WO2007057138A2 - Process for the production of biphenyls - Google Patents
Process for the production of biphenyls Download PDFInfo
- Publication number
- WO2007057138A2 WO2007057138A2 PCT/EP2006/010864 EP2006010864W WO2007057138A2 WO 2007057138 A2 WO2007057138 A2 WO 2007057138A2 EP 2006010864 W EP2006010864 W EP 2006010864W WO 2007057138 A2 WO2007057138 A2 WO 2007057138A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- alkyl
- palladium
- general formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- XOFOCFDXWDCHJH-UHFFFAOYSA-N Cc(cc1)ccc1-c(cccc1)c1[N+]([O-])=O Chemical compound Cc(cc1)ccc1-c(cccc1)c1[N+]([O-])=O XOFOCFDXWDCHJH-UHFFFAOYSA-N 0.000 description 1
- ZUDVWTVYJVGNBO-UHFFFAOYSA-N [O-][N+](c(cccc1)c1-c(cc1)ccc1-c(cc1)ccc1Br)=O Chemical compound [O-][N+](c(cccc1)c1-c(cc1)ccc1-c(cc1)ccc1Br)=O ZUDVWTVYJVGNBO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C201/00—Preparation of esters of nitric or nitrous acid or of compounds containing nitro or nitroso groups bound to a carbon skeleton
- C07C201/06—Preparation of nitro compounds
- C07C201/12—Preparation of nitro compounds by reactions not involving the formation of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C205/00—Compounds containing nitro groups bound to a carbon skeleton
- C07C205/07—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms
- C07C205/11—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms having nitro groups bound to carbon atoms of six-membered aromatic rings
- C07C205/12—Compounds containing nitro groups bound to a carbon skeleton the carbon skeleton being further substituted by halogen atoms having nitro groups bound to carbon atoms of six-membered aromatic rings the six-membered aromatic ring or a condensed ring system containing that ring being substituted by halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C209/00—Preparation of compounds containing amino groups bound to a carbon skeleton
- C07C209/30—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds
- C07C209/32—Preparation of compounds containing amino groups bound to a carbon skeleton by reduction of nitrogen-to-oxygen or nitrogen-to-nitrogen bonds by reduction of nitro groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C211/00—Compounds containing amino groups bound to a carbon skeleton
- C07C211/43—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton
- C07C211/54—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to two or three six-membered aromatic rings
- C07C211/56—Compounds containing amino groups bound to a carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings of the carbon skeleton having amino groups bound to two or three six-membered aromatic rings the carbon skeleton being further substituted by halogen atoms or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Definitions
- the present invention relates to a novel process for preparing 2-nitro-4'-bromo-biphenyl and its use for preparing 2-nitro- and 2-amino-4'-alkynyl-biphenyl compounds which may be used as intermediates for the manufacture of biphenyl fungicides of the type described in WO 2004/058723.
- the invention also includes a 'one-pot' process for preparing the 2-nitro-4'-alk- ynyl-biphenyl intermediates from 2-nitrobromobenzene and to certain of the intermediates themselves, which are novel compounds.
- the C i- 6 alkyl groups which R 1 and R 2 may be, are branched or unbranched alkyl groups containing from 1 to 6 carbon atoms and are, for example, methyl, ethyl, H-propyl, «-butyl, ⁇ -propyl, sec-butyl, wo-butyl, tert-butyl, w-pentyl or n-hexyl. Typically they are methyl, ethyl or iso-propyl.
- the C 2-3 alkylene group, which R 1 and R 2 may join together to form, is ethylene or propylene, optionally substituted by from 1 to 4 methyl or ethyl groups.
- An anhydride of the compound of formula (IH) is a product of the combination of two or more equivalents of the compound (111) with elimination of water, containing B-O-B bridges, for example the cyclic anhydride of the formula (Ilia):
- 4-bromophenyl boronic acid is employed.
- an alkyl ester it is conveniently the dimethyl, diethyl or di-zso-propyl ester.
- the amount of compound (III) used in the invention process is from 0.9 to 2 moles for each mole of compound (II), normally from 1.0 to 1.5 moles and preferably about 1.1 moles per mole of compound (II).
- the base used may be an organic base, such as a tertiary amine, for example, triethylamine or dimethylcyclohexylamine, but is preferably an alkali metal or alkaline earth metal hydroxide, carbonate, acetate or alkoxide or an alkali metal phosphate or bicarbonate, or mixtures thereof. Particularly suitable are the hydroxides or carbonates of sodium, potassium, lithium, calcium and barium and the phosphates of sodium and potassium.
- the amount of base used will depend on the particular base chosen, but for strong inorganic bases such as sodium or potassium hydroxide, it will normally be from 1 to 4, conveniently from 1.5 to 4 and typically about 3 moles per mole of compound (III).
- the invention process is carried out in the presence of a palladium catalyst which is either
- (triphenylphosphine)palladium and tetrakis[tri(o-tolyl)phosphine)palladium are particularly suitable.
- the palladium complexes having palladium in the oxidation state plus two di- (triphenylphosphine)palladium(II) acetate (Pd(O 2 CCH 3 ) 2 ([C 6 H 5 ] 3 P) 2 ) and di-(triphenyl)- phosphine)palladium(II) chloride (PdCl 2 ([C 6 Hs] 3 P) 2 ) are particularly suitable.
- a palladium(II) salt employed in the presence of a triarylphosphine ligand for example a triphenylphosphine or tri(o-tolyl)phosphine ligand, is suitably palladium(II) acetate or palladium dichloride.
- a triarylphosphine ligand for example a triphenylphosphine or tri(o-tolyl)phosphine ligand
- palladium(II) acetate or palladium dichloride is suitably palladium(II) acetate or palladium dichloride.
- from 2 to 6 equivalents of the triarylphosphine ligand is complex ed with one equivalent of the palladium salt or additionally used with the palladium-triarylphosphine complex.
- Metallic palladium is preferably used as a powder or on a support, for example, as palladium on activated carbon, palladium on aluminium oxide, palladium on barium carbonate, palladium on barium sulphate, palladium on calcium carbonate, palladium on aluminium silicates such as montmorillonite and palladium on silic, in each case having a - A -
- Such supported catalysts may additional contain further doping substances, for example, lead.
- the simultaneous use of a complexed ligand is beneficial, particularly the use of palladium on activated carbon in the presence of triphenylphosphine, tri(o-tolyl)phosphine or other triarylphoshine as complexed ligand, the aryl groups being suitably substituted with 1 to 3 sulphonate groups.
- 2 to 3 equivalents of these ligands are used for each equivalent of palladium metal.
- Preferred palladium catalysts are di-(triphenylphosphine)palladium(II) acetate, di- (triphenyl)phosphine)palladium(II) chloride and palladium(IT) acetate or palladium(IT) chloride in the presence of a triphenylphosphine or tri(o-tolyl)phosphine ligand.
- the palladium catalyst is employed in a ratio of from 0.01 to 10 mol %, preferably from 0.05 to 5 and especially from 0.1 to 3 mol %, based on compound
- Suitable organic solvents are, for example, ethers such as dimethoxyethane, diethylene glycol dimethyl ether, tetrahydrofuran (THF), dioxane and tert- butyl methyl ether; alcohols such as methanol, ethanol, 1-propanol, 2-butanol, ethylene glycol, 1-butanol, 2-butanol and tert-butanol; ketones such as acetone, ethyl methyl ketone and w ⁇ -butyl methyl ketone; and amides such as ⁇ iV-dimethylformamide, iVJV-dimethyl- acetamide and N-methylpyrrolidone. Mixtures of two or more of these solvents may be used, particularly where one of the
- the process may be carried out at a temperature of from 0 to 15O 0 C, normally from ambient (room) temperature to 15O 0 C. Usually, the reaction is carried out at the reflux temperature of the solvent system used.
- the reaction time will depend, inter alia, on the scale of the process, the proportion of catalyst and ligand used and the temperature, but will usually take from 1 to 48 hours, for example, from 6 to 24 hours, and typically from 10 to 20 hours.
- the process is conveniently carried out by mixing the compounds (II) and (III) in a water miscible organic solvent preferentially but not obligatorily under an inert gas atmosphere, most conveniently argon or nitrogen, adding the base and water, and then adding the palladium catalyst and ligand.
- a water miscible organic solvent preferentially but not obligatorily under an inert gas atmosphere, most conveniently argon or nitrogen, adding the base and water, and then adding the palladium catalyst and ligand.
- the order of addition is not critical.
- the crude product When the reaction is adjudged complete, for example, by gas chromatographic analysis of a sample of the reaction mixture, the crude product may be isolated by removing the palladium catalyst by filtration and freeing it of solvent. It may then be purified by standard laboratory techniques. The product, either in its crude or purified state, is a useful intermediate in, for example, the manufacture of biphenyl fungicides of the type described in WO 2004/058723.
- R 3 is H, Ci -6 alkyl [optionally substituted by one or more substituents each independently selected from halogen, hydroxy, C M alkoxy, C 1-4 haloalkoxy, Ci -4 alkylthio, C M haloalkylthio, Ci -4 alkylamino, di-(C M )alkylamino, Ci -4 alkoxycarbonyl, Ci -4 alkylcarbonyloxy, and tri-( Ci -4 )alkylsilyl)], C 2 ⁇ alkenyl [optionally substituted by one or more substituents each independently selected from halogen], C 3-7 cycloalkyl [optionally substituted by one or more substituents each independently selected from halogen, Ci -4 alkyl and Ci -4 haloalkyl] or tri-( in the presence of a base, a palladium catalyst as previously defined and a copper (I) salt to form a compound of the general formula (
- R J has the meaning given above.
- Alkyl groups and the alkyl moieties of alkoxy, alkylthio, alkylamino, etc. are as defined for the alkyl values of R 1 and R 2 above.
- Typical values of R 3 are H, methyl, wo-propyl, iso- butyl, tert-butyl, cyclopropyl, 1,1-dimethylpropyl, 2,2-dimethylpropyl, hydroxymethyl, hydroxyethyl, 1 -hydroxy- 1 -methylethyl, 1 -hydroxy- 1 -methylpropyl, 1 -hydroxymethyl- 1 -methylethyl, methoxymethyl, 1-methoxy-l -methylethyl, 1-methoxymethyl-l -methylethyl, 1-ethoxy-l -methylethyl, 1-iso-propyloxy-l -methylethyl, 2-methoxy-2-methylbutyl, 2,2,2-tri-
- the base used in this aspect of the invention is preferably an aliphatic or cycloaliphatic primary, secondary or tertiary amine such as piperidine, pyrrolidine, triethylamine, di- wopropyl ethyl amine, diethylamine or »-butylamine.
- the amount used will normally be from 1 to 4, conveniently from 1.5 to 4 and typically about 3 moles per mole of compound (IV).
- the palladium catalyst used may be any catalyst of the type defined in the process for the preparation of compound (I) and in similar amounts.
- the copper (I) salt is preferably cuprous iodide. The amount used will normally be from
- the amount of alkyne (IV) used is from 1 to 2 moles, typically from 1.1 to 1.5 moles for each mole of compound (I).
- the process to form the compound (V) is carried out in a similar solvent system to the one described for the preparation of the compound (I) at ambient or an elevated temperature, for example form 15 to 5O 0 C, typically up to about 4O 0 C, and optionally at a slightly elevated pressure.
- the crude reaction mixture is typically cooled to room temperature and neutralised by the addition of a dilute acid to a pH below 9, for example below 8, and typically to a pH between 6 and 8.
- the lower end of the pH range is not critical. However, if the reaction mixture is made too acid, more base than is necessary will need to be added at the subsequent Sonogashira stage.
- the acid used for neutralisation may be an organic or inorganic acid such as propionic acid or sulphuric acid, or, preferably, acetic acid or hydrochloric acid.
- cuprous salt and terminal alkyne (FV) are added sequentially and the reaction allowed to proceed, optionally at an elevated temperature and optionally at a slightly elevated pressure, as discussed earlier. Completion of reaction may be adjudged by standard chromatographic techniques.
- the reduction may be carried out by any suitable well-known literature method for reducing aromatic nitro compounds to anilines.
- Such methods which involve inter alia either catalytic or transfer hydrogenation or reduction with metals or metal salts, including methods which allow the presence of additional functional groups like halogens or unsaturated groups, are described in, for example, Houben Weyl: Methoden der organischen Chemie FV/lc, p. 506 et seq., p. 575 et seq.
- This aspect of the invention may be carried out in a similar fashion, using similar reagents and catalysts in similar proportions, to the Sonogashira process described above for preparing compound (V) from compound (I).
- R 3 is H, Ci -6 alkyl [optionally substituted by one or more substituents each independently selected from halogen, hydroxy, Ci -4 alkoxy, Cj -4 haloalkoxy, Ci -4 alkylthio, CM haloalkylthio, C 1-4 alkylamino, Ci -4 alkoxycarbonyl, Ci -4 alkyl- carbonyloxy, and tri-( C 2-4 alkenyl [optionally substituted by one or more substituents each independently selected from halogen], C 3-7 cycloalkyl [optionally substituted by one or more substituents each independently selected from halogen, Ci -4 alkyl and Ci -4 haloalkyl] or tri-( Ci -4 )alkylsilyl.
- R 3 is H, C 1-6 alkyl, C 3-6 cycloalkyl, Ci -4 haloalkyl, hydroxy(Ci -6 )alkyl, Ci -4 alkoxy(Ci-6)alkyl Ci -4 alkoxycarbonyl(Ci -6 )alkyl, C t-4 alkyl- carbonyloxy(Ci -6 )alkyl, alkylsilyl or tri-C ⁇ alkylsily ⁇ Ci ⁇ alkyl.
- Compounds (V) of especial interest are those where R 3 is tert-butyl, 1 -methyl- 1-meth- oxyethyl or 1 -methyl- 1-ethoxyethyl.
- a nitro compound of the general formula (V) may be reduced to form an amino compound of the general formula (VII) by a suitable reduction process of the type described above for the reduction of the compound of the formula (I) to the compound of the formula (VI).
- a suitable reduction process of the type described above for the reduction of the compound of the formula (I) to the compound of the formula (VI).
- R 3 has the same meaning.
- 2-Nitrobromobenzene (18.3g) and 4-bromophenyl-boronic acid (20.Og) were mixed in a mixture of THF (40 ml) and dimethoxyethane (60 ml) under an atmosphere of nitrogen gas.
- THF 40 ml
- dimethoxyethane 60 ml
- nitrogen gas 60 ml
- a solution of potassium carbonate (31.3g) and water (60 ml) was added. The temperature rose to 3O 0 C and the yellow emulsion was stirred for a few minutes.
- Tetrakis(triphenylphosphine)palladium (0.63g) was then added and the resulting mixture was refluxed overnight.
- the reaction mixture was cooled to room temperature, filtered and the filtrate was diluted with ethyl acetate.
- the organic phase was separated, the aqueous phase washed twice with ethyl acetate and the organic phase washed with water and brine and dried over sodium sulfate. After evaporation of the solvent the residue was chromato graphed over silica gel (hexane:ethyl acetate 19: 1).
- the unwanted bis-coupling product (A) is produced in low amounts.
- Applying the teaching of US Patent No. 6,087,542 to preparation methods of compounds of formula I leads typically to a significant formation of the unwanted bis-coupling product.
- ratios in the range of 1 : 1 of wanted compounds of formula I to unwanted bis-coupling products are observed.
- the amount of the unwanted product is substantially reduced.
- ratios in the range of at least 8:1 of compounds of formula I to unwanted bis-coupling products can be obtained, as it is mentioned in example 1.
- reaction mixture was cooled to room temperature, 2M hydrochloric acid solution was cautiously added until the pH reached 7.5, and triethylamine (50 ml) and cuprous iodide (0.043g) were added. 3,3-Dimethyl-l-butyne (5.5 ml) was added within one hour.
- the reaction mixture was then stirred at room temperature for 24 hours and at 4O 0 C for 15 hours. After cooling the solvents were removed under reduced pressure and the residue was dispersed in tert-butyl methyl ether and water. The organic phase was separated, back-washed with water and brine and the solvent evaporated. The remaining solid (J. Ig ) was dissolved in hot hexane and filtered while still hot.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description













Claims




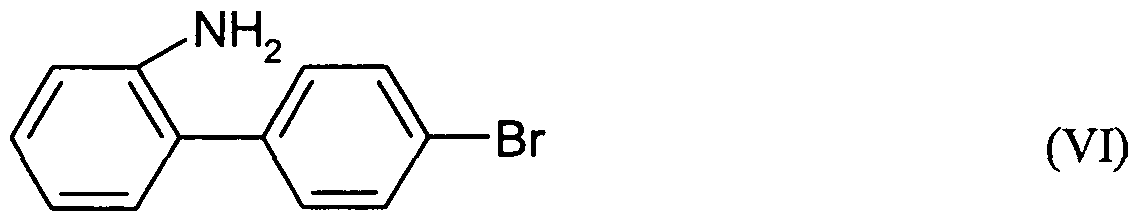




Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0618555-0A BRPI0618555A2 (en) | 2005-11-15 | 2006-11-13 | process for biphenyl production |
| CN200680042565XA CN101309893B (en) | 2005-11-15 | 2006-11-13 | Production method of biphenyl |
| US12/092,324 US20090030233A1 (en) | 2005-11-15 | 2006-11-13 | Process for the production of biphenyls |
| EP06829027A EP1957439A2 (en) | 2005-11-15 | 2006-11-13 | Process for the production of biphenyls |
| CA002628168A CA2628168A1 (en) | 2005-11-15 | 2006-11-13 | Process for the production of biphenyls |
| JP2008539355A JP4991744B2 (en) | 2005-11-15 | 2006-11-13 | Production method of biphenyls |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05024967.1 | 2005-11-15 | ||
| EP05024967 | 2005-11-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007057138A2 true WO2007057138A2 (en) | 2007-05-24 |
| WO2007057138A3 WO2007057138A3 (en) | 2007-08-09 |
Family
ID=37668244
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/010864 Ceased WO2007057138A2 (en) | 2005-11-15 | 2006-11-13 | Process for the production of biphenyls |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20090030233A1 (en) |
| EP (1) | EP1957439A2 (en) |
| JP (1) | JP4991744B2 (en) |
| KR (1) | KR20080067346A (en) |
| CN (1) | CN101309893B (en) |
| AR (1) | AR056809A1 (en) |
| BR (1) | BRPI0618555A2 (en) |
| CA (1) | CA2628168A1 (en) |
| GT (1) | GT200600485A (en) |
| TW (1) | TW200730473A (en) |
| WO (1) | WO2007057138A2 (en) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AR052930A1 (en) * | 2005-03-02 | 2007-04-11 | Basf Ag | PROCEDURE FOR THE PREPARATION OF REPLACED BIFENILES |
| ES2814952T3 (en) * | 2012-09-04 | 2021-03-29 | Celgene Corp | 3- (5-amino-2-methyl-4-oxoquinazolin-3 (4H) -yl) piperidine-2-6-dione isotopologues and methods of their preparation |
| CN104218068A (en) * | 2014-08-20 | 2014-12-17 | 京东方科技集团股份有限公司 | Light emitting structure, display device and light source device |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6087542B1 (en) | 1996-03-13 | 2000-07-11 | Basf Ag | Process for preparing nitrobiphenylene |
| US20030040538A1 (en) | 1999-12-16 | 2003-02-27 | Shiro Miyoshi | Novel substituted tricyclic compounds |
| WO2004058723A1 (en) | 2002-12-24 | 2004-07-15 | Syngenta Participations Ag | Biphenyl derivatives and their use as fungicides |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10007939A1 (en) * | 2000-02-22 | 2001-08-23 | Clariant Gmbh | Process for the preparation of substituted benzyl compounds and toluene derivatives |
| GB0322012D0 (en) * | 2003-09-19 | 2003-10-22 | Syngenta Participations Ag | Chemical compounds |
-
2006
- 2006-11-13 EP EP06829027A patent/EP1957439A2/en not_active Withdrawn
- 2006-11-13 BR BRPI0618555-0A patent/BRPI0618555A2/en not_active IP Right Cessation
- 2006-11-13 JP JP2008539355A patent/JP4991744B2/en not_active Expired - Fee Related
- 2006-11-13 CN CN200680042565XA patent/CN101309893B/en not_active Expired - Fee Related
- 2006-11-13 CA CA002628168A patent/CA2628168A1/en not_active Abandoned
- 2006-11-13 US US12/092,324 patent/US20090030233A1/en not_active Abandoned
- 2006-11-13 TW TW095141905A patent/TW200730473A/en unknown
- 2006-11-13 AR ARP060104960A patent/AR056809A1/en not_active Application Discontinuation
- 2006-11-13 KR KR1020087011637A patent/KR20080067346A/en not_active Abandoned
- 2006-11-13 WO PCT/EP2006/010864 patent/WO2007057138A2/en not_active Ceased
- 2006-11-14 GT GT200600485A patent/GT200600485A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6087542B1 (en) | 1996-03-13 | 2000-07-11 | Basf Ag | Process for preparing nitrobiphenylene |
| US20030040538A1 (en) | 1999-12-16 | 2003-02-27 | Shiro Miyoshi | Novel substituted tricyclic compounds |
| WO2004058723A1 (en) | 2002-12-24 | 2004-07-15 | Syngenta Participations Ag | Biphenyl derivatives and their use as fungicides |
Non-Patent Citations (16)
| Title |
|---|
| AN. R. SOC. ESP. FIS. QUIM., vol. 20, pages 476 |
| ANGEW. CHEM., vol. 105, 1993, pages 1589 |
| CHEM. BER., vol. 7, 1874, pages 53 |
| CHEM. ZENTRALBL., vol. 94, no. III, 1923, pages 1157 |
| GAZZ. CHIM. ITAL., vol. 68, 1938, pages 77,84 |
| J. AM. CHEM. SOC., vol. 64, 1942, pages 1848,1850,1851 |
| J. CHEM. SOC. PERKIN TRANS., vol. 1, 1972, pages 2555 - 2562 |
| J. CHEM. SOC., 1951, pages 2892,2902 |
| J. CHEM. SOC., vol. 4, 1957, pages 8 |
| J. HETEROCYCL. CHEM., vol. 38, no. 1, 2001, pages 11 - 24 |
| J. ORG. CHEM., vol. 37, 1972, pages 88 |
| JUSTUS LIEBIGS ANN. CHEM., vol. 174, 1874, pages 212 |
| JUSTUS LIEBIGS ANN. CHEM., vol. 207, 1881, pages 351 |
| ROCZ. CHEM., vol. 37, 1963, pages 153,155,158 |
| ROCZ. CHEM., vol. 47, 1973, pages 1483,1484,1486,1490,1491 |
| TETRAHEDRON LETT., 1969, pages 1623 |
Also Published As
| Publication number | Publication date |
|---|---|
| GT200600485A (en) | 2007-07-10 |
| CA2628168A1 (en) | 2007-05-24 |
| AR056809A1 (en) | 2007-10-24 |
| CN101309893A (en) | 2008-11-19 |
| BRPI0618555A2 (en) | 2011-09-06 |
| EP1957439A2 (en) | 2008-08-20 |
| JP2009515841A (en) | 2009-04-16 |
| CN101309893B (en) | 2012-03-28 |
| WO2007057138A3 (en) | 2007-08-09 |
| US20090030233A1 (en) | 2009-01-29 |
| KR20080067346A (en) | 2008-07-18 |
| TW200730473A (en) | 2007-08-16 |
| JP4991744B2 (en) | 2012-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR100451878B1 (en) | Process for Preparing Nitrobiphenylene | |
| JP2009526826A (en) | Process for producing substituted biphenyls | |
| WO2007057138A2 (en) | Process for the production of biphenyls | |
| US5306835A (en) | Method for producing octadienols | |
| US7153994B2 (en) | Manufacture of trimethylhydroquinone diacylates | |
| CN113620867A (en) | Synthesis method of fluopyram | |
| EP0808826B1 (en) | A method for preparing 3-amino substituted crotonates | |
| EP1002788B1 (en) | Process for preparing halogenated phenylmalonates | |
| JP5448572B2 (en) | Acetyl compound, method for producing the acetyl compound, and method for producing a naphthol compound using the acetyl compound | |
| US7038091B2 (en) | Process for producing acetylene compound | |
| CA2422457A1 (en) | Intermediates for use in the preparation of vitamin e | |
| WO1995030636A1 (en) | Method for preparing octadienols | |
| CN115260103B (en) | Preparation method of 4,5-dihalogen-1- (difluoromethyl) -1H-imidazole | |
| US5498725A (en) | Process for preparing 5-aminodihydropyrrole intermediate thereof and process for preparing said intermediate | |
| EP0872466B1 (en) | Process for the synthesis of 1,7-diaryl or 1,7-heteroarylheptan-4-ols and synthetic intermediates | |
| CN107721969B (en) | Preparation method of chiral catalyst ligand TADDOLs in asymmetric synthesis | |
| JPS6013015B2 (en) | Method for producing tetrakis[3-(3,5-dibutyl-4-hydroxyphenyl)propionyloxymethyl]methane | |
| US6096894A (en) | Production method of 2-(p-alkylphenyl)pyridine compound | |
| JP2558275B2 (en) | Method for producing geranylphenyl sulfone | |
| JP3579275B2 (en) | Method for producing benzoic acid derivative | |
| EP0005280B1 (en) | A process for the reduction of carboxylic acid halides to corresponding aldehydes | |
| CN115279725A (en) | Efficient and selective route for the synthesis of alkyl 2-benzoyl benzoates | |
| EP3224229A1 (en) | Process of production of 7,8-dihydro-c15-aldehyde | |
| CN121758496A (en) | Synthetic methods of arylphosphine compounds | |
| WO2000040534A1 (en) | Salicylaldoximes and method of preparation |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680042565.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2628168 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3833/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/005923 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2008539355 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006829027 Country of ref document: EP Ref document number: 1020087011637 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006829027 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12092324 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0618555 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080513 |