WO2007069814A1 - Purification process for chenodeoxycholic acid - Google Patents
Purification process for chenodeoxycholic acid Download PDFInfo
- Publication number
- WO2007069814A1 WO2007069814A1 PCT/KR2006/002972 KR2006002972W WO2007069814A1 WO 2007069814 A1 WO2007069814 A1 WO 2007069814A1 KR 2006002972 W KR2006002972 W KR 2006002972W WO 2007069814 A1 WO2007069814 A1 WO 2007069814A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- acid
- chenodeoxycholic acid
- crystallization
- process according
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C[C@](CC[C@@]1[C@@](C)(CCCC2)[C@]2C[C@]2C=*C)([C@@](CC3)[C@@]12N)[C@]3(CCCC(OC)=O)N Chemical compound C[C@](CC[C@@]1[C@@](C)(CCCC2)[C@]2C[C@]2C=*C)([C@@](CC3)[C@@]12N)[C@]3(CCCC(OC)=O)N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07J—STEROIDS
- C07J9/00—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane
- C07J9/005—Normal steroids containing carbon, hydrogen, halogen or oxygen substituted in position 17 beta by a chain of more than two carbon atoms, e.g. cholane, cholestane, coprostane containing a carboxylic function directly attached or attached by a chain containing only carbon atoms to the cyclopenta[a]hydrophenanthrene skeleton
Definitions
- the present invention relates to a process for purifying chenodeoxycholic acid
- Chenodeoxycholic acid is generally contained in bile of cow, swine, bear, or
- a typical process for preparing chenodeoxycholic acid comprises the steps of:
- Bile ,of poultry contains chenodeoxycholic acid, lithocholic acid, and a small
- chenodeoxycholic acid from chenodeoxycholic acid mixture derived from natural swine
- This process comprises the major steps of: pre-treatment to remove 3ohydroxy-6-
- oxo-5/3-cholic acid by saponification of bile; esterification of bile acid; acetylation of bile
- the object of the present invention is to provide
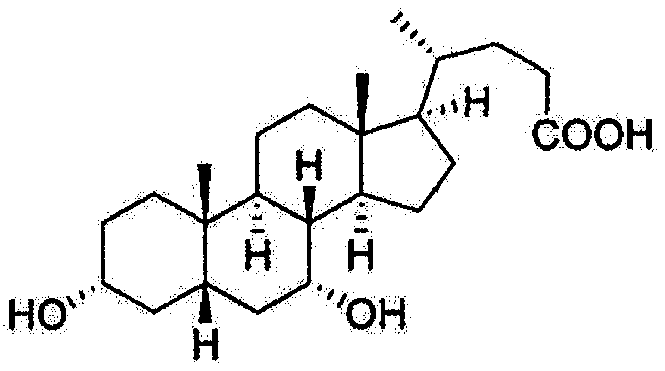
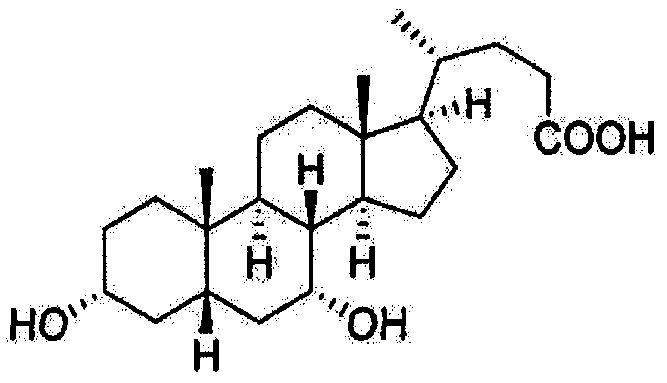
- the present invention provides a process for purifying the compound of formula I,
- IV represents 3 ⁇ -hydroxy-6-oxo-5/?-cholic acid(keto).
- Step 1 Pre-treatment of swine bile solid
- filter paper preferably
- Salt used in the present step can be optionally selected as long as it does not affect
- the salt is at least one selected from the group
- sodium sulfate more preferably sodium chloride.
- step is preferably 5 ⁇ 10 wt%, based on the amount of organic solvent. If the amount of
- salt is less than 5 wt%, water and insoluble materials (such as fatty acids, etc.) in the swine
- the organic solvent can be optionally selected from ones which can dissolve
- the solvent is ethyl acetate or acetone.
- Step 2 Esterification of chenodeoxycholic acid
- catalyst used in the present step is preferably sulfuric acid or para-toluenesulfonic acid
- PTSA sodium bicarbonate, sodium carbonate or potassium carbonate.
- Step 3 Acetylation of chenodeoxycholic acid ester
- reaction are removed by concentrating the reaction solution under reduced pressure, to
- present step is preferably anhydrous sodium acetate or pyridine, more preferably anhydrous sodium acetate.
- Non-polar solvent is added to the residue. The mixture is stirred with reflux
- HDCA-diAc-Me are crystallized and removed by filtration.
- filtered material is
- Step 5 Production of chenodeoxycholic acid-diacetate-methyl-ester
- alcohol solvent is added to the product obtained from the prior step, and then the
- compound of formula V is crystallized by standing the mixture for 2 ⁇ 3 hours at 0 ° C ⁇ 15 ° C , preferably 0 ° C ⁇ 5 ° C .
- the alcohol used in the crystallization is preferably lower alcohol
- methanol or isopropanol more preferably methanol or isopropanol, most preferably methanol, considering the
- crystallization is 0.5-3 times, preferably 1.5-3 times, to the amount of residue. If the amount is less than 0.5 times, the filtration is difficult since crystals coagulate each other.
- Step 6 Deprotection and crystallization of chenodeoxycholic acid
- reaction solution is adjusted to 4 or lower, preferably
- reaction solution stands at 35-45 ° C , preferably 35-40 ° C, to crystallize the compound of
- reaction solution is filtered, washed with water,
- deprotection is not specifically limited, but sodium hydroxide or potassium hydroxide is
- the acid used in neutralizing the reaction solution is also not specifically limited, but hydrochloric acid or sulfuric acid is preferred for the post-treatment step. Since the
- residue obtained from the prior step contains 98.5% or more of the compound of formula
- present invention is suitable for industrial manufacturing process since its melting point is
- HPLC HPLC was used to confirm the intermediate products separated form each step
- test conditions are as follows:
- Step 1 Pre-treatment of swine bile solid
- Step 2 Esterification of chenodeoxycholic acid
- Step 3 Acetylation of chenodeoxycholic acid ester
- Step 5 Production of chenodeoxycholic acid-diacetate-methyl-ester
- Step 6 Deprotection and crystallization of chenodeoxycholic acid
- Step 1 Pre-treatment of bile
- ketocholate of formula IV The filtrate was adjusted to pH 8 by using cone, sulfuric acid,
- Step 4 Acetylation of bile acid ester
- Step 6 Separation of the compound of formula V The residue obtained form the prior step was dissolved in 46ml of hot ethanol, and
- reaction solution was extracted by using ethyl acetate, and aqueous layer was
- the present invention can purify chenodeoxycholic acid of formula I from swine
- the present invention is suitable for industrial
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Steroid Compounds (AREA)
Abstract
The present invention relates to a process for purifying chenodeoxycholic acid (3α,7α-dihydroxy-5β-cholic acid). In particular, the present invention relates to a process for purifying chenodeoxycholic acid from low grade of chenodeoxycholic acid mixture in swine bile solid, with high yield and purity.
Description
PURIFICATION PROCESS FOR CHENODEOXYCHOLIC ACID
TECHNICAL FIELD
The present invention relates to a process for purifying chenodeoxycholic acid
(SαJα-dihydroxy-Sβ-cholic acid). In particular, the present invention relates to a process
for purifying chenodeoxycholic acid from low grade chenodeoxycholic acid mixture
contained in swine bile solid, with high yield and purity.
BACKGROUND ART
Chenodeoxycholic acid is generally contained in bile of cow, swine, bear, or
poultry such as chicken or goose, as well as in bile of human. Chenodeoxycholic acid is
used as starting material for the preparation of ursodeoxycholic acid which is effective to
alleviate biliary system diseases, hyperlipidemia, cholelithiasis, and chronic liver diseases,
and a typical process for preparing ursodeoxycholic acid known in the art is as follows.
A typical process for preparing chenodeoxycholic acid comprises the steps of:
esterifying cholic acid (3α,7α,12θ!-trihydroxy cholic acid) with methyl; protecting the
hydroxyl group of 3α and Ia position by acetylating them with anhydrous acetic acid;
oxidizing the hydroxyl group of 12α position to carbonyl group by using chromic acid,
and then removing the carbonyl group by Wolff-kichner reduction reaction; hydrolyzing
and deprotecting the obtained product to yield chenodeoxycholic acid. The above
process requires the reaction to be maintained at a high temperature of more than 200 °C ,
and the supply of raw material may be interrupted by bovine spongiform encephalopathy,
etc.
Bile ,of poultry contains chenodeoxycholic acid, lithocholic acid, and a small
amount of cholic acid. Thus, the process for separating chenodeoxycholic acid from
poultry is well known in the art, but is not economically reasonable due to the supply
decrease of raw material and low yield [see, Windhaus et al, I Physiol. Chem., 140,
177-185 (1924)].
US Patent No. 4,186,143 disclosed a process for purely separating and purifying
chenodeoxycholic acid from chenodeoxycholic acid mixture derived from natural swine
bile. This process comprises the major steps of: pre-treatment to remove 3ohydroxy-6-
oxo-5/3-cholic acid by saponification of bile; esterification of bile acid; acetylation of bile
acid ester; removal of intermediate product by using non-polar organic solvent;
crystallization of acetylated ester of formula I; deprotection; and production of the
compound of formula I by using crystallization in organic solvent. However, this patent
does not describe HPLC content for acetylated ester of formula I, and the purity of the
final product is very low since the specific rotatory power is [ofo25 +13.8° (c=l, CHCl3),
and the melting point is 119-121 °C [STD: [α]D 25 +15.2°(c=l, CHCl3), melting point
127- 129 "C]. Also, the crystallization for purifying the final product requires a very
long time (i.e., 16-48 hours), and the entire process is complex as eight (8) steps. Thus,
when purifying the compound of formula I by using the above process, the yield of the
final product becomes low, and the reaction time is as long as 12 days. Therefore, the
process is not economically reasonable.
In particular, when purifying the acetylated ester of formula I from the swine bile
solid having 5-35 wt% of chenodeoxycholic acid content used in the present invention by
using the above process, despite two times of recrystallization in ethanol solvent, the
content of the final product is as low as 80%.
To overcome the above problems, the object of the present invention is to provide
a new process for purifying the compound of formula I in high purity and yield, with
reducing the time required for the entire process.
DISCLOSURE OF THE INVENTION
The present invention provides a process for purifying the compound of formula I,
comprising the steps of pre-treatment of swine bile; esterification of bile acid; acetylation
of bile acid ester; removal of intermediate product by using non-polar organic solvent;
crystallization of acetylated bile acid ester; and deprotection; wherein the process is
characterized in
1) dissolving swine bile solid having 5—35 wt% of chenodeoxycholic acid content
in organic solvent containing salt, as pre-treatment step;
2) crystallizing the product obtained from the pre-treatment step in methanol or
isopropanol, within the temperature range of 0 ~ 15 °C , as crystallization step; and
3) deprotecting the product obtained from the crystallization step by adding base,
and crystallizing the deprotected product in the presence of water by adding acid, as
deprotection step.
[Formula I]
DETAILED DESCRIPTION OF THE INVENTION
In the present specification, the phrase "swine bile solid" represents solid derived
from swine bile, and contains the mixture of chenodeoxycholic acids of Formulae I~IV.
[Formula I]
[Formula II]

[Formula III]

[Formula IV]
 wherein, the compound of formula I represents chenodeoxycholic acid (3α,7α!-
wherein, the compound of formula I represents chenodeoxycholic acid (3α,7α!-
dihydroxy-5/3-cholic acid, CDCA); the compound of formula II represents hyodeoxycholic
acid (3θ!,6θ!-dihydroxy-5j8-cholic acid, HDCA); the compound of formula I represents
hyocholic acid (3α;6α;7α-trihydroxy-5/3-cholic acid, HCA); and the compound of formula
IV represents 3α-hydroxy-6-oxo-5/?-cholic acid(keto).
Hereinafter, each step for purifying chenodeoxycholic acid according to the
present invention will be exemplified in detail.
Step 1 : Pre-treatment of swine bile solid
To use swine bile solid having 5-35 wt% of chenodeoxycholic acid content in the
purification step, all of the swine bile solid is stirred and dissolved in organic solvent with
reflux. Then, the mixture is cooled to room temperature, and more stirred for 1-2 hours.
Then, insoluble materials are removed from the mixture with using filter paper, preferably
filter paper and diatomaceous earth. The organic solvent is removed under reduced
pressure to obtain residues (CDCA, HDCA, HCA and keto), which are used in the next
step. Salt used in the present step can be optionally selected as long as it does not affect
the compounds in the reactant. Preferably, the salt is at least one selected from the group
consisting of sodium chloride, anhydrous magnesium sulfate (MgSO4), and anhydrous
sodium sulfate, more preferably sodium chloride. The amount of salt used in the present
step is preferably 5~10 wt%, based on the amount of organic solvent. If the amount of
salt is less than 5 wt%, water and insoluble materials (such as fatty acids, etc.) in the swine
bile solid are not sufficiently removed, which makes the filtration difficult and reduces the
yield and velocity of esterification reaction. If the amount of salt is more than 10 wt%,
superfluous salt remains as impurity, which makes the purification difficult. Preferably,
the organic solvent can be optionally selected from ones which can dissolve
chenodeoxycholic acid of the swine bile solid and have no adverse effects thereto. More
preferably, the solvent is ethyl acetate or acetone.
Step 2: Esterification of chenodeoxycholic acid
Alcohol is added to the chenodeoxycholic acid mixture residue obtained from the
prior step, and then the solution is stirred with reflux before the residue is completely
dissolved. Then, the solution is cooled to 0-5 °C . Acid catalyst is added to the solution,
which is stirred with reflux at room temperature until the esterificaton reaction of
chenodeoxycholic acid mixture is completed. When the reaction is completed, the
solution is neutralized by adding base, and then filtered. The filtered material is washed
with alcohol and concentrated under reduced pressure to obtain chenodeoxycholic acid
ester mixture (CDCA-Me, HDCA-Me, HCA-Me and keto-Me) as residue. Alcohol used
in the present step is not specifically limited, but preferably lower alcohol having 1-4 of
carbon atoms, more preferably methanol, for easy esterification reaction. The acid
catalyst used in the present step is preferably sulfuric acid or para-toluenesulfonic acid
(PTSA), and the base is sodium bicarbonate, sodium carbonate or potassium carbonate.
Step 3: Acetylation of chenodeoxycholic acid ester
All the hydroxy groups in chenodeoxycholic acid ester mixture are acetylated by
adding anhydrous acetic and weak base to the residue obtained from the prior step with
reflux. When the reaction is completed, toluene is added to the reaction solution with
stirring at reflux. Then, the anhydrous acetic acid, acetic acid, and base remaining after
the reaction are removed by concentrating the reaction solution under reduced pressure, to
obtain a mixture of acetylated chenodeoxycholic acid ester (CDCA-diAc-Me, HDCA-
diAc-Me, HCA-triAc-Me and keto-Ac-Me) as residue. The weak base used in the
present step is preferably anhydrous sodium acetate or pyridine, more preferably
anhydrous sodium acetate.
Step 4: Removal of intermediate products of formulae III and IV
Non-polar solvent is added to the residue. The mixture is stirred with reflux
until all the residue is dissolved and cooled to room temperature. With maintaining the
temperature of solvent within 20~25 °C , intermediate products of formulae III and IV; and
a part of intermediate product of formula II (HCA-triAc-Me, keto-Ac-Me and part of
HDCA-diAc-Me) are crystallized and removed by filtration. Thus filtered material is
additionally washed with non-polar solvent, and then the filtered and washed solution is
concentrated under reduced pressure, and dried in vacuum. The non-polar solvent used
in the present step is preferably hexane, heptane, octane, isooctane and the like, more
preferably hexane or heptane.
Step 5 : Production of chenodeoxycholic acid-diacetate-methyl-ester
To produce chenodeoxycholic acid-diacetate-methyl-ester(CDCA-diAc-Me) of
formula V which is an intermediate product for preparing the compound of formula I,
alcohol solvent is added to the product obtained from the prior step, and then the
compound of formula V is crystallized by standing the mixture for 2~3 hours at 0 °C ~15 °C ,
preferably 0°C~5 °C .
[Formula V]

If the temperature is less than O °C , the content for the compound of formula V
decreases, and if the temperature is more than 15 °C , the crystallization is not done
sufficiently. When the compound of formula V is crystallized, the intermediate product
of formula II is removed from the solvent by using filtration. Thus filtered material is
washed with alcohol solvent, and dried in vacuum to obtain chenodeoxycholic acid-
diacetate-methyl-ester of formula V as crude product. To purify the compound of
formula V in high purity, recrystallization is performed until the content for the compound
of formula V becomes 98.5% or more, preferably 99% or more, under the same conditions.
To obtain the content of 99% or more, it is preferable to perform the recrystallizaton three
(3) times or more. The alcohol used in the crystallization is preferably lower alcohol,
more preferably methanol or isopropanol, most preferably methanol, considering the
content for the compound of formula V. The amount of alcohol used in the
crystallization is 0.5-3 times, preferably 1.5-3 times, to the amount of residue. If the
amount is less than 0.5 times, the filtration is difficult since crystals coagulate each other.
If the amount becomes more than 3 times, the content for the compound of formula V is
not affected by the amount.
Step 6: Deprotection and crystallization of chenodeoxycholic acid
The compound of formula V obtained from the prior step is deprotected in the
presence of base, and the pH of the reaction solution is adjusted to 4 or lower, preferably
2-3 in acid condition, to form chenodeoxycholic acid of formula I. Simultaneously, the
reaction solution stands at 35-45 °C , preferably 35-40 °C, to crystallize the compound of
formula I in the presence of water. The reaction solution is filtered, washed with water,
and dried in vacuum to refine chenodeoxycholic acid purely. The base used for the
deprotection is not specifically limited, but sodium hydroxide or potassium hydroxide is
preferred for the post-treatment step. If the pH is more than 4, crystals are not formed,
and if the pH is less than 2, the purity of the final product is reduced due to superfluous
acid. If the crystallization temperature in water is less than 35 °C , the purity of the
compound of formula I decreases, and if the temperature is more than 45 °C , the filtration
is difficult since crystals derived from the compound of formula I coagulate each other.
The acid used in neutralizing the reaction solution is also not specifically limited, but
hydrochloric acid or sulfuric acid is preferred for the post-treatment step. Since the
residue obtained from the prior step contains 98.5% or more of the compound of formula
V, preferably 99% or more, the compound of formula I can be purely refined without
additional crystallization using organic solvent since the content of impurities is low.
The product containing the compound of formula I crystallized in water according to the
present invention is suitable for industrial manufacturing process since its melting point is
about 20 °C higher, and has lower volume, than crystallized compound of formula I in
organic solvent.
The present invention will be more specifically explained in the following
examples. However, it should be understood that the following examples are intended to
illustrate the present invention, and cannot limit the scope of the present invention in any
manner.
Analytical method
HPLC was used to confirm the intermediate products separated form each step,
and the test conditions are as follows:
Column: Capcell pak UGl 20 Cl 8 (4.6 X 250mm, Shiseido)
Mobile phase: acetonitrile/water (85:15)
Detector: ultraviolet spectrometer (210nm)
Flow rate: l.Oml/min
Insertion: 20 μJi
Example
Step 1 : Pre-treatment of swine bile solid
15Og of swine bile solid having 30-35 wt% of chenodeoxycholic acid content, and 6Og of
sodium chloride in 600 ml of ethyl acetate were stirred with reflux for 1 hour, to dissolve
all the swine bile solid. Then, the mixture was cooled to 20-25 °C , stirred for 1 hour, and
filtered through diatomaceous earth, and thus filtered material was washed with 60 ml of
ethyl acetate. Organic solvent was removed by concentrating the filtered material under
reduced pressure to obtain chenodeoxycholic acid mixture (CDCA, HDCA, HCA and
keto) as residue.
Step 2: Esterification of chenodeoxycholic acid
To the residue obtained from the prior step was added 375ml of methanol, and the
mixed solution was stirred with reflux for 30 minutes until the residue was completely
dissolved. This solution was cooled to 0~10°C, 4.88ml of sulfuric acid was added to the
solution with stirring, and then the esterification reaction of chenodeoxycholic acid
mixture was completed by stirring at 20-25 °C for 2 hours. When the esterification
reaction is completed, the solution was neutralized with 53.9g of sodium bicarbonate, and
then filtered. Thus filtered material was washed with 150ml of methanol, and
concentrated under reduced pressure to obtain 134g of chenodeoxycholic acid ester
mixture (CDCA-Me, HDCA-Me, HCA-Me and keto-Me) as residue.
Step 3: Acetylation of chenodeoxycholic acid ester
To 134g of chenodeoxycholic acid ester mixture obtained from the prior step were
added 2Og of anhydrous sodium acetate and 200ml of anhydrous acetic acid. The mixed
solution was reflux ed at 120-140 °C for 5 hours, and then immediately concentrated under
reduced pressure. Anhydrous acetic acid and acetic acid were completely removed by
adding 25ml of toluene to the reaction solution, stirring with reflux for 15 minutes, and
concentrating under reduced pressure, to obtain acetylated chenodeoxycholic acid ester
mixture (CDCA-diAc-Me, HDCA-diAc-Me, HCA-triAc-Me and keto-Ac-Me) as residue.
HPLC result for the residue (RT): HCA-triAc-Me (8.76min), keto-Ac-Me (9.05min),
CDCA-diAc-Me (12.21min), and HDCA-diAc-Me (12.81min)
Step 4: Removal of intermediate products of formulae III and IV
To the residue obtained from the prior step was added non-polar solvent (400ml
of hexane), and then the mixed solution was stirred with reflux for 30 minutes. Then,
hexane solvent was cooled to 25-35 °C , stirred for 3 hours, and then filtered. Thus
filtered material (HCA-triAc-Me, keto-Ac-Me and part of HDCA-diAc-Me) was
additionally washed with 65ml of hexane, and the filtered and washed solution was
concentrated under reduced pressure to obtain the intermediate products of formulae I and
II (CDC A-di Ac-Me, HDCA-diAc-Me) as residue. HPLC result for the residue (RT):
CDCA-diAc-Me (12.21min) and HDCA-diAc-Me (12.81min).
Step 5: Production of chenodeoxycholic acid-diacetate-methyl-ester
To the residue obtained from the prior step was added 270ml of methanol, and the
mixed solution is stirred with reflux for 30 minutes, cooled to 0~10°C , more stirred for 2
hours, and then filtered. The filtered material (CDCA-diAc-Me) was washed with
methanol 70ml, and dried in vacuum at 60 "C to obtain 85% content of crude product.
Then, to the crude product was added 72ml of methanol, and recrystallization was
performed to the mixture at 0~5 °C for 2 hours. Recrystallization is additionally
performed to the mixture one more time to obtain 99% content of chenodeoxycholic acid-
diacetate-ester. The yield is 24.5g (19.5g+mother liquor 5g). m.p.: 128-129 °C .
HPLC result for the residue (RT): CDCA-diAc-Me (12.21 min).
Step 6: Deprotection and crystallization of chenodeoxycholic acid
To 220ml of water were added 24.5g of chenodeoxycholic acid-diacetate-ester
and 29.5g of sodium hydroxide, and then the solution was stirred with reflux for 4 hours.
To the solution was added 370ml of water. The solution's pH is adjusted to 2.0-3.0 by
using 59ml of hydrochloric acid. Then, the solution was stirred at 35-45 °C for 1 hour,
and then filtered. The filtered material was washed with 24.5ml of water and dried in
vacuum at 70 °C to obtain 19.5g of pure chenodeoxycholic acid, m.p.: 160-161 °C, [α]o25
+13.0°(c=l, CHCl3).
Comparative Example
Step 1 : Pre-treatment of bile
150g of concentrated swine bile was dissolved in 1000ml of hot water. Then,
lOOg of sodium hydroxide was added, and the solution was stirred with reflux for 20 hours.
This solution was cooled to 25 °C . 1500ml of water was added to the solution, which was
kept cool for one day. 1Og of diatomaceous earth was added to the reaction solution,
which was then stirred and filtered to remove precipitated sodium 3α-hydroxy-6-
ketocholate of formula IV. The filtrate was adjusted to pH 8 by using cone, sulfuric acid,
and then stirred for 15 minutes after adding 5g of sodium hydrosulfite. Then, 400ml of
ethyl acetate was added to the solution, which was then adjusted to pH 5 by using diluted
sulfuric acid. The solution was stirred for 30 minutes, and aqueous layer was removed
therefrom by layer separation. To the organic layer were added 7g of diatomaceous earth
and 7g of active carbon, which was then stirred for 30 minutes and filtered. Thus filtered
material was washed with 50ml of ethyl acetate, and concentrated under reduced pressure.
Step 2: Esterfication of bile acid
The residue obtained from the prior step was dissolved in 300ml of methanol.
Then, the solution was neutralized with sodium bicarbonate (pH 7), filtered, and then
concentrated under reduced pressure.
Step 3: Removal of methyl ester of formula II
The residue obtained from the prior step was dissolved in 320ml of hot benzene,
and the mixed solution was concentrated to 225ml, and kept cold for one day. Then, the
solution was filtered; thus filtered material (methyl ester benzene adduct of formula II)
was washed with benzene, and the benzene-filtered and washed solution was concentrated
under reduced pressure.
Step 4: Acetylation of bile acid ester
To the residue obtained from the prior step were added 75ml of anhydrous acetic
acid and 7.5g of anhydrous sodium acetate, and then the mixed solution was stirred with
reflux for 5 hours. The remaining anhydrous acetic acid was removed by distilling
anhydrous acetic acid, stirring with reflux for 15 minutes after adding 35ml of methanol,
and then distilling under reduced pressure.
Step 5: Removal of acetylated ester of formula IH
The residue obtained form the prior step was reflux ed in 200ml of hexane solvent,
and the solution was stored at 20 °C for one day, and filtered. Thus filtered material
(crude crystal of HDC A-tri Ac-Me) was washed with hexane, and the filtered and washed
solution was distilled under reduced pressure.
Step 6: Separation of the compound of formula V
The residue obtained form the prior step was dissolved in 46ml of hot ethanol, and
then kept cool for one day. This solution was filtered, and thus filtered material was
washed with 27ml of cold ethanol, and dried in vacuum at 60 °C. 21.5g of the compound
of formula V was recrystallized 3 times by using ethanol to obtain 18.5g of product, m.p.
119-1210C; [α]D 25 +10.4°(c=l, Dioxane); [α]D 25 +13.8°(c=l, CHCl3).
Step 7: Saponification and neutralization
To 185ml of water were added 18.5g of chenodeoxycholic acid-diacetate-methyl-
ester and 18.5g of sodium hydroxide, and then the mixed solution was stirred with reflux
for 14 hours. Then, the solution's pH was adjusted to 4.5 by using cone, sulfuric acid.
Step 8: Production of the compound of formula I
The reaction solution was extracted by using ethyl acetate, and aqueous layer was
discarded therefrom. Ethyl acetate layer in the solution was washed with 6% saline, and
the solution was distilled to about 90ml. This solution was cooled, kept cool for one day
after adding 90ml of hexane, and filtered. Thus filtered material was washed with 20ml
of hexane, and dried in vacuum at 60 °C to produce 12.7g of chenodeoxycholic acid. m.p.
142-1450C; [α]D 25 +13.0°(c=l, CHCl3).
INDUSTRIAL APPLICABILITY
The present invention can purify chenodeoxycholic acid of formula I from swine
bile solid in high yield and purity. Also, the present invention is suitable for industrial
purification by reducing the purification time.
Claims
1. A process for purifying the compound of formula I, comprising the steps of pre-
treatment of swine bile; esterification of bile acid; acetylation of bile acid ester; removal of
intermediate product by using non-polar organic solvent; crystallization of acetylated bile
acid ester; and deprotection; wherein the process is characterized in,
1) dissolving swine bile solid having 5—35 wt% of chenodeoxycholic acid content
in organic solvent containing salt, as pre-treatment step;
2) crystallizing the product obtained from the pre-treatment step in methanol or
isopropanol, within the temperature range of 0 ~ 15 °C , as crystallization step; and
3) deprotecting the product obtained from the crystallization step by adding base,
and crystallizing the deprotected product in the presence of water by adding acid, as
deprotection step.
[Formula I]

2. The process according to claim 1, wherein the salt used in the pre-treatment
step is at least one selected from the group consisting of sodium chloride, anhydrous magnesium sulfate, and anhydrous sodium sulfate.
3. The process according to claim 2, wherein the amount of salt is 5-10 wt%,
based on the total weight of organic solvent.
4. The process according to claim 1, wherein the product obtained from the
crystallization step contains 98.5% or more of the compound of formula V.
[Formula V]

5. The process according to claim 1, wherein the crystallization step is carried out
in the temperature range of 0-5 °C .
6. The process according to claim 1, wherein the amount of methanol or
isopropanol used in the crystallization step is 0.5-3 times the amount of residue obtained
from the step of removal of intermediate product.
7. The process according to claim 1, wherein the pH of the deprotection step is 4
or less.
8. The process according to claim 1, wherein the crystallization in the presence of
water is carried out in the temperature range of 35-45 °C .
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200680050155XA CN101351470B (en) | 2005-12-12 | 2006-07-28 | Purification process for chenodeoxycholic acid |
| JP2008545473A JP5107257B2 (en) | 2005-12-12 | 2006-07-28 | Method for purifying chenodeoxycholic acid |
| ES06783444.0T ES2533348T3 (en) | 2005-12-12 | 2006-07-28 | Chenodeoxycholic acid purification procedure |
| EP06783444.0A EP1960416B1 (en) | 2005-12-12 | 2006-07-28 | Purification process for chenodeoxycholic acid |
| SI200631886T SI1960416T1 (en) | 2005-12-12 | 2006-07-28 | Purification process for chenodeoxycholic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020050121605A KR100658511B1 (en) | 2005-12-12 | 2005-12-12 | Method for Purifying Kenodeoxycholic Acid |
| KR10-2005-0121605 | 2005-12-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007069814A1 true WO2007069814A1 (en) | 2007-06-21 |
Family
ID=37814712
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2006/002972 Ceased WO2007069814A1 (en) | 2005-12-12 | 2006-07-28 | Purification process for chenodeoxycholic acid |
Country Status (7)
| Country | Link |
|---|---|
| EP (1) | EP1960416B1 (en) |
| JP (1) | JP5107257B2 (en) |
| KR (1) | KR100658511B1 (en) |
| CN (1) | CN101351470B (en) |
| ES (1) | ES2533348T3 (en) |
| SI (1) | SI1960416T1 (en) |
| WO (1) | WO2007069814A1 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007078039A1 (en) | 2005-12-30 | 2007-07-12 | Daewoong Pharmaceutical Co., Ltd. | Purification process for chenodeoxycholic acid |
| CN103360454A (en) * | 2013-05-06 | 2013-10-23 | 广西大学 | Method for separating and purifying chenodeoxycholic acid from goose bile |
| EP2857414A1 (en) | 2013-10-03 | 2015-04-08 | Prodotti Chimici E Alimentari Spa | Polymorphous form of sodium hyodeoxycholate (NaHDC) and preparation process thereof |
| CN111285915A (en) * | 2020-03-26 | 2020-06-16 | 山东中京生物科技有限公司 | Novel process for extracting and refining chenodeoxycholic acid from poultry gall bladder |
| CN112010920A (en) * | 2020-09-22 | 2020-12-01 | 安徽科宝生物工程有限公司 | Method for preparing chenodeoxycholic acid by extraction complexation method |
| CN113234115A (en) * | 2021-05-31 | 2021-08-10 | 山东海钰生物技术股份有限公司 | Production process for extracting chenodeoxycholic acid from chicken bile |
| CN116355033A (en) * | 2023-03-29 | 2023-06-30 | 常德云港生物科技股份有限公司 | Method for removing allochenodeoxycholic acid from chenodeoxycholic acid from duck gall |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102372757B (en) * | 2011-10-31 | 2013-01-16 | 安徽科宝生物工程有限公司 | Method for preparing chenodeoxycholic acid in pig bile by esterification method |
| CN110845564B (en) * | 2019-11-14 | 2022-09-16 | 湖南九典制药股份有限公司 | Method for extracting chenodeoxycholic acid from chicken gall paste |
| CN111494424A (en) * | 2020-06-16 | 2020-08-07 | 重庆极泽生物科技有限公司 | Preparation method of poultry gall powder |
| CN116249536A (en) * | 2020-09-18 | 2023-06-09 | 首尔大学校产学协力团 | The batch production method of sodium taurodeoxycholate |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3919266A (en) | 1972-09-21 | 1975-11-11 | Intellectual Property Dev Corp | Production of bile acids |
| FR2429224A1 (en) * | 1978-06-19 | 1980-01-18 | Canada Packers Ltd | Chenodeoxycholic acid recovery from porcine bile - useful for dissolving gall stones in vivo |
| JPS60181096A (en) * | 1984-02-28 | 1985-09-14 | Tokyo Tanabe Co Ltd | Purification of bile acid |
| EP0386538A2 (en) | 1989-03-06 | 1990-09-12 | ERREGIERRE INDUSTRIA CHIMICA Spa | Process for preparing high purity 3-alpha-7-beta-dihydroxycholanic acid |
| JPH03227998A (en) * | 1990-02-02 | 1991-10-08 | Showa Denko Kk | Method for purifying chenodeoxycholic acid |
| CN1528779A (en) * | 2003-09-29 | 2004-09-15 | 华东理工大学 | A kind of preparation method of chenodeoxycholic acid |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1450939A (en) * | 1973-12-19 | 1976-09-29 | Intellectual Property | |
| US4186143A (en) * | 1977-06-20 | 1980-01-29 | Canada Packers Limited | Chenodeoxycholic acid recovery process |
| KR100658512B1 (en) * | 2005-12-30 | 2006-12-19 | 주식회사 대웅제약 | Method for Purifying Kenodeoxycholic Acid |
-
2005
- 2005-12-12 KR KR1020050121605A patent/KR100658511B1/en not_active Expired - Lifetime
-
2006
- 2006-07-28 EP EP06783444.0A patent/EP1960416B1/en active Active
- 2006-07-28 JP JP2008545473A patent/JP5107257B2/en active Active
- 2006-07-28 ES ES06783444.0T patent/ES2533348T3/en active Active
- 2006-07-28 WO PCT/KR2006/002972 patent/WO2007069814A1/en not_active Ceased
- 2006-07-28 SI SI200631886T patent/SI1960416T1/en unknown
- 2006-07-28 CN CN200680050155XA patent/CN101351470B/en active Active
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3919266A (en) | 1972-09-21 | 1975-11-11 | Intellectual Property Dev Corp | Production of bile acids |
| FR2429224A1 (en) * | 1978-06-19 | 1980-01-18 | Canada Packers Ltd | Chenodeoxycholic acid recovery from porcine bile - useful for dissolving gall stones in vivo |
| JPS60181096A (en) * | 1984-02-28 | 1985-09-14 | Tokyo Tanabe Co Ltd | Purification of bile acid |
| EP0386538A2 (en) | 1989-03-06 | 1990-09-12 | ERREGIERRE INDUSTRIA CHIMICA Spa | Process for preparing high purity 3-alpha-7-beta-dihydroxycholanic acid |
| JPH03227998A (en) * | 1990-02-02 | 1991-10-08 | Showa Denko Kk | Method for purifying chenodeoxycholic acid |
| CN1528779A (en) * | 2003-09-29 | 2004-09-15 | 华东理工大学 | A kind of preparation method of chenodeoxycholic acid |
Non-Patent Citations (2)
| Title |
|---|
| See also references of EP1960416A4 * |
| ZHANG ET AL.: "Isolation and purification of chenodeoxycholic acid from pig bile", SHENGWU HUAXUE YU SHENGWU WULI JINZHAN, no. 4, 1987, pages 68 - 71, XP008125239 * |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007078039A1 (en) | 2005-12-30 | 2007-07-12 | Daewoong Pharmaceutical Co., Ltd. | Purification process for chenodeoxycholic acid |
| EP1966228A4 (en) * | 2005-12-30 | 2009-11-11 | Daewoong Chemical Co Ltd | PROCESS FOR THE PURIFICATION OF CHENODESOXYCHOLIC ACID |
| CN103360454A (en) * | 2013-05-06 | 2013-10-23 | 广西大学 | Method for separating and purifying chenodeoxycholic acid from goose bile |
| CN103360454B (en) * | 2013-05-06 | 2015-12-09 | 广西大学 | A kind of method of separating-purifying Chenodiol from goose bile |
| EP2857414A1 (en) | 2013-10-03 | 2015-04-08 | Prodotti Chimici E Alimentari Spa | Polymorphous form of sodium hyodeoxycholate (NaHDC) and preparation process thereof |
| WO2015049657A1 (en) | 2013-10-03 | 2015-04-09 | Prodotti Chimici E Alimentari S.P.A. | Polymorphic form of sodium hyodeoxycholate (nahdc) and its preparation process |
| CN111285915A (en) * | 2020-03-26 | 2020-06-16 | 山东中京生物科技有限公司 | Novel process for extracting and refining chenodeoxycholic acid from poultry gall bladder |
| CN112010920A (en) * | 2020-09-22 | 2020-12-01 | 安徽科宝生物工程有限公司 | Method for preparing chenodeoxycholic acid by extraction complexation method |
| CN113234115A (en) * | 2021-05-31 | 2021-08-10 | 山东海钰生物技术股份有限公司 | Production process for extracting chenodeoxycholic acid from chicken bile |
| CN116355033A (en) * | 2023-03-29 | 2023-06-30 | 常德云港生物科技股份有限公司 | Method for removing allochenodeoxycholic acid from chenodeoxycholic acid from duck gall |
Also Published As
| Publication number | Publication date |
|---|---|
| KR100658511B1 (en) | 2006-12-19 |
| CN101351470B (en) | 2011-06-15 |
| EP1960416A4 (en) | 2009-10-21 |
| SI1960416T1 (en) | 2015-02-27 |
| EP1960416B1 (en) | 2014-12-31 |
| EP1960416A1 (en) | 2008-08-27 |
| JP2009518456A (en) | 2009-05-07 |
| ES2533348T3 (en) | 2015-04-09 |
| CN101351470A (en) | 2009-01-21 |
| JP5107257B2 (en) | 2012-12-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1960416B1 (en) | Purification process for chenodeoxycholic acid | |
| US5508453A (en) | Intermediate for use in the preparation of taurocholanic acids | |
| JP5799171B2 (en) | Method for producing 1-palmitoyl-3-acetylglycerol and method for producing 1-palmitoyl-2-linoleoyl-3-acetylglycerol using the same | |
| WO2014083512A1 (en) | Process for preparation of abiraterone acetate | |
| EP0124068A1 (en) | New derivatives of biliary acids, process for the production thereof and pharmaceutical compositions containing the same | |
| ES2550453T3 (en) | Procedure for the preparation of cholanic acids | |
| US4379093A (en) | Process for preparing high purity ursodeoxycholic acid | |
| US4186143A (en) | Chenodeoxycholic acid recovery process | |
| EP1966228B1 (en) | Purification process for chenodeoxycholic acid | |
| JP3746174B2 (en) | Method for producing mometasone furoate | |
| EP2221313A1 (en) | Process for the preparation of ursodeoxycholic acid disodium 3,7-disulfate | |
| US4277408A (en) | Novel purification process | |
| ES2363258T3 (en) | PROCESS FOR SELECTIVE INSULATION, PURIFICATION AND SEPARATION OF COMPOUNDS 3,17-MONOHIDROXYLED DICETOESTEROIDS. | |
| CN114133421A (en) | A kind of preparation method of β-mouse cholic acid | |
| US20210261599A1 (en) | Process for the Preparation of Obeticholic Acid and Intermediates Used In the Process Thereof | |
| JPS60181096A (en) | Purification of bile acid | |
| WO2002012267A1 (en) | Method for the production of fluorescein bile acid derivatives | |
| JPS58113202A (en) | Purification of bile acid | |
| WO2025090435A1 (en) | Alfaxalone synthesis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680050155.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008545473 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006783444 Country of ref document: EP |