WO2007000249A1 - Verfahren zur herstellung von substituierten halogenpyridinen - Google Patents
Verfahren zur herstellung von substituierten halogenpyridinen Download PDFInfo
- Publication number
- WO2007000249A1 WO2007000249A1 PCT/EP2006/005718 EP2006005718W WO2007000249A1 WO 2007000249 A1 WO2007000249 A1 WO 2007000249A1 EP 2006005718 W EP2006005718 W EP 2006005718W WO 2007000249 A1 WO2007000249 A1 WO 2007000249A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sub
- hydrogen
- reaction
- optionally substituted
- hydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- YKYIFUROKBDHCY-ONEGZZNKSA-N CCO/C=C/C(C(F)(F)F)=O Chemical compound CCO/C=C/C(C(F)(F)F)=O YKYIFUROKBDHCY-ONEGZZNKSA-N 0.000 description 1
- VVOYKYRKDGFJHQ-GQCTYLIASA-N CCO/C=C/C(CC#N)(C(F)(F)F)O Chemical compound CCO/C=C/C(CC#N)(C(F)(F)F)O VVOYKYRKDGFJHQ-GQCTYLIASA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/61—Halogen atoms or nitro radicals
Definitions
- 1, 3-dicarbonyl compounds and salts of substituted acetonitriles can be prepared.
- HX or substances / mixtures that produce HX (eg, with alcohols)
- Substituted pyridines are important substructures in a variety of chemical and pharmaceutical products. Especially attractive as
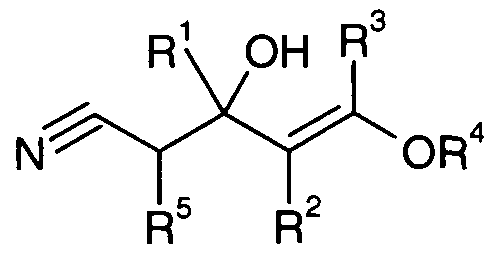
- the present invention solves this problem and relates to a process for preparing halogenopyridines (II) by reacting a ⁇ -hydroxy- ⁇ -acylbutyronitrile (I) or a suitable acyl-protected derivative with Hydrogen halides or substances or mixtures which can release hydrogen halides,
- R, R 4 is H, linear or branched alkyl radical, optionally substituted aryl radical, aralkyl radical, optionally substituted heteroaryl radical;
- R 1 , R 2 , R 3 is H, linear or branched alkyl radical, optionally substituted aryl, aralkyl, optionally substituted heteroaryl radical or one of the following radical C n H (2n + im) Xm, COOR, CN, where R 1 is in particular stands for a trifluoromethyl group;
- R 5 H, linear or branched alkyl radical, optionally substituted aryl radical, aralkyl, optionally substituted heteroaryl or one of the following radicals C n H ( 2n + im) Xm, COOR, CN 1 SO 2 R, SOR, PO (OR) 2 n : is a positive integer m: is a positive integer less than or equal to 2n + 1 X: is F, Cl, Br, I
- the required .beta.-hydroxy- ⁇ -acylbutyronitriles (I) can be conveniently generated under well reproducible conditions by reacting a 1,3-dicarbonyl compound (III) or a suitable monoprotected derivative with a metalated acetonitrile derivative (IV).
- M Li, Na, K, MgY o 1 Mg, 5, CaY, Ca o, 5, ZnY o 1 Zn, 5> CDY, Cd o, 5, Cu, AIY 2, TiY 3: This is the sought-halopyridine in accessible only 2 steps from the most easy to produce 1, 3-dicarbonyl compounds.
- first acetonitrile or a substituted derivative is metallated in a suitable solvent and the resulting salt (IV) then reacted with a 1, 3-dicarbonyl compound (IM) or a suitably monoprotected derivative.
- Suitable solvents for this reaction are all solvents which can be used for metallation reactions, in particular nonpolar, aprotic and protic solvents. These are especially ethers like Tetrahydrofuran, 2-methyltetrahydrofuran, diethyl ether, diisopropyl ether, di-n-butyl ether, dioxane, 1, 2-dimethoxyethane, Diethylengylcoldimethylether, Diethylenglcoldi-n-butyl ether, tetraethylene glycol dimethyl ether or mixtures of these solvents with each other or with an inert other solvent such as benzene, toluene , Xylo, cyciohexane or petroiethers
- Suitable metalating reagents are all bases which are sufficiently basic to abstract a hydrogen atom from the optionally substituted acetonitrile.
- bases which are sufficiently basic to abstract a hydrogen atom from the optionally substituted acetonitrile.
- acetonitrile itself or alkyl-substituted acetonitriles
- there are mainly very strong bases such as n-butyllithium, sec-butyllithium, t-butyllithium, n-hexyllithium, lithium N, N-diisopropylamide (LDA), lithium 2,2,6,6- tetramethylpiperidide (Li-TMP), lithium hexamethyldisilazane (LiHMDS), sodium hexamethyldisilazane (NaHMDS) or potassium hexamethyldisilazane (KHMDS).
- LDA lithium 2,2,6,6- tetramethylpiperidide
- LiHMDS lithium hex
- bases such as sodium amide, lithium hydride, sodium hydride or potassium hydride are suitable in addition to those mentioned above.
- alkoxides such as the lithium, sodium or potassium salts of methanol, Ethanol or t-butanol suitable as bases.
- reaction conditions to be complied with in the metallation depend on the acetonitriles used.
- R 5 alkyl or hydrogen
- the subsequent reaction with suitable 1,3-dicarbonyl compounds is best carried out at the same temperature as the metallation and is generally carried out by simple addition of the 1, 3-dicarbonyl compound (or a derivative) metallated acetonitrile derivative. However, the order of addition may be reversed.
- the reaction mixture is usually worked up by neutralizing the base it contains with a suitable acid (e.g., sulfuric acid, acetic acid, citric acid, hydrochloric acid) and removing the formed salt with water.
- a suitable acid e.g., sulfuric acid, acetic acid, citric acid, hydrochloric acid
- the resulting product is purified by conventional techniques such as distillation or crystallization or can often be used crude in the subsequent stage.
- the cyclization reaction of ⁇ -hydroxy- ⁇ -acylbutyronitriie to the halogenopyridines can be carried out either directly with hydrogen halides or with substances that form hydrogen halides with alcohols.
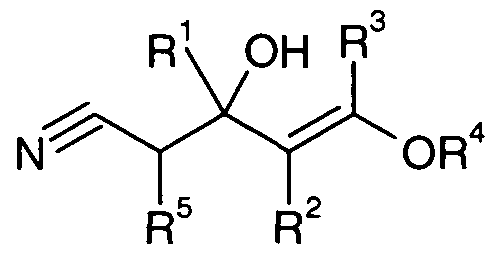
- R, R 4 hydrogen, alkyl, aryl, aralkyl, heteroaryl
- R 1 , R 2 , R 3 H, alkyl, aryl, aralkyl, heteroaryl, C n H (2 n + im) X m
- R 5 H, alkyl, aryl, aralkyl, heteroaryl, C n H (2 n + im) Xm, COOR, CN, SO 2 R, SOR, PO (oR) 2 n: positive integer number m: positive integer less than or equal to 2n +1
- X F, Cl, Br, I It is usually worked in a solvent when using HX.
- This solvent must be inert under the reaction conditions to the hydrogen halide used and should dissolve it sufficiently.
- Particularly suitable are, for example, acetic acid, acetic anhydride, dichloromethane, chloroform, carbon tetrachloride, 1, 2-dichloroethane or 1, 2-dibromoethane.
- Hydrogen halide is introduced into the reaction mixture in gaseous form under anhydrous conditions, forming the desired product directly.
- the required temperature depends, in addition to the substrate, above all on the hydrogen halide used.
- hydrogen bromide or iodine may generally be used at room temperature or slightly below room temperature, whereas the reaction with hydrogen chloride requires temperatures slightly above room temperature and usually does not start until 25 to 45 ° C.
- the gaseous hydrogen halides can advantageously be used in excess, as they are easy to remove after the reaction from the reaction mixture, without complicating the workup.
- at least 2 equivalents of hydrogen halide are preferably used, since 1 equivalent of the resulting pyridine can form a salt and the reaction is then no longer available.
- a second variant of the cyclization uses compounds as reagents capable of releasing hydrogen halides with alcohols. Suitable compounds are especially acid halides of inorganic acids, e.g. Thionyl chloride, sulfuryl chloride, phosphorus oxychloride, phosphorus trichloride, thionyl bromide, phosphoryl bromide or halides of organic acids such as
- Acetyl chloride, acetyl bromide, benzoyl chloride or benzoyl bromide The advantage of this process over that described above is that no gases have to be handled.
- the reactions are typically in the acid halide used as a solvent.
- the amount of acid halide is chosen so that the reaction can proceed completely and the mixture at the end of the reaction is still easy to stir. This generally requires at least one equivalent of acid halide, or preferably, 2 equivalents of acid halide are used. Larger amounts can also be used without negative effects on yield and product purity, but naturally complicate the work-up.
- the temperature depends on the used acid chloride and usually ranges from 0 to 130 0 C.
- thionyl chloride is, for example, preferably between 20 and 70 ° C worked while phosphorus oxychloride requires higher temperatures of 60 to 110 ° C to obtain a sufficiently rapid reaction to ensure.
- the workup of the reaction mixtures is carried out by aqueous quenching in a suitable pH range, which is mainly determined by the stability of the product. After quenching, the product is extracted with a suitable solvent and purified by distillation, chromatography or crystallization.
- the reaction of the 1, 3-dicarbonyl compound (III) with the metalated acetonitrile derivative (IV) and the subsequent reaction of the resulting ⁇ -hydroxy- ⁇ -acylbutyronitrils (I) with hydrogen halide HX or a substance or mixture that release hydrogen halides can be converted to a halopyridine (II) in a one-pot reaction.
- Step Example! 5 Preparation of 2-bromo-4- (trifluoromethyl) pyridine from 5-ethoxy-3-hydroxy-3- (trifluoromethyl) pent-4-enenitrile with HBr gas in dichloromethane
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pyridine Compounds (AREA)
Abstract
Description















Claims




Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0723334A GB2441915A (en) | 2005-06-27 | 2006-06-14 | Method for producing substituted halopyridines |
| US11/917,944 US20080214825A1 (en) | 2005-06-27 | 2006-06-14 | Method For Producing Substituted Halopyridines |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005030402.8A DE102005030402B4 (de) | 2005-06-27 | 2005-06-27 | Verfahren zur Herstellung von substituierten Halogenpyridinen |
| DE102005030402.8 | 2005-06-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007000249A1 true WO2007000249A1 (de) | 2007-01-04 |
Family
ID=37025232
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/005718 Ceased WO2007000249A1 (de) | 2005-06-27 | 2006-06-14 | Verfahren zur herstellung von substituierten halogenpyridinen |
Country Status (4)
| Country | Link |
|---|---|
| US (1) | US20080214825A1 (de) |
| DE (1) | DE102005030402B4 (de) |
| GB (1) | GB2441915A (de) |
| WO (1) | WO2007000249A1 (de) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2228366A1 (de) | 2009-03-12 | 2010-09-15 | Archimica GmbH | Verfahren zur Herstellung von 2-Amino-4-(halogenalkyl)pyridin-Derivaten durch Cyclisierung geeigneter Nitril-Vorstufen mit Stickstoff-Verbindungen |
| DE102009016374A1 (de) | 2009-04-07 | 2010-10-14 | Archimica Gmbh | Verfahren zur Herstellung von 2-Aminopyridin-4-(halogenalkyl)pyridin-Derivaten durch Cyclisierung geeigneter Nitril-Vorstufen mit Stickstoff-Verbindungen |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ES2939695T3 (es) * | 2018-12-27 | 2023-04-26 | Corteva Agriscience Llc | Preparación de intermedios de un proceso de herbicidas de sulfonamida |
| EP3902785A1 (de) * | 2018-12-27 | 2021-11-03 | Corteva Agriscience LLC | Herstellung von zwischenprodukten eines sulfonamid-herbizid-prozesses |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4321387A (en) * | 1980-03-21 | 1982-03-23 | Philip Morris, Incorporated | Process for the preparation of optically active nicotine analogs |
-
2005
- 2005-06-27 DE DE102005030402.8A patent/DE102005030402B4/de not_active Expired - Fee Related
-
2006
- 2006-06-14 US US11/917,944 patent/US20080214825A1/en not_active Abandoned
- 2006-06-14 WO PCT/EP2006/005718 patent/WO2007000249A1/de not_active Ceased
- 2006-06-14 GB GB0723334A patent/GB2441915A/en not_active Withdrawn
Non-Patent Citations (2)
| Title |
|---|
| F. COTTET, M. SCHLOSSER: "Trifluoro-Substituted Pyridines Through Displacement of Iodine by in situ Generated (Trifluoromethyl)copper", EUR. J. ORG. CHEM., 2002, pages 327 - 330, XP002401287 * |
| JIANG B ET AL: "CONVENIENT APPROACHES TO 4-TRIFLUOROMETHYLPYRIDINE", ORGANIC PROCESS RESEARCH AND DEVELOPMENT, CAMBRIDGE, GB, vol. 5, no. 5, 2001, pages 531 - 534, XP002357699 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2228366A1 (de) | 2009-03-12 | 2010-09-15 | Archimica GmbH | Verfahren zur Herstellung von 2-Amino-4-(halogenalkyl)pyridin-Derivaten durch Cyclisierung geeigneter Nitril-Vorstufen mit Stickstoff-Verbindungen |
| US8063226B2 (en) | 2009-03-12 | 2011-11-22 | Archimica Gmbh | Process for preparing 2-amino-4-(haloalkyl) pyridine derivatives by cyclizing suitable nitrile precursors with nitrogen compounds |
| DE102009016374A1 (de) | 2009-04-07 | 2010-10-14 | Archimica Gmbh | Verfahren zur Herstellung von 2-Aminopyridin-4-(halogenalkyl)pyridin-Derivaten durch Cyclisierung geeigneter Nitril-Vorstufen mit Stickstoff-Verbindungen |
Also Published As
| Publication number | Publication date |
|---|---|
| US20080214825A1 (en) | 2008-09-04 |
| DE102005030402B4 (de) | 2015-09-03 |
| GB2441915A (en) | 2008-03-19 |
| DE102005030402A1 (de) | 2006-12-28 |
| GB0723334D0 (en) | 2008-01-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE10109027A1 (de) | Rhodium- und Iridium-Komplexe | |
| WO1999058534A2 (de) | Epithilonderivate, verfahren zu deren herstellung und deren verwendung | |
| EP1480934A2 (de) | Herstellung von alkenonen | |
| DE102005030402B4 (de) | Verfahren zur Herstellung von substituierten Halogenpyridinen | |
| EP2228366B1 (de) | Verfahren zur Herstellung von 2-Amino-4-(halogenalkyl)pyridin-Derivaten durch Cyclisierung geeigneter Nitril-Vorstufen mit Stickstoff-Verbindungen | |
| EP0553778B1 (de) | Asymmetrische Hydrierung | |
| DE102007051694A1 (de) | Kupfer-Sauerstoff-Adduktkomplexe | |
| DE102018105179B4 (de) | Organometallischer Iridiumkomplex, synthetisches Verfahren dafür und organische, lichtemittierende Vorrichtung, die ihn verwendet | |
| DE60022684T2 (de) | Verfahren zur Herstellung von 6-Methyl-2-(4-methyl-phenyl)-imidazo [1,2-A]pyridin-3-(N,N-dimethyl-acetamid) und Zwischenprodukte | |
| DE10155064A1 (de) | Rhodium- und Iridium-Komplexe | |
| AT392784B (de) | Verfahren zur herstellung von derivaten der 2-thiophenessigsaeure | |
| WO2018007224A1 (de) | Verfahren zur herstellung halogenierter pyridinderivate | |
| EP0299277A2 (de) | Verfahren zur Herstellung von substituierten Pyridylalkylketonen | |
| CA1076133A (en) | Process for the preparation of esters | |
| WO2005097749A1 (de) | Alkylpyridiniumdicyanamide und verfahren zu ihrer herstellung | |
| DE3723069A1 (de) | 5-halogen-6-amino-nikotinsaeurehalogenide, ihre herstellung und ihre verwendung | |
| DE3531004C2 (de) | ||
| DE3873920T2 (de) | Oligopyridinliganden, verfahren zu ihrer herstellung und ihre verwendung als komplexbildnermittel. | |
| DE4340408A1 (de) | Derivate von Tricyclochinazolin und Verfahren zu ihrer Herstellung | |
| DE69127238T2 (de) | Verfahren zur Herstellung von Pyrido[1,2-a]pyrimidinderivaten | |
| WO1998043958A1 (de) | Verfahren zur herstellung von 2,6-dichlor-5-fluornicotinonitril und die chemische verbindung 3-cyano-2-hydroxy-5-fluorpyrid-6-on-mononatriumsalz sowie dessen tautomere | |
| AT413538B (de) | Verfahren zur herstellung von substituierten pyridin-n-oxid-verbindungen | |
| DE2604248C2 (de) | Verfahren zur Herstellung von Thienopyridinderivaten | |
| DE3534827A1 (de) | Verfahren zur herstellung von 5-(2-chlorobenzyl)-4,5,6,7-tetrahydrothieno(3,2-c)pyridin | |
| DE202024103926U1 (de) | Zusammensetzung und System zur Synthese von Zirkonocen-Dichlorid-Katalysator zur Gewinnung von Indiacen A und Indiacen B |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 0723334 Country of ref document: GB Kind code of ref document: A Free format text: PCT FILING DATE = 20060614 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 723334 Country of ref document: GB Ref document number: 0723334.9 Country of ref document: GB |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11917944 Country of ref document: US |
|
| REG | Reference to national code |
Ref country code: GB Ref legal event code: 789A Ref document number: 0723334 Country of ref document: GB |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06754360 Country of ref document: EP Kind code of ref document: A1 |