WO2007105253A2 - PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES - Google Patents
PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES Download PDFInfo
- Publication number
- WO2007105253A2 WO2007105253A2 PCT/IT2007/000185 IT2007000185W WO2007105253A2 WO 2007105253 A2 WO2007105253 A2 WO 2007105253A2 IT 2007000185 W IT2007000185 W IT 2007000185W WO 2007105253 A2 WO2007105253 A2 WO 2007105253A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- formula
- process according
- cephem
- oxa
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CSN=C([C@]1N2C(*)=C(*)C*1)C2=O Chemical compound CSN=C([C@]1N2C(*)=C(*)C*1)C2=O 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/02—Preparation
- C07D501/04—Preparation from compounds already containing the ring or condensed ring systems, e.g. by dehydrogenation of the ring, by introduction, elimination or modification of substituents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/02—Preparation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/18—7-Aminocephalosporanic or substituted 7-aminocephalosporanic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D503/00—Heterocyclic compounds containing 4-oxa-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxapenicillins, clavulanic acid derivatives; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D503/02—Preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D503/00—Heterocyclic compounds containing 4-oxa-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxapenicillins, clavulanic acid derivatives; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D503/10—Heterocyclic compounds containing 4-oxa-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxapenicillins, clavulanic acid derivatives; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D505/00—Heterocyclic compounds containing 5-oxa-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxacephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D505/02—Preparation
- C07D505/06—Preparation from compounds already containing the ring or condensed ring systems, e.g. by dehydrogenation of the ring, by introduction, elimination or modification of substituents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D505/00—Heterocyclic compounds containing 5-oxa-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxacephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D505/10—Heterocyclic compounds containing 5-oxa-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxacephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2
- C07D505/12—Heterocyclic compounds containing 5-oxa-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxacephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 substituted in position 7
- C07D505/14—Heterocyclic compounds containing 5-oxa-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. oxacephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring with a carbon atom having three bonds to hetero atoms with at the most one bond to halogen, e.g. an ester or nitrile radical, directly attached in position 2 substituted in position 7 with hetero atoms directly attached in position 7
- C07D505/16—Nitrogen atoms
Definitions
- the present invention concerns a process for the preparation of (1-oxa- or 1- thia-) 3-cephem derivatives which are useful intermediates in the preparation of (1-oxa- or l-thia-)cephalosporins. More particularly, the invention refers to a process for the preparation of a carboxy protected 7 ⁇ -amino-7 ⁇ -methoxy-3-[l- (alkyl- or ⁇ -hydroxy .
- the above compounds flomoxef and cefinetazole are 7 ⁇ -acylamino-7 ⁇ - methoxy-cephalosporin-type antibiotics used as antibacterial drugs.
- Flomoxef is an oxacephem, described in US 4,532,233, which is prepared by a multistep process in which the key intermediate is a carboxy-protected 7 ⁇ - amino-7 ⁇ -methoxy-3-[l-(2-hydroxyethyl)-lH-tetrazol-5-yl]thiomethyl-l-dethia- l-oxa-3-cephem-4-carboxylic acid.
- This compound is converted into flomoxef by protection of the primary hydroxyl group of the hydroxyethyl radical bound to the tetrazole ring, subsequent condensation with activated 2- (difluoromethylthio)acetic acid and removal of the hydroxy- and of the carboxy- protecting groups.
- Cefinetazole is a cephem, described in GB 1449420, which is prepared by reaction of the corresponding carboxy-protected 7 ⁇ -amino-7 ⁇ -methoxy-3-(l- methyl-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylic acid by condensation with activated 2-(cyanomethylthio)acetic acid and removal of the the carboxy- protecting groups.
- Each of flomoxef and cefinetazole is a 7 ⁇ -(substituted-thio)acetamido-7 ⁇ - methoxy-3-(l -substituted-l/f-tetrazol-5-yl)thiomethyl-(l -oxa- or 1 -thia-)3- cephem-4-carboxylic acid.
- an enantiomerically pure, carboxy-protected 7 ⁇ -amino-7 ⁇ - methoxy-3-(l-substituted-lH-tetrazol-5-yl)thiotnethyl-(l-oxa- or l-thia-)3- cephem-4-carboxylic acid may be prepared by reaction of a carboxy-protected 7- benzamido-3-(l-substituted-li7-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3- cephem-4-carboxylic acid with lithium methoxide in the presence of t-butyl hypochlorite (Kleemann et al.
- 7 ⁇ -methoxy-3-(l-substituted-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylic acid may be prepared by reaction of corresponding 7-methyl- or 7-(4-tolyl)- thioimino derivatives with methanol in the presence of triphenylphosphine and mercuric acetate in dichloromethane as described by E.M. Gordon et al. in J. Am. Chem. Soc. 1977, 99(16) 5504-5 and ibid. 1980, 102(5), 1690-1702.
- the presence of a mercuric salt in the reaction mixture renders the purification of the products difficult.
- the carboxy protected 7 ⁇ -amino-7 ⁇ -methoxy-3-[l-(alkyl- or ⁇ - hydroxyalkyl-) li- 7" -tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem-4- carboxylic acid thus obtained represents the key intermediate in the preparation of flomoxef (formula A) and of cefmetazole (formula B), or of their analogs, which are obtained by the classical condensation of carboxy protected 7 ⁇ -amino-7 ⁇ - methoxy-3-[l-(alkyl- or ⁇ -hydroxy alkyl-)lH-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem-4-caboxylic acid with the appropriately activated 2- (difluoromethylthio)acetic acid in the presence of silylating agents to obtain flomoxef after deprotection of the carb
- X represents oxygen or sulfur
- Y represents an alkyl group of from 1 to 3 carbon atoms or a ⁇ -hydroxyalkyl group of from 2 to 3 carbon atoms
- R° represents a carboxy-protecting group, or of a salt thereof, which comprises
- Y is an alkyl of from 1 to 3 carbon atoms or a ⁇ -hydroxyalkyl of from 2 to 3 carbon atoms, or with an alkaline metal salt thereof;
- step (c) sodium bicarbonate is preferably used as the base neutralizing aluminum trichloride.
- triarylphosphine or tri ⁇ -C ⁇ alkylphosphine designates phosphine wherein the three hydrogen atoms are replaced by an optionally substituted phenyl group such as phenyl or tolyl, by a monocyclic, aromatic heterocyclic group such as furyl or thienyl, or by a (C 1 -C 6 )Bl-CyI group such as rc-butyl.
- a 7-amino-3-chloromethyl-(l-oxa- or l-thia-)3-cephem-4-carboxylic acid esterified with an easily removable protecting group, especially in acidic medium, is used as starting material.
- Protective groups used according to the present invention and the methods for their removal are described, for example, by Theodora W. Greene, Peter G. V. Wuts in "Protective Groups in Organic Synthesis", III ed., John Wiley & Sons, Inc., 1999, pages 373-431.
- esters are 4-methoxybenzyl and diphenylmethyl (benzhydryl) esters which are easily removable with trifluoroacetic acid and anisole in dichloromethane or with aluminum trichloride and anisole in dichloromethane.
- step (a) the carboxy-protected 7-amino-3-chloromethyl-(l-oxa- or 1-thia- )3-cephem-4-carboxylic acid, as such or as the hydrochloride thereof, is reacted with a methyl- or aryl- sulfenyl chloride, in its turn separately prepared by reaction of the corresponding methyl- or aryl- disulfide with chlorine, in an inert organic solvent such as toluene or dichloromethane, at room temperature (20° ⁇ 30°C) and in the presence of hydrogen chloride acceptor such as, for example, 1,2-epoxypropane.
- a methyl- or aryl- sulfenyl chloride in its turn separately prepared by reaction of the corresponding methyl- or aryl- disulfide with chlorine, in an inert organic solvent such as toluene or dichloromethane, at room temperature (20° ⁇ 30°C) and in the presence of hydrogen chloride
- the 7-(alkyl- or aryl-)thioimino-3-chloromethyl-(l-oxa- or l-thia-)3- cephem of formula IV thus obtained is isolated according to conventional methods, for example by neutralization with a base such as an aqueous solution of sodium bicarbonate (NaHCO 3 ), removal of the salts and extraction using a suitable solvent such as dichloromethane.
- a base such as an aqueous solution of sodium bicarbonate (NaHCO 3 )
- step (b) the 7-(alkyl- or aryl- )thioimino-3-chloromethyl-(l-oxa- or 1- thia-)3-cephem of formula FV thus obtained is treated with a 1-alkyl- or l-( ⁇ - hydroxyalkyl)-lH-tetrazol-5-ylthiol of formula V, preferably in form of its sodium salt.
- the reaction is carried out at a temperature of 20° ⁇ 30°C in a biphasic system with an inert organic solvent, for example dichloromethane or toluene, and water in the presence of a quaternary ammonium salt such as tetra-r ⁇ - butylammonium bromide.
- 1 -Methyl- 1 H-tetrazol-5-ylthiol or 1 -(2-hydroxyethyl)- lH-tetrazol-5-ylthiol as a sodium salt thereof is preferably used as 1 -substituted- lH-tetrazol-5-ylthiol.
- Said sodium salt may be prepared according to known methods, for example by reaction of the l-substituted-lH-tetrazol-5-ylthiol with sodium 2-ethylhexanoate in acetone.
- step (c) the 7-(alkyl- or aryl-)thioimino-3-chloromethyl(l-oxa- or 1- thia)3-cephem of formula VI obtained at the end of step (b) is reacted with methanol in the presence of aluminum trichloride neutralized with a base and a triarylphosphine or ⁇ (Ci-C ⁇ alkylphosphine in an inert organic solvent such as dichloromethane or toluene.
- aluminum trichloride neutralized with a base and a triarylphosphine or ⁇ (Ci-C ⁇ alkylphosphine in an inert organic solvent such as dichloromethane or toluene.
- the methanol also contains sodium bicarbonate as the base neutralizing aluminum trichloride.
- the triarylphosphine or Ui(C 1 - C 6 )alkylphosphine is selected from the group consisting of triphenylphosphine, tri( ⁇ -tolyl)phosphine, tri(2-furyl)phosphine and tri( «-butyl)phosphine.
- a solution of aluminum trichloride in methanol previously prepared at a temperature of 10° ⁇ 15°C and neutralized with a base such as sodium bicarbonate, is added to a solution of the compound VI and of triphenylphosphine, tri(o-tolyl) phosphine, tri(2-furyl)phosphine or tri(r ⁇ -butyi) ⁇ hosphine in one of the above-mentioned solvents.
- the compound of formula I thus obtained is isolated by adding glacial acetic acid to the suspension obtained at the end of the reaction, subsequently separating of the phases, drying the organic phase and extracting the product of formula I with a solvent such as dichloromethane or toluene and optional addition of a salifying acid such as HCl in isopropanol, HBr in isopropanol, methanesulfonic acid, j?-toluene sulfonic acid or naphthalene-2- sulfonic acid to the organic solution.
- a solvent such as dichloromethane or toluene
- a salifying acid such as HCl in isopropanol, HBr in isopropanol, methanesulfonic acid, j?-toluene sulfonic acid or naphthalene-2- sulfonic acid
- alkaline metal methoxides such as lithium methoxide or sodium methoxide
- the compound of formula I is isolated as free base which may be purified according to known methods, for example by silica gel, an appropriate resin or by solution in a polar solvent such as dimethylacetamide and precipitation with an alcohol such as methanol or isopropanol.
- the compounds of formula II used as starting materials are known or may be easily prepared by treatment of the corresponding carboxy-protected 7- acylamido-3-chloromethyl-(l-oxa- or 1-thia-) 3-cephem-4-carboxylic acid by reaction with PCl 5 in the presence of pyridine, for example as described in DE
- the present invention provide a process as illustrated above, whereby, at the end of step (c), a compound of formula Y
- the present invention also provides a process as illustrated above, whereby, at the end of step (c), a compound of formula I"
- Y is ⁇ -hydroxyalkyl having from 2 to 3 carbon atoms, also in the presence of a silylating agent such as, for example, hexamethyldisilazane and/or trimethylchloro silane.
- a silylating agent such as, for example, hexamethyldisilazane and/or trimethylchloro silane.
- Y is methyl
- the diphenylmethyl or 4- methoxybenzyl ester of the 7 ⁇ -amino-7 ⁇ -methoxy-3-[l-(2-hydroxyethyl)-lH- tetrazol-5-yl]thiomethyl-l-dethia-l-oxa-3-cephem-4-carboxylic acid is treated with silylating agents, for example with hexamethyldisilazane and trimethylchlorosilane, then it is reacted with the 2-(difluoromethylthio)acetyl chloride (F 2 CH-S-CH 2 COCl) in the presence of pyridine in dichloromethane at a temperature of from -10° ⁇ - 25°C.
- silylating agents for example with hexamethyldisilazane and trimethylchlorosilane
- the product is isolated according to known techniques, for example by adding water to the reaction mixture, separating the phases, evaporating the organic one, taking up the residue, consisting of the diphenylmethyl or 4-methoxybenzyl ester of flomoxef, with dichloromethane and treating the solution thus obtained with trifluoroacetic acid and anisole.
- the flomoxef thus obtained, in a 98% purity, is isolated by adding water to the reaction mixture, separating the phases and recovering the product in acidic form by concentration of the organic phase or in form of the sodium or potassium salt thereof by treatment of the organic phase with sodium or potassium 2-ethylhexanoate.
- the sodium salt of flomoxef may be further purified by passing an aqueous solution at pH 5,8 ⁇ 6,2 through a column containing silica gel or a resin such as Amberlite ® XAD 1180, by eluting with deionized water or with a mixture of deionized water and of an alcohol, for example ethanol or isopropanol.
- the invention provides a process as illustrated above, whereby the compound of formula I" is further reacted and treated with activated 2-(cyanomethylthio)acetic acid and a compound of formula VII"
- the diphenylmethyl or 4- methoxy. benzyl 7 ⁇ -amino-7 ⁇ -methoxy-3-(l-methyl-lH " -tetrazol-5-yl) thiomethyl- 3-cephem-4-carboxylate is treated with 2-(cyanomethylthio)acetyl chloride (NCCH 2 SCH 2 COCI) in the presence of pyridine in dichloromethane at a temperature of from -10 to -25 0 C.
- NCH 2 SCH 2 COCI 2-(cyanomethylthio)acetyl chloride
- the product is isolated according to known methods, for example by adding water to the reaction mixture, separating the phases, evaporating the organic one, taking up the residue, consisting of the benzhydryl or 4-methoxybenzyl ester of cefmetazole, with dichloromethane and treating the solution thus obtained with trifluoroacetic acid and anisole or with aluminum trichloride and anisole.
- the cefmetazole thus obtained in a 99% purity is isolated by adding water to the reaction mixture, separating the phases and recovering the product in acidic form by concentration of the organic phase or as a sodium or potassium salt thereof by treatment of the organic phase with sodium or potassium 2-ethylhexanoate.
- X is oxygen
- Y is an alkyl group of from 1 to 3 carbon atoms or a ⁇ - hydroxyalkyl group of from 2 to 3 carbon atoms
- W is an alkyl having from 1 to 3 carbon atoms
- a benzyl group non-substituted or substituted with an alkyl having from 1 to 3 carbon atoms
- a phenyl group non-substituted or substituted with an alkyl having from 1 to 3 carbon atoms
- R° is a carboxy-protecting group.
- the alkyl substitution on the benzyl group is preferably in the benzene ring.
- Preferred compounds are those of formula VF wherein X' is oxygen, Y' is 2-hydroxyethyl, R° is benzhydryl or 4-methoxybenzyl and W is methyl, benzyl or j?-tolyl.
- X' is oxygen
- Y' is 2-hydroxyethyl
- R° is benzhydryl or 4-methoxybenzyl
- W is methyl, benzyl or j?-tolyl.
- step (b) To a solution of 11.5 g (0.026 m) of intermediate benzhydryl 7- methylthio imino-3 -chloromethyl- 1 -dethia- 1 -oxa-3 -cephem-4-carboxylate obtained in step (a) in 150 ml dichloromethane, a solution of 7 g (0.041 m) of sodium l-(2-hydroxyethyl)-li?-tetrazol-5-ylthiolate in 150 ml water is added at 2O 0 C. Then, 1.2 g of tetra- «-butyl ammonium bromide is added and the reaction mixture is stirred at a temperature of from 20°C to 25 0 C for 8 hours.
- step (c) To a solution of 51.4 g (0.093 m) of the intermediate benzhydryl 7- methylthio imino-3 - [ 1 -(2-hydroxyethyl)- lH-tetrazol-5 -yljthiomethyl- 1 -dethia- 1 - oxa-3 -cephem-4-carboxylate obtained in step (b) in 800 ml dichloromethane cooled to 5 0 C, 30 g of triphenylphosphine (0.1143 m) are added and the mixture is stirred 15 minutes at a temperature of from O 0 C to 5°C, then a solution of 13.12 g (0.097 m) of aluminum trichloride in 200 ml methanol, neutralized with 24.6 g (0.29 m) of sodium bicarbonate, is added thereinto.
- the mixture is stirred 30 minutes at 25°C, then it is cooled to a temperature of from 0°C to 5°C, diluted with 50 ml methanol and stirred 3 hours at 8 0 C ⁇ 1°C.
- the mixture is treated with 17 ml glacial acetic acid, stirred 15 minutes at a temperature of from 15°C to 2O 0 C, then treated with 250 ml water containing 5% sodium chloride and stirred for further 10 minutes.
- the phases are separated, the organic phase is washed with 250 ml of water containing 5% acetic acid and with 250 ml of water containing 5% of sodium chloride.
- the phases are separated, the organic phase is washed with 130 ml water containing 5% of acetic acid and with 130 ml water containing 5% of sodium chloride.
- the organic phase is dried and concentrated in vacuum to a volume of 145-155 ml.
- the residual solution is diluted with 210 ml methanol and the concentration in vacuum is started again until a volume of 210 - 230 ml.
- the mixture is let to crystallize at a temperature of from O 0 C to 5°C for 15 hours.
- the reaction mixture is stirred 30 minutes at a temperature of from — 10 0 C to -15°C, 440 ml water are added thereinto and stirring is continued for a further 15-minute period.
- the organic one is washed with 260 ml of 2N hydrochloric acid, then with a 5% aqueous solution of sodium bicarbonate and finally with 260 ml water.
- the organic phase is concentrated in vacuum to a dense oil which is taken up with methanol and left to crystallize for 15 hours.
- step (b) To a solution of 58 g (0.088 m) of the benzhydryl 7 ⁇ -[2- (difluoromethylthio) acetamido] -7 ⁇ -methoxy-3 -[I -(2-hydroxy ethyl)- lH-tetrazol- 5-yl]thiomethyl-l -dethia- 1 -oxa-3 -cephem-4-carboxylate intermediate obtained in step (a) in 250 ml of dichloromethane, cooled to a temperature of from -30 0 C to - 35 0 C 5 17.1 g (0.15 m) of trifluoroacetic acid and 32 g (0.29 m) of anisole are added.
- reaction mixture is stirred at the same temperature for 2-3 hours, then the temperature is let to rise to 20 o ⁇ 25°C.
- the mixture is washed with 45 ml of 5% HCl, then with 45 ml water.
- the separated organic phase is dried and concentrated in vacuum.
- the residue is crystallized with ethyl acetate and the solid is filtered to give 7 ⁇ -[2-(difluoromethylthio)acetamido]-7 ⁇ -methoxy-3-[l- (2-hydroxyethyl)- 1 H-tetrazol-5 -yl]thiomethyl- 1 -dethia- 1 -oxa-3 -cephem-4- carboxylic acid (flomoxef free acid).
- step (b) To 100 ml of anisole, 37.5 g (0.28 m) of aluminum trichloride are added at 20° ⁇ -25°C and the mixture is diluted with 250 ml of dichloromethane to obtain a solution. Separately, a solution of 50 g (0.078 m) of the benzhydryl 7 ⁇ -[2- (cyanomethylthio)acetamido] -7 ⁇ -methoxy-3 -(I -methyl- 1 /f-tetrazol-5 - yl)thiomethyl-3-cephem-4-carboxylate intermediate of step (a) in 800 ml of dichloromethane is prepared., cooled to -5 0 C and added with the solution obtained above.
- the obtained mixture is stirred 40 minutes at 0 0 C, then it is poured in a mixture of 500 ml water, 750 ml acetone and 25 ml of 35% HCl. After a 30-minute stirring, the phases are separated. To the aqueous phase, 28 g of NaCl and 75 ml of ethyl acetate are added. The organic phase is separated and washed with 200 ml of a 10% solution of NaCl. The collected organic phases are treated with 500 ml of 5% NaHCO 3 . The aqueous phase is collected, brought to pH 6 with 40 ml 35% HCl, then 15 g of activated alumina are added and the mixture is stirred for 30 minutes.
- Alumina is filtered and washed with water.
- the aqueous solution is added with 100 g of NaCl and 300 ml " of ethyl acetate, the pH is brought to 3.5-3.6 by addition of 50% H 3 PO 4 and 50 ml of methyl isobutyl ketone.
- the pH is slowly, in 2 hours, brought to 2.2 by addition of 50% H3PO4 and the product is left to crystallize for 15 hours in the cold.
- EXAMPLE 5 An amount of 40 g (0.742 m) of benzhydryl 7-methylthioimino-3-(l- methyl-li ⁇ ' -tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylate are added to 640 ml of dichloro methane under stirring at room temperature. The mixture is cooled to 5 0 C and 24 g (0.0914 m) of triphenylphosphine are added thereto under stirring. The mixture is kept at 0° ⁇ 5°C for 15 minutes whereby a clear solution is obtained (Solution A).
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Oncology (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Communicable Diseases (AREA)
- Cephalosporin Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
There is described a process for the preparation of carboxy-protected 7β-amino-7α-methoxy-(l-oxa- or l-thia-)3-(l-substituted-lH-tetrazol-5-yl)thiomethyl-3- cephem-4-carboxylic acid. Said process comprises (a) reacting a carboxy- protected 7-amino-3-chloromethyl-(l-oxa- or 1-thia-) 3-cephem-4-carboxylate with an alkyl- or aryl-sulfenyl chloride; (b) reacting the corresponding 7-alkyl- or aryl-thioimino derivative with a l-(alkyl- o ω-hydroxyalkyl-)lH-tetrazol-5- ylthiol; and (c) reacting the corresponding 7-(alkyl- or aryl-)thioimino-3-(l- substituted-lH-tetrazol-5-yl)Momethyl- (1-oxa- or l-thia-)3-cephem with methanol in the presence of triphenylphosphine and aluminum chloride neutralized with a base. The reaction of the obtained carboxy-protected 7β-amino- 7α-methoxy-(1-oxa- or 1 -thia-)3 -(1-substituted- 1H-tetrazol-5 -yl)thiomethyl-3 - cephem-4-carboxylic acid with activated 2-(cyanomethylthio)acetic or 2- (difiuoromethyl thio)acetic acid affords, after final removal of the protecting ester group, cefmetazole and fiomoxef, respectively. The 7-(alkyl- or aryl)thioimino-3- (1-substituted-1H-tetrazol-5-yl)thiomethyl-1-dethia-1-oxa-3-cephem intermediates are new compounds.
Description
PROCESS FOR THE PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES AND RELATED INTERMEDIATES
FIELD OF THE INVENTION
The present invention concerns a process for the preparation of (1-oxa- or 1- thia-) 3-cephem derivatives which are useful intermediates in the preparation of (1-oxa- or l-thia-)cephalosporins. More particularly, the invention refers to a process for the preparation of a carboxy protected 7β-amino-7α-methoxy-3-[l- (alkyl- or ω-hydroxy. alkyl-)li/-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3- cephem-4-carboxylic acid and for the conversion of said compound into (1-oxa- or l-thia-)cephalosporins of therapeutic interest, in particular of flomoxef of formula A


In the context of the present description and in order to simplify the nomenclature, the term "(1-oxa- or l-thia-)3-cephem" is used, even though improperly, instead of the more current expressions "(l-dethia-l-oxa)-3-cephem or 3-cephem" and "(5-oxa- or 5-thia-)l-azabicyclo[4.2.0]oct-2-ene", to designate the nucleus of structure (a)

The above compounds flomoxef and cefinetazole are 7β-acylamino-7α- methoxy-cephalosporin-type antibiotics used as antibacterial drugs.
Flomoxef is an oxacephem, described in US 4,532,233, which is prepared by a multistep process in which the key intermediate is a carboxy-protected 7β- amino-7α-methoxy-3-[l-(2-hydroxyethyl)-lH-tetrazol-5-yl]thiomethyl-l-dethia- l-oxa-3-cephem-4-carboxylic acid. This compound is converted into flomoxef by protection of the primary hydroxyl group of the hydroxyethyl radical bound to the tetrazole ring, subsequent condensation with activated 2- (difluoromethylthio)acetic acid and removal of the hydroxy- and of the carboxy- protecting groups. A multistep process for the preparation of flomoxef is depicted in A. Kleemann, J. Engel, "Pharmaceutical Substances", G. Thieme Verlag, 4 Ed., 2001, Flomoxef- 6515-S (A. Kleemenn et al).
Cefinetazole is a cephem, described in GB 1449420, which is prepared by reaction of the corresponding carboxy-protected 7β-amino-7α-methoxy-3-(l- methyl-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylic acid by condensation with activated 2-(cyanomethylthio)acetic acid and removal of the the carboxy- protecting groups. For the synthesis, see also Η. Nakao et al. J. Antibiot. (Tokyo), 1979, 32, 320-329. Each of flomoxef and cefinetazole is a 7β-(substituted-thio)acetamido-7α- methoxy-3-(l -substituted-l/f-tetrazol-5-yl)thiomethyl-(l -oxa- or 1 -thia-)3- cephem-4-carboxylic acid. Their synthesis involves the preparation of a carboxy- protected 7 β -amino-7α-methoxy-3 -(I -substituted- 1 H-tetrazol-5 -yl)thiomethyl-(l - oxa- or l-thia-)3-cephem-4-carboxylic acid and the condensation thereof with an activated (substituted-thio)acetic acid.
The synthesis of these two 7β-(substituted-thio)acetamido-7α-methoxy-3- (l-substituted-lH-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3-cephem-4- carboxylic acids must take into account the preparation of an enantiomerically pure, carboxy-protected 7β-amino-7α-methoxy-3-(l-substituted-liϊ-tetrazol-5- yl)thiomethyl-(l-oxa- or 1-thia-) 3-cephem-4-carboxylic acid.
Furthermore, the synthesis of flomoxef must take into account the presence of a 2-hydroxyethyl substitution on the tetrazole moiety, which involves a protection of the primary hydroxyl group during the condensation with the activated difluoromethyl thioacetic acid.
PRIOR ART
It is known that an enantiomerically pure, carboxy-protected 7β-amino-7α- methoxy-3-(l-substituted-lH-tetrazol-5-yl)thiotnethyl-(l-oxa- or l-thia-)3- cephem-4-carboxylic acid may be prepared by reaction of a carboxy-protected 7- benzamido-3-(l-substituted-li7-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3- cephem-4-carboxylic acid with lithium methoxide in the presence of t-butyl hypochlorite (Kleemann et al. and DE2806457). Another method for converting a 7-benzamido group into a 7β-amino-7α-methoxy function using chlorine or lithium methoxide is described in WO 2006/006290. It is also known that an enantiomerically pure, carboxy-protected 7β-amino-
7α-methoxy-3-(l-substituted-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylic acid may be prepared by reaction of corresponding 7-methyl- or 7-(4-tolyl)- thioimino derivatives with methanol in the presence of triphenylphosphine and mercuric acetate in dichloromethane as described by E.M. Gordon et al. in J. Am. Chem. Soc. 1977, 99(16) 5504-5 and ibid. 1980, 102(5), 1690-1702. However the presence of a mercuric salt in the reaction mixture renders the purification of the products difficult.
Finally, it is known that the condensation of the carboxy-protected 7β- arrnno-7a-methoxy-3-[l-(2-hydroxyethyl)-lJf-tetrazol-5-yl]thiomethyl-l-dethia- l-oxa-3-cephem-4-carboxylic acid with activated 2-(difluoromethylthio)acetic acid to prepare the flomoxef precursor involves the protection of the primary hydroxyl group on the tetrazole moiety as the 4-methylbenzoyloxycarbonyl ester thereof which is subsequently removed also using SnCl4 (Kleeman et al.).
SUMMARY OF THE INVENTION It has now been found that, starting from a carboxy-protected 7-amino-3- chloromethyl-(l-oxa- or l-thia-)3-cephem-4-carboxylic acid, its reaction with an alkyl- or aryl-sulfenyl chloride affords a corresponding 7-thioimino derivative which is reacted with the appropriate, l-(alkyl- or ω-hydroxyalkyl-)lH-tetrazol-5- ylthiol, or with a salt thereof, to obtain the corresponding, 7-(alkyl- or aryl- )iMoimino-3-[(l -substituted)- lH-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3- cephem which is transformed into the corresponding carboxy protected 7β-amino- 7α-methoxy-3-[l -(alkyl- or ω-hydroxyalkyl-) lH-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem-4-carboxylic acid by treatment with methanol in the presence of aluminum trichloride, triphenylphosphine and sodium bicarbonate.
The carboxy protected 7β-amino-7α-methoxy-3-[l-(alkyl- or ω- hydroxyalkyl-) li-7"-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem-4- carboxylic acid thus obtained represents the key intermediate in the preparation of flomoxef (formula A) and of cefmetazole (formula B), or of their analogs, which are obtained by the classical condensation of carboxy protected 7β-amino-7α- methoxy-3-[l-(alkyl- or ω-hydroxy alkyl-)lH-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem-4-caboxylic acid with the appropriately activated 2- (difluoromethylthio)acetic acid in the presence of silylating agents to obtain flomoxef after deprotection of the carboxyl, or with the appropriately activated 2- (cyanomethylthio)acetic acid to give cefmetazole after deprotection of the carboxyl.
DETAILED DESCRIPTION
Thus, it is an object of the present invention to provide a process for the preparation of a (1-oxa- or l-thia-)3-cephem derivative of formula I

wherein X represents oxygen or sulfur, Y represents an alkyl group of from 1 to 3 carbon atoms or a ω-hydroxyalkyl group of from 2 to 3 carbon atoms and R° represents a carboxy-protecting group, or of a salt thereof, which comprises
(a) treating a carboxy-protected 7-amino-3-chloromethyl-( 1-oxa- or 1-thia- )3-cephem-4-carboxylic acid of formula II

wherein X and R° are as defined above, or a salt thereof, with an (alkyl- or aryl)sulfenyl chloride of formula III
W-S-Cl (III)
wherein W is an alkyl of from 1 to 3 carbon atoms, a benzyl group, non- substituted or substituted with an alkyl of from 1 to 3 carbon atoms, or a phenyl group, non-substituted or substituted with an alkyl of from 1 to 3 carbon atoms;
(b) treating the corresponding carboxy-protected 7-(alkyl- or aryl- )thioimino-3-chloromethyl-(l-oxa- or l-thia-)3-cephem-4 carboxylic acid thus obtained of formula IV

wherein X, R° and W are as defined above, with a [1 -alkyl or l-(ω- hydroxy alkyl)]- lH-tetrazol-5-ylthiol of formula V

wherein Y is an alkyl of from 1 to 3 carbon atoms or a ω-hydroxyalkyl of from 2 to 3 carbon atoms, or with an alkaline metal salt thereof;
(c) treating the corresponding 7-(alkyl- or aryl-)thioimino-3-(l-substituted- lH-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3-cephem thus obtained of formula VI

wherein W, X, R° and Y are as defined above, with methanol in the presence of aluminum trichloride neutralized with a base and a triarylphosphine or M(C1- C6)alkylphosphine and isolating the compound of formula I as such or as an addition salt thereof which is optionally neutralized to free base. In formulas III and VI, the alkyl substitution on the benzyl group is preferably in the benzene ring. The compound of formula I thus obtained is in enantiomerically pure form..
In step (c), sodium bicarbonate is preferably used as the base neutralizing aluminum trichloride. In the same step (c), the expression "triarylphosphine or tri^-C^alkylphosphine" designates phosphine wherein the three hydrogen atoms are replaced by an optionally substituted phenyl group such as phenyl or tolyl, by a monocyclic, aromatic heterocyclic group such as furyl or thienyl, or by a (C1-C6)Bl-CyI group such as rc-butyl.
A 7-amino-3-chloromethyl-(l-oxa- or l-thia-)3-cephem-4-carboxylic acid esterified with an easily removable protecting group, especially in acidic medium, is used as starting material. Protective groups used according to the present invention and the methods for their removal are described, for example, by Theodora W. Greene, Peter G. V. Wuts in "Protective Groups in Organic Synthesis", III ed., John Wiley & Sons, Inc., 1999, pages 373-431. Preferred esters are 4-methoxybenzyl and diphenylmethyl (benzhydryl) esters which are easily removable with trifluoroacetic acid and anisole in dichloromethane or with aluminum trichloride and anisole in dichloromethane.
In step (a) the carboxy-protected 7-amino-3-chloromethyl-(l-oxa- or 1-thia- )3-cephem-4-carboxylic acid, as such or as the hydrochloride thereof, is reacted with a methyl- or aryl- sulfenyl chloride, in its turn separately prepared by reaction of the corresponding methyl- or aryl- disulfide with chlorine, in an inert organic solvent such as toluene or dichloromethane, at room temperature (20°÷30°C) and in the presence of hydrogen chloride acceptor such as, for example, 1,2-epoxypropane. Methylsulfenyl chloride (formula III, W = methyl), phenylsulfenyl chloride (formula III, W = phenyl) and. /7-tolylsulfenyl chloride (formula III, W = 4-methylphenyl) are preferred methyl- or aryl- sulfenyl chlorides. The 7-(alkyl- or aryl-)thioimino-3-chloromethyl-(l-oxa- or l-thia-)3- cephem of formula IV thus obtained is isolated according to conventional methods, for example by neutralization with a base such as an aqueous solution of sodium bicarbonate (NaHCO3), removal of the salts and extraction using a suitable solvent such as dichloromethane. In step (b), the 7-(alkyl- or aryl- )thioimino-3-chloromethyl-(l-oxa- or 1- thia-)3-cephem of formula FV thus obtained is treated with a 1-alkyl- or l-(ω- hydroxyalkyl)-lH-tetrazol-5-ylthiol of formula V, preferably in form of its sodium salt. The reaction is carried out at a temperature of 20°÷30°C in a biphasic system with an inert organic solvent, for example dichloromethane or toluene, and water in the presence of a quaternary ammonium salt such as tetra-rø-
butylammonium bromide. After 5-10 hours, the reaction is complete and the 7- (alkyl- or aryl-)thioimino-3-(l-substituted-liϊ-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3-cephem of formula VI thus obtained is isolated according to conventional methods, for example by separating the phases, washing the organic phase with water, drying, evaporating of the solvent and crystallizing of the residue for example with an alcohol such as methanol.
1 -Methyl- 1 H-tetrazol-5-ylthiol or 1 -(2-hydroxyethyl)- lH-tetrazol-5-ylthiol as a sodium salt thereof is preferably used as 1 -substituted- lH-tetrazol-5-ylthiol. Said sodium salt may be prepared according to known methods, for example by reaction of the l-substituted-lH-tetrazol-5-ylthiol with sodium 2-ethylhexanoate in acetone.
The 7-(alkyl- or aryl-)thioimino-3-chloromethyl(l-oxa- or l-thia-)3-cephem compounds of formula VI, wherein X is oxygen, obtained at the end of step (b) are novel, useful intermediates in the preparation of flomoxef and they also are a further obj ect of the present invention.
In step (c), the 7-(alkyl- or aryl-)thioimino-3-chloromethyl(l-oxa- or 1- thia)3-cephem of formula VI obtained at the end of step (b) is reacted with methanol in the presence of aluminum trichloride neutralized with a base and a triarylphosphine or ^(Ci-C^alkylphosphine in an inert organic solvent such as dichloromethane or toluene.
Generally, the methanol also contains sodium bicarbonate as the base neutralizing aluminum trichloride. Preferably, the triarylphosphine or Ui(C1- C6)alkylphosphine is selected from the group consisting of triphenylphosphine, tri(ø-tolyl)phosphine, tri(2-furyl)phosphine and tri(«-butyl)phosphine. In practice, a solution of aluminum trichloride in methanol, previously prepared at a temperature of 10°÷15°C and neutralized with a base such as sodium bicarbonate, is added to a solution of the compound VI and of triphenylphosphine, tri(o-tolyl) phosphine, tri(2-furyl)phosphine or tri(rø-butyi)ρhosphine in one of the above-mentioned solvents. The compound of formula I thus obtained is isolated by adding glacial acetic acid to the suspension obtained at the end of the reaction, subsequently separating of the phases, drying the organic phase and extracting the product of formula I with a solvent such as dichloromethane or toluene and optional addition of a salifying acid such as HCl in isopropanol, HBr in isopropanol, methanesulfonic acid, j?-toluene sulfonic acid or naphthalene-2- sulfonic acid to the organic solution.
Instead of sodium bicarbonate, also alkaline metal methoxides such as lithium methoxide or sodium methoxide and alkaline metal acetates such as sodium or potassium acetate are advantageous neutralizing bases.
By extraction of the product and evaporation of the solvent, the compound of formula I is isolated as free base which may be purified according to known methods, for example by silica gel, an appropriate resin or by solution in a polar solvent such as dimethylacetamide and precipitation with an alcohol such as methanol or isopropanol.
By isolation in form of a salt, for example as hydrochloride, hydrobromide, methanesulfonate, j?-toluenesulfonate or naphthalene-2-sulfonate, the thus salified compound of formula I is already in pure form and the corresponding, pure free base may be easily obtained by neutralization.
The compounds of formula II used as starting materials are known or may be easily prepared by treatment of the corresponding carboxy-protected 7- acylamido-3-chloromethyl-(l-oxa- or 1-thia-) 3-cephem-4-carboxylic acid by reaction with PCl5 in the presence of pyridine, for example as described in DE
2806457 (see also US 4,366,316).
In particular, the present invention provide a process as illustrated above, whereby, at the end of step (c), a compound of formula Y
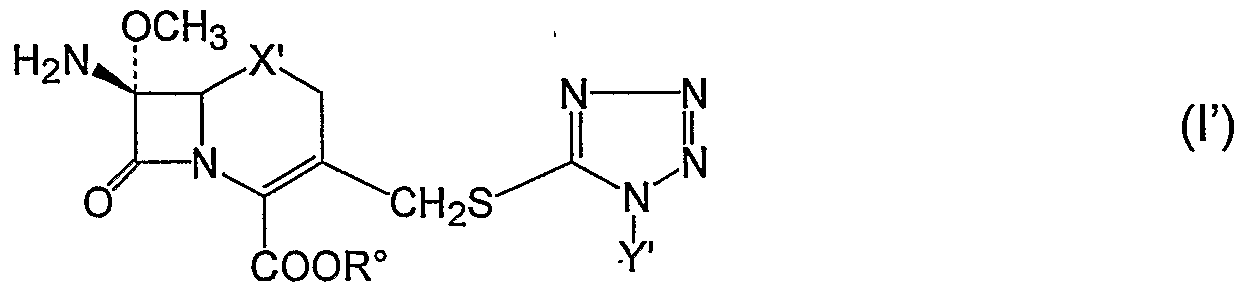
wherein X' is oxygen, Y' is 2-hydroxyethyl and R° is a carboxy-protecting group, preferably benzhydryl or 4-methoxybenzyl, is isolated. Compound F is obtained in enantiomerically pure form.
The present invention also provides a process as illustrated above, whereby, at the end of step (c), a compound of formula I"

The carboxy-protected 7β-amino-7α-methoxy-3-[l-alkyl- or l-(ω- hydroxyalkyl)-lH-tetrazol-5-yl]thiomethyl-(l-oxa- or l-thia-)3-cephem- 4- carboxylic acid thus obtained of formula I wherein R° is a carboxy-protecting group as illustrated above, preferably benzhydryl or 4-methoxybenzyl, may be easily transformed into flomoxef or into cefmetazole by condensation with appropriately activated 2-(difluoromethylthio)acetic acid or 2- (cyanomethylthio)acetic acid in the presence of a hydrogen chloride acceptor, for example a tertiary amine such as pyridine and, when in the compound of formula I
Y is ω-hydroxyalkyl having from 2 to 3 carbon atoms, also in the presence of a silylating agent such as, for example, hexamethyldisilazane and/or trimethylchloro silane. hi the case of the preparation of flomoxef the used intermediate compound has the formula I wherein X is oxygen and Y is 2-hydroxyethyl. In the caser of cefmetazole the intermediate compound has the formula I wherein X is sulfur and
Y is methyl.
Thus, it is a further object of the present invention to provide a process as illustrated above, wherein the compound of formula F is further reacted and treated with activated 2-(difluoromethylthio)acetic acid in the presence of a silylating agent to obtain the compound of formla VIF

wherein X' is oxygen, Y' is 2-hydroxyethyl and R° is a carboxy-protecting group which is removed to isolate flomoxef.
According to a preferred embodiment, the diphenylmethyl or 4- methoxybenzyl ester of the 7β-amino-7α-methoxy-3-[l-(2-hydroxyethyl)-lH- tetrazol-5-yl]thiomethyl-l-dethia-l-oxa-3-cephem-4-carboxylic acid is treated with silylating agents, for example with hexamethyldisilazane and trimethylchlorosilane, then it is reacted with the 2-(difluoromethylthio)acetyl chloride (F2CH-S-CH2COCl) in the presence of pyridine in dichloromethane at a
temperature of from -10°÷- 25°C. At the end of the reaction the product is isolated according to known techniques, for example by adding water to the reaction mixture, separating the phases, evaporating the organic one, taking up the residue, consisting of the diphenylmethyl or 4-methoxybenzyl ester of flomoxef, with dichloromethane and treating the solution thus obtained with trifluoroacetic acid and anisole. The flomoxef thus obtained, in a 98% purity, is isolated by adding water to the reaction mixture, separating the phases and recovering the product in acidic form by concentration of the organic phase or in form of the sodium or potassium salt thereof by treatment of the organic phase with sodium or potassium 2-ethylhexanoate.
The sodium salt of flomoxef may be further purified by passing an aqueous solution at pH 5,8÷6,2 through a column containing silica gel or a resin such as Amberlite® XAD 1180, by eluting with deionized water or with a mixture of deionized water and of an alcohol, for example ethanol or isopropanol. According to another embodiment, the invention provides a process as illustrated above, whereby the compound of formula I" is further reacted and treated with activated 2-(cyanomethylthio)acetic acid and a compound of formula VII"
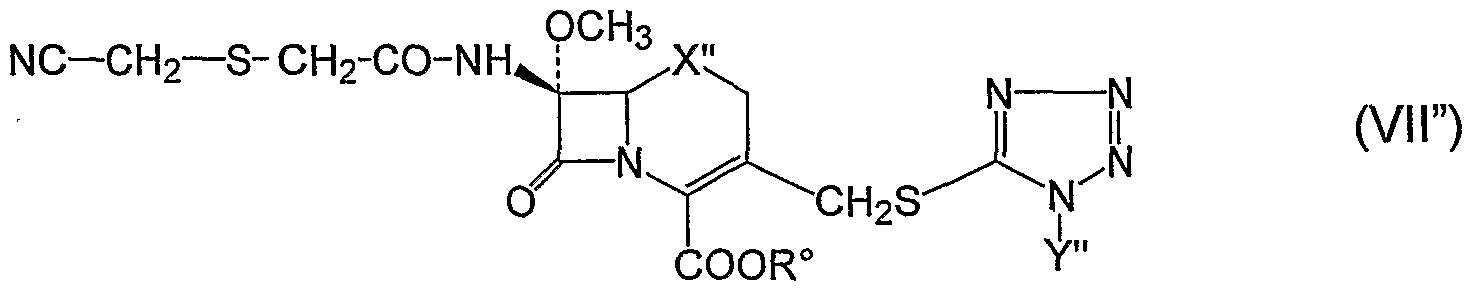
wherein X" is sulfur, Y" is methyl and R° is a carboxy-protecting group which is removed to isolate cefmetazole.
According to another preferred embodiment, the diphenylmethyl or 4- methoxy. benzyl 7β-amino-7α-methoxy-3-(l-methyl-lH"-tetrazol-5-yl) thiomethyl- 3-cephem-4-carboxylate is treated with 2-(cyanomethylthio)acetyl chloride (NCCH2SCH2COCI) in the presence of pyridine in dichloromethane at a temperature of from -10 to -250C. At the end of the reaction the product is isolated according to known methods, for example by adding water to the reaction mixture, separating the phases, evaporating the organic one, taking up the residue, consisting of the benzhydryl or 4-methoxybenzyl ester of cefmetazole, with dichloromethane and treating the solution thus obtained with trifluoroacetic acid and anisole or with aluminum trichloride and anisole. The cefmetazole thus
obtained in a 99% purity is isolated by adding water to the reaction mixture, separating the phases and recovering the product in acidic form by concentration of the organic phase or as a sodium or potassium salt thereof by treatment of the organic phase with sodium or potassium 2-ethylhexanoate.
Finally, it is a further object of the present invention to provide novel 7- (alkyl- -or aryl-)iMoiniino-3-(l-substiMed-lH-tetrazol-5-yl)thiometliyl-(l-oxa- or l-thia-)3-cephem compounds of formula VI

wherein X is oxygen, Y is an alkyl group of from 1 to 3 carbon atoms or a ω- hydroxyalkyl group of from 2 to 3 carbon atoms, W is an alkyl having from 1 to 3 carbon atoms, a benzyl group, non-substituted or substituted with an alkyl having from 1 to 3 carbon atoms, or a phenyl group, non-substituted or substituted with an alkyl having from 1 to 3 carbon atoms and R° is a carboxy-protecting group. The alkyl substitution on the benzyl group is preferably in the benzene ring.
Preferred compounds are those of formula VF wherein X' is oxygen, Y' is 2-hydroxyethyl, R° is benzhydryl or 4-methoxybenzyl and W is methyl, benzyl or j?-tolyl. The following examples illustrate the invention without, however, limiting it.
EXAMPLE 1
(a) To a solution of 60 g (0.138 m) of benzhydryl 7-amino-3-chloromethyl- l-dethia-l-oxa-3-cephem-4-carboxylate in 1800 ml of dichloromethane, cooled to -5°C, 96 g (1.65 m) of 1,2-epoxypropane are added in 5 minutes. Then, a solution of CH3SCl in dichloromethane, obtained by treatment of 26 g (0.276 m) of dimethyl sulfide in 800 ml dichloromethane at a temperature of from -50C to 00C with 200 ml of a 10% solution of chlorine in dichloromethane, is added thereinto. The mixture is stirred at a temperature of from — 5°C to 0°C for 30 minutes, then the temperature is brought to 20°C and the stirring is continued for one hour. The mixture is treated with a solution of 80 g of sodium bicarbonate in 1200 ml water, the phases are separated and the aqueous one is washed with 200 ml of dichloromethane. The phases are separated, the collected organic ones are washed
with 600 ml water, the aqueous phase is extracted with 200 ml dichloromethane, than the organic phases are collected and dried under reduced in vacuum. The residue is crystallized with 225 ml methanol to give the benzhydryl 7- methylthioimino-3 -chloromethyl- 1 -dethia- 1 -oxa-3 -cephem-4-carboxylate. Yield: 88% of the theoretical.
1H-RMN (CDCl3) δ p.p.m.: 2.9 (s, 3H); 4.4 (2d, 2H); 4.5 (2d, 2H); 5.2 (s, IH); 6.9 (s, IH); 7.2÷7.6 (m, 10H).
(b) To a solution of 11.5 g (0.026 m) of intermediate benzhydryl 7- methylthio imino-3 -chloromethyl- 1 -dethia- 1 -oxa-3 -cephem-4-carboxylate obtained in step (a) in 150 ml dichloromethane, a solution of 7 g (0.041 m) of sodium l-(2-hydroxyethyl)-li?-tetrazol-5-ylthiolate in 150 ml water is added at 2O0C. Then, 1.2 g of tetra-«-butyl ammonium bromide is added and the reaction mixture is stirred at a temperature of from 20°C to 250C for 8 hours. The phases are separated, the organic one is washed with 50 ml water. The organic phase is concentrated in vacuum and the residue is taken up with 30 ml methanol. The mixture is cooled and the solid product is filtered and dried to give benzhydryl 7- methylthioimino-3 - [ 1 -(2-hydroxyethyl)- 1 H-tetrazol-5 -yl]thiomethyl- 1 -dethia- 1 - oxa-3-cephem-4-carboxylate. Yield: 85% of the theoretical. 1H-RMN (CDCl3) δ p.p.m.: 2.90 (s, 3H); 3.9 (t,2H); 4.3 (t, 2H); 4.35 (s, 2H); 4.6 (2d, 2H); 5.2 (s, IH); 6.9 (s, IH); 7.4 (m, 10H).
(c) To a solution of 51.4 g (0.093 m) of the intermediate benzhydryl 7- methylthio imino-3 - [ 1 -(2-hydroxyethyl)- lH-tetrazol-5 -yljthiomethyl- 1 -dethia- 1 - oxa-3 -cephem-4-carboxylate obtained in step (b) in 800 ml dichloromethane cooled to 50C, 30 g of triphenylphosphine (0.1143 m) are added and the mixture is stirred 15 minutes at a temperature of from O0C to 5°C, then a solution of 13.12 g (0.097 m) of aluminum trichloride in 200 ml methanol, neutralized with 24.6 g (0.29 m) of sodium bicarbonate, is added thereinto. The mixture is stirred 30 minutes at 25°C, then it is cooled to a temperature of from 0°C to 5°C, diluted with 50 ml methanol and stirred 3 hours at 80C ± 1°C. At the end of the reaction the mixture is treated with 17 ml glacial acetic acid, stirred 15 minutes at a temperature of from 15°C to 2O0C, then treated with 250 ml water containing 5% sodium chloride and stirred for further 10 minutes. The phases are separated, the organic phase is washed with 250 ml of water containing 5% acetic acid and with 250 ml of water containing 5% of sodium chloride. The organic phase is collected, concentrated in vacuum to a small volume and the residue is taken up with a
methanol/dichloromethane mixture. The solid is filtered, washed with isopropyl ether and dried to give benzhydryl 7β-amino-7α-methoxy-3-[l-(2-hydroxyethyl)- lH-tetrazol-5-yl]thiomethyl- 1 -dethia- 1 -oxa-3 -cephem-4-carboxylate. Yield: 82% of the theoretical. 1H-RMN (DMSO-</6) δ p.p.m.: 3.45 (s, 3H); 3..7 (t, 2H); 4.2 and 4.3 (2d, 2H); 4.35 (m, 2H); 4.6 (2d, 2H); 5.1 (t, IH); 5.2 (s, IH); 6.9 (s, IH); 7,25-7,65 (m, 10H).
EXAMPLE 2 (a)(b) By operating as described in steps (a) and (b) of Example 1, under the same conditions, starting from benzhydryl 7-amino-3-chloromethyl-3-cephem-4- carboxylate, by reaction with CH3SCl there is obtained the benzhydryl 3- chloromethyl-7-methyl thioimino-3 -(I -methyl- 1 H"-tetrazol-5 -yl)thiomethyl-3 - cephem-4-carboxylate which, by reaction with sodium 1 -methyl- lH-tetrazol-5- ylthiolate, affords benzhydryl 7-methyl thioimino-3-(l-methyl-lH-tetrazol-5- yl)thiomethyl-3-cephem-4-carboxylate. Melting point: 211°÷212°C.
(c) To a stirred solution of 25.2 g (0.047 m) of the intermediate benzhydryl 7-methylthioimino-3 -(I -methyl- 1 if-tetrazol-5-yl)thiomethyl-3 -cephem-4- carboxylate obtained in the above steps (a)(b) in 400 ml dichloromethane, at 5°C, 15.18 g (0.058 m) of triphenylphosphine are added under stirring, then stirring is continued at a temperature of from 00C to 5°C for 15÷20 minutes to attain complete solution. To this solution, a solution of 6.64 g (0.097 m) of aluminum chloride in 105 ml of methanol, neutralized with 12.45 g (0.29 m) of sodium bicarbonate, is added. The mixture is cooled to a temperature of from 0°C to 50C, diluted with 25 ml methanol and stirred at 8°C + 1°C for 3 hours. At the end of the reaction (HPLC control), the mixture is treated with 8.6 ml glacial acetic acid, stirred 20 minutes at 20°÷25°C, then treated with 130 ml water containing 5% of sodium chloride and stirred for further 10 minutes. The phases are separated, the organic phase is washed with 130 ml water containing 5% of acetic acid and with 130 ml water containing 5% of sodium chloride. The organic phase is dried and concentrated in vacuum to a volume of 145-155 ml. The residual solution is diluted with 210 ml methanol and the concentration in vacuum is started again until a volume of 210 - 230 ml. The mixture is let to crystallize at a temperature of from O0C to 5°C for 15 hours. The solid product is filtered, washed with cold methanol and dried in vacuum at 30°C to give benzhydryl 7β-amino-7α-methoxy-
3-(l-methyl-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylate with a melting point of 127°÷128°C. Yield: 84% of the theoretical.
EXAMPLE 3
(a) A suspension of 80.7 g (0.15 m) of benzhydryl 7β-amino-7α-methoxy-3- [1 -(2-hydroxyethyl)- lH-tetrazol-5-yl]thiomethyl- 1 -dethia- 1 -oxa-3 -cephem-4- carboxylate in 250 ml of dichloromethane is prepared, then 103.3 g (0.64 m) of hexamethyldisilazane and 34.75 g (0.32 m) of trimethylchlorosilane are added thereinto.. The mixture is stirred for 30 minutes to obtain a clear solution which is cooled to a temperature of from -50C to -100C and treated with 30.2 g (0.38 m) of pyridine. To this mixture, a solution of 2-(difluoromethylthio)acetyl chloride, obtained by addition of 52.8 g (0.25 m) of phosphorus pentachloride (PCl5) to a solution of 32.8 g (0.213 m) of sodium 2-(difluoromethylthio)acetate in 625 ml of dichloromethane at 0°÷2°C, is added. The reaction mixture is stirred 30 minutes at a temperature of from — 100C to -15°C, 440 ml water are added thereinto and stirring is continued for a further 15-minute period. After separation of the phases, the organic one is washed with 260 ml of 2N hydrochloric acid, then with a 5% aqueous solution of sodium bicarbonate and finally with 260 ml water. The organic phase is concentrated in vacuum to a dense oil which is taken up with methanol and left to crystallize for 15 hours. The solid is filtered, washed with cold methanol and dried under vacuum to give benzhydryl 7β-[2- (difluoromethylthio) acetamido]-7α-methoxy-3-[l-(2-hydroxyethyl)-lH-tetrazol- 5-yl]thiomethyl-l -dethia- 1 -oxa-3 -cephem-4-carboxylate. Yield: 79% of the theoretical. 1H-RMN (DMSO-d6) δ p.p.m.: 3.45 (s, 3H); 3.65 (2d, 2H); 3.7 (t, 2H); 4.25 (2d, 2H); 4.3 (m, 2H); 4.6 (2d, 2H); 5.1 (t, IH); 5.2 (s, IH); 6.9 (s, IH); 7.35 (t, IH); 7.25-7.65 (m, 10H); 9.4 (s, IH).
(b) To a solution of 58 g (0.088 m) of the benzhydryl 7β-[2- (difluoromethylthio) acetamido] -7α-methoxy-3 -[I -(2-hydroxy ethyl)- lH-tetrazol- 5-yl]thiomethyl-l -dethia- 1 -oxa-3 -cephem-4-carboxylate intermediate obtained in step (a) in 250 ml of dichloromethane, cooled to a temperature of from -300C to - 350C5 17.1 g (0.15 m) of trifluoroacetic acid and 32 g (0.29 m) of anisole are added. The reaction mixture is stirred at the same temperature for 2-3 hours, then the temperature is let to rise to 20o÷25°C. The mixture is washed with 45 ml of 5% HCl, then with 45 ml water. The separated organic phase is dried and concentrated in vacuum. The residue is crystallized with ethyl acetate and the
solid is filtered to give 7β-[2-(difluoromethylthio)acetamido]-7α-methoxy-3-[l- (2-hydroxyethyl)- 1 H-tetrazol-5 -yl]thiomethyl- 1 -dethia- 1 -oxa-3 -cephem-4- carboxylic acid (flomoxef free acid). Yield: 92% of the theoretical. 1H-RMN (DMSO-d6) δ p.p.m.: 3.4 (s, 3H); 3.65 (s 2H); 3.7 (t, 2H); 4.2 (s, 2H); 4.3 (t, 2H); 4.5 (s, 2H); 5.1 (s, IH); 7.05 (t, IH); 9.2 (s, IH).
EXAMPLE 4
(a) A suspension of 60 g (0.113 m) of benzhydryl 7β-amino-7α-methoxy-3- [l-methyl-lH-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylate in 150 ml of dichloro methane is prepared, then 13.5 g (0.17 m) of pyridine are added and the mixture is cooled to 0°÷3°C. To this mixture, a solution of 2- (cyanomethylthio)acetyl chloride, obtained by addition, at 0°÷2°C, of 35 g (0.168 m) of phosphorus pentachloride (PCl5) to a solution of 25 g (0.213 m) of sodium 2-(cyanomethylthio)acetate in 150 ml of dichloromethane is added. The reaction mixture is stirred 30 minutes at a temperature of from -100C to -35°C, then 100 ml water are added thereinto. The phases are separated, the organic one is washed with a 10% aqueous solution of NaCl and subsequently with water. The dried organic phase is concentrated in vacuum to a residue which is taken up with 250 ml of methanol. The product is left to crystallize at 10°÷15°C for 15 hours, then the mixture is cooled to 5°C. The solid is filtered, washed with cold methanol and dried to give benzhydryl 7β-[2-(cyanomethylthio)acetamido]-7α-methoxy-3-(l- methyl-l/f-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylate: Yield: 86% of the theoretical
(b) To 100 ml of anisole, 37.5 g (0.28 m) of aluminum trichloride are added at 20°^-25°C and the mixture is diluted with 250 ml of dichloromethane to obtain a solution. Separately, a solution of 50 g (0.078 m) of the benzhydryl 7β-[2- (cyanomethylthio)acetamido] -7α-methoxy-3 -(I -methyl- 1 /f-tetrazol-5 - yl)thiomethyl-3-cephem-4-carboxylate intermediate of step (a) in 800 ml of dichloromethane is prepared., cooled to -50C and added with the solution obtained above. The obtained mixture is stirred 40 minutes at 00C, then it is poured in a mixture of 500 ml water, 750 ml acetone and 25 ml of 35% HCl. After a 30-minute stirring, the phases are separated. To the aqueous phase, 28 g of NaCl and 75 ml of ethyl acetate are added. The organic phase is separated and washed with 200 ml of a 10% solution of NaCl. The collected organic phases are treated with 500 ml of 5% NaHCO3. The aqueous phase is collected, brought to pH 6 with 40 ml 35% HCl, then 15 g of activated alumina are added and the mixture is
stirred for 30 minutes. Alumina is filtered and washed with water. The aqueous solution is added with 100 g of NaCl and 300 ml" of ethyl acetate, the pH is brought to 3.5-3.6 by addition of 50% H3PO4 and 50 ml of methyl isobutyl ketone. The pH is slowly, in 2 hours, brought to 2.2 by addition of 50% H3PO4 and the product is left to crystallize for 15 hours in the cold. The solid is filtered, washed and dried in vacuum to give 7β-[2-(cianomethylthio)acetamido]-7α- methoxy-3-(l-methyl-lH-tetrazol- 5-yl)thiomethyl-3-cephem-4-carboxylic acid (cefmetazole). Yield: 97% of the theoretical.
EXAMPLE 5 An amount of 40 g (0.742 m) of benzhydryl 7-methylthioimino-3-(l- methyl-liϊ'-tetrazol-5-yl)thiomethyl-3-cephem-4-carboxylate are added to 640 ml of dichloro methane under stirring at room temperature. The mixture is cooled to 50C and 24 g (0.0914 m) of triphenylphosphine are added thereto under stirring. The mixture is kept at 0°÷5°C for 15 minutes whereby a clear solution is obtained (Solution A). Separately, 16.4 g (0.078 m) of anhydrous aluminum trichloride are added portionwise, under nitrogen atmosphere, to 160 ml of methanol, previously cooled to 10°÷15°C. After a 5-minute stirring, 19.68 g (0.234 m) of sodium bicarbonate are added portionwise in 10-15 minutes at 10°÷15°C and the mixture is let to stand at 250C for 30 minutes under stirring to obtain a suspension (Suspension B). Suspension B is added to Solution A at 0°÷5°C and the reaction mixture is stirred at 80C ± 1°C for about 3 hours by following the course of the reaction by TLC (eluent: ethyl acetate/toluene 1/1). At the end of the reaction, the mixture is treated as described in Example 2(c) to give 32 g of benzhydryl 7β- amino-7α-methoxy-3-(l-methyl-lH-tetrazol-5-yl)thiomethyl-3-cephem-4- carboxylate as a crystalline powder. Melting point: 127°÷128°C.
Claims
1. A process for the preparation of a (1-oxa- or l-thia-)3-cephem derivative of formula I

wherein X represents oxygen or sulfur, Y represents an alkyl having from 1 to 3 carbon atoms or an ω-hydroxyalkyl having from 2 to 3 carbon atoms and R° represents a carboxy protecting group, or of a salt thereof, which comprises
(a) treating a carboxy-protected 7-amino-3-chloromethylr(l-oxa- or 1-thia- )3 -cephem-4-carboxylic acid of formula II

wherein X and R° are defined as above, or a salt thereof, with an (alkyl- or aryl-) sulfenyl chloride of formula III
W-S-Cl (111) wherein W is an alkyl having from 1 to 3 carbon atoms, a benzyl, non substituted or substituted with an alkyl having from 1 to 3 carbon atoms, or a phenyl, non substituted or substituted with an alkyl having from 1 to 3 carbon atoms; (b) treating the corresponding 7-(alkyl- or aryl-)thioimino-3-chloromethyl-
( 1-oxa- or l-thia-)3-cephem thus obtained of formula IV

wherein W, X and R° are as defined above, with a [1 -alkyl or l-(ω-hydroxyalkyl- )]-lH-tetrazol-5-ylthiol of formula V 
wherein Y is an alkyl of from 1 to 3 carbon atoms or a ω-hydroxyalkyl of from 2 to 3 carbon atoms, or with an alkaline metal salt thereof; (c) treating the corresponding 7-(alkyl- or aryl-)thioimino-3-(l-substituted-
IH- tetrazol-5-yl)thioniethyl-(l-oxa- or l-thia-)-3-cephem of formula VI

wherein W5 X, R° e Y are as defined above, with methanol in the presence of aluminum trichloride neutralized with a base and a triarylphosphine or Ui(C1- C6)alkylphosphine , to obtain the compound of formula I as such or in form of an acid addition salt thereof.
2. The process according to claim 1, wherein, in step (a), said alkyl- or aryl- sulfenyl chloride has the formula III, wherein W is methyl, phenyl or p-tolyl.
3. The process according to anyone of claims 1 and 2, wherein, in step (a), a carboxy-protected 7-amino-3-chlorornethyl-(l-oxa- or l-thia-)3-cephem-4- carboxylic acid of formula II, wherein X is oxygen or sulfur and R is benzhydryl or p-methoxybenzyl, or a salt thereof, is used.
4. The process according to claim 1, wherein, in step (b), a [1 -alkyl- or 1-
(ω-hydroxyalkyl-)]l/i-tetrazol-5-ylthiol of formula V3 wherein Y is methyl or 2- hydroxyethyl, is used.
5. The process according to claim 4, wherein said [1-alkyl- or l-(ω- hydroxyalkyl)-] l/i-tetrazol-5-ylthiol of formula V, wherein Y is methyl or 2- hydroxyethyl, is in form of its sodium salt.
6. The process according to claim I5 wherein, in step (b), a [1-alkyl- or 1- (ω-hydroxyalkyl-)]17f-tetrazol-5-ylthiol of formula V, wherein Y is methyl or 2- hydroxyethyl, in form of its sodium salt and a 7-(alkyl- or aryl-)thioimino-3- chloromethyl-(l-oxa- or l-thia-)3-cephem of formula IV, wherein X is oxygen or sulfur, R° is benzhydryl or 4-methoxybenzyl and W is methyl, phenyl or p-tolyl, are used.
7. The process according to claim 1, wherein, in step (c), a 7-(alkyl- or aryl-) thioimino-3-(l-substituted-lH-tetrazol-5-yl)thiomethyl-(l-oxa- or l-thia-)3- cephem of formula VI, wherein X is oxygen or sulfur, R° is benzhydryl or 4- methoxybenzyl, Y is methyl o 2-hydroxyethyl and W is methyl, phenyl or p-tolyl, is used.
8. The process according to claim 1, wherein, in step (c), a compound of formula I, wherein X is oxygen or sulfur, Y e methyl or 2-hydroxyethyl and R° is a carboxy protecting group, is isolated.
9. The process according to claim 1, wherein, in step (c), said triarylphosphine or  is selected from the group consisting of triphenylphosphine, tri(o-tolyl)phosphine, tri(2-furyl)phosphine and tή(n- butyl)phosphine.
is selected from the group consisting of triphenylphosphine, tri(o-tolyl)phosphine, tri(2-furyl)phosphine and tή(n- butyl)phosphine.
10. The process according to claim 8, wherein said group R° is benzhydryl.
11. The process according to claim 8, wherein said group R0 is 4- methoxybenzyl.
12. The process according to claim 1, wherein said base is sodium bicarbonate.
13. The process according to claim 1, wherein, in step (c), the compound of formula I is isolated in form of a salt thereof which is optionally neutralized to free base.
14. The process according to claim 13, wherein said salt is selected from the group consisting of hydrochloride, hydrobromide, methanesulfonate, p- toluenesulfonate and naphthalene-2-sulfonate salts.
15. The process according to claim 1, wherein, in step (c), a compound of formula I'

wherein X' is oxygen, Y' is 2-hydroxyethyl and R° is a carboxy protecting group, is isolated.
16. The process according to claim 1, wherein, in step (c), a compound of formula I" 
wherein X" is sulfur, Y" is methyl and R° is a carboxy protecting group, is isolated.
17. The process according claim 15, wherein said carboxy protecting group
R° is benzhydryl.
18. The process according to claim 15, wherein said carboxy protecting group R° is 4-methoxybenzyl.
19. The process according to claim 16, wherein said carboxy protecting group R° is benzhydryl.
20. The process according to claim 16, wherein said carboxy protecting group R° is 4-methoxybenzyl.
21. The process according to claim 16, wherein said compound of formula V is further reacted and treated with activated 2-(difluoromethylthio)acetic acid in the presence of a silylating agent to afford the compound of formula VIF

wherein X' is oxygen, Y' is 2-hydroxyethyl and R° is a carboxy protecting group which is removed by known methods to isolate flomoxef.
22. The process according to claim 21, wherein said silylating agent consists of hexamethyldisilazane and trimethylchlorosilane.
23. The process according to claim 21, wherein said carboxy protecting group R° is benzhydryl or 4-methoxybenzyl and the removal is carried out by treatment with trifluoroacetic acid and anisole in dichloroniethane.
24. The process according to claim 16, wherein said compound of formula I" is further reacted and treated with activated 2-(cyanomethyltliio)acetic acid to obtain the compound of formula VII" 
wherein X" is sulfur, R" is methyl and R0 is a carboxy protecting group which is removed by known methods to isolate cefmetazole.
25. The process according to claim 24, wherein said carboxy protecting group R° is benzhydryl or 4-methoxybenzyl and the removal is carried out by treatment with trifluoroacetic acid and anisole in dichloromethane.
26. The process according to claim 24, wherein said carboxy protecting group R° is benzhydryl or 4-methoxybenzyl and the removal is carried out by treatment with aluminum trichloride and anisole in dichloromethane.
27. A 7-(alkyl- or aryl-)tMoimino-l-detMa-l-oxa-3 -(I -substituted- IH- tetrazol-5-yl)thiomethyl-3-cephem of formula VF

wherein X' is oxygen, Y' is an alkyl having from 1 to 3 carbon atoms or a ω- hydroxyalkyl having 2 or 3 carbon atoms, W is an alkyl having from 1 to 3 carbon atoms optionally substituted with a phenyl group, or phenyl group non substituted or substituted with an alkyl having from 1 to 3 carbon atoms and R° is a carboxy protecting group.
28. The 7-(alkyl- or aryl-)thioimino-l-dethia-l-oxa-3 -(I -substituted- IH- tetrazol-5-yl)thiomethyl-3-cephem according to claim 27, of formula VF, wherein X' is oxygen, Y' is 2-hydroxyethyl, W is methyl, phenyl or p-tolyl, and R° is benzhydryl or 4-methoxybenzyl.
29. The benzhydryl 7-methylthioimino-3-[l-(2-hydroxyethyl)-lH-tetrazol-
5-yl] thiomethyl- 1 -dethia- 1 -oxa-3 -cephem-4-carboxylate.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2009500008A JP2009530268A (en) | 2006-03-15 | 2007-03-14 | Process for the preparation of (1-oxa- or 1-thia-) 3-cephem derivatives and related intermediates |
| EP07736690A EP1996595A2 (en) | 2006-03-15 | 2007-03-14 | PROCESS FOR THE PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES AND RELATED INTERMEDIATES |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ITMI20060466 ITMI20060466A1 (en) | 2006-03-15 | 2006-03-15 | PROCEDURE FOR THE PREPARATION OF 1-OXA-O 1-TIA-3-CEFEN DERIVATIVES AND RELATED INTERMEDIATES |
| ITMI2006A000466 | 2006-03-15 | ||
| ITMI20061096 ITMI20061096A1 (en) | 2006-06-06 | 2006-06-06 | PROCEDURE FOR THE PREPARATION OF (1-TIA-) 3-CEFEM DERIVATIVES |
| ITMI2006A001096 | 2006-06-06 |
Publications (4)
| Publication Number | Publication Date |
|---|---|
| WO2007105253A2 true WO2007105253A2 (en) | 2007-09-20 |
| WO2007105253A3 WO2007105253A3 (en) | 2007-11-01 |
| WO2007105253B1 WO2007105253B1 (en) | 2007-12-06 |
| WO2007105253A8 WO2007105253A8 (en) | 2008-10-30 |
Family
ID=38255802
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IT2007/000185 Ceased WO2007105253A2 (en) | 2006-03-15 | 2007-03-14 | PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP1996595A2 (en) |
| JP (1) | JP2009530268A (en) |
| KR (1) | KR20080111062A (en) |
| WO (1) | WO2007105253A2 (en) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101787040A (en) * | 2010-03-02 | 2010-07-28 | 哈药集团制药总厂 | Method for preparing cefmetazole sodium |
| CN101792454A (en) * | 2010-03-17 | 2010-08-04 | 河北九派制药有限公司 | Preparation method of 7-alpha amino 7-methoxyl-3-methly tetrazole sulfur methyl cafe-benzyl alkanoic acid |
| CN101792455A (en) * | 2010-03-17 | 2010-08-04 | 河北九派制药有限公司 | Preparation method of high-purity 7-alpha-amino-7-methoxy-3-methyltetrazole thiomethyl cephalosporin benzyl ester |
| CN102675342A (en) * | 2011-03-15 | 2012-09-19 | 四平市精细化学品有限公司 | Preparation method of 7 beta-amino-7 alpha-methoxy-3-((1-methyl-1H-tetrazole-5-group) sulfomethyl)-3-cephem-4-diphenylmethyl carboxylate |
| CN102850379A (en) * | 2012-08-30 | 2013-01-02 | 三峡大学 | Synthetic method for methoxy-cephalosporin intermediate 7-MAC |
| CN102952149A (en) * | 2012-11-09 | 2013-03-06 | 浙江新和成股份有限公司 | One-pot synthesis method of flomoxef intermediate |
| CN104151324A (en) * | 2014-09-03 | 2014-11-19 | 齐鲁天和惠世制药有限公司 | Method for preparing ampicillin sodium by menstruum crystallization method |
| CN104327100A (en) * | 2014-09-30 | 2015-02-04 | 华北制药河北华民药业有限责任公司 | Preparation technology of high-purity flomoxef sodium |
| CN104487445A (en) * | 2012-07-25 | 2015-04-01 | 第一药品株式会社 | Novel method for preparing 1-oxacephalosporin derivative |
| CN104557978A (en) * | 2014-12-31 | 2015-04-29 | 重庆福安药业(集团)股份有限公司 | Preparation method for cefmetazole sodium |
| CN105037393A (en) * | 2015-06-24 | 2015-11-11 | 浙江永宁药业股份有限公司 | Preparation method of flomoxef sodium |
| CN105399755A (en) * | 2015-11-03 | 2016-03-16 | 浙江永宁药业股份有限公司 | Method for synthesizing flomoxef acid |
| CN107722041A (en) * | 2017-11-12 | 2018-02-23 | 王龙 | The preparation method of cefmetazole acid |
| US20190077812A1 (en) * | 2017-09-13 | 2019-03-14 | Dongdo Co., Ltd. | METHOD FOR MANUFACTURING 7alpha-ALKOXYOXACEPHEM INTERMEDIATE COMPOUND |
| CN109608478A (en) * | 2018-11-15 | 2019-04-12 | 山东晶辉生物技术有限公司 | A kind of synthetic method of Flomoxef acid |
| CN109970766A (en) * | 2019-04-22 | 2019-07-05 | 山西千岫制药有限公司 | A kind of preparation method of Flomoxef acid |
| CN110003241A (en) * | 2019-04-23 | 2019-07-12 | 山西千岫制药有限公司 | A kind of preparation method of latamoxef parent nucleus |
| CN110804635A (en) * | 2019-11-11 | 2020-02-18 | 济南康和医药科技有限公司 | Synthesis method of latamoxef sodium |
| CN114292283A (en) * | 2022-01-24 | 2022-04-08 | 广州维奥康药业科技有限公司 | Preparation method of cefmetazole sodium impurity for injection |
| CN119979652A (en) * | 2025-02-10 | 2025-05-13 | 河南立诺制药有限公司 | A kind of synthetic method of fluoxetine |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN102391291B (en) * | 2011-09-21 | 2014-06-04 | 河北九派制药有限公司 | Cefmetazole acid preparation method |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5417755B2 (en) * | 1973-11-26 | 1979-07-02 | ||
| US4109084A (en) * | 1976-12-08 | 1978-08-22 | E. R. Squibb & Sons, Inc. | Thiooxime cephalosporin derivatives |
| JPS59139385A (en) * | 1982-12-23 | 1984-08-10 | Shionogi & Co Ltd | Fluoromethylthiooxacephalosporin |
| JPS6348286A (en) * | 1986-08-15 | 1988-02-29 | Shionogi & Co Ltd | Imino compound and production thereof |
| KR20000073152A (en) * | 1999-05-07 | 2000-12-05 | 조생현 | A Process for preparing 7-alpha-methoxy-cephalosporanic acid derivatives |
-
2007
- 2007-03-14 EP EP07736690A patent/EP1996595A2/en not_active Withdrawn
- 2007-03-14 WO PCT/IT2007/000185 patent/WO2007105253A2/en not_active Ceased
- 2007-03-14 KR KR1020087025168A patent/KR20080111062A/en not_active Withdrawn
- 2007-03-14 JP JP2009500008A patent/JP2009530268A/en not_active Ceased
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN101787040A (en) * | 2010-03-02 | 2010-07-28 | 哈药集团制药总厂 | Method for preparing cefmetazole sodium |
| CN101792454A (en) * | 2010-03-17 | 2010-08-04 | 河北九派制药有限公司 | Preparation method of 7-alpha amino 7-methoxyl-3-methly tetrazole sulfur methyl cafe-benzyl alkanoic acid |
| CN101792455A (en) * | 2010-03-17 | 2010-08-04 | 河北九派制药有限公司 | Preparation method of high-purity 7-alpha-amino-7-methoxy-3-methyltetrazole thiomethyl cephalosporin benzyl ester |
| CN101792455B (en) * | 2010-03-17 | 2012-07-04 | 河北九派制药有限公司 | Preparation method of high-purity 7-alpha-amino-7-methoxy-3-methyltetrazole thiomethyl cephalosporin benzyl ester |
| CN102675342A (en) * | 2011-03-15 | 2012-09-19 | 四平市精细化学品有限公司 | Preparation method of 7 beta-amino-7 alpha-methoxy-3-((1-methyl-1H-tetrazole-5-group) sulfomethyl)-3-cephem-4-diphenylmethyl carboxylate |
| CN104487445A (en) * | 2012-07-25 | 2015-04-01 | 第一药品株式会社 | Novel method for preparing 1-oxacephalosporin derivative |
| CN102850379B (en) * | 2012-08-30 | 2015-08-12 | 三峡大学 | The synthetic method of methoxy cephalosporin intermediate 7-MAC |
| CN102850379A (en) * | 2012-08-30 | 2013-01-02 | 三峡大学 | Synthetic method for methoxy-cephalosporin intermediate 7-MAC |
| CN102952149A (en) * | 2012-11-09 | 2013-03-06 | 浙江新和成股份有限公司 | One-pot synthesis method of flomoxef intermediate |
| CN104151324A (en) * | 2014-09-03 | 2014-11-19 | 齐鲁天和惠世制药有限公司 | Method for preparing ampicillin sodium by menstruum crystallization method |
| CN104327100A (en) * | 2014-09-30 | 2015-02-04 | 华北制药河北华民药业有限责任公司 | Preparation technology of high-purity flomoxef sodium |
| CN104557978B (en) * | 2014-12-31 | 2017-07-18 | 重庆福安药业(集团)股份有限公司 | A kind of preparation method of cefmetazole sodium |
| CN104557978A (en) * | 2014-12-31 | 2015-04-29 | 重庆福安药业(集团)股份有限公司 | Preparation method for cefmetazole sodium |
| CN105037393A (en) * | 2015-06-24 | 2015-11-11 | 浙江永宁药业股份有限公司 | Preparation method of flomoxef sodium |
| CN105399755A (en) * | 2015-11-03 | 2016-03-16 | 浙江永宁药业股份有限公司 | Method for synthesizing flomoxef acid |
| US20190077812A1 (en) * | 2017-09-13 | 2019-03-14 | Dongdo Co., Ltd. | METHOD FOR MANUFACTURING 7alpha-ALKOXYOXACEPHEM INTERMEDIATE COMPOUND |
| US10533019B2 (en) * | 2017-09-13 | 2020-01-14 | Dongdo Co., Ltd. | Method for manufacturing 7alpha-alkoxyoxacephem intermediate compound |
| CN107722041A (en) * | 2017-11-12 | 2018-02-23 | 王龙 | The preparation method of cefmetazole acid |
| CN109608478A (en) * | 2018-11-15 | 2019-04-12 | 山东晶辉生物技术有限公司 | A kind of synthetic method of Flomoxef acid |
| CN109970766A (en) * | 2019-04-22 | 2019-07-05 | 山西千岫制药有限公司 | A kind of preparation method of Flomoxef acid |
| CN110003241A (en) * | 2019-04-23 | 2019-07-12 | 山西千岫制药有限公司 | A kind of preparation method of latamoxef parent nucleus |
| CN110804635A (en) * | 2019-11-11 | 2020-02-18 | 济南康和医药科技有限公司 | Synthesis method of latamoxef sodium |
| CN110804635B (en) * | 2019-11-11 | 2021-08-17 | 济南康和医药科技有限公司 | Synthesis method of latamoxef sodium |
| CN114292283A (en) * | 2022-01-24 | 2022-04-08 | 广州维奥康药业科技有限公司 | Preparation method of cefmetazole sodium impurity for injection |
| CN119979652A (en) * | 2025-02-10 | 2025-05-13 | 河南立诺制药有限公司 | A kind of synthetic method of fluoxetine |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20080111062A (en) | 2008-12-22 |
| JP2009530268A (en) | 2009-08-27 |
| WO2007105253A8 (en) | 2008-10-30 |
| EP1996595A2 (en) | 2008-12-03 |
| WO2007105253A3 (en) | 2007-11-01 |
| WO2007105253B1 (en) | 2007-12-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1996595A2 (en) | PROCESS FOR THE PREPARATION OF (1-OXA- OR l-THIA-)3- CEPHEM DERIVATIVES AND RELATED INTERMEDIATES | |
| JPH0567632B2 (en) | ||
| EP0175610A2 (en) | New cephalosporin compounds and the production thereof | |
| EP1546155B1 (en) | Intermediate cefdinir salts | |
| US9139597B2 (en) | Method for the production of ceftobiprole medocaril | |
| DK155943B (en) | METHOD OF ANALOGUE FOR THE PREPARATION OF 7-D - (-) - ALFA-AMINO-ALFA- (P-ACETOXYPHENYL) ACETAMIDOCEPHALOSPORAN ACIDS | |
| FI63586C (en) | PROCEDURE FOR THE FRAMSTATION OF AV 7-BETA- (2-OXYIMINO-2-ARYLACETAMIDO) -3- (SULFOALKYLTETRAZOLE-5-YLTHOMETHYL) -3-CEFEM-4-CARBOXYLSYROR WITH ANTIBACTERIAL NETWORK | |
| JPH027315B2 (en) | ||
| CN102295655A (en) | Oxacephem antibiotic intermediate solvate and preparation method thereof | |
| DK148796B (en) | CEPHALOSPORANIC ACID DERIVATIVES USED AS INTERMEDIATES IN THE MANUFACTURE OF OTHER CEPHALOSPORANIC ACID DERIVATIVES | |
| CN1134447C (en) | Process for purification of cephalosporin derivative | |
| WO2011077217A1 (en) | An improved process for the preparation of cefpodoxime acid | |
| DK159447B (en) | 7BETA- (FLUORATED METHYLTHIO) ACETAMIDO-7ALFA-METHOXY-3- (1- (2-HYDROXYALKYL) -1H-TETRAZOL-5-YL) -THIOMETHYL-1-DETHIA-1-OXA-3-CEPHEM-4-CARBOXYL ANTIBACTERIAL AGENTS CONTAINING SUCH COMPOUNDS | |
| HU184882B (en) | Process for producing cepheme-carboxylic acid derivatives | |
| CY1618A (en) | Cephalosporanic acid derivatives | |
| HU221429B (en) | 7-acyl-3-(substituted carbamoyloxy)-methyl-cef-3-em-4-carboxylic acid derivatives, process for their preparation and pharmaceutical compositions comprising such compounds | |
| NO301766B1 (en) | Process for Preparation of a Cephalosporin Antibiotic Using the Syn-Isomer of a Thiazole Intermediate | |
| JPS5953277B2 (en) | Method for producing antibacterial agents | |
| JPH0134227B2 (en) | ||
| US4018921A (en) | Substituted phenylglycylcephalosporins | |
| JPH02138283A (en) | Novel o-substituted oxime derivative of 7-aminothiazolylacetamidocephalosporanic acid | |
| US4139701A (en) | 7-Amino-3-(sulfonic acid and sulfamoyl substituted tetrazolythiomethyl)cephalosporin intermediates | |
| JPH0733776A (en) | New production of cephalosporin antibiotic | |
| US3953439A (en) | Substituted phenylglycylcephalosporins | |
| WO1999020631A1 (en) | Process for producing 3-cephem compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07736690 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007736690 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009500008 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020087025168 Country of ref document: KR |