WO2007014524A1 - Erianin salts, their preparation methods and pharmaceutical compositions containing the same - Google Patents
Erianin salts, their preparation methods and pharmaceutical compositions containing the same Download PDFInfo
- Publication number
- WO2007014524A1 WO2007014524A1 PCT/CN2006/001918 CN2006001918W WO2007014524A1 WO 2007014524 A1 WO2007014524 A1 WO 2007014524A1 CN 2006001918 W CN2006001918 W CN 2006001918W WO 2007014524 A1 WO2007014524 A1 WO 2007014524A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- lanolin
- salt
- acid
- group
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C305/00—Esters of sulfuric acids
- C07C305/02—Esters of sulfuric acids having oxygen atoms of sulfate groups bound to acyclic carbon atoms of a carbon skeleton
- C07C305/04—Esters of sulfuric acids having oxygen atoms of sulfate groups bound to acyclic carbon atoms of a carbon skeleton being acyclic and saturated
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/06—Phosphorus compounds without P—C bonds
- C07F9/08—Esters of oxyacids of phosphorus
- C07F9/09—Esters of phosphoric acids
- C07F9/12—Esters of phosphoric acids with hydroxyaryl compounds
Definitions
- the present invention relates to a lanolin salt and a process for the preparation thereof.
- the invention also relates to pharmaceutical compositions comprising such compounds.
- An object of the present invention is to provide a pharmaceutically acceptable salt of a class of lanolin (hereinafter, referred to as a lanolin salt).
- the compounds provided by the present invention have better antitumor activity and have better water solubility than those of lanolin, thereby making the preparation convenient and having higher bioavailability in vivo.
- Another object of the present invention is to provide a process for the preparation of a lanolin salt.
- the present invention provides a class of lanolin salt which is a compound having the following formula (I):
- R is a salt formed by a monovalent acid group of an inorganic oxyacid and a metal, an ammonium salt or an organic amine;
- the metal is selected from the group consisting of metal-reducing and alkaline earth metals
- the inorganic oxyacid is selected from the group consisting of phosphoric acid, sulfuric acid, sulfurous acid, nitric acid, carbonic acid, hypochlorous acid, trifluoroacetic acid, and sulfonic acid.
- the sulfonic acid has the formula R-S0 3 H, wherein R is a hydrocarbyl group.
- the sulfonic acid includes, but is not limited to, benzenesulfonic acid, p-toluenesulfonic acid, 2-naphthalenesulfonic acid, mercaptotransacid, ethylsulfonic acid, 2-hydroxyethylsulfonic acid, and the like.
- the alkali metal includes sodium, potassium, and the like; the alkaline earth metal includes magnesium, calcium, and the like.
- the organic amine is selected from the group consisting of triethylamine, 3-(2-hydroxy)-amine, N-ethylpiperidine, N,N-diethyl-piperazine and the like.
- the lanolin salt is an esterification reaction of lanolin with the above acid, and then with a metal, an ammonium salt or an organic The salt formed by the amine reaction.
- Lanolin salt can be used as a prodrug of lanolin to increase water solubility and facilitate better absorption and utilization by the human body.
- the lanolin inorganic oxyacid ester salt of the present invention is a salt formed by reacting lanolin with an inorganic oxoacid and then reacting with a metal, an ammonium salt or an organic amine.
- the lanolin salt is a lanolin phosphate or a lanolin sulfate.
- R may be a sulphuric acid, and when R is a phosphoric acid, the lanolin salt is a lanolin phosphate salt, and the formula is as shown in (II):
- R 2 is Na, that is, the lanolin phosphate salt is disodium lanolin phosphate.
- R may be a sulfate, and when R is a sulfate, the crude salt is a lanolin sulfate, and the formula is as shown in (III):
- R 3 is Na, K or N3 ⁇ 4.
- R 3 is Na, that is, the lanolin sulfate salt is sodium lanolin sulfate. According to another object of the invention, the invention provides a process for the preparation of such compounds.
- the compounds of the above formula (I) can be prepared by those skilled in the art using conventional preparation means in the art using starting materials of lanolin.
- the preparation method provided by the present invention is as follows:
- the preparation method of the lanolin phosphate salt comprises the following steps:
- the reaction temperature is 25-80 °C
- the reaction solvent is an inert organic solvent
- reaction liquid obtained in the step A1 is directly reacted with a base to prepare a lanolin phosphate salt of the formula (II);
- the synthetic route is:
- R 5 in the formula (IV) is Obut, Cl, or Br or the like, wherein, represents a tert-butyl group; is Na, K or NH 4 , and is not H at the same time.
- the above two-step reaction can be carried out continuously, that is, the intermediate of the formula (V) obtained by the reaction of the step A1 can be directly subjected to the next reaction without further separation and purification.
- Al, lanolin and an excess of the phosphorylating agent are stirred under the action of an acid-binding agent for 2 to 12 hours to cause a phosphorylation reaction of the phenolic hydroxyl group to form a phosphorylation intermediate of the formula (IV).
- Body temperature The degree is 25 ⁇ 80 °C, and the reaction is carried out in an inert organic solvent;
- reaction liquid obtained in the step A1 is evaporated under reduced pressure to remove the solvent, and an inorganic base having a concentration of 1 to 6 mol/L is added dropwise to the reaction solution under stirring, and then stirring is continued at 0 to 90 ° C. ⁇ 24 hours, after removing the solvent under reduced pressure, the crude product of compound (II) is obtained, and the crude product is recrystallized from recrystallization solvent to obtain pure product of lanolin phosphate salt (yield 79-95 %, melting point 266-269 °C (decomposition)).
- step A1
- the phosphorylating agent is one or more pentavalent phosphorus halides selected from the group consisting of 0PC1 3 , OPBr 3 , ( ⁇ )) 2 ⁇ (0) ⁇ , and the like.
- the acid binding agent is one or more selected from the group consisting of pyridines, monoalkylamines, dialkylamines, trialkylamines and the like.
- the acid-binding agent was added dropwise to the reaction mixture under agitation. After the completion of the reaction, the mixture was washed with water and separated, and the organic layer was dried over anhydrous sodium sulfate.
- the inert organic solvent is one or more selected from the group consisting of dichloromethane, dichloroethane, benzene, carbon tetrachloride, acetonitrile and the like.
- reaction temperature of step A1 is room temperature.
- step ⁇ 2
- the base is selected from NaOH, or more, the concentration of a base KOH, NaOC3 ⁇ 4, KOCH 3, NaOCHbCI ⁇ KOCH2CH3 the like may be formulated according to the amount of 1 ⁇ 6mol L. and required volume
- the recrystallization solvent of the target product ( ⁇ ) is one or more selected from the group consisting of water, decyl alcohol, ethanol, isopropanol, acetone, and the like, and is preferably a methanol-acetone mixed solvent.
- the phosphorylating agent is OPC1 3 and the acid binding agent is triethylamine; in the step A2: the alkali is NaOH.
- an intermediate lanolin dibenzyl phosphate can be produced, which can be First, it reacts with Nal to form a phosphate ester of lanolin, and then reacts with a base to form a salt.
- a phosphorylating agent such as dibenzylphosphite ((BnO>P(0)H, bn represents a benzyl group)
- an intermediate lanolin dibenzyl phosphate can be produced, which can be First, it reacts with Nal to form a phosphate ester of lanolin, and then reacts with a base to form a salt.
- the present invention also provides a method for preparing another lanolin phosphate salt, which comprises the following steps: Bl, under the protection of inert gas, the reaction of lanolin and dibenzyl phosphite under the action of acid binding agent, phosphorylation of phenolic hydroxyl group, to prepare a compound of formula (V), the reaction temperature is - 40 "C ⁇ -10 ° C, the reaction is carried out in an inert organic solvent;
- bn represents a benzyl group; 1, 2 is 11, Na, K or N3 ⁇ 4, and 1 ⁇ and not simultaneously H. - In step 31:
- the dibenzyl phosphite is a phosphorylating agent.
- the acid binding agent is one or more selected from the group consisting of pyridines, monoalkylamines, dialkylamines, trialkylamines and the like.
- the acid binding agent is diisopropylethylamine and diaminopyridine (DMAP).
- the inert gas is argon gas, nitrogen gas, or carbon monoxide or the like.
- the inert gas is argon.
- the inert organic solvent is selected from the group consisting of dichlorosilane, dichloroethane, benzene, carbon tetrachloride, acetonitrile, and the like. One or more. In a specific embodiment of the invention, the inert organic solvent is acetonitrile and carbon tetrachloride.
- step B2
- the inert gas is argon, nitrogen, or carbon monoxide or the like. In a specific embodiment of the invention, the inert gas is argon.
- the inert organic solvent is one or more selected from the group consisting of dichlorosilane, dichloroethane, benzene, carbon tetrachloride, acetonitrile and the like.
- the inert organic solvent is acetonitrile.
- the reactive group protecting agent is trimethylchlorosilane or the like.
- step B3
- the base is one or more selected from the group consisting of NaOH, KOH, NaOCH 3 , KOCH 3 , NaOCHiCHs. KOCH2CH3 and the like.
- the base is sodium methoxide.
- the intermediate (V) when the intermediate (V) is synthesized from lanolin, under the protection of argon, the lanolin is dissolved in acetonitrile at -25 Torr, CCL 4 is added and stirred for 5 minutes, with a needle Diisopropylethylamine and dimethyl pyridine were added dropwise to the tube, and after 1 minute, dibenzyl phosphite was added dropwise to control the reaction temperature below -10 °C.
- the phosphate of the lanolin is formed from the intermediate (V)
- the intermediate (V) and Nal are dissolved in the acetonitrile solution under the protection of argon, and the trimethyl chlorosilane is slowly added under stirring, and stirred for 20 minutes.
- Preparation of lanolin acid ester (VI) The reaction of the lanolin phosphate (VI) with sodium methoxide gives colorless crystals (11).
- the preparation method of the lanolin sulfate salt comprises the following steps:
- reaction temperature is -5 ⁇ -
- the reaction solvent is an inert organic solvent at 10 ° C;
- the reaction liquid obtained in the step C1 is directly reacted with a base to prepare a sodium salt of the lanolin sulfate of the formula (III);
- the synthetic route is:
- the two steps included in the above preparation method are:
- step C2 Adjust the pH of the reaction solution obtained in step C1 to 8-12 with a concentration of 5-15 mol L. After cooling, filter, wash with anhydrous ether, and dry to dissolve the solid product in methanol. After passing through the column, the solvent is dried to obtain sodium salt of lanolin sulfate, and the reaction temperature is room temperature.
- the halosulfonic acid is chlorosulfonic acid or bromosulfonic acid.
- the acid binding agent is one or more selected from the group consisting of pyridines, monoalkylamines, dialkylamines, trialkylamines and the like. ,
- the inert organic solvent is one or more selected from the group consisting of dichloromethane, dichloroethane, benzene, carbon tetrachloride and the like.
- step C1 is: adding a lanolin, N, N-dimercaptoaniline to a round bottom flask, and adding an equal volume of dichloromethane with N, N-dimethylaniline in a water brine bath. After stirring (-5 ° - 10 ° C), chlorosulfonic acid was added dropwise, and after the addition, stirring was continued for 1 hour in a brine bath.
- step C2 is:
- the base is one or more selected from the group consisting of NaOH, KOH, NaOCH 3 , KOCH 3 , NaOCHaCH ⁇ KOCH2CH3, etc., and the concentration of the base can be adjusted accordingly according to the amount and volume required.
- step C2 is: removing the brine bath and stirring at room temperature for 24 hours; adjusting the pH of the reaction solution to 10 with 10 mol L of NaOH solution under stirring, filtering, washing with anhydrous ether, and drying.
- the solid product was dissolved in sterol and passed through a column, and the solvent was drained to obtain a sodium salt of lanolin sulfate.
- the halogen acid is chlorosulfonic acid
- the acid binding agent is N, N-dimethylaniline
- the inert organic solvent is dichloro Methane
- the base is sodium hydroxide
- the present invention provides a pharmaceutical composition comprising an effective therapeutic amount of a lanolin salt, and a pharmaceutically acceptable carrier and/or excipient.
- the pharmaceutical composition is an injection administration preparation or an oral administration preparation.
- a pharmaceutical preparation for use in the present invention can be prepared by using a lanolin salt as a medicinal ingredient, adding a pharmaceutically acceptable pharmaceutically acceptable excipient, and by a conventional method in the art.
- the dosage form of the pharmaceutical preparation may be: a dosage form for oral administration such as a tablet, a capsule (including a hard capsule, a soft capsule, an intestinal sol and a micro), a powder, a granule, and a syrup; a non-oral administration Dosage forms such as injections, suppositories, pills, gels, and patches.
- oral immediate release solid preparations such as tablets, granules, etc.
- sustained release preparations for oral or parenteral administration tablettes, granules, fine granules, pills, capsules
- syrups, emulsions, suspensions, solutions are used in the present invention, and these preparations can also be prepared by a conventional method.
- the formulations of the invention may be in coated or uncoated form, as desired.
- Preferred in the present invention is a dosage form in which a lanolin salt is used for administration by injection.
- the pharmaceutical excipients in the present invention include excipients, lubricants, binders, disintegrants, stabilizers, foaming agents, coating agents and the like for solid preparations, or for semisolid preparations, liquid preparations.
- other pharmaceutical additives such as preservatives, antioxidants, colorants, sweeteners and flavors may also be used as needed. Agents, etc.
- the dosage of the pharmaceutical preparation of the present invention can be appropriately selected depending on the subject to be treated, the dosage form, the administration method, and the like.
- the water solubility of the lanolin salt of the present invention is greatly improved, so that the preparation is easier, and the bioavailability in the body can be improved, and the anti-tumor effect can be better exerted.
- the cancer of the present invention generally refers to all malignant tumors.
- the pharmacodynamic test showed that: the disodium lanolin phosphate has obvious anti-tumor effect, and the effect of daily administration and the administration of the next day is also effective against the tumor, which indicates that the disodium lanolin phosphate is highly effective. , low toxicity anti-tumor compounds.
- the combination of lanolin phosphate disodium and other chemotherapeutic drugs can significantly increase the killing effect of chemotherapy drugs on tumors.
- the safety evaluation results showed that the LD 5Q value of the sodium lanolin phosphate was 1243.0377 mg kg, indicating that the disodium lanolin phosphate has good clinical safety.
- the lanolin salt of the present invention has better water solubility than lanolin, and thus is more convenient for the preparation, for example, a high concentration of the injection form can be prepared, and the bioavailability of the drug in the body can be improved.
- the lanolin salt of the present invention has an anti-cancer effect of high efficiency and low toxicity.
- the reagents used are common chemical industrial products which are cheap and easy to obtain; the synthesis route is short; the operation method is simple, and is suitable for industrial scale production.
- Figure 1 is a 1 H MR NMR image of the lanolin phosphate prepared by the present invention
- Figure 3 is an infrared spectrum of the lanolin phosphate prepared by the present invention.
- Figure 4 is a mass spectrum of the lanolin phosphate prepared in the present invention. DETAILED DESCRIPTION OF THE INVENTION
- the lanolin used in the present invention is provided by P ⁇ Company of Zhejiang Saier Biomedical Research, and the preparation method thereof can be selected from the following two methods; other test materials used in the present invention are commercially available unless otherwise specified. buy product.
- two methods for preparing lanolin are specifically described:
- the obtained product was subjected to the next reaction without further purification. If a pure product is desired, it can be distilled under reduced pressure, and a fraction of BP216 - 218 ° C / 12 mmHg is collected.
- the obtained product was subjected to the next reaction without further purification. If a pure product is desired, the solid can be washed with acetone to give a white powder solid.
- the organic phase was separated by shaking well; the organic layer was washed with water (50 ml ⁇ 2), and the aqueous layer was extracted with dichloromethane. The organic layer was combined and dried over anhydrous sodium sulfate overnight. The filtrate was filtered under reduced pressure to give a viscous liquid. Crude acid chloride.
- lanolin (20 g, 63.2 mmol) was dissolved in 200 ml of acetonitrile, stirred, cooled to -25 ° C, added CCl 4 (35 ml, 316 mmol), stirred for 5 minutes, used Diisopropylethylamine (23.13 ml, 133 mmol) was added dropwise to the syringe, then DMAP (772 mg, 6.32 mmol) was added dropwise. After 1 minute, dibenzyl phosphite (20.33 ml, 92 mmol) was added dropwise, and the reaction temperature was controlled.
- triamyl chlorosilane (9.22 ml, 71 mmol) was slowly added to a solution of dibenzyl phosphate (20.5 g, 35.6 mmol) and Nal (10. 7 g, 71 mmol) in 100 ml of acetonitrile with vigorous stirring. The mixture was stirred for 20 minutes, TLC was used to detect the absence of the starting point, water was added (just to dissolve the salt), 2% sodium thiosulfate 2 ml was added, the organic phase was separated, and the 7J layer was extracted with ethyl acetate 50 ml ⁇ 4. A yellow foam was obtained.
- EtOAc EtOAc EtOAc It is the disodium of lanolin phosphate.
- the physical properties of the disodium lanolin phosphate were measured in the same manner as in Example 1. From the results of the analysis, it was found that the finished product of the disodium lanolin phosphate was obtained according to the method of the present invention.
- mice were used, 10 in each group, male and female, and a total of 7 groups. Labeled with bitter acid.
- Each group was administered with 1300 mg/kg, 1250 mg/kg, 1200 mg/kg, 1150 mg/kg, llOOmg/kg, 1050 mg/kg, and 1000 mg/kg by tail vein.
- the confidence interval of AP LD50 1243.0377 mg/kg was: 1049.9757 ⁇ LD5 ( 1471.5984 There was no obvious lesion after dissection of the mice that died during the experiment; the surviving mice were sacrificed on the 14th day and pathological anatomy was performed without pathological changes.
- Sample Disodium phosphate of lanolin (AP, prepared as in Example 1) was prepared in physiological saline to the desired concentration.
- Control substance cyclophosphamide for injection (CTX), Shanghai Hualian Pharmaceutical Co., Ltd., batch number ⁇ 020806. It is dissolved in physiological saline during preparation.
- mice In S180 ascites mice, ascites was taken under aseptic conditions, and the cells were counted and diluted with physiological saline to 1-2 107 cells/ml. The mice were subcutaneously inoculated subcutaneously at 0.2 ml/mouse. The mice were randomly divided into 5 groups on the next day, 10 in each group.
- the reference cyclophosphamide was administered by intravenous route at a dose of 30 mg/kg x 9d.
- a blank control group was also set up.
- the animals were administered with body weight on the next day, and were intragastrically administered with 0.5 ml/20 g for 9 consecutive days. They were administered according to body weight, and sacrificed on the 10th day after inoculation. The tumor pieces were weighed and the tumor inhibition rate was calculated.
- the lanolin salt of the present invention has better water solubility than lanolin, and thus is more convenient for the preparation, can improve the bioavailability of the drug in vivo, and has an anti-cancer effect of high efficiency and low toxicity.
- the reagents used are all common chemical industrial products which are cheap and easy to obtain; the synthesis route is short; the operation method is simple, and it is suitable for industrial scale production.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Cosmetics (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
毛兰素盐及其制备方法和包含其的药物组合物
技术领域
本发明涉及毛兰素盐及其制备方法。
本发明还涉及包含该类化合物的药物组合物。 背景技术
癌症已成为仅次于心血管疾病的人类第二类杀手。目前常用抗癌化疗与放 疗的方法可造成对人体不利的严重毒副反应。国内外的专家已发现中药石斛具 有抗肿瘤、抗衰老及扩张血管等作用,其乙醇提取物及其联苄类化合物具有不 同程度的体内抗肿瘤活性作用。 石斛的活性成分已引起国内外的关注。
王天山等的研究结果(鼓槌石斛中化学成分对 K562舯瘤细胞株生长抑制 作用体外试验, 天然产物研究与开复, 1997, 9(2) , 1 ~ 3 )表明, 联苄类和菲 类化合物对体外培养的鼠类 L 。微管蛋白的聚集及有丝分裂、 P388细胞株、 多种 人体肿瘤细胞株如 A-549、 MCF- 7、 HT-29、 SKMEL-5 , MLM、 SK-OV-3、 HL-60 均有抑制作用。 鼓槌石斛中的二氢联苄类和菲类化合物对肿瘤细胞株 Κ 6的生 长具有不同程度的抑制作用, 尤以毛兰素(Erianin )的活性最强。 毛兰素的化 学结构式如下所示:

毛兰素
毛兰素对小鼠肝癌的作用最强, 抑瘤率为 50.82%。有关研究(中国药科大 学学报, 1994, 25 (3 ), 188-189 )还推测毛兰素的毒副作用远低于肿瘤化疗 药物 5FU。 李运曼等的研究表明 毛兰素诱导人白血病 HL-60细胞的凋亡, 中 国药理学报, 2001 , 22(11) ) , 毛兰素显著抑制人类白血病 HL-60细胞的生长, 其抑制作用可能是通过诱导细胞凋亡和改变 HL-60细胞的 bcl-2和 bax基因表达 而实现的。
由于毛兰素的水溶性较低, 在水中的溶解度仅为<111¾/1111 , 限制了其制剂 性能和临床应用, 因此有必要对能提高其水溶性的盐类进行研究。 目前, 尚没 有关于毛兰素盐及其制备方法的文献报道。 发明内容
本发明的一个目的在于提供一类毛兰素的药学上可接受的盐(以下, 简称 毛兰素盐)。 本发明所提供的化合物具有较好的抗肿瘤活性, 与毛兰素相比具 有更好的水溶性, 从而使得制剂方便, 并且在体内有更高的生物利用度。
本发明的另一个目的在于提供毛兰素盐的制备方法。
本发明的再一个目的在于提供包含毛兰素盐的药物组合物。
根据本发明的一个目的, 本发明提供了一类毛兰素盐, 所迷毛兰素盐为具 有如下通式(I ) 的化合物:

R为无机含氧酸的一价酸根与金属、 铵盐或有机胺所生成的盐;
所述金属选自减金属、 碱土金属; '
所述无机含氧酸选自磷酸、 硫酸、 亚硫酸、 硝酸、 碳酸、 次氯酸、 三氟乙 酸、 磺酸。
所述磺酸的通式为 R-S03H, 其中 R为烃基。 所述磺酸包括但不限于苯磺 酸、 对甲苯磺酸、 2-萘磺酸、 曱基横酸、 乙基磺酸、 2-羟乙基磺酸等。
所述碱金属包括钠、 钾等; 所述碱土金属包括镁、 钙等。
所述有机胺选自三乙胺、 3- ( 2-羟基) -胺、 N-乙基哌啶、 N, N-二乙基- 哌嗪等。
所述毛兰素盐为毛兰素与上述酸发生酯化反应后,再与金属、铵盐或有机
胺反应所生成的盐。
毛兰素盐可以作为毛兰素的前体药物, 以增加水溶性,便于更好的被人体 的吸收利用。
本发明所述的毛兰素无机含氧酸酯盐为毛兰素与上迷无机含氧酸发生酯 化反应后, 再与金属、 铵盐或有机胺反应所生成的盐。
更进一步的, 所迷毛兰素盐为毛兰素磷酸酯盐或毛兰素硫酸酯盐。
优选的, R可以为碑酸才艮, 当 R为磷酸^^时, 所述毛兰素盐为毛兰素磷酸 酯盐, 其通式如(II ) 所示:
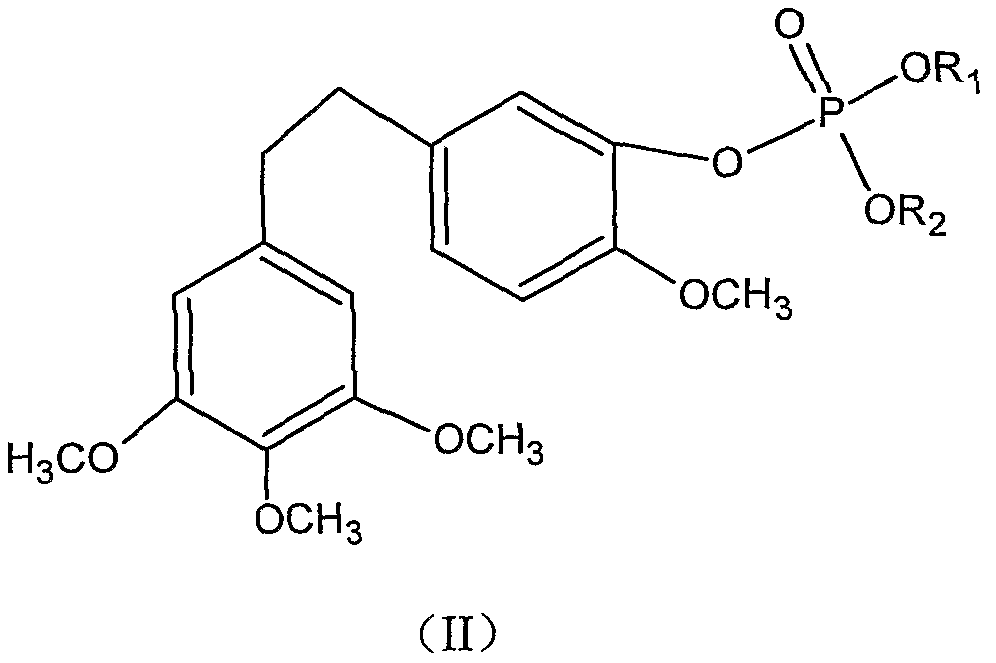
其中, 、 为11、 Na、 K或 ΝΉ4;
且 和 不同时为 Η。
更优选的, 、 R2均为 Na, 即所述毛兰素磷酸酯盐为毛兰素磷酸酯二钠。 优选的, R可以为硫酸根, 当 R为硫酸根时, 所述毛兰素盐为毛兰素硫酸 酯盐, 其通式如 ( III )所示:

(III)
其中, R3为 Na、 K或 N¾。
最优选的, R3为 Na, 即毛兰素硫酸酯盐为毛兰素硫酸酯钠。
根据本发明的另一个目的, 本发明提供了此类化合物的制备方法。
本领域技术人员可运用本领域的常规制备手段, 以毛兰素为原料,制备上 述通式(I ) 的化合物。 本发明提供的制备方法如下所述:
所迷毛兰素磷酸酯盐的制备方法, 包括下述步骤:
Al、 使毛兰素与磷酰化剂, 在縛酸剂的作用下, 发生酚羟基的磷酰化反 应, 生成式(IV ) 的毛兰素磷酰化中间体, 反应温度为 25 ~ 80°C , 反应溶剂 为惰性有机溶剂;
A2、 使由步骤 A1得到的反应液直接与碱反应, 制备得到式(II ) 的毛兰 素磷酸酯盐;
合成路线为:

式(IV ) 中的 、 R5为 Obut、 Cl、 或 Br等, 其中, 代表叔丁基; 、 为 Na、 K或 NH4, 且 和 不同时为 H。
以上两步反应可以是连续完成的, 即由步驟 A1反应得到的式(V ) 的中 间体无须实行进一步的分离和纯化 , 就可直接进行下一步反应。
进一步的, 上述制备方法的两个步骤为:
Al、 毛兰素与过量的磷酰化剂, 在缚酸剂的作用下, 充分搅拌 2 ~ 12 小 时, 发生酚羟基的磷酰化反应, 生成式(IV )的毛兰素磷酰化中间体, 反应温
度为 25 ~ 80 °C , 反应在惰性有机溶剂中进行;
A2、 将由步 A1 得到的反应液减压蒸去溶剂, 在搅拌下将浓度为 1 ~ 6mol/L的无机碱滴加到该反应液中, 然后在 0 ~ 90°C的条件下继续搅拌 2 ~ 24 小时, 减压除去溶剂后即得化合物(II )的粗产物, 粗产物经重结晶溶剂重结 晶后得毛兰素磷酸酯盐的纯品 (产率 79 ~ 95 % , 熔点 266-269°C (分解))。
在步骤 A1中:
所述磷酰化剂为选自 0PC13、 OPBr3、 (ΒιΛ))2Ρ(0)α等的一种或多种五价 磷卤化物。
所述缚酸剂为选自吡啶类、单烷基胺类、 双烷基胺类、 三烷基胺类等的一 种或多种。
进一步的, 縛酸剂在充分搅拌下逐滴加入反应液中, 反应完成后, 用水洗 涤, 分离, 有机层用无水硫酸钠干燥, 减压蒸馏除去溶剂。
所述惰性有机溶剂为选自二氯甲烷、 二氯乙烷、 苯、 四氯化碳、 乙腈等的 一种或多种。
在本发明的一个优选实施方式中, 步骤 A1的反应温度为室温。
在步骤 Α2中:
在充分搅拌下将碱滴加到该反应液中之后, 优选在 30 ~ 80°C的条件下继 续搅拌 8 ~ 10小时, 然后冷却反应液, 并经过滤除去不溶物, 减压下蒸餾除去 大部分溶剂等一系列后处理得到固体粗产物,经有机溶剂重结晶得到目标产物 ( 11 )。
所述碱为选自 NaOH、 KOH、 NaOC¾、 KOCH3、 NaOCHbCI^ KOCH2CH3 等的一种或多种, 碱的浓度可根据所需的量和体积配成 1 ~ 6mol L。
所述目标产物(Π )的重结晶溶剂为选用水、 曱醇、 乙醇、 异丙醇、 丙酮 等的一种或多种, 优选为甲醇-丙酮混合溶剂。
在本发明的一个优选实施方式中, 上述制备方法中, 在所述步骤 A1中: 磷酰化剂为 OPCl3, 縛酸剂为三乙胺; 在所述步骤 A2中: 碱为 NaOH。
当毛兰素与诸如二苄基亚磷酸酯((BnO>P(0)H, bn代表苄基)的磷酰化 剂反应时, 可生成中间体毛兰素磷酸二苄酯, 该中间体可先与 Nal反应, 生成 毛兰素的磷酸酯, 再与碱反应成盐。 即, 本发明还提供了另一种毛兰素磷酸酯 盐的制备方法, 其包括下述步骤:
Bl、 在惰性气体保护下, 使毛兰素与二苄基亚磷酸酯, 在縛酸剂的作用 下, 发生酚羟基的磷酰化反应, 制备得到式 (V ) 的化合物, 反应温度为 -40 "C ~ -10°C, 反应在惰性有机溶剂中进行;
B2、 在惰性气体保护下, 使式(V )的化合物与 Nal在活性基保护剂的作 用下发生反应 , 制备得到式( VI ) 的化合物, 反应在惰性有机溶剂中进行;
B3、 使式(VI ) 的化合物与碱反应, 制备得到式(Π ) 的化合物; 合成路线为:

式(V )中, bn代表苄基; 、 1 2为11、 Na、 K或 N¾, 且 1^和 不同 时为 H。 - 在步骤 31中:
所述二苄基亚磷酸酯为磷酰化剂。
所述缚酸剂为选自吡啶类、单烷基胺类、 双烷基胺类、 三烷基胺类等的一 种或多种。在本发明的一个具体实施方式中, 所述縛酸剂为二异丙基乙胺和二 曱氨基吡啶(DMAP )。
所述惰性气体为氩气、 氮气、或一氧化碳等。 在本发明的一个具体实施方 式中, 所述惰性气体为氩气。
所述惰性有机溶剂为选自二氯曱烷、 二氯乙烷、 苯、 四氯化碳、 乙腈等的
一种或多种。在本发明的一个具体实施方式中,所述惰性有机溶剂为乙腈和四 氯化碳。
在步骤 B2中:
所述惰性气体为氩气、 氮气、 或一氧化碳等。 在本发明的一个具体实施方 式中, 所述惰性气体为氩气。
所述惰性有机溶剂为选自二氯曱烷、 二氯乙烷、 苯、 四氯化碳、 乙腈等的 一种或多种。 在本发明的一个具体实施方式中, 所述惰性有机溶剂为乙腈。
所述活性基保护剂为三甲基氯硅烷等。
在步驟 B3中:
所述碱为选自 NaOH、 KOH、 NaOCH3、 KOCH3、 NaOCHiCHs. KOCH2CH3 等的一种或多种。 在本发明的一个具体实施方式中, 所述碱为甲醇钠。
在本发明的一个优选实施方式中, 由毛兰素合成中间体(V )时, 在氩气 保护下, 毛兰素溶于乙腈中, 在 -25Ό , 加入 CCL4并搅拌 5分钟, 用针筒滴加 二异丙基乙胺和二甲氣基吡啶, 1分钟后滴加二苄基亚磷酸酯, 控制反应温度 在 -10°C以下。由中间体(V )生成毛兰素的磷酸酯时,氩气保护下, 中间体(V ) 和 Nal溶于乙腈溶液中, 强烈搅拌下, 慢慢加入三曱基氯硅烷, 搅拌 20分钟, 制备得到毛兰素碑酸酯 (VI )。 使毛兰素磷酸酯(VI ) 与甲醇钠反应, 得到无 色晶体(11 )。
所述毛兰素硫酸酯盐的制备方法, 包括下述步骤:
Cl、 使毛兰素与卤磺酸, 在缚酸剂的作用下, 发生酚羟基的硫酰化反应, 生成式(VI )的毛兰素硫酰化中间体, 反应温度为 -5〜- 10°C, 反应溶剂为惰性 有机溶剂;
C2、 使由步骤 C1得到的反应液直接与碱反应, 制备得到式(III )的毛兰 素硫酸酯钠盐;
合成路线为:

. 式(VII ) 中的 为 CI或 Br等; R3为 Na、 K或 N 。
具体地说, 上述制备方法包括的两个步驟为:
Cl、 毛兰素与卤磺酸, 在缚酸剂的作用下, 充分搅拌 1 ~ 10 小时, 发生 酚羟基的硫酰化反应, 生成式 (V ) 的毛兰素硫酰化中间体, 反应温度为 -5~-10°C, 反应溶剂为惰性有机溶剂;
C2、 在充分搅拌下, 用浓度为 5 ~ 15mol L的碱将由步骤 C1得到的反应 液的 pH调至 8 ~ 12, 冷却后过滤, 用无水乙醚洗, 干燥后将固体产物溶解在 甲醇中过柱, 溶剂抽干后得毛兰素硫酸酯钠盐, 反应温度为室温。
关于步骤 C1:
所述卤磺酸为氯磺酸或溴磺酸。
所迷縛酸剂为选自吡啶类、 单烷基胺类、 双烷基胺类、 三烷基胺类等的一 种或多种。、
所述惰性有机溶剂为选自二氯甲烷、二氯乙烷、 苯、 四氯化碳等的一种或 多种。
进一步的, 步骤 C1为: 在圆底烧瓶中加入毛兰素, N、 N-二曱基苯胺, 并加入与 N、 N-二甲基苯胺等体积的二氯曱烷,在水盐水浴中搅拌( -5〜- 10°C ), 然后逐滴加入氯磺酸, 加完后在盐水浴中继续搅拌 1小时。
关于步驟 C2:
所述碱为选自 NaOH、 KOH、 NaOCH3、 KOCH3、 NaOCHaCH^ KOCH2CH3 等的一种或多种, 碱的浓度可才艮据所需的量和体积进行相应的调节。
进一步的, 步驟 C2为: 移去盐水浴, 在室温下搅拌 24小时; 用 10mol L 的 NaOH溶液在搅拌下将反应液的 pH值调至 10,冷却后过滤,用无水乙醚洗, 干燥后将固体产物溶解在曱醇中过柱, 溶剂抽干后得毛兰素硫酸酯钠盐。
在本发明的一个优选实施方式中, 上述制备方法中, 步骤 C1中: 所迷鹵 磅酸为氯磺酸、 所述缚酸剂为 N、 N-二甲基苯胺, 惰性有机溶剂为二氯甲烷; 步驟 C2中: 碱为氢氧化钠。
根据本发明的再一个方面,本发明提供了一种药物组合物,其包含有效治 疗量的毛兰素盐, 以及药学上可接受的载体和 /或赋形剂。
进一步的, 所述药物组合物为注射给药制剂或口服给药制剂。
以毛兰素盐为药效成分 ,添加药学上可接受的药用辅料并通过本领域的常 规方法可以制备用于本发明的药物制剂。本药物制剂的剂型可以是: 口服给药 的剂型诸如片剂、 胶嚢(包括硬胶嚢、 软胶嚢、 肠溶胶嚢和微嚢)、 粉剂、 颗 粒剂和糖浆剂; 非口服给药的剂型诸如注射剂、 栓剂、 丸剂、 凝胶剂和贴剂。 除这些常规剂型外, 还可以将口服的速释固体制剂(例如片剂、 颗粒剂等)和 用于口服或非口服给药的緩释制剂(片剂、 颗粒、 精细颗粒、 丸剂、 胶嚢、 糖 浆、 乳剂、 悬浮液、 溶液)用于本发明, 通过常规方法也可以制备这些制剂。 本发明中的制剂可以是包衣或未包衣的形式,视需要而定。本发明优选的是将 毛兰素盐用于注射给药的剂型。
本发明中的药用辅料包括用于固体制剂的赋形剂、 润滑剂、 粘合剂、 崩解 剂、 稳定剂、 发泡剂、 包衣剂等, 或用于半固体制剂、 液体制剂的溶剂、 增溶 剂、 悬浮剂、 等渗剂、 緩冲剂、 润肤剂、 乳化剂等, 此外, 也可以根据需要使 用其它药用添加剂诸如防腐剂、 抗氧化剂、 着色剂、 甜味剂和矫味剂等。
才艮据所治疗的对象、剂型、给药方法等可以对本发明药物制剂的剂量进行 合适的选择。
与毛兰素相比,本发明所述毛兰素盐的水溶性大大提高,因而制剂更容易, 并且可以提高体内的生物利用度,更好地发挥抗肿瘤作用。本发明所述的癌症 泛指所有恶性肿瘤。
药效学试验表明: 毛兰素磷酸酯二钠具有明显的抗肿瘤作用, 每天给药和 隔天给药的方式对抗肿瘤的作用同样有效,这说明毛兰素磷酸酯二钠是一种高 效,低毒的抗肿瘤化合物。毛兰素磷酸酯二钠与其它化疗药物联用可以明显增 加化疗药物对肿瘤的杀伤作用。
安全性评价结果表明: 毛兰素磷酸酯二钠的 LD5Q值为 1243.0377 mg kg, 说明毛兰素磷酸酯二钠具有很好的临床用药安全性。
本发明提供的毛兰素盐及其制备方法具有以下优点:
1、 本发明所述的毛兰素盐具有比毛兰素更好的水溶性, 因而对于制剂更 方便,例如可以制备高浓度的注射剂型, 并且可以提高药物在体内的生物利用 度。
2、 本发明所述的毛兰素盐具有高效、 低毒的抗癌作用。
3、 本发明的制备方法中, 所用试剂都是便宜易得的普通化学工业产品; 合成路线短; 操作方法简便, 适宜于工业规模生产。
为了更好地理解本发明的本质 ,下面结合附图通过对本发明较佳实施方式 的描述, 详细说明但不限制本发明。 附图的简要说明
图 1为本发明制备的毛兰素磷酸盐的 1H MR核磁共振图镨;
图 2为本发明制备的毛兰素磷酸盐的 13CNMR核磁共振图谱;
图 3为本发明制备的毛兰素磷酸盐的红外图谱;
图 4为本发明制备的毛兰素磷酸盐的质谱图谱。 发明的具体实施方式
本发明所用的毛兰素由浙江赛尔生物医学研究有 P艮公司提供,其制备方法 可从下述两种方法中加以选择; 本发明所用的其它试验材料如无特别说明, 均 为市售购买产品。 以下, 具体说明毛兰素的两种制备方法:
(一)从石斛中提取毛兰素的方法
参照中国专利申请 03115752.1 , 应用超临界 C02萃取和柱层析法以无水 乙醇、 甲醇、 丙酮等为夹带剂, 以 co2等为提取介质从兰科植物石斛中提取
抗肿瘤成分毛兰素; 对所得的粗提物再以石油醚: 醋酸乙酯溶液为洗脱剂进行 硅胶柱柱层析分离及重结晶等步骤获得精提物, 达到供人体注射用的标准。
(二)毛兰素的化学合成方法
参照中国专利申请 200510083055.4中描述的方法, 以 3,4,5-三甲氧基苯甲 醛(i )和异香兰素 (iv )为起始原料, 制备毛兰素的合成路线如下所示:

具体包括以下步骤:
1、 3,4,5-三甲氧基苯甲醇 ( ii )的制备
250ml的三颈瓶中, 加入 3,4,5-三甲氧基苯甲醛(i ) 15g ( 76.45mmol ), 200ml的无水乙醇, 加热 40°C溶解, 再加入硼氢化钠 1.48g ( 38.23mmol ), 加 热回流 45min后, TLC检测, 反应完全后, 冷却至室温, 加入去离子氷 10ml ( 555.8mmol ), 淬灭后, 抽滤, 滤渣用 20ml无水乙醇洗涤, 合并滤液后, 用 旋转蒸发仪浓缩至干, 加入 100ml二氯甲烷溶解, 用 2N氢氧化钠溶液 50ml x 2洗涤, 再用去离子水 50ml X 2洗涤, 加入适量的无水硫酸镁千燥过夜。 过
滤, 用 20ml二氯甲烷洗涤滤渣, 合并滤液, 用旋转蒸发仪浓缩至干, 得 3,4,5- 三甲氧基苯曱醇(ii ), 无色油状物 14.05g, 收率 92.72%。
得到的产物不需要进一步纯化, 即可进行下一步反应。 若想得到纯品可 以减压蒸餾, 收集 BP216 - 218°C/12mmHg的馏分。
2、 3,4,5_三甲氧基苄溴(iii ) 的制备
3,4,5-三甲氧基苯曱醇(11 ) 14.05§ ( 70.8911111101 ),用 100ml二氯甲烷溶解, 加入到 250ml的三颈瓶中, 三溴化磷 6.73ml ( 70.89mmol )溶于 25ml二氯甲 烷中, 緩慢滴加到反应液中, 室温反应 50min后, 水浴冷却, 緩慢滴加 18ml 去离子水 18ml ( l.Omol ), 淬灭反应。 然后再用 100 ml x 2去离子水洗涤, 无
 过滤, 用 20ml二氯曱烷洗涤滤渣, 合并滤液, 用旋转蒸发仪 浓缩至干, 真空干燥, 得 3,4,5-三甲氧基苄溴(iii ), 淡黄色固体 16.05g, 收率
过滤, 用 20ml二氯曱烷洗涤滤渣, 合并滤液, 用旋转蒸发仪 浓缩至干, 真空干燥, 得 3,4,5-三甲氧基苄溴(iii ), 淡黄色固体 16.05g, 收率
84.44%。
得到的产物不需要进一步纯化, 即可进行下一步反应。 若想得到纯品可 以用乙酸乙酯: 正己烷 =1: 3重结晶, 得到白色片状晶体。
3、 3,4,5-三曱氧基苄溴三苯基膦盐 (V ) 的制备
3,4,5-三甲氧基苄溴(iii ) 16.05g ( 61.47mmol )溶于 150ml甲苯中, 加入 到 250ml的三颈瓶中, 加入三苯基瞵 16.12g ( 61.47mmol ), 立即溶解, 加热 回流 lhr, 有白色固体析出, 冷却到室温, 抽滤, 滤饼用 30ml 甲苯洗涤, 真 空干燥后, 得 3,4,5-三曱氧基苄溴三苯基膦盐(v ), 白色粉末固体 27.81g, 收 率为 86.44%。
得到的产物不需要进一步純化, 即可进行下一步反应。若想得到純品可以 用丙酮洗涤固体, 得白色粉末固体。
4、 苄基保护异香兰素(vi ) 的制备
250ml三颈瓶中, 加入异香兰素( iv ) 15g ( 98.59mmol ), 200ml无水乙醇, 加热到 40 °C溶解, 加入碳酸钾 9g ( 65.07mmol ), 搅拌下加入氯化苄 15ml ( 130.13mmol ), 加热回流 lhr, TLC检测反应完全后, 冷却到 50°C , 趁热过 滤, 滤' 入水箱中冷却过夜, 晶体析出, 抽滤, 滤饼用 30ml无水乙醇洗涤, 真空干燥后,得苄基保护异香兰素(vi ),白色针状晶体 19.72g,收率为 82.56%。
得到的产物不需要进一步纯化, 即可进行下一步反应。 若想得到纯品可以 用无水乙醇重结晶, 得到白色柱状晶体。
5、 顺反异构体(vii ) 的制备
250ml 三颈瓶中, 加入 3,4,5-三甲氧基苄溴三苯基磷盐 (v ) 20.00g ( 38.21mmol ), 150ml四氢呋喃, 搅拌悬浮液, 苄基保护异香兰素( vi ) lO.OOg ( 41.27mmol )溶于 70ml四氢呋喃, 放入 100ml的滴液漏斗中。 反应瓶中加 入固体叔丁醇钾 7.46g ( 66.49mmol ), 反应体系变为血红色, 室温搅拌 5min, 水浴冷却,緩慢滴加苄基保护异香兰素的溶液, 20min滴完,再室温搅拌 30min, TLC检测反应完全后 , 倒入 500ml的分液漏斗中, 加入 140ml去离子水后, 溶液分层, 加入乙醚 300ml X 2萃取, 合并乙醚层, 用无水硫酸摸干燥, 过滤, 滤饼用 50ml乙醚洗涤, 滤液旋转蒸发仪浓缩至干, 得油状物 25g, 加入 20ml 无水乙醇固化, 抽滤得淡黄色固体 12.50g, 收率为 80.48%。
6、 顺反异构体(vii ) 的重结晶
50ml 的圆底烧瓶中, 加入顺反异构体(vii ) 12.50g ( 30.75mmol ), 加入 20ml无水乙醇, 加热部分固体溶解后, 室温搅拌, 抽滤, 滤饼用 10ml的无水 乙醚洗涤后, 红外灯千燥, 得纯顺反异构体(vii ) 9.27g, 淡黄色粉末固体, 收率为 74.16%。
7、 毛兰素的制备
250ml 的三颈瓶中, 加入纯顺反异构体 ( vii ) 5.14g ( 12.56mmol ), 溶于 100ml乙酸乙酯和 60ml无水乙醇中, 淡黄色溶液, 加入 5%钯炭 0.50g后, 搅 拌下通氢气, 室温搅拌 3hr, 过滤, 得无色滤液, 旋转蒸发仪浓缩至干, 得油 状物 4.05g, 毛兰素粗品, 收率为 100%。
8、 毛兰素的精制
500ml的圆底烧瓶中, 加入毛兰素粗品 4.05g ( 12.72mmol ), 用 20ml无 水乙醚溶解, 若有不溶物, 过滤除去, 室温静止, 有晶体析出, 放置过夜, 溶 剂挥发完全, 大量白色晶体析出。 过滤, 滤饼用乙醚洗涤, 白色晶体 3.56g, 收率为 100%。
毛兰素盐的制备
【实施例 1】 毛兰素磷酸酯二钠的制备 (直接转化成钠盐)
1、 磷酰化反应
在 100ml圆底烧瓶中,加入三氯氧嶙 4.4ml ( 47.4mmol )和二氯甲烷 25ml, 滴加入毛兰素 5g ( 15.7 mmol )于 10ml二氯甲烷的溶液。 滴完后, 搅拌 5分
钟。 滴加入三乙胺 3.3ml ( 23.8mmol )于 5ml二氯甲烷的溶液。 室温搅拌 3hr, TLC检测, 反应完全后, 加入 100ml冷水淬灭。 充分振荡分出有机相; 再用 水 50mlx2洗涤, 二氯曱烷萃取水层后合并有机层, 用适量无水硫酸纳干燥过 夜; 抽滤, 滤液减压蒸去溶剂, 得粘稠液体, 即为粗酰氯。
2、 成盐反症
用水浴冷却粗酰氯,搅拌下加入 2mol/L的 NaOH溶液,直到混合液 pH值在 8 ~ 10之间, 50°C ~ 80°C搅拌 8小时。 过滤除去不溶物, 减压蒸去大部分溶剂, 冷却析晶得白色固体, 为毛兰素磷酸酯二钠盐粗品。 粗品用乙醇加热溶解, 趁 热过滤除去不溶固体, 滤液冷却析晶得到毛兰素磷酸酯二钠盐纯品约 6.0克, 为白色结晶产品, 产率 86 % 。 m.p. 266-269°C (分解); 在水中的溶解度: 200mg/ml o
3、 物理性质鉴定: 制备得到的产物的核磁共振、 红外和质讲分析结果见 图 1一图 4。
lUNMR ( D2O ): 52.77(d,lH,J=13.7Hz,H-l α ') , 2.81(d,lH,J=13.7 Ηζ,Η-1 α ) , 3.67(s,3H,4'=OCH3), 3.69 ( s,6H,3,5-OCH3 ), 3.75(s,3H,4-OCH3), 6.47 ( s,2H,H-2,6 ) , 6.77(d,lH,J=8.36Hz,H-5,) , 6.79(d,lH,J=8.36Hz,H- 6,) ,
7.27(d,lH,J=1.8 Hz, H-2') o
13C MR(D20): 5152.27, 148.05, 148.00, 143.35, 139.54, 135.14, 134.71, 122.32, 120.63, 120.62, 112.89, 106.21 , 61.01, 56.30, 56.09, 37.590, 36.70;
IR(KBr): O3852W, 3385S, 2938m, 2839m, 1588m, 1509m, 1464m, 1420m,
1273m, 1124s, 987m, cm 。
ESIMS: m/z:396.9(M+). Calc. 441.97forCi8H22O PNa2。
【实施例 2】 毛兰素磷酸酯二钠的制备 (需生成磷酸酯中间体)
1、 毛兰素磷酸二苄酯的合成
在干燥的三颈瓶中, 氩气保护, 毛兰素(20g, 63.2mmol )溶于 200ml乙 腈中, 搅拌, 冷却到 -25°C, 加入 CCl4 ( 35ml, 316mmol ), 搅拌 5分钟, 用针 筒滴加二异丙基乙胺 (23.13ml , 133mmol ), 然后滴加 DMAP ( 772mg, 6.32mmol ), 1分钟后滴加二苄基亚磷酸酯( 20.33ml, 92mmol ), 控制反应温 度在- 10°C以下, lhr后用 TLC检测,若反应完全,加入 50ml 0.5M的 KH2P04,
用乙酸乙酯 100mlx3萃取, 合并有机层, 用 100ml水, 100ml饱和 NaCl洗, 干燥, 过滤。 抽干溶剂, 得到黄色油状物。 柱层析(乙酸乙酯: 己烷 =2: 3 ), 用乙酸乙酯己烷重结晶,得到无色针状物,即为毛兰素磷酸二苄酯。 m.p 73°C。
2、 毛兰素磷酸酯钠盐的制备
氩气保护下,毛兰素磷酸二苄酯( 20.5g, 35.6mmol )和 Nal( 10.7g, 71mmol ) 的 100ml乙腈溶液中,强烈搅拌下,慢慢加入三曱基氯硅烷( 9.02ml, 71mmol ), 搅拌 20分钟, TLC检测没有原料点, 加入水(刚好可以溶解盐), 加入 10% 硫代硫酸钠 2ml, 分离有机相, 7J层用乙酸乙酯 50mlx4萃取, 合并有机层, 真空浓缩, 得到黄色泡沫状物。
泡沫状物用 100ml甲醇溶解,加入 95%甲醇钠(4.1g, 71mmol ),搅拌 9h, 甲醇真空抽干, 固体用水-丙酮和甲醇-丙酮重结晶, 得到白色固体, m.p. 266-269 °C, 即为毛兰素磷酸酯二钠。
对毛兰素磷酸酯二钠的物理性质测定结果同实施例 1。 由分析结果可见, 按照本发明方法制备获得了毛兰素磷酸酯二钠的成品。
【实施例 3】 毛兰素硫酸酯盐的制备
在 250ml圆底烧瓶中加入毛兰素 lOmmol, N、 N-二曱基苯胺 75mmol, 并 加入与 N、 N-二甲基苯胺等体积的 CH2C12, 在水盐水浴中搅拌(-5~-10°C ), 然后逐滴加入氯横酸 12.5mmol。 加完后在盐水浴中继续搅拌 lh。 然后移去盐 水浴, 在室温下搅拌 24h。 用 10mol/I的 NaOH在搅拌下将反应液调 pH至 10, 冷却(或;氷根中冷却)后过滤。 并用无水乙醚洗。 干燥后将固体产物溶解在曱 醇中过柱。 溶剂抽干后得毛兰素硫酸酯钠盐。 白色固体(易吸水)。
物理性质鉴定:
1HNMR ( D20 ): 52.79(d,lH,J=13.7Hz,H-l α ') , 2.80(d,lH,J=13.7 Ηζ,Η-1 α ) , 3.67(s,3H,4,=OCH3), 3.74 ( s,6H ,5-OCH3 ) , 3.75(s,3H,4-OCH3), 6.55 ( s,2H,H-2,6 ) , 6.58(d,lH,J=8.36Hz,H-5,) , 6.85(d,lH,J=8.36Hz,H-6,) , 7.33(d5lH,J=1.8 Hz, H-2').
13CNMR(D20): 8152.27, 148.05, 148.00, 143.35, 139.54, 135.14, 134.71 , 122.32, 120.63, 114.33, 108.03, 106.21 , 61.01 , 56.30, 56.09, 37.590, 36.70;
IR(KBr): 3539S, 3433m, 3001w, 2939m, 2841m, 1589s, 1508s, 1466m,
1422m, 1330m, 1262m, 1242s, 1174m, 1131s, 1014m, 619m, cm .
ESIMS: m/z: 398.427 (M+). Calc. 421.46forCI8H2208SNa. 毛兰素盐的安全性评价
【实施例 4】毛兰素磷酸酯二钠的 LD50测定
一、 实—险材料
实臉动物: 昆明小鼠, 上海斯莱克实验动物中心提供, 体重 18 ~ 22g, 雌 雄各半。
试剂: 0.9 %NaCl, 毛兰素磷酸酯二钠: 浙江赛尔生物医学研究有限公司提 供, 依上述方法制备, 苦味酸
器材: 1ml—次性注射器
二、 实—检方法和步骤
1. 实验 选用 KM小鼠, 每组 10只, 雌雄各半, 共设 7组。 用苦 味酸标记。
2. 每组分别按体重以 1300 mg/kg、 1250 mg/kg、 1200 mg/kg、 1150 mg/kg、 llOOmg/kg. 1050mg/kg、 lOOOmg/kg尾静脉注射给药。
3. 给药后 0.25h、 0.5h、 lh、 2h、 4h、 24h分别观察一次, 记录死亡率。 以后每天观察 1次, 记录死亡率(表 1 ), 持续 14天, 第 15天处死未死亡的 小鼠, 进行病理解剖。
表 1 剂量一死亡率数据表
剂量 死亡数 总 死亡
( mg/kg ) 小时 天 率
0-4 1 2 3 4 5 6
1300 6 1 7
1250 1 3 2 6
1200 1 1 3
1150 1 1
1100 3 3
1050 1 1
1000 0
四、 实-脸结果:
AP LD50 = 1243.0377 mg/kg置信区间为: 1049.9757 < LD5( 1471.5984 对实验过程中死亡的小鼠进行解剖后没有明显病变; 14天处死存活小鼠 并进行病理解剖, 没有出现病理变化。
说明 3,4,5,4,-四甲氧基二苯乙烷 -3,- 0-磷酸二钠盐毒性很低,具有很好的临 床用药安全性。 毛兰素盐的抗肿瘤药效学试验
【实施例 5】 毛兰素磷酸酯二钠在抗肿瘤中的应用
1、 试验样品
样品: 毛兰素的磷酸酯二钠(AP, 按实施例 1 制得)用生理盐水配制为 所需浓度。
对照品:注射用环磷酰胺(CTX ),上海华联制药有限公司,批号 ·. 020806。 配制时用生理盐水溶解。
2、 动物和瘤株
昆明种小鼠 50只, 雌性, 体重 19±lg, 由上海医药工业研究院动物房提 供。
S180腹氷瘤小鼠 2只, 由上海医药工业研究院动物房提供。
3、 试验方法
S180腹水瘤小鼠, 无菌条件下抽取腹水, 记数细胞, 用生理盐水稀释为 1-2 107个 /ml, 按 0.2ml/只给小鼠腋下皮下接种。 次日将小鼠随机均分为 5 组, 每组 10只。
给药剂量(mg/kg./d ):
AP 25mg/kg 、 75 mg/kg 、 225 mg/kg
给药途径: 静脉注射
对照品环磷酰胺采用静脉注射途径给药, 剂量为 30mg/kg x 9d。
另设空白对照组。
动物接种次日开始按体重给药,灌胃 0.5ml/20g,连续 9天,根据体重给药, 接种后第 10天处死, 取瘤块称重, 计算抑瘤率。
结果判定根据以下公式:
对照组瘤重一给药组瘤重
对照组瘤重
4、 结果
实验期间各組均未见死亡。
表 2、 AP抑制 S180荷瘤小鼠实验结果
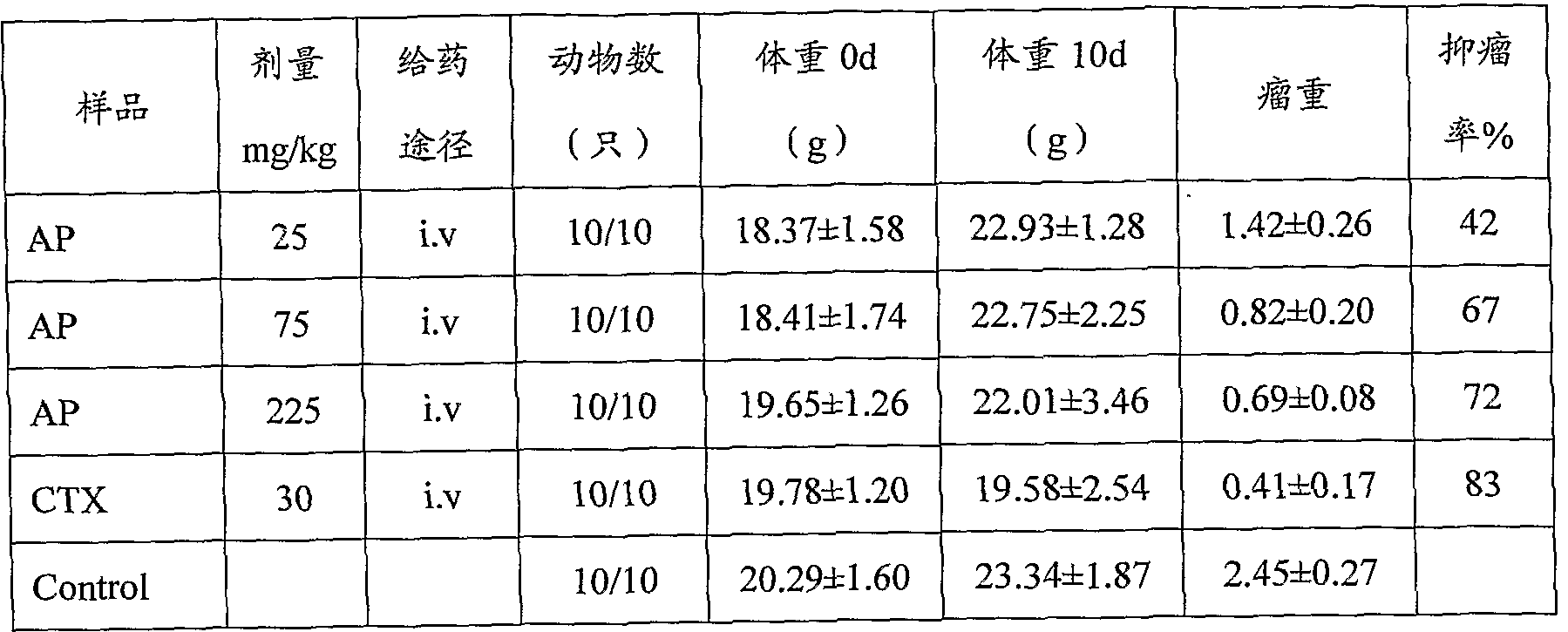
本发明所述的毛兰素盐具有比毛兰素更好的水溶性, 因而对于制剂更方 便, 可以提高药物在体内的生物利用度, 并且具有高效、 低毒的抗癌作用。
本发明所述的毛兰素盐的制备方法中,所用试剂都是便宜易得的普通化学 工业产品; 合成路线短; 操作方法简便, 适宜于工业规模生产。 以上对本发明较佳实施方式的描述并不限制本发明 ,本领域技术人员可以 根据本发明作出各种改变或变形,只要不脱离本发明的精神, 均应属于本发明 所附权利要求的范围。
Claims
权利要求书 类毛兰素盐, 其特征在于, 所述毛兰素盐为具有如下通式(I ) 的化

(I)
其中:
R为无机含氧酸的一价酸根与金属、 铵盐或有机胺所生成的盐; 所述金属选自碱金属、 碱土金属;
所述无机含氧酸选自磷酸、 硫酸、 亚硫酸、 硝酸、 碳酸、 次氯酸、 三氟 乙酸。
2、权利要求 1所述的毛兰素盐, 其特征在于, 所述毛兰素盐为毛兰素磷 酸酯盐或毛兰素硫酸酯盐。
3、权利要求 2所述的毛兰素盐, 其特征在于, 所述毛兰素盐为毛兰素磷 酸酯盐, 其通式如(II )所示:

其中, 、 为11、 Na、 K或 NH4;
且 和 不同时为 H。
4、 权利要求 3所述的毛兰素盐, 其特征在于, 式(II ) 中 R1为Na、 R:
为 Na, 所述毛兰素盐为毛兰素磷酸酯二钠。
5、权利要求 2所述的毛兰素盐, 其特征在于, 所述毛兰素盐为毛兰素石充 酸酯盐, 其通式如(III )所示:

(III)
其中, R3为 Na、 K或 ΝΉ4。
6、 权利要求 5所述的毛兰素盐, 其特征在于, 式(III )中 R3为Na, 所 述毛兰素盐为毛兰素硫酸酯钠。
7、 毛兰素盐的制备方法, 其特征在于, 包括下述步骤:
Al、 使毛兰素与磷酰化剂, 在縛酸剂的作用下, 发生酚 基的磷酰化反 应, 生成式(IV )的毛兰素磷酰化中间体, 反应温度为 25 ~ 80°C, 反应溶剂 为惰性有机溶剂;
A2、 使由步骤 A1得到的反应液直接与碱反应, 制备得到式(II ) 的毛 兰素碑酸酯盐;
合成路线为:

( II )
式(IV ) 中的 、 R5为 ObA Cl、 或 Br, 其中 代表叔丁基;
、 为11、 Na、 K或 NH4, 且 Ri和 R2不同时为11。
8、 权利要求 7所述的制备方法, 其特征在于,
在步骤 A1中:
所述磷酰化剂为选自 OPCl3、 OPBr3、 (ΒιΛ^Ρΐ 的一种或多种; 所述缚酸剂为选自吡啶类、 单烷基胺类、 双烷基胺类、 三烷基胺类的一 种或多种;
所述惰性有机溶剂为选自二氯曱烷、 二氯乙烷、 苯、 四氯化碳、 乙腈的 一种或多种;
在步骤 A2中:
所述碱为选自 NaOH、 KOH、 NaOCH3、 KOCH3、
 OCH2CH3 的一种或多种。
OCH2CH3 的一种或多种。
9、 权利要求 7所述的制备方法, 其特征在于,
在所述步骤 A1中: 磷酰化剂为 OPCl3, 缚酸剂为三乙胺;
在所述步骤 A2中: 碱为 NaOH。
10、 毛兰素盐的制备方法, 其特征在于, 包括下述步骤:
Bl、 在惰性气体保护下, 使毛兰素与二苄基亚磷酸酯, 在縛酸剂的作用 下, 发生酚羟基的磷酰化反应, 制备得到式 (V ) 的化合物, 反应温度为 -40 °C ~ -10°C , 反应在惰性有机溶剂中进行;
B2、 在惰性气体保护下, 使式(V ) 的化合物与 Nal在活性基保护剂的 作用下发生反应, 制备得到式(VI )的化合物, 反应在惰性有机溶剂中进行;
B3、 使式(VI ) 的化合物与碱反应, 制备得到式(Π ) 的化合物; 合成路线为:

式(V ) 中的 bn代表苄基; 、 R2为 H、 Na、 K或 Ν¾, 且 和 R: 不同时为 H。。
11、 权利要求 10所述的制备方法, 其特征在于,
在步骤 B1中:
所述缚酸剂为选自吡啶类、 单烷基胺类、 双烷基胺类、 三烷基胺类、 二 甲氨基吡啶的一种或多种;
所述惰性气体为氩气、 氮气、 或一氧化碳;
所述惰性有机溶剂为选自二氯曱烷、 二氯乙烷、 苯、 四氯化碳、 乙腈的 一种或多种;
在步驟 B2中:
所述惰性有机溶剂为选自二氯甲烷、 二氯乙烷、 苯、 四氯化碳、 乙腈的 一种或多种;
在步驟 B3中:
所述碱为选自 NaOH、 KOH、 NaOCH3、 KOCH3、 NaOCHaCHa. KOCH2CH3 的一种或多种。
12、 权利要求 10所述的制备方法, 其特征在于,
在所述步驟 B1 中: 所述缚酸剂为二异丙基乙胺和二甲氨基吡啶, 惰性 气体为氩气;
在所述步骤 B2中: 惰性气体为氩气, 活性基保护剂为三曱基氯硅烷, 惰性有机溶剂为乙腈和 /或四氯化碳;
在所述步驟 B3中: 碱为甲醇钠。
13、 毛兰素盐的制备方法, 其特征在于, 包括下述步骤:
Cl、使毛兰素与卤磺酸,在缚酸剂的作用下,发生酚羟基的硫酰化反应, 生成式 (VI )的毛兰素硫酰化中间体, 反应温度为 -5〜- 10 °C , 反应溶剂为惰 性有机溶剂;
C2、 使由步骤 C1得到的反应液直接与碱反应, 制备得到式(III ) 的毛 兰素 υ酸酯钠盐;
合成路线为:

式 (VII ) 中的 为 CI或 Br; R3为 Na、 * 或 ¾。
14、 权利要求 13所述的制备方法, 其特征在于,
在步骤 C1中:
所述卤磺酸为氯磺酸或溴磺酸;
所述缚酸剂为选自吡啶类、 单烷基胺类、 双烷基胺类、 三烷基胺类的一 种或多种;
所述惰性有机溶剂为选自二氯甲烷、 二氯乙烷、 苯、 四氯化碳的一种或 多种;
在步骤 C2中:
所述碱为选自 NaOH、 KOH、 NaOCH3、 KOCH3、 NaOCHbCHs KOCH2CH3 的一种或多种。
15、 权利要求 13所述的制备方法, 其特征在于,
在所述步骤 C1 中: 卤磺酸为氯磺酸, 縛酸剂为 >^、 N-二曱基苯胺, 惰 性有机溶剂为二氯甲烷;
在所述步骤 C2中: 碱为氢氧化钠。
16、 一种药物组合物, 其包含
治疗有效量的权利要求 1 ~ 6之一所述毛兰素盐; 以及
药学上可接受的载体和 /或赋形剂。
17、权利要求 16所述的药物组合物, 其特征在于, 所述药物组合物为注 射给药制剂或口服给药制剂。
18、 权利要求 1所述毛兰素盐在制备抗肿瘤药物中的应用。
Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06775252A EP1916251A4 (en) | 2005-08-02 | 2006-08-01 | ERIANINE SALTS, PROCESSES FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME |
| US11/997,595 US20080306028A1 (en) | 2005-08-02 | 2006-08-01 | Erianin Salts, Their Preparation Methods and Pharmaceutical Compositions Containing the Same |
| JP2008524345A JP2009502986A (ja) | 2005-08-02 | 2006-08-01 | エリアニン塩及びその調製方法、並びにそれを含む薬物組成物 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2005100890042A CN1907989B (zh) | 2005-08-02 | 2005-08-02 | 毛兰素盐及其制备方法和包含其的药物组合物 |
| CN200510089004.2 | 2005-08-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007014524A1 true WO2007014524A1 (en) | 2007-02-08 |
Family
ID=37699234
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/CN2006/001918 Ceased WO2007014524A1 (en) | 2005-08-02 | 2006-08-01 | Erianin salts, their preparation methods and pharmaceutical compositions containing the same |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20080306028A1 (zh) |
| EP (1) | EP1916251A4 (zh) |
| JP (1) | JP2009502986A (zh) |
| CN (1) | CN1907989B (zh) |
| WO (1) | WO2007014524A1 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012505841A (ja) * | 2008-10-15 | 2012-03-08 | シャンハイ エクスト バイオメディスン コンパニー リミテッド | エトキシジフェニルエタン誘導体ならびにその製造方法および使用 |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1907989B (zh) | 2005-08-02 | 2011-08-17 | 浙江天皇药业有限公司 | 毛兰素盐及其制备方法和包含其的药物组合物 |
| CN101235003B (zh) * | 2007-02-01 | 2010-09-15 | 浙江赛尔生物医学研究有限公司 | 3,4,5,4'-四甲氧基-α,β-二苯乙烷-3'-O-硫酸酯钠盐及其应用 |
| CN101735888B (zh) * | 2009-12-09 | 2012-09-26 | 浙江森宇实业有限公司 | 一种铁皮石斛花挥发油的提取方法 |
| JP5514973B2 (ja) * | 2010-04-30 | 2014-06-04 | 独立行政法人産業技術総合研究所 | ホスホン酸ジエステル誘導体の製造方法 |
| CN103980099B (zh) * | 2014-05-29 | 2016-01-13 | 云南金九地生物科技有限公司 | 一种从石斛中提取毛兰素的方法 |
| CN104782625B (zh) * | 2015-03-30 | 2016-09-14 | 上海应用技术学院 | 毛兰素及其衍生物作为农业杀菌剂的应用 |
| CN107382796B (zh) * | 2017-08-11 | 2020-10-09 | 浙江华理生物制药有限公司 | 一种ca-4类抗肿瘤药物、合成方法及其应用 |
| US20200163913A1 (en) * | 2017-07-25 | 2020-05-28 | Shanghai Huali Biomedical Co., Ltd. | CA-4 Antitumour Drug, Synthesis Method and Use Thereof |
| CN110958998B (zh) * | 2017-07-26 | 2022-09-23 | 香港大学 | 毛兰素衍生物和使用毛兰素衍生物的方法 |
| CN112480165A (zh) * | 2020-11-05 | 2021-03-12 | 上海应用技术大学 | 一种多取代二苯乙烷磷酸酯钠盐ebtp的a晶型及其制备方法 |
| CN117185908A (zh) * | 2023-09-09 | 2023-12-08 | 郑州大学 | 一种具有抗肿瘤活性的毛兰素衍生物、其药物组合物及用途 |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1435404A (zh) | 2003-03-12 | 2003-08-13 | 浙江天皇药业有限公司 | 一种从石斛中提取毛兰素的方法 |
| CN1907989A (zh) | 2005-08-02 | 2007-02-07 | 浙江赛尔生物医学研究有限公司 | 毛兰素盐及其制备方法和包含其的药物组合物 |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA1338645C (en) * | 1987-01-06 | 1996-10-15 | George R. Pettit | Isolation, structural elucidation and synthesis of novel antineoplastic substances denominated "combretastatins" |
| GB9106177D0 (en) * | 1991-03-22 | 1991-05-08 | Aston Molecules Ltd | Substituted diphenylethylenes and analogues or derivatives thereof |
| US5561122A (en) * | 1994-12-22 | 1996-10-01 | Arizona Board Of Regents Acting On Behalf Of Arizona State University | Combretastatin A-4 prodrug |
| JP2002500227A (ja) * | 1998-01-09 | 2002-01-08 | アリゾナ ボード オブ リーゼンツ | コンブレタスタチンa−4プロドラッグとそのトランス異性体 |
| US7078552B2 (en) * | 2000-04-27 | 2006-07-18 | Arizona Board Of Regents | Combretastatin A-1 phosphate and combretastatin B-1 phosphate prodrugs |
| US6670344B2 (en) * | 2000-09-14 | 2003-12-30 | Bristol-Myers Squibb Company | Combretastatin A-4 phosphate prodrug mono- and di-organic amine salts, mono- and di- amino acid salts, and mono- and di-amino acid ester salts |
| US6956126B2 (en) * | 2002-07-02 | 2005-10-18 | Wyeth | Preparation of 6-hydroxyequilenins |
| CN1312161C (zh) * | 2005-04-30 | 2007-04-25 | 中国科学院广州化学研究所 | 4,3’,4’,5’-四甲氧基联苄-3-o-磷酸酯盐及其组合物、制备方法和应用 |
| CN101235003B (zh) * | 2007-02-01 | 2010-09-15 | 浙江赛尔生物医学研究有限公司 | 3,4,5,4'-四甲氧基-α,β-二苯乙烷-3'-O-硫酸酯钠盐及其应用 |
-
2005
- 2005-08-02 CN CN2005100890042A patent/CN1907989B/zh not_active Expired - Fee Related
-
2006
- 2006-08-01 WO PCT/CN2006/001918 patent/WO2007014524A1/zh not_active Ceased
- 2006-08-01 EP EP06775252A patent/EP1916251A4/en not_active Withdrawn
- 2006-08-01 US US11/997,595 patent/US20080306028A1/en not_active Abandoned
- 2006-08-01 JP JP2008524345A patent/JP2009502986A/ja not_active Withdrawn
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1435404A (zh) | 2003-03-12 | 2003-08-13 | 浙江天皇药业有限公司 | 一种从石斛中提取毛兰素的方法 |
| CN1907989A (zh) | 2005-08-02 | 2007-02-07 | 浙江赛尔生物医学研究有限公司 | 毛兰素盐及其制备方法和包含其的药物组合物 |
Non-Patent Citations (6)
| Title |
|---|
| JOURNAL OF CHINA PHARMACEUTICAL UNIVERSITY, vol. 25, no. 3, 1994, pages 188 - 189 |
| LI YUNMAN: "Erianin Induces Apoptosis of Human leukemia HL-60 cells", ACTA PHARMACOLOGICA SINICA, vol. 22, no. LL, November 2001 (2001-11-01), pages 1018 - 1022 |
| NANDY P. ET AL.: "Quantitative structure-activity relationship analysis of combretastatins: a class of novel antimitotic agents", PHARMACEUTICAL RESEARCH, vol. 8, no. 6, 1991, pages 776 - 781, XP008076121 * |
| See also references of EP1916251A4 |
| WANG T. ET AL.: "In vitro inhibition of leukemia K562 cells growth by constituents from Dendrobium chrysotoxum", TIANRAN CHANWU YANJIU YU KAIFA, vol. 9, no. 2, 1997, pages 1 - 3, XP008076118 * |
| WANG TIANSHAN: "In vitro Inhibition Activities on the Growth of Tumor Cell Strain K256 by Constituents from Dendrobium Chrysotoxum", NATURAL PRODUCT RESEARCH AND DEVELOPMENT, vol. 9, no. 2, 1997, pages 1 - 3 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012505841A (ja) * | 2008-10-15 | 2012-03-08 | シャンハイ エクスト バイオメディスン コンパニー リミテッド | エトキシジフェニルエタン誘導体ならびにその製造方法および使用 |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009502986A (ja) | 2009-01-29 |
| CN1907989A (zh) | 2007-02-07 |
| CN1907989B (zh) | 2011-08-17 |
| EP1916251A4 (en) | 2010-09-08 |
| US20080306028A1 (en) | 2008-12-11 |
| EP1916251A1 (en) | 2008-04-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4617382B2 (ja) | アルコールおよびフェノールの水溶性ホスホノオキシメチル誘導体の調製法 | |
| WO2007014524A1 (en) | Erianin salts, their preparation methods and pharmaceutical compositions containing the same | |
| WO2010043180A1 (zh) | 一种乙氧基二苯乙烷衍生物及其制备方法和用途 | |
| EP2096924B1 (en) | Novel hydrophilic derivatives of 2-aryl-4-quinolones as anticancer agents | |
| CN102219817A (zh) | 活性偶联剂用于氟化嘧啶化合物烷氧羰酰化的方法 | |
| TW201212905A (en) | 1,5-diphenyl-penta-1,4-dien-3-one compounds | |
| EP2924042A1 (en) | Bis- -carboline compound and preparation method, pharmaceutical composition and use thereof | |
| CN108947949B (zh) | 抗焦虑氘代化合物及其医药用途 | |
| CN108947946B (zh) | 抗脑损伤氘代化合物及其医药用途 | |
| KR20090048504A (ko) | 에톡시콤브레타스타틴 및 해당 전구약물의 제조와 용도 | |
| CN107698521A (zh) | 一种5-氟尿嘧啶取代羧酸衍生物的制备及用途 | |
| CN101691384B (zh) | 含磷的二苯乙烯类化合物及其制备方法和用途 | |
| JP2018505184A5 (zh) | ||
| JP2006524672A (ja) | ウイルス感染の治療において使用するための複素環式化合物 | |
| CN106831397A (zh) | 一种蒽醌类化合物及其制备方法和医用用途 | |
| JP4249928B2 (ja) | 抗腫瘍剤としてのビス−(n,n’−ビス−(2−ハロエチル)アミノ)ホスホルアミデート | |
| CN117185908A (zh) | 一种具有抗肿瘤活性的毛兰素衍生物、其药物组合物及用途 | |
| CN102952151A (zh) | 3位双β-咔啉碱类化合物、其制法和其药物组合物与用途 | |
| JP2021504455A (ja) | トリプトリド誘導体およびその製造方法と使用 | |
| CN116925055B (zh) | 香豆素-哌嗪-呋喃酮杂化物及其制备方法和应用 | |
| CN114621135A (zh) | 一种lpa1小分子拮抗剂 | |
| CN105906665B (zh) | 咔唑磺酰胺衍生物前药或其可药用盐及其制备方法和应用 | |
| CN113549046B (zh) | 一种双联苄地钱素s衍生物及其制备方法和应用 | |
| CN120864998A (zh) | 具有抗肿瘤活性的新型硝酸酯修饰阿司匹林与boc-氨基酸偶联化合物的设计、合成 | |
| CN120943804A (zh) | 改进的金雀异黄酮类化合物及其制备方法和应用 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008524345 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006775252 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006775252 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11997595 Country of ref document: US |