WO2007106721A2 - Cannabinoid receptor antagonists/inverse agonists useful for treating obesity - Google Patents
Cannabinoid receptor antagonists/inverse agonists useful for treating obesity Download PDFInfo
- Publication number
- WO2007106721A2 WO2007106721A2 PCT/US2007/063631 US2007063631W WO2007106721A2 WO 2007106721 A2 WO2007106721 A2 WO 2007106721A2 US 2007063631 W US2007063631 W US 2007063631W WO 2007106721 A2 WO2007106721 A2 WO 2007106721A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tetrazole
- alkyl
- conr
- conh
- och
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(C(C1CC1)=C1c2ccccc2)=**1c1ccccc1 Chemical compound *C(C(C1CC1)=C1c2ccccc2)=**1c1ccccc1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/12—Antidiuretics, e.g. drugs for diabetes insipidus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/645—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having two nitrogen atoms as the only ring hetero atoms
- C07F9/6503—Five-membered rings
- C07F9/65031—Five-membered rings having the nitrogen atoms in the positions 1 and 2
Definitions
- the present invention provides cannabinoid receptor antagonists/inverse agonists and pharmaceutical compositions thereof and methods of using the same for treating obesity, diabetes, and/or cardiometabolic disorders. More particularly, the present invention relates to a novel method for treating obesity, diabetes, and/or cardiometabolic disorders using a pyrazole.
- Obesity is associated with an increase in the overall amount of adipose tissue (i.e., body fat), especially adipose tissue localized in the abdominal area. Obesity has reached epidemic proportions in the United States. The prevalence of obesity has steadily increased over the years among all racial and ethnic groups. The most recent data from the Centers for Disease Control and Prevention, and the National Center for Health Statistics report 66% of the adult population overweight (BMI, 25.0-29.9), 31% obese (BMI, 30-39.9), and 5% extremely obese (BMI, >40.0). Among children aged 6 through 19 years, 32% were overweight and 17% were obese. This translates to 124 million Americans medically overweight, and 44 million of these deemed obese.
- BMI body fat
- Obesity is responsible for more than 300,000 deaths annually, and will soon overtake tobacco usage as the primary cause of preventable death in the United States.
- Obesity is a chronic disease that contributes directly to numerous dangerous co-morbidities, including type 2 diabetes, cardiovascular disease, inflammatory diseases, premature aging, and some forms of cancer.
- Type 2 diabetes a serious and life-threatening disorder with growing prevalence in both adult and childhood populations, is currently the 7 th leading cause of death in the United States. Since more than 80% of patients with type 2 diabetes are overweight, obesity is the greatest risk factor for developing type 2 diabetes. Increasing clinical evidence indicates that the best way to control type 2 diabetes is to reduce weight.
- CNS appetite suppressants such as sibutramine
- gut lipase inhibitors such as orlistat.
- CNS appetite suppressants reduce eating behavior through activation of the 'satiety center' in the brain and/or by inhibition of the 'hunger center' in the brain.
- Gut lipase inhibitors reduce the absorption of dietary fat from the gastrointestinal (GI) tract.
- sibutramine and orlistat work through very different mechanisms, they share in common the same overall goal of reducing body weight secondary to reducing the amount of calories that reach the systemic circulation.
- these indirect therapies produce only a modest initial weight loss (approximately 5% compared to placebo) that is usually not maintained. After one or two years of treatment, most patients return to or exceed their starting weight.
- most approved anti-obesity therapeutics produce undesirable and often dangerous side effects that can complicate treatment and interfere with a patient's quality of life.
- the endocanabinoid system comprised of the canabinoid receptors (CBl and CB2) and their endogenous ligands (e.g., anandamide, 2-AG), plays a prominent role in the control of food intake and energy metabolism.
- CBl receptors are widely expressed in the brain, including cortex, hippocampus, amygdala, pituitary and hypothalamus.
- CB 1 receptors have also been identified in numerous peripheral organs and tissues, including thyroid gland, adrenal gland, reproductive organs, adipose tissue, liver, muscle, and gastrointestinal tract. CB2 receptors are localized almost exclusively in immune and blood cells [Endocrine Reviews 2006, 27, 73].
- ⁇ 9 -THC The plant-derived cannabinoid agonist ⁇ 9 -tetrahydrocannabinol ( ⁇ 9 -THC), the main psychoactive component of marijuana, binds to both CBl and CB2 receptors.
- ⁇ 9 -THC is widely reported to increase appetite and food intake (hyperphagia) in humans and in animals. This hyperphagic effect is largely blocked by pretreatment with selective CBl receptor antagonists/inverse agonists [e.g., rimonabant (SR141716A, Acomplia®)], strongly supporting the belief that CBl receptor activation mediates the hyperphagic effect of ⁇ 9 -THC, [Endocrine Reviews 2006, 27, 73].
- rimonabant produces a clinically meaningful weight loss in obese patients. Patients also experience improvements in associated cardiometabolic risk factors, including a decrease in fasting insulin levels and a decrease in triglyceride levels. Rimonabant also produces greater reductions in abdominal fat deposits, which are a known risk factor for diabetes and heart disease [Science 2006, 311, 323]. Taken together, these improvements in adiposity and cardiometabolic risk factors produce an overall decrease in the prevalence of the metabolic syndrome [Lancet 2005, 365, 1389 and N ⁇ JM 2005, 353, 2121].
- the CB 1 receptor is one of the most abundant and widely distributed G protein- coupled receptors in the mammalian brain. It is believed that the appetite-suppressant properties of CB 1 antagonists are mediated through an interaction with CB 1 receptors in the hypothalamus (regulation of food intake), and in the mesolimbic region (rewarding properties of food).
- CBl receptors are far more broadly distributed in brain (e.g., neocortex, hippocampus, thalamus, cerebellum, and pituitary), and while interacting with targeted CBl receptors in hypothalamus and mesolimbic regions, CBl antagonists have ready access to non-targeted CBl receptors that have little if any role in appetite control.
- CBl antagonist/inverse agonist rimonabant produces psychiatric and nervous system side effects. These include depressed mood, anxiety, irritability, insomnia, dizziness, and headache. These side effects are dose-related and are most pronounced at the most efficacious weight-reducing dose of rimonabant (JAMA 2006, 311, 323).
- the occurrence of therapeutic efficacy (appetite suppression) and side effects over the same dose range strongly suggest that both effects are mediated through concurrent antagonism of CBl receptors in both 'targeted' and 'non-targeted' brain regions.
- Brain-penetrant CBl antagonists/inverse agonists do not selectively target CBl receptors in efficacy brain regions, while ignoring CBl receptors in side effect brain regions.
- CBl receptor antagonists/inverse agonists with limited or no CNS adverse side effects, including mood disorders.
- CBl receptors in peripheral tissues (e.g., adipose tissue, liver, muscle, and gastrointestinal tract), while sparing CBl receptors in brain.
- peripheral tissues e.g., adipose tissue, liver, muscle, and gastrointestinal tract
- CNS side effects should be reduced or eliminated.
- This should provide a novel opportunity to develop safer agents for the prevention or treatment of obesity, diabetes, and cardiometabolic diseases (e.g., hypertension and dyslipidemias).
- the present invention provides novel pyrazoles or pharmaceutically acceptable salts thereof that are CBl receptor antagonists/inverse agonists.
- the present invention provides novel pharmaceutical compositions, comprising: a pharmaceutically acceptable carrier and a therapeutically effective amount of at least one of the compounds of the present invention or a pharmaceutically acceptable salt form thereof.
- the present invention provides novel methods for treating obesity, diabetes, and/or cardiometabolic disorders (e.g., hypertension, dyslipidemias, high blood pressure, and insulin resistance), comprising: administering to a mammal in need of such treatment a therapeutically effective amount of at least one of the compounds of the present invention or a pharmaceutically acceptable salt form thereof.
- the present invention provides processes for preparing novel compounds.
- the present invention provides novel compounds or pharmaceutically acceptable salts for use in therapy.
- the present invention provides the use of novel compounds for the manufacture of a medicament for the treatment of obesity, diabetes, and/or cardiometabolic disorders.
- the present invention is based on the finding that a CBl receptor antagonist/inverse agonist has beneficial effects on body weight, adiposity, and cardiometabolic risk factors such as high blood pressure, insulin resistance and eleveated levels of blood lipids that cannot be explained by weight loss derived from CNS- mediated appetite suppression alone and that this effect is mediated, at least in part, through interaction at peripheral receptors.
- the present invention provides compounds that are designed to preferentially target CB 1 receptors in peripheral tissues (e.g., adipose tissue, liver, muscle, and gastrointestinal tract), while sparing CBl receptors in brain. Peripherally-mediated beneficial effects of CB 1 antagonists/inverse agonists should be maintained, whereas CNS side effects should be reduced or eliminated.
- the compounds of the present invention have been designed to have reduced CNS exposure by virtue of their inability or limited ability to penetrate the blood-brain barrier (BBB) or by their participation in active transport systems, thus reducing centrally mediated side-effects, a potential problem with many anti-obesity agents. It is expected that the peripherally restricted compounds of the present invention will have no or very limited CNS effects. Thus, their peripherally mediated CBl antaonistic properties should provide therapeutic agents with greater safety.
- BBB blood-brain barrier
- a drug used in the treatment of obesity, diabetes, and/or cardiometabolic disorders e.g., hypertension, dyslipidemias, high blood pressure, and insulin resistance
- CNS side effects e.g., seizures, depression, anxiety, movement disorders, and hyperactivity
- incorporation of a peripherally restricting group in such a drug would lower the brain concentration of the drug relative to the concentration in the systemic circulation, thereby affording the opportunity to increase the dosage employed to treat the peripheral disorder.
- the increased dosage may provide greater therapeutic efficacy, as well as a more rapid onset of therapeutic action.
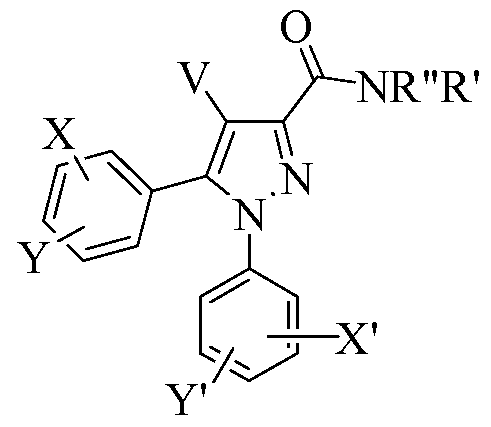
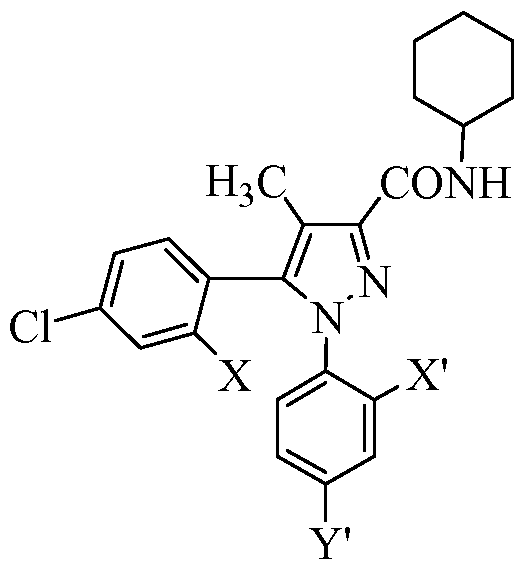
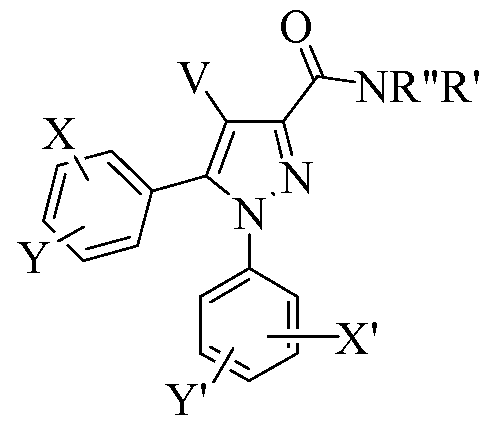
- the present invention provides novel compound AA or a stereoisomer or pharmaceutically acceptable salt thereof:
- X, Y, X', and Y' are independently selected from H, halogen, CF 3 , NO 2 , Ci_ 6 alkyl,
- R" is selected from H, Ci_ 6 alkyl, (CH 2 ) 0 - 6 aryl, and (CH 2 ) 0 - 6 heteroaryl;
- R' is selected from H, Ci_6 alkyl, and a 5-7 membered cyclic amine that is unsaturated, partially saturated, or fully saturated and is substituted with 0-4 groups selected from CF 3 , NO 2 , Ci_6 alkyl, benzyl, phenyl, OH, halogen, and Ci_6 alkoxy;
- ring R' is attached via its nitrogen atom to the amide nitrogen of AA; and
- at least one of X, Y, X', Y', V, R', or R" is suitably modified or replaced by a group capable of reducing or limiting the CNS (brain) levels of compound AA.
- the present invention provides novel compounds of formula I or a stereoisomer or
- A is selected from H, Ci_6 alkyl, (CH 2 ) m -C 3-6 -cycloalkyl, (CH 2 ) m -heteroaryl, and (CH 2 ) m -aryl, wherein each aryl and heteroaryl is substituted with 0-1 groups selected from CF 3, halogen, Ci_ 4 alkyl, -CN, CONR 2 , NO 2 , NR 2 , and OR; [0036] R' is selected from H, Ci_ 6 alkyl, CH(A)-(CH 2 ) m CO 2 R, CH(A)-(CH 2 ) m CONH 2 , CH(A)-(CH 2 ) m C(NH)NH 2, (CH 2 ) m -phenyl-(CH 2 ) m CO 2 R, (CH 2 ) m -pyridyl-(CH 2 ) m CO 2 R, (CH 2 ) m -phenyl-(CH 2 )
- R" is is selected from H, Ci_ 6 alkyl, (CH 2 ) 0 - 6 aryl, and (CH 2 ) 0 - 6 heteroaryl;
- Z is selected from H, Ci_ 6 alkyl, aryl, NR 2 , OR, -CN, (CH 2 ) m C(NH)NH 2 , CO 2 R,
- CH 2 OCH 2 CH CHCO 2 R, O(CH 2 ) n PO(OR) 2 , CH 2 O(CH 2 ) n PO(OR) 2 , O(CH 2 ) n C 6 H 4 CO 2 R,
- OCH 2 CH CHCONR 2
- CH 2 OCH 2 CH CHCONR 2
- Q is selected from N, CH, and CQ';
- Q' is selected from H, CO 2 R, (CH 2 ) n CO 2 R, CH 2 O(CH 2 ) n CO 2 R,
- CH 2 OCH 2 CH CHCO 2 R, CH 2 O(CH 2 ) n PO(OR) 2 , CONR 2 , (CH 2 ) n CONR 2 ,
- V is selected from H, Ci_6 alkyl, C 2 _ 6 alkenyl, CF 3 , aryl, -CN, (CH 2 ) m C(NH)NH 2 ,
- R is independently selected from H, C 1 ⁇ alkyl
- R a is independently selected from H, C 1 ⁇ alkyl, C 2 _6 alkenyl, and C 2 _6 alkynyl;
- m is selected from O, 1, 2, 3, and 4;
- n is selected from 1, 2, 3, and 4;
- p is selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12;
- At least one of X, Y, X', and Y' is other than H, halogen, Ci_ 6 alkyl, NO 2 , -CN, CF 3 , OR, and phenyl;
- R' is selected from CH(A)-(CH 2 ) m CO 2 R, CH(A)-(CH 2 ) m CONH 2 , CH(A)- (CH 2 ) m C(NH)NH 2 , (CH 2 ) m -phenyl-(CH 2 ) m CO 2 R, (CH 2 ) m -pyridyl-(CH 2 ) m CO 2 R, (CH 2 ) m -phenyl-(CH 2 ) m CONH 2 , (CH 2 ) m -pyridyl-(CH 2 ) m CONH 2 , (CH 2 ) m -phenyl-(CH 2 ) m C(NH)NH 2 , (CH 2 ) m -pyridyl-(CH 2 ) m (CN)NH 2 , (CH 2 ) m -phenyl-(CH 2 ) m -tetrazole, and (CH 2 ) )
- (c) Z is present and is other than H, Ci_6 alkyl, aryl, NR 2 , and OR;
- Q is present and is CQ' where Q' is other than H;
- V is other than H, -CN, CF 3 , C 2 _ 6 alkenyl, aryl, and Ci_ 6 alkyl.
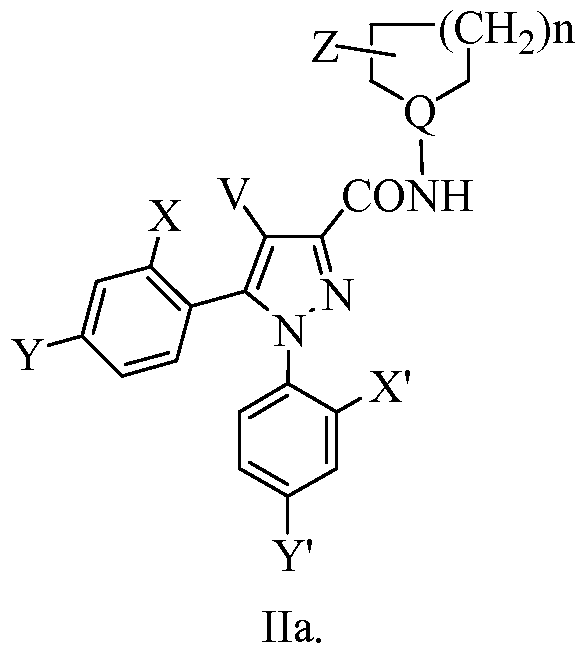
- the present invention provides novel compounds of formula II or a stereoisomer or pharmaceutically acceptable salt thereof:
- the present invention provides novel compounds of formula Ha or a stereoisomer or pharmaceutically acceptable salt thereof:
- X and Y are independently selected from H, halogen, C 1-4 alkyl, CF 3 , -CN, NO 2 ,
- X' and Y' are independently selected from H, halogen, Ci_ 4 alkyl, CF 3 , -CN, NO 2 ,
- Z is selected from (CH 2 ) m C(NH)NH 2 , (CH 2 ) m CO 2 R, O(CH 2 ) n CO 2 R,
- CH 2 OCH 2 CH CHCONR 2 , CH 2 NR a (CH 2 ) n tetrazole, CH 2 O(CH 2 ) n tetrazole, and
- Q is selected from N and CH;
- V is selected from H, Ci_6 alkyl, C 2 _ 6 alkenyl, CF 3 , aryl, and -CN;
- R is selected from H, C 1-4 alkyl, and C 2 _ 4 alkenyl;
- n is selected from 1 and 2.
- X and Y is other than H, halogen, C 1 ⁇ alkyl, -CN,
- X' and Y' are independently selected from H, halogen, Ci_ 4 alkyl, -CN, NO 2 , NR 2 , and OR;
- Z is selected from H, C 1-4 alkyl, and aryl;
- Q is selected from N and CH;
- V is selected from H, Ci_ 4 alkyl, and aryl;
- R is selected from H, C 1-4 alkyl, and C 2 _ 4 alkenyl;
- n is selected from 1 and 2.
- X and Y are independently selected from H, halogen, Ci_ 4 alkyl, -CN, NO 2 , NR 2 , and OR;
- X' and Y' is other than halogen, C 1-4 alkyl, -CN,
- Z is selected from H, Ci_ 4 alkyl, and aryl;
- Q is selected from N and CH;
- V is selected from H, Ci_ 4 alkyl, and aryl;
- R is selected from H, C 1-4 alkyl, and C 2 _ 4 alkenyl
- n is selected from 1 and 2;
- p is selected from 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11 and 12.
- X and Y are independently selected from H, CF 3 , halogen, Ci_ 4 alkyl, -CN, NO 2 ,
- X' and Y' are independently selected from H, CF 3, halogen, Ci_ 4 alkyl, -CN, NO 2 ,
- Z is selected from H, C 1-4 alkyl, and aryl;
- Q is selected from N and CH;
- V is selected from (CH 2 ) m CO 2 R, (CH 2 ) m CONR 2 , (CH 2 ) m C(NH)NH 2 , (CH 2 ) m - tetrazole, (CH 2 ) m CONR a CH(A)-(CH 2 ) m CO 2 R, (CH 2 ) m CONR a (CH 2 ) m
- A is selected from H, Ci_ 4 alkyl, and (CH 2 ) m -aryl, wherein each aryl is optionally substituted with 0-1 groups selected from CF 3 , halogen, Ci_ 4 alkyl, -CN, CONR 2 , NO 2 ,
- R is selected from H, Ci_ 4 alkyl, and C 2 _ 4 alkenyl;
- m is selected from 0, 1, and 2;
- n is selected from 1 and 2.
- X and Y are independently selected from H, CF 3 , halogen, Ci_ 4 alkyl, -CN, NO 2 ,
- X' and Y' are independently selected from H, CF 3 , halogen, Ci_ 4 alkyl, -CN, NO 2 ,
- R' is selected from CH(A)-(CH 2 ) m CO 2 R, CH(A)-(CH 2 ) m CONH 2 , CH(A)-
- A is selected from H, Ci_ 6 alkyl, (CH 2 ) m -C 3 _ 6 -cycloalkyl, (CH 2 ) m -phenyl,
- V is selected from H, Ci_ 4 alkyl, and aryl;
- R is selected from H, Ci_ 4 alkyl
- m is selected from 0, 1, and 2.
- the present invention provides novel pharmaceutical compositions, comprising: a pharmaceutically acceptable carrier and a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt form thereof.
- the present invention provides a novel method for treating a disease, comprising: administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt form thereof, wherein the disease is selected from obesity, diabetes, cardiometabolic disorders, and a combination thereof.
- the diabetes disorder is selected from Type 1 diabetes, Type 2 diabetes, inadequate glucose tolerance, and insulin resistance.
- the cardiometabolic disorder is selected from hypertension, dyslipidemias (e.g., undesirable blood lipid levels, elevated cholesterol levels, and lowered LDL levels), high blood pressure, and insulin resistance.
- the present invention provides a novel method for treating a co-morbidity of obesity, comprising: administering to a patient in need thereof a therapeutically effective amount of a compound of the present invention or a pharmaceutically acceptable salt form thereof.
- the co-morbidity is selected from diabetes,
- Metabolic Syndrome dementia, and heart disease.
- the co-morbidity is selected from hypertension; gallbladder disease; gastrointestinal disorders; menstrual irregularities; degenerative arthritis; venous statis ulcers; pulmonary hypoventilation syndrome; sleep apnea; snoring; coronary artery disease; arterial sclerotic disease; pseudotumor cerebri; accident proneness; increased risks with surgeries; osteoarthritis; high cholesterol; and, increased incidence of malignancies of the ovaries, cervix, uterus, breasts, prostrate, and gallbladder.
- the present invention also provides a method of preventing or reversing the deposition of adipose tissue in a mammal by the administration of a compound of the present invention.
- compound of the present invention are expected to reduce the incidence or severity of obesity, thereby reducing the incidence or severity of associated co-morbidities.
- the present invention provides a compound of the present invention for use in therapy.
- the present invention provides the use of the present invention for the manufacture of a medicament for the treatment of obesity, diabetes, cardiometabolic disorders, and a combination thereof.
- the compounds herein described may have asymmetric centers, geometric centers (e.g., double bond), or both. All chiral, diastereomeric, racemic forms and all geometric isomeric forms of a structure are intended, unless the specific stereochemistry or isomeric form is specifically indicated.
- Compounds of the present invention containing an asymmetrically substituted atom may be isolated in optically active or racemic forms. It is well known in the art how to prepare optically active forms, such as by resolution of racemic forms, by synthesis from optically active starting materials, or through use of chiral auxiliaries.
- cis and trans geometric isomers of the compounds of the present invention may also exist and may be isolated as a mixture of isomers or as separated isomeric forms. All processes used to prepare compounds of the present invention and intermediates made therein are considered to be part of the present invention. All tautomers of shown or described compounds are also considered to be part of the present invention.
- Alkyl includes both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- C 1 ⁇ alkyl for example, includes C 1 , C 2 , C3, C 4 , C5, and Ce alkyl groups.
- alkyl include methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl, t-butyl, n-pentyl, and s-pentyl.
- Alkenyl includes the specified number of hydrocarbon atoms in either straight or branched configuration with one or more unsaturated carbon-carbon bonds that may occur in any stable point along the chain, such as ethenyl and propenyl.
- C 2 - 6 alkenyl includes C 2 , C3, C 4 , C5, and Ce alkenyl groups.
- Alkynyl includes the specified number of hydrocarbon atoms in either straight or branched configuration with one or more triple carbon-carbon bonds that may occur in any stable point along the chain, such as ethynyl and propynyl.
- C 2 - 6 Alkynyl includes C 2 , C3, C 4 , C5, and Ce alkynyl groups.
- Cycloalkyl includes the specified number of hydrocarbon atoms in a saturated ring, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- C3-8 cycloalkyl includes C3, C 4 , C5, Ce, C 7 , and Cs cycloalkyl groups.
- Cyclic amine is a hydrocarbon ring wherein one carbon atom of the ring has been replaced by a nitrogen atom. The cyclic amine can be unsaturated, partially saturated, or fully saturated.
- the cyclic amine can also be bicyclic, tricyclic, and polycyclic.
- Examples of cyclic amine include pyrrolidine and piperdine.
- Halo or "halogen” refers to fluoro, chloro, bromo, and iodo.
- Counteririon is used to represent a small, negatively charged species, such as chloride, bromide, hydroxide, acetate, and sulfate.
- the group “C O H 4 " represents a phenylene.
- Aryl refers to any stable 6, 7, 8, 9, 10, 11, 12, or 13 membered monocyclic, bicyclic, or tricyclic ring, wherein at least one ring, if more than one is present, is aromatic.
- aryl include fluorenyl, phenyl, naphthyl, indanyl, adamantyl, and tetrahydronaphthyl.
- Heteroaryl refers to any stable 5, 6, 7, 8, 9, 10, 11, or 12 membered monocyclic, bicyclic, or tricyclic heterocyclic ring that is aromatic, and which consists of carbon atoms and 1, 2, 3, or 4 heteroatoms independently selected from the group consisting of N, O, and S. If the heteroaryl group is bicyclic or tricyclic, then at least one of the two or three rings must contain a heteroatom, though both or all three may each contain one or more heteroatoms. If the heteroaryl group is bicyclic or tricyclic, then only one of the rings must be aromatic.
- the N group may be N, NH, or N-substituent, depending on the chosen ring and if substituents are recited.
- the nitrogen and sulfur heteroatoms may optionally be oxidized (e.g., S, S(O), S(O) 2 , and N-O).
- the heteroaryl ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure.
- the heteroaryl rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- heteroaryl includes acridinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl, benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl, benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl, 2H,6H-l,5,2-dithiazinyl, dihydrofuro[2,3- ⁇ ]tetrahydrofuran, furanyl, furazanyl, imidazolyl, lH-indazolyl, indolenyl, indolinyl, indolizinyl, in
- “Mammal” covers warm blooded mammals that are typically under medical care (e.g., humans and domesticated animals). Examples of mammals include feline, canine, equine, bovine, and human, as well as just human.
- "Treating” or “treatment” covers the treatment of a disease-state in a mammal, and includes: (a) preventing the disease-state from occurring in a mammal, in particular, when such mammal is predisposed to the disease-state but has not yet been diagnosed as having it; (b) inhibiting the disease-state, i.e., arresting it development; and/or (c) relieving the disease-state, i.e., causing regression of the disease state until a desired endpoint is reached.
- “Pharmaceutically acceptable salts” refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include, but are not limited to, those derived from inorganic and organic acids selected from 1, 2-ethanedisulfonic, 2-acetoxybenzoic, 2-hydroxyethanesulfonic, acetic, ascorbic, benzenesulfonic, benzoic, bicarbonic, carbonic, citric, edetic, ethane disulfonic, ethane sulfonic, fumaric, glucoheptonic, gluconic, glutamic, glycolic, glycollyarsanilic, hexylresorcinic, hydrabamic, hydrobromic, hydrochloric, hydroiodide, hydroxymaleic, hydroxynaphthoic, isethionic, lactic, lactobionic, lauryl sulfonic, maleic, malic, mandelic, methanesulfonic, napsylic, nitric, oxalic, pamoic, pantothenic,
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are useful. Lists of suitable salts are found in Remington 's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Easton, PA, 1990, p 1445.
- Therapeutically effective amount includes an amount of a compound of the present invention that is effective when administered alone or in combination to treat obesity or another indication listed herein.
- “Therapeutically effective amount” also includes an amount of the combination of compounds claimed that is effective to treat the desired indication.
- the combination of compounds can be a synergistic combination. Synergy, as described, for example, by Chou and Talalay, Adv. Enzyme Regul. 1984, 22:27-55, occurs when the effect of the compounds when administered in combination is greater than the additive effect of the compounds when administered alone as a single agent. In general, a synergistic effect is most clearly demonstrated at sub-optimal concentrations of the compounds. Synergy can be in terms of lower cytotoxicity, increased effect, or some other beneficial effect of the combination compared with the individual components.
- Obesity is defined as having a body mass index (BMI) of 30 or above.
- the index is a measure of an individual's body weight relative to height.
- BMI is calculated by dividing body weight (in kilograms) by height (in meters) squared. Normal and healthy body weight is defined as having a BMI between 20 and 24.9. Overweight is defined as having a BMI > 25. Obesity has reached epidemic proportions in the U.S., with 44 million obese Americans, and an additional eighty million deemed medically overweight.
- Obesity is a disease characterized as a condition resulting from the excess accumulation of adipose tissue, especially adipose tissue localized in the abdominal area. It is desirable to treat overweight or obese patients by reducing their amount of adipose tissue, and thereby reducing their overall body weight to within the normal range for their sex and height. In this way, their risk for co-morbidities such as diabetes and cardiovascular disease will be reduced. It is also desirable to prevent normal weight individuals from accumulating additional, excess adipose tissue, effectively maintaining their body weights at a BMI ⁇ 25, and preventing the development of co-morbidities. It is also desirable to control obesity, effectively preventing overweight and obese individuals from accumulating additional, excess adipose tissue, reducing the risk of further exacerbating their co-morbidities.
- Cannabinoid receptors are located in a number of peripheral (non-CNS) tissues, including thyroid gland, adrenal gland, reproductive organs, adipose tissue, liver, muscle, and gastrointestinal tract.
- Cannabinoid receptor antagonists/inverse agonists being developed to treat obesity and smoking cessation, regardless of route of administration, enter the CNS from the systemic circulation. While present in the systemic circulation, such drugs have access to peripheral tissues.
- cannabinoid receptor antagonists/inverse agonists intended to enter the CNS from the systemic circulation in order to treat obesity and smoking cessation also have access to cannabinoid receptors in peripheral tissues.
- a cannabinoid receptor antagonist/inverse agonist useful for the present invention may have some access to the CNS from the systemic circulation.
- Drugs enter the CNS from the systemic circulation by crossing the blood- brain barrier (BBB).
- BBB blood- brain barrier
- the BBB is a highly specialized 'gate-keeper' that protects the brain by preventing the entry of many potentially harmful substances into the CNS from the systemic circulation. Much is known about the BBB, and of the physical-chemical properties required for compounds transported across it.
- Drugs that do not cross the BBB into the CNS or that are readily eliminated through transport mechanisms [J Clin Invest. 97, 2517(1996)] are known in the literature and have low CNS activity due to their inability to develop brain levels necessary for pharmacological action.
- the BBB has at least one mechanism to remove drugs prior to their accumulation in the CNS.
- P-Glycoproteins (P-gp) localized in plasma membrane of the BBB can influence the brain penetration and pharmacological activity of many drugs through translocation across membranes.
- P-gp P-Glycoproteins
- the lack of accumulation into the brain by some drugs can be explained by their active removal from the brain by P-gp residing in the BBB.
- the typical opioid drug loperamide clinically used as an antidiarrheal, is actively removed from the brain by P-gp, thus explaining its lack of opiate-like CNS effects.
- domperidone a dopamine receptor blocker that participates in the P-gp transport [J Clin Invest. 97, 2517(1996)].
- dopamine receptor blockers that cross the BBB can be used to treat schizophrenia
- the readily-eliminated domperidone can be used to prevent emesis, without the likelihood of producing adverse CNS effects.
- agents possessing structural characteristics that retard or prevent BBB penetration or contribute to participation in active elimination processes have been identified in various classes of therapeutics. These include antihistamines [Drug Metab. Dispos. 31, 312 (2003)], beta-adrenergic receptor antagonists (B-blockers) [Eur. J. Clin. Pharmacol. 28, Suppl: 21-3 (1985); Br. J. Clin.
- non-nucleoside reverse transcriptase inhibitors (NNRTIs) [J. Pharm ScL, 88(10) 950-954 (1999)]
- opioid antagonists This latter group has been tested in relation to their activity in the GI tract.
- NRTIs non-nucleoside reverse transcriptase inhibitors
- These peripherally selective opioid antagonists are described in various US patents as being useful in the treatment of non-CNS pathologies in mammals, in particular those of the GI tract [see US 5,260,542; US 5,434,171; US 5,159,081; and US 5,270,238].
- Other types of non-brain penetrant compounds can be prepared through the creation of a charge within the molecule.
- a methyl group to the tertiary amine functionality of the drugs scopolamine or atropine, unlike the parent molecules, prevents their passage across the BBB through the presence of a positive charge.
- the new molecules methyl-scopolamine and methyl-atropine retain their full anticholinergic pharmacological properties.
- these drugs can also be used to treat peripheral diseases, without the concern of adverse CNS effects.
- the quaternary ammonium compound methylnaltrexone is also used for the prevention and/or treatment of opioid and non-opioid induced side effects associated with opioid administration.
- minimal brain concentrations means levels that are too low to be therapeutically effective for the treatment of a CNS indication or too low to cause significant or measurable deleterious or undesired side effects.
- R, R , R , X, X 1 , V and Z is a group capable of reducing or limiting the CNS activity of compound AA. This reduced or limited CNS activity occurs via at least one of R, R , R , X, X 1 , V and Z being a group that either limits compound AA' s ability to cross the BBB relative to that of rimonabant or enables it to be actively removed from the brain at a rate greater than that of rimonabant.
- Examples of the amount of compound AA present in the brain can include (a) from 50, 55, 60, 65, 70, 75, 80, 85, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, to 100% lower than rimonabant, (b) from 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, to 100% lower than rimonabant, and (c) from 98, 99, to 100% lower than rimonabant, when administered at the same dosage.
- the compounds of the present invention are expected to be cannabinoid receptor antagonists or inverse agonists.
- An inverse agonist is compound that not only blocks the action of the endogenous agonist at the receptor, but also exhibits its own activity which is usually the opposite of that shown by the agonist. Inverse agonists are also effective against certain types of receptors (e.g. certain histamine receptors / GABA receptors) which have intrinsic activity without the interaction of a ligand upon them (also referred to as 'constitutive activity').
- Most methods of treating obesity are dependent on a significant reduction in energy intake, either by a decrease in food intake (e.g., sibutramine) or by inhibition of fat absorption (e.g., orlistat).
- adipose tissue may be reduced in the absence of a significant reduction in food intake.
- the weight loss comes from the treatment with a compound of the present invention, largely independent of, though not totally dissociated from, appetite and food intake.
- adipose tissue loss occurs while food intake is maintained, increased or (a) about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, or 20% below the normal range of the subject prior to being treated in accordance with the present invention (i.e., its pre-administration level), (b) about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15% below its pre-administration level, (c) about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10% below its pre-administration level, or (d) about 1, 2, 3, 4, or 5% below its pre- administration level.
- loss of adipose tissue can be accompanied by a concomitant loss of lean muscle mass. This is particularly evident in cancer patients who show a generalized wasting of body tissues, including adipose tissue and lean muscle mass. In the present invention, however, it can be desirable for body fat to be significantly reduced in the absence of a significant reduction in lean body mass. Adipose tissue loss comes from treatment with a compound of the present invention, independent of a significant change in lean body mass.
- adipose tissue loss can occur while lean body mass is maintained, increased, or (a) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30% below the normal range of the subject prior to being treated in accordance with the present invention (i.e., its pre-administration level), (b) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15% below pre-administration levels, (c) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10% below pre-administration levels, or (d) is no more than about 1, 2, 3, 4, or 5% below pre-administration levels.
- loss of adipose tissue can be accompanied by a concomitant loss of water mass. This is particularly evident with diet regimens that promote dehydration.
- it can be desirable for body fat to be significantly reduced in the absence of a significant reduction in water mass.
- adipose tissue loss comes from treatment with a compound of the present invention, independent of a significant change in water mass.
- adipose tissue loss occurs while water mass is maintained, increased, or (a) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, or 30% below the normal range of the subject prior to being treated in accordance with the present invention (i.e., its pre-administration level), (b) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, or 15% below pre-administration levels, (c) is no more than about 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10% below pre-administration levels, or (d) is no more than about 1, 2, 3, 4, or 5% below pre-administration levels.
- Sibutramine and orlistat are currently marketed for use in the treatment of obesity. These two compounds achieve weight loss through entirely different mechanisms. Sibutramine, a CNS appetite suppressant, inhibits the neuronal reuptake of serotonin and noradrenaline. Orlistat inhibits gut lipase enzymes that are responsible for breaking down ingested fat.
- Cannabinoid receptor antagonists/inverse agonists can promote weight loss through inhibition of peripheral cannabinoid receptors, a mechanism entirely different from appetite suppressants, gut lipase inhibitors, and other agents with similar indications (e.g., serotonin agonists, leptin, fatty acid synthase inhibitors, and monoamine oxidase (MAO) inhibitors).
- Co-administration of a cannabinoid receptor antagonist/inverse agonist together with one or more other agents that are useful for treating the indications described above e.g., obesity, diabetes, cardiometabolic disorders, and a combination thereof
- the present invention provides a method of treating obesity, diabetes, and/or cardiometabolic disorders, comprising administering a therapeutically effective amount of a compound of the present invention and a second component effective for treating the desired indication.
- anti-obesity agents include, but are not limited to: l) growth hormone secretagogues; 2) growth hormone secretagogue receptor agonists/antagonists; 3) melanocortin agonists; 4) Mc4r (melanocortin 4 receptor) agonists; 5) .beta.
- diabetes disorders include treating Type 1 diabetes, Type 2 diabetes, inadequate glucose tolerance, and insulin resistance.
- Examples of second components useful for treating diabetes include (a) insulin sensitizers including (i) PPAR- ⁇ agonists such as the glitazones (e.g. troglitazone, pioglitazone, englitazone, MCC-555, rosiglitazone), and compounds disclosed in WO97/27857, 97/28115, 97/28137, and 97/27847; and (ii) biguanides such as metformin and phenformin; (b) insulin or insulin mimetics; (c) sulfonylureas such as tolbutamide and glipizide, or related materials; (d) ⁇ -glucosidase inhibitors (e.g., acarbose); (e) cholesterol lowering agents such as (i) HMG-CoA reductase inhibitors (lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, rivastatin
- the compounds of the present invention are expected to be CBl receptor inhibitors/inverse agonists and are expected to be useful for treating diseases mediated by the CBi receptor.
- the compounds of the present are expected to possess an affinity in vitro for the central and/or peripheral cannabinoid receptors under the experimental conditions described by Devane et al, Molecular Pharmacology, 1988, 34, 605-613.
- the compounds according to the invention are also expected to possess an affinity for the cannabinoid receptors present on preparations of electrically stimulated isolated organs.
- CBl receptor affinities can be determined using membrane preparations of Chinese hamster ovary (CHO) cells in which the human cannabinoid CBl receptor is stably transfected (Biochem J. 1991, 279, 129-134) in conjunction with [3H]CP-55,940 as radioligand.
- IC50 values can be determined from at least three independent measurements.
- the compound(s) of the present invention can be administered in any convenient manner (e.g., enterally or parenterally).
- methods of administration include orally and transdermally.
- routes of administering the compounds of the present invention may vary significantly.
- sustained release compositions may be favored.
- Other acceptable routes may include injections (e.g., intravenous, intramuscular, subcutaneous, and intraperitoneal); subdermal implants; and, buccal, sublingual, topical, rectal, vaginal, and intranasal administrations.
- Bioerodible, non- bioerodible, biodegradable, and non-biodegradable systems of administration may also be used.
- oral formulations include tablets, coated tablets, hard and soft gelatin capsules, solutions, emulsions, and suspensions.
- the main active ingredient can be mixed with a pharmaceutical vehicle, examples of which include silica, starch, lactose, magnesium stearate, and talc.
- the tablets can be coated with sucrose or another appropriate substance or they can be treated so as to have a sustained or delayed activity and so as to release a predetermined amount of active ingredient continuously.
- Gelatin capsules can be obtained by mixing the active ingredient with a diluent and incorporating the resulting mixture into soft or hard gelatin capsules.
- a syrup or elixir can contain the active ingredient in conjunction with a sweetener, which is preferably calorie-free, an antiseptic (e.g., methylparaben and/or propylparaben), a flavoring, and an appropriate color.
- a sweetener which is preferably calorie-free, an antiseptic (e.g., methylparaben and/or propylparaben), a flavoring, and an appropriate color.
- Water-dispersible powders or granules can contain the active ingredient mixed with dispersants or wetting agents or with suspending agents such as polyvinylpyrrolidone, as well as with sweeteners or taste correctors. Rectal administration can be effected using suppositories, which are prepared with binders melting at the rectal temperature (e.g., cocoa butter and/or polyethylene glycols).
- Parenteral administration can be effected using aqueous suspensions, isotonic saline solutions, or injectable sterile solutions, which contain pharmacologically compatible dispersants and/or wetting agents (e.g., propylene glycol and/or polyethylene glycol).
- the active ingredient can also be formulated as microcapsules or microspheres, optionally with one or more carriers or additives.
- the active ingredient can also be presented in the form of a complex with a cyclodextrin, for example ⁇ -, ⁇ -, or ⁇ - cyclodextrin, 2-hydroxypropyl- ⁇ -cyclodextrin, and/or methyl- ⁇ -cyclodextrin.
- the dose of the compound of the present invention administered daily will vary on an individual basis and to some extent may be determined by the severity of the disease being treated (e.g., obesity, diabetes, and cardiometabolic disorders).
- the dose of the compound of the present invention will also vary depending on the compound administered. Examples of dosages of compounds of the present invention include from about 0.01, 0.02, 0.03, 0.04, 0.05, 0.06, 0.07, 0.08, 0.09, 0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 76, 80, 85, 90, 95, to 100 mg/kg of mammal body weight.
- the compound can be administered in a single dose or in a number of smaller doses over a period of time.
- the length of time during which the compound is administered varies on an individual basis, and can continue until the desired results are achieved (i.e., reduction of body fat, or prevention of a gain in body fat). Therapy could, therefore, last from 1 day to weeks, months, or even years depending upon the subject being treated, the desired results, and how quickly the subject responds to treatment in accordance with the present invention.
- a possible example of a tablet of the present invention is as follows.
- a possible example of a capsule of the present invention is as follows. Ingredient mg/T ablet
- the active ingredient has a suitable particle size.
- the crystalline lactose and the microcrystalline cellulose are homogeneously mixed with one another, sieved, and thereafter the talc and magnesium stearate are admixed.
- the final mixture is filled into hard gelatin capsules of suitable size.
- the compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis (e.g., see EP 0,658,546, J Med Chem 2002, 45, 2708).
- the compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below.
- the reactions are performed in a solvent appropriate to the reagents and materials employed and suitable for the transformations being effected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed.
- Scheme 1 shows how the condensation of a propiophenone with diethyl oxalate in the presence of a base such as lithium hexamethydilsilazide should afford, after acidification with hydrochloric acid solution, the diketoester (step a). Heating this ester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester along with the uncyclized imine (step b). These materials may be separated due to their solubility differences, or heated together in ethanolic hydroxide solution to cause further conversion of the imine to the pyrazole along with concomitant saponification of the ester to the carboxylic acid (step c).
- a base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with ethyl N- aminonipecotate should afford the hydrazide ester (step d). Hydrolysis of the ester with lithium hydroxide and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step e). Treatment of this acid with thionyl chloride followed by ammonia should afford the carboxamide (step f).
- the acid chloride generated from the product of step c can be treated with N-amino-2-pyrrolidine methanol 0-CH 2 CO 2 Et derivative to produce the hydrazide ester (step g). Subsequent hydrolysis of this ester with aqueous base should afford the carboxylic acid (step h).
- the acid chloride generated from the acid of Scheme 1 can be treated with 2-amino-cyclopentylmethanol 0-CH 2 CO 2 Et derivative to produce the amide ester (step c). Subsequent hydrolysis of this ester with aqueous base should afford the carboxylic acid (step d).
- Hydrolysis of the ester with lithium hydroxide in aqueous THF solution and acidification with dilute hydrochloric acid solution should yield the amide carboxylic acid (step b).
- Scheme 4 illustrates how the treatment of 4'-benzyloxypropiophenone with ethyl-t-butyl oxalate in the presence of an equivalent of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketoester (step a).
- base such as lithium hexamethydilsilazide
- step c Heating this diketo-diester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester along with the uncyclized imine (step c). These materials may be separated due to their solubility differences, or through flash column chromatography.
- the pyrazole upon treatment with trifluoroacetic acid in methylene chloride, should afford the carboxylic acid (step d). Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide ester (step e).
- Scheme 5 shows how the treatment of 3'-nitropropiophenone with diethyl oxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-ester (step a). Heating this ester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester along with the uncyclized imine (step b). These materials may be separated due to their solubility differences, or heated together in ethanolic hydroxide solution to cause further conversion of the imine to the pyrazole along with concomitant saponification of the ester to the carboxylic acid (step c).
- base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide (step d).
- the nitro compound can be reduced to the aniline using sodium dithionite in aqueous dioxane containing concentrated ammonium hydroxide solution to give the aniline (step e).
- Reaction of the aniline with ethyl 4- bromocrotonate in acetone at reflux in the presence of potassium carbonate should afford the ester (step f).
- Hydrolysis of the ester with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step g).
- the aniline can be treated with ethyl malonyl chloride in the presence of base to yield the ester (step h).
- the ester can then be hydrolyzed with lithium hydroxide in aqueous THF solution, and after acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step i).
- Scheme 7 depicts how the condensation of a propiophenone with ethyl-t- butyl oxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-ester (step a). Heating this diketo-ester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester which can be separated from the uncyclized imine via their solubility differences or through flash chromatography (step b). The pyrazole, upon treatment with trifluoroacetic acid in methylene chloride, should afford the carboxylic acid (step c).
- base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide ester (step d). Hydrolysis of the remaining ester with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step e). The carboxylic acid can then be treated with thionyl chloride followed by ammonia to produce the carboxamido compound (step f).
- Scheme 8 illustrates how the Reaction of a propiophenone with ethyl-t- butyl oxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-ester (step a). Heating this diketo-ester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester which can be separated from the uncyclized imine via their solubility differences or through flash chromatography (step b). The pyrazole, upon treatment with trifluoroacetic acid in methylene chloride, should afford the carboxylic acid (step c).
- base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide ester (step d). Hydrolysis of the remaining ester with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step e).
- the carboxylic acid can then be treated with thionyl chloride followed by ammonia, or with Boc anhydride (BOC 2 O) in THF in the presence of pyridine, followed by a solution of ammonia in THF at 0 degrees to ambient temperature to produce the benzamide (step f).
- Scheme 9 shows how the treatment of a propiophenone with ethyl-t-butyl oxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-ester (step a). Heating this diketo-ester with a hydrazine of an ethyl aryloxyacetate in a solvent such as ethanol should produce the pyrazole ester which can be separated from the uncyclized imine via their solubility differences or through flash chromatography (step b).
- base such as lithium hexamethydilsilazide
- the pyrazole upon treatment with trifluoroacetic acid in methylene chloride, should afford the carboxylic acid (step c).
- Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide ester (step d).
- Hydrolysis of the remaining ester with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step e).
- the carboxylic acid can then be treated with thionyl chloride followed by ammonia to produce the aryloxyacetamide (step f).
- Scheme 10 describes how the treatment of a propiophenone with diethyloxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-ester (step a). Heating this diketo-ester with a hydrazine of an arylnitrile in a solvent such as ethanol should produce the pyrazole ester which can be separated from the uncyclized imine via their solubility differences or through flash chromatography (step b). The pyrazole, upon treatment with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should afford the carboxylic acid (step c).
- base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydazide ester (step d).
- the nitrile can be treated with HCl gas in a solution of chloroform and methanol at about minus 15 to 0 degrees C to form the imidate ester which can immediately be converted to the carboxamidine by subsequent treatment with ammonium carbonate in methanol.
- Scheme 11 depicts how the reaction of ethyl benzoylacetate with ethyl-t- butyl oxalate in the presence of base, such as lithium hexamethydilsilazide, should afford, after acidification with hydrochloric acid solution, the diketo-diester (step a). Heating this diketo-diester with an aryl hydrazine in a solvent such as ethanol should produce the pyrazole ester which can be separated from the uncyclized imine via their solubility differences or through flash chromatography (step b). The pyrazole, upon treatment with trifluoroacetic acid in methylene chloride, should afford the carboxylic acid (step c).
- base such as lithium hexamethydilsilazide
- step d Subsequent conversion of the carboxylic acid to the acid chloride using thionyl chloride followed by treatment with N-aminopiperidine should afford the hydrazide ester (step d). Hydrolysis of the remaining ester with lithium hydroxide in aqueous THF solution, and acidification with dilute hydrochloric acid solution should yield the hydrazide carboxylic acid (step e).
- the carboxylic acid can then be treated with thionyl chloride followed by ammonia, or with Boc anhydride (BOC 2 O) in THF in the presence of pyridine, followed by a solution of ammonia in THF at 0 degrees to ambient temperature to produce the carboxamido hydrazide (step f).
- One stereoisomer of a compound of the present invention may be a more potent cannabinoid receptor antagonist than its counterpart(s).
- stereoisomers are included in the present invention.
- separation of the racemic material can be achieved by HPLC using a chiral column or by a resolution using a resolving agent such as described in Wilen, S. H. Tables of Resolving Agents and Optical Resolutions 1972, 308 or using enantiomerically pure acids and bases.
- a chiral compound of the present invention may also be directly synthesized using a chiral catalyst or a chiral ligand, e.g., Jacobsen, E. Ace. Chem. Res. 2000, 33, 421-431 or using other enantio- and diastereo-selective reactions and reagents known to one skilled in the art of asymmetric synthesis.
- +++ an IC50 ⁇ 100 nM.
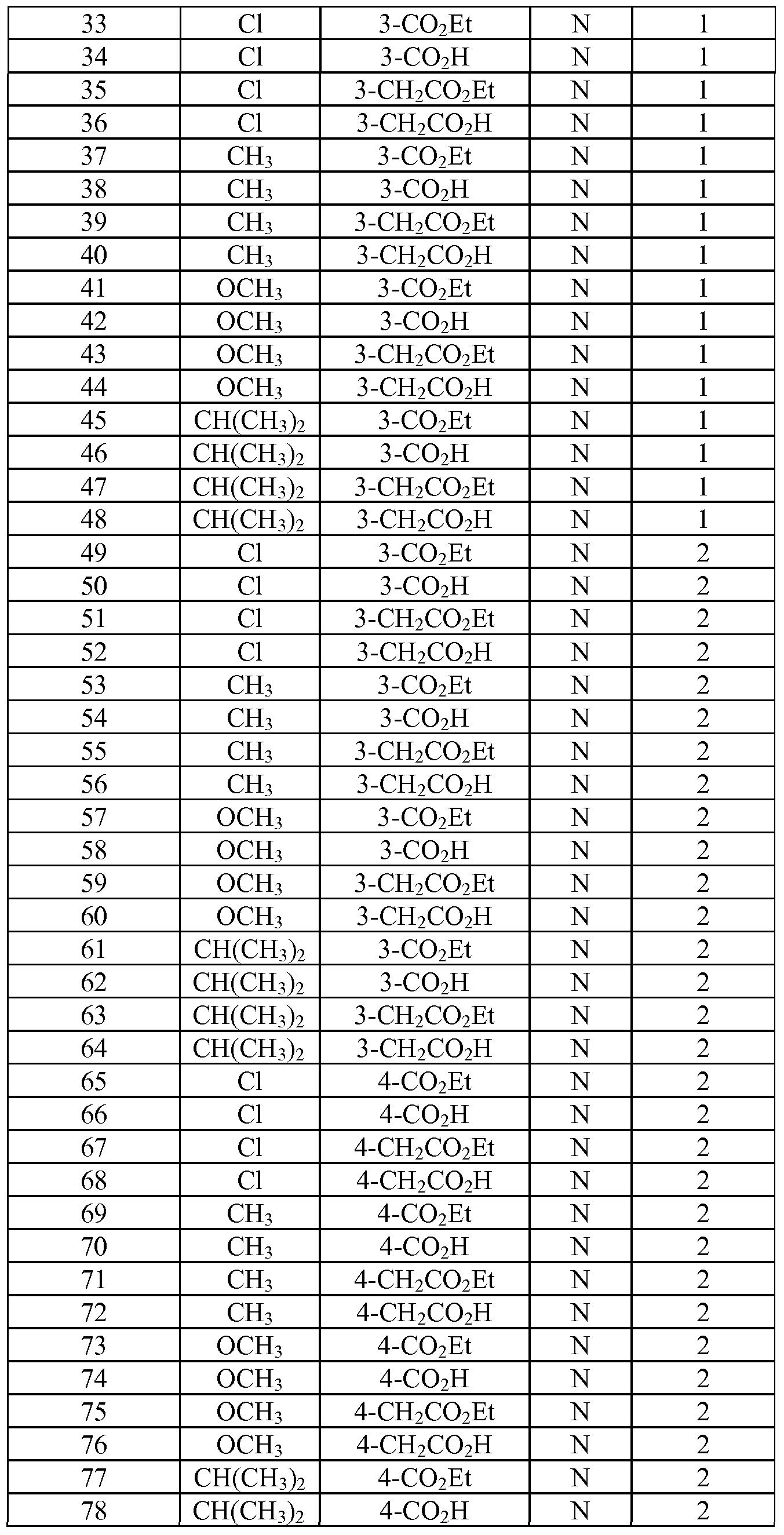
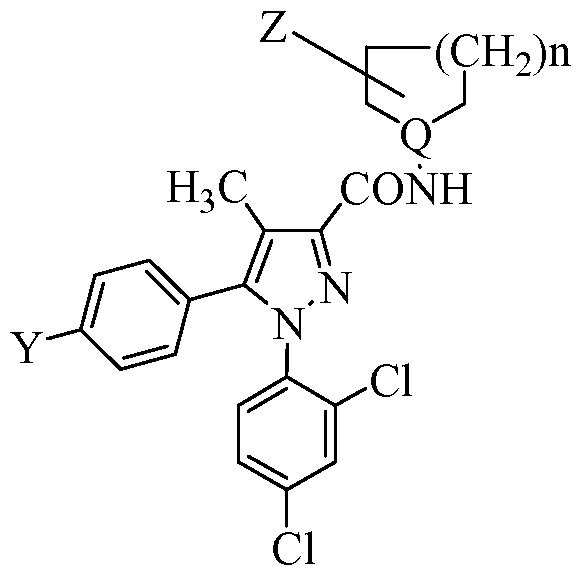
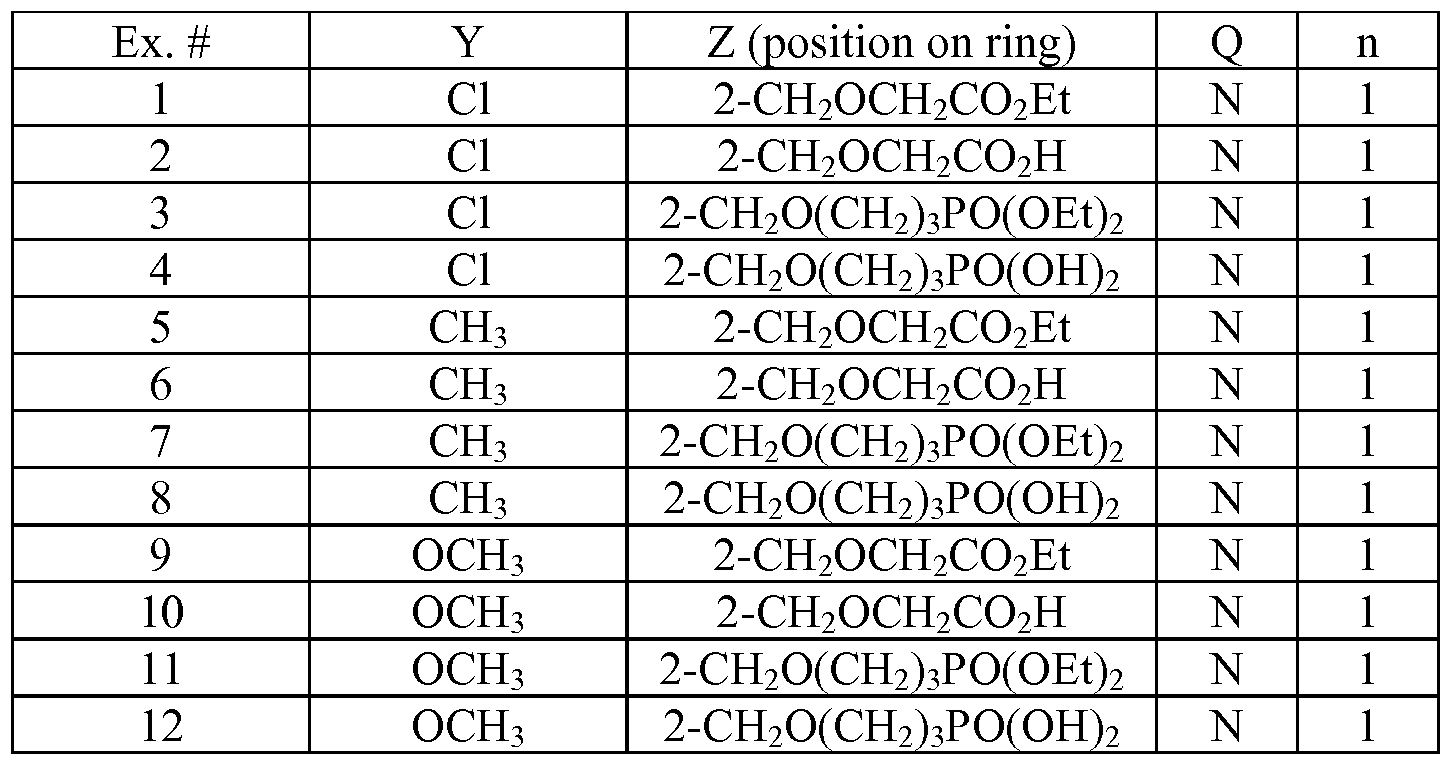
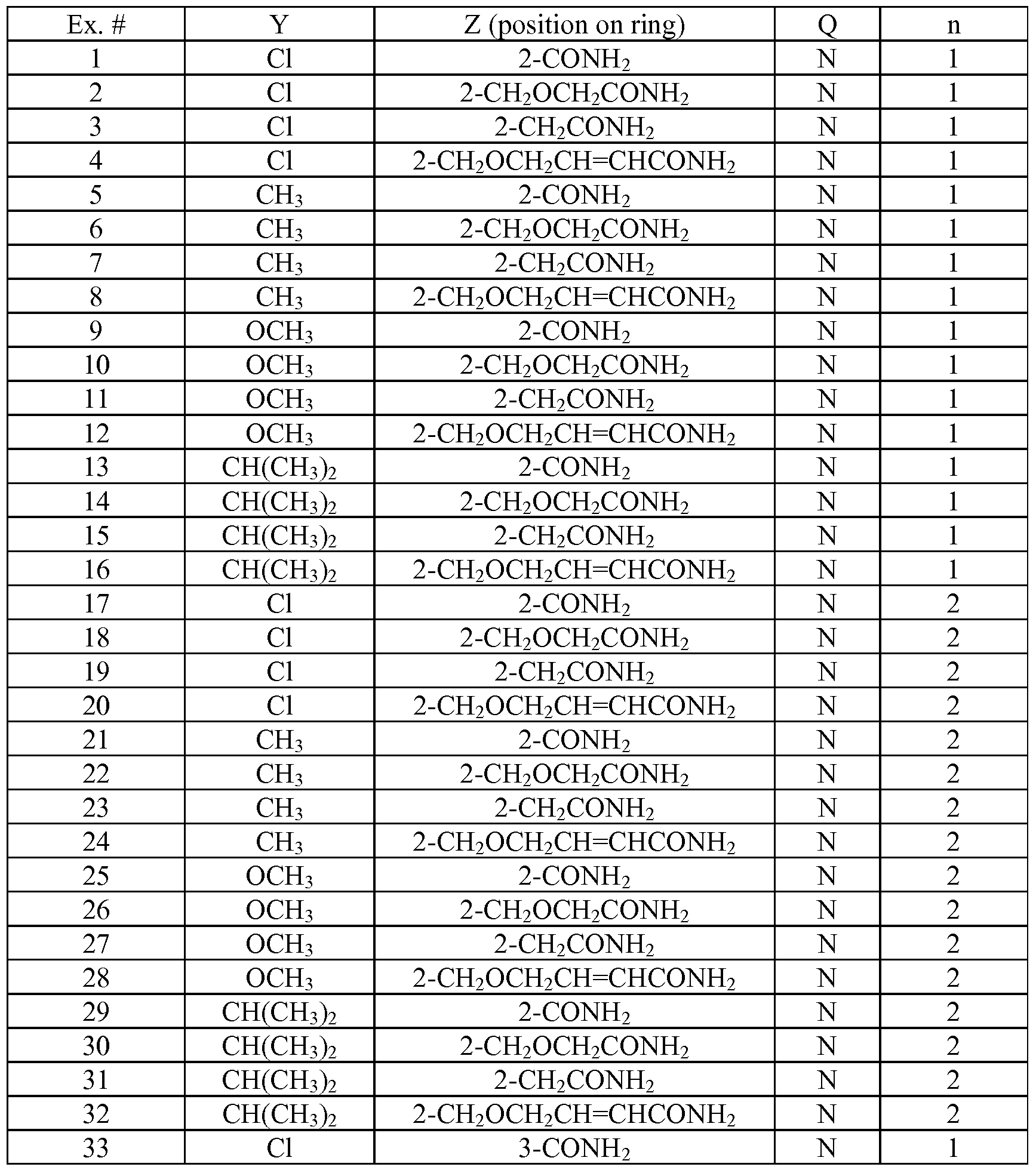
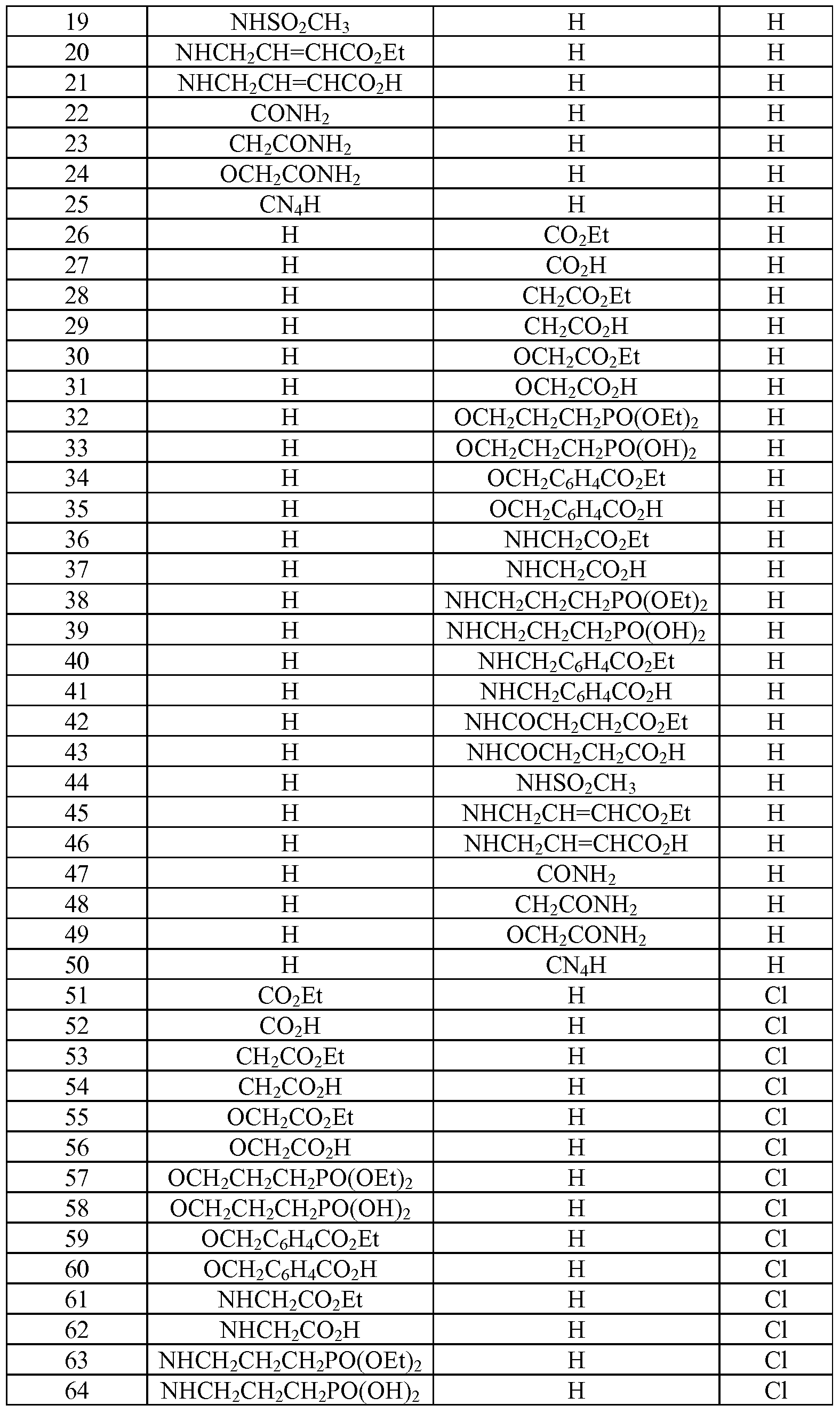
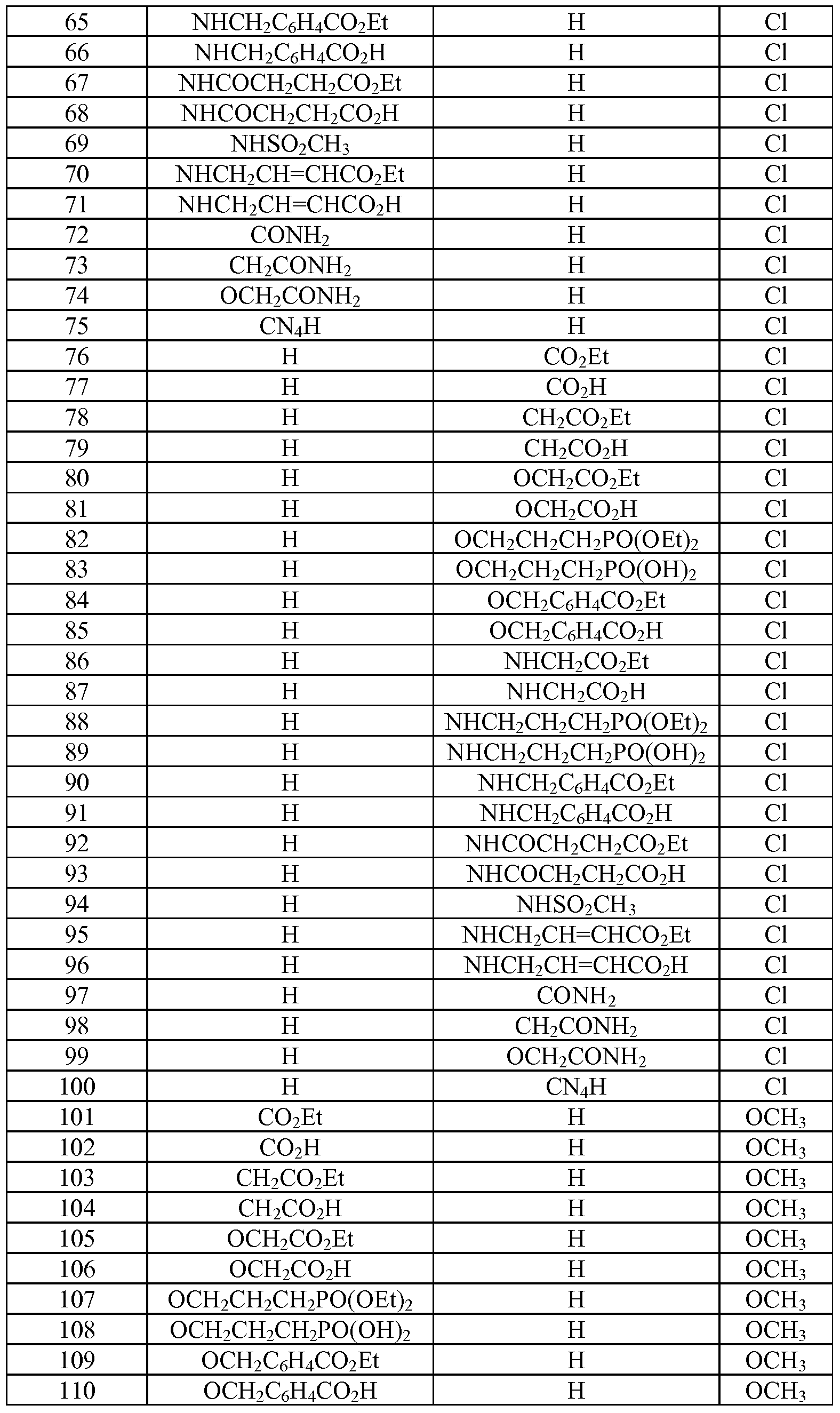
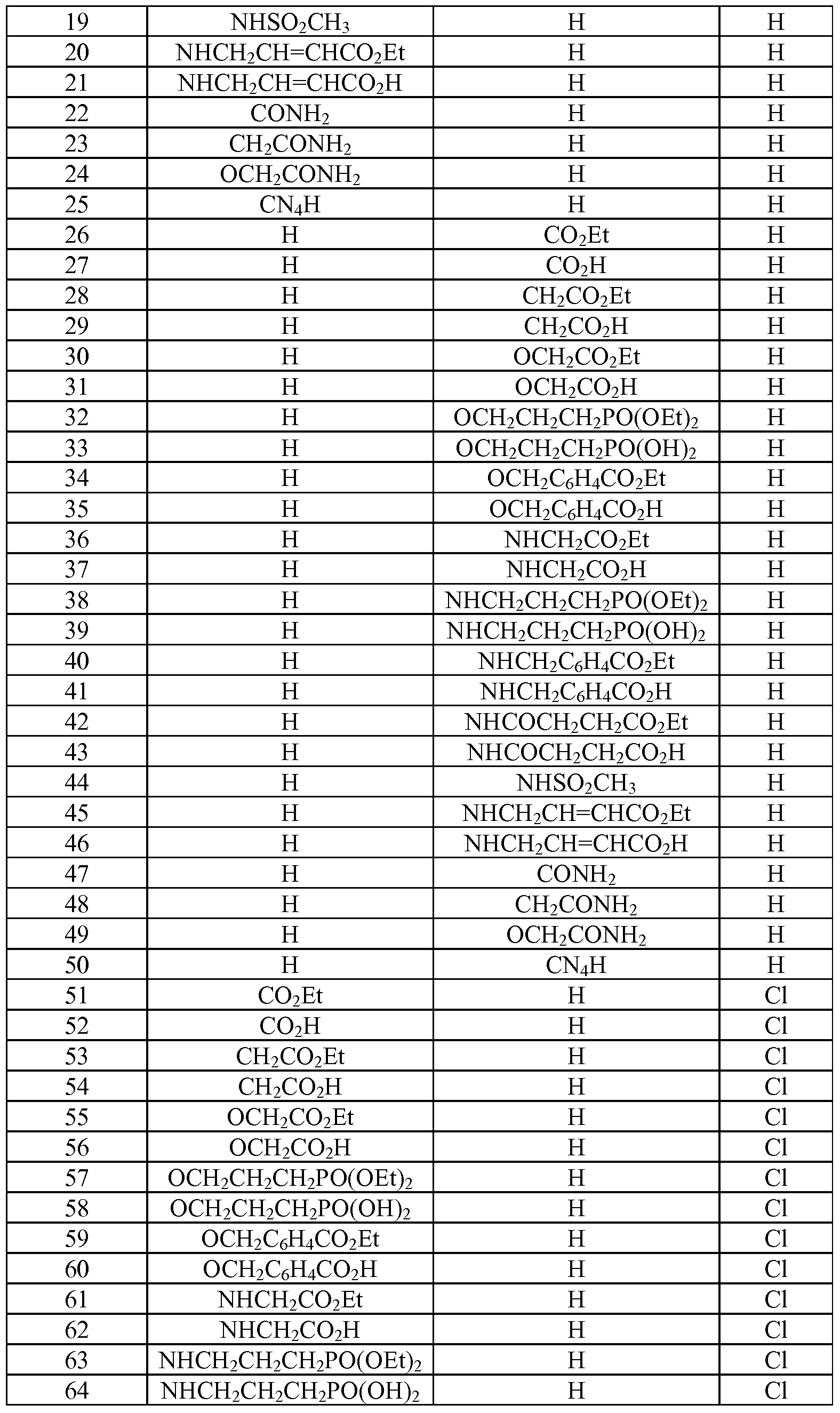
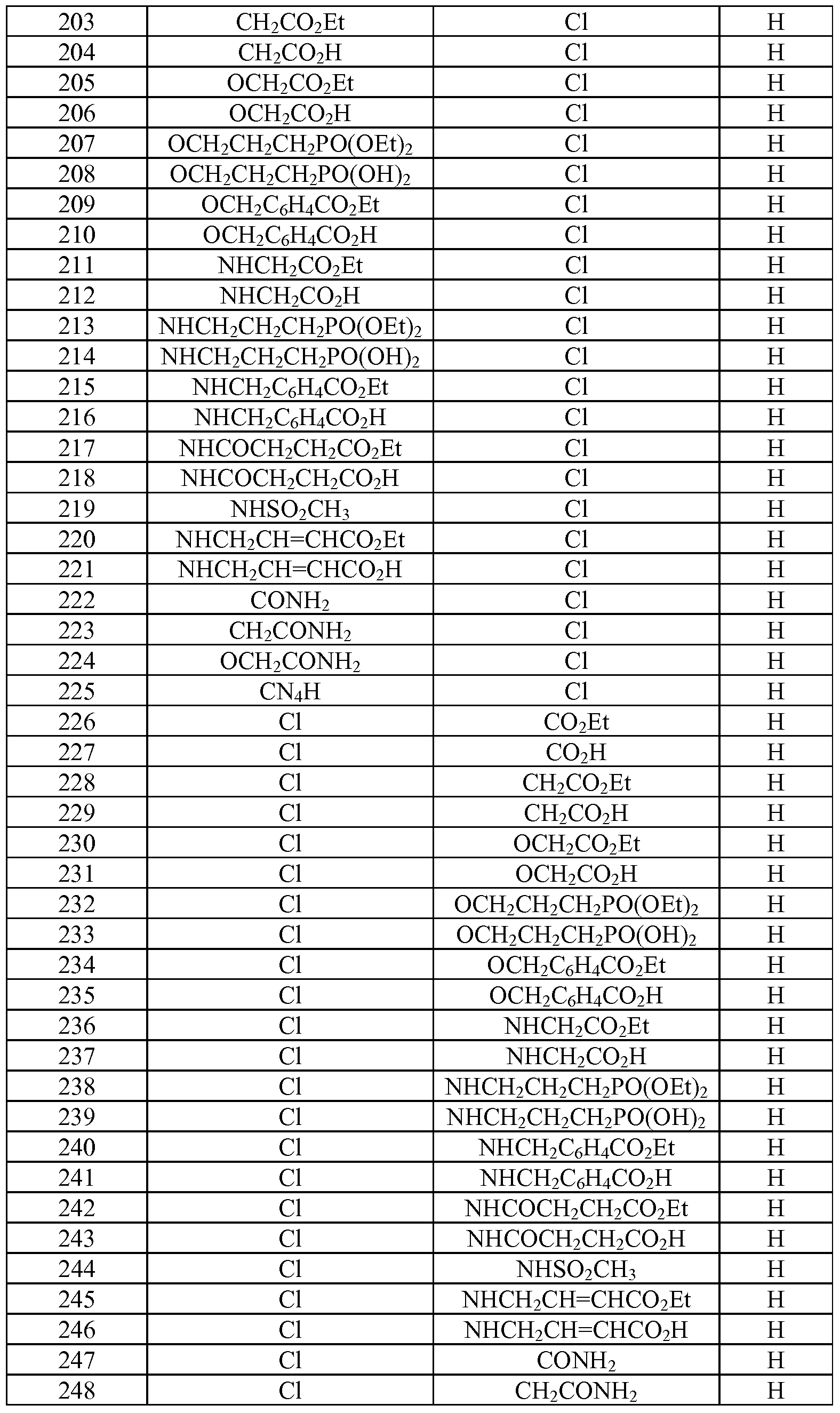
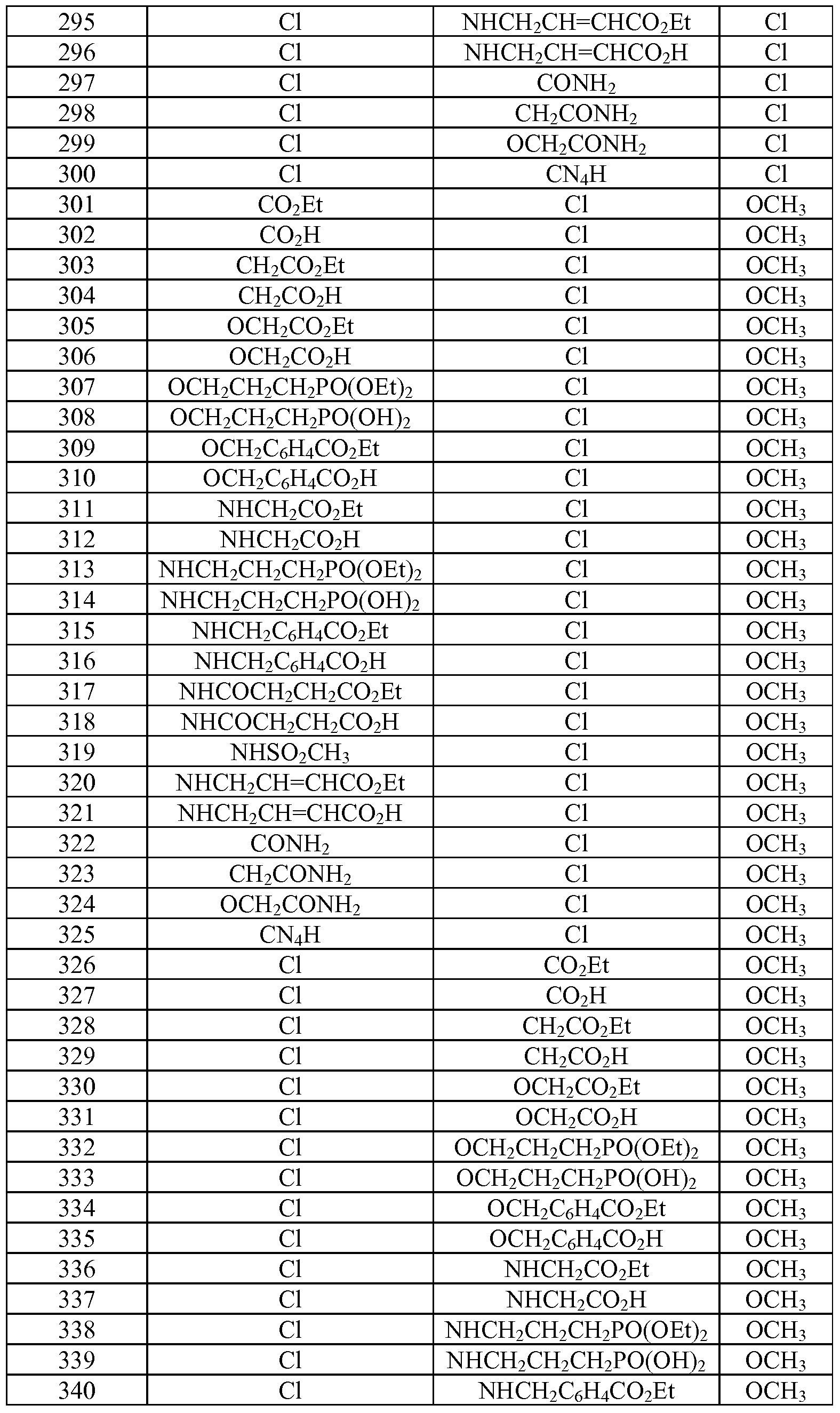
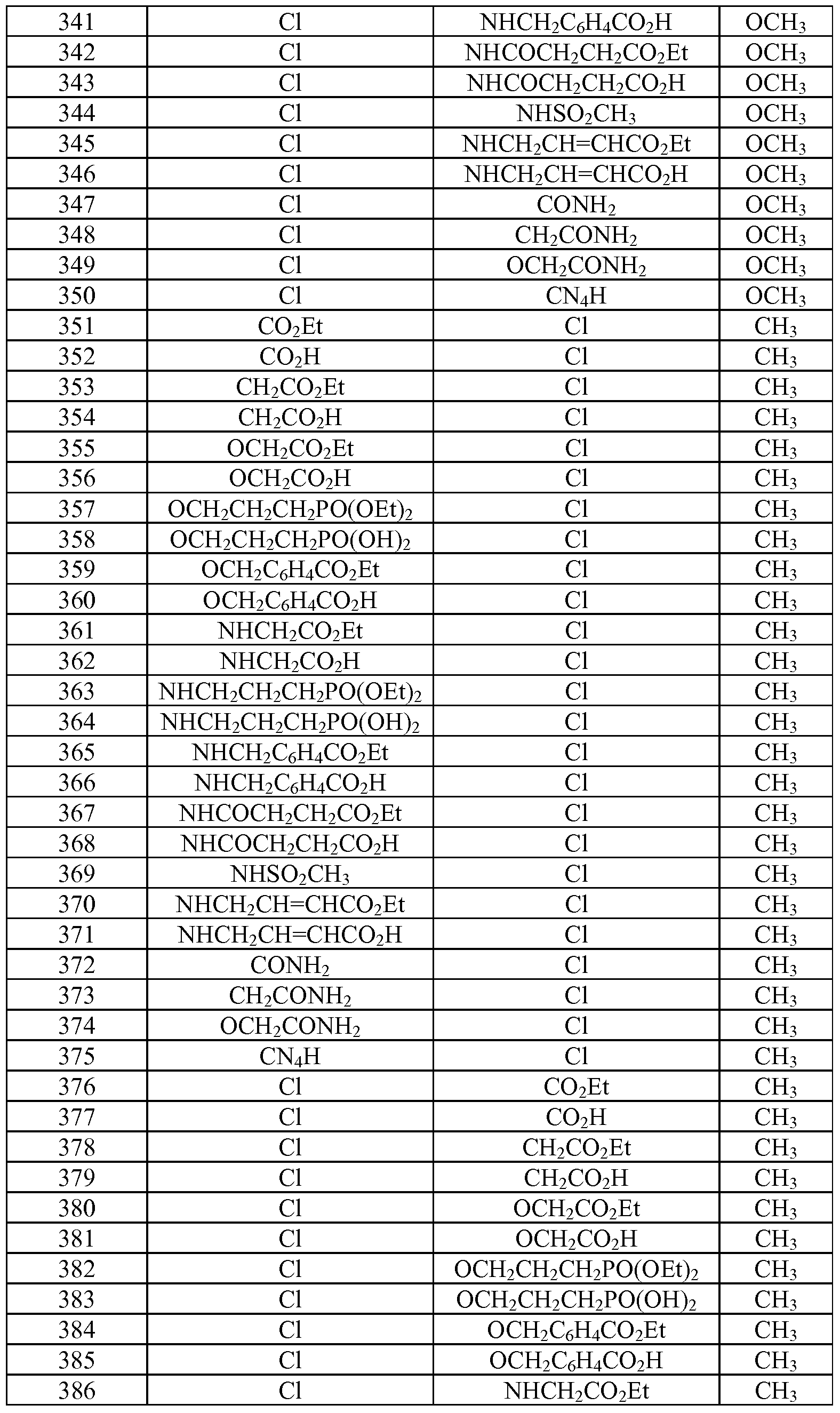
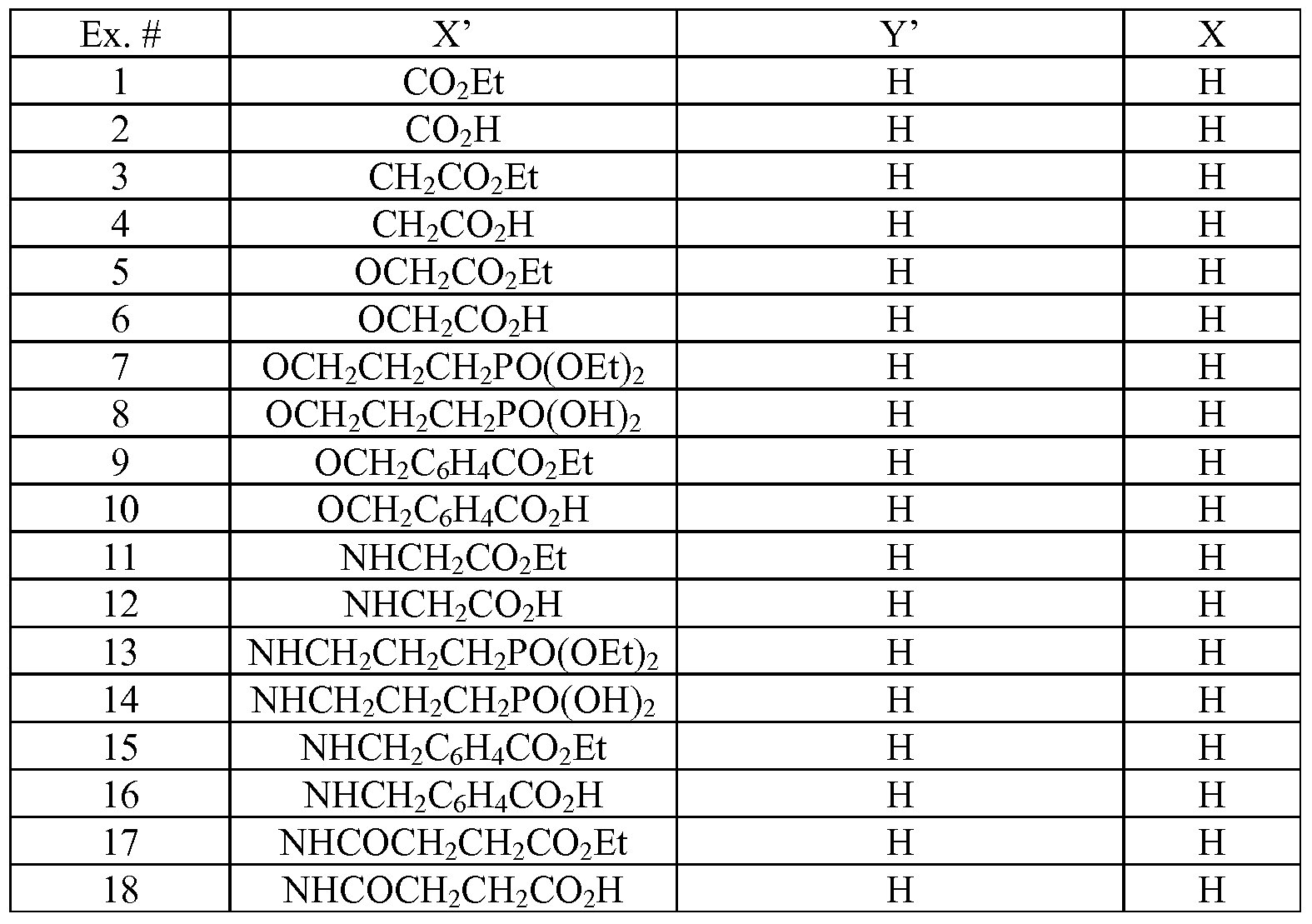
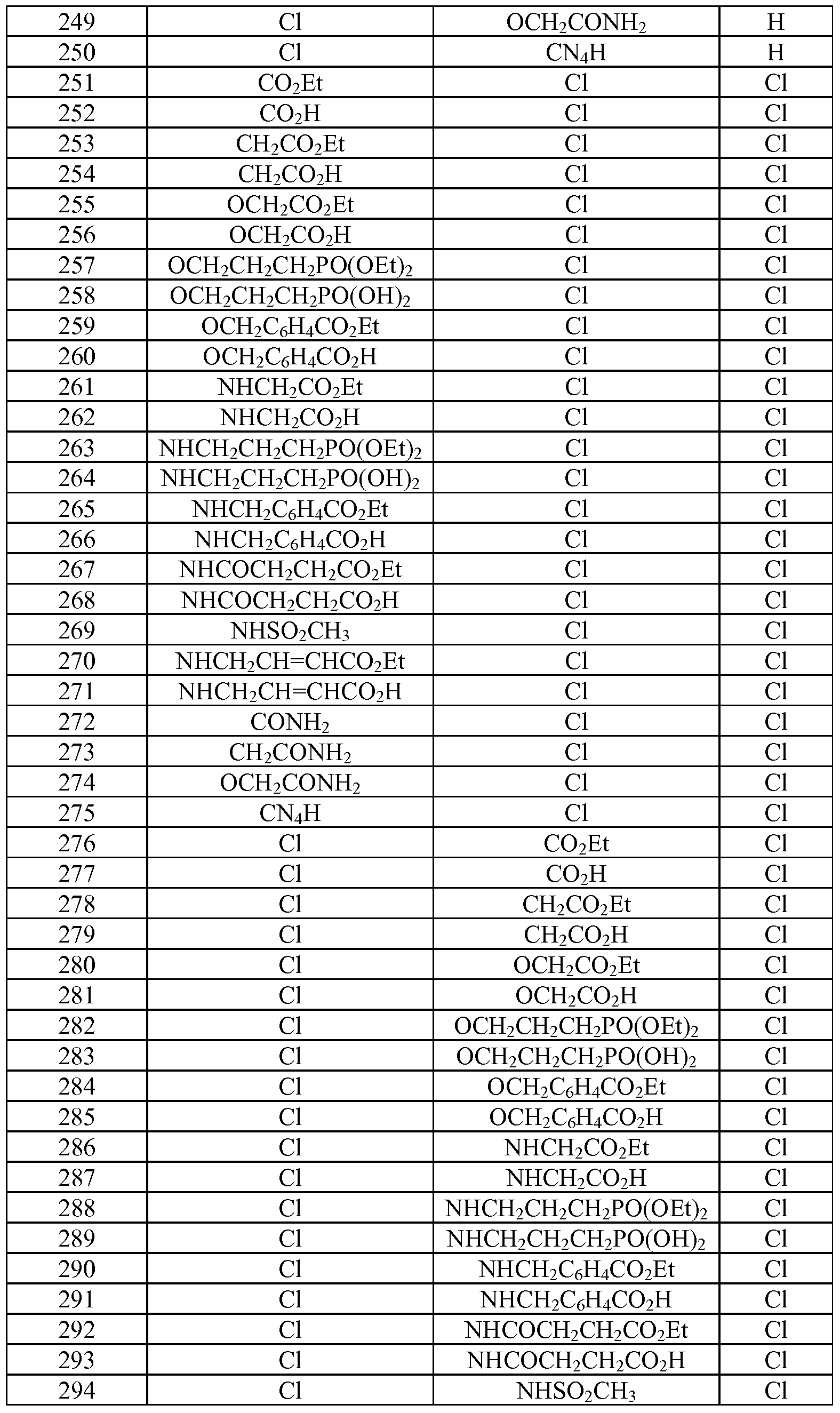
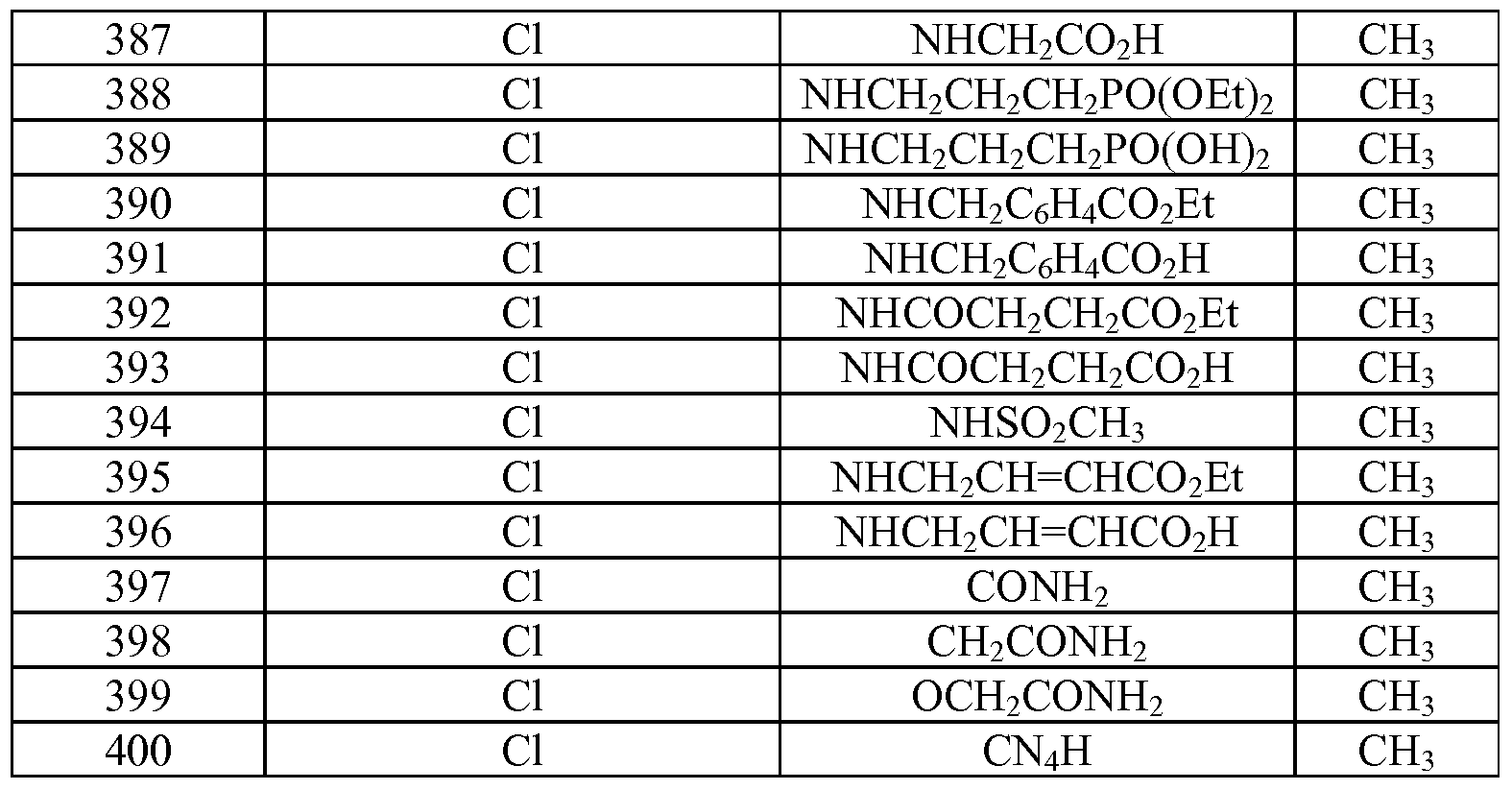
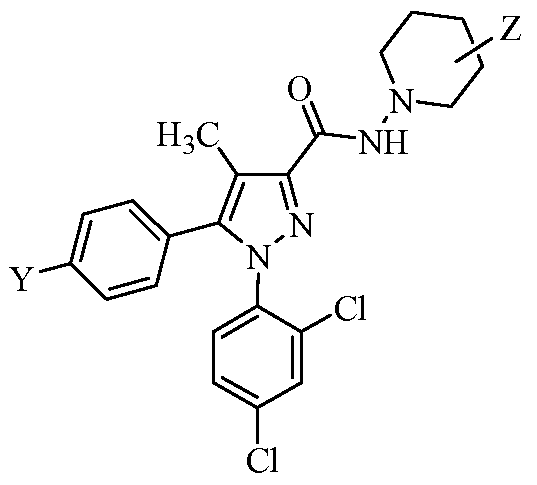
- Tables la-6b show representative examples of the compounds of the present invention. Each example in the tables represents an individual species of the present invention. Table Ia
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Obesity (AREA)
- Neurology (AREA)
- Endocrinology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Psychiatry (AREA)
- Child & Adolescent Psychology (AREA)
- Immunology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
- Vascular Medicine (AREA)
- Emergency Medicine (AREA)
- Reproductive Health (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description













































































































Claims












Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007226673A AU2007226673A1 (en) | 2006-03-10 | 2007-03-09 | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| JP2008558543A JP4851546B2 (en) | 2006-03-10 | 2007-03-09 | Cannabinoid receptor antagonist / inverse agonist useful for obesity treatment |
| CA002647031A CA2647031A1 (en) | 2006-03-10 | 2007-03-09 | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| EP07758206A EP1993560B1 (en) | 2006-03-10 | 2007-03-09 | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
| AT07758206T ATE538650T1 (en) | 2006-03-10 | 2007-03-09 | CANNABINOID RECEPTOR ANTAGONISTS / INVERSE AGONISTS FOR THE TREATMENT OF OBESITY |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US78148506P | 2006-03-10 | 2006-03-10 | |
| US60/781,485 | 2006-03-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007106721A2 true WO2007106721A2 (en) | 2007-09-20 |
| WO2007106721A3 WO2007106721A3 (en) | 2008-11-06 |
Family
ID=38510173
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/063631 Ceased WO2007106721A2 (en) | 2006-03-10 | 2007-03-09 | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US7687481B2 (en) |
| EP (1) | EP1993560B1 (en) |
| JP (1) | JP4851546B2 (en) |
| CN (1) | CN101437398A (en) |
| AT (1) | ATE538650T1 (en) |
| AU (1) | AU2007226673A1 (en) |
| CA (1) | CA2647031A1 (en) |
| WO (1) | WO2007106721A2 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008059207A1 (en) * | 2006-11-11 | 2008-05-22 | 7Tm Pharma A/S | Cannabinoid receptor modulators |
| JP2010513435A (en) * | 2006-12-18 | 2010-04-30 | 7ティーエム ファーマ エイ/エス | CB1 receptor modulator |
| DE102009036604A1 (en) | 2009-07-30 | 2011-02-03 | Aicuris Gmbh & Co. Kg | Substituted bis-arylpyrazolamides with terminal primary amide functionality and their use |
| EP2182807A4 (en) * | 2007-09-07 | 2011-02-23 | Jenrin Discovery | CANNABINOID RECEPTOR ANTAGONISTS / AGONISTS USEFUL FOR TREATING OBESITY |
| CN102250006A (en) * | 2011-05-12 | 2011-11-23 | 范如霖 | 3-Pyrazole carboxylic acid amide compound, its preparation method and its application in the preparation of drugs as CB1 receptor inhibitors |
| US8962845B2 (en) | 2011-09-30 | 2015-02-24 | National Health Research Institutes | Pyrazole compounds |
| WO2019150220A1 (en) * | 2018-01-30 | 2019-08-08 | Pi Industries Ltd. | Novel anthranilamides, their use as insecticide and processes for preparing the same. |
| US11180511B2 (en) | 2016-08-03 | 2021-11-23 | Friedrich-Alexander-Universität Erlangen-Nürnberg; Universitätsklinikum Erlangen | Diagnosis, treatment and prevention of neurotensin receptor-related conditions |
| US11401244B2 (en) | 2014-06-06 | 2022-08-02 | Research Triangle Institute | Apelin receptor (APJ) agonists and uses thereof |
| WO2022164239A1 (en) * | 2021-01-28 | 2022-08-04 | 주식회사 파미노젠 | Pyrazole-carboxamide derivative compound and use thereof |
| US11535630B2 (en) | 2015-12-09 | 2022-12-27 | Research Triangle Institute | Apelin receptor (APJ) agonists and uses thereof |
| RU2796538C2 (en) * | 2016-08-03 | 2023-05-25 | Фридрих-Александер-Универзитет Эрланген-Нюрнберг | Diagnosis, treatment and prevention of conditions associated with the neurotensin receptor |
| WO2024092205A1 (en) * | 2022-10-27 | 2024-05-02 | The Trustees Of Indiana University | Inhibition of ship1 as a therapeutic strategy for the treatment of alzheimer's disease |
| WO2025199092A1 (en) * | 2024-03-20 | 2025-09-25 | The Trustees Of Indiana University | Ship1 modulators and methods of treatment and uses thereof |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2008512364A (en) * | 2004-09-06 | 2008-04-24 | エフ.ホフマン−ラ ロシュ アーゲー | 4-Aminomethylbenzamidine derivatives and their use as factor Vila inhibitors |
| WO2007148062A1 (en) * | 2006-06-20 | 2007-12-27 | Astrazeneca Ab | Therapeutic agents |
| EP2025674A1 (en) | 2007-08-15 | 2009-02-18 | sanofi-aventis | Substituted tetra hydro naphthalines, method for their manufacture and their use as drugs |
| US7655685B2 (en) * | 2007-11-02 | 2010-02-02 | Jenrin Discovery, Inc. | Cannabinoid receptor antagonists/inverse agonists useful for treating metabolic disorders, including obesity and diabetes |
| RU2392943C2 (en) * | 2007-11-22 | 2010-06-27 | Людмила Александровна Орлова | Reducing diet therapy |
| NZ585704A (en) * | 2007-12-10 | 2012-08-31 | 7Tm Pharma As | Modulators of cannabinoid receptor CB1 for treating obesity |
| CA2709863A1 (en) * | 2007-12-18 | 2009-09-03 | Sanofi-Aventis | Azetidine derivatives, their preparation and their application in therapy |
| EP2242745A1 (en) * | 2008-02-07 | 2010-10-27 | Sanofi-Aventis | Novel phenyl-substituted imidazolidines, method for the production thereof, medicaments containing said compounds and use thereof |
| KR20230085944A (en) * | 2008-03-18 | 2023-06-14 | 아레나 파마슈티칼스, 인크. | Modulators of the prostacyclin (pgi2) receptor useful for the treatment of disorders related thereto |
| ES2349838B1 (en) * | 2009-05-04 | 2011-11-15 | Instituto Mediterraneo Para El Avance De La Biotecnologia Y La Investigacion Sanitaria (Fundacion Im | BIVALENT PIRAZOL DERIVATIVES AS INGESTA INHIBITORS |
| CN102260246B (en) * | 2010-05-28 | 2014-08-13 | 范如霖 | Low-toxicity CB1 receptor inhibitor, preparation method thereof and application thereof in preparation of medicaments for drug abstention, weight reduction or diabetes treatment |
| BR112013012958A2 (en) * | 2010-11-24 | 2016-07-12 | Univ Ohio State Res Found | kinase-linked integrin inhibitors |
| CN102603713B (en) * | 2011-01-25 | 2014-05-14 | 范如霖 | Chiral CB1 (cannabinoid) receptor inhibitor, and preparation method and medical application thereof |
| US8871758B2 (en) | 2011-03-08 | 2014-10-28 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| EP2683699B1 (en) | 2011-03-08 | 2015-06-24 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| US8828995B2 (en) | 2011-03-08 | 2014-09-09 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| EP2683705B1 (en) | 2011-03-08 | 2015-04-22 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| CN102212058B (en) * | 2011-04-14 | 2014-04-02 | 范如霖 | Single composite medicament (chiral nitrogenous heterocyclic ester) and synthesis method thereof as well as application of chiral nitrogenous heterocyclic ester in preparing low-toxicity CB1 receptor inhibitor |
| CN103554025B (en) * | 2011-05-12 | 2016-08-10 | 范如霖 | 3-arsenic triazole carboxylic acid's amides compound, its preparation method and preparation as the application in CB1 acceptor inhibitor medicine |
| US9133128B2 (en) | 2011-06-17 | 2015-09-15 | Research Triangle Institute | Pyrazole derivatives as cannabinoid receptor 1 antagonists |
| JP6021910B2 (en) | 2011-07-26 | 2016-11-09 | サノフイ | 3-Heteroaroylamino-propionic acid derivatives and their use as medicaments |
| BR102012017421A2 (en) | 2012-07-13 | 2015-04-14 | Proteimax Biotecnolgia Ltda | Peptide, pharmaceutical composition, cb receptor binder, method for modulating cb receptor function, use, method for treating obesity, and method for promoting aesthetic weight reduction in an individual |
| JP7104690B2 (en) * | 2016-10-12 | 2022-07-21 | リサーチ トライアングル インスティテュート | Heterocyclic apelin receptor (APJ) agonists and their use |
| IL273868B2 (en) * | 2016-10-13 | 2024-07-01 | Proteimax Biotecnologia Ltda | Use of a compound, a synthetic intermediate, a pharmaceutical preparation and a therapeutic method of neuromodulation |
| KR101974414B1 (en) * | 2017-09-12 | 2019-05-02 | 주식회사 티에스디라이프사이언스 | Composition for Preventing or Treating Fibrosis Comprising 1H-Pyrazole-3-Amide Compound Derivatives |
| US10897925B2 (en) | 2018-07-27 | 2021-01-26 | Joseph Pandolfino | Articles and formulations for smoking products and vaporizers |
| US20200035118A1 (en) | 2018-07-27 | 2020-01-30 | Joseph Pandolfino | Methods and products to facilitate smokers switching to a tobacco heating product or e-cigarettes |
| WO2020113094A1 (en) | 2018-11-30 | 2020-06-04 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| IL284752B2 (en) | 2019-01-15 | 2025-05-01 | Yissum Res Dev Co Of Hebrew Univ Jerusalem Ltd | Cb1r receptor blockers with acyclic backbones |
| WO2025175318A2 (en) * | 2024-02-18 | 2025-08-21 | Luminous Mind Inc. | Pharmacotherapies for improving treatment adherence after discontinuation of incretin-based therapies |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6432984B1 (en) | 1999-02-01 | 2002-08-13 | Sanofi-Synthelabo | Pyrazolecarboxylic acid derivatives, their preparation, pharmaceutical compositions containing them |
| US6509367B1 (en) | 2001-09-22 | 2003-01-21 | Virginia Commonwealth University | Pyrazole cannabinoid agonist and antagonists |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2665898B1 (en) | 1990-08-20 | 1994-03-11 | Sanofi | DERIVATIVES OF AMIDO-3 PYRAZOLE, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2692575B1 (en) | 1992-06-23 | 1995-06-30 | Sanofi Elf | NOVEL PYRAZOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| FR2714057B1 (en) | 1993-12-17 | 1996-03-08 | Sanofi Elf | New derivatives of 3-pyrazolecarboxamide, process for their preparation and pharmaceutical compositions containing them. |
| FR2741621B1 (en) | 1995-11-23 | 1998-02-13 | Sanofi Sa | NOVEL PYRAZOLE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THE SAME |
| FR2742148B1 (en) | 1995-12-08 | 1999-10-22 | Sanofi Sa | NOVEL PYRAZOLE-3-CARBOXAMIDE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM |
| US7393842B2 (en) | 2001-08-31 | 2008-07-01 | University Of Connecticut | Pyrazole analogs acting on cannabinoid receptors |
| US7145012B2 (en) * | 2003-04-23 | 2006-12-05 | Pfizer Inc. | Cannabinoid receptor ligands and uses thereof |
| FR2856683A1 (en) * | 2003-06-25 | 2004-12-31 | Sanofi Synthelabo | 4-CYANOPYRAZOLE-3-CARBOXAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
| JP2007536298A (en) * | 2004-05-10 | 2007-12-13 | エフ.ホフマン−ラ ロシュ アーゲー | Pyrrole or imidazole amide for the treatment of obesity |
| EP1831177A1 (en) | 2004-12-23 | 2007-09-12 | AstraZeneca AB | Therapeutic agents |
| WO2006133926A1 (en) | 2005-06-17 | 2006-12-21 | Carex Sa | Pyrazole derivates as cannabinoid receptor modulators |
| US7872006B2 (en) | 2005-10-21 | 2011-01-18 | Mitsubishi Tanabe Pharma Corporation | Pyrazole compounds having cannabinoid receptor (CB1) antagonizing activity |
| GB0622569D0 (en) | 2006-11-11 | 2006-12-20 | 7Tm Pharma As | Cannabinoid receptor modulators |
| BR112013030793A2 (en) | 2011-07-01 | 2016-12-06 | Inst De Pesquisas Tecnológicas Do Estado De São Paulo | nanoparticulate powder lanthanide metal-metalloid alloy recovery process with magnetic recovery and product |
-
2007
- 2007-03-09 WO PCT/US2007/063631 patent/WO2007106721A2/en not_active Ceased
- 2007-03-09 US US11/684,016 patent/US7687481B2/en not_active Expired - Fee Related
- 2007-03-09 CN CNA2007800165996A patent/CN101437398A/en active Pending
- 2007-03-09 EP EP07758206A patent/EP1993560B1/en not_active Not-in-force
- 2007-03-09 AT AT07758206T patent/ATE538650T1/en active
- 2007-03-09 AU AU2007226673A patent/AU2007226673A1/en not_active Abandoned
- 2007-03-09 JP JP2008558543A patent/JP4851546B2/en not_active Expired - Fee Related
- 2007-03-09 CA CA002647031A patent/CA2647031A1/en not_active Abandoned
-
2010
- 2010-02-06 US US12/701,528 patent/US8088798B2/en active Active
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6432984B1 (en) | 1999-02-01 | 2002-08-13 | Sanofi-Synthelabo | Pyrazolecarboxylic acid derivatives, their preparation, pharmaceutical compositions containing them |
| US6509367B1 (en) | 2001-09-22 | 2003-01-21 | Virginia Commonwealth University | Pyrazole cannabinoid agonist and antagonists |
Non-Patent Citations (1)
| Title |
|---|
| JAMA, vol. 311, 2006, pages 323 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008059207A1 (en) * | 2006-11-11 | 2008-05-22 | 7Tm Pharma A/S | Cannabinoid receptor modulators |
| JP2010513435A (en) * | 2006-12-18 | 2010-04-30 | 7ティーエム ファーマ エイ/エス | CB1 receptor modulator |
| EP2182807A4 (en) * | 2007-09-07 | 2011-02-23 | Jenrin Discovery | CANNABINOID RECEPTOR ANTAGONISTS / AGONISTS USEFUL FOR TREATING OBESITY |
| DE102009036604A1 (en) | 2009-07-30 | 2011-02-03 | Aicuris Gmbh & Co. Kg | Substituted bis-arylpyrazolamides with terminal primary amide functionality and their use |
| WO2011012630A1 (en) | 2009-07-30 | 2011-02-03 | Aicuris Gmbh & Co. Kg | Substituted bis-aryl pyrazole amides having terminal primary amide functionalization for treating retroviral diseases |
| CN102250006B (en) * | 2011-05-12 | 2014-03-05 | 范如霖 | 3-pyrazole carboxylic acid amide compound, its preparation method and its application in the preparation of drugs as CB1 receptor inhibitors |
| CN102250006A (en) * | 2011-05-12 | 2011-11-23 | 范如霖 | 3-Pyrazole carboxylic acid amide compound, its preparation method and its application in the preparation of drugs as CB1 receptor inhibitors |
| US8962845B2 (en) | 2011-09-30 | 2015-02-24 | National Health Research Institutes | Pyrazole compounds |
| US11401244B2 (en) | 2014-06-06 | 2022-08-02 | Research Triangle Institute | Apelin receptor (APJ) agonists and uses thereof |
| US11535630B2 (en) | 2015-12-09 | 2022-12-27 | Research Triangle Institute | Apelin receptor (APJ) agonists and uses thereof |
| USRE49594E1 (en) | 2015-12-09 | 2023-08-01 | Research Triangle Institute | Apelin receptor (APJ) agonists and uses thereof |
| US11180511B2 (en) | 2016-08-03 | 2021-11-23 | Friedrich-Alexander-Universität Erlangen-Nürnberg; Universitätsklinikum Erlangen | Diagnosis, treatment and prevention of neurotensin receptor-related conditions |
| RU2796538C2 (en) * | 2016-08-03 | 2023-05-25 | Фридрих-Александер-Универзитет Эрланген-Нюрнберг | Diagnosis, treatment and prevention of conditions associated with the neurotensin receptor |
| WO2019150220A1 (en) * | 2018-01-30 | 2019-08-08 | Pi Industries Ltd. | Novel anthranilamides, their use as insecticide and processes for preparing the same. |
| JP2021512089A (en) * | 2018-01-30 | 2021-05-13 | ピーアイ インダストリーズ リミテッドPi Industries Ltd | New anthranilamide, its use as an insecticide and its manufacturing method |
| WO2022164239A1 (en) * | 2021-01-28 | 2022-08-04 | 주식회사 파미노젠 | Pyrazole-carboxamide derivative compound and use thereof |
| WO2024092205A1 (en) * | 2022-10-27 | 2024-05-02 | The Trustees Of Indiana University | Inhibition of ship1 as a therapeutic strategy for the treatment of alzheimer's disease |
| WO2025199092A1 (en) * | 2024-03-20 | 2025-09-25 | The Trustees Of Indiana University | Ship1 modulators and methods of treatment and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US8088798B2 (en) | 2012-01-03 |
| EP1993560B1 (en) | 2011-12-28 |
| US20100144791A1 (en) | 2010-06-10 |
| ATE538650T1 (en) | 2012-01-15 |
| WO2007106721A3 (en) | 2008-11-06 |
| EP1993560A2 (en) | 2008-11-26 |
| JP2009529540A (en) | 2009-08-20 |
| EP1993560A4 (en) | 2010-06-02 |
| AU2007226673A1 (en) | 2007-09-20 |
| CN101437398A (en) | 2009-05-20 |
| JP4851546B2 (en) | 2012-01-11 |
| CA2647031A1 (en) | 2007-09-20 |
| US7687481B2 (en) | 2010-03-30 |
| US20070213302A1 (en) | 2007-09-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1993560B1 (en) | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity | |
| EP2023920B1 (en) | Cannabinoid receptor antagonists/inverse agonists | |
| AU2008318409B2 (en) | Cannabinoid receptor antagonists/inverse agonists useful for treating metabolic disorders, including obesity and diabetes | |
| WO2009033125A1 (en) | Cannabinoid receptor antagonists/inverse agonists useful for treating obesity | |
| AU2011329636B2 (en) | Cannabinoid receptor antagonists-inverse agonists useful for treating metabolic disorders, including obesity and diabetes | |
| US8236821B2 (en) | Substituted N-phenyl-5-phenyl-pyrazolin-3-yl amides as cannabinoid receptor antagonists/inverse agonists useful for treating obesity | |
| US8648072B2 (en) | Cannabinoid receptor antagonists/inverse agonists useful for treating metabolic disorders, including obesity and diabetes | |
| US8088926B2 (en) | Substituted 2-methyl-2-phenoxy-N-propyl-propionamides as cannabinoid receptor antagonists/inverse agonists useful for treating obesity | |
| US20090197857A1 (en) | Cannabinoid receptor antagonists/inverse agonists useful for treating metabolic disorders, including obesity and diabetes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008558543 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2647031 Country of ref document: CA Ref document number: 2007226673 Country of ref document: AU Ref document number: 2007758206 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007226673 Country of ref document: AU Date of ref document: 20070309 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2150/MUMNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780016599.6 Country of ref document: CN |