WO2007009913A1 - Pyridazinone derivatives as thyroid hormone receptor agonists - Google Patents
Pyridazinone derivatives as thyroid hormone receptor agonists Download PDFInfo
- Publication number
- WO2007009913A1 WO2007009913A1 PCT/EP2006/064093 EP2006064093W WO2007009913A1 WO 2007009913 A1 WO2007009913 A1 WO 2007009913A1 EP 2006064093 W EP2006064093 W EP 2006064093W WO 2007009913 A1 WO2007009913 A1 WO 2007009913A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- isopropyl
- phenyl
- dichloro
- pyridazin
- dihydro
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- ZKWUUZVSYHNCBF-UHFFFAOYSA-N CC(C)C1=CC(Cc(c(Cl)cc(N(C(NC2=O)=O)N=C2C#N)c2)c2Cl)=NNC1=O Chemical compound CC(C)C1=CC(Cc(c(Cl)cc(N(C(NC2=O)=O)N=C2C#N)c2)c2Cl)=NNC1=O ZKWUUZVSYHNCBF-UHFFFAOYSA-N 0.000 description 1
- AOXRZYHUTQBXCN-UHFFFAOYSA-N CC(C)C1=CC(Oc(c(Br)cc(CC(O)=O)c2)c2Br)=NNC1=O Chemical compound CC(C)C1=CC(Oc(c(Br)cc(CC(O)=O)c2)c2Br)=NNC1=O AOXRZYHUTQBXCN-UHFFFAOYSA-N 0.000 description 1
- JXYIMOHNAWTHCT-UHFFFAOYSA-N CC(C)C1=CC(S(c(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)(=O)=O)=NNC1=O Chemical compound CC(C)C1=CC(S(c(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)(=O)=O)=NNC1=O JXYIMOHNAWTHCT-UHFFFAOYSA-N 0.000 description 1
- PXGYZTQOCWWNLS-UHFFFAOYSA-N CC(C)C1=CC(S(c(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)=O)=NNC1=O Chemical compound CC(C)C1=CC(S(c(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)=O)=NNC1=O PXGYZTQOCWWNLS-UHFFFAOYSA-N 0.000 description 1
- FLCRQFBWKJIFKP-UHFFFAOYSA-N CC(C)C1=CC(S(c(c(Cl)cc(N(C(N2)=O)N=CC2=O)c2)c2Cl)(=O)=O)=NNC1=O Chemical compound CC(C)C1=CC(S(c(c(Cl)cc(N(C(N2)=O)N=CC2=O)c2)c2Cl)(=O)=O)=NNC1=O FLCRQFBWKJIFKP-UHFFFAOYSA-N 0.000 description 1
- URNDNHCCRPRCLG-UHFFFAOYSA-N CC(C)C1=CC(Sc(c(Cl)cc(CC(O)=O)c2)c2Cl)=NNC1=O Chemical compound CC(C)C1=CC(Sc(c(Cl)cc(CC(O)=O)c2)c2Cl)=NNC1=O URNDNHCCRPRCLG-UHFFFAOYSA-N 0.000 description 1
- UUHLODQDOSQOPR-UHFFFAOYSA-N CC(C)C1=CC(Sc(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)=NNC1=O Chemical compound CC(C)C1=CC(Sc(c(Cl)cc(CCc2nnn[nH]2)c2)c2Cl)=NNC1=O UUHLODQDOSQOPR-UHFFFAOYSA-N 0.000 description 1
- FXYVROILAKLOPS-UHFFFAOYSA-N CC(C)N(C(C=C1)=O)N=C1Oc(c(Br)cc(CC(O)=O)c1)c1Br Chemical compound CC(C)N(C(C=C1)=O)N=C1Oc(c(Br)cc(CC(O)=O)c1)c1Br FXYVROILAKLOPS-UHFFFAOYSA-N 0.000 description 1
- JRZDBBCBIQEJLA-UHFFFAOYSA-N CC(C)c(cc(nn1)Cl)c1Cl Chemical compound CC(C)c(cc(nn1)Cl)c1Cl JRZDBBCBIQEJLA-UHFFFAOYSA-N 0.000 description 1
- OGEKBXXDAFYZNN-UHFFFAOYSA-N CC(C)c(cc(nn1)Oc(c(Br)cc(CCCO)c2)c2Br)c1Cl Chemical compound CC(C)c(cc(nn1)Oc(c(Br)cc(CCCO)c2)c2Br)c1Cl OGEKBXXDAFYZNN-UHFFFAOYSA-N 0.000 description 1
- 0 CN(C(N1)=O)N=C(*)C1=O Chemical compound CN(C(N1)=O)N=C(*)C1=O 0.000 description 1
- JCNGURJWAXKZOE-UHFFFAOYSA-N OCCCc1cc(Br)c(C=O)c(Br)c1 Chemical compound OCCCc1cc(Br)c(C=O)c(Br)c1 JCNGURJWAXKZOE-UHFFFAOYSA-N 0.000 description 1
- YCCILVSKPBXVIP-UHFFFAOYSA-N OCCc(cc1)ccc1O Chemical compound OCCc(cc1)ccc1O YCCILVSKPBXVIP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/50—Pyridazines; Hydrogenated pyridazines
- A61K31/501—Pyridazines; Hydrogenated pyridazines not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
- A61P5/16—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4 for decreasing, blocking or antagonising the activity of the thyroid hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/14—Oxygen atoms
- C07D237/16—Two oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/06—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members
- C07D237/10—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D237/18—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention is directed to novel thyroid receptor ligands, particularly to pyridazinone analogs of formula (I)
- A is O, CH 2 , S, SO or SO 2 ;
- X and Y are each independently selected from the group consisting of Br, Cl and -CH 3 ;
- R 1 is selected from the group consisting of
- Z is H, or -C ⁇ N
- R 2 is lower alkyl having from 1 to 4 C atoms
- R 3 is H or lower alkyl; n is 1 or 2 ; p is 1 or 2; or a pharmaceutically acceptable salt or ester thereof.
- the invention is also directed to methods of preparing such compounds which are useful for treating metabolic diseases such as obesity, hyperlipidemia, hypercholesterolemia and diabetes and may be useful for other disorders and diseases such as NASH (nonalcoholic steatohepatitis), liver steatosis, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases. All documents cited or relied upon below are expressly incorporated herein by reference.
- Thyroid hormones are critical for normal growth and development and for maintaining metabolic homeostasis (Paul M. Yen Physiological reviews, Vol. 81(3): pp. 1097-1126 (2001)). Circulating levels of thyroid hormones are tightly regulated by feedback mechanisms in the hypothalamus/pituitary/thyroid (HPT) axis. Thyroid dysfunction leading to hypothyroidism or hyperthyroidism clearly demonstrates that thyroid hormones exert profound effects on cardiac function, body weight, metabolism, metabolic rate, body temperature, cholesterol, bone, muscle and behavior.
- HPT hypothalamus/pituitary/thyroid
- Thyroid hormone is produced by the thyroid gland and secreted into circulation as two distinct forms, 3,5,3',5'-tetra-iodo-L-thyronine (T4) and 3,5,3'-tri-iodo-L-thyronine (T3). While T4 is the predominant form secreted by the thyroid gland, T3 is the more biologically active form. T4 is converted to T3 by tissue specific deiodinases in all tissues but predominantly in the liver and kidney. The biological activity of thyroid hormones is mediated by thyroid hormone receptors (TRs) (M.A. Lazar Endocrine Reviews, Vol. 14: pp. 348-399 (1993)) TRs belong to the superfamily known as nuclear receptors.
- TRs thyroid hormone receptors
- TRs form heterodimers with the retinoid receptor that act as ligand-inducible transcription factors.
- TRs have a ligand binding domain, a DNA binding domain, and an amino terminal domain, and regulate gene expression through interactions with DNA response elements and with various nuclear co-activators and co-repressors.
- the thyroid hormone receptors are derived from two separate genes, ⁇ and ⁇ . These distinct gene products produce multiple forms of their respective receptors through differential RNA processing.
- the major thyroid receptor isoforms are ⁇ l, ⁇ 2, ⁇ l and B2.
- Thyroid hormone receptors ⁇ l, Bl and B2 bind thyroid hormone. It has been shown that the thyroid hormone receptor subtypes can differ in their contribution to particular biological responses.
- TR ⁇ l plays an important role in regulating TRH (thyrotropin releasing hormone) and on regulating thyroid hormone actions in the liver.
- TRB2 plays an important role in the regulation of TSH (thyroid stimulating hormone) (Abel et. al. J. Clin. Invest., VoI 104: pp. 291-300 (1999)).
- TR ⁇ l plays an important role in regulating heart rate (B. Gloss et. al. Endocrinology, Vol. 142: pp. 544-550 (2001); C. Johansson et. al. Am. J. Physiol., Vol. 275: pp. R640-R646 (1998)).
- thyroid hormones may be therapeutically beneficial if adverse effects can be minimized or eliminated (Paul M. Yen Physiological Reviews, Vol. 81(3): pp. 1097- 1126 (2001); Paul Webb Expert Opin. Investig. Drugs, Vol. 13(5): pp. 489-500 (2004)).
- thyroid hormones increase metabolic rate, oxygen consumption and heat production and thereby reduce body weight. Reducing body weight will have a beneficial effect in obese patients by ameliorating the co-morbidities associated with obesity, and may also have a beneficial effect on glycemic control in obese patients with Type 2 diabetes.
- LDL serum low density lipoprotein
- hypothyroidism is associated with low total serum cholesterol, which is attributed to thyroid hormone increasing hepatic LDL receptor expression and stimulating the metabolism of cholesterol to bile acids (JJ. Abrams et. al. J. Lipid Res., Vol. 22: pp 323-38 (1981)).
- Hypothyroidism in turn, has been associated with hypercholesterolemia and thyroid hormone replacement therapy is known to lower total cholesterol (M. Aviram et. al. CUn. Biochem., Vol.
- Thyroid hormone has been shown in animal models to have the beneficial effect of increasing HDL cholesterol and improving the ratio LDL to HDL by increasing the expression of apo A-I, one of the major apolipoproteins of HDL (Gene C. Ness et. al. Biochemical Pharmacology, Vol. 56: pp. 121-129 (1998); GJ. Grover et. al. Endocrinology, Vol. 145: pp. 1656-1661 (2004); GJ. Grover et. al. Proc. Natl. Acad. Sci. USA, Vol.
- thyroid hormone mimetics may yield desirable reductions in body weight, lipids, cholesterol, and lipoproteins, with reduced impact on cardiovascular function or normal function of the hypothalamus/pituitary/thyroid axis (A.H. Underwood et al. Nature, Vol. 324: pp. 425-429
- thyroid hormone analogs which avoid the undesirable effects of hyperthyroidism and hypothyroidism while maintaining the beneficial effects of thyroid hormones would open new avenues of treatment for patients with metaboUc diseases such as obesity, hyperUpidemia, hypercholesterolemia, diabetes and other disorders and diseases such as liver steatosis and NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer, thyroid diseases, and related disorders and diseases.
- novel thyroid hormone mimetics such as, for example, novel pyridazinone thyroid hormone mimetics, that have the beneficial effects of thyroid hormone while avoiding the undesirable effects.
- alkyl means, for example, a branched or unbranched, cyclic or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl group which may be substituted or unsubstituted.
- the alkyl group is preferably C3 to Cn, more preferably C 4 to Ci 0 , more preferably C 4 to C 7 .
- the alkyl group is preferably Ci to Cio, more preferably Ci to C 6 , more preferably methyl, ethyl, propyl (n-propyl or isopropyl), butyl (n-butyl, isobutyl or tertiary-butyl) or pentyl (including n-pentyl and isopentyl), more preferably methyl.
- alkyl as used herein includes alkyl (branched or unbranched), substituted alkyl (branched or unbranched), alkenyl (branched or unbranched), substituted alkenyl (branched or unbranched), alkynyl (branched or unbranched), substituted alkynyl (branched or unbranched), cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, cycloalkynyl and substituted cycloalkynyl.
- lower alkyl means, for example, a branched or unbranched, cyclic or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl group wherein said cyclic lower alkyl group is C 5 , C 6 or C 7 , and wherein said acyclic lower alkyl group is C 1 , C 2 , C3, C 4 , C5, or C 6 , preferably from 1 to 4 carbon atoms.
- lower alkyl groups include methyl, ethyl, propyl (n-propyl or isopropyl), butyl (n-butyl, isobutyl or tertiary-butyl), pentyl and hexyl.
- lower alkyl as used herein includes, for example, lower alkyl (branched or unbranched), lower alkenyl (branched or unbranched), lower alkynyl (branched or unbranched), cycloloweralkyl, cycloloweralkenyl and cycloloweralkynyl.
- lower alkyl as used herein may be divalent, e.g., -lower alkyl-COOH.
- Acyclic, branched or unbranchen lower alkyl groups are preferred.
- lower alkyl has from 1 to 4 carbon atoms.
- a preferred R 2 is lower alkyl having 3 carbon atoms. More preferred is isopropyl.
- aryl means, for example, a substituted or unsubstituted carbocyclic aromatic group, such as phenyl or naphthyl, or a substituted or unsubstituted heteroaromatic group containing one or more, preferably one, heteroatom, such as pyridyl, pyrrolyl, furanyl, thienyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl pyrazolyl, imidazolyl, triazolyl, pyrimidinyl pyridazinyl, pyrazinyl, triazinyl, indolyl, indazolyl, quinolyl, quinazolyl, benzimidazolyl, benzothiazolyl, benzisoxazolyl and benzisothiazolyl.
- alkyl and aryl groups may be substituted or unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substituents present, preferably 2 substituents.
- Substituents may include, for example: carbon-containing groups such as alkyl, aryl, arylalkyl (e.g. substituted and unsubstituted phenyl, substituted and unsubstituted benzyl); halogen atoms and halogen-containing groups such as haloalkyl (e.g. trifluoro methyl); oxygen-containing groups such as alcohols (e.g. hydroxyl, hydroxyalkyl, aryl(hydroxyl)alkyl), ethers (e.g.
- alkoxy, aryloxy, alkoxyalkyl, aryloxyalkyl aldehydes (e.g. carboxaldehyde), ketones (e.g. alkylcarbonyl, alkylcarbonylalkyl, arylcarbonyl, arylalkylcarbonyl, arycarbonylalkyl), acids (e.g. carboxy, carboxyalkyl), acid derivatives such as esters (e.g. alkoxycarbonyl, alkoxycarbonylalkyl, alkylcarbonyloxy, alkylcarbonyloxyalkyl), amides (e.g.
- aminocarbonyl mono- or di-alkylaminocarbonyl, aminocarbonylalkyl, mono-or di-alkylaminocarbonylalkyl, arylaminocarbonyl
- carbamates e.g. alkoxycarbonylamino, arloxycarbonylamino, aminocarbonyloxy, mono-or di- alkylaminocarbonyloxy, arylminocarbonloxy
- ureas e.g. mono- or di- alkylaminocarbonylamino or arylaminocarbonylamino
- nitrogen-containing groups such as amines (e.g.
- the lower alkyl groups may be substituted or unsubstituted, preferably unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substitutents present, preferably 2 substituents.
- alkoxy means, for example, alkyl-O- and "alkoyl” means, for example, alkyl-CO-.
- Alkoxy substituent groups or alkoxy-containing substituent groups may be substituted by, for example, one or more alkyl groups.
- halogen means, for example, a fluorine, chlorine, bromine or iodine group, preferably a chlorine or bromine group, and more preferably a chlorine group.
- “Pharmaceutically acceptable,” such as pharmaceutically acceptable carrier, excipient, etc., means pharmacologically acceptable and substantially non-toxic to the subject to whom the particular compound is administered.
- “Pharmaceutically acceptable salt” refers to conventional acid-addition salts or base- addition salts that retain the biological effectiveness and properties of the compounds of formula I and are formed from suitable organic or inorganic acids or organic or inorganic bases.
- Sample acid-addition salts include those derived from inorganic acids such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, phosphoric acid and nitric acid, and those derived from organic acids such as p- toluenesulfonic acid, salicylic acid, methanesulfonic acid, oxalic acid, succinic acid, citric acid, malic acid, lactic acid, fumaric acid, and the like.
- Sample base-addition salts include those derived from ammonium, potassium, sodium and, quaternary ammonium hydroxides, such as for example, tetramethylammonium hydroxide.
- “Pharmaceutically acceptable ester” refers to a conventionally esterified compound of formula I having a carboxyl group, which esters retain the biological effectiveness and properties of the compounds of formula I and are cleaved in vivo (in the organism) to the corresponding active carboxylic acid.
- ester groups which are cleaved (in this case hydrolyzed) in vivo to the corresponding carboxylic acids are those in which the hydrogen is replaced with -lower alkyl which is optionally substituted, e.g., with heterocycle, cycloalkyl, etc.
- substituted lower alkyl esters are those in which -lower alkyl is substituted with pyrrolidine, piperidine, morpholine, N-methylpiperazine, etc.
- the group which is cleaved in vivo may be, for example, ethyl, morpholino ethyl, and diethylamino ethyl.
- -CONH 2 is also considered an ester, as the -NH 2 may be cleaved in vivo and replaced with a hydroxy group, to form the corresponding carboxylic acid.
- A is O, CH 2 , S, SO or SO 2 ;
- X and Y are each independently selected from the group consisting of Br, Cl and -CH 3 ;
- Z is H, or -C ⁇ N; R 2 is lower alkyl; R 3 is H or lower alkyl; n is 1, 2 or 3; p is 1 or 2; or a pharmaceutically acceptable salt or ester thereof.
- Compounds of formula (I) are individually preferred and physiologically acceptable salts thereof are individually preferred and pharmaceutically acceptable esters thereof are individually preferred, with the compounds of formula (I) being particularly preferred.
- the compounds of formula (I) can have one or more asymmetric C or S atoms and can therefore exist as an enantiomeric mixture, mixture of stereoisomers or as optically pure compounds.
- Preferred compounds of formula (I) as described above are those, wherein X and Y are each Br. Other preferred compounds are those, wherein X and Y are each Cl. Still other preferred compounds are those, wherein X and Y are each -CH 3 . Furthermore, it is preferred that X is Cl, and Y is -CH 3 .
- R 1 is selected from the group consisting of:
- R 1 is -(CH 2 ) n COOH.
- n is 1.
- Other preferred compounds are those, wherein R 1 is:
- Z is CN
- R 2 is lower alkyl having from 1 to 4 C atoms. More preferably, R 2 is lower alkyl having from 1 to 3 C atoms.
- R 3 is CH 3 .
- Preferred are compounds as defined above, which have an ester group corresponding to R 1 which is -NHC( O)COOR, wherein the ester group corresponding to said "R" is NH 2 .
- Particularly preferred compounds of formula (I) as defined above are those selected from the group consisting of [4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)-3,5-dimethyl-phenyl]-acetic acid,
- the compounds of general formula (I) in this invention may be derivatised at functional groups to provide derivatives which are capable of conversion back to the parent compound in vivo.
- the novel compounds of the present invention have been found to be thyroid hormone analogs.
- the compounds of the present invention can therefore be used in the treatment and/or prophylaxis of diseases which are modulated by thyroid hormone analogs, particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and may be useful for other diseases such as NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- NASH nonalcoholic steatohepatitis
- An obese patient is a human with a body mass index of 25 or greater.
- the invention therefore also relates to pharmaceutical compositions comprising a compound as defined above and a pharmaceutically acceptable carrier and/or adjuvant.
- the invention likewise embraces compounds as described above for use as therapeutically active substances, especially as therapeutically active substances for the treatment and/or prophylaxis of diseases which are modulated by thyroid hormone analogs, particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- thyroid hormone analogs particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- the invention relates to a method for the therapeutic and/or prophylactic treatment of diseases which are modulated by thyroid hormone analogs, particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases, which method comprises administering a compound as defined above to a human being or animal.
- the amount of the compound administered is from about 0.01 mg/kg to about 50 mg/kg per day, more preferably from about 0.3 mg/kg to about 10 mg/kg per day, even more preferably from about 0.70 mg/kg to about 3.5 mg/kg per day.
- the invention also embraces the use of compounds as defined above for the therapeutic and/or prophylactic treatment of diseases which are modulated by thyroid hormone analogs, particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- metabolic diseases such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- the invention also relates to the use of compounds as described above for the preparation of medicaments for the therapeutic and/or prophylactic treatment of diseases which are modulated by thyroid hormone analogs, particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- thyroid hormone analogs particularly metabolic diseases, such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and NASH (nonalcoholic steatohepatitis), atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- Such medicaments comprise a compound as described above.
- Diabetes is more preferred, particularly non insulin dependent diabetes (type 2).
- Obesity also is more preferred.
- Hyperlipidemia also is more preferred, particularly hypercholesterolemia.
- Compound 6a was condensed with compound 7 using a base and a catalytic amount of an organometallic halide at elevated temperatures to afford 8a (see for example, Yuhpyng L. C, et.al. PCT Int. Appl. (1996) WO 9639388).
- Base hydrolysis of the methyl ester of 8a was performed via conventional procedures to afford compound 9a.
- Conversion of the chloropyridazine 9a to pyridizinone 10a was performed under acidic conditions (see for example, J. Chem. Soc. Perkin Trans. 1: Org. and Bioorg. Chem., 1988, 12, 3103-3111).
- Compound 10b was synthesized in the same manner as 10a starting from compound 6b.
- Compound 6b was synthesized in an analogous manner as 6a (scheme 1) starting from compound Ib.
- Compound 23 was synthesized following a series of reactions outlined in scheme 6. Bromination of 2-(4-hydroxyphenyl)ethanol to produce compound 20 was performed under conditions previously described. Compound 20 was condensed with compound 7 using potassium tert-butoxide in N,N-dimethylacetamide at high temperatures to produce compound 21. Compound 21 was oxidized to compound 22 by Jones oxidation (see for example, Bowden, K., et. al., J. Chem. Soc, 1946, 39-45). The chloropyridazine analog 22 was then converted to the pyridazinone 23 under conditions previously described.
- the 3,5- dichlorophenyl analog of 23, the 3,5-dimethylphenyl analog of 23, the 3-chloro-5- methylphenyl analog of 23, 3-bromo-5-methylphenyl analog of 23, and the 3-bromo-5- chlorophenyl analog of 23 can be synthesized in a similar manner.
- the 3,5-dibromophenyl analog of 26, the 3,5- dimethylphenyl analog of 26, the 3-chloro-5-methylphenyl analog of 26, 3-bromo-5- methylphenyl analog of 26, and the 3-bromo-5-chlorophenyl analog of 26 can be synthesized in a similar manner.
- Compound 29 was synthesized following a series of reactions outlined in scheme 8.
- Compound 24 was converted to compound 27 via acylation of the amine with methyl oxalyl chloride (see for example, Sellstedt. J.H., et. al, J. Med. Chem., 1975, 18(9), 926-933).
- Compound 27 was converted to compound 28 by hydrolysis of the ⁇ -keto ester to the OC- keto acid using standard conditions (see for example, Minisci, F., et. al., J. Org. Chem., 1995, 60, 5430-5433).
- the chloropyridazine 28 was converted to the pyridazinone 29 by the procedure described previously.
- the 3,5-dibromophenyl analog of 29, the 3,5- dimethylphenyl analog of 29, the 3-chloro-5-methylphenyl analog of 29, 3-bromo-5- methylphenyl analog of 29, and the 3-bromo-5-chlorophenyl analog of 29 can be synthesized in a similar manner.
- the 3,5-dimethylphenyl analog of 31, the 3-chloro-5-methylphenyl analog of 31, 3- bromo-5-methylphenyl analog of 31, and the 3-bromo-5-chlorophenyl analog of 31 can be synthesized in a similar manner.
- Compound 33 was synthesized following a series of reactions outlined in scheme 10.
- Compound 31 was converted to compound 32 by hydro lyzing the cyano group to a carboxylic acid via the procedure described by Carroll, R.D., et. al., J. Med. Chem., 1983, 26, 96-100.
- Compound 32 was then converted to compound 33 by decarboxylation following the procedure described by Carroll, R.D., et. al., J. Med. Chem., 1983, 26, 96- 100.
- the 3,5-dibromophenyl analog of 33, the 3,5-dimethylphenyl analog of 33, the 3- chloro-5-methylphenyl analog of 33, 3-bromo-5-methylphenyl analog of 33, and the 3- bromo-5-chlorophenyl analog of 33 can be synthesized in a similar manner.
- Compound 37 was synthesized following a series of reactions outlined in scheme 11.
- the chloropyridazine 17 was converted to the pyridazinone 34 by the procedure described previously.
- the pyridazinone 34 was alkylated using base and methyl iodide to produce compound 35 following the procedure similar to that described in J. Med. Chem., 1989, 32(10), 2381-2388.
- the acetate 35 was hydrolyzed to the alcohol 36 under standard basic conditions (see for example, Hauser, C. R., et. al., J. Amer. Chem. Soc, 1945, 67, 409- 412).
- Compound 36 was then oxidized to compound 37 by Jones oxidation.
- the 3,5- dibromophenyl analog of 37, the 3,5-dimethylphenyl analog of 37, the 3-chloro-5- methylphenyl analog of 37, 3-bromo-5-methylphenyl analog of 37, and the 3-bromo-5- chlorophenyl analog of 37 can be synthesized in a similar manner.
- Compound 48 was synthesized following a series of reactions outlined in scheme 13.
- the starting material, l,2,3-trichloro-5-nitrobenzene was converted to compound 43 by selectively displacing the chloro at the 2-position with the anion formed from tert-butyl cyanoacetate (see for example, Salturo, F., et. al., PCT WO 00/17204).
- the nitro group of compound 43 was reduced to an amine using standard conditions.
- the tert-butyl ester of 43 was hydrolyzed and decarboxylated to give compound 44 (see for example, Salturo, F., et. al., PCT WO 00/17204).
- the 3,5-dibromophenyl analog of 48, the 3,5-dimethylphenyl analog of 48, the 3-chloro-5-methylphenyl analog of 48, 3-bromo-5- methylphenyl analog of 48, and the 3-bromo-5-chlorophenyl analog of 48 can be synthesized in a similar manner.
- the bromide 51 was converted to the methyl ester 52 by palladium catalyzed carbonylation in methanol (see for example, Takatori, K., et. al., Tetrahedron, 1998, 54, 15861-15869).
- Compound 52 was reduced to compound 53 by standard reduction conditions, treatment with diisobutylaluminum hydride in tetrahydrofuran (see for example, Yoon, N.M., et. al., J. Org. Chem., 1985, 50, 2443-2450).
- the alcohol 53 was converted to bromide 54 using standard conditions (see for example, Lan, Aj. J. Y., et. al., J. Amer. Chem. Soc, 1987,
- the bromide 54 was displaced with sodium cyanide to produce nitrile 55 using the procedure described by Law, H., et. al. J. Med. Chem., 1998, 41, 2243-2251.
- the nitrile 55 was hydrolyzed to acid 56 by a conventional procedure to hydrolyze a nitrile to a carboxylic acid under aqueous acidic conditions (see for example, Wenner, O., Org. Synth.; CoU. Vol. IV, 1963, 760).
- the 3,5-dimethylphenyl analog of 56, the 3-chloro-5- methylphenyl analog of 56, 3-bromo-5-methylphenyl analog of 56, and the 3-bromo-5- chlorophenyl analog of 56 can be synthesized in a similar manner.
- Compound 64 was synthesized following a series of reactions outlined in scheme 16.
- the starting material methyl 3,5-dibromo-4-methylbenzoate, was brominated to produce compound 57 under standard bromination conditions (see for example, Buu-Hoy, .P., et. al., J. Org. Chem., 1953, 18, 649-652).
- Compound 57 was converted to compound 59 in two steps that were described previously.
- the alcohol 59 was converted to ether 60 by a conventional procedure to convert an alcohol to a tetrahydropyranyl ether (see for example, Miyashita, M., et. al., J. Org. Chem., 1977, 42, 3882-3774).
- the phthalimide protecting group of compound 66 was removed using butylamine in methanol at elevated temperatures to afford compound 67.
- Compound 67 was then converted to compound 68 using previously described methods.
- Compound 68 was converted to compound 69 using potassium acetate in N,N- dimethylacetamide at elevated temperatures.
- the 3,5-dibromophenyl analog of 69, the 3,5- dimethylphenyl analog of 69, the 3-chloro-5-methylphenyl analog of 69, 3-bromo-5- methylphenyl analog of 69, and the 3-bromo-5-chlorophenyl analog of 69 can be synthesized in a similar manner.
- Compound 76 was synthesized following a series of reactions outlined in scheme 18.
- the alcohol 16 was protected with a tert-butyldiphenylsilyl ether to produce compound 70 following a standard procedure as described in Chaudhary, S. K., et. al., Tet. Lett., 1979, 20(2), 99-102.
- the phenol 70 was converted to the thiophenol 73 using a three step procedure as described by D.M. Springer, et. al., Bioorg Med. Chem., 2003, 11, 265-279. Condensation of compound 7 and compound 73 was accomplished with potassium carbonate in dimethyl sulfoxide at high temperatures to produce compound 74.
- Compound 74 was converted to the pyridizinone acid 76 in two steps that have been previously described.
- the 3,5-dibromophenyl analog of 76, the 3,5-dimethylphenyl analog of 76, the 3-chloro-5-methylphenyl analog of 76, 3-bromo-5-methylphenyl analog of 76, and the 3- bromo-5-chlorophenyl analog of 76 can be synthesized in a similar manner.
- the 3,5-dibromophenyl analog of 77 and 78, the 3,5- dimethylphenyl analog of 77 and 78, the 3-chloro-5-methylphenyl analog of 77 and 78, 3- bromo-5-methylphenyl analog of 77 and 78, and the 3-bromo-5-chlorophenyl analog of 77 and 78 can be synthesized in a similar manner.
- the 3,5- dibromophenyl analog of 88, the 3,5-dimethylphenyl analog of 88, the 3-chloro-5- methylphenyl analog of 88, 3-bromo-5-methylphenyl analog of 88, and the 3-bromo-5- chlorophenyl analog of 88 can be synthesized in a similar manner.
- Compound 96 was synthesized following a series of reactions outlined in scheme 22.
- the amine 46 was treated with phthalic anhydride under acidic conditions at elevated temperatures to form the phthalimide 92 (see for example, Vera, L.M.S., et. al., Farmaco, 2003, 58(12), 1283-1288).
- the pyridizinone nitrogen of compound 92 was methylated under the conditions outlined in Sotelo, E., et. al., Synth. Commun., 2002, 32(11), 1675- 1680.
- the phthalimide protecting group of compound 93 was removed under acidic conditions at elevated temperatures to afford compound 94.
- Compound 94 was converted to compound 96 under conditions that have been previously described.
- the 3,5- dibromophenyl analog of 96, the 3,5-dimethylphenyl analog of 96, the 3-chloro-5- methylphenyl analog of 96, 3-bromo-5-methylphenyl analog of 96, and the 3-bromo-5- chlorophenyl analog of 96 can be synthesized in a similar manner.
- the alcohol 17 may be converted to bromide 97 using standard conditions (see for example, Lan, Aj. J. Y., et. al., J. Amer. Chem. Soc, 1987, 109, 2738-2745).
- the bromide of compound 97 can be displaced with sodium cyanide to produce nitrile 98 using a procedure similar to that of Law, H., et. al. J. Med. Chem., 1998, 41, 2243-2251.
- Nitriles such as 98 can be converted to tetrazoles like 98 using known procedures, for example by treatment of 98 with ammonium chloride and sodium azide at elevated temperature (see for example, Synthesis, 1998, 6, 910-914).
- Conversion of the chloropyridazine 99 to pyridizinone 100 may be accomplished under acidic conditions (see for example, J. Chem. Soc. Perkin Trans. 1: Org. and Bioorg. Chem., 1988, 12, 3103-3111).
- the 3,5-dibromophenyl analog of 100, the 3,5-dimethylphenyl analog of 100, the 3-chloro-5-methylphenyl analog of 100, 3- bromo-5-methylphenyl analog of 100, and the 3-bromo-5-chlorophenyl analog of 100 can be synthesized in a similar manner.
- ester of methyl 3,5-dichloro-4-hydroxybenzoate can be reduced to afford alcohol 101 using a reducing agent such as lithium aluminum hydride at low temperature (see for example, J. Org. Chem., 1998, 63, 5658-5661).
- a reducing agent such as lithium aluminum hydride at low temperature
- Compound 101 may then be condensed with compound 7 using a base at elevated temperatures to afford 102 (see for example,
- the alcohol 102 can be converted to compound 106 using conditions previously described.
- the bromide 54 may be homologated to ester 107 using conditions as illustrated in Can. J. Chem. 2001, 79(5-6), 752-759.
- Esters such as 107 can be converted to alcohols like 108 using known procedures, for example by treatment of 107 with diisobutylammonium hydride in tetrahydrofuran (see for example, Yoon, N.M., et.al., J. Org. Chem., 1985, 50, 2443-2450). Conversion of the alcohol 108 to tetrazole 111 may be accomplished under conditions as previously described.
- the 3,5-dibromophenyl analog of 111, the 3,5- dimethylphenyl analog of 111, the 3-chloro-5-methylphenyl analog of 111, 3-bromo-5- methylphenyl analog of 111, and the 3-bromo-5-chlorophenyl analog of 111 can be synthesized in a similar manner.
- Conversion of the nitrile 55 to tetrazole 112 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 112, the 3,5-dimethylphenyl analog of 112, the 3-chloro-5-methylphenyl analog of 112, 3-bromo-5-methylphenyl analog of 112, and the 3-bromo-5-chlorophenyl analog of 112 can be synthesized in a similar manner.
- Conversion of compound 74 to compound 116 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 116, the 3,5-dimethylphenyl analog of 116, the 3-chloro-5-methylphenyl analog of 116, 3-bromo-5-methylphenyl analog of 116, and the 3-bromo-5-chlorophenyl analog of 116 can be synthesized in a similar manner.
- Conversion of compound 116 to compounds 117 and 118 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 117 and 118, the 3,5- dimethylphenyl analog of 117 and 118, the 3-chloro-5-methylphenyl analog of 117 and 118, 3-bromo-5-methylphenyl analog of 117 and 118, and the 3-bromo-5-chlorophenyl analog of 117 and 118 can be synthesized in a similar manner.
- Conversion of compound 101 to compound 127 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 127, the 3,5-dimethylphenyl analog of 127, the 3-chloro-5-methylphenyl analog of 127, 3-bromo-5-methylphenyl analog of 127, and the 3-bromo-5-chlorophenyl analog of 127 can be synthesized in a similar manner.
- Conversion of compound 127 to compounds 128 and 129 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 128 and 129, the 3,5- dimethylphenyl analog of 128 and 129, the 3-chloro-5-methylphenyl analog of 128 and 129, 3-bromo-5-methylphenyl analog of 128 and 129, and the 3-bromo-5-chlorophenyl analog of 128 and 129 can be synthesized in a similar manner.
- the starting material, 2,6-dichloro-benzene-l,4-diol may be treated with bro mo-acetic acid methyl ester and base at elevated temperatures to produce compound 130 using a procedure similar to that of J. Het. Chem., 1994, 31(6), 1439-43.
- Conversion of compound 130 to compound 131 may be accomplished using conditions as previously described.
- Hydrolysis of ester 131 to compound 132 may be accomplished using standard aqueous basic conditions.
- Conversion of compound 132 to compound 133 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 133, the 3,5- dimethylphenyl analog of 133, the 3-chloro-5-methylphenyl analog of 133, 3-bromo-5- methylphenyl analog of 133, and the 3-bromo-5-chlorophenyl analog of 133 can be synthesized in a similar manner.
- Compound 46 may be converted to compound 134 under similar conditions as illustrated in Gattermann, L. (1914), Dietechnisch des organischen Chemikers (12), 228. Conversion of compound 134 to compound 136 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 136, the 3,5-dimethylphenyl analog of 136, the 3-chloro-5-methylphenyl analog of 136, 3-bromo-5-methylphenyl analog of 136, and the 3-bromo-5-chlorophenyl analog of 136 can be synthesized in a similar manner.
- Conversion of compound 130 to compound 141 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 141, the 3,5-dimethylphenyl analog of 141, the 3-chloro-5-methylphenyl analog of 141, 3-bromo-5-methylphenyl analog of 141, and the 3-bromo-5-chlorophenyl analog of 141 can be synthesized in a similar manner.
- Conversion of compound 141 to compounds 142 and 143 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 142 and 143, the 3,5- dimethylphenyl analog of 142 and 143, the 3-chloro-5-methylphenyl analog of 142 and 143, 3-bromo-5-methylphenyl analog of 142 and 143, and the 3-bromo-5-chlorophenyl analog of 142 and 143 can be synthesized in a similar manner.
- Conversion of compound 94 to compound 149 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 149, the 3,5-dimethylphenyl analog of 149, the 3-chloro-5-methylphenyl analog of 149, 3-bromo-5-methylphenyl analog of 149, and the 3-bromo-5-chlorophenyl analog of 149 can be synthesized in a similar manner.
- Conversion of compound 85 to compound 154 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 154, the 3,5-dimethylphenyl analog of 154, the 3-chloro-5-methylphenyl analog of 154, 3-bromo-5-methylphenyl analog of 154, and the 3-bromo-5-chlorophenyl analog of 154 can be synthesized in a similar manner.
- Conversion of compound 154 to compounds 155 and 156 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 155 and 156, the 3,5- dimethylphenyl analog of 155 and 156, the 3-chloro-5-methylphenyl analog of 155 and 156, 3-bromo-5-methylphenyl analog of 155 and 156, and the 3-bromo-5-chlorophenyl analog of 155 and 156 can be synthesized in a similar manner.
- Conversion of compound 152 to compound 162 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 162, the 3,5-dimethylphenyl analog of 162, the 3-chloro-5-methylphenyl analog of 162, 3-bromo-5-methylphenyl analog of 162, and the 3-bromo-5-chlorophenyl analog of 162 can be synthesized in a similar manner.
- Conversion of compound 162 to compounds 163 and 164 may be accomplished using conditions as previously described.
- the 3,5-dibromophenyl analog of 163 and 164, the 3,5- dimethylphenyl analog of 163 and 164, the 3-chloro-5-methylphenyl analog of 163 and 164, 3-bromo-5-methylphenyl analog of 163 and 164, and the 3-bromo-5-chlorophenyl analog of 163 and 164 can be synthesized in a similar manner.
- an effective amount of any one of the compounds of this invention or a combination of any of the compounds of this invention or a pharmaceutically acceptable salt or ester thereof is administered via any of the usual and acceptable methods known in the art, either singly or in combination.
- the compounds or compositions can thus be administered orally (e.g., buccal cavity), sublingually, parenterally (e.g., intramuscularly, intravenously, or subcutaneously), rectally (e.g., by suppositories or washings), transdermally (e.g., skin electroporation) or by inhalation (e.g., by aerosol), and in the form or solid, liquid or gaseous dosages, including tablets and suspensions.
- buccal cavity e.g., buccal cavity
- parenterally e.g., intramuscularly, intravenously, or subcutaneously
- rectally e.g., by suppositories or washings
- transdermally e.g., skin electroporation
- the administration can be conducted in a single unit dosage form with continuous therapy or in a single dose therapy ad libitum.
- the therapeutic composition can also be in the form of an oil emulsion or dispersion in conjunction with a lipophilic salt such as pamoic acid, or in the form of a biodegradable sustained-release composition for subcutaneous or intramuscular administration.
- Useful pharmaceutical carriers for the preparation of the compositions hereof can be solids, liquids or gases; thus, the compositions can take the form of tablets, pills, capsules, suppositories, powders, enterically coated or other protected formulations (e.g. binding on ion-exchange resins or packaging in lipid-protein vesicles), sustained release formulations, solutions, suspensions, elixirs, aerosols, and the like.
- the carrier can be selected from the various oils including those of petroleum, animal, vegetable or synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, and the like.
- formulations for intravenous administration comprise sterile aqueous solutions of the active ingredient(s) which are prepared by dissolving solid active ingredient(s) in water to produce an aqueous solution, and rendering the solution sterile.
- Suitable pharmaceutical excipients include starch, cellulose, talc, glucose, lactose, talc, gelatin, malt, rice, flour, chalk, silica, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, dried skim milk, glycerol, propylene glycol, water, ethanol, and the like.
- the compositions may be subjected to conventional pharmaceutical additives such as preservatives, stabilizing agents, wetting or emulsifying agents, salts for adjusting osmotic pressure, buffers and the like.
- Suitable pharmaceutical carriers and their formulation are described in Remington's Pharmaceutical Sciences by E. W. Martin. Such compositions will, in any event, contain an effective amount of the active compound together with a suitable carrier so as to prepare the proper dosage form for proper administration to the recipient.
- the pharmaceutical preparations can also contain preserving agents, solubilizing agents, stabilizing agents, wetting agents, emulsifying agents, sweetening agents, coloring agents, flavoring agents, salts for varying the osmotic pressure, buffers, coating agents or antioxidants. They can also contain other therapeutically valuable substances, including additional active ingredients other than those of formula I.
- the compounds of the present invention are useful as medicaments for the treatment of metabolic diseases such as obesity, hyperlipidemia, hypercholesterolemia and diabetes, and may be useful for other diseases such as NASH, atherosclerosis, cardiovascular diseases, hypothyroidism, thyroid cancer and related disorders and diseases.
- An obese patient is a human with a body mass index of 25 or greater.
- the therapeutically effective amount or dosage of a compound according to this invention can vary within wide limits and may be determined in a manner known in the art. Such dosage will be adjusted to the individual requirements in each particular case including the specific compound(s) being administered, the route of administration, the condition being treated, as well as the patient being treated. In general, in the case of oral or parenteral administration to adult humans weighing approximately 70 kg, a daily dosage of from about 0.01 mg/kg to about 50 mg/kg should be appropriate, although the upper limit may be exceeded when indicated. The dosage is preferably from about 0.3 mg/kg to about 10 mg/kg per day. A preferred dosage may be from about 0.70 mg/kg to about 3.5 mg/kg per day. The daily dosage can be administered as a single dose or in divided doses, or for parenteral administration it may be given as continuous infusion.
- the compounds of formula (I) are thyroid hormone analogs.
- the TR/RXR/GRIP Assay was used to test compounds of formula (I), as shown in the Examples below.
- the tested compounds are thyroid hormone receptor agonists, having EC50 of 1000 ⁇ M or less, with a preferred EC50 of 100 ⁇ M or less.
- Step 4 Preparation of (3,5-dimethyl-4-hydroxy-phenyl)-acetic acid (5a)
- the reaction mixture was heated to reflux for 2 d.
- the reaction mixture was concentrated under vacuum.
- the resulting solid was diluted with water (100 mL) and extracted with ethyl acetate (2 x 75 mL). The organic layers were discarded.
- Step 5 Preparation of (3,5-dimethyl-4-hydroxy-phenyl)-acetic acid methyl ester (6a)
- a solution of (3,5-dimethyl-4-hydroxy-phenyl)-acetic acid (5a) (12.4 g, 0.069 mmol) in methanol (300 mL) was treated with concentrated sulfuric acid (6.25 mL).
- the resulting solution was heated to 70 0 C overnight. At this time, the reaction was cooled to room temperature. The reaction was concentrated under vacuum.
- the residue was diluted with ethyl acetate (700 mL) and was washed with water (2 x 250 mL). The organics were dried over magnesium sulfate, filtered and concentrated under vacuum to give an orange solid.
- Step 6 Preparation of 3,6-dichloro-4-isopropyl pyridazine (7)
- the organic layer was saved.
- the aqueous layer was extracted with ethyl acetate (2 x 500 mL).
- the combined organics were washed with water (I x 400 mL) and a saturated aqueous sodium chloride solution (1 x 400 mL), dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 7 Preparation of [4-(6-Chloro-5-isopropyl-pyridazin-3-yloxy)-3,5-dimethyl-phenyl]- acetic acid methyl ester (8a)
- the reaction was cooled to room temperature and was poured onto a IN aqueous hydrochloric acid solution (200 mL) and ice.
- the aqueous layer was diluted with water (100 mL) and extracted with ethyl acetate (2 x 500 mL).
- the water layer was extracted again with ethyl acetate (I x 500 mL).
- the combined organics were washed with a saturated aqueous sodium chloride solution, dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 8 Preparation of [4-(6-Chloro-5-isopropyl-pyridazin-3-yloxy)-3,5-dimethyl-phenyl]- acetic acid (9a)
- Step 9 Preparation of [4-(5-Isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)-3,5-dimethyl- phenyl] -acetic acid (10a)
- Step 1 Preparation of 2-Chloro-4-dimethylaminomethyl-6-methyl-phenol (2b)
- a solution of 2-chloro-6-methyl phenol (5.0 g, 0.035 mol) in ethanol (25 mL) at room temperature was treated with dimethyl amine (3.95 mL of a 40% solution of dimethylamine in water, 0.035 mol) followed by formaldehyde (2.85 mL of a 37% solution of formaldehyde in water, 0.035 mol).
- the reaction was heated to reflux for 24 h. At this time, the reaction mixture was cooled to room temperature and concentrated under vacuum.
- the water layer was extracted with ethyl acetate (30OmL).
- the ethyl acetate layer was discarded.
- the organics were washed with a saturated aqueous sodium chloride solution, dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting oil was dried under high vacuum overnight to afford 2-chloro-
- Step 2 Preparation of 2-CMoro-6-methyl-4-trimethylaminomethylphenol (3b)
- the reaction mixture was stirred at room temperature overnight. At this time, the resulting solids were filtered and washed with ether. The solids were dried under high vacuum to afford 2-chloro-6-methyl-4-trimethylaminomethylphenol (3b) as an off-white solid which was used as is for the next step.
- Step 4 Preparation of (3-Chloro-4-hydroxy-5-methyl-phenyl)-acetic acid (5b)
- a suspension of (3-chloro-4-hydroxy-5-methyl-phenyl)-acetonitrile (4b) (1.3 g, 0.0071 mol) in water (2.06 mL) was treated with ethylene glycol dimethyl ether (13.93 mL, 0.133 mol) followed by potassium hydroxide (2.8 g, 0.0071 mol).
- the reaction mixture was heated to reflux for 24 h. At this time, the reaction mixture was concentrated under vacuum.
- the resulting solid was diluted with water (100 mL) and extracted with ethyl acetate (2 x 50 mL). The organic layers were discarded.
- Step 5 Preparation of (3-Chloro-4-hydroxy-5-methyl-phenyl)-acetic acid methyl ester (6b)
- a solution of (3-chloro-4-hydroxy-5-methyl-phenyl)-acetic acid (5b) (1.4 g, 6.98 mmol) in methanol (60 mL) was treated with concentrated sulfuric acid (0.5 mL) at room temperature under argon.
- the reaction mixture was heated to 70 0 C for 24 h. At this time, the reaction mixture was concentrated under vacuum.
- the resulting oil was diluted with ethyl acetate (100 mL). The organics were washed with water (2 x 50 mL), dried with magnesium sulfate, filtered and concentrated.
- Step 6 Preparation of [3-Chloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-5-methyl- phenyl]- acetic acid methyl ester (8b)
- Step 7 Preparation of [3-Chloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-5-methyl- phenyl]- acetic acid (9b)
- Step 8 Preparation of [3-Chloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)-5- methyl-phenyl] -acetic acid (10b)
- the water layer was acidified by the addition of a 3N aqueous hydrochloric acid solution and was extracted with ethyl acetate.
- the organic layers were combined, washed with water and a saturated aqueous sodium chloride solution, dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting solid was dissolved in methylene chloride and methanol and then was absorbed onto silica.
- the preabsorbed solid was purified by column chromatography using silica gel eluted with 20% ethyl acetate in hexanes containing 2% glacial acetic acid. The desired fractions were concentrated as several separate batches and placed under high vacuum for 15 min.
- the solid was diluted with a 1:1 methylene chloride:hexanes solution.
- Step 2 Preparation of 2-[3,5-Dibromo-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- ethanol (12)
- the reaction was cooled to room temperature, diluted with water (350 mL) and extracted with tert-butyl methyl ether (I x 600 mL) followed by isopropyl acetate (I x 600 mL).
- the organic layers were combined and washed with a IN aqueous sodium hydroxide solution (1 x 256 mL) and water (2 x 200 mL).
- the organic layer was distilled to about 300 mL.
- the residue was treated with heptane (300 mL). The mixture was stirred under reflux for 30 min and then was cooled to room temperature.
- Step 3 Preparation of [3,5-Dibromo-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)- phenyl] -acetic acid (14)
- reaction mixture was cooled to room temperature, diluted with toluene (200 mL) and concentrated under vacuum two times.
- the resulting residue was diluted with tetrahydrofuran (223 mL) and isopropyl acetate (892 mL) and was washed with water (3 x 150 mL).
- the organic layer was separated and concentrated under vacuum to about 600 mL.
- Isopropyl acetate (180 mL) was added and the mixture was concentrated under vacuum to about 450 mL.
- Heptane 300 mL was added in portions over 10 min. The mixture was stirred under reflux for 15 min and then was cooled to room temperature.
- the solid was dissolved in ethyl acetate (500 mL) and washed with water (1 x 125 mL). The organic layer was separated, dried with magnesium sulfate, filtered and concentrated under vacuum to give a white solid. The solid was dried overnight under high vacuum to afford (3,5-dichloro-4-hydroxy-phenyl)-acetic acid methyl ester (15) (18.81 g) as a white solid. The filtrate containing water and toluene were separated. The toluene layer was washed with water (150 mL), dried with magnesium sulfate, filtered and concentrated under vacuum to give a pale yellow solid.
- the mixture was then filtered.
- the solids were rinsed with ethyl acetate (2 x 500 mL).
- the organic layer was separated.
- the aqueous layer was extracted with ethyl acetate (2 x 200 mL).
- the organic layers were combined and washed with a 0.1N aqueous hydrochloric acid solution, water, and a saturated aqueous sodium chloride solution.
- the organic layer was dried with magnesium sulfate, filtered and concentrated to afford the first crop of solid.
- the resulting solid was placed under high vacuum overnight.
- the organic layers were combined and washed with water and a saturated aqueous sodium chloride solution.
- the organic layer was dried with magnesium sulfate, filtered and concentrated.
- Step 3 Preparation of 2-[3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- ethanol (17)
- the reaction was cooled to room temperature, diluted with water (30 mL) and extracted with tert-butyl methyl ether (1 x 60 mL) followed by isopropyl acetate (1 x 60 mL).
- the organic layers were combined and were washed with a IN aqueous sodium hydroxide solution (1 x 25 mL) and water (2 x 25 mL).
- the organic layer was separated and distilled to a volume of approximately 30 mL. This solution was treated with heptane (30 mL). The mixture was stirred under reflux for 30 min and then cooled to room temperature.
- Step 4 Preparation of [3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- acetic acid (18)
- Step 5 Preparation of [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)- phenyl]- acetic acid (19) A mixture of glacial acetic acid (200 mL), sodium acetate (6.1 g, 74.4 mmol) and [3,5- dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]-acetic acid (18) (8.3 g, 21.96 mmol) was heated to 125°C for 24 h. The reaction mixture was cooled to room temperature and concentrated.
- the resulting residue was diluted with methylene chloride (200 mL) and was washed with water (150 mL). The organic layer was separated. Hexanes (3 x 200 mL) were added to the residue in portions and then subsequently concentrated under vacuum. The resulting semi- solid was diluted with a minimum amount of ether, scratched, and slurried.
- Step 1 Preparation of 2,6-Dibromo-4-(3-hydroxy-propyl)-phenol (20)
- a solution of 4-(3-hydroxy-propyl)-phenol (10 g, 65.7 mmol) in glacial acetic acid (73.8 mL) was treated with a solution of bromine (7.4 mL, 144.5 mmol) in glacial acetic acid (7.3 mL).
- the reaction mixture was stirred at room temperature overnight.
- the solvent was removed under vacuum.
- the resulting residue was diluted with toluene (37 mL) and the solvent was again concentrated under vacuum.
- the resulting residue was dissolved in tetrahydrofuran (46 mL) and a 4N aqueous sodium hydroxide solution (64 mL) was added followed by water (27.5 mL). The reaction was stirred at room temperature for 2.5 h and the pH was adjusted to 5 by the addition of concentrated hydrochloric acid (14 mL). The layers were separated (the bottom layer is product). The aqueous layer was extracted with methyl tert-butyl ether (3 x 100 mL). The organic layers were combined, dried with sodium sulfate, filtered and concentrated under vacuum. The resulting residue was purified by column chromatography using silica gel eluted with a 1:1 ethyl acetate:hexanes solution.
- Step 2 Preparation of 3-[3,5-Dibromo-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- propan-1-ol (21)
- the reaction was cooled to room temperature, diluted with water (180 mL) and extracted with ethyl acetate (3 x 100 mL). The organic layers were combined and were washed with a IN aqueous sodium hydroxide solution (1 x 150 mL), followed by a saturated aqueous sodium chloride solution (1 x 150 mL). The organic layer was dried with sodium sulfate, filtered and concentrated. The resulting residue was purified by flash chromatography (Biotage 40M) eluted with 15% ethyl acetate in hexanes, followed by 25% ethyl acetate in hexanes, followed by 50% ethyl acetate in hexanes.
- the solvent was decanted.
- the solid was slurried in heptane, filtered and dried under high vacuum overnight.
- the filtrate was concentrated.
- the resulting solid was then diluted with a 1:1 mixture of isopropyl acetate: methyl tert-butyl ether (20 mL).
- the mixture was heated to reflux and then cooled to room temperature.
- the solvent was decanted and the solid was slurried in heptane and filtered to afford a second crop of solid which was dried under high vacuum overnight.
- Step 3 Preparation of 3-[3,5-Dibromo-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- propionic acid (22).
- Step 4 Preparation of 3-[3,5-Dibromo-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl] -propionic acid (23)
- the water layer was extracted with ethyl acetate (3 x 100 mL).
- the organic layers were combined, dried with sodium sulfate, filtered and concentrated under vacuum.
- the resulting solid was slurried in ethyl acetate (20 mL).
- Heptane (20 mL) was added to this mixture.
- the solvents were then concentrated under vacuum to a volume of approximately 20 mL.
- the solids were filtered and washed with a 1:1 ethyl acetate:heptane solution (2 x 10 mL).
- the water layer was diluted with ethyl acetate (1 x 500 mL), and the two phases were filtered over celite. The organic layer was separated. The celite was washed with ethyl acetate. The water layer was extracted again with ethyl acetate (I x 500 mL). The combined organics were then washed with a saturated aqueous sodium chloride solution (1 x 400 mL), dried with magnesium sulfate, filtered and concentrated under vacuum. The resulting residue was dissolved in chloroform and purified by column chromatography using silica gel eluted with 10-15% ethyl acetate in petroleum ether. The desired fractions were collected and concentrated under vacuum.
- the resulting solid was slurried in ether, filtered and rinsed with cold ether.
- the filtrate was concentrated under vacuum.
- the solid was diluted with ether and filtered to obtain a second crop. This contained a trace of the undesired isomer. It was re- slurried in ether and filtered and shown to be the pure desired isomer by 1 H NMR.
- the filtrate was concentrated and a third crop was obtained in the same manner.
- the filtrate was concentrated and was diluted with 60% ether in petroleum ether. Petroleum ether was added with scratching and a solid crystallized.
- the solid was collected and rinsed with 60% ether in petroleum ether.
- the four crops of solid were shown to be the pure desired isomer by 1 H NMR.
- Step 2 6-(4-Amino-2,6-dichloro-phenoxy)-4-isopropyl-2H-pyridazin-3-one (25)
- the water layer was extracted with ethyl acetate (1 x 250 mL).
- the organic layers were combined, dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting oil was diluted with methanol (20 mL) and was treated with a IN aqueous sodium hydroxide solution (20 mL, 20 mmol).
- the reaction mixture was heated to 120 0 C for 24 h.
- the reaction mixture was cooled to room temperature and the solvent was concentrated under vacuum.
- the residue was diluted with water (100 mL) and was extracted with ethyl acetate (200 mL).
- the residue was dissolved in chloroform and purified by flash chromatography (Biotage 40L) using silica gel eluted with a 1:1 ethyl acetate:hexanes solution with 0.5 % glacial acetic acid.
- the desired fractions were collected and concentrated under vacuum and dried under high vacuum at 37°C.
- the solid was slurried in diethyl ether (-10 mL) and petroleum ether (10 mL).
- the reaction mixture was then cooled to room temperature, filtered through celite, and rinsed with chloroform.
- a IN aqueous hydrochloric acid solution 150 mL was added to the filtrate.
- the resulting mixture was stirred for 30 min.
- the organic layer was separated.
- the water layer was extracted with chloroform (100 mL).
- the organic layers were combined, methanol (10 mL) was added to dissolve insoluble material and the organic layer was dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting solid was slurried with ether (3 mL) and diluted with petroleum ether (10 mL). The mixture was stirred for 45 min.
- the solids were collected by filtration and rinsed with petroleum ether.
- Step 1 Preparation of N-[3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- oxalamic acid methyl ester (27)
- the reaction mixture was poured onto water (IL) and was diluted with ethyl acetate (300 mL) and a saturated aqueous sodium chloride solution (100 mL). The organic layer was separated. The water layer was re-extracted with ethyl acetate (2 x 300 mL). The organic layers were combined and washed with a IN aqueous hydrochloric acid solution (1 x 150 mL) and a saturated aqueous sodium chloride solution (1 x 150 mL), dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 2 Preparation of N-[3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]- oxalamic acid (28)
- Step 3 Preparation of N-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl]-oxalamic acid (29)
- the solid was slurried in 1:1:2 isopropyl acetate: methyl tert-butyl ethe ⁇ hexanes (3 mL) and then was heated to reflux. The mixture was cooled and filtered. A slight amount of impurity was detected by TLC. The solid was slurried in isopropyl acetate (10 mL) and filtered.
- Step 1 Preparation of 2-Cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro- pyridazin-3-yloxy)-phenyl]-hydrazono ⁇ -acetyl)-carbamic acid ethyl ester (30)
- N-cyanoacetylurethane 73 mg, 0.46 mol
- water 9.4 mL
- pyridine 2.8 mL
- This reaction mixture was cooled to 0 0 C and the solution from the first reaction was quickly filtered and poured into the second reaction mixture. An orange precipitate formed and the suspension was stirred at 0 0 C for 30 min. The solid was filtered and rinsed with water followed by petroleum ether.
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile (31)
- the reaction mixture was heated to 120 0 C for 1.5 h. At this time, the reaction was cooled to 0 0 C, diluted with water (220 mL) and stirred for 30 min. The resulting solid was filtered and rinsed with water (3 x 100 mL) followed by petroleum ether (3 x 100 mL). The solid was air dried for 30 min. The solid was then diluted with hot acetonitrile (250 mL). The resulting red mixture was treated with neutral decolorizing carbon, filtered through celite and rinsed with acetonitrile (1 L) until no UV active material eluted. The yellow filtrate was concentrated under reduced pressure.
- the resulting solid was triturated with hot acetonitrile (50 mL), cooled for 15 min, diluted with water (100 mL) and filtered. The solid was triturated again with hot acetonitrile (10 mL), filtered and rinsed with acetonitrile, water, and petroleum ether.
- Step 1 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carboxylic acid (32)
- the reaction mixture was cooled to room temperature and was diluted with water (50 mL). At this time, the reaction was made basic by the addition of a IN aqueous sodium hydroxide solution and was extracted with ether (100 mL). The organic layer was discarded. The aqueous layer was acidified by the addition of a IN aqueous hydrochloric acid solution and was extracted with ethyl acetate (2 x 100 mL).
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl]-2H-[l,2,4]triazine-3,5-dione (33)
- the organic layer was separated and washed with a saturated aqueous sodium bicarbonate solution (2 x 100 mL) and a saturated aqueous sodium chloride solution (100 mL), dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting solid was dissolved in methylene chloride and was purified by flash chromatography (Biotage 40S) using silica gel eluted with 100% ethyl acetate to elute the impurity followed by 0.2 % glacial acetic acid in ethyl acetate to elute the desired product.
- the desired fractions were collected and concentrated under vacuum.
- the resulting solid was slurried in hot methanol (2 mL), filtered and rinsed with petroleum ether.
- Step 1 Preparation of Acetic acid 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro- pyridazin-3-yloxy)-phenyl] -ethyl ester (34)
- a solution of 2-[3,5-dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]-ethanol (17) (500 mg, 1.38 mmol) in glacial acetic acid (5 mL) was treated with sodium acetate (230 mg, 2.8 mmol) and heated to 114°C for 24 h. At this time, the reaction mixture was cooled to room temperature and was concentrated under vacuum.
- the resulting residue was diluted with methylene chloride (25 mL) and was washed with water (10 mL). The organic layer was separated and was washed with a saturated aqueous sodium bicarbonate solution (10 mL). The waters layers were combined and extracted with methylene chloride (10 mL).
- Step 2 Preparation of acetic acid 2-[3,5-dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6- dihydro-pyridazin-3-yloxy)-phenyl] -ethyl ester (35)
- the reaction mixture was concentrated and the resulting residue was partitioned between ethyl acetate (50 mL) and water (25 mL). The water layer was re- extracted with ethyl acetate (25 mL). The organic layers were combined, dried with sodium sulfate, filtered, and concentrated. The resulting residue was dissolved in 25% ethyl acetate in hexanes and was purified by flash chromatography (Biotage 40S) using silica gel eluted with 25-50% ethyl acetate in hexanes. The desired fractions were collected and concentrated under vacuum.
- Step 3 Preparation of 6-[2,6-Dichloro-4-(2-hydroxy-ethyl)-phenoxy]-4-isopropyl-2-methyl- pyridazin-3-one (36)
- a solution of 2-[3,5-dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl] -ethyl ester (35) (390 mg, 0.97 mmol) in methanol (4 mL) was treated with a IN aqueous sodium hydroxide solution (1.0 mL, 1.0 mmol) at room temperature. The reaction mixture was stirred for 24 h and was then concentrated under vacuum.
- Step 4 Preparation of [3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro-pyridazin- 3-yloxy)-phenyl] -acetic acid (37)
- the reaction mixture was then concentrated under vacuum.
- the resulting residue was diluted with ethyl acetate (25 mL) and water (20 mL) and was treated with sodium bisulfite (100 mg).
- the resulting mixture was shaken and turned from red to green.
- the aqueous layer was separated and was re- extracted with ethyl acetate (25 mL).
- the organic layers were combined and washed with a saturated aqueous sodium chloride solution.
- the organic layer was separated, dried with sodium sulfate, filtered, and concentrated under vacuum.
- Step 1 Preparation of 2-[3,5-Dibromo-4-(6-chloro-pyridazin-3-yloxy)-phenyl]-ethanol (38):
- This compound was prepared by a similar method to that described in Example 3, Step 2 except that 3,6-dichloro-pyridazine was used in place of 3,6-dichloro-4-isopropyl pyridazine (7).
- the reaction was heated at 140 0 C for 3.5 h. The work up was different than Example 3, Step 2.
- the reaction mixture was added slowly with stirring to a saturated aqueous sodium chloride solution.
- the mixture was extracted with methylene chloride.
- the organic layers were combined and washed with a saturated aqueous sodium chloride solution, dried over sodium sulfate, filtered, and concentrated.
- Step 2 Preparation of Acetic acid 2-[3,5-dibromo-4-(6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl] -ethyl ester (39)
- This compound was prepared by a similar method to that described in Example 10, Step 1 except that 2-[3,5-dibromo-4-(6-chloro-pyridazin-3-yloxy)-phenyl]-ethanol (38) was used in place of 2-[3,5-dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-yloxy)-phenyl]-ethanol (17) to afford acetic acid 2-[3,5-dibromo-4-(6-oxo-l,6-dihydro-pyridazin-3-yloxy)-phenyl]-ethyl ester (39) (92%) as a colorless oil; LRMS for Ci 4 Hi 2 Br 2 N
- This compound was prepared by a similar method to that described in Example 10, Step 2 except that isopropyl iodide was used in place of methyl iodide and acetic acid 2-[3,5- dibromo-4-(6-oxo-l,6-dihydro-pyridazin-3-yloxy)-phenyl]-ethyl ester (39) was used in place of acetic acid 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)- phenyl]-ethyl ester (34).
- the method was similar except that the reaction was heated to 50 0 C for 24 h.
- Step 4 Preparation of 6-[2,6-Dibromo-4-(2-hydroxy-ethyl)-phenoxy]-2-isopropyl- pyridazin-3-one (41)
- Step 5 Preparation of [3,5-Dibromo-4-(l-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)- phenyl]- acetic acid (42)
- This compound was prepared by a similar method to that described in Example 10, Step 4 except that 6-[2,6-dibromo-4-(2-hydroxy-ethyl)-phenoxy]-2-isopropyl-pyridazin-3-one (41) was used in place of 6-[2,6-dichloro-4-(2-hydroxy-ethyl)-phenoxy]-4-isopropyl-2-methyl- pyridazin-3-one (36).
- Step 1 Preparation of Cyano-(2,6-dichloro-4-nitro-phenyl)-acetic acid tert-butyl ester (43)
- a solution of l,2,3-trichloro-5-nitrobenzene (50.33 g, 222.26 mmol) in N,N- dimethylformamide (220 mL) at 25°C was treated with 2-butylcyano acetate (33 mL, 230.72 mmol) and potassium carbonate (61.44 g, 444.54 mmol).
- the reaction flask was fitted with a reflux condenser. It was then heated to 50 0 C for 18 h. At this time, the reaction was allowed to cool to 25°C and then was concentrated under vacuum.
- Step 2 Preparation of (4-amino-2,6-dichloro-phenyl)-acetonitrile (44): A solution of cyano-(2,6-dichloro-4-nitro-phenyl)-acetic acid tert-butyl ester (43) (78.69 g, crude) in ethanol (320 mL) at 25°C was treated with concentrated hydrochloric acid (160 mL). The reaction was fitted with a reflux condenser and then was heated to 75°C. At this time, the resulting homogeneous solution was treated portion wise with tin(II) chloride dihydrate (225.64 g, 1.0 mol).
- the reaction was warmed to 110-115 0 C where it was stirred for 3 h. At this time, the reaction was cooled to 25°C and then was diluted with ethyl acetate (1.5 L). The resulting solution was carefully neutralized with a saturated aqueous sodium carbonate solution (1.0 L). The resulting thick mixture was filtered through filter paper via vacuum filtration and was washed with ethyl acetate until no material was detected in the filtrate. The filtrate was then transferred to a separatory funnel where the resulting layers were separated.
- the organics were washed consecutively with a IN aqueous hydrochloric acid solution (1 x 250 mL), a saturated aqueous sodium carbonate solution (1 x 250 mL), a IN aqueous hydrochloric acid solution (1 x 250 mL), and a saturated aqueous sodium chloride solution (1 x 250 mL).
- the organics were then dried with magnesium sulfate, filtered, and concentrated under vacuum.
- Step 3 Preparation of (4-Amino-2,6-dichloro-phenyl)-(6-chloro-5-isopropyl-pyridazin-3- yl)-acetonitrile (45):
- the mixture was heated to 60 0 C for 45 min.
- the reaction was cooled to room temperature, transferred to a separatory funnel, diluted with ethyl acetate (500 mL) and was washed with a saturated aqueous sodium chloride solution (2 x 250 mL).
- the organic layer was separated, dried with magnesium sulfate, and was filtered.
- Silica gel 60 (70-230 mesh) was added to the filtrate and the solvent was concentrated under vacuum.
- the resulting mixture was purified by flash chromatography (Biotage 75L) using silica gel eluted with 15%-30% ethyl acetate in hexanes.
- the reaction was cooled to room temperature and the mixture was poured onto water (250 mL).
- the suspension was placed in the freezer for 15 min.
- the resulting solids were filtered and washed with water and petroleum ether.
- the solids were collected and dissolved in hot ethyl acetate.
- Silica gel 60 (70-230 mesh) was added and the solvent was concentrated under vacuum.
- Step 5 Preparation of (2-Cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro- pyridazin-3-ylmethyl)-phenyl]-hydrazono ⁇ -acetyl)-carbamic acid ethyl ester (47):
- Step 6 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile (48)
- This compound was prepared by a similar method to that described in Example 8, Step 2 except that (2-cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl)-phenyl]-hydrazono ⁇ -acetyl)-carbamic acid ethyl ester (47) was used in place of 2-cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yloxy)-phenyl]- hydrazonoj-acety ⁇ -carbamic acid ethyl ester (30) to afford 2-[3,5-dichloro-4-(5-isopropyl- 6-oxo-l,6-dihydro-pyridazin-3-ylmethyl)-phenyl]-3,5-dioxo-2,3,4,5-
- Step 1 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carboxylic acid (49)
- This compound was prepared by a similar method to that described in Example 9, Step 1 except that 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-ylmethyl)- phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile (48) was used in place of 2-[3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3-yl
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl) -phenyl] - [ 1 ,2,4] triazine-3,5-dione (50) :
- Step 1 Preparation of 6-(4-bromo-2,6-dichloro-benzyl)-4-isopropyl-pyridazin-3-one (51)
- a solution of 6-(4-amino-2,6-dichloro-benzyl)-4-isopropyl-pyridazin-3-one (46) (0.9 g, 2.88 mmol) in glacial acetic acid (16 mL) at room temperature was treated with concentrated sulfuric acid (4 mL).
- a solution of sodium nitrite (480 mg, 6.96 mmol) in water (5 mL) was added below the surface of the reaction slowly over 10 min. The reaction mixture was heated to 60 0 C for 1 h.
- Step 2 Preparation of 3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl)-benzoic acid methyl ester (52) A solution of 6-(4-bromo-2,6-dichloro-benzyl)-4-isopropyl-pyridazin-3-one (51) (194 mg,
- Step 3 Preparation of 6-(2,6-Dichloro-4-hydroxymethyl-benzyl)-4-isopropyl-pyridazin-3- one (53)
- Step 5 Preparation of [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl) -phenyl] -acetonitrile (55)
- a suspension of sodium cyanide (270 mg, 5.51 mmol) in dimethyl sulfoxide (2 mL) at room temperature was treated with concentrated sulfuric acid (0.1 mL, 1.88 mmol) and a solution of 6-(4-bromomethyl-2,6-dichloro-benzyl)-4-isopropyl-pyridazin-3-one (54) (70 mg, 0.180 mmol) in dimethyl sulfoxide (2 mL).
- the reaction mixture was stirred at room temperature for 30 min and then at 50 0 C for 1 h.
- the reaction was cooled to room temperature, poured into a saturated aqueous sodium bicarbonate solution (50 mL), was diluted with water (20 mL) and then extracted with ethyl acetate (3 x 75 mL). The organic layers were combined and washed with a saturated aqueous sodium chloride solution (50 mL), dried over magnesium sulfate, filtered, and concentrated under vacuum.
- Step 6 Preparation of [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl) -phenyl] -acetic acid (56)
- Step 1 Preparation of 3,5-Dibromo-4-bromomethyl-benzoic acid methyl ester (57)
- a solution of methyl 3,5-dibromo-4-methylbenzoate (5g, 16.24 mmol) in carbon tetrachloride (50 mL) was treated with N-bromosuccinamide (3.6 g, 20.23 mmol) and 2,2'- azobisisobutyronitrile (0.56 g, 3.410 mmol).
- the reaction mixture was heated to reflux for 24 h.
- the reaction was cooled to room temperature and concentrated under vacuum.
- Step 2 Preparation of (3,5-Dibromo-4-bromomethyl-phenyl)-methanol (58)
- a solution of 3,5-dibromo-4-bromomethyl-benzoic acid methyl ester (57) (5.2 g, 13.44 mmol) in tetrahydrofuran (50 mL) at 0 0 C was treated with a IM solution of diisobutylaluminum hydride in tetrahydrofuran (30 mL, 30 mmol).
- the reaction mixture was stirred for 2 h at 0 0 C. TLC revealed starting material was still present.
- This compound was prepared by a similar method to that described in Example 14, Step 5 except that (3,5-dibromo-4-bromomethyl-phenyl)-methanol (58) was used in place 6-(4- bromomethyl-2,6-dichloro-benzyl)-4-isopropyl-pyridazin-3-one (54).
- the product was purified by flash chromatography using silica gel eluted with a gradient of 3:1 to 1:1 hexanes:ethyl acetate.
- Step 4 Preparation of [2,6-Dibromo-4-(tetrahydro-pyran-2-yloxymethyl)-phenyl]- acetonitrile (60)
- reaction mixture was stirred for 45 min and was then treated with a saturated aqueous sodium bicarbonate solution (3 mL), a saturated aqueous sodium chloride solution (10 mL) and water (10 mL). This mixture was extracted with methylene chloride (3 x 50 mL). The organic layers were combined and washed with a saturated aqueous sodium chloride solution (30 mL), dried with magnesium sulfate, filtered and concentrated under vacuum.
- reaction was cooled to 25°C, diluted with water (50 mL), a saturated aqueous sodium chloride solution (50 mL) and a saturated aqueous sodium bicarbonate solution (20 mL). This mixture was extracted with ethyl acetate (200 mL). The organics were washed with a saturated aqueous sodium chloride solution (100 mL), dried with magnesium sulfate, filtered, and concentrated under vacuum.
- Step 6 Preparation of 6-(2,6-Dibromo-4-chloromethyl-benzyl)-4-isopropyl-pyridazin-3-one (62)
- a mixture of (6-chloro-5-isopropyl-pyridazin-3-yl)-[2,6-dibromo-4-(tetrahydro-pyran-2- yloxymethyl)-phenyl]-acetonitrile (61) (332.8 mg, 0.61 mmol) and sodium acetate (103.8 mg, 1.26 mmol) in glacial acetic acid (2.7 mL) was heated to reflux for 2.5 h.
- the reaction was then treated with concentrated hydrochloric acid (2.0 mL) and heated to reflux for 18 h.
- the reaction was treated with more concentrated hydrochloric acid (6.0 mL) and additional glacial acetic acid (3.0 mL) and heated to reflux for an additional 24 h.
- the reaction was then cooled to 25°C and diluted with ethyl acetate (150 mL). This solution was washed with a saturated aqueous sodium chloride solution (2 x 50 mL) and a saturated aqueous sodium carbonate solution (1 x 100 mL), dried with magnesium sulfate, filtered, and concentrated under vacuum.
- the resulting brown oil was treated with glacial acetic acid (1.0 mL) and concentrated hydrochloric acid (6.0 mL) and was heated to reflux for 3 d.
- reaction was cooled to 25°C and diluted with ethyl acetate (150 mL). This solution was washed with a saturated aqueous sodium chloride solution (2 x 50 mL) and a saturated aqueous sodium carbonate solution (1 x 100 mL), dried with magnesium sulfate, filtered, and concentrated under vacuum.
- a suspension of sodium cyanide (279 mg, 5.69 mmol) in dimethyl sulfoxide (2.0 mL) was treated with concentrated sulfuric acid (0.10 mL), 6-(2,6-dibromo-4-chloromethyl-benzyl)- 4-isopropyl-pyridazin-3-one (62) (346 mg, 0.79 mmol) and an addition rinse of dimethyl sulfoxide (3.0 mL).
- the reaction was stirred at 25°C for 5 min and at 40 0 C for 1 h. At this time, the reaction was heated to 60 0 C for 1 h and then was cooled to 25°C.
- Step 8 Preparation of [3,5-Dibromo-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylmethyl) -phenyl] -acetic acid (64)
- Example 16 Synthesis of 2-[3,5-dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-yloxy)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro[l,2,4]triazine-6-carbonitrile
- Step 1 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- yloxy)-phenyl]-isoindole-l,3-dione (65)
- a mixture of 4-amino-2,6-dichloro-phenol (50 g, 280.8 mmol) and potassium tert-butoxi ⁇ o (33.16 g, 280.8 mmol) in N,N-dimethylacetamide (200 mL) was heated to 90 0 C.
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-yloxy)-phenyl]-isoindole-l,3-dione (66)
- Step 3 Preparation of 6-(4-Amino-2,6-dichloro-phenoxy)-4-isopropyl-2-methyl-2H- pyridazin-3-one (67)
- Step 4 Preparation of (2-Cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6- dihydro-pyridazin-3-yloxy)-phenyl]-hydrazono ⁇ -acetyl)-carbamic acid ethyl ester
- reaction was stirred at 5-10 0 C for 30 min. At this time, the reaction was treated with N-cyanoacetylurethane (5.34 g, 33.52 mmol) followed by a solution of sodium acetate (7.5 g, 91.41 mmol) in water (22.5 mL). The reaction was stirred at 5-10 0 C for 30 min and then was diluted with water (50 mL).
- Step 5 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-yloxy)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile (69)
- Step 1 Preparation of 4-[2-(tert-Butyl-diphenyl-silanyloxy)-ethyl]-2,6-dichloro-phenol (70)
- a solution of 2,6-dichloro-4-(2-hydroxy-ethyl)-phenol (16) (2.0 g, 9.66 mmol) in methylene chloride (20 mL) at 25°C was treated with triethylamine (1.35 mL, 9.66 mmol) and tert- butyl diphenylsilylchloride (2.48 mL, 9.66 mmol). The reaction was stirred at 25°C for 18 h. At this time, the reaction was diluted with methylene chloride.
- Step 2 Preparation of Dimethyl-thiocarbamic acid O- ⁇ 4-[2-(te ⁇ t-butyl-diphenyl-silanyloxy)- ethyl]-2,6-dichloro-phenyl ⁇ ester (71)
- reaction was diluted with ethyl acetate and was then washed with a IN aqueous hydrochloric acid solution, water, and a saturated aqueous sodium chloride solution, dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 3 Preparation of Dimethyl-thiocarbamic acid S- ⁇ 4-[2-(te ⁇ t-butyl-diphenyl-silanyloxy)- ethyl]-2,6-dichloro-phenyl ⁇ ester (72)
- Step 4 Preparation of 2-(3,5-Dichloro-4-mercapto-phenyl)-ethanol (73)
- Step 5 Preparation of 2-[3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-ylsuh c anyl)- phenyl] -ethanol (74)
- a solution of 2-(3,5-dichloro-4-mercapto-phenyl)-ethanol (73) (620 mg, 2.78 mmol) in dimethyl sulfoxide (25 mL) was treated with 3,6-dichloro-4-isopropyl-pyridazine (7) (530 mg, 2.78 mmol) and potassium carbonate (1.54 g, 11.12). The resulting mixture was heated to 90 0 C for 18 h.
- the reaction was cooled to 25°C, poured into a solution of water (200 mL) and a IN aqueous hydrochloric acid solution (25 mL). The mixture was extracted with ethyl acetate (2 x 200 mL). The organics were then washed with water (1 x 100 mL) and a saturated aqueous sodium chloride solution (1 x 100 mL), dried with magnesium sulfate, filtered, and concentrated under vacuum.
- Step 6 Preparation of [3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-ylsuh c anyl)- phenyl]- acetic acid (75)
- Step 7 Preparation of [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylsulfanyl) -phenyl] -acetic acid (76)
- Step 1 Preparation of [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazine-3- sulfinyl) -phenyl] -acetic acid (77) and [3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro- pyridazine-3-sulfonyl)-phenyl]-acetic acid (78)
- Example 20 Synthesis of Synthesis of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6- dihydro-pyridazine-3 -sutfonyO-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-Cl ⁇ j ⁇ triazine-6- carbonitrile (88)
- Step 1 Preparation of Dimethyl-thiocarbamic acid O-(2,6-dichloro-4-nitro-phenyl) ester (79)
- a solution of 2,6-dichloro-4-nitro-phenol (3.0 g, 14.4 mmol) in N,N-dimethylformamide (70 mL) at 25°C was treated with 1,4-diazabicyclo [2.2.2] octane (3.16 mL, 28.8 mmol) and dimethylthiocarbamoyl chloride (2.85 g, 23.04 mmol). The reaction was stirred at 25°C for
- the solution was extracted with ethyl acetate (3 x 200 mL).
- the combined organics were dried with magnesium sulfate, filtered and concentrated under vacuum.
- the resulting residue was slurried with diethyl ether and was cooled in the freezer for 30 min.
- Step 4 Preparation of 4-Amino-2,6-dichloro-benzenethiol (82)
- Step 5 Preparation of 3,5-Dichloro-4-(6-chloro-5-isopropyl-pyridazin-3-ylsuh c anyl)- phenylamine (83)
- a solution of 4-amino-2,6-dichloro-benzenethiol (82) (1.0 g, 5.2 mmol) in N,N- dimethylformamide at 25°C was treated with 3,6-dichloro-4-isopropyl-pyridazine (7) (990 mg, 5.2 mmol) and potassium carbonate (2.16 g, 15.6 mmol). The resulting mixture was heated to 90 0 C for 18 h.
- the reaction was cooled to 25°C, poured onto a mixture of ice water and a IN aqueous hydrochloric acid solution (10 mL).
- the organics were washed with a saturated aqueous sodium chloride solution (1 x 50 mL), dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 7 Preparation of 6-(4-Amino-2,6-dichloro-phenylsulfanyl)-4-isopropyl-2H-pyridazin- 3-one (85)
- Step 8 Preparation of (2-Cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro- pyridazm-3-ylsulfany ⁇ -phenylJ-hydrazonoJ-acety ⁇ -carbamic acid ethyl ester (86) A mixture of 6-(4-amino-2,6-dichloro-phenylsuh c anyl)-4-isopropyl-2H-pyridazin-3-one (85) (3.40 g, 10.4 mmol) in water (135 mL) and concentrated hydrochloric acid (68 mL) cooled to 0 0 C was treated with a solution of sodium nitrite (853 mg, 12.36 mmol) in water (7.0 mL) via Pasteur pipette under the surface of the reaction mixture.
- Step 9 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylsuh c anyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile (87)
- the reaction was heated to 80 0 C for 3 d. An additional amount of the 30% aqueous hydrogen peroxide solution (0.28 mL, 0.98 mmol) was added each day. After 3 d, the reaction was diluted with ethyl acetate and water. The organics were washed with water, dried with magnesium sulfate, filtered and concentrated under vacuum.
- Step 1 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylsulfanyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carboxylic acid (89)
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazin-3- ylsulfanyl)-phenyl]-2H-[l,2,4]triazine-3,5-dione (90)
- This solid was dissolved in a IN aqueous sodium hydroxide solution (100 mL) and extracted with ethyl acetate (1 x 120 mL).
- the aqueous layer was neutralized by the addition of a IN aqueous hydrochloric acid solution (15 mL).
- Step 3 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-6-oxo-l,6-dihydro-pyridazine-3- sulfonyl)-phenyl]-2H-[l,2,4]triazine-3,5-dione (91)
- Example 22 Synthesis of 2-[3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-yhnethyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6- carbonitrile (96)
- Step 1 Preparation of [2,6-Dichloro-4-(l,3-dioxo-l,3-dihydro-isoindol-2-yl)-phenyl]- acetonitrile (92)
- a solution of 6-(4-amino-2,6-dichloro-benzyl)-4-isopropyl-2H-pyridazin-3-one (910 mg, 2.90 mmol) in glacial acetic acid (12 mL) was treated with phthalic anhydride (430 mg, 2.9 mmol). The reaction was then heated to 130 0 C for 3.5 h.
- Step 2 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-ylmethyl)-phenyl] -isoindole- 1 ,3-dione (93)
- Step 3 Preparation of 6-(4-Amino-2,6-dichloro-benzyl)-4-isopropyl-2-methyl-2H- pyridazin-3-one (94)
- Step 4 Preparation of (2-Cyano-2- ⁇ [3,5-dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6- dihydro-pyridazin-3-ylmethy ⁇ -phenyy-hydrazonoJ-acety ⁇ -carbamic acid ethyl ester (95) A mixture of 6-(4-amino-2,6-dichloro-benzyl)-4-isopropyl-2-methyl-2H-pyridazin-3-one
- Step 5 Preparation of 2-[3,5-Dichloro-4-(5-isopropyl-l-methyl-6-oxo-l,6-dihydro- pyridazin-3-ylmethyl)-phenyl]-3,5-dioxo-2,3,4,5-tetrahydro-[l,2,4]triazine-6-carbonitrile
- Example 23 TR/RXR/GRIP Assay
- a TR/RXR/GRIP assay was used to test representative compounds of formula (I).
- TR ⁇ and the ligand binding domain (amino acids 202-461) of thyroid hormone receptor ⁇ (H6-TR ⁇ ) were cloned into an E.coli expression vector pET28a (Novagen, Milwaukee,
- WI which contained a N-terminal hexaHis sequence.
- the resulting recombinant hexaHis tagged proteins were produced in E.coli BL21(DE3) cells.
- Cells were grown in Terrific Broth (in- house preparation in-house prepared medium of Bacto tryptone (3.3 %, w/v), Diflco yeast extract (2.0 %, w/v) and NaCl (0.5 %, w/v)) using shake flasks with a 24 hour induction in 0.2mM IPTG at 25° C, harvested and lysed with five volumes of Buffer A (0.05M Tris, 0.3M NaCL, 1%W/V Betaine, 0.01M imidazole, 0.02M b-mercapto ethanol, pH 8.0).
- Buffer A 0.05M Tris, 0.3M NaCL, 1%W/V Betaine, 0.01M imidazole, 0.02M b-mercapto ethanol, pH 8.0.
- Lysozyme (1.0mg/ml, Sigma) and Complete Protease Inhibitor Cocktail (Roche Diagnostics Gmbh) were added to slurry and solution sonicated for one min five times at 4° C.
- the suspension was centrifuged in a Ti45 Beckmann rotor for two hours at 127,300 RCF and the supernatant was loaded onto NI_ NTA Agarose (Quigen 30210) column. After washing with Buffer A, H6-TR ⁇ or H6-TR ⁇ was eluted with Buffer A containing 0.25M Imidazole.
- the ligand binding domain of human retinoid X receptor (amino acids 225-462) (RxRa) was engineered with N-terminal His6 and EE (EFMPME) tags, a thrombin cleavage site between the His6 and EE sequences, and cloned into pACYC vector.
- the resulting His6- EE-tagged protein was produced in E. coli cells. Cells were grown using shake flasks with an 18 hour induction in 0.ImM IPTG at 18° C, harvested and suspended with five volumes of Buffer B (0.025M Tris, 0.3M NaCl, 0.02 M imidazole, 0.01M ⁇ -mercaptoethanol, pH 8.0).
- Lysozyme (0.2 mg/ml, Sigma) and Complete Protease Inhibitor Cocktail (Roche Diagnostics Gmbh) were added and stirred for 30 min. at 4 0 C.
- the suspension was sonicated for 30 seconds, five times, at 4° C.
- the suspension was centrifuged for 20 min. at 12,000 RCF.
- the supernatant was filtered by 0.45 ⁇ m pore size membrane and 0.5% NP- 40 was added.
- the His6-tagged protein was bound to and eluted from NiNTA metal- affinity resin (QIAGEN, Valencia, CA). The protein was concentrated and dialyzed.
- the His6 tag was removed from EE-RxRa by thrombin digestion, using 10 units thrombin (Pharmacia, Piscataway, NJ) per mg protein and incubating for 2 hours at 25 0 C. Removal of thrombin was done batch- wise using Benzamidine-Sepharose 6B (Pharmacia,
- the protein was concentrated and dialyzed. This protein was used in the coactivator peptide recruitment assay.
- Europium-conjugated anti-hexa His antibody and APC-conjugated streptavidin were purchased from PerkinElmer Life and Analytical Sciences.
- H6-TR ⁇ 50 nM
- 50 mM Hepes, pH 7.0, ImM DTT, 0.05% NP40 and 0.2 mg/ml BSA (Binding Buffer) was mixed with an equal volume of EE-RxRa (50 nM) in Binding Buffer.
- Six microliters of T3 (0-14.8 uM) or test compound (0-1.2 mM) in DMSO was then added and the solution incubated at 37° C for 30 min.
- the assay protocol is essentially the same as that of TR ⁇ /RXR/GRIP coactivator peptide recruitment assay as described above except that 125 nM of H6-TR ⁇ , 125 nM of EE-RxRa and 250 nM of biotin-GRIP were used.
- the tested compounds are thyroid hormone receptor agonists, with EC50 values from the THR-beta/RXR/GRIP recruitment assay:
- Film coated tablets containing the following ingredients can be manufactured in a conventional manner:
- the active ingredient is sieved and mixed with microcristalline cellulose and the mixture is granulated with a solution of polyvinylpyrrolidone in water.
- the granulate is mixed with sodium starch glycolate and magesiumstearate and compressed to yield kernels of 120 or 350 mg respectively.
- the kernels are lacquered with an aqueous solution / suspension of the above mentioned film coat.
- Capsules containing the following ingredients can be manufactured in a conventional manner:
- the components are sieved and mixed and filled into capsules of size 2.
- Injection solutions can have the following composition:
- the active ingredient is dissolved in a mixture of polyethylene glycol 400 and water for injection (part).
- the pH is adjusted to 5.0 by acetic acid.
- the volume is adjusted to 1.0 ml by addition of the residual amount of water.
- the solution is filtered, filled into vials using an appropriate overage and sterilized.
- Soft gelatin capsules containing the following ingredients can be manufactured in a conventional manner:
- the active ingredient is dissolved in a warm melting of the other ingredients and the mixture is filled into soft gelatin capsules of appropriate size.
- the filled soft gelatin capsules are treated according to the usual procedures.
- Sachets containing the following ingredients can be manufactured in a conventional manner:
- Microcrystal ⁇ ne cellulose (AVICEL PH 102) 1400.0 mg
- Flavoring additives 1.0 mg
- the active ingredient is mixed with lactose, microcristalline cellulose and sodium carboxymethyl cellulose and granulated with a mixture of polyvinylpyrrolidone in water.
- the granulate is mixed with magnesium stearate and the flavouring additives and filled into sachets.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description


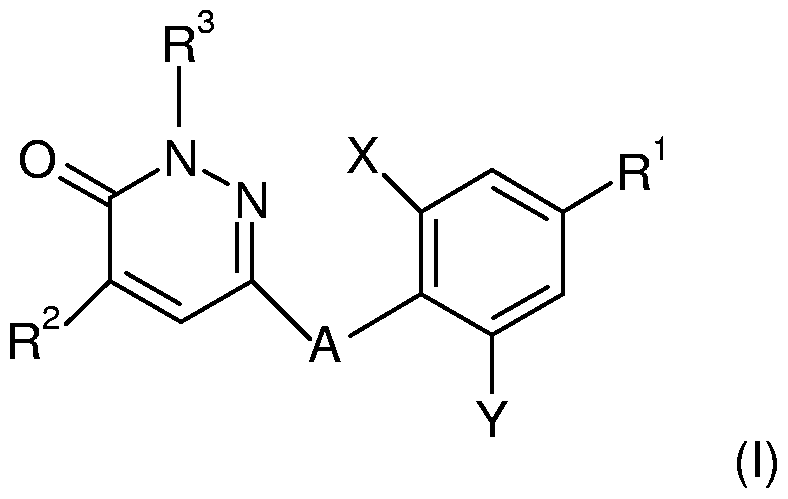


















































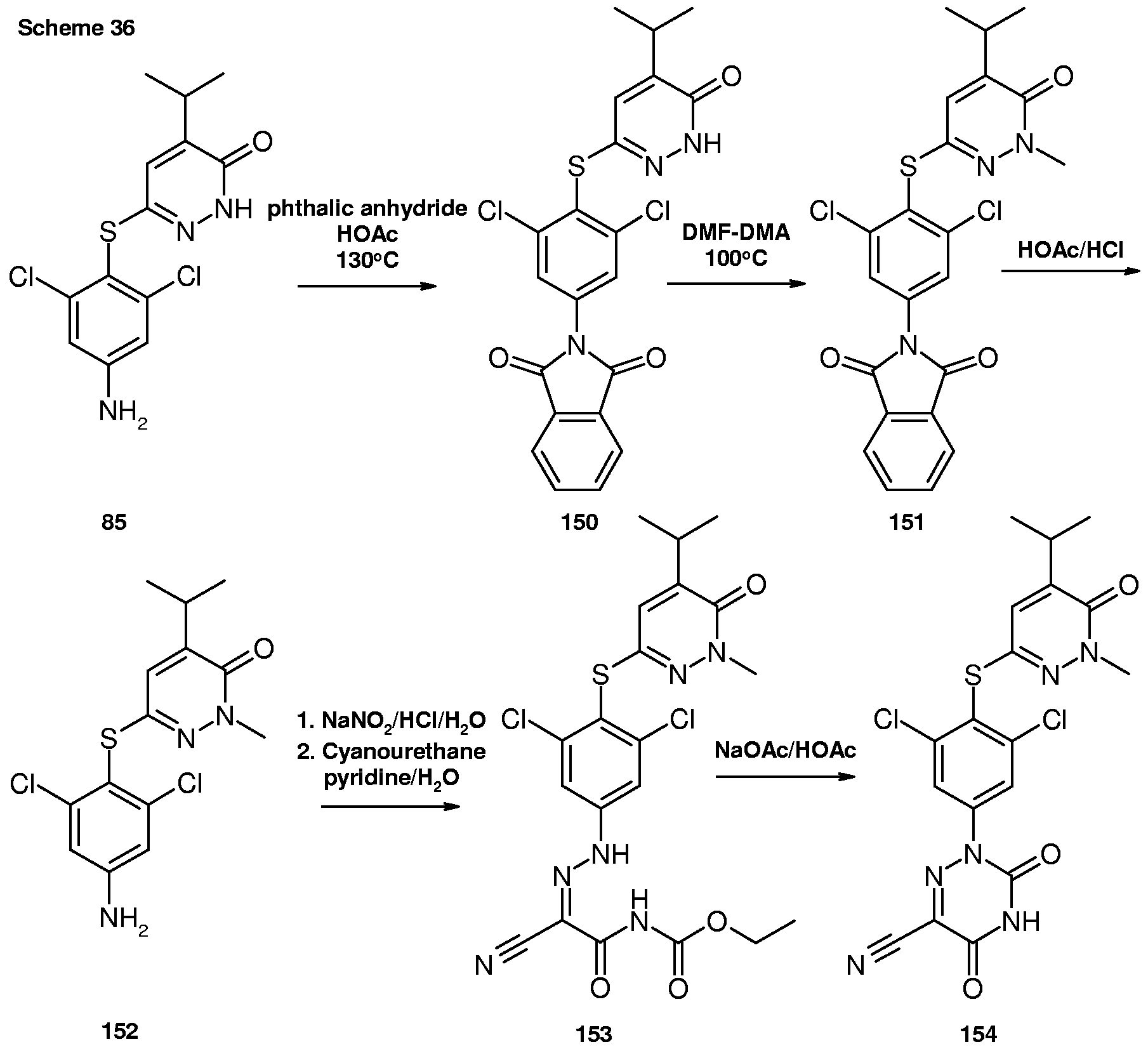










































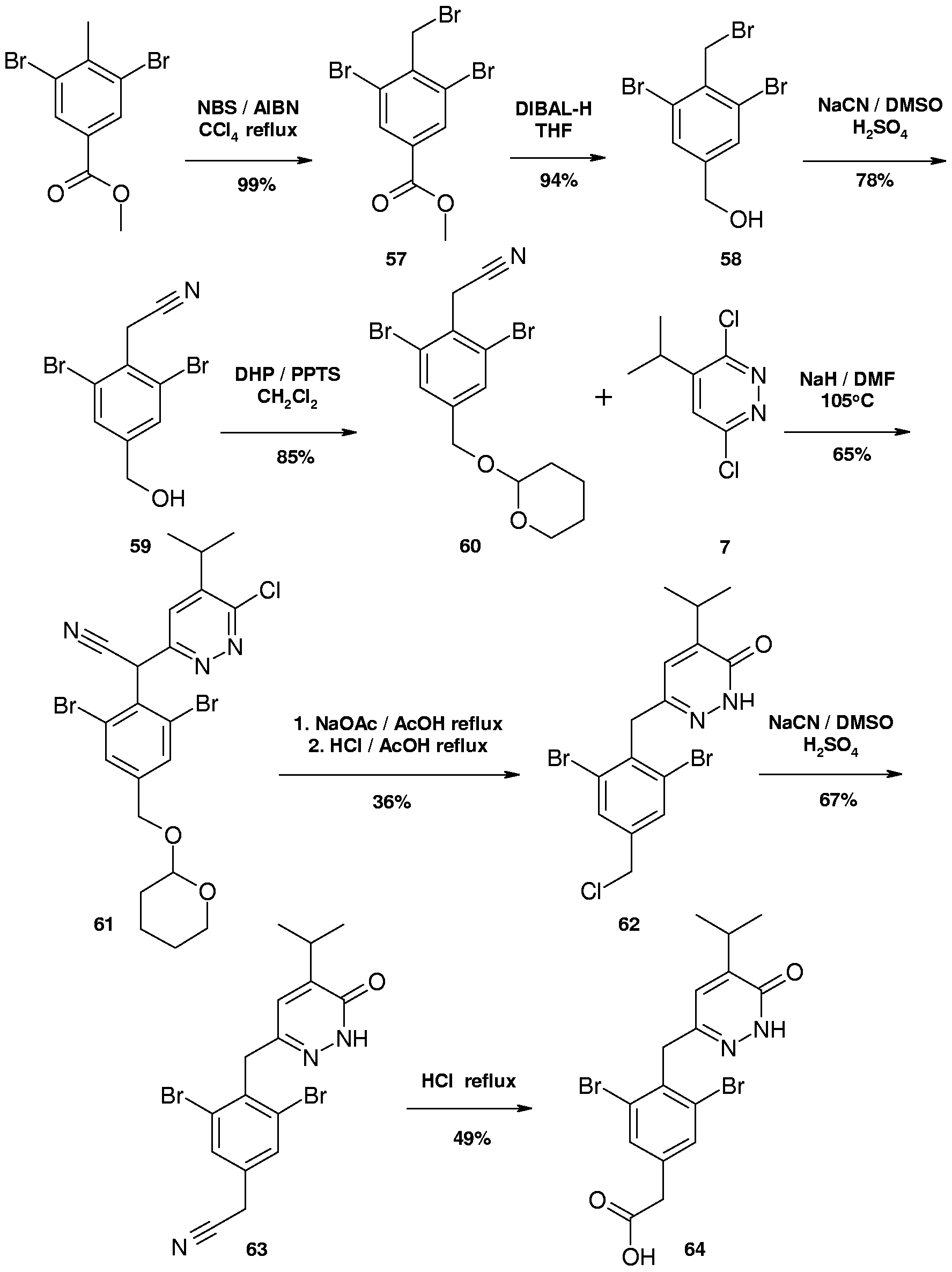

















Claims
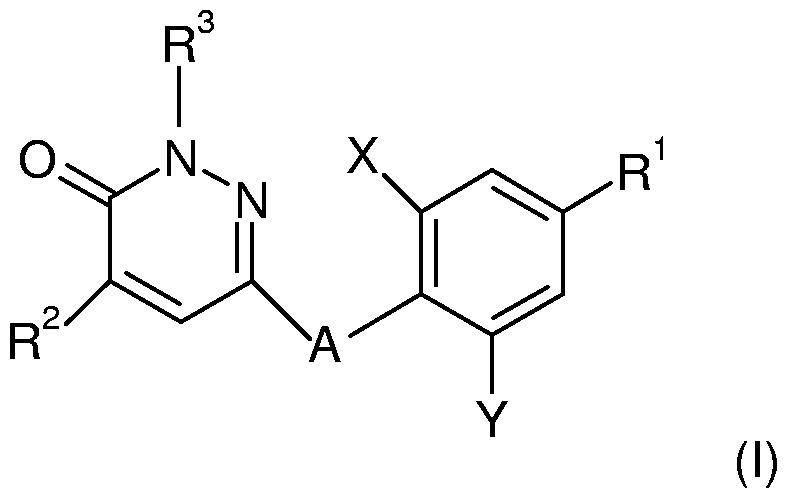









Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2006800267317A CN101228135B (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| DK06792493.6T DK1919878T3 (en) | 2005-07-21 | 2006-07-11 | Pyridazine derivatives as thyroid hormone receptor agonists |
| AU2006271721A AU2006271721C1 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| DE602006016821T DE602006016821D1 (en) | 2005-07-21 | 2006-07-11 | PYRIDAZINE DERIVATIVES AS THYROID HORMONE RECEPTOR AGONISTS |
| NZ565190A NZ565190A (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| BRPI0613754A BRPI0613754B8 (en) | 2005-07-21 | 2006-07-11 | compounds, ester of a compound, pharmaceutical compositions comprising the compound, method for the therapeutic and/or prophylactic treatment of diseases that are modulated by thyroid hormone analogues, and use of the compounds |
| EP06792493A EP1919878B1 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| SI200630817T SI1919878T1 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| HR20100633T HRP20100633T1 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| PL06792493T PL1919878T3 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| JP2008521935A JP5000649B2 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| HK08110278.4A HK1115129B (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivative as thyroid hormone receptor agonists |
| AT06792493T ATE480524T1 (en) | 2005-07-21 | 2006-07-11 | PYRIDAZINONE DERIVATIVES AS THYROID HORMONE RECEPTOR AGONISTS |
| CA2614529A CA2614529C (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| MX2008000818A MX2008000818A (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists. |
| IL188476A IL188476A (en) | 2005-07-21 | 2007-12-27 | Pyridazinone derivatives as thyroid hormone receptor agonists |
| NO20080058A NO340679B1 (en) | 2005-07-21 | 2008-01-04 | Pyridazine derivatives, pharmaceutical compositions comprising such and such compounds and preparations for use in therapeutically active substances for the treatment and prevention of diseases |
| ZA2008/00405A ZA200800405B (en) | 2005-07-21 | 2008-01-14 | Pyrisazinone derivatives as thyroid hormone receptor agonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US70121505P | 2005-07-21 | 2005-07-21 | |
| US60/701,215 | 2005-07-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007009913A1 true WO2007009913A1 (en) | 2007-01-25 |
Family
ID=37114368
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/064093 Ceased WO2007009913A1 (en) | 2005-07-21 | 2006-07-11 | Pyridazinone derivatives as thyroid hormone receptor agonists |
Country Status (31)
| Country | Link |
|---|---|
| US (3) | US7452882B2 (en) |
| EP (1) | EP1919878B1 (en) |
| JP (1) | JP5000649B2 (en) |
| KR (1) | KR100965006B1 (en) |
| CN (1) | CN101228135B (en) |
| AR (1) | AR054848A1 (en) |
| AT (1) | ATE480524T1 (en) |
| AU (1) | AU2006271721C1 (en) |
| BR (1) | BRPI0613754B8 (en) |
| CA (1) | CA2614529C (en) |
| CR (1) | CR9644A (en) |
| CY (1) | CY1110929T1 (en) |
| DE (1) | DE602006016821D1 (en) |
| DK (1) | DK1919878T3 (en) |
| EC (1) | ECSP088120A (en) |
| ES (1) | ES2349131T3 (en) |
| HR (1) | HRP20100633T1 (en) |
| IL (1) | IL188476A (en) |
| MA (1) | MA29731B1 (en) |
| MX (1) | MX2008000818A (en) |
| MY (1) | MY142861A (en) |
| NO (1) | NO340679B1 (en) |
| NZ (1) | NZ565190A (en) |
| PL (1) | PL1919878T3 (en) |
| PT (1) | PT1919878E (en) |
| RS (1) | RS51639B (en) |
| RU (1) | RU2379295C2 (en) |
| TW (1) | TWI330636B (en) |
| UA (1) | UA88104C2 (en) |
| WO (1) | WO2007009913A1 (en) |
| ZA (1) | ZA200800405B (en) |
Cited By (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2007138351A3 (en) * | 2006-05-31 | 2008-08-07 | Angeletti P Ist Richerche Bio | Pyridinone and pyridazinone derivatives as inhibitors of poly(adp-ribose)polymerase (parp) |
| WO2008149379A3 (en) * | 2007-06-06 | 2009-03-19 | Torrent Pharmaceuticals Ltd | Novel compounds |
| WO2009037172A1 (en) * | 2007-09-20 | 2009-03-26 | F. Hoffmann-La Roche Ag | Prodrugs to thyroid hormone analogs |
| US7514419B2 (en) | 2003-11-19 | 2009-04-07 | Metabasis Therapeutics, Inc. | Phosphorus-containing thyromimetics |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| JPWO2008133141A1 (en) * | 2007-04-24 | 2010-07-22 | 東洋紡績株式会社 | Osmotin recombinant protein, method for producing the same, and use thereof |
| WO2010122980A1 (en) | 2009-04-20 | 2010-10-28 | 田辺三菱製薬株式会社 | Novel thyroid hormone β receptor agonist |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| US8268827B2 (en) | 2007-11-15 | 2012-09-18 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa. | Pyridazinone derivatives as PARP inhibitors |
| WO2014043706A1 (en) * | 2012-09-17 | 2014-03-20 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US10130643B2 (en) | 2005-05-26 | 2018-11-20 | Metabasis Therapeutics, Inc. | Thyromimetics for the treatment of fatty liver diseases |
| WO2019054514A1 (en) | 2017-09-14 | 2019-03-21 | 国立研究開発法人理化学研究所 | Method for producing retinal tissues |
| WO2019240938A1 (en) | 2018-06-12 | 2019-12-19 | Fronthera U.S. Pharmaceuticals Llc | Thyroid hormone receptor agonists and uses thereof |
| WO2020010068A1 (en) * | 2018-07-02 | 2020-01-09 | Madrigal Pharmaceuticals, Inc. | Solid forms of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile |
| WO2020041741A1 (en) | 2018-08-24 | 2020-02-27 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2020073974A1 (en) * | 2018-10-12 | 2020-04-16 | Inventisbio Shanghai Ltd. | Thyroid hormone receptor agonists |
| WO2020123827A1 (en) | 2018-12-13 | 2020-06-18 | Terns, Inc. | Thrβ receptor agonist compound and preparation method and use thereof |
| WO2020169069A1 (en) | 2019-02-21 | 2020-08-27 | Nanjing Ruijie Pharma Co., Ltd. | Novel compounds and their uses as thyroid hormone receptor agonists |
| WO2020184720A1 (en) | 2019-03-13 | 2020-09-17 | 大日本住友製薬株式会社 | Method for evaluating quality of transplant neural retina, and transplant neural retina sheet |
| WO2020227549A1 (en) * | 2019-05-08 | 2020-11-12 | Aligos Therapeutics, Inc. | MODULATORS OF THR-β AND METHODS OF USE THEREOF |
| WO2020239076A1 (en) * | 2019-05-29 | 2020-12-03 | 南京明德新药研发有限公司 | Pyridazinone derivatives as thyroxine receptor-β agonists and application thereof |
| EP3725779A4 (en) * | 2018-01-23 | 2021-02-24 | Shenzhen TargetRx, Inc. | SUBSTITUTED PYRIDAZINONE COMPOUND |
| WO2021032218A1 (en) | 2019-08-19 | 2021-02-25 | 苏州闻天医药科技有限公司 | HETEROCYCLIC THR-β RECEPTOR AGONIST COMPOUND AND PREPARATION METHOD AND USE THEREFOR |
| WO2021041237A1 (en) | 2019-08-23 | 2021-03-04 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2021043185A1 (en) * | 2019-09-04 | 2021-03-11 | Sunshine Lake Pharma Co., Ltd. | A compound as a thyroid hormone beta receptor agonist and use thereof |
| WO2021050945A1 (en) * | 2019-09-12 | 2021-03-18 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| KR20210057758A (en) * | 2018-09-06 | 2021-05-21 | 갈메드 리서치 앤드 디벨롭먼트 리미티드 | Combination therapy to treat liver disease |
| WO2021104288A1 (en) | 2019-11-26 | 2021-06-03 | 昆药集团股份有限公司 | 1,2,4-triazine-3,5-dione compound, preparation method therefor, and application thereof |
| US11090308B2 (en) | 2016-10-18 | 2021-08-17 | Madrigal Pharmaceuticals, Inc. | Methods of treating liver disorders or lipid disorders with a THR-beta agonist |
| CN113698388A (en) * | 2020-05-20 | 2021-11-26 | 江苏恒瑞医药股份有限公司 | 6-oxo-1, 6-dihydropyridine derivatives, preparation method and application thereof in medicines |
| US11202789B2 (en) | 2016-11-21 | 2021-12-21 | Viking Therapeutics, Inc. | Method of treating glycogen storage disease |
| US11203587B2 (en) | 2018-10-12 | 2021-12-21 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2022041026A1 (en) * | 2020-08-27 | 2022-03-03 | InventisBio Co., Ltd. | Pyridazinone compounds |
| WO2022054925A1 (en) | 2020-09-11 | 2022-03-17 | 国立研究開発法人理化学研究所 | Complex containing neural retina-containing cell aggregates and matrix, and method for manufacturing same |
| WO2022054924A1 (en) | 2020-09-11 | 2022-03-17 | 大日本住友製薬株式会社 | Medium for tissue for transplantation |
| WO2022068915A1 (en) * | 2020-09-30 | 2022-04-07 | 江苏恒瑞医药股份有限公司 | 6-oxo-1,6-dihydropyridazin derivative, preparation method therefor, and application thereof in medicine |
| WO2022086894A1 (en) | 2020-10-19 | 2022-04-28 | Teva Pharmaceuticals International Gmbh | Solid state forms of resmetirom |
| WO2022099060A3 (en) * | 2020-11-06 | 2022-06-16 | Aligos Therapeutics, Inc. | Bicyclic pyridazinones as thyroid hormone receptor beta (tr-beta) agonists |
| WO2022143574A1 (en) | 2020-12-30 | 2022-07-07 | 昆药集团股份有限公司 | 2-pyridone derivative, and preparation method therefor and pharmaceutical application thereof |
| CN115073429A (en) * | 2021-03-15 | 2022-09-20 | 昆药集团股份有限公司 | Salt form and crystal form of 1,2, 4-triazine-3, 5-diketone compound and preparation method thereof |
| EP4019510A4 (en) * | 2019-08-21 | 2023-01-25 | Hinova Pharmaceuticals Inc. | HALOGEN SUBSTITUTED PHENYLATE COMPOUND AND USES THEREOF |
| CN115650928A (en) * | 2022-12-28 | 2023-01-31 | 凯思凯旭(上海)医药科技有限公司 | Polycyclic thyroid hormone beta receptor agonist and application thereof |
| CN116063296A (en) * | 2021-11-02 | 2023-05-05 | 凯思凯迪(上海)医药科技有限公司 | A compound as thyroid hormone beta receptor agonist and its use |
| WO2023090427A1 (en) | 2021-11-19 | 2023-05-25 | 国立研究開発法人理化学研究所 | Production method for sheet-like retinal tissue |
| US11707472B2 (en) | 2017-06-05 | 2023-07-25 | Viking Therapeutics, Inc. | Compositions for the treatment of fibrosis |
| RU2802282C2 (en) * | 2012-09-17 | 2023-08-24 | Ф. Хоффманн-Ля Рош Лтд. | Method for synthesis of thyroid hormone analogues and their polymorphs |
| US11780825B2 (en) | 2020-01-13 | 2023-10-10 | Eccogene (Shanghai) Co., Ltd. | Thyroid hormone receptor agonists and use thereof |
| US11786460B2 (en) | 2018-04-16 | 2023-10-17 | Ioulia Tseti | Pharmaceutical dry powder composition for inhalation comprising a thyroid hormone |
| EP4036090A4 (en) * | 2019-09-24 | 2023-11-15 | Sunshine Lake Pharma Co., Ltd. | CHEMICAL COMPOUND AS THYROID HORMONE BETA RECEPTOR AGONIST AND USE THEREOF |
| WO2024084491A1 (en) * | 2022-10-21 | 2024-04-25 | Mehta Nilesh Vipinchandra | Process for synthesis of res metirom and its intermediates thereof |
| WO2024120426A1 (en) | 2022-12-07 | 2024-06-13 | 昆药集团股份有限公司 | THYROID HORMONE β RECEPTOR AGONIST, CRYSTAL FORM, PREPARATION METHOD AND USE |
| WO2024141076A1 (en) * | 2022-12-29 | 2024-07-04 | Gannex Pharma Co., Ltd. | Prodrugs to thyroid hormone analogs and method of making and use thereof |
| US12102646B2 (en) | 2018-12-05 | 2024-10-01 | Viking Therapeutics, Inc. | Compositions for the treatment of fibrosis and inflammation |
| US12227533B2 (en) | 2018-03-22 | 2025-02-18 | Viking Therapeutics, Inc. | Crystalline forms and methods of producing crystalline forms of a compound |
| WO2025083699A2 (en) | 2023-10-16 | 2025-04-24 | Cipla Limited | Resmetirom polymorphs and process thereof |
| US12377104B1 (en) | 2024-02-06 | 2025-08-05 | Madrigal Pharmaceuticals, Inc. | Methods for treating a fatty liver disease |
| WO2025191457A1 (en) * | 2024-03-13 | 2025-09-18 | Morepen Laboratories Limited | Process for the preparation of pyridazinone derivative |
| WO2025230749A1 (en) | 2024-05-03 | 2025-11-06 | Aligos Therapeutics, Inc. | Methods of preparing 6-amino-2-[3,5-dichloro-4-[(5-isopropyl-6-oxo-1 h-pyridazin-3-yl)oxy]phenyl]-4h- 1, 2, 4-tri-azine-3, 5-dione |
| US12485118B2 (en) | 2023-04-07 | 2025-12-02 | Terns Pharmaceuticals, Inc. | Combinations of GLP-1R and THRβ agonists and methods of use thereof |
| WO2026074443A1 (en) | 2024-10-01 | 2026-04-09 | Assia Chemical Industries Ltd. | Solid state forms of resmetirom |
Families Citing this family (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1604988A1 (en) * | 2004-05-18 | 2005-12-14 | Sanofi-Aventis Deutschland GmbH | Pyridazinone derivatives, methods for producing them and their use as pharmaceuticals |
| PL1919878T3 (en) | 2005-07-21 | 2011-03-31 | Hoffmann La Roche | Pyridazinone derivatives as thyroid hormone receptor agonists |
| KR100981781B1 (en) | 2008-02-29 | 2010-09-10 | 재단법인 한국원자력의학원 | Collimator device for radiation therapy and radiation therapy device using the device |
| JP5847533B2 (en) * | 2010-10-19 | 2016-01-27 | 田辺三菱製薬株式会社 | Novel thyroid hormone beta receptor agonist |
| CN105272927A (en) * | 2015-11-17 | 2016-01-27 | 华中药业股份有限公司 | Novel method for preparing chlordiazepoxide |
| CN110627773B (en) * | 2018-06-22 | 2021-03-19 | 海创药业股份有限公司 | Deuterated MGL-3196 compound and application thereof |
| CN111592528A (en) * | 2019-02-20 | 2020-08-28 | 苏州泽璟生物制药股份有限公司 | Deuterated pyridazinone, derivatives thereof and pharmaceutical compositions |
| WO2020228577A1 (en) * | 2019-05-10 | 2020-11-19 | 深圳微芯生物科技股份有限公司 | Pyridazinone derivative and use thereof |
| CN112707892A (en) * | 2019-10-24 | 2021-04-27 | 苏州泽璟生物制药股份有限公司 | Pyridazinone or pyridazine compound and derivative and pharmaceutical composition thereof |
| WO2021129465A1 (en) * | 2019-12-26 | 2021-07-01 | 苏州科睿思制药有限公司 | Resmetirom crystal, preparation method for same, and uses thereof |
| CN113045551B (en) * | 2019-12-27 | 2024-07-09 | 广东东阳光药业股份有限公司 | A compound as a thyroid hormone beta receptor agonist and its use |
| CN113278013B (en) * | 2020-02-20 | 2022-09-23 | 昆药集团股份有限公司 | 2, 6-disubstituted 1,2, 4-triazine-3, 5-diketone compound and preparation method and application thereof |
| US20230257367A1 (en) | 2020-06-02 | 2023-08-17 | Chengdu Kanghong Pharmaceutical Co., Ltd. | Novel thyroid hormone beta receptor agonist |
| WO2022053028A1 (en) * | 2020-09-10 | 2022-03-17 | 南京明德新药研发有限公司 | 1,2,4-triazine-3,5(2h,4h)-diketone compound and application thereof |
| US12595254B2 (en) | 2020-09-10 | 2026-04-07 | Crystal Pharmaceutical (Suzhou) Co., Ltd. | Crystal form of Resmetirom, preparation method therefor, and use thereof |
| IL301821A (en) * | 2020-10-23 | 2023-06-01 | Madrigal Pharmaceuticals Inc | Process for the preparation of 6-(4-nitro-phenoxy)-2H-pyridazin-3-one and 6-(4-amino-phenoxy)-2H-pyridazin-3-one derivatives as intermediates of thyroid hormone analogues |
| CN114437042A (en) * | 2020-11-03 | 2022-05-06 | 深圳微芯生物科技股份有限公司 | Crystal form of phthalazine compound and preparation method thereof |
| WO2022099049A1 (en) * | 2020-11-06 | 2022-05-12 | Aligos Therapeutics, Inc. | Pyridazinone derivatives as thyroid receptor agonists and uses thereof |
| CN112645936B (en) | 2020-12-17 | 2022-03-01 | 山东第一医科大学(山东省医学科学院) | Substituted pyridazinone compound and use thereof |
| CN114907327A (en) * | 2021-02-10 | 2022-08-16 | 杭州领业医药科技有限公司 | Crystal form of Resmetirom and preparation method and application thereof |
| WO2023171556A1 (en) * | 2022-03-10 | 2023-09-14 | 積水化学工業株式会社 | Viral infection inhibitor, viral infection inhibiting product, viral infection inhibiting paint, and resin composition |
| CN116836158A (en) * | 2022-03-23 | 2023-10-03 | 凯思凯迪(上海)医药科技有限公司 | Compound serving as thyroid hormone beta receptor agonist and application thereof |
| CN116768802B (en) * | 2023-05-25 | 2025-11-21 | 杭州科巢生物科技有限公司 | Rasemet-teh Synthesis method of roxburgh |
| WO2025011259A1 (en) * | 2023-07-07 | 2025-01-16 | 苏州科睿思制药有限公司 | Crystal form of resmetirom, and preparation method therefor and use thereof |
| CN117263870B (en) * | 2023-09-21 | 2026-02-06 | 江西同和药业股份有限公司 | Preparation method of Resmetirom key intermediate III |
| WO2025171032A1 (en) | 2024-02-06 | 2025-08-14 | Madrigal Pharmaceuticals, Inc. | Methods for treating a fatty liver disease |
| CN119868564A (en) * | 2024-09-30 | 2025-04-25 | 海创药业股份有限公司 | Combined medicine for preventing and/or treating obesity |
| CN119798230B (en) * | 2025-03-13 | 2025-06-27 | 中国药科大学 | Indolinone compounds and their application in treating acute and chronic liver diseases and promoting tissue repair |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0188351A2 (en) * | 1985-01-18 | 1986-07-23 | Smith Kline & French Laboratories Limited | Chemical compounds |
| US5284971A (en) * | 1992-07-16 | 1994-02-08 | Syntex (U.S.A.) Inc. | 4-(3-cyclohexyl-4-hydroxy or-methoxy phenylsulfonyl) 3,5 dibromo phenyl acetic thyromimetic cholesterol-lowering agents |
| WO1999000353A1 (en) * | 1997-06-27 | 1999-01-07 | Karo Bio Ab | Novel thyroid receptor ligands and method |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4582842A (en) * | 1981-02-25 | 1986-04-15 | Merck & Co., Inc. | Anti-asthmatic 6H-dibenz-[b,e] [1,4]oxathiepin derivatives, compositions, and method of use therefor |
| AU4440596A (en) | 1995-02-10 | 1996-08-22 | Smithkline Beecham Corporation | Use of hcp SH2 specific compounds to enhance erythropoiesis |
| PT832067E (en) | 1995-06-06 | 2003-10-31 | Pfizer | PROCESS FOR THE CONVERSATION OF 2,4-DICHLOROPYRIDINES TO 2-ARYLOXY-4-CHLOROPYRIDINES |
| AU6480796A (en) | 1995-06-30 | 1997-02-05 | Smithkline Beecham Corporation | Use of stat 6 sh2 domain specific compounds to treat allergic reactions |
| US5883294A (en) | 1997-06-18 | 1999-03-16 | The Regeants Of The University Of California | Selective thyroid hormone analogs |
| CN1142148C (en) * | 1997-11-19 | 2004-03-17 | 兴和株式会社 | Novel pyridazine derivative and drug containing the same as active ingredient |
| AR023659A1 (en) | 1998-09-18 | 2002-09-04 | Vertex Pharma | AN INHIBITOR COMPOUND OF P38, A PHARMACEUTICAL COMPOSITION THAT INCLUDES IT AND THE USE OF SUCH COMPOSITION IN THE TREATMENT AND PREVENTION OF PATHOLOGICAL STATES |
| EA004229B1 (en) * | 1999-03-01 | 2004-02-26 | Пфайзер Продактс Инк. | Oxamic acids and derivatives as thyroid receptor ligands |
| WO2002051805A1 (en) * | 2000-12-27 | 2002-07-04 | Bayer Aktiengesellschaft | Indole derivatives as ligands of thyroid receptors |
| EP1358175A2 (en) * | 2001-02-08 | 2003-11-05 | Karo Bio Ab | Phenoxy substituted benzocondensed heteroaryl derivatives as thyroid receptor ligands |
| EP1236720B1 (en) * | 2001-02-28 | 2005-06-15 | Pfizer Products Inc. | Sulfonyl pyridazinone compounds useful as aldose reductase inhibitors |
| BRPI0408704A (en) * | 2003-03-24 | 2006-03-07 | Hoffmann La Roche | benzyl pyridazinones as reverse transcriptase inhibitors |
| CN1882327A (en) | 2003-11-19 | 2006-12-20 | 症变治疗公司 | Novel phosphorus-containing thyromimetics |
| WO2005090317A1 (en) * | 2004-03-23 | 2005-09-29 | F.Hoffmann-La Roche Ag | Non-nucleoside reverse transcriptase inhibitors |
| PL1919878T3 (en) | 2005-07-21 | 2011-03-31 | Hoffmann La Roche | Pyridazinone derivatives as thyroid hormone receptor agonists |
-
2006
- 2006-07-11 PL PL06792493T patent/PL1919878T3/en unknown
- 2006-07-11 JP JP2008521935A patent/JP5000649B2/en active Active
- 2006-07-11 BR BRPI0613754A patent/BRPI0613754B8/en active IP Right Grant
- 2006-07-11 DK DK06792493.6T patent/DK1919878T3/en active
- 2006-07-11 ES ES06792493T patent/ES2349131T3/en active Active
- 2006-07-11 PT PT06792493T patent/PT1919878E/en unknown
- 2006-07-11 KR KR1020087001480A patent/KR100965006B1/en active Active
- 2006-07-11 CN CN2006800267317A patent/CN101228135B/en active Active
- 2006-07-11 RU RU2008106058/04A patent/RU2379295C2/en active
- 2006-07-11 RS RS20100527A patent/RS51639B/en unknown
- 2006-07-11 EP EP06792493A patent/EP1919878B1/en active Active
- 2006-07-11 AU AU2006271721A patent/AU2006271721C1/en active Active
- 2006-07-11 AT AT06792493T patent/ATE480524T1/en active
- 2006-07-11 DE DE602006016821T patent/DE602006016821D1/en active Active
- 2006-07-11 NZ NZ565190A patent/NZ565190A/en unknown
- 2006-07-11 WO PCT/EP2006/064093 patent/WO2007009913A1/en not_active Ceased
- 2006-07-11 MX MX2008000818A patent/MX2008000818A/en active IP Right Grant
- 2006-07-11 HR HR20100633T patent/HRP20100633T1/en unknown
- 2006-07-11 CA CA2614529A patent/CA2614529C/en active Active
- 2006-07-18 US US11/488,870 patent/US7452882B2/en active Active
- 2006-07-18 TW TW095126242A patent/TWI330636B/en active
- 2006-07-19 AR ARP060103084A patent/AR054848A1/en active IP Right Grant
- 2006-07-19 MY MYPI20063456A patent/MY142861A/en unknown
- 2006-11-07 UA UAA200801933A patent/UA88104C2/en unknown
-
2007
- 2007-12-27 IL IL188476A patent/IL188476A/en active IP Right Grant
-
2008
- 2008-01-04 NO NO20080058A patent/NO340679B1/en unknown
- 2008-01-08 CR CR9644A patent/CR9644A/en unknown
- 2008-01-14 ZA ZA2008/00405A patent/ZA200800405B/en unknown
- 2008-01-18 EC EC2008008120A patent/ECSP088120A/en unknown
- 2008-02-01 MA MA30618A patent/MA29731B1/en unknown
- 2008-08-20 US US12/194,643 patent/US7807674B2/en active Active
-
2010
- 2010-11-22 CY CY20101101049T patent/CY1110929T1/en unknown
-
2015
- 2015-07-01 US US14/788,921 patent/USRE46024E1/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0188351A2 (en) * | 1985-01-18 | 1986-07-23 | Smith Kline & French Laboratories Limited | Chemical compounds |
| US5284971A (en) * | 1992-07-16 | 1994-02-08 | Syntex (U.S.A.) Inc. | 4-(3-cyclohexyl-4-hydroxy or-methoxy phenylsulfonyl) 3,5 dibromo phenyl acetic thyromimetic cholesterol-lowering agents |
| WO1999000353A1 (en) * | 1997-06-27 | 1999-01-07 | Karo Bio Ab | Novel thyroid receptor ligands and method |
Cited By (157)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7829552B2 (en) | 2003-11-19 | 2010-11-09 | Metabasis Therapeutics, Inc. | Phosphorus-containing thyromimetics |
| US7514419B2 (en) | 2003-11-19 | 2009-04-07 | Metabasis Therapeutics, Inc. | Phosphorus-containing thyromimetics |
| US10130643B2 (en) | 2005-05-26 | 2018-11-20 | Metabasis Therapeutics, Inc. | Thyromimetics for the treatment of fatty liver diseases |
| US10925885B2 (en) | 2005-05-26 | 2021-02-23 | Metabasis Therapeutics, Inc. | Thyromimetics for the treatment of fatty liver diseases |
| JP2009538896A (en) * | 2006-05-31 | 2009-11-12 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Pyridinone and pyridazinone derivatives as poly (ADP-ribose) polymerase (PARP) inhibitors |
| NO343950B1 (en) * | 2006-05-31 | 2019-07-22 | Msd Italia Srl | Pyridinone and pyridazinone derivatives as inhibitors of poly (ADP-ribose) polymerase (PARP), as well as pharmaceutical composition, and pharmaceutical combination thereof for use in therapy |
| WO2007138351A3 (en) * | 2006-05-31 | 2008-08-07 | Angeletti P Ist Richerche Bio | Pyridinone and pyridazinone derivatives as inhibitors of poly(adp-ribose)polymerase (parp) |
| US8188084B2 (en) | 2006-05-31 | 2012-05-29 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa. | Pyridinone and pyridazinone derivatives as inhibitors of poly (ADP-ribose) polymerase (PARP) |
| AU2007266836B2 (en) * | 2006-05-31 | 2013-09-26 | Msd Italia S.R.L. | Pyridinone and pyridazinone derivatives as inhibitors of poly(ADP-ribose)polymerase (PARP) |
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| JPWO2008133141A1 (en) * | 2007-04-24 | 2010-07-22 | 東洋紡績株式会社 | Osmotin recombinant protein, method for producing the same, and use thereof |
| JP2010529108A (en) * | 2007-06-06 | 2010-08-26 | トレント ファーマシューティカルズ リミテッド | New compounds |
| WO2008149379A3 (en) * | 2007-06-06 | 2009-03-19 | Torrent Pharmaceuticals Ltd | Novel compounds |
| EA019084B1 (en) * | 2007-06-06 | 2014-01-30 | Торрент Фармасьютикалс Лтд. | Novel thyroid compounds for treating diseases associated with metabolism |
| US8378118B2 (en) | 2007-06-06 | 2013-02-19 | Torrent Pharmaceuticals Ltd. | Pyrazole-based thyroid receptor compounds |
| KR101152965B1 (en) | 2007-06-06 | 2012-06-08 | 토렌트 파마슈티칼스 리미티드 | Novel compounds |
| US8143424B2 (en) | 2007-06-06 | 2012-03-27 | Torrent Pharmaceuticals Ltd. | Thyroid like compounds |
| CN101801960A (en) * | 2007-09-20 | 2010-08-11 | 霍夫曼-拉罗奇有限公司 | prodrugs to thyroid hormone analogs |
| WO2009037172A1 (en) * | 2007-09-20 | 2009-03-26 | F. Hoffmann-La Roche Ag | Prodrugs to thyroid hormone analogs |
| CN101801960B (en) * | 2007-09-20 | 2014-04-16 | 霍夫曼-拉罗奇有限公司 | Prodrugs to thyroid hormone analogs |
| KR101189040B1 (en) * | 2007-09-20 | 2012-10-08 | 에프. 호프만-라 로슈 아게 | Prodrugs to thyroid hormone analogs |
| US8076334B2 (en) | 2007-09-20 | 2011-12-13 | Hoffmann-La Roche Inc. | Prodrugs of thyroid hormone analogs |
| AU2008300649B2 (en) * | 2007-09-20 | 2013-05-02 | F. Hoffmann-La Roche Ag | Prodrugs to thyroid hormone analogs |
| JP2010539209A (en) * | 2007-09-20 | 2010-12-16 | エフ.ホフマン−ラ ロシュ アーゲー | Prodrug of thyroid hormone analog |
| US8268827B2 (en) | 2007-11-15 | 2012-09-18 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa. | Pyridazinone derivatives as PARP inhibitors |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US8791266B2 (en) | 2009-04-20 | 2014-07-29 | Mitsubishi Tanabe Pharma Corporation | Thyroid hormone β receptor agonist |
| KR101396606B1 (en) | 2009-04-20 | 2014-05-16 | 미쓰비시 타나베 파마 코퍼레이션 | NOVEL THYROID HORMONE β RECEPTOR AGONIST |
| WO2010122980A1 (en) | 2009-04-20 | 2010-10-28 | 田辺三菱製薬株式会社 | Novel thyroid hormone β receptor agonist |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| KR20150056630A (en) * | 2012-09-17 | 2015-05-26 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US11986481B2 (en) | 2012-09-17 | 2024-05-21 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| AU2013315017C1 (en) * | 2012-09-17 | 2017-11-23 | F. Hoffmann-La Roche Ltd. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US9968612B2 (en) | 2012-09-17 | 2018-05-15 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| RU2668960C2 (en) * | 2012-09-17 | 2018-10-05 | Мадригал Фармасьютикалз, Инк. | Method of synthesis of analogues of hormones of the thyroid gland and their polymorphs |
| EP2895466A4 (en) * | 2012-09-17 | 2016-08-10 | Madrigal Pharmaceuticals Inc | METHOD FOR SYNTHESIZING ANALOGUES OF THYROID HORMONE AND ITS POLYMORPHS |
| KR20180131647A (en) * | 2012-09-17 | 2018-12-10 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| KR102363776B1 (en) * | 2012-09-17 | 2022-02-15 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US9266861B2 (en) | 2012-09-17 | 2016-02-23 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US10376517B2 (en) | 2012-09-17 | 2019-08-13 | Madrigal Pharmaceuticals, Inc. | Methods of synthesizing thyroid hormone analogs and polymorphs thereof |
| KR101966490B1 (en) * | 2012-09-17 | 2019-08-14 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| EP4691557A3 (en) * | 2012-09-17 | 2026-03-18 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| EP4023641A3 (en) * | 2012-09-17 | 2022-09-14 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| EP4406594A3 (en) * | 2012-09-17 | 2024-11-06 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| IL288133B1 (en) * | 2012-09-17 | 2024-08-01 | F Hoffmann La Roche Ltd | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| AU2013315017B2 (en) * | 2012-09-17 | 2017-07-27 | F. Hoffmann-La Roche Ltd. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| KR102138750B1 (en) | 2012-09-17 | 2020-07-29 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| EP3689853A1 (en) * | 2012-09-17 | 2020-08-05 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| KR20210083381A (en) * | 2012-09-17 | 2021-07-06 | 마드리갈 파마슈티칼스, 인크. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| WO2014043706A1 (en) * | 2012-09-17 | 2014-03-20 | Madrigal Pharmaceuticals, Inc. | Method of synthesizing thyroid hormone analogs and polymorphs thereof |
| US10894050B2 (en) | 2012-09-17 | 2021-01-19 | Madrigal Pharmaceuticals, Inc. | Methods of synthesizing thyroid hormone analogs and polymorphs thereof |
| RU2802282C2 (en) * | 2012-09-17 | 2023-08-24 | Ф. Хоффманн-Ля Рош Лтд. | Method for synthesis of thyroid hormone analogues and their polymorphs |
| US11564926B2 (en) | 2012-09-17 | 2023-01-31 | Madrigal Pharmaceuticals, Inc. | Methods of synthesizing thyroid hormone analogs and polymorphs thereof |
| US11806353B2 (en) | 2016-10-18 | 2023-11-07 | Madrigal Pharmaceuticals, Inc. | Methods of treating liver disorders or lipid disorders with a THR-beta agonist |
| US11090308B2 (en) | 2016-10-18 | 2021-08-17 | Madrigal Pharmaceuticals, Inc. | Methods of treating liver disorders or lipid disorders with a THR-beta agonist |
| US11202789B2 (en) | 2016-11-21 | 2021-12-21 | Viking Therapeutics, Inc. | Method of treating glycogen storage disease |
| US11707472B2 (en) | 2017-06-05 | 2023-07-25 | Viking Therapeutics, Inc. | Compositions for the treatment of fibrosis |
| WO2019054514A1 (en) | 2017-09-14 | 2019-03-21 | 国立研究開発法人理化学研究所 | Method for producing retinal tissues |
| EP3725779A4 (en) * | 2018-01-23 | 2021-02-24 | Shenzhen TargetRx, Inc. | SUBSTITUTED PYRIDAZINONE COMPOUND |
| US11485729B2 (en) | 2018-01-23 | 2022-11-01 | Shenzhen Targetrx, Inc. | Substituted pyridazinone compound |
| US12227533B2 (en) | 2018-03-22 | 2025-02-18 | Viking Therapeutics, Inc. | Crystalline forms and methods of producing crystalline forms of a compound |
| US11786460B2 (en) | 2018-04-16 | 2023-10-17 | Ioulia Tseti | Pharmaceutical dry powder composition for inhalation comprising a thyroid hormone |
| US11964964B2 (en) | 2018-06-12 | 2024-04-23 | Xizang Haisco Pharmaceutical Co., Ltd. | Thyroid hormone receptor agonists and uses thereof |
| EP3807267B1 (en) | 2018-06-12 | 2025-03-19 | Xizang Haisco Pharmaceutical Co., Ltd. | Thyroid hormone receptor agonists and uses thereof |
| KR20200143711A (en) * | 2018-06-12 | 2020-12-24 | 프론테라 유에스 파마슈티컬스 엘엘씨 | Thyroid hormone receptor agonists and uses thereof |
| KR102531771B1 (en) * | 2018-06-12 | 2023-05-11 | 스촨 하이스코 파마수티컬 씨오., 엘티디 | Thyroid hormone receptor agonists and uses thereof |
| US12358899B2 (en) | 2018-06-12 | 2025-07-15 | Xizang Haisco Pharmaceutical Co., Ltd. | Thyroid hormone receptor agonists and uses thereof |
| AU2019287679B2 (en) * | 2018-06-12 | 2022-04-21 | Xizang Haisco Pharmaceutical Co., Ltd. | Thyroid hormone receptor agonists and uses thereof |
| EP4574208A3 (en) * | 2018-06-12 | 2025-10-22 | Xizang Haisco Pharmaceutical Co., Ltd. | Thyroid hormone receptor agonists and uses thereof |
| WO2019240938A1 (en) | 2018-06-12 | 2019-12-19 | Fronthera U.S. Pharmaceuticals Llc | Thyroid hormone receptor agonists and uses thereof |
| EP4552643A3 (en) * | 2018-07-02 | 2025-07-02 | Madrigal Pharmaceuticals, Inc. | Solid forms of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile |
| WO2020010068A1 (en) * | 2018-07-02 | 2020-01-09 | Madrigal Pharmaceuticals, Inc. | Solid forms of 2-(3,5-dichloro-4-((5-isopropyl-6-oxo-1,6-dihydropyridazin-3-yl)oxy)phenyl)-3,5-dioxo-2,3,4,5-tetrahydro-1,2,4-triazine-6-carbonitrile |
| AU2019325656B2 (en) * | 2018-08-24 | 2025-04-24 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| TWI837169B (en) * | 2018-08-24 | 2024-04-01 | 美商拓臻股份有限公司 | Thyroid hormone receptor beta agonist compounds |
| US10800767B2 (en) | 2018-08-24 | 2020-10-13 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2020041741A1 (en) | 2018-08-24 | 2020-02-27 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| IL281244B2 (en) * | 2018-09-06 | 2025-05-01 | Galmed Res And Development Ltd | Combination therapy for the treatment of liver disease |
| US12239712B2 (en) | 2018-09-06 | 2025-03-04 | Galmed Research And Development Ltd | Combination therapy for the treatment of liver disease |
| IL281244B1 (en) * | 2018-09-06 | 2025-01-01 | Galmed Res And Development Ltd | Combination therapy for the treatment of liver disease |
| KR20210057758A (en) * | 2018-09-06 | 2021-05-21 | 갈메드 리서치 앤드 디벨롭먼트 리미티드 | Combination therapy to treat liver disease |
| KR102876077B1 (en) * | 2018-09-06 | 2025-10-23 | 갈메드 리서치 앤드 디벨롭먼트 리미티드 | Combination therapy for treating liver disease |
| EP3846821A4 (en) * | 2018-09-06 | 2022-05-25 | Galmed Research and Development Ltd. | POLYTHERAPY FOR THE TREATMENT OF HEPATIC DISEASE |
| US20210355110A1 (en) * | 2018-10-12 | 2021-11-18 | InventisBio Co., Ltd. | Thyroid hormone receptor agonists |
| CN112739692A (en) * | 2018-10-12 | 2021-04-30 | 益方生物科技(上海)股份有限公司 | Thyroid hormone receptor agonists |
| US12391677B2 (en) | 2018-10-12 | 2025-08-19 | InventisBio Co., Ltd. | Thyroid hormone receptor agonists |
| CN112739692B (en) * | 2018-10-12 | 2024-11-22 | 益方生物科技(上海)股份有限公司 | Thyroid hormone receptor agonists |
| US11203587B2 (en) | 2018-10-12 | 2021-12-21 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2020073974A1 (en) * | 2018-10-12 | 2020-04-16 | Inventisbio Shanghai Ltd. | Thyroid hormone receptor agonists |
| US12102646B2 (en) | 2018-12-05 | 2024-10-01 | Viking Therapeutics, Inc. | Compositions for the treatment of fibrosis and inflammation |
| US12459926B2 (en) | 2018-12-13 | 2025-11-04 | Terns, Inc. | THRβ receptor agonist compound and preparation method and use thereof |
| EP4529954A2 (en) | 2018-12-13 | 2025-04-02 | Terns, Inc. | Thrb receptor agonist compound and preparation method and use thereof |
| US12365669B2 (en) | 2018-12-13 | 2025-07-22 | Terns, Inc. | THRβ receptor agonist compound and preparation method and use thereof |
| WO2020123827A1 (en) | 2018-12-13 | 2020-06-18 | Terns, Inc. | Thrβ receptor agonist compound and preparation method and use thereof |
| EP4574821A1 (en) | 2018-12-13 | 2025-06-25 | Terns, Inc. | Thrb receptor agonist compound and preparation method and use thereof |
| US11034676B2 (en) | 2018-12-13 | 2021-06-15 | Terns, Inc. | THRB receptor agonist compound and preparation method and use thereof |
| US11084802B2 (en) | 2018-12-13 | 2021-08-10 | Terns, Inc. | THRβ receptor agonist compound and preparation method and use thereof |
| US12338232B2 (en) | 2018-12-13 | 2025-06-24 | Terns, Inc. | THRβ receptor agonist compound and preparation method and use thereof |
| RU2809040C2 (en) * | 2018-12-13 | 2023-12-06 | Тернс, Инк. | THRβ RECEPTOR AGONIST AND METHOD OF ITS OBTAINING AND USE |
| CN113194958B (en) * | 2018-12-13 | 2023-05-23 | 拓臻股份有限公司 | THR beta receptor agonist compound, preparation method and application thereof |
| CN113194958A (en) * | 2018-12-13 | 2021-07-30 | 拓臻股份有限公司 | THR beta receptor agonist compound and preparation method and application thereof |
| US12338206B2 (en) * | 2019-02-21 | 2025-06-24 | Nanjing Ruijie Pharma Co., Ltd. | Compounds and their uses as thyroid hormone receptor agonists |
| US20220162161A1 (en) * | 2019-02-21 | 2022-05-26 | Nanjing Ruijie Pharma Co., Ltd. | Novel compounds and their uses as thyroid hormone receptor agonists |
| WO2020169069A1 (en) | 2019-02-21 | 2020-08-27 | Nanjing Ruijie Pharma Co., Ltd. | Novel compounds and their uses as thyroid hormone receptor agonists |
| WO2020184720A1 (en) | 2019-03-13 | 2020-09-17 | 大日本住友製薬株式会社 | Method for evaluating quality of transplant neural retina, and transplant neural retina sheet |
| US12180192B2 (en) | 2019-05-08 | 2024-12-31 | Aligos Therapeutics, Inc. | Modulators of THR-β and methods of use thereof |
| EP4523692A3 (en) * | 2019-05-08 | 2025-06-04 | Aligos Therapeutics, Inc. | Modulators of thr-ss and methods of use thereof |
| US11091467B2 (en) | 2019-05-08 | 2021-08-17 | Aligos Therapeutics, Inc. | Modulators of THR-β and methods of use thereof |
| AU2020267576B2 (en) * | 2019-05-08 | 2025-03-13 | Aligos Therapeutics, Inc. | Modulators of THR-β and methods of use thereof |
| WO2020227549A1 (en) * | 2019-05-08 | 2020-11-12 | Aligos Therapeutics, Inc. | MODULATORS OF THR-β AND METHODS OF USE THEREOF |
| WO2020239076A1 (en) * | 2019-05-29 | 2020-12-03 | 南京明德新药研发有限公司 | Pyridazinone derivatives as thyroxine receptor-β agonists and application thereof |
| CN114174282A (en) * | 2019-05-29 | 2022-03-11 | 南京明德新药研发有限公司 | Pyridazinone derivatives as thyroxine receptor-beta agonists and uses thereof |
| WO2021032218A1 (en) | 2019-08-19 | 2021-02-25 | 苏州闻天医药科技有限公司 | HETEROCYCLIC THR-β RECEPTOR AGONIST COMPOUND AND PREPARATION METHOD AND USE THEREFOR |
| US12378268B2 (en) | 2019-08-19 | 2025-08-05 | Hepagene Therapeutics (HK) Limited | Heterocyclic THR-β receptor agonist compound and preparation method and use therefor |
| EP4019510A4 (en) * | 2019-08-21 | 2023-01-25 | Hinova Pharmaceuticals Inc. | HALOGEN SUBSTITUTED PHENYLATE COMPOUND AND USES THEREOF |
| CN114430743A (en) * | 2019-08-23 | 2022-05-03 | 拓臻制药公司 | Thyroid hormone receptor beta agonist compounds |
| US12528791B2 (en) | 2019-08-23 | 2026-01-20 | Terns Pharmaceuticals, Inc. | Thyroid hormone receptor beta agonist compounds |
| EP4017875A4 (en) * | 2019-08-23 | 2023-05-03 | Terns Pharmaceuticals, Inc. | THYROID HORMONE BETA RECEPTOR AGONIST COMPOUNDS |
| WO2021041237A1 (en) | 2019-08-23 | 2021-03-04 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| WO2021043185A1 (en) * | 2019-09-04 | 2021-03-11 | Sunshine Lake Pharma Co., Ltd. | A compound as a thyroid hormone beta receptor agonist and use thereof |
| WO2021050945A1 (en) * | 2019-09-12 | 2021-03-18 | Terns, Inc. | Thyroid hormone receptor beta agonist compounds |
| US12398127B2 (en) | 2019-09-12 | 2025-08-26 | Terns Pharmaceuticals, Inc. | Thyroid hormone receptor beta agonist compounds |
| EP4036090A4 (en) * | 2019-09-24 | 2023-11-15 | Sunshine Lake Pharma Co., Ltd. | CHEMICAL COMPOUND AS THYROID HORMONE BETA RECEPTOR AGONIST AND USE THEREOF |
| WO2021104288A1 (en) | 2019-11-26 | 2021-06-03 | 昆药集团股份有限公司 | 1,2,4-triazine-3,5-dione compound, preparation method therefor, and application thereof |
| EP4083024A4 (en) * | 2019-11-26 | 2023-11-01 | KPC Pharmaceuticals, Inc. | COMPOUND OF 1,2,4-TRIAZINE-3,5-DIONE, ITS PREPARATION METHOD AND ITS USE |
| US12291518B2 (en) | 2020-01-13 | 2025-05-06 | Eccogene Inc. | Thyroid hormone receptor agonists and use thereof |
| US11780825B2 (en) | 2020-01-13 | 2023-10-10 | Eccogene (Shanghai) Co., Ltd. | Thyroid hormone receptor agonists and use thereof |
| CN113698388B (en) * | 2020-05-20 | 2022-11-22 | 江苏恒瑞医药股份有限公司 | 6-oxo-1, 6-dihydropyridine derivative, preparation method and application thereof in medicine |
| CN113698388A (en) * | 2020-05-20 | 2021-11-26 | 江苏恒瑞医药股份有限公司 | 6-oxo-1, 6-dihydropyridine derivatives, preparation method and application thereof in medicines |
| WO2022041026A1 (en) * | 2020-08-27 | 2022-03-03 | InventisBio Co., Ltd. | Pyridazinone compounds |
| WO2022054925A1 (en) | 2020-09-11 | 2022-03-17 | 国立研究開発法人理化学研究所 | Complex containing neural retina-containing cell aggregates and matrix, and method for manufacturing same |
| WO2022054924A1 (en) | 2020-09-11 | 2022-03-17 | 大日本住友製薬株式会社 | Medium for tissue for transplantation |
| WO2022068915A1 (en) * | 2020-09-30 | 2022-04-07 | 江苏恒瑞医药股份有限公司 | 6-oxo-1,6-dihydropyridazin derivative, preparation method therefor, and application thereof in medicine |
| WO2022086894A1 (en) | 2020-10-19 | 2022-04-28 | Teva Pharmaceuticals International Gmbh | Solid state forms of resmetirom |
| US11858913B2 (en) | 2020-11-06 | 2024-01-02 | Aligos Therapeutics, Inc. | Bicyclic pyridazinones and methods of use thereof |
| WO2022099060A3 (en) * | 2020-11-06 | 2022-06-16 | Aligos Therapeutics, Inc. | Bicyclic pyridazinones as thyroid hormone receptor beta (tr-beta) agonists |
| WO2022143574A1 (en) | 2020-12-30 | 2022-07-07 | 昆药集团股份有限公司 | 2-pyridone derivative, and preparation method therefor and pharmaceutical application thereof |
| CN115073429A (en) * | 2021-03-15 | 2022-09-20 | 昆药集团股份有限公司 | Salt form and crystal form of 1,2, 4-triazine-3, 5-diketone compound and preparation method thereof |
| WO2022194073A1 (en) * | 2021-03-15 | 2022-09-22 | 昆药集团股份有限公司 | Salt form and crystal form of 1,2,4-triazine-3,5-dione compound, and preparation method therefor |
| CN116063296A (en) * | 2021-11-02 | 2023-05-05 | 凯思凯迪(上海)医药科技有限公司 | A compound as thyroid hormone beta receptor agonist and its use |
| WO2023090427A1 (en) | 2021-11-19 | 2023-05-25 | 国立研究開発法人理化学研究所 | Production method for sheet-like retinal tissue |
| WO2024084491A1 (en) * | 2022-10-21 | 2024-04-25 | Mehta Nilesh Vipinchandra | Process for synthesis of res metirom and its intermediates thereof |
| WO2024120426A1 (en) | 2022-12-07 | 2024-06-13 | 昆药集团股份有限公司 | THYROID HORMONE β RECEPTOR AGONIST, CRYSTAL FORM, PREPARATION METHOD AND USE |
| EP4631943A1 (en) | 2022-12-07 | 2025-10-15 | Kpc Pharmaceuticals, Inc | THYROID HORMONE ß RECEPTOR AGONIST, CRYSTAL FORM, PREPARATION METHOD AND USE |
| CN115650928A (en) * | 2022-12-28 | 2023-01-31 | 凯思凯旭(上海)医药科技有限公司 | Polycyclic thyroid hormone beta receptor agonist and application thereof |
| WO2024141076A1 (en) * | 2022-12-29 | 2024-07-04 | Gannex Pharma Co., Ltd. | Prodrugs to thyroid hormone analogs and method of making and use thereof |
| US12485118B2 (en) | 2023-04-07 | 2025-12-02 | Terns Pharmaceuticals, Inc. | Combinations of GLP-1R and THRβ agonists and methods of use thereof |
| WO2025083699A2 (en) | 2023-10-16 | 2025-04-24 | Cipla Limited | Resmetirom polymorphs and process thereof |
| US12377104B1 (en) | 2024-02-06 | 2025-08-05 | Madrigal Pharmaceuticals, Inc. | Methods for treating a fatty liver disease |
| WO2025191457A1 (en) * | 2024-03-13 | 2025-09-18 | Morepen Laboratories Limited | Process for the preparation of pyridazinone derivative |
| WO2025230749A1 (en) | 2024-05-03 | 2025-11-06 | Aligos Therapeutics, Inc. | Methods of preparing 6-amino-2-[3,5-dichloro-4-[(5-isopropyl-6-oxo-1 h-pyridazin-3-yl)oxy]phenyl]-4h- 1, 2, 4-tri-azine-3, 5-dione |
| WO2026074443A1 (en) | 2024-10-01 | 2026-04-09 | Assia Chemical Industries Ltd. | Solid state forms of resmetirom |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1919878B1 (en) | Pyridazinone derivatives as thyroid hormone receptor agonists | |
| US8076334B2 (en) | Prodrugs of thyroid hormone analogs | |
| TWI241295B (en) | Pyridazine derivative and medicine containing the same as effect component | |
| TWI249522B (en) | Compounds that modulate PPAR activity and methods for their preparation | |
| CN101772487B (en) | Thyroid compounds | |
| TWI532731B (en) | New pyridazinone and pyridone compounds | |
| TW200530199A (en) | 1-piperazine- and 1-homopiperazine-carboxylate derivatives, preparation and therapeutic application thereof | |
| CN117186077A (en) | 3-triazolylmethyl-1, 3, 5-triazine-2, 4-dione compound and preparation method and application thereof | |
| HK1115129B (en) | Pyridazinone derivative as thyroid hormone receptor agonists | |
| CN105418580A (en) | New trelagliptin preparation process |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006792493 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 188476 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2614529 Country of ref document: CA Ref document number: CR2008-009644 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08002797 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006271721 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/000818 Country of ref document: MX Ref document number: 565190 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008500147 Country of ref document: PH Ref document number: 1020087001480 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680026731.7 Country of ref document: CN Ref document number: 2008010108 Country of ref document: EG Ref document number: 2008521935 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 658/DELNP/2008 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2006271721 Country of ref document: AU Date of ref document: 20060711 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006271721 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008106058 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006792493 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2010/0527 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: PI0613754 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080121 |