WO2007084622A2 - Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof - Google Patents
Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof Download PDFInfo
- Publication number
- WO2007084622A2 WO2007084622A2 PCT/US2007/001345 US2007001345W WO2007084622A2 WO 2007084622 A2 WO2007084622 A2 WO 2007084622A2 US 2007001345 W US2007001345 W US 2007001345W WO 2007084622 A2 WO2007084622 A2 WO 2007084622A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- mmol
- heteroaryl
- thieno
- reaction
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 **(CC(*)(*)*(*)(*I)C(C1=C(*2)*CCCCIC(CII)C*C1)=C2C1(*)*)C1(*)O Chemical compound **(CC(*)(*)*(*)(*I)C(C1=C(*2)*CCCCIC(CII)C*C1)=C2C1(*)*)C1(*)O 0.000 description 1
- FOIXJIRKUYJBOI-UHFFFAOYSA-N CCS(c1cc(CCNCC2)c2[s]1)(=O)=O Chemical compound CCS(c1cc(CCNCC2)c2[s]1)(=O)=O FOIXJIRKUYJBOI-UHFFFAOYSA-N 0.000 description 1
- BFUSEPHZJSMUCK-UHFFFAOYSA-N O=S(c1cc(CCNCC2)c2[s]1)(c1ccccc1)=O Chemical compound O=S(c1cc(CCNCC2)c2[s]1)(c1ccccc1)=O BFUSEPHZJSMUCK-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N43/00—Biocides, pest repellants or attractants, or plant growth regulators containing heterocyclic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/06—Peri-condensed systems
Definitions
- the present invention generally relates to a series of compounds, to pharmaceutical compositions containing the compounds, and to use of the compounds and compositions as therapeutic agents. More specifically, compounds of the present invention are thiophenyl and pyrrolyl azepine compounds. These compounds are serotonin receptor (5-HT2 C ) ligands and
- Serotonin has been implicated in a number of diseases, disorders, and conditions that originate in the central nervous system, including diseases, disorders, and conditions related to, for example, sleeping, eating, perceiving pain, controlling body temperature, controlling blood pressure, depression, anxiety, addiction and schizophrenia.
- Serotonin also plays an important role in peripheral systems, such as the gastrointestinal system, where it has been found to mediate a variety of contractile, secretory, and electrophysiologic effects.
- antagonists of serotonergic systems are of interest for the treatment of a wide range of disorders, including anxiety, depression, hypertension, migraine, obesity, compulsive disorders, schizophrenia, autism, neurodegenerative disorders (e.g., Alzheimer's disease, Parkinsonism, and Huntington's chorea), and chemotherapy-induced vomiting.
- disorders including anxiety, depression, hypertension, migraine, obesity, compulsive disorders, schizophrenia, autism, neurodegenerative disorders (e.g., Alzheimer's disease, Parkinsonism, and Huntington's chorea), and chemotherapy-induced vomiting.
- the major classes of serotonin receptors (5-HTi -7 ) contain one to seven
- the 5-HT 2 family of receptors contains 5-HT 2a , 5-HT 2b , and 5-
- HT 2 C subtypes which have been grouped together on the basis of primary structure, secondary messenger system, and operational profile. All three 5-HT2 subtypes are G-protein coupled, ATH101037P00101PC
- the 5-HT 2b and 5-HT 28 receptors are widely distributed in the peripheral nervous system, with 5-HT 2a also found in the brain .
- the 5-HT 20 receptor has been found only in the central nervous system, being highly expressed in many regions of the human brain. See G. Baxter, et al. Trends in Pharmacol. ScL 1995, 16, 105-110.
- Subtype 5-HT 23 has been associated with effects including vasoconstriction, platelet aggregation, and bronchoconstriction, as well as certain CNS effects, while subtype 5- HT 2 Q has been associated with diseases that include depression, anxiety, obsessive compulsive disorder, addiction, panic disorders, phobias, psychiatric syndromes, and obesity. Very little is known about the pharmocologic role of the 5-HT 2b receptor. See F. Jenck, et al., Exp. Opin. Invest. Drugs, 1998, 7, 1587-1599; M. Bos, et al., J. Med. Chenu, 1997, 40, 2762-2769; J.R.
- 4,575,504 discloses thienothiazole derivatives
- U.S. Patent No. 5,258,378 discloses certain pyrroloazepine compounds
- U.S. Patent Nos. 4,414,225 and 4,904,653 disclose certain azepine derivatives
- WO 2005/019179 discloses certain benzazepines, WO 2005/003096, WO 2005/042490, and WO 2005/042491 disclose benzazepine derivatives
- WO 96/11201 discloses furan derivatives
- WO 2005/040169 discloses certain fused pyrrole- and pyrazole- containing heterocyclic compounds which are serotonin modulators
- WO 2004/024065 discloses substituted bicyclic thiophene derivatives. None of these patents or patent applications disclose compounds of the instant invention.
- the present invention is directed to compounds of the formula:
- Ri and R 2 taken together with the atoms to which they are attached can form a 5- 7- member carbocycle or heterocycle optionally substituted with up to two substituents selected from alkyl, CF 3 , and -OR 8 ;
- R 3 is selected from the group consisting of H 3 C t-8 alkyl, OR 8 , aryl and heteroaryl;
- R 3a is H or Ci -S alkyl; or R 3 and R 3a taken together are -CH 2 CH 2 -;
- R 2 and R 3 taken together form a 5-7-member carbocycle or heterocycle optionally substituted with up to two substituents selected from alkyl, CF 3 , and -OR 8 ;
- R 4 is H, C t-8 alkyl, or OR 8 ;
- R 42 is H, C I -8 alkyl; or R 4 and R 4a taken together are -CH 2 CH 2 -;
- R 5 is selected from the group consisting of H, Cj -8 alkyl, -Cj -8 alkyl-O-Ci -8 alkyl, Ci -8 alkylaryl or heteroaryl, and -Ci -8 alkyl-O-aryl or heteroaryl;
- Rs a is H or -Ci -8 alkyl; g
- Rg is selected from the group consisting of H,- -C I -8 alkyl, Ci-g alkyl-O-Ci-salkyl, Ci_ 9 alkylaryl or heteroaryl, and -Ci -8 alkyl-O-aryl or heteroaryl;
- R 63 is H or -Ci -8 alkyl;
- R 7 is selected from the group consisting of H, -Ci -8 alkyl, and — Ci -8 alkylaryl or heteroaryl;
- R 8 , Rg are independently selected from the group consisting of H 3 -Ci -8 alkyl, -C 2-8 alkenyl, -C 2-8 alkynyl, aryl or heteroaryl, -Ci -8 alkylaryl or heteroaryl, 15 -C i- 8 alkyl-O-Ci- 8 alkyl, and -C i_s alkyl — O-aryl or heteroaryl;
- R 8 and R9 taken together with the atom to which they are attached form a 5-7-member heterocycle
- 20 Rio is selected from the group consisting of -Cj -8 alky], -C 2-8 alkenyl, -C 2-8 alkynyl, aryl or heteroaryl, -Ci -8 alkylaryl or heteroaryl, -Cj -8 alkyl-O-C ⁇ -8 alkyl, and -C ⁇ _ 8 alkyl-O-aryl or heteroaryl;
- Ri 1 is selected from the group consisting of H, -Ci -8 alkyl, -Ci -8 alkyl-O-Ci-s alkyl, 25 -SO2R10J aryl, and heteroaryl, or Ci -8 alkylaryl or heteroaryl;
- Ri 1 and R together with the atoms to which they are attached may form a 5-7- membered heterocycle optionally substituted with up to two substituents selected from -Ci -8 alkyl, CF3, and -OR 8 ; and 30
- Rn and R 4 together with the atoms to -which they are attached may form a 5-7- membered heterocycle optionally substituted with up to two substituents selected from -C 1 - S alkyl, CF 3 , and -OR 8 ;
- aryl and heteroaryl are optionally substituted with up to two substituents selected from -Ci -8 alkyl. halogen, CN, and alkoxy, and the pharmaceutically acceptable salts thereof. &m «
- Another embodiment of the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- Still another embodiment of the present invention provides a method of treating a disease, disorder and/or condition in a mammal (e.g., animal or human), wherein a 5-HT 2 C receptor is implicated and modulation of a 5-HT 20 function is desired.
- the method comprises administering a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, to the mammal.
- Yet another embodiment of the present invention comprises a method of modulating 5 -HT receptor function with an effective amount of compound of Formula (I), or a pharmaceutically acceptable salt thereof.
- a further embodiment of the present invention provides a method of treating or preventing diseases, disorders, and/or conditions of the central nervous system.
- the method comprises administering a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt thereof, to the mammal.
- Formula (I) may have activity include obesity, depression, schizophrenia, anxiety, obsessive compulsive disorder, addiction, panic disorders, sleep disorders, migraine, Type II diabetes, epilepsy, phobias and psychiatric syndromes.
- alkyl includes straight chained and branched hydrocarbon groups containing the indicated number of carbon atoms, typically methyl, ethyl, and straight chain and branched propyl and butyl groups.
- alkyl also encompasses cycloalkyl, i.e., a cyclic C 3 -C 8 hydrocarbon group, such as cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- alkenyl refers to a substituted or unsubstituted straight-chain or substituted or unsubstituted branched-chain alkenyl radical containing from 2 to 10 carbon atoms. Examples of such radicals include, but are not limited to, ethenyl, E- and Z-penteny], decenyl and the like. > i : ! i ! ! I ! ' ! J H I i i ! i i ! ! ! : ! i ! :Tl ill : ! ! ft i iifii IiMi iMi 1 ⁇ -ll !]]!: i i ⁇ J i i : i ! > I i i i i i :
- alkynyl refers to a substituted or unsubstituted straight or substituted or unsubstituted branched chain alkynyl - - radical containing from 2 to 10 carbon atoms.
- examples of such radicals include, but are not limited to, ethynyl, propynyl, propargyl, butynyl, hexynyl, decynyl and the like.
- alkoxy refers to an alkyl
- alkyl is as defined above.
- suitable alkyl ether radicals include, but are not limited to, methoxy, ethoxy, n-propoxy, iso-propoxy, n-butoxy, iso-butoxy, sec-butoxy, tert-butoxy and the like.
- halo is defined herein to include fluoro, chloro, bromo, or iodo.
- halogen is defined herein to include fluorine, chlorine, bromine, and 15 iodine.
- amino alone or in combination, includes the group -NH 2 or -NR a R b wherein R 3 and R b are independently hydrogen, alkyl, alkylaryl, or aryl.
- R a and R b are independently hydrogen, .
- aryl denotes a phenyl group, or an ortho-fused bicyclic 25 carbocyclic group having nine to ten ring atoms in which at least one ring is aromatic (e.g. naphthyl or tetrahydronaphthyl).
- aromatic e.g. naphthyl or tetrahydronaphthyl.
- aryl also is abbreviated in the various chemical structures as "Ar.”
- heteroaryl is defined herein as a monocyclic, bicyclic, or tricyclic ring system containing one, two, or three aromatic rings and containing at least one nitrogen, 30 oxygen, or sulfur atom in an aromatic ring, and which can be unsubstituted or substituted, for example, with one or more, and in particular one to three, substiruents, like halo, alkyl, hydroxy, hydroxyalkyl, alkoxy, alkoxyalkyl, haloalkyl, nitro, amino, alkylamino, acylamino, alkylthio, alkylsulfonyl, and alkylsulfonyl.
- heteroaryl groups include, but are not limited to, 2H-pyrrolyl, 3H-indolyl, 4H-quinolizinyl, 4H-carbazolyl, acr ⁇ dinyl, 35 benzo[b]thienyl, benzothiazolyl, 13-carbolinyl, carbazolyl, chromenyl, cinnaolinyl, dibenzo[b,d]furanyl, furazanyl, furyl, imidazolyl, imidizolyl, indazolyl, indolisinyl, indolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridinyl, naptho[2,3- b], oxazolyl, perimidinyl, phenanthridinyl, phenanthrolinyl, phenarsazinyl, phenazin
- heteroaryl denotes a monocyclic aromatic ring containing five or six ring atoms containing carbon and 1, 2, 3, or 4 heteroatoms independently selected from
- heteroaryl denotes an ortho-fused b ⁇ cyclic heterocycle of about eight to ten ring atoms derived therefrom, particularly a benz- derivative or one derived by fusing a propylene, or tetramethylene diradical thereto.
- Het or “heterocycle” generally represents a heterocyclic group
- Het is a monocyclic, bicyclic, or tricyclic group containing one or more heteroatoms selected from the group consisting of oxygen, nitrogen, and sulfur.
- 20 of Het groups include 1,3-dihydrobenzofuran, 1,3-dioxolane, 1,4-dioxane, 1,4-dithiane, 2H- pyran, 2-pyrazoline, 4H-pyran, chromanyl, imidazolidinyl, imidazolinyl, indolinyl, isochromanyl, isoindolinyl, morpholine, piperazinyl, piperidine, piperidyl, pyrazolidine, pyrazolidinyl, pyrazolinyl, pyrrolidine, pyrroline, quinuclidine, and thiomorpholine.
- Certain compounds of the invention may exist in different isomeric (e.g. enantiomers and distereoisomers) forms.
- the invention contemplates all such isomers both in pure form and in admixture, including racemic mixtures. Enol forms are also included.
- the compounds of the invention can exist in unsolvated as well as solvated forms, including hydrated forms, e.g., hemi-hydrate. In general, the solvated forms, with pharmaceutically acceptable solvents such as water, ethanol, and the like are equivalent to the unsolvated forms for the purposes of the invention.
- Certain compounds of the invention also form pharmaceutically acceptable salts, e.g., acid addition salts.
- the nitrogen atoms may form salts with acids.
- suitable acids for salt formation are hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic, malonic, salicylic, malic, furmaric, succinic, ascorbic, maleic, methanesulfonic and other mineral carboxylic acids well known to those in the art.
- the salts are prepared by contacting the free base form with a sufficient amount of the desired acid to produce a salt in the conventional manner.
- the free base forms may be regenerated by treating the salt with a suitable dilute aqueous base solution such as dilute aqueous hydroxide potassium carbonate, ammonia, and sodium bicarbonate.
- a suitable dilute aqueous base solution such as dilute aqueous hydroxide potassium carbonate, ammonia, and sodium bicarbonate.
- the free base forms differ from their respective salt forms somewhat in certain physical properties, such as solubility in polar solvents, but the acid salts are equivalent to their respective free base forms for purposes of the invention. (See, for example S. M. Berge, et al., "Pharmaceutical Salts," J. Pharm. Sci., 66: 1 -19 (1977) which is incorporated herein by reference.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which
- the compounds of the present invention can be used in the form of pharmaceutically acceptable salts derived from inorganic or organic acids.
- pharmaceutically acceptable salt means those salts which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like and are commensurate with a reasonable benefit/risk ratio.
- Pharmaceutically acceptable salts are well-known in the art. For example, S. M. Berge et al. describe pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences J 977, 66: 1 et seq.
- the salts can be prepared in situ during the final isolation and purification of the compounds of the invention or separately by reacting a free base function with a suitable organic acid.
- Representative acid addition salts include, but are not limited to acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate, digluconate, glycerophosphate, hemisulfate, heptanoate, hexanoate, fumarate, hydrochloride, hydrobromide, hydroiodide, 2- hydroxyethansulfonate (isothionate), lactate, maleate, methanesulfonate, nicotinate, 2- naphthalenesulfonate, oxalate, palmitoate, pectinate, persulfate, 3-phenylpropionate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, phosphate, glutamate, bicarbonate, p-toluenesulfonate and undecano
- the basic nitrogen-containing groups can be quaternized with such agents as lower alkyl halides such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates; long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides; arylalkyl halides like benzyl and phenethyl bromides and others. Water or oil-soluble or dispersible products are thereby obtained.
- lower alkyl halides such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates
- long chain halides such as decyl
- acids which can be employed to form pharmaceutically acceptable acid addition salts include such inorganic acids as hydrochloric acid, hydrobromic acid, sulphuric acid and phosphoric acid and such organic acids as oxalic acid, maleic acid, succinic acid and citric acid.
- Basic addition salts can be prepared in situ during the final isolation and purification of compounds of this invention by reacting a carboxylic acid-containing moiety with a suitable base such as the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine.
- suitable bases such as the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine.
- Pharmaceutically acceptable salts include, but are not limited to, cations based on alkali metals or alkaline earth metals such as lithium, sodium, potassium, calcium, magnesium and aluminum salts and the like and nontoxic quaternary ammonia and amine cations including 134J
- Dosage forms for topical administration of a compound of this invention include powders, sprays, ointments and inhalants.
- the active compound is mixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives, buffers or propellants which can be required.
- Opthalmic formulations, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- compositions of this invention can be varied so as to obtain an amount of the active compound(s) which is effective to achieve the desired therapeutic response for a particular patient, compositions and mode of administration.
- the selected dosage level will depend upon the activity of the particular compound, the route of administration, the severity of the condition being treated
- the compound 25 of one of the compounds of the present invention can be employed in pure form or, where such forms exist, in pharmaceutically acceptable salt, ester or prodrug form.
- the compound can be administered as a pharmaceutical composition containing the compound of interest in combination with one or more pharmaceutically acceptable excipients.
- the phrase "therapeutically effective amount" of the compound of the invention is a pharmaceutical composition containing the compound of interest in combination with one or more pharmaceutically acceptable excipients.
- the total daily dose of the compounds of this invention administered to a human or lower animal may range from about 0.0001 to about 1000 mg/kg/day.
- more preferable doses can be in the range of from about 0.001 to about 5 mg/kg/day.
- the effective daily dose can be divided into multiple doses for purposes of administration; consequently, single dose compositions may contain such amounts or submultiples thereof to make up the daily dose.
- the present .invention also provides pharmaceutical compositions that comprise compounds of the present invention formulated together with one or more non-toxic pharmaceutically acceptable carriers.
- the pharmaceutical compositions can be specially formulated for oral administration in solid or liquid form, for parenteral injection or for rectal administration.
- compositions of this invention can be administered to humans and other mammals orally, rectally, parenterally , intracisternally, intravaginally, intraperitoneally, topically (as by powders, ointments or drops), bucally or as an oral or nasal spray.
- parenterally refers to modes of administration which include intravenous, intramuscular, intraperitoneal, intrasternai, subcutaneous and intraarticular injection and infusion.
- the present invention provides a pharmaceutical composition comprising a component of the present invention and a physiologically tolerable diluent.
- the present invention includes one or more compounds as described above formulated into compositions together with one or more non-toxic physiologically tolerable or acceptable diluents, carriers, adjuvants or vehicles that are collectively referred to herein as diluents, for parenteral injection, for intranasal delivery, for oral administration in solid or liquid form, for rectal or topical administration, among others.
- compositions can also be delivered through a catheter for local delivery at a target site, via an intracoronary stent (a tubular device composed ' of a fine wire mesh), or via a biodegradable polymer.
- the compounds may also be complexed to ligands, such as antibodies, for targeted delivery.
- compositions suitable for parenteral injection may comprise physiologically acceptable, sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions ATH101037P00101PC
- aqueous and nonaqueous carriers, diluents, solvents or vehicles examples include water, ethanol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and the like), vegetable oils (such as olive oil), injectable organic esters such as ethyl oleate, and suitable mixtures thereof.
- These compositions can also contain adjuvants such as preserving, wetting, emulsifying, and dispensing agents.
- adjuvants such as preserving, wetting, emulsifying, and dispensing agents.
- Prevention of the action of microorganisms can be ensured by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example sugars, sodium chloride and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monoste
- Suspensions in addition to the active compounds, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances, and the like.
- suspending agents as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances, and the like.
- the absorption of the drug in order to prolong the effect of the drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection. This can be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the drug then depends upon its rate of dissolution which, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered drug form is accomplished by dissolving or suspending the drug in an oil vehicle.
- Injectable depot forms are made by forming microencapsule matrices of the drug in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio of drug to polymer and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions which are compatible with body tissues.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium just prior to use.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules.
- the active compound may be mixed with at least one inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalcium phosphate and/or a) fillers or extenders such as starches, lactose, sucrose, glucose, mannitol and silicic acid; b) binders such as carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidone, sucrose and acacia; c) humectants such as glycerol; d) disintegrating agents such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates and sodium carbonate; e) solution retarding agents such as paraffin; f) absorption accelerators such as quaternary ammonium compounds; g) wetting agents such as cetyl alcohol and glycerol monostearate; h)
- compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polyethylene glycols and the like.
- the solid dosage forms of tablets, dragees, capsules, pills and granules can be prepared with coatings and shells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition such that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- coatings and shells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition such that they release the active ingredient(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- embedding compositions which can be used include polymeric substances and waxes.
- the active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluents commonly used in the art such as, for example, water or other solvents, solubilizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, dimethyl formamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan and mixtures thereof.
- the oral compositions may also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfumingagents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfumingagents.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the active compound.
- Liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multi -lamellar hydrated liquid crystals which are dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and metabolizable lipid capable of forming liposomes can be used.
- the present compositions in liposome form can contain, in addition to a compound of the present invention, stabilizers, preservatives, excipients and the like.
- the preferred lipids are natural and synthetic phospholipids and phosphatidyl cholines (lecithins) used separately or together.
- prodrugs of the compounds of the present invention represents those prodrugs of the compounds of the present invention which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, and the like, commensurate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwitterionic forms, where possible, of the compounds of the invention.
- Prodrugs of the present invention may be rapidly transformed in vivo to the parent compound of the above formula, for example, by hydrolysis in blood. A thorough discussion is provided in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Svstems,V. 14 of the A.C.S. Symposium Series, and in Edward B.
- HPLC analysis and purification was performed using a Waters 2525 binary gradient pump, Waters 2767 sample manager, waters 2487 UV detector (220 and 254 nM), and Waters Micromass ZQ electrospray mass spec detector.
- Analytical HPLC analysis was performed as follows:
- step (a) The crude material from step (a) (165 mmol, ⁇ 47g) was dissolved in EtOH (700 mL) and treated with 600 mL of IM NaOH. After stirring overnight, the reaction was acidified with concentrated HCl to pH ⁇ l . The crude reaction was diluted with EtOAc (400 mL) and washed with water. The water was back-extracted with EtOAc. The combined organic extracts were washed with water (2x) and dried over MgSO 4 . Concentration and evaporation from toluene (2x) gave the sub-title product as a solid.
- step (b) The product of step (b) (-165 mmol, ⁇ 42g) was dissolved in IL of DCM. DMF (100 uL) was added followed slowly by oxalyl chloride (21.7 mL, 247 mmol). After 1 hour, the reaction was concentrated to dryness and the crude material was re-dissolved in DCE (IL). AlCl 3 (55g, 410 mmol) was carefully added and the reaction was stirred at room temperature for VT. hour. The crude reaction was quenched with ice, washed with water (3x), and dried over MgSO 4 . The title product was purified by silica gel chromatography (30% EtOAc in
- step (d) The product of step (d) (1.45g, 6.44 mmol) and NaHCO 3 (3.2g, 38.6 mmol) were stirred in 60 mL cyclohexane. Bromine (1.0 mL, 19.3 mmol) was added slowly and the reaction was stirred in the dark for 15 minutes. The reaction was quenched with 5% Na 2 SO 3 and stirred rapidly for 15 minutes. The sub-title compound was extracted into EtOAc (2x). Drying over MgSO 4 and concentration gave the sub-title compound (2.6g) as a yellow oil.
- step (e) The product of step (e) (1.3g, 3.39 mmol) was dissolved in 1 :1 HOAc:water (40 mL), treated with Zn dust (0.44 g, 6.79 mmol), and heated to reflux for lhour. The reaction was cooled, diluted with water and extracted 2x EtOAc. The organic extracts were dried over MgSO 4 and concentrated to give 0.76g of the sub-title compound.
- step (f) 3-Methyl-4,5,7.8-tetrahydro-thieno[23-d "
- the product of step (f) (375 mg, 1.23 mmol) was dissolved in 4 mL dioxane and treated with Me 2 Zn (1.25 mL of 2M in toluene) and Pd(dppf) 2 Cl 2 . After heating to 100° C for 3h, the reaction was cooled, quenched with water, filtered through silica gel (washing with EtOAc), and concentrated to give 337 mg of the sub-title compound as an oil.
- step (g) The product of step (g) (337 mg, 1.4 mmol) was dissolve in 10 mL of 1 :1 CHC ⁇ HOAc and treated with NBS (301 mg, 1.7 mmol). After stirring for Vz hour , the reaction was diluted wmm
- step (h) The product of step (h) (60 mg, 0.19 mmol) was dissolve in 2 mL THF and cooled to -78° C. Butyl lithium (0.15 mL of 1.6M) was added and the reaction was stirred for 5 minutes. Trimethylacetyl chloride (36 uL, 0.3 mmol) was added and the reaction was warmed to room temperature. The reaction was quenched with water (5 mL) and the product was extracted into DCM (2x). The extracts were concentrated to dryness and treated with 4 mL of 1 :1 EtOH:40%KOH (aq) and heated to 100° C for 14 hours. The reaction was cooled and diluted with water. The product was extracted into DCM (2x5mL).
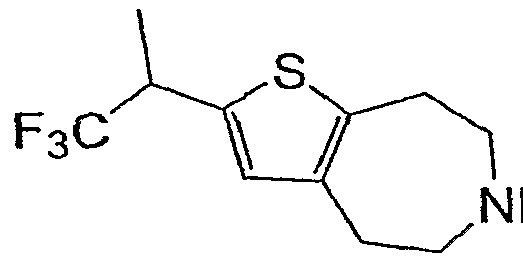
- step (e) 200 mg, 0.52 mmol was dissolved in 5 mL THF, cooled to -78° C and treated with BuLi (0.33 mL of 1.6 M). After stirring for 15 minutes at -78° C, trifluoroacetyl 2,2,2-trifluoroethanol (132 uL, 0.68 mmol) was added and the reaction was allowed to warm to room temperature. The reaction was quenched with water (50 uL) and concentrated. The residue was purified by silica gel chromatography to give 73 mg of the sub-title compound.
- step (a) The product of step (a) (73 mg, 0.18 mraol) was stirred in 3 mL EtOH and treated with NaBH 4 (20 mg, 0.5 mmol). After stirring for 20 minutes, the reaction was quenched with
- step (b) The product of step (b) (0.18 mmol) was dissolved in CHCl 3 (3 mL) and treated with TMSI (1 mmol, 200 wL). After heating for V 2 hour at 70° C, another 200 uL of TMSI was added and heating was continued for Vz hour. The reaction was cooled and carefully quenched with 0.5 mL each of EtOH and water. The reaction was diluted with IM NaOH (3 mL) and the product was extracted into DCM (2x5mL). The organic extracts were concentrated and the
- step (d) The product of Example 1, step (d) (75mg, 0.33 mmol) was dissolved in 2 mL DCE and
- step (d) The product of Example 1, step (d) (410 mg, 1.82 mmol) was dissolved in 20 mL DCE and treated with trifluoroacetic anhydride (510 ⁇ L, 3.64 mmol) and AlCl 3 (484 mg, 3.64 mmol).
- Triphenyl phosphonium bromide (393 mg, 1.1 mmol) was stirred in 7 mL THF.
- KHMDS • (199 mg, 1.0 mmol) was added and the yellow solution was stirred for 30 minutes at room temperature.
- the product of step (a) (180 mg, 0.56 mmol) was dissolved in in 7 mL THF and added to the above reaction. The solution was allowed to stir for 1 hour at room temperature, then diluted with EtOAc (25 mL) and washed with water (2x20 mL).
- the crude product was ATH101O37POO1O1PC
- step (b) The product of step (b) (40 mg, 0.12 mmol) was dissolved in 5 mL EtOH and treated with 10 mg of 10% Pd/C (wet, Degussa grade ElOl). The reaction was stirred rapidly under an atmosphere of hydrogen for 14 hours. Filtration through celite and concentration gave 37 mg of the sub-title compound.
- step (c) The product of step (c) (37 mg, 0.11 mmol) was dissolved in 1 mL CHCl 3 and treated with TMSI (47 uL, 0.35 mmol). After heating to 60° C for 2 hours, the reaction was cooled and concentrated. Purification of the crude residue by preparative HPLC-MS gave the title compound.
- Example I 5 step (d) The product of Example I 5 step (d) (80 mg, 0.35 mmol) was dissolved in 2 mL of 1 : 1 CHCl 3 :HO Ac and treated with NBS (62 mg, 0.35 mmol). After 15 minutes, the reaction was concentrated to dryness, dissolved in a minimal amount of EtOAc and filtered through a pad of silica gel. The filtrate was evaporated to give 105 mg of the sub-title compound.
- step (a) 2-Ethylsulfanyl-4,5,7,8-tetrahydro-thienor2,3-d]azepine-6-carb ⁇ xylic acid ethyl ester:
- the product of step (a) (105 mg, 0.35 mmol) was dissolved in 1 rnL NMP and treated with NaSEt (59 mg, 0.7 mmol). KI (5 mg, 0.03 mmol), and CuO (14 mg, 0.18 mmol). The reaction was heated to 120° C for 24 hours. Additional NaSEt (59 mg, 0.7 mmol), KI (5 mg, 0.03 mmol), and CuO (14 mg, 0.18 mmol) was added and the heating was continued for 24 hours.
- the reaction was diluted with water and DCM ( ⁇ 5 mL each) and filtered to remove the dark powdery precipitate.
- the crude product was extracted into DCM (3x 5 mL) and concentrated to -ImL.
- the crude residue was diluted with DCM (2 mL) and treated with Et 3 N (140 uL, 1.05 mmol) and ethyl chloroformate (50 ⁇ L, 0.52 mmol). After stirring overnight, the reaction was diluted with EtOAc (5 mL) and washed with water (5x) in order to remove the residual NMP.
- the organic solution was concentrated to give 80 mg of the sub-title compound which was used without further purification.
- step (b) The product of step (b) (80 mg, 0.28 mmol) was dissolved in 3 mL HOAc and treated with H 2 O 2 (300 ⁇ L of 30%, ⁇ 3 mmol). After stirring at room temperature for 3 days, the reaction was diluted with EtOAc and washed 4x with water. The organic extract was dried over MgS ⁇ 4 and concentrated to give 62 mg of the sub-title compound which was used without further purification.
- step (c) The product of step (c) (21 mg, 0.066 mmol) was dissolved in 2 mL EtOH and treated with 2 mL of 40% KOH and subsequently heated to 100°C in a sealed vessel overnight. The reaction was cooled and diluted with water. The title compound was extracted into DCM (3x) and purified by preparative HPLC-MS.
- Triethylphosphonoacetate (56 mg, 0.25 mmol) and the product of Example 4, step (a) (40 mg, 0.12 mmol) were stirred in 2 mL THF.
- the reaction was treated with LiHMDS (0.2 mL of IM) and stirred at room temperature for 1 hour.
- the reaction was quenched with 5 mL water and the product was extracted into DCM (2x5mL) and dried over MgSO 4 .
- the organic extract was concentrated to dryness and the residue was dissolved in 5 mL EtOH and treated with ⁇ 10 mg of 10% Pd/C (wet, Degussa grade ElOl).
- the reaction was filtered through celite and treated with IM NaOH (1 mL). After stirring for 2 hours at 60° C, the reaction was diluted with water and the product was extracted into DCM (3x5 mL) to give 45 mg of the sub-title compound which was used without further purification.
- step (a) The product of step (a) (45 mg, 0.12 mmol) was dissolved in DCE (2 mL) and treated with oxalyl chloride (43 ⁇ L, 0.49 mmol) and 1 drop of DMF. After stirring for 5 minutes at room temperature, the reaction was concentrated to dryness and dissolved in 2 mL DCM. AlCl 3 (66 mg, 0.50 mmol) was added and the reaction was stirred for 5 minutes. The reaction was quenched with water and the crude product was extracted into DCM (2x) to give 32 mg of a dark oil that was used without further purification. The enantiomers of the sub-title compound could be separated using a Chiralpak® AD-RH® 20 x 250 mm column from
- step (b) The product of step (b) (racemic, 32 mg, 0.10 mmol) was dissolved in DCE (ImL) and treated with ZnI 2 (64 mg, 0.2 mmol) and NaCNBH 3 (44 mg, 0.7 mmol). A thick slurry formed after stirring overnight. The reaction was filtered and the solid was washed with DCM. The combined filtrates were washed with water (lx5mL) and concentrated to dryness. The residue was dissolved in CHCl 3 (2 mL) and treated with TMSI (70 uL, 0.5 mmol). After heading to 60° C for 1 hour, an additional 50 «L of TMSI was added and heating was continued for 14 hours.
- step (b) (racemic, 100 mg, 0.29 mmol) was added as a solution in 3 mL DCM.
- the reaction was warmed to 0" C and stirred for 3 hours.
- the solution was poured over ice and the product was extracted into DCM (2x10 mL).
- the organic extract was dried over MgSO 4 and concentrated to give 84 mg of the sub-title compound, which was used without further purification.
- step a) The product of step a) (37 mg, 0.10 mmol) was dissolved in CHCl 3 (2 mL) and treated with TMSI (1 mmol,- 140 wL). After heating to 60° C for 2 hours, the reaction was quenched with MeOH and concentrated to dryness. The title compound was obtained after purification of the crude residue by preparative HPLC-MS.
- step (a) (40 mg, 0.125 mmol) was added as a solution in 3 mL DCM.
- the reaction was warmed to 0° C for 1 hour then to room temperature for 6 hours.
- the reaction was quenched over ice and extracted into DCM (2x5 mL). The organic extracts were dried over MgSO 4 and concentrated to give 34 mg of the sub-title compound which was used without further purification.
- step (a) The product of step (a) (34 mg, 0.1 mmol) was dissolved in 2 mL CHCl 3 and treated with TMSI (140 uL, 1 mmol). After heating to 60° C for 2 hours, the reaction was concentrated to dryness and the title compound was purified by preparative HPLC-MS.
- 1 H NMR (CD 3 OD) ⁇ 6.91 (s, IH), 3.39-3.32 (m, 4H), 3.21-3.17 (m, 2H), 3.09-3.05 (m, 2H), 1.55 (s, 6H); MS: ESI (positive): 264 (M+H).
- Methyl triphenylphosphonium bromide (6.3g, 17.6 mmol) was dissolved in 150 mL THF and cooled to 0° C. KHMDS (3.2g, 16.2 mmol) was added portionwise and the reaction was stirred for 1 A hour.
- the product of Example 1, step (c) (3.0g, 12.5 mmol) was added as a solution in 25 mL THF. The reaction was warmed to room temperature and stirred for 1 hour. The mixture was concentrated and the title product was purified by silica gel chromatography (0% to 40% EtOAc in hexanes) to give 2.6g of the sub-title compound.
- step (a) The product of step (a) was dissolved in 100 mL EtOH and treated with 0.5g of 10% Pd/C (wet, Degussa type ElOl). After stirring rapidly for 14 hours under an atmosphere of hydrogen, the reaction was filtered through celite and concentrated to give 2.3g of the subtitle compound as a clear oil. MS: ESI (positive): 240 (M+H).
- step (b) The product of step (b) (5.6g, 23.4 mmol) was dissolved in 250 mL cyclohexane and treated with NaHCO 3 (11.8g, 140 mmol). Bromine (3.6 mL, 70.3 mmol) was added slowly and the reaction was stirred for 1 A hour at room temperature after which time it was quenched with Na 2 S0 3 (180 mL of 5% aqueous). After stirring rapidly for 15 minutes. EtOAc was added
- step (c) The product of step (c) (enantiomer 2, 60 mg, 0.15 mmol) was dissolved in 2 mL dry THF and cooled to -78° C. Butyl lithium (0.11 mL of 1.6 M) was added and the sol ⁇ tion was stirred for 5 minutes then quenched with trifluoroacetyl 2,2,2-trifluoroethanol (50 uL, 0.24 mmol). After warming to room temperature, the reaction was filtered through a pad of silica gel (washing with EtOAc). The filtrate was evaporated to give 62 mg of the sub-title compound that was used without further purification.
- step (d) The product of step (d) (62 mg, 0.15 mmol) was dissolved in 2 mL EtOH and treated with NaBH 4 (0.6 mmol). After 15 minutes, the reaction was quenched with HOAc till the bubbling ceased and diluted with water. The product was extracted into DCM (2x5 mL), dried over MgSO 4 and concentrated. The crude residue was dissolved in 3 mL of 1 :1 HOAcxoncentrated HCl and treated with SnCl 2 (225 mg, 1 mmol). The reaction was heated to 70° C for 1 hour and then stirred at room temperature for 3 days. The reaction was diluted with EtOAc (10 mL) and washed with water (2x) and IM NaOH (2x). The organic solution was concentrated to dryness giving 40 mg of the sub-title compound that was used without further purification.
- step (e) The product of step (e) (40 mg, 0.10 mmol) was deprotected and purified according to the procedure described for Example 2, step (c).
- 1 H NMR (CD3OD) ⁇ 3.75 (q, J 10.4 Hz, 2H),
- step (c) enantiomer 2, 0.75g, 1.9 mmol
- Zn (0.25g, 3.8 mmol
- the product of Example 9, step (c) (enantiomer 2, 0.75g, 1.9 mmol) and Zn (0.25g, 3.8 mmol) were heated to reflux in 20 mL each water and HOAc. After 1 A hour, the reaction was cooled, diluted with EtOAc, and washed 2x with water. The organic layer was dried over MgSO 4 and concentrated to give 490 mg of the sub-title compound as an oil.
- step (a) The product of step (a) (150 mg, 0.47 mmol) was dissolved in 3 mL dioxane and treated with Me 2 Zn (0.47 mL of 2M in toluene) and Pd(ddf) 2 Cl 2 (1 1 mg, 0.014 mmol). After heating to 100° C for 3 hours, the reaction was quenched with water and filtered. The filtrate was partitioned between EtOAc and water (7 mL each). The organic layer was dried over MgSO 4 and concentrated to give 92 mg of the sub-title compound, which was used without further purification.
- step (b) The product of step (b) (92 mg, 0.36 mmol) was dissolved in 4 mL of 1 : 1 HOAC/CHCI 3 and treated with NBS (67 mg, 0.38 mmol). After stirring for 1 A hour, the reaction was diluted with EtOAc (70 mL) was washed with water (3x30 mL) and IM NaOH (2x30 mL).
- EtOAc 70 mL
- IM NaOH 2x30 mL
- step (c) The product of step (c) (60 mg, 0.18 mmol) was dissolved in 2 mL NMP and treated with NaSEt (45 mg, 0.54 mmol), KI (3 mg, 0.018 mmol), and CuO (7 mg, 0.09 mmol). After heating for 2 days at 120° C, an additional quantity of NaSEt (45 mg, 0.54 mmol), KI (3 mg, 0.018 mmol), and CuO (7 mg, 0.09 mmol) was added and heating was continued for 3 days.
- step (d) The product of step (d) (55 mg, 0.17 mmol) was dissolved in 2 mL HOAc and treated with 25 30% H 2 O 2 (220 ML, 2 mmol). After heating to 70° C for 1 hour, the reaction was diluted with water (8 mL) and the product was extracted into DCM (3x5 mL). The crude reside was dissolved in 4 mL of 1 :1 EtOH:40% KOH (aq) and heated to 100° C for 14 hours. The reaction was cooled and diluted with water. The product was extracted into DCM (2x5 mL) and purified by preparative HPLC-MS to give the title compound.
- step (b) The product of Example 9, step (b) (racemic, 80 mg, 0.35 mmol) was dissolved in 2 mL of 1 : 1 CHCI 3 /HOAC. N-Bromo-succinamide (62 mg, 0.35 mmol) was added and the reaction was stirred for 15 minutes. Concentration and purification by silica gel chromatography gave the sub-title compound as a yellow oil.
- step (a) The product of step (a) (55 mg, 0.17 mmol) was dissolved in THF (2 mL) and cooled to -78° C. Butyl lithium (0.16 mL of 1.6M) was added and the reaction was stirred for 15 minutes. After quenching with trimethyl acetyl chloride (41 uL, 0.34 mmol) the reaction was warmed to room temperature and concentrated. The crude residue was dissolved in 2 mL EtOH arid treated with 2 mL of 40% KOH (aq). After heating overnight to 100° C, the reaction was cooled and diluted with water. The product was extracted into DCM (2x) and the title compound was purified by preparative HPLC-MS.
- step (c) enantiomer 2, 200 mg, 0.50 mmol was dissolved in 5 10 mL THF and cooled to -78° C. Butyl lithium (0.31 mL of 1.6M) was added and the reaction was stirred for 15 minutes. After quenching with trimethyl acetyl chloride (90 uL, 0.75 mmol) the reaction was warmed to room temperature and concentrated. Purification of the crude residue by silica gel chromatography gave 170 mg of the sub-title compound.
- step (a) The product of step (a) (56 mg, 0.14) was stirred in 2 mL CHCl 3 and treated with TMSI (57 uL, 0.42 mmol). After heating to 70° C for lhour, another 0.42 mmol of TMSI was added 20 and the heating was continued for 1 hour. The reaction was cooled and partitioned between IM NaOH and CHCl 3 . The organic layer was concentrated and purified by HPLC-MS gave the title compound.
- step (a) The product of step (a) (2.47 g, 8 mmol) was stirred with 2.Og of 10% Pd/C (wet, Degussa grade ElOl) in methanol (8 mL) under H 2 (latm) for 72 hours. The reaction was filtered over celite and concentrated to dryness to give the sub-title compound as an oil, which was used without further purification. MS: ESI (positive): 312 (M+H).
- step (b) The product of step (b) (2.47 g, 8 mmol) was stirred in ethanol (60 mL) with IM NaOH (30 25 mL) at ambient temperature overnight. The reaction acidified with IM HCl and partitioned between DCM and water. The organic layer was washed with water, dried over MgSO 4 , and concentrated to dryness to give the 2.13g of the sub-title compound as a yellow oil. MS: ESI (positive): 284 (M+H).
- the sub-title compound was prepared by the method of Example 1, step (h) using the product from step (e) and was used in crude form without purification.
- Trifluoroacetic anhydride 750 ⁇ L, 5.36 mmol
- AlCl 3 600 mg, 4.51 mmol
- the sub-title compound was prepared by the method of example 5, step (a) using the product from step (a) and was used in crude form without purification.
- step (c) The product of Example 1, step (c) (1.06g, 4.4 mmol) was dissolved in 20 mL THF and treated with LHMDS (5.3 mL of IM in THF). After stirring for 1 hour at room temperature, MeI (326 uL, 5.3 mmol) was added and the reaction was stirred 1 hour at room temperature. 5 The reaction was evaporated onto silica gel and purified by silica gel chromatography (10% to 30% EtOAc in hexanes) to give 175 mg of the sub-title compound.
- step (a) The product of step (a) (75 mg, 0.30 mmol) was dissolved in DCE (3 mL) and treated with ZnI 2 (143 mg, 0.45 mmol) followed by NaCNBH3 (132 mg, 2.1 mmol). After stirring for 3 days at room temperature, the reaction was filtered and the filtrate was washed with DCM and discarded. The organic washes were combined, washed with water (2 x 10 mL), and dried over MgSO ⁇ Concentration gave 22 mg of a mixture of the sub-title compound and the 5 alcohol corresponding to mono-reduction of the ketone starting material. The mixture was carried directly into the subsequent steps.
- step (b) The product of step (b) (22 mg, 0.092 mmol) was dissolved in 2 mL of 1 : 1 HOAc:CHCl 3 and treated with NBS (25 mg, 0.14 mmol). After stirring for Y 2 hour, the reaction was concentrated to dryness and treated with 2 mL EtOH and 2 mL of 40% KOH. After heating to 100° C for 24 hours and then to 120° C for an additional 24 hours, the reaction was cooled, diluted with water, and extracted 3x with DCM. Purification by preparative HPLC-MS gave 5 the title compound. MS: ESI (positive): 248 (M+H). P rC ⁇ T I//UUSO2Z0U0U7///0U0U1 I3O4H-*;
- step (c) enantiomer 2, 1.Og 3 . 2.51 mmol
- EtOH aqueous ethanol
- Ig of Pd/C wet, Degussa grade ElOl
- the reaction was filtered through celite and concentrated to give 600 mg of the sub-title compound.
- step (a) The product of step (a) was dissolved in DCE (100 mL) and treated with AlCl 3 (3.3 g, 25 mmol) followed by triflouroacetic anhydride (3.5 mL, 25 mmol). After heating to 80° C for 2 hours, another portion OfAlCl 3 and trifluoroacetic anhydride was added (25 mmol each). After stirring for 1 hour at 80° C, the reaction was quenched over ice and carefully made basic by the cautious addition OfEt 3 N. The thick precipitate was filtered and discarded. The resulting mixture was extracted with DCM (3x100 mL). After concentration of the extracts to approximately 100 mL, 0.5 mL each of ethyl chloroformate and DIEA was added. After stirring for X A hour, the solution was washed with water, concentrated and purified by silica gel chromatography (25% EtO Ac/Hex) to give 460 mg of the sub-litle compound.
- step (b) The product of step (b) (460 mg, 1.37 mrnol) was dissolved in 25 mL THF and treated with triethylphosphonacetate (550 uL, 2.74 mmol) and LHMDS (2.2 mL of IM in THF). After • stirring for 15 minutes, the reaction was quenched with water (100 mL) and the product was extracted into DCM (2x50mL). After drying the extracts over MgSO4 and concentration, the crude residue was dissolved in EtOH (50 mL) and treated with 300 mg Pd/C (10%, wet, Degussa grade ElOl). After stirring under 1 atm of H 2 for 2 hours, the reaction was filtered through celite and treated with IM NaOH (10 mL).
- step (c) 500 mg, 1.3 mmol was dissolved in DCM (20 mL) and treated with DMF (50 uL) followed by oxalyl chloride (240 uL, 1.45 mmol). After stirring for 1 A hour, an additional quantity of oxalyl chloride (240 uL) was added. After stirring for an additional 1 hour, the reaction was concentrated to dryness and the residue was re-dissolved in DCM (5 mL). AICI 3 (360 mg, 2.7 mmol) was added and the reaction was stirred for 1 hour at room temperature. The reaction was quenched with ice water (10 mL) and the product was extracted into DCM/EtOH (4:1, 2x30 mL).
- step (d) mixture of diastereomers, 234 mg, 1.54 mmol was dissolved in 5 mL EtOH and treated with NaBH 4 (117 mg, 3.1 mmol). After stirring for 15 minutes, the reaction was quenched with HOAc and diluted with water (10 mL). The product was extracted into DCM (2x25 mL) and the extracts were concentrated to dryness. The crude residue was dissolved in HOAc (8 mL) and concentrated HCl (4 mL) and treated with SnCl 2 r ⁇ mw
- step (e) The product of step (e) (diastereomer 1 and diastereomer 2, separately, 20 mg, 0.06 mmol) was dissolved in 3 mL CHCl 3 and treated with TMSI (40 uL, 0.29 mmol). After heating to 70° C overnight, the reaction was quenched with MeOH (5 mL) and 1 M NaOH (2 mL). The title compound was extracted into CHCl 3 (2x10 mL) and subsequently purified by preparative HPLC-MS.
- Diastereomer 1 1 H NMR (CD 3 OD) ⁇ 4.01-3.90 (m, IH), 3.43-2.95 (m, 8H), 2.87-2.64 (m, 3H), 2.56-2.44 (m, IH), 1.33-1.31 (m, 3H). MS: ESI (positive): 276 (M+H).
- Diastereomer 2 1 H NMR (CD 3 OD) ⁇ 4.01-3.90 (m, IH) 5 3.42-2.94 (m, 8H), 2.84-2.64 (m, 3H), 2.55-2.45 (m, IH), 1.32-1.29 (m, 3H). MS: ESI (positive): 276 (M+H).
- Example 28 The original "Examples” referred to Example 28, which is clearly a typo since there are only 19 example. We think it should be Example 16, but we are not 100% sure.
- step (f) The bismesylate dioxane solution generated in step (f) was transferred to a 3 -neck reaction flask equipped with a dropping funnel and condenser. Anhydrous potassium carbonate (4.93 g, 35.7 mmol) was added and the contents were heated to reflux. Next, a solution of benzylam ⁇ ne (2.71g, 25.3 mmol) in anhydrous dioxane (27 ml) was added dropwise over 45 minutes and heating was continued for 16 hours. The salts were filtered off and the solvent was concentrated. The crude was purified by silica gel chromatography
- step (g) The product from step (g) (109 mg, 0.35 mmol) in anhydrous THF (5 ml) was cooled to -78°C under a nitrogen atmosphere and treated with 1.6 M n-butyi lithium in hexane (0.25 ml). The reaction mixture was stirred at -78 0 C for 5 minutes followed by the addition of trimethyl acetyl chloride (0.22 ml, 1.75 mmol). After 10 additional minutes at -78°C, the reaction mixture was allowed to warm to room temperature. The reaction was quenched with 10 saturated NaHCO 3 (20 ml) and extracted with ethyl acetate (3 x 20 ml).
- HEK 293 EBNA expressing the human 5HT 20 receptor (VNV Isoform) (Burns et al., NATURE 387:30308, 1997 Fitzgerald et al., NEUROPSYCHO-PHARMACOLOGY 21 : 825-905, 1999) were grown in DMEM containing 10% dialysed FBS, 9 ⁇ g/ml blasticid ⁇ n at 37°C in 5% CO 2 atmosphere.
- 5 HEK 293 EBNA cells expressing human 5HT 2c receptor (2x10 4 /well) were seeded in black 384-well collagen coated plates and incubated overnight at 37 0 C in a 5% CO2/95% atmosphere. After removing medium, cells were treated with HBSS buffer (137 mM NaCl, 5.4 raM KCl, 5.5 mM Glucose, 20 mM Hepes, pH 7.5, 2.1 mM MgCl 2 , 0.3 mM CaCl 2, 0.02mM MgSO 4 , 3.OmM NaHCO 3 , and 0.64mM KH 2 PO 4 ) containing the Calcium3
- HBSS buffer 137 mM NaCl, 5.4 raM KCl, 5.5 mM Glucose, 20 mM Hepes, pH 7.5, 2.1 mM MgCl 2 , 0.3 mM CaCl 2, 0.02mM MgSO 4 , 3.O
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Obesity (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Hematology (AREA)
- Epidemiology (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Anesthesiology (AREA)
- Addiction (AREA)
- Endocrinology (AREA)
- Agronomy & Crop Science (AREA)
- Pest Control & Pesticides (AREA)
- Plant Pathology (AREA)
- Dentistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Abstract
Description


































Claims

Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0707183-3A BRPI0707183A2 (en) | 2006-01-19 | 2007-01-17 | compound, pharmaceutical composition, and method of treating a disease, disorder and / or condition in a patient |
| CA2636992A CA2636992C (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof |
| KR1020147012091A KR101503681B1 (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof |
| MX2008009178A MX2008009178A (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof. |
| JP2008551392A JP5188986B2 (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolylazepines as ligands for serotonin 5-HT2C receptors and uses thereof |
| NZ569554A NZ569554A (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-HT2C receptor ligands and uses thereof |
| AU2007207508A AU2007207508B2 (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-HT2c receptor ligands and uses thereof |
| EP07718077.6A EP1981337B1 (en) | 2006-01-19 | 2007-01-17 | Thiophenyl azepines as serotonin 5-ht2c receptor ligands and uses thereof |
| IL192744A IL192744A0 (en) | 2006-01-19 | 2008-07-10 | Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof |
| NO20083481A NO20083481L (en) | 2006-01-19 | 2008-08-12 | Thiophenyl and pyrrolylazepines as serotonin 5-HT 2C receptor ligands and uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US76024006P | 2006-01-19 | 2006-01-19 | |
| US60/760,240 | 2006-01-19 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007084622A2 true WO2007084622A2 (en) | 2007-07-26 |
| WO2007084622A3 WO2007084622A3 (en) | 2007-12-21 |
Family
ID=38288237
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/001345 Ceased WO2007084622A2 (en) | 2006-01-19 | 2007-01-17 | Thiophenyl and pyrrolyl azepines as serotonin 5-ht2c receptor ligands and uses thereof |
Country Status (17)
| Country | Link |
|---|---|
| US (3) | US7893051B2 (en) |
| EP (1) | EP1981337B1 (en) |
| JP (1) | JP5188986B2 (en) |
| KR (2) | KR20080107375A (en) |
| CN (1) | CN101404882A (en) |
| AR (1) | AR059076A1 (en) |
| AU (1) | AU2007207508B2 (en) |
| BR (1) | BRPI0707183A2 (en) |
| CA (1) | CA2636992C (en) |
| IL (1) | IL192744A0 (en) |
| MX (1) | MX2008009178A (en) |
| NO (1) | NO20083481L (en) |
| NZ (1) | NZ569554A (en) |
| RU (1) | RU2434872C2 (en) |
| TW (1) | TWI402272B (en) |
| WO (1) | WO2007084622A2 (en) |
| ZA (1) | ZA200805915B (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009063992A1 (en) | 2007-11-15 | 2009-05-22 | Takeda Pharmaceutical Company Limited | Condensed pyridine derivative and use thereof |
| WO2011071136A1 (en) | 2009-12-11 | 2011-06-16 | アステラス製薬株式会社 | Therapeutic agent for fibromyalgia |
| WO2012030953A1 (en) | 2010-09-01 | 2012-03-08 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists in the treatment of disorders ameliorated by reduction of norepinephrine level |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2013133325A1 (en) | 2012-03-06 | 2013-09-12 | 武田薬品工業株式会社 | Tricyclic compound |
| EP2571888B1 (en) * | 2010-05-18 | 2014-12-31 | Corning Incorporated | Methods of making fused thiophenes |
| WO2015066344A1 (en) | 2013-11-01 | 2015-05-07 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists and compositions and methods of use |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9309262B2 (en) | 2013-03-13 | 2016-04-12 | Abt Holding Company | Thienylindole azepines as serotonin 5-HT2C receptor ligands and uses thereof |
Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4414225A (en) | 1981-02-18 | 1983-11-08 | Dr. Karl Thomae Gesellschaft Mit Beschrankter Haftung | Azepine derivatives and their anti-thrombotic compositions and methods |
| US4575504A (en) | 1982-08-18 | 1986-03-11 | Dr. Karl Thomae Gmbh | Thienothiazole derivatives |
| US4904653A (en) | 1988-01-15 | 1990-02-27 | Lilly Industries Limited | Thieno[2,3-O]azepine compounds and CNS affecting use thereof |
| WO1993013105A1 (en) | 1991-12-26 | 1993-07-08 | Yoshitomi Pharmaceutical Industries, Ltd. | Condensed thiophene compound and pharmaceutical use thereof |
| US5258378A (en) | 1990-11-28 | 1993-11-02 | Lilly Industries Limited | Pyrroloazepine compounds useful as dopaminergic agents |
| WO1996011201A1 (en) | 1994-10-11 | 1996-04-18 | Takeda Chemical Industries, Ltd. | Substituted heterobicyclic alkyl amines and their use as squalene oxide cyclase inhibitors |
| US5532240A (en) | 1991-12-26 | 1996-07-02 | Yoshitomi Pharmaceutical Industries, Ltd. | Condensed thiophene compound and pharmaceutical use thereof |
| WO2004024065A2 (en) | 2002-09-12 | 2004-03-25 | Merck & Co., Inc. | Substituted bicyclic thiophene derivatives, compostions containing such compounds and methods of use |
| WO2005003096A1 (en) | 2003-06-17 | 2005-01-13 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives useful for the treatment of 5ht2c receptor associated diseases |
| WO2005019179A2 (en) | 2003-06-17 | 2005-03-03 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives useful for the treatment of 5ht2c receptor associated diseases |
| WO2005040169A2 (en) | 2003-09-17 | 2005-05-06 | Janssen Pharmaceutica, N.V. | Fused heterocyclic compounds as serotonin receptor modulators |
| WO2005042490A1 (en) | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| WO2005042491A1 (en) | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| WO2006004931A2 (en) | 2004-06-30 | 2006-01-12 | Athersys, Inc. | Substituted azepine derivatives as serotonin receptor modulators |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3652588A (en) * | 1969-10-23 | 1972-03-28 | Upjohn Co | 6-alkyl-1 2 3 4 5 6-hexahydroazepino(4 5-b)indoles |
| CH649553A5 (en) * | 1982-05-06 | 1985-05-31 | Sandoz Ag | AZEPINOINDOLS AND METHOD FOR THE PRODUCTION THEREOF. |
| PE20010052A1 (en) * | 1999-04-23 | 2001-01-27 | Upjohn Co | AZEPININDOL TETRACYCLIC COMPOUNDS AS AGONISTS OR ANTAGONISTS OF THE 5-HT RECEPTOR |
| US6407092B1 (en) * | 1999-04-23 | 2002-06-18 | Pharmacia & Upjohn Company | Tetracyclic azepinoindole compounds |
| MXPA01012969A (en) * | 1999-06-15 | 2003-10-14 | Bristol Myers Squibb Pharma Co | Substituted heterocycle fused gamma-carbolines. |
| AU2001292898A1 (en) * | 2000-09-20 | 2002-04-02 | Brad A. Acker | Substituted azepino[4,5b)indole derivatives |
| PT1345609E (en) * | 2000-12-22 | 2005-08-31 | Hoffmann La Roche | TETRA-HYDRO- (BENZO OR TIENO) -AZEPINE-PYRAZINE DERIVATIVES AS MGLUR ANTAGONISTS 1 |
-
2007
- 2007-01-17 BR BRPI0707183-3A patent/BRPI0707183A2/en not_active IP Right Cessation
- 2007-01-17 AU AU2007207508A patent/AU2007207508B2/en not_active Ceased
- 2007-01-17 EP EP07718077.6A patent/EP1981337B1/en not_active Not-in-force
- 2007-01-17 RU RU2008133980/04A patent/RU2434872C2/en not_active IP Right Cessation
- 2007-01-17 KR KR1020087020239A patent/KR20080107375A/en not_active Ceased
- 2007-01-17 KR KR1020147012091A patent/KR101503681B1/en not_active Expired - Fee Related
- 2007-01-17 WO PCT/US2007/001345 patent/WO2007084622A2/en not_active Ceased
- 2007-01-17 CN CNA2007800097927A patent/CN101404882A/en active Pending
- 2007-01-17 CA CA2636992A patent/CA2636992C/en not_active Expired - Fee Related
- 2007-01-17 NZ NZ569554A patent/NZ569554A/en not_active IP Right Cessation
- 2007-01-17 JP JP2008551392A patent/JP5188986B2/en not_active Expired - Fee Related
- 2007-01-17 MX MX2008009178A patent/MX2008009178A/en active IP Right Grant
- 2007-01-17 US US11/654,979 patent/US7893051B2/en not_active Expired - Fee Related
- 2007-01-18 AR ARP070100226A patent/AR059076A1/en unknown
- 2007-01-19 TW TW096102154A patent/TWI402272B/en not_active IP Right Cessation
-
2008
- 2008-07-07 ZA ZA200805915A patent/ZA200805915B/en unknown
- 2008-07-10 IL IL192744A patent/IL192744A0/en unknown
- 2008-08-12 NO NO20083481A patent/NO20083481L/en not_active Application Discontinuation
-
2011
- 2011-01-12 US US13/005,395 patent/US8716276B2/en not_active Expired - Fee Related
-
2014
- 2014-03-19 US US14/219,304 patent/US20140206672A1/en not_active Abandoned
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4414225A (en) | 1981-02-18 | 1983-11-08 | Dr. Karl Thomae Gesellschaft Mit Beschrankter Haftung | Azepine derivatives and their anti-thrombotic compositions and methods |
| US4575504A (en) | 1982-08-18 | 1986-03-11 | Dr. Karl Thomae Gmbh | Thienothiazole derivatives |
| US4904653A (en) | 1988-01-15 | 1990-02-27 | Lilly Industries Limited | Thieno[2,3-O]azepine compounds and CNS affecting use thereof |
| US5258378A (en) | 1990-11-28 | 1993-11-02 | Lilly Industries Limited | Pyrroloazepine compounds useful as dopaminergic agents |
| US5532240A (en) | 1991-12-26 | 1996-07-02 | Yoshitomi Pharmaceutical Industries, Ltd. | Condensed thiophene compound and pharmaceutical use thereof |
| WO1993013105A1 (en) | 1991-12-26 | 1993-07-08 | Yoshitomi Pharmaceutical Industries, Ltd. | Condensed thiophene compound and pharmaceutical use thereof |
| US5691330A (en) | 1991-12-26 | 1997-11-25 | Yoshitomi Pharmaceutical Industries, Ltd. | Condensed thiophene compound and pharmaceutical use thereof |
| WO1996011201A1 (en) | 1994-10-11 | 1996-04-18 | Takeda Chemical Industries, Ltd. | Substituted heterobicyclic alkyl amines and their use as squalene oxide cyclase inhibitors |
| WO2004024065A2 (en) | 2002-09-12 | 2004-03-25 | Merck & Co., Inc. | Substituted bicyclic thiophene derivatives, compostions containing such compounds and methods of use |
| WO2005003096A1 (en) | 2003-06-17 | 2005-01-13 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives useful for the treatment of 5ht2c receptor associated diseases |
| WO2005019179A2 (en) | 2003-06-17 | 2005-03-03 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives useful for the treatment of 5ht2c receptor associated diseases |
| WO2005040169A2 (en) | 2003-09-17 | 2005-05-06 | Janssen Pharmaceutica, N.V. | Fused heterocyclic compounds as serotonin receptor modulators |
| WO2005042490A1 (en) | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| WO2005042491A1 (en) | 2003-10-22 | 2005-05-12 | Arena Pharmaceuticals, Inc. | Benzazepine derivatives and methods of prophylaxis or treatment of 5ht2c receptor associated diseases |
| WO2006004931A2 (en) | 2004-06-30 | 2006-01-12 | Athersys, Inc. | Substituted azepine derivatives as serotonin receptor modulators |
Non-Patent Citations (12)
| Title |
|---|
| A. DEKEYNE ET AL., NEUROPHANNACOLOGY, vol. 38, 1999, pages 415 - 423 |
| BURNS ET AL., NATURE, vol. 387, 1997, pages 30308 |
| D. HOYER ET AL., PHANNACOL. REV., vol. 46, 1994, pages 157 - 203 |
| F. JENCK ET AL., EXP. OPIN. INVEST. DRUGS, vol. 7, 1998, pages 1587 - 1599 |
| FITZGERALD ET AL., NEUROPSYCHO-PHARMACOLOGY, vol. 21, 1999, pages 825 - 905 |
| G. BAXTER ET AL., TRENDS IN PHARMACOL. SCI., vol. 16, 1995, pages 105 - 110 |
| G.A. KENNETT, DRUGS, vol. 1, no. 4, 1998, pages 456 - 470 |
| GLENNON ET AL., NEUROSCIENCE AND BEHAVIORAL REVIEWS, vol. 14, 1990, pages 35 |
| J.R. MARTIN ET AL., THE JOURNAL OFPHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 286, 1998, pages 913 - 924 |
| M. BOS ET AL., J. MED. CHEM, vol. 40, 1997, pages 2762 - 2769 |
| M. BROMIDGE ET AL., I . MED. CHEM., vol. 41, 1998, pages 1598 - 1612 |
| See also references of EP1981337A4 |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2789338A2 (en) | 2007-11-15 | 2014-10-15 | Takeda Pharmaceutical Company Limited | Condensed pyridine derivate and use thereof |
| WO2009063992A1 (en) | 2007-11-15 | 2009-05-22 | Takeda Pharmaceutical Company Limited | Condensed pyridine derivative and use thereof |
| WO2011071136A1 (en) | 2009-12-11 | 2011-06-16 | アステラス製薬株式会社 | Therapeutic agent for fibromyalgia |
| EP2571888B1 (en) * | 2010-05-18 | 2014-12-31 | Corning Incorporated | Methods of making fused thiophenes |
| WO2012030953A1 (en) | 2010-09-01 | 2012-03-08 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists in the treatment of disorders ameliorated by reduction of norepinephrine level |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013133325A1 (en) | 2012-03-06 | 2013-09-12 | 武田薬品工業株式会社 | Tricyclic compound |
| US9556200B2 (en) | 2012-03-06 | 2017-01-31 | Takeda Pharmaceutical Company Limited | Tricyclic compound |
| WO2015066344A1 (en) | 2013-11-01 | 2015-05-07 | Arena Pharmaceuticals, Inc. | 5-ht2c receptor agonists and compositions and methods of use |
Also Published As
| Publication number | Publication date |
|---|---|
| JP5188986B2 (en) | 2013-04-24 |
| BRPI0707183A2 (en) | 2011-04-26 |
| EP1981337B1 (en) | 2015-09-02 |
| CN101404882A (en) | 2009-04-08 |
| AR059076A1 (en) | 2008-03-12 |
| US7893051B2 (en) | 2011-02-22 |
| NO20083481L (en) | 2008-08-12 |
| JP2009523809A (en) | 2009-06-25 |
| AU2007207508A1 (en) | 2007-07-26 |
| KR20140062185A (en) | 2014-05-22 |
| TWI402272B (en) | 2013-07-21 |
| ZA200805915B (en) | 2009-04-29 |
| CA2636992A1 (en) | 2007-07-26 |
| NZ569554A (en) | 2011-10-28 |
| EP1981337A2 (en) | 2008-10-22 |
| IL192744A0 (en) | 2009-02-11 |
| TW200738732A (en) | 2007-10-16 |
| MX2008009178A (en) | 2008-12-05 |
| US8716276B2 (en) | 2014-05-06 |
| RU2008133980A (en) | 2010-02-27 |
| EP1981337A4 (en) | 2010-08-11 |
| US20140206672A1 (en) | 2014-07-24 |
| RU2434872C2 (en) | 2011-11-27 |
| AU2007207508B2 (en) | 2011-08-18 |
| US20110112072A1 (en) | 2011-05-12 |
| US20070191342A1 (en) | 2007-08-16 |
| CA2636992C (en) | 2015-10-06 |
| WO2007084622A3 (en) | 2007-12-21 |
| KR101503681B1 (en) | 2015-03-19 |
| KR20080107375A (en) | 2008-12-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2007207508B2 (en) | Thiophenyl and pyrrolyl azepines as serotonin 5-HT2c receptor ligands and uses thereof | |
| US20100190772A1 (en) | Substituted Azepine Derivatives As Serotonin Receptor Modulators | |
| US7981919B2 (en) | Tricyclic heteroaryl piperazines, pyrrolidines and azetidines as serotonin receptor modulators | |
| NZ552806A (en) | Tricyclic indeno-pyrrole derivatives as serotonin receptor modulators | |
| MXPA06013392A (en) | Substituted azepine derivatives as serotonin receptor modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 569554 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007207508 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 192744 Country of ref document: IL Ref document number: 12008501638 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2636992 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2864/KOLNP/2008 Country of ref document: IN Ref document number: 2007718077 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/009178 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008551392 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007207508 Country of ref document: AU Date of ref document: 20070117 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2008133980 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087020239 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780009792.7 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: PI0707183 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080721 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020147012091 Country of ref document: KR |