WO2007070433A2 - 2-arylthiazole derivatives as cxcr3 receptor modulators - Google Patents
2-arylthiazole derivatives as cxcr3 receptor modulators Download PDFInfo
- Publication number
- WO2007070433A2 WO2007070433A2 PCT/US2006/047065 US2006047065W WO2007070433A2 WO 2007070433 A2 WO2007070433 A2 WO 2007070433A2 US 2006047065 W US2006047065 W US 2006047065W WO 2007070433 A2 WO2007070433 A2 WO 2007070433A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- thiazol
- tert
- butyl
- piperidin
- oxoethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C1C*CCCNCC1 Chemical compound C1C*CCCNCC1 0.000 description 12
- JFJOKKHSMPZWJZ-UHFFFAOYSA-N C(CNCC1)C1c1c[s]c(-c2ccncc2)n1 Chemical compound C(CNCC1)C1c1c[s]c(-c2ccncc2)n1 JFJOKKHSMPZWJZ-UHFFFAOYSA-N 0.000 description 1
- RWFMICIKXIVKFA-UHFFFAOYSA-N CC(C)(C)c(cc(cc1C(C)(C)C2CCCCC2)-c2ncc(C(CC3)CCN3C(C[n]3c(C)nc(Cl)c3C)=O)[s]2)c1OC Chemical compound CC(C)(C)c(cc(cc1C(C)(C)C2CCCCC2)-c2ncc(C(CC3)CCN3C(C[n]3c(C)nc(Cl)c3C)=O)[s]2)c1OC RWFMICIKXIVKFA-UHFFFAOYSA-N 0.000 description 1
- NBLNRQMYMIHMSH-UHFFFAOYSA-N CC(C)(C)c1cc(-c2ncc(C(CC3)CCN3C(CN(CCN3c4ccccc4)C3=O)=O)[s]2)cc(C(C)(C)C)c1OC Chemical compound CC(C)(C)c1cc(-c2ncc(C(CC3)CCN3C(CN(CCN3c4ccccc4)C3=O)=O)[s]2)cc(C(C)(C)C)c1OC NBLNRQMYMIHMSH-UHFFFAOYSA-N 0.000 description 1
- WCNALMYAIKFDLC-UHFFFAOYSA-N CC(C)(C)c1cc(-c2ncc(C(CC3)CCN3C(C[n]3c(C(F)(F)F)ncc3)=O)[s]2)cc(C(C)(C)C)c1OC Chemical compound CC(C)(C)c1cc(-c2ncc(C(CC3)CCN3C(C[n]3c(C(F)(F)F)ncc3)=O)[s]2)cc(C(C)(C)C)c1OC WCNALMYAIKFDLC-UHFFFAOYSA-N 0.000 description 1
- YPHCZPICPFHZGW-UHFFFAOYSA-N CC(C)(C)c1cc(-c2ncc(C3CCNCC3)[s]2)cc2c1OCCC2(C)C Chemical compound CC(C)(C)c1cc(-c2ncc(C3CCNCC3)[s]2)cc2c1OCCC2(C)C YPHCZPICPFHZGW-UHFFFAOYSA-N 0.000 description 1
- BJOZXCDOLVXFSU-UHFFFAOYSA-N CC(C)(C)c1cc(C(Cl)=O)cc2c1OCCC2(C)C Chemical compound CC(C)(C)c1cc(C(Cl)=O)cc2c1OCCC2(C)C BJOZXCDOLVXFSU-UHFFFAOYSA-N 0.000 description 1
- DBGYOLLVMZDGKA-WAYWQWQTSA-N CC(C)N/C(/C)=C(\N)/Cl Chemical compound CC(C)N/C(/C)=C(\N)/Cl DBGYOLLVMZDGKA-WAYWQWQTSA-N 0.000 description 1
- CXAXBADUCTZITE-UHFFFAOYSA-N CC(CC=C1C(C)(C)C)C(C(C)(C)C)=C1OC Chemical compound CC(CC=C1C(C)(C)C)C(C(C)(C)C)=C1OC CXAXBADUCTZITE-UHFFFAOYSA-N 0.000 description 1
- ZVTAEZXHLXWEAU-UHFFFAOYSA-N CC(CN=C1)c2c1nc[n]2CC(N(CC1)CCC1c1cnc(-c2cc(C(F)(F)F)cc(C(F)(F)F)c2)[s]1)=O Chemical compound CC(CN=C1)c2c1nc[n]2CC(N(CC1)CCC1c1cnc(-c2cc(C(F)(F)F)cc(C(F)(F)F)c2)[s]1)=O ZVTAEZXHLXWEAU-UHFFFAOYSA-N 0.000 description 1
- YCVVXKAHYCZLNT-UHFFFAOYSA-N CC(N1CC(N(CC2)CCC2c2cnc(C)[s]2)=O)=CN(C)C1=O Chemical compound CC(N1CC(N(CC2)CCC2c2cnc(C)[s]2)=O)=CN(C)C1=O YCVVXKAHYCZLNT-UHFFFAOYSA-N 0.000 description 1
- WLYZNMZZCLNAPB-UHFFFAOYSA-N CC(N1CC(O)=O)=CN(C)C1=O Chemical compound CC(N1CC(O)=O)=CN(C)C1=O WLYZNMZZCLNAPB-UHFFFAOYSA-N 0.000 description 1
- AVRBNZMLWXGVKZ-UHFFFAOYSA-N CCN(C=CN1)C1=O Chemical compound CCN(C=CN1)C1=O AVRBNZMLWXGVKZ-UHFFFAOYSA-N 0.000 description 1
- MGLXFROMWBOVKG-UHFFFAOYSA-N COC(C[n]1c(SC)ncc1)=O Chemical compound COC(C[n]1c(SC)ncc1)=O MGLXFROMWBOVKG-UHFFFAOYSA-N 0.000 description 1
- NZSDDQGIZSFYFD-UHFFFAOYSA-N Cc([nH]1)cnc1SC Chemical compound Cc([nH]1)cnc1SC NZSDDQGIZSFYFD-UHFFFAOYSA-N 0.000 description 1
- JESBJYBJWCRPPB-DHDCSXOGSA-N Cc1c(CC(O)=O)nc[n]1CC(N(CC1)CCC1/C(/SC)=C/NC)=O Chemical compound Cc1c(CC(O)=O)nc[n]1CC(N(CC1)CCC1/C(/SC)=C/NC)=O JESBJYBJWCRPPB-DHDCSXOGSA-N 0.000 description 1
- UZMIWZFUPRJXEZ-UHFFFAOYSA-N Cc1c[s]c(C(OC)=O)c1O Chemical compound Cc1c[s]c(C(OC)=O)c1O UZMIWZFUPRJXEZ-UHFFFAOYSA-N 0.000 description 1
- FQLFGQKWJNALEN-UHFFFAOYSA-N Cc1cnc[n]1CC(O)=O Chemical compound Cc1cnc[n]1CC(O)=O FQLFGQKWJNALEN-UHFFFAOYSA-N 0.000 description 1
- WNMCUAIWCYFHLK-UHFFFAOYSA-N Clc1cc(-c2nc(C3CCNCC3)c[s]2)cc(Cl)c1 Chemical compound Clc1cc(-c2nc(C3CCNCC3)c[s]2)cc(Cl)c1 WNMCUAIWCYFHLK-UHFFFAOYSA-N 0.000 description 1
- JAKZRAQZYGSIAQ-UHFFFAOYSA-N O=C(C[n]1c(cncc2)c2nc1)N(CC1)CCC1c1cnc(-c2cc(C(F)(F)F)cc(C(F)(F)F)c2)[s]1 Chemical compound O=C(C[n]1c(cncc2)c2nc1)N(CC1)CCC1c1cnc(-c2cc(C(F)(F)F)cc(C(F)(F)F)c2)[s]1 JAKZRAQZYGSIAQ-UHFFFAOYSA-N 0.000 description 1
- YRGIRZFYHYKXAX-LJQANCHMSA-N O=C(C[n]1c(nccc2)c2nc1)N(CC1)CCC1[C@@H]1SC(c2cc(C(F)(F)F)cc(C(F)(F)F)c2)=NC1 Chemical compound O=C(C[n]1c(nccc2)c2nc1)N(CC1)CCC1[C@@H]1SC(c2cc(C(F)(F)F)cc(C(F)(F)F)c2)=NC1 YRGIRZFYHYKXAX-LJQANCHMSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- chemokines are a family of small (70-120 amino acids), pro-inflammatory cytokines, with potent chemotactic activities. As their name implies, one function of chemokines, which are released by a wide variety of cells at sites of inflammation, is to attract leukocytes , including monocytes, macrophages, T lymphocytes, eosinophils, basophils and neutrophils and to promote their migration through endothelial layers, (reviewed in Schall, Cytokine. 3, 165-183 (1991) and Murphy, Rev. Immun.. 12, 593-633 (1994)).
- chemokines play a role in a number of other biological processes including cellular proliferation, hematopoiesis, angiogenesis, tumor metastasis and host defense.
- polypeptides were originally defined as having four conserved aminoterminal cysteines , and divided into two major and two minor subfamilies based on the spacing arrangement of the first cysteine pair.
- the two major subfamilies consist of the CXC (or ⁇ ) and CC (or ⁇ ) chemokines.
- CXC-chemokine family which includes CXCLl (MGSA or GRO ⁇ ), CXCL7 (NAP-2), CXCL8 (interleukin-8 or 1L-8), CXCL9 (MIG), CXCLlO (lP-10) and CXCLl 1 (I-TAC), these two cysteines are separated by a single amino acid
- CC-chemokine family which includes CCL5 (RANTES), CCL2 (monocyte chemotactic protein- 1 or MCP-I), CCL8 (MCP-2), CCL7 (MCP-3), CCL3 (MEP- l ⁇ ), CCL4 (MIP- IB) and CCLl 1 (eotaxin), these two residues are adjacent.
- CXC-chemokines such as CXCLl , CXCL7 and CXCL-8 are chemotactic primarily for neutrophils while another subset of CXC chemokines , including CXCL9, CXCL10 and CXCLl 1, are chemotactic primarily for T- lymphocytes.
- the CC_chemokines such as CCL5, CCL3, CCL4, CCL2, CCL8, CCL7and CCLl 1 , are more broad in their action and are chemotactic for macrophages, monocytes, T- lymphocytes, eosinophils and basophils (Deng, et al., Nature. 381, 661-666 (1996), Murphy et al. Pharmacol Revw. 52(1) 145-176, (2000).).
- chemokines bind to specific G-protein coupled receptors (GPCRs) present on leukocytes and other cells, (reviewed in Horuk, Trends Pharm. Sci., 15, 159-165 (1994), Murphy et al. Pharmacol Revw. 52(1) 145-176, (2000).)
- GPCRs G-protein coupled receptors
- chemokine receptors Upon interaction with their cognate ligands, chemokine receptors transduce an intracellular signal though their associated heterotrimeric G proteins, resulting in a rapid cellular responses, including an increase in intracellular calcium concentration.
- GPCRs G-protein coupled receptors
- chemokine receptors are more selectively expressed on subsets of leukocytes.
- generation of specific chemokines provides a mechanism for recruitment of particular leukocyte subsets.
- the restricted expression and defined function of the chemokine receptors has focused attention on intervention in the chemokine signaling pathways as a method for highly selective intervention in pathological immunological and inflammatory processes.
- Chemokine receptors such as CCRl , CCR2A, CCR2B, CCR3, CCR4, CCR5, CXCR3,
- CXCR4 have been implicated as important mediators of inflammatory diseases and immunoregulatory disorders, including asthma, allergic rhinitis and and atherosclerosis. They are also purported to play a role in the pathogenesis of autoimmune disorders such as rheumatoid arthritis, psoriasis, multiple sclerosis. An extensive review of the role of chemokines in disease is provided by in Seminars in Immunology.. 15(1), 1-55 (2003).
- chemokines are potent chemoattractants for lymphocytes.
- CXCR3 CD 183 is expressed in activated T lymphocytes, some B lymphocytes and NK cells. Expression and receptor responsiveness are both increased by activation of the T lymphocytes.
- the potent inflammatory cytokines CXCLlO and CXCLl t are chemoattractant for T lymphocytes and tumor infiltrating lymphocytes.
- the relatively restricted expression of the CXCR3 expression on these proinflammatory cell types mark CXCR3 as a very promising target for selective intervention in the inflammatory process.
- a connection with disease processes, particularly Th-I mediated processes, is indicated by the presence of the CXCR3 on most activated T lymphocytes within inflamed joint synovium in rheumatoid arthritis as well as within inflamed tissue present in other inflammatory disorders including ulcerative colitis, Graves' disease, MS and rejecting graft tissues.
- agents which inhibit or modulate the function of chemokine receptors such as the CXCR3 receptor would be useful in treating or preventing such disorders and diseases.
- Data from animal models of inflammation further supports the hypothesis regarding the effectiveness of chemokine blockade, specifically CXCR3 inhibition, in diseases with clear T -lymphocyte mediated tissue damage such as transplant rejection, graft versus host disease, multiple sclerosis, optic neuritis and rheumatoid or psoriatic arthritis.
- Many other diseases are characterized by T lymphocyte infiltrates, and by inference are therefore also good candidates for interventions which prevent the migration of T lymphocytes.
- These diseases include psoriasis and other chronic inflammatory diseases of the skin such as atopic dermatitis, lichen planus and bullous pemphigoid, inflammatory bowel diseases such as ulcerative colitis and Crohn's disease and autoimmune diseases such as systemic and cutaneous lupus erythematosus, Behcet's disease, type ⁇ diabetes or Graves' disease.
- inflammatory lung diseases such as chronic obstructive pulmonary disease, hypersensitivity pneumonitis, chronic eosinophilic pneumonia, pulmonary sarcoidosis, bronchiolitis obliterans syndrome, asthma, kidney diseases such as glomerulonephritis, pathogenesis of chronic HCV infection and atherosclerosis show a dependence on T lymphocytes and are promising targets for agents which modulate the function of chemokine receptors such as the CXCR3 receptor.
- CXCR3 in some B cell tumors indicates that intervention in CXCR3 function could have beneficial effects in these cancers, particularly in suppressing metastasis.
- chemokine receptor function Several methods are under investigation for modulation of chemokine receptor function. These include antibodies binding to and neutralizing the chemokine ligands, antibodies binding to and modulating the function of the chemokine receptors and small molecules which bind to and inhibit function of the chemokine receptor.
- the ideal method for intervention in CXCR3 mediated chemotaxis is the binding of orally bioavailable small molecules which prevent the function of the receptor. Molecules with affinity for the CXCR3 chemokine receptor and ability to modulate the function of the receptor are described here.
- the invention encompasses compounds of Formula I
- the invention encompasses a genus of compounds of Formula I
- D is CR4 or N
- R3 is selected from the group consisting of: H, halo, C]_4alkyl, -CF3, -OCF3 and -S(O) n CF3, wherein n is 0 or 2;
- R4 is selected from the group consisting of: H, halo, -OH 5 Ci_4alkyl, -OCH3, -OCH2CF3 and -CF3;
- R3 and R4 may be joined together with the carbon atoms to which they are attached to form a five- or six-membered monocyclic ring, said rings containing oxygen or tetra-substituted with methyl groups as follows:
- R5 is selected from the group consisting of: -H 5 halo, Ci_4alkyl, C3_6cycloalkyl, CF3, -CF2CH3, -OCF3 and -SCF3;
- R-6 is selected from the group consisting of — H and -OCH3,
- R5 and R ⁇ may be joined together with the carbon atoms to which they are attached to form a monocyclic 5-membered ring, said ring di-substituted with methyl as follows:
- p 0, 1 or 2.
- the invention encompasses a sub-genus of compounds of Formula I wherein:
- R"2, R"3, R"4 and R"5 are independently selected from the group consisting of: -H, carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, C]-3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy,
- R"6 is H or OH
- the invention encompasses a sub-genus of compounds of Formula I wherein D is N.
- the invention encompasses a sub-genus of compounds of Formula
- the invention encompasses a class of compounds of Formula I wherein: R-3 and R4 are joined together with the carbon atoms to which they are attached to form a six-membered monocyclic ring as follows:
- the invention encompasses a class of compounds of Formula I wherein:
- R3 is selected from the group consisting of: H, halo, Ci_4alkyl and -CF3;
- R4 is selected from the group consisting of: H, halo, Ci_4alkyl, -OCH3 and -CF3;
- R5 is selected from the group consisting of: H, halo, Ci_4alkyl, CF3 and -SCF3; and
- R6 is H.
- the invention encompasses a sub-class of compounds of Formula I wherein:
- R3 is selected from the group consisting of: H, Cl, Br, tert-buty ⁇ and -CF3;
- R4 is selected from the group consisting of: H, Cl, Br, tert-buty], -OCH3 and -CF3;
- R5 is selected from the group consisting of: H, CI, Br, tert-b ⁇ tyl, CF3 and -SCF3;
- R6 is H.
- Another embodiment of the invention encompasses a sub-genus of compounds of Formula I within the above-described genus wherein R ⁇ is -H.
- Another embodiment of the invention encompasses a sub-genus of compounds of Formula 1 within the above-described genus wherein:
- D is CR4
- R3 is selected from the group consisting of: H, Cl, Br, tert-butyl and -CF3;
- R4 is selected from the group consisting of: H, Cl, Br, tert-butyl, -OCH3 and -CF3;
- R3 and R4 may be joined together with the carbon atoms to which they are attached to form a six- membered monocyclic ring as follows:
- R5 is selected from the group consisting of: H, Cl, Br, tert-buty ⁇ , CF3 and -SCF3;
- R6 is -H.
- the invention encompasses a class of compounds of Formula I wherein:
- R3 is /erf-butyl, R4 is H and R5 is tert-butyl;
- R3 is tert-butyl
- R4 is -OCH3
- R5 is tert-butyl
- R3 is -CF3, R4 is -H and R5 is -CF3; (4) R3 is H, R4 is -OCH3 and R5 is H;
- R3 is H, R4 is tert-butyl and R5 is H;
- R3 is H, R4 is Br and R5 is H;
- R3 is H, R4 is -CF3 and R5 is H; (9) R3 is H, R4 is H and R5 is Cl;
- R3 is Cl
- R4 is H
- R5 is Cl
- the invention encompasses a class of compounds of Formula
- R"2, R"3, R"4 and R"5 are independently selected from the group consisting of: -H, carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, Ci_3alkoxy and Ci_3alkyl, said C]_3alkyl optionally substituted with carboxy or hydroxy,
- R"6 is H or OH
- the invention encompasses a class of compounds of Formula I wherein:
- D is CR4
- R3 is tert-buty ⁇
- R4 is Hor-OCH3
- R-6 is H.
- the invention encompasses a compound selected from the following group:
- (21) 1 -(2- ⁇ 4-[2-(3,5-di-tert-buty 1-4-methoxyphenyl)- 1 ,3-thiazol-4-yl]piperidin- 1 -yl ⁇ -2- oxoethyl)-3-methyl-l,3-dihydro-2H-benzimidazol-2-one;
- (22) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(2-ethyl-4-methyl-lH- imidazol-l-yl)acetyl]piperidine;
- (32) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyl-lH-l,2,4- triazol-l-yl)acetyl]piperidine;
- the invention also encompasses a pharmaceutical composition comprising a compound of Formula I in combination with a pharmaceutically acceptable carrier.
- the invention also encompasses a method for treating a disease or condition mediated by the CXCR3 chemokine receptor comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound of Formula I.
- the invention also encompasses a method for treating a disease or condition mediated by the CXCR3 chemokine receptor comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound of Formula T, wherein the disease or condition is selected from the group consisting of: acute and chronic transplant rejection, psoriasis, rheumatoid arthritis and multiple sclerosis.
- halogen or "halo” includes F, Cl, Br, and I.
- alkyl means linear or branched structures and combinations thereof, having the indicated number of carbon atoms.
- Ci-6alkyl includes methyl, ethyl, propyl, 2- propyl, s- and t-butyl, butyl, pentyl, hexyl and 1,1-dimethylethyl.
- cycloalkyl means mono-, bi- or tri-cyclic structures, optionally combined with linear or branched structures, having the indicated number of carbon atoms.
- cycloalkyl groups include cyclopropyl, cyclopentyl, cycloheptyl, adamantyl, cyclododecylmethyl, 2-ethyl-l- bicyclo[4.4.0]decyl, cyclobutylmethyl, cyclopropylmethyl 1-methylcyclopropyl and the like.
- tautomers embraces the standard meaning of the term, i.e. a type of isomerism in which two or more isomers are rapidly interconverted so that they ordinarily exist together in equilibrium.
- Tautomers include, e.g., compounds that undergo facile proton shifts from one atom of the compound to another atom of the compound.
- Some of the compounds described herein may exist as tautomers with different points of attachment of hydrogen. Such an example might be a ketone and its enol form known as keto- enol tautomers or an amide and its hydroxy imine tautomer.
- the individual tautomers of the compounds of Formula I, as well as mixtures thereof, are included in the scope of this invention.
- tautomers included in this definition include, but are not limited to:
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, mo ⁇ holine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
- the compounds of the present invention are modulators of CXCR3 chemokine receptor function and are of use in antagonizing chemokine mediated cell signalling and in particular are of use in the prophylaxis and/or treatment of diseases or disorders involving inappropriate T-cell trafficking.
- the invention extends to such a use and to the use of the compounds of Formula I for the manufacture of a medicament for treating such diseases and disorders.
- diseases include inflammatory, autoimmune and immunoregulatory disorders.
- mammals including, but not limited to, cows, sheep, goats, horses, dogs, cats, guinea pigs, rats or other bovine, ovine, equine, canine, feline, rodent or murine species can be treated.
- the method can also be practiced in other species, such as avian species (e.g., chickens).
- Diseases or conditions of humans or other species which can be treated with compounds of Formula I include, but are not limited to: autoimminue mediated inflammatory or allergic diseases and conditions, including respiratory diseases such as asthma, particularly bronchial asthma, systemic lupus erythematosus, ankylosing spondylitis, systemic sclerosis, autoimmune diseases, such as rheumatoid arthritis, psoriatic arthritis, multiple sclerosis, systemic lupus erythematosus, myasthenia gravis, juvenile onset diabetes", glomerulonephritis, autoimmune thyroiditis, Behcet's disease; acute and chronic graft rejection (e.g., in transplantation), including allograft rejection or graft-versus-host disease; inflammatory bowel diseases, such as Crohn's disease and ulcerative colitis; spondyloarthropathies; scleroderma; psoriasis (including T-cell mediated psoriasis);
- Other diseases or conditions in which undesirable inflammatory responses are to be inhibited can be treated, including, but not limited to, reperfusion injury, atherosclerosis, certain hematologic malignancies, and polymyositis.
- the compounds of the present invention are accordingly useful in treating, preventing, ameliorating, controlling or reducing the risk of a wide variety of inflammatory and immunoregulatory disorders and diseases as well as autoimmune pathologies.
- the present invention is directed to the use of the subject compounds for treating, preventing, ameliorating, controlling or reducing the risk of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, psoriasis or psoriatic arthritis.
- the instant invention may be used to evaluate putative specific agonists or antagonists of chemokine receptors, including CXCR3. Accordingly, the present invention is directed to the use of these compounds in the preparation and execution of screening assays for compounds which modulate the activity of chemokine receptors.
- the compounds of this invention are useful for isolating receptor mutants, which are excellent screening tools for more potent compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other compounds to chemokine receptors, e.g., by competitive inhibition.
- the compounds of the instant invention are also useful for the evaluation of putative specific modulators of the chemokine receptors, including CXCR3.
- CXCR3 putative specific modulators of the chemokine receptors
- the present invention is further directed to a method for the manufacture of a medicament for treating CXCR3 mediated diseases in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- a subject compound may be used in a method of inhibiting the binding of a chemokine to a chemokine receptor, such as CXCR3, of a target cell, which comprises contacting the target cell with an amount of the compound which is effective at inhibiting the binding of the chemokine to the chemokine receptor.
- the subject treated in the methods above is a mammal, preferably a human being, male or female, in whom modulation of chemokine receptor activity is desired.
- “Modulation” as used herein is intended to encompass antagonism, agonism, partial antagonism, inverse agonism and/or partial agonism. In a preferred aspect of the present invention, modulation refers to antagonism of chemokine receptor activity.
- therapeutically effective amount means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment.
- treatment refers both to the treatment and to the prevention or prophylactic therapy of the aforementioned conditions.
- prophylactic or therapeutic dose of a compound of Formula I will, of course, vary with the nature and severity of the condition to be treated, and with the particular compound of Formula 1 used and its route of administration.
- the dose will also vary according to the age, weight and response of the individual patient.
- the daily dose range lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- a suitable dosage range is from about 0.01 mg to about 25 mg (preferably from 0.1 mg to about 10 mg) of a compound of Formula I per kg of body weight per day.
- a suitable dosage range is, e.g. from about 0.01 mg to about 100 mg of a compound of Formula I per kg of body weight per day, preferably from about 0.1 mg to about 10 mg per kg.
- a suitable dosage range is from 0.01 mg to about 25 mg (preferably from 0.1 mg to about 5 mg) of a compound of Formula I per kg of body weight per day.
- compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier.
- composition is intended to encompass a product comprising the active ingredients), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of Formula I, additional active ingredient(s), and pharmaceutically acceptable excipients.
- Any suitable route of administration may be employed for providing a mammal, especially a human with an effective dosage of a compound of the present invention.
- oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed.
- Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
- compositions of the present invention comprise a compound of Formula I as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
- the compounds of the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulizers.
- the compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device.
- the preferred delivery systems for inhalation are metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellants, such as fluorocarbons or hydrocarbons and dry powder inhalation (DPI) aerosol, which may be formulated as a dry powder of a compound of Formula 1 with or without additional excipients.
- MDI metered dose inhalation
- DPI dry powder inhalation
- Suitable topical formulations of a compound of formula I include transdermal devices, aerosols, creams, ointments, lotions, dusting powders, and the like.
- the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, capsules and tablets, with the solid oral preparations being preferred over the liquid preparations. Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or non-aqueous techniques.
- compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient, as a powder or granules or as a solution or a suspension in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion or a water-in-oil liquid emulsion.
- compositions may be prepared by any of the methods of pharmacy but all methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more necessary ingredients.
- the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation.
- a tablet may be prepared by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent. Desirably, each tablet contains from about 1 mg to about 500 mg of the active ingredient and each cachet or capsule contains from about 1 to about 500 mg of the active ingredient.
- Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula T are useful. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I- When a compound of Formula I is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula 1.
- Examples of other active ingredients that may be combined with a compound of Formula I, either administered separately or in the same pharmaceutical compositions include, but are not limited to: (a) VLA-4 antagonists such as those described in US 5,510,332, WO97/03094, WO97/02289, WO96/40781, WO96/22966, WO96/20216, WO96/01644, WO96/06108, WO95/15973 and WO96/31206, as well as natalizumab; (b) steroids such as beclomethasone, methyl prednisolone, betamethasone, prednisone, dexamethasone, and hydrocortisone; (c) immunosuppressants such as cyclosporin, tacrolimus, rapamycin and other FK-506 type immunosuppressants; (d) immunomodulaltory antibody therapies including anti-TNF therapies such as Etanercept (Enbrel®), Infliximab (Remicade®), Adalim
- piroxicam sudoxicam and tenoxican
- salicylates acetyl salicylic acid, sulfasalazine, olsalazine, mesalamine and balsalazide
- pyrazolones acetyl salicylic acid, sulfasalazine, olsalazine, mesalamine and balsalazide
- pyrazolones acetyl salicylic acid, sulfasalazine, olsalazine, mesalamine and balsalazide
- pyrazolones cyclooxygenase-2 (COX-2) inhibitors such as celecoxib, rofecoxib, and parecoxib
- inhibitors of phosphodiesterase type IY PDE-TV
- antagonists of the other chemokine receptors especially CCRl, CCR2, CCR5 and CCR3
- cholesterol lowering agents such as
- the weight ratio of the compound of the Formula I to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the Formula I is combined with an NSAID the weight ratio of the compound of the Formula I to the NSAID will generally range from about 1000: 1 to about 1:1000, preferably about 200:1 to about 1 :200. Combinations of a compound of the Formula I and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
- Ac is acetyl [CH3C(O)-]; Ac2 ⁇ is acetic anhydride; 9-BBN is 9-borabicyclo[3.3.1]nonane; Bn is benzyl;
- substituted thioamides 3 are prepared from their corresponding benzoic acids 1 or benzonitriles 2 as shown in Scheme 1.
- Benzoic acids 1 are converted to their acid chlorides and ammonolyzed to the amide.
- the amide is converted to the th ⁇ oamide with Lawesson's reagent.
- Benzonitiles 2 are converted to their amides by partial hydrolysis and then to the thioamides 3 as above.
- Bromoketone 6 is prepared from N-FMOC isonipecotic acid 4 as shown in Scheme 2. Acid 4 is converted to its acid chloride and exposed to trimethylsilyldiazomethane, giving the diazoketone 5. Decomposition of 5 with 48% HBr gives the bromoketone 6.
- heterocyclic acetic acids 9 are prepared from the corresponding known heterocycles 7 as shown in Scheme 3. Exposure of 7 to a bromoacetic acid ester in the presence of alkali or alkaline metal bases in polar aprotic solvents affords the esters 8. Basic hydrolysis, reduction or solvolysis as appropriate gives the desired 9.
- Step B gg-fluoren-g-ylmethvI ⁇ -Cdiazoacety ⁇ piperidine-l-carboxYlate
- Step G 9Jy-fluoren-9-ylmethyl-4-r2-(3,5-di-terf-butyl-4-methoxyphenvn-13-thiazol-4- yll piperidine-1-carboxylate
- Step H 4-f2-(3,5-di--'gr-'-butvl-4-inethoxvphenvn-1.3-thiazol-4-vIlpiperidine
- Step K 3J? " -i ⁇ nidazof4,5-Alpyridine-3-vIacetic acid
- Step L 3-(2-(4-r2-(3,5-di-fer/'-but ⁇ l-4-methoxyphenv ⁇ -li3-thiazol-4-yllpiperidin-l-vU-2-oxoethyl>- 3H-ini idazo f 4,5-&l p v rid ine
- Step A 2,4-dimethyI-l/7-imidazol-l-yIacetic acid teri-butyl ester and 2,5-dimethyl-l/* r -imidazol-l- ylacetic acid tert-butyl ester
- Step B 2,4-dimethvI-l/y-imidazoI-l-ylacetic acid hydrotrifluoroacetate
- Step C 4-f2-f3,5-di-/er/-butvI-4-methoxyphenvn-l,3-thiazol-4-vH-l-ff2.4-dimethvI-l.g-imidazol-l- vDacetyll piperidine
- Step E 4-f2-f3,5-di-l'erAbutyl-4-methoxyphenvn-l,3-tIiiazol-4-vH-l-rf2,5-dimethvI-l/r-i ⁇ nidazol-l- vDacetyll piperidine
- a solution of dihydroxyacetone (1.905g; 21.16mmol) in aqueous ammonia (28%; 15mL) was placed in a microwavable vessel.
- Acetamidine hydrochloride (2.0Og; 21.16mmol) was added, the vessel sealed and the solution heated via microwave irradiation at 120° for lOmin.
- the solution was lyophilized.
- the derived residue was digested in refluxing acetone (10OmL), cooled to ambient temperature and filtered. The filtrate was evaporated and the residue dissolved in aqueous HCl (5OmL; IN).
- the solution was lyophilized to a heavy oil.
- Step B f4-fhydroxymethvO-2-methyl-l//-imidazol-l-v ⁇ acet.c acid methyl ester and f5- (hvdroxymethyl)-2-methyl-l//-iinidazol-l-vIlaeetie acid methyl ester
- Step D ri-f2-f4-f2-f3.5-di-ter/-butyl-4-methoxyphenyl)-l,3-thiazol-4- ⁇ piperidin-l-yl>-2-oxoethvn-
- Step F H-(2- ⁇ 4-r2-(3,5-di-te/-f-butvI-4-methoxyphenvn-l,3-thiazol-4-ynpiperidin-l-vU-2-oxoethvn- 2-methyl-l//-imidazol-5-v ⁇ methanol
- Step A l-ethyl-l «3-dihvdro-2 J fl r -imidazol-2-one
- Step J) l-f2- ⁇ 4-r2-r3.5-di-fer ⁇ -butyl-4-methoxyphenvn-13-thiazoI-4-vHpiperidin-l-vn-2-oxoethvI>-
- Step E (3-methyl-2-oxo-2,3-dihvdro-liy-imidazol-l-yl)acetic acid fe/7-buty! ester
- Step B Using the method of Example 4, Step B with l-methyl-l,3-dihydro-2/7-imidazol-2-one as the starting material the title compound was obtained.
- Step F (3-methyl-2-oxo-2 «3-dihvdro-l.fl r -imidazol-l-vOacetic acid
- Step G l-(2-(4-r2-(3 ⁇ 5-di-te ⁇ -butyl-4-methoxyphenylVl.,3-thiazoI-4-yllpiperidin-l-vU-2-oxoethvn- 3-methyl-l,3-dihvdro-2/f-imidazol-2-one
- Step B 1.4-dimethyl-1.3-dihydro-2 J fir-imidazol-2-one
- Step C O.S-dimethvI-l-oxo-Z ⁇ -dihydro-lJy-i ⁇ iidazot-l-vnacetic acid ferf-buty. ester
- Step D (3,5-dimethyl-2-oxo-23-dihydro-li?-imidazol-l-vI)acetic acid
- Step E 3-(2-(4-r2-f3,5-di-ferf-butvI-4-methoxyphenv ⁇ -l,3-thiazol-4-v ⁇ piperidin-l-vU-2-oxoethyl)- l,4-dimethyl-l,3-dihvdro-2/y-iinidazol-2-one
- Step D teri-butyl methyl 2,2'-(4-KiCtI-V-- l.ff-pyrazole-l,,3-diyl)diacetate
- Step E r3-f2-methoxy-2-oxoethyl)-4-methvI-lfl r -pyrazol-l-vHacetic acid hydrotrifluoroacetate
- Step F ri-(2-f4-f2-(3,5-di-te/-f-butvI-4-methoxyphenyl)-l,3-thiazoI-4-yllpiperidin-l-yU-2-oxoethv ⁇ - 4-methvI-lfl r -pyrazol-3-vHacetie acid methyl ester
- Step G ri-(2- ⁇ 4-f2-(3,5-di-ter/-butvI-4-methoxyphenvI)-l,3-thiazol-4-vnpiperidin-l-vU-2-oxoethvn- 4-methyl-l ⁇ r-pyrazo--3-yl
- Step B r4-methvI-2-(methvIthio)-l/7-imidazol-l-yl]acetic acid methyl ester
- Step E 4-r2-f3,5-di-ferr-buryl-4-methoxyphenv ⁇ -l,3-thiazol-4-yll-l- ⁇ f4-methyl-2-(methylsuIfonylV l-g-imidazol-l-yllacetvUpiperidine
- Step A Using the method of Example 4, Step A with ethyl isocyanatoacetate in place of ethyl isocyanate as one of the starting materials the title compound was obtained.
- Step B Potassium (2-oxo-2,3-dihvdro-l ⁇ r-iinidazol-l-v0aeetate
- Step C l-f2-f4-[2-(3,5-dl-i'gr/-butvI-4-methoxyphenyl)-l ,3-thiazol-4-yll piperidin-l-yl ⁇ -2-oxoethv ⁇ - l,3-dihydro-2Jy-iniidazol-2-one
- Step B Using the method of Example 4, Step B with the product of Example 8, Step A as starting material the title compound was obtained.
- Silica gel chromatography was carried out using 40:1 CH2Cl2/MeOH.
- Step B f3-f2-ethoxy-2-oxoethyl)-2-oxo-2,3-dihvdro-lJy-imidazoI-l-vHacet ⁇ c acid
- Step C r3-f2-f4-f2-f3,5-di-tert-butyl-4-methoxyphenvn-1.3-thiazol-4-vHpiperidin-l-vU-2-oxoethyl)- 2-oxo-2.3-dihvdro-LH r -imidazoI-l-vHacetic acid ethyl ester
- Step D Potassium r3-r2-f4-r2-f3,5-di-tert-butvi-4-methoxyphenvn-l,3-thiazol-4-vUpiperidin-l-yl)- 2-oxoethyl * )-2-oxo-2,3-dihydro-l/r-imidazoI-l-vHacetate
- Step B (l-oxo-S-phenylimidazolidin-l-vDacetic acid tert-butyl ester
- Step B Using the method of Example 4, Step B with the product of Example 10, Step A the title compound was obtained. Isolation was effected by silica gel chromatography (40:1 CH2Cl2/MeOH).
- Step B Using the method of Example 2, Step B and the product of Example 10, Step B the title compound was obtained.
- Step D l-(2-(4-r2-(3,5-di-? g rl-butyl-4-methoxyphenvI)-l,3-thiazol-4-vIlpiperidi ⁇ i-l-vI ⁇ -2-oxoethvI)- 3-phenylimidazolidin-2-one
- Step K 9Jy-fluoren-9-ylmethyl-4-(2-l3-tert-butyl-5-r(trinuoromethvnthiolphenyl>-l,3-thiazoI-4- vQpiperidine-l-carboxylate
- Step L 4-(2-(3-ter ⁇ '-butyl-5-r(trifluoroniethyl)thiolphenvO-l,3-thiazol-4-vnpiperidine
- Step M 3-f2-r4-(2-f3-terf-butyr-5-rftrifluoromethvnthiolphenvn-l,3-th ⁇ azol-4-vnpiperidin-l-yll-2-
- Step B (4-methoxy-l ⁇ T-iinidazol-l- ⁇ I)acetic acid methyl ester
- Step D 4-[2--S-di-tert-butyl- ⁇ inethoxyphenvn-l ⁇ -thiazoM-yn-l-g-rf ⁇ methoxy-l-iniidazol-l- vQacetyl 1 piperidine
- Step B 3-/grf-butyl-4f(3-methylbut-2-en-l-yl)oxylbenzoic acid methyl ester
- Step C 8-fer/-buty.-4,4-dimethyIchroina ⁇ e-6-carboxylic acid methyl ester
- Step A Using the method of Example 1, Step A and the product of Example 13, Step D (309mg; 1.20mmol) as starting material the title compound was obtained.
- Step H 9Jy-fluoren-9-ylmethyl4-r2-(8-fgrf-butvI-4,4-dimethyl-3,4-dihvdro-2 ⁇ r -chroinen-6-vn-l,3- thiazoI-4-yIlpiperidine-l-carboxvlate
- Step G Using the method of Example 1, Step G with the products of Example 1, Step C and Example 13, Step G as starting materials the title compound was obtained.
- Step J 4-r2-(8-/er/-butyl-4,4-diinethvI-3,4-diIivdro-2.-y-chromen-6-vn-1.3-thiazol-4-vIlpiperidine
- Step K 4-12-(8-ter/-butyl-4,4-dimethvL-3.4-dihvdro-2 J H r -chromen-6-vn-lJ-thiazol-4-vIM-r(2,4- dimethvI-177-imidazoI-l-vDacetvIlpiperidine
- Step B (4-chIoro-2-inethyl-l.H-imidazol-l-yl)acetic acid tert-butyl ester
- Step D l-[f4-chloro-2-methyI-l£?-iniidazol-l-vI)acctvIl-4-r2-f3,S-di-ferf-butvI-4-methoxyptienvn- 1.3-thiazol-4-yllpiper.dine
- Step F (4-chIoro-2,5-dimethyl-lJ? ' -iinidazol-l-vnacetic acid /erf-butyl ester
- Step G (4-chloro-2,5-dimethvI-l/r-imidazol-l-yl)acetic acid hydrotrifluoroacetate
- Step H l-[f4-ch1oro-2.5-dimethyl-l/r-imidazol-l-vnacetyll-4-r2-f3.5-di-terf-butyl-4- methoxyplienvI)-1..3-thiazol-4-vIlpiperidine
- Step D 9fl r -fluoren-9-ylmethvI-4-r2-f3.5-di-ferf-butylphenvn-1.3-thiazol-4-vIlpiperidine-l- carboxylate
- Step E 4-[2-f3,5-di-fer/-butylphenv ⁇ -1.3-thiazol-4-vIlpiperidine
- Step F l-(chIoroacetyl)-4-f2-(3.,5-di-ter/-butvIphenv ⁇ -1.3-thiazoI-4-yllpiperidine
- Step G 3-(2- ⁇ 4-f2-f3,5-di-rgrf-butylphenyl)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoethv ⁇ -3Jy- imidazof4,5-c]pyridine and l-(2- ⁇ 4-[2-(3,5-di-terf-butylphenvI)-l,3-thiazol-4-yllpiperidin-l-yl ⁇ -2- oxoethvO-l.fl r -iinidazof4,5-clpyridine
- Step B (3-ethyl-5-methyl-2-oxo-2,3-dihydro-liy-imidazol-l-yl)acetic acid ethyl ester
- Step D 3-f2-f4-f2-(3,5-di-/gr ⁇ -butyl-4-inethoxyphenv ⁇ -l,3-thiazol-4-vnpiperidin-l-vU-2-oxoethyl)- l-ethyl-4-methvI-l,3-dihvdro-2fl r -iinidazol-2-one
- Step B l-eth y l-S-methvI-l,3-dihvdro-2.H r -imidazoI-2-one
- Step C P-ethyl ⁇ -methyl-Z-oxo ⁇ -dihydro-liy-imidazot-l-vOacetic acid tert-butyl ester
- Step P (3-ethyl-4-methyl-2-oxo-23-dihvdro-l//-imidazol-l-yl)acetic acid
- Step E l-(2-(4-[2-(3,5-di-tert-but ⁇ I-4- ⁇ nethoxyphenyl)-13-thiazol-4-ylTpiperidin-l-vU-2-oxoethv ⁇ - 3-ethyl-4-methvI-l,3-dihvdro-2/7-imidazol-2-one
- Step F l-(2-f4-r2-(3,5-di-terif-butylphenvn-l,3-thiazoI-4-v ⁇ piperidin-l-vn-2-oxoethvn-3-ethyl-4- methyl-13-dihvdro-2/r-imidazoI-2-one
- Step B (2-oxo-23-dihvdro-l/- f -benzimidazol-l-v0acetic acid
- Step C l-f2- ⁇ 4-12-f3.5-di-ter/-butvI-4-methoxyphenvn-13-thiazol-4-vnpiperidin-l-vU-2-oxoethyl)- l,3-dihydro-2.fl-benzimidazol-2-one
- Step D C3-methyI-2-oxQ-2,3-dihvdro-lfl ' -benzimidazoI-l-vnacetic acid tert-butvl ester
- Step F l-f2-(4-r2-f3,5-di-fert-butyl-4-methoxyphenvn-13-thiazol-4-vnpiperidin-l-vI ⁇ -2-oxoethvn- 3-methyl-13-dihvdro-2/- r -benzimidazol-2-one
- Step A 2-ethyl-4-methyIimidazole as starting material the title compound was obtained.
- the title compound was purified by silica gel chromatography (preparative TLC; 100:2.5:1 CH2Cl2/MeOH/Et3N).
- Step B 2-ethyl-4-meth ⁇ I- 1H-imidazol-l-ylacetic acid hydrotrifluoroaeetate
- Step C 4-[2-(3,5-tert-butyl- ⁇ methoxyphenvn-l ⁇ -thiazoI- ⁇ vil-l-f ⁇ -ethvM-methyl-lJy- imidazol-l-vOaeetyllpiperidine
- Step D 9iH r -fluoren-9-ylmethyl-4-f2-f3,5-di-ter ⁇ '-butvI-4-hvdroxyphenv0-13-thiazol-4-yllpiperidine- 1-carboxvlate
- Step A r4-(hvdroxymethvi)-5-methyl-l/r-imidazol-l-v ⁇ acetic acid methyl ester and f5- fliydroxymethvD-4-methyl-l/j r -iniidazol-l-yl1acetic acid methyl ester
- Step B Using the method of Example 3, Step B and 4-(hydroxymethyl)-5-methyl-lH-imidazole as starting material the title compounds were obtained.
- the crude product was adsorbed onto silica gel and eluted without fractionation (10:1 C ⁇ 2Cl2 ⁇ MeO ⁇ ).
- Step B Potassium f4-fhvdroxymethvD-5-methyl-l/-r-imidazoI-l-v ⁇ acetate
- Step C fl-(2- ⁇ 4-r2-(3,5-di-fg/-f-but ⁇ -4-methoxyphenyl)-l,3-thiazol-4- ⁇ llpiperidin-l-vU-2-oxoethvn- 5-methyl-l//-imidazol-4-v.lmethanol
- Step C Using the method of Example 3, Step C and [5-(hydroxymethyl)-4-methyl-li/-imidazol-l-yl]acetic acid methyl ester (Example 23, Step A) as starting material the title compound was obtained.
- Step E ri-f2-(4-f2-f3,5-di-ferf-butyl-4-methoxyphenyl)-1.3-thiazol-4-v]lpiperidii ⁇ -l-vI>-2-oxoethvI>- 4-methyl-l//-imidazol-5-yl ⁇ methanoI
- Step B f2-(methylthiQ)-lJy-imidazol-l-yllacetic acid methyl ester
- Step C [2-(metlrylthioyi//-imidazol-l-yl]acetic acid hydrochloride
- Step D 4-r2-(3,5-di-terf-butyl-4-methoxyphenvn-l,3-thiazol-4-vIl-l-f2-(methylthio)-l J fi r -imidazoI-l- yll acetyl? piperid ine
- Step E fl-fmethylsulfonvD-lZr-imidazo.-l-v ⁇ acetic acid methyl ester
- Step F r2-fmethylsuIfonvD-l//-iinidazol-l-yl]acetie acid hydrochloride
- Step G 4-r2-f3.5-di-tert-butyI-4-niethoxyphenvn-l.,3-thiazoI-4-vIl-l- ⁇ 2-fmethvIsuIfonv ⁇ -lg- imidazol-l-yllacetyl ⁇ piperidine
- Step A /erf-butyl methyl 2,2'-(4-methyl-l.ff-imidazole-l,5-divDdiaeetate and fer/-butyl methyl 2,2 r - (S-methyl-l/T-imidazole-l ⁇ -diyDdiaeetate
- Step B r4-(2-methoxy-2-oxoethviy5-methyl-l/y-imidazoI-l-yllacetic acid hvdrotrifluoroacetate
- Example 25 Using the method of Example 2, Step B and fer/-butyl methyl 2,2'-(5-rnethyl-I//-imidazoIe-l ,4- diyl)diacetate (Example 25, Step A) as starting material the title compound was obtained.
- Step C fl-f2-f4-[2-(3,5-di-ter/-butvI-4-methoxyphenyl)-l ⁇ -thiazol-4-yllpiperidin-l-vU-2-oxoethv ⁇ - 5-meth ⁇ I-lJy-imidazoI-4-yllacetic acid methyl ester
- Step D fl-(2- ⁇ 4-r2-(3,5-di-fgrf-butyl-4-methoxyphenvn-l.,3-thiazol-4-yl]piperidi ⁇ i-l-vU-2-oxoethyl) 5-methyl-li?-iinidazol-4-vIlacetic dihydrochloride
- Step G ri-r2-f4-12-(3,5-di- ⁇ 'er/-butyl-4-methoxyphenv»-13-tfaiazoi-4-vHpiperidin-l-yl ⁇ -2-oxoethv ⁇ -
- Step A 4-f2-(3,5-di-/grf-butyl-4-methoxyphenvO-l ⁇ -thiazol-4-vIl-l-K3,5-dimethyI-l/f-l,2,4-triazol- 1-vQaeetyll piperidi ne
- Step A (4-methvI-l.H ' -imidazoI-l-yI)acetic acid tert-butyl ester and f5-methYl-l/T-imidazoI-l- vDacetic acid tert-butyl ester
- Step A with 4-methyl imidazole as starting material the title compounds were obtained.
- Step B (4-methyl-l//-imidazol-l-vI)aeetic acid hydrotrifluoroacetate
- Step C 4-r2-f3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yll-l-r(4-niethyl-l-i ⁇ nidazoI-l- vDacetyll piperidine
- Step E 4-[2-(3,5-di-tert-butvI-4-methoxyphenvn-13-thiazoI-4-vn-l-rf5-methyl-l-imidazoI-l- vDacetyllpiperidine
- Step B O-methvI-Z-oxoimidazolidin-l-vDacetic acid
- Step C l-(2-f4-f2-(3,S-di-terf-butyl-4-methoxyphenvO-l,3-thiazol-4-yllpiperidin-l-Yl ⁇ -2-oxoethvn-
- Step F 3-f2- ⁇ 4-r2-(3,5-di-/gr/-butvI-4-methoxyphenyl)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoeth ⁇ l)- 1 ,3-oxazolidin-2-one
- Step A 4-[2-(3,5-di-ter- ⁇ -butyl-4-methoxyphenyl)-1 ⁇ 3-thiazol-4-yll-l-r(3.5-dimethyl-lig-pyrazol-l- vDacetyllpiperidine
- Step B 4-r2-(3,5-di-fer/-butvphenvn-l,3-thiazol-4-ylM-r(3,5-dimethyl-l.H-pyrazol-l- yDacetyli piperidi ⁇ e
- Step B lH- -pyrrolo[2,3- ⁇ 1pyridin-l-ylacetic acid hvdrotrifluoroacetate
- Step C l-(2-(4-[2-f3,5-di--'g/ i -'-butyl-4-methoxyphenyl)-l,3-thiazol-4-vIlpiperidin-l-yl ⁇ -2-oxoethyl)- l J H r -pyrroloF23- ⁇ lpyridine
- Step B l-r2- ⁇ 4-r2-f3,5-di-ter/-butyl-4-methoxyphenylV13-thiazol-4-yllpiperidin-l-vU-2-oxoethvn- 2-methyl-l/f-benzimidazole
- Step C l-(2-f4-(2-(3-ter/-butyl-5-r(trifluoromethyl)thio1phenvU-l,3-thiazol-4-yl)piperidin-l- ⁇ U-2- oxoetliyU-l/J-benzimidazole
- Step D l-f2-I4-(2- ⁇ 3-tert-butyl-5-rftrifluoromethyl)thio1phenyl ⁇ -13-thiazol-4-vnpiperidin-l-yll-2- oxoethyl ⁇ -2-methyl-liJ r -benzimidazole
- Step B f5-methyl-3-(trifluoroniethyl)-l// ' -pyrazoI-l-vHacetic acid hydrotrifluoroacetate
- Step B Using the method of Example 2, Step B with the product of Example 32, Step A as the starting material the title compound was obtained.
- Step C 4-f2-(3,5-di-ferf-butylphenyl)-l 3-thiazoI-4-yl1-l- ⁇ r5-methv.-3-(trifluoromethyl)-Lg- pyrazol-1-vUacetvUpiperidine
- Step D l-fr3,5-bis(trifluoroinethvn-ljy-pyrazol-l-vnacetvU-4-f2-f3,5-di-/er/-butylphenyl)-l,3- thiazol-4-yl 1 piperid i ne
- Step F l-rf4-iodo-3,5-dimethyl-ljy-Pyrazol-l-vnacetv ⁇ -4-f2-(3.5-di-/gr/-butyIphenvn-13-thiazol-4- yllpiperidine
- Step B r ⁇ -oxoimidazolidin-l-vDacetic acid
- Step B Using the method of Example 2, Step B and the product of Example 33, Step A as the starting material the title compound was obtained.
- Step C l-(2- ⁇ 4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-13-thiazoI-4-yllpiperidin-l-vU-2- oxoethyl)imidazolidin-
- the silica gel was then eluted without fractionation (100:10:1 CH2Cl2/MeOH/Et3N) to recover the title compound.
- Step B Potassium r2-(hvdroxymethyl)-l//-imidazol-l-yllacetate
- Step C Using the method of Example 3, Step C and the product of Example 34, Step A as the starting material the title compound was obtained.
- Step C ll-(2- ⁇ 4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-1.3-thiazoI-4-vIlpiperidin-l- ⁇ yl>-2-oxoethvI>- l//-imidazol-2- ⁇ l] ⁇ nethanol
- Step B Using the method of Example 3, Step B and lH-benzimidazol-2-ylmethanol as the starting material the title compound was obtained as the sole product. Purification was performed by silica gel chromatography (preparative TLC; 20:1 CH2Cl2/MeOH).
- Step C Using the method of Example 3, Step C and the product of Example 34, Step D as the starting material the title compound was obtained.
- Step F fl-(2-(4-f2-(3,5-di-ter ⁇ 1 -butyl-4-methoxyphenyl)-l,3-thiazoI-4-vnpiperidin-l-vU-2-oxoethvn- liy-benzimidazoI-2-v ⁇ iethanol
- Step A 4-[2-(3.5-di-te/- ⁇ '-butvI-4-methoxyphenvI)-l,3-thiazoI-4-yll-l-(f2-(trifluoroinethvn-l/r- i midazol-l-yl lacetyl) piperid ine
- Step B 4-r2-r3.5-di-ter/-butyl-4-metho ⁇ yphenvn-l,3-thiazoI-4-vn-l-(r2-niethyl-l J H r -iinidazol-l- yli acetyl ⁇ pi perid ine
- Step C 4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-13-thiazol-4-yll-l-fl.fi r -imidazol-l- ylacetvDpiperidine
- Step B f5-methyl-l//-pyrazol-3-yI)acetic acid methyl ester
- Step C benzyl methyl 2,2 > -f5-methyl-Lfl r -pyrazole-l,3-diyl)diaeetate
- Step L Using the method of Example 1, Step L with the products of 15, Step E and Example 36, Step D as the starting materials the title compound was obtained.
- Step F fl-(2- ⁇ 4-r2-f3,5-di-/grf-butylphenv ⁇ -l,3-thiazol-4-yl
- Step C Using the method of Example 3, Step C with the product of Example 36, Step E as starting material the title compound was obtained. Isolation was effected by partitioning the reaction mixture between isopropyl acetate and ⁇ H4 phthalate buffer. The organic was dried over MgS ⁇ 4, filtered and evaporated.
- Step G fl-l2-r4-f2-0-ferf-butyl-5-rrtrifluoromethvnthiolphenyl ⁇ -1.3-thiazol-4-yl)piperidin-l-vIl-2- oxoethvU-5-methyl-l/ir-pyrazol-3-yl)acetic acid methyl ester
- Step H Potassium fl-f2-F4-(2- ⁇ 3-fer/-butvI-5-f(trifluoromethyl)thiolphen ⁇ l>-l,3-thiazol-4- vDpiperidin-l-v ⁇ -2-oxoeth ⁇ I)-5-methyI-l/j r .-pyrazol-3- ⁇ l)acetate
- Step A 4-f2-(3.5-di-ter/'-butylphenvn-l,3-thiazol-4-vn-l-rf2,4-dimethvi-l£r-iinidazol-l- vOacetyllpiperidine
- Step B l-f2-f4-r2-(3.5-di-ter/-butylphenvn-13-thiazol-4-vnpiperidin-l-yll-2-oxoethvn-3-ethvI-l,3- dihydro-2/?-imidazol-2-one
- Step D 9 ⁇ r-fluoren-9-vImethvI4- ⁇ 2-f3,5-bis(trifluoroinethyl)phenv ⁇ -1.3-thiazol-4-vI>piperidine-l- carboxylate
- Step G Using the method of Example 1, Step G with the products of Example 1, Step C and Example 38, Step C as the starting materials the title compound was obtained.
- Step E 4-f2-f3,5-bisftrifluoromcthyl)phenv ⁇ -13-thia2oI-4-vUpiperidine
- Step F 3-f2-(4-f2-r3,5-bisftrifluoro ⁇ nethvnphenvn-l,3-thiazol-4-vUpiperidin-l-vI)-2-oxoethyll-3/f- imidazof4,5-A]pyridine
- Step G 4-(2-r3,5-bisftrifluoromethvnphenyll-1.3-thiazol-4-yl)-l-rf3 ⁇ 5-dimethyl-l/r-1.2,4-triazol-l- vDacetylipiperidine
- Step H 4-
- Step J 3-[2-f4- ⁇ 2-r3.5-bisftrifluoromethyl)phenvI1-1.3-thiazol-4-vUpiperidin-l-yl)-2-oxoethvn-3Jy- imidazof4,5-cl pyridine and 142-(4-f243,5-p.s(trifluoromethy0pheny ⁇ -13-thiazoI-4-vUpiperidin- l-yl)-2-oxoethy ⁇ -liy-imidazo[4,5-clp-yridine
- Step K l-f2-(4-(2-f3.5-bisftrifluoromethyl)phenvU-l,3-thiazoI-4-vUpiperidin-l-vn-2-oxoethylI-2- methyl-lg-iinidazo[4.5-clpyridine
- Step B f2-(ethoxycarbonyl)-l ⁇ r-imidazol-l-vU acetic acid hvdrotrifluoroacetate
- Step B Using the method of Example 2, Step B with the product of Example 39, Step A as the starting materia! the title compound was obtained.
- Step C l-f2-l4-r2-f3.5-di-fer/-butvI-4-methoxyphenyl)-1.3-thiazol-4-vIlpiperidin-l-vU-2-oxoethvO- l/7-imidazoIe-2-carboxyHc acid ethyl ester
- Step D l-(2-(4-r2-(3,5-di-ferf-butyl-4-methoxyphenyl)-13-thiazoI-4-vnpiperidin-l-vU-2-oxoethvI)- lH-imidazole-2-carboxylic acid dihvdrochloride
- Step C l-ethyl-3- ⁇ 2-oxo-2-14-(2-phenvI-l,3-thiazoI-4-vnpiperidin-l-vnethvU-l,3-dih ⁇ dro-2/r- imidazol-2-one
- Step D 9H-fl uo ren-9-yI methyl 4-[2-(4-methoxyphenv-)-l,3-thiazol-4-yI]piperidine-l-carboxvlate
- Step E 4-[2-(4-methoxyphenyl)-l.,3-thiazol-4-yllpiDeridine
- Step F l-ethyl-3-(2-f4-f2-f4-methox ⁇ phenyl)-l.,3-thiazol-4-vHpiperidin-l-yl ⁇ -2-oxoethv ⁇ -l,3- dihydro-2//-irnidazol-2-one
- Step G 9./y-fluoren-9-ylmethvI 4-f2-(4-ferf-butyIphenvO-1.3-thiazol-4-v ⁇ piperidine-l-carboxvlate
- Step J l-(2-
- Step K 9/7-fluoren-9-ylinethvI 4-f2"(4-chlorophenylM,3-thiazoI-4-yl1piperidine-l-carboxylate
- Step M l-(2-H-r2-(4-chIorophenvI)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoethyl)-3-ethyl-13- dihydro-2 J H r -imidazol-2-one .
- Step N 9// r -fluoren-9-ylmethy.4-f2-(4-bromophenyI)-1.3-thiazoI-4-vHpi ⁇ eridine-l-carboxvlate
- Step G 4-f2-f4-bromophen ⁇ -l,3-thiazol-4-yl1piperidine
- Step Q l-(2-f4-f2-f4-bromophenv0-13-thiazol-4-yllpiperidin-l-yl>-2-oxoethvI)-3-ethyl-13- dihydro-2//-imidazol-2-one
- Step R g/Z-fluoren-P-ylmethyl 4-f2-(4-trifluoromethylphenvO-l,3-thiazol-4-y ⁇ piperidine-l- carboxylate
- Step S 4-[2-f4-trifluorometh ⁇ lphenvI)-13-thiazol-4-vl]piperidine
- Step T l-f2-f4-r2-f4-trifluoromethylphenyl>-l,3-thiazol-4-vHpiperidin-l-yl ⁇ -2-oxoethyl)-3-eth ⁇ l- 13-dihvd ro-2iy-imidazol-2-one
- Step W l-(2-(4-F2-(3-chlorophenvn-l,3-thiazoI-4-yIlpiperidin-l-vn-2-oxoethvn-3-ethyl-l,3- dihydro-2jy-imidazoI-2-one
- Step X 9iy-fluoren-9- ⁇ lmethyI 4-f2-(3-bromophenyl)-13-thiazol-4-v ⁇ piperidine-l-carboxvlate
- Step Z l-(2- ⁇ 4-f2-f3-bromophenv ⁇ -1.3-thiazol-4-vIlpiperidin-l- ⁇ U-2-oxoethyl)-3-ethyi-13- dihvdro-2i7-imidazol-2-one
- Step AA 9/f-fIuoren-9-'ylinethyl 4-[2-(3-trifluoromethylphenyiyi,3-thiazol-4-vI1piperidine-l- carboxvlate
- Step BB 4- [ 2- ( 3-trifluoromethvlphenvlM3-thiazol-4- ⁇ -l p i p eridine
- Step H Using the method of Example 1, Step H with the product of Example 40, Step AA as starting material the title compound was obtained.
- Step CC l-(2-(4-f2-(3-trifluoromethylphenvI)-l,3-thiazol-4-yl1piperidin-l-vU-2-oxoethylV3-ethyl- l.S-dihvdro-l/y-imidazol- ⁇ -one
- Step DD 9g-fluoren-9-ylmethyl 4-f2-f3,5-dichlorophenyl)-l,3-thiazoI-4-yllpiperidine-t- carboxylate
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Immunology (AREA)
- Transplantation (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
The invention encompasses compounds of Formula I or pharmaceutically acceptable salts thereof, which are modulators of the CXCR3 chemokine receptor function useful for the treatment or prevention of pathogenic inflammatory processes, autoimmune diseases or graft rejection processes. Methods of use and pharmaceutical compositions are also encompassed.
Description
TITLE OF THE INVENTION
2-ARYLTHIAZOLE DERTV ATIVES AS CXCR3 RECEPTOR MODULATORS
BACKGROUND OF THE INVENTION The chemokines are a family of small (70-120 amino acids), pro-inflammatory cytokines, with potent chemotactic activities. As their name implies, one function of chemokines, which are released by a wide variety of cells at sites of inflammation, is to attract leukocytes , including monocytes, macrophages, T lymphocytes, eosinophils, basophils and neutrophils and to promote their migration through endothelial layers, (reviewed in Schall, Cytokine. 3, 165-183 (1991) and Murphy, Rev. Immun.. 12, 593-633 (1994)). In addition to their well characterized role in leukocyte trafficking, it is now also appreciated that that chemokines play a role in a number of other biological processes including cellular proliferation, hematopoiesis, angiogenesis, tumor metastasis and host defense.
These polypeptides were originally defined as having four conserved aminoterminal cysteines , and divided into two major and two minor subfamilies based on the spacing arrangement of the first cysteine pair. The two major subfamilies consist of the CXC (or α) and CC (or β) chemokines. In the CXC-chemokine family, which includes CXCLl (MGSA or GROα), CXCL7 (NAP-2), CXCL8 (interleukin-8 or 1L-8), CXCL9 (MIG), CXCLlO (lP-10) and CXCLl 1 (I-TAC), these two cysteines are separated by a single amino acid, while in the CC-chemokine family, which includes CCL5 (RANTES), CCL2 (monocyte chemotactic protein- 1 or MCP-I), CCL8 (MCP-2), CCL7 (MCP-3), CCL3 (MEP- lα), CCL4 (MIP- IB) and CCLl 1 (eotaxin), these two residues are adjacent.
Some CXC-chemokines, such as CXCLl , CXCL7 and CXCL-8 are chemotactic primarily for neutrophils while another subset of CXC chemokines , including CXCL9, CXCL10 and CXCLl 1, are chemotactic primarily for T- lymphocytes. In comparision, the CC_chemokines, such as CCL5, CCL3, CCL4, CCL2, CCL8, CCL7and CCLl 1 , are more broad in their action and are chemotactic for macrophages, monocytes, T- lymphocytes, eosinophils and basophils (Deng, et al., Nature. 381, 661-666 (1996), Murphy et al. Pharmacol Revw. 52(1) 145-176, (2000).).
The chemokines bind to specific G-protein coupled receptors (GPCRs) present on leukocytes and other cells, (reviewed in Horuk, Trends Pharm. Sci., 15, 159-165 (1994), Murphy et al. Pharmacol Revw. 52(1) 145-176, (2000).) Upon interaction with their cognate ligands, chemokine receptors transduce an intracellular signal though their associated heterotrimeric G proteins, resulting in a rapid cellular responses, including an increase in intracellular calcium concentration. These chemokine receptors form a sub-family of GPCRs, which, at present, consists of a number of well characterized members with known ligands as well as a number of orphans. Unlike receptors for promiscuous classical chemoattractants such as C5a, fMLP, PAF, and LTB4, chemokine receptors are more selectively
expressed on subsets of leukocytes. Thus, generation of specific chemokines provides a mechanism for recruitment of particular leukocyte subsets. The restricted expression and defined function of the chemokine receptors has focused attention on intervention in the chemokine signaling pathways as a method for highly selective intervention in pathological immunological and inflammatory processes. Chemokine receptors, such as CCRl , CCR2A, CCR2B, CCR3, CCR4, CCR5, CXCR3,
CXCR4, have been implicated as important mediators of inflammatory diseases and immunoregulatory disorders, including asthma, allergic rhinitis and and atherosclerosis. They are also purported to play a role in the pathogenesis of autoimmune disorders such as rheumatoid arthritis, psoriasis, multiple sclerosis. An extensive review of the role of chemokines in disease is provided by in Seminars in Immunology.. 15(1), 1-55 (2003).
A subset of chemokines are potent chemoattractants for lymphocytes. For example CXCR3 (CD 183) is expressed in activated T lymphocytes, some B lymphocytes and NK cells. Expression and receptor responsiveness are both increased by activation of the T lymphocytes. The potent inflammatory cytokines CXCLlO and CXCLl t are chemoattractant for T lymphocytes and tumor infiltrating lymphocytes. The relatively restricted expression of the CXCR3 expression on these proinflammatory cell types mark CXCR3 as a very promising target for selective intervention in the inflammatory process. A connection with disease processes, particularly Th-I mediated processes, is indicated by the presence of the CXCR3 on most activated T lymphocytes within inflamed joint synovium in rheumatoid arthritis as well as within inflamed tissue present in other inflammatory disorders including ulcerative colitis, Graves' disease, MS and rejecting graft tissues. (Oin. J. Clin.
Invest.. 101 (4), 746-754 (1998), Garcia-Lopez, Lab. Investig. 81(3), 409-418 (2001), Balashov, PNAS. 96, 6873-6878 (1999), DeVries, Seminars in Immunology, 15(1), 33-48 (2003)) A similar but somewhat less pronounced association is shown with the CCR5 receptor and its ligand CCL5
Accordingly, agents which inhibit or modulate the function of chemokine receptors such as the CXCR3 receptor would be useful in treating or preventing such disorders and diseases. Data from animal models of inflammation further supports the hypothesis regarding the effectiveness of chemokine blockade, specifically CXCR3 inhibition, in diseases with clear T -lymphocyte mediated tissue damage such as transplant rejection, graft versus host disease, multiple sclerosis, optic neuritis and rheumatoid or psoriatic arthritis. Many other diseases are characterized by T lymphocyte infiltrates, and by inference are therefore also good candidates for interventions which prevent the migration of T lymphocytes. These diseases include psoriasis and other chronic inflammatory diseases of the skin such as atopic dermatitis, lichen planus and bullous pemphigoid, inflammatory bowel diseases such as ulcerative colitis and Crohn's disease and autoimmune diseases such as systemic and cutaneous lupus erythematosus, Behcet's disease, type \ diabetes or Graves' disease.
Many inflammatory lung diseases such as chronic obstructive pulmonary disease, hypersensitivity pneumonitis, chronic eosinophilic pneumonia, pulmonary sarcoidosis, bronchiolitis obliterans syndrome, asthma, kidney diseases such as glomerulonephritis, pathogenesis of chronic HCV infection and atherosclerosis show a dependence on T lymphocytes and are promising targets for agents which modulate the function of chemokine receptors such as the CXCR3 receptor.
The expression of CXCR3 in some B cell tumors indicates that intervention in CXCR3 function could have beneficial effects in these cancers, particularly in suppressing metastasis.
Several methods are under investigation for modulation of chemokine receptor function. These include antibodies binding to and neutralizing the chemokine ligands, antibodies binding to and modulating the function of the chemokine receptors and small molecules which bind to and inhibit function of the chemokine receptor. The ideal method for intervention in CXCR3 mediated chemotaxis is the binding of orally bioavailable small molecules which prevent the function of the receptor. Molecules with affinity for the CXCR3 chemokine receptor and ability to modulate the function of the receptor are described here.
SUMMARY OF THE INVENTION
The invention encompasses compounds of Formula I

DETAILED DESCRIPTION OF THE INVENTION
The invention encompasses a genus of compounds of Formula I

I or a pharmaceutically acceptable salt thereof, wherein:
D is CR4 or N;
R3 is selected from the group consisting of: H, halo, C]_4alkyl, -CF3, -OCF3 and -S(O)nCF3, wherein n is 0 or 2;
R4 is selected from the group consisting of: H, halo, -OH5 Ci_4alkyl, -OCH3, -OCH2CF3 and -CF3;
or R3 and R4 may be joined together with the carbon atoms to which they are attached to form a five- or six-membered monocyclic ring, said rings containing oxygen or tetra-substituted with methyl groups as follows:

R5 is selected from the group consisting of: -H5 halo, Ci_4alkyl, C3_6cycloalkyl, CF3, -CF2CH3, -OCF3 and -SCF3;
R-6 is selected from the group consisting of — H and -OCH3,
or R5 and Rβ may be joined together with the carbon atoms to which they are attached to form a monocyclic 5-membered ring, said ring di-substituted with methyl as follows:

is a 5 membered non-aromatic or aromatic ring or a 9 membered fused bicyclic partially
 aromatic or aromatic ring, each ring containing at least 1 nitrogen atom and optionally up to 3 additional heterotaoms selected from S, O and N, said rings optionally substituted with 1 to 3 substituents independently selected from the group consisting of: oxo, hydroxy, carboxy, -CF3, halo, -S(O)p-CH3, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy; and
aromatic or aromatic ring, each ring containing at least 1 nitrogen atom and optionally up to 3 additional heterotaoms selected from S, O and N, said rings optionally substituted with 1 to 3 substituents independently selected from the group consisting of: oxo, hydroxy, carboxy, -CF3, halo, -S(O)p-CH3, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy; and
p is 0, 1 or 2.
Within this genus, the invention encompasses a sub-genus of compounds of Formula I wherein:
is selected from the group consisting of:


R"2, R"3, R"4 and R"5 are independently selected from the group consisting of: -H, carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, C]-3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy,
R"6 is H or OH, and
is an optional double bond.
Also within this genus, the invention encompasses a sub-genus of compounds of Formula I wherein D is N.
Also within this genus, the invention encompasses a sub-genus of compounds of Formula
I wherein D is CR4.
Within this sub-genus, the invention encompasses a class of compounds of Formula I wherein:
R-3 and R4 are joined together with the carbon atoms to which they are attached to form a six-membered monocyclic ring as follows:

Also within this sub-genus, the invention encompasses a class of compounds of Formula I wherein:
R3 is selected from the group consisting of: H, halo, Ci_4alkyl and -CF3;
R4 is selected from the group consisting of: H, halo, Ci_4alkyl, -OCH3 and -CF3;
R5 is selected from the group consisting of: H, halo, Ci_4alkyl, CF3 and -SCF3; and
R6 is H.
Within this class, the invention encompasses a sub-class of compounds of Formula I wherein:
R3 is selected from the group consisting of: H, Cl, Br, tert-buty\ and -CF3;
R4 is selected from the group consisting of: H, Cl, Br, tert-buty], -OCH3 and -CF3;
R5 is selected from the group consisting of: H, CI, Br, tert-bυtyl, CF3 and -SCF3; and
R6 is H.
Another embodiment of the invention encompasses a sub-genus of compounds of Formula I within the above-described genus wherein Rβ is -H.
Another embodiment of the invention encompasses a sub-genus of compounds of Formula 1 within the above-described genus wherein:
D is CR4;
R3 is selected from the group consisting of: H, Cl, Br, tert-butyl and -CF3;
R4 is selected from the group consisting of: H, Cl, Br, tert-butyl, -OCH3 and -CF3;
or R3 and R4 may be joined together with the carbon atoms to which they are attached to form a six- membered monocyclic ring as follows:

R5 is selected from the group consisting of: H, Cl, Br, tert-buty\, CF3 and -SCF3; and
R6 is -H.
Within this sub-genus, the invention encompasses a class of compounds of Formula I wherein:
(1) R3 is /erf-butyl, R4 is H and R5 is tert-butyl;
(2) R3 is tert-butyl, R4 is -OCH3 and R5 is tert-butyl;
(3) R3 is -CF3, R4 is -H and R5 is -CF3; (4) R3 is H, R4 is -OCH3 and R5 is H;
(5) R3 is H, R4 is tert-butyl and R5 is H;
(6) R3 is H, R4 is Cl and R5 is H;
(7) R3 is H, R4 is Br and R5 is H;
(8) R3 is H, R4 is -CF3 and R5 is H; (9) R3 is H, R4 is H and R5 is Cl;
(10) R3 is H, R4 is H and R5 is Cl;
(11) Ra is H, R4 is H and R5 is -CF3; or
(12) R3 is Cl, R4 is H and R5 is Cl.
Also within this sub-genus, the invention encompasses a class of compounds of Formula
I wherein
is selected from the group consisting of:


R"2, R"3, R"4 and R"5 are independently selected from the group consisting of: -H, carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, Ci_3alkoxy and Ci_3alkyl, said C]_3alkyl optionally substituted with carboxy or hydroxy,
R"6 is H or OH, and
is an optional double bond.
Within this class, the invention encompasses a sub-class of compounds of Formula I wherein:

Also within this sub-genus, the invention encompasses a class of compounds of Formula I wherein:
D is CR4;
R3 is tert-buty\;
R4 is Hor-OCH3;
Rs is tert-buty\; and
R-6 is H.
In another embodiment, the invention encompasses a compound selected from the following group:
(1) 3-(2-{4-[2-(3,5-dUerf-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3H-imidazo[4,5-6]pyridine;
(2) 4-[2-(355-di-/e^-butyI-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(2,5-dimethyl-lH-imidazol- l-yl)acetyl]piperidine;
(3) [1 -(2- {4-[2-(3,5-di-ter/-butyl-4-methoxyphenyl)-l ,3-thiazol-4-yl]piperidin- 1 -yl}-2- oxoethyl)-2-methyi-lH-imidazol-5-yl]rnethanol;
(4) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3-methyl-l,3-dihydro-2H-imidazol-2-one;
(5) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)- 1 ,4-dimethyl-l ,3-dihydro-2H-imidazol-2-one;
(6) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-4-methyl-lH-pyrazol-3-yl]acetate bis(hydrotrifluoroacetate);
(7) 4-[2-(3,5-di-tert-buty 1-4-methoxyphenyl)- 1 ,3-th iazol-4-yl]- 1 -{ [4-methyl-2- (methylsulfonyl)- lH-imidazol- 1 -yl]acetyl}piperidine;
(8) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l ,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyI)-l,3-dihydro-2H-imidazol-2-one;
(9) Potassium_[3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l- yl}-2-oxoethyl)-2-oxo-2,3-dihydro- lH-imidazol- l-yl]acetate;
(10) l-(2-{4-[2-(3,5-di-tert-butyI-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3-phenylimidazolidin-2-one;
(11) 3-{2-[4-(2-{3-tert-butyl-5-[(trifluoroniethyl)thio]phenyl}-l,3-thiazol-4-yl)piperidin-l- yl]-2-oxoethyl}-3H-imidazo[4,5-δ]pyridine;
(12) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thϊazol-4-yl]-lH-[(4-methoxy-l-imidazol-l- yl)acetyl]piperidine;
(13) 4-[2-(8-tert-butyl-4,4-dimethyl-3,4-dihydro-2H-chromen-6-yl)-l,3-thiazol-4-yl]-l-[(2,4- dimethyl-lH-imidazol-l-yl)acetyl]piperidine;
(14) l-[(4-chloro-2,5-dimethyl-l/-'-imidazol-l-yl)acetyl]-4-[2-(3,5-di-/er/-butyl-4- methoxyphenyl)-l,3-thiazol-4-yl]piperidine;
(15) 3-(2-{4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3H- imidazo[4,5-c]pyridine;
(16) l-(2-{4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-lH- imidazo[4,5-c]pyridine;
(17) 3-(2-{4-[2-(3,5-di-tert -butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3H- imidazo[4,5-6]pyridinel;
(18) l-(2-{4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyI)-lH- imidazo[4,5-Z>]pyridine;
(19) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazoI-4-yl]piperidin-l-yl}-2- oxoethyl)- 1 -ethyl-4-methyl- 1 ,3-dihydro-2H-imidazol-2-one;
(20) l-(2-{4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]ρiperidin-l-yl}-2-oxoethyl)-3-ethyl- 4-methyl-l,3-dihydro-2H-imidazol-2-one;
(21) 1 -(2- {4-[2-(3,5-di-tert-buty 1-4-methoxyphenyl)- 1 ,3-thiazol-4-yl]piperidin- 1 -yl} -2- oxoethyl)-3-methyl-l,3-dihydro-2H-benzimidazol-2-one;
(22) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(2-ethyl-4-methyl-lH- imidazol-l-yl)acetyl]piperidine;
(23) 2,6-di-tert-butyl-4-{4-[l-(l /^-imidazo^^-cJpyridin-l-ylacetylpiperidin^-yl]-!^- thiazol-2-yl}phenoI;
(24) 2,6-di-tert-butyl-4-{4-[l-(3H-innidazo[4,5-c]pyridin-3-ylacetyl)piperidin-4-yl]-l,3- thiazol-2-yl}phenol;
(25) 2,6-di-tert-butyl-4-{4-[l-(3H-imidazo[4,5-δ]pyridin-3-ylacetyl)piperidin-4-yl]-l,3- thiazol-2-yl}phenol;
(26) 2,6-di-tert-butyl-4-(4-{l-[(3,5-dimethyl-lH-l,2,4-triazol-l-yl)acetyl]piperidin-4-yl}-l,3- thiazol-2-yl)phenol;
(27) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-4-methyl-lH-irnidazoI-5-yl]rnethanol;
(28) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-{2-(methylthio)-lH- imidazol-l-yl]acetyl}piperidine;
(29) 4-[2-(355-di-tert-butyl-4-methoxyphenyI)-l,3-thiazol-4-yl]-l-{2-(methylsulfonyl)-lH- imidazol-l-yl]acetyl}piperidine;
(30) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl) 5-methyl-lH-imidazol-4-yl]acetic dihydrochloride;
(31) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyρhenyl)-l,3-thiazol-4-yl]piperidin-l-y]}-2- oxoethyl^-methyl-ljff-imidazol-S-yljacetic dihydrochloride;
(32) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyl-lH-l,2,4- triazol-l-yl)acetyl]piperidine;
(33) 4-[2-(3,5-di-tert-butylphenyl)- 1 ,3-thiazol-4-yl]- 1-[(3 ,5-dimethyl- IH- 1 ,2,4-triazol- 1 - yl)acetyl]piperidine;
(34) 4-[2-(3J5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(4-methyl-l-imidazol-l- yl)acetyl]piperidine;
(35) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l33-thiazol-4-yl]-l-[(5-methyl-l-imidazol-l- yl)acetyl]piperidine;
(36) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3-methylimidazolidin-2-one;
(37) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-l ,3-oxazolidin-2-one;
(38) 4-[2-(3;5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyl-lH:-pyrazol- l-yl)acetyl]piperidine;
(39) 4-[2-(3,5-di-tert-butyphenyl)-l,3-thiazol-4-yl]-l-[(3:>5-dimethyl-lH-pyrazol-l- yl)acetyl]piperidine;
(40) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l ,3-thiazoI-4-yl]piperidin-l-yl}-2- oxoethyl)-lH-pyrrolo[253-6]pyridine;
(41) l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazoI-4-yl)piperidin-l- yl]-2-oxoethyl}-lH-pyrrolo[2,3-ό]pyridine;
(42) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-lH-benzimidazole;
(43) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l :>3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-2-methyl-l H-benzimidazole;
(44) 2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l53-thiazol-4-yl)piperidin-l-yl]-2- oxoethyl}-lH-benzimidazole;
(45) l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazol-4-yl)piperidin-l- yl]-2-oxoethyl}-2-methyl-lH-benzimidazole;
(46) 4-[2-(3 ,5-di-tert-butylphenyl)- 1 ,3-thiazol-4-yl]- 1- { [5-methyl-3-(trifluorornethyI)- 1 H- pyrazol- 1 -y 1] acetyl) piperidine;
(47) l-iP^-bis^ifluoromethyO-lH-pyrazol-l-y^acety^^-^^^-di-tert-butylphenyO-l^- thiazol-4-yl]piperϊdine;
(48) 4-bromo-3,5-dimethyl-li7-pyrazol-l-yl)acetyl]-4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol- 4-yl]piperidine;
(49) l-[(4-iodo-3,5-dimethyl-lH-pyrazol-l-yl)acetyl]-4-[2-(3,5-di-tert -butylphenyl)-l,3- thiazol-4-yl]piperidine;
(50) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l53-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)imidazolidin-2-one;
(51) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l:,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-lH-imidazol-2-yl]metanol;
(52) [l-(2-{4-[2-(3,5-dϊ-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl>-2- oxoethyl)-lH-benzimidazol-2-yl]metanol;
(53) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l ,3-thiazol-4-yl]-l-{[2-(trifluoromethyl)-lH- imidazol- 1 -yljacetyl} piperϊdine;
(54) 4-[2-(335-di-tert-butyl-4-methoxyphenyl)-lJ3-thiazol-4-yl]-l-{[2-methyl-lH-imidazol-l- yl]acetyl}piperidine;
(55) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-(lH-imidazol-l- ylacetyl)piperidine;
(56) [l-(2-{4-[2-(3,5-di-tert-butylphenyl)-l ,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-5- methyl- 1 H-pyrazol-3 -yl]acetic acid;
(57) Potassium (l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazol-4- yl)piperidin- 1 -yl]-2-oxoethyl} -5-methyl- lH-pyrazol-3-y I)acetate;
(58) 4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yI]-l-[(2,4-dimethyl-lH-imidazol-l- yl)acetyl]piperidine;
(59) l-(2-{4-[2-(3,5-di-rer/-butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethy!)-3-ethyl- 1 ,3 -dihydro-2H-imidazol-2-one;
(60) 3-[2-(4-{2-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]- 3H-imidazo[4,5-δ]pyridme;
(61) 4-{2-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-4-yl}-l-[(3,5-dimethyl-lH-l>2,4- triazol-l-yl)acetyl]piperidine;
(62) 3-[2-(4-{2-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]-
3H-imidazo[4,5-c]pyridine;
(63) l-[2-(4-{2-[3,5-bis(trifluoromethyl)phenyl]-l,3-thiazol-4-yl}ρiperidin-l-yl)-2-oxoethyl]- lH-imidazo[4,5-c]pyridine;
(64) l-[2-(4-{2-[3,5-bis(trifluoromethyl)phenyI]-l:,3-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]- 2-methyl-lH-imidazo[4,5-c]pyridine;
(65) l-(2-{4-[2-(3,5-di-ferf-butyl-4-methoxyphenyl)-l,3-thiazol-4-yI]piperidin-l-yl}-2- oxoethyl)-lH-imidazo!e-2-carboxylic acid dihydrochloride;
(66) l-ethyl-3-{2-oxo-2-[4-(2-phenyl-l ,3-thiazol-4-yl)piperidin-l-yl]ethyl}- l,3-dihydro-2i7- imidazol-2-one;
(67) l-ethyl-3-(2-{4-[2-(4-methoxyphenyl)-l,3-thiazol-4-yl]ρiperidin-l-yt}-2-oxoethyl)-l,3- dihydro-2H-imidazol-2-one;
(68) l-(2-{4-[2-(4-/ert-butylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-l ,3- dihydro-2H-imidazol-2-one;
(69) l-(2-{4-[2-(4-chlorophenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-l,3- dihydro-2H-imidazol-2-one;
(70) l-(2-{4-[2-(4-bromophenyI)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-l ,3- dihydro-2H-imidazol-2-one;
(71) l-(2-{4-[2-(4-trifluoromethylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3- ethyl- 1 ,3-dihydro-2H-imidazol-2-one;
(72) l-(2-{4-[2-(3-chlorophenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-lJ3- dihydro-2H-imidazol-2-one;
(73) 1 -(2- {4-[2-(3 -bromopheny I)- 1 ,3 -thiazol-4-y 1] piperidin- 1 -y 1 } -2-oxoethy l)-3 -ethyl- 1,3- dihydro-2H-imidazol-2-one;
(74) l-(2-{4-[2-(3-trifluoromethylphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3- ethyl-l,3-dihydro-2H-imidazol-2-one;
(75) l-(2-{4-[2-(335-dichlorophenyl)-l,3-thiazol-4-yl]piperϊdin-l-yI}-2-oxoethyl)-3-ethyl-l,3- dihydro-2H-imidazol-2-one; and
(76) l-ethyl-3-{2-oxo-2-[4-(2-pyridin-4-yl-l,3-thiazol-4-yl)piperidϊn-l-yl]ethyl}-l ,3-dihydro- 2H-imidazol-2-one.
The invention also encompasses a pharmaceutical composition comprising a compound of Formula I in combination with a pharmaceutically acceptable carrier.
The invention also encompasses a method for treating a disease or condition mediated by the CXCR3 chemokine receptor comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound of Formula I.
The invention also encompasses a method for treating a disease or condition mediated by the CXCR3 chemokine receptor comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound of Formula T, wherein the disease or condition is selected from the group consisting of: acute and chronic transplant rejection, psoriasis, rheumatoid arthritis and multiple sclerosis.
The term "halogen" or "halo" includes F, Cl, Br, and I.
The term "alkyl" means linear or branched structures and combinations thereof, having the indicated number of carbon atoms. Thus, for example, Ci-6alkyl includes methyl, ethyl, propyl, 2- propyl, s- and t-butyl, butyl, pentyl, hexyl and 1,1-dimethylethyl. The term "cycloalkyl" means mono-, bi- or tri-cyclic structures, optionally combined with linear or branched structures, having the indicated number of carbon atoms. Examples of cycloalkyl groups include cyclopropyl, cyclopentyl, cycloheptyl, adamantyl, cyclododecylmethyl, 2-ethyl-l- bicyclo[4.4.0]decyl, cyclobutylmethyl, cyclopropylmethyl 1-methylcyclopropyl and the like.
Optical Isomers - Diastereomers - Geometric Isomers - Tautomers
Some of the compounds described herein may exists as mixtures of tautomers. The terra "tautomers" embraces the standard meaning of the term, i.e. a type of isomerism in which two or more isomers are rapidly interconverted so that they ordinarily exist together in equilibrium. Tautomers include, e.g., compounds that undergo facile proton shifts from one atom of the compound to another atom of the compound. Some of the compounds described herein may exist as tautomers with different points of attachment of hydrogen. Such an example might be a ketone and its enol form known as keto- enol tautomers or an amide and its hydroxy imine tautomer. The individual tautomers of the compounds of Formula I, as well as mixtures thereof, are included in the scope of this invention. By way of illustration, tautomers included in this definition include, but are not limited to:

Salts
The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'- dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine,
ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, moφholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like. When the compound of the present invention is basic, salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids. Such acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like. Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, and tartaric acids.
It will be understood that, as used herein, references to the compounds of Formula I are meant to also include the pharmaceutically acceptable salts.
Utilities
The compounds of the present invention are modulators of CXCR3 chemokine receptor function and are of use in antagonizing chemokine mediated cell signalling and in particular are of use in the prophylaxis and/or treatment of diseases or disorders involving inappropriate T-cell trafficking. The invention extends to such a use and to the use of the compounds of Formula I for the manufacture of a medicament for treating such diseases and disorders. Particular diseases include inflammatory, autoimmune and immunoregulatory disorders.
In addition to primates, such as humans, a variety of other mammals can be treated according to the method of the present invention. For instance, mammals including, but not limited to, cows, sheep, goats, horses, dogs, cats, guinea pigs, rats or other bovine, ovine, equine, canine, feline, rodent or murine species can be treated. However, the method can also be practiced in other species, such as avian species (e.g., chickens).
Diseases or conditions of humans or other species which can be treated with compounds of Formula I, include, but are not limited to: autoimminue mediated inflammatory or allergic diseases and conditions, including respiratory diseases such as asthma, particularly bronchial asthma, systemic lupus erythematosus, ankylosing spondylitis, systemic sclerosis, autoimmune diseases, such as rheumatoid arthritis, psoriatic arthritis, multiple sclerosis, systemic lupus erythematosus, myasthenia gravis, juvenile onset diabetes", glomerulonephritis, autoimmune thyroiditis, Behcet's disease; acute and chronic graft rejection (e.g., in transplantation), including allograft rejection or graft-versus-host disease; inflammatory bowel diseases, such as Crohn's disease and ulcerative colitis; spondyloarthropathies; scleroderma;
psoriasis (including T-cell mediated psoriasis); vasculitis (e.g., necrotizing, cutaneous, and hypersensitivity vasculitis); cancers with leukocyte infiltration of the skin or organs. Other diseases or conditions in which undesirable inflammatory responses are to be inhibited can be treated, including, but not limited to, reperfusion injury, atherosclerosis, certain hematologic malignancies, and polymyositis. The compounds of the present invention are accordingly useful in treating, preventing, ameliorating, controlling or reducing the risk of a wide variety of inflammatory and immunoregulatory disorders and diseases as well as autoimmune pathologies. In a specific embodiment, the present invention is directed to the use of the subject compounds for treating, preventing, ameliorating, controlling or reducing the risk of autoimmune diseases, such as rheumatoid arthritis, multiple sclerosis, psoriasis or psoriatic arthritis.
In another aspect, the instant invention may be used to evaluate putative specific agonists or antagonists of chemokine receptors, including CXCR3. Accordingly, the present invention is directed to the use of these compounds in the preparation and execution of screening assays for compounds which modulate the activity of chemokine receptors. For example, the compounds of this invention are useful for isolating receptor mutants, which are excellent screening tools for more potent compounds.
Furthermore, the compounds of this invention are useful in establishing or determining the binding site of other compounds to chemokine receptors, e.g., by competitive inhibition. The compounds of the instant invention are also useful for the evaluation of putative specific modulators of the chemokine receptors, including CXCR3. As appreciated in the art, thorough evaluation of specific agonists and antagonists of the above chemokine receptors has been hampered by the lack of availability of non-peptidyl
(metabolically resistant) compounds with high binding affinity for these receptors. Thus the compounds of this invention are commercial products to be sold for these purposes.
The present invention is further directed to a method for the manufacture of a medicament for treating CXCR3 mediated diseases in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
In a preferred aspect of the present invention, a subject compound may be used in a method of inhibiting the binding of a chemokine to a chemokine receptor, such as CXCR3, of a target cell, which comprises contacting the target cell with an amount of the compound which is effective at inhibiting the binding of the chemokine to the chemokine receptor. The subject treated in the methods above is a mammal, preferably a human being, male or female, in whom modulation of chemokine receptor activity is desired. "Modulation" as used herein is intended to encompass antagonism, agonism, partial antagonism, inverse agonism and/or partial agonism. In a preferred aspect of the present invention, modulation refers to antagonism of chemokine receptor activity. The term "therapeutically effective amount" means the amount of the subject compound that
will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
The term "composition" as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts. By "pharmaceutically acceptable" it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
The terms "administration of and or "administering a" compound should be understood to mean providing a compound of the invention to the individual in need of treatment. As used herein, the term "treatment" refers both to the treatment and to the prevention or prophylactic therapy of the aforementioned conditions.
Dose Ranges
The magnitude of prophylactic or therapeutic dose of a compound of Formula I will, of course, vary with the nature and severity of the condition to be treated, and with the particular compound of Formula 1 used and its route of administration. The dose will also vary according to the age, weight and response of the individual patient. In general, the daily dose range lie within the range of from about 0.001 mg to about 100 mg per kg body weight of a mammal, preferably 0.01 mg to about 50 mg per kg, and most preferably 0.1 to 10 mg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
For use where a composition for intravenous administration is employed, a suitable dosage range is from about 0.01 mg to about 25 mg (preferably from 0.1 mg to about 10 mg) of a compound of Formula I per kg of body weight per day.
In the case where an oral composition is employed, a suitable dosage range is, e.g. from about 0.01 mg to about 100 mg of a compound of Formula I per kg of body weight per day, preferably from about 0.1 mg to about 10 mg per kg.
For use where a composition for sublingual administration is employed, a suitable dosage range is from 0.01 mg to about 25 mg (preferably from 0.1 mg to about 5 mg) of a compound of Formula I per kg of body weight per day.
Pharmaceutical Compositions
Another aspect of the present invention provides pharmaceutical compositions which comprises a compound of Formula I and a pharmaceutically acceptable carrier. The term "composition", as in pharmaceutical composition, is intended to encompass a product comprising the active
ingredients), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of Formula I, additional active ingredient(s), and pharmaceutically acceptable excipients.
Any suitable route of administration may be employed for providing a mammal, especially a human with an effective dosage of a compound of the present invention. For example, oral, rectal, topical, parenteral, ocular, pulmonary, nasal, and the like may be employed. Dosage forms include tablets, troches, dispersions, suspensions, solutions, capsules, creams, ointments, aerosols, and the like.
The pharmaceutical compositions of the present invention comprise a compound of Formula I as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids.
The compositions include compositions suitable for oral, sublingual, rectal, topical, parenteral (including subcutaneous, intramuscular, and intravenous), ocular (ophthalmic), pulmonary (aerosol inhalation), or nasal administration, although the most suitable route in any given case will depend on the nature and severity of the conditions being treated and on the nature of the active ingredient. They may be conveniently presented in unit dosage form and prepared by any of the methods well-known in the art of pharmacy.
For administration by inhalation, the compounds of the present invention are conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulizers. The compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device. The preferred delivery systems for inhalation are metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellants, such as fluorocarbons or hydrocarbons and dry powder inhalation (DPI) aerosol, which may be formulated as a dry powder of a compound of Formula 1 with or without additional excipients.
Suitable topical formulations of a compound of formula I include transdermal devices, aerosols, creams, ointments, lotions, dusting powders, and the like.
In practical use, the compounds of Formula I can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding
techniques. The carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous). In preparing the compositions for oral dosage form, any of the usual pharmaceutical media may be employed, such as, for example, water, glycols, oils, alcohols, flavoring agents, preservatives, coloring agents and the like in the case of oral liquid preparations, such as, for example, suspensions, elixirs and solutions; or carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents and the like in the case of oral solid preparations such as, for example, powders, capsules and tablets, with the solid oral preparations being preferred over the liquid preparations. Because of their ease of administration, tablets and capsules represent the most advantageous oral dosage unit form in which case solid pharmaceutical carriers are obviously employed. If desired, tablets may be coated by standard aqueous or non-aqueous techniques.
In addition to the common dosage forms set out above, the compounds of Formula I may also be administered by controlled release means and/or delivery devices such as those described in U.S. Patent Nos. 3,845,770; 3,916,899; 3,536,809; 3,598,123; 3,630,200 and 4,008,719. Pharmaceutical compositions of the present invention suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient, as a powder or granules or as a solution or a suspension in an aqueous liquid, a non-aqueous liquid, an oil-in-water emulsion or a water-in-oil liquid emulsion. Such compositions may be prepared by any of the methods of pharmacy but all methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more necessary ingredients. In general, the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product into the desired presentation. For example, a tablet may be prepared by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent. Desirably, each tablet contains from about 1 mg to about 500 mg of the active ingredient and each cachet or capsule contains from about 1 to about 500 mg of the active ingredient.
Combination Therapy
Compounds of Formula I may be used in combination with other drugs that are used in the treatment/prevention/suppression or amelioration of the diseases or conditions for which compounds of Formula T are useful. Such other drugs may be administered, by a route and in an amount commonly
used therefor, contemporaneously or sequentially with a compound of Formula I- When a compound of Formula I is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of Formula I is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of Formula 1. Examples of other active ingredients that may be combined with a compound of Formula I, either administered separately or in the same pharmaceutical compositions, include, but are not limited to: (a) VLA-4 antagonists such as those described in US 5,510,332, WO97/03094, WO97/02289, WO96/40781, WO96/22966, WO96/20216, WO96/01644, WO96/06108, WO95/15973 and WO96/31206, as well as natalizumab; (b) steroids such as beclomethasone, methyl prednisolone, betamethasone, prednisone, dexamethasone, and hydrocortisone; (c) immunosuppressants such as cyclosporin, tacrolimus, rapamycin and other FK-506 type immunosuppressants; (d) immunomodulaltory antibody therapies including anti-TNF therapies such as Etanercept (Enbrel®), Infliximab (Remicade®), Adalimumab (Humira®) or other TNF peptide or receptor sequestrants; Efalizumab (Raptiva®), Daclizumab (Zenapax®), Basiliximab (Simulect ®), Rituximab (Rituxan®), visilizumab (Nuvion®), Abatacept (Orencia®) or other interleukin peptide or receptor binding antibodies; (e) antihistamines (Hl -histamine antagonists) such as bromopheniramine, chlorpheniramine, dexchiorpheniramine, triprolidine, clemastine, diphenhydramine, diphenylpyraline, tripelennamine, hydroxyzine, methdilazine, promethazine, trimeprazine, azatadine, cyproheptadine, antazoline, pheniramine pyrilamine, astemizole, terfenadine, loratadine, cetirizine, fexofenadine, descarboethoxyloratadine, and the like; (f) non-steroidal anti-asthmatics such as β2-agonists (terbutaline, metaproterenol, fenoterol, isoetharine, albuterol, bitolterol, salmeterol and pirbuterol), theophylline, cromolyn sodium, atropine, ipratropium bromide, leukotriene antagonists (zafirlukast, montelukast, pranlukast, iralukast, pobilukast, SKB-106,203), leukotriene biosynthesis inhibitors (zileuton, BAY- 1005); (g) non-steroidal antiinflammatory agents (NSATDs) such as propionic acid derivatives (alminoprofen, benoxaprofen, bucloxic acid, carprofen, fenbufen, fenoprofen, fluprofen, flurbiprofen, ibuprofen, indoprofen, ketoprofen, miroprofen, naproxen, oxaprozin, pirprofen, pranoprofen, suprofen, tiaprofenic acid, and tioxaprofen), acetic acid derivatives (indomethacin, acemetacin, alclofenac, clidanac, diclofenac, fenclofenac, fenclozic acid, fentiazac, furofenac, ibufenac, isoxepac, oxpinac, sulindac, tiopinac and zidometacin), fenamic acid derivatives (flufenamic acid, meclofenamic acid, mefenamic acid, niflumic acid and tolfenamic acid), biphenylcarboxylic acid derivatives (diflunisal and flufenisal), oxicams (isoxicam. piroxicam, sudoxicam and tenoxican), salicylates (acetyl salicylic acid, sulfasalazine, olsalazine, mesalamine and balsalazide) and the pyrazolones (apazone, bezpiperylon, feprazone, mofebutazone, oxyphenbutazone, phenylbutazone); (h) cyclooxygenase-2 (COX-2) inhibitors such as celecoxib, rofecoxib, and parecoxib; (i) inhibitors of phosphodiesterase type IY (PDE-TV); (j)
antagonists of the other chemokine receptors, especially CCRl, CCR2, CCR5 and CCR3; (k) cholesterol lowering agents such as HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and other statins), sequestrants (cholestyramine and colestipol), nicotinic acid, fenofibric acid derivatives (gemfibrozil, clofibrat, fenofibrate and benzafibrate), and probucol; (1) anti-diabetic agents such as insulin, sulfonylureas, biguanides (metformin), a-glucosidase inhibitors (acarbose), glitazars (muraglitazar) and glitazones (troglitazone, pioglitazone, englitazone, MCC-555, BRL49653 and the like); (m) preparations of interferon beta (Avonex®, Rebif®, interferon beta- Ia5 Betaseron®, interferon beta-lb); (n) anticholinergic agents such as muscarinic antagonists (ipratropium and tiatropium); (o) current treatments for multiple sclerosis, including prednisolone, glatiramer, deoxyadenosine, mitoxantrone, methotrexate, and cyclophosphamide; (p) p38 kinase inhibitors; (q) DMARDs 5 such as methotrexate, leflunamide or plaquenil; (r) other compounds such as 5- aminosalicylic acid and prodrugs thereof, antimetabolites such as azathioprine, mycophenolate and 6- mercaptopurine, cytotoxic cancer chemotherapeutic agents and cytokine sequestrants.
The weight ratio of the compound of the Formula I to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the Formula I is combined with an NSAID the weight ratio of the compound of the Formula I to the NSAID will generally range from about 1000: 1 to about 1:1000, preferably about 200:1 to about 1 :200. Combinations of a compound of the Formula I and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used.
Methods of Synthesis
The following abbreviations are used in the synthetic schemes:
Ac is acetyl [CH3C(O)-]; Ac2θ is acetic anhydride; 9-BBN is 9-borabicyclo[3.3.1]nonane; Bn is benzyl;
DIAD is diisopropylazodicarboxylate; DIBAL is dϊisobutylalumϊnum hydride; DMF is N,N- dimethylformamide; DMSO is dimethyl sulfoxide; EDAC (or EDC) is l-ethyl-3-[3- (dimethylamino)propyl]-carbodiimide HCl; Et3N is triethylamine; Et is ethyl; EtOAc is ethyl acetate;EtOH is ethanol; HCl is hydrochloric acid; HOBt is 1-hydroxybenzotriazole; HPLC is high performance liquid chromatography; LG is leaving group; M is molar; mmol is millimole; Me is methyl; MeOH is methanol; MsCl methanesulfonyl chloride; N is normal; NaHMDS is sodium hexamethyldisiliazide; NaOAc is sodium acetate; NaOtBu is sodium /ert-butoxide; NMO is N- methylmorpholine N oxide; PG is protecting group; Pd(dba)2 is fm(dibenzylideneacetone)dipaUadium;
PdCl2(Ph3P)2 is dichloro δώ-(triphenylphosphine) palladium; Ph is phenyl; PhMe is toluene; PPh3 is triphenylphosphine; PMB is /rara-methoxybenzyl; RT is room temperature; TBAF is tetrabutyl ammonium fluoride; TBS is tør*-butyldimethylsilyl; tBu is tert-buty\; Tf is triflate; TFA is trifluoroacetic acid; THF is tetrahydrofuran; TLC is thin layer chromatography; TMS is trimethylsilyl; TPAP is tetrapropylammonium perruthenate;
GENERAL SCHEMES
These compounds are prepared by the convergent assembly of three molecular fragments. The synthesis of these fragments is depicted in Schemes 1 , 2 and 3 below.
Scheme 1

a) oxalyl chloride, DMF (cat), CH2CI2 b) NH3, dioxane c) Lawessons's Reagent, dioxane d) Naθ2H, aq. EtOH
Using known methods substituted thioamides 3 are prepared from their corresponding benzoic acids 1 or benzonitriles 2 as shown in Scheme 1. Benzoic acids 1 are converted to their acid chlorides and ammonolyzed to the amide. The amide is converted to the thϊoamide with Lawesson's reagent. Benzonitiles 2 are converted to their amides by partial hydrolysis and then to the thioamides 3 as above.
Scheme 2

a) oxalyl chloride, DMF(cat), CH2CI2 b) (CH3)3 S 1CHN2, toluene/hex c) 48%HBr, THF
Bromoketone 6 is prepared from N-FMOC isonipecotic acid 4 as shown in Scheme 2. Acid 4 is converted to its acid chloride and exposed to trimethylsilyldiazomethane, giving the diazoketone 5. Decomposition of 5 with 48% HBr gives the bromoketone 6.
Scheme 3

a) alkali/alkaline metal carbonate/alkoxide/hydride, alkyl bromoacetate, DMF/DMSO b) (R=Me) K2CO3, aq. MeOH; (R=Et) KOH, aq. EtOH c) H2, 50psi, 10%Pd/C, EtOH d) TFA, CH2CI2
The heterocyclic acetic acids 9 are prepared from the corresponding known heterocycles 7 as shown in Scheme 3. Exposure of 7 to a bromoacetic acid ester in the presence of alkali or alkaline metal bases in polar aprotic solvents affords the esters 8. Basic hydrolysis, reduction or solvolysis as appropriate gives the desired 9.
Antagonists I are assembled as shown in Scheme 4.
Scheme 4

a) THF or dioxane, then aq. NaHCO3/iPrOAc workup b) dioxane, reflux c) nPrNH2 d) EDC, HOBT, N-Me moφholinβ, DMF.
Combination of 6 and 3 in ethereal solvents such as THF or dioxane followed by basic workup affords the imino-thioether 10. Thermal cyclization gives the thiazole 11. Removal of the FMOC protecting group gives the piperidine 12. Amide formation using acids 9 affords the target antagonists I.
Specific examples are described below.
Example 1 Step A 9iy-f1uoren-9-yImethyl-4-(chlorocarboiryQpiperidine-l-carboxylate

A 0° solution of l-[(9H-fluoren-9-yImethoxy)carbonyI]piperidine-4-carboxylic acid (FMOC-isonipecotic acid; 25.01g; 71.17mmol) in dry CΗ2CI2 (225mL) was treated dropwise with oxalyl chloride (8.69mL;
99.64mmol). The cold bath was removed and the solution stirred at ambient temperature for 3h. The
solution was recovered and evaporated to an oil, then flushed with toluene (2x50mL). The residue was used without further purification.
Step B gg-fluoren-g-ylmethvI^-Cdiazoacetyπpiperidine-l-carboxYlate

A solution of 9H-fluoren-9-ylmethyl-4-(chlorocarbonyI)piperidine-l-carboxylate in toluene (125mL) was added dropwise to a 0° solution of trimethylsϊlyldiazomethane (144mL; 2M in hexane; 288mmol). The cold bath was removed and the solution stirred at ambient temperature for 16h. The solution was recovered and evaporated to a residue that was used without further purification. lH NMR (CDCI3): δ 7.80 (d, 2H5 J = 7.5Hz), 7.60 (d, 2H, J = 7.5Hz), 7.43 (t, 2H, J= 7.3Hz)5 7.35 (dt, 2H, Jt = 7.6 Hz, Jd = 0.9Hz), 4.47 (bs, 2H)5 4.28 (t, I H5 J = 6.8Hz), 4.23 (vbs, IH), 4.17 (vbs, IH), 2.91 (vbs, 3H), 2.77 (vbs, IH), 1.92-1.60 (bmult, 4H).
Step C 9/7-fluoren-9-vImethyl-4-(bromoacet l i eridine-l-carboxyIate
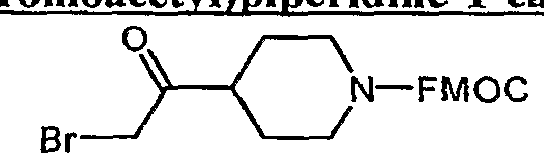
A 0° solution of 9H-fluoren-9-ylmethyl-4-(diazoacetyl)piρeridine-l-carboxyIate in TΗF (15OmL) was treated dropwise with 48% HBr (5OmL). The mixture was warmed to ambient temperature and partitioned between water (10OmL) and isopropyl acetate (50OmL). The organic was dried over MgSOzj, filtered and evaporated to a heavy oil. The crude product was chromatographed over silica gel (50% to 100% CH2Cl2/hexane; linear gradient), affording the title bromoketone as a white solid (27.88g). *H NMR (CDCI3): δ 7.81 (d, 2H, J= 7.4Hz), 7.60 (d, 2H, J= 7.4Hz), 7.44 (t, 2H, J= 7.3Hz), 7.37 (t, 2H, J = 7.2 Hz), 4.50 (vbs, 2H), 4.26 (t, J= 6.7Hz), 4.22 (vbs, IH), 4.04 (vbs, IH), 4.00 (s, 2H), 2.93 (bmult, 3H), 1.91 (vbs, 2H), 1.60 (vbs, 2H).
Step D 3.5-di-fert-butyl-4-methoxybenzonitriIe

A solution of 3,5-di-rer/-butyl-4-hydroxybenzonitrile (15.13g; 65.40mmol) in dry DMF (20OmL) was sequentially treated with methyl iodide (12.22mL; 196.2mmol) and cesium carbonate (29.83g;
91.55mmol). The mixture was stirred for 8h. The reaction mixture was diluted with isopropyl acetate (50OmL) and washed with water (3x200mL). The organic was dried over MgSθ4, filtered and evaporated to a solid (16.12g) that was used without further purification. ^H NMR (CDCI3): δ 7.80 (s, 2H), 3.79 (s, 3H), 1.49 (s, 18H).
Step E 3,5-di-terf-butvt-4-methoxybenzainide

A solution of 3,5-di-?erϊ-butyl-4-methoxybenzonitrile (16.12g; 65.40mmol based on 100% yield above) in ethanol (30OmL) was treated with an aqueous solution of sodium hydroperoxide (65.4mL; 2M; 130.8mmol). The solution was stirred at ambient temperature for 16h, during which a white solid precipitated. The reaction mixture was diluted with isopropyl acetate (50OmL) and washed with water (10OmL). The organic was dried over MgSO4, filtered and evaporated to a white solid. The solid was digested in methylcyclohexane (10OmL) with stirring at reflux, then cooled to ambient. Filtration afforded the title compound as a white solid (15.97g). lH NMR (CDCI3): δ 7.78 (s, 2H)5 6.02 (vbs,
IH)5 5.71 (vbs, IH), 3.76 (s, 3H), 1.48 (s, 18H).
Step F 3,5-di-ter/-butyl-4-methoxybenzthioamide

A solution of 3,5-di-ter*-butyl-4-methoxybenzamide (22.35g; 84.86mmol) in dioxane (25OmL) was treated with Lawesson's Reagent (51.49g; 127.3mmol). The mixture was stirred at ambient temperature for 16h. The now yellow mixture was filtered and the filtrate concentrated to an oil. The crude product was chromatographed over silica gel (0% to 100% CH2Cl2/hexane; linear gradient). The yellow
product fractions were combined and evaporated to give the title compound as a yellow solid (23.44g). IH NMR(CDCb): δ 7.80 (s, 2H), 7.62 (vbs, IH)5 7.17 (vbs, IH), 3.74 (s, 3H), 1.42 (s, 18H).
Step G 9Jy-fluoren-9-ylmethyl-4-r2-(3,5-di-terf-butyl-4-methoxyphenvn-13-thiazol-4- yll piperidine-1-carboxylate

A solution of 3,5-di-/erf-butyl-4-methoxybenzthioamide (15.26g; 56.61mmol) in dioxane (425mL) was treated in portions with 9H-fluoren-9-ylmethyl-4-(bromoacetyl)piperidine-l-carboxylate (23.4Og; 54.63mmol) at ambient temperature. The bromoketone dissolved in 15min. The reaction mixture was stirred for 3h during which it became a thick suspension of solid. The solid was recovered by filtration and pull-dried in the filtering funnel until it was sufficiently granular to enable facile removal. The solid was transferred to a 5L motor stirred flask containing isopropyl acetate (2L). The mixture was vigorously stirred for Ih until the mixture was a fine solid suspension. Saturated aqueous sodium bicarbonate (IL) was added and the mixture stirred until dissolution was complete (about lOmin). The organic was recovered and dried over MgSO4. Filtration and concentration afforded a heavy oil
(33.4Og). The crude product (consisting of a mixture of the title compound and its imino thioether precursor) was dissolved in dioxane (25OmL) and refluxed for 45min. The solvent was evaporated to give the title compound (32.04g) which was used without further purification. 1H NMR (CD3OD): δ 7.84 (s, 2H), 7.81 (d, 2H, J= 7.4Hz), 7.64 (d, 2H, J= 7.4Hz), 7.41 (bt, 2H5 J= 7.3Hz)5 7.37 (bt, 2H, J= 7.2Hz)5 7.13 (S5 IH)5 4.61 (vbs, 3H), 4.29 (t, IH5 J - 6.7Hz), 3.76 (s5 3H), 3.68 (s5 2H)5 3.00 (vbt, IH)5 2.93 (bmult, IH), 2.13-1.90 (vbmult, 2H, 1.49 (s, 18H). LRMS calc: 608.3 obs: 609.4 (M+H).
Step H 4-f2-(3,5-di--'gr-'-butvl-4-inethoxvphenvn-1.3-thiazol-4-vIlpiperidine

Step J 3//-imidazoF4,5-61pyridine-3-ylacetic acid benzyl ester

A solution of 4-azabenzimidazole (13.68g; 114.8mmol) in dry DMF (35OmL) was sequentially treated with benzyl bromoacetate (18.0ImL; 1 14.8mmol) and cesium carbonate (37.4Og; 114.8mmol) at ambient temperature. The mixture was stirred for 1.5h. The reaction was diluted with isopropyl acetate (70OmL) and filtered. The filtrate was washed with water (3x350mL). The organic was dried over MgSθ4, filtered and evaporated to an oil. The crude product was chromatographed over silica gel (70OmL c.v.; EtOAc) affording the title compound (12.7Og). lH NMR (CDCI3): δ 8.40 (dd, IH, J = 4.6, 1.3Hz), 8.13 (s, IH), 8.11 (dd, IH, J= 7.9, 1.3Hz), 7.39-7.35 (mult, 3H), 7.34-7.32 (mult, 2H), 7.28 (dd, IH, J= 8.0, 4.7Hz), 5.23 (s, 2H), 5.13 (s, 2H).
Step K 3J?"-iιnidazof4,5-Alpyridine-3-vIacetic acid

IH, J= 8.0, 4.7Hz), 5.19 (s, 2H).
Step L 3-(2-(4-r2-(3,5-di-fer/'-butγl-4-methoxyphenvπ-li3-thiazol-4-yllpiperidin-l-vU-2-oxoethyl>- 3H-ini idazo f 4,5-&l p v rid ine

A solution of 4-[2-(3,5-di-/er/-butyl-4-methoxyphenyl)-l,3-thiazoI-4-yl]piperidine (5.7Og; 14.71mmol) in dry DMF (5OmL) was treated sequentially with 3H-imidazo[4,5-ό]pyridine-3-ylacetic acid (3.91g; 22.06mmol), 4-methylmorpholine (3.23mL; 29.41mmol), 1-hydroxybenzotriazole hydrate (3.97g; 29.41mmol) and EDC (5.64g; 29.41mmol). The solution was stirred at ambient temperature for 3h. Saturated aqueous sodium bicarbonate (75mL) was added dropwise to the reaction with vigorous stirring. The precipitated solid was recovered by filtration (8.67g). The solid was added to vigorously stirred water (45OmL) and heated to reflux momentarily. Cooling to ambient and filtration afforded the title compound (7.95g). 1 H NMR (CD3OD): δ 8.37 (dd, IH, J= 4.8, 1.3Hz)5 8.35 (s, IH), 8.10 (dd, IH, J= 8.0, 1.3Hz), 7.81 (s, 2H), 7.35 (dd, IH, J= 8.0, 4.8Hz), 7.16 (s, IH), 5.42 ('/2AB, IH5 J= 17.0Hz), 5.36 O/2AB, IH, J= 16.9Hz), 4.55 (bd, IH, J= 13.3Hz), 4.20 (bd, IH, J= 13.4Hz), 3.71 (s, 3H), 3.42 (bt, IH, J= 12.2Hz), 3.15 (tt, IH, J = 11.6, 3.2Hz), 2.90 (bt, IH, J= 11.9Hz), 2.24 (bd, IH, J= 13.2Hz), 2.12 (bd, IH, J= 13.3Hz), 1.94 (dquart, IH, Jq = 12.7, Jd = 3.7Hz), 1.73 (dquart, IH, Jq = 12.6, Jd = 3.6Hz),
1.47 (s, 18H). LRMS calc: 545.3 obs: 546.4 (M+H).
Example 2
Step A 2,4-dimethyI-l/7-imidazol-l-yIacetic acid teri-butyl ester and 2,5-dimethyl-l/*r-imidazol-l- ylacetic acid tert-butyl ester

A solution of 2,4-dimethylimidazole (8.4Og; 87.38mmol) in dry DMF (25OmL) was treated with potassium ter/-butoxide (9.806g; 87.38mmol). The mixture was stirred at ambient temperature until homogenous. tert-Butyϊ bromoacetate (15.29mL; 104.9mmo!) was added dropwise. The solution was stirred for 15min, then diluted with isopropyl acetate (40OmL) and washed with pH7 phosphate buffer (3x250mL). The organic was dried over MgSθ4, filtered and evaporated to an oil. The crude product
(consisting of the two title compounds) was chromatographed over silica gel (0% to 10% MeOH/CH2Cl2; linear gradient). All fractions containing the two products were combined. The residue was rechromatographed on Chiralcel OD stationary phase (Daicel Chemical Industries Ltd., Chiralcel Technologies Inc.; 10%ethaπo I/heptane; λ=220nM). The more mobile 2,5 isomer and less mobile 2,4 isomer were obtained. lHNMR (CDCl3): (2,4 isomer) δ 6.49 (s, IH), 4.39 (s, 2H), 2.24 (s,
3H), 2.17 (s, 3H), 1.42 (s, 9H); (2,5 isomer) δ 6.67 (s, IH), 4.41 (s, 2H), 2.35 (s, 3H), 2.15 (s, 3H), 1.48 (s, 9H).
Step B 2,4-dimethvI-l/y-imidazoI-l-ylacetic acid hydrotrifluoroacetate

A solution of 2,4-dirnethyl-lH-imidazol-l-yIacetic acid tert-butyl ester (1.07g; 5.09mmol) in CH2CI2 (15mL) was treated with trifluoroacetic acid (3OmL) at ambient temperature. The solution was stirred for Ih. The volatiles were removed and the residue flushed with toluene (2x15mL) affording the title compound as an oil (t.32g). lH NMR (CD3OD): δ 7.20 (s, IH), 5.03 (s, 2H), 2.59 (s, 3H)3 2.32 (s, 3H).
Step C 4-f2-f3,5-di-/er/-butvI-4-methoxyphenvn-l,3-thiazol-4-vH-l-ff2.4-dimethvI-l.g-imidazol-l- vDacetyll piperidine
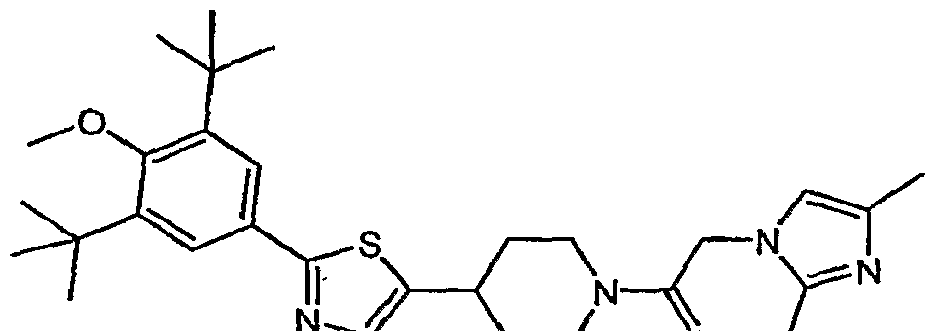
Using the method of Example 1, Step L and starting w the products of Example 1, Step H and Example 2, Step B the title compound was obtained. lH NMR (CD3OD): δ 7.88 (s, 2H)5 7.21 (s, IH)3
6.67 (bs, IH), 5.03 (!Z2AB, IH, J= 17.0Hz)5 4.96 ('/2AB3 IH. J= 17.0Hz), 4.59 (bd, IH5 J= 13.4Hz)5 4.11 (bd, IH5 J= 12.9Hz), 3.76 (s, 3H)5 3.37 (bt5 IH3 J= 12.9Hz)5 3.15 (tt5 IH5 J= 11.4, 3.8Hz)5 2.88 (bt, IH5 J = 12.8Hz), 2.26 (s5 3H)5 2.21 (bd5 IH. J = 12.9Hz)5 2.16 (bd, I H, J = 15.0Hz)5 2.11 (s, 3H)3 1.84 (dquart, IH, Jq = 12.5, Jd = 3.5Hz), 1.72 (dquart, IH5 Jq = 12.7, Jd = 3.7Hz), 1.49 (s, 18H). LRMS calc: 522.3 obs: 523.4 (M+H).
Step P 2,5-dimethyl-l/y-iiϊiidazoI-l-yIacetic acid hydrotrifluoroacetate

Step E 4-f2-f3,5-di-l'erAbutyl-4-methoxyphenvn-l,3-tIiiazol-4-vH-l-rf2,5-dimethvI-l/r-iιnidazol-l- vDacetyll piperidine
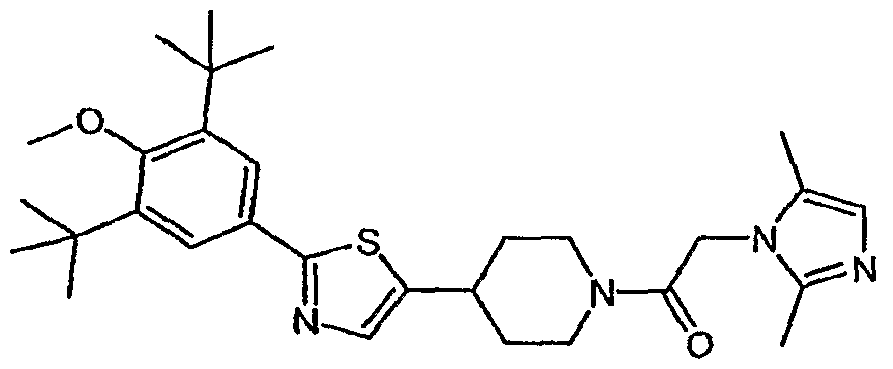
Using the method of Example 1, Step L and starting with the products of Example I5 Step Η and Example 2, Step D the title compound was obtained. ^H NMR (CDCI3): δ 7.79 (s3 2Η), 6.85 (s, IH)5 6.73 (vbs, IH), 4.72 (vbs, 2H), 4.64 (bd, IH, J= 13.6Hz), 3.98 (bd, IH, J= 13.5Hz)5 3.70 (s, 3H), 3.34 (bt, IH5 J= 12.3Hz)5 3.11 (bt, IH, J= 1 1.6Hz), 2.85 (bt, IH5 J= 12.0Hz), 2.40 (bs, 3H), 2.30 (bd5 IH5 J=
13.2Hz), 2.20-2.15 (bs overlapping bd, 4H)5 1.76 (bpent, 2H, J= 12.6Hz), 1.47 (s, 18H). LRMS calc: 522.3 obs: 523.4 (M+H).
Example 3 Step A f2-methyl-l/r-imidazol-5-vπmethanoI hydrochloride

A solution of dihydroxyacetone (1.905g; 21.16mmol) in aqueous ammonia (28%; 15mL) was placed in a microwavable vessel. Acetamidine hydrochloride (2.0Og; 21.16mmol) was added, the vessel sealed and the solution heated via microwave irradiation at 120° for lOmin. The solution was lyophilized. The derived residue was digested in refluxing acetone (10OmL), cooled to ambient temperature and filtered. The filtrate was evaporated and the residue dissolved in aqueous HCl (5OmL; IN). The solution was lyophilized to a heavy oil. The oil was flushed with methanol (2x20mL), then diluted with acetone (10OmL) and allowed to stand at -10° for 8 hours. The supernatant was decanted and the crystallized solid rinsed with cold acetone, affording the title compound (l.25g). lH NMR (CD3OD): δ 7.30 (s, IH), 4.61 (s= 2H), 2.62 (s, 3H).
Step B f4-fhydroxymethvO-2-methyl-l//-imidazol-l-vπacet.c acid methyl ester and f5- (hvdroxymethyl)-2-methyl-l//-iinidazol-l-vIlaeetie acid methyl ester

A solution of (2-methyl-lH-imidazol-5-yl)methanol hydrochloride (990mg; 6.66mmol) in dry DMF (2OmL) was sequentially treated with cesium carbonate (5.43g; 16.66mmol) and methyl bromoacetate (633 μL; 6.66mmol). The mixture was stirred at ambient temperature for 16h. The reaction was diluted with isopropyl acetate (10OmL) and filtered. The filtrate was concentrated and flushed with xylene (3x50mL; lmm vacuum; 60° bath). The residue was chromatographed on Chiralcel OJ stationary phase (15% ethanol/heptane, λ=220nM). The more mobile 5-hydroxymethyl and less mobile 4-hydroxymethyl title isomers were obtained. lH NMR (CD3OD): δ 6.93 (bs, IH), 4.82 (s, 2H), 4.46 (s, 2H), 3.77 (s,
3H), 2.29 (s, 3H).
Step C Potassium f4-(hvdroxymethyl)-2-methvI-lg-imidazol-l-yllacetate

A solution of [4-(hydroxymethyl)-2-methyl-l//'-imidazol-l-yl]acetic acid methyl ester (58mg; 0.315mmol) in methanol (2mL) was treated with aqueous potassium carbonate (315μL; IM; 0.315mmol). The solution was stirred at ambient temperature for 24h. All volatiles were removed and the resultant solid digested in hot ethanol (5mL). The mixture was cooled to ambient and filtered. The filtrate was evaporated to give the title compound. lH NMR (CD3OD): δ 6.87 (bs, IH), 4.44 (s, 2H)5 4.42 (s, 2H), 2.29 (s, 3H).
Step D ri-f2-f4-f2-f3.5-di-ter/-butyl-4-methoxyphenyl)-l,3-thiazol-4-γπpiperidin-l-yl>-2-oxoethvn-

6.99 (s, IH), 5.09 ('/2AB5 IH, J= 17.4Hz)5 5.03 (!Z2AB5 IH, J= 17.3Hz)5 4.58 (bd, IH, J = 12.6Hz), 4.49
(bs, 2H)5 4.03 (bd, IH5J= 13.2Hz)5 3.72 (s, 3H)53.34 (bt, IH5J= 12. IHz), 3.13 (tt, IH5 J= 11.8, 4.0Hz),
2.90 (bt5 IH5 J = 11.8Hz), 2.34 (s, 3H)5 2.20 (bd, IH5 J = 12.6Hz)5 2.13 (bd, IH, J = 13.2Hz)5 1.85 (dquart, IH, Jq = 12.6, 3.7Hz), 1.71 (dquart, IH, Jq = 12.8, Jd = 3.5Hz), 1.46 (s, 18H). LRMS calc:
538.3 obs: 539.3 (M+H).
Step E Potassium F5-(hvdroxyinethyl)-2-methγl-l^-iiiiidazol-l-yllacetate

Step F H-(2-{4-r2-(3,5-di-te/-f-butvI-4-methoxyphenvn-l,3-thiazol-4-ynpiperidin-l-vU-2-oxoethvn- 2-methyl-l//-imidazol-5-vΗmethanol

Using the method of Example 1, Step L and starting with the products of Example 1, Step H and Example 3, Step E the title compound was obtained. *H NMR (CD3OD): δ 7.81 (s, 2H), 7.16 (s, I H),
6.77 (s, IH), 5.08 ('ΛAB, IH, J= 17.6Hz), 5.01 ('/2AB3 IH, J= 17.4Hz), 4.57 (bd, IH, J= 13.7Hz), 4.47 (bs, 2H), 4.03 (bd, IH5 J = 12.8Hz), 3.72 (s, 3H)3 3.37 (bt5 IH, J== 13.6Hz), 3.14 (bt, IH, J= 11.7Hz),
2.90 (bt, IH, J = 12.4Hz), 2.29 (s, 3H), 2.22 (bd, IH5 J = 12.6Hz), 2.14 (bd, IH, J = 12.5Hz)5 1.87
(dquart, IH, Jq = 12.7, Jd = 4.0Hz), 1.71 (dquart, IH, Jq = 12.8, Jd = 4.0Hz), 1.47 (s, 18H). LRMS calc: 538.3 obs: 539.3 (M+H).
Example 4
Step A l-ethyl-l«3-dihvdro-2Jflr-imidazol-2-one

A solution of aminoacetaldehyde dimethyl acetal (2.72g; 22.86mmol) in dry THF (5OmL) was treated with ethyl isocyanate (1.8ImL; 22.86mmol). The solution was stirred at ambient temperature for 15min. The reaction was evaporated and the residue dissolved in dry benzene (5OmL). Trifluoroacetic acid (1OmL) was added and the solution refluxed for 2h. Cooling and evaporation gave a residue that was flushed with toluene (2xl0mL). The crude product was adsorbed onto silica gel (CH2CI2) and eluted without fractionation (100:5:1 CH2θ2/MeOH/Et3N) to give the title compound (1.92g). ^H NMR (CD3OD): δ 6.49 (d, IH, J= 2.9Hz), 6.41 (d, IH, J= 3.0Hz), 3.68 (quart, 2H, J= 7.3Hz), 1.29 (t, 3H, J = 7.3Hz).
Step B (3-ethyl-2-oxo-2.3-dihγdro-l/y-imidazol-l-yr)acetic acid tert-butyl ester

A solution of l-ethyl-l,3-dihydro-2H-imidazol-2-one (4.26g; 37.99mmol) in dry DMF (10OmL) was sequentially treated with fer/-butyl bromoacetate (5.54mL; 37.99mmol) and cesium carbonate (12.38g; 37.99mmol). The reaction was stirred at ambient temperature for 16h. The reaction mixture was diluted with isopropyl acetate (30OmL) and washed with pH7 phosphate buffer (3xl00mL). The organic was dried over MgSO4, filtered and evaporated to a residue. The crude product was chromatographed over silica gel (50% to 100% MTBE/hexane; linear gradient) to give the title compound (4.97g). ^H NMR (CD3OD): δ 6.52 (d, IH3 J= 2.7Hz)3 6.48 (d3 IH3 J= 2.7Hz), 4.33 (s, 2H), 3.67 (quart, 2H, J= 7.3Hz),
1.47 (s, 9H), 1.27 (t, 3H, J= 7.3Hz).
Step C (3-ethvI-2-oxo-23-dihvdro-1//-imidazoI-l-vQacetic acid

Using the method of Example 2, Step B with the product of Example 4, Step B as the starting material the title compound was obtained. ^H NMR (CD3OD): δ 6.52 (d, IH3 J = 3.0Hz)3 6.49 (d, IH, J = 2.9Hz), 4.41 (s, 2H), 3.67 (quart, 2H3 J= 7.2Hz), 1.27 (t, 3H, J= 7.2Hz).
Step J) l-f2-{4-r2-r3.5-di-fer<-butyl-4-methoxyphenvn-13-thiazoI-4-vHpiperidin-l-vn-2-oxoethvI>-

Using the method of Example I, Step L and starting with the products of Example 1, Step H and Example 4, Step C the title compound was obtained. lH NMR (CD3OD): δ 7.84 (s, 2H), 7.18 (s, IH),
6.55 (d, IH, J = 3.0Hz), 6.48 (d, IH3 J = 3.0Hz)3 4.68 ('ΛAB, IH3 J = 16.7Hz), 4.59 (1AAB, IH3 J = 16.8Hz)3 4.60 (bd, IH, J= 13.0Hz)3 4.09 (bd, IH, J= 13.9Hz), 3.70 (quart, 2H, J= 7.3Hz), 3.31 (bt, IH, J = 12.0Hz), 3.15 (tt, IH, J = 11.7, 3.2Hz), 2.90 (bt, IH, J = 11.8Hz), 2.22-2.14 (bmult, 2H), 1.83 (dquart, IH, Jq = 12.6, Jd = 3.7Hz), 1.73 (dquart, IH, Jq = 12.6, Jd = 3.8Hz)3 1.50 (s, 18H), 1.30 (t, 3H, J = 7.2Hz). LRMS calc: 538.3 obs: 539.4 (M+H).
Step E (3-methyl-2-oxo-2,3-dihvdro-liy-imidazol-l-yl)acetic acid fe/7-buty! ester

Using the method of Example 4, Step B with l-methyl-l,3-dihydro-2/7-imidazol-2-one as the starting material the title compound was obtained.
Step F (3-methyl-2-oxo-2«3-dihvdro-l.flr-imidazol-l-vOacetic acid

Using the method of Example 2, Step B with the product of Example 4, Step E as the starting material the title compound was obtained.
Step G l-(2-(4-r2-(3<5-di-te^-butyl-4-methoxyphenylVl.,3-thiazoI-4-yllpiperidin-l-vU-2-oxoethvn- 3-methyl-l,3-dihvdro-2/f-imidazol-2-one

Using the method of Example I3 Step L and starting with the products of Example 1, Step H and Example 4, Step F the title compound was obtained. 'H NMR (CD3OD): δ 7.80 (s, 2H), 7.14 (s, IH)3
6.45 (d, IH, J= 3.0Hz), 6.43 (d, IH, J = 3.0Hz), 4.63 (1AAJB, IH3 J = 16.9Hz)3 4.55 (14AB, IH, J = 16.8Hz), 4.56 (bd, IH, J= 13.1Hz)3 4.04 (bd, IH3 J= 13.7Hz), 3.71 (s, 3H), 3.31 (dt, IH, Jt = 12.9, Jd =
2.5Hz), 3.25 (s, 3H), 3.10 (tt, IH, /= 11.7, 3.6Hz), 2.85 (dt, IH5 Jt = 12.9, JA = 2.6Hz)5 2.16 (bd, IH5 J= 13.5Hz)5 2.10 (bd, IH, J= 13.3Hz)5 1.80 (dquart, IH5 Jq = 12.5, Jd = 4.2Hz)5 1.68 (dquart, IH, Jq = 12.6, Jd = 4.3Hz), 1.46 (s, 18H). LRMS calc: 524.3 obs: 525.3 (M+H).
Example 5
Step A N-Methylaminoacetone hydrochloride

A solution of chloroacetone (1.07mL; OmL) was sequentially treated with N- methyl benzylamine (1.63g; 13.42mmol), triethylamine (2.07mL; 14.76mmol) and tetrabutylammonium bromide (21 1mg; 0.654mmol). The solution was stirred at ambient temperature for 72h. The now heterogenous mixture was filtered and evaporated to an oil (2.6Ig). The oil was dissolved in ethanol
(25mL). Concentrated HCl was added (3mL) followed by 10%Pd/C hydrogenation catalyst (560mg).
The mixture was shaken under a hydrogen atmosphere (50psi) for 4h. The mixture was filtered through Celite and the filtrate evaporated, then allowed to stand under high vacuum for 1 hour. The residue was flushed with acetone (2x30mL) causing a solid to precipitate. Trituration from acetone gave the title compound (1.29g).
Step B 1.4-dimethyl-1.3-dihydro-2Jfir-imidazol-2-one

A solution of N-methylaminoacetone hydrochloride (462mg; 3.74mmol) in methanol (1OmL) was treated with KOCN (334mg; 4.11mmol). The mixture was refluxed Ih. After cooling to ambient temperature, the mixture was filtered and the filtrate evaporated. The residue was adsorbed onto silica gel (CH2CI2) and eluted without fractionation (20: 1 OH^C^/MeOH). Evaporation of the eluant gave the title compound (35 lmg).
Step C O.S-dimethvI-l-oxo-Z^-dihydro-lJy-iπiidazot-l-vnacetic acid ferf-buty. ester

Step D (3,5-dimethyl-2-oxo-23-dihydro-li?-imidazol-l-vI)acetic acid

Using the method of Example 2, Step B with the product of Example 5, Step C the title compound was obtained.
Step E 3-(2-(4-r2-f3,5-di-ferf-butvI-4-methoxyphenvπ-l,3-thiazol-4-vπpiperidin-l-vU-2-oxoethyl)- l,4-dimethyl-l,3-dihvdro-2/y-iinidazol-2-one

6.19 (d, IH, J= LlHz)5 4.65 (!Z2AB, IH, J = 17.2Hz), 4.58 (V2AB, IH5 J = 17.0Hz)5 4.56 (bd, IH5 J = 13.1Hz), 4.10 (bd, IH, J= 13.9Hz)5 3.73 (s, 3H)5 3.31 (dt, IH5 Jt = 12.9, Jd = 2.5Hz)5 3.22 (s5 3H), 3.13 (tt, IH, J= 11.7, 3.7Hz), 2.88 (dt, IH, Jt = 12.6, Jd = 2.3Hz), 2.20 (bd, 1 H, J= 12.5Hz), 2.13 (bd, IH, J= 12.6Hz), 1.84 (dquart, IH, Jq = 12.6, Jd = 4.1Hz)5 1.71 (dquart, IH, Jq = 12.6, Jd = 4.3Hz), 1.48 (s, 18H). LRMS: calc. 538.3 obs: 539.4 (M+H).
Example 6 Step A 3,3-dimethoxy-2-methylpropionic acid methyl ester

Step B 3-hvdroxy-4-methylthiophene-2-carboxylic acid methyl ester

3,3-dimethoxy-2-methylpropionic acid methyl ester (4.02g; 24.79mmol) and methyl mereaptoacetate (5.26g; 49.57mmol) were combined and treated with BF3 etherate (94μL; 0.74mmol). A short path distillation head was attached and the flask immersed in a 130° bath. Heating was continued until methanol stopped distilling (approx. lOmin). The residue was cooled to ambient and dissolved in methanolic sodium methoxide (62mL; IM). The solution was stirred for 16h. The reaction was evaporated to a residue and partitioned between isopropyl acetate (20OmL) and aq. HCl (10OmL; IN). The organic was dried over MgSθ4, filtered and concentrated. The crude product was chromatographed over silica gel (50% to 100% CH2Cl2/hexane; linear gradient) to give the title compound (3.15g).
Step C (4-methyl-lJy-pyrazol-3-vI)acetic acid methyl ester hydrochloride

A solution of the product of Example 6, Step B (3.15g; 18.29mmol) in n-butanol (5OmL) was treated with hydrazine monohydrate (8.9OmL; 183mmol). The solution was refluxed for 16h. The reaction was concentrated to a residue and dissolved in aq. NaOH (75mL; 2.5N). The solution was refluxed for 8h. The solution was lyophilized and the resultant solids digested in hot ethanol (5OmL). The mixture was cooled to ambient and filtered. The filtrate was concentrated and dissolved in methanol (5OmL). Dry gaseous HCl was bubbled into the solution causing the precipitation of a solid. The mixture was stirred
until it returned to ambient temperature, then filtered and the filtrate concentrated to afford the title compound (3.04g).
Step D teri-butyl methyl 2,2'-(4-KiCtI-V-- l.ff-pyrazole-l,,3-diyl)diacetate

A solution of the product of Example 6, Step C (3.04g; 15.95mmol) in dry DMSO (5OmL) was treated with tert-hu\y\ bromoacetate (3.49mL; 23.92mmol) followed by cesium carbonate (10.39g; 31.89mmol). The mixture was stirred at ambient temperature for 24h. The mixture was diluted with isopropyJ acetate (15OmL) and filtered. The filtrate was washed with pH7 phosphate buffer solution (3x50mL). The organic was dried over MgSθ4, filtered and concentrated to a residue. The crude product, consisting of the title compound and its regioisomer, was chromatographed on Chiralcel OJ stationary phase (10% ethanol/heptane; λ=230nM). The less mobile, major isomer was isolated affording the title compound (1.2Ig).
Step E r3-f2-methoxy-2-oxoethyl)-4-methvI-lflr-pyrazol-l-vHacetic acid hydrotrifluoroacetate

Using the method of Example 2, Step B and the product of Example 6, Step D the title compound was obtained.
Step F ri-(2-f4-f2-(3,5-di-te/-f-butvI-4-methoxyphenyl)-l,3-thiazoI-4-yllpiperidin-l-yU-2-oxoethvπ- 4-methvI-lflr-pyrazol-3-vHacetie acid methyl ester

Step G ri-(2-^4-f2-(3,5-di-ter/-butvI-4-methoxyphenvI)-l,3-thiazol-4-vnpiperidin-l-vU-2-oxoethvn- 4-methyl-lϋr-pyrazo--3-yl|aeetate bis(hydrotrifluoroacetate)

A solution of the product of Example 6, Step F (127mg, 0.219mmol) in methanol (4mL) was treated with aq. K2CO3 (590μL; IM; 0.59mmol). The solution was stirred at ambient temperature for 24h. All volatiles were removed and the residue digested in hot ethanol (5mL). The mixture was cooled to ambient temperature and filtered. The filtrate was evaporated and the derived solid purified on C8 reverse phase HPLC (CH3CN/H2θ/0.1%TFA). Evaporation of the appropriate fractions afforded the title compound (120mg). lH NMR (CD3OD): δ 7.81 (s, 2H)3 7.47 (vbs, IH)5 7.21 (s, IH), 5.15 (1Z2AB, IH, J= 16.4Hz), 5.06 (1AAB, 1 H, J= 16.6Hz), 4.57 (bd, IH, J= 12.9Hz), 4.04 (bd, IH, J= 13.3Hz), 3.71 (s, 3H), 3.64 (!4AB, IH, J = 15.7Hz), 3.59 (1AAB, IH, J= 15.5Hz), 3.31 (bt, IH, J= 11. OHz). 3.13 (tt, IH, J= 3 1.8, 3.6Hz), 2.85 (bt, IH, J= 11.9Hz), 2.14 (bd, IH, J = 15.5Hz), 2.11 (bd, IH, J = 15.7Hz)3 2.04 (s3 3H), 1.76 (dquart, IH3 Jq = 12.6, Jd = 3.6Hz)3 1.66 (dquart, IH5 Jq = 12.4, Jd = 3.3Hz), 1.46 (s,
3H). LRMS calc: 566.3 obs: 567.4 (M+H).
Example 7
Step A 4-methyl-2-(methylthio)-l//-imidazole

A solution of 2-mercapto-4-methyI-lH-imidazole hydrochloride (prepared using the method of Smith, et. al. JCS 1951, p. 2217: 320mg; 2.80mmol) in dry acetone (6mL) was treated with potassium carbonate (774mg; 5.60mmol) followed by methyl iodide (174μL; 2.80mmol). The mixture was stirred at ambient temperature for 8h. The mixture was diluted with isopropyl acetate (2OmL) and washed with pH7
phosphate buffer solution (2OmL). The organic was dried over MgSθ4, filtered and evaporated to give the title compound (352mg).
Step B r4-methvI-2-(methvIthio)-l/7-imidazol-l-yl]acetic acid methyl ester

A solution of 4-methyl-2-(methylthio)-lH-imidazoIe (352mg; 2.75mmol) in dry DMF (8mL) was treated sequentially with cesium carbonate (895mg; 2.75mmol) and methyl bromoacetate (261 μL; 2.75mmol). The mixture was stirred at ambient temperature for 16h. The reaction mixture was diluted with isopropyl acetate (3OmL) and filtered. The filtrate was washed with pH7 phosphate buffer solution (3x1 OmL). The organic was dried over MgSO.4, filtered and evaporated to a residue. The crude product was chromatographed over silica gel (30:1 CH2Cl2/MeOH). The derived mixture of isomers (163mg) was rechromatographed on Chiralcel OJ stationary phase (10% ethanol/heptane; λ=220nM). The major, more mobile product was isolated affording the title compound (80mg).
Step C [4-methyl-2-(inethylsuIfonyO-liy-imidazol-l-y.lacetic acid methyl ester

A solution of [4-methyI-2-(methylthio)-lH-imidazol-l-yl]acetic acid methyl ester (80mg; 0.40mmol) in CH2CI2 (3mL) was treated with mCPBA (276mg; titer=75wt%; 1.20mmol). The mixture was stirred at ambient temperature for 16h. The mixture was filtered and evaporated. The derived solids were slurried in methanol and filtered. The filtrate was evaporated to give a residue (132mg) which was chromatographed over silica gel (30:1 ClrføCtø/MeOH). The major product was recovered, dissolved in isopropyl acetate and washed once with aq. sodium bicarbonate. Drying, filtration and evaporation afforded the title compound (51mg).
Step D f4-methyl-2-(methylsulfonyl')-Lflr-imidazoI-l-yllacetic acid hydrochloride

A solution of [4-methyl-2-(methylsulfonyl)-l//1imidazol-l-yl]acetic acid methyl ester (51mg; 0.22mmol) in methanol (2mL) was treated with aq. potassium carbonate (220μL; 0.22mmol). The solution was stirred at ambient temperature for 16h. The volatiles were removed and the resultant solids digested in hot ethanol (5mL). The mixture was allowed to cool to ambient and filtered, then diluted with aq. HCl (2mL; IN). Lyophilization afforded the title compound (46mg).
Step E 4-r2-f3,5-di-ferr-buryl-4-methoxyphenvπ-l,3-thiazol-4-yll-l-{f4-methyl-2-(methylsuIfonylV l-g-imidazol-l-yllacetvUpiperidine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 7, Step D as starting materials the title compound was obtained. lH NMR (CDCI3): δ 7.81 (s, 2H), 6.88 (s, IH),
6.75 (s, IH), 5.20 C/2AB, IH5 J= 16.5Hz), 5.11 (V2AB, IH, J = 16.4Hz), 4.62 (bd, IH, J= 13.2Hz), 3.86 (bd, IH, J- 13.3Hz), 3.72 (s, 3H), 3.32 (bt, IH, J= 12.1Hz)5 3.26 (s, 3H), 3.13 (vbt, IH5 J= 11.3Hz), 2.89 (bt, IH5 J= 11.8Hz), 2.27-2.25 (s overlapping bd, 4H total), 2.21 (bd, IH5 J= 11.8Hz), 2.19 (bd, 1 H, J= 12.8Hz), 1.84 (dquart, IH, Jq = 12.6, Jd = 3.5Hz), 1.74 (dquart, IH, Jq = 12.6, Jd = 3.5Hz), 1.48 (s, 18H). LRMS calc: 586.3 obs: 587.4 (M+H).
Example 8 Step A (2-oxo-2,3-dihvdro-l/-r--midazol-l-vDacetic acid ethyl ester

Step B Potassium (2-oxo-2,3-dihvdro-lϋr-iinidazol-l-v0aeetate
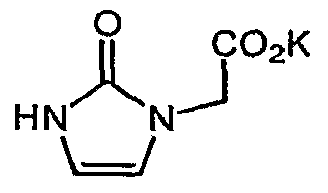
A solution of (2-oxo-2,3-dihydro-lH-imidazol-l-yl)acetic acid ethyl ester (180mg; 1.06mmol) in ethanol (5mL) was treated with aq. KOH (1.06mL; IM). The solution was stirred at ambient temperature for 6h. All volatiles were removed to give the title compound.
Step C l-f2-f4-[2-(3,5-dl-i'gr/-butvI-4-methoxyphenyl)-l ,3-thiazol-4-yll piperidin-l-yl}-2-oxoethvπ- l,3-dihydro-2Jy-iniidazol-2-one
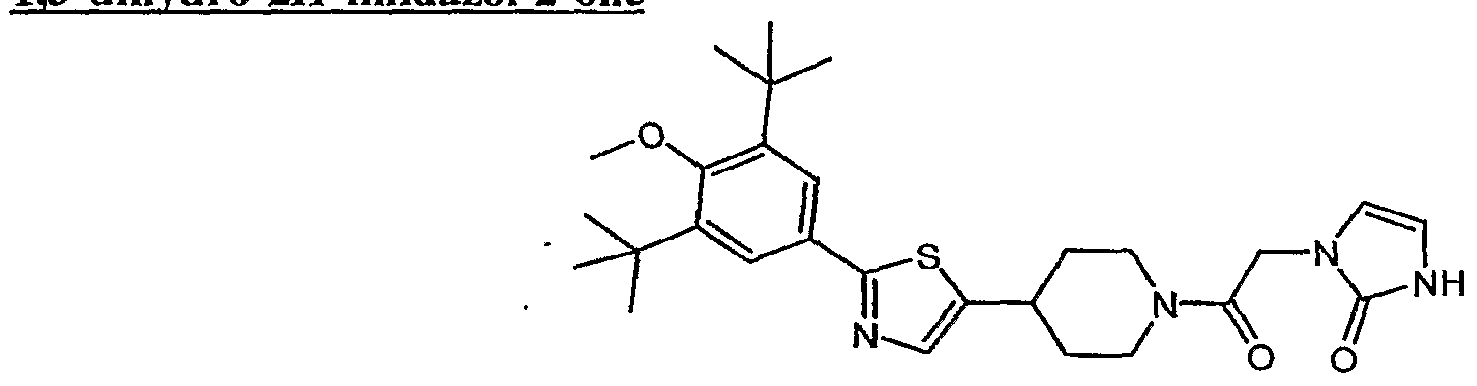
6.43 (d, IH, J = 3.0Hz), 6.41 (d, IH, J = 3.0Hz), 4.64 ('/2AB3 IH, J = 16.7Hz), 4.56 ('/2AB, IH, J = 16.7Hz), 4.60 (bd, IH, J= 13.1Hz), 4.07 (bd, IH, J= 13.2Hz), 3.73 (s, 3H), 3.31 (dt, IH, Jt = 12.9, Jd = 2.5Hz), 3.12 (tt, IH, J= 11.6, 3.6Hz), 2.87 (dt, IH, Jt = 13.0, Jd = 2.5Hz), 2.18 (bd, IH, J= 12.7Hz), 2.13 (bd, IH, J= 12.6Hz), 1.81 (dquart, IH, Jq = 12.5, Jd = 4.2Hz), 1.70 (dquart, IH, Jq = 12.6, Jd = 4.3Hz), 1.49 (s, 18H). LRMS calc: 510.3 obs: 511.3 (M+H).
Example 9 Step A tert-butyl ethyl 2,2'-(2-oxo-lg-iro.idazole-1.3-diyl)diacetate

Step B f3-f2-ethoxy-2-oxoethyl)-2-oxo-2,3-dihvdro-lJy-imidazoI-l-vHacetιc acid

Using the method of Example 2, Step B with the product of Example 9, Step A as starting material the title compound was obtained.
Step C r3-f2-f4-f2-f3,5-di-tert-butyl-4-methoxyphenvn-1.3-thiazol-4-vHpiperidin-l-vU-2-oxoethyl)- 2-oxo-2.3-dihvdro-LHr-imidazoI-l-vHacetic acid ethyl ester

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 9, Step B as starting materials the title compound was obtained.
Step D Potassium r3-r2-f4-r2-f3,5-di-tert-butvi-4-methoxyphenvn-l,3-thiazol-4-vUpiperidin-l-yl)- 2-oxoethyl*)-2-oxo-2,3-dihydro-l/r-imidazoI-l-vHacetate

A solution of the product of Example 9, Step C (52mg; 0.087mmol) in ethanol/DMF (2.5mL; 4:1) was treated with aq. KOH (122μL; IM). The solution was stirred at ambient temperature for 16h. The solution was flushed with xylene (3x1 OmL; lmm vacuum; 60° bath). The derived solids were digested in
hot ethanol (SmL), allowed to cool and filtered. The filtrate was evaporated and the resulting solid triturated with MTBE (1OmL) affording the title compound (47mg). 1H NMR (CD3OD): 5 7.82 (s,
2H), 7.16 (s, IH), 6.48 (d, IH, J = 2.8Hz), 6.43 (d, IH, J = 2.9Hz), 4.66 (1AAB, IH, J = 16.7Hz), 4.58 ('/2AB5 IH, J= 16.8Hz), 4.60 (bd, IH, J= 13.1Hz), 4.20 (s, 2H), 4.07 (bd, IH, J= 14.4Hz), 3.73 (s, 3H), 3.31 (dt, IH, Jt = 12.9, Jd = 2.5Hz), 3.12 (tt, IH, J= 11.7, 3.6Hz), 2.87 (dt, IH, Jt = 13.0, Jd = 2.3Hz), 2.18 (bd, IH, J = 13.5Hz), 2.12 (bd, I H, J = 12.0Hz), 1.82 (dquart, IH, Jq = 12.9, Jd = 4.2Hz), 1.70 (dquart, IH, Jq = 12.6, Jd = 4.3Hz), 1.48 (s, 18H). LRMS calc: 568.3 obs: 569.3 (M+H).
Example 10 Step A l-phenylimidazolidin-2-one

A solution of 2-chloroethylamine hydrochloride (270mg; 2.33mmol) in dry DMF (3mL) was treated sequentially with phenyl isocyanate (254μL; 2.33mmol) and cesium carbonate (758mg; 2.33mmol). The mixture was stirred at ambient temperature for 16h. The mixture was treated with potassium tert- butoxide (261mg; 2.33mmol) and stirred an additional 2h. The reaction was diluted with pH7 phosphate buffer (1OmL) and filtered. The derived solid was chromatographed over silica gel (40:1 CH2Cl2/MeOH) to afford the title compound (195mg).
Step B (l-oxo-S-phenylimidazolidin-l-vDacetic acid tert-butyl ester

Using the method of Example 4, Step B with the product of Example 10, Step A the title compound was obtained. Isolation was effected by silica gel chromatography (40:1 CH2Cl2/MeOH).
Step C (2-oxo-3-phenylimidazolidin-l-γl)acetic acid

Step D l-(2-(4-r2-(3,5-di-?grl-butyl-4-methoxyphenvI)-l,3-thiazol-4-vIlpiperidiιi-l-vI}-2-oxoethvI)- 3-phenylimidazolidin-2-one

Using the method of Example 1, Step L and the products of Example 1, Step H and Example 10, Step C as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.82 (s, 2H), 7.55 (d,
2H, J= 8.0Hz), 7.33 (t, 2H, J = 7.9Hz), 7.17 (s, IH), 7.05 (t, IH, J= 7.5Hz), 4.62 (bd, IH, J= 13.1Hz), 4.28 ('/2AB, IH, J= 16.9HzX 4.19 (!Z2AB, IH, J = 16.7Hz), 4.06 (bd, IH, J= 13.9Hz)5 3.94 (t, 2H, ./ = 8.IHz)5 3.74 (s, 3H), 3.64 (t, 2H5 J= 8.4Hz), 3.30 (dt, IH5 Jt = 14.8, Jd = 2.3Hz), 3.13 (tt, IH, J= 11.9,
3.6Hz)5 2.88 (bt, IH, J= 11.9Hz), 2.19 (bd, IH, J= 13.1Hz), 2.14 (bd, IH, J= 12.9Hz), 1.82 (dquart, IH, Jq = 12.6, Jd = 3.6Hz), 1.72 (dquart, IH, Jq = 12.4, Jd = 3.5Hz), 1.48 (s, 18H). LRMS calc: 588.3 obs:
589.3 (M+H).
Example 11
Step A 2-iodo-4-fer;-butylaniHne

A solution of 4-terϊ-butylaniline (75.0Og; 0.503mol) in methanol (14OmL) was treated with water (14OmL) and potassium carbonate (72.93g; 0.528mol). A solution of iodine rnonochloride (528mL; IM in CH2CI2) was added dropwise to the reaction mixture and stirred at ambient temperature for 2h. The dark solution was recovered and evaporated. Isopropyl acetate (2L) was added and the organic washed with water until the aqueous layer was clear. The organic was dried over MgSθ4, filtered and evaporated onto silica gel. The silica gel was exhaustively eluted (3:1 hexane/MTBE) without
fractionation. The eluant was evaporated giving the partially purified title compound which was used as is (121.4g).
Step B 2-iodo-4-fgrt-butylnitrobenzene

A solution of 2-iodo-4-/erf-burylaniIine (121.4g; 0.442mol) in CH2CI2 (4.2L) was treated at ambient temperature with mCPBA (305g; titer=75wt%; 1.326mol) in portions. The mixture was stirred for 16h. The thick suspension was filtered and evaporated. Isopropyl acetate (3L) was added and the organic washed with saturated aq. sodium bicarbonate until all mCBA was removed from the organic (by LC analysis; 5x750mL). The organic was dried over MgSO.4, filtered and evaporated onto silica gel. The silica gel was exhaustively eluted (6:1 hexane/MTBE) without fractionation. The eluant was evaporated giving the partially purified title compound which was used as is (82.46g).
Step C 2-trifluoromethylthio-4-fer/-burylnitrobenzene
A solution of 2-iodo-4-tert-butylnitrobenzene (76.89g; 0.252mol) in dry NMP (83OmL) was treated with CuSCF3 (31.08g; 0.189mol). The mixture was heated to 160° for Ih. After cooling to ambient, the reaction mixture was diluted with isopropyl acetate (2L) and filtered. The filtrate was washed with pH7 phosphate buffer solution (3x1 L). The organic was dried over MgSCH, filtered and evaporated onto silica gel. The silica gel was exhaustively eluted (10:1 hexane/MTBE) without fractionation. The eluant was evaporated giving the partially purified title compound which was used as is (57.28g).
Step D 2-trifluoromethvIthio-4-ferf-butvIaniline

A solution of 2-trifluoromethylthio-4-tert-butylnitrobenzene (57.28g; 0.205mol) in methanol (57OmL) was treated with powdered zinc (134.9g; 2.05mol). Acetic acid (145mL) was added dropwise through an attached condenser. The mixture was stirred for Ih at ambient temperature. The reaction mixture was filtered through Celite and the filtrate concentrated. The resulting residue was slurried in CH2CI2
(50OmL) and refiltered through Celite. The filtrate was evaporated onto silica gel. The silica gel was exhaustively eluted (10:1 hexane/MTBE) without fractionation. The eluant was concentrated to an oil. The oil was chromatographed over silica gel (2L c.v.; 20:1 hexane/MTBE) to give the title compound (16.04g).
Step E 2-trifluoromethylthio-4-/grt-butvI-6-bromoaniline

Step F 3-trifluoromethvIthio-5-/gr/-butyI-bromobenzene

A 70° solution of isoamyl nitrite (17.2mL; 129.2mmol) in DMF (1OmL) was treated dropwise with a solution of 2-trifluoromethylthio-4-tørt-butyl-6-bromoaniline (21.2g; 64.6mmol) in DMF (11OmL) over a Ih period. Heating was continued for an additional 1.5h. The reaction was diluted with isopropyl acetate (50OmL) and washed with pH7 phosphate buffer solution (3x200mL). The organic was dried over MgSO-J, filtered and evaporated onto silica gel. The silica gel was placed atop a packed silica gel column and eluted (80OmL c.v.; hexane) to give the title compound as a pale rose colored oil (13.Og). Step G 3-trifluoromethvIthio-5-ferf-butyl-benzonitrile
Step H 3-tr-fluoromethyIthio-5-/er/-butγl-benzamide

Step J 3-trifluoromethylthio-5-ferf-butyl-benzthioamide

Step K 9Jy-fluoren-9-ylmethyl-4-(2-l3-tert-butyl-5-r(trinuoromethvnthiolphenyl>-l,3-thiazoI-4- vQpiperidine-l-carboxylate

Using the method of Example 1, Step G and the products of Example 1, Step C and Example 11, Step J as the starting materials the title compound was obtained.
Step L 4-(2-(3-terι'-butyl-5-r(trifluoroniethyl)thiolphenvO-l,3-thiazol-4-vnpiperidine

Step M 3-f2-r4-(2-f3-terf-butyr-5-rftrifluoromethvnthiolphenvn-l,3-thιazol-4-vnpiperidin-l-yll-2-

Using the method of Example 1, Step L with the products of Example 1, Step K and Example 11, Step L as starting materials the title compound was obtained. lH NMR (CD3OD): δ 8.39 (dd, IH5 J = 4.8,
1.1Hz), 8.38 (s, IH)5 8.15 (t, IH, J = 1.6Hz)3 8.12 (dd, IH, J = 8.O5 1.3Hz), 8.07 (s, IH), 7!8O (s, IH), 7.37 (dd, IH, J = 8.0, 4.8Hz), 7.32 (s, IH), 5.45 (V2AB, IH, J= 17.0Hz), 5.39 ('/2AB3 IH, J= 16.9Hz)3 4.58 (bd, IH, J= 13.3Hz), 4.23 (bd, IH3 J= 13.5Hz)3 3.46 (dt, IH, Jt = 12.4, Jd = 2.5Hz), 3.21 (tt, IH, J = 11.5, 3.6Hz), 2.95 (dt, IH, Jt = 12.8, Jd = 2.5Hz), 2.28 (bd, IH, J= 12.0Hz), 2.15 (bd, IH, J= 12.5Hz)3 1.94 (dquart, IH, Jq = 12.6,' Jd = 3.6Hz), 1.79 (dquart, IH, Jq = 12.6, Jd = 3.6Hz), 1.42 (s, 18H). LRMS calc: 559.2 obs: 560.3 (M+H).
Example 12 Step A 4-methoxy-l/r-imidazole

A solution of potassium carbonate (7.5g, 54.3mrnol) in water (2OmL) was treated with aminoacetonitrile hydrochloride (9.Og, 97.3mmol) in portions with stirring. After effervescence stopped, the solution was extracted in a liq/liq continuous extractor for 2h (EtOAc). The organic was dried over MgSθ4, filtered and concentrated to a residue (2.7Og). Sodium sulfate (7.Og; 50mmol) was placed in a 3-neck flask followed by trimethylorthoformate (7OmL) and con. H2SO4 (2 drops). A variable take-off distillation head was attached and the mixture heated to reflux. Aminoacetonitrile (2.7g, 50mmol) was added dropwise via syringe over 5min. Methanol was distilled at a reflux ratio of 10:1 for 20min. The solution was cooled, filtered and concentrated to a residue (4.9g). The crude methyl (cyanomethyl)imidoformate was dissolved in methanolic NaOCH3 (5OmL; IM). The solution was refluxed for 2h. The now black solution was neutralized with con HCl (4mL). The mixture was filtered and evaporated to a residue which was adsorbed onto silica gel (CH2CI2). The silica gel was eluted (50:50:1 CH2Cl2/acetone/trϊethyIamine) without fractionation to give the title compound (1.16g).
Step B (4-methoxy-l^T-iinidazol-l-γI)acetic acid methyl ester

Step C (4-methoxy-lH -imidazol-l-yl)acetic acid hydrochloride

Using the method. of Example 7, Step D and the product of Example 12, Step B the title compound was obtained.
Step D 4-[2--S-di-tert-butyl-^inethoxyphenvn-l^-thiazoM-yn-l-g-rf^methoxy-l-iniidazol-l- vQacetyl 1 piperidine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 12, Step C as starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.80 (s, 2H), 7.26 (s, IH),
7.13 (s, IH), 6.41 <s, IH), 5.01 (1Z2AB, IH, J = 16.7Hz), 4.95 (1Z2AB, IH5 J = 16.7Hz), 4.59 (bd, IH, J = 12.8Hz), 4.09 (bdd, IH, J= 13.0, 9.3Hz), 3.73 (s, 3H), 3.71 (s, 3H), 3.30 (bt, IH, ./= 12.0Hz), 3.11 (dt, IH, Jt = 11.8, Jd = 3.6Hz), 2.87 (bt, IH, J = 12.9Hz), 2.17 (bd, IH, J = 13.2Hz), 2.1 1 (bd, I H, J = 12.5Hz), 1.79 (dquart, IH, Jq = 12.5, Jd = 3.4Hz), 1.70 (dquart, IH, Jq = 12.7, Jd = 4.2Hz), 1.46 (s, 18H). LRMS 524.3 obs: 525.3 (M+H).
Example 13

A solution of 3-fe?*/-butyI-4-hydroxybenzoic acid (10.Og; 51.5mmol) in methanol (10OmL) was treated with con. H2SO4 (ImL) and refluxed for Ih. The reaction was partitioned between water (15OmL) and isopropyl acetate (20OmL). The organic was dried over MgSO-J, filtered and concentrated to afford the title compound (10.7g).
Step B 3-/grf-butyl-4f(3-methylbut-2-en-l-yl)oxylbenzoic acid methyl ester

A solution of S-Zerz'-butyl^-hydroxybenzoic acid methyl ester (10.7g; 51.3mmol) and l-bromo-3- methy!but-2-ene (6.0OmL; 51.3mmol) in DMF (15OmL) was treated with CS2CO3 (30.1 Og; 92.3mmol).
The mixture was stirred for 48h at ambient temperature. The reaction was partitioned between pH4 phthalate buffer solution (20OmL) and isopropyl acetate (20OmL). The organic was washed with water (2xl50mL), dried over MgSO4, filtered and concentrated to a residue. A portion of the crude (5g) was chromatographed on Siθ2 (1:1 hexane/CH2Cl2). The derived material was flushed with xylene (3xl0mL) affording the title compound (1.38g).
Step C 8-fer/-buty.-4,4-dimethyIchroinaπe-6-carboxylic acid methyl ester

Step D 8-/grf-butyl-4,4-dimethylchromane-6-carboxyh'c acid
Step E 8-tert-butyl-4,4-ditnethylchromane-6-carbonyl chloride

Using the method of Example 1, Step A and the product of Example 13, Step D (309mg; 1.20mmol) as starting material the title compound was obtained.
Step F 8-tert-butyl-4.4-dimethvIchromane-6-carboxamide
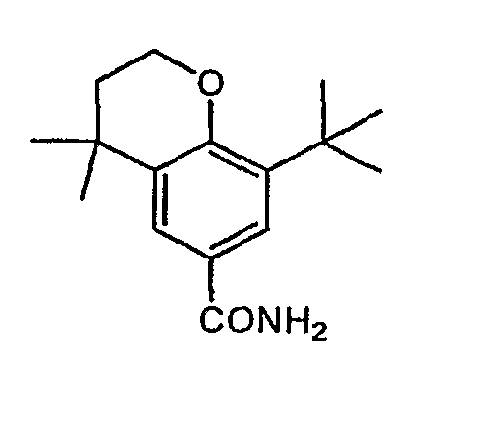
Step G 8-tert-butyl-4,4-dimethylchromane-6-carboxthioainide

Using the method of Example 1, Step F with the product of Example 13, Step F as starting material the title compound was obtained.
Step H 9Jy-fluoren-9-ylmethyl4-r2-(8-fgrf-butvI-4,4-dimethyl-3,4-dihvdro-2^r-chroinen-6-vn-l,3- thiazoI-4-yIlpiperidine-l-carboxvlate

Using the method of Example 1, Step G with the products of Example 1, Step C and Example 13, Step G as starting materials the title compound was obtained.
Step J 4-r2-(8-/er/-butyl-4,4-diinethvI-3,4-diIivdro-2.-y-chromen-6-vn-1.3-thiazol-4-vIlpiperidine

Step K 4-12-(8-ter/-butyl-4,4-dimethvL-3.4-dihvdro-2JHr-chromen-6-vn-lJ-thiazol-4-vIM-r(2,4- dimethvI-177-imidazoI-l-vDacetvIlpiperidine

Using the method of Example 1, Step L with the products of Example 13, Step J and Example 2, Step B as starting materials the title compound was obtained. lH NMR (CDCI3): δ 7.63 (s, IH), 7.52 (s, IH),
6.78 (s, IH), 6.59 (bs, IH), 4.72 (vbs, 2H), 4.64 (bd, IH, J = 13.0Hz)5 3.87 (bd, IH3 J = 13.3Hz), 3.72- 3.60 (bmult, 2H)3 3.29 (bt, IH, J= 12.4Hz)3 3.09 (vbt, IH, J= 10.6Hz), 2.87-2.84 (bmult, 2H), 2.40 (bs, 3H)3 2.30-2.22 (bs overlapping bd, 4H total), 2.16 (bd, IH, J= 10.5Hz), 1.83 (bt, IH, J= 6.8Hz), 1.74 (vbmult, 2H), 1.41 (s, 9H), 1.38 (s, 6H). LRMS calc: 520.3 obs: 521.4 (M+H).
Example 14 Step A 4-chloro-2-methyl-l/_r-imidazoIe

A solution of 2-methyl-lH-imidazoIe (1.05g, 12.84mmol) in acetonitrile (35mL) was treated with N- chlorosuccinimide (1.7 Ig, 12.84mmol). The reaction was stirred 8h and then evaporated. The crude product was digested in CΗ2CI2 (2OmL) and the solution evaporated to a brown residue. The residue was adsorbed onto silica gel and eluted (100:2.5:1 CH2Cl2/MeOH/triethylamine) without fractionation. The eluant was concentrated and the residue chromatographed on silica gel (preparative TLC; 100:5:1 CH2Cl2/methanol/triethylamine). Part of the derived mixture of mono and dichloroimidazoles was rechromatographed on Chiralcel AS stationary phase (5% ethanol/heptane; λ=220nM) to afford the less mobile title compound (109mg).
W
Step B (4-chIoro-2-inethyl-l.H-imidazol-l-yl)acetic acid tert-butyl ester
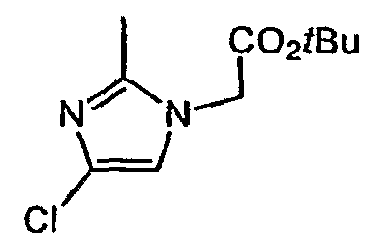
Using the method of Example 2, Step A and the product of Example 14, Step A the title compound was obtained.
Step C H-chloro^-methyl-l/y-imidazol-l-yDacetic acid hydrotrifluoroacetate

Using the method of Example 2, Step B and the product of Example 14, Step B as starting material the title compound was obtained.
Step D l-[f4-chloro-2-methyI-l£?-iniidazol-l-vI)acctvIl-4-r2-f3,S-di-ferf-butvI-4-methoxyptienvn- 1.3-thiazol-4-yllpiper.dine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 14, Step C as the starting materials the title compound was obtained. ^H NMR (CDCI3): δ 7.81 (s, 2H), 6.87 (s,
IH), 6.75 (s, IH), 4.70-4.67 (bs overlapping bd, 3H), 3.87 (bd, IH, J= 13.7Hz), 3.74 (s, 3H), 3.34 (bt,
IH, /= 11.9Hz), 3.12 (tt, IH, J= 11.4, 3.4Hz), 2.90 (bt, 1H,7= 11.7Hz), 2.34 (s, 3H), 2.30 (bd, IH5 J= 11.7Hz), 2.20 (bd, IH, J= 12.5Hz), 1.78 (bdpent, 2H, Jp = 12.5, Jd = 3.3Hz), 1.50 (s, 18H). LRMS calc: 542.3 obs: 543.3 (M+H).
Step E 4-chIoro-2,5-dimethyl-l/r-imidazole

Using the method of Example 14, Step A and 2,4-dimethylimidazole as starting material the title compound was obtained.
Step F (4-chIoro-2,5-dimethyl-lJ?'-iinidazol-l-vnacetic acid /erf-butyl ester

Using the method of Example 2, Step A and the product of Example 14, Step E the title compound was obtained.
Step G (4-chloro-2,5-dimethvI-l/r-imidazol-l-yl)acetic acid hydrotrifluoroacetate

Using the method of Example 2, Step B and the product of Example 14, Step F as starting material the title compound was obtained.
Step H l-[f4-ch1oro-2.5-dimethyl-l/r-imidazol-l-vnacetyll-4-r2-f3.5-di-terf-butyl-4- methoxyplienvI)-1..3-thiazol-4-vIlpiperidine

IH), 4.65 (bd, IH, J= 13.2Hz), 4.59 (V2AB, IH, J= 16.7Hz), 4.55 ('/2AB3 IH, J= 16.7Hz), 3.91 (bd, IH, J = 13.4Hz), 3.71 (s, 3H), 3.33 (bt, IH, J= 12.1Hz), 3.13-3.06 (mult, 2H), 2.87 (bt, IH3 J= 12.0Hz), 2.31-2.28 (s overlapping with bd, 4H total), 2.17 (bd, IH, J= 13.3Hz), 2.09 (s, 3H), 1.76 (bquart, 2H, J= 11. IHz), 1.47 (s, 18H). LRMS calc: 556.3 obs: 557.4 (M+H).
Example 15 Step A 3.5-di-fgrt-butylbenzoyl chloride

Using the method of Example 1, Step A and 3,5-di-tø-/-butylbenzoic acid as the starting material the title compound was obtained.
Step B 3.5-di-fert-butyIbenzamide

Using the method of Example 13, Step F with the product of Example 15, Step A as the starting material the title compound was obtained.
Step C 3,5-di-fgrf-butyIbenzthioamide

Using the method of Example 1, Step F with the product of Example 15, Step B as the starting material the title compound was obtained.
Step D 9flr-fluoren-9-ylmethvI-4-r2-f3.5-di-ferf-butylphenvn-1.3-thiazol-4-vIlpiperidine-l- carboxylate

Using the method of Example 1, Step G with the products of Example 1, Step C and Example 15, Step C as the starting materials the title compound was obtained.
Step E 4-[2-f3,5-di-fer/-butylphenvπ-1.3-thiazol-4-vIlpiperidine

Using the method of Example 1, Step H with the product of Example 15, Step D as the starting material the title compound was obtained.
Step F l-(chIoroacetyl)-4-f2-(3.,5-di-ter/-butvIphenvπ-1.3-thiazoI-4-yllpiperidine

Step G 3-(2-{4-f2-f3,5-di-rgrf-butylphenyl)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoethvπ-3Jy- imidazof4,5-c]pyridine and l-(2-{4-[2-(3,5-di-terf-butylphenvI)-l,3-thiazol-4-yllpiperidin-l-yl}-2- oxoethvO-l.flr-iinidazof4,5-clpyridine


A TEDF solution of the product of Example 15, Step F was added to a solution of 5-azabenzimidazole (48mg; 0.405mmol) in dry DMF (ImL). Cesium carbonate (132mg; 0.405mmol) was added and the mixture stirred for 2h. The reaction was diluted with isopropyl acetate (1OmL) and washed with pH7 phosphate buffer solution (3x1 OmL). The organic was dried over MgSθ4, filtered and evaporated to a residue. Chromatography over silica gel (prep. TLC; 10:1 CH2Cl2/MeOH) afforded the more mobile 3- (3H) title compound and the less mobile 1-(IH) title compound. *Η NMR (CD3OD): δ 8.88 (s, IH), 8.37 (s, IH), 8.36 (d, IH, J = 4.0Hz)5 7.76 (d, 2H, J = 1.8Hz)5 7.73 (d, IH, J = 5.8Hz), 7.57 (t, IH, J = 1.7Hz), 7.21 (s, IH), 5.55 (14AB, IH, J = 17.2Hz), 5.49 ('/2AB5 IH, J = 17.1Hz), 4.58 (bd, IH, J = 13.3Hz), 4.16 (bd, IH, J= 13.5Hz), 3.43 (dt, IH, Jt = 13.6, Jd = 2.3Hz), 3.18 {iϋ, IH, J= 11.8, 3.4Hz), 2.93 (dt, IH, Jt = 12.9, Jd = 2.3Hz), 2.26 (bd, IH, J= 13.3Hz), 2.14 (bd, IH, J= 13.2Hz), 1.95 (dquart, IH, Jq = 12.7, Jd = 4.0Hz), 1.77 (dquart, IH, Jq = 12.5, Jd = 4.0Hz), 1.37 (s, 18H). LRMS calc: 515.3 obs: 516.4. lH NMR (CD3OD): δ 8.94 (s, IH), 8.36 (d, IH, J= 5.8Hz), 8.31 (s, IH), 7.76 (d, 2H3 J= 1.7Hz), 7.64 (d, I H, J= 5.8Hz), 7.57 (t, IH, J= 1.7Hz), 7.21 (s, IH), 5.48 ('/2AB5 IH, J= 17.3Hz), 5.42
(!4AB, IH, J= 17.2Hz), 4.57 (bd, I H5 /= 13.3Hz), 4.16 (bd, IH, J= 13.5Hz), 3.43 (dt5 IH, Jt = 14.0, Jd
= 2.5Hz), 3.18 (tt, IH5 J= 11.5, 3.3Hz)5 2.93 (bt, IH5 J= 12.7Hz)5 2.26 (bd, IH, J= 13.2Hz), 2.14 (bd, IH, J= 13.2Hz), 1.95 (dquart, IH, Jq = 12.6, Jd = 3.6Hz)5 1.77 (dquart, IH, Jq = 12.8, Jd = 3.8Hz)5 1.37
(s, 18H). LRMS calc: 515.3 obs: 516.4 (M+H).
Example 16
Step 3-f2-{4-f2-f3,5-di-terf-butylphenvn-l,3-thiazoI-4-yllρiperidin-l-vU-2-oxoethyl)-3/r-
Jinidazof4,5-61pyridine and l-(2-(4-f2-(3.,5-di-fer/'-butylphenyl)-l^-thiazol-4-vnpiperidin-l-vU-2- oxoethvD-l/y-imidazof4,5-6]pyridine
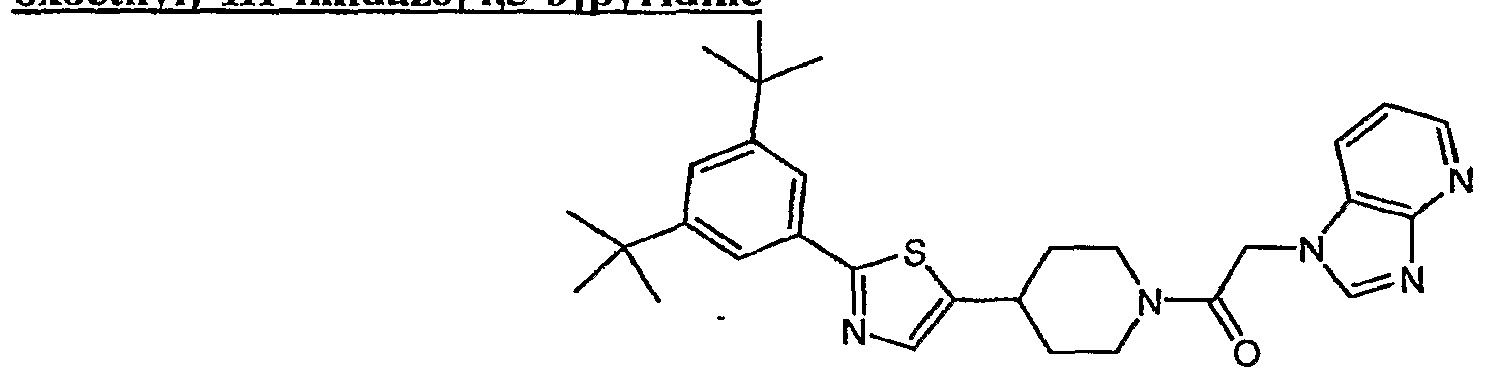

A THF solution of the product of Example 15, Step F was added to a solution of 4-azabenzimidazole (774mg; 6.50mmol) in dry DMF (6mL). Cesium carbonate (2.12g; 6.50mmol) was added and the mixture stirred for 2h. The reaction was diluted with isopropyl acetate (5OmL) and washed with brine (3x15mL). The organic was dried over MgSO4, filtered and evaporated to a residue. Chromatography over silica gel (prep. TLC; 15:1 CH2Cl2/MeOH) afforded the more mobile 3-(3H) and the less mobile 1- (IH) title compounds (20mg and 80mg respectively). lΗ NMR (CD3OD): δ 8.46 (dd, IH5 J = 4.8, Ll Hz), 8.40 (s, IH), 8.03 (dd, IH, J= 8.2, 1.3Hz), 7.78 (d, 2H5 J= 1.6Hz), 7.59 (t, IH, J= 1.6Hz), 7.37 (dd, IH, J= 8.2, 4.8Hz)5 7.22 (s, IH)5 5.49 ('/2AB5 IH, J= 17.2Hz), 5.43 ('/2AB5 IH5 J= 17.1Hz)5 4.59 (bd, IH5 J = 13.0Hz), 4.18 (bd, IH, J = 13.7Hz), 3.44 (dt, IH, Jt = 13.3, Jd = 2.5Hz)5 3.19 (tt, IH, J =
1 L95 3.7Hz), 2.94 (bt, IH, J = 13.1Hz), 2.26 (bd, IH, J = 12.8Hz), 2.15 (bd, IH5 J = 12.6Hz)5 1.96 (dquart, IH5 Jq = 12.5, Jd = 3.8Hz), 1.77 (dquart, IH, Jq = 12.6, Jd = 3.8Hz), 1.39 (s, 18H). LRMS calc: 515.3 obs: 516.4 (M+H). lH NMR (CD3OD): δ 8.39 (dd, IH, J= 5.1, 1.4Hz), 8.38 (s, IH), 8.12
(dd, IH, J = 8.2, 1.4Hz), 7.78 (d, 2H3 J = 1.8Hz), 7.58 (t, IH, J = 1.8Hz), 7.37 (dd, IH, J= 8.1, 4.9Hz)5 7.22 (s, IH), 5.44 (V2AB, IH3 J= 16.9Hz), 5.38 ('/zAB, I H3 J= 16.9Hz), 4.58 (bd, IH3 J= 13.3Hz), 4.23 (bd, IH, J= 13.5Hz), 3.45 (dt, IH, Jt = 13.0, Jd = 2.5Hz), 3.19 (tt, IH, J= 11.4, 3.7Hz)5 2.93 (dt, IH5 Jt = 12.8, Jd = 2.5Hz), 2.28 (bd, IH, J= 13.0Hz), 2.14 (bd, IH, J= 12.8Hz), 1.98 (dquart, IH3 Jq = 12.5, Jd = 4.1Hz), 1.77 (dquart, IH, Jq = 12.6, Jd = 4.0Hz), 1.39 (s, 18H). LRMS calc: 515.3 obs: 516.3 (M+H).
Example 17 Step A N-Ethylaminoacetone hydrochloride
Using the method of Example 5, Step A with N-ethyl benzyl amine in place of N-methyl benzyl amine as starting material the title compound was obtained.
Step B (3-ethyl-5-methyl-2-oxo-2,3-dihydro-liy-imidazol-l-yl)acetic acid ethyl ester

A solution of N-ethylaminoacetone hydrochloride (503mg; 3.66mmol) in dry DMF (1OmL) was treated with ethyl isocyanatoacetate (472mg; 3.66mτnol) followed by cesium carbonate (1.19g; 3.66mmol). The mixture was stirred at ambient temperature for 16h. The mixture was filtered and the filtrate flushed with xylene (3x30mL; lmm vacuum; 60° bath). The derived residue was dissolved in dry benzene (2OmL) and treated with TFA (4mL). The solution was refluxed 30min. The reaction was evaporated and flushed with toluene (2x30mL). The crude product was adsorbed onto silica gel and eluted (MTBE) without fractionation. The eluant was evaporated to a residue and chromatographed over silica gel (preparative TLC3 MTBE) to give the title compound (429mg).
Step C Potassium (3-ethvl-5-methyl-2-oxo-2.3-dihvd ro-liZ-imidazol-l-yDacetate

Step D 3-f2-f4-f2-(3,5-di-/gr^-butyl-4-inethoxyphenvπ-l,3-thiazol-4-vnpiperidin-l-vU-2-oxoethyl)- l-ethyl-4-methvI-l,3-dihvdro-2flr-iinidazol-2-one

Using the method of Example 1, Step L with the products of Example I, Step H and Example 17, Step C as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.84 (s, 2H), 7.18 (s,
IH), 6.26 (s, 1 H), 4.69 ('/2AB5 IH, J = 17.2Hz), 4.61 ('/2AB5 IH, J= 17.1Hz), 4.60 (bd, IH5 J= 13.0Hz), 4.15 (bd, IH, J= 13.5Hz), 3.75 (s, 3H), 3.66 (quart, 2H, J = 7.3Hz), 3.36 (bt, IH, J= 12.0Hz), 3.15 (tt,
IH, J = 11.7, 3.2Hz)5 2.91 (bt, IH5 J = 1 1.8HzX 2.22 (bd, IH, J = 12.5Hz), 2.15 (bd, IH, J= 12.7Hz),
1.85 (dquart, IH, Jq = 12.3, Jd = 3.6Hz), 1.73 (dquart, IH, Jq = 12.5, Jd = 3.4Hz), 1.50 (s, 18H), 1.27 (t,
3H, J= 7.2Hz). LRMS calc: 552.3 obs: 553.3 (M+H).
Example 18
Step A Aminoacetone hydrochloride
A solution of chloroacetone (800μL; 10.05mmol) in dry d^-deuteroacetone (1OmL) was treated with sodium azide (790mg; 12.14mmol). The mixture was stirred for 18h (progress followed by NMR). The mixture was filtered and concentrated to a residue (850mg). The residue was dissolved in ethanol (1OmL) and treated with con. HCl (2mL). The solution was combined with 10% Pd/C hydrogenation catalyst (175mg) and shaken under a hydrogen atmosphere (50psi) for 2h. The mixture was filtered through Celite and evaporated to give the title compound (707mg).
Step B l-ethyl-S-methvI-l,3-dihvdro-2.Hr-imidazoI-2-one

Step C P-ethyl^-methyl-Z-oxo^^-dihydro-liy-imidazot-l-vOacetic acid tert-butyl ester

A solution of l-ethyl-5-methyl-l,3-dihydro-2H-imidazol-2-one (250mg; 1.98mmol) in dry DMF (6mL) was treated with /erf-butyl bromoacetate (289μL; 1.98mmol). Sodium hydride (59mg; 80%; 1.98mmol) was added and the mixture stirred at ambient temperature for Ih. The reaction mixture was diluted with isopropyl acetate (3OmL) and washed with pH7 phosphate buffer (3xl0mL). The organic was dried over MgSθ4, filtered and evaporated to a residue. The crude product was chromatographed over silica gel (preparative TLC; 30:1 CKføCtø/MeOH) to give the title compound (172mg).
Step P (3-ethyl-4-methyl-2-oxo-23-dihvdro-l//-imidazol-l-yl)acetic acid

Using the method of Example 2, Step B and the product of Example 18, Step C as starting material the title compound was obtained.
Step E l-(2-(4-[2-(3,5-di-tert-butγI-4-ιnethoxyphenyl)-13-thiazol-4-ylTpiperidin-l-vU-2-oxoethvπ- 3-ethyl-4-methvI-l,3-dihvdro-2/7-imidazol-2-one

1.4Hz), 4.62 (V2AB, IH5 J= 16.7Hz)5 4.60 (bd, IH5 J= 13.0Hz)5 4.52 ('/2AB5 IH5 J= 16.8Hz)5 4.06 (bd, IH5 J= 13.5Hz)5 3.73 (s, 3H)5 3.70 (quart, 2H, J = 7.3Hz), 3.30 (bt, IH5 J = 11.4Hz)5 3.12 (tt, IH5 J = 11.5, 3.4Hz), 2.87 (dt, IH5 Jt = 11.8, Jd = 2.7Hz), 2.19 (bd, IH, J= 13.3Hz)5 2.14-2.11 (bd overlapping d, 4H total, Jd = 1.3Hz), 1.81 (dquart, IH5 Jq = 12.6, Jd = 3.7Hz), 1.70 (dquart, IH, Jq = 12.5, Jd = 3.8Hz)5 1.48 (s5 18H)5 1.23 (t5 3H5 J= 7.2Hz). LRMS calc: 552.3 obs: 553.4 (M+H).
Step F l-(2-f4-r2-(3,5-di-terif-butylphenvn-l,3-thiazoI-4-vπpiperidin-l-vn-2-oxoethvn-3-ethyl-4- methyl-13-dihvdro-2/r-imidazoI-2-one

Using the method of Example 1, Step L with the products of Example 15, Step E and Example 18, Step D as the starting materials the title compound was obtained. 1H NMR (CD3OD): δ 7.75 (d5 2H5 J = 1.6Hz)5 7.55 (t, IH, J= 1.7Hz), 7.17 (s, IH)5 6.16 (s, IH), 4.59 (MB overlapping bd, 2H total, JAB = 16.7Hz), 4.51 ('/2AB, IH, J= 16.6Hz), 4.05 (bd, IH, J= 13.1Hz), 3.69 (quart, 2H, J= 7.2Hz), 3.31 (bt, IH5 J = 12.0Hz), 3.12 (tt, IH, J = 1 1.7, 3.5Hz), 2.86 (bt, IH5 J = 12.8Hz), 2.17 (bd, IH, J = 12.9Hz), 2.12-2.09 (s overlapping bd, 4H total), 1.80 (dquart, IH5 Jq = 12.2, Jd = 3.8Hz), 1.70 (dquart, IH, Jq = 12.3, Jd = 3.8Hz), 1.36 (s, 18H)5 1.21 (t5 3H5 J= 7.3Hz). LRMS calc: 522.3 obs: 524.4 (M+H).
Example 19
Step A (2-oxo-2,3-dihvdro-liy-benzimidazol-l-vOaeetic acid fer/-butvl ester
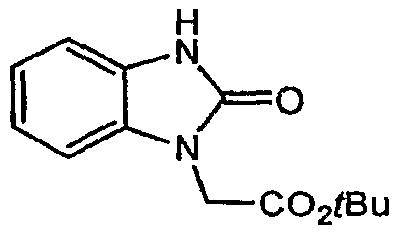
Using the method of Example 18, Step C and l,3-dihydro-2H-benzimidazol-2-one as starting material the title compound was obtained.
Step B (2-oxo-23-dihvdro-l/-f-benzimidazol-l-v0acetic acid

Using the method of Example 2, Step B with the product of Example 19, Step A as starting material the title compound was obtained.
Step C l-f2-{4-12-f3.5-di-ter/-butvI-4-methoxyphenvn-13-thiazol-4-vnpiperidin-l-vU-2-oxoethyl)- l,3-dihydro-2.fl-benzimidazol-2-one

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 19, Step B as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.83 (s, 2H), 7.15 (s,
I H), 7.07 (bmult, 4H), 4.89 (MB, IH, J = 17.1Hz), 4.81 ('ΛAB, IH, J = 17.0Hz), 4.58 (bd, IH, J = 13.5Hz), 4.19 (bd, IH5 J= 13.4Hz), 3.73 (s, 3H)5 3.38 (bt, IH5 J= 12.3Hz), 3.14 (tt, IH5 J= 11.7, 3.7Hz)5 2.89 (bt, IH, J= 12.3Hz)5 2.22 (bd, IH, J= 13.2Hz), 2.12 (bd, IH5 J= 13.3Hz), 1.85 (dquart, IH, Jq = 12.5, Jd = 3.7Hz), 1.70 (dquart, IH, Jq = 12.6, Jrf = 3.6Hz), 1.48 (s, 18H). LRMS calc: 560.3 obs: 561.4 (M+H).
Step D C3-methyI-2-oxQ-2,3-dihvdro-lfl'-benzimidazoI-l-vnacetic acid tert-butvl ester

Using the method of Example 18, Step C with the product of Example 19, Step A and methyl iodide as the starting materials the title compound was obtained.
tep E (3-metfayI-2-oxo-2^3-dihydro-lJ?-benzimidazoI-l-vπacetic acid

Using the method of Example 2, Step B with the product of Example 19, Step D as starting material the title compound was obtained.
Step F l-f2-(4-r2-f3,5-di-fert-butyl-4-methoxyphenvn-13-thiazol-4-vnpiperidin-l-vI}-2-oxoethvn- 3-methyl-13-dihvdro-2/-r-benzimidazol-2-one

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 19, Step E as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.81 (s, 2H), 7.15-7.07
(mult, 5H), 4.92 (1MB, IH, J = 17.2Hz), 4.85 (14AB, IH, J= 17.0Hz), 4.56 (vbs, IH), 4.19 (bd, IH, J = 13.0Hz), 3.72 (s, 3H), 3.44 (s, 3H), 3.38 (bt, IH, J= 13.1Hz), 3.14 (tt, IH, J= 11.5, 3.9Hz), 2.89 (dt, IH, Jt = 12.5, Jd = 2.2Hz), 2.22 (bd, IH, J= 13.2Hz), 2.12 (bd, IH3 J= 13.3Hz), 1.86 (dquart, IH, Jq = 12.5, Jd = 3.7Hz), 1.70 (dquart, IH, Jq = 12.6, Jd = 3.6Hz), 1.47 (s, 18H). LRMS calc: 574.3 obs: 575.4 (M+H).
Example 20 Step A 2-ethvl-4-methv--l;Hr-imidazol-l-vlacet.e acid έeri-butyl ester

Using the method of Example 2, Step A and 2-ethyl-4-methyIimidazole as starting material the title compound was obtained. The title compound was purified by silica gel chromatography (preparative TLC; 100:2.5:1 CH2Cl2/MeOH/Et3N).
Step B 2-ethyl-4-methγI- 1H-imidazol-l-ylacetic acid hydrotrifluoroaeetate

Using the method of Example 2, Step B and the product of Example 20, Step A as starting material the title compound was obtained.
Step C 4-[2-(3,5-tert-butyl-^methoxyphenvn-l^-thiazoI-^vil-l-f^-ethvM-methyl-lJy- imidazol-l-vOaeetyllpiperidine

Using the method of Example 1, Step L and starting with the products of Example 1, Step H and Example 20, Step B the title compound was obtained. 1H NMR (CDCI3): δ 7.81 (s, 2H), 6.86 (s, IH),
6.56 (bs, IH)5 4.74-4.56 (bmult, 3H), 3.89 (bd, IH, J= 12.6Hz), 3.73 (s, 3H), 3.31 (bt, IH, J= 12.8Hz), 3.11 (vbt, IH, J = 10.0Hz), 2.87 (bt, IH, J = 11.6Hz)5 2.64 (quart, 2H, J = 7.5Hz), 2.33-2.15 (s overlapping bmult, 5H), 1.82-1.62 (bmult, 2H), 1.49 (s5 18H)5 1.35 (t, 3H, J = 7.6Hz). LRMS calc: 536.3 obs: 537.4 (M+H).
Example 21 Step A 3,5-di-fert-butyl-4-hvdroxybenzoyl chloride

Using the method of Example 1, Step A and 3,5-di-/ert-butyl-4-hydroxybenzoic acid as the starting material the title compound was obtained.
Step B 3,5-di-ferf-butyl-4-hvdroxybenzamide

Using the method of Example 13, Step F with the product of Example 21, Step A as the starting material the title compound was obtained.
Step C 3,5-di-fer<'-butvI-4-livdroxybenzthioaπiide

Using the method of Example 1, Step F with the product of Example 21, Step B as the starting material the title compound was obtained.
Step D 9iHr-fluoren-9-ylmethyl-4-f2-f3,5-di-ter<'-butvI-4-hvdroxyphenv0-13-thiazol-4-yllpiperidine- 1-carboxvlate

Using the method of Example 1, Step G with the products of Example 1, Step C and Example 21, Step C as the starting materials the title compound was obtained.
tep E 2,6-di-tert-butyl-4-(4-piperidin-4-yl-l^-thiazoI-2-vnphenol

Using the method of Example 1, Step H with the product of Example 21, Step D as the starting material the title compound was obtained.
Step F 2,6-di-tert-butyl-4-f4-ri-('chloroacetyl)piperidin-4-vU-1.3-thiazol-2-vi} phenol

Using the method of Example 15, Step F with the product of Example 21, Step E as the starting material the title compound was obtained.
Step G 2,6-di-tert-butyl-4-H-H-(liy-imidazo[4.t5-c]pyridin-t-ylacetyl)piperidin-4-yH-l<3-thiazol-2- yllphenol and 2,6-di-tert-butvI-4-(4-ri-(3 H-- imidazor4.5-g1pyridin-3-ylacetyl)piperidin-4-vn-l,3- thiazoI-2-yll phenol

Using the method of Example 15, Step G with the product of Example 21, Step F as starting material the title compounds were obtained.lH NMR (CD3OD): δ 8.93 (s, IH)3 8.36 (d, IH, J = 5.7Hz), 8.31 (s,
IH), 7.72 (s, IH), 7.64 (d. IH, J= 5.7Hz), 7.1 1 (s, IH), 5.47 ('/2AB5 IH5 J= 17.3Hz)5 5.41 ('/2AB5 IH5 J=
17.1Hz)5 4.57 (bd, IH5 J= 10.4Hz)54.15 (bd, IH, J= 12.2Hz), 3.42 (bt, IH, J= 13.0Hz)5 3.17 (tt, IH5 J= 11.5, 3.2Hz)5 2.93 (dt, IH, Jt = 13.2, Jd = 2.4Hz)5 2.24 (bd, IH, J= 12.7Hz)5 2.13 (bd, IH5 J= 12.3Hz)5
1.92 (dquart, IH, Jq = 12.I5 Jd = 3.6Hz), 1.74 (dquart, IH, Jq = 12.5, Jd = 3.8Hz), 1.47 (s, 18H). LRMS calc: 531.3 obs: 532.3 (M+H). lH NMR (CD3OD): δ 8.88 (s, IH)5 8.37 (s overlapping d5 2H total, Jd
= 5.7Hz), 7.73 (s overlapping d, 3H total, Jd = 6.5Hz), 7.11 (s, IH), 5.54 (y2AB, IH5 J= 17.1HZ)5 5.48
(1AAB, IH5 J= 17.2Hz)5 4.57 (bd, IH5 J= 13.8Hz), 4.15 (bd, IH5 J= 12.4Hz), 3.43 (bt, IH5 J= 13.2Hz), 3.14 (tt, IH5 J = 11.7, 3.6Hz)5 2.92 (bt, IH5 J = .12.6Hz)5 2.24 (bd, IH, J= 12.4Hz)5 2.12 (bd, IH5 J =
12.0Hz)5 1.92 (dquart, IH5 Jq = 12.6, Jd = 3.8Hz)5 1.74 (dquart, IH5 Jq = 12.6, Jd = 4.0Hz)5 1.47 (s5
18H). LRMS calc: 531.3 obs: 532.2 (M+H).
Example 22 Step A 2,6-di-fgjrf-butvI-4-(4-fl-(3jy-imidazor4,5-^lpyridin-3-ylacetyl)piperidin-4-vH-13-thiazol-2- vU phenol

Using the method of Example 1 , Step L with the products of Example I5 Step K and Example 21, Step E as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 8.37 (dd, IH5 J= 4.8,
1.3Hz)5 8.36 (S5 IH)5 8.10 (dd5 IH5 /= 8.0, 1.3Hz)5 7.72 (s, 2H)5 7.35 (dd, IH5 J = 7.9, 4.8Hz), 7.10 (s5 IH), 5.42 ('/2AB5 IH5 J= 17.1Hz), 5.36 (KzAB, IH5 J= 17.0Hz), 4.56 (bd, IH5 J= 11.4Hz)5 4.20 (bd, IH5 J= 11.4Hz), 3.42 (bt5 IH, J= 11.9Hz)5 3.13 (btt, IH5 J= 11.4, 3.2Hz), 2.91 (bt, IH, J= 12.8Hz), 2.25 (bd, IH, J= 12.7Hz)5 2.12 (bd, IH, J= 12.9Hz)5 1.93 (dquart, IH, Jq = 12.7, Jd = 3.5Hz)3 1.73 (dquart, IH, Jq = 12.6, Jd = 3.6Hz), 1.47 (s5 18H). LRMS calc: 531.3 obs: 532.2 (M+H).
Step B 2,6-di-fer/-butvI-4-f4-a-rf3,5-dimethyl-l.jy-l^,4-triazol-l-vI)acetvilpiperidin-4-yll-1.3- thiazoI-2-yl)phenol

Using the method of Examp e 1, Step L with the products of Example 21, Step E and (3,5-dimethyl-lH- 1,2,4-triazol-l-yl)acetic acid as the starting materials the title compound was obtained. lΗ NMR
(CD3OD): δ 7.73 (S, 2Η), 7.10 (s, IH), 5.20 ('/2AB5 IH, J= 17.2Hz), 5.14 (V-AB, IH, J= 17.0Hz), 4.58
(bd, IH3 J= 13.3Hz), 4.08 (bd, IH, J= 14.1Hz), 3.34 (bt, IH5 J= 11.4Hz), 3.13 (tt, IH5 J= 11.9, 3.5Hz), 2.91 (dt, IH, Jt = 12.8, Jd = 2.3Hz), 2.38 (s, 3H), 2.28 (s, 3H), 2.22 (bd, IH, J= 12.8Hz), 2.14 (bd, 1 H, J = 12.8Hz), 1.84 (dquart, IH, Jq = 12.8, Jd = 4.1Hz), 1.71 (dquart, IH, Jq = 12.6, Jd = 4.1Hz), 1.48 (s, 18H). LRMS calc: 509.3 obs: 510.3 (M+H).
Example 23
Step A r4-(hvdroxymethvi)-5-methyl-l/r-imidazol-l-vπacetic acid methyl ester and f5- fliydroxymethvD-4-methyl-l/jr-iniidazol-l-yl1acetic acid methyl ester

Using the method of Example 3, Step B and 4-(hydroxymethyl)-5-methyl-lH-imidazole as starting material the title compounds were obtained. The crude product was adsorbed onto silica gel and eluted without fractionation (10:1 CΗ2Cl2^MeOΗ). The eluant was evaporated and the derived residue chromatographed on Chiralcel OJ stationary phase (15%EtOH/heptane; λ=215nM). The more mobile 5- hydroxymethyl and the less mobile 4-hydroxymethyl title isomers were isolated.
Step B Potassium f4-fhvdroxymethvD-5-methyl-l/-r-imidazoI-l-vπacetate

Step C fl-(2-{4-r2-(3,5-di-fg/-f-butγϊ-4-methoxyphenyl)-l,3-thiazol-4-γllpiperidin-l-vU-2-oxoethvn- 5-methyl-l//-imidazol-4-v.lmethanol

Using the method of Example 1, Step L and starting with the products of Example 1, Step H and Example 23, Step B the title compound was obtained. lH NMR (CD3OD): δ 7.84 (s, 2H), 7.53 (s, IH),
7.19 (s, IH), 5.09 C/2AB, IH, J= 17.1Hz), 5.01 (1Z2AB, IH, J= 17.3Hz), 4.62 (bd, IH, J= 11.5Hz), 4.52 (bs, 2H), 4.1 1 (bd, IH, J= 13.7Hz), 3.75 (s, 3H), 3.38 (bt, IH, J= 14.4Hz), 3.17 (bdt, IH, Jt= 11.7, Jd = 3.7Hz), 2.93 (bt, IH5 J= 13.0Hz), 2.26-2.15 (s overlapping bmult, 5H total), 1.87 (dquart, IH, Jq = 12.6, Jd = 3.2Hz), 1.75 (dquart, IH, Jq = 12.5, Jd = 4.0Hz), 1.50 (s, 18H). LRMS calc: 538.3 obs: 539.3 (M+H).
Step D Potassium r5-fhvdroxymethyl)-4-nietfavI-l/r-imidazol-l-vH acetate

Using the method of Example 3, Step C and [5-(hydroxymethyl)-4-methyl-li/-imidazol-l-yl]acetic acid methyl ester (Example 23, Step A) as starting material the title compound was obtained.
Step E ri-f2-(4-f2-f3,5-di-ferf-butyl-4-methoxyphenyl)-1.3-thiazol-4-v]lpiperidiiπ-l-vI>-2-oxoethvI>- 4-methyl-l//-imidazol-5-ylϊmethanoI

Using the method of Example 1, Step L and starting with the products of Example 1, Step H and Example 23, Step D the title compound was obtained. ΪH NMR (CD3OD): δ 7.80 (s, 2H)5 7.59 (s, IH),
7.15 (s, IH), 5.14 (V2AB, IH, J = 17.0Hz), 5.07 ('/2AB, IH, J= 17.1Hz), 4.59 (bd, IH5 J= 13.3Hz)5 4.49 (s, 2H), 4.07 (bd, IH, J= 13.9Hz), 3.71 (s, 3H), 3.35 (dt, IH, Jt = 14.1, Jd = 2.6Hz), 3.13 (tt, IH, J = 1 1.7, 3.7Hz), 2.89 (dt, 1 H, Jt = '3-0, Jd = 2.3Hz), 2.21-2.16 (s overlapping bd, 4H total), 2.12 (bd, IH, J = 13.8Hz), 1.86 (dquart, IH, Jq = 12.6, Jd = 3.7Hz), 1.71 (dquart, IH, Jq = 12.6, Jd = 3.8Hz), 1.46 (s, 18H). LRMS calc: 538.3 obs: 539.3 (M+H).
Example 24
Step A 2-methylthio-liy-imidazole

Using the method of Example 7, Step A and 2-mercaptoimidazole as starting material the title compound was obtained.
Step B f2-(methylthiQ)-lJy-imidazol-l-yllacetic acid methyl ester

Using the method of Example 7, Step B and the product of Example 24, Step A as starting material the title compound was obtained as the sole product.
Step C [2-(metlrylthioyi//-imidazol-l-yl]acetic acid hydrochloride

Using the method of Example 7, Step D and the product of Example 24, Step B as starting material the title compound was obtained.
Step D 4-r2-(3,5-di-terf-butyl-4-methoxyphenvn-l,3-thiazol-4-vIl-l-f2-(methylthio)-lJfir-imidazoI-l- yll acetyl? piperid ine

Using the method of Example I5 Step L with the products of Example 1, Step H and Example 24, Step C as starting materials the title compound was obtained. I H NMR (CD3OD): δ 7.81 (s, 2H), 7.15 (s, IH),
7.14 (d, IH, J = L2Hz), 7.03 (d, IH, J = 1.3Hz), 5.14 (Y2AB, IH, J = 16.9Hz), 5.06 ('ΛAB, IH, J = 16.7Hz), 4.57 (bd, IH5 J= 13.3Hz), 4.09 (bd, IH, J= 13.5Hz), 3.72 (s, 3H), 3.36 (bt, IH, J= 11.8Hz),
3.14 (vbt, IH, J= 12.0Hz), 2.90 (bt, IH, J= 12.9Hz), 2.45 (s, 3H), 2.21 (bd, IH3 J= 11.8Hz)5 2.13 (bd,
IH, J= 12.2Hz), 1.85 (dquart, IH, Jq = 12.6, Jd = 3.7Hz), 1.71 (dquart, IH, Jq = 13.0, Jd = 3.5Hz), 1.47
(s, 18H). LRMS calc: 540.3 obs: 541.4 (M+H).
Step E fl-fmethylsulfonvD-lZr-imidazo.-l-vπ acetic acid methyl ester

Using the method of Example 7, Step C and the product of Example 24, Step B as starting material the title compound was obtained.
Step F r2-fmethylsuIfonvD-l//-iinidazol-l-yl]acetie acid hydrochloride

Step G 4-r2-f3.5-di-tert-butyI-4-niethoxyphenvn-l.,3-thiazoI-4-vIl-l-{2-fmethvIsuIfonvπ-lg- imidazol-l-yllacetyl}piperidine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 24, Step F as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.81 (s, 2H), 7.32 (s,
IH), 7.20 (s, IH), 7.14 (s, IH), 5.47 (1AAB, IH, J= 16.8Hz), 5.36 (»/2AB, IH, J= 16.6Hz), 4.57 (bd, IH, J= 13.9Hz), 4.00 (bd, IH, J = 13.7Hz), 3.71 (s, 3H), 3.34 (bt, IH, J = 13.1Hz), 3.23 (s, 3H), 3.14 (bt, IH, J= 11.6Hz), 2.90 (bt, IH, J= 11.9Hz), 2.19 (bd, IH, J= 13.1Hz), 2.12 (bd, IH, J= 12.9Hz), 1.89 (dquart, IH, Jq = 12.4, Jd = 3.3Hz), 1.71 (dquart, IH, Jq = 12.5, J& = 3.5Hz), 1.46 (s, 18H). LRMS calc: 572.3 obs: 573.4 (M+H).
Example 25
Step A /erf-butyl methyl 2,2'-(4-methyl-l.ff-imidazole-l,5-divDdiaeetate and fer/-butyl methyl 2,2r- (S-methyl-l/T-imidazole-l^-diyDdiaeetate

Step B r4-(2-methoxy-2-oxoethviy5-methyl-l/y-imidazoI-l-yllacetic acid hvdrotrifluoroacetate

Using the method of Example 2, Step B and fer/-butyl methyl 2,2'-(5-rnethyl-I//-imidazoIe-l ,4- diyl)diacetate (Example 25, Step A) as starting material the title compound was obtained.
Step C fl-f2-f4-[2-(3,5-di-ter/-butvI-4-methoxyphenyl)-l^-thiazol-4-yllpiperidin-l-vU-2-oxoethvπ- 5-methγI-lJy-imidazoI-4-yllacetic acid methyl ester

Using the method of Example 1, Step L with the product of Example 1, Step H and the product of Example 25, Step B as starting materials the title compound was obtained.
Step D fl-(2-{4-r2-(3,5-di-fgrf-butyl-4-methoxyphenvn-l.,3-thiazol-4-yl]piperidiιi-l-vU-2-oxoethyl) 5-methyl-li?-iinidazol-4-vIlacetic dihydrochloride


Using the method of Example 25, Step D with the product of Example 25, Step F as starting material the title compound was obtained. ^H NMR (CD3OD): δ 8.87 (s, IH), 7.96 (s, 2H), 7.69 (s, I H), 5.48
(V2AB, IH, J = 16.9Hz), 5.34 (!4AB, IH, J = 17.0Hz), 4.62 (bd, IH, J = 12.6Hz), 4.10 (bd, IH, J = 12.8Hz), 3.79 (s, 2H), 3.77 (s, 3H), 3.41 (vbquart, 2H, J= 10.5Hz), 2.95 (bt, IH, J= 12.0Hz), 2.37 (bs, 3H), 2.28 (vbd, IH, J = 9.0Hz), 2.20 (vbd, IH, J = 12.2Hz), 2.02 (vbquart, IH, J = 10.5Hz), 1.82 (vbquart, IH, J= 10-5Hz), 1.51 (s, 18H). LRMS calc: 566.3 obs: 567.4 (M+H).
Example 26
Step A 4-f2-(3,5-di-/grf-butyl-4-methoxyphenvO-l^-thiazol-4-vIl-l-K3,5-dimethyI-l/f-l,2,4-triazol- 1-vQaeetyll piperidi ne

Using the method of Example 1, Step L with the product of Example ] , Step H and (3,5-dimethyl-lH- l,2,4-triazol-l-yl)acetic acid as starting materials the title compound was obtained. ^H NMR (CD3OD): δ 7.83 (s, 2Η), 7.17 (s, IH), 5.22 (V2AB, IH, J= 17.2Hz), 5.14 (!Z2AB, IH, J= 17.0Hz), 4.59 (bd, IH, J= 12.8Hz), 4.09 (bd, IH, J= 12.8Hz), 3.74 (s, 3H), 3.36 (dt, IH, Jt = 13.4, Jd = 2.2Hz), 3.16 (tt, IH, J =
11.7, 3.0Hz), 2.93 (bt, IH, J= 12.8Hz), 2.39 (s, 3H), 2.30 (s, 3H), 2.23 (bd, IH, J= 13.0Hz), 2.16 (bd, IH, J= 14.0Hz), 1.86 (dquart, IH, Jq = 12.6, Jj = 3.7Hz), 1.73 (dquart, IH, Jq = 12.6, Jd = 3.9Hz), 1.49
(s, 18H). LRMS calc: 523.3 obs: 524.4 (M+H).
Step B 4-r2-f3,5-di-ι'erf-butvIphenyl)-13-thiazol-4-yll-l-r(3,5-diιnethyl-l.H-l,2,4-triazol-l- vDacetyllpiperidine

Using the method of Examp 1, Step L with the product of Example 15, Step E and (3,5-dimethyl-lH- l,2,4-triazol-l-yl)acetic acid as starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.77 (d, 2H, J= 1.6Hz), 7.58 (t, IH, J= 1.6Hz), 7.21 (s, IH)3 5.21 (V2AB, IH, J= 17.2Hz), 5.14 (1AAB, IH, J = 17.0Hz), 4.59 (bd, IH5 J = 13.5Hz), 4.09 (bd, IH, J = 13.9Hz)5 3.36 (dt5 IH5 Jx = 13.7, Jd = 2.5Hz), 3.17 (tt, IH3 J= 11.4, 3.4Hz)3 2.92 (dt3 IH3 Jt = 13-O3 Jy = 2.7Hz)5 2.38 (s, 3H)5 2.28 (s, 3H), 2.23 (bdd, IH3 J = 14.2, 1.5Hz)5 2.16 (bd, IH5 J = 12.3Hz), 1.87 (dquart, IH3 Jq = 13.0, Jd = 3.9Hz)3 1.74 (dquart, lH, Jq = 12.6, Jd = 4.1Hz)3 1.39 (s3 18H). LRMS calc: 493.3 obs: 494.3 (M+H).
Example 27
Step A (4-methvI-l.H'-imidazoI-l-yI)acetic acid tert-butyl ester and f5-methYl-l/T-imidazoI-l- vDacetic acid tert-butyl ester

Using the method of Example 2, Step A with 4-methyl imidazole as starting material the title compounds were obtained. The mixture of isomers was chromatographed on Chiralcel OJ stationary phase (10% ethanol/heptane; λ=220nM). The more mobile 4-methyl and less mobile 5-methyl title isomers were isolated. Step B (4-methyl-l//-imidazol-l-vI)aeetic acid hydrotrifluoroacetate

Step C 4-r2-f3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yll-l-r(4-niethyl-l-iιnidazoI-l- vDacetyll piperidine

Using the method of Example 1, Step L with the products of Example 1, Step Η and Example 27, Step B as starting materials the title compound was obtained. lΗ NMR (CD3OD): δ 7.80 (s, 2Η), 7.73 (bs,
IH), 7.14 (s, IH), 6.87 (bs, IH), 5.10 (!4AB, IH, J= 16.8Hz), 5.04 (1MB, IH, J= 17.0Hz).4.57 (bd, IH, J = 12.8Hz), 4.01 (bd, IH, J= 13.0Hz), 3.71 (s, 3H), 3.31 (bt, IH, J= 12.2Hz), 3.12 (btt, IH, J= 11.6, 3.4Hz), 2.88 (bt, IH, J= 12.8Hz), 2.20 (s, 3H), 2.21 (s overlapping bd, 3H), 2.11 (bd, IH, J = 13.1Hz), 1.80 (dquart, IH3 Jq = 12.2, Jd = 3.6Hz), 1.71 (dquart, IH, Jq = 12.2, Jd = 3.6Hz)3 1.46 (s, 18H). LRMS calc: 508.3 obs: 509.4 (M+H).
Step D (S-methyl-l/T-iinidazol-l-yDacetic acid hydrotrifluoroacetate

Using the method of Example 2, Step B with (5-methyl-lH-imidazol-l-yl)acetic acid tert-butyl ester (Example 27, Step A) as starting material the title compound was obtained.
Step E 4-[2-(3,5-di-tert-butvI-4-methoxyphenvn-13-thiazoI-4-vn-l-rf5-methyl-l-imidazoI-l- vDacetyllpiperidine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 27, Step D as starting materials the title compound was obtained. IH NMR (CDSOD): δ 7.80 (s, 2H), 7.52 (s, IH),
7.15 (s, IH)5 6.69 (bs, IH)5 5.05 (Z2AB5 IH5 J= 17.3Hz), 4.98 (/2AB3 IH5 J= 17.1Hz)5 4.58 (bd, IH5 J= 11.8Hz), 4.09 (bd, IH, J= 13.4Hz)3 3.71 (s, 3H), 3.34 (dt, IH5 Jt = 13.5, Jd = 2.2Hz), 3.13 (tt, IH, J= 11.8, 3.2Hz), 2.89 (bt5 IH5 J = 12.9Hz), 2.20 (bd, IH5 J = 13.3Hz), 2.13 (s, 3H)5 2.11 (bd, IH5 J = 13.5Hz), 1.82 (dquart, IH, Jq = 12.5, Jd = 3.6Hz), 1.71 (dquart, IH, Jq = 12.6, Jd = 3.8Hz), 1.46 (s,
18H). LRMS calc: 508.3 obs: 509.4 (M+H).
Example 28 Step A (3-methyI-2-oxoiπiidazolidin-l-vI)aeetic acid tert-butyl ester

Using the method of Example 18, Step with l-methylϊmidazolϊdin-2-one as starting material the title compound was obtained.
Step B O-methvI-Z-oxoimidazolidin-l-vDacetic acid

Using the method of Example 2, Step B with the product of Example 28, Step A as starting material the title compound was obtained.
Step C l-(2-f4-f2-(3,S-di-terf-butyl-4-methoxyphenvO-l,3-thiazol-4-yllpiperidin-l-Yl}-2-oxoethvn-

Using the method of Example 1, Step L with the products of Example I5 Step H and Example 28, Step B as starting materials the title compound was obtained. lHNMR (CD3OD): δ 7.81 (s, 2H), 7.14 (s, IH),
4.59 (bd, IH3 J = 13.0Hz), 4.15 (1AAB, IH, ./= 16.7Hz)5 4.06 (1AAB5 IH5 J= 16.7Hz)5 4.01 (bd, IH5 J=
13.9Hz), 3.72 (s, 3H)5 3.47 (mult, 2H), 3.41 (mult, 2H), 3.25 (dt, IH, Jt = 14.8, Jd = 2.3Hz), 3.12 (tt, IH, J = 11.7, 3.4Hz), 2.83 (dt, IH, Jt = 12.8, Jd = 2.3Hz), 2.79 (s, 3H), 2.14 (bt, 2H, J = 16.6Hz)3 1.78 (dquart, IH, Jq = 11.9, Jd = 3.6Hz), 1.68 (dquart, IH, Jq = 12.0, Jd = 3.5Hz), 1.47 (s, 18H). LRMS calc: 526.3 obs: 527.3 (M+H).
Step P (2-oxo-1.3-oxazoIidin-3-yl)acetic acid tert-butyl ester
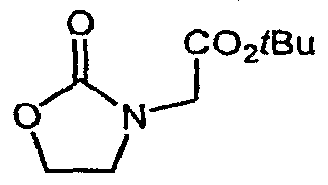
Using the method of Example 18, Step C with l,3-oxazolidin-2-one as starting material the title compound was obtained.
Step E (Z-oxo-LS-oxazolidin-S-vOacetic acid
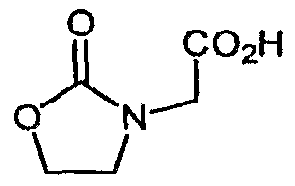
Using the method of Example 2, Step B with the product of Example 28, Step D as starting material the title compound was obtained.
Step F 3-f2-{4-r2-(3,5-di-/gr/-butvI-4-methoxyphenyl)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoethγl)- 1 ,3-oxazolidin-2-one

4.62 (bd, IH, J= 13.3Hz), 4.43 (t, 2H, J= 8.2Hz), 4.27 (MB, IH, J= 17.2Hz), 4.20 (!4AB, IH, J = 17.0Hz), 4.00 (bd, IH, J= 13.5Hz), 3.74 (s, 3H), 3.73 (t, 2H, J= 8.1Hz), 3.29 (bt, IH, J= 12.5Hz), 3.13 (tt, 1H, J= 11.7, 3.4Hz), 2.89 (dt, IH5 Jt = 12.8, Jd = 2.3Hz), 2.17 (bt, 2H, J= 17.2Hz), 1.81 (dquart, IH, Jq = 12.6, Jj = 3.9Hz), 1.72 (dquart, IH, Jq = 12.5, Jd = 3.7Hz), 1.49 (s, 18H). LRMS calc: 513.3 obs: 514.4 (M+H).
Example 29
Step A 4-[2-(3,5-di-ter-<-butyl-4-methoxyphenyl)-1^3-thiazol-4-yll-l-r(3.5-dimethyl-lig-pyrazol-l- vDacetyllpiperidine

Using the method of Example 1, Step L with the product of Example 1, Step H and (3,5-dimethyI-lH- pyrazol-l-yl)acetic acid as starting materials the title compound was obtained. *H NMR (CDCI3): δ
7.84 (s, 2H), 7.31 (s, IH), 6.86 (s, IH), 4.99 ('/2AB5 IH, J= 16.2Hz), 4.92 QAAB, IH, J= 16.4Hz), 4.70
(bd, IH, J= 13.4Hz), 4.09 (bd, IH, J= 13.4Hz), 3.75 (s,.3H), 3.28 (bt, IH, J= 12.4Hz), 3.10 (tt, IH, J= 11.7, 3.7Hz), 2.86 (dt, IH, Jt = 12.8, Jd = 2.1Hz), 2.29 (s, 3H)5 2.27 (s, 3H), 2.22 (bd, IH3 J = 13.0Hz),
2.18 (bd, IH, J= 13.0Hz), 1.80-1.64 (mult, 2H), 1 .52 (s, 18H). LRMS calc: 522.3 obs: 523.3 (M+H).
Step B 4-r2-(3,5-di-fer/-butvphenvn-l,3-thiazol-4-ylM-r(3,5-dimethyl-l.H-pyrazol-l- yDacetyli piperidiπe

Using the method of Examp 1 , Step L w the product of ExamYple 15, Step E and (3,5-dimethyl-lH- pyrazol-l-yl)acetic acid as starting materials the title compound was obtained. lH NMR (CD3OD): δ
7.77 (d, 2H5 J= 1.6Hz), 7.58 (t, IH, J= 1.6Hz), 7.21 (s, IH), 6.40 (s, IH), 5.21 ('/2AB5 IH5 J= 17.2Hz), 5.14 QAAB, IH5 J= 17.0Hz), 4.59 (bd, IH5 J= 13.5Hz), 4.09 (bd, IH, J = 13.9Hz), 3.36 (dt, IH, Jt = 13.7, Jd = 2.5Hz)5 3.17 (tt, IH, J= 11.4, 3.4Hz), 2.92 (dt, IH5 Jt = 13.0, Jd = 2.7Hz), 2.38 '(S5 3H), 2.28 (s, 3H)5 2.23 (bdd, IH, J= 14.2, 1.5Hz), 2.16 (bd, IH, J = 12.3Hz)5 1.87 (dquart, IH, Jq = 13.0, Jd = 3.9Hz), 1.74 (dquart, IH, Jq = 12.6, Jd = 4.1Hz), 1.39 (s, 18H). LRMS calc: 493.3 obs: 494.4 (M+H).
Example 30 Step A Ijy-pyrrolo[2v3-61pyridin-l-ylacetic acid tert-buty. ester

Using the method of Example 4, Step B and lH-pyrrolo[2,3-b]pyridine as the starting material the title compound was obtained.
Step B lH- -pyrrolo[2,3-^1pyridin-l-ylacetic acid hvdrotrifluoroacetate

Using the method of Example 2, Step B and the product of Example 3O5 Step A as starting material the title compound was obtained.
Step C l-(2-(4-[2-f3,5-di--'g/i-'-butyl-4-methoxyphenyl)-l,3-thiazol-4-vIlpiperidin-l-yl}-2-oxoethyl)- lJHr-pyrroloF23-^lpyridine

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 30, Step B as starting materials the title compound was obtained. 1H NMR (CD3OD): δ 8.22 (d, IH, J = 3.7Hz)5 8.02 (dd, IH5 J = 6.8, 3.4Hz)5 7.85 (s, 2H), 7.39 (quart, IH3 J= 3.3Hz), 7.19 (bs, IH), 7.15 (dd, IH, J = 7.7, 4.8Hz), 6.58 (t, IH, J= 3.3Hz), 5.37 ('/2AB5 IH, J= 16.9Hz), 5.28 ('/2AB5 IH5J= 16.9Hz), 4.61 (bd, IH5 J= 13.3Hz), 4.25 (bd, IH, J= 13.4Hz), 3.76 (s, 3H), 3.42 (bt, IH, J= 12.0Hz), 3.17 (vbt, IH, J = 12.1Hz), 2.92 (bt, IH, J= 13.0Hz), 2.23 (bd, IH5 J= 12.8Hz), 2.14 (bd, IH5 J= 12.9Hz), 1.91 (dquart, IH, Jq = 12.3, Jd = 3.0Hz)5 1.76 (dquart, IH, Jq = 12.2, Jj = 3.1Hz), 1.51 (s, 18H). LRMS calc: 544.3 obs: 545.3 (M+H).
Step D l-{2-f4-f2-|3--'grf-butvI-5-rftrifluoroinethvnthiolphenvU-1.3-thiazoI-4-vnpiperidin-l-vIl-2- oxoethyl}-l//-pyrrolo [23-61 pyridine

Using the method of Example 1, Step L with the products of Example 1 1, Step L and Example 30, Step B as starting materials the title compound was obtained. *H NMR (CD3OD): δ 8.23 (dd, IH, J = 4.8,
1.4Hz), 8.17 (t, IH, J= 1.6Hz), 8.08 (s, IH), 8.03 (dd, IH, J = 7.8, 1.4Hz), 7.82 (s, IH)5 7.40 (d, IH, J= 3.6Hz), 7.32 (s, IH), 7.15 (dd, IH, J= 7.7, 4.8Hz), 6.57 (d, IH, J= 3.5Hz), 5.38 ('/2AB5 IH, J= 16.9Hz), 5.29 O/2AB, IH, J= 16.9Hz), 4.61 (bd, IH, J= 13.2Hz), 4.26 (bd, IH3 J= 13.7Hz), 3.43 (dt, IH, Jt = 12.5, Jd = 2.6Hz), 3.21 (vbt, IH, J= 11.7Hz), 2.93 (bt, IH, J= 1 1.8Hz), 2.24 (bd, IH, J= 12.8Hz), 2.16 (bd, IH, J= 12.8Hz), 1.95 (dquart, IH, Jq = 12.8, Jd = 3.7Hz), 1.79 (dquart, IH, Jq = 12.5, Jd = 3.6Hz), 1.44 (s, 9H). LRMS calc: 558.2 obs: 559.2 (M+H).
Example 31 Step A l-(2-{4-r2-(3,5-di-ferf-butyI-4-methoxyphenyl)-l,3-thiazoI-4-yllpiperidin-l-vU-2-oxoethvIV 1/7-benzimidazole
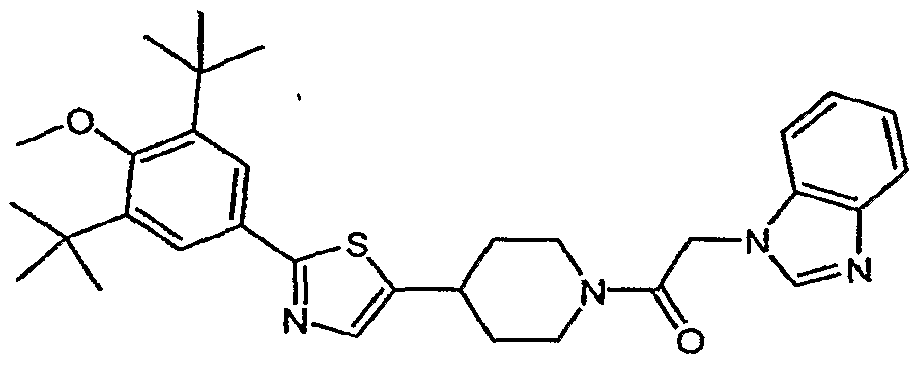
Using the method of Example I, Step L with the product of Example 1, Step H and lH-benzimidazol-1- ylacetic acid as the starting materials the title compound was obtained. lΗ NMR (CD3OD): δ 8.12 (s,
IH), 7.81 (s5 2H), 7.67 (d, IH, J= 7.5Hz), 7.48 (d, IH, J= 8.4Hz), 7.32-7.25 (mult, 2H), 7.16 (s, IH), 5.39 C/2AB, IH, J= 17.3Hz), 5.33 (1^AB, IH, J= 17.3Hz), 4.58 (bd, IH, J= 12.7Hz), 4.19 (bd, IH, J = 12.6Hz), 3.72 (s, 3H), 3.41 (bt, IH, J= 13.1Hz), 3.15 (vbt, IH, J == 11.8Hz), 2.91 (bt, I H5 J= 1 1.6Hz), 2.24 (bd, IH, J = 12.9Hz), 2.12 (bd, IH3 J = 13.6Hz), 1.88 (dquart, I H, Jq = 13.1, Jd = 2.8Hz), 1.73 (dquart, IH, Jq = 12.9, Jd = 4. IHz), 1.47 (s, 18H). LRMS calc: 544.3 obs: 545.4 (M+H).
Step B l-r2-{4-r2-f3,5-di-ter/-butyl-4-methoxyphenylV13-thiazol-4-yllpiperidin-l-vU-2-oxoethvn- 2-methyl-l/f-benzimidazole

Using the method of Example 1, Step L with the product of Example 1, Step H and (2-methyl-lH- benzimidazol-l-yl)acetic acid as the starting materials the title compound was obtained. lH NMR (CD3OD); δ 7.82 (s, 2H), 7.54 (d, IH, J = 8.5Hz), 7.38 (d, IH5 J= 8.5Hz), 7.23-7.19 (mult, 2H), 7.18
(s, IH), 5.31 (1Z2AB, IH, J= 17.6Hz), 5.33 (1AAB, IH, J= 17.7Hz), 4.56 (bd, IH, J= 11.0Hz), 4.22 (bd, IH, J= 13.0Hz), 3.72 (s, 3H), 3.44 (dt, IH, Jt = 11.9, Jd = 2.4Hz), 3.16 (bt, IH, J= 11.9Hz), 2.92 (dt, IH, Jt = 12.6, Jd = 2.6Hz), 2.52 (s, 3H), 2.26 (bd, IH, J = 11.0Hz), 2.13 (bd, IH, J = 1 1.5Hz), 1.92 (dquart, I H, Jq = 11.5, Jd = 3.6Hz), 1.74 (dquart, IH, Jq = 12.4, Jd = 4.1Hz), 1.47 (s, 18H). LRMS calc: 558.3 obs: 559.4 (M+H).
Step C l-(2-f4-(2-(3-ter/-butyl-5-r(trifluoromethyl)thio1phenvU-l,3-thiazol-4-yl)piperidin-l-γU-2- oxoetliyU-l/J-benzimidazole

Using the method of Example 1, Step L with the product of Example 11, Step L and l/f-benzimidazol-l- ylacetic acid as starting materials the title compound was obtained. ^H NMR (CD3OD): δ 8.60 (bs, IH), 8.13 (s, IH), 8.05 (s, IH), 7.79 (s, IH), 7.74 (d, IH, J= 7.5Hz), 7.64 (d, IH, J= 8.0Hz), 7.45-7.40 (mult, 2H), 7.30 (s, IH), 5.52 ('/2AB5 IH, J= 17.3Hz), 5.45 ('/aAB, IH, J= 17.4Hz), 4.58 (bd, IH, J = 13.0Hz), 4.18 (bd, IH, J= 14.3Hz), 3.44 (dt, IH, Jt = 14.7, Jd = 2.3Hz), 3.20 (tt, IH, J= 11.7, 3.6Hz)3 2.94 (dt, IH, Jt = 13.0, Jd = 2.6Hz), 2.26 (bd, IH, J = 13.6Hz), 2.16 (dd, IH, J = 12.8, 5.5Hz), 1.95 (dquart, IH, Jq = 12.6, Jd = 3.7Hz), 1.78 (dquart, IH, Jq = 12.4, Jd = 3.7Hz), 1.40 (s, 9H). LRMS calc: 558.2 obs: 559.2 (M+H).
Step D l-f2-I4-(2-{3-tert-butyl-5-rftrifluoromethyl)thio1phenyl}-13-thiazol-4-vnpiperidin-l-yll-2- oxoethyl}-2-methyl-liJr-benzimidazole

Using the method of Example 1, Step L with the product of Example 11, Step L and (2-methyl-li/- benzimidazol-l-yl)acetic acid as the starting materials the title compound was obtained. IR NMR (CD3OD): δ 8.16 (s, IH), 8.07 (s, IH), 7.81 (s, IH), 7.57 (dd5 IH5 J= 8.5, 1.6Hz), 7.40 (dd, IH, J= 8.4,
1.4Hz), 7.33 (s, IH), 7.26-7.21 (mult, 2H), 5.32 (Z2AB, IH5 J= 17.8Hz), 5.25 QΔAB, IH3 J= 17.7Hz)5 4.58 (bd, IH, J = 12.1Hz)5 4.24 (bd, IH, J= 13.2Hz), 3.46 (bt, IH, J= 11.9Hz)5 3.21 (tt, IH, J= 11.7, 3.5Hz), 2.91 (dt, IH, Jt = 12.8, Jd = 1.8Hz)5 2.54 (s, 3H)5 2.29 (bd, IH, J= 12.1 Hz), 2.16 (bd, IH, J = 13.2Hz), 1.97 (dquart, IH, Jq = 12.8, J<χ = 3.8Hz), 1.78 (dquart, IH, Jq = 12.8, Jd = 3.9Hz), 1.42 (s, 9H). LRMS calc: 572.2 obs: 573.3 (M+H).
Example 32 Step A f5-methyI-3-(trifluorotnethγl)-l^r-pγrazoI-l-yllacetic acidtert-butyl ester

Using the method of Example 4, Step B with 5-methyl-3-(trifluoromethyl)-lH-pyrazole as the starting material the title compound was obtained as the sole product.
Step B f5-methyl-3-(trifluoroniethyl)-l//'-pyrazoI-l-vHacetic acid hydrotrifluoroacetate
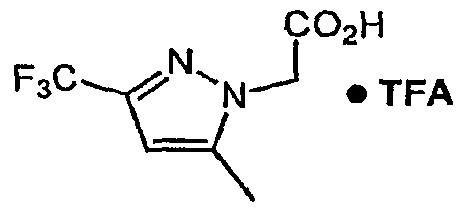
Using the method of Example 2, Step B with the product of Example 32, Step A as the starting material the title compound was obtained.
Step C 4-f2-(3,5-di-ferf-butylphenyl)-l 3-thiazoI-4-yl1-l-{r5-methv.-3-(trifluoromethyl)-Lg- pyrazol-1-vUacetvUpiperidine

Using the method of Example 1 , Step L with the products of Example 15, Step E and Example 32, Step B as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.79 (d, 2H, J =
1.8Hz), 7.60 (t, IH, J = 1.7Hz), 7.23 (s, IH), 6.44 (s, IH)3 5.29 (1AAB, IH5 J= 17.0Hz), 5.21 (V2AB, IH, J = 17.0Hz), 4.59 (bs, IH), 4.14 (bd, IH, J= 13.7Hz), 3.40 (bt, IH, J= 11.8Hz), 3.19 (tt, IH, J= 11.9, 3.5Hz), 2.95 (bt, IH, J= 11.7Hz), 2.33 (s, 3H), 2.25 (bd, IH, J= 13.5Hz), 2.18 (bd, IH, J = 13.0Hz), 1.88 (dquart, IH, Jq = 12.6, Jd = 4.1Hz), 1.76 (dquart, IH, Jq = 12.6, Jd = 4.1Hz), 1.40 (s, 18H). LRMS calc: 546.3 obs: 547.4 (M+H).
Step D l-fr3,5-bis(trifluoroinethvn-ljy-pyrazol-l-vnacetvU-4-f2-f3,5-di-/er/-butylphenyl)-l,3- thiazol-4-yl 1 piperid i ne

Using the method of Example 1, Step L with the product of Example 15, Step E and [3,5- bis(trifluoromethyl)-lH-pyrazol-l-yl]acetic acid as starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.78 (bs, 2H), 7.58 (bs, IH), 7.23 (s, IH), 7.22 (s, IH), 5.51 QAAB, IH, J =
16.7Hz), 5.44 (fcAB, IH, J = 16.9Hz), 4.59 (bs, IH), 4.07 (bd, IH, J = 13.7Hz), 3.39 (bt, IH, J = 12.6Hz), 3.18 (bt, IH, J= 11.9Hz)5 2.94 (bt, IH, J= 12.1Hz), 2.24 (bd, IH3 J= 13.3Hz), 2.16 (bd, IH, J = 13.0Hz)5 1.86 (dquart, IH, Jq = 12.3, Jd = 4.0Hz), 1.75 (dquart, IH, Jq = 12.2, Jd = 3.8Hz), 1.39 (s,
18H). LRMS calc: 600.2 obs: 601.3 (M+H).
Step E l-ff4-bromo-3,5-dimethvI-lJy-pyrazol-l-vnacetvH-4-r2-(3,5-di-fert-bHtylphenyl)-13-thiazol- 4-yITpiperidine
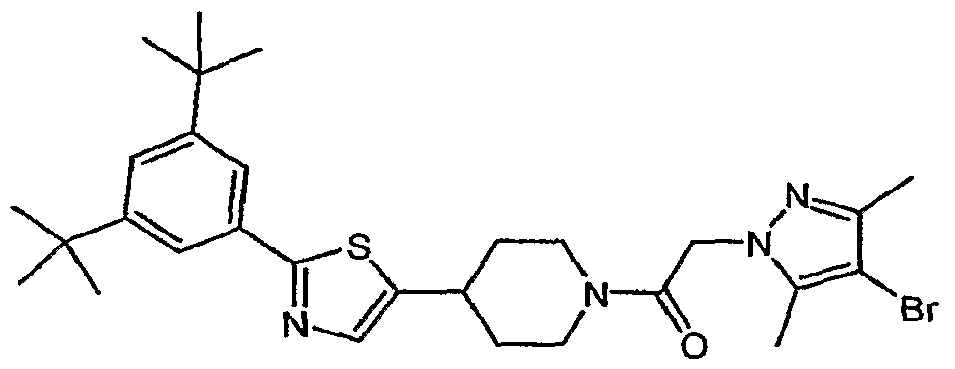
Using the method of Example 1, Step L with the product of Example 15, Step E and (4-bromo-3,5- dimethyl- lH-pyrazol-l-yl)acetic acid as starting materials the title compound was obtained. lΗ NMR
(CD3OD): δ 7.79 (bs, 2Η), 7.59 (bs, IH)5 7.22 (s, IH), 5.16 (V2AB, IH, J= 17.1Hz), 5.09 ('/2AB5 IH, J
= 17.0Hz), 4.60 (bd, IH, J= 11.5Hz), 4.12 (bd, IH, J= 12.8Hz), 3.37 (bt, IH, J= 12.2Hz), 3.18 (bt, IH5 J = 11.5Hz)5 2.92 (bt, IH, J= 12.6Hz), 2.24-2.15 (2s overlapping 2bd, 8H total), 1.87 (dquart, IH, Jq = 11.4, Jd = 2.9Hz), 1.75 (dquart, IH, Jq = 11.8, Jd = 3.2Hz), 1.40 (s, 18H). LRMS calc: 570.2 obs: 571.3 (M+H).
Step F l-rf4-iodo-3,5-dimethyl-ljy-Pyrazol-l-vnacetvπ-4-f2-(3.5-di-/gr/-butyIphenvn-13-thiazol-4- yllpiperidine


Using the method of Example 18, Step C and imidazolidin-2-one as the starting material the tile compound was obtained. Purification was carried out on Chiralcel AS stationary phase ( 10%EtOH/heptane; λ = 21 OnM).
Step B rø-oxoimidazolidin-l-vDacetic acid

Using the method of Example 2, Step B and the product of Example 33, Step A as the starting material the title compound was obtained.
Step C l-(2-{4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-13-thiazoI-4-yllpiperidin-l-vU-2- oxoethyl)imidazolidin-

IH), 4.58 (bd, IH, J= 13.0Hz), 4.14 ('/2AB5 IH, J= 16.7Hz), 4.05 (V2AB, IH, J= 16.7Hz), 4.00 (bd, IH, J= 13.1Hz)3 3.71 (s, 3H), 3.56 (dpent, 2H3 Jp = 8.2, J& = 4.6Hz), 3.44 (t, 2H, J= 8.0Hz), 3.24 (dt, IH, Jx = 14.3, Jd = 2.4Hz), 3.09 (tt, IH, J= 11.7, 3.6Hz), 2.83 (dt, IH, Jt = 13.0, Jd = 2.7Hz), 2.12 (bmult, 2H), 1.77 (dquart, IH, Jq = 12.4, Jd = 3.7Hz), 1.67 (dquart, IH, Jq = 12.3, Jd = 3.5Hz), 1.46 (s, 18H). LRMS calc: 512.3 obs: 513.3 (M+H).
Example 34 Step A β-fhvdroxymethvO-l/y-imidazoI-l-yllacetic acid methyl ester

A solution of lH-ϊmidazol-2-ylmethanol hydrochloride (986mg; 7.33mmol) in dry DMSO (2OmL) was treated with methyl bromoacetate (696μL; 7.33mmol) and cesium carbonate (5.97g; 18.32mmol). The mixture was stirred at ambient temperature for 16h. The reaction mixture was diluted with isopropyl acetate (10OmL) and filtered. The filtrate was evaporated onto silica gel. The silica gel was eluted (40:1 CΗ2Cl2/MeOΗ) without fractionation to desorb DMSO. The silica gel was then eluted without fractionation (100:10:1 CH2Cl2/MeOH/Et3N) to recover the title compound. The second eluant was evaporated and chromatographed over Chiralcel AD stationary phase (15%EtOH/heptane; λ = 22OnM). The second eluting peak was recovered, affording the title compound (109mg).
Step B Potassium r2-(hvdroxymethyl)-l//-imidazol-l-yllacetate

Using the method of Example 3, Step C and the product of Example 34, Step A as the starting material the title compound was obtained.
Step C ll-(2-{4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-1.3-thiazoI-4-vIlpiperidin-l-^yl>-2-oxoethvI>- l//-imidazol-2-γl]ιnethanol

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 34, Step B as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.84 (s, 2H), 7.17 (s,
IH), 7.08 (bs, IH), 6.93 (bs, IH)3 5.23 (V2AB, IH, J = 17.2Hz), 5.16 (V2AB, IH, J = 17.3Hz), 4.62 (s overlapping bd, 3H total, Jd = 14.5Hz), 4.09 (bd, IH, J= 13.9Hz), 3.74 (s, 3H), 3.36 (dt, IH, Jt = 14.2,
Jd = 2.1Hz), 3.15 (tt, IH5 J = 11.9, 3.4Hz), 2.91 (dt, IH, Jt = 12.8, Jd = 2.3Hz), 2.22 (bd, IH, J = 12.8Hz), 2.15 (bd, IH3J= 14.2Hz), 1.87 (dquart, IH, Jq = 12.6, Jd = 3.6Hz), 1.74 (dquart, IH, Jq = 12.6, Jd = 3.9Hz), 1.49 (s, 18H). LRMS calc: 524.3 obs: 525.3 (M+H).
Step P f2-(hvdroxymethyI)-l/?-benzimidazol-1-vIlacetic acid methyl ester

Using the method of Example 3, Step B and lH-benzimidazol-2-ylmethanol as the starting material the title compound was obtained as the sole product. Purification was performed by silica gel chromatography (preparative TLC; 20:1 CH2Cl2/MeOH).
Step E Potassium r2-(hydroxymethvO-lli/-benzimidazol-l-γπaeetate

Step F fl-(2-(4-f2-(3,5-di-ter<1-butyl-4-methoxyphenyl)-l,3-thiazoI-4-vnpiperidin-l-vU-2-oxoethvn- liy-benzimidazoI-2-vππiethanol

Using the method of Example 1, Step L with the products of Example 1, Step H and Example 34, Step E as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.84 (s, 2H), 7.64 (d,
I H, J= 7.8Hz), 7.46 (d, IH, J= 7.8Hz), 7.31-7.24 (mult, 2H), 7.18 (s, IH), 5.47 (1AAB, IH, J= 18.0Hz),
5.39 O/2AB, IH, J= 17.8Hz), 4.86 (s, 2H), 4.57 (bd, IH, J= 13.5Hz), 4.22 (bd, IH, J= 13.9Hz), 3.74 (s, 3H), 3.43 (bt, IH, J= 11.9Hz), 3.17 (tt, IH, J= 1 1.7, 3.4Hz), 2.92 (bt, IH, J= 11.9Hz), 2.26 (bd, IH, J
= 12.6Hz), 2.14 (bd, IH5 J = 13.0Hz)5 1.93 (dquart, IH5 Jq = 12.3, Jd = 3.4Hz), 1.73 (dquart, IH5 Jq = 12.8, Jd = 3.9Hz)5 1.49 (s5 18H). LRMS calc: 574.3 obs: 575.5 (M+H).
Example 35
Step A 4-[2-(3.5-di-te/-<'-butvI-4-methoxyphenvI)-l,3-thiazoI-4-yll-l-(f2-(trifluoroinethvn-l/r- i midazol-l-yl lacetyl) piperid ine

Using the method of Example 1, Step L with the product of Example 1, Step H and [2-(trifluoromethyl)- lH-imidazol-l-yl]acetic acid as the starting materials the title compound was obtained. 1H NMR (CDCI3): δ 7.73 (s, 2H), 7.19 (dd, IH, J= 3.4, 0.9Hz), 7.13 (dd, IH, J= 3.6, 1.0Hz)5 6.96 (s, IH)5 5.01
(/2AB, IH5 16.7Hz)5 4.90 ('AAB5 IH, J= 16.9Hz), 4.73 (bd, IH5 J= 13.9Hz), 3.88 (bd, IH3 J= 14.4Hz), 3.75 (s, 3H)5 3.42 (bt, IH5 J= 12.2Hz), 3.20 (bt5 IH3 J= 11.6Hz)5 2.91 (bt5 IH5 J= 11.8Hz)5 2.48 (bd, IH, J= 13.1Hz)5 2.23 (bd5 IH5 J= 13.5Hz)5 1.89-1.70 (vbmult, 2H)5 1.52 (s, 18H). LRMS calc: 562.3 obs: 563.4 (M+H).
Step B 4-r2-r3.5-di-ter/-butyl-4-methoχyphenvn-l,3-thiazoI-4-vn-l-(r2-niethyl-lJHr-iinidazol-l- yli acetyl^ pi perid ine

Using the method of Example 1, Step L with the product of Example 1, Step H and [2-methyl-lH- imidazol-l-yl]acetic acid as the starting materials the title compound was obtained. lΗ NMR (CDCI3): δ 7.83 (s, 2Η), 6.99 (bs, IH)5 6.87 (bs5 2H)5 4.75-4.70 (bs overlapping bd, 3H)5 3.91 (bd, IH5 J = 14.8Hz)5 3.75 (s5 3H)5 3.33 (bt, IH, J = 12.5Hz)5 3.13 (vbt, IH5 J= 11.6Hz)5 2.90 (bt5 IH, J = 11.8Hz),
2.40 (bs, 3H)5 2.29 (bd, IH, J = 13.0Hz), 2.21 (bd, IH, J = 13.1Hz), 1.77 (bdhex, 2H3 Jh = 12.6, Jd = 4.0Hz), 1.51 (s, 18H). LRMS calc: 508.3 obs: 509.4 (M+H).
Step C 4-r2-(3,5-di-ferf-butyl-4-methoxyphenvn-13-thiazol-4-yll-l-fl.fir-imidazol-l- ylacetvDpiperidine

Using the method of Example 1, Step L with the product of Example 1, Step H and [l/f-imidazol-l- yl]acetic acid as the starting materials the title compound was obtained. *H NMR (CD3OD): δ 7.80 (s,
2H), 7.62 (s, IH), 7.14 (s, IH), 7.08 (s, IH), 6.97 (s, IH), 5.15 (V2AB, IH, J= 16.9Hz), 5.07 (V2AB, IH, J = 17.1Hz), 4.58 (bd, IH, J= 14.2Hz), 4.04 (bd, IH5 J= 13.9Hz), 3.72 (s, 3H), 3.33 (bt, ] H5 J= 13.5Hz),
3.12 (vbt, IH, J = 11.8Hz), 2.88 (bt, IH, J = 12.8Hz), 2.19 (bd, IH, J = 12.6Hz), 2.12 (bd, IH, J =
12.0Hz), 1.81 (dquart, IH, Jq = 12.6, Jd = 3.3Hz), 1.70 (dquart, IH, Jq = 12.6, Jd = 4.0Hz), 1.46 (s,
18H). LRMS calc: 494.3 obs: 495.4 (M+H).
Example 36
Step A methyl 3,5-dioxohexanoate
A solution of dehydroacetic acid (20.60 g; 168mmol) in methanol (40OmL) was treated with a solution of magnesium methoxide (6wt% in methanol; 350 mL; 184mmol) at ambient temperature. The reaction was refluxed for 5h. The solvent was removed and the residue added to aq. HCl (IL; IN). The aqueous was extracted (EtOAc, 2x500mL). The organic was dried over MgSO4, filtered and evaporated to give the title compound (17.0 g).
Step B f5-methyl-l//-pyrazol-3-yI)acetic acid methyl ester

Step C benzyl methyl 2,2>-f5-methyl-Lflr-pyrazole-l,3-diyl)diaeetate

A solution of the product of Example 36, Step B (462 mg; 3.0mmol) in dry DMF (8mL) was treated with potassium carbonate (415 mg; 3.0mmol) at ambient temperature. The mixture was warmed to 50°C. Benzyl 2-bromoacetate (687 mg; 3.0mmoi) was added dropwϊse. The reaction mixture was stirred for 4h then cooled to ambient and stirred for 16h. The mixture was diluted with water and extracted (EtOAc). The organic was dried over MgSO-J, filtered and evaporated to a residue. Silica gel chromatography (EtOAc/hexanes; 1:2) gave a mixture of two isomers. The mixture was chromatographed on Chiralcel OJ stationary phase (60% ethanol/heptane; λ=220nM) to give the title compound (194 mg).
Step D [S-rø-methoxy-Z-oxoethyO-S-methvI-l-fl'-pyrazol-l-yllacetic acid

A solution of the product of Example 36, Step C (194 mg; 0.64mmol) in methanol (5OmL) was treated with 10% palladium on carbon hydrogenation catalyst (60 mg). The mixture was shaken under a hydrogen atmosphere (latm) for 2h. The mixture was filtered through Celite and the filtrate concentrated to give the title compound (118 mg).
Step E ri-(2-{4-f2-(3,5-di-terf-butylphenyl)-1.3-thiazol-4-yllpiperidin-l-yl}-2-oxoethyl)-5-methvt- l/y-pyrazoI-S-vU acetic acid methyl ester

Using the method of Example 1, Step L with the products of 15, Step E and Example 36, Step D as the starting materials the title compound was obtained.
Step F fl-(2-{4-r2-f3,5-di-/grf-butylphenvπ-l,3-thiazol-4-yl|piperidin-l-vU-2-oxoethyl)-5-methyI- l/y-pyrazol-3-vU acetic acid

Step G fl-l2-r4-f2-0-ferf-butyl-5-rrtrifluoromethvnthiolphenyl}-1.3-thiazol-4-yl)piperidin-l-vIl-2- oxoethvU-5-methyl-l/ir-pyrazol-3-yl)acetic acid methyl ester

Step H Potassium fl-f2-F4-(2-{3-fer/-butvI-5-f(trifluoromethyl)thiolphenγl>-l,3-thiazol-4- vDpiperidin-l-vπ-2-oxoethγI)-5-methyI-l/jr.-pyrazol-3-γl)acetate

Using the method of Example 3, Step C with the product of Example 36, Step G as starting material the title compound was obtained. Isolation was effected by removal of volatiles and digestion of the derived solids in hot ethanol. The mixture was filtered and the filtrate evaporated to give the title compound. lH NMR (CD3OD): δ 8.12 (bdd, IH, J= 5.1, 1.6Hz), 8.03 (bd, IH, J = 4.4Hz), 7.77 (bd, IH, J= 4.6Hz),
7.28 (bd, IH, J = 5.8Hz), 6.03 (bd, IH, J = 8.1Hz), 5.07 (V2AB, IH, J = 17.2Hz), 5.00 (MB, IH, J = 17.4Hz), 4.58 (bd, IH, J= 13.3Hz), 4.10 (vbs, IH), 3.41 (vbd, 2H, J= 5.5Hz), 3.33 (bt, IH, J= 12.2Hz), 3.15 (vbs, IH), 2.88 (bt, IH3 J = 12.7Hz), 2.21-2.17 (s overlapping bd, 4H total), 2.12 (bd, IH, J = 12.9Hz), 1.83 (vbquart, IH, J= 12.5Hz), 1.72 (vbquart, IH, J= 12.5Hz), 1.39 (s, 4.5H), 1.38 (s, 4.5H). LRMS calc: 580.2 obs: 581.3 (M+H).
Example 37
Step A 4-f2-(3.5-di-ter/'-butylphenvn-l,3-thiazol-4-vn-l-rf2,4-dimethvi-l£r-iinidazol-l- vOacetyllpiperidine

Using the method of Example 1, Step L with the products of Example 15, Step F and Example 2, Step B as starting materials the title compound was obtained. *H NMR (CDCI3): δ 7.78 (bs, 2H), 7.54 (bs,
IH), 6.90 (s, IH)54.73 (bd, IH, J= 13.2Hz), 4.73-4.63 (AB, 2H3 J= 16.9Hz), 3.90 (bd, IH5J= 13.2Hz), 3.32 (bt, IH, J= 12.1Hz), 3.14 (bt, IH, J= 11.3Hz), 2.89 (bt, IH, J= 11.8Hz), 2.35 (bs, 3H), 2.28 (bd,
IH5 J = 13.7Hz)5 2.21-2.16 (bs overlapping bd5 4H total), 1.84-1.72 (bniult, 2H), 1.41 (s, 18H). LRMS calc: 492.3 obs: 493.4 (M+H).
Step B l-f2-f4-r2-(3.5-di-ter/-butylphenvn-13-thiazol-4-vnpiperidin-l-yll-2-oxoethvn-3-ethvI-l,3- dihydro-2/?-imidazol-2-one

Using the method of Example 1, Step L with the products of Example 15, Step F and Example 4, Step C as starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.79 (d, 2H5 J = 1.8Hz),
7.59 (t, IH, J = 1.7Hz), 7.21 (s, IH), 6.55 (d, IH, J= 2.8Hz)5 6.48 (d, IH, J= 2.8Hz), 4.68 (!Z2AB, IH5 J = 16.7Hz), 4.60 ('/2AB5 IH, J = 16.6Hz), 4.60 (bd, IH, J = 13.1Hz), 4.09 (bd, IH, J = 13.5Hz), 3.70 (quart, 2H, J= 7.3Hz)5 3.31 (bt, I H5 J= 12.0Hz), 3.15 (tt, IH, J= 1 1.9, 3.4Hz)5 2.90 (dt, IH, Jt = 13.0, Jd = 2.5Hz), 2.22 (bd, IH, J = 13.0Hz)5 2.16 (bd, IH, J = 12.8Hz), 1.85 (dquart, IH, Jq = 12.8, Jd = 3.6Hz), 1.74 (dquart, IH, Jq = 12.6, Jd = 3.7Hz), 1.40 (s, 18H), 1.30 (t, 3H, J = 7.3Hz). LRMS calc: 508.3 obs: 509.3 (M+H).
Example 38 Step A 3,5-bisftrifluoromethyl)benzoyl chloride

Step B 3,5-bis(trifiuorometb.vPbenzaιnide

Using the method of Example 13, Step F with the product of Example 38, Step A as the starting material the title compound was obtained.
Step C 3.5-bis(ϊrifluoromethvDbenzenecarbothioamide

Using the method of Example 1, Step F with the product of Example 38, Step B as the starting material the title compound was obtained.
Step D 9^r-fluoren-9-vImethvI4-{2-f3,5-bis(trifluoroinethyl)phenvπ-1.3-thiazol-4-vI>piperidine-l- carboxylate

Using the method of Example 1, Step G with the products of Example 1, Step C and Example 38, Step C as the starting materials the title compound was obtained.
Step E 4-f2-f3,5-bisftrifluoromcthyl)phenvπ-13-thia2oI-4-vUpiperidine

Using the method of Example 1, Step H with the product of Example 38, Step D as starting material the title compound was obtained.
Step F 3-f2-(4-f2-r3,5-bisftrifluoroιnethvnphenvn-l,3-thiazol-4-vUpiperidin-l-vI)-2-oxoethyll-3/f- imidazof4,5-A]pyridine

Using the method of Example 1, Step L with the products of Example 38, Step E and Example 1, Step K as the starting materials the title compound was obtained. lH NMR (CDCI3): δ 8.42 (d, IH, J= 4.1Hz),
8.37 (s, 2H), 8.13 (d, IH5 J = 7.4Hz),.7.92 (s, I H)3 7.30 (bmult, 2H), 7.07 (s, IH), 5.31-5.23 (vbmult, 2H), 4.70 (bd, IH3 J= 13.5Hz), 4.15 (bd3 IH3 J= 13.7Hz), 3.40 (bt, IH, J= 12.6Hz), 3.14 (bt, IH, J = 11.6Hz)3 2.89 (bt, IH3 J = 12.8Hz)3 2.28 (bd, IH3 J= 13.0Hz)3 2.17 (bd3 IH, J = 12.9Hz), 1.88 (bquart, IH3 J= 12.5Hz)3 1.79 (bdquart, lH, Jq = 12.5, ./d = 3.6Hz). LRMS calc: 539.1 obs: 540.1 (M+H).
Step G 4-(2-r3,5-bisftrifluoromethvnphenyll-1.3-thiazol-4-yl)-l-rf3<5-dimethyl-l/r-1.2,4-triazol-l- vDacetylipiperidine

Using the method of Example 1, Step L with the products of Example 38, Step E and (3,5-dimethyl-l/f- 1 ,2,4-triazoI- 1 -yl)acetic acid as the starting materials the title compound was obtained. lH NMR
(CD3OD): δ 8.50 (bs, 2H)3 8.08 (bs, IH)3 7.45 (s, IH)3 5.23 ('/2AB3 IH, J= 16.9Hz), 5.16 (/2AB3 IH3 J
= 17.0Hz), 4.60 (bd, IH, J= 13.0Hz)3 4.11 (bd, IH3 J= 13.5Hz), 3.40 (bt, IH3 J= 12.9Hz)3 3.23 (vbt, IH, J= 11.7Hz), 2.95 (bt, IH, J= 12.7Hz)3 2.40. (d, 3H3 J= LlHz), 2.30 (d3 3H, J= I. IHz), 2.25 (bd, IH3 J= 13.2Hz)3 2.17 (bd3 IH3 J= 13.0Hz)3 1.94 (dquart, IH, Jq = 12.4, Jd = 2.8Hz), 1.79 (dquart, 1 H3 Jq = 12.5, Jd = 3.2Hz). LRMS calc: 517.1 obs: 518.2 (M+H).
Step H 4-|2-f3,5-bis(trifluoromethvnphenvn-l,3-thiazol-4-vn-l-fchIoroacetyl)piperidine

Step J 3-[2-f4-{2-r3.5-bisftrifluoromethyl)phenvI1-1.3-thiazol-4-vUpiperidin-l-yl)-2-oxoethvn-3Jy- imidazof4,5-cl pyridine and 142-(4-f243,5-p.s(trifluoromethy0phenyπ-13-thiazoI-4-vUpiperidin- l-yl)-2-oxoethyπ-liy-imidazo[4,5-clp-yridine

Using the method of Example 15, Step G with the product of Example 38, Step H as starting material the title compounds were obtained. *H NMR (CD3OD): δ 8.88 (s, IH), 8.45 (bs, 2H) 8.33 (s, I H), 8.31 (d,
IH, J = 4.0Hz), 8.13 (bs, IH)3 7.77 (d, IH, J = 5.8Hz), 7.19 (s, IH), 5.67 ('/2AB5 IH, J= 17.0Hz), 5.59 ('ΛAB, IH, ./= 16.9Hz), 4.60 (bd, IH5 J= 13.0Hz), 4.13 (bd, IH, J= 13.0Hz), 3.43 (dt, lH, Jt = 13.3, Jd = 2.1Hz), 3.13 (tt, IH, J= 11.8, 3.5Hz), 2.91 (dt, IH, Jt = 12.6, Jd = 2.2Hz), 2.26 (bd, IH, J= 13.0Hz), 2.15 (bd, IH, J= 13.0Hz), 1.90 (dquart, IH, Jq = 12.9, Jd = 4.0Hz), 1.79 (dquart, IH, Jq = 12.7, Jd = 4.1Hz), 1.41 (s, 18H). LRMS calc: 539.1 obs: 540.2. IH NMR (CD3OD): δ 8.91 (s, I H), 8.41 (bs, 2HX 8.36 (d, I H, .7 = 5.5Hz), 8.30 (s, IH), 8.17 (bs, IH), 7.69 (d, IH, J = 5.6Hz), 7.20 (s, IH), 5.55 (/2AB, IH, J = 17.1 Hz), 5.47 (V2AB, IH, J = 17.2Hz), 4.59 (bd, IH, J = 12.9Hz), 4.14 (bd, IH, J = 13.0Hz), 3.45 (dt, IH, Jt = 13.0, Jd = 2.3Hz), 3.12 (tt, IH, J= 11.9, 3.3Hz), 2.89 (bt, IH, J = 12.6Hz), 2.19 (bd, IH, J = 12.8Hz), 2.14 (bd, IH, J = 12.7Hz), 1.95 (dquart, IH, Jq = 12.7, Jd = 3.5Hz), 1.77 (dquart, IH5 Jq = 12.8, Jd = 3.8Hz), 1.37 (s, 18H). LRMS calc: 539.1 obs: 540.1 (M+H).
Step K l-f2-(4-(2-f3.5-bisftrifluoromethyl)phenvU-l,3-thiazoI-4-vUpiperidin-l-vn-2-oxoethylI-2- methyl-lg-iinidazo[4.5-clpyridine
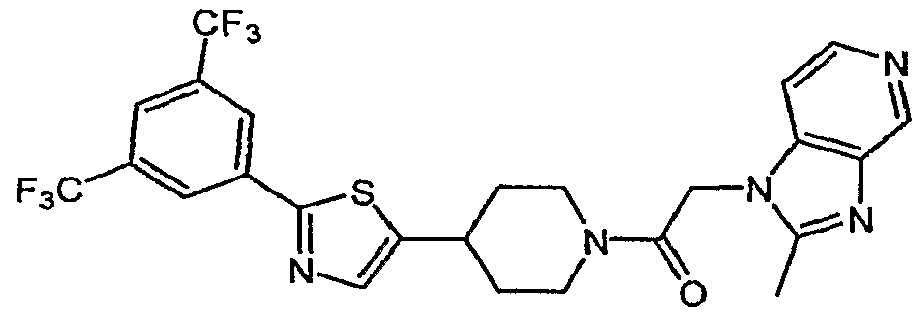
Using the method of Example 15, Step G with the product of Example 38, Step H and 2-methyl-5- azabenzimidazole as starting materials the title compound was obtained. lH NMR (CD3OD): δ 8.80 (s,
IH), 8.48 (s, IH), 8.30 (d, IH, J= 5.6Hz), 8.05 (s, IH), 7.55 (d, IH, J= 5.7Hz), 7.44 (s, IH), 5.41 ('/2AB5 IH, J= 17.6Hz), 5.34 (1AAB, IH, J= 17.5Hz), 4.56 (vbs, IH), 4.20 (bd, IH, J= 7.1Hz), 3.46 (bt, IH, J=
11.7Hz), 3.23 <tt, IH, J= 11.8, 3.7Hz), 2.95 (bt, IH, J= 11.7Hz), 2.58 (s, 3H), 2.29 (bd, IH, J= 14.0Hz), 2.15 (bd, IH, J = 13.8Hz), 2.00 (dquart, IH, Jq = 12.0, Jd = 2.4Hz), 1.79 (dquart, IH, Jq = 12.2, Jrf = 3.8Hz). LRMS calc: 553.1 obs: 554.1 (M+H).
Example 39 Step A l-(2-ferl-butoxy-2-oxoethyI)-li/-imidazole-2-carboxyKc acid ethyl ester

Using the method of Example 4, Step B with lH-imidazoIe-2-carboxylic acid ethyl ester as the starting material the title compound was obtained.
Step B f2-(ethoxycarbonyl)-l^r-imidazol-l-vU acetic acid hvdrotrifluoroacetate

Using the method of Example 2, Step B with the product of Example 39, Step A as the starting materia! the title compound was obtained.
Step C l-f2-l4-r2-f3.5-di-fer/-butvI-4-methoxyphenyl)-1.3-thiazol-4-vIlpiperidin-l-vU-2-oxoethvO- l/7-imidazoIe-2-carboxyHc acid ethyl ester

Using the method of Example I, Step L with the products of Example 1, Step H and Example 39, Step B as the starting materials the title compound was obtained.
Step D l-(2-(4-r2-(3,5-di-ferf-butyl-4-methoxyphenyl)-13-thiazoI-4-vnpiperidin-l-vU-2-oxoethvI)- lH-imidazole-2-carboxylic acid dihvdrochloride

A solution of the product of Example 39, Step C (21mg; 0.038mmol) in ethanol (ImL) was treated with aq. potassium hydroxide (76μL; IM; 0.076mmol). The solution was stirred at ambient temperature for 48h. The ethanol was evaporated and the remaining residue dissolved in methanol (0.5mL) and diluted with aq. HCl (1OmL; IN). Lyophilization afforded a solid that was digested in hot ethanol (3mL). The mixture was filtered and the filtrate evaporated to give the title compound (20mg). 1H NMR (CD3OD): δ 7.95 (s, 2H), 7.87 (bs, IH), 7.78 (s, IH), 7.63 (s, IH), 5.78 (Y2AB, IH, J = 15.5Hz), 5.67 (IZ2AB, IH, J = 15.7Hz), 4.62 (bd, IH, J= 12.6Hz), 4.12 (bd, IH5 J= 11.9Hz), 3.79 (s, 3H), 3.47 (vbt, IH, J- 11.0Hz), 3.39 (vbs, IH), 2.99 (bt, IH, J= 12.2Hz)5 2.32 (bd, IH, J = 10.0Hz), 2.23 (bd, IH, J = 11.5Hz), 2.01 (vbd, IH, J = 9.6Hz), 1.77 (vbquart, IH, J= 10.3Hz), 1.52 (s, 18H). LRMS calc: 538.3 obs: 539.4 (M+H).
Example 40 Step A 9flr-fluoren-9-vlmethvl 4-f2-phenvI-1.3-thiazol-4-vπpiperidine-l-carboxylate

Step B 4-f2-phenvI-l,3-thiazoI-4-yl)piperidine

Using the method of Example 1, Step H with the product of Example 40, Step A as starting material the title compound was obtained.
Step C l-ethyl-3-{2-oxo-2-14-(2-phenvI-l,3-thiazoI-4-vnpiperidin-l-vnethvU-l,3-dihγdro-2/r- imidazol-2-one

Using the method of Example 1, Step L with the products of Example 40, Step B and Example 4, Step C as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 7.91 (mult, 2H)5 7.45 (mult, 3H)5 7.19 (s, IH), 6.51 (d, IH, J = 2.8Hz), 6.44 (d, I H, J= 2.9Hz), 4.64 ('/2AB5 IH, J= 16.7Hz), 4.56 (14AB overlapping bs, 2H total, JAB = 16.8Hz), 4.04 (bd, IH, J = 13.0Hz), 3.67 (quart, 2H, J =
7.3Hz), 3.31 (bt, 1H, J= 12.0Hz), 3.11 (bt, IH5 J= 12.2Hz)5 2.87 (bt, IH5 J= 12.0Hz)5 2.16 (bd, IH5 J = 12.9Hz), 2.10 (bd, IH5 J= 12.9Hz), 1.81 (dquart, IH5 Jq = 12.6, Jd = 3.7Hz), 1-73 (dquart, IH5 Jq = 12.6, Jd = 3.8Hz), 1.26 (1, 3H, ^= 7.2Hz). LRMS calc: 396.2 obs: 397.2 (M+H).
Step D 9H-fl uo ren-9-yI methyl 4-[2-(4-methoxyphenv-)-l,3-thiazol-4-yI]piperidine-l-carboxvlate

Using the method of Example 1, Step G with the product of Example 1, Step C and 4- methoxybenzenecarbothioamide as the starting materials the title compound was obtained.
Step E 4-[2-(4-methoxyphenyl)-l.,3-thiazol-4-yllpiDeridine

Using the method of Example 1, Step H with the product of Example 40, Step D as starting material the title compound was obtained.
Step F l-ethyl-3-(2-f4-f2-f4-methoxγphenyl)-l.,3-thiazol-4-vHpiperidin-l-yl}-2-oxoethvπ-l,3- dihydro-2//-irnidazol-2-one
Using the method of Example 1, Step L with the products of Example 40, Step E and Example 4, Step C as the starting materials the title compound was obtained. I H NMR (CD3OD): δ 7.84 (d, 2H, J = 8.8Hz), 7.09 (s, IH), 6.99 (d, 2H, J = 8.8Hz), 6.51 (d, IH, J = 2.8Hz), 6.44 (d, IH, J= 2.9Hz), 4.63 (1MB, IH, J= 16.6Hz)5 4.55 (V2AB overlapping bs, 2H total, JAB = 16.6Hz), 4.03 (bd, IH, J= 13.2Hz),
3.83 (s, 3H), 3.67 (quart, 2H, J= 7.3Hz), 3.31 (bt, IH, J= 12.0Hz), 3.08 (tt, IH, J= 11.7, 3.6Hz), 2.86 (dt, I H, Jt = 12.7, Jd = 2.1Hz), 2.15 (bd, IH3 J= 11.9Hz), 2.09 (bd, IH3 J= 12.0Hz), 1.80 (dpent, IH, Jp
= 12.2, Jd = 3.5Hz), 1.69 (dpent, IH, Jp = 12.4, Jd = 3.6Hz), 1.26 (t, 3H, J= 7.2Hz). LRMS calc: 426.2 obs: 427.3 (M+H).
Step G 9./y-fluoren-9-ylmethvI 4-f2-(4-ferf-butyIphenvO-1.3-thiazol-4-vπpiperidine-l-carboxvlate

Using the method of Example 1, Step G with the product of Example 1, Step C and A-tert- butylbenzenecarbothioamide as the starting materials the title compound was obtained.
Ill
W
Step H 442-f4-ter/4>utvlphenvI)-l^-thiazoI-4-vllpiperidine

Using the method of Example 1, Step H with the product of Example 40, Step G as starting material the title compound was obtained.
Step J l-(2-|4-[2-(4-.'ejr/-butvIphenyl>-l,3-thiazoI-4-vnpipcridin-l-vIt-2-oxoethyl)-3-ethvI-l^-

Using the method of Example 1, Step L with the products of Example 40, Step H and Example 4, Step C as the starting materials the title compound was obtained. ^H NMR (CD3OD): δ 7.83 (d, 2H5 J =
8.5Hz)3 7.49 (d, 2H, ./ = 8.6Hz), 7.15 (s, I H)5 6.51 (d, IH, J = 2.8Hz), 6.44 (d, IH, J = 2.9Hz), 4.63 ('/2AB, IH, J= 16.6Hz)5 4.56 QAAB overlapping bs, 2H total, JAB = 16.7Hz), 4.04 (bd, IH, J= 12.7Hz), 3.67 (quart, 2H, J= 7.3Hz), 3.33 (bt, 1 H, J= 12.0Hz), 3.10 (tt, IH, J= 11.5, 3.5Hz), 2.86 (bt, IH, J = 11.6Hz), 2.15 (bd, IH5 J= 13.0Hz), 2.10 (bd, IH5 J- 12.5Hz), l.80 (dquart, IH5 Jq = 12.4, Jd = 3.5Hz), 1.69 (dquart, IH5 Jq = 12.4, Jd = 3.6Hz)5 1.34 (s, 9H)5 1.26 (t, 3H, J= 7.2Hz). LRMS calc: 452.2 obs: 453.3 (M+H).
Step K 9/7-fluoren-9-ylinethvI 4-f2"(4-chlorophenylM,3-thiazoI-4-yl1piperidine-l-carboxylate

Using the method of Example I5 Step G with the product of Example I5 Step C and 4- chlorobenzenecarbothioamide as the starting materials the title compound was obtained.
Step L 4-f2-(4-chIorophenyl>1.3-thiazol-4-vπpiperidine

Using the method of Example 1, Step H with the product of Example 40, Step K as starting material the title compound was obtained.
Step M l-(2-H-r2-(4-chIorophenvI)-l,3-thiazol-4-yllpiperidin-l-vU-2-oxoethyl)-3-ethyl-13- dihydro-2JHr-imidazol-2-one .

Using the method of Example 1, Step L with the products of Example 40, Step L and Example 4, Step C as the starting materials the title compound was obtained. Ifϊ NMR (CD3OD): δ 7.91 (d, 2H3 J = 8.5Hz), 7.46 (d, 2H, J = 8.6Hz), 7.22 (s, IH), 6.51 (d, I H, J = 2.8Hz), 6.44 (d, IH, J = 2.9Hz), 4.63 (!4AB, IH, J= 16.8Hz), 4.56 QAAB overlapping bs, 2H total, JAB = 16.8Hz), 4.04 (bd, IH, J= 13.9Hz), 3.67 (quart, 2H, J = 7.3Hz), 3.31 (bt, IH, J= 11.8Hz), 3.11 (tt, IH, J= 1 1.6, 4.0Hz), 2.87 (dt, IH, Jt = 12.9, Jd = 2.2Hz), 2.15 (bd, IH5 J= 13.0Hz), 2.09 (bd, IH, J= 13.1Hz), 1.81 (dquart, 1 H, Jq = 12.6, Jd == 3.4Hz), 1.70 (dquart, IH, Jq = 12.3, Jd = 3.7Hz), 1.26 (t, 3H, J = 7.2Hz). LRMS calc: 430.1 obs: 431.2 (M+H).
Step N 9//r-fluoren-9-ylmethy.4-f2-(4-bromophenyI)-1.3-thiazoI-4-vHpiρeridine-l-carboxvlate

Using the method of Example 1, Step G with the product of Example 1, Step C and 4- bromobenzenecarbothioamide as the starting materials the title compound was obtained.
Step P 4-f2-f4-bromophenγπ-l,3-thiazol-4-yl1piperidine

Using the method of Example 1, Step H with the product of Example 40, Step N as starting material the title compound was obtained.
Step Q l-(2-f4-f2-f4-bromophenv0-13-thiazol-4-yllpiperidin-l-yl>-2-oxoethvI)-3-ethyl-13- dihydro-2//-imidazol-2-one

Using the method of Example 1, Step L with the products of Example 40, Step P and Example 4, Step C as the starting materials the title compound was obtained. JH NMR (CD3OD): δ 7.89 (d, 2H, J =
8.4Hz)5 7.66 (d, 2H, J = 8.5Hz), 7.26 (s, IH), 6.54 (d, IH, J = 2.9Hz), 6.47 (d, IH, J = 3.0Hz), 4.68 O/2AB, IH, J= 16.7Hz), 4.59 (1AAB overlapping bs, 2H total, JAB = 16.6Hz), 4.07 (bd, IH, J= 13.0Hz),
3.70 (quart, 2H, J = 7.3Hz), 3.31 (bt, IH, J= 11.8Hz), 3.15 (tt, IH, J= 11.5, 3.6Hz), 2.90 (dt, IH, Jt =
13.0, Jd = 2.1Hz), 2.19 (bd, IH, J= 12.6Hz), 2.13 (bd, IH, J= 12.6Hz), 1.85 (dquart, IH, Jq = 12.4, Jd =
3.7Hz), 1.74 (dquart, IH, Jq = 12.3, J<j = 3.9Hz), 1.30 (t, 3H, J = 7.2Hz). LRMS calc: 474.1 obs:
475.2 (M+H).
Step R g/Z-fluoren-P-ylmethyl 4-f2-(4-trifluoromethylphenvO-l,3-thiazol-4-yπpiperidine-l- carboxylate

Using the method of Example 1, Step G with the product of Example 1, Step C and 4- trifluoromethylbenzenecarbothioamide as the starting materials the title compound was obtained.
Step S 4-[2-f4-trifluoromethγlphenvI)-13-thiazol-4-vl]piperidine

Using the method of Example 1 , Step H with the product of Example 40, Step R as starting material the title compound was obtained.
Step T l-f2-f4-r2-f4-trifluoromethylphenyl>-l,3-thiazol-4-vHpiperidin-l-yl}-2-oxoethyl)-3-ethγl- 13-dihvd ro-2iy-imidazol-2-one

Using the method of Example 1, Step L with the products of Example 40, Step S and Example 4, Step C as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 8.11 (d, 2H, J = 8.1Hz)5 7.76 (d, 2H, J = 8.3Hz), 7.31 (s, IH), 6.51 (d, IH, J = 2.8Hz)3 6.44 (d, I H, J = 2.9Hz), 4.64 C'/aAB, IH, J= 16.8Hz), 4.56 QAAB overlapping bs, 2H total, JAB = 16.8Hz), 4.05 (bd, IH, J= 13.0Hz),
3.67 (quart, 2H, J= 7.3Hz), 3.33 (bt, IH, J= 12.2Hz), 3.14 (tt, IH, J= 11.6, 3.4Hz), 2.88 (bt, IH, J = 11.8Hz), 2.17 (bd, IH3 J= 12.8Hz), 2.11 (bd, IH5 J= 12.4Hz), 1.83 (dquart, IH, Jq = 12.6, Jd = 3.9Hz), 1.73 (dquart, IH5 Jq = 12.7, Jd = 4.0Hz), 1.26 (t, 3H, J = 7.4Hz). LRMS calc: 464.2 obs: 465.2 (M+H).
Step U 9//-fluoren-9-vImethvl 4-f2-(3-chIorophenvD-l,3-thiazoI-4-yI1piperidine-l-carboxvlate

Using the method of Example 1, Step G with the product of Example 1, Step C and 3- chlorobenzenecarbothioamide as the starting materials the title compound was obtained.
tep V 4-12-(3-chIorophenvr)-l,3-thiazol-4-yllpiperidine

Using the method of Example 1 , Step H with the product of Example 40, Step U as starting material the title compound was obtained.
Step W l-(2-(4-F2-(3-chlorophenvn-l,3-thiazoI-4-yIlpiperidin-l-vn-2-oxoethvn-3-ethyl-l,3- dihydro-2jy-imidazoI-2-one

Using the method of Example 1, Step L with the products of Example 40, Step V and Example 4, Step C as the starting materials the title compound was obtained. IH NMR (CDSOD): δ 7.94 (bs, IH), 7.82-
7.80 (bmult, IH)5 7.43-7.41 (bmult, 2H), 7.24 (s, I H), 6.51 (d, IH, J = 2.8Hz), 6.44 (d, IH, J= 2.9Hz), 4.63 (!Z2AB, IH, J = 16.8Hz), 4.56 (14AB overlapping bs, 2H total, JAB = 16.8Hz), 4.04 (bd, IH5 J =
13.3Hz), 3.67 (quart, 2H, J= 7.4Hz), 3.31 (bt, IH, J= 12.2Hz)5 3.11 (tt, IH, J= 11.5, 3.7Hz), 2.86 (bt, IH5 J= 11.6Hz), 2.15 (bd, IH, J= 13.1Hz), 2.09 (bd, IH5 J= 13.2Hz), 1.81 (dquart, lH, Jq = 12.3, Jd = 3.8Hz), 1.71 (dquart, IH, Jq = 12.2, Jd = 3.8Hz), 1.26 (t, 3H, J = 7.2Hz). LRMS calc: 430.1 obs: 431.2 (M+H).
Step X 9iy-fluoren-9-γlmethyI 4-f2-(3-bromophenyl)-13-thiazol-4-vπpiperidine-l-carboxvlate

Using the method of Example I5 Step G with the product of Example 1, Step C and 3- bromobenzenecarbothioamide as the starting materials the title compound was obtained.
Step Y 4-f2-(3-bromophenvI)-l,3-thiazol-4-vπpiperidine

Step Z l-(2-{4-f2-f3-bromophenvπ-1.3-thiazol-4-vIlpiperidin-l-γU-2-oxoethyl)-3-ethyi-13- dihvdro-2i7-imidazol-2-one

Using the method of Example 1, Step L with the products of Example 40, Step Y and Example 4, Step C as the starting materials the title compound was obtained. IH NMR (CDSOD): δ 8.10 (bs, IH), 7.86 (d,
IH, J = 7.7Hz), 7.58 (d, IH, J= 7.9Hz), 7.37 (t, IH, J= 7.8Hz), 7.25 (s, IH), 6.51 (d, IH, J = 2.7Hz), 6.44 (d, IH, J = 2.7Hz)5 4.64 ("4AB, IH, J = 16.8Hz), 4.56 (1^AB overlapping bs, 2H total, JAB =
16.8Hz), 4.04 (bd, IH, J= 12.3Hz), 3.67 (quart, 2H, J= 7.2Hz), 3.33 (bt, IH3 J= 12.5Hz), 3.12 (bt, IH, J = 11.9Hz), 2.87 (bt, IH, J= 1 1.9Hz), 2.15 (bd, IH, J= 13.3Hz), 2.09 (bd, IH, J= 13.0Hz), 1.83 (dquart, IH, Jq = 12.7, Jd = 3.4Hz), 1.73 (dquart, IH, Jq = 12.5, Jd = 3.6Hz), 1.26 (t, 3H, J = 7.3Hz). LRMS calc: 474.1 obs: 475.1 (M+H).
Step AA 9/f-fIuoren-9-'ylinethyl 4-[2-(3-trifluoromethylphenyiyi,3-thiazol-4-vI1piperidine-l- carboxvlate

Step BB 4-[2-(3-trifluoromethvlphenvlM3-thiazol-4-γ-lpiperidine

Using the method of Example 1, Step H with the product of Example 40, Step AA as starting material the title compound was obtained.
Step CC l-(2-(4-f2-(3-trifluoromethylphenvI)-l,3-thiazol-4-yl1piperidin-l-vU-2-oxoethylV3-ethyl- l.S-dihvdro-l/y-imidazol-Σ-one

Using the method of Example I, Step L with the products of Example 40, Step BB and Example 4, Step C as the starting materials the title compound was obtained. IH NMR (CDSOD): δ 8.22 (bs, IH), 8.15
(d, IH3 J = 7.6Hz), 7.73 (d, IH, J= 7.7Hz), 7.66 (t, IH, J- 7.8Hz), 7.29 (s, IH), 6.51 (d, IH, J= 2.8Hz), 6.44 (d, IH, J = 2.7Hz), 4.64 (MB, IH, J = 16.8Hz), 4.56 (!Z2AB overlapping bs, 2H total, JAB =
16.8Hz), 4.05 (bd, IH, J= 13.0Hz), 3.67 (quart, 2H, J= 7.2Hz), 3.33 (bt, IH, J= 13.0Hz), 3.14 (tt, IH, J = 11.8, 3.6Hz), 2.87 (dt, 1 H, Jt = 12.8, Jd = 2.0Hz), 2.17 (bd, IH, J= 13.2Hz), 2.1 1 (bd, IH, J= 13.3Hz), 1.84 (dquart, IH, Jq = 12.7, Jd = 3.7Hz), 1.72 (dquart, IH, Jq = 12.3, Jd = 3.8Hz), 1.26 (t, 3H, J = 7.4Hz). LRMS calc: 464.2 obs: 465.2 (M+H).
Step DD 9g-fluoren-9-ylmethyl 4-f2-f3,5-dichlorophenyl)-l,3-thiazoI-4-yllpiperidine-t- carboxylate

Using the method of Example 1, Step G with the product of Example 1, Step C and 3,5- dichlorobenzenecarbothioamide as the starting materials the title compound was obtained.
Step EE 4-f2-f3,5-diehIorophenviyi,3-thiazoI-4-yllpiperidine

Using the method of Example 1, Step H with the product of Example 40, Step DD as starting material the title compound was obtained.
Step FF l-f2-(4-r2-(3,5-dichlorophenyl)-l.,3-thiazol-4-vnpiperidin-l-vn-2-oxoethvn-3-ethvI-1.3- dihvdro-2/y-imidazoI-2-one

I.6H2), 7.51 (d, IH5 J = 1.6Hz)3 7.31 (s, IH), 6.51 (d, IH, J = 2.8Hz), 6.44 (d, IH, J = 2.8Hz), 4.64 (Y2AB, IH, J= 16.4Hz), 4.56 (/2AB overlapping bs, 2H total, JAB = 16.5Hz), 4.05 (bd, IH, J= 13.3Hz), 3.67 (quart, 2H, J= 7.4Hz), 3.30 (bt, IH, J= 12.0Hz)3 3.12 (tt3 IH3 J= 11.5, 3.4Hz)5 2.87 (dt, IH, Jt = 12.6, Jd = 2.2Hz)3 2.15 (bd, IH. J= 12.5Hz), 2.09 (bd, IH3 J= 12.7Hz)3 1.83 (dquart, IH3 Jq = 12.4, Jd = 3.7Hz), 1.71 (dquart, IH, Jq = 12.5, Jd = 3.8Hz)3 1.26 (t, 3H, J = 7.2Hz). LRMS calc: 464.1 obs: 465.2 (M+H).
Step NN 9/ιr-fluoren-9-vImethvI 4-(2-pyridin-4-yl-l,3-thiazot-4-vI)piperidine-l-carboxylate
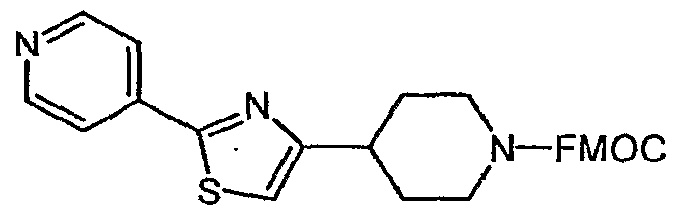
Using the method of Example 1, Step G with the product of Example 1, Step C and 4-pyridin-4- ylbenzenecarbothioamide as the starting materials the title compound was obtained.
Step PP 4-(4-piperidin-4-yl-1.3-thiazo--2-yl)pyr-dine

Using the method of Example 1, Step H with the product of Example 4O3 Step NN as starting material the title compound was obtained.
Step QQ l-ethvI-3-^2-oxo-2-r4-(2-pyridin-4-yl-l,3-thiazoI-4-yI)piperidin-l-vUethvU-13-dihvdro- 2/T-imidazoI-2-one

Using the method of Example 1, Step L with the products of Example 40, Step PP and Example 4, Step C as the starting materials the title compound was obtained. lH NMR (CD3OD): δ 8.63 (dd, 2H, J= 4.7,
1.5Hz), 7.94 (dd, 2H, J = 4.8, 1.5Hz), 7.42 (s, IH), 6.52 (d, IH, J = 2.9Hz), 6.44 (d, IH, J= 2.9Hz), 4.65 (1AAB, IH, J= 16.8Hz), 4.56 (1AAB overlapping bs, 2H total, JAB = 16.7Hz), 4.05 (bd, IH, J= 13.8Hz), 3.67 (quart, 2H, J= 7.3Hz), 3.31 (bt, IH, J= 11.9Hz), 3.16 (bt, IH, J= 11.7Hz), 2.88 (dt, IH, Jx = 12.6, Jd = 2.2Hz), 2.17 (bd, IH, J = 1 1.5Hz), 2.11 (bd, IH, J = 13.1Hz), 1.84 (dquart, IH, Jq = 12.7, Jd = 3.8Hz), 1.73 (dquart, IH, Jq = 12.4, Jd = 3.9Hz), 1.26 (t, 3H, J = 7.5Hz). LRMS calc: 397.2 obs: 398.2 (M+H).
Assays for Determining Biological Activity
HUMAN CXCR3 RECEPTOR. The compounds claimed here are assayed for affinity and functional potency at the
CXCR3 receptor using the assays described below.
Since the expression of CXCR3 on naive T cells is low, PBMCs were cultured in the presence of a mixture of superantigens to provide primary cells with sufficient CXCR3 expression to use routinely in binding and functional assays. Briefly, mononuclear cells were enriched from buffy coats obtained from a local blood bank by centrifugation over FicoH-Hypaque. Residual red blood cells were lysed in hypotonic buffer, (ACK), cells were washed with PBS and resuspended in media (RPMI containing 10% FBS, 2 mM glutamine, MEM non essential amino acids and sodium pyruvate) containing 500 Units/ml of BL-2 and 0.5 ng/ml SE cocktail (containing equal amounts of SEA, SEB, SECl, SED and SEE all from Toxin Technology). After several days in culture, cells were switched to fresh media containing 500 units/ml of IL-2 and cultures were maintained at 2-4 million cells /ml for up to 21 days.
BENDING ASSAY.
Inhibition of binding of CXCLlO or CXCLl 1 to human CXCR3 was measured in whole cells, using superantigen activated T cells (SE-T) at day 7-14 post stimulation. Binding of 125i-τp_io (2200 Ci/mmol, typically 20 pM) in the presence of unlabeled Iigands was initiated by adding intact T cells (200,000 cells/assay) in a total assay volume of 250 μl containing 50 mM HEPES, pH 7.2, 5 mM MgC12, 1 mM CaC12 and 0.5% BSA. Binding of 125i_i_TAC (2200 Ci/mmol, 2OpM) was performed as
described for IP- 10 except for the addition of 0.15M NaCl to the binding buffer. After incubation at room temperature for 2 hours with shaking, the reaction was terminated by filtering through a 0.1% polyethylenimine (Sigma) soaked GF/C filter plate (Packard) using a Packard Filtermate cell harvester and the plate washed with approximately 750 μl of 50 mM HEPES (Sigma), pH 7.2, 500 mM NaCl chilled to 4°C. The plates were dried; scintillant added and counted on a Packard TopCount. Nonspecific binding was measured in the presence of 1 μM ligand (IP-10 or I-TAC). Binding results were analyzed using Microsoft Excel and GraphPad Prism software.
The Examples disclosed herein were tested in the above receptor binding assay and demonstrated an IC50 ranging from I to 1200 nM against 125i-]p-io. The range of IC50 values is similar against 125i-i-χ AC.
FUNCTIONAL ASSAYS.
The functional potency of the claimed compounds was assessed by measuring inhibition of the chemotaxis of leukocytes in response to CXCR3 ligands. A modified Boyden chamber chemotaxis system (ChemoTxTM, NeuroProbe, Gaithersburg, MD), consisting of a 96-well microplate and a filter (6.0-mm diameter, 5-μ pore size), coated on the bottom with fibronectin (50 μl of a 10 μg/ml solution, then air-dried), was used for chemotaxis measurements. Briefly, aliquots of human T cells (day 14 to day 17 post activation) were washed and resuspended at 1 x 107cells/ml in warm (37°C) Hanks' balanced saline solution (HBSS)/bovine serum albumin [(BSA); HBSS without phenol red, calcium, or magnesium (Mediatec)+0.01% BSA] and loaded with Calcein-AM (Molecular Probes) at a concentration of 2 μM for 30 min at 37°C. The cells were washed again in HBSS/BSA and resuspended in RPMI/BSA [RPMI without phenol red (Mediatec)+0.5% BSA+1% dimethyl sulfoxide] to a concentration of 6 x 106celIs/mI. To initiate the chemotaxis, chemokines were diluted in warm (37°C) RPMI/BSA and added in 30 μl to the bottom of the microplate before affixing the filter to the unit. Aliquots (50 μl) of the Calcein-loaded T cells were then added to the top of the filter over each individual well. The microplates were subsequently incubated for 1 h at 37°C. Remaining cells were suctioned off the top of the filter. The filter was rinsed with PBS and wiped with a rubber squeegee. The plate with filter intact was read in a CytofluorTM II fluorometer (PerSeptive Biosystems, Foster City, CA). For assay of antagonists, compounds were diluted in DMSO and added to both cells and ligand in a final DMSO concentration of 0.5%.
The Examples disclosed herein were tested in the above assay against both IP-10 and I- TAC. The Examples demonstrated an IC50 ranging from 0.5 to 600 nM against EP-IO and typically a somewhat higher IC50 ranging from 25 to 1700 nM against I-TAC.
Claims
1. A compound of Formula I

I or a pharmaceutically acceptable salt thereof, wherein:
D is CR4 or N;
R.3 is selected from the group consisting of: H, halo, Cχ_4alkyl, -CF3, -OCF3 and -S(O)nCF3, wherein n is 0 or 2;
R4 is selected from the group consisting of: H, halo, -OH, Ci_4alkyl, -OCH3, -OCH2CF3 and -CF3;
or R3 and R4 may be joined together with the carbon atoms to which they are attached to form a five- or six-membered monocyclic ring, said rings containing oxygen or tetra-substituted with methyl groups as follows:

R5 is selected from the group consisting of: -H, halo, Ci-4alkyl, C3_6cycloalkyl, CF3, -CF2CH3, -OCF3 and -SCF3;
Rg is selected from the group consisting of — H and -OCH3, or R5 and R.6 may be joined together with the carbon atoms to which they are attached to form a monocyclic 5-membered ring, said ring di-substituted with methyl as follows:

is a 5 membered non-aromatic or aromatic ring or a 9 membered fused bicyclic partially  aromatic or aromatic ring, each ring containing at least 1 nitrogen atom and optionally up to 3 additional heterotaoms selected from S, O and N, said rings optionally substituted with 1 to 3 substituents independently selected from the group consisting of: oxo, hydroxy, carboxy, -CF3, halo, -S(O)p-CH3, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy; and
aromatic or aromatic ring, each ring containing at least 1 nitrogen atom and optionally up to 3 additional heterotaoms selected from S, O and N, said rings optionally substituted with 1 to 3 substituents independently selected from the group consisting of: oxo, hydroxy, carboxy, -CF3, halo, -S(O)p-CH3, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy; and
p is 0, 1 or 2.
2. The compound according to Claim 1 wherein:
is selected from the group consisting of: 

R"2> TR-"3? R"4 and R"5 are independently selected from the group consisting of: -H5 carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy,
R"6 is H or OH5 and
is an optional double bond.
3. The compound according to Claim 1 wherein D is N.
4. The compound according to Claim 1 wherein D is CR4.
5. The compound according to Claim 4 wherein:
R3 and R4 are joined together with the carbon atoms to which they are attached to form a six-membered monocyclic ring as follows:

6. The compound according to Claim 4 wherein:
R3 is selected from the group consisting of: H, halo, Ci_4alkyl and -CF3;
R4 is selected from the group consisting of: H, halo, Ci_4alkyl, -OCH3 and -CF3;
R5 is selected from the group consisting of: H, halo, C] _4alkyl, CF3 and -SCF3; and
Rζ is H.
7. The compound according to Claim 6 wherein:
R3 is selected from the group consisting of: H, Cl, Br5 tert-butyl and -CF3;
R4 is selected from the group consisting of: H, Cl, Br, tert-butyl, -OCH3 and -CF3;
R5 is selected from the group consisting of: H, Cl, Br5 fer/-butyl, CF3 and -SCF3; and
R6 is H.
8. The compound according to Claim 1 wherein R6 is -H.
9. The compound according to Claim 1 wherein:
D is CR4;
R3 is selected from the group consisting of: H, Cl, Br, tert-butyl and -CF3; W
R4 is selected from the group consisting of: H, Cl, Br3 tert-butyl, -OCH3 and -CF3;
or R3 and R4 may be joined together with the carbon atoms to which they are attached to form a six- membered monocyclic ring as follows:

R5 is selected from the group consisting of: H, CI, Br, tert-butyl, CF3 and -SCF3; and
R6 ΪS -H.
10. The compound according to Claim 9 wherein:
(I) R3 is ter t-butyl, R4 is H and R5 is tert-butyl; (2) R3 is tert-butyl, R4 is -OCH3 and R5 is tert-buty\;
(3) R3 is -CF3, R4 is -H and R5 is -CF3;
(4) R3 is H, R4 is -OCH3 and R5 is H;
(5) R3 is H, R4 is rerΛ-butyl and R5 is H;
(6) R3 is H5 R4 is Cl and R5 is H; (7) R3 is H, R4 is Br and R5 is H;
(8) R3 is H, R4 is -CF3 and R5 is H;
(9) R3 is H, R4 is H and R5 is Cl;
(10) R3 is Ff, R4 is H and R5 is Cl;
(I I) R3 is H, R4 is H and R5 is -CF3; or (12) R3 is Cl, R4 is H and R5 is Cl.
11. The compound according to Claim 9 wherein


R"2, R"3, R"4 and R"5 are independently selected from the group consisting of: -H, carboxy, -CF3, halo, methylthio, methylsulfonyl, phenyl, Ci_3alkoxy and Ci_3alkyl, said Ci_3alkyl optionally substituted with carboxy or hydroxy,
R"6 is H or OH, and
is an optional double bond.
12. The compound according to Claim 11 wherein:

13. The compound according to Claim 9 wherein:
D is CR4;
R3 is tert-butyl;
R4 is H or -OCH3;
R5 is tert-butyl; and
R6 is H.
14. A compound according to Claim 1 selected from the following group:
(1) 3-(2-{4-[2-(3,5-di-tert-bu1yl-4-methoxyphenyl)-l53-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3H-imidazo[4;5-ό]pyridine;
(2) 4-[2-(3,5-di-tert-butyI-4-methoxyphenyl)-I,3-thiazol-4-yl]-l-[(255-dimethyI-lH-imidazol- 1 -yl)acetyl]piperidine;
(3) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l:,3-thiazol-4-yI]piperidin-l-yl}-2- oxoethy l)-2-methyl- 1/f-imidazo 1-5-y l]methano 1 ;
(4) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l53-thiazol-4-yl]piperidin-l-y]}-2- oxoethy l)-3 -methyl- 1 ,3 -d ihydro-2H-im idazol-2-one;
(5) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-ylJpϊperidin-l-yl}-2- oxoethyl)-l,4-dimethyl-l,3-dihydro-2H-imidazoI-2-one;
(6) [l-(2-{4-[2-(3,5-di-tert-butyI-4-methoxyphenyl)-l,3-thiazol-4-yl3piperidin-l-yl}-2- oxoethyl)-4-methyl-lH"-pyrazol-3-yl]acetate bis(hydrotrifluoroacetate); (7) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-{[4-methyI-2- (methylsulfonyl)-lH-imϊdazol-l-yI]acetyl}piperidine;
(8) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-] ,3-dihydro-2//-imidazol-2-one;
(9) Potassium_[3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l- yI}-2-oxoethyl)-2-oxo-2,3-dihydro-lH-imidazol-l-yl]acetate;
(10) l-(2-{4-[2-(3;5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-3-phenylimidazolidin-2-one;
(11) 3-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazol-4-yl)piperidin-l- yl]-2-oxoethyl}-3H-imidazo[4.,5-ό]pyridine;
( 12) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)- 1 ,3-thiazol-4-yl]- lH-[(4-methoxy- 1 -imidazol- 1 - yl)acetyl]piperidine;
(13) 4-[2-(8-tert-butyl-4,4-dimethyl-3,4-dihydro-2H-chromen-6-yl)-I,3-thiazol-4-yl]-l-[(2,4- dimethyl-1 H-imidazol-l-yl)acetyl]piperidine;
(14) l-[(4-chloro-2,5-dimethyl-lH-imidazol-l-yl)acetyl]-4-[2-(3,5-di-/e -butyl-4- methoxyphenyl)-l,3-thiazol-4-yl]piperidine;
(15) 3-(2-{4-[2-(3,5-di-/e/-f-butylphenyl)-l,3-thiazol-4-yI]piperidin-l-yl}-2-oxoethyl)-3H- imidazo[435-c]pyridine;
(16) l-(2-{4-[2-(3,5-di-tert--butyIphenyl)-l,3-thiazol-4-yl]piperidJn-l-yl}-2-oxoethyl)-lH- imidazo[4.5-c]pyridine;
(17) 3-(2-{4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazoI-4-yl]piperidin-l-yl}-2-oxoethyl)-3H- imidazo[4,5-b]pyridinel; (18) l-(2-{4-[2-(3;5-di-tert-butyIphenyl)-l,3-thiazol-4-yl]piperidin-l-yI}-2-oxoethyl)-l/T- imidazo[4,5-ά]pyridine;
(19) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yI]piperidin-l-yl}-2- oxoethyl)-l-ethyl-4-methyl-l,3-dihydro-2i/-imidazol-2-one;
(20) l-(2-{4-[2-(3,5-di-tert-butylphenyl )-l,3-thiazoI-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyI- 4-methyl-l,3-dihydro-2i?-imidazoI-2-one;
(21) l-(2-{4-[2-(3,5-di-tert-butyI-4-raethoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyO-S-methyl-l^-dihydro^H^benzimidazol^-one;
(22) 4-[2-(3,5-di-tert-butyI-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(2-ethyI-4-methyI-lH- imidazol-l-yl)acetyl]piperidine;
(23) 2,6-di-tert-butyl-4-{4-[l-(lH-imidazo[4,5-c]pyridin-l-ylace1yl)piperidin-4-yl]-l,3- thiazoI-2-yl}phenol;
(24) 2,6-di-tert-butyl-4-{4-[l-(3H-imidazo[4,5-c]pyridin-3-ylacetyl)piperidin-4-yl]-l :,3- thiazol-2-yl} phenol;
(25) 2,6-di-tert-butyI-4-{4-[l-(3H -imidazo[4,5-b]pyridin-3-ylacetyI)piperidin-4-yl]-l,3- thiazol-2-yl}phenol;
(26) 2,6-di-tert-butyl-4-(4-{l-[(3,5-dimethyl-1H -l,2,4-triazol-l-yl)acetyl]pipendin-4-yI}-l,3- thiazo!-2-yI)phenol;
(27) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-4-methyl-lZf-imidazol-5-yl]methanol;
(28) 4-[2-(3,5-di-tert-butyI-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-{2-(methylthio)-lH- imidazol-1 -yl]acety]} piperidine; (29) 4-[2-(3,5-di-tert-butyI-4-methoxyphenyI)-l ,3-thiazol-4-yI]-l -{2-(methylsulfonyl)-lH- imidazol-l-yl]acetyl}pϊperidine;
(30) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]pipβridin-1 -yl}-2- oxoethyl) 5-methyl-lH-imidazol-4-yl]acetic dihydrochloride;
(31) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazoI-4-yl]piperidin-l-yl}-2- oxoethyl)-4-methyl-l H-imidazol-5-yl]acetic dihydrochloride;
(32) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyl-lH-l ,2,4- triazol-l-yl)aceryl]piperidine;
(33) 4-[2-(3,5-di-tert-butyIphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyl-lH'-l,2,4-triazol-l- yl)acetyl]piperidine;
(34) 4-[2-(3,5-di-tert-butyl-4-methoxypheny I)- 1 ,3-thiazol-4-yl]- 1 -[(4-methy 1- 1 -imidazol- 1- yl)acetyl]piperidine;
(35) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(5-methyl-l-imidazol-l- yl)acetyl]piperidine;
(36) 1 -(2- {4-[2-(3,5-di-tert butyI-4-methoxyphenyl)- 1 ,3-thiazol-4-yl]piperidiπ- 1 -yl } -2- oxoethyl)-3-methylimidazolidin-2-one;
(37) 3-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyρhenyI)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-l,3-oxazolidin-2-one;
(38) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-[(3,5-dimethyI-lH-pyrazoI- 1 -yl)acety l]piperidine;
(39) 4-[2-(3,5-di-tert-butyphenyl)-l,3-thiazol-4-y]]-l-[(3,5-dimethyl-lH-pyrazoI-l- yl)acety]]piperidine; (40) l-(2-{4-[2-(355-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-lH-pyrrolo[2,3-ά]pyridine;
(41) l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l:,3-thiazol-4-yI)piperidin-l- yl]-2-oxoethyI}-lH-pyrrolo[2,3-Z>]pyridine;
(42) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)-lH~benzimidazole;
(43) 1 -(2-{4-[2-(355-di-tert-butyl-4-methoxyρhenyl)-l,3-thϊazol-4-yI]piperidin-l -yl}-2- oxoethyl)-2-methyI-l//-benzimidazole;
(44) 2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazol-4-yl)piperidin-l-yl]-2- oxoethyI}-lH-benzimidazole;
(45) l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l ,3-thiazol-4-yI)piperidin- 1 - yl]-2-oxoethyl }-2-methyl- lH-benzimidazole;
(46) 4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]-l -{[5-methyl-3-(trifluoromethyI)- IH- pyrazol-l-yl]acetyi}pϊρeridine;
(47) l-fp.S-bis^rifluoromethyO-lH-pyrazol-l-yljacetyll^-P^^-di-tert-butylphenyO-l^- thiazol-4-y l]pi peridine;
(48) 4-bromo-3,5-dimethyl-lH-pyrazol-l-yl)acety]]-4-[2-(3,5-di-tert-butyIphenyI)-l,3-thiazol-
4-yl]piperidine;
(49) 1 -[(4-iodo-3,5-dimethyl- lH"-pyrazoI- 1 -yI)acetyl]-4-[2-(3,5-di-tert-butylphenyl)-l ,3- thiazoI-4-yl]piperidine;
(50) l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyl)imidazol i d in-2-one; (51) [1 -(2- {4-[2-(3,5-di-tert butyl-4-methoxyphenyI)- 1 ,3 -thiazoI-4-yl] piperidin- 1 -yl} -2- oxoethyl)- 1 H-im idazol-2-yl] metanol;
(52) [l-(2-{4-[2-(3,5-di-tert-butyl-4-methoxyphenyI)-l,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethy I)- li7-benzimidazol-2-yl]metanol;
(53) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yI]-l-{[2-(trifluoromethyl)-lH- i m idazo 1- 1 -y l]acetyl } p iperid ine;
(54) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-{[2-methyI-lH"-imidazol-l- y l]acety 1 } piperidine;
(55) 4-[2-(3,5-di-tert-butyl-4-methoxyphenyl)-l,3-thiazol-4-yl]-l-(lH-imidazol-l- ylacetyl)piperidine;
(56) [l-(2-{4-[2-(3,5-di-tert-butyIphenyl)-l:,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-5- methyl-lH-ρyrazol-3-yl]acetic acid;
(57) Potassium (l-{2-[4-(2-{3-tert-butyl-5-[(trifluoromethyl)thio]phenyl}-l,3-thiazol-4- yl)piperidin-l-yl]-2-oxoethyl}-5-methy!-lH-pyrazol-3-yl)acetate;
(58) 4-[2-(3,5-di-tert-butylphenyl)-l,3-thiazol-4-yl]-l-[(2,4-dimethyl-lH-imidazol-l- yl)acetyl]piperidine;
(59) 1 -(2-{4-[2-(3,5-di-tert butylphenyl)-l ,3-thiazol-4-yI]piperidin-l-y]}-2-oxoethyl)-3-ethyl-
1 ,3-dihydro-2//-imidazol-2-one;
(60) 3-[2-(4-{2-[3,5-bis(trifluoromethyl)pheπyl]-l,3-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]- 3H-imidazo[4,5-έ]pyridine;
(61) 4-{2-[3,5-bis(trifluoromethyl)pheny0-l,3-thtazol-4-yl}-l-[(355-dimethyl-lH-l52,4- triazol-1 -yl)acetyl]piperidine; (62) 3-[2-(4-{2-[355-bis(trifluoromethyl)phenyl]-l,3-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]- 3H-imidazo[4,5-c]pyridine;
(63) l-[2-(4-{2-[3,5-bis(trifluoromethyI)phenyl]-l53-thiazoI-4-yl}piperidin-l-yl)-2-oxoethyl]- lH-imidazo[4,5-c]pyridine;
(64) l-[2-(4-{2-[335-bis(trifluoromethyl)phenyI]-l53-thiazol-4-yl}piperidin-l-yl)-2-oxoethyl]- 2-methyl-lH-irnidazo[4,5-c]pyridine;
(65) l-(2-{4-[2-(3,5-di-re^-butyl-4-methoxyphenyl)-i,3-thiazol-4-yl]piperidin-l-yl}-2- oxoethyI)-lH-imϊdazole-2-carboxylic acid dihydrochloride;
(66) l-ethyl-3-{2-oxo-2-[4-(2-phenyl-l :,3-thiazol-4-yI)piperidin-l-yI]ethyI}-l,3-dihydro-2/-r- imidazol-2-one;
(67) l-ethyl-3-(2-{4-[2-(4-methoxyphenyl)-l,3-thiazoI-4-yl]piperidin-l-yl}-2-oxoethyl)-l,3- dihydro-2i7-imidazol-2-one;
(68) l-(2-{4-[2-(4-fert-butylphenyl)-l,3-thiazol-4-yl]piperidin-l -yl}-2-oxoethyl)-3-ethyl-l,3- dihydro-2if-imidazol-2-one;
(69) l-(2-{4-[2-(4-chlorophenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-l,3- dihydro-2H-imidazol-2-one;
(70) l-(2-{4-[2-(4-bromophenyI)-l,3-thiazoI-4-yI]pϊperidin-l-yI}-2-oxoethyl)-3-ethyI-l33- dihydro-2H-imidazol-2-one;
(71) l-(2-{4-[2-(4-trifluoromethy]phenyl)-l,3-thiazol-4-yl]piperidm-l-yl}-2-oxoethyl)-3- ethyl-l^-dihydro^H-imidazol^-one;
(72) l-(2-{4-[2-(3-chlorophenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3-ethyl-l,3- dihydro-2H-imidazol-2-one; (73) l-(2-{4-[2-(3-bromophenyl)-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyI)-3-ethyl-l,3- dihydro-2H-imidazol-2-one;
(74) l-(2-{4-[2-(3-trifluoromethylpheny])-l,3-thiazol-4-yl]piperidin-l-yl}-2-oxoethyl)-3- ethyl-l,3-dihydro-2H"-imidazol-2-one;
(75) l-(2-{4-[2-(3,5-dichlorophenyl)-l,3-thiazol-4-yl]piperidin-l-yI}-2-oxoethyl)-3-ethyl-l,3- dihydro-2H-imidazol-2-one; and
(76) l-ethyI-3-{2-oxo-2-[4-(2-pyridin-4-yl-l,3-thiazol-4-yl)piperidin-l-yl]ethyl}-l,3-dihydro-
2H-imidazo!-2-one.
15. A pharmaceutical composition comprising a compound according to Claim 1 in combination with a pharmaceutically acceptable carrier.
16. A method for treating a disease or condition mediated by the CXCR3 chemokine receptor comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound according to Claim 1.
17. The method according to Claim 16 wherein the disease or condition is selected from the group consisting of: acute and chronic transplant rejection, psoriasis, rheumatoid arthritis and multiple sclerosis.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06839263A EP1962605A2 (en) | 2005-12-12 | 2006-12-08 | 2-arylthiazole derivatives as cxcr3 receptor modulators |
| US12/086,361 US20100286191A1 (en) | 2005-12-12 | 2006-12-08 | 2-Arylthiazole Derivatives as CXCR3 Receptor Modulators |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US74941905P | 2005-12-12 | 2005-12-12 | |
| US60/749,419 | 2005-12-12 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007070433A2 true WO2007070433A2 (en) | 2007-06-21 |
| WO2007070433A3 WO2007070433A3 (en) | 2008-02-21 |
Family
ID=38163445
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/047065 Ceased WO2007070433A2 (en) | 2005-12-12 | 2006-12-08 | 2-arylthiazole derivatives as cxcr3 receptor modulators |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20100286191A1 (en) |
| EP (1) | EP1962605A2 (en) |
| WO (1) | WO2007070433A2 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008013622A3 (en) * | 2006-07-27 | 2008-03-27 | Du Pont | Fungicidal azocyclic amides |
| ES2335849A1 (en) * | 2008-10-03 | 2010-04-05 | Consejo Superior De Investigaciones Cientificas (Csic) | Chroman derivatives and uses thereof as inhibitors of the activity of decoupling proteins |
| WO2011098776A1 (en) | 2010-02-15 | 2011-08-18 | Cambridge Enterprise Limited | 5-ht receptor modulators |
| JP2012510520A (en) * | 2008-12-02 | 2012-05-10 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | Fungicidal heterocyclic compounds |
| CN102464620A (en) * | 2010-11-16 | 2012-05-23 | 上海药明康德新药开发有限公司 | Preparation method of 4-methyl-2-thiomethyl-1 (3) H-imidazole |
| WO2012123298A1 (en) | 2011-03-11 | 2012-09-20 | F. Hoffmann-La Roche Ag | Antiviral compounds |
| CN101928259B (en) * | 2009-06-18 | 2012-10-03 | 南京海辰药业有限公司 | 2-arylthiazole derivative and medicament composition thereof |
| WO2013053657A1 (en) | 2011-10-10 | 2013-04-18 | F. Hoffmann-La Roche Ag | Antiviral compounds |
| JP2013518823A (en) * | 2010-02-08 | 2013-05-23 | バイオウタ サイエンティフィック マネジメント プロプライエタリー リミテッド | Compounds for treating respiratory syncytial virus infection |
| US8449898B2 (en) | 2007-10-23 | 2013-05-28 | E I Du Pont De Nemours And Company | Fungicidal mixtures |
| WO2013087743A1 (en) | 2011-12-16 | 2013-06-20 | F. Hoffmann-La Roche Ag | Inhibitors of hcv ns5a |
| WO2013114332A1 (en) * | 2012-02-02 | 2013-08-08 | Actelion Pharmaceuticals Ltd | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| WO2015011099A1 (en) * | 2013-07-22 | 2015-01-29 | Actelion Pharmaceuticals Ltd | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| US9090604B2 (en) | 2006-07-27 | 2015-07-28 | E I Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| WO2015152254A1 (en) * | 2014-04-01 | 2015-10-08 | 大日本住友製薬株式会社 | Five-membered ring heteroaryl derivative |
| WO2016113346A1 (en) * | 2015-01-15 | 2016-07-21 | Actelion Pharmaceuticals Ltd | (r)-2-methyl-piperazine derivatives as cxcr3 receptor modulators |
| WO2016113344A1 (en) | 2015-01-15 | 2016-07-21 | Actelion Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| WO2018011265A1 (en) | 2016-07-13 | 2018-01-18 | Idorsia Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| US9951063B2 (en) | 2014-03-24 | 2018-04-24 | Idorsia Pharmaceuticals Ltd | 8-(piperazin-1-yl)-1,2,3,4-tetrahydro-isoquinoline derivatives |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111848521A (en) * | 2020-08-26 | 2020-10-30 | 韶远科技(上海)有限公司 | Preparation method of 2-substituted-4-alkoxy imidazole compound |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE9902765D0 (en) * | 1999-07-21 | 1999-07-21 | Astra Pharma Prod | Novel compounds |
| SE9902987D0 (en) * | 1999-08-24 | 1999-08-24 | Astra Pharma Prod | Novel compounds |
| GB0203994D0 (en) * | 2002-02-20 | 2002-04-03 | Celltech R&D Ltd | Chemical compounds |
-
2006
- 2006-12-08 EP EP06839263A patent/EP1962605A2/en not_active Withdrawn
- 2006-12-08 WO PCT/US2006/047065 patent/WO2007070433A2/en not_active Ceased
- 2006-12-08 US US12/086,361 patent/US20100286191A1/en not_active Abandoned
Cited By (55)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008013925A3 (en) * | 2006-07-27 | 2008-04-03 | Du Pont | Fungicidal azocyclic amides |
| US9920030B2 (en) | 2006-07-27 | 2018-03-20 | E I Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| US9604962B2 (en) | 2006-07-27 | 2017-03-28 | E I Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| US9090604B2 (en) | 2006-07-27 | 2015-07-28 | E I Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| WO2008013622A3 (en) * | 2006-07-27 | 2008-03-27 | Du Pont | Fungicidal azocyclic amides |
| US8642634B2 (en) | 2006-07-27 | 2014-02-04 | E I Du Pont De Nemours And Company | Fungicidal azocyclic amides |
| AU2007277157B2 (en) * | 2006-07-27 | 2013-01-31 | Corteva Agriscience Llc | Fungicidal azocyclic amides |
| US8449898B2 (en) | 2007-10-23 | 2013-05-28 | E I Du Pont De Nemours And Company | Fungicidal mixtures |
| ES2335849A1 (en) * | 2008-10-03 | 2010-04-05 | Consejo Superior De Investigaciones Cientificas (Csic) | Chroman derivatives and uses thereof as inhibitors of the activity of decoupling proteins |
| WO2010037886A1 (en) * | 2008-10-03 | 2010-04-08 | Consejo Superior De Investigaciones Científicas (Csic) | Chroman derivatives and uses thereof as inhibitors of the activity of decoupling proteins |
| ES2335849B1 (en) * | 2008-10-03 | 2011-03-22 | Consejo Superior De Investigaciones Cientificas (Csic) | CHROMAN DERIVATIVES AND THEIR USES AS INHIBITORS OF THE ACTIVITY OF DECOOPLANT PROTEINS. |
| JP2012510520A (en) * | 2008-12-02 | 2012-05-10 | イー・アイ・デュポン・ドウ・ヌムール・アンド・カンパニー | Fungicidal heterocyclic compounds |
| CN101928259B (en) * | 2009-06-18 | 2012-10-03 | 南京海辰药业有限公司 | 2-arylthiazole derivative and medicament composition thereof |
| JP2013518823A (en) * | 2010-02-08 | 2013-05-23 | バイオウタ サイエンティフィック マネジメント プロプライエタリー リミテッド | Compounds for treating respiratory syncytial virus infection |
| WO2011098776A1 (en) | 2010-02-15 | 2011-08-18 | Cambridge Enterprise Limited | 5-ht receptor modulators |
| CN102464620A (en) * | 2010-11-16 | 2012-05-23 | 上海药明康德新药开发有限公司 | Preparation method of 4-methyl-2-thiomethyl-1 (3) H-imidazole |
| WO2012123298A1 (en) | 2011-03-11 | 2012-09-20 | F. Hoffmann-La Roche Ag | Antiviral compounds |
| WO2013053657A1 (en) | 2011-10-10 | 2013-04-18 | F. Hoffmann-La Roche Ag | Antiviral compounds |
| WO2013087743A1 (en) | 2011-12-16 | 2013-06-20 | F. Hoffmann-La Roche Ag | Inhibitors of hcv ns5a |
| CN104093715A (en) * | 2012-02-02 | 2014-10-08 | 埃科特莱茵药品有限公司 | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| WO2013114332A1 (en) * | 2012-02-02 | 2013-08-08 | Actelion Pharmaceuticals Ltd | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| JP2015506957A (en) * | 2012-02-02 | 2015-03-05 | アクテリオン ファーマシューティカルズ リミテッドActelion Pharmaceuticals Ltd | 4- (Benzimidazol-2-yl) -thiazole compounds and related aza derivatives |
| US9266876B2 (en) | 2012-02-02 | 2016-02-23 | Actelion Pharmaceuticals Ltd. | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| KR101800607B1 (en) | 2012-02-02 | 2017-11-23 | 액테리온 파마슈티칼 리미티드 | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| EA027595B1 (en) * | 2012-02-02 | 2017-08-31 | Актелион Фармасьютиклз Лтд. | 4-(benzoimidazol-2-yl)thiazole compounds and related aza derivatives |
| CN104093715B (en) * | 2012-02-02 | 2017-04-26 | 埃科特莱茵药品有限公司 | 4-(benzoimidazol-2-yl)-thiazole compounds and related aza derivatives |
| EA030067B1 (en) * | 2013-07-22 | 2018-06-29 | Идорсиа Фармасьютиклз Лтд | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)ethanone derivatives |
| TWI630205B (en) * | 2013-07-22 | 2018-07-21 | 瑞士商愛杜西亞製藥有限公司 | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivative |
| KR102295416B1 (en) | 2013-07-22 | 2021-08-30 | 이도르시아 파마슈티컬스 리미티드 | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| US10259807B2 (en) | 2013-07-22 | 2019-04-16 | Idorsia Pharmaceuticals Ltd. | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| CN105658649A (en) * | 2013-07-22 | 2016-06-08 | 埃科特莱茵药品有限公司 | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| CN105658649B (en) * | 2013-07-22 | 2019-03-22 | 爱杜西亚药品有限公司 | 1-(Piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivative |
| AU2014295123B2 (en) * | 2013-07-22 | 2018-08-02 | Idorsia Pharmaceuticals Ltd | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| JP2016525137A (en) * | 2013-07-22 | 2016-08-22 | アクテリオン ファーマシューティカルズ リミテッドActelion Pharmaceuticals Ltd | 1- (Piperazin-1-yl) -2-([1,2,4] triazol-1-yl) -ethanone derivatives |
| KR20160033218A (en) * | 2013-07-22 | 2016-03-25 | 액테리온 파마슈티칼 리미티드 | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| WO2015011099A1 (en) * | 2013-07-22 | 2015-01-29 | Actelion Pharmaceuticals Ltd | 1-(piperazin-1-yl)-2-([1,2,4]triazol-1-yl)-ethanone derivatives |
| US9951063B2 (en) | 2014-03-24 | 2018-04-24 | Idorsia Pharmaceuticals Ltd | 8-(piperazin-1-yl)-1,2,3,4-tetrahydro-isoquinoline derivatives |
| WO2015152254A1 (en) * | 2014-04-01 | 2015-10-08 | 大日本住友製薬株式会社 | Five-membered ring heteroaryl derivative |
| AU2016207988B2 (en) * | 2015-01-15 | 2019-08-15 | Idorsia Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as CXCR3 receptor modulators |
| WO2016113346A1 (en) * | 2015-01-15 | 2016-07-21 | Actelion Pharmaceuticals Ltd | (r)-2-methyl-piperazine derivatives as cxcr3 receptor modulators |
| WO2016113344A1 (en) | 2015-01-15 | 2016-07-21 | Actelion Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| CN107250133A (en) * | 2015-01-15 | 2017-10-13 | 爱杜西亚药品有限公司 | It is used as the hydroxyalkyl bridged piperazine derivatives of CXCR3 receptor modulators |
| CN107207491A (en) * | 2015-01-15 | 2017-09-26 | 爱杜西亚药品有限公司 | It is used as (R) 2 methyl piperazine derivate of CXCR3 receptor modulators |
| US10047080B2 (en) | 2015-01-15 | 2018-08-14 | Idorsia Pharmaceuticals Ltd. | (R)-2-methyl-piperazine derivatives as CXCR3 receptor modulators |
| US10053457B2 (en) | 2015-01-15 | 2018-08-21 | Idorsia Pharmaceuticals Ltd. | Hydroxyalkyl-piperazine derivatives as CXCR3 receptor modulators |
| KR20170102010A (en) * | 2015-01-15 | 2017-09-06 | 이도르시아 파마슈티컬스 리미티드 | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| KR101967083B1 (en) | 2015-01-15 | 2019-04-08 | 이도르시아 파마슈티컬스 리미티드 | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| JP2018502161A (en) * | 2015-01-15 | 2018-01-25 | イドーシア ファーマシューティカルズ リミテッドIdorsia Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as CXCR3 receptor modulators |
| JP2018502162A (en) * | 2015-01-15 | 2018-01-25 | イドーシア ファーマシューティカルズ リミテッドIdorsia Pharmaceuticals Ltd | (R) -2-methyl-piperazine derivatives as CXCR3 receptor modulators |
| EA032927B1 (en) * | 2015-01-15 | 2019-08-30 | Идорсия Фармасьютиклз Лтд | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
| RU2706232C2 (en) * | 2015-01-15 | 2019-11-15 | Идорсиа Фармасьютиклз Лтд | (r)-2-methyl-piperazine derivatives as modulators of cxcr3 receptor |
| AU2016207990B2 (en) * | 2015-01-15 | 2020-01-30 | Idorsia Pharmaceuticals Ltd | (R)-2-methyl-piperazine derivatives as CXCR3 receptor modulators |
| CN107207491B (en) * | 2015-01-15 | 2020-08-25 | 爱杜西亚药品有限公司 | (R)-2-Methyl-piperazine derivatives as CXCR3 receptor modulators |
| CN107250133B (en) * | 2015-01-15 | 2020-09-15 | 爱杜西亚药品有限公司 | Hydroxyalkyl piperazine derivatives as CXCR3 receptor modulators |
| WO2018011265A1 (en) | 2016-07-13 | 2018-01-18 | Idorsia Pharmaceuticals Ltd | Hydroxyalkyl-piperazine derivatives as cxcr3 receptor modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| US20100286191A1 (en) | 2010-11-11 |
| WO2007070433A3 (en) | 2008-02-21 |
| EP1962605A2 (en) | 2008-09-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007070433A2 (en) | 2-arylthiazole derivatives as cxcr3 receptor modulators | |
| US10647696B2 (en) | Substituted benzimidazoles and benzopyrazoles as CCR(4) antagonists | |
| US20090030012A1 (en) | Pyridine, Pyrimidine and Pyrazine Derivatives as Cxcr3 Receptor Modulators | |
| WO2007064553A2 (en) | Thiazole derivatives as cxcr3 receptor modulators | |
| EP1720545B1 (en) | Bicyclic and bridged nitrogen heterocycles | |
| EP1615699B1 (en) | Benzoxazinyl-amidocyclopentyl-heterocyclic modulators of chemokine receptors | |
| US7514431B2 (en) | Piperidinyl cyclopentyl aryl benzylamide modulators of chemokine receptor activity | |
| RS62855B1 (en) | Piperidine cxcr7 receptor modulators | |
| CZ20032990A3 (en) | N-aroyl cyclic amines | |
| BRPI0717722A2 (en) | COMPOUND OR A SALT OF THE SAME, PRODUCT, GLYCOKINASE ACTIVATOR, PHARMACEUTICAL AGENT, METHODS TO ACTIVATE A GLYCHOKINASE IN A MAMMAL AND FOR PROPHYLAXIS OR TREATMENT OF DIABETES OR OBESITY, OR USE OF THE MAMMAL DRUG THIS | |
| WO2007022257A2 (en) | Monocyclic and bicyclic compounds and methods of use | |
| EP1979346A1 (en) | Thiazole derivatives and use thereof | |
| EP1983992A2 (en) | 2-imino-benzimidazoles | |
| CA2366944A1 (en) | Pyrrolidine modulators of chemokine receptor activity | |
| US20180282304A1 (en) | Rorc2 inhibitors and methods of use thereof | |
| CA2503715A1 (en) | Kinase inhibitors | |
| WO2013010453A1 (en) | Chemoking receptor antagonists | |
| WO2003030898A1 (en) | Modulators of ccr5 chemokine receptor activity | |
| WO2004041777A2 (en) | Heteroarylpiperidine modulators of chemokine receptor activity | |
| KR20070007345A (en) | Piperidine Derivatives as Regulators of Chemokine Receptors CCR5 | |
| US20250179061A1 (en) | Rip1 modulators, preparations, and uses thereof | |
| HK1202866B (en) | Substituted benzimidazoles and benzopyrazoles as ccr(4) antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006839263 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12086361 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |