other is hydrogen or alkyl, or R 9B and R 9C are both alkyl, in which case the nitrogen to which they are attached is quaternary and carries a positive charge; or R 9B and R 9C may be joined together with the nitrogen to which they are attached to form a ring;
- R 10A is hydrogen or alkyl;
- R 10B and R 10G are independently hydrogen or alkyl, or alternatively R 10B and R 10C may be joined together to form a ring; m2 is 1-3;
- a 2 is selected from one of the following groups
- Ar 1 , Ar 2 are independently an aryl or heteroaryl group; or a pharmaceutically acceptable salt, solvate or N-oxide thereof.
- Compounds of the invention may be useful in the treatment or prevention of diseases in which HNE is implicated, for example chronic obstructive pulmonary disease (COPD), chronic bronchitis, lung fibrosis, pneumonia, acute respiratory distress syndrome (ARDS), pulmonary emphysema, smoking-induced emphysema, severe asthma, sarcoidosis, bronchiectasis, cystic fibrosis, inflammatory bowel disease; ulcerative colitis and Crohn's disease.
- COPD chronic obstructive pulmonary disease
- COPD chronic obstructive pulmonary disease
- ARDS acute respiratory distress syndrome
- pulmonary emphysema smoking-induced emphysema
- severe asthma sarcoidosis
- bronchiectasis cystic fibrosis
- inflammatory bowel disease ulcerative colitis and Crohn's disease.
- Another aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention and a pharmaceutically acceptable carrier or excipient.
- Another aspect of the invention is the use of a compound of the invention for the manufacture of a medicament for the treatment or prevention of a disease or condition in which HNE is implicated.
- compounds of the invention may be used in a method of therapy, for the treatment of a patient suffering from a condition or disease as defined above.
- Alkylcarbonyl means a -CO-alkyl group in which the alkyl group is as described herein.
- exemplary acyl groups include -COCH 3 and -COCH(CH 3 ) 2 .
- Acylamino means a -NR-acyl group in which R and acyl are as described herein.
- exemplary acylamino groups include -NHCOCH 3 and -N(CH 3 )COCH 3 .
- Alkenoxy means an -O-alkenyl group in which alkenyl is as described below.
- Alkenoxycarbonyl means a -COO-alkenyl group which alkenyl is as described below.
- Exemplary groups includes -C(O)O-allyl.
- Alkoxy and “alkyloxy” means an -O-alkyl group in which alkyl is as described below.
- exemplary alkoxy groups include methoxy (-OCH 3 ) and ethoxy (-OC 2 H 5 ).
- Alkoxycarbonyl means a -COO-alkyl group in which alkyl is as defined below.
- exemplary alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
- Alkyl or “lower alkyl”, as a group or part of a group, refers to a straight or branched chain saturated hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms, in the chain.
- exemplary alkyl groups include methyl, ethyl, 1 -propyl and 2- propyl.
- alkenyl as a group or part of a group refers to a straight or branched chain hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms and one carbon- carbon double bond in the chain.
- alkenyl groups include ethenyl, 1 -propenyl, and 2-propenyI.
- Alkylamino means a -NH-alkyl group in which alkyl is as defined above.
- exemplary alkylamino groups include methylamino and ethylamino.
- Alkylene means an -alkyl- group in which alkyl is as defined previously.
- exemplary alkylene groups include -CH 2 -, -(CH 2 ) 2 - and -C(CH 3 )HCH 2 -.
- Alkenylene means an -alkenyl- group in which alkenyl is as defined previously.
- Alkylthio means a -S-alkyl group in which alkyl is as defined above.
- Exemplary alkylthio groups include methylthio and ethylthio.
- Amino means a -NR 1 R 2 group where R 1 and R 2 may be independently a hydrogen atom, alkyl, aryl, arylalkyl, alkenyl, alkynyl, heteroaryl or heterocycloalkyl group, (i.e. The amino group may be primary, secondary or tertiary). Exemplary amino groups include -NH 2 , NHCH 3 , -NHPh, -N(CH 3 ) 2 , etc.
- Aminocarbonyl means a -CO-NRR group in which R is as herein described.
- exemplary aminocarbonyl groups include -CONH 2 , -CONHCH 3 and -CONH-phenyl.
- Ammonium means a quarternary nitrogen group -N + R 1 R 2 R 3 where R 1 , R 2 and
- R 3 are alkyl, aryl, alkenyl, arylalkyl, heteroaryl, heterocycloalkyl, and the nitrogen atom carries a formal positive charge.
- Aryl as a group or part of a group denotes an optionally substituted monocyclic or multicyclic aromatic carbocyclic moiety of from 6 to 14 carbon atoms, preferably from 6 to 10 carbon atoms, such as phenyl or naphthyl.
- the aryl group may be substituted by one or more substituent groups.
- Arylalkyl means an aryl-alkyl- group in which the aryl and alkyl moieties are as previously described.
- Exemplary arylalkyl groups include benzyl, phenethyl and naphthlenemethyl.
- Arylalkyloxy means an aryl-alkyloxy- group in which the aryl and alkyloxy moieties are as previously described. Preferred arylalkyloxy groups contain a C 1 4 alkyl moiety.
- Exemplary arylalkyl groups include benzyloxy.
- exemplary groups include benzoyl (-C(O)Ph).
- Aryloxy means an -O-aryl group in which aryl is described above.
- Exemplary aryloxy groups include phenoxy.
- Cyclic amine means an optionally substituted 3 to 8 membered monocyclic cycloalkyl ring system where one of the ring carbon atoms is replaced by nitrogen, and which may optionally contain an additional heteroatom selected from O, S or NR (where R is as described herein).
- Exemplary cyclic amines include pyrrolidine, piperidine, morpholine, piperazine and ⁇ /-methylpiperazine.
- the cyclic amine group may be substituted by one or more substituent groups.
- Cycloalkyl means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms.
- Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclopentyl, cyclohexyl and cycloheptyl.
- the cycloalkyl group may be substituted by one or more substituent groups.
- Cyloalkylene means means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms, as a bivalent radical.
- Exemplary cycloalkylene groups include cyclohexane-1 ,4-diyl.
- Cycloalkylalkyl means a cycloalkyl-alkyl- group in which the cycloalkyl and alkyl moieties are as previously described.
- Exemplary monocyclic cycloalkylalkyl groups include cyclopropylmethyl, cyclopentylmethyl, cyclohexylmethyl and cycloheptylmethyl.
- “Dendrimer” means a multifunctional core group with a branching group attached to each functional site. Each branching site can be attached to another branching molecule and this process may be repeated multiple times.
- Halo or halogen means fluoro, chloro, bromo, or iodo.
- Haloalkoxy means an -O-alkyl group in which the alkyl is substituted by one or more halogen atoms. Exemplary haloalkyl groups include trifluoromethoxy and difluoromethoxy.
- Haloalkyl means an alkyl group which is substituted by one or more halo atoms.
- Exemplary haloalkyl groups include trifluoromethyl.
- Heteroaryl as a group or part of a group denotes an optionally substituted aromatic monocyclic or multicyclic organic moiety of from 5 to 14 ring atoms, preferably from 5 to 10 ring atoms, in which one or more of the ring atoms is/are element(s) other than carbon, for example nitrogen, oxygen or sulfur.
- Examples of such groups include benzimidazolyl, benzoxazolyl, benzothiazolyl, benzofuranyl, benzothienyl, furyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, oxazolyl, oxadiazolyi, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyl, tetrazolyl, 1 ,3,4-thiadiazolyl, thiazolyl, thienyl and triazolyl groups.
- the heteroaryl group may be substituted by one or more substituent groups.
- the heteroaryl group may be attached to the remainder of the compound of the invention by any available carbon or nitrogen atom.
- Heteroarylcarbonyl means a heteroaryl group attached to a carbonyl group - C(O)-.
- exemplary groups are pyridine-2-carbonyl, thiophene-2-carbonyl.
- Heteroaryloxy means a heteroaryloxy- group in which the heteroaryl is as previously described.
- exemplary heteroaryloxy groups include pyridyloxy.
- Heterocycloalkyl means: (i) an optionally substituted cycloalkyl group of from 4 to 8 ring members which contains one or more heteroatoms selected from O, S or NR; (ii) a cycloalkyl group of from 4 to 8 ring members which contains CONR and CONRCO
- heterocycloalkyl group may be substituted by one or more substituent groups.
- the heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- Heterocycloalkylalkyl means a heterocycloalkyl-alkyl- group in which the heterocycloalkyl and alkyl moieties are as previously described.
- Hydrocarbonyl means a group -COOH.
- “Pharmaceutically acceptable salt” means a physiologically or toxicologically tolerable salt and include, when appropriate, pharmaceutically acceptable base addition salts and pharmaceutically acceptable acid addition salts.
- pharmaceutically acceptable base addition salts that may be formed include sodium, potassium, calcium, magnesium and ammonium salts, or salts with organic amines, such as, diethylamine, ⁇ /-methyl-glucamine, diethanolamine or amino acids (e.g.
- a compound of the invention contains a basic group, such as an amino group
- pharmaceutically acceptable acid addition salts that may be formed include hydrochlorides, hydrobromides, phosphates, acetates, citrates, lactates, tartrates, malonates, methanesulphonates and the like.
- “Pharmaceutically acceptable salt” also means quaternary ammonium salts. In this case, the acceptable salts may be chlorides, bromides, iodides, mesylates, tosylates, succinates and the like.
- Prodrug refers to a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis, reduction or oxidation) to a compound of the invention.
- metabolic means e.g. by hydrolysis, reduction or oxidation
- an ester prodrug of a compound of the invention containing a hydroxy group may be convertible by hydrolysis in vivoto the parent molecule.
- Suitable esters of compounds of the invention containing a hydroxy group are for example acetates, citrates, lactates, tartrates, malonates, oxalates, salicylates, propionates, succinates, fumarates, maleates, methylene-bis- ⁇ -hydroxynaphthoates, gentisates, isethionates, di-p-toluoyltartrates, methanesulfonates, ethanesulfonates, benzenesulfonates, p-toluenesulfonates, naphthalene bis sulfonates, cyclohexylsulfamates and quinates.
- ester prodrug of a compound of the invention containing a carboxy group may be convertible by hydrolysis in v/Voto the parent molecule.
- ester prodrugs are those described by F. J. Leinweber, Drug Metab. Res., 1987, 18, 379.
- “Saturated” pertains to compounds and/or groups which do not have any carbon- carbon double bonds or carbon-carbon triple bonds.
- Suitable optional substituent groups include acyl (e.g. -COCHJ 1 alkoxy (e.g., -OCHJ, alkoxycarbonyl (e.g. -COOCH 3 ), alkylamino (e.g. -NHCH 3 ), alkylsulfinyl (e.g. -SOCH 3 ), alkylsulfonyl (e.g. -SO 2 CH 3 ), alkylthio (e.g.
- -SCH 3 ), -NH 2 , aminoacyl (e.g. -CON(CH 3 ) 2 ), aminoalkyl (e.g. -CH 2 NH 2 ), arylalkyl (e.g. -CH 2 Ph Or -CH 2 -CH 2 -Ph), cyano, dialkylamino (e.g. -N(CH g ) 2 ), halo, haloalkoxy (e.g. -OCF 3 or -OCHF 2 ), haloalkyl (e.g. -CF 3 ), alkyl (e.g.
- -CH 3 or -CH 2 CH 3 -OH, -CHO, -NO 2 , aryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heteroaryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heterocycloalkyl, aminoacyl (e.g. -CONH 2 , - CONHCH 3 ), aminosulfonyl (e.g. -SO 2 NH 2 , -SO 2 NHCH 3 ), acylamino (e.g. -NHCOCH 3 ), sulfonylamino (e.g.
- heteroarylalkyl cyclic amine (e.g. morpholine), aryloxy, heteroaryloxy, arylalkyloxy (e.g. benzyloxy) and heteroarylalkyloxy.
- Alkylene or alkenylene groups may be optionally substituted. Suitable optional substituent groups include alkoxy (e.g., -OCH 3 ), alkylamino (e.g. -NHCH 3 ), alkylsulfinyl
- alkylsulfonyl e.g. -SO,CH J, alkylthio (e.g. -SCHJ, -NH 2 , aminoalkyl (e.g. - CH NHJ, arylalkyl (e.g. -CH 9 Ph or -CH -CH -Ph), cyano, dialkylamino (e.g. -N(CHJJ, halo, haloalkoxy (e.g. -OCF 3 or -OCHF 2 ), haloalkyl (e.g. -CF 3 ), alkyl (e.g. -CH 3 or - CH 0 CHJ, -OH, -CHO, and -NO 2 .
- alkyl e.g. -CH 3 or - CH 0 CHJ, -OH, -CHO, and -NO 2 .
- Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to cis- and frans-forms, E- and Z-forms, R-, S- and meso-forms, keto-, and enol-forms. Unless otherwise stated a reference to a particular compound includes all such isomeric forms, including racemic and other mixtures thereof. Where appropriate such isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate such isomers may be prepared by the application of adaptation of known methods (e.g. asymmetric synthesis).
- G may be a group of any of formulae (V) to (VII) or a dendrimer.
- groups of formulae (V) to (VII) include, but are not limited to phenoxyphenyl, biphenyl, bipyridyl, ethylenediamino, propylenediamino and the like. It is to be understood that the number of possible attachment points is dictated by the valency of the groups present, so that for example, biphenyl can contain up to 10 possible attachments (5 on each phenyl ring), and ethylenediamine can possess up to 4 possible attachments (2 on each terminal amine).
- An example of a dendrimer suitable for use in the invention is:
- each M is the same or different and is a group of formula (II) as defined herein.
- formula (II) the arrow denotes the point of attachment of M to the linker L.
- L is a group of Formula (III) as defined herein.
- compounds are of Formula (I).
- A is a phenyl ring.
- R 1 is a cyano group and R 2 is a hydrogen atom.
- D is an oxygen atom.
- the groups M have the stereochemistry shown below;
- R 6 is a haloalkyl group.
- Y 1 -Y 5 are carbon atoms.
- L a is a group C(O).
- R 7 and L b are a bond.
- W is a radical
- W is the radical -N(R 9A ) - R 8B - N(R 9A ) - R 8B -
- R 5 is an alkyl group. In yet another preferred embodiment R 5 is a methyl group. In one embodiment R 4 is a hydrogen atom In a further embodiment R 4 is a group of Formula (VIII)
- Preferred compounds of the invention include those of the Examples, e.g. of Examples 1 , 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 , 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 , 22, 23, 24, 25, 26, 27, 28, 29, 30, 32, 34, 36, 37 and 38.
- the therapeutic utility of the present compounds is pertinent to any disease that is known to be at least partially mediated by the action of human neutrophil elastase.
- the present compounds may be beneficial in the treatment of chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), acute respiratory, distress syndrome (ARDS), pulmonary emphysema, pneumonia and lung fibrosis.
- COPD chronic obstructive pulmonary disease
- CF cystic fibrosis
- ARDS acute respiratory, distress syndrome
- pulmonary emphysema pulmonary emphysema
- pneumonia and lung fibrosis lung fibrosis.
- the present invention is also concerned with pharmaceutical formulations comprising, as an active ingredient, a compound of the invention.
- Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung.
- the present invention is also concerned with pharmaceutical compositions for preventing and treating inflammatory diseases of the lung comprising a therapeutically effective amount of a compound of the invention and one or more other therapeutic agents.
- Suitable therapeutic agents for a combination therapy with compounds of the invention include: (1 ) a corticosteroid, for example fluticasone, ciclesonide or budesonide; (2) a ⁇ 2-adrenoreceptor agonist, for example salmeterol, indacaterol orformeterol; (3) a leukotriene modulator, for example montelukast or pranlukast; (4) muscarinic-3 (M3) receptor antagonists such as tiotropium bromide; (5) bronchodilators that possess both M3 receptor antagonism and ⁇ 2-adrenoreceptor agonism in a single molecule (6) phosphodiesterase-IV (PDE-IV) inhibitors, for example roflumilast or cilomilast; (7) an antitussive agent, such as codeine or dextramorphan;(8) a non-steroidal antiinflammatory agent (NSAID), for example ibuprofen or ketoprofen; (9)
- the weight ratio of the first and second active ingredients may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used.
- prophylactic or therapeutic dose of a compound of the invention will, of course, vary with the nature of the severity of the condition to be treated and with the particular compound and its route of administration. It will also vary according to the age, weight and response of the individual patient. In general, the daily dose range will lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- compositions which comprise a compound of the invention and a pharmaceutically acceptable carrier.
- composition is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the invention, additional active ingredient(s), and pharmaceutically acceptable excipients.
- compositions of the present invention comprise a compound of the invention as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention.
- the active compound may be administered by any convenient, suitable or effective route.
- Suitable routes of administration are known to those skilled in the art, and include oral, intravenous, rectal, parenteral, topical, ocular, nasal, buccal and pulmonary. Delivery by inhalation is preferred.
- compositions suitable for administration by inhalation are known, and may include carriers and/or diluents that are known for use in such compositions.
- the composition may contain 0.01-99% by weight of active compound.
- a unit dose comprises the active compound in an amount of 1 ⁇ g to 10 mg.
- the most suitable dosage level may be determined by any suitable method known to one skilled in the art. It will be understood, however, that the specific amount for any particular patient will depend upon a variety of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment.
- the active compound is preferably in the form of microparticles. They may be prepared by a variety of techniques, including spray-drying, freeze-drying and micronisation.
- a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI).
- PMDI pressurised metered dose inhaler
- Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, HFA-134a, HFA- 227, HCFC-22 (CCI2F2) and HFA-152 (CH4F2 and isobutane).
- a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI).
- DPI dry powder inhaler
- Microparticles for delivery by administration may be formulated with excipients that aid delivery and release.
- microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung.
- Suitable carrier particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 ⁇ m.
- a preferred composition is:
- Compounds of the invention may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which present compounds are useful. Such other drugs may be administered, by a route and in an amount commonly used therefore, contemporaneously or sequentially with a compound of the invention. When a compound of the invention is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of the invention is preferred.
- compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the invention.
- Aerosol generation can be carried out using, for example, pressure-driven jet atomizers or ultrasonic atomizers, preferably using propellant-driven metered aerosols or propellant- free administration of micronized active compounds from, for example, inhalation capsules or other "dry powder" delivery systems.
- the active compounds may be dosed as described depending on the inhaler system used.
- the administration forms may additionally contain excipients, such as, for example, propellants (e.g. Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavorings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
- the compounds of the invention of the present invention can be prepared according to the procedures of the following schemes and examples, using appropriate materials, and are further exemplified by the following specific examples. Moreover, by utilising the procedures described with the disclosure contained herein, one of ordinary skill in the art can readily prepare additional compounds of the present invention claimed herein.
- the compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention.
- the examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the compounds of the invention may be isolated in the form of their pharmaceutically acceptable salts, such as those described previously herein above.
- the free acid form corresponding to isolated salts can be generated by neutralisation with a suitable acid such as acetic acid and hydrochloric acid and extraction of the liberated free acid into an organic solvent followed by evaporation.
- a suitable acid such as acetic acid and hydrochloric acid
- the free acid form isolated in this manner can be further converted into another pharmaceutically acceptable salt by dissolution in an organic solvent followed by addition of the appropriate base and subsequent evaporation, precipitation, or crystallisation.
- reaction Schemes illustrate how compounds of the invention, in particular the Example compounds, may be prepared. It will be understood that the processes detailed below are solely for the purpose of illustrating the invention and should not be construed as limiting. A process utilising similar or analogous reagents and/or conditions known to one skilled in the art may also be used to obtain a compound of the invention. The following reaction schemes illustrate how compounds of the invention, in particular the Example compounds, may be prepared. It will be understood that the processes detailed below are solely for the purpose of illustrating the invention and should not be construed as limiting. A process utilising similar or analogous reagents and/or conditions known to one skilled in the art may also be used to obtain a compound of the invention.
- the monomers may be joined together by a range of standard chemistries, for example, reaction with a suitable bifunctional linker molecule in the presence of a suitable reagent and optional solvent, Scheme 1 (Route A).
- An alternative methodology involves attachment of a spacer group incorporating a second functional group, which subsequently allows attachment of a bidentate linker molecule, Scheme 1 (Route B).
- the linker part of the dimer may be modified after dimerisation.
- X nucleophilic group e.g. NHR, OH
- G reactive group e.g. CHO, CO 2 H, NH 2 etc.
- reaction of the monomer with a suitable diamine or diol can be effected in the presence of a base and coupling reagent, for example HATU, and optional solvent.
- a base and coupling reagent for example HATU
- Reaction of the monomer with a protected aminoaldehyde (protected as an acetal), in the presence of a suitable base and coupling reagent, and optional solvent, followed by deprotection leads to an intermediate that can be reacted with a suitable bidentate species, such as a diamine, to generate compounds of the invention, Scheme 2.
- the monomers of Formula (II) may be prepared as racemates according to methods described in WO2004024700, WO2004024701 , GB2392910, WO200508263 and WO2005082864.
- the monomers may be separated by chiral HPLC into their enantiomers.
- the monomer (as a carboxylic acid) may be resolved by formation of diastereomeric salts with a suitable chiral base, such as norephedrine, followed by fractional recrystallisation, Scheme 3.
- N-3 may be functionalised via alkylation with a suitably activated alkane in the presence of a suitable base, for example a metal hydride, and optional solvent. Suitable leaving groups include halogen and sulfonate.
- N-3 of the monomer may be acylated with an acid halide under similar conditions. This group (R in Schemes 1-4) may be modified further in subsequent steps. Prior protection of the carboxylic acid by, for example, conversion to the corresponding allyl ester, is required to prevent unwanted side- reactions.
- Compounds that are quaternary ammonium salts can be generated by reaction of the parent amine with a suitably activated alkane.
- Suitable leaving groups include halogen and sulfonate.
- 'Isolute SPE Si cartridge' refers to a pre-packed polypropylene column containing unbonded activated silica with irregular particles with average size of 50 ⁇ m and nominal 6OA porosity.
- 'lsolute SCX-2 cartridge' refers to a pre-packed polypropylene column containing a non end-capped propylsulphonic acid functionalised silica strong cation exchange sorbent. All solvents and commercial reagents were used as received. After HPLC purification, fractions containing product were combined and freeze-dried to give the product as a white or off-white solid. In some cases, where the compound contained a basic centre, the product was obtained as the formate salt.
- Phenyl hexyl column (250 x 21.20 mm Luna column with 5 ⁇ m particle size), eluting with a gradient of A: water + 0.1 % TFA; B: acetonitrile + 0.1 % TFA at a flow rate of 5 ml/min with UV detection at 254 nm.
- Amylose tris(3,5-dirnethylphenylcarbamate) (250 x 20 mm CHIRALPAK IA column with 5 ⁇ m particle size), eluting with an isocratic mixture of ethanol (15%) in n-heptane +
- DIPEA di-isopropylethylamine
- EDCI 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- HATU O-(7-Azabenzotriazol-1 -yl)-N,N,N',N'- tetramethyluroniumhexafluorophosphate.
- TFA trifluoroacetic acid
- Example 2 was prepared from intermediate 3 (100 mg) and tetra(ethylene) glycol by a similar procedure to that used in Example 1 , and purified using HPLC (System 2).
- Example 4 was prepared from Intermediate 3 and 4,9-dioxa-1 ,12-dodecanediamine by a similar method to that used in the synthesis of Example 3.
- Example 15 was prepared from Intermediate 10 and 3,3'-diamino-N-methyldipropylamine using a procedure similar to that used in the synthesis of Example 5.
- the crude product was dissolved in MeOH and loaded onto an SCX-2 cartridge (10 g) which had been pre- treated with MeOH.
- the cartridge was flushed with MeOH and then the product was eluted with 2M NH 3 in MeOH.
- Intermediate 15 was obtained as a pale yellow foam.
- Example 16 was prepared from Intermediate 4 and triethylenetetramine using a similar procedure to that used in the synthesis of Example 5.
- the crude product was purified on an lsolute SPE Si Il cartridge (10 g) eluting with 0-50% MeOH in EtOAc and isolated as a white solid.
- a small sample was further purified using HPLC (System 1).
- Example 15 (212 mg, 0.201 mmol) was treated with 1 M NaOH (25 ml) and MeOH (15 ml). The reaction mixture was stirred at RT for 1.5 h. The mixture was acidified using 1 M HCI (50 ml) and extracted with EtOAc (3 x 60 ml). The organic extracts were combined, washed with brine (50 ml), and dried (Na 2 SO 4 ). Evaporation gave the diacid as a white solid.
- Example 18 was prepared from Example 17 and 4 equivalents of 3,3'-diamino-N- methyldipropylamine using conditions to those used in the synthesis of Example 5. Purification was achieved using HPLC (System 1) and example 18 was obtained as a white solid.
- Example 15 (150 mg, 0.142 mmol) was dissolved in DCM (20 ml) and iodomethane (5 ml) was added. The solution was allowed to stand at RT for 60 h. The volatiles were evaporated.
- Example 20 was prepared from example 19 using a similar method to that used in the synthesis of example 17. Purification was achieved by trituration with Et 2 CVDCM (5:1) giving the diacid as a pale yellow solid.
- Example 21 was prepared from Example 20 and 4 equivalents of 3,3'-diamino-N- methyldipropylamine using conditions to those used in the synthesis of Example 5. Purification was achieved using HPLC (System 1) and example 21 was obtained as a pale cream solid.
- Example 16 (102 mg, 0.112 mmol) was dissolved in DCM (10 ml) and 1 ,1'- thiocarbonyldipyridone (13 mg, 0.0559 mmol) was added. The solution was heated under reflux for 4 h and then allowed to stand at RT for 3 days. The DCM was evaporated and the residue was chromatographed on an lsolute SPE Si Il cartridge (5 g) eluting with 0- 10% MeOH in EtOAc. The product-containing fractions were combined and eluted through an lsolute SPE SCX-2 cartridge, flushing with MeOH. Evaporation gave a white solid.
- Example 31 (66 mg, 0.0691 mmol) and iodomethane (2 ml) were dissolved in IMS (6 ml) and the solution was allowed to stand at RT for 24 h. The volatiles were evaporated and the residue was dissolved in 2M NH 3 in EtOH (5 ml). The solution was heated at 50 3 C for 2 h, after which time the solvent was evaporated and the product was purified using HPLC (System 1). Example 32 was obtained as a white solid.
- Example 33 A solution of Example 33 (190 mg, 0.189 mmol) in MeOH (10 ml) was treated with a solution of potassium carbonate (261 mg, 1.89 mmol) in water (4 ml). After stirring for 30 min the solution was diluted with water (70 ml) and extracted with EtOAc (100 ml). The organic extract was dried (Na 2 SO 4 ) and evaporated. Purification was achieved using HPLC (System 1) giving a white solid.
- Example 35 was prepared from Intermediate 4 and Intermediate 12 by a method similar to that used in the synthesis of Example 33. Yield: 70% LC-MS (Method 4): Rt 3.61 min, m/z 1036 [MH]+ Example 36
- Example 36 was synthesized from Example 35 using a procedure similar to that used in the preparation of Example 34. Purification was achieved using HPLC (System 1).
- Example 6 (131 mg, 0.144 mmol) was dissolved in DCM (15 ml) and iodomethane (4 ml) was added. The reaction mixture was allowed to stand for 65 h before evaporation of the volatile materials. The mixture was purified using HPLC (System 1).
- Example 38 was synthesized from Example 8 using a procedure similar to that used in the preparation of Example 37.
- Assays were performed in 96-well plates at a total assay volume of 100 ⁇ l.
- the final concentration of the enzyme human leukocyte elastase, Sigma E8140
- a peptide substrate (MeO-Suc-Ala-Ala-Pro-ValAMC, Calbiochem #324745) was used, at the final concentration of 100 ⁇ M.
- the final concentration of DMSO was 1 % in the assay buffer (0.05M Tris.HCI, pH 7.5, 0.1 M NaCI; 0.1 M CaCI2; 0.0005% brij-35).
- the enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT and after 30mins stopped by adding 50 ⁇ l soybean trypsin inhibitor (Sigma T-9003) at a final concentration of 50 ⁇ g/well. Fluorescence was read on the FLEXstation (Molecular Devices) using 380 nm excitation and 460 nm emission filters. The potency of the compounds was determined from a concentration series of 10 concentrations in range from 1000 nM to 0.051 nM. The results are means of two independent experiments, each performed in duplicate.
- the enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT and read after 120 minutes. Fluorescence was read on the
- FLEXstation (Molecular Devices) using 485 nm excitation and 530 nm emission filters.
- the potency of the compounds was determined from a concentration series of 10 concentrations in range from 2500OnM to 1 nM. The results are means of two independent experiments, each performed in duplicate.
- a common, generic substrate was used for all proteases: fluorescently labelled casein (Molecular Probes, E-6639), at the final concentration of 20 ⁇ g/ml (Cathepsin G and Chymotrypsin), 10 ⁇ g/ml (Plasmin and Thrombin) or 5 ⁇ g/ml (Proteinase 3 and Trypsin).
- the final concentration of the substrate was close to the respective K m values as determined for this substrate.
- the final concentration of DMSO was 5% in the assay buffer (0.05M Tris.HCI, pH 7.5, 0.1 M NaCI; 0.1 M CaCI2; 0.0005% brij-35). The enzymatic reaction was started by adding the enzyme.
- the enzymatic reaction was performed at RT for 60min. Fluorescence was read on the FLEXstation (Molecular Devices) using 589 nm excitation and 617 nm emission filters. The potency of the compounds was determined from a concentration series of 8 concentrations in range from 500 ⁇ M to 0.2 ⁇ M. The results are means of two independent experiments, each performed in duplicate.
- the compounds tested showed selectivities for a range of proteases fromi to >300 fold.
- Intracellular Elastase (controlled with lysed cell elastase) PMNs were isolated as described previously. PMNs were added to 96-well polypropylene plates and DMSO or compound added to each well to give 150 ⁇ l volume. The plate was incubated at 37°C for 30minutes. Cells were washed by centrifugation and lysed with HBSS containing 0.04% triton. Cell debris was pelleted and the supernatant transferred to a fresh pate, with compounds or DMSO. Fluorogenic AAPV substrate was added to all wells and the plate was read using a Spectramax Gemini Ex 380nm Em 460 for 30minutes at 37°C.
- Neutrophil Released Elastase Activity Assay Human, Mouse, Guinea Pig
- Guinea pigs were treated with an LPS aerosol. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN. Bronchoalveolar lavage fluid (BAL) was spun at 40Og for 10minutes and the cells resuspended in HBSS. 10 ⁇ M cytocholasin B was added to the cell suspension and incubated at 370C for 5 minutes after which 1 ⁇ M fMLP was added for a further 5 minutes. Cells were centrifuged at 40Og for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube.
- BAL Bronchoalveolar lavage fluid
- mice were anaesthetised and treated with LPS i.n. Animals were left for 4 hours, euthanized and the lungs lavaged to recover PMN. Bronchoalveolar lavage fluid (BAL) was centrifuged at 40Og for 10minutes and the cells resuspended in 1 ml of HBSS.10 ⁇ M cytocholasin B was added to the cell suspension and incubated at 370C for 5 minutes after which 1 ⁇ M fMLP was added for a further 5 minutes. Cells were centrifuged at 40Og for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. Generation of Human Released Neutrophil Elastase, from Humans
- Human PMN were isolated as described previously. 10 ⁇ M cytocholasin B was added to the cell suspension and incubated at 370C for 5 minutes after which 1 ⁇ M fMLP was added for a further 5 minutes. Cells were centrifuged at 40Og for 10 minutes. 'Elastase rich supernatant' was transferred to a fresh tube. To a clear bottomed 96-well plate compounds were added and incubated for 5 minutes at 37)C with 'elastase rich' supernatant. Fluorogenic AAPV substrate was added to all wells and the plate read using a Spectramax Gemini Ex 380nm Em 460 for 30 minutes at 37°C. For comparison, an activity matched control of human elastase was also run. HNE induced lung haemorrhage in the rat
- HNE human neutrophil elastase
- the vehicle used was 1 % DMSO/Saline. Inhibitors were dissolved in 1 % DMSO before the addition of 0.9% saline.
- HNE human neutrophil elastase
- Animals either received vehicle or compound at a dose volume of 0.5ml/kg. Animals that had been allowed to recover after dosing were terminally anaesthetised with hypnorm:hypnovel:water (1.5:1 :2 at 2.7ml/kg). Once sufficiently anaesthetised, HNE (600units/ml) or sterile saline was administered by transoral tracheal instillation at a volume of 100 ⁇ l using a Penn Century microsprayer. Animals were kept warm in a temperature controlled box and given top up doses of anaesthetic as required to ensure continuous anaesthesia until termination.
- HNE 600units/ml
- sterile saline was administered by transoral tracheal instillation at a volume of 100 ⁇ l using a Penn Century microsprayer. Animals were kept warm in a temperature controlled box and given top up doses of anaesthetic as required to ensure continuous anaesthesia until termination.
- the BALF was centrifuged at 1000 r.p.m. for 10 minutes in a centrifuge cooled to between 4 and 10oC. The supernatant was discarded and the cell pellet resuspended in 1 ml 0.1 % CETAB/PBS to lyse the cells. Cell lysates were frozen until spectrophotometric analysis for blood content could be made. Standards were prepared by making solutions of whole rat blood in 0.1% CETAB/PBS.
- a standard curve was constructed by measuring the OD (at 415nm) of different concentrations of blood in 0.1% CETAB/PBS (30, 10, 7, 3, 1 , 0.3, 0.1 ⁇ l/ml).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description



















































Claims






Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06755623A EP1893584A1 (en) | 2005-06-24 | 2006-06-26 | Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors |
| JP2008517602A JP2008543920A (en) | 2005-06-24 | 2006-06-26 | Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors |
| US11/993,699 US20090105268A1 (en) | 2005-06-24 | 2006-06-26 | Dihydropyrimidone Multimers and Their Use as Human Neutrophil, Elastase Inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0512940.8A GB0512940D0 (en) | 2005-06-24 | 2005-06-24 | Compounds and their use |
| GB0512940.8 | 2005-06-24 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006136857A1 true WO2006136857A1 (en) | 2006-12-28 |
Family
ID=34856116
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2006/002337 Ceased WO2006136857A1 (en) | 2005-06-24 | 2006-06-26 | Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20090105268A1 (en) |
| EP (1) | EP1893584A1 (en) |
| JP (1) | JP2008543920A (en) |
| CN (1) | CN101243055A (en) |
| GB (1) | GB0512940D0 (en) |
| WO (1) | WO2006136857A1 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008003412A1 (en) * | 2006-07-01 | 2008-01-10 | Bayer Healthcare Ag | The use of 1,4-diaryl-dihydropyrimidine-2-on derivatives for treating pulmonary arterial hypertension |
| WO2008030158A1 (en) * | 2006-09-04 | 2008-03-13 | Astrazeneca Ab | Multimeric heterocyclic compounds useful as neutrophil elastase inhibitors |
| WO2009060158A1 (en) * | 2007-11-07 | 2009-05-14 | Argenta Discovery Limited | 4- (4-cyanophenyl) -1- (3-trifluoromethylphenyl) -3,4, 6, 7-tetrahydro-1h-pyrrolo [3, 4- d] pyrimidine-2, 5-dione derivatives and their use as human neutrophil elastase inhibitors |
| DE102007061766A1 (en) | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | New 4-(4-cyano-2-thioaryl)-dihydro-pyrimidinone compounds are human neutrophil elastase inhibitor, useful for the treatment or prevention of e.g. pulmonary arterial hypertonia, acute lung injury and diseases of the cardiovascular system |
| DE102008022521A1 (en) | 2008-05-07 | 2009-11-12 | Bayer Schering Pharma Aktiengesellschaft | 1,4-Diaryl-pyrimidopyridazine-2,5-diones and their use |
| DE102008052013A1 (en) | 2008-10-17 | 2010-04-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-(4-cyano-2-thioaryl)dihydropyrimidinone compounds are neutrophil elastase inhibitors useful to treat or prevent e.g. pulmonary arterial hypertension, chronic obstructive pulmonary disease, acute lung injury, or cystic fibrosis |
| DE102009016553A1 (en) | 2009-04-06 | 2010-10-07 | Bayer Schering Pharma Aktiengesellschaft | Sulfonamide- and sulfoximine-substituted diaryldihydropyrimidinones and their use |
| US7998984B2 (en) | 2006-05-08 | 2011-08-16 | Astrazeneca Ab | 2-pyridone derivatives for the treatment of disease or condition in which inhibition of neutrophil elastase activity is beneficial |
| US8063073B2 (en) | 2003-09-18 | 2011-11-22 | Astrazeneca Ab | 2-pyridone derivatives as neutrophil elastase inhibitors and their use |
| DE102010030187A1 (en) | 2010-06-16 | 2011-12-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-cyano-2-sulfonylphenyl-pyrazolyl-substituted pyridinones and pyrazinones compounds are human neutrophil elastase inhibitors, useful to treat and prevent e.g. pulmonary arterial hypertension and chronic obstructive pulmonary disease |
| US8114881B2 (en) | 2006-05-08 | 2012-02-14 | Astrazeneca Ab | 2-pyrazinone derivatives for the treatment of disease or condition in which inhibition of neutrophil elastase activity is beneficial |
| US8232296B2 (en) | 2009-02-20 | 2012-07-31 | Astrazeneca Ab | Salt 628 |
| US8288402B2 (en) | 2007-12-20 | 2012-10-16 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and their use |
| US8436024B2 (en) | 2009-10-02 | 2013-05-07 | Astrazeneca Ab | 2-pyridone compounds |
| US8466284B2 (en) | 2007-11-06 | 2013-06-18 | Astra Zeneca Ab | Some 2-pyrazinone derivatives and their use as inhibitors of neutrophile elastase |
| WO2014122160A1 (en) | 2013-02-06 | 2014-08-14 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| WO2014135414A1 (en) | 2013-03-04 | 2014-09-12 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9290457B2 (en) | 2014-07-31 | 2016-03-22 | Boehringer Ingelheim International Gmbh | Substituted dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9440930B2 (en) | 2014-07-31 | 2016-09-13 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9458113B2 (en) | 2014-07-31 | 2016-10-04 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9475779B2 (en) | 2014-07-31 | 2016-10-25 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9657015B2 (en) | 2014-07-31 | 2017-05-23 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US10316001B2 (en) | 2015-03-18 | 2019-06-11 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrohydro pyrimidine-5-carbonitrile |
| USRE47493E1 (en) | 2014-02-20 | 2019-07-09 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| WO2021053058A1 (en) | 2019-09-17 | 2021-03-25 | Mereo Biopharma 4 Limited | Alvelestat for use in the treatment of graft rejection, bronchiolitis obliterans syndrome and graft versus host disease |
| WO2021209740A1 (en) | 2020-04-16 | 2021-10-21 | Mereo Biopharma 4 Limited | Methods involving neutrophil elastase inhibitor alvelestat for treating coronavirus infection |
| WO2023067103A1 (en) | 2021-10-20 | 2023-04-27 | Mereo Biopharma 4 Limited | Neutrophil elastase inhibitors for use in the treatment of fibrosis |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004024700A1 (en) * | 2002-09-10 | 2004-03-25 | Bayer Healthcare Ag | Pyrimidinone derivatives as therapeutic agents against acute and chronic inflammatory, ischaemic and remodelling processes |
-
2005
- 2005-06-24 GB GBGB0512940.8A patent/GB0512940D0/en not_active Ceased
-
2006
- 2006-06-26 JP JP2008517602A patent/JP2008543920A/en not_active Withdrawn
- 2006-06-26 CN CNA2006800303313A patent/CN101243055A/en active Pending
- 2006-06-26 WO PCT/GB2006/002337 patent/WO2006136857A1/en not_active Ceased
- 2006-06-26 EP EP06755623A patent/EP1893584A1/en not_active Withdrawn
- 2006-06-26 US US11/993,699 patent/US20090105268A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004024700A1 (en) * | 2002-09-10 | 2004-03-25 | Bayer Healthcare Ag | Pyrimidinone derivatives as therapeutic agents against acute and chronic inflammatory, ischaemic and remodelling processes |
Non-Patent Citations (1)
| Title |
|---|
| HANDL H L ET AL: "Hitting multiple targets with multimeric ligands", EXPERT OPINION ON THERAPEUTIC TARGETS, vol. 8, no. 6, 2004, pages 565 - 586, XP009069513 * |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8501784B2 (en) | 2003-09-18 | 2013-08-06 | Astrazeneca Ab | 2-pyridone derivatives as neutrophil elastase inhibitors and their use |
| US8063073B2 (en) | 2003-09-18 | 2011-11-22 | Astrazeneca Ab | 2-pyridone derivatives as neutrophil elastase inhibitors and their use |
| US7998984B2 (en) | 2006-05-08 | 2011-08-16 | Astrazeneca Ab | 2-pyridone derivatives for the treatment of disease or condition in which inhibition of neutrophil elastase activity is beneficial |
| US8114881B2 (en) | 2006-05-08 | 2012-02-14 | Astrazeneca Ab | 2-pyrazinone derivatives for the treatment of disease or condition in which inhibition of neutrophil elastase activity is beneficial |
| WO2008003412A1 (en) * | 2006-07-01 | 2008-01-10 | Bayer Healthcare Ag | The use of 1,4-diaryl-dihydropyrimidine-2-on derivatives for treating pulmonary arterial hypertension |
| WO2008030158A1 (en) * | 2006-09-04 | 2008-03-13 | Astrazeneca Ab | Multimeric heterocyclic compounds useful as neutrophil elastase inhibitors |
| US8466284B2 (en) | 2007-11-06 | 2013-06-18 | Astra Zeneca Ab | Some 2-pyrazinone derivatives and their use as inhibitors of neutrophile elastase |
| WO2009060158A1 (en) * | 2007-11-07 | 2009-05-14 | Argenta Discovery Limited | 4- (4-cyanophenyl) -1- (3-trifluoromethylphenyl) -3,4, 6, 7-tetrahydro-1h-pyrrolo [3, 4- d] pyrimidine-2, 5-dione derivatives and their use as human neutrophil elastase inhibitors |
| DE102007061766A1 (en) | 2007-12-20 | 2009-06-25 | Bayer Healthcare Ag | New 4-(4-cyano-2-thioaryl)-dihydro-pyrimidinone compounds are human neutrophil elastase inhibitor, useful for the treatment or prevention of e.g. pulmonary arterial hypertonia, acute lung injury and diseases of the cardiovascular system |
| US9174997B2 (en) | 2007-12-20 | 2015-11-03 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and use thereof |
| US8288402B2 (en) | 2007-12-20 | 2012-10-16 | Bayer Intellectual Property Gmbh | 4-(4-cyano-2-thioaryl)dihydropyrimidinones and their use |
| US8580800B2 (en) | 2008-05-07 | 2013-11-12 | Bayer Intellectual Property Gmbh | 1,4-diaryl-pyrimidopyridazine-2,5-diones and their use |
| DE102008022521A1 (en) | 2008-05-07 | 2009-11-12 | Bayer Schering Pharma Aktiengesellschaft | 1,4-Diaryl-pyrimidopyridazine-2,5-diones and their use |
| DE102008052013A1 (en) | 2008-10-17 | 2010-04-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-(4-cyano-2-thioaryl)dihydropyrimidinone compounds are neutrophil elastase inhibitors useful to treat or prevent e.g. pulmonary arterial hypertension, chronic obstructive pulmonary disease, acute lung injury, or cystic fibrosis |
| US8232296B2 (en) | 2009-02-20 | 2012-07-31 | Astrazeneca Ab | Salt 628 |
| US8691817B2 (en) | 2009-04-06 | 2014-04-08 | Bayer Intellectual Property Gmbh | Sulfonic amide and sulfoximine-substituted diaryl-dihydropyrimidinones and usage thereof |
| WO2010115548A1 (en) | 2009-04-06 | 2010-10-14 | Bayer Schering Pharma Aktiengesellschaft | Sulfonic amide and sulfoximine-substituted diaryl-dihydropyrimidinones and usage thereof |
| DE102009016553A1 (en) | 2009-04-06 | 2010-10-07 | Bayer Schering Pharma Aktiengesellschaft | Sulfonamide- and sulfoximine-substituted diaryldihydropyrimidinones and their use |
| US8436024B2 (en) | 2009-10-02 | 2013-05-07 | Astrazeneca Ab | 2-pyridone compounds |
| DE102010030187A1 (en) | 2010-06-16 | 2011-12-22 | Bayer Schering Pharma Aktiengesellschaft | New 4-cyano-2-sulfonylphenyl-pyrazolyl-substituted pyridinones and pyrazinones compounds are human neutrophil elastase inhibitors, useful to treat and prevent e.g. pulmonary arterial hypertension and chronic obstructive pulmonary disease |
| WO2014122160A1 (en) | 2013-02-06 | 2014-08-14 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| WO2014135414A1 (en) | 2013-03-04 | 2014-09-12 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9115093B2 (en) | 2013-03-04 | 2015-08-25 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| USRE47493E1 (en) | 2014-02-20 | 2019-07-09 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9475779B2 (en) | 2014-07-31 | 2016-10-25 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9458113B2 (en) | 2014-07-31 | 2016-10-04 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9440930B2 (en) | 2014-07-31 | 2016-09-13 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9657015B2 (en) | 2014-07-31 | 2017-05-23 | Boehringer Ingelheim International Gmbh | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US9290457B2 (en) | 2014-07-31 | 2016-03-22 | Boehringer Ingelheim International Gmbh | Substituted dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| EP3539952A1 (en) | 2014-07-31 | 2019-09-18 | Boehringer Ingelheim International GmbH | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| EP3604308A1 (en) | 2014-07-31 | 2020-02-05 | Boehringer Ingelheim International GmbH | Substituted bicyclic dihydropyrimidinones and their use as inhibitors of neutrophil elastase activity |
| US10316001B2 (en) | 2015-03-18 | 2019-06-11 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrohydro pyrimidine-5-carbonitrile |
| US10676443B2 (en) | 2015-03-18 | 2020-06-09 | Ph Pharma Co., Ltd. | Method for producing (4S)-4-[4-cyano-2-(methylsulfonyl)phenyl]-3,6-dimethyl-2-oxo-1-[3-(trifluoromethyl)phenyl]-1,2,3,4-tetrahydro pyrimidine-5-carbonitrile |
| WO2021053058A1 (en) | 2019-09-17 | 2021-03-25 | Mereo Biopharma 4 Limited | Alvelestat for use in the treatment of graft rejection, bronchiolitis obliterans syndrome and graft versus host disease |
| WO2021209740A1 (en) | 2020-04-16 | 2021-10-21 | Mereo Biopharma 4 Limited | Methods involving neutrophil elastase inhibitor alvelestat for treating coronavirus infection |
| WO2023067103A1 (en) | 2021-10-20 | 2023-04-27 | Mereo Biopharma 4 Limited | Neutrophil elastase inhibitors for use in the treatment of fibrosis |
Also Published As
| Publication number | Publication date |
|---|---|
| GB0512940D0 (en) | 2005-08-03 |
| EP1893584A1 (en) | 2008-03-05 |
| US20090105268A1 (en) | 2009-04-23 |
| CN101243055A (en) | 2008-08-13 |
| JP2008543920A (en) | 2008-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1856059B1 (en) | Multimers of pyrimidinone derivatives and their use as human neutrophil elastase inhibitors | |
| WO2006136857A1 (en) | Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors | |
| JP4999920B2 (en) | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors | |
| CN102858774B (en) | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as COPD | |
| AU2011225903B2 (en) | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as COPD | |
| US9023855B2 (en) | Compounds | |
| US8198288B2 (en) | Tetrahydropyrrolopyrimidinediones and their use in therapy | |
| WO2007107706A2 (en) | Dimers of heterocyclic compounds for the treatment of copd | |
| WO2009013444A1 (en) | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors | |
| WO2009037413A1 (en) | Dimers of 5- [ (4-cyanophenyl) sulfinyl] -6-methyl-2-oxo-1- [3- (trifluoromethyl)phenyl] -1,2-dihydropyridine-3-carboxamide as inhibitors of human neutrophil elastase for treating respiratory diseases | |
| WO2009060158A1 (en) | 4- (4-cyanophenyl) -1- (3-trifluoromethylphenyl) -3,4, 6, 7-tetrahydro-1h-pyrrolo [3, 4- d] pyrimidine-2, 5-dione derivatives and their use as human neutrophil elastase inhibitors | |
| WO2007042815A1 (en) | Compounds containing more than one human neutrophil elastase inhibiting moiety for use in the treatment of respiratory diseases | |
| CN101479270B (en) | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors | |
| HK1120790A (en) | Dihydropyrimidone multimers and their use as human neutrophil elastase inhibitors | |
| HK1130794B (en) | Tetrahydropyrrolopyrimidinediones and their use as human neutrophil elastase inhibitors | |
| HK1180322B (en) | Pyrimidine derivatives and their use in the treatment of respiratory diseases such as copd | |
| HK1116773A (en) | Multimers of pyrimidinone derivatives and their use as human neutrophil elastase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008517602 Country of ref document: JP Ref document number: 2006755623 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10018/DELNP/2007 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680030331.3 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006755623 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11993699 Country of ref document: US |