WO2007075872A2 - Nitroimidazole compounds - Google Patents
Nitroimidazole compounds Download PDFInfo
- Publication number
- WO2007075872A2 WO2007075872A2 PCT/US2006/048763 US2006048763W WO2007075872A2 WO 2007075872 A2 WO2007075872 A2 WO 2007075872A2 US 2006048763 W US2006048763 W US 2006048763W WO 2007075872 A2 WO2007075872 A2 WO 2007075872A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- substituted
- halogen
- formula
- group
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *CC[C@]1COC2=*C(*(O)=O)=C*2C1 Chemical compound *CC[C@]1COC2=*C(*(O)=O)=C*2C1 0.000 description 12
- FDCIYMPHZLSRDV-GLIQTQJLSA-N C#C/C=C(\C=C/CCOC1COc2nc([N+]([O-])=O)c[n]2C1)/Br Chemical compound C#C/C=C(\C=C/CCOC1COc2nc([N+]([O-])=O)c[n]2C1)/Br FDCIYMPHZLSRDV-GLIQTQJLSA-N 0.000 description 1
- OFXIRUZCOXQHPB-UHFFFAOYSA-N N#Cc1ccc(COC2COc3nc([N+]([O-])=O)c[n]3C2)cc1 Chemical compound N#Cc1ccc(COC2COc3nc([N+]([O-])=O)c[n]3C2)cc1 OFXIRUZCOXQHPB-UHFFFAOYSA-N 0.000 description 1
- ZLHZLMOSPGACSZ-UHFFFAOYSA-N [O-][N+](c1c[n](CC(CO2)OCc(cc3)ccc3OC(F)(F)F)c2n1)=O Chemical compound [O-][N+](c1c[n](CC(CO2)OCc(cc3)ccc3OC(F)(F)F)c2n1)=O ZLHZLMOSPGACSZ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Definitions
- W is O and V is absent; one of R2 and R4 is -L(B) n -(Z) p , -(L-B) q -(Z) P or -Y-(B) q -Z, and the other is H; and
- R1 and R3 are both H; wherein L is an atom group having of the formula -O-R5- where R5 is a lower alkylene, -C(O)-, lower alkylene-C(O)-, -C(O)-lower alkylene, lower alkylene-C(O)-NH-, lower alkytene-NH-; B is a cycloalkyl, heterocyclic, aryl or heteroaryl ring which is optionally further substituted with one or more substituents; n is 1 or 2; and Z is halogen, lower alkyl substituted with at least one halogen, lower alkoxy substituted with at least one halogen or lower thioalkyl substituted with at least one halogen; and Y is -NHC(O)-; n is 1 or 2; p is O 1 1 or 2; and q is 1 or 2;
- R2 or R4 is -L-(B) n -(Z) p wherein n is 1 , B is phenyl and L is
- V is a benzyl group optionally substituted with one or more methoxy groups. It is further preferred that one of R2 and R4 is a methoxy group. Where the compound is a compound of formula (I) wherein (c) above applies, it is preferred that one of R1 and R3 is an ethyl, pentyl or phenyl group.
- the compound is a compound of formula (I) wherein (d) above applies, and wherein one of R2 and R4 is -Y-(B) q -Z it is preferred that B is a piperidine, pyrimidine or phenyl group. It is also preferred that p is 1.
- the compound is a compound of formula (I) wherein (d) above applies, and wherein one of R2 and R4 is -(L-B) q -(Z) P it is preferred that B is phenyl or cyclohexyl. It is further preferred that L is -O-lower alkylene, more preferably -O-CH 2 -.
- L is -OCH 2 C(O)-, -OCH 2 C(O)NH-, -OCH 2 C(O)N-, -OCH 2 C(O)NHCH 2 -, -OCH 2 - or -OCH 2 CH 2 -. More preferably L is -OCH 2 C(O)-.
- B is a 4 to 12, preferably 5 or 6 membered cycloalkyl, heterocyclic, aryl or heteroaryl ring.
- B is a cyclic ring selected from the group consisting of cyclopentyl, cyclohexyl, phenyl, morpholinyl, piperazinyl, piperidinyl, pyridyl, pyrrolidinyl, pyrazinyl, pyrimidinyl, purinyl, pyranyl, benzimidazolyl, benzoxazolyl, benzthiazolyl, naphthyl, indolyl, indolinyl, quinolinyl, isoquinolinyl, 1,2,3,4-tetrahydroquinolinyl, thiazolyl, imidazolyl, benzotriazolyl, indanyl, oxadiazolyl, pyrazolyl, triazo
- B is a cyclic ring selected from the group consisting of piperazinyl, phenyl, pyridyl, benzimidazolyl, benzthiazolyl, benzoxazolyl, thiazolyl.
- Z is halogen, lower alkyl substituted with at least one halogen or lower alkoxy substituted with at least one halogen, such as e.g. halomethyl, dihalomethyl, trihalomethyl, pentahaloethyl, halomethoxy, dihalomethoxy, trihalomethoxy, pentahaloethyl or pentahaloethoxy.
- the compound is a compound of formula (II), or a pharmaceutically acceptable salt, ester or prodrug thereof:
- L is an atom group having of the formula -OR5- wherein R5 is a lower alkylene, -C(O)-, lower alkylene-C(O)-, -C(O)-lower alkylene, lower alkylene-C(O)-NH-, lower alkylene-NH-; In a preferred embodiment L is selected from the group consisting of -OCH 2 C(O)-, -OCH 2 C(O)NH-, -OCH 2 C(O)N-, -OCH 2 C(O)NHCH 2 -, -OCH 2 - and -OCH 2 CH 2 -;
- B is a 4 to 12, preferably 5 or 6 membered cycloalkyl, heterocyclic, aryl or heteroaryl ring.
- the ring can optionally be further substituted with one or more substituents, preferably selected from the group consisting of lower alkyl, halogen, hydroxy, amino or lower alkoxy.
- B is a cyclic ring selected from the group consisting of cyclopentyl, cyclohexyl, phenyl, morpholinyl, piperazinyl, piperidinyl, pyridyl, pyrrolidinyl, pyrazinyl, pyrimidinyl, purinyl, pyranyl, benzimidazolyl, benzoxazolyl, benzthiazolyl, naphthyl, indolyl, indolinyl, quinolinyl, isoquinolinyl, 1 ,2,3,4-tetrahydroquinolinyl, thiazolyl, imidazolyl, benzotriazolyl, indanyl, oxadiazolyl, pyrazolyl, triazolyl, or tetrazolyl.
- Z is halogen, lower alkyl substituted with at least one halogen or lower alkoxy substituted with at least one halogen, such as e.g. halomethyl, dihalomethyl, trihalomethyl, pentahaloethyl, halomethoxy, dihalomethoxy, trihalomethoxy, pentahaloethy! or pentahaloethoxy.
- the halogen is fluoro or chloro, fluoro being the most preferred halogen.
- B is not phenyl or if n is 1 and B is phenyl then L is -OCH 2 C(O)NH-Or -OCH 2 C(O)NHCH 2 -.
- the present invention relates to a compound, or a pharmaceutically acceptable salt, ester or prodrug thereof, of the formula:
- L and Z are as defined above for the compound of formula (II) and wherein each ring can independently be further substituted by 1 , 2, 3 or more substituents, e.g. selected from the group consisting of lower alkyl, halogen, hydroxy, amino or lower alkoxy.
- L is selected from the group consisting of -OCH 2 C(O)-, -OCH 2 C(O)NH-, -OCH 2 C(O)NHCH 2 -, -OCH 2 CH 2 - and -OCH 2 - and Z is selected from the group consisting of -F, -CF 3 , or -OCF 3 .
- Z is in the 3-position, more preferably in the 4-position.
- L and Z are as defined above for the compound of formula (II) and wherein Y is S or N and wherein each ring can independently be further substituted by 1 , 2, 3 or more substituents, e.g. selected from the group consisting of lower alkyl, halogen, hydroxy, amino or lower alkoxy.
- L is selected from the group consisting of -OCH 2 C(O)-, -OCH 2 C(O)NH-, -OCH 2 C(O)NHCH 2 -, -OCH 2 CH 2 - and -OCH 2 - and Z is selected from the group consisting of -F, -CF 3 , or -OCF 3 .
- Z is in position 4 or 7, more preferably in position 5 and 6.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), (II) or (V), or a compound of any of formulae (Ia) to (Ii) as defined above, or a pharmaceutically acceptable salt, ester or prodrug thereof in combination with a pharmaceutically acceptable excipient, diluent or carrier.
- the present invention provides a compound of formula (I), (II) or (V) 1 or a compound of any of formulae (Ia) to (Ii) as defined above, or a pharmaceutically acceptable salt, ester or prodrug thereof for use as a medicine.
- the present invention provides the use of a compound of formula (I), (II) or (V), or a compound of any of formulae (Ia) to (Ii) as defined above, or a pharmaceutically acceptable salt, ester or prodrug thereof, for the manufacture of a medicament for the treatment and/or prevention of a disease caused by a pathogenic microbe such as Mycobacterium tuberculosis.
- a pathogenic microbe such as Mycobacterium tuberculosis.
- R1 and R3 are both H; wherein L is an atom group having of the formula -O-R5- where R5 is a lower alkylene, -C(O)-, lower alkylene-C(O)-, -C(O)-lower alkylene, lower alkylene-C(O)-NH-, lower alkylene-NH-; B is a cycloalkyl, heterocyclic, aryl or heteroaryl ring which is optionally further substituted with one or more substituents; and Z is halogen, lower alkyl substituted with at least one halogen, lower alkoxy substituted with at least one halogen or lower thioalkyl substituted with at least one halogen; and Y is -NHC(O)-; n is 1 or 2; p is 0, 1 or 2; and q is 1 or 2.
- nitrogen heterocyclic compound in free or salt form, is represented by a compound of formula (IV)
- R 4 H, alkyl, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl
- R 5 H, alky!, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl
- R 6 trimethylsilyl, triethylsilyl, f- butyldimethylsilyl, dibutylmethylsilyl, diphenylmethylsilyl, phenyldimethylsilyl or diphenyl-f- butylsilyl.
- the molar ratio of the non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.55 to 0.95:1 , more preferably in the range of 0.6 to 0.9:1 , still more preferably in the range of 0.65 to 0,85:1 , still more preferably in the range of 0.65 to 0.8:1, still more preferably in the range of 0.7 to 0.85:1 , still more preferably in the range of 0.7 to 0.8:1.
- the non-sterically hindered substituted epoxide is reacted with a haloimidazole compound, to form the adduct with an alcohol functional group at a temperature range of 45-105 0 C, more preferably 55-95 0 C, still more preferably 65-85 0 C 1 still more preferably 60- 80 0 C.
- the haloimidazole compound contains a halogen substituent selected from the group consisting of chloro or bromo.
- the catalyst is pyridinium-p-toluene sulfonate. It is also preferred that the cyclizing agent is selected from the group consisting of anhydrous TBAF, anhydrous TBABr or NaH.
- the alcohol-protected adduct is treated with a cyclizing agent to form the nitrogen heterocyclic compound in vacuo.
- the nitrogen heterocyclic compound is a 3-alkyloxy-6-nitro-2H-3,4-dihydro-[2- 1b]imidazopyran or a 3-aryloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran. More preferably the 3-alkyloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran or the 3-aryloxy-6-nitro-2H-3,4- dihydro-[2-1b]imidazopyran is an (S)- or an (R)-isomer.
- the nitrogen heterocyclic compound is 3(S)- tetrahydropyranyloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran. In another preferred embodiment the nitrogen heterocyclic compound is 3(R)- tetrahydropyranyloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran.
- Ri nitro, acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo or alkoxycarbonyl;
- R 2 2-tetrahydropyranyl, 2-ethoxyethyl, trityl, methyl, ethyl, ally!, trimethylsilylethoxymethyl, 2,2,2-t ⁇ chloroethyl, benzyl, trimethylsilyl, f-butyldimethylsilyl, phenyldimethylsilyl, tri/sopropylsilyl or thexyldimethylsilyl;
- R3 H, acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo or alkoxycarbonyl;
- R 4 H, alkyl, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl;
- R 5 H 1 alkyl, alkenyl,
- the cyclizing agent is selected from the group consisting of anhydrous TBAF, anhydrous TBABr or NaH. It is also preferred that the alcohol-protected adduct is treated under microwave conditions under elevated pressure.
- the invention also provides a method as described above, further comprising reacting a compound represented by the formula (IV)
- Ri nitro
- R 2 2-tetrahydropyranyl, 2-ethoxyethyl, trityl, methyl, ethyl, allyl, trimethylsilylethoxymethy], 2,2,2-trichloroethyl, benzyl, trirhethylsilyl, /-butyldimethylsilyl, phenyldimethylsilyl, tri/sopropylsilyl or thexyldimethylsilyl;
- R 3 H; with 4-(trifluoromethoxy)benzyl halide
- the 4-(trifluoromethoxy)benzyl halide is selected from the group of consisting of 4-(trifluoromethoxy)benzyl bromide, 4-(trifluoromethoxy)benzyl chloride and 4- (trifluoromethoxy)benzyl iodide.
- Ri nitro
- R 2 2-tetrahydropyranyl, 2-ethoxyethyl, trityl, methyl, ethyl, allyl, trimethylsilylethoxymethyl, 2,2,2-trichloroethyl, benzyl, trimethylsilyl, f-butyldimethylsilyl, phenyldimethylsilyl, tri/sopropylsilyl or thexyldimethylsilyl;
- R 3 H;
- the alcohol-deprotecting agent is selected from the group comprising of acetic acid, TBAF, TBABr.
- the nitrogen heterocyclic compound is an (R)- or (S)-isomer.
- lower alkyl refers to branched or straight chain alkyl groups comprising 1 to 5 carbon atoms, preferably 1 to 3 carbon atoms such as e.g., methyl, ethyl, propyl, isopropyl, n-propyl, n-butyl, sec-butyl, t-butyl.
- lower alkoxy refers to -OR wherein R is lower alkyl as defined above. Examples of lower alkoxy groups include e.g. methoxy, ethoxy, t-butoxy.
- alkenyl as used herein includes straight chain or branched alkenyl, which may be, for example, C 2 - C 12 alkenyl in all its isomeric forms.
- alkoxycarbonyl means a group RCO wherein R is an alkoxy group, for example, a Ci- C 12 -alkoxy group, in all its isomeric forms.
- Halo or halogen means F, Cl, Br or I, preferably F or Cl.
- alkyl halogen or “haloalkyl” refers to an alkyl group as defined above to which at least one halogen as defined above is attached. Examples are e.g. fluoromethyl, difluoromethyl, trjfluoromethyl, pentafluoroethyl.
- lower alkyl halogen or “lower haloalkyl” has a corresponding meaning to the term"(!ower alkyl" as defined above.
- lower alkoxy halogen or “lower haloalkoxy” refers to a lower alkoxy group as defined above to which at least one halogen as defined above is attached. Examples are e.g. fluoromethoxy, difluoromethoxy, trifluoromethoxy, pentafluoroethoxy.
- cycloalkyl refers to a saturated or partially saturated (non-aromatic) cyclic ring which is optionally further substituted, e.g. with lower alkyl, halogen, hydroxy, amino. Examples include e.g. cyclopentyl, cyclohexyl, methylcyclohexyl.
- the cycloalkyl ring is preferably a 5 or 6 membered cyclic ring.
- aryl refers to an aromatic monocyclic or fused bicyclic ring structure which can contain from 4 to 12 carbon atoms, preferably 5 or 6 carbon atoms for monocyclic rings and 8, 9 or 10 carbon atoms for fused bicyclic rings.
- the aryl group is optionally further substituted, e.g. with lower alkyl, halogen, hydroxy, amino.
- An aryl group may be e.g. phenyl or naphthyl, preferably phenyl.
- haloaryl means an aryl group substituted with one or more halogens as defined above, preferably one or more fluoro groups.
- alkylaryl means -R-aryl where R is an alkyl group as defined above and aryl is as defined above. An example is benzyl.
- heteroaryl refers to an aromatic heterocyclic ring, e.g. a 5 or 6 membered aromatic heterocyclic ring, optionally condensed to 1 or 2 benzene rings and/or to a further heterocyclic ring and optionally further substituted on a ring C or ring heteroatom with e.g. a lower alkyl, halogen, hydroxy, amino group.
- heterocyclic and heteroaryl groups include e.g.
- nitrogen heterocyclic compound refers to a cyclic structure containing an sp 2 -hybridized nitrogen in a ring of the structure.
- non-sterically hindered substituted epoxide refers to substituted epoxides where the substituents do not ste ⁇ cally hinder the underlying reaction and includes the implicit proviso that the substitution is in accordance with permitted valence of the substituted atom.
- substituted nitroimidazole refers to an imidazole nucleus which carries both a nitro substituent as well as another substituent on the imidazole nucleus, commonly at an imidazole ring carbon position or at an imidazole ring nitrogen position.
- protecting group refers to temporary or permanent chemical moieties which protect a potentially reactive functional group from unintended chemical transformations.
- protecting groups include esters of carboxylic acids, silyl ethers of alcohols, and acetals and ketals of aldehydes and ketones, respectively.
- the field of protecting group chemistry is known in the art; examples of protecting groups include conventionally used protective groups which can be found, for example, in "Protective Groups in Organic Synthesis," T. W. Greene, P. M. Wuts, John Wiley and sons 1991, pp. 10-142).
- masked alcohol moiety refers to any group commonly used for the temporary protection of the hydroxyl functional group, resulting in masking the reactivity of the free alcohol group.
- suitable protecting groups for hydroxyl functional groups include but are not limited to the following examples: alkoxycarbonyl, acyl, alkylsilyl or alkylarylsilyl groups, and alkoxyalkyl groups.
- deprotecting or “deprotection” as used herein is known in the art of organic synthesis and refers to conditions for the removal of protecting groups or chemical moieties that mask the underlying reactivity of the functional group, leaving the unprotected functional group. Examples of suitable deprotecting agents or conditions for various functional groups can be found, for example in “Protective ⁇ roups in Organic Synthesis,” T. W. Greene, P. M. Wuts, John Wiley and sons 1991 ).
- free form refers to non-salt forms of the compounds. Additionally, the compounds of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms, crystalline forms and polymorphic forms.
- electron-withdrawing group is recognized in the art, referring to the tendency of a substituent to attract valence electrons from neighboring atoms, i.e., the substituent is electronegative with respect to neighboring atoms.
- exemplary electron-withdrawing groups include nitro, acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo, and the like.
- microwave conditions refers to the use of technology used to produce or simulate microwave irradiation. Examples for the use of microwave irradiation in organic synthesis can be found, for example in Tetrahedron, 57:9225-9283 (2001 ) and Ace. Chem. Soc, 82:14-19 (2004).
- the present invention also encompasses enantiomers, racemates, diastereoisomers and mixtures of the compounds of the invention.
- the compounds of the invention contain asymmetric carbon atoms. It should be understood, therefore, that the individual stereoisomers are contemplated as being included within the scope of the invention.
- the terms "R” and “S” for chemical configuration as used herein, are as defined by IUPAC in "Recommendations for Section E, Fundamental Stereochemistry", Pure Appt. Chem., 45:13-30 (1976).
- the compounds of the present invention are useful in the treatment and/or prevention of infections by a pathogen.
- the pathogen is preferably a bacterium or a protozoan, in particular a Mycobacterium, Clostriduim, Cryptosporidium, Helicobacter, Trypanosoma, Leishmania or Plasmodium. More specifically the bacterium or protozoan can be Mycobacterium tuberculosis (in particular multi-drug resistant Mycobacterium tuberculosis), Clostridium difficile, Cryptosporidium pan/um, Helicobacter pylori, T. brucei rhodesiense, T.
- the compounds of the present invention in free form or in pharmaceutically acceptable salt form, exhibit valuable pharmacological properties, e.g.. as antimicrobial agents, for example, as indicated by the tests of the Examples and are therefore indicated for therapy.
- the compounds of the present invention exhibit IC 5 O against Leishmania donovani that are below 5 ⁇ M, preferably below 4 ⁇ M, more preferably below 3 ⁇ M, even more preferably below 2 ⁇ M, even more preferably below 1 ⁇ M, even more preferably below 0.5 ⁇ M and still more preferably below 0.1 ⁇ M.
- the compounds of the present invention exhibit IC50 against Trypanosoma cruzi that are below 5 ⁇ M, preferably below 4 ⁇ M, more preferably below 3 ⁇ M, even more preferably below 2 ⁇ M, even more preferably below 1 ⁇ M and still more preferably below 0.5 ⁇ M.
- the compounds show a MIC against Mycobacterium tuberculosis that is preferably lower than 0.8 ⁇ M, more preferably lower than 0.5 ⁇ M, more preferably lower than 0.1 ⁇ M, more preferably lower than 0.05 ⁇ M, more preferably lower than 0.01 ⁇ M, more preferably lower than 0.005 ⁇ M, more preferably lower than 0.001 ⁇ M, more preferably lower than 0.0005 ⁇ M.
- the compounds of the present invention may exist in free form or in salt form, e.g. addition salts with e.g. organic or inorganic acids, for example trifluoroacetic acid or hydrochloride acid, or salts obtainable when they comprise a carboxy group, e.g. with a base, for example alkali salts such as sodium, potassium, or substituted or ⁇ nsubstituted ammonium salts.
- addition salts with e.g. organic or inorganic acids for example trifluoroacetic acid or hydrochloride acid
- salts obtainable when they comprise a carboxy group e.g. with a base, for example alkali salts such as sodium, potassium, or substituted or ⁇ nsubstituted ammonium salts.
- the compounds of the present invention may be administered as the sole active ingredient or as one active component in a combination tablet containing several active components, e.g. antibiotics.
- the required dosage for pharmaceutical use will of course vary depending on the mode of administration, the particular condition to be treated and the effect desired. In general, satisfactory results are indicated to be obtained systemically at daily dosages of from about 0.03 to 2.5 mg/kg per body weight.
- An indicated daily dosage in the larger mammal, e.g. humans, is in the range from about 0.5 mg to about 100 mg, conveniently administered, for example, in divided doses up to four times a day or in retard form.
- Suitable unit dosage forms for oral administration comprise from ca. 1 to 100 mg active ingredient.
- X is Cl, Br or I
- W is H, Li, Na, K, CO 2 H, CO 2 " , f-butoxycarbonyl, N 1 N- dimethylaminosulfonyl, p-toluenesulfonyl, or tri/sopropylsilyl.
- the present invention provides a method for the preparation of a nitrogen heterocyclic compound.
- the method comprises reacting a non-sterically hindered substituted epoxide with a haloimidazole compound, wherein the molar ratio of the non- sterically hindered substituted epoxide to the haloimidazole compound is less than or equal to 1 :1, to form an adduct with an alcohol functional group; protecting the alcohol functional group on the adduct to form an alcohol-protected adduct; and treating the alcohol-protected add ⁇ ct with a cyclizing agent to form the nitrogen heterocyclic compound.
- the present invention provides furthermore a method to convert the bisprotected alcohol adduct to the mono deprotected primary alcohol adduct which is reacted with a cyclizing agent to form the nitrogen heterocyclic compound.
- the use of explosive dinitroimidazoles are circumvented altogether by the use of haloimidazoles containing an electron-withdrawing substituent.
- the method of the present invention provides an efficient method to the preparation of imidazopyrans while requiring fewer labour-intensive purification steps. The overall process is more efficacious and more amenable to large-scale synthesis than, for example, the method disclosed in US 6,087,358.
- the step-wise yield as well as the overall yield of the synthesis may be increased. Additionally, with the use of an appropriate molar ratio of the starting materials, the laborious need to purify the product formed is obviated.
- the method of this invention may further expedite the efficiency of synthesis by incorporating a cyclization step, to form the nitroimidazopyran product in high yield. Cyclization may be effected in preferred embodiments under microwave conditions, under elevated pressure or a combination of both microwave conditions and elevated pressure.
- Cyclization may be effected in preferred embodiments under microwave conditions, under elevated pressure or a combination of both microwave conditions and elevated pressure.
- microwave conditions refers to the use of technology used to produce or simulate microwave irradiation. Examples for the use of microwave irradiation in organic synthesis can be found, for example in Tetrahedron, 57:9225-9283 (2001) and Ace. Chem. Soc, 82:14-19 (2004).
- the present invention provides a process for the conversion of compound (Ir) to compound (IV) using microwave irradiation.
- the haloimidazole compound is represented by a compound of formula (Ip)
- the haloimidazole compound is a nitrogen heterocyclic compound containing the imidazole nucleus which carries a halogen substituent such as chloro, bromo, iodo and the like.
- the imidazole nucleus may be further substituted, commonly at the carbon or nitrogen ring atoms.
- Substituents in the representative classes such as acyl, formyl, sulfonyl, silyl, trifluoromethyl, cyano, halo, nitro or alkoxycarbonyl may be used.
- the substituent at a carbon ring atom includes an electron- withdrawing group such as acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo, nitro or alkoxycarbonyl.
- the substituent at an imidazole ring carbon atom is a nitro group.
- the substituent is a nitro group in the 4-position on the imidazole nucleus.
- substituents on the epoxide may include silyl-protecting groups such as trimethylsilyl, triethylsilyl, f-butyldimethylsilyl, dibutylmethylsilyl, diphenylmethylsilyl, phenyldimethylsilyl, diphenyl-f-butylsilyl and analogous alkylated silyl radicals.
- the non-sterically hindered substituted epoxide further comprises a masked amine moiety.
- Representative amino protecting groups include, formyl, acetyl, trifluoroacetyl, benzyl, benzyloxycarbonyl, f-butoxycarbonyl, trimethyl silyl, 2- trimethylsilyl-ethanesulfonyl, trityl and substituted trityl groups, allyloxycarbonyl, 9- fluorenylmethyloxycarbonyl, nitro-veratryloxycarbonyl, and the like.
- non-sterically hindered substituted epoxide is represented by the compound of formula (Iq)
- R 5 is H 1 alky!, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl
- Re is H, alkyl, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl
- R 7 Js trimethylsilyl, triethylsilyl, t- butyldimethylsilyl, dibutylmethylsilyl, diphenylmethylsilyl, phenyldimethylsilyl or diphenyl-f- butylsilyl.
- One aspect of the invention relates to the use of a molar ratio of an amount of the non- sterically hindered substituted epoxide to the haloimidazole compound.
- the molar ratio of the non-sterically hindered substituted epoxide is less than or equal to 1 :1.
- the molar ratio of the non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.55 to 0.95:1.
- the molar ratio of a non-sterically hindered substituted epoxide to haloimidazole compound is in the range of 0.6 to 0.9:1.
- the molar ratio of a non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.65 to 0.85:1. In another embodiment, the molar ratio of a non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.65 to 0.8:1. In another embodiment, the molar ratio of a non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.7 to 0.85:1. In yet another embodiment, the molar ratio of a non-sterically hindered substituted epoxide to the haloimidazole compound is in the range of 0.7 to 0.8:1.
- the non-sterically hindered substituted epoxide is reacted with a haloimidazole compound, to form the adduct with an alcohol functional group at a temperature range of about 55-95 0 C.
- the non-sterically hindered substituted epoxide is reacted with a haloimidazole compound, to form the adduct with an alcohol functional group at a temperature range of about 65-85 0 C.
- the non-sterically hindered substituted epoxide is reacted with a haloimidazole compound, to form the adduct with an alcohol functional group at a temperature range of about 60-80 0 C.
- the haloimidazole compound reacts with a non-sterically hindered substituted epoxide to give a reaction product which, upon isolation in ways known to one skilled in the art, yields the adduct with the alcohol functional group as the end product.
- An example of the workup to form the end product includes filtration, removal of solvents, extraction between aqueous and organic phases using conventional organic solvents such as ethyl acetate, diethyl ether, chloroform, methylene chloride and the like, followed by drying the organic phase over conventional drying agents to give, upon removal of solvents, the adduct with an alcohol functional group.
- the adduct formed by reaction of the non-sterically hindered epoxide with the haloimidazole contains an alcohol functional group derived by nucleophilic ring opening of the epoxide.
- the resulting adduct containing the alcohol functional group is isolated in its protonated form and is obtained with a purity greater than 90% and in yields equal to or greater than 90% based on the epoxide.
- the adduct wi,th an alcohol functional group can be prepared on a large scale and is used in the subsequent step of the method without the need for further purification.
- the unreacted haloimidazole compound used as starting material in the reaction can be recovered from the aqueous layer and recycled for reaction, making the overall process even more cost-effective, efficient and amenable to large scale synthesis.
- the alcohol-protected adduct is represented by a compound of formula
- R ⁇ is nitro, acyl, formyl, sulfonyl, trifl ⁇ oromethyl, cyano, halo or alkoxycarbonyl;
- R 3 is a protection group, e.g.
- the stereochemistry of the method of the present invention is determined by the enantiomer selected for use for the non-sterically hindered substituted epoxide. Accordingly, the enantiomer afforded by the method of the present invention can be either the (S)- or the (R)-enantiomer, depending on the choice of the enantiomer used in the epoxide starting material.
- regioisomers are another aspect of this invention.
- one or the other regioisomer is isolated.
- only one regioisomer is favoured and is isolated from the reaction.
- the other regioisomer has been undetectable by the methods of detection using LC-MS and LC-UV spectroscopy.
- Another step of the method of the present invention comprises protecting the alcohol adduct, preferably in the presence of a catalyst, to form an alcohol-protected adduct.
- the method of this invention comprises the step of treating the alcohol adduct with a protecting group to convert the alcohol adduct to the corresponding alcohol-protected adduct.
- alcohol protecting groups include but are not limited to dihydropyranyl-, 2-tetrahydropyranyl, 2-ethoxyethyl, trityl, methyl, ethyl, allyl, trimethylsilylethoxymethyl, 2,2,2-trichloroethyl, benzyl, trimethylsilyl, t- butyidimethylsilyl, phenyldimethylsilyl, tri/sopropylsilyl and thexyldimethylsilyl.
- the 3,4-dihydro-2H-pyran is used to convert the alcohol adduct to the corresponding dihydropyranyl-protected alcohol adduct. Freshly-distilled 3,4-dihydro-2H- pyran obtained for example from a Kugelrohr distillation apparatus is preferred.
- mild reaction conditions are employed to avoid cleavage of any reactive groups or transiently masked reactive groups derived from the non-sterically hindered epoxide.
- the mild conditions include stirring the reaction at room temperatures ranging from 15 0 C to 35 0 C, for a time period of approximately 20-30 hours.
- a catalyst may be added.
- Such catalysts are known in the art and include acyl groups, benzyl and trityl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers and allyl ethers.
- the use of catalysts such as para- toluenesulfonic acid has been found to effect cleavage of the f-butylidimethylsilyl (TBDMS) protecting group.
- the catalyst is pyridinium-p-toluene sulfonate.
- the reaction may be stopped, for example, by quenching the reaction, with saturated aqueous sodium bicarbonate solution and the like.
- the aqueous layer is extracted several times with a volatile organic solvent such as dichloromethane, diethylether and ethyl acetate.
- the organic layers obtained from extraction are combined, washed with water, brine and dried over conventional drying agents such as magnesium sulfate. Removal of the solvents in vacuo gives a residue which is filtered over silica to remove any residual catalyst which adheres to the column. Elution with 50% EtOAc in hexanes and removal of the solvents in vacuo yields the alcohol- protected adduct which can be subsequently used without the need for further purification.
- Another aspect of the invention relates to the cyclization of the alcohol-protected adduct to form the nitrogen heterocyclic compound.
- the alcohol-protected adduct is cyclized in the presence of a cyclizing agent, wherein the cyclizing agent is selected from the group consisting of anhydrous TBAF and anhydrous TBABr.
- the primary unprotected alcohol adduct is treated with a cyclizing agent to form the nitrogen heterocyclic compound under microwave conditions.
- the primary unprotected alcohol adduct is treated with a cyclizing agent under elevated pressure to form the nitrogen heterocyclic compound.
- the primary unprotected alcohol-protected adduct is treated with a cyclizing agent to form the nitrogen heterocyclic compound under microwave conditions under elevated pressure.
- the alcohol-protected adduct is dissolved in an anhydrous aprotic solvent such as THF before addition of a cyclizing agent.
- an autosampler to prepare several 20 to 30 mL reaction vessels containing anhydrous THF expedites as well as scales up the process of preparing large quantities of anhydrous THF for use.
- the cyclizing agent is anhydrous TBAF.
- Commercially available TBAF Aldrich, 1M, THF solution
- anhydrous TBAF is degassed, preferably with nitrogen or argon, prior to use.
- the reaction vessels containing the ether in anhydrous solvent as well as the cyclizing agent are sealed before exposure to microwave conditions under elevated pressure at a temperature range from 100 0 C to 160 0 C, for a time period of 10-30 minutes.
- microwave conditions can be generated using the Biotage system. (http://www.biotagedcg.com/)
- removal of the solvent from the reaction vessel yields a residue which upon purification on silica gel column chromatography, gives the desired nitrogen heterocyclic compound in yields equal or greater than 70%.
- alternative synthetic routes for the synthesis of N- substituted-4-nitro-haloimidazoles and nitroimidazopyrans as disclosed in WO 2004/035547 report yields of about 50%.
- the nitrogen heterocyclic compound formed is an imidazopyran carrying a alcohol functional group at the 3-position on the pyran ring.
- the nitrogen heterocyclic compound, in free or salt form is represented by a compound of formula (Ii)
- R 1 nitro, acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo or alkoxycarbonyl
- R 2 2-tetrahydropyranyl, 2-ethoxyethyl, trityl, methyl, ethyl, allyl, trimethylsitylethoxymethyl, 2,2,2-trichloroethyl, benzyl, trimethylsilyl, /- butyldimethylsilyl, phenyldimethylsilyl, tri/sopropylsilyl or thexyldimethylsilyl;
- R 3 H, acyl, formyl, sulfonyl, trifluoromethyl, cyano, halo or alkoxycarbonyl.
- the nitrogen heterocyclic compound contains a nitro substituent at the 6-position.
- the nitrogen heterocyclic compound formed is an (S)- isomer.
- the nitrogen heterocyclic compound formed is an (S)- isomer, with a nitro substituent at the 6-position.
- Another aspect of the invention provides a method for the preparation of a nitrogen heterocyclic compound, wherein the nitrogen heterocyclic compound is an alkyloxy-6-nitro- 2H-3,4-dihydro-[2-1b]imidazopyran or aryloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran.
- a method for the preparation of a nitrogen heterocyclic compound of formula (I), wherein the nitrogen heterocyclic compound is 3- alkyloxy-6-nitro-2H-3,4-dihydro-[2-1b]imidazopyran or the 3-aryloxy-6-nitro-2H-3,4-dihydro- [2-1b]imidazopyran.
- the nitrogen heterocyclic compound may be an (S)- or (R)-isomer.
- Another aspect of the invention further comprises converting a compound as represented by the formula (IV)
- the present invention provides a process for preparing the compounds of the present invention, comprising:
- R 2 is defined as above for compounds (Ip)-(Ir);
- R 4 is defined as above is defined as above for compounds (Ip)-(Ir);
- Re is H, alkyl, alkenyl, aryl, heteroalkyl, heteroalkenyl or heteroaryl and wherein X is Cl, Br, I, -OCOO-isobutenyl, lower alkyl, phenyl;
- Y is N or CH;
- R 9 is H 1 alkyl, alkenyl, aryl, heteroalkyl, heteroalkenyl, heteroaryl, ortho-, meta-or para substituted trifluoro-, trifl ⁇ oromethoxy-, fluroro- phenyl, biphenyl, heteroaryl, benzyl.
- R ⁇ is defined as above for compounds (Ip)-(Ir);
- R 4 is defined as above for compounds (Ip)-(Ir); and
- R 3 is H or a counterion as Li, Na 1 K, Mg, Zn, Ca;
- X is a halogen, Cl, Br, I;
- Ri 0 is H, alkyl, alkenyl, aryl, heteroalkyl, fluoroalkyl, fluoroalkenyl, difluoroalky, trifluoroalkyl, pentafluoroethyl, hepafluoropropyl, nonafl ⁇ orobutyl, heteroalkenyl, heteroaryl, ortho-meta-or para substituted trifluoro-, trifluoromethoxy-, fluroro- phenyl, biphenyl, heteroaryl, benzimidazolyl, benzothiazolyl, benzoxazolyl, benzyl and wherein X is a halogen, Cl, Br, I.
- Scheme 1 illustrates two important intermediates 6 and 7.
- Scheme 2 illustrates the preparation of a compound of formula (If), scheme 3 for a compound of formula (Ig), scheme 4 for a compound of formula (i), (ii), (Ia), (Ic), (Ie), scheme 5 and scheme 6 for a compound of formula (Id).
- Scheme 1.1 illustrates two important intermediates 6 and 7.
- Scheme 2 illustrates the preparation of a compound of formula (If), scheme 3 for a compound of formula (Ig), scheme 4 for a compound of formula (i), (ii), (Ia), (Ic), (Ie), scheme 5 and scheme 6 for a compound of formula (Id).
- the reaction mixture is heated to 70 0 C for 6-10 h.
- the solvent is then removed in vacuo and the reaction mixture is taken up in EtOAc.
- the organic layer is washed several times with water, 0.5 N HCI, water, brine and the solvent is removed in vacuo to give the crude alcohol as a yellowish solid.
- the solid is suspended in diethyl ether and filtrated to give the final compound as a colorless powder. The remaining filtrate is concentrated and the process of precipitating the product with diethyl ether is repeated twice.
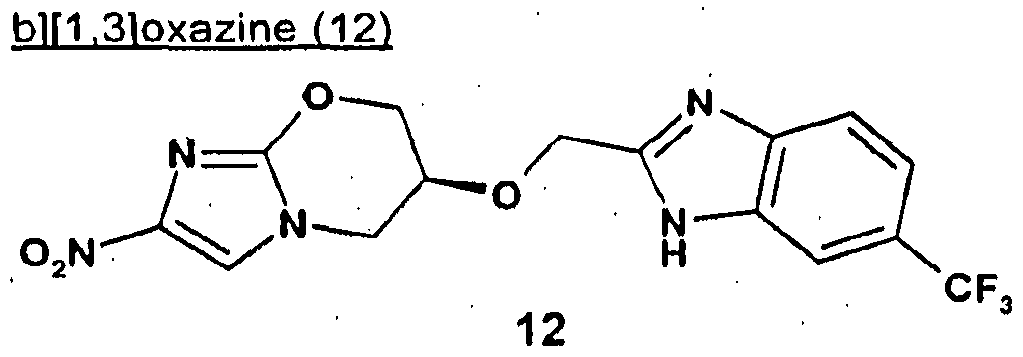
- Compound 32 is prepared as described in example 25 from 2-nitro-6,7-dihydro-5H- imidazo[2,1-b][1 ,3]oxazin-6-ol (6) and i-chloromethyl-2-trifluoromethoxybenzene. MS: M + 360.3
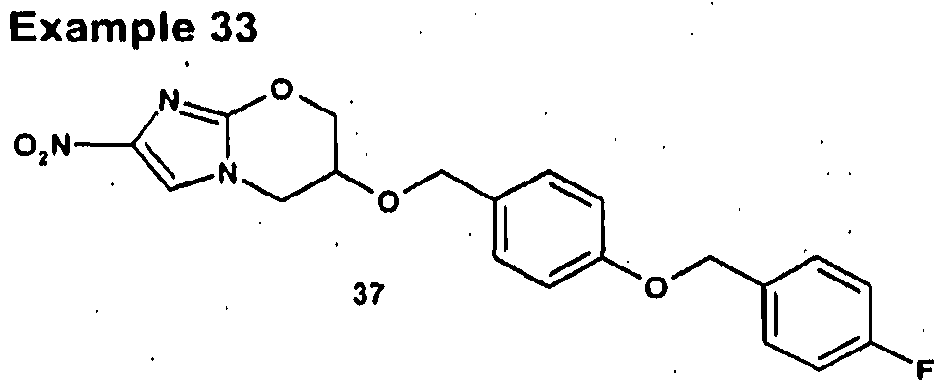
- Compound 37 is prepared as described in example 25 from 2-nitro-6,7-dihydro-5H- imidazo[2,1-b][1 ,3]oxazin-6-ol (29) and 1-chloromethyl-4-(4-fluorobenzy!oxy)benzene). Work up provided 37 as a pale yellow solid.
- the crude compound is purified over silica gel (60-120 mesh) using 15% to 25% gradient of ethyl acetate: pet ether as eluent to afford 45 mg of desired compound 44.
- reaction mixture is diluted with diethyl ether (80 ml), stirred for 10 minutes, filtered through celite, washed several times with ether and the solvent is removed in vacuo to give crude 2,2-dimethyl- [1,3]dioxane-5-carbaldehyde, which is used in the next step without further purification.
- 2,2-dimethyl-1 ,3-dioxane-5-carbaldehyde (3.52 g, 24.4 mmol) is dissolved in 1 ,2- dichloroethane (250 ml) and 1-[4-(trifluoromethoxy)phenyl]piperazine (6.01 g , 24.4 mmol) in 1 ,2-dichloroethane (50 ml) is added.
- the reaction mixture is maintained at room temperature for 1 h.
- sodiumtriacetoxyborohydride (20.72g, 97.7 mmol) in small portions.
- the reaction mixture is allowed to stir at room temperature for 9h. Water is added to the reaction mixture and is extracted with chloroform. The organic layer is dried and concentrated to give1-(2,2-dimethyl-[1 ,3]dioxan-5-ylmethyl)-4-(4- trifluoromethoxy-pheny!-piperazine.
- the piperazine derivative (7.09 g, 18.9 mmol) is dissolved in methanol (80 ml), water is added (3 ml), followed by p-toluenesulfonic acid (3.91 g, 22.7mol) and the reaction mixture is heated under reflux at 6O 0 C for 3hrs.
- the reaction mixture is concentrated and neutralized with 10% aq. NaHCO 3 solution, followed by extraction with chloroform.
- the solvent is removed in vacuo to give 2-[4-(4-trifluoromethoxy-phenyl)-piperazin-1-ylmethyl]-propane- 1 ,3-diol which is used in the next step without further purification.
- a suspension of sodium hydride (2.58 g, 64.6 mmo! in dry DMF (100 ml) is cooled to -20 0 C and the diol (4.5 g, 16.1 mmol) in DMF (25 ml) is added to the reaction mixture and stirred for 1h.
- a solution of t-butyldimethylsilyl chloride (2.92 g, 19.3 mmol) in dry DMF is added to the reaction mixture drop wise and stirred for 1h.
- the reaction mixture is quenched with ice- cold water and extracted with ethyl acetate.
- the MIC values of the test and standard compounds are tested against two reference organisms Mycobacterium bovis Bacillus Calmette Guerin (BCG) Pasteur (ATCC 35745) and Mycobacterium tuberculosis H37Rv (ATCC 27294).
- BCG Mycobacterium bovis Bacillus Calmette Guerin
- ADS 0.81% NaCI 1 5% BSA fraction V (Roche, Mannheim, Germany) and 2% glucose]
- 0.2% glycerol 0.2% glycerol, 0.05% Tween-80.
- the drug susceptibility testing is carried out in flat-bottom 96-well plate using the microdilution broth method (NCCLS, National Committee for Clinical Laboratory Standards. 2003 Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard. Sixth Edition), with some modifications. Streptomycin and PA-824 are used as the standard drugs. Actively growing mycobacterium cultures (OD600-0.2) are diluted in complete 7H9 broth to obtain an optical density of OD600-0.04 (approximately 106 CFU/ml). Equal volume (100 ⁇ L) of the diluted culture is added to wells containing serially diluted drugs (100 ⁇ L).
- the MIC microplates are sealed to prevent "evaporation and are incubated at 37 0 C for 4-7 days.
- the growth of the bacteria is quantified either by using redox dye Alamar blue (Serotec Ltd., Oxford, UK) or by optical density (OD600) measurement.
- MIC is defined as the lowest drug concentration which yield a RFU reading of ⁇ 15000.
- MIC is defined as the lowest drug concentration which yield an absorbance reading of ⁇ 1/10 to the value obtained for the antibiotic-free growth control. Both MIC assays give consistent and reproducible results; for standard drug streptomycin, the MIC values are 06-0.13 and 0.25 ⁇ g/mL for M. bovis BCG and M. tuberculosis H37Rv, respectively.
- Example 53 Activity against Trypanosoma cruzi
- the Trypanosoma cruzi Tulahuen C2C4 strain is used.
- the infective amastigote and trypomastigote stages are cultivated in L-6 cells (a rat skeletal myoblast cell line) in RPMI 1640 medium supplemented with 2 mM L-glutamine and 10% heat-inactivated foetal bovine serum in 12,5 cm ⁇ tissue culture flasks.
- Amastigotes develop intracellular!/, differentiate into trypomastigotes and leave the host cell.
- These trypomastigotes infect new L-6 cells and are the stages used to initiate an infection in the assay. All 'cultures and assays are conducted at 37°C under an atmosphere of 5% CO2 in air.
- DMSO dimethy ⁇ sulfoxide
- Assays are performed in sterile 96-well microtiter plates, each well containing 100 ⁇ L medium with 2x1 O ⁇ L-6 cells. After 24 hours, 50 ⁇ L of a trypanosome suspension containing
- CPRG/Nonidet chlorophenolred-D-galactopyranoside (CPRG, Roche Diagnostics Ltd; 15.19mg) plus 250 ⁇ L Nonidet P-40 are dissolved in 100 mL sterile phosphate-buffered saline (pH 7.2), giving 5x the final desired concentration of CPRG in 0.25% Nonidet P-40/PBS), 50 ⁇ L is added to all wells. A colour reaction becomes visible within 2-6 hours and can be read photometrically at 540 nm. Data are transferred into a graphics programme, sigmoidal inhibition curves determined and IC 50 values calculated.
- Example 54 Activity against Leishmania donovani
- the Leishmania donovani strain MHOM/ET/67/L82 (obtained from Dr. S. Croft, London School of Hygiene and Tropical Medicine) is used. The strain is maintained in the Syrian Golden hamster. Amastigotes are collected from the spleen of an infected hamster. Amastigotes are grown in axenic culture at 37°C in SM medium (Cunningham I., J. Protozool. 24, 325-329, 1977) at pH 5.4 supplemented with 10% heat-inactivated foetal bovine serum (FBS) under an atmosphere of 5% CO 2 in air.
- FBS heat-inactivated foetal bovine serum
- DMSO dimethylsulfoxide
- Assays are performed in 96-well flat-bottom microtiter plates (Costar, Corning Inc.), each well containing 100 ⁇ L of culture medium with 10 5 amastigotes from axenic culture with or without a serial drug dilution. Concentration of amastigotes is determined in a CASY cell analysing system (Scharfe System, Reutlingen, Germany). Before the amastigotes are counted, the parasite culture is passed twice through a 22 gauge needle to break up clusters of amastigotes.
- the highest concentration for the test compounds is 90 ⁇ g/mL. Seven 3-fold dilutions are used, covering a range from 30 ⁇ g/mL to 0.041 ⁇ g/mL. Each compound is tested in duplicate. Active compounds are tested twice for confirmation. After 72 hours of incubation, the plates are inspected under an inverted microscope to assure growth of the controls and sterile conditions.
- nitroimidazole compounds of the present invention have useful pharmaceutical properties.
- the compounds are useful in the treatment and/or prevention of infections such as those caused by Mycobacterium tuberculosis, Trypanosoma cruzi or Leish mania donovani.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pulmonology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description



























































































Claims




Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006331676A AU2006331676A1 (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole compounds |
| BRPI0620251-9A BRPI0620251A2 (en) | 2005-12-23 | 2006-12-22 | nitroimidazole compounds |
| US12/097,976 US20080275035A1 (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole Compounds |
| EP06847903A EP1984369A2 (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole compounds |
| JP2008547540A JP2009521464A (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole compounds |
| CA002631661A CA2631661A1 (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75376305P | 2005-12-23 | 2005-12-23 | |
| US75378105P | 2005-12-23 | 2005-12-23 | |
| US60/753,763 | 2005-12-23 | ||
| US60/753,781 | 2005-12-23 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007075872A2 true WO2007075872A2 (en) | 2007-07-05 |
| WO2007075872A3 WO2007075872A3 (en) | 2007-12-06 |
Family
ID=38069269
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/048763 Ceased WO2007075872A2 (en) | 2005-12-23 | 2006-12-22 | Nitroimidazole compounds |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20080275035A1 (en) |
| EP (1) | EP1984369A2 (en) |
| JP (1) | JP2009521464A (en) |
| KR (1) | KR20080079280A (en) |
| AU (1) | AU2006331676A1 (en) |
| BR (1) | BRPI0620251A2 (en) |
| CA (1) | CA2631661A1 (en) |
| RU (1) | RU2008129726A (en) |
| WO (1) | WO2007075872A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011014774A1 (en) | 2009-07-31 | 2011-02-03 | Global Alliance For Tb Drug Development | Nitroimidazooxazines and their uses in anti-tubercular therapy |
| WO2011014776A1 (en) | 2009-07-31 | 2011-02-03 | Global Alliance For Tb Drug Development | Nitroimidazooxazine and nitroimidazooxazole analogues and their uses |
| JP2011515487A (en) * | 2008-03-26 | 2011-05-19 | グローバル、アライアンス、フォア、ティービー、ドラッグ、ディベロップメント | Bicyclic nitroimidazoles covalently linked to substituted phenyloxazolidinones |
| KR101112992B1 (en) * | 2009-10-28 | 2012-02-24 | 한국화학연구원 | Nitroimidazole compounds, process for the preparation thereof, and pharmaceutical composition for treating tuberculosis comprising the same |
| WO2011087995A3 (en) * | 2010-01-13 | 2013-03-21 | Clifton Barry | Nitro - imidazo - oxazine derivatives and their use in the treatment of mycobacterial infections |
| WO2013072903A1 (en) | 2011-11-17 | 2013-05-23 | Ithemba Pharmaceuticals (Proprietary) Limited | Nitroimidazoxadiazocine compounds |
| US9198913B2 (en) | 2009-07-31 | 2015-12-01 | Global Alliance For Tb Drug Development | Nitroimidazooxazines and their uses in anti-tubercular therapy |
| KR20160052048A (en) | 2014-11-04 | 2016-05-12 | 한국화학연구원 | Imidazo oxazine derivatives, pharmaceutically acceptable salts thereof or optical isomer thereof and pharmaceutical composition containing the same as an active ingredient |
| US9725427B2 (en) | 2012-03-16 | 2017-08-08 | Biohaven Pharmaceutical Holding Company Limited | Prodrugs of riluzole and their method of use |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
| US10227362B2 (en) | 2015-01-29 | 2019-03-12 | Medshine Discovery Inc. | Anti-pulmonary tuberculosis nitroimidazole derivative |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP5770096B2 (en) | 2009-10-26 | 2015-08-26 | 富士フイルムRiファーマ株式会社 | Infectious disease diagnostic agent |
| CN102234287B (en) * | 2010-04-26 | 2015-08-05 | 上海阳帆医药科技有限公司 | Nitro glyoxaline compound, Preparation Method And The Use |
| KR101252632B1 (en) * | 2010-12-02 | 2013-04-09 | 한국화학연구원 | Nitroimidazole compounds, process for the preparation thereof, and pharmaceutical composition for treating tuberculosis comprising the same |
| KR101650716B1 (en) * | 2012-11-22 | 2016-08-24 | 한국화학연구원 | Bicyclic nitroimidazole derivatives, preparation method thereof and pharmaceutical composition for prevention or treatment of tuberculosis containing the same as an active ingredient |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5668127A (en) * | 1995-06-26 | 1997-09-16 | Pathogenesis Corporation | Nitroimidazole antibacterial compounds and methods of use thereof |
| JP4761756B2 (en) * | 2003-10-31 | 2011-08-31 | 大塚製薬株式会社 | 2,3-Dihydroimidazo [2,1-b] oxazole compound |
| PT1678185E (en) * | 2003-10-31 | 2009-01-13 | Otsuka Pharma Co Ltd | 2,3-dihydro-6-nitroimidazo [2,1-b] oxazole compounds for the treatment of tuberculosis |
-
2006
- 2006-12-22 US US12/097,976 patent/US20080275035A1/en not_active Abandoned
- 2006-12-22 KR KR1020087015156A patent/KR20080079280A/en not_active Ceased
- 2006-12-22 RU RU2008129726/04A patent/RU2008129726A/en not_active Application Discontinuation
- 2006-12-22 WO PCT/US2006/048763 patent/WO2007075872A2/en not_active Ceased
- 2006-12-22 JP JP2008547540A patent/JP2009521464A/en active Pending
- 2006-12-22 AU AU2006331676A patent/AU2006331676A1/en not_active Abandoned
- 2006-12-22 BR BRPI0620251-9A patent/BRPI0620251A2/en not_active IP Right Cessation
- 2006-12-22 EP EP06847903A patent/EP1984369A2/en not_active Withdrawn
- 2006-12-22 CA CA002631661A patent/CA2631661A1/en not_active Abandoned
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011515487A (en) * | 2008-03-26 | 2011-05-19 | グローバル、アライアンス、フォア、ティービー、ドラッグ、ディベロップメント | Bicyclic nitroimidazoles covalently linked to substituted phenyloxazolidinones |
| CN102753558B (en) * | 2009-07-31 | 2014-10-15 | 全球结核病药物研发联盟 | Nitroimidazoxazines and their application in anti-tuberculosis therapy |
| WO2011014774A1 (en) | 2009-07-31 | 2011-02-03 | Global Alliance For Tb Drug Development | Nitroimidazooxazines and their uses in anti-tubercular therapy |
| US9198913B2 (en) | 2009-07-31 | 2015-12-01 | Global Alliance For Tb Drug Development | Nitroimidazooxazines and their uses in anti-tubercular therapy |
| CN102753558A (en) * | 2009-07-31 | 2012-10-24 | 全球结核病药物研发联盟 | Nitroimidazoxazines and their application in anti-tuberculosis therapy |
| JP2013500996A (en) * | 2009-07-31 | 2013-01-10 | グローバル、アライアンス、フォア、ティービー、ドラッグ、ディベロップメント | Nitroimidazooxazine and its use in anti-tuberculosis therapy |
| AU2010278777B2 (en) * | 2009-07-31 | 2015-05-07 | Global Alliance For Tb Drug Development | Nitroimidazooxazines and their uses in anti-tubercular therapy |
| WO2011014776A1 (en) | 2009-07-31 | 2011-02-03 | Global Alliance For Tb Drug Development | Nitroimidazooxazine and nitroimidazooxazole analogues and their uses |
| KR101112992B1 (en) * | 2009-10-28 | 2012-02-24 | 한국화학연구원 | Nitroimidazole compounds, process for the preparation thereof, and pharmaceutical composition for treating tuberculosis comprising the same |
| WO2011087995A3 (en) * | 2010-01-13 | 2013-03-21 | Clifton Barry | Nitro - imidazo - oxazine derivatives and their use in the treatment of mycobacterial infections |
| WO2013072903A1 (en) | 2011-11-17 | 2013-05-23 | Ithemba Pharmaceuticals (Proprietary) Limited | Nitroimidazoxadiazocine compounds |
| US10844026B2 (en) | 2012-03-16 | 2020-11-24 | Biohaven Pharmaceutical Holding Company Ltd. | Prodrugs of riluzole and their method of use |
| US9725427B2 (en) | 2012-03-16 | 2017-08-08 | Biohaven Pharmaceutical Holding Company Limited | Prodrugs of riluzole and their method of use |
| US12172974B2 (en) | 2012-03-16 | 2024-12-24 | Biohaven Pharmaceutical Holding Company Ltd. | Prodrugs of riluzole and their method of use |
| US11440893B2 (en) | 2012-03-16 | 2022-09-13 | Biohaven Pharmaceutical Holding Company Ltd. | Prodrugs of riluzole and their method of use |
| US10562870B2 (en) | 2012-03-16 | 2020-02-18 | Biohaven Pharmaceutical Holding Company Ltd. | Prodrugs of riluzole and their method of use |
| KR20160052048A (en) | 2014-11-04 | 2016-05-12 | 한국화학연구원 | Imidazo oxazine derivatives, pharmaceutically acceptable salts thereof or optical isomer thereof and pharmaceutical composition containing the same as an active ingredient |
| US10227362B2 (en) | 2015-01-29 | 2019-03-12 | Medshine Discovery Inc. | Anti-pulmonary tuberculosis nitroimidazole derivative |
| WO2018033455A1 (en) | 2016-08-15 | 2018-02-22 | Bayer Cropscience Aktiengesellschaft | Condensed bicyclic heterocycle derivatives as pest control agents |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009521464A (en) | 2009-06-04 |
| CA2631661A1 (en) | 2007-07-05 |
| BRPI0620251A2 (en) | 2011-11-08 |
| RU2008129726A (en) | 2010-01-27 |
| EP1984369A2 (en) | 2008-10-29 |
| WO2007075872A3 (en) | 2007-12-06 |
| KR20080079280A (en) | 2008-08-29 |
| US20080275035A1 (en) | 2008-11-06 |
| AU2006331676A1 (en) | 2007-07-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN111032034B (en) | Spiro compounds and methods of making and using the same | |
| WO2007075872A2 (en) | Nitroimidazole compounds | |
| KR20210010475A (en) | MAGL inhibitor | |
| WO2009001060A2 (en) | Use of compounds for preparing anti-tuberculosis agents | |
| EP2767531B1 (en) | Cyclic n,n'-diarylthioureas and n,n'-diarylureas as androgen receptor antagonists, anti-cancer agent, method for producing and using same | |
| KR102756049B1 (en) | Dual magl and faah inhibitors | |
| JP2017510564A (en) | Novel heteroaromatic derivatives and their use as pharmaceuticals | |
| CA2861150A1 (en) | Morphinan derivative | |
| JP5355551B2 (en) | Quinolone compounds and pharmaceutical compositions | |
| EP3057422A1 (en) | Quinolinyl modulators of ror(gamma)t | |
| ES2942761T3 (en) | Heterocyclic compounds useful as antibacterial agents and method for the production thereof | |
| KR20250086658A (en) | Phosphoramidate compounds and uses thereof | |
| JP5769504B2 (en) | Medicine | |
| JP6838045B2 (en) | Compound | |
| JP2019519605A (en) | Heterocyclic compounds useful as antibacterial agents and method for producing the same | |
| EP3743419B1 (en) | Novel compounds for the treatment of parasitic infections | |
| CN101341150A (en) | Nitroimidazole compound | |
| ES2952989T3 (en) | Antibacterial heterocyclic compounds and their synthesis | |
| MX2008008195A (en) | Nitroimidazole compounds | |
| EP3296298A1 (en) | 7-substituted 1-aryl-naphthyridin-3-carboxamides and their use | |
| EP2487157A1 (en) | Enantioselective synthesis method of 4-aminoalcoholquinoline derivatives and the use | |
| HK40092567A (en) | Aminoheteroaryl kinase inhibitors | |
| HK40043248A (en) | Substituted oxazolidinones for the treatment of mammalian infections |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680048054.9 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 3982/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2631661 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006331676 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12097976 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/008195 Country of ref document: MX Ref document number: 1020087015156 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008547540 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006331676 Country of ref document: AU Date of ref document: 20061222 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008129726 Country of ref document: RU Ref document number: 2006847903 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0620251 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080623 |