WO2007027248A2 - 3', 5' - cyclic nucleoside analogues for treatment of hcv - Google Patents
3', 5' - cyclic nucleoside analogues for treatment of hcv Download PDFInfo
- Publication number
- WO2007027248A2 WO2007027248A2 PCT/US2006/019114 US2006019114W WO2007027248A2 WO 2007027248 A2 WO2007027248 A2 WO 2007027248A2 US 2006019114 W US2006019114 W US 2006019114W WO 2007027248 A2 WO2007027248 A2 WO 2007027248A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- methyl
- cycloalkyl
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
Definitions
- This invention relates to cyclic nucleoside analogues and to the use of such compounds in the treatment of Hepatitis C infection.
- HCV infection presents a worldwide health problem that affects approximately 170 million people, with about 30,000 new cases in the United States each year. Because HCV is not easily cleared by immunological defenses, as many as 85% of the people who are infected with HCV become chronically infected (Hoofnagle, J. H., 1997, Hepatology 26: 15S-20S). A substantial portion of these infected individuals will slowly progress into severe liver diseases, including cirrhosis, liver failure, and hepatocellular carcinoma (Lauer, G. M.; Walker B. D. 2001, N. Engl. J. Med. 345: 41-52).
- HCV Liver failure due to HCV is the leading cause of liver transplantation in the United States and Europe (Seeff, L. B.2002, Hepatology 36(Suppl 1): 35S-46S; Adam R. et al.2000, Lancet 356:621-627).
- HCV is a single-stranded, positive-sense RNA virus belonging to the hepacivirus genus of the Flaviviridae family (Choo, Q. L. et ah, 1989, Science 244:359-364).
- the 9.6 kb genome of HCV encodes a single polyprotein which is cleaved co- and post-translationally by cellular and viral proteases into at least four structural (C, El, E2, and p7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins.
- structural C, El, E2, and p7
- non-structural NS2, NS3, NS4A, NS4B, NS5A and NS5B
- NS5B the RNA- dependent RNA polymerase
- HCV forms membrane-associated replication complexes in catalyzing RNA synthesis during viral RNA replication.
- These replication complexes contain viral non-structural proteins (NS3, NS4A, NS4B, NS5A and NS5B), viral RNA and unidentified host cellular proteins.
- compositions and methods for effective treatment of viral infections especially for effective treatment of HC V infections.
- nucleoside and nucleotide analogues for the treatment of HCV is accepted clinical practice, in particular, the combination of the pyrimidine analogue ribavirin (l- ⁇ -D-ribofuranosyl-lH-l ,2,4-triazole-3-carboxamide) and ⁇ -interferon is the current therapy of choice.
- ribavirin l- ⁇ -D-ribofuranosyl-lH-l ,2,4-triazole-3-carboxamide
- ⁇ -interferon is the current therapy of choice.
- the search for more potent analogues with fewer side effects continues.
- Sommoadossi and LaColla described a group of purine and pyrimidine nucleosides and nucleotides derived from 2-methyl ribofuranose in WO 01/90121 and WO 01/92282 (now U.S. Patent No. 6,812,219 to Idenix).
- the compounds disclosed are those with 6-alkylamino, 6-alkylamino-8-amino, and 6-alkylamino-2- amino substitution patterns.
- Another group of purine nucleotides and nucleosides for the treatment of HCV can be found in WO 02/18404 by Devos et al. (to Hoffmann-LaRoche).
- the nucleotides disclosed by Davos include purines with 2-amino-6-substituted amino bases and having 2',2'-difluoro- or 2'-deoxy ribofuranose, but not 2' methyl ribofuranose.
- Stuyver and Watanabe make a generic disclosure that includes but does not specifically describe 2'-methyl ribofuranose- containing nucleosides.
- Several 2'-methyl ribonucleotides with 7-deazapurine and 7- substituted-purine moieties are disclosed by Bhat et al. in U.S. Patent No. 6,777,395. Bhat et al.
- An et al. disclose a group of 2'-m ethyl ribofuranosyl purine nucleoside derivatives in WO 03/062256, which is commonly owned with the present application.
- the purine derivatives described by An et al. contain substitutions — including hydrazinyl, methylhydrazinyl and methylsulfonylhydrazinyl — at the 6- position.
- Modified nucleotides and nucleosides have been widely used to treat not only HCV and other viral infections.
- the active drug is the nucleotide triphosphate.
- the compound administered to the patient is a prodrug; the active drug results from intracellular phosphorylation to yield the triphosphate.
- nucleotide Because it is unstable in plasma, and because it does not penetrate the cell membrane (due to its being charged at physiological pH), the nucleotide itself is never administered to the patient.
- the effectiveness of modified nucleotides as anti-virals depends both on the selectivity and affinity of the active drug for the viral polymerase and on the efficiency of the in vivo phosphorylation of the prodrug form that is administered.
- This invention provides a compound according to formula 1
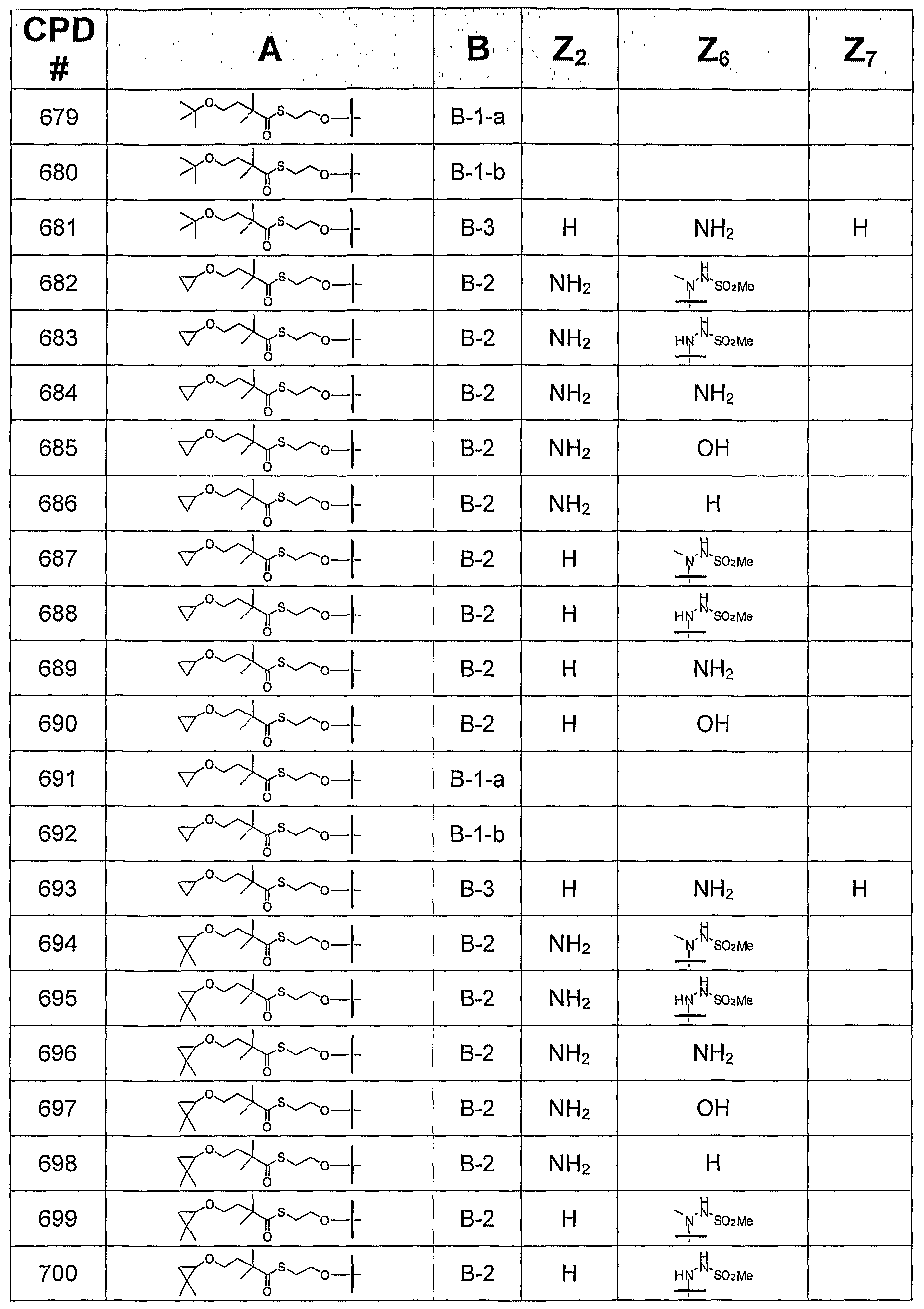
- B is a 6-membered monocyclic nitrogen-containing heteroaryl group or a 5 + 6 fused bicyclic nitrogen-containing heteroaryl group, and A is selected from Groups (1), (2), (3), and (4)
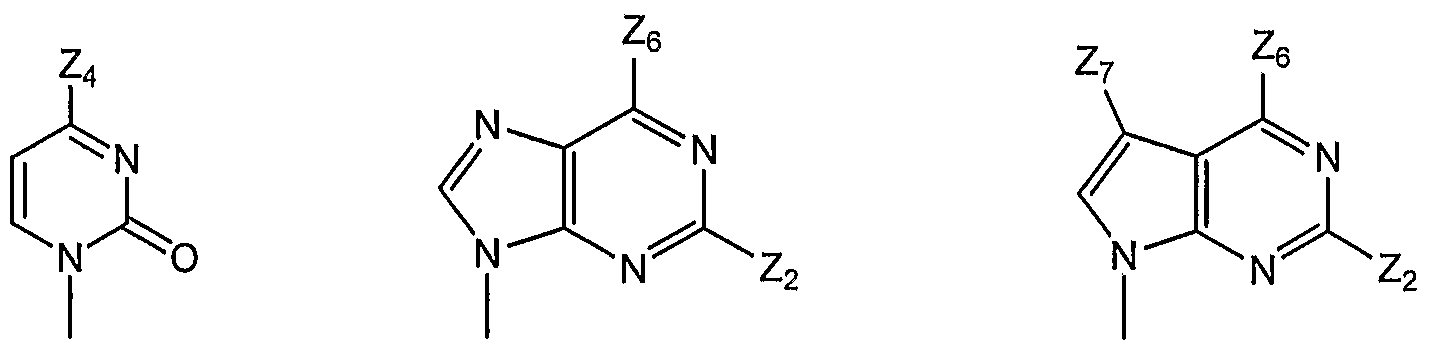
- this invention provides a compound of formula I, in which B is selected from bases B-I, B-2, and B-3,
- Z 2 is H, NH 2 , NHMe, or NMe 2 ;
- Z 4 is -NH 2 or -OH;
- Z 6 is H, OH, OMe, OEt, SCH 3 , thienyl, furyl, or NR 3 R 4 , where R 3 is H, C 1 -C 3 alkyl, or cyclopropyl, and R 4 is H or NHR 5 , R 5 is H, C 1 -C 4 alkyl, or SO 2 R 6 , and R 6 is C t -C 4 alkyl; and
- Z 7 is H, halogen, or CN. All tautomeric forms are included in these definitions.
- this invention provides a compound of formula I, in which B is B-I and is selected from B-l-a and B-l-b:
- this invention provides a compound of formula I, in which B is B-2.
- this invention provides a compound of formula I, in which B is B-2, where Z 2 is NHMe or NMe 2 .
- this invention provides a compound of formula I, in which B is B-2, where Z 2 is H or NH 2 .
- this invention provides a compound of formula I, in which B is B-2, and Z 6 is H, OH, OCH 3 , OEt, or SCH 3 .
- this invention provides a compound of formula I, in which B is B-2, and Z 6 is thienyl or furyl.
- this invention provides a compound of formula I, in which B is B-2, and Z 6 is NR 3 R 4 , where R 3 is H, C 1 -C 3 alkyl, or cyclopropyl; R 4 is H or NHR 5 ; R5 is H, C 1 -C 4 alkyl, or SO 2 R 6 ; and R 6 is C 1 -C 4 alkyl.
- this invention provides a compound of formula I, in which B is B-2 and Z 6 is H, NH 2 , or NHCH 3 .
- this invention provides a compound of formula I, in which B is B-2 and Z 6 is NR 3 R 4 , where R 3 is H, methyl, ethyl, n- propyl, isopropyl, or cyclopropyl; R 4 is NHR 5 ; R 5 is H, C 1 -C 4 alkyl, or SO 2 R 6 ; and R 6 is methyl or ethyl.
- this invention provides a compound of formula I, in which B is B-3.
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is NHMe, or NMe 2 .
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 . In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 and Z 6 is OH, OCH 3 , OEt, or SCH 3 .
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 and Z 6 is thienyl or furyl.
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or -NH 2 and Z 6 is NR 3 R 4 , where R 3 is H, C 1 -C 3 alkyl, or cyclopropyl; R 4 is H or NHR 5 ; R 5 is H, C 1 -C 4 alkyl, or SO 2 R 6 ; and R 6 is C 1 -C 4 alkyl.
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 and Z 6 is H, NH 2 or NHCH 3 .
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 ; Z 6 is H, NH 2 or NHCH 3 ; and Z 7 is H, F, or CN.
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or NH 2 and Z 6 is NR 3 R 4 , where R 3 is H, methyl, ethyl, or cyclopropyl; R 4 is H Or NHR 5 ; R 5 is H, C 1 -C 4 alkyl, or SO 2 R 6 ; and R 6 is methyl or ethyl.
- this invention provides a compound of formula I, in which B is B-3, where Z 2 is H or -NH 2 and Z 6 is NR 3 R 4 , where R 3 is H, methyl, ethyl, or cyclopropyl; R 4 is H or NHR 5 ; R 5 is H, C 1 -C 4 alkyl, or SO 2 R 6 ; R 6 is methyl or ethyl; and Z 7 is H, F, or CN.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1).
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2).
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3). In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4).
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where R, is C 3 -C 6 cycloalkyl-O-(CH 2 ) m -, C 3 -C 6 cycloalkyl-(CH 2 ) n -O-
- this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where R 1 ' and R
- " are both methyl and where Ri is selected from CH 2 Cl, CH 2 CH 2 Cl, CH 2 CH 2 CH 2 Cl, CH 2 Cl-O-(CH 2 ) m , ClCH 2 CH 2 -O-(CH 2 ) m , or ClCH 2 CH 2 CH 2 -O-(CH 2 ) m , where m 0 to 2.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, ethyl, or isopropyl and R 2 " is H or methyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl or isopropyl, R 2 " is H or methyl, and R 2 is C 1- C 6 alkyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is C 3- C 5 cycloalkyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is phenyl, benzyl, or xylyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is fluorophenyl, chlorophenyl, or o-, m-, or p-tolyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is mono-halo benzyl or di- halo benzyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is mono-alkyl benzyl or di- alkyl benzyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R 2 ' is methyl, R 2 " is H or methyl, and R 2 is trifluoromethyl phenyl or trifluoromethyl benzyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is C 1- C 6 alkyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is methyl, ethyl, or isopropyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is C 3 .Cs cycloalkyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is phenyl, benzyl, or xylyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is trifluoromethyl phenyl or trifluoromethyl benzyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is mono-alkyl phenyl, di-alkyl phenyl, mono-alkyl benzyl or di- alkyl benzyl.
- the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R 2 is mono-halo phenyl, di-halo phenyl, mono-halo benzyl, or di- halo benzyl.
- the invention provides a compound of formula I, where B is B-2 or B-3, where Z 2 is H or NH 2 , and Z 6 is N(CH 3 )NHSO 2 (CH 3 ) Or NHNHSO 2 (CH 3 ).
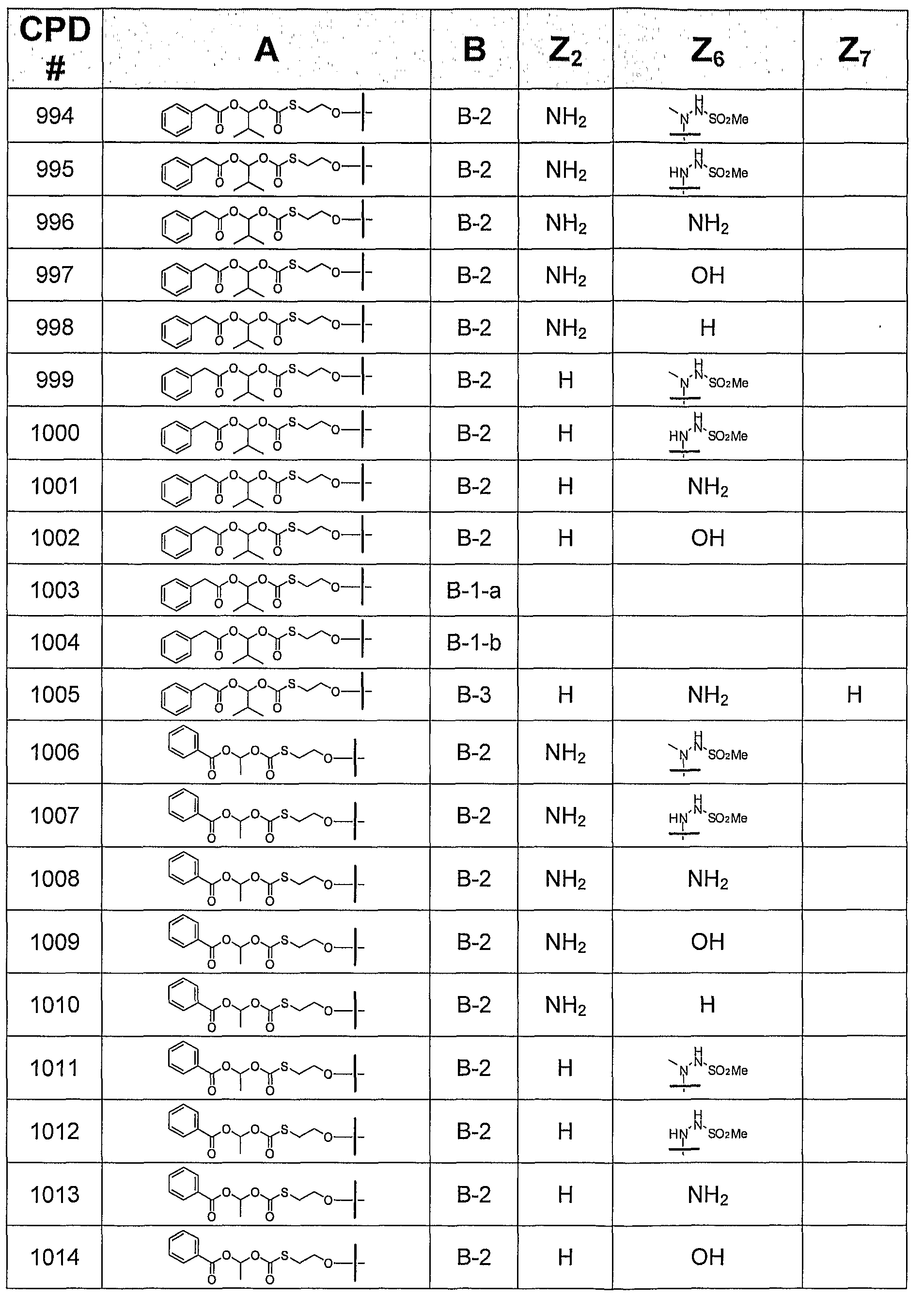
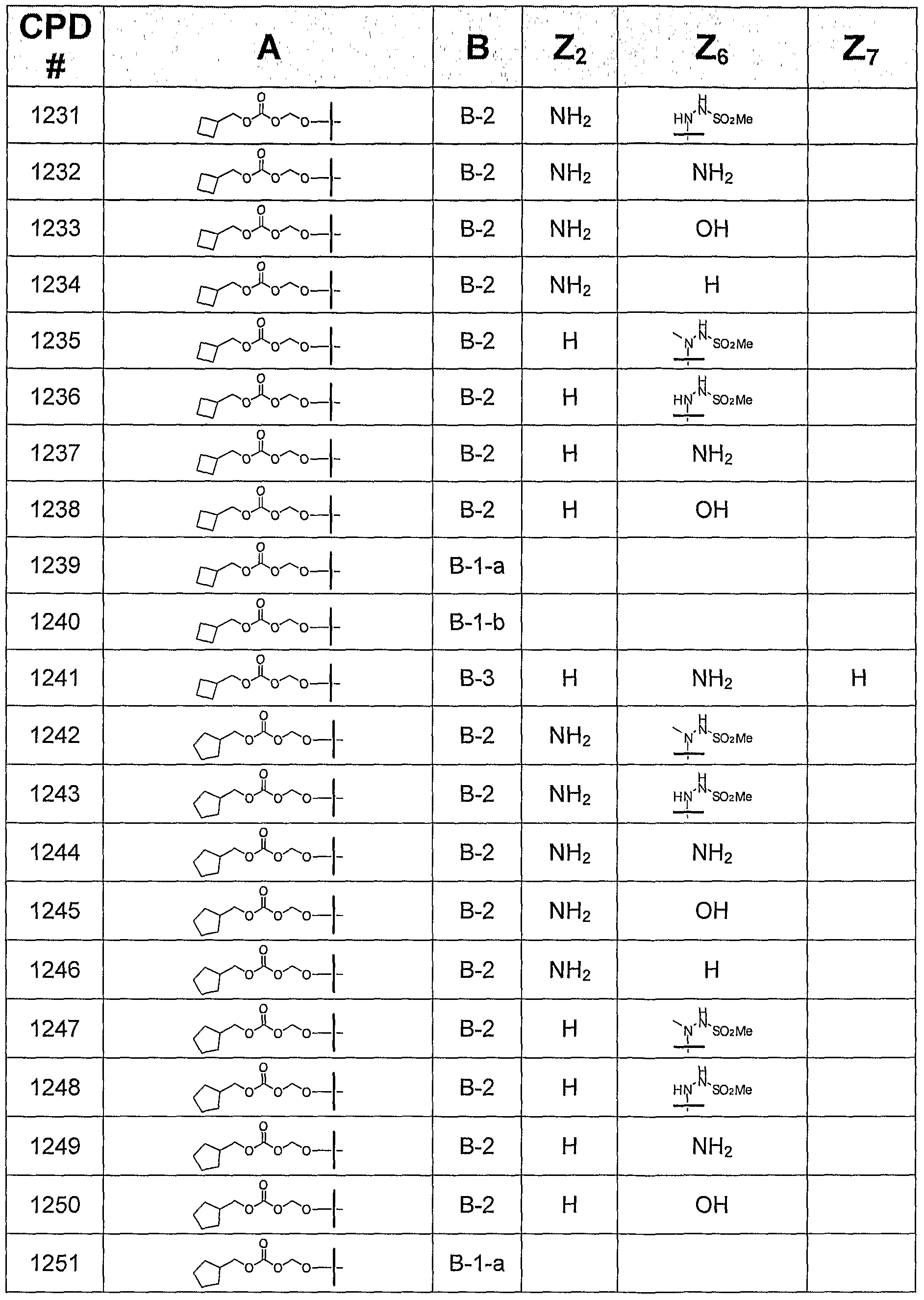
- the invention provides or contemplates a compound of formula I, where B is B-2 and is selected from the following bases:
- this invention provides or contemplates a compound of formula I, in which B is B-3 and is selected from the following bases:
- the present invention is directed to various substituted 3', 5'- cyclic monophosphate esters of 2'-methyl ribofuranosyl derivatives of heterocyclic bases, as depicted above.
- heterocyclic bases are commercially available, and many others may be readily synthesized from commercially available precursors via standard published procedures. (See, for example, Vorbruggen, H. and Ruh-Pohlenz, C, Handbook of Nucleoside Synthesis, New York, Wiley-Interscience; ISBN:0471093831). Additionally, a number of novel derivatives of heterocyclic bases are disclosed in the commonly owned, co-pending PCT Application titled "6- Hydrazinopurine-2 '-methyl Ribonucleosides and Nucleotides for Treatment of HCV," by E. Gunic and F. Rong, filed May 10, 2006 (Attorney Docket Number 6319-3032). Additional synthetic schemes are shown below.
- SATE S- acetylthioethyl
- MSNT l-(2-mesitylenesulfonyl)-3-nitro-l,2,4-triazole).
- a carbonic acid ester derivative of the cyclic nucleotide analogue can be prepared as depicted in synthetic Scheme 4 below:
- the compounds of scheme 4 can be made using any chloromethyl alkyl carbonate.
- a pivaloyloxymethyl (POM) cyclic nucleotide analogue can be prepared as shown in synthetic Scheme 5 below:
- one or more hydrogens of the methyl groups of the pivaloyl group may be replaced by halo, alkoxy, or alkyl groups.
- one of the methyl groups of the pivaloyl group can be replaced by a C 1 -C 4 alkoxy group.
- the nucleoside above (403 mg, 1.0 mmole) was dried over P 2 O 5 and dissolved in dry trimethyl phosphate (5.0 ml) by heating. The solution was cooled to 0 °C under argon. To this cold (0 °C) stirred solution was added POCl 3 (186 ⁇ L, 2.0 mmol) and the mixture stirred at 0 °C for 6 hr. The mixture was added drop by drop into a cold (0 °C) stirred solution of 0.08 M KOH in H 2 CVCH 3 CN (4:6, 120 ml) mixture during 10 minutes period. After 15 minutes, aq. HCl solution was added to pH 5 (0 °C), and the volume was reduced to 20 ml by evaporation.
- the reaction mixture was stirred at room temperature for 2 days and at 65 °C for 12 h under argon. It was evaporated to dryness. The residue was partitioned between EtOAc (500 ml) and water (300 ml), neutralized with acetic acid to pH 6 and extracted in EtOAc. The organic layer was washed with brine (200 ml), dried, and concentrated to dryness to give 14 g of crude product.
- reaction mixture was evaporated to dryness and the residue was purified by silica column using as the eluent methanol in chloroform. The fractions having the required product were pooled, and the mixture was concentrated to dryness. This purified material was again purified by HPLC to give 12.6 mg of pure product. The structure of the product was confirmed by 1 H NMR and 31 P NMR.
- 2-amino-6-(N-(methanesulfonyIhydrazidyl)-9-(2-C-methyl- ⁇ -D- ribofuranosyl)adenine (2): 2-(2-amino-6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)-3-methyl tetrahydrofuran-3,4-diol 1 (2.0 g, 6.35 mmol) and methane sulfonylhydrazide (4.0 g, 36.36 mmol) in a mixture of ethyl alcohol (100 ml) and dioxane (20 ml) was heated at 92 °C under argon for 2 days. Reaction mixture was evaporated to dryness.
- the anti-HCV activities of the exemplary compounds were tested in two biological assays — a cell-based HCV replicon assay and a cytotoxicity assay.
- a human hepatoma cell line (Huh-7) containing replicating HCV Conl subgenomic replicon with a luciferase reporter gene (luc-ubi-neo) was used to evaluate anti-HCV activity of the compounds. In this assay, the level of luciferase signal correlates directly with the viral RNA replication.
- the HCV repl icon-reporter cell line (NK/luc-ubi-neo) was cultured in DMEM medium supplemented with 10 % fetal bovine serum and 0.5 mg/ml Geneticin (G418). Cells were maintained in a subconfluent state to ensure high levels of HCV replicon RNA synthesis.
- a Huh-7 cell line carrying a luciferase reporter gene (driven by a HIV LTR promoter) stably integrated into the chromosome was used to analyze the cytotoxic effect of the selected compounds.
- This cell line (LTR-luc) was maintained in DMEM medium with 10 % FBS.
- Design of the cytotoxicity assay was similar to that of the HCV replicon assay. Reduction of luciferase activity in the treated cells correlated with the cytotoxic effect of the test compound and was used to calculate the CC 50 value (concentration that inhibited cell growth by 50%).
- the biological activities and cytotoxicity of the selected compounds are summarized in Table 4.
- Anti-HCV and anti-BVDV activities (EC 50 values) of the test compounds were categorized into three different groups: A (1 to 50 nM), B (50 to 500 ⁇ M) and C (500 to 1000 nM).
- the cytotoxicity (CC50 values) of the compounds are indicated in ⁇ M.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
Abstract
This invention provides compounds according to formula (I) where B is a 6-membered monocyclic nitrogen-containing heteroaryl group or a 5 + 6 fused bicyclic nitrogen-containing heteroaryl group, and A is selected from Groups (1), (2), (3), and (4) where Groups (1), (2), (3), and (4), are defined herein. These compounds are useful in the treatment of Hepatitis C infection.
Description
3',5'-CYCLIC NUCLEOSIDEANALOGUESFORTREATMENT OFHCV
Cross Reference to Related Applications This application claims priority to U.S. Provisional Application Set. No.
60/681,332, filed May 16, 2005; U.S. Provisional Application Ser. No. 60/748,130, filed December 6, 2005; and U.S. Provisional Application Ser. No. 60/785,238, filed March 22, 2006. All are incorporated by reference herein in their entirety.
Field of the Invention
This invention relates to cyclic nucleoside analogues and to the use of such compounds in the treatment of Hepatitis C infection.
Background of the Invention Hepatitis C virus (HCV) infection presents a worldwide health problem that affects approximately 170 million people, with about 30,000 new cases in the United States each year. Because HCV is not easily cleared by immunological defenses, as many as 85% of the people who are infected with HCV become chronically infected (Hoofnagle, J. H., 1997, Hepatology 26: 15S-20S). A substantial portion of these infected individuals will slowly progress into severe liver diseases, including cirrhosis, liver failure, and hepatocellular carcinoma (Lauer, G. M.; Walker B. D. 2001, N. Engl. J. Med. 345: 41-52). Liver failure due to HCV is the leading cause of liver transplantation in the United States and Europe (Seeff, L. B.2002, Hepatology 36(Suppl 1): 35S-46S; Adam R. et al.2000, Lancet 356:621-627). The Centers for Disease Control and Prevention estimate that chronic hepatitis C virus infection is responsible for 10,000 to 12,000 deaths in the United States annually. Without effective intervention, this number is expected to triple in the next 10 to 20 years.
HCV is a single-stranded, positive-sense RNA virus belonging to the hepacivirus genus of the Flaviviridae family (Choo, Q. L. et ah, 1989, Science 244:359-364). The 9.6 kb genome of HCV encodes a single polyprotein which is cleaved co- and post-translationally by cellular and viral proteases into at least four structural (C, El, E2, and p7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins. One of these nonstructural proteins is NS5B, the RNA- dependent RNA polymerase, plays a central role in viral RNA replication and is therefore an attractive target for the development of antiviral intervention.
Similar to the replication of most positive-strand RNA viruses, it is believed that HCV forms membrane-associated replication complexes in catalyzing RNA synthesis during viral RNA replication. These replication complexes contain viral non-structural proteins (NS3, NS4A, NS4B, NS5A and NS5B), viral RNA and unidentified host cellular proteins. Recently, several groups have demonstrated the in vitro replication activity of HCV replicase complexes in a crude membrane fraction isolated from the HCV subgenomic replicon cells (AIi, N. et al. 2002, J. Virol.
76: 12001-12007; Hardy, R. W. et al. 2003, J. Virol. 77:2029-2037; Lai, V. C. H. et al. 2003, J. Virol. 77:2295-2300). The success to replicate HCV RNA using authentic replicase complexes in vitro will facilitate molecular dissection of the replication process and provide a system to evaluate potential antiviral drugs against the entire replicase complex of HCV.
The development of effective vaccines for prophylaxis and treatment of HCV infection have been unsuccessful due to various virus-specific difficulties, especially immune evasion. Nonetheless, treatment of chronic hepatitis C has achieved significant advances in recent years. The current standard therapy for chronic hepatitis C consists of a combination of pegylated interferon-alpha (IFNα) and ribavirin. This therapy confers an overall sustained viral response (SVR) of around 54-56% among treated patients but is less effective against infection by HCV genotype 1. The limited effectiveness and adverse side effects of the current therapy underscores the urgent need for development of more effective and HCV-specific antiviral therapeutics.
Thus, in view of the prevalence of HCV infection and the limited effectiveness and adverse side effects of the current therapies, there is a need for compositions and
methods for effective treatment of viral infections, especially for effective treatment of HC V infections.
The use of nucleoside and nucleotide analogues for the treatment of HCV is accepted clinical practice, in particular, the combination of the pyrimidine analogue ribavirin (l-β-D-ribofuranosyl-lH-l ,2,4-triazole-3-carboxamide) and α-interferon is the current therapy of choice. The search for more potent analogues with fewer side effects continues. These efforts have involved not only modification of the native heterocyclic base as in ribavirin, but also modification of the ribofuranose moiety. Sommoadossi and LaColla described a group of purine and pyrimidine nucleosides and nucleotides derived from 2-methyl ribofuranose in WO 01/90121 and WO 01/92282 (now U.S. Patent No. 6,812,219 to Idenix). Among the compounds disclosed are those with 6-alkylamino, 6-alkylamino-8-amino, and 6-alkylamino-2- amino substitution patterns. Another group of purine nucleotides and nucleosides for the treatment of HCV can be found in WO 02/18404 by Devos et al. (to Hoffmann-LaRoche). The nucleotides disclosed by Davos include purines with 2-amino-6-substituted amino bases and having 2',2'-difluoro- or 2'-deoxy ribofuranose, but not 2' methyl ribofuranose. Stuyver and Watanabe (WO 02/32920 to Pharmasset) make a generic disclosure that includes but does not specifically describe 2'-methyl ribofuranose- containing nucleosides. Several 2'-methyl ribonucleotides with 7-deazapurine and 7- substituted-purine moieties are disclosed by Bhat et al. in U.S. Patent No. 6,777,395. Bhat et al. also disclose the use of two neutral phosphate ester derivatives of nucleotides ~ acetyl-SATE (S-acyl-2-thioethyl) and pivaloyl-SATE ~ as prodrugs of the ribonucleotide analogues. SATE prodrugs are disclosed generically in U.S. Patent No. 5,770,725.
An et al. disclose a group of 2'-m ethyl ribofuranosyl purine nucleoside derivatives in WO 03/062256, which is commonly owned with the present application. The purine derivatives described by An et al. contain substitutions — including hydrazinyl, methylhydrazinyl and methylsulfonylhydrazinyl — at the 6- position.
Modified nucleotides and nucleosides have been widely used to treat not only HCV and other viral infections. In each case, the active drug is the nucleotide triphosphate. The compound administered to the patient is a prodrug; the active drug results from intracellular phosphorylation to yield the triphosphate. Because it is unstable in plasma, and because it does not penetrate the cell membrane (due to its being charged at physiological pH), the nucleotide itself is never administered to the patient. The effectiveness of modified nucleotides as anti-virals depends both on the selectivity and affinity of the active drug for the viral polymerase and on the efficiency of the in vivo phosphorylation of the prodrug form that is administered.
Brief Description of the Invention
This invention provides a compound according to formula 1

I wherein B is a 6-membered monocyclic nitrogen-containing heteroaryl group or a 5 + 6 fused bicyclic nitrogen-containing heteroaryl group, and A is selected from Groups (1), (2), (3), and (4)
(1) (2)


where, in Groups (1) and (2), where Ri is H, CpCg alkyl, C3-C6 cycloalkyl-(CH2)m-,
C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloalkyl-(CH2)n-O-(CH2)m-> or C1-C8 alkyl-O- (CH2)m-, where the alkyl and cycloalkyl groups are optionally substituted with one or two groups selected independently from halogen, CF3, and C1-C3 alkyl, where m = zero to 4, and where n = zero to 2; and Ri' and R]" are, independently, H, C3-C5 cycloalkyl, or Q-C4 alkyl; and, in Groups (3) and (4), R2 is C1-C6 alkyl, C3-C5
cycloalkyl-(CH2)n, or the following group
 where n = 0 to 2, and where substituents Q and Q' are selected independently from H, F, Cl, Br, and Ct-C3 alkyl, which C1-C3 alkyl group or groups are optionally substituted with one or more halogen atoms; R2' is H, C3-C5 cycloalkyl, or C1-C4 alkyl; and R2" is H or C1-C4 alkyl. In one generic embodiment, this invention provides a compound of formula I, in which B is selected from bases B-I, B-2, and B-3,
where n = 0 to 2, and where substituents Q and Q' are selected independently from H, F, Cl, Br, and Ct-C3 alkyl, which C1-C3 alkyl group or groups are optionally substituted with one or more halogen atoms; R2' is H, C3-C5 cycloalkyl, or C1-C4 alkyl; and R2" is H or C1-C4 alkyl. In one generic embodiment, this invention provides a compound of formula I, in which B is selected from bases B-I, B-2, and B-3,

B-I B-2 B-3 where Z2 is H, NH2, NHMe, or NMe2; Z4 is -NH2 or -OH; Z6 is H, OH, OMe, OEt, SCH3, thienyl, furyl, or NR3R4, where R3 is H, C1-C3 alkyl, or cyclopropyl, and R4 is H or NHR5, R5 is H, C1-C4 alkyl, or SO2R6, and R6 is Ct-C4 alkyl; and Z7 is H, halogen, or CN. All tautomeric forms are included in these definitions.
In one subgeneric embodiment, this invention provides a compound of formula I, in which B is B-I and is selected from B-l-a and B-l-b:

B-l-a B-l-b
In another subgeneric embodiment, this invention provides a compound of formula I, in which B is B-2.
In a more specific embodiment, this invention provides a compound of formula I, in which B is B-2, where Z2 is NHMe or NMe2.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-2, where Z2 is H or NH2.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-2, and Z6 is H, OH, OCH3, OEt, or SCH3.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-2, and Z6 is thienyl or furyl.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-2, and Z6 is NR3R4, where R3 is H, C1-C3 alkyl, or cyclopropyl; R4 is H or NHR5; R5 is H, C1-C4 alkyl, or SO2R6; and R6 is C1-C4 alkyl.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-2 and Z6 is H, NH2, or NHCH3.
In another still more specific embodiment, this invention provides a compound of formula I, in which B is B-2 and Z6 is NR3R4, where R3 is H, methyl, ethyl, n- propyl, isopropyl, or cyclopropyl; R4 is NHR5; R5 is H, C1-C4 alkyl, or SO2R6; and R6 is methyl or ethyl.
In another subgeneric embodiment, this invention provides a compound of formula I, in which B is B-3.
In a more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is NHMe, or NMe2.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2 and Z6 is OH, OCH3, OEt, or SCH3.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2 and Z6 is thienyl or furyl.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or -NH2 and Z6 is NR3R4, where R3 is H, C1-C3 alkyl, or cyclopropyl; R4 is H or NHR5; R5 is H, C1-C4 alkyl, or SO2R6; and R6 is C1-C4 alkyl.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2 and Z6 is H, NH2 or NHCH3.
In a still more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2; Z6 is H, NH2 or NHCH3; and Z7 is H, F, or CN.
In another more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or NH2 and Z6 is NR3R4, where R3 is H, methyl, ethyl, or cyclopropyl; R4 is H Or NHR5; R5 is H, C1-C4 alkyl, or SO2R6; and R6 is methyl or ethyl.
In a still more specific embodiment, this invention provides a compound of formula I, in which B is B-3, where Z2 is H or -NH2 and Z6 is NR3R4, where R3 is H, methyl, ethyl, or cyclopropyl; R4 is H or NHR5; R5 is H, C1-C4 alkyl, or SO2R6; R6 is methyl or ethyl; and Z7 is H, F, or CN.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1).
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2).
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3).
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4).
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where R, is C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloalkyl-(CH2)n-O-
(CH2)m-, or C1-C8 alkyl-O-(CH2)m-, where m = 0 to 4, where n = 1 or 2, and where R|' and Ri" are, independently, H, C3-C5 cycloalkyl, or C1-C4 alkyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I , B-2, and B-3, and where A is Group (1), where R, is H, C1-C8 alkyl, or C3-C6 cycloalkyl-(CH2)m-, where m = 0 to 4, and where Ri' and Ri" are, independently, H, C3-C5 cycloalkyl, or C1-C4 alkyl.
In a still more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where Ri is C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloalkyl-(CH2)n-O- (CH2)m-, or C1-C8 alkyl-O-(CH2)m-, where m = 0 to 3, where n = 1 or 2, and where R, ' and Ri" are identical and are C1-C3 alkyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where Ri is H, C1-C8 alkyl, or C3-C6 cycloalkyl-(CH2)m-, where m = 0 to 2, and where Ri' and R]" are identical and are C1-C3 alkyl.
In a still more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where Rt is C3-C6 cycloalkyl-O-(CH2)m- or C3-C6 cycloalkyl-(CH2)n-0- (CH2)m-, or C3-C6 cycloalkyl-(CH2)m-, where the cycloalkyl group is substituted with methyl or halo, where m = 0 to 3, where n = 1 or 2, and where Ri' and Ri" are identical and are C1-C3 alkyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where Ri is H, C1-C8 alkyl, where m = 0 to 2, and where Ri' and Ri" are identical and are C1-C3 alkyl.
In another, even more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (1), where R1 is C1-C6 alkyl or C1-C6 alkyl-O-(CH2)m-, where the C,-C6 allcyl group is substituted with one or two halogens, where m = 0 to 2, and where R]' and R1" are both methyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2), where R1 is C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloaIkyl-(CH2)n-O-(CH2)m-, or C1- C6 alkyl-O-(CH2)m-, where m = 0 to 4, where n = 1 or 2, and where R|' and R1" are, independently, H, C3-C5 cycloalkyl, or C1-C4 alkyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2), where R1 is H, C1-C6 alkyl, or C3-C6 cycloalkyl-(CH2)m-, where m = 0 to 4, and where R1' and R1" are, independently, H, C3-C5 cycloalkyl, or C1-C4 alkyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2), where R1 is C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloalkyl-(CH2)n-0-(CH2)m-, or C1- C6 alkyl-O-(CH2)m-, where m = 0 to 2, where n = 0 or 2, and where R1 ' and R1 " are identical and are C1-C3 alkyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2), where R1 is H, C1-C6 alkyl, or C3-C6 cycloalkyl-(CH2)m-, where m = 0 to 2, and where Ri' and R1" are identical and are C1-C3 alkyl.
In an even more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (2), where R1 is C1-C6 alkyl, or C1-C6 alkyl-O-(CH2)m-, where m = 0 to 2, and where R1' and Ri" are both methyl.
In another more specific embodiment, this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where R1 is C5-C8 alkyl or C5-C8 alkyl-O-(CH2)m-, where m = 0 to 2, and where R1' and R1" are both methyl.
In another more specific embodiment, this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where Ri' and R]" are both methyl and where R1 is selected from CH2F, CH2CH2F, CH2CH2CH2F, CH2F-O- -(CH2)m, FCH2CH2-O-(CH2)m, or FCH2CH2CH2-O-(CH2)m, where m = 0 to 2.
In another more specific embodiment, this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where Ri' and R]" are both methyl and where R1 is selected from CH2CH2CH2CH2F, CH2CH2CH(CH2F)2, CH2CH2CH2CH2CF3, CH2CH2CH2CHCF3, CH2CH2CH2F, CH3CH2F-O-(CH2)m, CF3CH2-O-(CH2)m, or CF3CH2CH2-O-(CH2)m, where m = 0 to 2.
In another more specific embodiment, this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where R1' and R|" are both methyl and where Ri is selected from CH2Cl, CH2CH2Cl, CH2CH2CH2Cl, CH2Cl-O-(CH2)m, ClCH2CH2-O-(CH2)m, or ClCH2CH2CH2-O-(CH2)m, where m = 0 to 2.
In another more specific embodiment, this invention provides or contemplates a compound of formula I, where A is Group (1) or Group (2), where R1' and R1" are both methyl and where R1 is selected from CH2CH2CH2CH2Cl, CH2CH2CH(CH2Cl)2, CH2CH2CH2Cl, CH2Cl-O-(CH2)m, ClCH2CH2-O-(CH2)m, or ClCH2CH2CH2-O- (CH2)m, where m = 0 to 2.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, ethyl, or isopropyl and R2" is H or methyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl or isopropyl, R2" is H or methyl, and R2 is C1-C6 alkyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is C3-C5 cycloalkyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is phenyl, benzyl, or xylyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is fluorophenyl, chlorophenyl, or o-, m-, or p-tolyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is mono-halo benzyl or di- halo benzyl.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is mono-alkyl benzyl or di- alkyl benzyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (3), where R2' is methyl, R2" is H or methyl, and R2 is trifluoromethyl phenyl or trifluoromethyl benzyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is C1-C6 alkyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is methyl, ethyl, or isopropyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is C3.Cs cycloalkyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is phenyl, benzyl, or xylyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is trifluoromethyl phenyl or trifluoromethyl benzyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is mono-alkyl phenyl, di-alkyl phenyl, mono-alkyl benzyl or di- alkyl benzyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is selected from B-I, B-2, and B-3, and where A is Group (4), where R2 is mono-halo phenyl, di-halo phenyl, mono-halo benzyl, or di- halo benzyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is B-2 or B-3, where Z2 is H or NH2, and Z6 is N(CH3)NHSO2(CH3) Or NHNHSO2(CH3).
In another subgeneric embodiment, the invention provides a compound of formula I, where B is B-2 or B-3, where Z2 is H or NH2; Z6 is N(CH3)NHSO2(CH3) or NH-NHSO2(CH3); and A is Group (1) or Group (2), with R,1 = R1 " = methyl.
In another subgeneric embodiment, the invention provides a compound of formula I, where B is B-2 or B-3, where Z2 is H or NH2; Z6 is N(CH3)NHSO2(CH3) or NHNHSO2(CH3); and A is Group (3) or Group (4), with R2' = methyl and R2 = C1-C6 alkyl, phenyl, benzyl, mono-substituted phenyl, mono-substituted benzyl, C3-5 cycloalkyl or C3-5 cycloalkyl-(CH2)-.
In a more specific subgeneric embodiment, the invention provides a compound of formula I, where B is B-2 or B-3, where Z2 is H or NH2; Z6 is N(CH3)NHSO2(CH3) Or NHNHSO2(CH3); and A is Group (3) or Group (4), with R2'
= methyl and R2 = o-tolyl, o-chloro phenyl, o-fluoro phenyl, o-trifluoromethyl phenyl, o-methyl benzyl, o-chloro benzyl, o-fluoro benzyl, o-trifluoromethyl benzyl.
In another more specific subgeneric embodiment, the invention provides a compound of formula I, where B is B-2 or B-3, where Z2 is H Or NH2; Z6 is N(CH3)NHSO2(CH3) or NHNHSO2(CH3); and A is Group (3) or Group (4), with R2' = methyl and R2 = m-tolyl, m-chloro phenyl, m-fluoro phenyl, m-trifluoromethyl phenyl, m-methyl benzyl, m-chloro benzyl, m-fluoro benzyl, m-trifluoromethyl benzyl.
In a still more specific subgeneric embodiment, the invention provides or contemplates a compound of formula I, where B is B-2 and is selected from the following bases:




In another more specific embodiment, this invention provides or contemplates a compound of formula I, in which B is B-3 and is selected from the following bases:


The present invention is directed to various substituted 3', 5'- cyclic monophosphate esters of 2'-methyl ribofuranosyl derivatives of heterocyclic bases, as depicted above.
Compounds of this invention may be prepared by a variety of procedures. An exemplary synthetic procedure is shown in Scheme 1 below, where B is a heterocyclic base as previously defined, typically in the form B-H. In Scheme 1 a suitably protected 2-methyl ribose is first coupled to the heterocyclic base B (optionally substituted), which is then further modified to produce the desired compounds.

POCI3 PO(OMe)3, O 0C

Scheme 1
Numerous heterocyclic bases are commercially available, and many others may be readily synthesized from commercially available precursors via standard published procedures. (See, for example, Vorbruggen, H. and Ruh-Pohlenz, C, Handbook of Nucleoside Synthesis, New York, Wiley-Interscience; ISBN:0471093831). Additionally, a number of novel derivatives of heterocyclic bases are disclosed in the commonly owned, co-pending PCT Application titled "6- Hydrazinopurine-2 '-methyl Ribonucleosides and Nucleotides for Treatment of HCV," by E. Gunic and F. Rong, filed May 10, 2006 (Attorney Docket Number 6319-3032).
Additional synthetic schemes are shown below. For example, a SATE-cyclic nucleotide analogue can be prepared as illustrated in Scheme 2 below. (SATE = S- acetylthioethyl, MSNT = l-(2-mesitylenesulfonyl)-3-nitro-l,2,4-triazole).
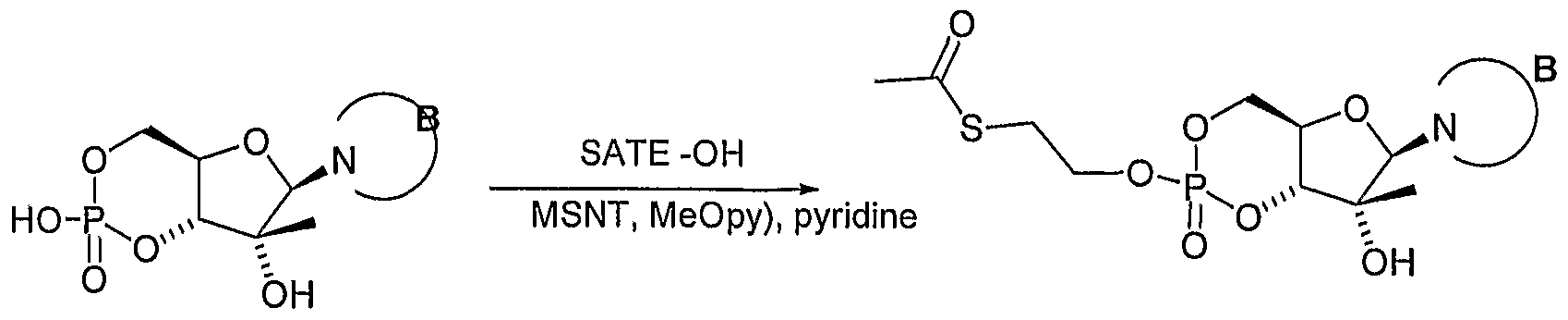
Scheme 2
In another variation, a derivative where A = Group (3) can be prepared as shown in Scheme 3 below:

Scheme 3
In another variation, a carbonic acid ester derivative of the cyclic nucleotide analogue can be prepared as depicted in synthetic Scheme 4 below:

Scheme 4
More generally, the compounds of scheme 4 can be made using any chloromethyl alkyl carbonate.
In another variation, a pivaloyloxymethyl (POM) cyclic nucleotide analogue can be prepared as shown in synthetic Scheme 5 below:

In variations of this scheme, one or more hydrogens of the methyl groups of the pivaloyl group may be replaced by halo, alkoxy, or alkyl groups. Alternatively, one of the methyl groups of the pivaloyl group can be replaced by a C1-C4 alkoxy group.
In the following chemical structures, if the stereochemical configuration of C
4 is not indicated, it should be understood that in each case a ribonucleotide is intended.
2-Amino-6-[N-Methyl-N-(methanesulfonylhydrazidyI]-9-[3,5-(O,O'-cycIic-O"-{S- 2-ethyl-2,2-dimethyl-3-propoxypropanethioate}phosphoryl)-2-C-methyI-β-D- ribofuranosyl]adenine

2-Amino-6-(N-methyl-N-(methanesulfonyIhydrazidyI)-9-(3,5-O,O'-cycIic- phosphoryl-2-C-methyl-β-D-ribofuranosyl)adenine triethylammonium salt
The nucleoside above (403 mg, 1.0 mmole) was dried over P2O5 and dissolved in dry trimethyl phosphate (5.0 ml) by heating. The solution was cooled to 0 °C under argon. To this cold (0 °C) stirred solution was added POCl3 (186 μL, 2.0 mmol) and the mixture stirred at 0 °C for 6 hr. The mixture was added drop by drop into a cold (0
°C) stirred solution of 0.08 M KOH in H2CVCH3CN (4:6, 120 ml) mixture during 10 minutes period. After 15 minutes, aq. HCl solution was added to pH 5 (0 °C), and the volume was reduced to 20 ml by evaporation. The solution was then extracted with CHCl3 (2 x 50 ml) and evaporated to dryness. The residue was suspended in MeOH (50 ml) to precipitate insoluble salts, which were removed by filtration. The filtrate was evaporated to dryness, and the residue was dissolved in water (10 ml) and loaded onto a Sephadex column (bicarbonate form). The column was eluted with water (250 ml), followed by triethylammonium bicarbonate (TEAB, pH ~ 8.0) buffer 0.2 M (250 ml) and 0.4 M (250 ml). The fractions having the required product (mass M+ 567 and M" 565) were collected and lyophilized to give ( 290 mg, 51%). 31P NMR (DMSO- dό): -2.39 PPM. [HPLC Purification: The filtrate was fractionated by C8 reverse phase column chromatography with 0.2 M triethylammonium formate buffer in MeOH. Lyophilization of the product containing fraction yields the cyclic phosphate.] 1HNMR (DMSO-d6): δ 0.86 (s, 3H), 1.18 (s, 9H), 3.0 (s, 9H), 3.67 (s, 3H), 4.1 (m, 4H), 5.6 (s, IH), 5.84 (s, IH), 6.32 (s, 2H), 7.8 (s, IH), 9.51 (s, IH), 10.1 (bs, IH).
S-2-hydroxy ethyl 2,2-dimethyI-3-propoxypropanethioate: To a stirred cold solution of methyl 2,2-dimtheyl-3-hydroxypropionate (25.0 g, 189.4 mmol) in dry THF (300 ml) at 0 °C in under argon atmosphere was added NaH (60% in oil, 8.0 g, 200.0 mmol) during 30 minutes period. After the addition of NaH, the reaction mixture was stirred at 0 °C for 1 h and at room temperature for 5 h. The reaction mixture was cooled to 0 °C and 1-iodopropane (42.25 g, 250.0 mmol) was added. The reaction mixture was stirred at room temperature for 2 days and at 65 °C for 12 h under argon. It was evaporated to dryness. The residue was partitioned between EtOAc (500 ml) and water (300 ml), neutralized with acetic acid to pH 6 and extracted in EtOAc. The organic layer was washed with brine (200 ml), dried, and concentrated to dryness to give 14 g of crude product.
The above crude product (14 g) was dissolved in THF (50 ml) and treated with 5% KOH in methanol/water (300:25, 325 ml). The reaction mixture was heated at 45 °C for 2 days and evaporated to dryness. The residue was partitioned between EtOAc (200 ml) and IN KOH (100 ml) and extracted in KOH solution. The EtOAc extract was discarded. The aqueous KOH solution was made acidic with acetic acid and extracted into EtOAc (2 x 100 ml). The organic extract was washed with brine (100ml), dried, and evaporated to dryness to give 4.85 g of the acid.
To a stirred solution of the acid (4.85 g, 30.0 mmol) in dry CH2Cl2 (100 ml) at 5 °C under argon was added l,l'-carbonyldiimidazole (5.25 g, 32.5 mmol) and allowed to stir at room temperature for 4h. The protected ethanethiol, 2-{tert- butyldimethylsilyloxy) ethanethiol (5.74 g, 30.0 mmol) was added, and the solution was stirred at room temperature for 12 h. Diluted with CHCl3 (100 ml), washed with IN HCl (70 ml), water (100 ml) and brine (50 ml). The organic extract was dried and concentrated to dryness. Residue was purified over silica column using ethyl acetate in hexane as the eluent. Pure fractions were pooled and evaporated dryness to give 9.0 g (89%) of product. To a stirred solution of protected alcohol (9.0 g, 26.95 mmol) in THF (50 ml) was added tetrabutylammonium fluoride (1 M, 35.0 ml, pH was adjusted to 6 with acetic acid) at room temperature. The reaction mixture was stirred at room temperature for 12 h and evaporated to dryness. The residue was partitioned between EtOAc (300 ml) and water (300 ml) and extracted in EtOAc. The organic extract was washed with brine (100ml), dried, and concentrated to dryness. The residue was purified over silica gel using ethyl acetate in hexane as the eluent. Pure fractions were pooled and evaporated dryness to give 3.0 g (51%) of alcohol. 1HNMR (CDCl3): δ 0.85 (t, 3H), 1.21 (s, 9H), 1.50 (m, 2H), 3.1 (t, 2H), 3.34 (t, 2H), 3.42 (s, 2H), 3.70 (t, 2H).
2-Amino-6-(N-Methyl-iV-(inethanesulfonylhydrazidyl)-9-(3,5-O,O'-cycIic- phosphoryl-2-C-methyl-β-D-ribofuranosyl)adenine triethylammonium salt (114 mg, 0.2 mmole) was dried over P2Os and dissolved in dry pyridine (5.0 ml) and allowed to stir at room temperature under argon atmosphere. To the stirred solution was added the SATE alcohol (0.44 g, 2.0 mmol) followed by 1 -(2-mesitylene-2- sulfonyl-3-nitro)-l,2,4-triazole (Aldrich, 0.36 g, 1.2 mmole) under argon. The mixture was stirred at room temperature under argon and protected from light for 3 days. Evaporated to dryness and dissolved in CHCl3 (100 ml). The chloroform solution was washed with water (50 ml) and brine (50 ml). Dried and evaporated to dryness. The residue was purified on a silica gel chromatography using methanol in chloroform as the eluent. The required fractions having the required product (mass M+ 667) were collected and evaporated below 35 °C to give 19.5 mg (15%). 31P NMR (DMSO-d6): -5.25 PPM. 1HNMR (DMSO-d6): δ 0.79 (m, 3H), 0.96 (s, 3H), 1.10 (m, 9H), 1.4 (m,
2H), 3.0 (s, 3H), 3.68 (s, 3H), 3.96 (s, IH)5 4.1 (m, 2H), 4.2 (m, IH), 4.6 (m, 2H), 5.9 (s, IH), 6.2 (s, IH), 6.4 (s, 2H), 8.1 (s, IH), 9.5 (s, IH).

Preparation of chloromethyl isopropyl carbonate: To an ice cold stirred solution of 2-propanol (15.0 g, 250.0 mmol) in dry pyridine (19.8g, 250.0 mmol) and dry ether (100 ml) under argon was added a solution of chloromethyl chloroformate (29.0g, 226.6 mmol) in dry ether (100 ml) during 30 min period. After the addition, the reaction mixture was stirred at room temperature for 12 hr. Precipitated solid was filtered and washed with dry ether (50 ml) and the filtrate was evaporated to dryness. The residue was distilled at 5 mm and temperature 30 °C.
2-Amino-6-(N-methyI-N-(methanesuIfonyIhydrazidyI)-9-(3,5-O,O'-cycIic- phosphoryl-2-C-methyl-β-D-ribofuranosyl)adenine triethylanimonium salt (260 mg, 0.4 mmol) was allowed to stir in dry DMF (10 ml) at room temperature under argon. To the stirred solution was added N,N-diisopropylethylamine (0.103g, 0.8 mmol) followed by chloromethyl isopropyl carbonate (0.122g, 0.8 mmol). The reaction was stirred at room temperature for 15 min and at 60 °C for 10 hr. The reaction mixture was evaporated to dryness and the residue was purified by silica column using as the eluent methanol in chloroform. The fractions having the required product were pooled, and the mixture was concentrated to dryness. This purified material was again purified by HPLC to give 12.6 mg of pure product. The structure of the product was confirmed by 1H NMR and 31P NMR.
2-Amino-6-[N-Methyl-N-(methanesulfonyIhydrazidy!]-9-[3,5-(O,O'-cyclic-0"- {S-acetoxyethyIoxycarbonylthioeth-l-yl}phosphoryl)~2-C~methyl-β-D~
ribofuranosyl]adenine

To an ice-cold solution of 4-nitrophenol (2.0 g, 15 mmol) and triethylamine (1.6 g, 16 mmol) in dichloromethane (20 ml) was added a solution of 1-chloroethyl chloroformate (2.1 g, 19 mmol) in dichloromethane, and the mixture was stirred for 15 min at 0 °C and then overnight at room temperature. The mixture was extracted with dichloromethane (50 ml), washed successively with water (50 ml), 0.5 N sodium hydroxide (50 ml), saturated aqueous sodium chloride solution (50 ml), and water (3 x 50 ml), and dried over Na2SO4. The dichloromethane solution was filtered and evaporated under reduced pressure, and the residue was purified by silica gel column chromatography with chloroform (100%) as eluent to furnish pure 1-chloroethyl 4- nitrophenyl carbonate, as a white solid in 85% yield.
To a solution of 1-chloroethyl 4-nitrophenyl carbonate (2.0 g, 8.2 mmol) in glacial acetic acid (50 ml) at room temperature was added mercuric acetate (3.8 g, 12 mol) and the mixture was stirred for 40 h. Water (100) was added slowly to the mixture, which was then extracted with diethyl ether (2 x 65 ml). The ether phase was
washed with 0.5 N NaOH (30 ml), saturated NaCl solution (30 ml), and water (2 x 50 ml) and dried over anhydrous Na2SO4. The solution was filtered, concentrated under reduced pressure, and purified by silica gel column chromatography to afford pure 1- acetoxyethyl 4-nitrophenyl carbonate in 89% yield as a colorless liquid.
To a solution of 2-mercaptoethanol (5.0 g, 64 mmol) in dichloromethane (25 ml) was added imidazole (10.5 g, 154 mmol). The mixture was stirred and then added tert -butyl dimethyl silyl chloride (10.61 g, 70.4 mmol) in dichloromethane (15 ml). The mixture was stirred continuously at room temperature for 2 hours and at 35 °C for 2 hours. The mixture was evaporated under reduced pressure to dryness. The residue was purified by silica gel column chromatograph using 15% ethyl acetate / hexane as eluent to afford pure 2-mercaptoethanoxy tert -butyldimethylsilane in 85% yield as a colorless liquid.
4. To a solution of 2-mercaptoethanoxy tert-bn\y\ dimethylsilane (2.0 g, 10.4 mmol) in dimethylformamide (25 ml) at room temperature was added 1-acetoxyethyl 4-nitrophenyl carbonate (2.8 g, 10.4 mmol) in dimethylformamide ( 5 ml). The mixture was then heated to 75 °C for 8 hours. The mixture was then evaporated under reduced pressure to dryness. The residue was purified by silica gel column chromatography using 10% EtOAc/hexane as eluent to afford pure 1-acetoxyethyl t- butyl dimethylsilyloxyethyl thiacarbonate in 65% yield.
5. To a solution of l-((2-(tert-butyldimethylsilyloxy)ethylthio)carbonyloxy)ethyl acetate (2.0 g, 6.2 mmol) in 25 ml THF was added dropwise at O°C 1.0 M tetrabutyl ammonium fluoride in THF (6.2 ml, 6.2 mmol). The mixture was stirred continuously at room temperature overnight. The mixture was then evaporated to dryness under reduced pressure. The residue was purified by silica gel column chromatography using 5% methanol in chloroform as eluent to give the product l-((2- hydroxyethylthio)carbonyloxy)ethyl acetate in 75% yield.
To a solution of 2-amino-6-(N-methyI-N-(methanesuIfonyIhydrazidyI)-9- (3,5-0,(?'-cycIic-phosphoryl-2-C-methyl-b-D-ribofuranosyl)adenine (100 mg,
0.21 mmol) in DMF (5 ml) was added l-(2-mesitylenesulfonyl)-3-nitro-l,2,4-triazole (62 mg, 0.21 mmol) in DMF (1 ml) and l-((2-hydroxyethylthio)carbonyloxy)ethyI
acetate (134 mg, 0.64 mmol) in DMF (0.5 ml). The mixture was stirred continuously at room temperature for 10 hours and at 65 °C for 2 days. The mixture was then evaporated under reduced pressure to dryness. The residue was purified by silica gel column chromatograph using 3% methanol in chloroform as eluent to give the final product, 2'-methyl-3'-5'cyclic -N6-methyl-N6-(methanesulfonylhydrazido)-2-amino- adenosine-2-[S-(l-acetoxyethyloxythiacarbonyl)]-eth-l-yl phosphotri ester in 35% yield.
2-Amino-6-[ N-(methanesulfonylhydrazidyl]-9-[3,5-(O,O'-cyclic- O"-{S-2-ethyl- 2,2-dimethyl-3-propoxypropanethioate }phosphoryI)-2-C-methyl-β-D- ribofuranosyl]adenine

2-amino-6-(N-(methanesulfonyIhydrazidyl)-9-(2-C-methyl-β-D- ribofuranosyl)adenine:
(2): 2-(2-amino-6-chloro-9H-purin-9-yl)-5-(hydroxymethyl)-3-methyl tetrahydrofuran-3,4-diol 1 (2.0 g, 6.35 mmol) and methane sulfonylhydrazide (4.0 g, 36.36 mmol) in a mixture of ethyl alcohol (100 ml) and dioxane (20 ml) was heated at 92 °C under argon for 2 days. Reaction mixture was evaporated to dryness. The residue was purified over silica gel using methanol in chloroform as the eluent. Pure fractions were pooled and evaporated dryness to give 1.0 g (40%) of product 2. 1HNMR (DMSOd6): δ 0.78 (s, 3H), 3.02 (s, 3H), 3.60 (d, IH), 3.74 (m, 2H)5 4.00 (d, IH), 5.78 (s, IH), 6.20 (s, sH), 7.72 (s, IH), 8.13 (s, IH).
2-Amino-6-(N-(methanesulfonylhydrazidyl)-9-(3,5-O,O'-cyclic-phosphoryl-2-C- methyl-β-D-ribofuranosyl)adenine triethylammonium salt (3): Nucleoside 2 ( 0.9 mg, 2.3 mmole) was dried over P2O5 and dissolved in dry trimethyl phosphate (5.0 ml) by heating. The solution was cooled to 0 °C under argon. To this cold (0 °C) stirred solution was added POCl3 (1.0 ml, 10.8 mmol). The mixture was stirred at 0 °C for 6 hr. then added dropwise into a cold (0 °C) stirred solution of 0.08 M KOH in H2OZCH3CN (4:6, 600 ml) mixture over 30 minutes. After 15 minutes, the mixture was neutralized to pH 5 (0 °C) with aq. HCl solution, and its volume was reduced by evaporation to 20 ml. The aqueous solution was extracted with CHCl3 (2 x 100 ml) and discarded. The aqueous solution was evaporated to dryness. The residue was suspended in MeOH (100 ml) to precipitate insoluble salts and filtered off. The
MeOH filtrate was evaporated to dryness. The residue was dissolved in water (10 ml) and loaded on top of Sephadex column (bicarbonate form). The column was eluted with water (500 ml) followed by triethylammonium bicarbonate (TEAB, pH ~ 8.0) buffer 0.2 M (500 ml) and 0.4 M (500 ml). The fractions having the required product (mass M+ 452 and M" 450) were collected and lyophilized to give ( 500 mg, 51%). 31P NMR (DMSO-de): -2.39 PPM. [HPLC Purification: The filtrate was fractionated by C8 reverse-phase chromatography (25 cm x 3 cm) with 0.2 M triethylammonium formate buffer in MeOH. Lyophilization of the product containing fraction yields the cyclic phosphate.] 1HNMR (DMSO-d6): δ 0.86 (s, 3H), 1.18 (s, 9H), 3.0 (s, 9H), 4.1 (m, 4H), 5.6 (s, IH), 5.84 (s, IH), 6.32 (s, 2H), 7.8 (s, IH), 9.51 (s, IH), 10.1 (bs, IH).
2-Amino-6-[7V-(methanesuIfonyIhydrazidyl]-9-[3,5-(O,O'-cyclic-0"-{S-2-ethyl- 2,2-dimethyI-3-propoxypropanethioate}phosphoryI)-2-C-methyl-β-D-
ribofuranosyl]adenine (4): Cyclic phosphate TEA salt 3 ( 220 mg, 0.4 mmole) was dried over P2O5 and dissolved in dry pyridine (5.0 ml) and allowed to stir at room temperature under argon atmosphere. To the stirred solution was added S-2- hydroxyethyl 2,2-dimethyl-3-propoxypropanethioate (0.88 g, 4.0 mmol) followed by l -(2-mesitylene-2-sulfonyl-3-nitro)-l,2,4-triazole (Aldrich, 0.72 g, 2.4 mmole) under argon. The mixture was stirred at room temperature under argon and protected from light for 3 days. Evaporated to dryness and dissolved in CHCb (100 ml). The chloroform solution was washed with water (50 ml) and brine (50 ml). Dried and evaporated to dryness. The residue was purified on a silica gel chromatography using methanol in chloroform as the eluent. (Note: Purification might require 2 times on silica column). The required fractions having the required product (mass M+ 653) were collected and evaporated below 35 °C to give 19.5 mg (15%). 31P NMR (DMSO-de): -5.17 PPM and -3.99 PPM. 1HNMR (DMSO-d6): δ 0.78 (m, 3H), 0.96 (s, 3H), 1.13 (m, 6H), 1.4 (m, 2H), 3.0 (s, 3H), 3.38 (m, 2H), 4.1 (m, 3H), 4.2 (m, IH), 4.6 (m, 3H), 5.9 (s, IH), 6.06 (s, IH), 6.26 (m, 2H), 8.1 (s, IH), 9.06 (s, IH), 9.62 (s, IH).
Additional prophetic examples are shown in Table 2 below
TABLE 2 ADDITIONAL CONTEMPLATED COMPOUNDS OF FORMULA

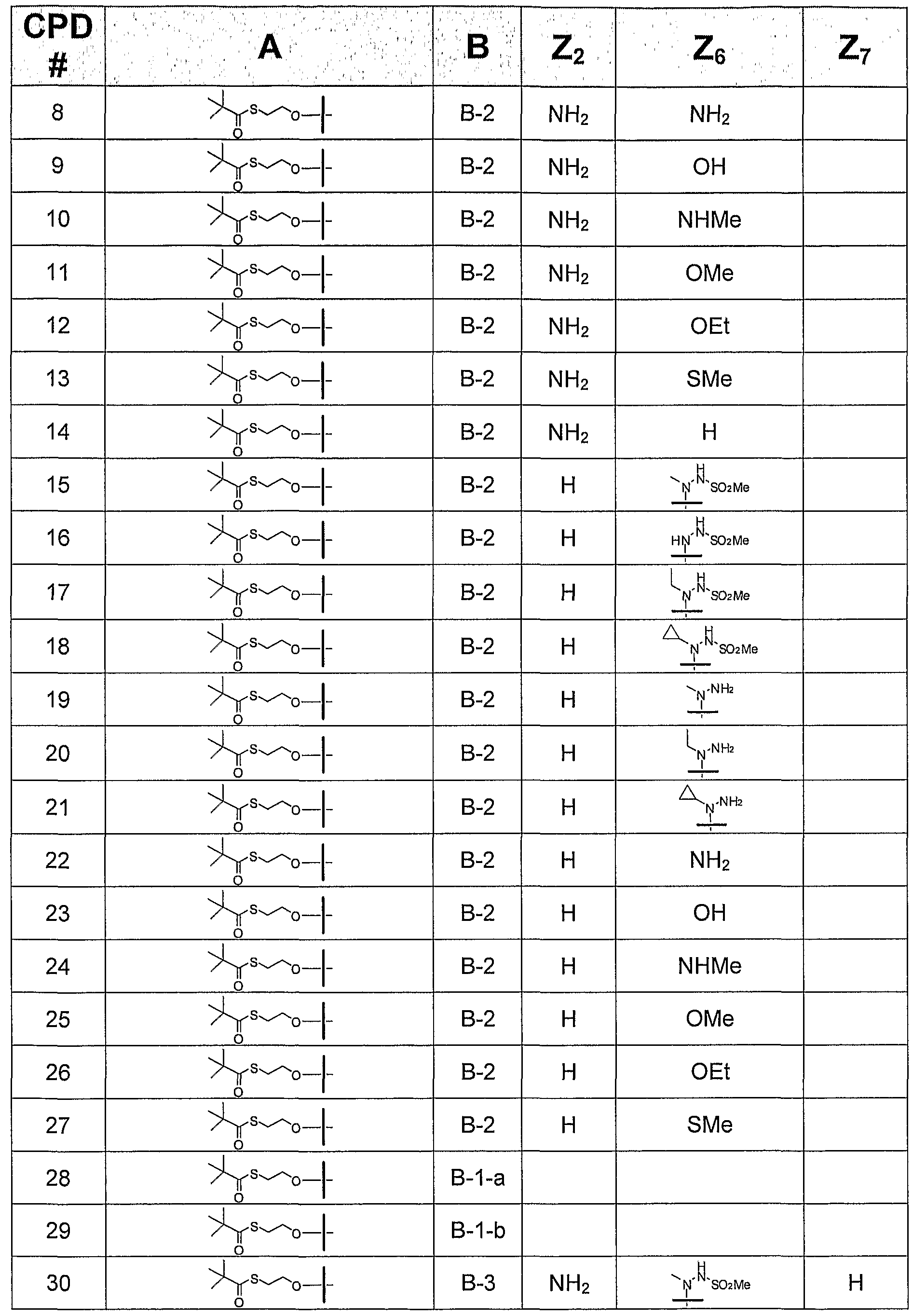




























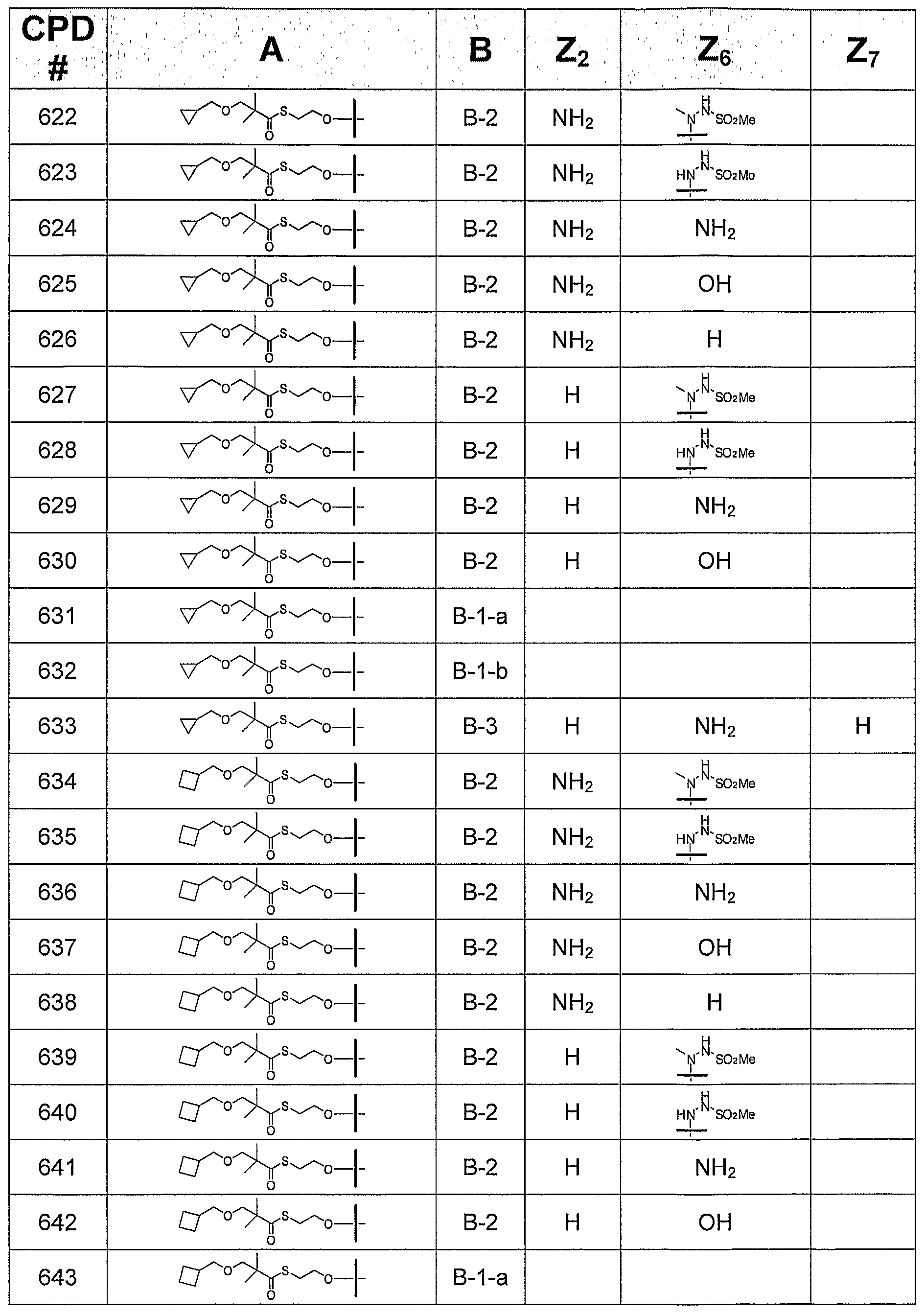















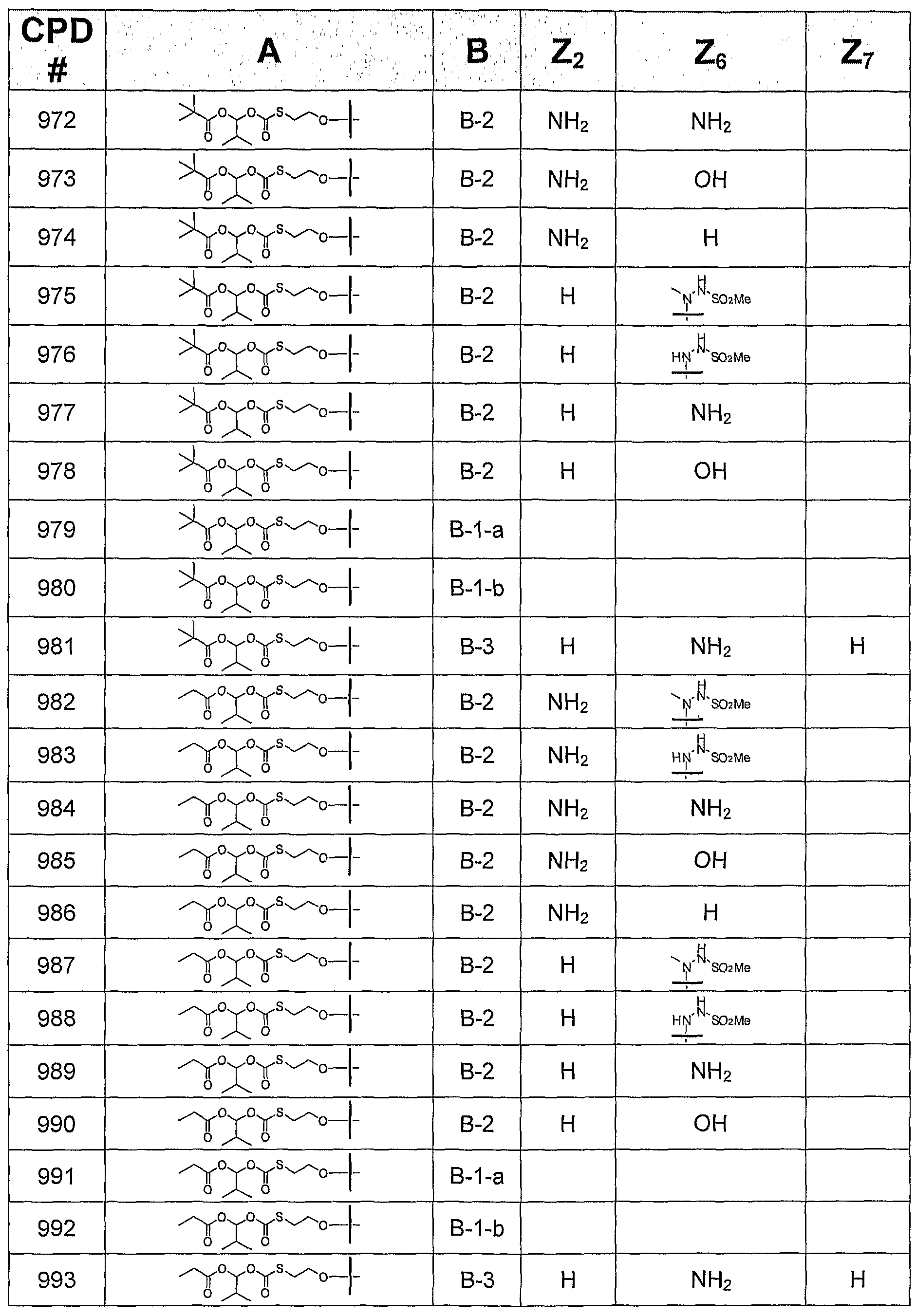













Biological activity
The anti-HCV activities of the exemplary compounds were tested in two biological assays — a cell-based HCV replicon assay and a cytotoxicity assay.
1. HCV Replicon Assay
A human hepatoma cell line (Huh-7) containing replicating HCV Conl subgenomic replicon with a luciferase reporter gene (luc-ubi-neo) was used to evaluate anti-HCV activity of the compounds. In this assay, the level of luciferase signal correlates directly with the viral RNA replication. The HCV repl icon-reporter cell line (NK/luc-ubi-neo) was cultured in DMEM medium supplemented with 10 % fetal bovine serum and 0.5 mg/ml Geneticin (G418). Cells were maintained in a subconfluent state to ensure high levels of HCV replicon RNA synthesis.
To evaluate the antiviral activity of compounds, serial dilutions were prepared with concentrations ranging from 0.14 to 300 μM. Diluted compounds were transferred to a 96-well plate followed by the addition of replicon cells (6000 cells per
well). Cells were incubated with the compounds for 48 hours after which luciferase activity was measured. Reduction of luciferase signal reflected the decrease of HCV replicon RNA in the treated cells and used to determine the EC50 value (concentration which yielded a 50 % reduction in luciferase activity).
2, Cytotoxicity Assay
A Huh-7 cell line carrying a luciferase reporter gene (driven by a HIV LTR promoter) stably integrated into the chromosome was used to analyze the cytotoxic effect of the selected compounds. This cell line (LTR-luc) was maintained in DMEM medium with 10 % FBS. Design of the cytotoxicity assay was similar to that of the HCV replicon assay. Reduction of luciferase activity in the treated cells correlated with the cytotoxic effect of the test compound and was used to calculate the CC50 value (concentration that inhibited cell growth by 50%).
The biological activities and cytotoxicity of the selected compounds are summarized in Table 4. Anti-HCV and anti-BVDV activities (EC50 values) of the test compounds were categorized into three different groups: A (1 to 50 nM), B (50 to 500 μM) and C (500 to 1000 nM). The cytotoxicity (CC50 values) of the compounds are indicated in μM.



Claims
1. A compound of formula I

B-I B-2 B-3 where Z2 is H, NH2, NHMe, or NMe2; Z4 is NH2 or OH; Z6 is H, OH, OCH3, SCH3, thienyl, furyl, or NR3R4, where R3 is H, C1-C3 alkyl, or cyclopropyl, and R4 is H or NHR5, R5 is H, C1-C4 alkyl, or SO2R6, and R6 is C1-C4 alkyl; and Z7 is H, halogen, or CN; and A is selected from Groups (1), (2), (3) and (4)


(3) (4)
where Ri is H, C-C8 alkyl, C3-C6 cycloalkyl-(CH2)m-, C3-C6 cycloalkyl-O-(CH2)m-, C3-C6 cycloalkyl-(CH2)n-O-(CH2)m-, or C1-C8 alkyl-O-(CH2)m-, where the alkyl and cycloalkyl groups are either unsubstituted or are optionally substituted with one or two C1-C3 alkyl groups, with one or two trifluoromethyl groups, and with one or two halogen atoms, where m = zero to 4, and where n = zero to 2; and Ri' and Ri" are, independently, H, C3-C5 cycloalkyl, or C1-C4 alkyl; R2 is C1-6 alkyl or C3-5 cycloalkyl- (CHa)n or

2. The compound of claim 1, where B is B-I and A is (1) or (2).
3. The compound of claim 1, where B is B-I and A is (3) or (4)
4. The compound of claim 2, where Ri' and Ri" are both methyl and Ri is C1-C6 alkyl or C1-C6 alkyl-O-(CH2)m, where m = zero to 4.
5. The compound of claim 2, where R]' and R] " are both methyl and R] is C3-C6 cycloalkyl-(CH2)m- or C3-C6 cycloalkyl-(CH2)n-O-(CH2)m-, where n = zero to 2 and m = zero to 4.
6. The compound of claim 1, where B is B-2 or B-3, where Z2 is H, NH2, or NHMe; Z6 is H or NR3R4; and Z7 is H, CN, or halogen.
7. The compound of claim 6, where R3 is H, methyl, ethyl, or cyclopropyl.
8. The compound of claim 7, where Z6 is NR3NHR5; and Z7 is H, F, or CN.
9. The compound of claim 8, where Z2 is H or NH2; Z6 is NR3NHSO2R6; and Z7 is H, F, or CN.
10. The compound of claim 9, where R3 is H, methyl, or cyclopropyl; R6 is methyl or ethyl; and Z7 is H, F, or CN.
11. The compound of claim 6 or claim 8, where A is (1) or (2), where R]' and Ri" are both methyl and Ri is C1-C6 alkyl or C1-C6 alkyl-O-(CH2)m, where m = zero to 2.
12. The compound of claim 10, where R1 is C3-C6 cycloalkyl-(CH2)n-O-(CH2)m-, said cycloalkyl optionally substituted as described in claim 1, where m = 1 or 2 and n = zero or 1.
13. The compound of claim 1 1, where A is (2), R3 is H or methyl, R6 is methyl, and R, is C1-C6 alkyl or C1-C6 alkyl-O-(CH2)-.
14. The compound of claim 13, where Z7 is H, Ri is Y or Y-O-(CH2)-, where Y is methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, or isohexyl.
15. The compound of claim 11, where A is (1), R3 is H or methyl, R6 is methyl, and R1 is C1-C6 alkyl or C1-C6 alkyl-O-(CH2)-.
16. The compound of claim 15, where Z7 is H, R1 is Y or Y-O-(CH2)-, where Y is methyl, ethyl, 77-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, ter/-butyl, n-pentyl, isopentyl, neopentyl, n-hexyl, or isohexyl.
17. The compound of claim 14 or claim 16, where B is B-2.
18. The compound of claim 14 or claim 16, where B is B-3.
19. The compound of claim 7, where R5 is H or C1-C4 alkyl; A is (1) or (2); R1' and R1" are both methyl; and R1 is C1-C6 alkyl, C1-C6 alkyl-O-(CH2)m, or C3-C6 cycloalkyl-(CH2)n-O-(CH2)m-, where m = zero to 2 and n = zero to 2.
20. The compound of claim 19, where, R3 is H or methyl, and R5 is H or C1-C4 alkyl.
21. The compound of claim 9, where A is (3) or (4), wherein, if A is (3), R2' is methyl, ethyl, or isopropyl and R2" is H or methyl, and where R2 is C1-6 alkyl or C3-5 cycloalkyl-(CH2)n, where n = 0 to 2.
22. The compound of claim 21, where R2 is C1-4 alkyl; R3 is H or methyl; and R6 is methyl.
23. The compound of claim 9, where A is (3) or (4), wherein, if A is (3), R2' is methyl, ethyl, or isopropyl, and R2" is H or methyl, and where R2 is phenyl or benzyl, wherein said phenyl group or the phenyl moiety of said benzyl group is substituted with one or two halogen atoms or with one or two C1-C3 alkyl groups.
24. The compound of claim 22 or claim 23, where A is (3).
25. The compound of claim 22 or claim 23, where A is (4).
26. The compound of claim 6, where B is B-2, Z2 is NH2, and Z6 is NH-NH-SO2Me, N(Me)-NH-SO2Me, N(cyclopropyl)-NH-SO2Me, or N(Et)-NH-SO2Me.
27. The compound of claim 6, where B is B-2, Z2 is NH2, and Z6 is NH2, NHMe, N(Me)-NH2, N(cyclopropyl)-NH2, Or N(Et)-NH2.
28. The compound of claim 26, where A is (2) and Ri' and Ri" are both methyl.
29. The compound of claim 27, where A is (2) and Ri' and Ri" are both methyl.
30. The compound of claim 28 or claim 29, where Ri is C1-C6 alkyl or C1-C6 alkyl-O- (CH2)m-, where the C1 -C6 alkyl group is substituted with one or two halogens, where m = 0 to 2.
31. The compound of claim 28 or claim 29, where Ri is C5-C8 alkyl or C5-C8 alkyl-O- (CH2)m-, where the C5-C8 alkyl group is substituted with one or two halogens, where m = 0 to 2.
32. The compound of claim 28 or claim 29, where R1 is C1-C6 alkyl or C1-C6 alkyl-O- (CH2)m-, where the C1-C6 alkyl group is substituted with one or two trifluoromethyl groups, and where m = 0 to 2.
31. The compound of claim 28 or claim 29, where Ri is C1-C6 alkyl or C1-C6 alkyl-O- (CH2)m-! where the C1-C6 alkyl group is unsubstituted, and where m = 0 to 2.
32. The compound of claim 28 or claim 29, where Ri is C1-C6 alkyl or C1-C6 alkyl-O, where the C1-C6 alkyl group is unsubstituted.
33. The compound of claim 28 or claim 29, where Ri is C1-C6 alkyl or C1-C6 alkyl-O- (CH2)m~, where the C1-C6 alkyl group is unsubstituted, and where m = 2.
33. The compound of claim 28 or claim 29, where Ri is C1-C6 alkyl or C1-C6 alkyl-O- (CH2)-, where the C1-C6 alkyl group is unsubstituted.
34. The compound of claim 1, which is selected from the following




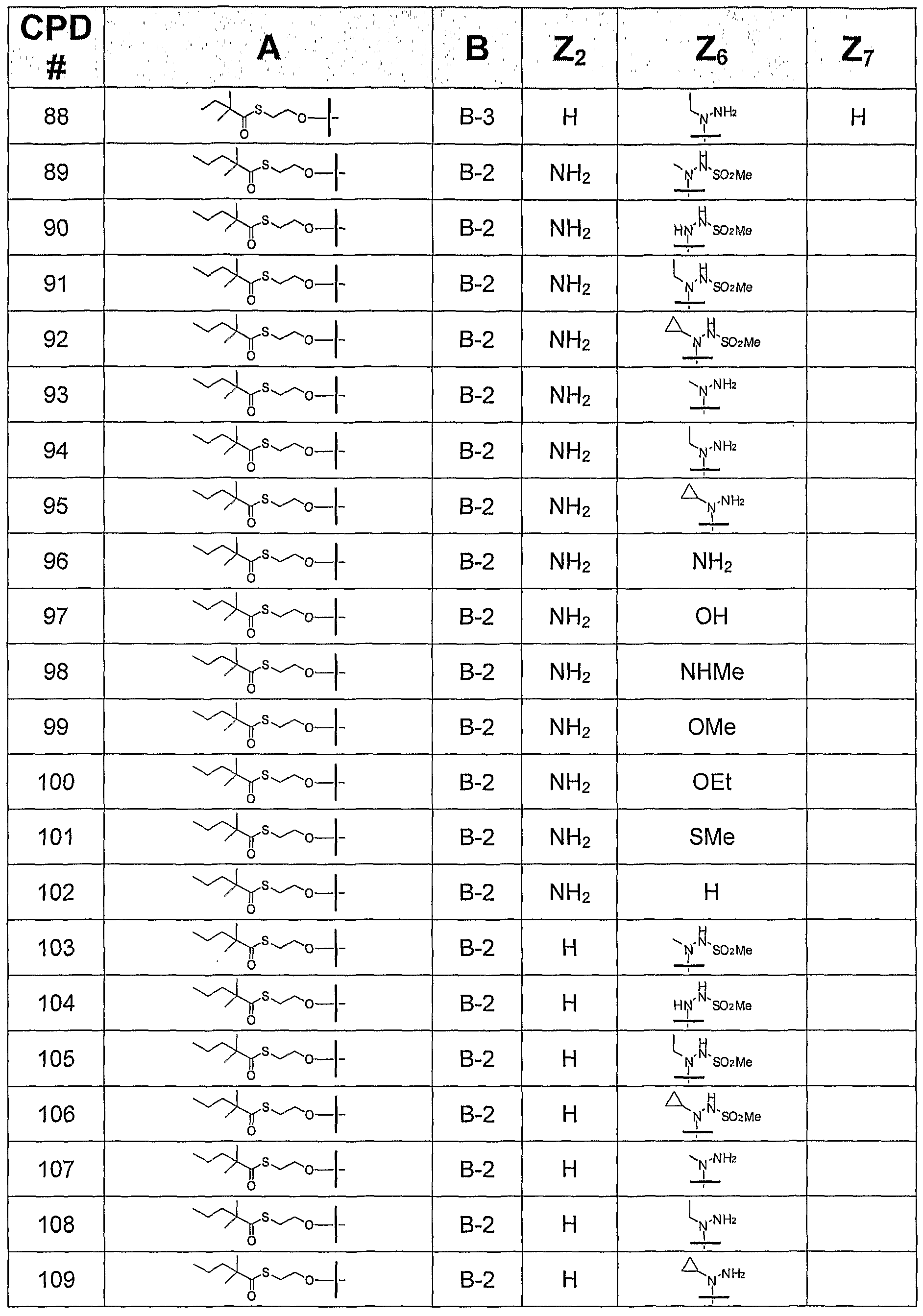














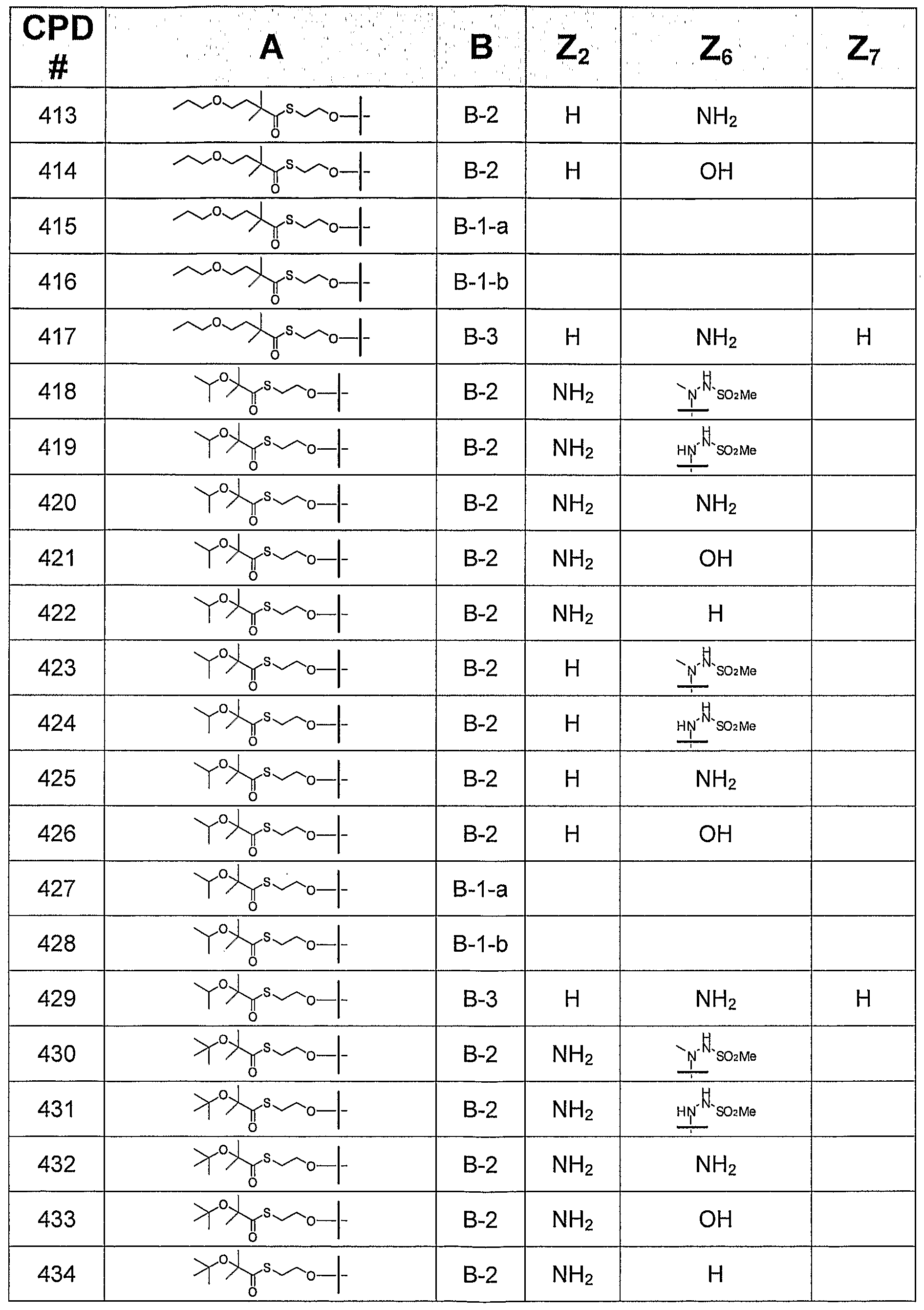








































Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US68133205P | 2005-05-16 | 2005-05-16 | |
| US60/681,332 | 2005-05-16 | ||
| US74813005P | 2005-12-06 | 2005-12-06 | |
| US60/748,130 | 2005-12-06 | ||
| US78523806P | 2006-03-22 | 2006-03-22 | |
| US60/785,238 | 2006-03-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007027248A2 true WO2007027248A2 (en) | 2007-03-08 |
| WO2007027248A3 WO2007027248A3 (en) | 2007-11-29 |
Family
ID=37809326
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/019114 Ceased WO2007027248A2 (en) | 2005-05-16 | 2006-05-16 | 3', 5' - cyclic nucleoside analogues for treatment of hcv |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007027248A2 (en) |
Cited By (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008117046A1 (en) * | 2007-03-27 | 2008-10-02 | Astrazeneca Ab | Pyrazolo [4, 3-d] pyrimidines as antibacterial compounds |
| WO2010075554A1 (en) * | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2011035231A1 (en) | 2009-09-21 | 2011-03-24 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| WO2012039791A1 (en) | 2010-09-20 | 2012-03-29 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| WO2012039787A1 (en) | 2010-09-20 | 2012-03-29 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| WO2012088155A1 (en) | 2010-12-22 | 2012-06-28 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| WO2013056046A1 (en) * | 2011-10-14 | 2013-04-18 | Idenix Pharmaceuticals, Inc. | Substituted 3',5'-cyclic phosphates of purine nucleotide compounds and pharmaceutical compositions for the treatment of viral infections |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| US8580765B2 (en) | 2007-03-30 | 2013-11-12 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| WO2013177195A1 (en) * | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphate prodrugs for hcv infection |
| WO2013177188A1 (en) * | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| US8609627B2 (en) | 2009-02-06 | 2013-12-17 | Rfs Pharma, Llc | Purine nucleoside monophosphate prodrugs for treatment of cancer and viral infections |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8629263B2 (en) | 2009-05-20 | 2014-01-14 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| CN103687866A (en) * | 2011-05-19 | 2014-03-26 | Rfs制药公司 | Purine monophosphate prodrugs for the treatment of viral infections |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| US8871737B2 (en) | 2010-09-22 | 2014-10-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US8916538B2 (en) | 2012-03-21 | 2014-12-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| EP2313422B1 (en) * | 2008-06-11 | 2015-03-04 | Gilead Pharmasset LLC | Nucleoside cyclicphosphates |
| US8980865B2 (en) | 2011-12-22 | 2015-03-17 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US9187515B2 (en) | 2013-04-01 | 2015-11-17 | Idenix Pharmaceuticals Llc | 2′,4′-fluoro nucleosides for the treatment of HCV |
| US9192621B2 (en) | 2012-09-27 | 2015-11-24 | Idenix Pharmaceuticals Llc | Esters and malonates of SATE prodrugs |
| US9211300B2 (en) | 2012-12-19 | 2015-12-15 | Idenix Pharmaceuticals Llc | 4′-fluoro nucleosides for the treatment of HCV |
| US9243025B2 (en) | 2011-03-31 | 2016-01-26 | Idenix Pharmaceuticals, Llc | Compounds and pharmaceutical compositions for the treatment of viral infections |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| US9339541B2 (en) | 2013-03-04 | 2016-05-17 | Merck Sharp & Dohme Corp. | Thiophosphate nucleosides for the treatment of HCV |
| US9403863B2 (en) | 2011-09-12 | 2016-08-02 | Idenix Pharmaceuticals Llc | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US20160250342A1 (en) * | 2012-11-16 | 2016-09-01 | Redwood Bioscience, Inc. | Hydrazinyl-Indole Compounds and Methods for Producing a Conjugate |
| US9447132B2 (en) | 2013-04-12 | 2016-09-20 | Achillion Pharmaceuticals, Inc. | Highly active nucleoside derivative for the treatment of HCV |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10005779B2 (en) | 2013-06-05 | 2018-06-26 | Idenix Pharmaceuticals Llc | 1′,4′-thio nucleosides for the treatment of HCV |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US10202411B2 (en) | 2014-04-16 | 2019-02-12 | Idenix Pharmaceuticals Llc | 3′-substituted methyl or alkynyl nucleosides nucleotides for the treatment of HCV |
| US10231986B2 (en) | 2013-03-13 | 2019-03-19 | Idenix Pharmaceuticals Llc | Amino acid phosphoramidate pronucleotides of 2′-cyano, azido and amino nucleosides for the treatment of HCV |
| US10238680B2 (en) | 2013-08-01 | 2019-03-26 | Idenix Pharmaceuticals Llc | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US10513534B2 (en) | 2012-10-08 | 2019-12-24 | Idenix Pharmaceuticals Llc | 2′-chloro nucleoside analogs for HCV infection |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US10682368B2 (en) | 2017-03-14 | 2020-06-16 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US10717758B2 (en) | 2012-05-22 | 2020-07-21 | Idenix Pharmaceuticals Llc | D-amino acid compounds for liver disease |
| US10723754B2 (en) | 2012-10-22 | 2020-07-28 | Idenix Pharmaceuticals Llc | 2′,4′-bridged nucleosides for HCV infection |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US10988498B2 (en) | 2009-09-21 | 2021-04-27 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| JP2022512751A (en) * | 2018-10-17 | 2022-02-07 | リャオ,シビン | 6-Mercaptopurine nucleoside analog |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US12102689B2 (en) | 2015-11-09 | 2024-10-01 | R.P. Scherer Technologies, Llc | Anti-CD22 antibody-maytansine conjugates and methods of use thereof |
| US12275726B2 (en) | 2019-12-03 | 2025-04-15 | Wigen Biomedicine Technology (shanghai) Co., Ltd. | Aurora kinase inhibitors and use thereof |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
| US12552820B2 (en) | 2020-04-21 | 2026-02-17 | Ligand Pharmaceuticals Incorporated | Benzyloxy phosph(on)ate compounds |
| EP4232456A4 (en) * | 2020-10-21 | 2026-04-01 | Ligand Pharm Inc | ANTIVIRAL PRODRUG COMPOUNDS |
| AU2020263283B2 (en) * | 2019-04-22 | 2026-04-23 | Ligand Pharmaceuticals Incorporated | Cyclic phosphate compounds |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080261913A1 (en) | 2006-12-28 | 2008-10-23 | Idenix Pharmaceuticals, Inc. | Compounds and pharmaceutical compositions for the treatment of liver disorders |
| AU2011336632B2 (en) | 2010-11-30 | 2015-09-03 | Gilead Pharmasset Llc | Compounds |
-
2006
- 2006-05-16 WO PCT/US2006/019114 patent/WO2007027248A2/en not_active Ceased
Non-Patent Citations (3)
| Title |
|---|
| ANDERSON ET AL.: 'Studies with the cyclic adenosine monophosphate receptor and stimulation of in vitro transcription of the Gal operon' JOURNAL OF BIOLOGICAL CHEMISTRY vol. 249, no. 11, 1974, pages 3592 - 3596 * |
| BERES ET AL.: 'Synthesis, structure, and antitumor and antiviral activities of a series of 5-halouridine cyclic 3',5'-monophosphates' J. MED. CHEM. vol. 29, 1986, pages 488 - 493 * |
| GUNIC ET AL.: 'Cyclic monophosphate prodrugs of base-modified 2'-C-methyl ribonucleosides as potent inhibitors of hepatitis C virus RNA replication' BIOORGANIC & MEDICINAL CHEMISTRY LETTERS vol. 17, 2007, pages 2452 - 2455 * |
Cited By (137)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008117046A1 (en) * | 2007-03-27 | 2008-10-02 | Astrazeneca Ab | Pyrazolo [4, 3-d] pyrimidines as antibacterial compounds |
| US8735372B2 (en) | 2007-03-30 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US10183037B2 (en) | 2007-03-30 | 2019-01-22 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8580765B2 (en) | 2007-03-30 | 2013-11-12 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US12121529B2 (en) | 2007-03-30 | 2024-10-22 | Gilead Sciences, Inc. | Nucleoside phosphoramidate prodrugs |
| US11642361B2 (en) | 2007-03-30 | 2023-05-09 | Gilead Sciences, Inc. | Nucleoside phosphoramidate prodrugs |
| US9585906B2 (en) | 2007-03-30 | 2017-03-07 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US9085573B2 (en) | 2007-03-30 | 2015-07-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8906880B2 (en) | 2007-03-30 | 2014-12-09 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8853171B2 (en) | 2008-04-23 | 2014-10-07 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| US8318682B2 (en) | 2008-04-23 | 2012-11-27 | Gilead Sciences, Inc. | 1′substituted carba-nucleoside analogs for antiviral treatment |
| US8012941B2 (en) | 2008-04-23 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8008264B2 (en) | 2008-04-23 | 2011-08-30 | Gilead Sciences, Inc. | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| USRE46762E1 (en) | 2008-04-23 | 2018-03-27 | Gilead Sciences, Inc | 1′-substituted carba-nucleoside analogs for antiviral treatment |
| EP2937350A1 (en) | 2008-04-23 | 2015-10-28 | Gilead Sciences, Inc. | 1' -substituted carba-nucleoside analogs for antiviral treatment |
| EP2313422B1 (en) * | 2008-06-11 | 2015-03-04 | Gilead Pharmasset LLC | Nucleoside cyclicphosphates |
| EA019295B1 (en) * | 2008-12-23 | 2014-02-28 | Джилид Фармассет, Ллс. | Synthesis of purine nucleosides and process for preparing them |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| WO2010075554A1 (en) * | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2671888A1 (en) * | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| US9173893B2 (en) | 2009-02-06 | 2015-11-03 | Cocrystal Pharma, Inc. | Purine nucleoside monophosphate prodrugs for treatment of cancer and viral infections |
| US8609627B2 (en) | 2009-02-06 | 2013-12-17 | Rfs Pharma, Llc | Purine nucleoside monophosphate prodrugs for treatment of cancer and viral infections |
| US8012942B2 (en) | 2009-02-10 | 2011-09-06 | Gilead Sciences, Inc. | Carba-nucleoside analogs for antiviral treatment |
| US8633309B2 (en) | 2009-05-20 | 2014-01-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9637512B2 (en) | 2009-05-20 | 2017-05-02 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9206217B2 (en) | 2009-05-20 | 2015-12-08 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8629263B2 (en) | 2009-05-20 | 2014-01-14 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9284342B2 (en) | 2009-05-20 | 2016-03-15 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8735569B2 (en) | 2009-05-20 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8642756B2 (en) | 2009-05-20 | 2014-02-04 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP3150608A1 (en) | 2009-09-21 | 2017-04-05 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US10988498B2 (en) | 2009-09-21 | 2021-04-27 | Gilead Sciences, Inc. | Processes and intermediates for the preparation of 1′-substituted carba-nucleoside analogs |
| WO2011035231A1 (en) | 2009-09-21 | 2011-03-24 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| US9090642B2 (en) | 2010-07-19 | 2015-07-28 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US9487544B2 (en) | 2010-07-19 | 2016-11-08 | Gilead Sciences, Inc. | Methods for the preparation of diasteromerically pure phosphoramidate prodrugs |
| US11492353B2 (en) | 2010-07-22 | 2022-11-08 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US10065958B2 (en) | 2010-07-22 | 2018-09-04 | Gilead Sciences, Inc. | Methods and compounds for treating Paramyxoviridae virus infections |
| US12509466B2 (en) | 2010-07-22 | 2025-12-30 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| US10696679B2 (en) | 2010-07-22 | 2020-06-30 | Gilead Sciences, Inc. | Methods and compounds for treating paramyxoviridae virus infections |
| WO2012039791A1 (en) | 2010-09-20 | 2012-03-29 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| WO2012039787A1 (en) | 2010-09-20 | 2012-03-29 | Gilead Sciences, Inc. | 2' -fluoro substituted carba-nucleoside analogs for antiviral treatment |
| US8871737B2 (en) | 2010-09-22 | 2014-10-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US9278990B2 (en) | 2010-09-22 | 2016-03-08 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| AU2011349278B2 (en) * | 2010-12-22 | 2016-08-04 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| WO2012088155A1 (en) | 2010-12-22 | 2012-06-28 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| US8772474B2 (en) | 2010-12-22 | 2014-07-08 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| US9365605B2 (en) | 2010-12-22 | 2016-06-14 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| AU2011349278C1 (en) * | 2010-12-22 | 2017-01-19 | Alios Biopharma, Inc. | Cyclic nucleotide analogs |
| US9243025B2 (en) | 2011-03-31 | 2016-01-26 | Idenix Pharmaceuticals, Llc | Compounds and pharmaceutical compositions for the treatment of viral infections |
| EP2710023A4 (en) * | 2011-05-19 | 2014-12-03 | Rfs Pharma Llc | PURINE MONOPHOSPHATE PRODRUGS FOR TREATING VIRAL INFECTIONS |
| CN103687866A (en) * | 2011-05-19 | 2014-03-26 | Rfs制药公司 | Purine monophosphate prodrugs for the treatment of viral infections |
| US9403863B2 (en) | 2011-09-12 | 2016-08-02 | Idenix Pharmaceuticals Llc | Substituted carbonyloxymethylphosphoramidate compounds and pharmaceutical compositions for the treatment of viral infections |
| WO2013056046A1 (en) * | 2011-10-14 | 2013-04-18 | Idenix Pharmaceuticals, Inc. | Substituted 3',5'-cyclic phosphates of purine nucleotide compounds and pharmaceutical compositions for the treatment of viral infections |
| US8507460B2 (en) | 2011-10-14 | 2013-08-13 | Idenix Pharmaceuticals, Inc. | Substituted 3′,5′-cyclic phosphates of purine nucleotide compounds and pharmaceutical compositions for the treatment of viral infections |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US8980865B2 (en) | 2011-12-22 | 2015-03-17 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US9605018B2 (en) | 2011-12-22 | 2017-03-28 | Alios Biopharma, Inc. | Substituted nucleotide analogs |
| US8916538B2 (en) | 2012-03-21 | 2014-12-23 | Vertex Pharmaceuticals Incorporated | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US9856284B2 (en) | 2012-03-21 | 2018-01-02 | Alios Biopharma, Inc. | Solid forms of a thiophosphoramidate nucleotide prodrug |
| US9012427B2 (en) | 2012-03-22 | 2015-04-21 | Alios Biopharma, Inc. | Pharmaceutical combinations comprising a thionucleotide analog |
| WO2013177188A1 (en) * | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphoramidate prodrugs for hcv infection |
| WO2013177195A1 (en) * | 2012-05-22 | 2013-11-28 | Idenix Pharmaceuticals, Inc. | 3',5'-cyclic phosphate prodrugs for hcv infection |
| US9109001B2 (en) | 2012-05-22 | 2015-08-18 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphoramidate prodrugs for HCV infection |
| US9296778B2 (en) | 2012-05-22 | 2016-03-29 | Idenix Pharmaceuticals, Inc. | 3′,5′-cyclic phosphate prodrugs for HCV infection |
| US10717758B2 (en) | 2012-05-22 | 2020-07-21 | Idenix Pharmaceuticals Llc | D-amino acid compounds for liver disease |
| US9845336B2 (en) | 2012-05-25 | 2017-12-19 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9422323B2 (en) | 2012-05-25 | 2016-08-23 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US10301347B2 (en) | 2012-05-25 | 2019-05-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10774106B2 (en) | 2012-05-25 | 2020-09-15 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10544184B2 (en) | 2012-05-25 | 2020-01-28 | Janssen Sciences Ireland Unlimited Company | Uracyl spirooxetane nucleosides |
| US10040814B2 (en) | 2012-05-25 | 2018-08-07 | Janssen Sciences Ireland Uc | Uracyl spirooxetane nucleosides |
| US9192621B2 (en) | 2012-09-27 | 2015-11-24 | Idenix Pharmaceuticals Llc | Esters and malonates of SATE prodrugs |
| US10513534B2 (en) | 2012-10-08 | 2019-12-24 | Idenix Pharmaceuticals Llc | 2′-chloro nucleoside analogs for HCV infection |
| US10723754B2 (en) | 2012-10-22 | 2020-07-28 | Idenix Pharmaceuticals Llc | 2′,4′-bridged nucleosides for HCV infection |
| AU2013344644B2 (en) * | 2012-11-16 | 2017-09-28 | Redwood Bioscience, Inc. | Hydrazinyl-indole compounds and conjugates |
| US9833515B2 (en) | 2012-11-16 | 2017-12-05 | Redwood Bioscience, Inc. | Hydrazinyl-indole compounds and methods for producing a conjugate |
| US20160250342A1 (en) * | 2012-11-16 | 2016-09-01 | Redwood Bioscience, Inc. | Hydrazinyl-Indole Compounds and Methods for Producing a Conjugate |
| US10888623B2 (en) | 2012-11-16 | 2021-01-12 | Redwood Bioscience, Inc. | Hydrazinyl-indole compounds and methods for producing a conjugate |
| US11426465B2 (en) | 2012-11-16 | 2022-08-30 | Redwiid Bioscience, Inc. | Hydrazinyl-indole compounds and methods for producing a conjugate |
| US10314919B2 (en) | 2012-11-16 | 2019-06-11 | Redwood Bioscience, Inc. | Hydrazinyl-indole compounds and methods for producing a conjugate |
| US9211300B2 (en) | 2012-12-19 | 2015-12-15 | Idenix Pharmaceuticals Llc | 4′-fluoro nucleosides for the treatment of HCV |
| US9339541B2 (en) | 2013-03-04 | 2016-05-17 | Merck Sharp & Dohme Corp. | Thiophosphate nucleosides for the treatment of HCV |
| US9309275B2 (en) | 2013-03-04 | 2016-04-12 | Idenix Pharmaceuticals Llc | 3′-deoxy nucleosides for the treatment of HCV |
| US10231986B2 (en) | 2013-03-13 | 2019-03-19 | Idenix Pharmaceuticals Llc | Amino acid phosphoramidate pronucleotides of 2′-cyano, azido and amino nucleosides for the treatment of HCV |
| US9187515B2 (en) | 2013-04-01 | 2015-11-17 | Idenix Pharmaceuticals Llc | 2′,4′-fluoro nucleosides for the treatment of HCV |
| US9447132B2 (en) | 2013-04-12 | 2016-09-20 | Achillion Pharmaceuticals, Inc. | Highly active nucleoside derivative for the treatment of HCV |
| US10005779B2 (en) | 2013-06-05 | 2018-06-26 | Idenix Pharmaceuticals Llc | 1′,4′-thio nucleosides for the treatment of HCV |
| US10238680B2 (en) | 2013-08-01 | 2019-03-26 | Idenix Pharmaceuticals Llc | D-amino acid phosphoramidate pronucleotides of halogeno pyrimidine compounds for liver disease |
| US11707479B2 (en) | 2013-08-27 | 2023-07-25 | Gilead Sciences, Inc. | Combination formulation of two antiviral compounds |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US10202411B2 (en) | 2014-04-16 | 2019-02-12 | Idenix Pharmaceuticals Llc | 3′-substituted methyl or alkynyl nucleosides nucleotides for the treatment of HCV |
| US11344565B2 (en) | 2014-10-29 | 2022-05-31 | Gilead Sciences, Inc. | Methods for the preparation of ribosides |
| US11266666B2 (en) | 2014-10-29 | 2022-03-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10251898B2 (en) | 2014-10-29 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US9724360B2 (en) | 2014-10-29 | 2017-08-08 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US9949994B2 (en) | 2014-10-29 | 2018-04-24 | Gilead Sciences, Inc. | Methods for treating Filoviridae virus infections |
| US10695357B2 (en) | 2014-10-29 | 2020-06-30 | Gilead Sciences, Inc. | Methods for treating filoviridae virus infections |
| US10251904B2 (en) | 2015-09-16 | 2019-04-09 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US11382926B2 (en) | 2015-09-16 | 2022-07-12 | Gilead Sciences, Inc. | Methods for treating Arenaviridae and Coronaviridae virus infections |
| US10695361B2 (en) | 2015-09-16 | 2020-06-30 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US11007208B2 (en) | 2015-09-16 | 2021-05-18 | Gilead Sciences, Inc. | Methods for treating arenaviridae and coronaviridae virus infections |
| US12102689B2 (en) | 2015-11-09 | 2024-10-01 | R.P. Scherer Technologies, Llc | Anti-CD22 antibody-maytansine conjugates and methods of use thereof |
| US11260070B2 (en) | 2017-03-14 | 2022-03-01 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US10682368B2 (en) | 2017-03-14 | 2020-06-16 | Gilead Sciences, Inc. | Methods of treating feline coronavirus infections |
| US10836787B2 (en) | 2017-05-01 | 2020-11-17 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5- (4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy)(phenoxy) phosphoryl)amino)propanoate |
| US11597742B2 (en) | 2017-05-01 | 2023-03-07 | Gilead Sciences, Inc. | Crystalline forms of (S)-2-ethylbutyl 2-(((S)-(((2R,3S,4R,5R)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US12030906B2 (en) | 2017-05-01 | 2024-07-09 | Gilead Sciences, Inc. | Crystalline forms of (s)-2-ethylbutyl 2-(((s)-(((2r,3s,4r,5r)-5-(4-aminopyrrolo[2,1-f] [1,2,4]triazin-7-yl)-5-cyano-3,4-dihydroxytetrahydrofuran-2-yl)methoxy) (phenoxy) phosphoryl)amino)propanoate |
| US11266681B2 (en) | 2017-07-11 | 2022-03-08 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US10675296B2 (en) | 2017-07-11 | 2020-06-09 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| US11975017B2 (en) | 2017-07-11 | 2024-05-07 | Gilead Sciences, Inc. | Compositions comprising an RNA polymerase inhibitor and cyclodextrin for treating viral infections |
| EP3866932A4 (en) * | 2018-10-17 | 2022-11-30 | Xibin Liao | 6-MERCAPTOPURIN NUCLEOSIDE ANALOGUES |
| JP2022512751A (en) * | 2018-10-17 | 2022-02-07 | リャオ,シビン | 6-Mercaptopurine nucleoside analog |
| US11535645B2 (en) | 2018-10-17 | 2022-12-27 | Xibin Liao | 6-mercaptopurine nucleoside analogues |
| AU2020263283B2 (en) * | 2019-04-22 | 2026-04-23 | Ligand Pharmaceuticals Incorporated | Cyclic phosphate compounds |
| US12275726B2 (en) | 2019-12-03 | 2025-04-15 | Wigen Biomedicine Technology (shanghai) Co., Ltd. | Aurora kinase inhibitors and use thereof |
| US11660307B2 (en) | 2020-01-27 | 2023-05-30 | Gilead Sciences, Inc. | Methods for treating SARS CoV-2 infections |
| US12012431B2 (en) | 2020-03-12 | 2024-06-18 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11613553B2 (en) | 2020-03-12 | 2023-03-28 | Gilead Sciences, Inc. | Methods of preparing 1′-cyano nucleosides |
| US11701372B2 (en) | 2020-04-06 | 2023-07-18 | Gilead Sciences, Inc. | Inhalation formulations of 1'-cyano substituted carba-nucleoside analogs |
| US12552820B2 (en) | 2020-04-21 | 2026-02-17 | Ligand Pharmaceuticals Incorporated | Benzyloxy phosph(on)ate compounds |
| US11903953B2 (en) | 2020-05-29 | 2024-02-20 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11975012B2 (en) | 2020-05-29 | 2024-05-07 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11491169B2 (en) | 2020-05-29 | 2022-11-08 | Gilead Sciences, Inc. | Remdesivir treatment methods |
| US11939347B2 (en) | 2020-06-24 | 2024-03-26 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US12404289B2 (en) | 2020-06-24 | 2025-09-02 | Gilead Sciences, Inc. | 1′-cyano nucleoside analogs and uses thereof |
| US12297226B2 (en) | 2020-08-27 | 2025-05-13 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11926645B2 (en) | 2020-08-27 | 2024-03-12 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11814406B2 (en) | 2020-08-27 | 2023-11-14 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| EP4232456A4 (en) * | 2020-10-21 | 2026-04-01 | Ligand Pharm Inc | ANTIVIRAL PRODRUG COMPOUNDS |
| US12180217B2 (en) | 2022-03-02 | 2024-12-31 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12448383B2 (en) | 2022-03-02 | 2025-10-21 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US11780844B2 (en) | 2022-03-02 | 2023-10-10 | Gilead Sciences, Inc. | Compounds and methods for treatment of viral infections |
| US12357577B1 (en) | 2024-02-02 | 2025-07-15 | Gilead Sciences, Inc. | Pharmaceutical formulations and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007027248A3 (en) | 2007-11-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007027248A2 (en) | 3', 5' - cyclic nucleoside analogues for treatment of hcv | |
| EP1928475B1 (en) | Antiviral phosphoramidates of 4'-c-azido-substituted pronucleotides | |
| CN100503628C (en) | Modified fluorinated nucleoside analogues | |
| US7632821B2 (en) | Ribonucleoside cyclic acetal derivatives for the treatment of RNA-dependent RNA viral infection | |
| US20120010164A1 (en) | Antiviral agents | |
| US8148349B2 (en) | Nucleoside cyclic phosphoramidates for the treatment of RNA-dependent RNA viral infection | |
| AU2009257647C1 (en) | Nucleoside cyclicphosphates | |
| US20110306573A1 (en) | Nucleoside derivatives as inhibitors of viral polymerases | |
| US9186369B2 (en) | Purine nucleoside analogues for treating flaviviridae including hepatitis C | |
| KR102456417B1 (en) | 2'-substituted-n6-substituted purine nucleotides for rna virus treatment | |
| US20100234316A1 (en) | Nucleoside Aryl Phosphoramidates for the Treatment of RNA-Dependent RNA Viral Infection | |
| KR20140033446A (en) | Purine monophosphate prodrugs for treatment of viral infections | |
| WO2003062256A1 (en) | 2'-beta-modified-6-substituted adenosine analogs and their use as antiviral agents | |
| JP2006507235A (en) | Compounds having bicyclo [4.2.1] nonane system for the treatment of Flaviviridae virus infection | |
| US7189724B2 (en) | Quinoxaline derivatives having antiviral activity | |
| MXPA06001017A (en) | Purin nucleoside analogues for treating flaviviridae including hepatitis c |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| NENP | Non-entry into the national phase in: |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06770513 Country of ref document: EP Kind code of ref document: A2 |