WO2007042178A1 - Diacylindazol-derivate als inhibitoren von lipasen und phospholipasen - Google Patents
Diacylindazol-derivate als inhibitoren von lipasen und phospholipasen Download PDFInfo
- Publication number
- WO2007042178A1 WO2007042178A1 PCT/EP2006/009577 EP2006009577W WO2007042178A1 WO 2007042178 A1 WO2007042178 A1 WO 2007042178A1 EP 2006009577 W EP2006009577 W EP 2006009577W WO 2007042178 A1 WO2007042178 A1 WO 2007042178A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compounds
- heterocycle
- formulas
- phenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- XCUGDGATSFHWMJ-CFGPIGJWSA-N CC(C)(C)NC(C(CC1)(CCN1C([C@@H](Cc(cc1)ccc1F)NC(C(C1)C2(C3)N(C)C[C@@H]1C3C2)=O)=O)C1CCCCC1)=O Chemical compound CC(C)(C)NC(C(CC1)(CCN1C([C@@H](Cc(cc1)ccc1F)NC(C(C1)C2(C3)N(C)C[C@@H]1C3C2)=O)=O)C1CCCCC1)=O XCUGDGATSFHWMJ-CFGPIGJWSA-N 0.000 description 1
- YDGSHFJUSTYFRE-FTJBHMTQSA-N CC(C)(C)NC(C(CC1)(CCN1C([C@@H](Cc(cc1)ccc1F)NC([C@H]1NCCN(C)C1)=O)=O)C1CCCCC1)=O Chemical compound CC(C)(C)NC(C(CC1)(CCN1C([C@@H](Cc(cc1)ccc1F)NC([C@H]1NCCN(C)C1)=O)=O)C1CCCCC1)=O YDGSHFJUSTYFRE-FTJBHMTQSA-N 0.000 description 1
- FXFBGLBXECAHAB-JGJGAQJBSA-N CC(C1)(C2O[C@H](COC(C=C3)=CCC3Cl)CN2C(c2c(C)cccc2)=O)C=CC(C)=C1Cl Chemical compound CC(C1)(C2O[C@H](COC(C=C3)=CCC3Cl)CN2C(c2c(C)cccc2)=O)C=CC(C)=C1Cl FXFBGLBXECAHAB-JGJGAQJBSA-N 0.000 description 1
- YBWLTKFZAOSWSM-UHFFFAOYSA-N Cc(cc1)cnc1S(Nc1nc(-c2ccncc2)nc(OC)c1Oc(cccc1)c1OC)(=O)=O Chemical compound Cc(cc1)cnc1S(Nc1nc(-c2ccncc2)nc(OC)c1Oc(cccc1)c1OC)(=O)=O YBWLTKFZAOSWSM-UHFFFAOYSA-N 0.000 description 1
- 0 Cc1cc(O*([C@](C2=O)O)O[C@](COC(OC)=O)[C@]2O)c(C(CCc2ccc3[o]ccc3c2)=O)c(O)c1 Chemical compound Cc1cc(O*([C@](C2=O)O)O[C@](COC(OC)=O)[C@]2O)c(C(CCc2ccc3[o]ccc3c2)=O)c(O)c1 0.000 description 1
- XJFABANLLCWIFH-UHFFFAOYSA-N Cc1cccc(S(Nc2nc(CC(N3CCN(C)CC3)=O)c[s]2)(=O)=O)c1C Chemical compound Cc1cccc(S(Nc2nc(CC(N3CCN(C)CC3)=O)c[s]2)(=O)=O)c1C XJFABANLLCWIFH-UHFFFAOYSA-N 0.000 description 1
- HLCHESOMJVGDSJ-LOYHVIPDSA-N O=C([C@@H](Cc(cc1)ccc1Cl)NC([C@@H](C1)NCc2c1cccc2)=O)N(CC1)CCC1(C[n]1ncnc1)C1CCCCC1 Chemical compound O=C([C@@H](Cc(cc1)ccc1Cl)NC([C@@H](C1)NCc2c1cccc2)=O)N(CC1)CCC1(C[n]1ncnc1)C1CCCCC1 HLCHESOMJVGDSJ-LOYHVIPDSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/416—1,2-Diazoles condensed with carbocyclic ring systems, e.g. indazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/16—Central respiratory analeptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/12—Keratolytics, e.g. wart or anti-corn preparations
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- Diacylindazole derivatives as inhibitors of lipases and phospholipases
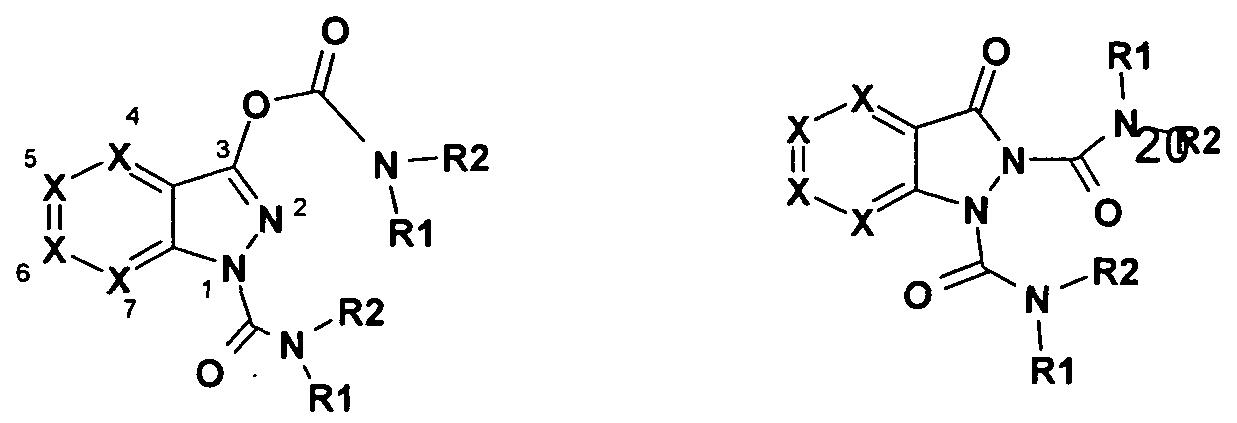
- the present invention relates to diacylindazole derivatives of the general formulas I or II, their pharmaceutically acceptable salts and their use as medicaments.
- the object of the present invention is to provide alternative compounds which cause an inhibition of hormone-sensitive lipase or endothelial lipase.
- the invention relates to diacylindazole derivatives of the general formulas I or II,
- R identically or differently hydrogen, halogen, (CrC ⁇ alkyl, (Ci-C 3) - alkyloxy (Ci-C 3) -alkylene, (C r C 3) haloalkyl, hydroxy, (CrC 6) -
- R1 are identical or different (C 5 -C 6) alkyl, (C3-Ci2) cycloalkyl, Y-aryl, Y-
- bicyclic heterocycle said cycloalkyl, aryl, heterocycle or bicyclic by halogen, (CrC 6) alkyl, (Ci-C3) -alkyloxy, hydroxy, (C r C6) -alkylmercapto, amino, (CrC 6) alkylamino , di- (C 2 -C 2) -alkylamino, mono- (Ci-C6) -alkylaminocarbonyl, di- (C 2 -C 8) alkylaminocarbonyl, (C 1 - C 6) alkyloxycarbonyl, (Ci-C 6 ) -alkylcarbonyl, cyano, trifluoromethyl,
- Trifluoromethyloxy, (Ci-C ⁇ J-alkylsulfonyl, aminosulfonyl, nitro may be monosubstituted or polysubstituted;
- Y (Ci-C 3) alkyl which (C r C 3) may be alkyl, hydroxy or trifluoromethyl, one or more times by halogen;
- R2 hydrogen
- R 3 is hydrogen, (C 1 -C 6 ) -alkyl, benzyl
- R4, R5 are identical or different (Ci-C 6) -alkyl, halogen, trifluoromethyl, phenyl, heterocyclic, COOR 3, (C 3 -C 2) cycloalkyl;
- R 6, R 7 are identical or different hydrogen, (C 1 -C 6 ) -alkyl, phenyl, benzyl, (C 3 -
- R is identical or different hydrogen, halogen, (C 1 -C 6 -alkyl, hydroxy,
- bicyclic heterocycle said cycloalkyl, phenyl, heterocycle or bicyclic by halogen, (dC 6) alkyl, (Ci-C3) -alkyloxy, hydroxy, (C 1 - C 6) -alkylmercapto, amino, (CrC 6) - Alkylamino, di (C 2 -C 12 ) alkylamino, mono (C 1 -C 6 ) alkylaminocarbonyl, di (C 2 -C 8 ) alkylaminocarbonyl, (Cr
- C 6 ) -alkyloxycarbonyl, (C 1 -C 6 ) -alkylcarbonyl, cyano, trifluoromethyl, trifluoromethyloxy, (C 1 -C 6 ) -alkylsulfonyl, aminosulfonyl, nitro may be monosubstituted or polysubstituted;
- Y is -CH 2 -, which may be monosubstituted by fluorine, methyl or hydroxy;
- R2 hydrogen
- Units from the series -O-, -S- may not be adjacent;
- R 3 is hydrogen, (C 1 -C 6 ) -alkyl, benzyl
- R 4 are identical or different (C 1 -C 6 ) -alkyl, halogen, trifluoromethyl, COOR 3,
- R 6, R 7 are identical or different hydrogen, (C 1 -C 6 ) -alkyl, phenyl, benzyl, (C 3 -
- R are identical or different hydrogen, halogen, hydroxy, (C 1 -C 6 ) -alkyl,
- R 1 is identical or different (C 6 -C 10 ) -alkyl, (C 6 -C 12 ) -cycloalkyl, Y-phenyl, Y-
- Heterocycle where cycloalkyl, phenyl, or heterocycle may be monosubstituted or polysubstituted by halogen, (C 1 -C 6 ) -alkyl, (C 1 -C 3 ) -alkyloxy, hydroxyl, amino, trifluoromethyl, trifluoromethyloxy;
- Y is -CH 2 -;
- R2 hydrogen
- R3 is hydrogen, (C 1 -C 4) alkyl, benzyl;
- R 4 are identical or different (C 1 -C 6 ) -alkyl, halogen, trifluoromethyl, COOR 3,
- R is identical or different hydrogen, halogen, hydroxy, (C 1 -C 6 ) -alkyl,
- R1 are identical or different (C 6 -C 0) alkyl or Y-phenyl substituted by
- Halogen, (Ci-C 6 ) -alkyl, (Ci-C 3 ) -alkyloxy or trifluoromethyl can be substituted;
- R2 hydrogen
- R1 and R2 together with the nitrogen atom bearing them form a monocyclic, saturated 5- to 6-membered ring system whose individual members are selected from one to three atoms or atomic groups from the series -CHR4-, -NR4-, -O-, -S- be replaced with the proviso that two units from the series -O-, -S- may not be adjacent;
- R4 is (Ci-C ⁇ J-alkyl, cyclopropyl, trifluoromethyl or phenyl;
- NR1 R2 represents a monocyclic saturated 5- to 6-membered ring system containing in the 4 position an atom or atomic member of the series -CHR4-, -
- CR4R5-, - (C R4) -, -NR4-, -0-, -S- contains.
- R is the same or different hydrogen, F, Cl, hydroxy, methyl,
- R 1 is identical or different (C 6 -C 10) -alkyl or Y-phenyl, which may be substituted by methyl;
- R2 hydrogen; or R1 and R2 together with the nitrogen atom bearing them form a monocyclic, saturated 5- to 6-membered ring system whose individual members are selected from one to three atoms or atomic groups from the series -CHR4-, -NR4-, -O-, -S- be replaced with the proviso that two units from the series -O-, -S- may not be adjacent; or form a bicyclic, partially unsaturated 9- to 10-membered ring system in which one member may be replaced by -S-;
- R 4 is methyl, trifluoromethyl or phenyl which may be monosubstituted or disubstituted by methyl or Cl;
- R 1 and R 2 together with the nitrogen atom bearing them, can form a monocyclic, saturated 5- to 6-membered ring system or a bicyclic saturated or partially unsaturated 9- to 10-membered ring system whose individual members of the ring systems are selected from an atom or an atomic group -CHR4-, -NR4, can be replaced.
- NR1 R2 represents piperidine which contains the atomic member -CHR4- in the 4 position. Preference is furthermore given to compounds of the formulas I or II in which
- a further preferred embodiment are compounds of the formulas I or II, in which
- R is not hydrogen
- the invention relates to compounds of the formulas I or II, in the form of their salts, racemates, racemic mixtures and pure enantiomers, and to their diastereomers and mixtures thereof.
- alkyl radicals in the substituents R, R1, R2, R3, R4, R5, R6 and R7 can be both straight-chain and branched.
- Halogen is fluorine, chlorine, bromine or iodine, especially fluorine or chlorine.
- Haloalkyl are alkyl radicals in which one, several or all hydrogen atoms are replaced by halogen, preferably fluorine.
- aryl radical is understood as meaning a phenyl or naphthyl radical.
- the aryl radicals may be substituted one or more times by suitable groups, for example: F, Cl, Br, I 1 CF 3, NO 2, CN, COOH, COO (CrC 6) alkyl, CONH 2, CONH (C 1 -C 6 ) alkyl, CON [(Ci-C 6) alkyl] 2 ⁇ , (C 3 -C 0) cycloalkyl, (C r Cio) alkyl, (C 2 -C 6) alkenyl, (C 2 -C 6) Alkynyl, O- (C r C ⁇ ) -alkyl O-CO- (C r C 6 ) -alkyl, O-CO- (C r C 6 ) -aryl, PO 3 H 2 , SO 3 H, SO 2 -NH 2 , SO 2 NH (C 1 -C 6 ) -alkyl, SO 2
- (Heterocycle) 2lN (Ary I) -CO- (C 1 -C 4 alkyl, N (heterocyclic) -CO- (C 1 -C 6 ) alkyl, N (aryl) -COO- (C r C 6 ) - Alkyl, N (heterocycle) -COO- (C 1 -C 6 ) -alkyl, N (aryl) -CO-aryl, N (heterocycle) -CO-aryl, N (aryl) -COO-aryl, N (heterocycle) -COO-aryl, N (aryl) -CO- NH- (Ci-C 6) -alkyl), N (heterocycle) -CO-NH- (C 1 -C 6) alkyl), N (aryl) -CO -NH-aryl, N (heterocycle) -CO-NH- (C 1 -C 6) alkyl), N (aryl)
- Heterocycle is a mono- or bicyclic ring system having 5 to 12 ring members, wherein at least one atom in the ring system is a heteroatom from the series N, O and S. Also included in this definition are ring systems in which the heterocycle is fused to a benzene nucleus.
- Cs-CrJ heterocycle is a monocyclic, (Ce- C 12 ) heterocycle, a bicyclic ring system.
- Suitable "heterocyclic rings” or “heterocyclic radicals” are azocinyl, benzimidazolyl, benzofuryl, benzothienyl, benzothiophenyl, benzoxazolyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl,
- N-oxides of these compounds e.g. 1- Oxy-2-, 3- or 4-pyridyl.
- heterocyclic rings or heterocyclic radicals may be monosubstituted or polysubstituted by suitable groups, for example: F, Cl, Br, I, CF 3 , NO 2 , N 3 , CN, COOH, COO (C r C 6 ) alkyl, CONH 2, CONH (C 1 -Ce) AIKyI 1 CON [(Ci-C 6) alkyl] 2, (Ci-C 6) -alkyl, (C 2 - C 6) alkenyl, (C 2 -C 6) Alkynyl, O- (C 1 -C 6 ) -alkyl, where in the alkyl radicals one, more, or all hydrogen (s) may be replaced by fluorine;
- suitable groups for example: F, Cl, Br, I, CF 3 , NO 2 , N 3 , CN, COOH, COO (C r C 6 ) alkyl, CONH 2, CONH (C 1 -Ce) AIKyI 1
- a cycloalkyl radical is to be understood as meaning a ring system containing one or more rings which is saturated or partially unsaturated (having one or two double bonds), which is composed exclusively of carbon atoms, e.g. Cyclopropyl, cyclopentyl, cyclopentenyl, cyclohexyl or adamantyl.
- the cycloalkyl radicals may be substituted one or more times with suitable groups, such as: F, Cl, Br, I 1 CF 3 , NO 2 , N 3 , CN, COOH 1 COO (C 1 -C 6 ) AIKyI 1 CONH 2 , CONH (C 1 -C 6) AIKyI, CONI (C 1 -C 6) AIKyI] 2, cycloalkyl, (C 1 -C 1O) -alkyl 1 (C 2 -C 6) -alkenyl, (C2 -Ce) - AIKIfIyI 1 0- (C 1 -Ce) -AlKyI 0-CO- (C 1 -Ce) -Alkyl 1 0-CO- (C 1 -Ce) -A ⁇ I, O-CO- (C 1 -C 6 ) - heterocycle ,; PO 3 H 2 , SO 3 H, SO 2 -NH 2 , SO 2 NH
- Heterocyclic radical may be monosubstituted to trisubstituted by F 1 Cl 1 Br, I, OH, CF 3 , NO 2 , CN, OCF 3 , O- (C 1 -C 6 ) -alkyl, (C 1 -C 6 -alkyl, NH 2 , NH (C 1 -Ce) -AlkVl 1 Nt (C 1 -C 6 ) alkyl) 2) SO 2 -CH 3 , COOH 1 COO (C 1 -Ce) -AlkVl, CONH 2 .
- Bicyclic is a partially unsaturated bicyclic ring system with 8 to 14 ring members, which has only carbon atoms as ring members.
- tetrahydronaphthyl alpha- or beta-tetralonic, indanyl or indan-1-onyl radical.
- Preferred bicyclic radicals are tetrahydronaphthyl and indanyl.
- the bicyclic radicals may be monosubstituted or polysubstituted by suitable groups, such as, for example: F 1 Cl, Br, I 1 CF 3 , NO 2 , N 3 , CN, COOH, COO (C r C 6 ) alkyl, CONH 2 , CONH ( C 1 - C 6) alkyl, CON [(Ci-C 6) alkyl] 2, cycloalkyl, (C r Cio) alkyl, (C 2 -C 6) alkenyl, (C 2 -C 6) - alkynyl, O- (C r C6) -alkyl O-CO- (C r C6) -alkyl, O-CO- (C r C6) -aryl, 0-CO- (C1-C6) - heterocycle, ;
- suitable groups such as, for example: F 1 Cl, Br, I 1 CF 3 , NO 2 , N 3 , CN, COOH, COO (C
- Suitable pharmaceutically acceptable acid addition salts of the compounds of the present invention are salts of inorganic acids such as hydrochloric, hydrobromic, phosphoric, metaphosphoric, nitric and sulfuric and organic acids, e.g.
- Suitable pharmaceutically acceptable basic salts are ammonium salts, alkali metal salts (such as sodium and potassium salts) and alkaline earth salts (such as magnesium and calcium salts) and salts of trometamol (2-amino-2-hydroxymethyl-1,3-propanediol), diethanolamine, lysine or ethylenediamine.
- Salts with a non-pharmaceutically acceptable anion are also useful within the scope of the invention.
- physiologically functional derivative denotes any physiologically acceptable derivative of a compound of the formula I according to the invention or II, for example an ester which, when administered to a mammal, such as man, is able to form (directly or indirectly) compounds of formulas I or II or an active metabolite thereof.
- the physiologically functional derivatives also include prodrugs of the compounds according to the invention, as described, for example, in H. Okada et al., Chem. Pharm. Bull. 1994, 42, 57-61. Such prodrugs can be metabolized in vivo to a compound of the invention. These prodrugs may or may not be effective.
- the compounds of the invention may also be in various polymorphic forms, e.g. as amorphous and crystalline polymorphic forms. All polymorphic forms of the compounds of the invention are within the scope of the invention and are a further aspect of the invention.
- the compounds of the general formulas I or II according to the invention have a surprising inhibitory effect on the hormone sensitive lipase, HSL 1 an allosteric enzyme in adipocytes, which is inhibited by insulin and for the
- the compounds of the general formulas I or II according to the invention may have an inhibitory effect on the endothelial lipase (EL).
- EL endothelial lipase
- the antiatheroskleotically effective HDL is the preferred substrate for EL. Lowering the HDL level leads to the progression of atherosclerosis and her
- Consequences such as metabolic syndrome and coronary heart disease. An inhibition of EL should thus lead to the prevention of atherosclerotic diseases.
- Insulin sensitivity disorders of muscle, fat and liver cells insulin resistance
- Diabetes mellitus especially type 2 diabetes, including the prevention of associated sequelae. Special aspects are the
- Dyslipidaemias and their consequences such as atherosclerosis, coronary heart disease, cerebrovascular diseases etc, especially those (but not limited to) characterized by one or more of the following factors: high plasma triglyceride, high postprandial plasma triglyceride
- High blood pressure heart failure e.g. (but not limited to) at state after
- Atherosclerosis e.g. (but not limited to) coronary sclerosis incl.
- Fat cell carcinomas such as liposarcomas solid tumors and neoplasias such as (but not limited to) carcinomas of the gastrointestinal tract, liver, biliary tract and pancreas, endocrine tumors, carcinomas of the lung, kidney and urinary tract organs, the genital tract, prostate carcinomas etc.
- Erythemato-squamous dermatoses e.g. Psoriasis (psoriasis) - acne vulgaris
- Dermatitides e.g. seborrheic dermatitis or light dermatitis - keratitis and keratoses, e.g. seborrheic keratoses, senile keratoses, actinic keratosis, photo-induced keratoses or follicular keratosis
- HPV Human papilloma viral infections, e.g. venereal papillomata, viral warts, e.g. Molluscum contagiosum, leukoplakia
- - skin cancer e.g. Basal cell carcinomas, melanomas or cutaneous T-cell lymphomas
- PCOS Polycystic Ovarian Syndrome
- ARDS - Acute Respiratory Distress Syndrome
- the amount of compound of the invention required to achieve the desired biological effect is dependent upon a number of factors, eg, the specific compound chosen, the intended use, the mode of administration, and the clinical condition of the patient.
- the daily dose ranges from 0.3 mg to 100 mg (typically from 3 mg to 50 mg) per day per kilogram of body weight, eg, 3 -10 mg / kg / day.
- an intravenous dose may range from 0.3 mg to 1.0 mg / kg, which may suitably be administered as an infusion of 10 ng to 100 ng per kilogram per minute.
- Suitable infusion solutions for these purposes may contain, for example, from 0.1 ng to 10 mg, typically from 1 ng to 10 mg per milliliter.
- Single doses may contain, for example, from 1 mg to 10 g of the active ingredient.
- injectable ampoules, and orally administrable unit dose formulations such as tablets or capsules, may contain, for example, from 0.05 to 1000 mg, typically from 0.5 to 600 mg.
- the compounds according to formulas I or II themselves can be used as compound, but they are preferably present with a compatible carrier in the form of a pharmaceutical composition.
- the carrier must of course be compatible in the sense that it is compatible with the other ingredients of the composition and is not harmful to the patient.
- the carrier may be a solid or a liquid or both and is preferably formulated with the compound as a single dose, for example as a tablet, which may contain from 0.05% to 95% by weight of the active ingredient.
- Other pharmaceutically active substances may also be present, including other compounds of the invention.
- the pharmaceutical compositions according to the invention can be prepared by one of the known pharmaceutical methods, which consist essentially in that the ingredients are mixed with pharmacologically acceptable carriers and / or excipients.
- compositions according to the invention are those which are suitable for oral, rectal, topical, peroral (eg sublingual) and parenteral (eg subcutaneous, intramuscular, intradermal or intravenous) administration, although the most suitable mode of administration in each individual case is the type and severity of the treatment to be treated State and on the nature of the compounds used in each case according to formulas I or II is dependent. Also coated formulations and coated slow release formulations are within the scope of the invention. Preference is given to acid and enteric formulations. Suitable enteric coatings include cellulose acetate phthalate, polyvinyl acetate phthalate, hydroxypropylmethylcellulose phthalate and anionic polymers of methacrylic acid and methyl methacrylate.
- Suitable pharmaceutical preparations for oral administration may be in separate units, such as capsules, cachets, lozenges or tablets, each containing a certain amount of the compound according to formulas I or II; as a powder or granules; as a solution or
- compositions may be prepared by any suitable pharmaceutical method comprising a step of contacting the active ingredient and the carrier (which may consist of one or more additional ingredients).
- the compositions become uniform and homogeneous Mixing the active ingredient with a liquid and / or finely divided solid carrier, after which the product is shaped if necessary.
- a tablet can be made by compressing or molding a powder or granules of the compound, optionally with one or more additional ingredients.
- Pressed tablets may be prepared by tableting the compound in free-flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, and / or one or more surface active / dispersing agents in a suitable machine. Molded tablets may be prepared by shaping the powdered compound moistened with an inert liquid diluent in a suitable machine.
- compositions suitable for peroral (sublingual) administration include lozenges containing a compound of formulas I or II with a flavoring, usually sucrose and gum arabic or tragacanth, and lozenges containing the compound in an inert base such as gelatin and glycerol or sucrose and gum arabic.
- Suitable pharmaceutical compositions for parenteral administration preferably comprise sterile aqueous preparations of compounds according to formulas I or II which are preferably isotonic with the blood of the intended recipient. These preparations are preferably administered intravenously, although the administration may also be subcutaneous, intramuscular or intradermal as an injection. These preparations may preferably be prepared by mixing the compound with water and rendering the resulting solution sterile and isotonic with the blood. Injectable compositions of the invention generally contain from 0.1% to 5% by weight of the active compound.
- Suitable pharmaceutical compositions for rectal administration are preferably as single dose suppositories. These can be made by mixing a compound according to formulas I or II with one or more conventional solid carriers, for example cocoa butter, and shaping the resulting mixture.
- Suitable pharmaceutical compositions for topical application to the skin are preferably as an ointment, cream, lotion, paste, spray, aerosol or oil.
- Vaseline, lanolin, polyethylene glycols, alcohols, and combinations of two or more of these substances can be used as the carrier.
- the active ingredient is generally present at a level of from 0.1% to 15% by weight of the composition, for example from 0.5% to 2%.
- Suitable pharmaceutical compositions for transdermal applications may exist as single patches suitable for long-term close contact with the epidermis of the patient. Such patches suitably contain the active ingredient in an optionally buffered aqueous solution, dissolved and / or dispersed in an adhesive or dispersed in a polymer.
- a suitable active ingredient concentration is about 1% to 35%, preferably about 3% to 15%.
- the active ingredient can be released by electrotransport or iontophoresis as described, for example, in Pharmaceutical Research, 2 (6): 318 (1986).
- the compounds of the formulas I and II are distinguished by favorable effects on metabolic disorders. They have a positive influence on the fat and sugar metabolism, they reduce in particular the triglyceride level and are suitable for the prevention and treatment of type II diabetes and atherosclerosis as well as their diverse secondary diseases.
- the compounds according to the invention can be administered alone or in combination with one or more further pharmacologically active substances.
- the compounds according to the invention can be administered with active substances which have a similar pharmacological action as they themselves have.
- they may be administered in combination with drugs that have beneficial effects on metabolic disorders or commonly associated diseases. Examples of such medications are
- hypoglycaemic drugs antidiabetics
- Agents for the treatment of hypertension 9. agents for the treatment of heart failure and
- Agents for the treatment of diseases of the central nervous system 13. Active substances for the treatment of drug, nicotine and alcohol dependence
- the administration of the drug combination may be either by separate administration of the drugs to the patient or in the form of combination products where multiple drugs are present in a pharmaceutical preparation.
- Antidiabetics include insulin and insulin derivatives, such as Lantus ® (see www.lantus.com) or HMR 1964 or those as described in WO2005005477 (Novo Nordisk) fast-acting insulins (see US 6,221, 633), inhalable insulins such as, z. B. Exubera ® or oral insulins such as.
- GLP-1 derivatives such as exenatide, liraglutide or those described in WO 98/08871 or WO2005027978 of Novo Nordisk A / S, in WO 01/04156 by Zealand or WO 00/34331 to Beaufour-Ipsen, Pramlintide Acetate (Symlin; Amylin Pharmaceuticals), as well as orally active hypoglycemic agents.
- the active ingredients preferably comprise
- Glucosidase inhibitors inhibitors of glycogen phosphorylase
- Glucose Transporter 4 Modulators GLUT4
- Potassium channel opener e.g. those described in WO 97/26265 and WO 99/03861 of
- PTP1 B protein tyrosine phosphatase 1 B
- Modulators of the sodium-dependent glucose transporter 1 or 2 (SGLT1, SGLT2), fat metabolism-altering compounds such as antihyperlipidemic agents and antilipidemic agents,
- HMGCoA reductase inhibitor such as simvastatin, fluvastatin
- WO2005042692 MD-0727 (Microbia Inc., WO2005021497) or with compounds as described in WO2002066464 (Kotobuki Pharmaceutical Co. Ltd.), WO2005062824 (to Merck & Co.). Co.) or WO2005061451 and WO2005061452 (AstraZeneca AB).
- the compound of the formula I or II is administered in combination with a PPAR gamma agonist, e.g. Rosiglitazone, pioglitazone, JTT-501, Gl 262570, R-483 or CS-011 (rivoglitazone).
- a PPAR gamma agonist e.g. Rosiglitazone, pioglitazone, JTT-501, Gl 262570, R-483 or CS-011 (rivoglitazone).
- the compound of formula I or II is administered in combination with a PPAR alpha agonist, e.g. GW9578, GW-590735, K-111, LY-674, KRP-101 or DRF-10945.
- a PPAR alpha agonist e.g. GW9578, GW-590735, K-111, LY-674, KRP-101 or DRF-10945.
- the compound of formula I or II is used in combination with a mixed PPAR alpha / gamma agonist, e.g. Muraglitazar, Tesaglitazar, Naveglitazar, LY-510929, ONO-5129, E-3030 or as in WO00 / 64888, WO00 / 64876, WO03 / 020269, WO2004075891, WO2004076402, WO2004075815, WO2004076447, WO2004076428, WO2004076401, WO2004076426, WO2004076427, WO2006018118, WO2006018115, and WO2006018116 or described in JP Berger et al., TRENDS in Pharmacological Sciences 28 (5), 244-251, 2005.
- a mixed PPAR alpha / gamma agonist e.g. Muraglitazar, Tesaglitazar, Naveglitazar, LY-5109
- the compound of formula I or II is used in combination with a PPAR delta agonist, e.g. GW-501516, or as described in WO2005097762, WO2005097786, WO2005097763, WO2006029699.
- a PPAR delta agonist e.g. GW-501516, or as described in WO2005097762, WO2005097786, WO2005097763, WO2006029699.
- the compound of formula I or II is administered in combination with metaglidases or with MBX-2044 or other partial PPAR gamma agonist / antagonist. In one embodiment of the invention, the compound of formula I or II is administered in combination with a fibrate, such as fenofibrate, clofibrate or bezafibrate.
- the compound of formula I or II is administered in combination with an MTP inhibitor, e.g. Implitapide, BMS-201038, R-103757 or those described in WO2005085226.
- an MTP inhibitor e.g. Implitapide, BMS-201038, R-103757 or those described in WO2005085226.
- the compound of formula I or II is administered in combination with a CETP inhibitor, e.g. Torcetrapib or JTT-705.
- a CETP inhibitor e.g. Torcetrapib or JTT-705.
- the compound of formula I or II is used in combination with a bile acid resorption inhibitor (see, for example, US 6,245,744, US 6,221,897 or WO00 / 61568), e.g. HMR 1741 or as described in DE 10 2005 033099.1 and DE 10 2005 033100.9.
- a bile acid resorption inhibitor see, for example, US 6,245,744, US 6,221,897 or WO00 / 61568
- HMR 1741 e.g. HMR 1741 or as described in DE 10 2005 033099.1 and DE 10 2005 033100.9.
- the compound of formula I or II is administered in combination with a polymeric bile acid adsorber, such as e.g. Cholestyramine or colesevelam.
- a polymeric bile acid adsorber such as e.g. Cholestyramine or colesevelam.
- the compound of formula I or II is used in combination with an LDL receptor inducer (see US 6,342,512), e.g. HMR1171, HMR1586 or as described in WO2005097738.
- an LDL receptor inducer see US 6,342,512
- the compound of formula I or II is administered in combination with Omacor® (omega-3 fatty acids, high-concentration ethyl esters of eicosapentaenoic acid and docosahexaenoic acid).
- Omacor® omega-3 fatty acids, high-concentration ethyl esters of eicosapentaenoic acid and docosahexaenoic acid.
- the compound of the formula I or II is administered in combination with an ACAT inhibitor, such as avasimibe. In one embodiment of the invention, the compound of formula I or II is administered in combination with an antioxidant such as OPC-14117, probucol, tocopherol, ascorbic acid, ⁇ -carotene or selenium.
- an ACAT inhibitor such as avasimibe.
- an antioxidant such as OPC-14117, probucol, tocopherol, ascorbic acid, ⁇ -carotene or selenium.
- the compound of formula I or II in combination with a vitamin such as. As vitamin B6 or vitamin B12 administered.
- the compound of the formula I or II is administered in combination with a lipoprotein-lipase modulator, e.g. Ibrolipim (NO-1886).
- a lipoprotein-lipase modulator e.g. Ibrolipim (NO-1886).
- the compound of formula I or II is used in combination with an ATP citrate lyase inhibitor, e.g. SB-204990 administered.
- an ATP citrate lyase inhibitor e.g. SB-204990 administered.
- the compound of formula I or II is used in combination with a squalene synthetase inhibitor, e.g. BMS-188494 or as described in WO2005077907.
- a squalene synthetase inhibitor e.g. BMS-188494 or as described in WO2005077907.
- the compound of formula I or II is used in combination with a lipoprotein (a) antagonist, e.g. Gemcabene (CI-1027).
- a lipoprotein (a) antagonist e.g. Gemcabene (CI-1027).
- the compound of formula I or II in combination with an HM74A receptor agonist such as e.g. Nicotinic acid administered.
- the compound of formula I or II is administered in combination with a lipase inhibitor such as orlistat or cetilistat (ATL-962). In one embodiment of the invention, the compound of the formula I or II is administered in combination with insulin.
- the compound of formula I or II is used in combination with a sulfonylurea, e.g. Tolbutamide, glibenclamide, glipizide or glimepiride.
- a sulfonylurea e.g. Tolbutamide, glibenclamide, glipizide or glimepiride.
- the compound of formula I or II is used in combination with a biguanide, e.g. Metformin, administered.
- a biguanide e.g. Metformin
- the compound of formula I or II is used in combination with a meglitinide, e.g. Repaglinide or nateglinide.
- a meglitinide e.g. Repaglinide or nateglinide.
- the compound of formula I or II is used in combination with a thiazolidinedione, e.g. Troglitazone, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. med. Reddy's Research Foundation disclosed compounds, particularly 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy) phenyl] methyl] -2,4-thiazolidinedione.
- a thiazolidinedione e.g. Troglitazone, ciglitazone, pioglitazone, rosiglitazone or those described in WO 97/41097 by Dr. med. Reddy's Research Foundation disclosed compounds, particularly 5 - [[4 - [(3,4-dihydro-3-methyl-4-oxo-2-quinazolinylmethoxy) phen
- the compound of formula I or II is administered in combination with an ⁇ -glucosidase inhibitor, e.g. Miglitol or acarbose, administered.
- an ⁇ -glucosidase inhibitor e.g. Miglitol or acarbose
- the compound of formula I or II is administered in combination with an agent which acts on the ATP-dependent potassium channel of the beta cells, e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- an agent which acts on the ATP-dependent potassium channel of the beta cells e.g. Tolbutamide, glibenclamide, glipizide, glimepiride or repaglinide.
- the compound of the formula I or II is used in combination with more than one of the abovementioned compounds, e.g. in
- the compound of formula I or II is used in combination with an inhibitor of glycogen phosphorylase, e.g. PSN-357 or FR-258900 or those described in WO2003084922, WO2004007455, WO2005073229-31 or WO2005067932.
- an inhibitor of glycogen phosphorylase e.g. PSN-357 or FR-258900 or those described in WO2003084922, WO2004007455, WO2005073229-31 or WO2005067932.
- the compound of formula I or II is used in combination with glucagon receptor antagonists, e.g. A-770077, NNC-25-2504 or as described in WO2004100875 or WO2005065680.
- glucagon receptor antagonists e.g. A-770077, NNC-25-2504 or as described in WO2004100875 or WO2005065680.
- the compound of formula I or II in combination with activators of glucokinase such as. RO-4389620, LY-2121260 (WO2004063179), PSN-105, PSN-110, GKA-50, or those as described e.g. B. from
- the compound of formula I or II in combination with an inhibitor of gluconeogenesis, such as. FR-225654.
- the compound of the formula I or II is administered in combination with inhibitors of fructose-1, 6-bisphosphatase (FBPase), such as, for example, CS-917.
- FBPase fructose-1, 6-bisphosphatase
- the compound of formula I or II is used in combination with modulators of glucose transporter-4 (GLUT4), e.g. KST-48 (D.O. Lee et al .: Arzneim.-Forsch. Drug Res. 54 (12), 835 (2004)).
- GLUT4 glucose transporter-4
- KST-48 D.O. Lee et al .: Arzneim.-Forsch. Drug Res. 54 (12), 835 (2004).
- the compound of the formula I or II in combination with inhibitors of glutamine-fructose-6-phosphate amidotransferase (GFAT), as described, for. As described in WO2004101528 administered.
- GFAT glutamine-fructose-6-phosphate amidotransferase
- the compound of formula I or II in combination with inhibitors of dipeptidyl peptidase-IV such as. Vildagliptin (LAF-237), sitagliptin (MK-0431), saxagliptin ((BMS-477118), GSK-823093, PSN-IV
- DPP-IV dipeptidyl peptidase-IV
- the compound of the formula I or II in combination with inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 such. B. BVT-2733 or those, such as. B. in WO200190090-94, WO200343999, WO2004112782, WO200344000, WO200344009, WO2004112779, WO2004113310, WO2004103980, WO2004112784, WO2003065983, WO2003104207, WO2003104208, WO2004106294, WO2004011410, WO2004033427, WO2004041264, WO2004037251, WO2004056744, WO2004065351, WO2004089367, WO2004089380, WO2004089470-71 , WO2004089896, WO2005016877 or WO2005097759.
- 11-beta-hydroxysteroid dehydrogenase-1 11-beta-hydroxysteroid dehydrogenase-1
- the compound of formula I or II in combination with inhibitors of protein tyrosine phosphatase-1 B as described, for.
- PTP1 B protein tyrosine phosphatase-1 B
- PCT / EP2005 / 005311, PCT / EP2005 / 005321, PCT / EP2005 / 007151, PCT / EP2005 / 01294 or DE 10 2004 060542.4 As described in WO200119830-31, WO200117516, WO2004506446, WO2005012295, PCT / EP2005 / 005311, PCT / EP2005 / 005321, PCT / EP2005 / 007151, PCT / EP2005 / 01294 or DE 10 2004 060542.4.
- the compound of formula I or II is used in combination with modulators of the sodium-dependent glucose transporter 1 or 2 (SGLT1, SGLT2), e.g. KGA-2727, T-1095 and SGL-0010 or as they are e.g. In WO2004007517, WO200452903, WO200452902, WO2005121161, WO2005085237, JP2004359630 or by A.L. Handion in Expert Opin. Ther. Patents (2005) 15 (11), 1531-1540.
- modulators of the sodium-dependent glucose transporter 1 or 2 SGLT1, SGLT2
- HSL hormone-sensitive lipase
- the compound of the formula I or II in combination with inhibitors of acetyl-CoA carboxylase such as those described in WO199946262, WO200372197, WO2003072197 or WO2005044814.
- ACC acetyl-CoA carboxylase
- the compound of formula I or II is used in combination with an inhibitor of phosphoenolpyruvate carboxykinase (PEPCK), e.g. such as described in WO2004074288 administered.
- PPCK phosphoenolpyruvate carboxykinase
- the compound of the formula I or II in combination with an inhibitor of glycogen synthase kinase-3 beta such as.
- an inhibitor of glycogen synthase kinase-3 beta such as.
- GSK-3 beta glycogen synthase kinase-3 beta
- GSK-3 beta glycogen synthase kinase-3 beta
- the compound of the formula I or II in combination with an inhibitor of protein kinase C beta such as. B. Ruboxistaurin administered.
- PLC beta protein kinase C beta
- the compound of formula I or II in combination with an endothelin A receptor antagonists such as. B. avosentan (SPP-301).
- the compound of the formula I or II is administered in combination with inhibitors of the "I-kappaB kinase" (IKK inhibitors), as described, for example, in WO2001000610, WO2001030774, WO2004022553 or WO2005097129.
- IKK inhibitors inhibitors of the "I-kappaB kinase"
- the compound of the formula I or II in combination with modulators of the glucocorticoid receptor as described, for.
- modulators of the glucocorticoid receptor as described, for.
- WO2005090336 administered.
- the compound of the formula I or II is administered in combination with CART modulators (see “cocaine-amphetamine-regulated transcript-influenced transient-energy metabolism, anxiety and gastric emptying in mice" Asakawa, A. et al .: Hormone and Metabolism Research (2001), 33 (9), 554-558); NPY antagonists such as naphthalene-1-sulfonic acid ⁇ 4 - [(4-amino-quinazolin-2-ylamino) -methyl] -cyclohexylmethyl ⁇ -amide hydrochloride (CGP 71683A); Peptide YY 3-36 (PYY3-36) or analogous compounds, such as.
- CART modulators see “cocaine-amphetamine-regulated transcript-influenced transient-energy metabolism, anxiety and gastric emptying in mice” Asakawa, A. et al .: Hormone and Metabolism Research (2001), 33 (9), 554-558); NPY antagonists such as na
- CJC-1682 PYY3-36 conjugated to human serum albumin via Cys34
- CJC-1643 derivative of PYY3-36 conjugated to serum albumin in vivo
- Cannabinoid receptor 1 antagonists such as rimonabant, SR147778 or those as described in, for example, US Pat. B.
- MC4 agonists eg 1-amino-1,2,3,4-tetrahydronaphthalene-2-carboxylic acid [2- (3-benzyl-2-methyl-3-oxo-2,3,3a, 4,6, 7-hexahydro-pyrazolo [4,3-c] pyridin-5-yl) -1- (4-chlorophenyl) -2-oxo-ethyl] -amide (WO 01/91752) or LB53280, LB53279, LB53278 or THIQ, MB243, RY764, CHIR-785, PT-141 or those as described in WO2005060985, WO2005009950, WO2004087159, WO2004078717, WO2004078716, WO2004024720, US20050124652, WO2005051391, WO2004112793, WO20050222014, US20050176728, US20050164914, US20050124636, US200501
- Orexin receptor antagonists eg, 1- (2-methyl-benzoxazol-6-yl) -3- [1,5] naphthyridin-4-yl-urea hydrochloride (SB-334867-A) or those as described, for example, in US Pat in WO200196302, WO200185693, WO2004085403 or WO2005075458); Histamine H3 receptor agonists (eg, 3-cyclohexyl-1- (4,4-dimethyl-1,4,6,7-tetrahydro-imidazo [4,5-c] pyridin-5-yl) -propane-1 oxalic acid salt (WO 00/63208) or those as described in WO200064884, WO2005082893);
- Histamine H3 receptor agonists eg, 3-cyclohexyl-1- (4,4-dimethyl-1,4,6,7-tetrahydro-imidazo [4,5-c] pyridin-5-yl)
- CRF antagonists eg [2-methyl-9- (2,4,6-trimethyl-phenyl) -9H-1,3,9-triaza-fluoren-4-yl] -dipropyl-amine (WO 00/66585) );
- CRF BP antagonists e.g., urocortin
- Urocortin agonists such as 1- (4-chloro-3-methanesulfonylmethyl-phenyl) -2- [2- (2,3-dimethyl-1H-indol-6-yloxy) -ethyl-amino] -ethanol hydrochloride (WO 01 / 83451)); MSH (melanocyte-stimulating hormone) agonists;
- MCH (melanin-concentrating hormone) receptor antagonists such as NBI-845,
- CCK-A agonists such as ⁇ 2- [4- (4-chloro-2,5-dimethoxy-phenyl) -5- (2-cyclohexyl-ethyl) -thiazol-2-ylcarbamoyl] -5,7-dimethyl- indol-1-yl ⁇ acetic acid trifluoroacetic acid salt (WO 99/15525), SR-146131 (WO 0244150) or SSR-125180);
- Serotonin reuptake inhibitors e.g., dexfenfluramines
- mixed serotonin and noradrenergic compounds e.g., WO 00/71549
- 5-HT receptor agonists e.g. 1- (3-Ethylbenzofuran-7-yl) piperazine oxalic acid salt
- 5-HT2C receptor agonists such as APD-356, BVT-933 or those as described in U.S. Pat
- 5-HT6 receptor antagonists as e.g. in WO2005058858 are described;
- Bombesin receptor agonists BRS-3 agonists
- Galanin receptor antagonists BRS-3 agonists
- BRS-3 agonists Bombesin receptor agonists
- Galanin receptor antagonists Galanin receptor antagonists
- Growth hormone eg, human growth hormone or AOD-9604
- Growth hormone releasing compounds (6-Benzyloxy-1- (2-diisopropylamino-ethylcarbamoyO-S ⁇ -dihydro-1H-isoquinoline) -carboxylic acid tertiary butyl ester (WO
- Growth Hormone Secretagogue Receptor Antagonists such as B. A-778193 or those as described in WO2005030734;
- TRH agonists see, e.g., EP 0 462 884; decoupling protein 2- or 3-modulators;
- Leptin agonists See, e.g., Lee, Daniel W., Leinung, Matthew C; Rozhavskaya Arena,
- DA agonists bromocriptine or doprexin
- Lipase / amylase inhibitors (as described, for example, in WO 00/40569);
- DGATs diacylglycerol O-acyltransferases
- FAS fatty acid synthase
- thyroid hormone receptor agonists such as. B: KB-2115 or those as described in WO20058279, WO200172692, WO200194293,
- the further active ingredient is leptin; see, e.g. "Perspectives in the therapeutic use of leptin", Salvador, Javier; Gomez-Ambrosi, Javier; Fruhbeck, Gema, Expert Opinion on Pharmacotherapy (2001), 2 (10), 1615-1622.
- the further active ingredient is dexamphetamine or amphetamine. In one embodiment of the invention, the further active ingredient is fenfluramine or dexfenfluramine.
- the further active ingredient is sibutramine.
- the further active ingredient is mazindol or phentermine.
- the compound of formula I or II in combination with bulking agents preferably insoluble bulking agents (see, for example, carob / Caromax ® (Zunft HJ; et al, Carob pulp preparation for treatment of hypercholesterolemia, ADVANCES IN THERAPY (2001 September -Oct), 18 (5), 230-6).
- Caromax is a carob-containing product of the company Nutrinova, Nutrition Specialties & Food Ingredients GmbH, Industriepark availability, 65926 Frankfurt / Main) administered. Combination with Caromax ® is possible in one preparation or by separate administration of compounds of the formula I or Il and Caromax ®. Caromax ® can also be administered in the form of food, such as in baked goods or muesli bars.
- the compound of the formula I or II in combination with PDE inhibitors as described, for.
- PDE inhibitors phosphodiesterase
- the compound of formula I or II in combination with NAR-1 Natural Acid Receptor
- NAR-1 Natural Acid Receptor
- agonists as described, for.
- WO2004094429 administered.
- the compound of formula I or II in combination with CB2 (cannabinoid receptor) agonists as described, for. As described in US2005 / 143448 administered. In one embodiment of the invention, the compound of formula I or II in combination with histamine-1 agonists, as described, for. As described in WO2005101979 administered.
- CB2 cannabinoid receptor
- the compound of the formula I or II is administered in combination with bupropion, as described in WO2006017504.
- the compound of the formula I or II in combination with opioid antagonists as described, for.
- opioid antagonists as described, for.
- the compound of the formula I or II in combination with neutral endopeptidase inhibitors as described, for.
- neutral endopeptidase inhibitors as described, for.
- the compound of the formula I or II in combination with NPY inhibitors (neuropeptide Y), as described, for. As described in WO2002047670 administered.
- the compound of the formula I or II in combination with sodium / hydrogen exchange inhibitors as described, for.
- sodium / hydrogen exchange inhibitors as described, for.
- WO2003092694 administered As described in WO2003092694 administered.
- the compound of the formula I or II in combination with modulators of the glucocorticoid receptor as described, for.
- modulators of the glucocorticoid receptor as described, for.
- WO2005090336 administered.
- the compound of formula I or II in combination with nicotine receptor agonists as described, for. As described in WO2004094429 administered.
- the compound of the formula I or II in combination with NRIs Norepinephrine reuptake inhibitors
- NRIs Norepinephrine reuptake inhibitors
- the compound of formula I or II is used in combination with MOA (E-beta-methoxyacrylate), e.g. Segeline or how they z. As described in WO2002053140 administered.
- MOA E-beta-methoxyacrylate
- the compound of formula I or II in combination with antithrombotic agents such.
- Isolated rat fat cells are obtained from epididymal adipose tissue from untreated male rats (Wistar, 220-250 g) by collagenase treatment according to published procedures (eg S. Nilsson et al., Anal. Biochem., 158, 1986, 399-407, G. Fredrikson et Chem. 256, 1981, 6311-6320, H. Tornquist et al., J. Biol. Chem. 251, 1976, 813-819).
- the fat cells from 10 rats are washed three times by flotation with in each case 50 ml of homogenization buffer (25 ml Tris / HCl, pH 7.4, 0.25 M sucrose, 1 mM EDTA, 1 mM DTT, 10 ⁇ g / ml leupeptin, 10 ⁇ g / ml antipain, 20 ⁇ g / ml pepstatin) and finally taken up in 10 ml homogenization buffer.
- the fat cells are homogenized in a Teflon-in-glass homogenizer (Braun-Melsungen) by 10 strokes at 1500 rpm and 15 0 C.
- the homogenate is centrifuged (Sorvall SM24 tube, 5000 rpm, 10 min, 4 ° C).
- the shelter between the fat layer above and the pellet is removed and the centrifugation repeated.
- the resulting shelter is centrifuged again (Sorvall SM24 tubes, 20000 rpm, 45 min, 4 0 C).
- the shelter is removed and washed with 1 g of heparin-Sepharose (Pharmacia Biotech, CL-6B, 5x 25 mM Tris / HCl, pH 7.4, 150 mM NaCl).
- the precipitates are (rpm Sorvall SS34, 12000, 10 min, 4 0 C) collected by centrifugation and sucrose in 2.5 ml 20 mM Tris / HCl, pH 7.0, 1 mM EDTA, 65 mM NaCl, 13%, 1 mM DTT, 10 ⁇ g / ml leupeptin / pepstatin / antipain suspended. The suspension is dialyzed overnight at 4 ° C.
- the HSL is eluted with a volume of equilibration buffer containing 0.5 M potassium phosphate, then dialyzed (see above) and concentrated 5-10 fold by ultrafiltration (Amicon Diaflo PM 10 filter) at 4 ° C.
- the partially purified HSL can be stored for 4 to 6 weeks at -7O 0 C.
- 100 ⁇ l substrate solution are prepared to 100 ⁇ l HSL solution (HSL as above, diluted in 20 mM KPi, pH 7.0, 1 mM EDTA, 1 mM DTT, 0.02% BSA, 20 ⁇ g / ml pepstatin, 10 ⁇ g / ml Leupeptin) and incubated for 30 min at 37 ° C. After addition of 3.25 ml of methanol / chloroform / heptane (10: 9: 7) and of 1.05 ml of 0.1 M K 2 CO 3, 0.1 M boric acid (pH 10.5) is mixed well and finally centrifuged (800 xg, 20 min). After phase separation becomes Equivalent of the upper phase (1 ml) and the radioactivity determined by liquid scintillation measurement.
- Substances are usually tested in four independent approaches.
- the inhibition of the enzymatic activity of HSL by a test substance is determined by comparison with a non-inhibited control reaction.
- the IC 50 value is calculated using an inhibition curve with at least 10 concentrations of the test substance.
- the software package GRAPHIT, Elsevier BIOSOFT is used for the analysis of the data.
- EL is released as a secretory protein from recombinant cell lines (CHO, HEK293) in high concentration in cell culture medium (conditioned medium). This is used after concentration as an enzyme solution.
- the phospholipase-specific substrate becomes 1,2-bis- (4,4-difluoro- ⁇ J-dimethyl-bora- Sa-a-diaza-s-indacene-S -undecanoyO-sn-glycero-S-phosphocholine, (manufacturer: Molecular Probes)
- Hydrolysis of the A1 ester bond of this phospholipid by the enzyme releases the fluorescent dye Bodipy, which after separation by thin-layer chromatography on an HPTLC plate (Kieselgel 60, Merck) or directly in the reaction vessel by measuring the fluorescence, to prepare the substrate solution, 100 ⁇ g of 1,2-bis (4,4-difluoro-5,7-dimethyl-4-bora-3a, 4a-diaza-s indacene-3-undecanoyl) -sn-glycero-3-phospho-choline (manufact
- the subsequent enzyme reaction takes place for 60 Minutes at 37 ° C.
- 45 .mu.l of the substrate solution with 1 .mu.l of inhibitor corresponding concentration (dissolved in DMSO, to control pure DMSO solution is used) and 5 .mu.l of enzyme solution (conditioned medium) incubated.
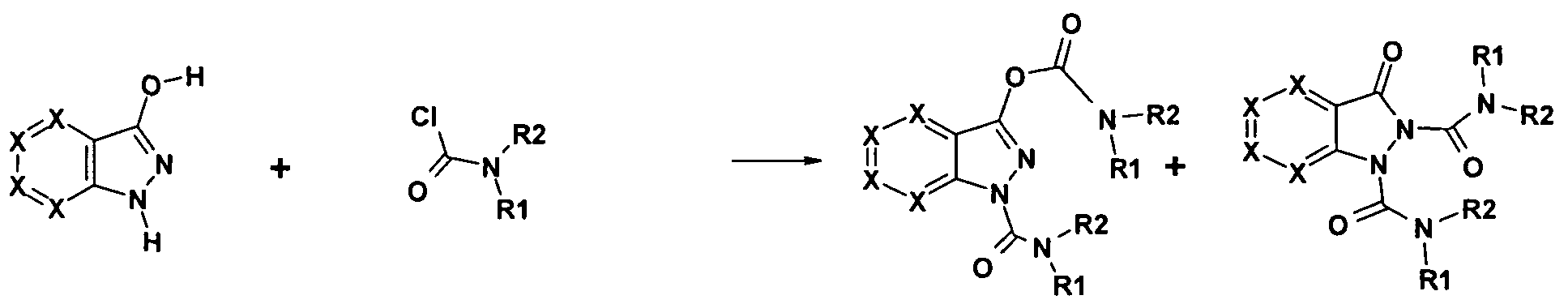
- the compounds of general formulas I or II according to the invention are prepared by methods known per se, for example by acylation of substituted or unsubstituted indazole derivatives III with carbamoyl chlorides IV (method A) 1 or in two stages by reaction of indazole derivatives III with phosgene or equivalents such as trichloromethyl chloroformate, carboxylic acid trichloromethyl ester or 4-nitrophenyl chloroformate and further reaction of the indazole-carboxylic acid derivative obtained with an amine V (method B).
- R2 is hydrogen
- indazole derivatives or corresponding aza-substituted derivatives used as starting compounds III are commercially available or can be prepared by literature processes (for example L. Baiocchi, G. Corsi Synthesis (1978), 633-648, I. Sekikawa et al Het. Chem. (1973), 931-932).
- the compounds of the formula I and II obtained by the above-described methods can be prepared by known separation methods, such as, for example, chromatographic
- Example 1 4-Phenyl-piperazine-1-carboxylic acid 6-fluoro-1- (4-phenyl-piperazine-1-carbonyl) -1H-indazol-3-yl ester (1a) and 6-fluoro-1,2 bis- (4-phenyl-piperazine-1-carbonyl) -1,2-dihydro-indazol-3-one (1b)
- Example 6 4-Methyl-piperidine-1-carboxylic acid 4,6-difluoro-1- (4-methylpiperidine-1-carbonyl) -1 H-indazol-3-yl ester and 4,6-difluoro-1, 2-bis- (4-methylpiperidine-1-carbonyl) -1,2-dihydro-indazol-3-one
- Example 7 Analogously to Example 1, 60 mg (0.35 mmol) of 4,6-difluoro-1H-indazol-3-ol were reacted with 85.6 mg (0.53 mmol) of 4-methyl-piperidine-1-carbonyl chloride. Yield: 73 mg (49%), M + H +: 421.35 4-methylpiperidine-1-carboxylic acid 4,6-difluoro-1- (4-methylpiperidine-1-carbonyl) -1 H -indazol-3-one yl ester and 3 mg (2%), M + H +: 421.34 4,6-difluoro-1,2-bis (4-methylpiperidine-1-carbonyl) -1,2-dihydro-indazol-3-one , Example 7:
- Example 9 6-Fluoro-1,2-bis (4-methylpiperidine-1-carbonyl) -1,2-dihydro-indazol-3-one (9a) and 4-methylpiperidine-1-carboxylic acid 6 -fluoro-1 - (4-methyl-piperidine-1-carbonyl) -1H-indazol-3-yl ester (9b)
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Cardiology (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Dermatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Obesity (AREA)
- Pulmonology (AREA)
- Endocrinology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Emergency Medicine (AREA)
- Physical Education & Sports Medicine (AREA)
- Vascular Medicine (AREA)
- Urology & Nephrology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Ophthalmology & Optometry (AREA)
- Epidemiology (AREA)
- Psychology (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description










Claims





Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008534900A JP2009511517A (ja) | 2005-10-12 | 2006-10-04 | リパーゼおよびホスホリパーゼのインヒビターとしてのジアシルインダゾール誘導体 |
| EP06806017.7A EP1937648B1 (de) | 2005-10-12 | 2006-10-04 | Diacylindazol-derivate als inhibitoren von lipasen und phospholipasen |
| AU2006301591A AU2006301591A1 (en) | 2005-10-12 | 2006-10-04 | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| BRPI0617222-9A BRPI0617222A2 (pt) | 2005-10-12 | 2006-10-04 | derivados de diacilindazol como inibidores de lipase e fosfolipase |
| CA002625546A CA2625546A1 (en) | 2005-10-12 | 2006-10-04 | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| IL190672A IL190672A0 (en) | 2005-10-12 | 2008-04-07 | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| US12/099,337 US7772268B2 (en) | 2005-10-12 | 2008-04-08 | Diacyl indazole derivatives as lipase and phospholipase inhibitors |
| NO20081878A NO20081878L (no) | 2005-10-12 | 2008-04-18 | Diacylindazolderivater som lipase- og fosfolipaseinhibitorer |
| US12/839,863 US8357698B2 (en) | 2005-10-12 | 2010-07-20 | Diacyl indazole derivatives as lipase and phospholipase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE102005048897.8 | 2005-10-12 | ||
| DE102005048897A DE102005048897A1 (de) | 2005-10-12 | 2005-10-12 | Diacylindazol-derivate als Inhibitoren von Lipasen und Phospholipasen |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/099,337 Continuation US7772268B2 (en) | 2005-10-12 | 2008-04-08 | Diacyl indazole derivatives as lipase and phospholipase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007042178A1 true WO2007042178A1 (de) | 2007-04-19 |
Family
ID=37813518
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/009577 Ceased WO2007042178A1 (de) | 2005-10-12 | 2006-10-04 | Diacylindazol-derivate als inhibitoren von lipasen und phospholipasen |
Country Status (18)
| Country | Link |
|---|---|
| US (2) | US7772268B2 (de) |
| EP (1) | EP1937648B1 (de) |
| JP (1) | JP2009511517A (de) |
| KR (1) | KR20080048062A (de) |
| CN (1) | CN101282939A (de) |
| AR (1) | AR056686A1 (de) |
| AU (1) | AU2006301591A1 (de) |
| BR (1) | BRPI0617222A2 (de) |
| CA (1) | CA2625546A1 (de) |
| DE (1) | DE102005048897A1 (de) |
| IL (1) | IL190672A0 (de) |
| MA (1) | MA29856B1 (de) |
| NO (1) | NO20081878L (de) |
| RU (1) | RU2008114360A (de) |
| TW (1) | TW200728285A (de) |
| UY (1) | UY29853A1 (de) |
| WO (1) | WO2007042178A1 (de) |
| ZA (1) | ZA200802105B (de) |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010044441A1 (ja) | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有する酢酸アミド誘導体 |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| KR20110060901A (ko) * | 2008-09-23 | 2011-06-08 | 겐쿄텍스 에스에이 | Nadph 옥시다아제 억제제로서 피라졸로 피리딘 유도체 |
| WO2011074560A1 (ja) | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有するオキサジアゾール誘導体 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012031383A1 (zh) * | 2010-09-06 | 2012-03-15 | 中国科学院广州生物医药与健康研究院 | 酰胺类化合物 |
| US8173635B2 (en) | 2007-11-02 | 2012-05-08 | Vertex Pharmaceuticals Incorporated | Kinase inhibitors |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013048942A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Quinolinone carboxamide inhibitors of endothelial lipase |
| WO2013048982A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013049096A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013048930A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| WO2013049104A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013048928A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013151923A1 (en) | 2012-04-03 | 2013-10-10 | Bristol-Myers Squibb Company | Pyrimidinone carboxamides as inhibitors of endothelial lipase |
| WO2013151877A1 (en) | 2012-04-03 | 2013-10-10 | Bristol-Myers Squibb Company | Pyrimidinedione carboxamide inhibitors of endothelial lipase |
| WO2014011461A1 (en) | 2012-07-09 | 2014-01-16 | Bristol-Myers Squibb Company | Amide or urea substituted benzothiazole derivatives as inhibitors of endothelial lipase |
| WO2014011513A1 (en) | 2012-07-09 | 2014-01-16 | Bristol-Myers Squibb Company | Sulfonyl containing benzothiazole inhibitors of endothelial lipase |
| WO2014015088A1 (en) | 2012-07-19 | 2014-01-23 | Bristol-Myers Squibb Company | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| WO2015105749A1 (en) | 2014-01-07 | 2015-07-16 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| US9169240B2 (en) | 2012-09-11 | 2015-10-27 | Bristol-Myers Squibb Company | Ketone linked benzothiazole inhibitors of endothelial lipase |
| WO2017214005A1 (en) | 2016-06-06 | 2017-12-14 | Bristol-Myers Squibb Company | 2-(benzothiazol-2-yl)-2-cyano-acetamide derivatives and their use as endothelial lipase inhibitors |
| US9856253B2 (en) | 2015-04-17 | 2018-01-02 | Abbvie, Inc. | Tricyclic modulators of TNF signaling |
| US9879016B2 (en) | 2015-04-17 | 2018-01-30 | Abbvie Inc. | Indazolones as modulators of TNF signaling |
| US10160748B2 (en) | 2015-04-17 | 2018-12-25 | Abbvie Inc. | Indazolones as modulators of tnf signaling |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2002835A1 (de) | 2007-06-04 | 2008-12-17 | GenKyo Tex | Pyrazolopyridinderivate als NADPH-Oxidasehemmer |
| EP2000176A1 (de) | 2007-06-04 | 2008-12-10 | GenKyo Tex | Tetrahydroindol-Derivate als NADPH-Oxidaseinhibitoren |
| EP2166010A1 (de) | 2008-09-23 | 2010-03-24 | Genkyo Tex Sa | Pyrazolopyridinderivate als NADPH-Oxidasehemmer |
| EP2165707A1 (de) | 2008-09-23 | 2010-03-24 | Genkyo Tex Sa | Pyrazolopyridinderivate als NADPH-Oxidasehemmer |
| EP2166008A1 (de) | 2008-09-23 | 2010-03-24 | Genkyo Tex Sa | Pyrazolopyridinderivate als NADPH-Oxidasehemmer |
| EP2305679A1 (de) | 2009-09-28 | 2011-04-06 | GenKyoTex SA | Pyrazolindionderivate als NADPH-Oxidasehemmer |
| EP3479843A1 (de) | 2017-11-01 | 2019-05-08 | GenKyoTex Suisse SA | Verwendung von nox-inhibitoren zur behandlung von krebs |
| CN110526898A (zh) * | 2018-05-25 | 2019-12-03 | 北京诺诚健华医药科技有限公司 | 3-吲唑啉酮类化合物、其制备方法及其在医药学上的应用 |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1164421A1 (de) * | 2000-06-13 | 2001-12-19 | Eastman Kodak Company | Photothermographisches Bildaufzeichnungselement mit verbessertem Kontrast und Verfahren zur Bildentwicklung |
| WO2003035005A2 (en) * | 2001-10-26 | 2003-05-01 | University Of Connecticut | Heteroindanes: a new class of potent cannabimimetic ligands |
| WO2004035550A1 (de) * | 2002-10-12 | 2004-04-29 | Aventis Pharma Deutschland Gmbh | Neue bicyclische inhibitoren der hormon sensitiven lipase |
| WO2004037814A1 (en) * | 2002-10-25 | 2004-05-06 | Vertex Pharmaceuticals Incorporated | Indazolinone compositions useful as kinase inhibitors |
| WO2004093872A1 (en) * | 2003-03-31 | 2004-11-04 | Eli Lilly And Company | 3-oxo-1, 3-dihydro-indazole-2-carboxylic acid amide derivatives as phospholipase inhibitors |
| WO2005073199A1 (de) * | 2004-02-02 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | Indazolderivate als inhibitoren der hormon sensitiven lipase |
-
2005
- 2005-10-12 DE DE102005048897A patent/DE102005048897A1/de not_active Withdrawn
-
2006
- 2006-10-04 AU AU2006301591A patent/AU2006301591A1/en not_active Abandoned
- 2006-10-04 JP JP2008534900A patent/JP2009511517A/ja not_active Abandoned
- 2006-10-04 KR KR1020087008736A patent/KR20080048062A/ko not_active Withdrawn
- 2006-10-04 WO PCT/EP2006/009577 patent/WO2007042178A1/de not_active Ceased
- 2006-10-04 CA CA002625546A patent/CA2625546A1/en not_active Abandoned
- 2006-10-04 RU RU2008114360/04A patent/RU2008114360A/ru not_active Application Discontinuation
- 2006-10-04 CN CNA2006800376850A patent/CN101282939A/zh active Pending
- 2006-10-04 BR BRPI0617222-9A patent/BRPI0617222A2/pt not_active IP Right Cessation
- 2006-10-04 EP EP06806017.7A patent/EP1937648B1/de active Active
- 2006-10-05 TW TW095136995A patent/TW200728285A/zh unknown
- 2006-10-10 AR ARP060104444A patent/AR056686A1/es not_active Application Discontinuation
- 2006-10-11 UY UY29853A patent/UY29853A1/es not_active Application Discontinuation
-
2008
- 2008-03-06 ZA ZA200802105A patent/ZA200802105B/xx unknown
- 2008-04-07 IL IL190672A patent/IL190672A0/en unknown
- 2008-04-08 MA MA30823A patent/MA29856B1/fr unknown
- 2008-04-08 US US12/099,337 patent/US7772268B2/en not_active Expired - Fee Related
- 2008-04-18 NO NO20081878A patent/NO20081878L/no not_active Application Discontinuation
-
2010
- 2010-07-20 US US12/839,863 patent/US8357698B2/en not_active Expired - Fee Related
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1164421A1 (de) * | 2000-06-13 | 2001-12-19 | Eastman Kodak Company | Photothermographisches Bildaufzeichnungselement mit verbessertem Kontrast und Verfahren zur Bildentwicklung |
| WO2003035005A2 (en) * | 2001-10-26 | 2003-05-01 | University Of Connecticut | Heteroindanes: a new class of potent cannabimimetic ligands |
| WO2004035550A1 (de) * | 2002-10-12 | 2004-04-29 | Aventis Pharma Deutschland Gmbh | Neue bicyclische inhibitoren der hormon sensitiven lipase |
| WO2004037814A1 (en) * | 2002-10-25 | 2004-05-06 | Vertex Pharmaceuticals Incorporated | Indazolinone compositions useful as kinase inhibitors |
| WO2004093872A1 (en) * | 2003-03-31 | 2004-11-04 | Eli Lilly And Company | 3-oxo-1, 3-dihydro-indazole-2-carboxylic acid amide derivatives as phospholipase inhibitors |
| WO2005073199A1 (de) * | 2004-02-02 | 2005-08-11 | Sanofi-Aventis Deutschland Gmbh | Indazolderivate als inhibitoren der hormon sensitiven lipase |
Cited By (64)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (de) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituierte imidazolidin-2,4-dione, verfahren zu ihrer herstellung, diese verbindungen enthaltende arzneimittel und ihre verwendung |
| WO2009021740A2 (de) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituierte tetrahydronaphthaline, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US8367697B2 (en) | 2007-11-02 | 2013-02-05 | Vertex Pharmaceuticals Incorporated | Kinase inhibitors |
| US8173635B2 (en) | 2007-11-02 | 2012-05-08 | Vertex Pharmaceuticals Incorporated | Kinase inhibitors |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| KR20110060901A (ko) * | 2008-09-23 | 2011-06-08 | 겐쿄텍스 에스에이 | Nadph 옥시다아제 억제제로서 피라졸로 피리딘 유도체 |
| KR101716511B1 (ko) | 2008-09-23 | 2017-03-14 | 겐쿄텍스 에스에이 | Nadph 옥시다아제 억제제로서 피라졸로 피리딘 유도체 |
| WO2010044441A1 (ja) | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有する酢酸アミド誘導体 |
| JPWO2010044441A1 (ja) * | 2008-10-17 | 2012-03-15 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有する酢酸アミド誘導体 |
| US8957219B2 (en) | 2008-10-17 | 2015-02-17 | Shionogi & Co., Ltd. | Acetic acid amide derivative having inhibitory activity on endothelial lipase |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US8754113B2 (en) | 2009-12-15 | 2014-06-17 | Shionogi & Co., Ltd. | Oxadiazole derivative having endothelial lipase inhibitory activity |
| WO2011074560A1 (ja) | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | 血管内皮リパーゼ阻害活性を有するオキサジアゾール誘導体 |
| WO2011107494A1 (de) | 2010-03-03 | 2011-09-09 | Sanofi | Neue aromatische glykosidderivate, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2011157827A1 (de) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2011161030A1 (de) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclisch substituierte methoxyphenylderivate mit oxogruppe, verfahren zu ihrer herstellung und ihre verwendung als gpr40 rezeptor modulatoren |
| WO2012004270A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclisch substituierte 1,3-propandioxidderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012004269A1 (de) | 2010-07-05 | 2012-01-12 | Sanofi | ( 2 -aryloxy -acetylamino) - phenyl - propionsäurederivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| WO2012010413A1 (de) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylen-substituierte hydroxy-phenyl-hexinsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| GB2497476A (en) * | 2010-09-06 | 2013-06-12 | Guangzhou Inst Biomed & Health | Amide Compounds |
| US9238643B2 (en) | 2010-09-06 | 2016-01-19 | Guangzhou Institutes Of Biomedicine And Health, Chinese Academy Of Sciences | Amide compounds |
| GB2497476B (en) * | 2010-09-06 | 2018-01-10 | Guangzhou Inst Biomed & Health | Amide Compounds |
| WO2012031383A1 (zh) * | 2010-09-06 | 2012-03-15 | 中国科学院广州生物医药与健康研究院 | 酰胺类化合物 |
| WO2012120054A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120055A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Di- und trisubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120052A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Mit carbozyklen oder heterozyklen substituierte oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung |
| WO2012120056A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituierte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2012120053A1 (de) | 2011-03-08 | 2012-09-13 | Sanofi | Verzweigte oxathiazinderivate, verfahren zu deren herstellung, ihre verwendung als medikament sowie sie enthaltendes arzneimittel und deren verwendung |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9249096B2 (en) | 2011-09-27 | 2016-02-02 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| US8952180B2 (en) | 2011-09-27 | 2015-02-10 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US9120794B2 (en) | 2011-09-27 | 2015-09-01 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013048928A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| US9493412B2 (en) | 2011-09-27 | 2016-11-15 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013049096A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013048982A1 (en) | 2011-09-27 | 2013-04-04 | Bristol-Myers Squibb Company | Pyrrolinone carboxamide compounds useful as endothelial lipase inhibitors |
| WO2013048930A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| US8933235B2 (en) | 2011-09-30 | 2015-01-13 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| US8946430B2 (en) | 2011-09-30 | 2015-02-03 | Bristol-Myers Squibb Company | Quinolinone carboxamide inhibitors of endothelial lipase |
| WO2013048942A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Quinolinone carboxamide inhibitors of endothelial lipase |
| US8993557B2 (en) | 2011-09-30 | 2015-03-31 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| WO2013049104A1 (en) | 2011-09-30 | 2013-04-04 | Bristol-Myers Squibb Company | Pyridinedione carboxamide inhibitors of endothelial lipase |
| WO2013151923A1 (en) | 2012-04-03 | 2013-10-10 | Bristol-Myers Squibb Company | Pyrimidinone carboxamides as inhibitors of endothelial lipase |
| WO2013151877A1 (en) | 2012-04-03 | 2013-10-10 | Bristol-Myers Squibb Company | Pyrimidinedione carboxamide inhibitors of endothelial lipase |
| US9394260B2 (en) | 2012-04-03 | 2016-07-19 | Bristol-Myers Squibb Company | Pyrimidinone carboxamide inhibitors of endothelial lipase |
| US9199946B2 (en) | 2012-04-03 | 2015-12-01 | Bristol-Myers Squibb Company | Pyrimidinone carboxamide inhibitors of endothelial lipase |
| US9139578B2 (en) | 2012-07-09 | 2015-09-22 | Bristol-Myers Squibb Company | Amide or urea containing benzothiazole inhibitors of endothelial lipase |
| US8680090B2 (en) | 2012-07-09 | 2014-03-25 | Bristol-Myers Squibb Company | Sulfonyl containing benzothiazole inhibitors of endothelial lipase |
| WO2014011513A1 (en) | 2012-07-09 | 2014-01-16 | Bristol-Myers Squibb Company | Sulfonyl containing benzothiazole inhibitors of endothelial lipase |
| WO2014011461A1 (en) | 2012-07-09 | 2014-01-16 | Bristol-Myers Squibb Company | Amide or urea substituted benzothiazole derivatives as inhibitors of endothelial lipase |
| WO2014015088A1 (en) | 2012-07-19 | 2014-01-23 | Bristol-Myers Squibb Company | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| US8987314B2 (en) | 2012-07-19 | 2015-03-24 | Bristol-Myers Squibb Company | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| US9169240B2 (en) | 2012-09-11 | 2015-10-27 | Bristol-Myers Squibb Company | Ketone linked benzothiazole inhibitors of endothelial lipase |
| US10173991B2 (en) | 2014-01-07 | 2019-01-08 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| WO2015105749A1 (en) | 2014-01-07 | 2015-07-16 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| US9856253B2 (en) | 2015-04-17 | 2018-01-02 | Abbvie, Inc. | Tricyclic modulators of TNF signaling |
| US9879016B2 (en) | 2015-04-17 | 2018-01-30 | Abbvie Inc. | Indazolones as modulators of TNF signaling |
| US10160748B2 (en) | 2015-04-17 | 2018-12-25 | Abbvie Inc. | Indazolones as modulators of tnf signaling |
| US10266532B2 (en) | 2015-04-17 | 2019-04-23 | Abbvie Inc. | Tricyclic modulators of TNF signaling |
| US10273238B2 (en) | 2015-04-17 | 2019-04-30 | Abbvie Inc. | Indazolones as modulators of TNF signaling |
| WO2017214005A1 (en) | 2016-06-06 | 2017-12-14 | Bristol-Myers Squibb Company | 2-(benzothiazol-2-yl)-2-cyano-acetamide derivatives and their use as endothelial lipase inhibitors |
| WO2020033919A1 (en) | 2018-08-10 | 2020-02-13 | Diapin Therapeutics, Llc | Tri-peptides and treatment of metabolic, cardiovascular and inflammatory disorders |
Also Published As
| Publication number | Publication date |
|---|---|
| US8357698B2 (en) | 2013-01-22 |
| UY29853A1 (es) | 2007-05-31 |
| MA29856B1 (fr) | 2008-10-03 |
| AR056686A1 (es) | 2007-10-17 |
| JP2009511517A (ja) | 2009-03-19 |
| US20080287448A1 (en) | 2008-11-20 |
| US20100286133A1 (en) | 2010-11-11 |
| CA2625546A1 (en) | 2007-04-19 |
| TW200728285A (en) | 2007-08-01 |
| EP1937648B1 (de) | 2014-11-26 |
| BRPI0617222A2 (pt) | 2011-07-19 |
| IL190672A0 (en) | 2008-11-03 |
| CN101282939A (zh) | 2008-10-08 |
| EP1937648A1 (de) | 2008-07-02 |
| KR20080048062A (ko) | 2008-05-30 |
| US7772268B2 (en) | 2010-08-10 |
| AU2006301591A1 (en) | 2007-04-19 |
| DE102005048897A1 (de) | 2007-04-19 |
| NO20081878L (no) | 2008-04-18 |
| RU2008114360A (ru) | 2009-10-20 |
| ZA200802105B (en) | 2009-09-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1937648B1 (de) | Diacylindazol-derivate als inhibitoren von lipasen und phospholipasen | |
| EP1940838B1 (de) | Triazolopyridin-derivate als inhibitoren von lipasen und phospholipasen | |
| EP2152711B1 (de) | 2 -heteroaryl- pyrrolo [3, 4-c]pyrrol- derivate und deren verwendung als scd inhibitoren | |
| EP2114937B1 (de) | Phenothiazin derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2328898B1 (de) | 2-heteroaryl-pyrrolo[3,4-c]pyrrol- derivate und ihre verwendung als scd inhibitoren | |
| EP2024347B1 (de) | Phenylamino-benzoxazol substituierte carbonsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2024340B1 (de) | 4,5-diphenyl-pyrimidinyl-amino substituierte carbonsäuren, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP2144885B1 (de) | Imidazolidin-carbonsäureamid-derivate als inhibitoren von lipasen und phospholipasen | |
| DE102006021872B4 (de) | 4,5-Diphenyl-pyrimidinyl-oxy oder -mercapto substituierte Carbonsäuren, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP2203448B1 (de) | Phenothiazin-derivate mit doppelbindung, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1940807B1 (de) | Carbamoylbenzotriazol-derivate als inhibitoren von lipasen und phospholipasen | |
| EP2144891B1 (de) | 5-oxo-isoxazole als inhibitoren von lipasen und phospholipasen | |
| US20100240580A1 (en) | Azoloarine derivatives, method for the production thereof, pharmaceuticals containing these compounds and the use thereof | |
| EP2683702B1 (de) | Neue substituierte phenyl-oxathiazinderivate, verfahren zu deren herstellung, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| EP2683698B1 (de) | Mit adamantan- oder noradamantan substituierte benzyl-oxathiazinderivate, diese verbindungen enthaltende arzneimittel und deren verwendung | |
| DE102007005045B4 (de) | Phenothiazin Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| HK1125363A (en) | Triazolopyridine derivatives as inhibitors of lipases and phospholipases | |
| HK1141027A (en) | Imidazolidine carboxamide derivatives as lipase and phospholipase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680037685.0 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2008000140 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008500602 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006806017 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 190672 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2625546 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/004679 Country of ref document: MX Ref document number: 08035548 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006301591 Country of ref document: AU Ref document number: 1802/CHENP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008534900 Country of ref document: JP Ref document number: 2008114360 Country of ref document: RU Ref document number: 1020087008736 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 567434 Country of ref document: NZ |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006301591 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006806017 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0617222 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080410 |